cell contact–dependent priming and fc interaction with ... journal of immunology cell...
TRANSCRIPT

of June 23, 2018.This information is current as
Cytokine ResponseContribute to the TGN1412-Triggered
Immune Cells+Interaction with CD32Dependent Priming and Fc−Cell Contact
Hengel, John Castle, Thomas Hünig and Ulrich KalinkeChuvpilo, Dmitry Y. Tyrsin, Alexey Matskevich, HartmutValesca Boisguerin, Paula S. Römer, Paula Tabares, Sergey Patrick Bartholomaeus, Linda Y. Semmler, Thomas Bukur,
http://www.jimmunol.org/content/192/5/2091doi: 10.4049/jimmunol.1302461January 2014;
2014; 192:2091-2098; Prepublished online 27J Immunol
MaterialSupplementary
1.DCSupplementalhttp://www.jimmunol.org/content/suppl/2014/01/25/jimmunol.130246
Referenceshttp://www.jimmunol.org/content/192/5/2091.full#ref-list-1
, 13 of which you can access for free at: cites 37 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2014 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on June 23, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on June 23, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
Cell Contact–Dependent Priming and Fc Interaction withCD32+ Immune Cells Contribute to the TGN1412-TriggeredCytokine Response
Patrick Bartholomaeus,* Linda Y. Semmler,* Thomas Bukur,†,‡ Valesca Boisguerin,†
Paula S. Romer,x,{ Paula Tabares,x Sergey Chuvpilo,{ Dmitry Y. Tyrsin,{
Alexey Matskevich,{ Hartmut Hengel,‖ John Castle,† Thomas H€unig,x and
Ulrich Kalinke*
Following inconspicuous preclinical testing, the superagonistic anti-CD28 mAb TGN1412 was applied to six study participants who
all developed a devastating cytokine storm. We verified that TGN1412 treatment of fresh PBMCs induced only moderate responses,
whereas restoration of tissue-like conditions by high-density preculture (HDC) allowed vigorous cytokine production. TGN1412
treatment of T cells isolated fromHDC-PBMCs induced moderate cytokine responses, which upon additional anti-IgG crosslinking
were significantly boosted. Moreover, coincubation of TGN1412-treated T cells with B cells expressing the intermediate affinity Fcg
receptor IIB (CD32B), or coincubation with CD32B+ transfectants, resulted in robust T cell activation. This was surprising because
TGN1412 was expressed as an Ig of the subclass 4 (IgG4), which was shown before to exhibit only minor affinity to FcgRs.
Transcriptome analysis of TGN1412-treated T cells revealed that similar gene signatures were induced irrespective of whether
T cells derived from fresh or HDC-PBMCs were studied. Collectively, these data indicate that HDC-PBMCs and HDC-PBMC–
derived T cells mount rapid TGN1412 responses, which are massively boosted by FcgR crosslinking, in particular by CD32-
expressing B cells. These results qualify HDC-PBMCs as a valuable in vitro test system for the analysis of complex mAb
functions. The Journal of Immunology, 2014, 192: 2091–2098.
In rodent preclinical models, superagonistic anti-CD28 mAbtreatment ameliorates various different autoimmune diseasessuch as collagen-induced arthritis and experimental autoim-
mune encephalomyelitis (1–5). These data encouraged the de-velopment of a human CD28-specific superagonistic mAb as acandidate for the treatment of many different autoimmune con-ditions in humans. Toward this end, the CDRs of a superagonisticmouse anti-human CD28 mAb were transplanted into a human
IgG4k mAb and preclinical studies of that reagent were initiated.The whole preclinical program was inconspicuous, and becauserepeated dose toxicity testing of nonhuman primates treated withTGN1412 at doses up to 50 mg/kg body weight on 4 consecutiveweeks did not reveal toxicity signals, the reagent was assumed to besafe (6). The nonhuman primate test species Macaca fascicularisused in the repeated-dose toxicity study was considered a predic-tive model because it showed extracellular CD28 sequences iden-tical with the human ones (7, 8). Unexpectedly, on March 13, 2006all six study participants treated with 0.1 mg/kg body weight ofTGN1412 developed a life-threatening cytokine storm within hoursafter the intervention (9). In the aftermath of this incident no de-viation of existing regulations could be identified (10). Althoughmeanwhile it is clear that the nonhuman primate studies weremisleading owing to subtle species differences of CD28 expressionon T cell subsets (11), until today it is not fully understood why thein vivo potential of TGN1412 to induce massively deregulatedcytokine responses was not detected during the preclinical pro-gram. As an immediate reaction to the incident, regulatory bodiesimplemented a new guideline for risk mitigation of early clinicaltrials (12), which turned out to be helpful. Nevertheless, the pre-dictive value of experiments with nonhuman primates is still beingdebated (11, 13–15) and the need to establish new experimentalsettings based on human cells is evident.In previous studies we and others found that TGN1412-mediated
triggering of isolated T cells was not sufficient to induce robustcytokine responses (8, 16). Instead, stimulating TGN1412 hadeither to be immobilized on plastics, or TGN1412 bound to T cellshad to be crosslinked by an anti-IgG to obtain considerable T cellactivation (8, 16, 17). Thus, it is likely that also under in vivoconditions crosslinking by some FcgR-expressing cell is needed toconfer full T cell activation. Additionally, a recent study showed
*Institute for Experimental Infection Research, TWINCORE, Center for Exper-imental and Clinical Infection Research, Helmholtz Centre for Infection Re-search, Braunschweig, and Hannover Medical School, 30625 Hannover, Germany;†Translational Oncology at the University Medical Center Mainz, Johannes Guten-berg University of Mainz, 55131 Mainz, Germany; ‡Department of Internal Medi-cine III, University Medical Center, Johannes Gutenberg University of Mainz,55122 Mainz, Germany; xInstitute for Virology and Immunobiology, Julius MaximiliansUniversity Wuerzburg, 97070 Wuerzburg, Germany; {TheraMAB, 121069 Moscow,Russia; and ‖Institute of Virology, Albert Ludwigs University Freiburg, 79104 Freiburg,Germany
Received for publication September 12, 2013. Accepted for publication December 21,2013.
This work was supported by European Union Grant MRTN-CT-2005-019248 (toH.H.), German Research Council Grants SFB 854, B15 (to U.K.) and SFB-TR 52 (toT.H.), Bundesministerium f€ur Bildung, Wissenschaft, Forschung und Technologie Grant0315498B (to U.K.), the Helmholtz Association Viral Strategies of Immune EvasionProgram (to H.H. and U.K.), and the Center for Infection Biology, Hannover, Germany.
Address correspondence and reprint requests to Prof. Dr. Ulrich Kalinke, Institute forExperimental Infection Research, TWINCORE, Center for Experimental and ClinicalInfection Research, Helmholtz Centre for Infection Research, Braunschweig, andHannover Medical School, Feodor-Lynen-Strasse 7, D-30625 Hannover, Germany.E-mail address: [email protected]
The online version of this article contains supplemental material.
Abbreviations used in this article: HDC-PBMC, high-density precultured PBMC;TNFRSF, TNFR superfamily.
