catalysis effects of water molecules and of charge on intramolecular proton transfer of uracil
TRANSCRIPT

Catalysis Effects of Water Molecules and of Charge on Intramolecular Proton Transfer ofUracil
Dejie Li and Hongqi Ai*School of Chemistry and Chemical Engineering, UniVersity of Jinan, 250022 P.R. China
ReceiVed: April 07, 2009; ReVised Manuscript ReceiVed: June 23, 2009
In this work, the three most stable uracil isomers (U1, U2, and U3) and their neutral, positive, and negativecharged multihydrates are chosen as research objects to investigate the tautomeric process between the moststable uracil, U1, and its two minor stable isomers, U2 and U3. By the study, deeper insight can be obtainedregarding point mutations induced by uracil deformation. Toward the target, the activation energies of theintramolecular proton transfer (tautomeric process) as well as the catalysis effects of water molecules and ofcharges attached are investigated using density functional theory (DFT) calculations by means of the B3LYPexchange and correlation functions. Results reveal that water molecules hold a stronger catalysis effect onthe proton transfer in these negative charged uracil hydrates than in the neutral counterparts. The optimalnumber of water molecules needed to catalyze the proton transfer is determined as two in the neutral hydratedsystems, whereas it is three in the negative charged systems. Positive charge attachment, however, hindersthe intramolecualr proton transfer of uracil, and the charge and the proton of uracil will transfer to the waterclusters if water molecules are attached. Then the positive charged hydrates look more like U1a/b+[(H2O)n+H+]species in structure. Analysis reveals that it is the acceptance process of the last proton to determine theimpossibility of proton transfer and result in the failure of tautomeric processes from cat-U1a-nw to cat-U2-nw and from cat-U1b-nw to cat-U3-nw. Detailed structural parameters and energy changes are discussed forthe above different processes.
Introduction
Different tautomers of nucleic acid (NA) bases can beobtained when one of its active hydrogen atoms binds differentpositions of the base.1 In vivo, those rare tautomers will beinvolved in various biochemical processes.2 Moreover, theexistence of rare tautomeric forms of NA bases increases thepossibility of mispairing of purines and pyrimidines that maylead to the spontaneous point mutations.3-5 Thus, a large numberof theoretical and experimental studies have been devoted tothe tautomerism of NA bases.6-17
Several factors, such as excitation,18 chemical modification,19
metal cation interaction,20,21 electron attachment,22 irradiation,23
etc., had been found to be responsible for the tautomericequilibrium between the “normal” and the “rare” forms of NAbases. For example, the attachment of a low-energy electron tothe NA bases or excitation of an electron from the NA baseswould cause a series of property changes of these NA basesand their hydrated systems, such as the changes of relativestability of tautomers,24-26 incremental probability of protonintramolecular transfer to form new tautomers,27 the rearrange-ment of surrounding water molecules,28 etc. In addition,hydration also plays a significant role in the tautomeric process.Studies revealed that a water molecule might influence thestability of different tautomeric forms of NA bases throughhydrogen bonding interactions.29,30 Meanwhile, water-assistedproton transfer will greatly increase the populations of the raretautomers by lowering the activation energy barrier of theintramolecular proton transfer.29-31
As one of the important bases, uracil has also been paid muchattention in recent years.12,13,32-40 Among thirteen possible
tautomers of uracil,13 three lowest-energy tautomers are fre-quently employed. One is the most stable “ketone” uracil (U1).Another two are U2 and U3, respectively, which are at least10.8 kcal/mol higher in energy than the ketone one.13,38
Therefore, the three most stable uracil isomers are selected asresearch objects to investigate the hydration effect on theirrelative stability and the intramolecular proton transfer in thepresent paper. Their structures are shown in Figure 1. Obviously,if H10 of U1 transfers to its O8 site, then U1 will become U2.This indicates that intramolecular H10 transfer of U1, in fact,corresponds to the tautomeric process U1 f U2. Correspond-ingly, if H9 transfers to the O7 site, then the process will beU1 f U3. In the gas phase the two transfer processes aredifficult due to a high activation energy barrier, while thehydration case will improve it.12,13,32,33 Despite the importanceof hydration on the tautomeric process, the optimal watermolecule number, which will minimize the activation energyof the intramolecular proton transfer in the tautomeric process,remains unknown.
In addition, Shukla et al. reported that a charge effect greatlyreduced the activation energy of proton transfer in the guanine* E-mail: [email protected].
Figure 1. Three most stable uracil isomers.
J. Phys. Chem. B 2009, 113, 11732–1174211732
10.1021/jp9031833 CCC: $40.75 2009 American Chemical SocietyPublished on Web 07/31/2009
system or even resulted in it being barrierless.34 Li et al. showedthat attachment of an electron rather than detachment of anelectron from a GC pair would favor the intramolecular protontransfer.35 A photoelectron spectroscopy experiment indicatedthat the transformation from a dipole-bound uracil anion to avalence-bound one took place when the radical anions experi-enced solvation and multibody interactions in the condensedphase.36 Another experiment on the electron binding to thepyrimidine nucleobases in the presence of water clusters foundthat the electron affinities were almost linearly proportional tothe number of solvent water molecules.37 Thus, under thecondition of negative or positive charge, it would be of greatinterest to conduct the investigation on the optimal watermolecule number required to induce the imtramolecular protontransfer of the “ketone” uracil and correspondingly to form itsother two (U2 and U3) isomers. This is our other aim, in whichthe mechanism of the charge effect will be elucidated and theconcerted effect between the charge and hydration will also bediscussed. Correspondingly, in the following statement, ani-U1aor cat-U1b will be used to refer to the charged uracil isomers,where the prefixes “ani” and “cat” represent the complexes witha negative (anion) and a positive (cation) charge, respectively.The subscripts “a” and “b” represent the areas of H10 transferto O8 (then U1 becomes U2) and of H9 transfer to O7 (thenU1 becomes U3), respectively, where water molecules will beattached.
Due to the importance of hydration and the charge effect onthe tautomeric processes of U1 f U2/U3, in this investigationwe attempt to answer the following questions: (1) How does anegative or a positive charge influence the relative stability ofthe uracil tautomers? (2) What is the preferred position for theproton transfer to take place in these neutral and charged uracilisomer systems with and without hydration? (3) What is theoptimal number of water molecules assisting proton transfer inthese neutral and charged systems? (4) How do the two differentquality charges affect the intramolecular transfer proton (i.e.,tautomeric processes of U1 f U2 and of U1 f U3)? etc.
Computational Methods
Studies had confirmed that B3LYP and MP2 could give verysimilar results for the geometrical and vibrational features ofNA bases38 and DFT is an excellent compromise betweencomputational cost and reasonable results. Therefore, in thisarticle the B3LYP in combination with a considerably large basisset 6-311++G(d,p) is adopted thoroughly. The computedstationary points have been characterized as minima or transitionstates (TSs) by diagonalizing the Hessian matrix and analyzingthe vibrational normal modes. In this way, the stationary pointscan be classified as minima if no imaginary frequencies areobserved or as TSs if only one imaginary frequency isobtained.41,42 The particular natures of these TSs are determinedby analyzing the motion described by the eigenvector associatedwith the imaginary frequency.
After geometry optimization and frequency calculations(without scaling), zero-point energies (ZPEs) and the sum ofelectronic and thermal free energies (G) could be obtained. Theeffect of basis set superposition error was not taken into accountbecause it had been predicted to be trivial in the uracil-watersystem,13,43 especially when such a considerably large basis setwas used. Adiabatic potential energy surface (PES) changesalong the N-H bond stretch were calculated using the optimiza-tion keyword opt)z-matrix, with the S action code in theadditional input (N-H distance). Correspondingly, analysis onthe charge/spin distributions and molecular orbital information
along with the N-H changes was also made. Net atomic chargeswere obtained using the natural population analysis (NPA) ofWeinhold et al.44
All calculations were performed with the GAUSSIAN 0345
suite of packages.
