c copyright 2012 mary ann liebert notice changes ... · (djavan et al., 2003). thus, ......
TRANSCRIPT

This is the author’s version of a work that was submitted/accepted for pub-lication in the following source:
Adamson, R.E., Frazier, A.A., Evans, H., Chambers, Karen F., Schenk, E.,Essand, M., Birnie, R., Mitry, R.R., Dhawan, A., & Maitland, N.J. (2012) Invitro primary cell culture as a physiologically relevant method for preclinicaltesting of human oncolytic adenovirus. Human Gene Therapy, 23(2), pp.218-230.
This file was downloaded from: http://eprints.qut.edu.au/55851/
c© Copyright 2012 Mary Ann Liebert
Notice: Changes introduced as a result of publishing processes such ascopy-editing and formatting may not be reflected in this document. For adefinitive version of this work, please refer to the published source:
http://dx.doi.org/10.1089/hum.2011.021

In Vitro Primary Cell Culture as a PhysiologicallyRelevant Method for Preclinical Testing
of Human Oncolytic Adenovirus
R.E. Adamson,1 A.A. Frazier,2 H. Evans,1 K.F. Chambers,1 E. Schenk,3 M. Essand,4
R. Birnie,2 R.R. Mitry,5 A. Dhawan,5 and N.J. Maitland1,2
Abstract
Ad[I/PPT-E1A] is an oncolytic adenovirus that specifically kills prostate cells via restricted replication by aprostate-specific regulatory element. Off-target replication of oncolytic adenoviruses would have serious clinicalconsequences. As a proposed ex vivo test, we describe the assessment of the specificity of Ad[I/PPT-E1A] viralcytotoxicity and replication in human nonprostate primary cells. Four primary nonprostate cell types were selectedto mimic the effects of potential in vivo exposure to Ad[I/PPT-E1A] virus: bronchial epithelial cells, urothelial cells,vascular endothelial cells, and hepatocytes. Primary cells were analyzed for Ad[I/PPT-E1A] viral cytotoxicity inMTS assays, and viral replication was determined by hexon titer immunostaining assays to quantify viral hexonprotein. The results revealed that at an extreme multiplicity of infection of 500, unlikely to be achieved in vivo,Ad[I/PPT-E1A] virus showed no significant cytotoxic effects in the nonprostate primary cell types apart from thehepatocytes. Transmission electron microscopy studies revealed high levels of Ad[I/PPT-E1A] sequestered in thecytoplasm of these cells. Adenoviral green fluorescent protein reporter studies showed no evidence for nuclearlocalization, suggesting that the cytotoxic effects of Ad[I/PPT-E1A] in human primary hepatocytes are related toviral sequestration. Also, hepatocytes had increased amounts of coxsackie adenovirus receptor surface protein.Active viral replication was only observed in the permissive primary prostate cells and LNCaP prostate cell line,and was not evident in any of the other nonprostate cells types tested, confirming the specificity of Ad[I/PPT-E1A]. Thus, using a relevant panel of primary human cells provides a convenient and alternative preclinical assayfor examining the specificity of conditionally replicating oncolytic adenoviruses in vivo.
Introduction
The field of gene therapy has rapidly expanded in thelast 20 years, with particular efforts focused on cancer
treatment (Vorberg and Hunt, 2002). Viral vectors remain themost efficient vehicles for transferring therapeutic genes intohuman cells, and various approaches have been employed,including suicide-gene therapy, oncolytic adenovirus therapy,immunogene therapy, and vaccine-based strategies. Of the1,644 gene-therapy clinical protocols reported (see Wiley Website: www.wiley.com/legacy/wileychi/genmed/clinical/),29% use adenovirus vectors or adeno-associated vectors todeliver therapeutic or marker genes. The human serotype 5adenovirus is often the vector of choice, because it is able to
transduce cells at high efficiency and independently of celldivision. Oncolytic gene therapy for cancer has evolved fromutilizing replication-defective adenoviruses containing a sin-gle therapeutic gene, to conditionally replicating oncolyticadenoviruses (CRAds), which allow preferential replication intarget cancer cells. Such CRAds can produce hundreds of viralprogeny per cell and result in an amplified therapeutic gene-targeted response as they spread locally, infecting largernumbers of neighboring cells until all susceptible cells areeliminated. Thus, infection by a small number of CRAd par-ticles could theoretically eliminate a large tumor burden.In humans, both replication-competent and replication-incompetent adenoviruses have shown low toxicity, withspecific antitumor activity in preclinical tumor models and
1YCR Cancer Research Unit, Department of Biology, University of York, Heslington, York YO10 5DD, United Kingdom.2Pro-Cure Therapeutics Ltd., York Science Park, York, YO10 5NY, United Kingdom.3Department of Urology, Erasmus MC, Rotterdam 3015CE, The Netherlands.4Department of Immunology, Genetics and Pathology, Uppsala University, Uppsala SE-75185, Sweden.5Hepatocyte Biology and Transplantation Group, Institute of Liver Studies, King’s College Hospital, London SE5 9RS, United Kingdom.
HUMAN GENE THERAPY 22:1–14 (XXXXX 2011)ª Mary Ann Liebert, Inc.DOI: 10.1089/hum.2011.021
1

humans (Kim et al., 1994; Freytag et al., 1998, 2002; Rogulskiet al., 2000). Human adenovirus-based vectors are hampered,however, by the lack of a good animal model in which toassay off-target effects and to generate high viral titers tostudy human adenoviral serotypes ( Jogler et al., 2006). Un-fortunately, human adenoviruses are at best only semi-permissive even in nonhuman primates (Maitland et al.,2010). Animal adenoviruses have been studied in theircorresponding host cells, to allow safety and efficacy issuesto be addressed, but systems for extrapolating such con-clusions to humans are limited. In 1999, the use of adeno-virus vectors as a gene-delivery system generated muchcontroversy when a patient died from the injections of anornythine transcarbamylase–encoding adenovirus. The im-munocompromised patient received the highest dose everadministered in clinic [3.6 · 1013 virus particles (VP)]. Thiswas the first reported death resulting as a direct conse-quence of viral gene therapy (Carmen, 2001). As a conse-quence, the National Institutes of Health and the U.S. Foodand Drug Administration issued tighter guidelines andregulations, and new standards were set to improve thequality and safety of adenoviral clinical trials. Thus, thereis a requirement for the generation of more relevantand convenient preclinical testing protocols for oncolyticadenoviruses.
Within the European Union GIANT consortium (http://www.york.ac.uk/biology/units/cru/giant/welcome.htm),a prostate-specific CRAd is due to be examined in a phase Idose-escalating clinical trial in patients with localized pros-tate cancer (Schenk et al., 2010). Prostate cancer is the mostcommonly diagnosed cancer in men aged 65 and over andthe second most common cause of cancer death in men in theUnited Kingdom. Treatment of localized disease is possi-ble by radical prostatectomy, but in 35% of cases micro-metastases can persist after primary treatment and oftendevelop into an incurable androgen-insensitive disease(Djavan et al., 2003). Thus, there is an urgent need for im-proved localized and systemic treatments that provide long-lasting effects. The prostate-specific CRAd to be used in theGIANT clinical trial, Ad[I/PPT-E1A], contains a complexchimeric promoter (Cheng et al., 2004), which combinesthe T-cell receptor c-chain alternate reading frame protein(TARP) promoter (Cheng et al., 2003), with the prostate-specific PSA- and PSMA- (prostate-specific membrane anti-gen) enhancer elements, to drive adenoviral replication inprostate cells only (Cheng et al., 2006). Ad[I/PPT-E1A] hasthe added advantage that it is active in both the presence andabsence of androgens, so this virus should also be suitablefor the treatment of patients with castration-resistant pros-tate cancer. In both in vitro and animal studies, Ad[I/PPT-E1A] has specifically demonstrated destruction of prostatetumor cells, for example, by enhancing the survival of micebearing tumors (Cheng et al., 2004, 2006). In the proposedtrial, immunocompetent patients with localized prostatecancer will be treated by a single intraprostatic injection ofAd[I/PPT-E1A], ranging from 1 · 1011 to 5 · 1012 VP, prior toradical prostatectomy. According to the International Con-ference on Harmonisation of Technical Requirements forRegistration of Pharmaceuticals for Human Oncolytic Viru-ses (September 17, 2009), the regulatory authorities requireevidence that an oncolytic virus to be tested in a clinical trialcannot replicate in nontarget cells and cause damage to un-
desired organs and tissues. Thus, they must undergo rigor-ous testing in preclinical toxicology studies to show relevantoncolytic efficacy and safety.