Copyright� 2014 by The American Association of Immunologists, Inc. 0022-1767/14/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1302461
by guest on June 23, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

that ICOS/LICOS interaction of T cells and cytokine-activatedHUVECs induced proliferation and cytokine expression of TGN1412-decorated T cells (18).More recently some of us described that the failure of freshly
isolated PBMCs to efficiently respond to soluble TGN1412 iscorrected by preculture of PBMCs at high density (19). Evidencewas presented that during PBMC precultivation, T cells scanningMHC class I and II molecules of neighboring cells via their TCRsreceived a “tonic” preactivation signal that led to a higher sensi-tivity to foreign stimuli (20–22). These observations prompted usto address whether T cells enriched from high-density preculturedPBMC required Fcg-mediated interactions to display full TGN1412-mediated T cell activation. Although TGN1412 treatment of high-density precultured PBMC (HDC-PBMC)–derived T cells aloneinduced some cytokine production, which was also detected upontreatment with TGN1412-F(ab9)2, massive enhancement of respon-ses depended on crosslinking of TGN1412-decorated T cells.
Materials and MethodsCell purification and high-density precultivation of PBMCs
PBMCs were isolated from buffy coats by Ficoll (Biochrom) densitygradient centrifugation. For HDC, 1.5 3 107 PBMCs were incubated in1.5 ml X-VIVO 15 (Lonza) or RPMI 1640 medium (supplemented as inRef. 19) per well in 24-well culture plates (BD Biosciences or Greiner Bio-One) for 2 d. Afterward, cells were harvested with ice-cold medium. CD3+
T cells and NK cells were negatively isolated using the human Pan CD3+
T cell isolation kit II or a human NK cell isolation kit (both MiltenyiBiotec), whereas B cells and monocytes were purified by positive selectionusing CD19-specific MicroBeads or CD14-specific MicroBeads (bothMiltenyi Biotec), following the manufacturer’s instructions. Purity of MACS-enriched cell subsets usually exceeded 97%.
In vitro stimulation of immune cells
T cells were seeded at 2 3 105 cells per 96-well plate (BD Biosciences) in200 ml X-VIVO 15 (Lonza) or RPMI 1640 medium (supplemented as inRef. 19) and treated as indicated. As controls, T cells were treated withanti-CD3/anti-CD28 Dynabeads (Invitrogen), Tysabri (natalizumab, IgG4subclass; Biogen Idec, Hillerød, Denmark), Orthoclone OKT3 (Janssen-Cilag), or conventional anti-CD28 mAb (BioLegend, clone CD28.2). ForTGN1412 crosslinking a mouse anti-human IgG4 mAb (BD Pharmingen,clone G17-4) was added at duplicated concentration of TGN1412 (0.2 mg/mlto 20 mg/ml). For FcgR-blocking experiments, 2 3 105 cells were incu-bated with 10 mg/ml mouse anti-human CD32 mAb (AbD Serotec), whichspecifically binds FcgRIIA (CD32A) as well as FcgRIIB (CD32B), for30 min prior to treatment with TGN1412.
F(ab9)2 fragment preparation
The bivalent F(ab9)2 fragment was prepared by using the Pierce F(ab9)2preparation kit (Thermo Scientific), following the manufacturer’s instruc-tions. In brief, purified TGN1412 was added to a spin column containingequilibrated immobilized pepsin (Thermo Scientific). Upon 6.5 h incubationat 37˚C, the solid phase was separated from the digest by centrifugation(5000 3 g for 1 min). The column was washed twice with PBS and thewashes were collected together with the digest. For removal of undigestedIgG, a protein A (Thermo Scientific) purification was performed. The purityof the separated F(ab9)2 fragment was checked by reducing SDS-PAGE inwhich the H and L chain of the native mAb migrated at 55 and 33 kDa,respectively, whereas the F(ab9)2 fragment consisted of the truncated H chainand the L chain migrating between 25 and 33 kDa.
Flow cytometric analysis and intracellular cytokine staining
Cell surface marker stainings were carried out for 15 min at room tem-perature using the following mAbs: anti–CD3-PerCP (clone UCHT1), anti–CD4-PerCP (clone OKT4), anti–CD19-Pacific Blue (clone HIB19) (allBiolegend), anti–CD3-allophycocyanin (clone UCHT1), anti–CD19-PE(clone HIB19), anti–CD14-Pacific Blue (clone MfP9.1), and anti–CD28-PE-Cy7 (all BD Pharmingen). Early cytokine responses were assessed after6 h stimulation by intracellular cytokine staining using anti–TNF-a-PE-Cy7(BD Pharmingen or BioLegend, clone MAb11), anti–IFN-g-PE (clone 4S.B3), and anti–IL-2-Pacific Blue (clone MQ1-17H12) (all BioLegend) Abs.Cells were FACS analyzed using an LSR II flow cytometer (Becton Dick-inson) with FACSDiva and FlowJo analysis software.
ELISPOT
For ELISPOT determination of the frequency of cytokine-producing cells,96-well plates with nitrocellulose bottoms were coated with TNF-a–spe-cific mAb (BD Biosciences). Upon blocking of nonspecific binding withRPMI 1640 medium containing 10% human AB serum, HDC-PBMCswere added and stimulated with TGN1412 or TGN1412-F(ab9)2 and cul-tured overnight. Following washing with PBS, bound TNF-a was detectedwith biotinylated anti–TNF-a Ab (BD Biosciences) and streptavidin-alkaline phosphatase (Genemed Biotechnologies) and subsequent addi-tion of the substrate NBT/5-bromo-4-chloro-3-indolyl phosphate (Sigma-Aldrich). ELISPOTs were counted by an automated counter (CTL).
Whole-transcriptome gene expression by next-generation RNAsequencing
Total RNA has been used for RNA sequencing library preparation using theTruSeq RNA sample prep kit v.2 (Illumina, San Diego, CA). All cleanupswere done using Agencourt AMPure XP magnetic beads (BeckmannCoulter, Danvers, MA). Quality control of the prepared sequencing librarieswas done quantitatively by Qubit 2.0 using the Qubit high-sensitivity DNAkit (both Life Technologies, Carlsbad, CA) and qualitatively by Bioanalyzer2100 on DNA 1000 chips (Agilent Technologies, Santa Clara, CA). Libraryconcentrations ranged from 4.3 to 31.2 ng/ml and their sizes from 269 to301 base pairs. Barcoded sequencing libraries were clustered on the Illu-mina cBot using the TruSeq single-read cluster kit v.3. Sequencing wasdone on the Illumina HiSeq 2000 using the Illumina TruSeq SBS v.3 kit.Raw data were filtered and demultiplexed using Illumina’s softwareCASAVA (v.1.8.2). Sequence reads were aligned to the human referencegenome hg19 (23) using Bowtie (24), allowing two mismatches and re-trieving best matches in case of ambiguous alignment. Unaligned readswere retained for a second round of alignment to a database of all possibleexon–exon junction sequences of the University of California Santa Cruzgenes (25). Coordinates of both alignments were compared with exoncoordinates of the University of California Santa Cruz transcripts and readswere counted per transcript. Read counts were normalized to reads perkilobase of exon model per million mapped reads (26). Further down-stream analysis was performed using the statistical language R (27). Dif-ferential gene expression was calculated using the package DESeq (28) forcomparisons of sample groups with default options, that is, parametricnormalization of count data and Benjamini–Hochberg adjusted p values.Gene sets for downregulation and upregulation were processed indepen-dently. Gene sets with a p value of ,0.01 were considered significant.