Results and Discussion
1. Stability Orders of Neutral, Negative, and PositiveCharged U1, U2, and U3. The geometries of the uracil andmicrosolvated uracil complexes were fully optimized at theB3LYP/6-311++G** level of theory. Our optimized geo-metrical parameters of uracil isomers listed in Table 1 are ingood agreement not only with the recent theoretical results13
obtained at the B3LYP/6-31++G** level (the maximumdispersion is less than 0.002 Å) but also with those experimentalones obtained by X-ray38 and electron diffraction.46
The results of various calculations on relative free energies(∆G) and dipole moments (DMs) as well as vertical electrondissociation energies (VDEs) are compiled in Table 2. Resultsreveal that their stability order (in kcal/mol) is U1(0.0) > U2(11.4) > U3(12.1), in good agreement with theoretical predic-tions (U1(0.0) > U2 (10.9) > U3(11.8), obtained at the B3LYP/6-31+G** level) by Kryachko et al.13 Moreover, the energyvalues of U2 and U3 tautomers are very similar to each other.With a more compact and keto structure, U1 is, thus, more stablethan the two enol forms U2 and U3.47 Both U2 and U3molecules, however, are characterized by two conjugated doublebonds, CdC and CdN, in the ring and a hydroxyl linking onthe ring (Figure 1).
Table 2 reveals that the three tautomers have different valuesof dipole moment. The larger value would indicate considerablestabilization when the tautomer is attached to an electron orexposed to a polar solvent such as water.48 Comparisons revealthat U3 has the largest dipole moment (5.0) and would be mostfavorable to bind an excess electron in a dipole-bound orvalence-bound state. Calculations reveal that the orbital of theU3 anion has a π-character and the attached electron is locatedon the molecular frame of the complex. Its diffuse characterand orbital shape also confirm that the calculations describe avalence-bound anion (not shown). The stability order of anionicU2 (ani-U2) and U3 (ani-U3) also confirms that ani-U3 becomesthe second stable tautomer and its energy is 4.9 kcal/mol lessthan that of ani-U2. This may be closely related to the VDEdifference (4.0 kcal/mol) between ani-U2 (10.1) and ani-U3(14.1), in which the VDE is calculated by the anionic UB3LYP
TABLE 1: Optimized Bond Distances (Å) of the Three MostStable Uracil Isomers
U1
bond present theory a exptb exptc U2 U3
N1-C2 1.384 1.385 1.377 1.399 1.354 1.380C2-N3 1.393 1.394 1.371 1.399 1.302 1.423N3-C4 1.377 1.377 1.359 1.399 1.375 1.358C4-C5 1.352 1.352 1.340 1.343 1.365 1.362C5-C6 1.459 1.459 1.430 1.462 1.448 1.430C6-N1 1.412 1.412 1.371 1.399 1.423 1.308N1-H9 1.014 1.015 0.877 1.002 1.015 2.253C2-O8 1.220 1.220 1.215 1.212 1.342 1.221N3-H10 1.010 1.012 0.836 1.002 2.306 1.012C4-H11 1.084 1.085 0.957 1.072 1.086 1.085C5-H12 1.081 1.082 0.970 1.072 1.083 1.081C6-O7 1.223 1.223 1.245 1.212 1.224 1.344
a Calculated at the B3LYP/6-31++G** level.13 b From X-ray.38
c From electron diffraction.46
Catalysis Effects on Proton Transfer of Uracil J. Phys. Chem. B, Vol. 113, No. 34, 2009 11733

energies minus B3LYP energies of the corresponding neutralat the optimized anion geometries. The remaining energy(4.9-4.0 ) 0.9 kcal/mol) may be attributed to the deformationenergy.49 In the meantime, we also calculate the adiabaticelectron affinity (AEA) values by using the neutral speciesenergy minus the corresponding anion one at their respectiveoptimized geometries. Table 2 reveals that our results are ingood agreement with those available theoretical ones. The slightdifference may be derived from the different methods and basissets used.37,50-53 When an electron is attached, the planar ringstructures of U1 and U3 still remain, but that of U2 changes.Observation shows that N1 of U2 is exposed outside of the ringplane. So this change may consume part of the energy. In detail,the dihedral angles O7-C6-N1-H9 and O7-C6-N1-C2 are0.0 and 180.0° in neutral U2, respectively, whereas the samedihedral angles become 22 and 163° in anionic U2, respectively.Thus, it is not surprising that the energy of anionic U2 is higherthan that of anionic U1 and U3. Strong torsion of molecularframework of ani-U2 is characteristic of the covalence-boundanionic state, not of the dipole-bound one.54
The stability ordering of the three cationic (cat) tautomers isthe same as the neutral one. Compared to the neutral tautomers,the energies increase in the order 213.3, 215.6, and 219.8 kcal/mol for cat-U1, cat-U2, and cat-U3, respectively (Table 2). Thisindicates that ionizing an electron from these neutral moleculesis very difficult.
2. Tautomeric Process. 2.1. Tautomeric Processes fromIsolated U1 to Two Tautomers U2 and U3. Figure 2 showsthe proton transfer processes from U1 or its hydrates to thetautomers U2 and U3 or their corresponding hydrates via TSs.The tautomeric process of U1 f U2, in fact, corresponds tothe proton (H10) intermolecular transfer from the N3 site tothe O8 site of U1. Optimizations show the distances of N3-H10bonds are 1.010, 1.328, and 2.306 Å in U1, TS12, and U2,respectively, suggesting a process of intramolecular protontransfer. Meanwhile, the C2-O8 bond of U1 varies from 1.220Å to 1.283 Å. The activation energy in the process is 41.8 kcal/mol, indicating that the transfer is hard to achieve. The case issimilar for the U1 f U3 process because of its high energybarrier of 40.8 kcal/mol. With a smaller basis set, the resultsobtained by Hu et al.38 and Iwona et al.55 are 42.7 (B3LYP/6-31G**) and 44.0 kcal/mol (B3LYP/6-31++G**), respec-tively, in good agreement with our conclusion. Interestingly,attachment of an electron can effectively lower the activationenergy either in the tautomeric process of U1f U2 (22.2 kcal/mol) or of U1 f U3(6.4 kcal/mol). Ionization of an electroncan also lower the energy from 42.8 (neutral) to 35.7 kcal/mol(cationic) in the U1 f U2 process; however, it can increasethe energy from 41.7 to 45.5 kcal/mol in the U1fU3 process.
So attachment instead of ionization of an electron in the twoprocesses is more effective for the proton transfer and tautomericprocess.
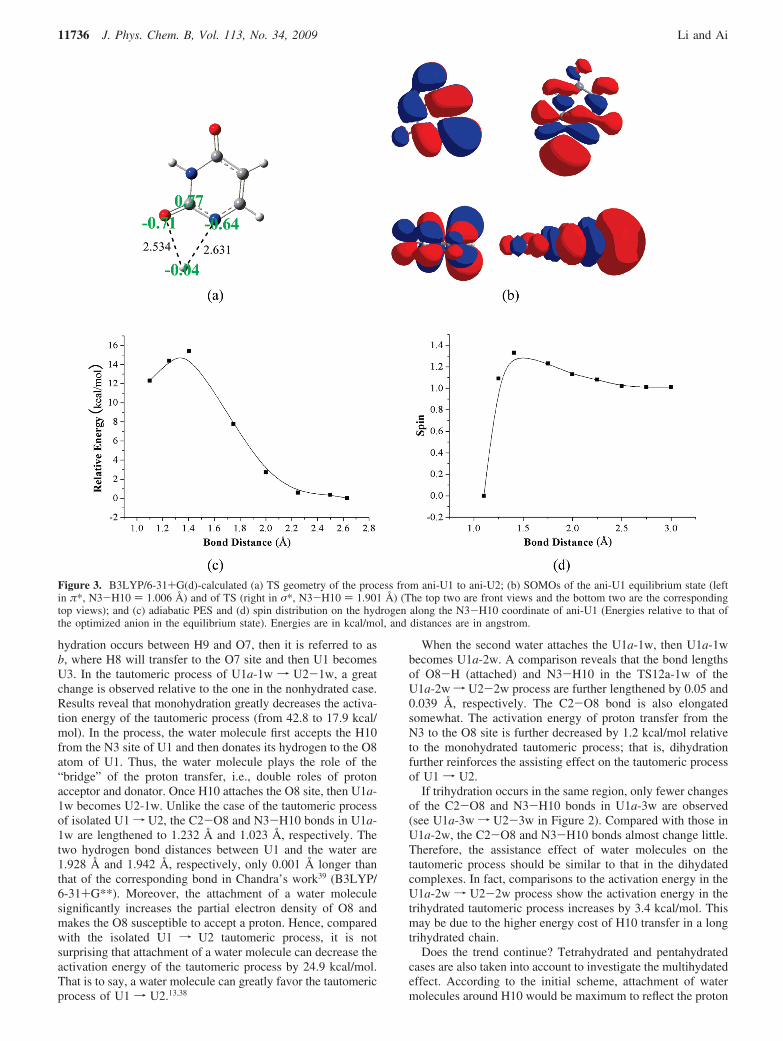
Meanwhile another interesting case is observed in thetautomeric process of ani-U1 f ani-U2. As shown for the TSgeometry in the process of ani-U1 f ani-U2 in Figure 3a, thecaptured electron in the TS will mainly localize on H10 (∼spin:1.01), while on other adjacent atoms the spin is small (C2 ∼spin:0.06, O8 ∼spin: 0.00, N3 ∼spin: -0.03). This result indicatesthat the tautomeric process, in fact, corresponds to the coopera-tive transfer processes of H10 and an electron. The correspond-ing charge distributions in some of the atoms are as follows:H10, -0.04; C2, 0.77; O8, -0.71; and N3, -0.64. So theH10-O8 and H10-N3 bonds are elongated by 2.534 and 2.631Å, respectively, due to the electrostatic repulsion.