As an alternative preclinical approach to animal testing, wehave used human nonprostate primary cells in in vitro cellculture to test whether Ad[I/PPT-E1A] replication is limitedonly to the prostate target cells by assessing the specificity andsensitivity of Ad[I/PPT-E1A] replication and cytolytic killingin vitro. The primary cell types were chosen for study, becausethe parental organs are either close to the site of adenoviraladministration or should be exposed to the highest percentageof cardiac output (blood flow) and would likely have in-creased contact with any circulating Ad[I/PPT-E1A] virusin vivo in the phase I GIANT human clinical trial. They in-cluded human vascular endothelial cells, bronchial epithelialcells, urothelial cells, and hepatocytes. In all cases, no repli-cation of the retargeted Ad[I/PPT-E1A] virus was seen, de-spite retention of the ability to attach to and penetrate thevarious cell types. We conclude that in vitro preclinical testingprocedures using a relevant panel of human primary cells is afeasible approach for the preclinical analysis of the efficacyand specificity of oncolytic adenoviruses, and in this respectwill have significant additional value next to animal models.
Materials and Methods
Maintenance of primary cells and cell lines
LNCaP cells (malignant human prostate adenocarcinomacell line derived from a lymph-node metastasis; EuropeanCollection of Animal Cell Cultures, Porton Down, UK) werecultured in RPMI (Invitrogen, UK) supplemented with 10%heat-inactivated fetal calf serum (FCS; PAA, UK) and 2 mmol/LL-glutamine (Invitrogen, UK). HEK293 cells (human embryonickidney cell line; ATCC, Manassas, VA) were cultured inDulbecco’s modified Eagle’s medium (DMEM; GIBCO, UK)supplemented with 10% heat-inactivated FCS (PAA) and2 mmol/L L-glutamine (Invitrogen). The following humanprimary cells were maintained in their associated media: pri-mary human bronchial epithelial cells (HBEC; TCS/Cellworks,UK) were cultured in bronchial epithelial cell growth mediumwith supplements (TCS/CellWorks, UK); primary humanumbilical vein endothelial cells (HUVEC; Lonza, USA) werecultured in Endothelial Cell Basal Medium with supplements(Lonza, USA); primary human urothelial cells (HUC; kindlydonated by Professor J. Southgate, Jack Birch Unit for MolecularCarcinogenesis, University of York, UK) were cultured in Pri-maria flasks (BD Biosciences, UK) in keratinocyte serum-freemedium (KSFM; Invitrogen, UK) supplemented with 2 mmol/L L-glutamine, epidermal growth factor (EGF), and bovinepituitary extract (GIBCO, UK); primary human hepatocytes(kindly provided by Prof. A. Dhawan and Dr. R. Mitry,Hepatocyte Biology and Transplantation Group, King’s CollegeHospital, UK) were cultured in Eagle’s minimum essentialmedium, supplemented with 10% heat-inactivated FCS,2 mmol/L L-glutamine; primary human prostate cells wereisolated with consent, from thick-needle cores of patient tissueremoved by radical prostatectomy (York District Hospital,York, UK; Castle Hill Hospital, Hull, UK). Prostate cancer wasconfirmed by a uropathologist, by histology of adjacent tissue.Undifferentiated prostate cells were maintained in completeKSFM supplemented with EGF (Invitrogen), bovine pituitaryextract (Invitrogen), 2 ng/ml leukemia inhibitory factor (Sigma,
2 ADAMSON ET AL.

UK), 2 ng/ml stem cell factor (Sigma, UK), 1 ng/ml Gm, and100 ng/ml cholera toxin (Sigma, UK). Irradiated STO cells(mouse embryonic fibroblasts) were routinely added as feeders.Differentiated primary prostate cells were cultured in 1:1DMEM:Ham’s F-12 medium (vol/vol; DMEM Culture Med-ium, Gibco, UK; Ham’s F-12 Culture Medium, Lonza, UK),2 mmol/L L-glutamine, 10% heat-inactivated FCS, and 10 nMdihydrotestosterone (Innovative Research of America, USA). Allcell culture was performed without the addition of antibiotics.
Adenoviral samples
Ad[I/PPT-E1A] was manufactured at the GMP Facilities atthe Center for Cell and Gene Therapy at Baylor College ofMedicine (Houston, TX), specifically for the European UnionGIANT project. The virus was supplied at a concentration of1 · 1011 VP/ml in 20 mM Tris, pH 8.0, 25 mM NaCl, 2.5%glycerol. As a positive viral control, wild-type adenovirus(AdWT, serotype 5) was used (sourced from ATCC) (batch no.001504, purity > 99% in buffer: 20 mM Tris, pH 8.0, 25 mMNaCl, 2.5% glycerol). Viral titer was 5.8 · 1011 VP/ml. As anegative viral control, a replication-incompetent adenovirus(AdMock) was used (Carlsson et al., 2003). This virus has anE1-deleted and E3-deleted replication-deficient adenoviralvector of serotype 5. It contains no extra transgene and wasproduced by recombination of pShuttle and pAdEasy1 (Heet al., 1998). AdMock was sourced from Professor MagnusEssand (University of Uppsala, Sweden) (batch no. 20090505,purity 99%, in buffer: 10 mM Tris-HCl, pH 7.9, 1 mM MgCl2, 4%sucrose). The viral titer was 4.1 · 1012 VP/ml. Two replication-incompetent reporter adenoviruses, AdCMV-gfp and AdI/PPT-gfp, were also sourced from Professor Essand.
MTS assays
LNCaP and primary cells were seeded in triplicate at80% confluency in 48-well plates, 500 ll/well cell culturemedium. The following day, cells were infected at roomtemperature (RT) for 1–2 hr with gentle agitation with 0, 50,500, and/or 5,000 multiplicity of infection (MOI) (VP/cell) ofeither Ad[I/PPT-E1A] or the AdWT. Virus was prepared inthe appropriate cell culture medium for each primary celltype assayed. After viral incubation, the virus was removed.The cells were washed with medium and incubated for up to14 days, at 37�C, 5% CO2. Medium was changed on days 3, 6,7, 10, and 11. On days 0, 3, 6, 10, and 14, 60 ll/well MTSreagent (CellTiter 96 AQueous One Solution Cell ProliferationAssay; Promega, UK) was added and incubated for 2 hr at37�C, 5% CO2, followed by ultraviolet irradiation for 15 min.The absorbance at 485 nm and 620 nm was determined foreach sample on a POLARstar OPTIMA plate reader (BMGLabtech, UK). Average final absorbance values for the tripli-cate wells were calculated and standard error propagationdetermined by using Microscoft Excel (Redmond, WA). Thepercent cell viability for each time point was determined bynormalization against MOI 0, plotted with SigmaPlot(Hounslow, London, UK) and presented as bar graphs.
Adenoviral replication assays using hexon titration
Primary cells and cell lines were seeded at 80% confluencyin 12-well plates. Twenty-four hours later, cells were infectedin triplicate with Ad[I/PPT-E1A] or AdWT at MOI 0 and 500
for 1–2 hr at RT, with gentle agitation. Virus was removed bywashing the cells with culture medium, and subsequentlythe cells were incubated at 37�C, 5% CO2 for 14 days. Freshmedium was added every 3 days to maintain cell viability.Culture media from the cells were collected on days 3, 6, 10,and 14 post infection and stored at or below - 75�C. On days7 and 11, medium was changed and discarded. Cells wereharvested on day 14 where possible, and total cell lysateswere prepared by repeated freeze/thaw. Hexon titer assayswere performed in triplicate on 2.5 · 105 HEK293 cells(ATCC) using serially diluted viral-infected medium sam-ples, following the Adeno-X Rapid Titre Kit procedure(Clontech, UK). By using this kit, adenoviral plaques weredetected by immunostaining using an anti-hexon antibody(Clontech, UK), and viral plaques were counted under lightmicroscopy. Mean viral titers for each time point and cor-responding standard deviations were calculated with Mi-crosoft Excel. Bar graphs were plotted in SigmaPlot withstandard errors shown for each sample.