Statistical analysis
The statistical evaluation was performed using Prism v.5 software(GraphPad Softward) as stated in the figure legends. A p value ,0.05 wasconsidered significant.
ResultsPurified human T cells treated with soluble TGN1412 do notmount cytokine responses
T cells enriched from freshly isolated PBMCs are not strongly acti-vated upon treatment with soluble superagonistic anti-CD28 mAbTGN1412 (8, 16). To induce early cytokine responses, TGN1412-decorated T cells have either to be treated with crosslinking anti-IgG4, or T cells have to be added to wells coated with TGN1412(Fig. 1A and Refs. 8, 11, 16, 17). Interestingly, application of T cellsto wells coated with TGN1412 induced significantly enhanced cy-tokine responses when compared to TGN1412-decorated T cellstreated with crosslinking anti-IgG4 (Fig. 1A). Under the condi-tions tested, CD4+ T cells were the main responders (Fig. 1B). Theseresults raised the question whether coincubation of TGN1412-decorated T cells with some immune cell subset was similarly ableto confer crosslinking and to trigger robust T cell activation.
Coincubation of TGN1412-decorated T cells withFcgR-expressing immune cells induces early cytokine responses
To assess the crosslinking potential of different immune cellsubsets, we next coincubated TGN1412-decorated T cells withMACS-enriched B cells, monocytes, or NK cells. Coincubationwith NK cells expressing high levels of low-affinity FcgRIII
2092 TGN1412-MEDIATED T CELL ACTIVATION
by guest on June 23, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

(CD16) (29) (Fig. 2A) and coincubation with T cells expressing nosignificant levels of FcgRs (29) did not induce cytokine responsesof TGN1412-decorated T cells (Fig. 2A). On the contrary, coin-cubation with FcgRIIB+ (CD32B+) B cells or with monocytesexpressing FcgRI (CD64), FcgRIIA (CD32A), and also low levelsof FcgRIIB (CD32B) (30, 31) induced cytokine expression(Fig. 2A). Overall, monocytes induced less TNF-a–producingT cells than B cells, that is, ∼1 versus 2.2%, respectively (Fig. 2B).To address whether monocytes conferred trogocytosis (32) ofTGN1412 bound to CD28 and thus triggered reduced cytokineresponses compared with B cells, we performed CD3 and CD28cell surface marker stainings of monocytes and B cells duringcoculture experiments. Under such conditions no CD3 or CD28transfer from T cells to monocytes or B cells was detected (datanot shown). Therefore, we conclude that trogocytosis of CD28 didnot account for weaker cytokine responses of TGN1412-decoratedT cells coincubated with monocytes compared with coincubationwith B cells.Next we studied whether FcgRII CD32 and FcgRI CD64, or
some other molecular interaction with B cells or monocytes, werecritically involved in the induction of early TNF-a responses ofTGN1412-decorated T cells. To this end, stable transfectantsexpressing chimeric proteins consisting of the mouse TCRz-transmembrane and cytoplasmic domains fused to the extra-cellular domains of human CD32A, CD32B/C, or CD64 wereused (33). Although staining with different Abs does not allowcomparison of the expression levels of the different FcgRs, andthe FcgR expression levels of the transfectants may differ fromthose of immune cells, cytofluorometric analysis of the trans-fectants revealed that all recombinant receptors were expressed onthe cell surface (data not shown). In coincubation experiments ofTGN1412-decorated T cells with CD32A-, CD32B/C-, or CD64-expressing transfectants, a significantly enhanced proportion ofintracellular TNF-a+ T cells was induced when compared withuntreated T cells, whereas more dominant effects were detectedwith CD64-expressing transfectants than with CD32-expressingones (Fig. 2C). Because of the above considerations, these
results should be interpreted qualitatively, that is, that CD32 andCD64 can confer crosslinking of TGN1412 bound to T cells,whereas quantitative conclusions about the strength of CD64- andCD32-mediated crosslinking are difficult to be drawn. Taken to-gether, the above results were surprising, because TGN1412 wasdesigned as an IgG4k molecule that so far was thought not toshow significant FcgR interactions, particularly not with the in-termediate affinity FcgRII CD32.To next validate the role of CD32 in B cell–mediated cross-
linking of TGN1412-decorated T cells, TGN1412-treated T cellswere coincubated with B cells in the presence of an anti-CD32blocking Ab. To assure maximal CD32 inhibition, the level ofanti-CD32–blocking Ab was increased up to 10 mg/ml. Undersuch conditions, early induction of intracellular TNF-a was re-duced (Fig. 3). These data indicated that B cells, and to a lesserextent monocytes, may confer crosslinking of TGN1412 bound toT cells, thus boosting the induction of early cytokine responses.Overall, B cells induced stronger responses than monocytes andother immune cells. These analyses revealed that coincubation ofTGN1412-decorated T cells with syngeneic immune cell subsetsrather reflected in vivo conditions than experiments in which pu-rified T cells were added to TGN1412-coated wells.
Upon TGN1412 treatment T cells isolated from high-densityprecultured PBMCs showed enhanced TNF-a responses
A recent study showed that T cell responsiveness to TGN1412 canbe restored when PBMCs were precultured at high density for 2 dprior to superagonistic CD28 stimulation (19). We therefore nextprepared fresh PBMCs and HDC-PBMCs and treated the cellswith soluble TGN1412. As expected, after 6 h incubation HDC-PBMCs showed a greatly enhanced proportion of TNF-a+ T cellswhen compared with freshly isolated PBMCs (Fig. 4A and Ref.19). To rigorously examine whether T cells present in fresh orHDC-PBMCs can respond to TGN1412 without the involvementof Fc crosslinking, we next prepared the F(ab9)2 fragment ofTGN1412 (Fig. 4B). HDC-PBMCs treated with native TGN1412showed stronger TNF-a responses than TGN1412-F(ab9)2–treated
FIGURE 1. TGN1412-decorated T cells show enhanced
cytokine responses upon crosslinking of the Fcg moiety
of TGN1412 bound to CD28. (A) MACS-enriched CD3+
T cells (2 3 105) seeded in X-VIVO 15 medium were
stimulated with anti-CD3/anti-CD28 DynaBeads (Invi-
trogen) (CD3/CD28), or TGN1412 was added at the indicated
final concentrations (mg/ml) and where indicated anti-IgG4
was added at duplicate concentration of TGN1412. As a
positive control, TGN1412 was coated to plastics at a
concentration of 1 mg/ml and subsequently T cells were
added. After 6 h incubation at 37˚C, CD3+ T cells were
stained with anti–CD4-PerCP and intracellular TNF-a,
IFN-g, and IL-2 expression of CD3+ T cells was deter-
mined cytofluorometrically. One representative experiment
out of five similar ones is shown. (B) Statistical analysis
(Mann–Whitney test, *p # 0.05, **p # 0.01) of experi-
ments shown in (A) for CD4+ or CD8+ T cells with immune
cells of five different donors.