Along the increase of the N3-H10 distance, i.e., thetransformation from ani-U1 to ani-U2, the bonding property ofthe N3-H10 single occupied molecular orbital (SOMO) gradu-ally becomes a σ* bond from its initial π* bond and then adipole-bound contribution will become dominant. The processcan be visually observed from Figure 3b, in which the SOMOsof the equilibrium-state uracil anion (mostly π* state, N3-H10) 1.006 Å) and the TS anion (σ*-state, N3-H10 ) 1.901 Å)are displayed. Both pure π* and σ* states have a separate PES,respectively. One planar π* state corresponds to a shortN1-H10 distance, whereas one planar localized σ* statecorresponds to a long N1-H10 distance. They branch at aprojected localized antibonding orbital, and for the σ* state(based on a symmetry conserved calculation), a dipole-boundcontribution becomes dominant at the long distance (see Figure3b and c). The energy of this state is too high to be realisticbecause the basis set used is not sufficient for describing a truedipole-bound state. Although the basis set limitation, the dipole-bound character of the N3-H10 bond at the ani-U1 TS hasnoticeably emerged. Moreover, the spin distribution shown inFigure 3d also confirms that almost one atomic unit of the spindensity locates over H10 when H10 is about 1.4 Å far awayN3. This indicates that now anionic hydrogen H10 is dipole-bound to the U1 radical.5 Of most importance is that the bondingcharacter of N1-H10 will remind us of the real hydratedenvironment, i.e., if the character in a hydrated system will befurther toned up and favor U1 isomerization.
2.2. Tautomeric Processes between Hydrated Systems. Fig-ure 2 also lists the tautomeric processes of hydrated U1a-nwf U2-nw and of U1b-nw f U3-nw, where “nw” representsthe number of water molecules attached and n ) 1-5, whilea/b denotes two different positions of hydration in U1, asmentioned above. Obviously, if hydration is at the range betweenH10 and O8 of U1, then the hydration will affect the tautomericprocess of U1f U2 and we define the range as a. Similarly, if
TABLE 2: ZPVE-Corrected Relative Free Energies (∆G in kcal/mol),a Dipole Moments (DM in Debye), and VDE/AEA (inkcal/mol)
neutral uracil anionic uracil cationic uracil
U1 U2 U3 ani-U1 ani-U2 ani-U3 cat-U1 cat-U2 cat-U3
∆G 0.0 11.4 12.1 -5.4 10.7 5.8 213.3 215.6 219.8DM 4.6 3.3 5.0 3.9 3.9 3.5 6.1 3.0 5.5VDE 13.5 10.1 14.1 217.8 209.4 210.5AEA 5.4, 5.5,b 4.2,c 1.6,d 3.2,e 3.5 ( 2.8f 0.7 6.4
a Note that the ∆G values are the differences between all these neutral/charged uracil complexes and the neutral uracil (U1). b DZP++ basisset used in ref 50. c 6-311+G(2df,p) basis set used in ref 51. d 6-311++G basis set used without ZPVE in ref 52. e TZVP basis set used in ref53. f Extrapolated values from photoelectron spectra of nucleobase(H2O)n clusters in ref 37. All computed values in the present paper arezero-point corrected unless otherwise noted.
11734 J. Phys. Chem. B, Vol. 113, No. 34, 2009 Li and Ai
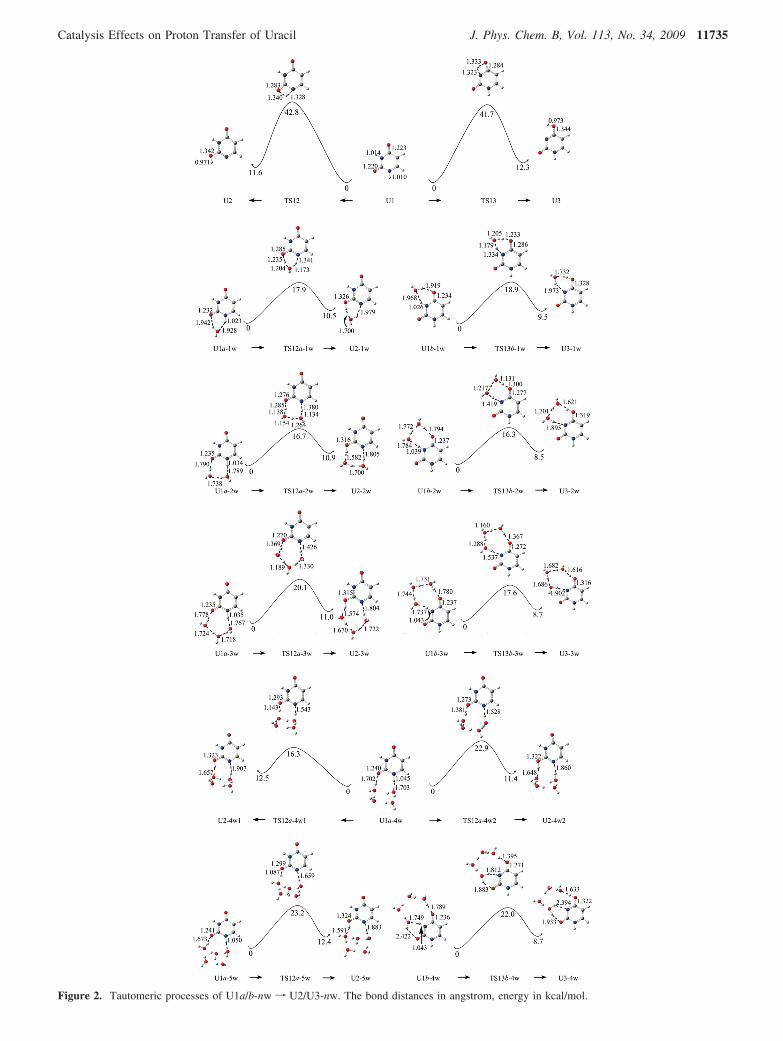
Figure 2. Tautomeric processes of U1a/b-nw f U2/U3-nw. The bond distances in angstrom, energy in kcal/mol.
Catalysis Effects on Proton Transfer of Uracil J. Phys. Chem. B, Vol. 113, No. 34, 2009 11735
hydration occurs between H9 and O7, then it is referred to asb, where H8 will transfer to the O7 site and then U1 becomesU3. In the tautomeric process of U1a-1w f U2-1w, a greatchange is observed relative to the one in the nonhydrated case.Results reveal that monohydration greatly decreases the activa-tion energy of the tautomeric process (from 42.8 to 17.9 kcal/mol). In the process, the water molecule first accepts the H10from the N3 site of U1 and then donates its hydrogen to the O8atom of U1. Thus, the water molecule plays the role of the“bridge” of the proton transfer, i.e., double roles of protonacceptor and donator. Once H10 attaches the O8 site, then U1a-1w becomes U2-1w. Unlike the case of the tautomeric processof isolated U1f U2, the C2-O8 and N3-H10 bonds in U1a-1w are lengthened to 1.232 Å and 1.023 Å, respectively. Thetwo hydrogen bond distances between U1 and the water are1.928 Å and 1.942 Å, respectively, only 0.001 Å longer thanthat of the corresponding bond in Chandra’s work39 (B3LYP/6-31+G**). Moreover, the attachment of a water moleculesignificantly increases the partial electron density of O8 andmakes the O8 susceptible to accept a proton. Hence, comparedwith the isolated U1 f U2 tautomeric process, it is notsurprising that attachment of a water molecule can decrease theactivation energy of the tautomeric process by 24.9 kcal/mol.That is to say, a water molecule can greatly favor the tautomericprocess of U1 f U2.13,38
When the second water attaches the U1a-1w, then U1a-1wbecomes U1a-2w. A comparison reveals that the bond lengthsof O8-H (attached) and N3-H10 in the TS12a-1w of theU1a-2wf U2-2w process are further lengthened by 0.05 and0.039 Å, respectively. The C2-O8 bond is also elongatedsomewhat. The activation energy of proton transfer from theN3 to the O8 site is further decreased by 1.2 kcal/mol relativeto the monohydrated tautomeric process; that is, dihydrationfurther reinforces the assisting effect on the tautomeric processof U1 f U2.