Ad[I/PPT] promoter expression in primary humanhepatocytes using green fluorescentprotein (gfp) reporter adenovirus
Primary prostate and hepatocyte cells were infected witheither AdCMV-gfp or AdI/PPT-gfp virus at MOI 500 for2 hr. Virus was removed by washing the cells with culturemedium, and the cells were incubated at 37�C, 5% CO2 for48 hr. After this time, gfp fluorescent images were taken at· 60 magnification, under oil emersion, using a Nikon EclipseTE300 fluorescent microscope.
Quantitative PCR (qPCR) analysis of adenoviralgenome replication
Primary hepatocytes were infected in triplicate with Ad-Mock and AdWT at MOI 500 and with Ad[I/PPT-E1A] atMOI 5, 50, and 500 for 2 hr at RT. Virus was removed bywashing the cells with culture medium, and the cells wereincubated at 37�C, 5% CO2. Cells were harvested at 2, 24, and48 hr, and genomic DNA was extracted using a DNeasyBlood and Tissue kit (Qiagen). qPCR was performed on anABI Step-One-Plus Real Time PCR machine. Amplification ofan 84-bp fragment of the adenovirus fiber gene was carriedout using the primers 5’ TGGCTGTTAAAGGCAGTTTGG 3’and 5’ GCACTCCATTTTCGTCAAATCTT 3’ with detectionof amplified sequences by a Taqman probe (5’ TCCAATATCTGGAACAGTTCAAGTGCTCATCT 3’), which was la-beled at the 5’ end with the FAM fluorophore and at the 3’end with the TAMRA quencher. Primers and probe werepurchased from Sigma Genosys, UK. Reactions were carriedout in a volume of 20 ll in ABI Taqman Fast Master mix(Applied Biosystems, UK), containing primers and probe atconcentrations of 1.0 lM and 0.1 lM, respectively. Thermo-cycling parameters were 2 min at 50�C, 10 min at 95�C fol-lowed by 40 cycles of 95�C (30 sec) and 60�C (2 min).Analysis of the data was carried out using the softwareprovided, and test samples were compared to standards ofknown viral DNA content. Standard curves were preparedby spiking serial dilutions of virus into hepatocyte genomicDNA. Data are presented as bar graphs plotted in SigmaPlotwith standard errors shown for each sample.
PRECLINICAL TESTING OF HUMAN ADENOVIRUS 3

Localization of adenovirus in primary cellswith immunogold labeling of the hexon capsid proteinusing transmission electron microscopy (TEM)
Primary cells were seeded in appropriate cell culturemedia, onto Thermanox plastic coverslips (NUNC, UK)coated in a thin layer of Matrigel (Invitrogen). The cells wereinfected with MOI 500 of either AdWT, Ad[I/PPT-E1A], orAdMock for 1 hr. The virus was removed, and the cells werecultured for a further 36 hr. Cells were then fixed in 2%glutaraldehyde for 20 min. After washing in 100 mM sodiumphosphate buffer, adherent cells were osmicated and dehy-drated in a series of graded alcohols. A final solution of 50%TAAB resin (TAAB Laboratories Ltd., Australia) and 50%absolute ethanol was applied with warming to evaporate offall traces of alcohol. Resin-filled capsules were invertedover the coverslips and left to polymerize overnight at 60�C.Ultrathin (70 nm) sections were cut and collected on nickelgrids. Adenovirus-infected cells were observed on an FEITecnai G2 transmission electron microscope, operated at120 kV. Images were captured on an SIS MegaView IIcamera.
To localize the hexon capsid protein by immunogold la-beling of virus particles, primary cells were infected withAd[I/PPT-E1A] at MOI 500 for 1 hr at RT. The virus wasremoved, and the cells were cultured for a further 60 hr. Cellswere washed in PBS, removed from the well surface using acell scraper, and collected in a 1.5-ml Eppendorf tube. Cellswere then fixed and processed for cryosectioning followingthe method of Tokuyasu (1986). Immunogold labeling ofinfected adenoviruses within the primary cells using a rabbitpolyclonal anti-hexon antibody to adenovirus serotype 5(catalog no. ab24240; Abcam, UK) was performed followingthe procedure of Mitry et al. (2000). Adenovirus within pri-mary cells was observed on an FEI Tecnai G2 transmissionelectron microscope, operated at 120 kV. Images were cap-tured on an SIS MegaView II camera.
Immunocytochemical detection of coxsackieadenovirus receptor (CAR) protein in nonprostateprimary cells
CAR (green) was detected in nonprostate primary cellsand prostate cell lines by immunofluorescent staining using aprimary anti-CAR mouse monoclonal antibody (clone RmcB;Millipore Ltd.) (at 1:50) and a goat anti-mouse Alexa 488secondary antibody (at 1:500). For cell morphology, actin(red) was detected using an anti-actin rabbit monoclonalantibody (clone EP184E; Millipore Ltd.) (at 1:300) and a goatanti-rabbit Alexa 568 secondary antibody (at 1:500). Cellswere mounted in VECTORSHIELD containing DAPI (VectorLaboratories, Inc.) to visualize the nuclei (blue). Fluorescentimages were taken at · 60 magnification, under oil, using aNikon Eclipse TE300 fluorescent microscope.
Results
Cytotoxicity of Ad[I/PPT-E1A] infectionof human primary cells
Using a colorimetric MTS assay to determine cytotoxicity,cell viability was measured to determine whether the pros-tate-specific Ad[I/PPT-E1A] could kill nonprostate primarycells. The cytotoxicity data for the infection of primary cells
with a positive control adenovirus, AdWT, compared withthe prostate-specific adenovirus, Ad[I/PPT-E1A], are shownin Fig. 1. AdWT virus was cytotoxic to all primary cells in adose- and time-dependent manner, and at all concentrationstested all cells were dead by the final day (day 14) of theassay. In two positive control cell types (prostate cells), Ad[I/PPT-E1A] showed significant cytotoxic effects at MOI 500 and5,000 in LNCaP cells and in basal primary prostate cells byday 14 (Fig. 1A and B). This confirmed that the MTS assaywas able to detect cell death caused by Ad[I/PPT-E1A].
There was no significant cytotoxicity associated withAd[I/PPT-E1A] infections in bronchial, endothelial, or ur-othelial cells at any virus concentration (Fig. 1C–E and Table 1).In primary human hepatocytes, there was no significant cy-totoxicity associated with Ad[I/PPT-E1A] infections at lowervirus concentration (MOI 50). However, at higher viralamounts (MOI 500 and 5,000), a large number of dead he-patocytes were observed at day 6 with Ad[I/PPT-E1A] virus(Fig. 1F and G and Table 1). One possible mechanism ofaction for the cytotoxicity of Ad[I/PPT-E1A] in the fragilehepatocytes is an excessive virus uptake and overloadingof the cell with virus particles. A replication-incompetentadenovirus (AdMock) was used to transduce primary he-patocytes to examine the potential of viral load for initiatingthe observed cytotoxic effects (Fig. 1G). These results showedno cytotoxicity associated with the replication-incompetentadenovirus, indicating that the cytotoxicity observed in he-patocytes infected with Ad[I/PPT-E1A] was most likely notdue to nonspecific adenoviral particle overloading. How-ever, digital images of hepatocytes in culture throughout theduration of the assay showed that the uninfected cells weredying by day 6, indicating their fragile nature when main-tained in prolonged in vitro cell culture conditions (Fig. 2).