The Journal of Immunology 2093
by guest on June 23, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

ones, whereas in fresh PBMCs this effect was basically invisible(Fig. 4C). Of note, the control IgG4 Tysabri as well as a conven-tional anti-CD28 Ab (clone 28.2) did not as effectively induceTNF-a responses as TGN1412 (Fig. 4C). To next determine thefrequency of T cells that were activated either by TGN1412 orTGN1412-F(ab9)2, an ELISPOT analysis was performed. Whereasincubation of HDC-PBMCs with $0.1 mg/ml native Ab induced∼7000 TNF-a–producing CD4+ T cells, at a concentration of
0.1 mg/ml the corresponding F(ab9)2 fragment induced 3000 TNF-a–producing CD4+ T cells (Fig. 4D). The response to the F(ab9)2fragment reached an optimum at estimated equimolarity with CD28(∼0.5 mg/ml) and then declined (Fig. 4D), suggesting that at higherdoses monomeric binding dominated above the bivalent one, therebyreducing bivalent CD28 binding. Interestingly, native Ab inducedenhanced frequencies of TNF-a+ cells also at elevated TGN1412concentrations (Fig. 4D), which presumably was due to additionalFcg/FcgR interactions contributed by other immune cells.Upon treatment with soluble TGN1412, purified T cells enriched
from freshly isolated PBMCs did not mount significant TNF-aresponses, whereas T cells isolated from HDC-PBMCs showedlow but distinct TNF-a production (Fig. 5A, 5B). This indicatedthat high-density precultivation conferred an intrinsic effect onT cells, whereas additional crosslinking with anti-IgG4 stronglyenhanced TNF-a responses of T cells isolated from fresh PBMCsor HDC-PBMCs (Fig. 5A, 5B). Of note, under such conditions theoverall responsiveness of HDC-PBMC–derived T cells was su-perior compared with T cells of freshly prepared PBMCs (Fig. 5B).Previously it was shown that during the precultivation step en-hancement of overall T cell reactivity was mediated primarilyby monocytes, whereas B cells did not play a critical role (19).Therefore, we next addressed whether B cells conferred FcgR-mediated enhancement of TGN1412-induced responses. Towardthis end, TGN1412-decorated T cells derived from fresh or HDC-PBMCs were coincubated with syngeneic B cells. Coincubationwith syngeneic B cells induced significant TNF-a (Fig. 5C), IFN-g,and IL-2 (Supplemental Fig. 1) responses, whereas again effectswith HDC-PBMC–derived T cells were more pronounced thanwith T cells enriched from fresh PBMCs (Fig. 5C, SupplementalFig. 1). Furthermore, OKT3-decorated T cells derived from fresh orHDC-PBMCs and coincubated with syngeneic B cells showedsignificantly reduced TNF-a and IL-2, but enhanced IFN-g pro-duction, when compared with TGN1412-treated T cells from thesame donors (Fig. 5C, Supplemental Fig. 1). Because Eastwoodet al. (34) showed that IL-2 is a marker associated with the in-duction of a cytokine storm, we addressed whether under the aboveconditions T cells were present that secreted TNF-a, IFN-g, andIL-2 in parallel. Irrespective of whether T cells derived from PBMCsor HDC-PBMCs, polyfunctional T cells were detected (Fig. 5D).Cultivation of HDC-PBMCs in medium supplemented with humanAB serum did not significantly affect TGN1412-triggered TNF-aresponses, whereas addition of Polyglobin reduced TNF-a re-sponses without completely inhibiting them (Fig. 6A). These datawere in agreement with the ability of TGN1412-F(ab9)2 fragmentsto trigger residual TNF-a responses (Fig. 4D). Addition of anti-CD32 plus AB serum only moderately further reduced TGN1412induced TNF-a responses (Fig. 6B).
TGN1412- and anti-IgG4–treated naive CD4+ T cells isolatedfrom fresh PBMCs or HDC-PBMCs show similar genesignatures
To next study whether TGN1412-treated T cells isolated from freshPBMC or HDC-PBMCs showed similar activation signatures, we
FIGURE 2. Analysis of early TNF-a responses of TGN1412-treated
T cells coincubated with B cells, monocytes, or NK cells. (A) CD3+ T cells
(2 3 105) stimulated with 1 mg/ml TGN1412 were coincubated with 2 3105 syngeneic B cells, monocytes, or NK cells or they were left untreated
and after 6 h incubation intracellular TNF-a expression was determined.
For optimal resolution, a responder with a particularly high TNF-a re-
sponse is shown. (B) Statistical analysis of experiments as shown in (A)
with cells of 16 different donors. (C) T cells stimulated with 1 mg/ml
TGN1412 were coincubated with BW5147 transfectants expressing fusion
receptors displaying the extracellular portion of human CD32A (BW:
FcgRIIA-z), CD32B/C (BW:FcgRIIB-z), or CD64 (BW:FcgRI-z) and af-
ter 6 h incubation intracellular TNF-a was determined. Experiments
with T cells of 8–11 different donors are shown. Statistical analyses in (B)
and (C) were performed using a paired one-tailed Student t test. *p# 0.05,
**p # 0.01, ***p # 0.001.
FIGURE 3. Specific blockade of CD32 inhibits B cell–mediated
early TNF-a induction of TGN1412-treated T cells. (A) B cells
(2 3 105) were incubated in the presence or absence of a CD32
blocking Ab for 30 min and subsequently 2 3 105 TGN1412-
decorated CD3+ T cells were added. After 6 h incubation, intra-
cellular TNF-a expression of CD3+ T cells was determined
cytofluorometrically. (B) Experiments with T cells of a total of six
different donors are shown. Statistical analysis was performed
using a paired one-tailed Student t test. **p# 0.01, ***p # 0.001.
2094 TGN1412-MEDIATED T CELL ACTIVATION
by guest on June 23, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

performed whole-transcriptome gene expression analysis using thenext-generation RNA sequencing assay. Toward this end, naiveCD4+ T cells isolated either from fresh PBMCs or HDC-PBMCswere treated with 1 mg/ml TGN1412 and anti-IgG4. After 6 hincubation the cells were harvested and total RNAwas isolated forRNA sequencing library preparation. For comparison of gene ex-pression levels of untreated T cells and T cells stimulated withTGN1412 plus anti-IgG, the 100 most abundantly upregulatedgenes were further analyzed (Fig. 7A, 7B). Interestingly, 48 ofthese 100 genes were detected in the analyses of T cells derivedfrom fresh PBMCs and HDC-PBMCs (Fig. 7A, 7B, marked inred) and comprised genes such as IL-2R a-chain (CD25), IL-2,TNFR superfamily (TNFRSF)4 (OX40 or CD134), TNFRSF18
(glucocorticoid-induced TNFR-related protein GITR), C-X-Cmotif chemokine 10 (CXCL10), and immature microRNAs (e.g.,microRNA-155 and microRNA-146) (Fig. 7A, 7B). Comparedwith untreated T cells derived from fresh PBMCs, T cells treatedwith TGN1412 without the addition of anti-IgG4 showed a meanfold change of the 100 most abundantly upregulated genes of 1.86,whereas additional anti-IgG4 further increased the mean foldchange to a value of 105.21 (Fig. 7C). This indicated an ∼55-foldincrease of the 100 most abundantly upregulated genes upon anti-IgG4 treatment of TGN1412-decorated PBMC-derived T cells. Incontrast, TGN1412 treatment of HDC-PBMC–derived T cellsinduced a mean fold change of 2.02, whereas upon TGN1412plus anti-IgG4 treatment this value reached 185.67 (Fig. 7C),
FIGURE 4. High-density precultured PBMCs show enhanced
intracellular TNF-a expression upon treatment with TGN1412, but
not with TGN1412-F(ab9)2 fragment. (A) Freshly isolated PBMCs
(d) or HDC-PBMCs (n) were stimulated with 1 mg/ml TGN1412
and intracellular TNF-a expression of T cells was determined
cytofluorometrically. Experiments with cells of 16 different donors
are shown. (B) A 12.5% reducing SDS-PAGE analysis. Lane 1,
native TGN1412 Ab; lanes 2 and 3, two different preparations
of TGN1412-F(ab9)2 fragment. (C) Fresh and HDC-PBMCs
were stimulated with either 1 mg/ml TGN1412, TGN1412-F(ab9)2fragment, Tysabri, or conventional anti-CD28 Ab. Experiments
with cells of six different donors are shown. (D) HDC-PBMCs
(1 3 106) were incubated overnight with native TGN1412 (s) or the
corresponding F(ab9)2 fragment (n) at the indicated concentrations.
