carcinoma papilÍfero familiar de ... - medicina.ufrj.br · estudo de corte transversal, no qual...
TRANSCRIPT

CARCINOMA PAPILÍFERO FAMILIAR DE TIREÓIDE: AVALIAÇÃO
DE PREVALÊNCIA E ANORMALIDADES FUNCIONAIS, AUTO-
IMUNES E MORFOLÓGICAS DA TIREÓIDE
Mestranda: Elaine Maria dos Santos Gomes
Dissertação de Mestrado apresentada ao
Programa de Pós-graduação em
Endocrinologia, da Universidade Federal do
Rio de Janeiro, como parte dos requisitos
necessários à obtenção do título de Mestre em
Endocrinologia.
Orientador: Prof. Alexandru Buescu
Março/2008

2
CARCINOMA PAPILÍFERO FAMILIAR DE TIREÓIDE: AVALIAÇÃO DA
PREVALÊNCIA E ANORMALIDADES FUNCIONAIS, AUTO-IMUNES E
MORFOLÓGICAS DA TIREÓIDE
Elaine Maria dos Santos Gomes
Orientador: Prof. Alexandru Buescu
Dissertação de Mestrado submetida ao Programa de Pós-graduação em
Endocrinologia, da Universidade Federal do Rio de Janeiro – UFRJ, como parte
dos requisitos necessários à obtenção do título de Mestre em Endocrinologia.
Aprovada por:
_________________________________________
Presidente, Prof. Mario Vaisman
_____________________________________
Profa. Rossana Corbo
_____________________________________
Profa. Flávia Lúcia Conceição
Rio de Janeiro
Março/2008

3
Gomes, Elaine Maria dos Santos Carcinoma papilífero familiar de tireóide: avaliação de prevalência e anormalidades funcionais, auto-imunes e morfológicas da tireóide / Elaine Maria dos Santos Gomes. – Rio de Janeiro: UFRJ / Faculdade de Medicina, 2008. xiv, 75 f. : il. ; 31 cm. Orientador: Alexandru Buescu Dissertação (mestrado) – UFRJ/Faculdade de Medicina, Programa de Pós-Graduação em Medicina, Endocrinologia, 2008. Referências bibliográficas: f. 45-57 1. Neoplasias da glândula tireóide - epidemiologia. 2. Nódulo da glândula tireóide - epidemiologia. 3. Glândula tireóide - imunologia. 4. Glândula tireóide – ultra-sonografia. 5. Glândula tireóide - citologia. 6. Humano. 7. Prevalência. 8. Endocrinologia – Tese. I. Buescu, Alexandru. II. Universidade Federal do Rio de Janeiro, Faculdade de Medicina, Programa de Pós-Graduação em Medicina, Endocrinologia. III. Título.

4
RESUMO
CARCINOMA PAPILÍFERO FAMILIAR DE TIREÓIDE: AVALIAÇÃO DA
PREVALÊNCIA E ANORMALIDADES FUNCIONAIS, AUTO-IMUNES E
MORFOLÓGICAS DA TIREÓIDE
Elaine Maria dos Santos Gomes
Orientador: Prof. Alexandru Buescu
Resumo da Dissertação de Mestrado submetida ao Programa de Pós-
graduação em Endocrinologia, da Universidade Federal do Rio de Janeiro – UFRJ,
como parte dos requisitos necessários à obtenção do título de Mestre em
Endocrinologia.
O carcinoma não-medular da tireóide é o tipo mais comum de câncer de
tireóide e sua forma familiar tem sido cada vez mais aceita como uma entidade
clínica distinta. Atualmente admite-se uma prevalência de 5% das formas
familiares entre os CNMT. Os objetivos deste estudo foram: avaliar a prevalência
de carcinoma papilífero de tireóide entre os familiares de primeiro grau de
portadores deste tumor; verificar a existência de relação entre a incidência de
carcinoma papilífero familiar de tireóide com disfunção e auto-imunidade
tireoideana e a prevalência de alteração estrutural tireoideana. Realizamos um
estudo de corte transversal, no qual incluímos 143 familiares de 52 pacientes em
acompanhamento ambulatorial por carcinoma papilífero de tireóide, sendo a média
de familiares de 2,75 por caso índice. Foram realizadas dosagens séricas de TSH,

5
T4l e ATPO bem como realizadas ultra-sonografias de tireóide nestes pacientes.
Os casos que apresentaram nódulos maiores que 1 cm e os menores suspeitos
foram encaminhados para PAAF, seguido de cirurgia no caso de citopatológico de
malignidade ou suspeito. No subgrupo de pacientes portadores de CPFT, 75%
eram do sexo feminino e 25% masculino, o valor do TSH foi de 1,2 ± 0,2 mUl/L, do
T4L de 1,3 ± 0,3 ng/dl e o ATPO esteve positivo em 25% dos casos. Dos 18
pacientes que não realizaram ultra-sonografia de tireóide, 8 foram submetidos à
tireoidectomia antes do início do estudo. Os exames foram alterados em 68
(54,4%): 49 (39,2%) tinham doença nodular, dos quais 24 (19,2%) apresentavam
nódulo único e 25 (20%) bócio multinodular. 11 casos foram classificados como
CPFT, 7 eram previamente tireoidectomizados e os outros 4 apresentaram bócio
multinodular. A prevalência de CPFT foi de 7,7% nos indivíduos e 19,2% quando
analisamos por famílias, com um viés de seleção. Nenhuma relação entre a
presença de CPFT com disfunção ou auto-imunidade tireoideana foi estabelecida.
.
Rio de Janeiro
Março/2008

6
FAMILIAL PAPILLARY CARCINOMA OF THE THYROID: ASSESSMENT OF
ITS PREVALENCE AND ASSOCIATED FUNCTIONAL, MORPHOLOGICAL AND
AUTOIMMUNE ABNORMALITIES.
Elaine Maria dos Santos Gomes
Orientador: Prof. Alexandru Buescu
Abstract da Dissertação de Mestrado submetida ao Programa de Pós-
graduação em Endocrinologia, da Universidade Federal do Rio de Janeiro – UFRJ,
como parte dos requisitos necessários à obtenção do título de Mestre em
Endocrinologia.
Papillary carcinoma is the most common type of non-medullary thyroid
cancer and its familial form has been increasingly reported with an estimated
prevalence of about 5%.This study’s objectives were: to evaluate the prevalence of
papillary carcinoma among the first degree relatives of patients with this tumor;
verify the existence of any relationship with thyroid dysfunction or autoimmunity
and the prevalence of thyroid structural abnormalities.A transverse study was
performed with the inclusion of 143 first degree relatives of 52 patients followed for
papillary carcinoma of the thyroid in the outpatient clinic of the HUCFF ( average of
2.75 relatives per index case). Serum TSH, free T4 and TPOAb were determined
and thyroid ultrasonography (US) was performed. Any nodule larger than 1 cm and
all smaller but suspicious nodules were sent to FNAB. Malignant or suspicious
cytologies were indication for surgery. Of the patients harboring the familial form of

7
papillary carcinoma (FPC) 75% were females and 25% males. Average serum
TSH was 1.2±0.2 mU/L, free T4 1.3±0.3 ng/dL and TPOAb were positive in 25%.
Of the 18 patients who were not submitted to US, 8 had undergone
thyroidectomies previously. The remaining exams were abnormal in 68 cases
(54.4%): 49 (39.2%) had nodular disease of which 24 were solitary (19.2%) and 25
multinodular (20%).11 cases were classified as FPC: 7 had been operated
previously and the other 4 had multinodular glands. We found a prevalence of FPC
of 7.7% in the examined relatives and 19.2% in the examined families with an
obvious selection bias.No relationship between thyroid dysfunction or autoimmunity
and FPC could be established. There was a higher prevalence of multinodular
goiters.
Rio de Janeiro
Março/2008

8
Ao meu marido José Mauro, pelo grande companheirismo, incentivo e
compreensão, fundamentais para a conquista deste título de mestrado.

9
AGRADECIMENTOS
Inicialmente, agradeço a toda a minha família por ter conseguido chegar até
aqui. Aos meus pais, Maria Rosa e Paulo Roberto pelo constante incentivo e
confiança durante toda a minha vida. Aos meus irmãos, Roberta e Paulo Gustavo,
pela ajuda e imenso companheirismo, fundamentais para minhas conquistas. A
minha tia Maria Helena, que é minha segunda mãe, cujos ensinamentos ajudam
muito em minha formação. Ao meu marido, José Mauro, pelo seu grande apoio,
paciência e compreensão.
Ao meu orientador, Professor Alexandru Buescu, importante na minha
formação durante a residência médica e no mestrado. Sua atenção e críticas
foram fundamentais na construção deste trabalho, bem como sua compreensão
em relação às dificuldades de horários.
Ao Professor Mario Vaisman, um exemplo de chefe. Sempre nos
incentivando a obter a melhor formação profissional possível e nos mostrando a
importância da pesquisa e da ética médica.
Agradeço, também, a todos os professores do Serviço de Endocrinologia do
HUCFF: Prof Alice Helena Dutra Violante, Prof Flávia Lúcia Conceição, Prof Maria
Lúcia Fleiuss de Farias, Prof Marília Martins Guimarães e Prof Mônica Roberto
Gadelha, por seus ensinamentos e orientações durante todo o período da
residência médica e mestrado.
À Prof. Rossana Corbo, pelo apoio na realização do projeto e pelos
ensinamentos durante o período da residência médica e mestrado.

10
À Dra Heloísa Maria Pereira Freitas, pela sua dedicação e empenho na
realização dos exames de ultra-sonografia e PAAFs, indispensáveis para
realização deste trabalho.
À doutora Patrícia de Fátima dos Santos Teixeira, por sua ajuda
fundamental na análise estatística.
Às Dras. Sabrina, Heloísa, Vanessa e Sheila, do ambulatório de câncer de
tireóide, pela grande ajuda na seleção dos pacientes.
Às secretárias Nádia (do Serviço de Endocrinologia) e Tânia, Denise e
Maria Cristina (do Serviço de Ultra-sonografia). A primeira, pelo constante apoio,
paciência e importância na formação dos residentes e pós-graduandos. As
demais, pela organização e ajuda na marcação dos exames deste projeto, sem o
que, o andamento do trabalho seria muito dificultado.
Aos meus companheiros de residência médica e do Mestrado, em especial
a Dra. Carla Amaral pelo constante incentivo e ajuda na coleta dos dados, e aos
meus amigos que muito contribuíram na minha formação pessoal e profissional.
Elaine Maria dos Santos Gomes

11
LISTA DE SIGLAS E ABREVIATURAS
CNMT – carcinoma não-medular de tireóide
CNMFT – carcinoma não-medular familiar de tireóide
CPT – carcinoma papilífero de tireóide
CPFT – carcinoma papilífero de tireóide
BMN – bócio multinodular
PAAF – punção aspirativa por agulha fina
PAF – polipose adenomatosa familiar
TSH – tireotrofina
T4l – tiroxina livre
US – ultra-sonografia
ATPO – anticorpo anti-peroxidase
OR – odds ratio
HUCFF – Hospital Universitário Clementino Fraga Filho
UFRJ – Universidade Federal do Rio de Janeiro
MHz – mega Hertz
NEM 2A – Neoplasia Endócrina Múltipla tipo 2A
CI – Intervalo de Confiança

12
SUMÁRIO
I. Introdução ........................................................................................................p.1
II. Revisão da Literatura
II.1. Carcinoma não-medular de tireóide
II.1.1. Contexto atual ........................................................................p.3
II.1.2. Características clínicas ..........................................................p.5
II.1.3. Ultra-sonografia e PAAF ......................................................p.11
II.1.4. Prognóstico e tratamento .....................................................p.14
II.1.5. Genética ..............................................................................p.17
II.2. Síndromes familiares associadas com carcinoma não-medular de
tireóide ................................................................................................................p.20
III. Objetivos .......................................................................................................p.24
IV. Casuística e métodos
IV.1. Características do estudo e considerações éticas............................p.25
IV.2. Seleção de indivíduos ......................................................................p.25
IV.3. Exames laboratoriais .......................................................................p.26
IV.4. Ultra-sonografia da tireóide ..............................................................p.27
IV.5. PAAF................................................................................................p.28
IV.6. Realização de cirurgias ...................................................................p.29
IV.7. Anatomia Patológica.........................................................................p.29
V. Análise estatística ........................................................................................p.31
VI. Resultados
VI.1. Características gerais da amostra ...................................................p.32