The cytotoxicity seen in hepatocytes was further investi-gated by adenovirus gfp reporter assays, genome-replicationstudies, adenoviral replication assays, and TEM studies todetermine whether the Ad[I/PPT-E1A] virus was activelyreplicating, or whether this observation was due to anothereffect (see below).
Ad[I/PPT-E1A] replication in human primary cells
The replication capacity of Ad[I/PPT-E1A] in vascularendothelial cells, bronchial epithelial cells, urothelial cells,and hepatocytes was assessed by determining the presenceof active viral particles secreted into the cell culture media atvarious times after viral infection, and in cell lysates at thefinal time point of the assay. Primary prostate cells alongwith a prostate-derived cell line, LNCaP, were used as pos-itive cell-type controls for Ad[I/PPT-E1A] replication. Thecollected supernatants and cell lysates were titered by in-fection of HEK293 cells, and the infectious plaques resultingfrom active Ad[I/PPT-E1A] virus were detected by im-munostaining for adenovirus hexon protein (Fig. 3). As areference for nonspecific viral replication, wild-type adeno-virus (AdWT) was included in each assay. The infectivity ofthe cell-culture supernatants collected at the different timepoints was compared with the level of infectivity of the inputvirus (AdWT or Ad[I/PPT-E1A]), namely, the ‘‘infection titer.’’To define the infection titers, HEK293 cells (2.5 · 105) weredirectly infected with the same concentrations (in VP/ml) ofAdWT or Ad[I/PPT-E1A] as were used to infect the prostate
4 ADAMSON ET AL.
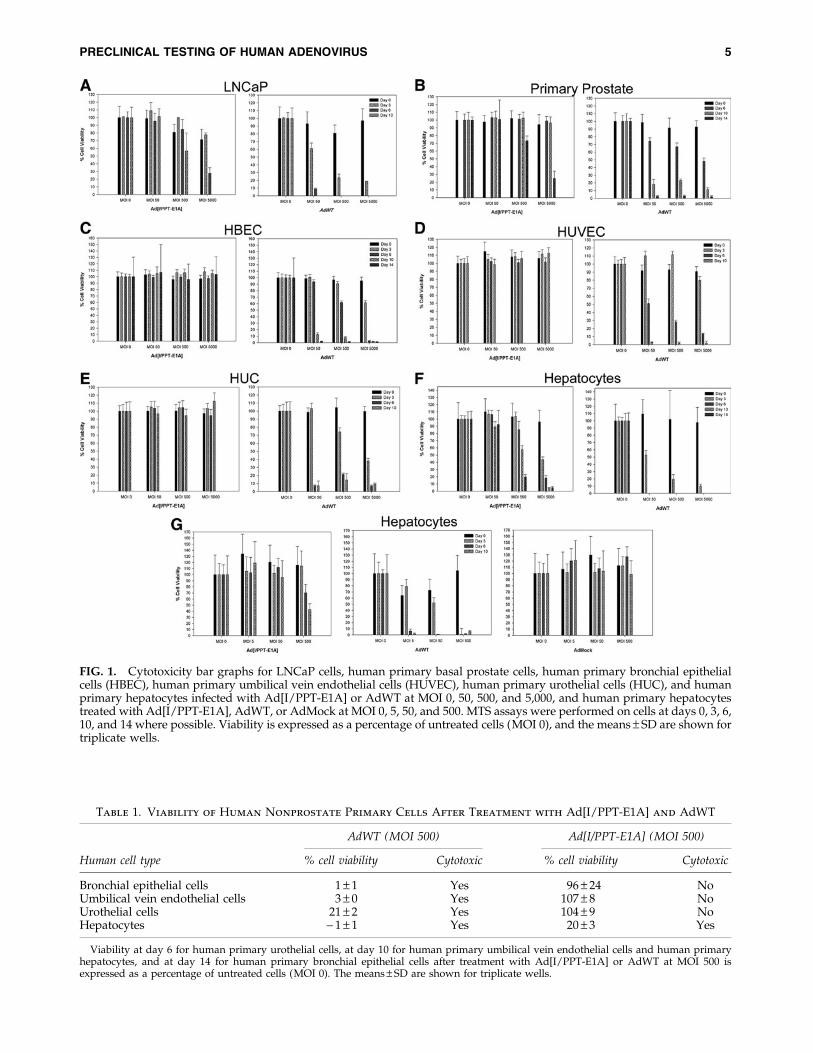
FIG. 1. Cytotoxicity bar graphs for LNCaP cells, human primary basal prostate cells, human primary bronchial epithelialcells (HBEC), human primary umbilical vein endothelial cells (HUVEC), human primary urothelial cells (HUC), and humanprimary hepatocytes infected with Ad[I/PPT-E1A] or AdWT at MOI 0, 50, 500, and 5,000, and human primary hepatocytestreated with Ad[I/PPT-E1A], AdWT, or AdMock at MOI 0, 5, 50, and 500. MTS assays were performed on cells at days 0, 3, 6,10, and 14 where possible. Viability is expressed as a percentage of untreated cells (MOI 0), and the means – SD are shown fortriplicate wells.
Table 1. Viability of Human Nonprostate Primary Cells After Treatment with Ad[I/PPT-E1A] and AdWT
AdWT (MOI 500) Ad[I/PPT-E1A] (MOI 500)
Human cell type % cell viability Cytotoxic % cell viability Cytotoxic
Bronchial epithelial cells 1 – 1 Yes 96 – 24 NoUmbilical vein endothelial cells 3 – 0 Yes 107 – 8 NoUrothelial cells 21 – 2 Yes 104 – 9 NoHepatocytes - 1 – 1 Yes 20 – 3 Yes
Viability at day 6 for human primary urothelial cells, at day 10 for human primary umbilical vein endothelial cells and human primaryhepatocytes, and at day 14 for human primary bronchial epithelial cells after treatment with Ad[I/PPT-E1A] or AdWT at MOI 500 isexpressed as a percentage of untreated cells (MOI 0). The means – SD are shown for triplicate wells.
PRECLINICAL TESTING OF HUMAN ADENOVIRUS 5

cancer cell line and human primary cells. These concentrationsdiffered between the various cell types (see Table 2). In caseswhere the data showed viral titers above the infection titer, weconcluded that the virus was actively replicating in these cells(Fig. 3A–C). Viral titers below the infection titer suggested thatthe virus had been sequestered internally, and active viralreplication was not occurring. Throughout the duration of theassay, cells undergoing apoptosis released their sequesteredvirus into the medium, which was observed in titers at day 6,10, and 14 in bronchial epithelial cells, urothelial cells, andhepatocytes (Fig. 3D, F, and G).
To establish the sensitivity and time parameters for theassay, infections of permissive LNCaP cells with AdWT andAd[I/PPT-E1A] were carried out (Fig. 3A). The data revealedthat both AdWT and Ad[I/PPT-E1A] actively replicated inLNCaP cells. The replication level titers were 10-fold less forAd[I/PPT-E1A], and there was also a slight delay comparedwith the AdWT virus (Fig. 3A). Similar replication assayswere carried out with cultures from primary prostate tissue,both undifferentiated (basal cells) and cultures induced todifferentiate to a luminal phenotype, both at MOI 500 (VP/cell). This MOI value was chosen to eliminate even inefficientattachment and replication as a possibility in vivo, althoughsuch viral titers would be impossible to achieve except inclose proximity to the needle-injection site. We observedactive replication of Ad[I/PPT-E1A] in basal prostate epi-thelial cells, increasing to > 3.13 · 107 infectious units (ifu)/ml by day 14 (ifu > 795% of the viral load) compared with a
peak titer of 6.72 · 106 ifu/ml at day 14 in differentiatedprostate epithelia (ifu 172% of the viral load) (Fig. 3B and C).Furthermore, the rate of replication of Ad[I/PPT-E1A] indifferentiated prostate epithelial cells was much slower thanthat in both LNCaP cells and basal prostate epithelial cells,suggesting that viral replication in these cells is attenuated.