The number of TNF-a–producing cells was determined in an
ELISPOT assay. Statistical analysis in (A) and (C) was performed
using a paired one-tailed Student t test. **p # 0.01, ***p # 0.001.
FIGURE 5. TGN1412-treated T cells isolated from high-density
precultured PBMCs show enhanced intracellular TNF-a expres-
sion after anti-IgG crosslinking or coincubation with B cells. (A)
PBMC- (d) and HDC-PBMC (n)–derived T cells were treated
with TGN1412 and anti-IgG4 and 6 h later intracellular TNF-a
was determined. Experiments with cells of 10–11 different donors
are shown (paired one-tailed Student t test: *p# 0.05, **p# 0.01,
***p# 0.001). (B) PBMC- and HDC-PBMC–derived T cells were
treated as indicated and 6 h later intracellular TNF-a was deter-
mined. Experiments with cells of five different donors are shown.
Statistical analysis of PBMC- or HDC-PBMC–derived T cells
(black aterisks): one-way ANOVA (Bonferroni posttest: ***p #
0.001). Comparison of PBMC- and HDC-PBMC–derived T cells
treated with TGN1412 and anti-IgG4 (gray asterisks): paired one-
tailed Student t test (**p # 0.01). (C) PBMC- (d) and HDC-
PBMC (n)–derived T cells were treated with TGN1412 or OKT3
and coincubated with B cells from PBMCs or HDC-PBMCs, re-
spectively. After 6 h incubation, intracellular TNF-a was deter-
mined. Experiments with cells of 8–14 different donors are shown
(paired one-tailed student t test: **p # 0.01, ***p # 0.001). (D)
PBMC- and HDC-PBMC–derived T cells were treated as indicated
in (C) and TGN1412-treated TNF-a+ T cells [second and fifth
columns in (C)] were analyzed for IFN-g and/or IL-2 expression.
Shown are the mean values (%) 6 SEM from experiments with
cells of 10 different donors.
The Journal of Immunology 2095
by guest on June 23, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
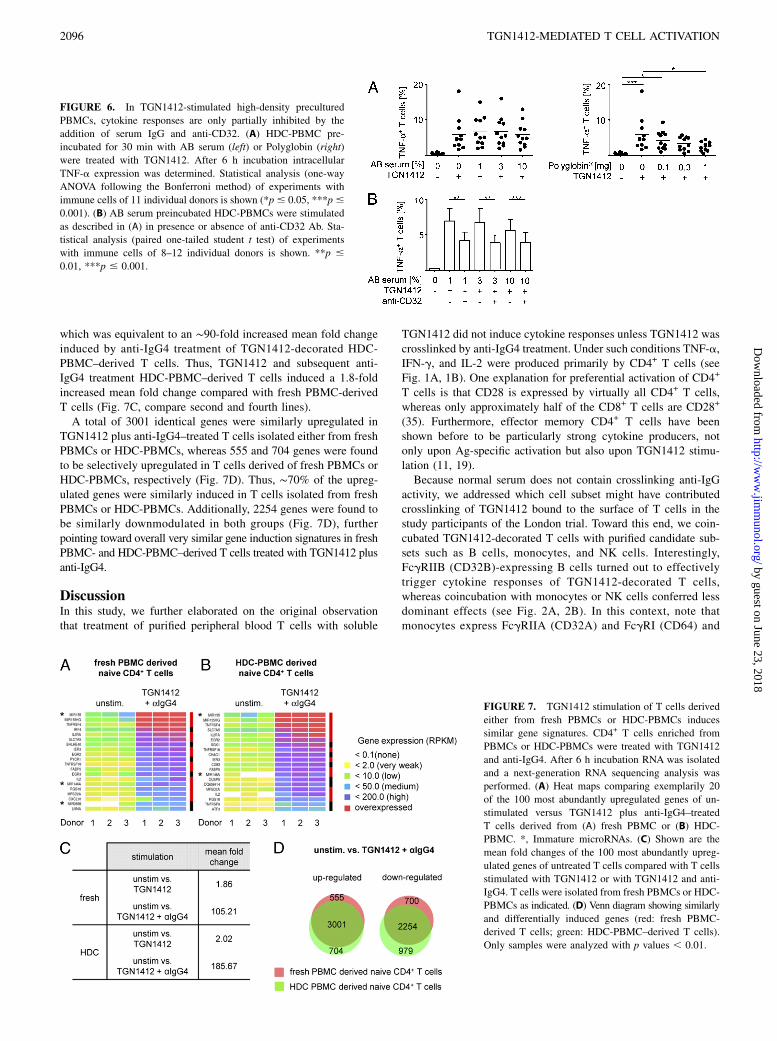
which was equivalent to an ∼90-fold increased mean fold changeinduced by anti-IgG4 treatment of TGN1412-decorated HDC-PBMC–derived T cells. Thus, TGN1412 and subsequent anti-IgG4 treatment HDC-PBMC–derived T cells induced a 1.8-foldincreased mean fold change compared with fresh PBMC-derivedT cells (Fig. 7C, compare second and fourth lines).A total of 3001 identical genes were similarly upregulated in
TGN1412 plus anti-IgG4–treated T cells isolated either from freshPBMCs or HDC-PBMCs, whereas 555 and 704 genes were foundto be selectively upregulated in T cells derived of fresh PBMCs orHDC-PBMCs, respectively (Fig. 7D). Thus, ∼70% of the upreg-ulated genes were similarly induced in T cells isolated from freshPBMCs or HDC-PBMCs. Additionally, 2254 genes were found tobe similarly downmodulated in both groups (Fig. 7D), furtherpointing toward overall very similar gene induction signatures in freshPBMC- and HDC-PBMC–derived T cells treated with TGN1412 plusanti-IgG4.