13
VI.2 – Análise comparativa entre os portadores e os não-portadores de
CPFT ...................................................................................................................p.33
VI.3. Resultados da ultra-sonografia de tireóide.......................................p.34
VI.4 Resultados da cirurgia ......................................................................p.36
VI.5 Resultados da anatomia patológica ..................................................p.36
VI.6. Análise de prevalência .....................................................................p.38
VII. Discussão ....................................................................................................p.39
VIII. Conclusão ..................................................................................................p.44
IX. Referências bibliográficas ..........................................................................p.45
X. Tabelas
Tabela 1 – Comparação de variáveis clínicas e laboratoriais entre os portadores e
não-portadores de CPFT.....................................................................................p.33
Tabela 2 – Comparação de variáveis laboratoriais entre os portadores e não-
portadores de CPFT com indicação de PAAF.....................................................p.34
Tabela 3 – Alterações mais frequentes encontradas nas ultra-sonografias
realizadas em familiares de primeiro grau de portadores de CPT ......................p.35
Tabela 4 – Resultados citopatológicos realizadas em familiares de primeiro grau
de portadores de CPT ........................................................................................p.37
Tabela 5 – Resultados da análise da prevalência CPFT por famílias ................p.38
XI. Anexos
Anexo I – Aprovação pela Comissão de Ética e Pesquisa do HUCFF ..............p.58
Anexo II – Termo de consentimento livre e esclarecido ....................................p.59
Anexo III – Ficha de avaliação clínica - caso índice ..........................................p.62
Anexo IV – Ficha de avaliação clínica - familiar de primeiro grau.....................p.64

14
Anexo V – Perfil clínico e laboratorial dos familiares dos portadores de
CPT......................................................................................................................p.66
Anexo VI – Resultados das ultra-sonografias dos familiares de portadores de
CPT..................................................................................................................…p.70
Anexo VII – Resultados da anatomia patológica dos familiares de portadores de
CPT......................................................................................................................p.73
Anexo VIII – Resultados da pesquisa de carcinoma papilífero por família ........p.74

15
I – INTRODUÇÃO
O câncer de tireóide apesar de raro (cerca de 1% de todos os
cânceres), é a neoplasia maligna endócrina mais frequente1.
Aproximadamente 95% dos casos correspondem a cânceres originados de
células foliculares da tireóide, sendo os 5% restantes oriundos das células
parafoliculares, classificados como carcinomas medulares2. Entre os
carcinomas medulares, sabe-se que 80% dos casos são esporádicos,
enquanto 20 a 25% são familiares1. Desde a identificação do proto-oncogene
RET como gene causador do carcinoma medular hereditário, o conhecimento
acumulado sobre a patogênese do carcinoma medular e suas neoplasias
associadas tem sido significativo3-7.
Denominamos carcinoma não-medular da tireóide os casos oriundos de
células foliculares, bem diferenciados (papilífero, folicular e células de Hürtle)
ou não (anaplásicos). Desde o primeiro relato de caso de carcinoma papilífero
familiar de tireóide (CPFT), publicado em 19558, vários trabalhos vêm
reforçando a existência de uma nova entidade clínica, o carcinoma não-
medular familiar da tireóide (CNMFT)2, 9-11.
Atualmente estima-se que existe uma origem familiar em 3,5 - 6,2% dos
pacientes com CNMT9,12-16, sendo a grande maioria composta do subtipo
histológico papilífero, presente em mais de 90% dos casos 17-19.
Define-se como CNMFT a existência de dois ou mais familiares de
primeiro grau afetados pelo câncer da tireóide, descartando-se os casos de

16
associações com síndromes familiares e a presença de exposição a outros
fatores ambientais, sabidamente predisponentes a esta neoplasia14,18,20,21.
As características mais frequentemente encontradas nos CNMFT são
uma maior precocidade no aparecimento da doença nos casos familiares,
como ocorre na maioria das síndromes familiares11,22-24, a associação com
outras doenças tireoideanas não-malignas 25,26, multifocalidade 14,
envolvimento ganglionar 23 e recorrência 15.
Diferente do que ocorre com os carcinomas medulares, a genética dos
CTNM ainda não está bem compreendida. Até o presente momento vários
estudos estão sendo realizados e alguns genes têm sido envolvidos como
possíveis responsáveis pelo desenvolvimento dos CNMFT. Entre estes se
encontram o TCO, PRN1 e NMTC118. Todavia, nenhum destes genes
responde pela maior parcela dos casos de CNMFT.
Neste trabalho, avaliamos a prevalência de carcinoma papilífero de
tireóide entre familiares de primeiro grau de portadores desta patologia. Além
disso, analisamos a existência de relação entre a incidência de carcinoma
papilífero familiar de tireóide com disfunção e auto-imunidade tireoideana bem
como a prevalência de alteração estrutural da glândula.

17
II – REVISÃO DA LITERATURA
II. 1 – CARCINOMA NÃO-MEDULAR FAMILIAR DE TIREÓIDE
II. 1. 1 – CONTEXTO ATUAL
O câncer de tireóide é considerado o oitavo mais comum nos Estados
Unidos da América, com uma estimativa de 30.000 novos diagnósticos em 2006
com 1500 mortes associadas27. Entre estes, aproximadamente 95% dos casos
correspondem a cânceres originados de células foliculares da tireóide, sendo os
5% restantes oriundos das células parafoliculares, classificados como carcinomas
medulares2. Existem 4 subtipos histológicos de carcinoma não-medular de
tireóide(CNMT): papilífero (85%), folicular (11%), células de Hürtle (3%) e
anaplásico (1%)17,28. Estes tumores são em sua grande maioria esporádicos.
Em 1955 Robinson e Orr8 descreveram o primeiro caso de carcinoma
papilífero familiar de tireóide (CPFT), em gêmeos idênticos de 24 anos de idade.
Desde então diversos trabalhos têm sido publicados reforçando a existência de
uma nova entidade clínica, o carcinoma não-medular familiar de tireóide
(CNMFT)2,9-11. Atualmente estima-se que existe uma origem familiar em 3,5 - 6,2%
dos pacientes com CNMT9,12-16. Entre estes, a grande maioria é composta do
subtipo histológico papilífero, presente em mais de 90% dos casos17-19. A origem
familiar também pode ocorrer em carcinomas de células de Hürtle, mas é raro em
carcinoma folicular14.

18
Define-se como CNMFT a existência de dois ou mais familiares de primeiro
grau afetados pelo câncer da tireóide (papilífero, folicular, células de Hurtle ou
anaplásico). A associação a outras síndromes familiares, como complexo de
Carney, polipose adenomatosa familiar, síndrome de Gardner, doença de Werner
e doença de Cowden, bem como a presença de exposição a outros fatores
ambientais (por exemplo, deficiência ou excesso de iodo) sabidamente
predisponentes a esta neoplasia, invalidam o diagnóstico14,18,20,21. Pelo fato de não
sabermos ainda se o CNMFT ocorre somente por alteração genética ou pela
combinação entre esta com o meio ambiente, a exposição a doses baixas de
radiação não deve excluir o diagnóstico de CNMFT9,14,17,.
Ao longo dos anos as evidências epidemiológicas foram sedimentando a
existência de uma predisposição familiar do CNMT, como descrito anteriormente.
Vários estudos de diferentes metodologias evidenciaram um aumento do risco no
desenvolvimento de CNMT em parentes de primeiro grau de portadores desta
patologia, podendo chegar até a cinco vezes9,11,12,15,22,29.
Pal e cols avaliaram 339 pacientes com CNMT e constataram que 17 (5%)
apresentavam ao menos um familiar de primeiro grau afetado por câncer de
tireóide, sendo o subtipo histológico papilífero encontrado em todos estes casos 9.
Observaram ainda que o risco relativo de um familiar de primeiro grau desenvolver
câncer de tireóide foi de 10,3% (95% CI, 2,2 - 47,6). Estes achados são
consistentes com trabalhos prévios populacionais de carcinoma papilífero de
tireóide nos quais os casos familiares têm uma prevalência de 6% 2,30.
Em 2005, Hemminki e cols22 realizaram um grande estudo populacional no
qual identificaram maior risco de carcinoma papilífero de tireóide familiar quando

19
pais ou irmãos apresentavam diagnóstico de câncer de tireóide: de 3,21 e 6,24
respectivamente. Percebeu-se também uma aparente predileção por gênero,
particularmente entre irmãs, cujo risco foi de 11,19.
Estima-se que se dois membros de uma família são afetados pelo CNMFT,
existe uma chance de 53% de que este tumor seja de origem familiar e 47% de
chance de ser esporádico. Entretanto, se três ou mais familiares forem afetados, a
chance da doença ser familiar é de 99,9%17,31,32. Isto ocorre devido à alta
prevalência de câncer de tireóide na população em geral.
II. 1. 2 – CARACTERÍSTICAS CLÍNICAS
A distribuição por sexo nos CNMFT seguiu a mesma orientação que
os casos esporádicos, com maior incidência no sexo feminino15,33. Entretanto, em
relação à idade, ainda não existe um consenso. A maioria dos autores percebeu
em seus estudos uma média de idade dos pacientes com CNMFT semelhante aos
casos esporádicos9,14,15,22,32,34. Todavia, outros descreveram uma maior
precocidade no aparecimento da doença nos casos familiares, como ocorre na
maioria das síndromes familiares18,22-24. Uma meta-análise evidenciou que
pacientes com CNMFT apresentam a doença uma década antes, quando
comparados com os casos esporádicos36,37, com pico de incidência na quarta
década 2. Hemminki e cols22 acharam que a média de idade do aparecimento foi
dois anos antes para câncer papilífero em descendentes e cinco anos antes para
irmãos afetados. Já Musholt e cols 23, em sua série de casos de CNMFT, não

20
encontrou diferença entre as idades entre os membros afetados por ocasião do
diagnóstico .
Estudos têm demonstrado a presença de doenças tireoideanas não
malignas como adenoma folicular, tireoidite de Hasimoto, bócio multinodular
(BMN), hiper e hipotireoidismo com bastante freqüência (57%) em familiares de
portadores do subtipo papilífero do CNMFT9,10.
Em 2002, Uchino e cols15 encontraram uma maior incidência de múltiplos
nódulos benignos nos familiares com CNMFT que nos casos esporádicos (41,5%
vs 29,8%; P<0,001). Não houve diferença entre os casos familiares e esporádicos
em relação aos nódulos únicos benignos e tireoidite crônica.
Em 2000, Musholt e cols23 realizaram uma meta-análise da literatura sobre
carcinoma papilífero familiar de tireóide. Neste estudo, eles analisaram dentre
outras coisas o número de indivíduos afetados em famílias com maior ocorrência
de carcinoma papilífero de tireóide (CPT) ou BMN (ou ambos). Encontraram um
total de 160 famílias com um ou mais membros portadores de CPT (com ou sem
BMN) descritos na literatura de 1958 a 1999. Uma alta incidência de BMN,
usualmente aparecendo em idades menores, e doenças tireoideanas como
hipo/hipertireoidismo, tireoidite imunomediada e adenomas com aparecimento na
adolescência foram achados comuns nos familiares de CPFT. Baseados nestas
conclusões desenvolveram critérios para identificar famílias de risco para
suscetibilidade hereditária para CPFT. Critérios de exclusão (exposição à radiação
previa, associação com outras malignidades, mutações oncogênicas somáticas)
foram criados para distinguir as famílias com CPFT dos CPT presentes em
famílias com exposição a agentes ambientais e outras síndromes neoplásicas.

21
Critérios primários e secundários foram escolhidos baseados nas conclusões da
meta-análise. Primários: CPT em dois ou mais familiares de primeiro grau com
CPT e BMN em pelo menos 3 familiares de primeiro ou segundo grau de
portadores de CPT. Secundários: diagnóstico em pacientes menores que 33 anos,
CPT multifocal ou bilateral, crescimento tumoral ultrapassando os limites da
glândula (T4), metástases para linfonodos (N1) ou à distância (M1) e acumulo de
doenças tireoideanas em adolescentes. Foi considerada predisposição hereditária
ao CPFT quando ambos os critérios primários estão presentes ou quando há um
critério primário mais três secundários.
Pacientes com CNMFT também podem apresentar um maior risco para o
desenvolvimento de câncer de mama, rim, colon, bexiga, assim como melanomas
e linfomas11,38.
Os CNMFT apresentam diferentes características em relação aos casos
esporádicos conforme já relatado em algumas séries, em especial seu
aparecimento mais precoce, alta incidência de nódulos benignos, multifocalidade,
envolvimento ganglionar e recorrência.
Dentre os CNMFT o subtipo mais prevalente é o papilífero, correspondendo
a mais de 90% dos casos na maioria das séries publicadas17-19.
Grossman e cols14 fizeram uma análise retrospectiva de 14 pacientes com
CNMFT entre 1980-1994 e encontraram que o tamanho dos tumores era
ligeiramente maior. Neste mesmo estudo, o CNMFT foi multifocal em 93% e
bilateral em 43% dos casos. Contrastando estes resultados, os carcinomas
papilíferos esporádicos foram multifocais em 20-32% e bilaterais em 19% dos
casos39,40.

