by ladislav vyklicky jr*, morris benveniste 1
TRANSCRIPT

Journal of Physiology (1990), 428, pp. 313-331 313With 7 figuresPrinted in Great Britain
MODULATION OF N-METHYL-D-ASPARTIC ACID RECEPTORDESENSITIZATION BY GLYCINE IN MOUSE CULTURED
HIPPOCAMPAL NEURONES
BY LADISLAV VYKLICKY JR*, MORRIS BENVENISTEAND MARK L. MAYERt
From the Unit of Neurophysiology and Biophysics, Laboratory of DevelopmentalNeurobiology, NICHD, Building 36, Room 2A21, National Institutes of Health,
Bethesda, MD 20892, USA
(Received 17 October 1989)
SUMMARY
1. Responses to N-methyl-D-aspartic acid (NMDA) were recorded from mouseembryonic hippocampal neurones in dissociated culture, using the tight-seal, whole-cell, patch-clamp technique for voltage clamp. A rapid perfusion system, with anexchange time constant of less than 10 ms, was used to apply NMDA underconditions which minimized slow, calcium-sensitive desensitization. With no addedglycine, responses to 100 ,uM-NMDA applied for 1-5 s declined by greater than 90%,due to an additional component of desensitization of time constant 250 ms.
2. Adding glycine to the extracellular solution, over the range 30 nm to 3/tM,both potentiated responses to NMDA and to L-glutamate, and reduced fastdesensitization. In the presence of 3 ,uM-glycine responses to NMDA declined by only10 %. Similar potentiation and reduction of desensitization was obtained with 3 /tMconcentrations of the glycine analogues D-alanine and D-serine.
3. Analysis of dose-response curves for the action of glycine on responses to100 ,tM-NMDA revealed a 3-fold higher potency of glycine for potentiation of peakversus steady-state responses, with concentrations for half-activation of 134 and382 nm, respectively. The competitive glycine antagonist 7-chlorokynurenic acidproduced a similar shift of both the peak and steady-state dose-response curves forglycine, consistent with an equilibrium dissociation constant of 280 nm for interactionof 7-chlorokynurenic acid with the glycine binding site on NMDA receptors.
4. In the presence of 100 nM-glycine, 10 ,tM-7-chlorokynurenic acid producednearly complete block of the response to 3 mM-NMDA, suggesting that glycine isabsolutely required for activation of the NMDA receptor channel complex.
5. In some neurones responses to NMDA showed essentially no desensitization inthe presence of 3 /tM-glycine. Under these conditions, 7-chlorokynurenic acidproduced a concentration-dependent block of both the initial and equilibriumresponse to NMDA, with a 4-fold greater sensitivity for block of the steady-state
* Present address: Institute of Physiology, Czechoslovak Academy of Sciences, 142 20 Prague4, Vfdenska 1083, Czechoslovakia.
t Author for correspondence.MS 8020

L. VYKLICKY JR, M. BENVENISTE AND M. L. MAYER
current (IC50 = 2 25 /Lm) than for block of the peak current (IC50 = 8 96 /IM). As aresult, in the presence of 7-chlorokynurenic acid, responses to NMDA showed strongdesensitization, even in the presence of 3 /am-glycine.
6. Our results show that glycine-evoked potentiation of NMDA receptor activityis accompanied by reduced desensitization. Because responses to NMDA measured atequilibrium require higher concentrations of glycine for expression of activity thanresponses measured early after the application of agonist, the reduction ofdesensitization by glycine is an important mechanism for promoting responses tosustained applications of NMDA receptor agonists.