In the cases of bronchial, endothelial, and urothelial cells,there was no detectable replication of Ad[I/PPT-E1A] whencompared with the input infection titers (Fig. 3D–F). How-ever, there was evidence of sequestered viral releasethroughout the different time points of the assay, as the cellsnaturally died in culture. In contrast, efficient replication ofAdWT was observed in these cell types.
We examined four independent donor primary hepato-cyte samples, all of which showed high levels of AdWTreplication. Although significant amounts of Ad[I/PPT-E1A]were retained by the cells, no net viral increase was ob-served, suggesting that Ad[I/PPT-E1A] does not replicate inhepatocytes (Fig. 3G). The data indicated that hepatocytescan ingest both AdWT and Ad[I/PPT-E1A]. Table 3 sum-marizes viral yields per cell for each cell type studied, as wellas the maximum percentage viral load. When compared withactive replication in primary prostate samples and LNCaPcells, the replication data for Ad[I/PPT-E1A] indicate agreatly reduced number of virus particles per cell in thenonprostate primary cells, including hepatocytes, after in-fection with the high MOI 500, indicating the presence ofresidual, nonreplicating virus.
FIG. 2. Digital images of cultures of uninfected primary hepatocytes (no virus) and hepatocytes infected with AdWT orAd[I/PPT-E1A] at MOI 500. Images were taken on days 3, 6, and 10 post infection. · 10 and · 20 refer to the microscopemagnification of the images.
6 ADAMSON ET AL.

FIG. 3. Viral replication bargraphs for LNCaP cells,human primary basal pros-tate cells, human primarydifferentiated prostate cells,human primary bronchialepithelial cells (HBEC), hu-man primary umbilical veinendothelial cells (HUVEC),human primary urothelialcells (HUC), and humanprimary hepatocytes infectedwith Ad[I/PPT-E1A] andAdWT at MOI 500. Infectedculture media collected ondays 3, 6, 10, and 14 (wherepossible) were titered inhexon titer assays, and themeans – SD are shown fortriplicate wells. The linesindicate the input load ofinfectious virus: solid line,AdWT; dashed line, Ad[I/PPT-E1A]. Black bars, AdWTvirus; gray bars, Ad[I/PPT-E1A] virus. *Viral titer isgreater than maximum titermeasured in the hexon titra-tion assay, i.e., > 3.13 · 107
ifu/ml or > 1.1 · 109 ifu/ml.
PRECLINICAL TESTING OF HUMAN ADENOVIRUS 7

I/PPT transcriptional activity in primaryhepatocytes and prostate cells
To establish the possibility of active Ad[I/PPT-E1A] rep-lication in primary hepatocytes, we examined I/PPT pro-moter activity in both primary hepatocytes and primaryprostate cells using serotype 5 adenoviruses that express gfp.The primary cells were infected for 48 hr with MOI 500 AdI/PPT-gfp or AdCMV-gfp. Figure 4 shows gfp expression fromAdI/PPT-gfp virus in prostate cells, but not in hepatocytes,whereas the control AdCMV-gfp virus expressed gfp in bothcell types after this time.
Ad[I/PPT-E1A] genome replication in hepatocytes
qPCR studies were performed in primary hepatocytes tomonitor any increase in adenoviral genome content between2 and 48 hr post infection (Fig. 5). Hepatocyte cultures wereinfected in triplicate with AdMock or AdWT at MOI 500 andwith Ad[I/PPT-E1A] at MOI 5, 50, and 500. Genomic DNAwas extracted from the cells at 2, 24, and 48 hr, and adeno-viral genome quantities were analyzed by qPCR targeting an84-bp fragment of the adenoviral fiber gene. At 48 hr, at MOIvalues under 500, which are not associated with a cytotoxiceffect in hepatocytes, Ad[I/PPT-E1A] genome-replicationlevels were equivalent to those of the replication-incompetent
AdMock, i.e., 1.21 · 10–3 ng compared with 1.36 · 10–3 ng forMOI 5, respectively. This level was 3,600 times lower thanlevels with AdWT and comparable to the hepatocyte hexontiter replication data. However, at the much higher MOI 500,although the Ad[I/PPT-E1A] genomic DNA quantity hadincreased, it was still significantly lower (45 times) than thatof the AdWT control. The data suggest that at extreme MOIvalues at or above 500, Ad[I/PPT-E1A] genomic DNA rep-lication occurs, but as the replication assays do not showactive viral particle production even at MOI 500, it is unlikelythat these genomes are packaged into infective viral particlesthat could actively establish secondary infections. TEMstudies were performed to examine this finding further.
Intracellular localization of Ad[I/PPT-E1A] particlesin human primary hepatocytes by TEMand immunogold TEM
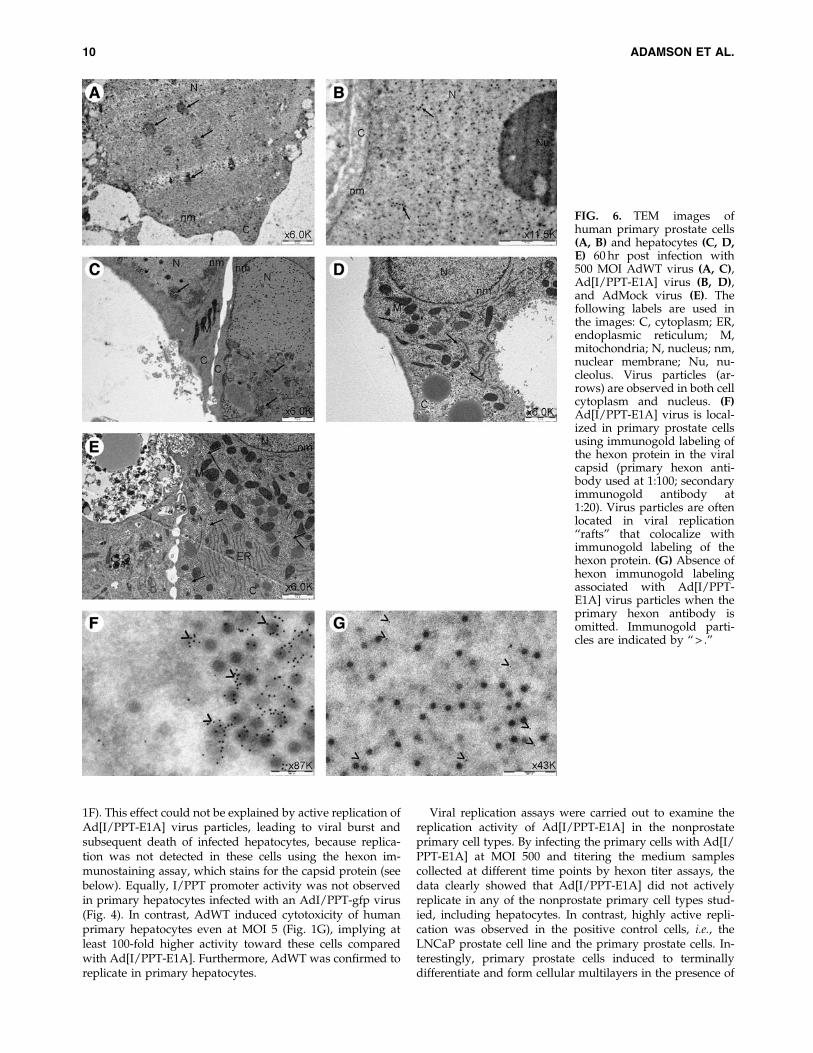
We examined the location of adenovirus particles in in-fected basal prostate cells and hepatocytes initially by TEM.TEM images showed replicated AdWT in ‘‘rafts’’ within thenuclei of both primary prostate cells and primary hepato-cytes by 60 hr post infection (Fig. 6A and C). We also ob-served replicated Ad[I/PPT-E1A] in similar rafts in thenuclei of primary prostate cells (Fig. 6B). Replication raftswere not observed in the nuclei of primary hepatocytes with
Table 2. Viral Titer Counts for the Input Virus and the Infection Titers of the Prostate
LNCaP Cell Line and the Human Nonprostate Primary Cell Types
Humancell types
Numberof cells plated
per well MOI
AdWTinput virus
(VP/ml)
AdWT infectiontiter counts
(ifu/ml)
Ad[I/PPT-E1A]input virus
(VP/ml)
Ad[I/PPT-E1A]infection titer
counts (ifu/ml)
LNCaP cells 240,000 500 6.0 · 107 1.4 · 107 6.0 · 107 5.6 · 106
Undifferentiated prostate cells 70,000 500 3.5 · 107 4.4 · 106 3.5 · 107 3.9 · 106
Differentiated prostate cells 140,000 500 7.0 · 107 4.8 · 106 7.0 · 107 3.3 · 106
Bronchial epithelial cells 70,000 500 3.5 · 107 4.4 · 106 3.5 · 107 3.9 · 106
Umbilical vein endothelial cells 70,000 500 3.0 · 107 4.3 · 106 3.0 · 107 3.1 · 106
Urothelial cells 60,000 500 2.5 · 107 3.6 · 106 2.5 · 107 2.6 · 106
Hepatocytes 800,000 500 4.0 · 108 1.1 · 108 4.0 · 108 4.0 · 107
Human primary cell and LNCaP cell input viral titer counts (in VP/ml) and directly assayed active viral infection titers (in ifu/ml) weremeasured by hexon titer assays for AdWT and Ad[I/PPT-E1A]. The mean values are shown for triplicate wells.