22
Em 1997, Loh2 realizou uma revisão da literatura e identificou 178 pacientes
com CNMFT. Nesta revisão, a média de idade para o aparecimento de CNMFT foi
de 10 anos antes quando comparados aos casos esporádicos e a taxa de
multifocalidade foi de 49%.
O maior estudo sobre características gerais dos pacientes com CNMFT foi
realizado em 2002 por Uchino e cols15 no qual foram comparados casos de
CNMFT com carcinomas papilíferos e foliculares esporádicos para avaliar os
aspectos característicos e histológicos desta patologia familiar. Foram
comparados 258 pacientes de 154 famílias de CNMFT com um grupo de
pacientes com tumores foliculares ou papilíferos esporádicos (grupo controle). Os
resultados obtidos foram: 4% de incidência entre a população de pacientes com
CNMT; a disseminação intraglandular do tumor foi mais freqüente nos casos
familiares que nos esporádicos (40,7% vs 28,5% ;p <0,0001); múltiplos nódulos
benignos foram mais frequentes nos familiares que nos esporádicos (41,5% vs
29,8%%; p<0,001), assim como a recorrência tumoral (16,3% vs 9,6%; p=0,005);
A recorrência locorregional foi maior nos casos familiares que nos esporádicos,
sem diferença em relação às metástases à distância, e o período de sobrevida
livre de doença foi menor nos casos familiares (p=0,0041pelo teste de Wilcoxon).
Não houve diferença estatística entre os dois grupos em relação a gênero, idade,
tamanho do tumor, aderência ou invasão de tecidos vizinhos, metástases
macroscópicas observadas na cirurgia, histologia, presença de nódulos únicos
benignos, presença de tiroidite crônica, metástases ganglionares microscópicas,
taxa de metástases ganglionares, tempo de seguimento e número de mortes
relacionadas à doença.

23
Em relação à apresentação do CPFT, Musholt e cols23 concluíram, que as
principais características do CPFT eram: início precoce, comportamento mais
agressivo que os CPT esporádicos, multifocalidade e metástases ganglionares e a
distância, mesmo em microcarcinomas, além da alta incidência de BMN
previamente mencionado.
Vários grupos já estudaram o comportamento do CNMFT. A maioria
considera tratar-se de uma doença de maior agressividade. Um dos “defensores”
desta característica do CNMFT é Grossman14 que em 1995 junto com seus
colaboradores reviu 14 pacientes com CNMFT e encontrou 57% de metástases
ganglionares ao diagnóstico e invasão extracapsular em 57% dos pacientes. Ainda
neste trabalho, a taxa de recorrência foi de 50%. Séries de CNMT esporádicos
demonstram uma taxa menor destas alterações, sendo 38-43% de metástases
ganglionares e 5-14% de invasão extracapsular39,41. Quanto à recorrência, esta
gira em torno de 5-15% dos casos esporádicos14.
Alsanea e cols24 realizaram um estudo multicêntrico de caso-controle, no
qual avaliaram 48 pacientes com CNMFT. Neste estudo, a taxa de recorrência foi
de 44% para os casos familiares e de apenas 17% no grupo controle. Os
pacientes com CNMFT também tiveram uma sobrevida livre de doença menor que
quando comparados com o grupo controle.
Lupoli e cols35 examinaram 119 pacientes com microcarcinoma papilífero e
compararam estes com um subgrupo com características de CNMFT. Apenas 7
dos 119 casos eram familiares, mas as diferenças encontradas foram importantes.
Nos casos familiares, a despeito da média do diâmetro do tumor ser de 5,9mm,
eles foram multifocais em 71% (vs 19%), bilaterais em 43% (vs 8%),

24
apresentavam metástases ganglionares em 57% (vs 28%) e invasão vascular em
43% (vs 5%). A taxa de recorrência também foi significativamente maior, sendo
43% contra 5% dos casos esporádicos. Este estudo mostra a natureza agressiva
dos CNMFT, presente mesmo nos menores tamanhos, ao contrário dos casos
esporádicos, nos quais os microcarcinomas tipicamente apresentam um
prognóstico favorável39,40.
Apesar dos trabalhos acima referidos indicarem um comportamento mais
agressivo do CNMFT, há autores que não confirmam esta teoria como Loh:2 em
sua meta-análise realizada em 1997. Em seu trabalho revisou 15 séries
identificando 178 pacientes com CNMFT. Devido à variação metodológica entre os
estudos, sua análise foi apenas descritiva, não tendo sido realizada estatística.
Comparando os resultados com as grandes séries de carcinoma papilífero,
concluiu que os casos familiares não eram mais agressivos. Entretanto, ao ser
revisto seu estudo, apesar de não haver diferenças claras no número de pacientes
com metástases ganglionares ou a distância e mortes, existe quase que o dobro
da incidência de invasão local e recorrência nos casos familiares. Este dado é
relevante, pois aproximadamente um terço dos pacientes (mesmo os de baixo
risco) com carcinoma papilífero familiar de tireóide que desenvolvem recorrência
morrem da própria doença42.
Maxwell e cols43 num estudo pequeno de caso-controle não demonstraram
nenhuma diferença na agressividade entre os casos de CNMT familiares e
esporádicos. Vinte e quatro pacientes foram identificados retrospectivamente e
comparados com grupo controle. Os autores não encontraram aumento de
incidência de multifocalidade ou recorrência, sendo a taxa desta última variável de

25
4,5%, que pode ser atribuída pelo curto tempo de seguimento e pelo uso de
exame físico como única ferramenta para avaliar recorrência.
Sippel e cols44 em seu estudo publicado em 2007 acreditam que há
evidências suficientes para apoiar a idéia de que os CNMFT são mais agressivos.
Há, porém, uma grande dificuldade em provar esta afirmação pela inabilidade de
separar completamente os casos esporádicos dos familiares. Em famílias com
apenas dois membros afetados por CNMFT, apesar de preencherem critérios para
a definição, até 69% dos casos podem corresponder a esporádicos32. Entre os
casos com três ou mais pacientes afetados, onde um componente genético é
quase certo, a agressividade do tumor é mais facilmente demonstrada15,21,24.
II. 1. 3 – ULTRA-SONOGRAFIA E PAAF
O uso da ultra-sonografia da região cervical na triagem de pacientes
assintomáticos tem aumentado nos últimos tempos. A incidência de detecção
ultrassonográfica de nódulos tireoideanos em adultos saudáveis já foi
documentada variando de 19 a 67%45 .
Um grande estudo foi realizado por Uchino e cols46 em 2004 para avaliar a
indicação de ultra-sonografia de tireóide em familiares de CNMFT como método
de triagem. Neste experimento eles selecionaram 149 familiares assintomáticos de
53 famílias portadoras de CNMFT para realizarem uma triagem. Todos os
pacientes foram submetidos à ultra-sonografia de tireóide e quando um nódulo
suspeito era identificado, realizava-se PAAF e análise citológica do nódulo. Se a
citologia fosse compatível com suspeita de malignidade, indicava-se

26
tireoidectomia. Os resultados foram: pelo menos um nódulo foi encontrado em 77
dos 149 pacientes (51,7%); cirurgia foi realizada em 18 pacientes dos quais 15
tinham câncer, todos do subtipo papilífero sendo que em um paciente havia além
do CPT um carcinoma folicular associado; a incidência foi maior em mulheres
(p<0,05); CPT múltiplo foi encontrado em 7 pacientes (47%) e solitário em 8 além
de ter sido associado a bócio adenomatoso em 8 pacientes (53%) e a doença de
Graves em 1; metástases intraglandulares foram encontradas em 7 pacientes (
47%) e metástases ganglionares microscópicas em 6(43%). Concluíram que a
triagem ulttrassonográfica de membros de famílias com CNMFT pode habilitar o
diagnóstico de câncer de tireóide assintomático e ela é recomendada pela alta
incidência de metástases intratireoideanas e ganglionares.
Sippel e cols44, recomendam que apenas os familiares de primeiro grau de
CNMFT devam ser submetidos a triagem através de história e exame físico. Se
ao exame físico houver dúvida ou encontrar alteração deve-se realizar ultra-
sonografia de tireóide. Além disto, multifocalidade e BMN são achados frequentes
em CNMFT, e se estes forem identificados em um parente afetado, deve-se
recomendar a realização de triagem familiar, incluindo ultra-sonografia da região
cervical, mesmo se não houver outros membros afetados46.
A triagem para indivíduos com dois ou mais parentes com diagnóstico de
CNMFT deve incluir sempre ultra-sonografia de região cervical. Se a história
familiar, exame físico e ultra-sonografia forem negativos, recomenda-se um
seguimento anual regular21.
A avaliação citológica de material coletado de PAAF é um procedimento
pré-operatório de rotina na propedêutica de nódulos tireoideanos suspeitos na

27
maioria dos centros médicos que dispõem de patologistas experientes47. A PAAF
é considerada o melhor custo-benefício em exame atualmente disponível para
avaliação dos nódulos tireoideanos. Geralmente é um procedimento seguro e
acurado para a determinação de um nódulo tireoideano como benigno, maligno ou
suspeito48,49. A PAAF não é tão preditiva em pacientes com nódulos tireoideanos
que foram expostos a baixas doses de radiação ionizante devido à presença tanto
de nódulos benignos quanto de malignos. Os tumores tireoideanos podem ser
mutifocais e os nódulos dominantes podem ser benignos em pacientes com
tumores malignos de tireóide situados em outros locais da própria glândula.
Pessoas de famílias com CNMFT têm geralmente tanto nódulos benignos quanto
malignos na mesma glândula50,51.
Vriens e cols34 em 1999 publicaram um estudo no qual 27 pacientes de 24
famílias com CNMFT tiveram suas tireóides analisadas histologicamente após
análise citológica pré-operatória. Os resultados obtidos foram: 17 dos 27 (63%)
apresentavam múltiplos nódulos; 25 dos 27 tinham câncer (92,6%); câncer de
tireóide foi diagnosticado pela PAAF em 22 dos 25 pacientes (88%) com história
familiar e em 26 dos 27 (96,3%) do grupo controle, gerando um falso negativo de
12% nos casos de CNMFT e de 3,7% grupo controle. Nestes pacientes o tumor
estava situado em outros nódulos pequenos ou foi multifocal e não no nódulo
index ou mais suspeito. Conclui-se que a confiabilidade da PAAF em pacientes
com CNMFT parece ser menor que nos demais pacientes pela alta incidência de
câncer multifocal e coexistência com nódulos benignos.

28
II. 1. 4 – PROGNÓSTICO E SEGUIMENTO
O CNMFT tem algumas características que naturalmente são associadas a
um pior prognóstico, em especial a agressividade, maiores riscos de metástases
para linfonodos e taxas de recorrência.
Alguns estudos têm tentado avaliar o impacto do CNMFT no tempo de
sobrevida livre de doença14,15,21. Estes estudos são limitados pelo fato do taxa
geral de mortalidade por CNMT persistir baixa, necessitando de uma grande
população e longo tempo de seguimento para mostrar alguma diferença entre os
subgrupos familiares e esporádicos.
Recentemente Triponez e cols21 publicaram uma análise de expectativa de
vida e sobrevida onde comparam 139 pacientes portadores CNMFT com 757
membros da família não afetados, por um tempo de seguimento de 9,4 ± 11,7
anos. Dez pacientes morreram de câncer de tireóide durante o seguimento. A
expectativa de vida foi semelhante nos dois grupos. A taxa de sobrevida foi
significativamente menor para pacientes com 3 ou mais membros afetados e para
pacientes com diagnóstico anterior ao reconhecimento da etiologia familiar (caso
índice). Adicionalmente, o carcinoma anaplásico pareceu ser mais comum e, em
todas as situações, foi associado com declínio na sobrevida. Este estudo foi muito
importante porque sugere que um tratamento precoce aumenta a sobrevida.
Uchino e cols15 também avaliaram a sobrevida em uma grande série de
casos e encontraram que o período de sobrevida livre de doença foi menor nos
casos familiares. Entretanto a taxa de sobrevida não foi diferente nos dois grupos.
Também observaram que sobrevida livre de doença foi menor nos casos com 3 ou
mais familiares afetados.