INTRODUCTION
The L-glutamate receptor subtype selectively activated by N-methyl-D-asparticacid (NMDA) plays an important role in regulating neuronal excitability andsynaptic plasticity in many areas of the mammalian CNS (e.g. Mayer & Westbrook,1987; Lodge, 1988). The NMDA receptor ion channel complex displays unusuallycomplicated behaviour, and its activity is modulated by many different drugs andions (e.g. Lodge, 1988; Mayer, Vyklicky & Sernagor, 1989a). Recent electro-physiological experiments on embryonic mouse neurones by Johnson & Ascher(1987 a) showed striking stimulation ofNMDA receptor activity by glycine. This hasprompted a large number of experiments utilizing a variety of techniques to studythe interaction ofglycine with NMDA receptors. Estimates ofequilibrium dissociationconstants for glycine action range from 100 to 700 nM (Bristow, Bowery & Woodruff,1986; Johnson & Ascher, 1987 b; Kleckner & Dingledine, 1988; Kushner, Lerma,Zukin & Bennett, 1988; Snell, Morter & Johnson, 1988). Several glycine antagonistshave been discovered; these competitively displace glycine from its binding site, andact as non-competitive NMDA antagonists (Ascher, Henderson & Johnson, 1988;Kemp, Foster, Leeson, Priestley, Tridget, Iversen & Woodruff, 1988; Mayer, West-brook & Vyklicky, 1988; Foster & Kemp, 1989; Huettner, 1989; Kessler, Terramani,Lynch & Baudry, 1989). Structure-activity relationships for neutral amino acidsdifferentiate the glycine binding site linked to NMDA receptors from strychnine-sensitive receptors coupled to anion-permeable channels, which mediate inhibitorysynaptic responses in the spinal cord (Bristow et al. 1986; Snell et al. 1988). Theaugmentation by glycine of the binding of use-dependent NMDA antagonists, whichare thought to act within the ion channel portion of the NMDA receptor complex,suggests that glycine promotes conformational transitions of the NMDA receptorchannel to open states (Reynolds, Murphy & Miller, 1987; Snell et al. 1988).Potentiation of NMDA receptor responses by low concentrations of glycine in intacttissues from adult animals has also been detected, validating the results from studieson cultured neurones as important for the normal behaviour of NMDA receptorsin vivo (Larson & Betz, 1988; Thomson, Walker & Flynn, 1989).The molecular mechanism by which glycine modulates NMDA receptor responses
has not been established, but does not appear to involve large changes in receptoraffinity for excitatory amino acid agonists, a prolongation of single-channel opentimes, or an increase in single-channel conductance (Johnson & Ascher, 1987a;Huettner, 1989). Recently we described a reduction by glycine of NMDA receptor
314

AIODUTLATIONV OF NAIDA RECEPTOR DESE.NTSITIZATION3
desensitization (Mayer, Vyklicky & Clements, 1989 b), and report here furtherexperiments on this observation. Our results suggest that, under the conditions usedfor whole-cell recording, block of desensitization by glycine is a major mechanismresponsible for the generation of sustained NMDA receptor activity during prolongedapplications of agonist.
METHODS
Cell culturePrimary dissociated nerve cell cultures were prepared by dissociating the hippocampi of 16- to
17-day C57BI/6 mouse embryos, and plating the resulting cell suspension onto confluenthippocampal glial cell feeder layer cultures. Prior to removal of the fetuses the mothers were killedby cervical dislocation. The growth medium contained MEM, 5% horse serum, and a nutrientsupplement containing transferrin, insulin, selenium, corticosterone, triidothyronine, progesterone,and putrescine (Guthrie, Brenneman & Neale, 1987); no antibiotics were used. Complete details aregiven in Mayer, Vyklickv & Westbrook (1989c).
Experimental solutionsThe extracellular solution contained (mM): NaCl, 160; KCl, 2-5; CaCl2, 2 or 0-2; MgCl2, I or 2;
HEPES, 10; glucose, 10; phenol red, 0-01 mg ml-'; pH adjusted to 7-3 with NaOH. Tetrodotoxin,400 nm, was added to block action potentials and synaptic activity, and bicuculline methochloride,5 /tM, added to block spontaneous inhibitory postsynaptic potentials. Magnesium was omitted fromsolutions containing NMDA to avoid ion channel block. Glycine, excitatory amino acids, andantagonists were added to the extracellular solution as required. The intracellular solutioncontained (mM): CsMeSO3, 125; CsCl, 15; HEPES, 10; 1-2-bis(o-aminophenoxy)ethane-N7N7N'N'-tetracetic acid (BAPTA), 5; CaCl2, 0 5; MgCl2, 2 or 3; MgATP, 2; pH adjusted to 7-2 with CsOH;osmolarity, 300 mosm. Salts, biochemicals, excitatory amino acids and antagonists were purchasedfrom Aldrich, Fluka, Molecular Probes, Sigma and Tocris Neuramin.