Table 3. Viral Replication Status for Ad[I/PPT-E1A] and AdWT in the LNCaPCell Line and in the Human Nonprostate Primary Cell Types
AdWT (MOI 500) Ad[I/PPT-E1A] (MOI 500)
Human cell type
Peak virustiter
(ifu/ml)
ifu per cell(ifu as %of input)a
Viralreplication
status
Peak virustiter
(ifu/ml)
ifu per cell(ifu as %of input)a
Viralreplication
status
LNCaP 1.6 · 109 7.5 · 103 (13,333) Active 7.3 · 107 304 (1,304) ActiveUndifferentiated
(basal prostate)> 3.1 · 107 > 443 ( > 705) Active > 3.1 · 107 > 443 ( > 795) Active
Differentiated prostate 1.1 · 109 7.9 · 103 (22,917) Active 6.7 · 106 48 (203) AttenuatedBronchial epithelial > 3.1 · 107 > 443 ( > 721) Active 4.5 · 104 0.64 (1.15) ResidualUmbilical vein endothelial > 3.1 · 107 > 517 ( > 721) Active 8.5 · 103 0.14 (0.27) ResidualUrothelial > 3.1 · 107 > 620 ( > 861) Active 9.4 · 104 1.88 (3.62) ResidualHepatocytes 2.8 · 108 3.5 · 103 (255) Active 1.9 · 104 0.22 (0.05) Residual
Replication status of AdWT and Ad[I/PPT-E1A] in the primary cell types and LNCaP cell line is shown with the peak viral titers, thenumber of infectious viral particles per cell, and the active virus represented as a percentage of the input virus load.
aThe number of infectious units (ifu) per cell calculated by dividing the peak viral titer by the number of cells available for infection at day 0.
8 ADAMSON ET AL.

either Ad[I/PPT-E1A] or AdMock virus. However, therewere large numbers of Ad[I/PPT-E1A] and AdMock virusparticles sequestered, only in the hepatocyte cytoplasm, in-dicating no evidence for viral replication in these cells. (Fig.6D and E). To validate that the particles observed within thecell cytoplasm, as well as the raft formations in the nuclei,were adenovirus, we performed immunogold labeling of thehexon protein located in the viral capsid, using a rabbitpolyclonal antibody linked to immunogold nanoparticles. Inprimary prostate cells, the virus particles situated in the
nuclear rafts colocalized with the hexon immunogold nano-particles (Fig. 6F and G).
CAR levels in nonprostate primary cells
Finally, the protein levels of the primary adenovirus re-ceptor CAR (green) were measured by immunocytochemistryin primary nonprostate bronchial epithelial cells (HBEC),vascular endothelial cells (HUVEC), and hepatocytes andcompared with those of the prostate cell line LNCaP and theadenoviral replication-competent HEK293 cells. Figure 7 con-firms that CAR is expressed at high levels in HEK293 cells andLNCaP cells, particularly at lateral cell junctions where it co-localizes (yellow) with actin (red). In contrast, very little CARwas detected in the bronchial epithelial cells and vascularendothelial cells. Interestingly, higher CAR levels were seen inthe cytoplasm and at the cell surface of primary hepatocytes,suggesting a possible route of viral entry into these cells.
Discussion
We have applied in vitro cell culture techniques as a pre-clinical testing approach to examine the ability of Ad[I/PPT-EA], a human prostate-specific conditionally replicatingadenovirus, to infect, replicate, and produce cytotoxic effectsin nonprostate primary cells from humans. Initially, we ex-amined the cytotoxic effects of Ad[I/PPT-E1A] at a range ofMOI values in primary bronchial epithelial cells, urothelialcells, vascular endothelial cells, and hepatocytes using anMTS assay. We compared these data with viral cytotoxicityin two permissive cell types: the LNCaP prostate cell line andprimary prostate cells isolated from a prostate cancer patient.The data revealed high levels of cytotoxicity of Ad[I/PPT-E1A] in both prostate cell types studied, and confirmed thatno significant cytotoxic effects were observed in all thenonprostate cells, apart from the human hepatocytes (Fig.
FIG. 5. Adenoviral genome replication bar graph forhuman hepatocytes, measured by qPCR. Hepatocyte cellswere infected in triplicate with AdMock or AdWT at MOI500 and with Ad[I/PPT-E1A] at MOI 5, 50, and 500. Cellswere harvested at 2, 24, and 48 hr, and qPCR to detect viralDNA quantities was performed using a probe/primer mixtargeted to an 84-bp fragment of the adenovirus fiber gene.The means – SEM are shown for triplicate wells.
FIG. 4. Fluorescent imagesshowing I/PPT-specific pro-moter activity in primaryhuman prostate cells, but notin human hepatocytes, 48 hrafter infection with MOI 500AdCMV-gfp virus or Ad[I/PPT]-gfp virus. Images weretaken at · 60 magnificationunder oil.
PRECLINICAL TESTING OF HUMAN ADENOVIRUS 9
1F). This effect could not be explained by active replication ofAd[I/PPT-E1A] virus particles, leading to viral burst andsubsequent death of infected hepatocytes, because replica-tion was not detected in these cells using the hexon im-munostaining assay, which stains for the capsid protein (seebelow). Equally, I/PPT promoter activity was not observedin primary hepatocytes infected with an AdI/PPT-gfp virus(Fig. 4). In contrast, AdWT induced cytotoxicity of humanprimary hepatocytes even at MOI 5 (Fig. 1G), implying atleast 100-fold higher activity toward these cells comparedwith Ad[I/PPT-E1A]. Furthermore, AdWT was confirmed toreplicate in primary hepatocytes.
Viral replication assays were carried out to examine thereplication activity of Ad[I/PPT-E1A] in the nonprostateprimary cell types. By infecting the primary cells with Ad[I/PPT-E1A] at MOI 500 and titering the medium samplescollected at different time points by hexon titer assays, thedata clearly showed that Ad[I/PPT-E1A] did not activelyreplicate in any of the nonprostate primary cell types stud-ied, including hepatocytes. In contrast, highly active repli-cation was observed in the positive control cells, i.e., theLNCaP prostate cell line and the primary prostate cells. In-terestingly, primary prostate cells induced to terminallydifferentiate and form cellular multilayers in the presence of
FIG. 6. TEM images ofhuman primary prostate cells(A, B) and hepatocytes (C, D,E) 60 hr post infection with500 MOI AdWT virus (A, C),Ad[I/PPT-E1A] virus (B, D),and AdMock virus (E). Thefollowing labels are used inthe images: C, cytoplasm; ER,endoplasmic reticulum; M,mitochondria; N, nucleus; nm,nuclear membrane; Nu, nu-cleolus. Virus particles (ar-rows) are observed in both cellcytoplasm and nucleus. (F)Ad[I/PPT-E1A] virus is local-ized in primary prostate cellsusing immunogold labeling ofthe hexon protein in the viralcapsid (primary hexon anti-body used at 1:100; secondaryimmunogold antibody at1:20). Virus particles are oftenlocated in viral replication‘‘rafts’’ that colocalize withimmunogold labeling of thehexon protein. (G) Absence ofhexon immunogold labelingassociated with Ad[I/PPT-E1A] virus particles when theprimary hexon antibody isomitted. Immunogold parti-cles are indicated by ‘‘ > .’’