29
É muito difícil de realizar uma análise de sobrevivência em uma doença
relativamente rara e com baixa mortalidade. Porém, a avaliação da sobrevida livre
de doença talvez seja o objetivo mais apropriado a ser buscado e existem
evidências, em estudos bem conduzidos, que portadores de CNMFT têm um
tempo de sobrevida livre de doença menor.
Independente do tamanho do tumor, todos os pacientes com biópsia
positiva para CNMFT (carcinomas diferenciados de células foliculares) devem ser
submetidos à tireoidectomia total. Esta tem sido a recomendação da grande
maioria dos autores15,18,23,34,55. Mesmo as lesões pequenas podem estar
relacionadas com multifocalidade, invasão local, metástases ganglionares e alta
taxa de recorrência35. Sippel e cols44, entre outros autores15,18, recomendam uma
cirurgia inicial mais extensa, com dissecção profilática da região central do
pescoço (linfonodos ipsilaterais ou centrais bilaterais) associada a tireoidectomia
em qualquer paciente com tumor maior que 1cm. A taxa de recorrência é maior e
todos os passos devem ser seguidos na primeira cirurgia para evitar nova
exploração cervical e seus riscos associados de morbidade52.
Não existem estudos clínicos que demonstrem a melhor estratégia de
conduta do CNMFT. Existe muito debate em relação à indicação de tireoidectomia
profilática para os pacientes com risco de CNMFT. Pela menor confiabilidade da
PAAF nos casos de CNMFT34 e pelos estudos que sugerem maior agressividade,
alguns autores têm sugerido esta abordagem.
A tireoidectomia profilática proporciona o tratamento antes que a doença se
torne clinica, radiológica ou citologicamente evidente na triagem. Sippel e cols44
sugerem que a abordagem deva ser semelhante a dos casos de carcinoma

30
medular familiar, onde o tratamento precoce prolonga o tempo de sobrevida livre
de doença. Sua estratégia de tratamento seria realizar tiroidectomia total em todos
os pacientes com forte história familiar de câncer de tireóide e com nódulo de
tireóide. Estas recomendações estariam baseadas nos seguintes fatos: todas as
células foliculares da tireóide estariam sob risco de malignização pela
predisposição genética; existe um aumento do risco da doença ser multifocal e
bilateral no CNMFT; facilita o tratamento e o seguimento com iodo radioativo; e
facilita o uso da tireoglobulina sérica como marcador de atividade de doença no
pós operatório.
O seguimento ideal para estes pacientes com CNMFT ainda não esta bem
definido. Como todos são portadores de câncer de tireóide, inicialmente devem-se
seguir os padrões internacionalmente recomendados para cada estágio da
doença.
A alta taxa de recorrência encontrada nestes pacientes e o notável risco de
morte a ela relacionada42 leva a crer que seria razoável a administração de dose
ablativa de iodo radioativo no pós-operatório, seguida de supressão com
hormônio tireoideano para todos os pacientes44 . Alsanea e cols17 recomendam
que todos os pacientes submetidos a tireoidectomia devam ser tratados com dose
ablativa de radioiodo, independente do tamanho do tumor, com posterior
supressão do TSH com hormônio tireoideano para valores < 0,1mlU/l nos
pacientes de risco moderados e <0,05 nos de alto risco.
Mesmo os pacientes que em outras situações seriam considerados como
baixo risco (tumores <1 cm) devem ser tratados de forma mais rigorosa, pois
existem evidências de comportamento mais agressivo35.

31
Nos casos inicialmente não tratados com tireoidectomia total, esta deve ser
considerada a seguir. Se este procedimento não for realizado, estes pacientes
devem ser submetidos a avaliações anuais com ultra-sonografia e dosagens dos
níveis de tireoglobulina, pois eles permanecem num grupo de risco de recorrência.
II. 1. 5 – GENÉTICA
Ao contrário do que ocorre com os carcinomas medulares, a genética dos
CNMT ainda não está bem compreendida. A NEM 2A e o carcinoma medular
familiar estão associados a mutações no oncogene RET. Desde que o RET foi
identificado53 a triagem, o diagnóstico e o tratamento precoce resultaram e maior
sobrevida e percentual de cura54. Para Sturgeon e cols18 uma descoberta desta
no CNMFT traria benefícios de importância e magnitudes semelhantes.
Até o presente momento vários estudos estão sendo realizados e vários
genes têm sido envolvidos como possíveis responsáveis pelo desenvolvimento
dos CNMFT. Entre estes se encontram o TCO, PRN1 e NMTC118.
O gen TCO, localizado no lócus 19p13.2, foi identificado em uma família
francesa com tumores tireoideanos oxifílicos (TCO) num estudo realizado em 1998
por Canzian e cols55. Um dado interessante neste estudo foi o fato de que tanto os
tumores tireoideanos malignos quanto os benignos possuíam alguma medida de
células oxifílicas, o que até então não havia sido descrito em CNMFT. Bevan e
cols56 encontraram evidências que apóiam esta linhagem em famílias isoladas. Em
estudos prévios de CNMFT, o grupo de Sturgeon e cols18 analisaram o genótipo
de membros de duas famílias com carcinoma de células de Hürtle usando cinco

32
marcadores numa região de 10 cM. Em uma destas famílias houve evidência
compatível de vinculação ao gene que predispõem ao câncer tireoideano oxifílico
familiar (gen TCO) no cromossomo 19p13.218. Estes achados foram de acordo
com os encontrados por Canzian e cols55.
Uma síndrome tumoral hereditária composta de CPFT, doença nodular
tireoideana e neoplasia papilífera renal foi caracterizada por Malchoff e cols57. O
gen predisponente PRN1 foi mapeado no cromossomo 1q21.
Outro gene candidato a causador do CNMFT é o gene NMTC1, localizado
no cromossomo 2q21. Este gen foi encontrado numa grande família da Tasmânia
e vem sendo associado com o desenvolvimento da variante folicular do carcinoma
papilífero familiar de tiróide, e foi confirmado em 80 pedigrees adicionais de
CNMFT58 Também existem evidências recentes que os lócus do TCO e NTMC1
podem interagir para aumentar o risco de câncer de tireóide em um subgrupo de
pedigrees de CNMFT 59. Entretanto, parece pouco provável que esta interação
seja comum na maioria dos casos de CNMFT18.
Todavia, nenhum destes genes responde pela maior parcela dos casos de
CNMFT. A maioria dos estudos de vinculação para CNMFT renderam escores de
LOD (logaritmo de odds) menores do que três, falharam ao identificar o mesmo
lócus ou não mostraram vinculação 41,55-58,60. Além do mais, a maior parte dos
experimentos foi realizada em uma ou poucas famílias, não podendo ser
generalizado para a população de alto risco18.
Um modo de herança autossômica dominante, com penetrância variável
aparece na grande parte das famílias com CNMTF. Esta conclusão baseia-se em
relatórios de famílias com três ou mais membros afetados, transmissões

33
sucessivas homem a homem e apresentação horizontal em irmãos10,61. Herança
poligênica também é uma hipótese plausível, especialmente nos casos de apenas
dois membros da família afetados. Alguns destes casos podem estar associados a
exposição a antígenos carcinogênicos da tireóide, como a radiação ionizante,
deficiência ou excesso de iodo62-64.
As alterações genéticas encontradas nos casos esporádicos como os
rearranjos RET/PTC, TRK, RAS e MET não parecem estar associados com o
CNMFT65-67, bem como mutações BRAF e translocações PAX8-PPARg66,67.
Outros genes relacionados às síndromes familiares já foram previamente
mencionados nesta dissertação.
Como a causa genética permanece incerta não se recomenda testes
genéticos para triagem de CNMFT, com já está estabelecido nos casos de
carcinomas medulares.
A caracterização dos genes envolvidos no surgimento de CNMFT
possivelmente trará novas formas de manuseio deste grupo de pacientes,
possibilitando diagnósticos mais precoces e talvez até um melhor prognóstico.

34
II.2 – SÍNDROMES FAMILIARES ASSOCIADAS COM CARCINOMA NÃO-MEDULAR DE
TIREÓIDE
Algumas síndromes familiares estão classicamente associadas à presença
de carcinoma de tireóide não-medular. Entretanto, a presença delas invalida o
diagnóstico de CNMFT e serão descritas a seguir:
- Polipose adenomatosa familiar (PAF) – Caracteriza-se pelo desenvolvimento de
múltiplos pólipos adenomatosos na mucosa do trato gastrointestinal,
particularmente no colon, com potencial de transformação maligna. É uma doença
autossômica dominante, causada por uma mutação germinativa inativadora do
gene de supressão tumoral APC (localizado no cromossomo 5q21). Um aumento
do risco de carcinoma papilífero de tireóide ocorre em algumas famílias com PAF.
Estes tumores tireoideanos tipicamente apresentam um padrão histológico
cribiforme e ocorrem mais comumente em jovens (menores que 30 anos) e em
mulheres68-70,71. A maioria das mulheres com PAF e carcinoma papilífero de
tireóide tem a mutação somática RET/PTC assim como a mutação germinativa do
gene APC38,73-74. O risco cumulativo do desenvolvimento de câncer de tireóide
antes dos 60 anos é de 2,8%75. As mulheres com idade entre 25 e 35 anos
representam o grupo de maior risco de desenvolver câncer de tireóide e o risco
aumenta para 6%76. Soravia e cols74 determinaram que a perda de função do
gene APC num paciente com PAF não aumenta o risco de carcinoma papilífero de
tireóide familiar, ao menos que venha associado com ganho de função secundário
a mutações somáticas nas isoformas RET/PTC1 ou RET/PTC23. A taxa de

35
sobrevida em 5 e 10 anos de PAF associada a carcinoma papilífero familiar de
tireóide é de 90% e 77%, respectivamente76.
- Síndrome de Gardner: É uma variante da PAF, na qual além dos pólipos existem
as manifestações extra-colônicas, como displasia fibrosa do crânio, osteomas de
mandíbula, dentes supranumerários, fibromas, tumores desmóides, cistos
epiteliais, hamartomas do trato gastrointestinal, hepatoblastomas e tumores
tireoideanos77,78. Os tumores de tireóide ocorrem mais comumente em jovens
(menores de 30 anos) e em mulheres. O risco de desenvolvimento de câncer de
tireóide é de aproximadamente 2%79.
- Doença de Cowden: Caracterizada pelos múltiplos hamartomas e tumores de
mama, cólon, endométrio e cérebro. Os tumores de tireóide podem ser benignos
ou malignos – predominantemente foliculares e representam a manifestação
extra-cutânea mais comum da doença, podendo ocorrer em até dois terços dos
casos. Esta enfermidade tem herança autossômica dominante e esta associada
com mutação no gen de supressão tumoral PTEN, localizado no cromossomo
10q22-2380-82. Os pacientes desenvolvem neoplasias de tireóide do subtipo papilar
e de células de Hürtle em sua maioria e numa idade mais avançada, que pode
significar uma progressão do tumor, uma vez que 85% destes pacientes possuíam
bócio multinodular anteriormente83.
- Doença de Werner: Tem herança autossômica recessiva e tem sido ligada a
mutações no gene WRN no cromossomo 8p11-21. Caracteriza-se por
envelhecimento prematuro, alterações cutâneas esclerodérmicas, catarata,
calcificações subcutâneas, atrofia muscular, diabetes mellitus e maior risco de
neoplasias. Os tumores de tireóide ocorrem numa idade mais precoce e são

36
predominantemente do subtipo folicular. Os papilíferos e anaplásicos também são
comuns84.
- Complexo de Carney: É uma patologia de caráter autossômico dominante
causada por mutação no PPKAR1α no cromossomo 17q24. Há uma associação
de mixomas de partes moles, pigmentação mucosa e da pele, schwannomas,
tumores de adrenais, hipófise e testículos. As patologias da tireóide são comuns
(aproximadamente 11% dos pacientes) e incluem hiperplasia adenomatosa, cistos
e carcinomas papilíferos e foliculares85,86.
Existem outras síndromes que estão possivelmente associadas com o
desenvolvimento de CNMT, porém a ligação entre estas patologias é muito menos
estabelecida do que as anteriormente citadas. Por exemplo:
- Ataxia-telangectasia ( Síndorme de Louis-Bar’s ): É uma doença de transmissão
autossômica recessiva na qual ocorre ataxia cerebelar e nistagmo, telangectasias
oculocutâneas, imunodeficiência humoral e celular, infecções bacterianas
recorrentes do trato respiratório e câncer de origem linforeticular. Associações
com carcinomas de tireóide foram documentadas em relatos de casos87-89.
- Síndrome de Peutz-Jeghers: Faz parte da síndrome hereditária de hamartoma
polipose, que inclui a doença de Cowden, síndrome de polipose juvenil dentre
outras. É de origem autossômica dominante e se caracteriza por hamartomas do
intestino delgado excesso de pigmentação da pele e mucosas. Esta relacionada a
mutações no gene LKB1 ( cromossomo 19p13.3). Também há relatos de casos
desta patologia associada com câncer de tireóide90,91.
- Síndrome de Wermer (neoplasia endócrina múltiplas tipo 1 [NEM1]):
Caracterizada por tumores da hipófise, paratireóide e ilhotas pancreáticas.