If trihydration occurs in the same region, only fewer changesof the C2-O8 and N3-H10 bonds in U1a-3w are observed(see U1a-3w f U2-3w in Figure 2). Compared with those inU1a-2w, the C2-O8 and N3-H10 bonds almost change little.Therefore, the assistance effect of water molecules on thetautomeric process should be similar to that in the dihydatedcomplexes. In fact, comparisons to the activation energy in theU1a-2w f U2-2w process show the activation energy in thetrihydrated tautomeric process increases by 3.4 kcal/mol. Thismay be due to the higher energy cost of H10 transfer in a longtrihydrated chain.
Does the trend continue? Tetrahydrated and pentahydratedcases are also taken into account to investigate the multihydatedeffect. According to the initial scheme, attachment of watermolecules around H10 would be maximum to reflect the proton
Figure 3. B3LYP/6-31+G(d)-calculated (a) TS geometry of the process from ani-U1 to ani-U2; (b) SOMOs of the ani-U1 equilibrium state (leftin π*, N3-H10 ) 1.006 Å) and of TS (right in σ*, N3-H10 ) 1.901 Å) (The top two are front views and the bottom two are the correspondingtop views); and (c) adiabatic PES and (d) spin distribution on the hydrogen along the N3-H10 coordinate of ani-U1 (Energies relative to that ofthe optimized anion in the equilibrium state). Energies are in kcal/mol, and distances are in angstrom.
11736 J. Phys. Chem. B, Vol. 113, No. 34, 2009 Li and Ai

transfer. Therefore, further optimization for the tetrahydratedU1 shows that two different structures can be obtained. One isthat U1 interacts directly with only two water molecules andthe other two water molecules interact with the former twothrough intermolecular H-bonds, resulting in two eight-membered ring structures. Moreover, the two rings are almostperpendicular to each other (∼) (see U1a-4w in U1a-4w1 fU2-4w1 of Figure 2). Thus intramolecular proton (H10) transfershould be mainly assisted by two inside water molecules andlike the dihydrated case discussed above. The structure showsthe outside two water molecules are H-bonding with the insideones. The calculated activation energy of the proton transfer inthis process is 16.3 kcal/mol, which is further less by 0.4 kcal/mol than that of the dihydrated process. This indicates that theeffect of second-shell water molecules on the intramolecularproton transfer is still favorable, although it is subordinate.Another tetrahydrated structure covers the intramolecular protontransfer via all four water molecules (see U1a-4w f U2-4w2in Figure 2). The activation energy of this process is 22.9 kcal/mol, more than that of the trihydrated case. The result illu-minates again that the activation energy is not always decreasingwith the number increase of water molecules. According to thefirst tetrahydrated case, the fifth water is attached at anotherside of the inside two water molecules to probe if the thirdsecond-shell water molecule can further decrease the activationenergy of proton (H10) transfer. Results reveal that the activationenergy increases a lot.
These results indicate that the long-range proton transferassisted directly by four or even more water molecules is notoptimal; that is, all the water molecules do not directlyparticipate in and favor the proton transfer process56 due to thelimited space and increased activation energy. Of course, moresophisticated investigations are required to further verify thispoint soon. With the increase in the number of water moleculesfrom 1 to 5, the activation energies of proton transfer in theseuracil hydrates are not always reduced. The dihydrated complexis a milestone; that is, dihydration minimizes the activationenergy in the tautomeric processes of U1a-nw f U2-nw. Insummary, the order of the activation energies (kcal/mol) in thesedifferent hydrated systems can be listed as follows: U1a-4w1f U2-4w1(16.3) ≈ U1a-2w f U2-2w (16.7) < U1a-1w fU2-1w (17.9) < U1a-3w f U2-3w (20.1) < U1a-4w fU2-4w(22.9) ≈ U1a-5w f U2-5w(23.2) < U1 f U2(42.8).
Like the discussion on the tautomeric processes of U1a-nwf U2-nw, we also probe the tautomeric process of U1 f U3with and without hydration (Figure 2). Results reveal that theactivation energy in the tautomeric process of U1b-1w fU3-1w is 18.9 kcal/mol, much less than that of the nonhydratedU1 f U3 process (41.7 kcal/mol) and a little larger (1.0 kcal/mol) than that of the U1a-1w f U2-1w process. The C6-O7bond in U1b-1w is lengthened to 1.234 Å, and the N1-H9 bondin U1b-1w is 0.014 Å also longer than that of U1. The bonddistance between the oxygen atom of water and H9 is 1.968Å.39,40 Thus, the monohydrated effect in the tautomeric processof U1b-1wf U3-1w is also remarkable. Further hydration leadsto the longer C6-O7 and N1-H9 distances in U1b-2w. In thetransition state (TS13b-2w) of the U1b-2w f U3-2w process,the N1-H9 bond is lengthened by about 0.085 Å. Accordingly,dihydration further reinforces the catalysis effect on the tauto-meric process of U1 f U3, which is confirmed by the loweractivation energy of 16.3 kcal/mol. The calculated results forthe trihydrated tautomeric process reveal that the catalysis effectof hydration on the tautomeric process also decreases. Incomparison with the activation energy of the U1b-2w f U3-
2w process, the energy of the trihydrated process increases by1.3 kcal/mol, which is less than the energy difference (3.4 kcal/mol) between U1a-3w f U2-3w and corresponding U1a-2wf U2-2w. To verify the further increasing trend of the activationenergy in the multihydated circumstance, tetrahydrated com-plexes are also considered. Optimization shows that the interac-tion of the uracil with four water molecules forms a twelve-membered-ring structure due to the coexistence of twoneighboring electronegative groups (N1 and O8). A hydrogenatom of a water molecule neighboring to H9 (see U1b-4w fU3-4w in Figure 2) forms a hydrogen bond with O8. Theactivation energy of the tautomeric process is 21.9 kcal/mol,higher by 3.1 kcal/mol than that of the trihydrated process. Theresults further confirm that the catalysis of water molecules alsoexists in the tautomeric process of U1 f U3 and the optimalnumber of water molecules needed for the assisting protontransfer is two. Correspondingly, the minimum activation energyis 16.3 kcal/mol, less by 0.4 kcal/mol than that of U1a-2w fU2-2w. The value is equal to that (16.3 kcal/mol) of the U1a-4w1 f U2-4w1 process. This implies that in aqueous solutionthe tautomeric processes of U1 f U2 and U1 f U3 arecompetitive, and the transfer rates in the two processes wouldbe equivalent.
Different from the case of U1a-nw f U2-nw, the activationenergy of U1b-3w f U3-3w is less than that of U1b-1w fU3-1w but more than that of U1b-2w f U3-2w. An analysisreveals the bond length of N1-H9 in U1-3W is greatlyelongated, which is propitious to proton transfer. With theincrease in the number of water molecules from 1 to 3, theassisting ability should be reinforced. Possibly due to the largerenergy cost for proton transfer in a longer chain, the activationenergy of the U1b-3wf U3-3w process also begins to increase.The proton-transfer case in more than pentahydrated systemsis not taken into account because there is an increased trendfor the activation energy after attaching more than two watermolecules. The order of the activation energy (kcal/mol) forthese different hydrated U1 f U3 tautomeric processes can besummarized as follows: U1b-2w f U3-2w (16.3) < U1b-3wf U3-3w (17.6) < U1b-1wf U3-1w (18.9) < U1b-4wf U3-4w (22.0). From the ordering, we can also observe that theactivation energy of U1b-3w f U3-3w is less than that ofU1b-1wf U3-1w, which is different from the correspondinghydrated U1 f U2 processes.