10 ADAMSON ET AL.

serum and 10 nM dihydrotestosterone showed delayed viralreplication with Ad[I/PPT-E1A], indicative of attenuatedviral replication. This may be because superficial differenti-ated luminal-like cells can mask the CAR located on theunderlying basal cell surface, which is required for adeno-viral infection. Evidence for a reduction in CAR expression indifferentiated primary prostate tissue has been previouslyreported by IF studies (Maitland et al., 2010). This was notobserved for the AdWT virus; however, at high viral titers,AdWT may also transduce cells via other surface receptorsand by endocytosis mechanisms (Carlisle et al., 2009).
The transcriptional activity of the prostate-specific I/PPTpromoter was examined in primary hepatocytes using a gfp-reporter adenovirus (Fig. 4). After 48 hr, no evident gfp ex-pression was observed in hepatocytes infected with MOI 500AdI/PPT-gfp. However, the I/PPT promoter was transcrip-tionally active in primary prostate cells by this time.
To examine this further, we used a highly sensitive qPCRassay to quantify Ad[I/PPT-E1A] genome replication in he-patocytes at MOI 5, 50, and 500 and compared the levels withthose of AdMock and AdWT at MOI 500. At MOI < 500, thequantity of Ad[I/PPT-E1A] genomic DNA was comparableto that of the replication-incompetent AdMock. Only at thehighest MOI used (MOI 500), was there an increase in ge-nome quantity, indicative of genome replication. This maybe due to ‘‘leaky’’ TARP promoter activity within the ade-novirus genome. In this context, the TARP promoter is sur-rounded by less condensed chromatin compared with thehuman genome context (on chromosome 7), where highlycondensed chromatin tightly regulates TARP gene expres-sion. However, the hepatocyte replication data indicatedthat active viral replication due to viral burst, cell death,and reinfection was not occurring in these cells, suggestingthat these genomes were not packaged into infective, rep-lication-competent adenovirus. Electron microscopy imageswere taken of AdWT-, AdMock-, and Ad[I/PPT-E1A]-infected prostate cells and human hepatocytes. The dataconfirmed the hypothesis that Ad[I/PPT-E1A] virus did notactively replicate in hepatocytes, but was sequestered inlarge numbers in the cell cytoplasm and was not presentin the nucleus. This observation was confirmed with thereplication-incompetent virus, which was also present inhigh numbers in the hepatocyte cytoplasm. In contrast,AdWT was organized in rafts in the nuclei of both primaryprostate cells and primary hepatocytes, which is indicativeof replication.
Taking the above observations into consideration, thereplication data (Fig. 3) for hexon immunostaining, theadenoviral-gfp reporter assay, and the outcome of the elec-tron microscopy analyses, all indicate that Ad[I/PPT-E1A]does not actively replicate in human primary hepatocytes.The cytotoxic effect observed in these cells upon treatmentwith Ad[I/PPT-E1A] (Fig. 1F and G) might be explained bycytoplasmic sequestration of viral particles in combinationwith a fragile condition and short life span of human pri-mary hepatocytes in in vitro culture (Fig. 2). The increasedCAR levels observed in the hepatocyte cytoplasm and on thesurface membranes (Fig. 7), when compared with bronchialepithelial cells and vascular endothelial cells, could clearlyresult in higher levels of adenovirus being sequesteredwithin the hepatocyte cytoplasm.
It is well known that culture conditions, including seedingdensity, matrix conditions, and medium supplements, affectthe morphological development, expression of metabolicenzymes, response to drugs, and survival of cultured humanprimary hepatocytes. To the best of our knowledge, the effectof oncolytic adenoviruses on cultured human primary he-patocytes has not been reported. Therefore, it remains to beclarified whether this phenomenon is an artifact of the modelsystem used. When an extrapolation of the in vitro conditionsin which cytotoxicity in cultured human primary hepato-cytes, as observed here, is made to the in vivo situation, andassuming a worst case scenario of maximal viral progenygeneration within the prostate, substantial leakage of virusfrom the prostate into the circulation, and lack of any neu-tralization of systemic virus in the circulation, it is clear thatthe in vitro upper safety margin threshold (MOI 50) willnever be achieved in a liver of a patient treated locally withAd[I/PPT-E1A] in the planned clinical trial. It has to be taken
FIG. 7. Immunocytochemical detection of CAR protein(green) in HEK293 cells, LNCaP cells, primary humanbronchial cells (HBEC), primary human umbilical vein en-dothelial cells (HUVEC), and human hepatocytes. Actin (red)stained for cell morphology, and DAPI (blue) was used tovisualize the nuclei. Images were taken at · 60 magnifica-tion, under oil.
PRECLINICAL TESTING OF HUMAN ADENOVIRUS 11

into account that the human body is capable of efficientneutralization of systemic adenovirus by binding to eryth-rocytes via the complement receptor CR1 and CAR ex-pressed on the surface of these cells (Carlisle et al., 2009) andby the immune system. These neutralization effects are themost likely explanation for the fact that systemic adenovirusinfection in immunocompetent individuals, such as the pa-tients to be recruited in the planned trial, is not associatedwith serious effects, and that to date, oncolytic adenoviruseshave been well tolerated by patients even upon systemicadministration. For example, intravenous administration ofCG7870, a PSA-selective oncolytic adenovirus expressing theE3 region proteins that are involved in controlling the hostimmune response, was well tolerated up to 6.0 · 1012 VP inpatients with hormone-refractory prostate cancer (Smallet al., 2006). In addition, only 20% of the patients with met-astatic colorectal cancer treated with up to eight infusions inthe hepatic artery from 2.0 · 108 to 2.0 · 1012 VP of Onyx-015experienced transient grade I/II hepatic toxicity (Au et al.,2007). Finally, mild and transient dose-dependent transami-nitis was observed after repeated intravenous injections withOnyx-015 at doses of 2.0 · 1012 VP and higher in 10 patientswith metastatic solid tumors (Nemunaitis et al., 2001). Thesystemic levels in these three trials are comparable to thelevels that will be locally administered in our trial (1.0 · 1011
to 5.0 · 1012 VP). Thus, if a part of the locally administeredAd[I/PPT-E1A] does leak into the circulation, no adverseeffects are to be expected. Based on results from previousprostate cancer trials on oncolytic adenoviruses, leakage ofAd[I/PPT-E1A] to the circulation upon intraprostatic ad-ministration is expected to be minimal. DeWeese et al. (2001)showed by qPCR that after a single intraprostatic treatmentat 20–80 deposits up to 1.0 · 1013 VP with the prostate-specific oncolytic adenovirus CV706, 0.007–0.129% of theadministered dose could be detected in blood after 30 min inthe majority of the patients. This was followed by a secondviral DNA peak in 13 out of 16 patients between days 2 and8. In four patients, this peak was higher than the first peakwith the highest peak at < 2% of dose. It was not reported ifthe PCR signal was derived from viable virus. In the trialsconducted by Freytag et al. (2002, 2003), using a single in-traprostatic injection up to 1.0 · 1012 VP with the non-prostate-specific oncolytic adenovirus Ad5-CD/TKrep, viralDNA was found in blood up to day 76, but infectious viruscould not be demonstrated.