37
Também pode apresentar tumores do córtex adrenal e outros tecidos. A
anormalidade genética está no gene MENIN, no cromossomo11q13. Doenças
tireoideanas (especialmente tumores foliculares) são encontrados em 26% dos
pacientes com NEM192.
- Síndrome de McCune-Albright: Causada pela mutação no gene da GNAS1
localizado no cromossomo 20q13. Tem como achados típicos: displasia fibrosa
poliostótica, manchas café-com-leite e hiperfunção endócrina. As glândulas mais
habitualmente acometidas são: hipófise, tireóide, adrenal e gônadas. Doença
adenomatosa da tireóide é comum e carcinoma folicular já foi previamente
relatado93.
- Síndrome de bócio multinodular familiar (SBMNF): A análise de uma grande
família do Canadá com SBMNF resultou no mapeamento gene MNG1, localizado
no cromossomo 14q como alteração genética responsável pela doença. Neste
estudo, 2 dos 18 pacientes apresentavam lesões sugestivas de CPT, sendo
razoável considerar que genes que predispõem a SBMNF possam ser candidatos
a CNMFT41. Como resultado da associação de BMN e CPT, Bignell e cols41
examinaram 37 famílias para vinculação com MNG1 e não estabeleceram esta
relação em nenhuma destas famílias.

38
III – OBJETIVOS
Principal: avaliar a prevalência de carcinoma papilífero de tireóide entre os
familiares de primeiro grau de portadores desta patologia.
Secundários: � verificar se há relação entre a incidência de carcinoma
papilífero familiar de tireóide com disfunção (através da dosagem de T4l e TSH),
bem como com a presença de auto-imunidade tireoideana (através da dosagem
de ATPO);
� verificar a prevalência de alteração estrutural tireoideana
(bócio e/ou nódulo) em familiares de primeiro grau de portadores de carcinoma
papilífero de tireóide.

39
IV – CASUÍSTICA E MÉTODOS
IV.1 – CARACTERÍSTICAS DO ESTUDO E CONSIDERAÇÕES ÉTICAS
Realizamos um estudo de corte transversal, avaliando a função, a presença
de auto-imunidade e a estrutura da glândula tireóide em familiares de primeiro
grau de portadores de carcinoma papilífero de tireóide.
Este estudo faz parte do projeto de pesquisa “Carcinoma Papilífero
Familiar de Tireóide”, tendo sido observadas as Normas Éticas Internacionais
para Pesquisas Biomédicas Envolvendo Seres Humanos do CIOMS (Conselho
para Organizações Internacionais de Ciências Médicas), OMS, Genebra –
1993. Além disso, foi aprovado pelo Comitê de Ética em Pesquisa do Hospital
Universitário Clementino Fraga Filho (HUCFF) / Faculdade de Medicina, sob o
número 066/06, em 17/08/06, conforme parecer em anexo (anexo I).
IV.2 - SELEÇÃO DE INDIVÍDUOS
Os indivíduos selecionados para este trabalho são familiares de primeiro
grau de pacientes portadores de carcinoma papilífero de tireóide – considerados
como casos índices, em acompanhamento nos ambulatórios dos Serviços de
Endocrinologia do HUCFF, da Universidade Federal do Rio de Janeiro (UFRJ). Os
ambulatórios de Endocrinologia prestam atendimento especializado em
endocrinologia, especialmente em doenças da glândula tireóide, e recebem

40
pacientes já acompanhados em outros serviços do mesmo hospital ou
referenciados pelos postos de saúde.
Todos, após esclarecimento verbal, assinaram o termo de consentimento
livre esclarecido (anexo II), recebendo uma cópia deste.
CRITÉRIOS DE INCLUSÃO:
� indivíduos com idade entre 15 e 80 anos;
� ter no mínimo um familiar portador de carcinoma papilífero de tireóide em
acompanhamento ambulatorial no serviço de endocrinologia do HUCFF.
� o número de familiares por cada caso índice não é pré-estabelecido. Todos, de
primeiro grau, foram convidados a participar.
CRITÉRIOS DE EXCLUSÃO:
� História familiar de síndromes familiares associadas ao carcinoma não-medular
de tireóide (polipose adenomatosa familiar, doença de Cowden, doença de
Carney, síndrome de Wermer etc)
Após avaliação clínica dos pacientes considerados casos índices e seus
familiares, foram preenchidas as fichas de avaliação clínica (anexo III e IV,
respectivamente).
IV.3 EXAMES LABORATORIAIS
As amostras para a análise laboratorial foram obtidas através da coleta de
sangue venoso periférico, por médicos do serviço de Endocrinologia do HUCFF,
não sendo necessário jejum. Para verificação de disfunção tireoideana foram
dosados TSH e T4L, e para avaliação de auto-imunidade o ATPO. As amostras

41
foram direcionadas para dosagem hormonal e ATPO pelo Laboratório de
Hormônios do HUCFF.
O TSH foi dosado por ensaio imunométrico por quimioluminescência de
terceira geração, com kit para dosagem DPC (Diagnostic Products Corporation) e
aparelho automático Immulite 2000®. Os valores de referência (VR) são de 0,4 a
4,0 mUI/L, sensibilidade de 0,002 mUI/L, coeficiente de variação (CV) intra-ensaio
de 3,8% a 12,5% e inter-ensaio, de 4,6% a 12,5%.
O T4l foi dosado com ensaio imunoenzimático por quimioluminescência,
com kit para dosagem DPC® (Diagnostic Products Corporation) e aparelho
automático Immulite 2000®. Os VR são de 0,8 a 1,9 ng/dL, sensibilidade de 0,15
ng/dL, CV intra-ensaio de 4,4% a 7,5% e inter-ensaio, de 4,8% a 9,0%.
Os anticorpos ATPO foram dosados por ensaio imunométrico por
quimioluminescência, com kit para dosagem DPC® (Diagnostic Products
Corporation) e aparelho automático Immulite 2000®. São considerados normais
valores iguais ou inferiores a 35 UI/mL. O CV intra-ensaio é de 4,3% a 5,6% e o
inter-ensaio, de 7,8% a 10,5%.
IV.4 – ULTRA-SONOGRAFIA DA TIREÓIDE
As ultra-sonografias de tireóide para avaliação morfológica da glândula
foram realizadas utilizando-se um aparelho de ultra-som (SIEMENS, MCMDO1AA)
com transdutor linear multifrequência de 11 MHz. Os exames foram feitos, em sua
maioria, pelo mesmo observador do Serviço de Radiodiagnóstico do HUCFF. Os

42
demais exames foram feitos de acordo com a disponibilidade de cada paciente,
em diversos locais do estado do Rio de Janeiro, com aparelhos semelhantes.
Os pacientes com história prévia de tireoidectomia total não realizaram
exame da região cervical.
IV.5 – PAAF DE TIREÓIDE
Os nódulos maiores que 1 cm e os menores com pelo menos três critérios
ultra-sonográficos sugestivos de malignidade, que ainda não tinham sido
estudados previamente ou que apresentaram mudança evolutiva foram
submetidos a PAAF.
As características evidenciadas à ultra-sonografia que são associadas a
um maior risco de malignidade são: hipocogenicidade, microcalcificações,
margens irregulares, fluxo sanguíneo intranodular aumentado ao doppler, aumento
do diâmetro antero-posterior em relação ao transverso em nódulos não
palpáveis94,95 e, especialmente, a detecção de adenomegalia regional.
As PAAFs foram realizadas pelo mesmo operador do serviço de
radiodiagnóstico do HUCFF, guiadas por USG com o mesmo aparelho utilizado
para a realização dos exames ultra-sonográficos descrito acima. As amostras
coletadas foram enviadas em seguida para o laboratório de anatomia patológica
do mesmo hospital, e analisadas pelo mesmo citopatologista.
O Doppler colorido não foi utilizado em conjunto com a USG devido a
indisponibilidade deste no decorrer do trabalho.

43
IV.6 – REALIZAÇÃO DE CIRURGIAS
As indicações de cirurgia durante o estudo foram definidas com base nos
critérios clínicos, laboratoriais e citológicos. Foram encaminhados para cirurgia os
pacientes com laudos citológicos de malignidade e padrão indeterminado na
citopatologia e um caso com nódulos grandes associados a sintomas
compressivos.
A técnica cirúrgica empregada ficou a cargo dos cirurgiões do serviço,
sendo individualizada, de acordo com cada caso.
Os indivíduos selecionados para procedimento cirúrgico foram
encaminhados para o serviço de cirurgia geral do HUCFF, para sua apreciação.
Posteriormente, seguiram o fluxo habitual do hospital que inclui uma lista de
espera para agendamentos de cirurgias.
Os pacientes puderam optar por nova avaliação clínica e cirúrgica, de
acordo com a sua conveniência, em outros serviços públicos ou privados.
IV.7 – ANATOMIA PATOLOGICA
As amostras de aspirado de tireóide, após serem preparadas para análise
citológica à microscopia óptica, foram avaliadas pelo mesmo observador do
serviço de anatomia patológica do HUCFF. A preparação dos espécimes seguiu a
rotina de coloração do hospital.

44
Os resultados citológicos possíveis em amostra de material adequado são:
benignidade, malignidade ou indeterminado. Além disso, podem ocorrer amostras
com material inadequado para o diagnóstico citopatológico96 -98.
Nos casos em que existiam resultados citopatológicos prévios, estes laudos
foram utilizados para análise.
As peças cirúrgicas dos pacientes submetidos à tireoidectomia no HUCFF
durante o estudo foram avaliadas pelo serviço de anatomia patológica deste
hospital. Manteve-se a rotina de preparação do material habitualmente utilizada.
Os laudos histopatológicos oriundos de cirurgias realizadas em outros
hospitais, bem como das realizadas previamente ao início do estudo foram
utilizados para análise.

45
V – ANÁLISE ESTATÍSTICA
Foram utilizados os programas SPSS, versão 10 e epi-info versão 6.0 nas
análises estatísticas. Através de análise descritiva, obtiveram-se as frequências
das variáveis de interesse e foi realizada a comparação das proporções através
dos testes Qui-quadrado e exato de Fisher. Os dados referentes às variáveis
contínuas foram expressos como média ± desvio padrão. As comparações das
médias entre dois grupos de estudo foram feitas utilizando-se o teste de Mann-
Whitney, (exceto para idade, que teve distribuição normal ). Foi realizado o cálculo
do risco relativo de pacientes com história familiar de carcinoma papilífero e
ATPO de apresentarem carcinoma papilífero. O nível de significância estatístico
aceito foi de 5%.

46
VI – RESULTADOS
VI.1 – CARACTERÍSTICAS GERAIS DA AMOSTRA
Foram incluídos 143 familiares de 52 pacientes em acompanhamento
ambulatorial por carcinoma papilífero de tireóide, dos quais 7 eram sabidamente
portadores de CPFT. A média de familiares foi de 2,75 (variação: 1 a 7), por
paciente índice. Destes 96 (67,1%) eram do sexo feminino e 47 (32,9%) do
masculino, com média de idade de 42,4 ± 16,2 anos ( variação: 15 a 75).
A média de T4l foi de 1,348 ± 0,275 ng/dL, e a do TSH foi 2,36 ± 3,47
mUI/L. Entretanto 5 familiares apresentavam alteração do TSH secundária ao
tratamento com levotiroxina. Entre eles, dois estavam em supressão e uma em
hipotireoisismo para tratamento do CPFT e dois por reposição hormonal
suprafisiológica, para tratamento de hipotireoidismo. Após a exclusão destes
pacientes, a média do TSH foi de 2,15 ± 1,4 mUI/L. Para as análises
comparativas realizadas a seguir, foram excluídos os cinco pacientes com as
alterações do TSH mencionadas acima.
Foi observado que 8,4% dos pacientes apresentavam positividade, para os
ATPO. Porém, este anticorpo não foi dosado em 4 pacientes (2,8%), todos estes
portadores de CPFT. O risco relativo dos familiares de primeiro grau de portadores
de CPT que apresentam ATPO positivo e US com nódulos suspeitos (com
indicação de PAAF) desenvolverem CPFT foi de 3,38 (p = 0,300).