3. Hydrated Uracil Anions. 3.1. Geometries and Energies.Figure 4 shows the selected geometry parameters of these ani-U1a/b-nw complexes. Their activation energies, TSs, andproduct in the tautomeric processes are listed in Figure S1 ofthe Supporting Information (SI). The most significant changesin the geometry of the anionic U1a-1w complex are the dramaticelongation of the H10 · · ·O13 distance. The H10 · · ·O13 distancevaries from 1.937 Å in the neutral U1a-1w to 2.508 Å in ani-U1a-1w. As expected, the proton donor N3 of uracil is closerto the proton (the N3-H10 bond distance decreases by 0.014Å compared to the neutral species); the O8 · · ·H15 bond lengthin ani-U1a-1w is also shorter by 0.235 Å than that in thecorresponding neutral species U1a-1w. Since the attachment ofan extra electron on the monohydrated uracil, the activationenergy of the tautomeric process, ani-U1a-1w f ani-U2-1w,increases by 0.2 kcal/mol instead of decreases. By comparison,when an extra electron attaches to U1b-1w, the activation energyof its tautomeric process, ani-U1b-1w f ani-U3-1w, reducesto 13.9 kcal/mol. The the H9 · · ·O13 distance of ani-U1b-1welongates by 0.645 Å, while its O7 · · ·H14 distance is shortenedby 0.258 Å relative to that in the neutral U1b-1w.
Catalysis Effects on Proton Transfer of Uracil J. Phys. Chem. B, Vol. 113, No. 34, 2009 11737

According to the geometric features of the neutral species,the dihydrated anionic uracil complexes can be classified intotwo types, ani-U1a-2w and ani-U1b-2w (Figure 4). The ani-U1a-2w structure looks like its neutral counterpart U1a-2w.Notice that the H10 · · ·O16 bond (2.137 Å) and the H-bondbetween the two water molecules (1.771 Å) in ani-U1a-2w aregreatly elongated compared to those in the corresponding neutralspecies (1.789 and 1.738 Å). The H15 · · ·O8 (1.613 Å) distanceis, however, far shorter than that (1.790 Å) in the neutral species.The structural feature implies that anionic U1a-2w should bemore prone to transfer H10 to produce the ani-U2-2w. Theactivation energy result, which is about 1.7 kcal/mol lower thanthe neutral tautomeric process, confirms the prediction. Differentfrom neutral U1b-2w, the ani-U1b-2w structure changes greatly.That is, two water molecules are respectively attached at theO7 and O8 sites of the uracil with 1.659 and 2.044 Å H-bonddistances. Meanwhile they also H-bond to each other with a2.177 Å distance. The bifurcated weak H-bond O13 · · ·H9 · · ·O15is significantly longer than that of the normal H-bond. In other
words, only a water molecule acts as donor and acceptor of theproton (H9) transfer in the dihydrated ani-U1b-2w complex,which is just like the case in the monohydrated ani-U1b-1wcomplex. The optimized TS listed in Figure S1 confirms theprediction. Moreover, the activation energy of the process ani-U1b-2w f ani-U3-2w is 10.4 kcal/mol, lower than those inthe neutral (17.6 kcal/mol) and anionic (13.9 kcal/mol) mono-hydrate tautomeric processes, indicating that the second attachedwater also actually assists the proton transfer although it doesnot act as a bridge of proton transfer. Thus, dihydrate is morefavorable to the transfer of a proton as compared to amonohydrate. Relative to the neutral dihydrate process, thetautomeric process is also advantageous due to less activationenergy (lower by 6.3 kcal/mol), further implying the importanceof an extra electron for the proton transfer or tautomeric process.
The trihydrated anion structure of ani-U1a-3w shows thatthere are only two water molecules directly H-bonding to theO8 atom of the uracil. The third water molecule locates at thesecond shell, H-bonding to the two internal water molecules.
Figure 4. Optimized geometries of anionic and cationic U1 hydrates. Distances in angstrom.
11738 J. Phys. Chem. B, Vol. 113, No. 34, 2009 Li and Ai
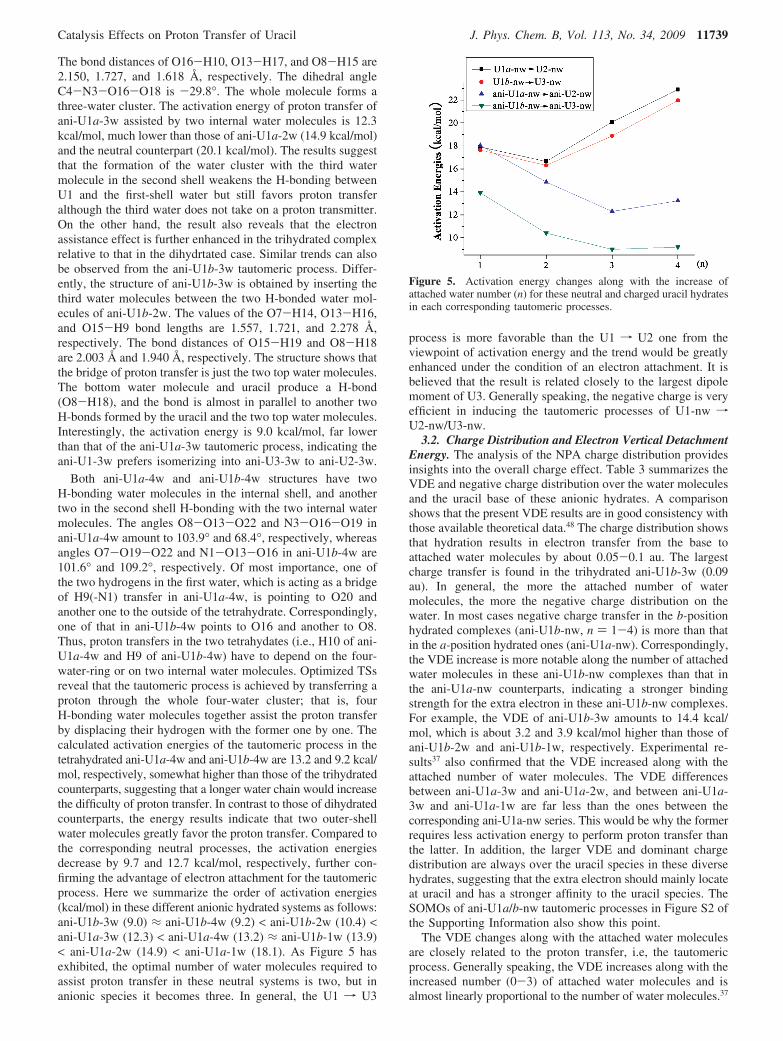
The bond distances of O16-H10, O13-H17, and O8-H15 are2.150, 1.727, and 1.618 Å, respectively. The dihedral angleC4-N3-O16-O18 is -29.8°. The whole molecule forms athree-water cluster. The activation energy of proton transfer ofani-U1a-3w assisted by two internal water molecules is 12.3kcal/mol, much lower than those of ani-U1a-2w (14.9 kcal/mol)and the neutral counterpart (20.1 kcal/mol). The results suggestthat the formation of the water cluster with the third watermolecule in the second shell weakens the H-bonding betweenU1 and the first-shell water but still favors proton transferalthough the third water does not take on a proton transmitter.On the other hand, the result also reveals that the electronassistance effect is further enhanced in the trihydrated complexrelative to that in the dihydrtated case. Similar trends can alsobe observed from the ani-U1b-3w tautomeric process. Differ-ently, the structure of ani-U1b-3w is obtained by inserting thethird water molecules between the two H-bonded water mol-ecules of ani-U1b-2w. The values of the O7-H14, O13-H16,and O15-H9 bond lengths are 1.557, 1.721, and 2.278 Å,respectively. The bond distances of O15-H19 and O8-H18are 2.003 Å and 1.940 Å, respectively. The structure shows thatthe bridge of proton transfer is just the two top water molecules.The bottom water molecule and uracil produce a H-bond(O8-H18), and the bond is almost in parallel to another twoH-bonds formed by the uracil and the two top water molecules.Interestingly, the activation energy is 9.0 kcal/mol, far lowerthan that of the ani-U1a-3w tautomeric process, indicating theani-U1-3w prefers isomerizing into ani-U3-3w to ani-U2-3w.