To reduce hepatotoxicity further, the use of microRNA-regulated gene expression systems to enhance the safetyand efficacy of viral gene-therapy approaches has been de-scribed (Sakurai et al., 2011). By inserting four copies of thehepatocyte-specific microRNA, mir122, into the 3’ untrans-lated region of the adenoviral E1A transcription cassette,Cawood et al. (2011) were able to genetically reduce hepaticadenovirus replication and liver pathology. If required, suchan approach could be applied to further enhance Ad[I/PPT-E1A] in future clinical trials.
The data presented here confirm the specificity of Ad[I/PPT-E1A] and are part of the clinical dossier submitted forregulatory approval. The risk of adverse events in patientstreated with Ad[I/PPT-E1A] related to the in vitro observa-tion of cytotoxicity of cultured human primary hepatocytesby clinically unrealistically high amounts of Ad[I/PPT-E1A]is minimal and will be controlled by recruiting only immu-
nocompetent patients. As a prelude to clinical trials, the useof multiple primary cultures from a variety of normal humancell types should provide a convenient assay for testing on-colytic adenovirus specificity in vivo, to limit and predict anyundesired off-target effects.
Acknowledgments
We thank Mr. Mike Stower (York District Hospital, York,UK) and Mr. Matthew Simms (Castle Hill Hospital, Hull, UK)for providing prostate tissue samples; Professor Jenny South-gate ( Jack Birch Unit, University of York, York, UK) for do-nating primary human urothelial cells; Berith Nilsson forprovision of AdWT genomic DNA; Meg Stark for technicalassistance with electron microscopy; and members of theYorkshire Cancer Research Unit for their technical advice. Thiswork was supported by the European Union through the SixthFramework Programme Integrated Project GIANT (contractno. LSHB-CT-2004-512087). Norman Maitland also receivedinvaluable core support from Yorkshire Cancer Research.
Author Disclosure Statement
Rachel Adamson, April Frazier, Helen Evans, KarenChance, Ellen Schenk, Magnus Essand, Richard Birnie, RagaiMitry, Anil Dhawan, and Norman Maitland declare nocompeting financial interests.
References
Au, T., Thorne, S., Korn, W.M., et al. (2007). Minimal hepatictoxicity of Onyx-015: spatial restriction of coxsackie-adenoviralreceptor in normal liver. Cancer Gene Ther. 14, 139–150.
Carlisle, R.C., Di, Y., Cerny, A.M., et al. (2009). Human eryth-rocytes bind and inactivate type 5 adenovirus by presentingCoxsackie virus-adenovirus receptor and complement recep-tor 1. Blood. 13, 1909–1918.
Carlsson, B., Cheng, W.S., Totterman, T.H., and Essand M.(2003). Ex vivo stimulation of cytomegalovirus (CMV)-specificT cells using CMV pp65-modified dendritic cells as stimula-tors. Br. J. Haematol. 121, 428–438.
Carmen, I.H. (2001). A death in the laboratory: the politics of theGelsinger aftermath. Mol. Ther. 3, 425–428.
Cawood, R., Wong, S.-L., Di, Y., et al. (2011). MicroRNA con-trolled adenovirus mediates anti-cancer efficacy without af-fecting endogenous microRNA activity. PLoS One 6, e16152.
Cheng, W.S., Giandomenico, V., Pastan, I., and Essand, M.(2003). Characterization of the androgen-regulated prostate-specific T cell receptor c-chain alternate reading frame protein(TARP) promoter. Endocrinology. 144, 3433–3440.
Cheng, W.S., Kraaij, R., Nilsson, B., et al. (2004). A novel TARP-promoter-based adenovirus against hormone-dependent andhormone-refractory prostate cancer. Mol. Ther. 10, 355–364.
Cheng, W.S., Dzojic, H., Nilsson, B., et al. (2006). An oncolyticconditionally replicating adenovirus for hormone-dependentand hormone-independent prostate cancer. Cancer Gene Ther.13, 13–20.
DeWeese, T.L., van der Poel, H., Li, S., et al. (2001). A phase Iclinical trial of CV706, a replications-competent, PSA selectiveoncolytic adenovirus for the treatment of locally recurrentprostate cancer following radiation therapy. Cancer Res. 61,7464–7472.
Djavan, B., Judd, M.W., Zlotta, A., et al. (2003). PSA progressionfollowing radical prostatectomy and radiation therapy: newstandards in the new millenium. Eur. Urol. 43, 12–27.
12 ADAMSON ET AL.

Freytag, S.O., Rogulski, K.R., Paielli, D.L., et al. (1998). A novelthree-pronged approach to kill cancer cells selectively: con-comitant viral, double suicide gene, and radiotherapy. Hum.Gene Ther. 9, 1323–1333.
Freytag, S.O., Khil, M., Stricker, H., et al. (2002). Phase I study ofreplication-competent adenovirus-mediated double suicidegene therapy for the treatment of locally recurrent prostatecancer. Cancer Res. 62, 4968–4976.
Freytag, S.O., Stricker, H., Pegg, J., et al. (2003). Phase I study ofreplication-competent adenovirus-mediated double-suicidegene therapy in combination with conventional-dose three-dimensional conformal radiation therapy for the treatment ofnewly diagnosed, intermediate- to high-risk prostate cancer.Cancer Res. 63, 7497–7506.
He, T.-C., Zhou, S., da Costa, L.T., et al. (1998). A simplifiedsystem for generating recombinant adenoviruses. Proc. Natl.Acad. Sci. U.S.A. 95, 2509–2514.
Jogler, C., Hoffman, D., Theegarten, D., et al. (2006). Replicationproperties of human adenovirus in vivo and in cultures ofprimary cells from different animal species. J. Virol. 80, 3549–3558.
Kim, J.H., Kim, S.H., Brown, S.L., and Freytag, S.O. (1994). Se-lective enhancement by an antiviral agent of the radiation-induced cell killing of human glioma cells transduced withHSV-tk gene. Cancer Res. 54, 6053–6056.
Maitland, N., Chamber, K., Georgopoulos, L., et al.; GIANT FP6Consortium. (2010). Gene transfer vectors targeted to humanprostate cancer: do we need better preclinical testing systems?Hum. Gene Ther. 21, 815–827.
Mitry, R.R., Sarraf, C.E., Havlık, R., and Habib, N.A. (2000).Detection of adenovirus and initiation of apoptosis in hepato-cellular carcinoma cells after Ad-p53 treatment. Hepatology. 31,885–889.
Nemunaitis, J., Cunningham, C., Buchanan, A., et al. (2001). In-travenous infusion of a replication-selective adenovirus
(ONYX-015) in cancer patients: safety, feasibility and biologi-cal activity. Gene Ther. 8, 746–759.
Rogulski, K.R., Freytag, S.O., Zhang, K., et al. (2000). In vivoantitumor activity of ONYX-015 is influenced by p53 statusand is augmented by radiotherapy. Cancer Res. 60, 1193–1196.
Sakurai, F., Katayama, K., and Mizuguchi, H. (2011). Micro-RNA-regulated transgene expression systems for gene therapyand virotherapy. Front. Biosci. 17, 2389–2401.
Schenk, E., Essand, M., Bangma, C.H., et al. (2010) Clinical ad-enoviral gene therapy for prostate cancer. Hum. Gene Ther.21, 807–813.
Small, E.J., Carducci, M.A., Burke, J.M., et al. (2006). A phase Itrial of intravenous CG7870, a replication-selective, prostate-specific antigen targeted oncolytic adenovirus, for the treat-ment of hormone-refractory, metastatic prostate cancer. Mol.Ther. 14, 107–117.
Tokuyasu, K.T. (1986). Cryosection for immunochemistry. J.Electron Microsc. (Tokyo) 35, 1977–1978.
Vorberg, S.A., and Hunt, K.K. (2002). Production of adenoviralgene therapy. Oncologist. 7, 46–59.
Address correspondence to:Dr. Rachel Adamson
YCR Cancer Research UnitDepartment of Biology
University of YorkHeslington, York, YO10 5DD
United Kingdom
E-mail: [email protected]
Received for publication February 11, 2011;accepted after revision August 5, 2011.
Published online: August 8, 2011.
PRECLINICAL TESTING OF HUMAN ADENOVIRUS 13