47
Do total da amostra, 125 pacientes fizeram ultra-sonografia de tireóide.
Dentre estes, 17 pacientes foram submetidos à análise citopatológica e em 4 o
diagnóstico foi de CPFT. Entre os 18 que não realizaram o exame, 8 familiares
eram previamente tireoidectomizados, sendo que 7 deles eram portadores de
CPFT. Portanto encontramos no total 11 pacientes com CPFT ao final do estudo.
Detalhes da análise destes resultados serão descritos a seguir.
Os dados clínicos e laboratoriais de cada indivíduo participante do estudo,
bem como a distribuição por famílias (de acordo com o caso índice) estão
resumidos no anexo V.
VI.2 – ANÁLISE COMPARATIVA ENTRE OS PORTADORES E OS NÃO-PORTADORES DE
CPFT
Para fins de comparações entre os dois grupos acima referidos,
consideramos como critério diagnóstico de CPFT os resultados obtidos no cito e
histopatológicos somados. Os resultados encontrados na comparação das
variáveis sexo, TSH, T4l e ATPO estão descritos em seguida, na tabela 1.
Tabela 1 – Comparação de variáveis clínicas e laboratoriais entre os portadores e
não-portadores de CPFT.
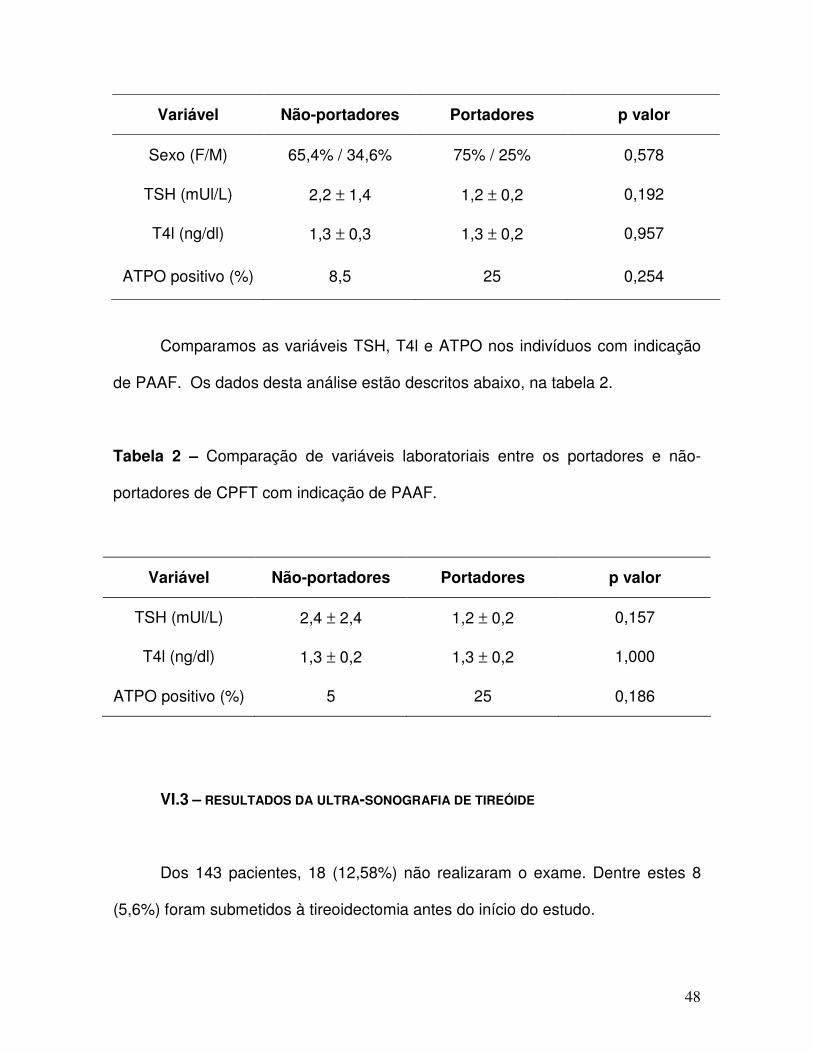
48
Variável Não-portadores Portadores p valor
Sexo (F/M) 65,4% / 34,6% 75% / 25% 0,578
TSH (mUl/L) 2,2 ± 1,4 1,2 ± 0,2 0,192
T4l (ng/dl) 1,3 ± 0,3 1,3 ± 0,2 0,957
ATPO positivo (%) 8,5 25 0,254
Comparamos as variáveis TSH, T4l e ATPO nos indivíduos com indicação
de PAAF. Os dados desta análise estão descritos abaixo, na tabela 2.
Tabela 2 – Comparação de variáveis laboratoriais entre os portadores e não-
portadores de CPFT com indicação de PAAF.
Variável Não-portadores Portadores p valor
TSH (mUl/L) 2,4 ± 2,4 1,2 ± 0,2 0,157
T4l (ng/dl) 1,3 ± 0,2 1,3 ± 0,2 1,000
ATPO positivo (%) 5 25 0,186
VI.3 – RESULTADOS DA ULTRA-SONOGRAFIA DE TIREÓIDE
Dos 143 pacientes, 18 (12,58%) não realizaram o exame. Dentre estes 8
(5,6%) foram submetidos à tireoidectomia antes do início do estudo.

49
Entre os 125 pacientes que fizeram ultra-sonografia, os exames foram
normais em 57 (45,6%) indivíduos e alterados em 68 (54,4%). Identificamos a
presença de doença nodular em 49 (39,2%) pacientes, dos quais 24 (19,2%)
apresentavam nódulo único.
Os achados mais frequentes nos exames considerados alterados estão
descritos logo abaixo, na tabela 3.
Tabela 3 – Alterações mais frequentes encontradas nas ultra-sonografias
realizadas em familiares de primeiro grau de portadores de CPT.
USG N. Pacientes Percentual (%)
Alterados 68 100
Textura Heterogênea 6 8,8
Cistos simples 13 19,11
Nódulos <1 cm 24 35,3
Nódulos >1 cm 22 32,3
Nódulos suspeitos 3 4,4
Identificamos 25 (20%) pacientes com dois ou mais nódulos (bócio
multinodular) entre os indivíduos avaliados por ultra-sonografia. Dentre os 4
pacientes com CPFT que fizeram o exame todos (100%; p= 0,001) apresentaram
BMN . Entre os que possuíam BMN ao USG, 16% tinham CPFT (p=0,001).

50
Dos 125 pacientes que fizeram ultra-sonografia, 25 (20%) foram
encaminhados para PAAF, havendo BMN em 15 deles. Entre os familiares com
indicação de PAAF, 15 (60%) realizaram o procedimento, 5 (20%) não
compareceram para fazer o exame, em 2 (8%) pacientes não foi possível a
realização do exame por dificuldade técnica na abordagem do nódulo e 1(4%) foi
encaminhado direto para cirurgia, sem PAAF pré-operatória. Em 2 (8%) não foi
necessário a repetição do procedimento pela estabilidade do nódulo e laudo
citopatológico prévio.
Os resultados ultra-sonográficos de cada indivíduo participante do estudo,
bem como a distribuição por famílias (de acordo com o caso índice) estão
resumidos no anexo VI.
VI.4 – RESULTADOS DA CIRURGIA
No decorrer do estudo, dois pacientes foram submetidos à tireoidectomia
total, sendo que em um dos casos o procedimento foi realizado no Instituto
Nacional do Câncer (INCA). Em ambos os casos não houve intercorrências no peri
e pós-operatório.
Sete pacientes aguardam para realização de cirurgia, sendo quatro com
diagnóstico de CPFT e três com laudo citológico indeterminado ou suspeito.
VI.5 – RESULTADOS DA ANATOMIA PATOLÓGICA
Os resultados citopatológicos dos 17 pacientes avaliados estão descritos
abaixo, na tabela 4.

51
Tabela 4 – Resultados citopatológicos realizadas em familiares de primeiro grau
de portadores de CPT.
PAAF N. Pacientes Percentual (%)
Carcinoma papilífero 4 23,5
Benignidade 4 23,5
Indeterminado 3 17,6
Inadequado 6 35,6
Entre os 10 pacientes com BMN que realizaram a PAAF, os resultados
foram: 4 (40%) carcinoma papilífero, 2 (20%) indeterminados ou suspeitos, 2
(20%) benignos e 2 (20%) amostras inadequadas.
Um total de 8 pacientes possuíam laudos histopatológicos de cirurgias
realizadas previamente ao estudo. Destes, 1 com benignidade e 7 com carcinoma
papilífero.
Durante o estudo dois pacientes foram submetidos à tireoidectomia, sendo
em um diagnosticado que carcinoma folicular (operado por sintomas
compressivos) e no outro, benignidade (operado por PAAF indeterminada).
Os dados da anatomia patológica de cada indivíduo participante do
estudo, bem como a distribuição por famílias (de acordo com o caso índice) estão
resumidos no anexo VIII.

52
VI.6 – ANÁLISE DE PREVALÊNCIA DE CPFT
A análise dos resultados de prevalência de CPFT foi realizada
considerando-se como diagnóstico de CPT os resultados de cito e histopatológico
somados e apenas com os diagnósticos histopatológicos.
Quando avaliamos por indivíduos, 11 (7,7%) receberam o diagnóstico de
CPFT. Levando-se em conta somente os casos com laudo histopatológico, 6
(4,89%) dos pacientes possuíam a patologia.
Os resultados obtidos na análise de prevalência de CPFT estão descritos
em seguida, na tabela 5.
Tabela 5 – Resultados da análise da prevalência CPFT por famílias.
Variável Número Percentual
Famílias com CPFT - laudos cito +histopatológicos 10 19,23
Famílias com CPFT - laudos histopatológicos 6 11,53
Entre as 52 famílias participantes apenas uma tinha CPFT em pelo menos
três membros. A prevalência de CPFT nestes familiares foi de 1,39%.

53
VII – DISCUSSÃO
Encontramos uma prevalência de CPFT entre os familiares de primeiro grau
de portadores de CPT semelhante aos relatos da literatura, tanto quando
utilizamos como critério diagnóstico o somatório dos laudos cito e histopatológicos
positivos para CPT (7,7%), quanto ao avaliarmos apenas o segundo (4,89%).
Musholt e cols23 em sua meta-análise sobre CPFT encontraram uma prevalência
de 1,92 a 10,5%30, com média de 5%. Quando analisamos por famílias,
identificamos uma alta prevalência do CPFT (19,23%), com significância
estatística (p valor de 0,003 e <0,001 para análise de cito e histopatológico
somados e histopatológico apenas, respectivamente), traduzindo a relevância
clínica do CPFT.
Se considerarmos como CPFT apenas os casos com três ou mais
familiares acometidos, como propõe Charkes32, a incidência cai bruscamente para
1,39% dos familiares, visto que em apenas uma família encontramos três pessoas
acometidas. Neste caso estaríamos diminuindo muito o risco (< 6%) de tratar-se
de um caso esporádico, pois como já mencionado anteriormente, estima-se que
se dois membros de uma família são afetados pelo CNMFT, existe uma chance de
47% de este tumor ser esporádico17,31,32. Ainda neste estudo, o autor propõe,
através de análises matemáticas, que a investigação sobre a atividade clínica e a
genética envolvida nos CNMFT deva ser limitada aos casos onde existam três ou
mais familiares de primeiro grau acometidos pela doença. Mas isto ainda é motivo
de controvérsia, e até o presente momento, permanece a definição de dois ou
mais membros acometidos para diagnóstico de CNMFT.

54
A média de familiares por caso índice foi de 2,75, porém a média de
familiares de primeiro grau vivos que potencialmente poderiam fazer parte da
pesquisa foi de 4,59. Os principais motivos desta não participação foram a
distância entre a moradia e o HUCFF (muitos moravam em outras cidades ou
estados) e a incompatibilidade dos horários das consultas e exames com a rotina
de trabalho individual. De fato, pode ter ocorrido alguma seleção, na qual os mais
interessados ou mais preocupados foram os avaliados. Entretanto, este possível
viés faz parte do modelo de estudo em questão.
A amostra foi composta em sua maioria por pacientes do sexo feminino e
com a média de idade em torno de 42 anos. Entre os que possuíam ou não o
diagnóstico de CPFT a maioria era de mulheres.
As mulheres também foram maior número em outros estudos
realizados15,46. Uchino e cols46 propuseram uma triagem por ultra-sonografia em
todos os familiares de primeiro grau de portadores de carcinoma papilífero, em
especial as mulheres, pela maior incidência da doença neste grupo.
Em geral, as publicações comparam os casos familiares de CNMT com os
esporádicos, e os relatos são similaridade entre as idades9,14,15,32,34 ou o
aparecimento da patologia em idades mais precoces nos casos familiares18,22,23,24.
Não avaliamos esta variável neste estudo por impossibilidade de determinar em
todos os pacientes com CPFT a idade do diagnóstico da doença.
Vários estudos têm demonstrado a presença de doenças tireoideanas não
malignas com bastante freqüência (57%) em pacientes com o subtipo papilífero do
CNMFT25,26. Em particular, uma maior incidência de múltiplos nódulos benignos
nos familiares de CPFT que nos casos esporádicos (41,5% vs 29,8%; P<0,001)15 .

55
Em relação à análise morfológica da tireóide, realizada através de ultra-
sonografia, encontramos um número de exames alterados discretamente superior
aos normais (54,4% vs 46,5%). Todavia se considerarmos apenas os casos de
doença nodular, o percentual de exames alterados cai para 39,2%, dos quais
19,2% com nódulo único e 20% com BMN. OS resultados encontrados estão de
acordo com as variações descritas na literatura, de 19 a 67%45. Uchino e cols15
não encontraram diferença estatística em relação à presença de nódulos únicos
benignos ao comparar os casos de CTNM familiares com esporádicos, mas esta
foi significativamente maior para presença de BMN.
Quando analisamos apenas o subgrupo CPFT a prevalência de BMN foi de
100%, com significância estatística, maior do que a descrita anteriormente na
literatura. Ressalta – se que entre os 11 pacientes com CPFT apenas 4 fizeram
USG, pois os demais eram tireoidectomizados previamente ao estudo (não tinham
ultra-sonografias pré-operatórias disponíveis para análise). É, portanto um número
muito pequeno de pacientes sendo necessária muita cautela na interpretação
destes resultados. No entanto, do ponto de vista clínico é uma informação
importante.
Vale lembrar que dos 15 pacientes com BNM e indicação de punção, 10
foram submetidos à PAAF e em 3 a análise do material obtido na PAAF foi
insuficiente para o diagnóstico. Ou seja, em mais da metade dos pacientes (3 com
material inadequado e 5 que não fizeram o exame) não foi possível a análise
citopatológica, o que certamente prejudicou o julgamento dos dados encontrados,
sendo um ponto bastante negativo em nosso trabalho. Diante destes fatos é
prudente não extrapolarmos nossos resultados para a população geral.