Both ani-U1a-4w and ani-U1b-4w structures have twoH-bonding water molecules in the internal shell, and anothertwo in the second shell H-bonding with the two internal watermolecules. The angles O8-O13-O22 and N3-O16-O19 inani-U1a-4w amount to 103.9° and 68.4°, respectively, whereasangles O7-O19-O22 and N1-O13-O16 in ani-U1b-4w are101.6° and 109.2°, respectively. Of most importance, one ofthe two hydrogens in the first water, which is acting as a bridgeof H9(-N1) transfer in ani-U1a-4w, is pointing to O20 andanother one to the outside of the tetrahydrate. Correspondingly,one of that in ani-U1b-4w points to O16 and another to O8.Thus, proton transfers in the two tetrahydates (i.e., H10 of ani-U1a-4w and H9 of ani-U1b-4w) have to depend on the four-water-ring or on two internal water molecules. Optimized TSsreveal that the tautomeric process is achieved by transferring aproton through the whole four-water cluster; that is, fourH-bonding water molecules together assist the proton transferby displacing their hydrogen with the former one by one. Thecalculated activation energies of the tautomeric process in thetetrahydrated ani-U1a-4w and ani-U1b-4w are 13.2 and 9.2 kcal/mol, respectively, somewhat higher than those of the trihydratedcounterparts, suggesting that a longer water chain would increasethe difficulty of proton transfer. In contrast to those of dihydratedcounterparts, the energy results indicate that two outer-shellwater molecules greatly favor the proton transfer. Compared tothe corresponding neutral processes, the activation energiesdecrease by 9.7 and 12.7 kcal/mol, respectively, further con-firming the advantage of electron attachment for the tautomericprocess. Here we summarize the order of activation energies(kcal/mol) in these different anionic hydrated systems as follows:ani-U1b-3w (9.0) ≈ ani-U1b-4w (9.2) < ani-U1b-2w (10.4) <ani-U1a-3w (12.3) < ani-U1a-4w (13.2) ≈ ani-U1b-1w (13.9)< ani-U1a-2w (14.9) < ani-U1a-1w (18.1). As Figure 5 hasexhibited, the optimal number of water molecules required toassist proton transfer in these neutral systems is two, but inanionic species it becomes three. In general, the U1 f U3
process is more favorable than the U1 f U2 one from theviewpoint of activation energy and the trend would be greatlyenhanced under the condition of an electron attachment. It isbelieved that the result is related closely to the largest dipolemoment of U3. Generally speaking, the negative charge is veryefficient in inducing the tautomeric processes of U1-nw fU2-nw/U3-nw.
3.2. Charge Distribution and Electron Vertical DetachmentEnergy. The analysis of the NPA charge distribution providesinsights into the overall charge effect. Table 3 summarizes theVDE and negative charge distribution over the water moleculesand the uracil base of these anionic hydrates. A comparisonshows that the present VDE results are in good consistency withthose available theoretical data.48 The charge distribution showsthat hydration results in electron transfer from the base toattached water molecules by about 0.05-0.1 au. The largestcharge transfer is found in the trihydrated ani-U1b-3w (0.09au). In general, the more the attached number of watermolecules, the more the negative charge distribution on thewater. In most cases negative charge transfer in the b-positionhydrated complexes (ani-U1b-nw, n ) 1-4) is more than thatin the a-position hydrated ones (ani-U1a-nw). Correspondingly,the VDE increase is more notable along the number of attachedwater molecules in these ani-U1b-nw complexes than that inthe ani-U1a-nw counterparts, indicating a stronger bindingstrength for the extra electron in these ani-U1b-nw complexes.For example, the VDE of ani-U1b-3w amounts to 14.4 kcal/mol, which is about 3.2 and 3.9 kcal/mol higher than those ofani-U1b-2w and ani-U1b-1w, respectively. Experimental re-sults37 also confirmed that the VDE increased along with theattached number of water molecules. The VDE differencesbetween ani-U1a-3w and ani-U1a-2w, and between ani-U1a-3w and ani-U1a-1w are far less than the ones between thecorresponding ani-U1a-nw series. This would be why the formerrequires less activation energy to perform proton transfer thanthe latter. In addition, the larger VDE and dominant chargedistribution are always over the uracil species in these diversehydrates, suggesting that the extra electron should mainly locateat uracil and has a stronger affinity to the uracil species. TheSOMOs of ani-U1a/b-nw tautomeric processes in Figure S2 ofthe Supporting Information also show this point.
The VDE changes along with the attached water moleculesare closely related to the proton transfer, i.e, the tautomericprocess. Generally speaking, the VDE increases along with theincreased number (0-3) of attached water molecules and isalmost linearly proportional to the number of water molecules.37
Figure 5. Activation energy changes along with the increase ofattached water number (n) for these neutral and charged uracil hydratesin each corresponding tautomeric processes.
Catalysis Effects on Proton Transfer of Uracil J. Phys. Chem. B, Vol. 113, No. 34, 2009 11739

The VDE values, however, keep fewer changes between theanionic trihydrates and tetrahydrates. Correspondingly, theactivation energy of proton transfer of trihydrates is theminimum. An exception is ani-U1a-2w, which has a lower VDEand activation energy than those of ani-U1a-1w. Why? It shouldderive from the fact that the dihydrate has a more advantageousspatial configuration than the monohydrate for proton transfer.That is, two water molecules in the dihydrate reduce the distancebetween the proton and water molecule and favor the transfer.In addition, the activation energies of neutral U1a-3w and U1b-3w are somewhat larger than those of their correspondingdihydrated counterparts. However, there is a zooming VDE forthe anionic trihydrates; that is, there is a larger differencebetween ani-U1a/b-3w and each corresponding anionic dihy-drate, which results in the lower activation energy of protontransfer in the anionic trihydrate.
4. Cationic Uracil Hydrates. 4.1. Geometries and ProtonTransfer. To further investigate the positive charge effect onthe tautomeric process, we also calculate positively charged U1,U2, and U3 without and with hydration. Unexpectedly, theoptimized geometries in Figure 4 show that a single watermolecule only attaches to the adjacent hydrogen of U1 ratherthan H-bonds to the adjacent oxygen (see cat-U1a-1w and cat-U1b-1w). Thus, monohydration does not favor the intramo-lecular proton transfer, i.e. tautomeric process. Since there is ahydrogen atom of water pointing to the adjacent carboxyloxygen of cat-U1a-1w, we hope that the second attached watercan link the first water and the carboxyl oxygen and even offera bridge of proton (H10) transfer. Optimization shows that thehydrogen of the second water is further far from the carboxyloxygen (O8) of the uracil relative to that of the first water incat-U1a-1w. This is, dihydration at the (N3)-H10 site cannotassist the proton transfer to isomerize into cat-U3a-2w.
The structure of cat-U1b-2w shows a potential for the protontransfer because O7-O15-O13-N1 forms an eight-memberedring, in which three hydrogen atoms link them. However, thedistance between H15 and O7 is 2.528 Å, far longer than aregular H-bond distance. In addition, NPA analysis reveals thatthe whole electron spin is mainly located on the uracil speciesand the net charge on the base is equal to 1 au. Thus, cat-U1b-2w is better described as cat-U1b++2w. Alpha molecular orbitalcoefficients indicate that there are two strong antibondinginteractions between O7 and H14, and between O13 and H9,respectively. Therefore, in such a way, proton transfer assistedby dihydration also seems hard to achieve.