DiscussionIn this study, we further elaborated on the original observationthat treatment of purified peripheral blood T cells with soluble
TGN1412 did not induce cytokine responses unless TGN1412 wascrosslinked by anti-IgG4 treatment. Under such conditions TNF-a,IFN-g, and IL-2 were produced primarily by CD4+ T cells (seeFig. 1A, 1B). One explanation for preferential activation of CD4+
T cells is that CD28 is expressed by virtually all CD4+ T cells,whereas only approximately half of the CD8+ T cells are CD28+
(35). Furthermore, effector memory CD4+ T cells have beenshown before to be particularly strong cytokine producers, notonly upon Ag-specific activation but also upon TGN1412 stimu-lation (11, 19).Because normal serum does not contain crosslinking anti-IgG
activity, we addressed which cell subset might have contributedcrosslinking of TGN1412 bound to the surface of T cells in thestudy participants of the London trial. Toward this end, we coin-cubated TGN1412-decorated T cells with purified candidate sub-sets such as B cells, monocytes, and NK cells. Interestingly,FcgRIIB (CD32B)-expressing B cells turned out to effectivelytrigger cytokine responses of TGN1412-decorated T cells,whereas coincubation with monocytes or NK cells conferred lessdominant effects (see Fig. 2A, 2B). In this context, note thatmonocytes express FcgRIIA (CD32A) and FcgRI (CD64) and
FIGURE 6. In TGN1412-stimulated high-density precultured
PBMCs, cytokine responses are only partially inhibited by the
addition of serum IgG and anti-CD32. (A) HDC-PBMC pre-
incubated for 30 min with AB serum (left) or Polyglobin (right)
were treated with TGN1412. After 6 h incubation intracellular
TNF-a expression was determined. Statistical analysis (one-way
ANOVA following the Bonferroni method) of experiments with
immune cells of 11 individual donors is shown (*p# 0.05, ***p#
0.001). (B) AB serum preincubated HDC-PBMCs were stimulated
as described in (A) in presence or absence of anti-CD32 Ab. Sta-
tistical analysis (paired one-tailed student t test) of experiments
with immune cells of 8–12 individual donors is shown. **p #
0.01, ***p # 0.001.
FIGURE 7. TGN1412 stimulation of T cells derived
either from fresh PBMCs or HDC-PBMCs induces
similar gene signatures. CD4+ T cells enriched from
PBMCs or HDC-PBMCs were treated with TGN1412
and anti-IgG4. After 6 h incubation RNA was isolated
and a next-generation RNA sequencing analysis was
performed. (A) Heat maps comparing exemplarily 20
of the 100 most abundantly upregulated genes of un-
stimulated versus TGN1412 plus anti-IgG4–treated
T cells derived from (A) fresh PBMC or (B) HDC-
PBMC. *, Immature microRNAs. (C) Shown are the
mean fold changes of the 100 most abundantly upreg-
ulated genes of untreated T cells compared with T cells
stimulated with TGN1412 or with TGN1412 and anti-
IgG4. T cells were isolated from fresh PBMCs or HDC-
PBMCs as indicated. (D) Venn diagram showing similarly
and differentially induced genes (red: fresh PBMC-
derived T cells; green: HDC-PBMC–derived T cells).
Only samples were analyzed with p values , 0.01.
2096 TGN1412-MEDIATED T CELL ACTIVATION
by guest on June 23, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

variable levels of CD32B, depending on the donor analyzed (30,31), whereas NK cells only express FcgRIII (CD16) (29). Theseresults were surprising because generally it was assumed that Absof the IgG4k subclass, the format in which TGN1412 was engi-neered for the use in humans (6), would not show significant in-teraction with FcgR (29, 36–38), whereas few studies speculatedthat IgG4k may show some interaction with CD64 and very lowinteraction with CD32 (29, 37).Moreover, coincubation of TGN1412-treated T cells with
transfectants expressing FcgR CD32A, CD32B, or CD64 fusionreceptors showed significant T cell activation, whereas strongestT cell responses were induced by CD64-expressing transfectants(see Fig. 2C). Although staining with different Abs does not allowcomparison of levels of surface expression of different FcgRs,cytofluorometric analysis revealed that all recombinant receptorswere expressed (33). FcgR expression levels of transfectantsmight deviate from those of immune cell subsets. Therefore, thecapacity of CD64 and CD32 transfectants to booster TNF-aresponses of TGN1412-decorated T cells should be interpretedonly qualitatively, and no quantitative conclusions of CD32- andCD64-mediated crosslinking should be made. The observation thatTGN1412-treated T cells coincubated with transfectants expressingFcgRIIB fusion receptors showed enhanced T cell activation wasfurther corroborated by experiments with a CD32-blocking mAbin which B cell–mediated triggering of TGN1412-decorated T cellswas inhibited (see Fig. 3A, 3B).Recently, Rossi et al. (32) published that monocytes were able to
induce trogocytosis of multiple B cell surface markers upon CD22targeting with the mAb epratuzumab. Because coincubation ofTGN1412-decorated T cells with monocytes induced weaker cy-tokine responses than coincubation with B cells, we addressedwhether monocyte-induced trogocytosis of CD28 or CD3 mighthave played a role. Under the conditions tested no CD3 or CD28transfer from TGN1412-decorated T cells to monocytes was ob-served. Therefore, we conclude that trogocytosis does not playa role in coculture experiments of TGN1412-decorated T cells andmonocytes.In human blood, the leukocyte population comprises ∼7–24%
T cells and 1–7% B cells, whereas in secondary lymphoid tissuessuch as tonsils, T cells and B cells represent ∼18 and 33%, re-spectively (39). Furthermore, in a recent study it was found thatT cells from secondary lymphoid tissue showed some state of pre-activation and therefore were more readily activated upon TGN1412treatment (19). These observations implied that in TGN1412-treated subjects cytokine release of TGN1412-decorated T cellsrather took place in secondary lymphoid organs and other tissues,but not in the peripheral blood.To more easily study TGN1412-mediated stimulation of T cells
with an activation status resembling that of tissue-derived T cells,Romer et al. (19) developed a method resetting the activationstatus of PBMC-derived T cells to that of tissue-derived ones. Forthis purpose PBMCs were precultivated for 2 d at high density,which then contained T cells showing enhanced cytokine pro-duction upon treatment with soluble TGN1412 (Fig. 4A). Inter-estingly, restoration of T cell responsiveness by preculture wasdependent on the presence of the predominant population of APCsin PBMC, that is, the monocytes, whereas precultured PBMCsdepleted of B cells still showed enhanced [3H]thymidine incor-poration after TGN1412 stimulation. These observations promp-ted us to study whether T cells enriched from HDC-PBMCs alsoshowed enhanced cytokine responses upon TGN1412 treatmentand subsequent crosslinking. Our experiments indicate that additionalanti-IgG crosslinking significantly enhanced cytokine responses,irrespective of whether T cells enriched from freshly isolated
PBMCs or HDC-PBMCs were tested. Of note, addition of B cellsto TGN1412-decorated T cells enriched from HDC-PBMCs sim-ilarly conferred this boosting effect (see Fig. 5C). These results arein accordance with the study by Romer et al. (19) in whichmonocytes were identified as the immune cell subset that con-ferred TCR-dependent preactivation of T cells, whereas our resultsshow that B cells are one cell subset that may confer crosslinkingof TGN1412-decorated T cells.Ball et al. (40) recently described that removal of the Fc part of