56
Em dois grandes estudo americanos, que avaliaram mais de 16500
amostras de PAAF de tireóide, 6 a 21% (média de15,7%) foram sem diagnóstico
ou insatisfatórias99,100. Vilar e cols1 relatam que em mais de 5000 exames
realizados em aproximadamente 10 anos, 11 a 21% (média de 15,5%) foram sem
diagnóstico ou insatisfatórios.
A experiência de quem realiza a coleta de amostras e o rigor dos critérios
utilizados pelo citopatologista na classificação dos espécimes são outros fatores
determinantes das taxas variáveis de ocorrência de resultados insatisfatórios1.
Provavelmente a discrepância entre o percentual de amostras inadequadas
que encontramos e os relatos descritos na literatura pode ser justificada pelas
razões acima mencionadas.
Sabemos que com a repetição da PAAF, geralmente se consegue um
diagnóstico em 50% casos das amostras consideradas insatisfatórias para o
diagnóstico96,101,102, razão pela qual os seis pacientes que receberam este
diagnóstico foram encaminhados para a repetição do exame (os resultados ainda
não estavam disponíveis).
A análise do estudo foi prejudicada pela não realização de exames
importantes como ultra-sonografia e PAAF por alguns participantes, independente
do motivo que os levou ao não comparecimento.
Não encontramos maior incidência de disfunção tireoideana tanto na
amostra total quanto no subgrupo de portadores de CPFT. Todavia, o TSH foi
mais baixo quando comparamos os portadores da doença com os não-portadores,
sendo quase o dobro. Este achado permanece quando acrescentamos outro
critério nesta análise, que foi a indicação de PAAF. Apesar de não ter sido

57
demonstrado significância estatística, este dado é no mínimo curioso. Ressalta-se
que foram tomados os cuidados de excluir os pacientes com alterações do TSH
induzidas pelo uso da levotiroxina.
Quanto à presença de auto-imunidade, esta foi maior no grupo de CPFT
quando comparados com os não-portadores da patologia. Apesar de não termos
encontrado significância estatística, o fato de ter sido quase três vezes maior no
grupo do CPFT é clinicamente relevante. Esta análise pode estar até subestimada,
pois em 4 pacientes com CPFT este exame não foi realizado. Quanto ao risco
relativo para o desenvolvimento de CPT nos familiares com nódulos suspeitos ao
US, este não foi maior nos portadores de ATPO positivo.
Em resumo, nosso estudo encontrou 11 familiares de primeiro grau
acometidos por CPFT em 52 famílias analisadas. Houve uma prevalência de BMN
nestes familiares de 20%. Não encontramos mais disfunção tireoideana ou auto-
imunidade do que o esperado na população geral.
Estudos prospectivos de investigação de familiares dos familiares de
primeiro grau de pacientes com CNMT são necessários para uma real
determinação da prevalência da forma familiar de CNMFT.

58
VIII – CONCLUSÕES
1. A prevalência de carcinoma de tireóide foi de 7,7% entre os familiares de
primeiro grau de portadores desta patologia.
2. Não houve relação entre a presença de carcinoma papilífero familiar de
tireóide com a presença de disfunção ou auto-imunidade tireoideana.
3. Houve uma prevalência de 39,2% de doença nodular tireoideana, sendo
19,2% de nódulo único e 20% de bócio multinodular, entre os familiares de
primeiro grau de portadores de carcinoma papilífero de tireóide que fizeram
ultra-sonografia de tireóide.

59
IX – REFERÊNCIAS BIBLIOGRÁFICAS
1. Vilar L. Endocrinologia clínica. 3.ed – Rio de Janeiro: Guanabara Koogan,
2006.
2. Loh KC. Familial nonmedullary thyroid carcinoma: a meta-review of case
series. Thyroid 1997; 7:107.
3. Takahashi M, Ritz J, Cooper GM. Activation of a novel human transforming
gene, ret, by DNA rearrangement. Cell 1985;42(2):581-8.3.
4. Mulligan LM, Kwok JB, Healey CS, Elsdon MJ, Eng C, Gardner E, et al.
Germ-line mutations of the RET proto-oncogene in multiple endocrine
neoplasia type 2A. Nature 1993;363(6428):458-60.
5. Donis-Keller H, Dou S, Chi D, Carlson KM, Toshima K, Lairmore TC, et al.
Mutations in the RET proto-oncogene are associated with MEN 2A and
FMTC. Hum Mol Genet 1993;2(7):851-6.
6. Hofstra RM, Landsvater RM, Ceccherini I, Stulp RP, Stelwagen T, Luo Y, et
al. A mutation in the RET proto-oncogene associated with multiple endocrine
neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature
1994;367(6461):375-6.
7. Santoro M, Carlomagno F, Melillo RM, Billaud M, Vecchio G, Fusco A.
Molecular mechanisms of RET activation in human neoplasia. J Endocrinol
Invest 1999;22(10):811-9.
8. Robinson DW, Orr TG. Carcinoma of the thyroid and other diseases of the
thyroid in identical twins. A.M.A. Arch. Surg. 1955;70:923–928
9. Pal T, Vogl FD, Chappuis PO, et al. Increased risk for nonmedullary thyroid
cancer in the first degree relatives of prevalent cases of nonmedullary

60
thyroid cancer: a hospital-based study. J Clin Endocrinol Metab.
2001;86:5307-5312.
10. Goldgar DE, Easton DF, Cannon-Albright LA, Skolnick MH. Systematic
population-based assessment of cancer risk in first-degree relatives of
cancer probands. J Natl Cancer Inst 1994;86:1600-8.
11. Ron E, Kleinerman RA, LiVolsi VA, Fraumeni JF. Familial nonmedullary
thyroid cancer. Oncology 1991;48:309-11.
12. Fischer DK, Groves MD, Thomas SJ Jr, et al. Papillary carcinoma of the
thyroid: additional evidence in support of a familial component. Cancer
Invest. 1989;7:323-325.
13. Stoffer SS, Van Dyke DL, Bach JV, et al. Familial papillary carcinoma of the
thyroid. Am J Med Genet. 1986;25:775-782.
14. Grossman RF, Tu SH, Duh QY, et al. Familial nonmedullary thyroid cancer:
an emerging entity that warrants aggressive treatment. Arch Surg.
1995;130:892-899.
15. Uchino S, Noguchi S, Kawamoto H, Yamashita H, Watanabe S, Shuto S.
Familial nonmedullary thyroid carcinoma characterized by multifocality and a
high recurrence rate in a large study population. Word J Surg 2002;26:897-
902.
16. Kwok CG, Mc Dougall IR. Familial differentiated carcinoma of the thyroid:
report of five pairs of sibilins. Thyroid 1955;5;395-397.
17. Alsanea O, Clark OH. Familial thyroid cancer. Curr. Opin. Oncol. 2001;13:44
18. Sturgeon C, Clark O. Familial nonmedullary thyroid cancer. Thyroid
2005;15:588-593.

61
19. Sippel SR, Caron RN, Clark OH. An evidence-based approach to familial
nonmedullary thyroid cancer:screening, clinical management, and follow-up.
Word J Surg 2007;31:924-933.
20. Nódulos de tireóide e cancer diferenciadode tireóide: Consenso brasileiro.
Arq Bras Endocrinol Metab 2007;51/5:867-93.
21. Triponez F, Wong M, Sturgeon C, Caron N, Ginzinger DG, Segal MR, et al.
Does familial non-medullary thyroid cancer adversely affect survival? World
J Surg 2006;30:787-93.
22. Hemminki K, Charis E, Chen B. Familial risks for nonmedullary thyroid
cancer. J Clin Endocrinol Metab 2005;90:5747-53.
23. Musholt TJ, Musholt PB, Petrich T, Oetting G, Knapp WH, Klempnauer J.
Familial papillary thyroid carcinoma: genetics, criteria for diagnosis, clinical
features and surgical treatment. World J Surg 2000;24:1409-17.
24. Alsanea O. Familial nommedullary thyroid cancer. Curr Treat Options Oncol
2000;1:345-51.
25. Cetta F, Curia MC, Montalto G, et al. Thyroid carcinoma usually occurs in
patients with familial adenomatous polyposis in the absence of biallelic
inactivation of the adenomatous polyposis coli gene. J Clin Endocrinol
Metab. 2001;86:427-432.
26. Stratakis CA, Courcoutsakis NA, Abati A, et al. Thyroid gland abnormalities
in patients with the syndrome of spotty skin pigmentation, myxomas,
endocrine overactivity, and schwannomas (Carney complex). J Clin
Endocrinol Metab. 1997;82:2037-2043.

62
27. Jemal A, Siegal R, Ward E, et al. Cancer statistics,2006. Ca Cancer J clin
2006;56;106-130.
28. Hemminki K, li X. Familial risk of cancer by site and histopathology. Int J
Cancer 2003;103:105-109.
29. Galanti MR, Ekbom A, Grimelius L, et al. Parental cancer and risk of
papillary and follicular thyroid carcinoma. Br J Cancer. 1997;75:451-456.
30. Kraimps JL, Bouin-Pineau MH, Amati P, et al. Familial papillary carcinoma of
the thyroid. Surgery 1997;121:715
31. Charkes ND. On the prevalence of familial nommedullary thyroid cancer in
multiply affected kindreds. Thyroid 2006;16:181-6.
32. Charkes ND. On the prevalence of familial nonmedullary thyroid cancer.
Thyroid 1998;8:857
33. Uchino S, Noguchi S, Yamashita H, Murakami T, Watanabe S, Ogawa T, et
al. Detection of asymptomatic differentiated thyroid carcinoma by neck
ultrasonographic screening for familial nonmedullary thyroid carcinoma.
World J Surg 2004;28:1099-102.
34. Vriens MR, Sabanci U, Epstein HD, et al.: Reliability of fine-needle aspiration
in patients with familial nonmedullary thyroid cancer. Thyroid 1999; 9:1011–
1016.
35. Lupoli G, Vitale G, Caraglia M, Fittipaldi MR, Abbruzzese A, Tagliaferri P, et
al. Familial papillary thyroid microcarcinoma: a new clinical entity. Lancet
1999;353:637-9.
36. Segal K, Friedental R, lubin E, et al. Papillary carcinoma of the thyroid.
Otolaryngol Head Neck Surg 1995;113:356-363.

63
37. McConahey WM, Hay ID, Woolner LB, et al. Papillary thyroid cancer treated
at the Mayo Clinic, 1946 through 1970: initial manifestation, pathologic
findings, therapy, and outcome. Mayo Clin proc 1986;61:978-996.
38. Cetta F, Montalto G, Gori M, et al. Germline mutations of the APC gene in
patients with familial adenomatous polyposis-associated thyroid carcinoma:
results from a European cooperative study. J Clin Endocrinol Metab.
2000;85:286-292.
39. Mazzaferri EL. Papillary thyroid carcinoma: factores influencing prognosis
and current therapy. Semin Oncol 1987;14:315-332.
40. Hay ID. Papillary thyroid carcinoma. Endocrinol Metab Clin North Am
1990;19:545-576.
41. Bignell GR, Canzian F, Shayeghi M, et al. Familial nontoxic multinodular
thyroid goiter locus maps to chromosome 14q but does not account for
familial nonmedullary thyroid cancer. Am J Hum Genet. 1997;61:1123-1130
42. Sanders LE, Cady B. Differentiated thyroid cancer:reexamination of risk
groups and outcome of treatment. Arch Surg 1998;133:419-425.
43. Maxwell EL, Hall FT, Freeman JL. Familial non-medullary thyroid cancer: a
matched-case control study. Laryngoscope. 2004;114:2182-2186.
44. Sippel SR, Caron RN, Clark OH. An evidence-based approach to familial
nonmedullary thyroid cancer:screening, clinical management, and follow-up.
Word J Surg 2007;31:924-933.
45. Tan GH, Gharib H. Thyroid incidentalomas: management approaches to
nonpalpable nodules discovered incidentally on thyroid imaging. Ann Intern
Med 1997;126:226-31.