How about the trihydrated case? Maybe the third water canlink the second water and O8 of cat-U1a-2w or O7 of cat-U1b-2w to form a ring structure and, thus, improve the potential ofproton transfer. According to the idea, we do get two ring-structure trihydrates, cat-U1a-3w and cat-U1b-3w. However,our several attempts to optimize the TS of proton transfer fail,
i.e., the cat-U2-3w and cat-U3-3w cannot be formed. So do,for the two tetrahydrated complexes, cat-U1a-4w and cat-U1b-4w. To investigate the positive charge effect, we calculate theNPA of these hydrates and list the charge in Table 3. Note thatthe charge of the nH2O species in the cat-U1a-3/4w includesthat of H10 because H10 has almost transferred to the waterclusters (see the N3-H distances in Figure 4), whereas that ofH9 in the cat-U1b-3/4w does not due to shorter N1-H distances.However, there is a fast growth tendency for the N1-H distancealong with the number increase of attached water molecules.So these cationic multihydrates can be regarded as cat-U1a/b+[(H2O)n+H+], especially for the cat-U1a-nw series. Mean-while we also optimize the change relationships of PESs alongthe N3-H10 and O8-H (the hydrogen of the last water, whichis pointing to the O8) for cat-U1a-3/4w, as well as along theN1-H9 and O7-H (the hydrogen of the last water, which ispointing to the O7)) for cat-U1b-3/4w, respectively. The resultsare depicted in Figure 6.
It is surprising that the activation energies of H9 atomdissociation (from the N1 site to the linked water) in both cat-U1b-3w (1.5 kcal/mol) and cat-U1b-4w (3.6 kcal/mol) are quitelow, whereas that of H10 dissociation (from the N3 site to linkedwater) in both cat-U1a-3w and cat-U1a-4w become barrierless(see Figure 6I). This indicates that the two hydrogen atoms arevery readily dissociated by the oxygen of adjacent water. Figure4 confirms that the hydrogen in the reactants (from cat-U1b-1w to cat-U1b-4w) runs to the adjacent water and the trendbecomes more and more obvious along with the number increaseof water molecules attached. The conclusion can also be drawnfrom the gradually shortened H9-O13 distance along with theincrease of water molecule number. That is, the H9-O13distances are 1.667, 1.540, 1.481, and 1.465 Å in cat-U1b-1w,cat-U1b-2w, cat-U1b-3w, and cat-U1b-4w, respectively. Similarphenomenon can also be observed from these different cat-U1a-nw complexes. Differently, H10 in these hydrates, especiallyin the triand tetrahydrates, has completely separated from theuracil and transferred to the adjacent water molecule. Thisindicates that it is the proton acceptance processes (O7-H orO8-H) instead of the proton donation process (N1-H orN3-H) that is crucial for the tautomeric process. Z-matrixoptimized O7-H/O8-H distance-PES results in Figure 6-IIreveal that the determining step of proton transfer indeed locatesat the proton acceptance process. That is, all these PESs of cat-U1a-3w, cat-U1a-4w, cat-U1b-3w, and cat-U1b-4w increasegradually as the proton of water transfers to the O8/O7 of theuracil, which results in the proton transfer being unavailable.In the proton transfer process (from the nearest water moleculeto the O8 site of cat-U1a-3/4w or to the O7 site of cat-U1b-3/4w), there is no local energy minimum found and the energiesin these PESs almost keep increasing as the proton transfers(see Figure 6-II), which further confirms the impossibility of
TABLE 3: NPA Charge Distribution (au), VDE, and AEA (kcal/mol) of Charged Hydrates (n ) 1-4)
charge charge
hydrate U nH2O VDE AEA hydrate U nH2O
ani-U1a-1w -0.95 -0.05 25.7, 22.6a 8.6, 8.1a cat-U1a-1w 0.93 0.07ani-U1a-2w -0.94 -0.06 24.1, 20.5a 7.4, 6.9a cat-U1a-2w 0.89 0.11ani-U1a-3w -0.92 -0.08 34.2 7.7 cat-U1a-3w 0.07 0.93ani-U1a-4w -0.91 -0.09 36.1 8.4 cat-U1a-4w 0.06 0.94ani-U1b-1w -0.94 -0.06 28.3, 25.8a 12.2, 11.5a cat-U1b-1w 0.95 0.05ani-U1b-2w -0.92 -0.08 33.6, 29.1a 12.0, 11.3a cat-U1b-2w 0.90 0.10ani-U1b-3w -0.91 -0.09 38.7 15.2 cat-U1b-3w 0.89 0.12ani-U1b-4w -0.92 -0.08 37.8 13.6 cat-U1b-4w 0.88 0.12
a DZP++ basis set used in ref 48. All computed values are zero-point corrected unless otherwise noted.
11740 J. Phys. Chem. B, Vol. 113, No. 34, 2009 Li and Ai

the transfer of a proton. This further confirms that the productscat-U2-3/4w and cat-U3-3/4w are hard to produce due to theirhigh activation energies.
Conclusions
To investigate the catalysis effects of water molecules, charge,or their union on the intramolecular proton transfer of uracil,24 uracil hydrates have been discussed. Series of significantTSs in the related proton-transfer processes have also beenstudied. On the basis of the results obtained from our calcula-tions, the following conclusions can be drawn.
The optimal water molecule number needed for favoring theproton transfer is determined. Initially, intramolecular protontransfer of the most stable uracil (U1) has a high energy barrier.That is, it would need much more activation energy to completethe tautomeric processes of U1 f U2 and U1 f U3. But theenergies will decrease considerably when water molecules arebound to the U1. Comparisons can get the following order ofactivation energy (kcal/mol) of proton transfer in differenttautomeric processes: U1b-2w f U3-2w (16.3) < U1a-2w fU2-2w (16.7) < U1b-3w f U3-3w (17.6) < U1a-1w fU2-1w (17.9) < U1b-1wfU3-1w (18.9) < U1a-3wfU2-3w(20.1) < U1b-4wf U3-4w (22.0) < U1a-4wfU2-4w (22.9)< U1a-5wf U2-5w (23.2). The result indicates that two watermolecules are optimal for catalyzing the intramolecular protontransfer of the neutral U1. Moreover, the U1b-nw f U3-nwprocess seems more readily available than the U1a-nw f U2-nw one due to the larger dipole moment of U3. Proton transferoccurred at two different sites, N1 and N3, of U1, in fact,corresponding to the tautomeric processes of U1a-nw f U2-nw and U1b-nw f U3-nw, respectively. For example, if H9transfers from N1 to the O7 site, then U1 becomes U3.
An extra electron attachment to the uracil would dramaticallydecrease the activation energy of the above tautomeric processes.The order of activation energies (kcal/mol) in these differentanionic hydrated systems is as follows: ani-U1b-3w (9.0) ≈ ani-U1b-4w (9.2) < ani-U1b-2w (10.4) < ani-U1a-3w (12.3) < ani-U1a-4w (13.2) ≈ ani-U1b-1w (13.9) < ani-U1a-2w (14.9) <ani-U1a-1w (18.1). This indicates that the concerted effect ofelectron and hydration is pivotal to the proton transfer. Incontrast with the case of the neutral hydrates, the optimal numberof water molecules to assist proton transfer in these anioniccomplexes has become three and the activation energies ofproton transfer in these ani-U1b-nw complexes seem lower thanthose in the corresponding ani-U1a-nw counterparts. Moreover,the electron attachment also influences greatly the structures of
the hydrates. For example, attachment of an extra electron tothe uracil-water complex results in a significant decrease inthe O · · ·H · · ·O · · ·H bond lengths (typical bond length amountsto 1.540-1.680 Å, about 0.200 Å shorter than those in theneutral counterpart) and a great increase in the NH · · ·OH atomicdistances.
Comparisons reveal that hydration is more efficacious toreduce the activation energy of proton transfer than the electronattachment does, whereas electron attachment can furtherdecrease the activation energy somewhat on the basis ofhydration. The concerted effect is more obvious for thosesystems with larger dipole moments, such as U3 in the presentstudy. Due to the electron mainly locating at the uracil species,the number increase of hydrated water molecules can strengthenthe effect, which is confirmed by the increased VDE. Cor-respondingly, the VDE is also closely related to the activationenergy of proton transfer. That is, both hydration and electronattachment favor the tautomeric processes from the normal U1to the rare U2 or U3, i.e., increase the rate of mutations.2
Addition of a positive charge will hinder the proton transfer.NPA and the PES changes along with the H-bond distance revealthat in these cationic hydrates spin is mainly located on uracilspecies, whereas the positive charge as well as the proton willtransfer to the water cluster. Moreover, the trend becomes moreobvious along with the increasing number of water molecules.Total cationic hydrates can approximately be regarded as cat-U1+ [(H2O)n+H+] structures. Calculations reveal that protontransfer from the nitrogen site of the uracil hydrates to its watercluster is effortless. The key question, which will determinethe tautomeric process, is the last step. That is, it is difficultand almost impossible for the proton transfer of a water clusterto the uracil. As a result, we cannot observe the proton transferin these cationic U1 hydrates. In a word, hydration, electronattachment, or their concerted effect, but the electron ionization,would induce the mutation of the most stable U1 to its tworare U2 and U3 isomers.