TGN1412 curtailed or fully abolished PBMC activation. There-fore we rigorously examined whether restored T cells respondedto the F(ab9)2 fragment of TGN1412. We found that indeed restoredT cells mounted residual cytokine responses upon TGN1412-F(ab9)2treatment. Determination of the frequency of TNF-a–producingT cells revealed that the response to the F(ab9)2 fragment wentthrough an optimum at the estimated equimolarity with CD28(∼0.5 mg/ml), suggesting that reduced crosslinking occurred athigher doses. Interestingly, the response to intact Ab remainedelevated even at higher concentrations (see Fig. 4D), probably dueto the contribution of Fcg/FcgR interactions.Hanke et al. (38) as well as others discussed that IgG4 is ex-
pected to show moderate interaction with CD64 and weak or nointeraction with other FcgRs (29, 36, 37). This raised the questionabout the biological relevance of the CD32/Fcg4 interaction. Ofnote, in presence of 10% AB serum TGN1412-decorated T cellsisolated from fresh PBMCs and coincubated with B cells showedreduced cytokine responses. On the contrary, high-density pre-cultured PBMCs showed only moderately reduced early cytokineresponses in the presence of AB serum or Polyglobin (see Fig 6A),and also further addition of anti-CD32 only moderately reducedTNF-a production (see Fig. 6B). Therefore, it is possible thatunder such conditions in addition to crosslinking of TGN1412bound to T cells, other FcgR-independent mechanisms play a role(18). Importantly, currently neither mathematical nor experimentalmodels exist describing dynamic interactions of different FcgRswith different polyclonal or monoclonal IgG isotypes in the blood-stream or in the lymph. Thus, it is difficult to make predictions aboutthe IgG binding capacity of low- to high-affinity FcgRs expressedby immune cells that recirculate in the blood or the lymph stream,or that reside in secondary lymphoid organs.The markedly increased TGN1412 responsiveness of HDC-PBMC–
derived T cells further prompted us to closer study whether overallsimilar signaling pathways were triggered upon TGN1412 stim-ulation and subsequent anti-IgG treatment of T cells derived eitherfrom fresh PBMCs or HDC-PBMCs. Therefore we analyzed genesignatures of both T cell types, which turned out to be overall verysimilar. Interestingly, among the most abundantly upregulatedcommon genes not only genes for cytokines or their receptors suchas IL-2R a-chain (CD25) and IL-2 were found, but also costim-ulatory molecules such as TNFRSF4 (OX40) or TNFRSF18 (GITR)(Fig. 7A, 7B). In a recent study the severity of the TGN1412-induced cytokine storm was correlated with the IL-2 release,suggesting that IL-2 production might be used as a prognosticfactor in upcoming trials of new biologicals (34). Thus, in additionto cytokine secretion, analysis of costimulatory molecules ex-pression may be also informative for the evaluation of new bio-logicals. Futhermore, several studies revealed the importance ofmicroRNAs in the regulation of the immune system. For ex-ample, microRNAs play a central role in Th cell activation aswell as subset differentiation (41, 42). Therefore, microRNAexpression analysis should be taken into consideration in thecontext of future studies.In conclusion, in this study, we found that the rapid cytokine
response to TGN1412 of PBMCs reset to tissue-like conditions by
The Journal of Immunology 2097
by guest on June 23, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

high-density preculture is not strictly dependent on, but is furthermassively boosted by, FcgR-mediated crosslinking, in particularby CD32-expressing B cells. These observations reveal a morecomplex mode of action of TGN1412 than originally anticipatedand further illustrate the need to develop new advanced in vitroexperimental test systems involving human immune cells for thedetailed analysis of new biologicals.
AcknowledgmentsWe thank Susanne Berr for excellent technical assistance and Thorsten
Volgmann from Blutbank Springe for provision of buffy coats.
DisclosuresP.S.R., S.C., D.Y.T., and A.M. are employed by TheraMAB, the company
owning the reagent TGN1412. T.H. is a consultant of TheraMAB. H.H.
and U.K. hold a patent on FcgR-expressing transfectants.
References1. H€unig, T., and K. Dennehy. 2005. CD28 superagonists: mode of action and
therapeutic potential. Immunol. Lett. 100: 21–28.2. Beyersdorf, N., S. Gaupp, K. Balbach, J. Schmidt, K. V. Toyka, C. H. Lin,
T. Hanke, T. H€unig, T. Kerkau, and R. Gold. 2005. Selective targeting of reg-ulatory T cells with CD28 superagonists allows effective therapy of experimentalautoimmune encephalomyelitis. J. Exp. Med. 202: 445–455.
3. Rodrıguez-Palmero, M., A. Franch, M. Castell, C. Pelegrı, F. J. Perez-Cano,C. Kleinschnitz, G. Stoll, T. H€unig, and C. Castellote. 2006. Effective treatmentof adjuvant arthritis with a stimulatory CD28-specific monoclonal antibody.J. Rheumatol. 33: 110–118.
4. Schmidt, J., K. Elflein, M. Stienekemeier, M. Rodriguez-Palmero, C. Schneider,K. V. Toyka, R. Gold, and T. H€unig. 2003. Treatment and prevention of ex-perimental autoimmune neuritis with superagonistic CD28-specific monoclonalantibodies. J. Neuroimmunol. 140: 143–152.
5. Beyersdorf, N., T. Hanke, T. Kerkau, and T. H€unig. 2006. CD28 superagonistsput a break on autoimmunity by preferentially activating CD4+CD25+ regulatoryT cells. Autoimmun. Rev. 5: 40–45.
6. TeGenero, A. G. TGN1412 investigational medicinal product dossier. Availableat: www.circare.org/foia5/tgn1412dossier.pdf.
7. Gibbs, R. A., J. Rogers, M. G. Katze, R. Bumgarner, G. M. Weinstock,E. R. Mardis, K. A. Remington, R. L. Strausberg, J. C. Venter, R. K. Wilson,et al; Rhesus Macaque Genome Sequencing and Analysis Consortium. 2007.Evolutionary and biomedical insights from the rhesus macaque genome. Science316: 222–234.
8. Waibler, Z., L. Y. Sender, C. Merten, R. Hartig, S. Kliche, M. Gunzer,P. Reichardt, U. Kalinke, and B. Schraven. 2008. Signaling signatures andfunctional properties of anti-human CD28 superagonistic antibodies. PloS One3: e1708.
9. Suntharalingam, G., M. R. Perry, S. Ward, S. J. Brett, A. Castello-Cortes,M. D. Brunner, and N. Panoskaltsis. 2006. Cytokine storm in a phase 1 trial ofthe anti-CD28 monoclonal antibody TGN1412. N. Engl. J. Med. 355: 1018–1028.
10. Duff, G. 2006. Expert group on phase one clinical trials: final report. Availableat: http://webarchive.nationalarchives.gov.uk/20130107105354/http://www.dh.gov.uk/en/Publicationsandstatistics/Publications/PublicationsPolicyAndGuidance/DH_063117.
11. Eastwood, D., L. Findlay, S. Poole, C. Bird, M. Wadhwa, M. Moore, C. Burns,R. Thorpe, and R. Stebbings. 2010. Monoclonal antibody TGN1412 trial failureexplained by species differences in CD28 expression on CD4+ effector memoryT-cells. Br. J. Pharmacol. 161: 512–526.
12. European Medicines Agency. 2007. Guideline on strategies to identify andmitigate risk for first-in human clinical trials with investigational medicinalproducts. Available at: http://www.emea.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002988.pdf.
13. Rogers, K. A., F. Scinicariello, and R. Attanasio. 2006. IgG Fc receptor IIIhomologues in nonhuman primate species: genetic characterization and ligandinteractions. J. Immunol. 177: 3848–3856.