Recording and perfusion techniquesExperiments were performed at room temperature (25-27 °C), 10-20 days after cultures were
plated. Voltage clamp was achieved using whole-cell patch-clamp recording, and an AxonInstruments 'Axoclamp 2' discontinuous amplifier set at a gain of 2-4 nA mV-', with a switchingfrequency of approximately 10 kHz. The series resistance was usually kept below 10 MQi. Therecording chamber was perfused at 05-2 ml per minute with extracellular solution, and a flow pipearray similar to that described by Johnson & Ascher (1986) was used for faster perfusion aroundindividual neurones as described below.The fast perfusion system consisted of an array of nine glass tubes (Yellen, 1982), each tube of
external diameter 400 ,um, wall thickness approximately 30,uM. The array was mounted on ahydraulic manipulator driven by a stepping motor and positioned within 200 ,um of neuronal cellbodies using a mechanical manipulator. A pump was used to drive solution through the tubes atapproximately 150 ,um ms-', and 3-way latching solenoid valves used to direct the solution backto reservoirs, or onto nerve cells. Cells were always bathed in a rapidly flowing stream of controlsolution, except during application of NMDA. Rapid solution changes were achieved as follows:one barrel of the perfusion system was positioned above the soma of a selected neurone, and thevalve connected to this barrel opened to perfuse the area around the nerve cell; the stepper motorthen moved the flow pipe such that an adjacent barrel was positioned above the neurone (traversetime 140 ms); on reaching this new position the valve controlling solution flow through the barrelpreviously centred above the soma was closed and, simultaneously, solution flow onto the neuronewas started through the newly positioned barrel. In well-isolated neurones, with clearly defineddendritic trees, the solution exchange time constant was less than 10 ms, as measured using sodiumion concentration jumps in the presence of kainic acid (Fig. 1). The junction potential changemeasured with a cell-free patch electrode was faster, time constant less than 1 ms. The slowersolution change recorded in whole-cell experiments probably reflects perfusion of the much largerarea of the dendritic tree of hippocampal neurones compared to faster exchange around the tinyopening of a patch pipette.
315

L. VYKLICKY JR, M. BEN 7ENISTE AND M. L. MA YER
In the majority of our experiments the control solution contained 1 mM-Mg2+; as a result thekinetics of the 'off' response to N-MDA, recorded following termination of the application of agonistby a rapid perfusion of control solution, was determined in part by the kinetics of onset of Mg2+block. Similarly, one would expect the rising phase of the response to NMDA to be reduced in
A B
50 /M-kainic acid Patch pipette current
165 mM Control5mM [Nalo 1/10
5 nA 'r ~~~~~~~~2nAT=93msl |~~5nA r=028ms t|n,r 9.3 ms
100 ms 5 ms
Fig. 1. Fast solution changes recorded using sodium concentration jumps. A, response ofa hippocampal neurone bathed in a solution containing 50 ,SM-kainic acid, and voltageclamped at -60 mV during a sodium concentration jump from 5 to 165 mm. Therelaxation is due to the increased driving force for inward sodium current flowing throughion channels activated by kainic acid, and is fitted with a single exponential of timeconstant 9 3 ms. Sucrose was used as a sodium substitute. B, change in current recordedusing an open path pipette voltage clamped at 0 mV, during a switch from extracellularsolution diluted by 90% with water to normal extracellular solution. The relaxation isdue to a change in junction potential, and is fitted with an exponential of time constant0-28 ms.
amplitude by the presence of Mg2+ during the switch from control solution with 1 mM-Mg2+ to Mg2+-free solution with NMDA. Experiments with Mg2+-free control solutions did in fact reveal a smalldepression of the peak response to NMDA by Mg2+, when responses were recorded in the same cellswith control solutions containing either 0 or 1 mM-Mg2+. In the presence of 1 /uM-glycine responseswith Mg2+ control solution were 0-81 times the peak amplitude of those recorded with Mg2+-freecontrol solution. As a result the glycine dose-response curve of the peak response to NMDA has ashallower slope and lower affinity EC50 value than would be recorded using Mg2+-free solutions.
RESULTS
Results are presented as means + standard deviation unless indicated differently.All experiments were performed at a membrane potential of -60 mV, except duringstudy of the voltage dependence of glycine-sensitive NMDA receptor desensitization.No voltage dependence of either the degree of desensitization or its time constant ofonset was detected over the membrane potential range -80 to + 60 mV.