Acknowledgment. This work is supported by NS-FC(20573047) and NSF(Y2008B56) of Shandong Province andFoundations for doctoral start-up by Jinan University (B0418).
Supporting Information Available: Ani-U1a/b-nw tauto-meric processes and relative energies (Figure S1) and SOMOsof ani-U1a/b-nw tautomeric processes (Figure S2). This infor-mation is available free of charge via the Internet at http://pubs.acs.org.
Figure 6. PES changes along with (I) N3-H10 (cat-U1a-3/4w)/N1-H9 (cat-U1b-3/4w) and (II) H-O8 (cat-U1a-3/4w)/H-O7 (cat-U1b-3/4w),calculated at the B3LYP/ 6-31+G(d) level.
Catalysis Effects on Proton Transfer of Uracil J. Phys. Chem. B, Vol. 113, No. 34, 2009 11741

References and Notes
(1) Hanus, M.; Ryjacek, F.; Kabelac, M.; Kubar, T. J. Am. Chem. Soc.2003, 125, 7678.
(2) Florian, J.; Leszczynski, J. J. Am. Chem. Soc. 1996, 118, 3010.(3) Watson, J. D.; Crick, F. H. C. Nature 1953, 171, 737.(4) Barrios, R.; Skurski, P.; Simons, J. J. Phys. Chem. B 2002, 106,
7991.(5) Li, X.; Sevilla, M. D.; Sanche, L. J. Phys. Chem. B 2004, 108,
5472.(6) Hobza, P.; Sponer, J. Chem. ReV. 1999, 99, 3247.(7) Fogarosi, G. J. Phys. Chem. A 2002, 106, 1381.(8) van Mourik, T.; Benoit, D. M.; Price, S. L.; Clary, D. C. Phys.
Chem. Chem. Phys. 2000, 2, 1281.(9) Clary, D. C.; Benoit, D. M.; van Mourik, T. Acc. Chem. Res. 2000,
33, 441.(10) Kobayashi, R. J. Phys. Chem. A 1998, 102, 10813.(11) Sambrano, J. R.; Souza, A. R.; Queralt, J. J.; Andres, J. Chem.
Phys. Lett. 2000, 317, 437.(12) Kryachko, E. S.; Nguyen, M. T.; Zeegers-Huyskens, T. J. Phys.
Chem. A 2001, 105, 1288.(13) Kryachko, E. S.; Nguyen, M. T.; Zeegers-Huyskens, T. J. Phys.
Chem. A 2001, 105, 1934.(14) Chandra, A. K.; Nguyen, M. T.; Zeegers-Huyskens, T. J. Mol.
Struct. 2000, 519, 1.(15) Carles, S.; Lecomte, F.; Schermann, J. P.; Desfrancois, C. J. Phys.
Chem. A 2000, 104, 10662.(16) Gorb, L.; Leszczynski, J. Int. J. Quantum Chem. 1998, 70, 855.(17) Luque, F. J.; Sponer, J.; Hobza, P. Phys. Chem. Chem. Phys. 2002,
4, 4192.(18) Bertran, J.; Oliva, A.; et al. J. Am. Chem. Soc. 1998, 120, 8159.(19) Hu, X.; Li, H.; Liang, W.; Han, S. Chem. Phys. Lett. 2006, 417,
320.(20) Schllhorn, H.; Thewalt, U.; Lippen, B. J. Am. Chem. Soc. 1989,
111, 7213.(21) Gutle, C.; Salpin, J.-Y.; Cartailler, T.; et al. J. Phys. Chem. A 2006,
110, 41.(22) Roehrig, G. H.; Oyler, N. A.; Adamowicz, L. Chem. Phys. Lett.
1994, 225, 265.(23) Colson, A. O.; Sevila, M. D. J. Phys. Chem. 1996, 100, 4420.(24) Samijlenko, S. P.; Krechkivska, O. M. J. Mol. Struct. 2004, 708,
97.(25) Fogarasi, G. J. Phys. Chem. A 2002, 106, 1381.(26) Mazurkiewicz, K.; Bachorz, R. A.; Gutowski, M.; Rak, J. J. Phys.
Chem. B 2006, 110, 24696.(27) Nilles, J. M.; Bowen, K. H.; Phys, J. Chem. B 2003, 107, 7889.(28) Bao, X.; Liang, G.; Wang, N.; Gu, J. J. Phys. Chem. A 2007, 111,
666.
(29) Gu, J.; Leszczynski, J. J. Phys. Chem. A 1999, 103, 577.(30) Gorb, L.; Leszczynski, J. J. Am. Chem. Soc. 1998, 120, 5024.(31) Antonczak, S.; Ruiz-Lopez, M. F.; Rivail, J. L. J. Am. Chem. Soc.
1994, 116, 3912.(32) Morpugo, S.; Bossa, M.; Morpugo, G. O. Chem. Phys. Lett. 1997,
280, 233.(33) Hobza, P.; Sponer, J. Chem. ReV. 1999, 99, 3247.(34) Shukla, M. K.; Leszczynski, J. J. Phys. Chem. A 2005, 109, 7775.(35) Li, X.; Cai, Z.; Sevilla, M.; Sevilla., D. J. Phys. Chem. B 2001,
105, 10115.(36) Hendricks, J. H.; Lyapustina, S. A.; de Clercq, H. L.; Bowen, K. H.
J. Chem. Phys. 1998, 108, 8.(37) Schiedt, J.; Weinkauf, R.; Neumark, D.; Schlag, E. Chem. Phys.
1998, 239, 511.(38) Hu, X.; Li, H.; Liang, W.; Han, S. J. Phys. Chem. B 2004, 108,
12999.(39) Chandra, A. K.; Uchimaru, T.; Zeegers-Huyskens, T. J. Mol. Struct.
2002, 605, 213.(40) Di Laudo, M.; Whittleton, S. R.; Wetmore, S. D. J. Phys. Chem. A
2003, 107, 10406.(41) Malick, D. K.; Petersson, G. A. J. Chem. Phys. 1998, 108, 5704.(42) Peiro-Garcia, J.; Nebot-Gil, I. Chem. Phys. Chem. 2003, 4, 843.(43) Nguyen, M. T.; Chandra, A. K.; Zeegers-Huyskens, T. J. Chem.
Soc. 1998, 94, 1277.(44) Weinhold, F.; Carpenter, J. E. Plenum: New York, 1988.(45) Frisch, M. J., et al. Gaussian 03, revision C.02; Gaussian, Inc.:
Pittsburgh, PA, 2003.(46) Gu, J.; Leszczynski, J. J. Phys. Chem. A 1999, 103, 2744.(47) Orozco, M.; Hernandez, B.; Luque, F. J. J. Phys. Chem. B 1998,
102, 5228.(48) Bao, X.; Li, G.; Wang, N.; Gu, J. J. Phys. Chem. B 2006, 110,
5865.(49) Rejnek, J.; Hanus, M.; Kabela, M.; Ryjaek, F.; Hobza, P. Phys.
Chem. Chem. Phys. 2005, 7, 2006.(50) Wesolowski, S. S.; Leininger, M. L.; Pentchev, P. N.; Schaefer,
H. F. J. Am. Chem. Soc. 2001, 123, 4023.(51) Wetmore, S. D.; Boyd, R. J.; Eriksson, L. A. Chem. Phys. Lett.
2000, 322, 129.(52) Desfrancuois, C.; Periquet, V.; Bouteiller, Y.; Schermann, J. P. J.
Phys. Chem. A 1998, 102, 1274.(53) Russo, N.; Toscano, M.; Grand, A. J. J. Comput. Chem. 2000, 21,
1243.(54) Haranczyk, M.; Gutowski, M. J. Chem. Soc. 2005, 127, 699.(55) Dabkowska, I.; Gutowski, M.; Rak, J. J. Chem. Soc. 2005, 127,
2238.(56) Li, P.; Bu, Y. J. Phys. Chem. B 2004, 108, 18088.
JP9031833
11742 J. Phys. Chem. B, Vol. 113, No. 34, 2009 Li and Ai