14. Jacobsen, F. W., R. Padaki, A. E. Morris, T. L. Aldrich, R. J. Armitage,M. J. Allen, J. C. Lavallee, and T. Arora. 2011. Molecular and functionalcharacterization of cynomolgus monkey IgG subclasses. J. Immunol. 186: 341–349.
15. Warncke, M., T. Calzascia, M. Coulot, N. Balke, R. Touil, F. Kolbinger, andC. Heusser. 2012. Different adaptations of IgG effector function in human andnonhuman primates and implications for therapeutic antibody treatment. J.Immunol. 188: 4405–4411.
16. Stebbings, R., L. Findlay, C. Edwards, D. Eastwood, C. Bird, D. North,Y. Mistry, P. Dilger, E. Liefooghe, I. Cludts, et al. 2007. “Cytokine storm” in thephase I trial of monoclonal antibody TGN1412: better understanding the causesto improve preclinical testing of immunotherapeutics. J. Immunol. 179: 3325–3331.
17. Findlay, L., D. Eastwood, R. Stebbings, G. Sharp, Y. Mistry, C. Ball, J. Hood,R. Thorpe, and S. Poole. 2010. Improved in vitro methods to predict the in vivotoxicity in man of therapeutic monoclonal antibodies including TGN1412. J.Immunol. Methods 352: 1–12.
18. Weissm€uller, S., L. Y. Semmler, U. Kalinke, S. Christians, J. M€uller-Berghaus,and Z. Waibler. 2012. ICOS-LICOS interaction is critically involved inTGN1412-mediated T-cell activation. Blood 119: 6268–6277.
19. Romer, P. S., S. Berr, E. Avota, S. Y. Na, M. Battaglia, I. ten Berge, H. Einsele,and T. H€unig. 2011. Preculture of PBMCs at high cell density increases sensi-tivity of T-cell responses, revealing cytokine release by CD28 superagonistTGN1412. Blood 118: 6772–6782.
20. Randriamampita, C., G. Boulla, P. Revy, F. Lemaitre, and A. Trautmann. 2003.T cell adhesion lowers the threshold for antigen detection. Eur. J. Immunol. 33:1215–1223.
21. Stefanova, I., J. R. Dorfman, and R. N. Germain. 2002. Self-recognition promotesthe foreign antigen sensitivity of naive T lymphocytes. Nature 420: 429–434.
22. Germain, R. N., I. Stefanova, and J. Dorfman. 2002. Self-recognition and theregulation of CD4+ T cell survival. Adv. Exp. Med. Biol. 512: 97–105.
23. International Human Genome Sequencing Consortium. 2004. Finishing the eu-chromatic sequence of the human genome. Nature 431: 931–945.
24. Langmead, B., C. Trapnell, M. Pop, and S. L. Salzberg. 2009. Ultrafast andmemory-efficient alignment of short DNA sequences to the human genome.Genome Biol. 10: R25.
25. Hsu, F., W. J. Kent, H. Clawson, R. M. Kuhn, M. Diekhans, and D. Haussler.2006. The UCSC known genes. Bioinformatics 22: 1036–1046.
26. Mortazavi, A., B. A. Williams, K. McCue, L. Schaeffer, and B. Wold. 2008.Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Meth-ods 5: 621–628.
27. R Development Core Team. 2011. R: A language and environment for statisticalcomputing. Available at: http://www.R-project.org/
28. Anders, S., and W. Huber. 2010. Differential expression analysis for sequencecount data. Genome Biol. 11: R106.
29. Bruhns, P. 2012. Properties of mouse and human IgG receptors and their con-tribution to disease models. Blood 119: 5640–5649.
30. Boruchov, A. M., G. Heller, M. C. Veri, E. Bonvini, J. V. Ravetch, andJ. W. Young. 2005. Activating and inhibitory IgG Fc receptors on human DCsmediate opposing functions. J. Clin. Invest. 115: 2914–2923.
31. Veri, M. C., S. Gorlatov, H. Li, S. Burke, S. Johnson, J. Stavenhagen, K. E. Stein,E. Bonvini, and S. Koenig. 2007. Monoclonal antibodies capable of discrimi-nating the human inhibitory Fcg-receptor IIB (CD32B) from the activating Fcg-receptor IIA (CD32A): biochemical, biological and functional characterization.Immunology 121: 392–404.
32. Rossi, E. A., D. M. Goldenberg, R. Michel, D. L. Rossi, D. J. Wallace, andC. H. Chang. 2013. Trogocytosis of multiple B-cell surface markers by CD22targeting with epratuzumab. Blood 122: 3020–3029.
33. Corrales-Aguilar, E., M. Trilling, H. Reinhard, E. Merce-Maldonado, M. Widera,H. Schaal, A. Zimmermann, O. Mandelboim, and H. Hengel. 2013. A novelassay for detecting virus-specific antibodies triggering activation of Fcg recep-tors. J. Immunol. Methods 387: 21–35.
34. Eastwood, D., C. Bird, P. Dilger, J. Hockley, L. Findlay, S. Poole, S. J. Thorpe,M. Wadhwa, R. Thorpe, and R. Stebbings. 2013. Severity of the TGN1412 trialdisaster cytokine storm correlated with IL-2 release. Br. J. Clin. Pharmacol. 76:299–315.
35. Rajasekaran, K., V. Xiong, L. Fong, J. Gorski, and S. Malarkannan. 2010.Functional dichotomy between NKG2D and CD28-mediated co-stimulation inhuman CD8+ T cells. PloS One 5: e12635.
36. Hansel, T. T., H. Kropshofer, T. Singer, J. A. Mitchell, and A. J. George. 2010. Thesafety and side effects of monoclonal antibodies. Nat. Rev. Drug Discov. 9: 325–338.
37. Bruhns, P., B. Iannascoli, P. England, D. A. Mancardi, N. Fernandez, S. Jorieux,and M. Daeron. 2009. Specificity and affinity of human Fcgamma receptors andtheir polymorphic variants for human IgG subclasses. Blood 113: 3716–3725.
38. Hanke, T., M. Trischler, and C. Guntermann, inventors. Nucleic acids encodingsuperagonistic anti-CD28 antibodies. United States patent no. 7,585,960. 2008Sep 9.
39. Sada-Ovalle, I., A. Talayero, L. Chavez-Galan, L. Barrera, A. Castorena-Maldonado, A. Soda-Merhy, and L. Torre-Bouscoulet. 2012. Functionality ofCD4+ and CD8+ T cells from tonsillar tissue. Clin. Exp. Immunol. 168: 200–206.
40. Ball, C., B. Fox, S. Hufton, G. Sharp, S. Poole, R. Stebbings, D. Eastwood,L. Findlay, P. W. Parren, R. Thorpe, et al. 2012. Antibody C region influencesTGN1412-like functional activity in vitro. J. Immunol. 189: 5831–5840.
41. O’Connell, R. M., D. Kahn, W. S. Gibson, J. L. Round, R. L. Scholz,A. A. Chaudhuri, M. E. Kahn, D. S. Rao, and D. Baltimore. 2010. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T celldevelopment. Immunity 33: 607–619.
42. Baumjohann, D., and K. M. Ansel. 2013. MicroRNA-mediated regulation ofT helper cell differentiation and plasticity. Nat. Rev. Immunol. 13: 666–678.
2098 TGN1412-MEDIATED T CELL ACTIVATION
by guest on June 23, 2018http://w
ww
.jimm
unol.org/D
ownloaded from