64
46. Uchino S, Noguchi S, Yamashita H, Murakami T, Watanabe S, Ogawa T, et
al. Detection of asymptomatic differentiated thyroid carcinoma by neck
ultrasonographic screening for familial nonmedullary thyroid carcinoma.
World J Surg 2004;28:1099-102.
47. Akerman M, Tennval J, biörklund A, Martensson, MöllerT. Sensivity and
specificity of fine needle aspiration cytology in the diagnosis of tumors of the
thyroid gland. Acta Cytol1985; 29:850-855.
48. Mc Henry CR, Walfish PG, Rosen IB. 1993 Non diagnostic fine needle
aspiration biopsy: A dilemma in management of nodular thyroid disease. Am
Surg 1993;59:415-419.
49. Löwhagen T,Granberg PO, Lundell G, Skinnari P, Sundblad R, Willwms JS.
Aspiration biopsycytology (ABC) in nodules of thyroid gland suspected to be
malignant. Surg Clin North Am 1979;59:3-18.
50. Ozaki O, Ito K, Kobayashi K, Suzuki A, ManabeY, Hosoda Y. familial
occurrence of differentiated, nonmedullary thyroid carcinoma. World J Surg
1988; 12:565-571.
51. Kobayashi K,Tanaka y, Ishiguro S, Mori T, Mitani Y, Shigemasa C. Family
with nonmedullary thyroid neoplasmas. J Surg Onc 1995; 58:274-277.
52. Zarnegar R, Brunaud L, Clarck OH. Prevention, evaluation, and
management of complications following thyroidecomy for thyroid carcinoma.
Endocrinol Metabol Clin North Am 2003;32:483-502.
53. Donis-Keller H, Dou S, Chi D, Carlson KM, Toshima K, Lairmore TC, Howe
JR, Moley JF, Goodfellow P, Wells SA Jr . Mutations in the ret proto-

65
oncogene are associated with men 2a and fmtc. Hum Mol Genet 1993;
2:851–856.
54. Wells SA Jr, Chi DD, Toshiima K, Dehner LP, Coffin CM, Dowton SB,
Ivanovich JL, DeBenedetti MK, Dilley WG, Moley JF, et al. Predictive DNA
testing and prophylactic thyroidectomy in patients at risk for multiple
endocrine neoplasia type 2a. Ann Surg 1994; 220:237–247; discussion 247–
250.
55. Canzian F, Amati P, Harach HR. A gene predisposing to familial thyroid
tumors with cell oxyphilia maps to chromosome 19p13.2. Am J Hum Genet
1998; 63:1743–1748.
56. Bevan S, Pal T, Greenberg CR, Kraimps JL, Lesueur F, Barbier J, Levillain
P, Romeo G, Bonneau D. A comprehensive analysis of mng1, tco1, fptc,
pten, tshr, and trka in familial nonmedullary thyroid cancer: Confirmation of
linkage to tco1. J Clin Endocrinol Metab 2001; 86:3701–3704.
57. Malchoff CD, Sarfarazi M, Tendler B, Forouhar F, Whalen G, Joshi V, Arnold
A, Malchoff DM. Papillary thyroid carcinoma associated with papillary renal
neoplasia: Genetic linkage analysis of a distinct heritable tumor syndrome. J
Clin Endocrinol Metab 2000; 85:1758–1764.
58. McKay JD, Lesueur F, Jonard L, Pastore A, Williamson J, Hoffman L,
Burgess J, Duffield A, Papotti M, Stark M, Sobol H, Maes B, Murat A,
Kaariainen H, Bertholon-Gregoire M, Zini M, Rossing MA, Toubert ME,
Bonichon F, Cavarec M, Bernard AM, Boneu A, Leprat F, Haas O, Lasset C,
Schlumberger M, Canzian F, Goldgar DE, Romeo G. Localization of a

66
susceptibility gene for familial nonmedullary thyroid carcinoma to
chromosome 2q21. Am J Hum Genet 2001; 69: 440–446.
59. McKay JD, Thompson D, Lesueur F, Stankov K, Pastore A, Watfah C, Strolz
S, Rickabona G, Moncayo R, Romeo G, Goldgar DE. Evidence for
interaction between the tco and nmtc1 loci in familial nonmedullary thyroid
cancer. J Med Genet 2004;41:407–412.
60. Lesueur F, Stark M, Tocco T, Ayadi H, Delisle MJ, Goldgar DE,
Schlumberger M, Romeo G, Canzian F. Genetic heterogeneity in familial
nonmedullary thyroid carcinoma: Exclusion of linkage to ret, mng1, and tco
in 56 families. Nmtc consortium. J Clin Endocrinol Metab 1999;84:2157–
2162.
61. Burgess JR, Duffield A, Wilkinson SJ, Ware R, Greenaway TM, Percival J,
Hoffman L. Two families with an autosomal dominant inheritance pattern for
papillary carcinoma of the thyroid. J Clin Endocrinol Metab 1997;82:345–
348.
62. Kazakov VS, Demidchik EP, Astakhova LN. Thyroid cancer after
chernobyl. Nature 1992;359:21.
63. Bounacer A, Wicker R, Caillou B, Cailleux AF, Sarasin A, Schlumberger M,
Suarez HG. High prevalence of activating ret proto-oncogene
rearrangements, in thyroid tumors from patients who had received external
radiation. Oncogene 1997;15:1263–1273.
64. Bacher-Stier C, Riccabona G, Totsch M, Kemmler G, Oberaigner W,
Moncayo R. Incidence and clinical characteristics of thyroid carcinoma after
iodine prophylaxis in an endemic goiter country. Thyroid 1997;7:733–741.

67
65. Learoyd DL, Messina M, Zedenius J, Robinson BG. Molecular genetics of
thyroid tumors and surgical decisionmaking. World J Surg 2000; 24:923–
933.
66. Xing M. The T1799A BRAF mutation is not a germline mutation in familial
non-medullary thyroid cancer. Clin Endocrinol 2005;63:263-6.
67. McKay JD, Williamson J, Leseur F, Stark M, Duffield S, Canzian F, et al. At
least three genes account for familial papillary thyroid carcinoma: TCO and
MNG1 excluded as susceptibility loci from a large Tasmanian family. Eur J
Endocrinol 1999;141:122-5.
68. Plail RO, Bussey HJ, Glazer G, Thomson JP. Adenomatous polyposis: An
association with carcinoma of the thyroid. Br J Surg 1987;74:377–380.
69. Iwama T, Mishima Y, Utsunomiya J. The impact of familial adenomatous
polyposis on he tumorigenesis and mortality at the several organs. Its
rational treatment. Ann Surg 1993;217:101–108.
70. Harach HR, Williams GT, Williams ED. Familial adenomatous polyposis
associated thyroid carcinoma: A distinct type of follicular cell neoplasm.
Histopathology 1994;25:549–561.
71. Lee S, Hong SW, Shin SJ, Kim YM, Rhee Y, Jeon BI, et al. Papillary thyroid
carcinoma associated with familial adenomatous polyposis: molecular
analysis of pathogenesis and review of the literature. Endocr J
2004;51(3):317-23.
72. Cetta F, Pelizzo MR, Curia MC, Barbansi A. Genetics and clinicopathological
findings in thyroid carcinomas associated with familial adenomatous
polyposis. Am J Pathol 1999;155:7–9.

68
73. Cetta F, Chiapetta G, Melillo RM. The ret/ptc1 oncogene is activated in
familial adenomatous polyposis-associated thyroid papillary carcinomas. J
Clin Endocrinol Metab 1998;83:1003–1006.
74. Soravia C, Sugg SL, Berk T, Petracci M, Montasto G, Santoro M, Fusco A.
Familial adenomatous polyposis-associated thyroid cancer: A clinical,
pathological, and molecular genetics study. Am J Pathol 1999 ;154:127–135.
75. van der Linde K, Vasen HF, van Vliet AC: Occurrence of thyroid carcinoma
in Dutch patients with familial adenomatous polyposis. An epidemiological
study and report of new cases. Eur J Gastroenterol Hepatol 1998, 10:777–
781.
76. Perrier ND, van Heerden JA, Goellner JR, et al. Thyroid cancer in patients
with familial adenomatous polyposis. World J Surg 1998, 22:738–742;
discussion, 743.
77. Bell B, Mazzaferri EL. Familial adenomatous polyposis (Gardner’s
syndrome) and thyroid carcinoma. A case report and review of the literature.
Dig Dis Sci 1993;38:185–190.
78. Camiel MR, Mule JE, Alexander LL, Benninghoff DL. Association of thyroid
carcinoma with Gardner’s syndrome in siblings. N Engl J Med
1968;278:1056–1058.
79. Houlston RS, Stratton MR. Genetics of non-medullary thyroid cancer. Q J
Med 1995;88:685–693.
80. Mallory SB. Cowden syndrome (multiple hamartomas syndrome). Dermatol
Clin 1995;13:27–31.

69
81. Haggitt RC, Reid BJ. Hereditary gastrointestinal polyposis syndromes. Am J
Surg Pathol 1986;10:871–887.
82. Kovich O, Cohen D. Cowden’s syndrome. Dermatol Online J 2004;10:3.
83. Harach HR, Soubeyran I, Brown A, et al.: Thyroid pathologic findings in
patients with Cowden disease. Ann Diagnost Pathol 1999; 3:331–140.
84. Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, Matthews S,
Nakura J, Miki T, Ouasis S, Martin GM, Mulligan J, Schellenberg GD 1996
Positional cloning of the Werner’s syndrome gene. Science 272:258–262.
85. Stratakis CA, Courcoutsakis NA, Abati A, Filie A, Doppman JL, Carney JA,
Shawker T. Thyroid gland abnormalities in patients with the syndrome of
spotty skin pigmentation, myxomas, endocrine overactivity, and
schwannomas (carney complex). J Clin Endocrinol Metab 1997;82:2037–
2043.
86. Bertherat J. Carney complex. Orphanet J Rare Dis 2006;1:21-6.
87. Ohta S, Katsura T, Shimada M, Shima A, Chishiro H, Matsubara H. Ataxia-
telangiectasia with papillary carcinoma of the thyroid. Am J Pediatr Hematol
Oncol 1986; 8:255–257.
88. Sandoval C, Schantz S, Posey D, Swift M. Parotid and thyroid gland cancers
in patients with ataxia-telangiectasia. Pediatr Hematol Oncol 2001;18:485–
490.
89. Narita T, Takagi K. Ataxia-telangiectasia with dysgerminoma of right ovary,
papillary carcinoma of thyroid, and adenocarcinoma of pancreas. Cancer
1984;54:1113–1116.

70
90. Reed MW, Harris SC, Quayle AR, Talbot CH. The association between
thyroid neoplasia and intestinal polyps. Ann R Coll Surg Engl 1990; 72:357–
359.
91. Yamamoto M, Hoshino H, Onizuka T, Ichikawa M, Kawakubo A, Hayakowa
S. Thyroid papillary adenocarcinoma in a woman with peutz-jeghers
syndrome. Intern Med 1992;31:1117–1119.
92. Brandi M-L. Pituitary and less common abnormalities in multiple endocrine
neoplasia type 1. In: Doherty GM, Skogseid B (eds) Surgical Endocrinology.
Lippincott Williams & Wilkins, Philadelphia; 2001pp 525–529.
93. Yang GC, Yao JL, Feiner HD, Roses DF, Kumar A, Mulder JE. Lipid-rich
follicular carcinoma of the thyroid in a patient with mccune-albright
syndrome. Mod Pathol 1999;12: 969–973.
94. Peccin S, de Castsro JA, Furlanetto TW, Furtado AP, Brasil BA,
Czepielewski MA. Ultrasonography: is it useful in the diagnosis of cancer in
thyroid nodules? J Endocrinol Invest 2002;25:39-43.
95. Tomimori EK, Bisi H, Medeiros-Neto G, Camargo RY. [Ultrasonographic
evaluation of thyroid nodules: comparison with cytologic and histologic
diagnosis]. Arq Bras Endocrinol Metab 2004;48:105-13.
96. Burch HB. Evaluation and management of the solid thyroid nodule.
Endocrinol Metab Clin North Am 1995;24:663-710.
97. Baloch ZW, LiVolsi VA. Fine-needle aspiration of thyroid nodules: past,
present, and future.Endocr Pract 2004;10:234-41.
98. Yokozawa T. Cancer de tireóide detectado por agulha fina guiada pelo ultra-
som. Arq Bras Endocrinol Metab 1998;42:296-8.

71
99. Hegedus L. The Thyroid nodule. N Engl J Med 2004;351:1764-71.
100. Gharib H. Manegement of thyroid nodules: another look. Thyroid
Today 1997 ;20:1-1.
101. Hegedus L, St BOnnema SJ et al. Manegement of simple nodular
goiter:current status and future perspectives. Endoc Rev 2003;24:102-32.
102. Baloch Z, LiVolsi VA, Jain P et al. Role of repeat fine-needle
aspiration biopsy (FNAB) in the manegement of thyroid nodules. Diagn
Cutopathol 2003;29:203-6.