Calcium-8ensitive desensitizationPrevious experiments have suggested a role for calcium in mediating desensi-
tization at NMDA receptors (Mayer & Westbrook, 1985; Mayer, MacDermott,Westbrook, Smith & Barker, 1987; Zorumski, Yang & Fischbach, 1989). The actionof glycine on NMDA receptor responses could be studied in isolation of calcium-dependent desensitization if precautions were taken which would be expected tominimize NMDA-evoked intracellular calcium transients. Our routine experimental
316

MODULATION OF NMDA RECEPTOR DESENSITIZATION
conditions included the use of 5 mM-BAPTA (a fast calcium buffer) in theintracellular solution, and the application of NMDA in solutions containing only0-2 mM-extracellular calcium. Under these conditions, responses to near saturatingconcentrations of NMDA and glycine showed almost no decrease in amplitude forapplications 1-5 s in duration (Fig. 2), and ifMgATP was included in the intracellularsolution, the peak amplitudes in response to successive applications were relativelystable (e.g. Mody, Salter & MacDonald, 1988). If the extracellular calciumconcentration was raised to 2 mm, responses to NMDA showed time-dependentinactivation, which recovered on returning to 0-2 mM-calcium (Fig. 2). If EGTA, acalcium chelator with slower Ca2+-binditig kinetics, was substituted for BAPTA,then responses to NMDA showed marked time-dependent inactivation and, inaddition, gradually declined in amplitude with repeated applications. Because thedesensitization of responses to NMDA recorded with BAPTA and 2 mM-extracellularcalcium is much slower than glycine-sensitive desensitization, it is likely that theyare mediated by different molecular mechanisms. Although the use of intracellularsolutions with strongly buffered calcium ion activity, together with low extracellularconcentrations of calcium, is not physiological, this approach has the advantage thatit allows the mechanism of action of glycine to be studied in isolation.
Modulation of responwe8 to NMDA and L-glutamate by glycine, D-serine and D-alanineWe recorded from 206 neurones, all of which gave results consistent with
moderately fast desensitization (time constant approximately 250 ms) to 100 ,UM-NMDA, at low (10-100 nM) but not high (3-10 iM) concentrations of glycine (Fig. 3).A similar reduction of desensitization by glycine also occurred with responses to10 ,M-L-glutamate (Fig. 3). These concentrations of NMDA and L-glutamate areapproximately 3 and 5 times greater than those required for 50% activation ofNMDA receptors and thus produce large responses. With 100 nM-glycine the ratiosteady-state/peak current was: NMDA 0-31 + 0-05, and L-glutamate 0-39 + 0-02 (tenobservations of four cells). With 3 ,uM-glycine, the peak response to both NMDA andL-glutamate was potentiated and, when compared to responses recorded in the samecells with 100 nM-glycine, responses with 3 /SM-glycine showed less desensitization:the ratio steady-state/peak current for NMDA was 0X86+ 0 05, and for L-glutamate0-88 + 0-01. As a result, on raising the glycine concentration from 0 I to 3 ,M, thepotentiation of responses evoked by NMDA receptor activation was much largerwhen measured at steady state (NMDA, 5-33 ± 0-69; L-glutamate, 4.45 ± 0 74), thanat the peak of the agonist-evoked response (NMDA, 1-91 +0-14; L-glutamate,1-96+0'38).Because L-glutamate is a mixed agonist, with activity at both NMDA and non-
NMDA receptors (Mayer & Westbrook, 1984), the above experiments would bedifficult to interpret if part of the response to L-glutamate was due to activation ofquisqualate receptors, which also show strong desensitization. We have shownpreviously that glycine has no effect on desensitization at quisqualate receptors(Mayer et al. 1989b), and that glycine does not alter responses to kainate (see alsoJohnson & Ascher, 1987 a). In addition, dose-response curve analysis for theactivation of NMDA and quisqualate receptors by L-glutamate shows the potency ofL-glutamate for activation ofNMDA receptors to be more than 100 times higher than
317

318 L. VY-KLICKY JR. AM. BENVENISTE AND M. L. MAYER
its potency for activation of rapidly desensitizing responses at quisqualate receptors(Mayer. 1989). As a result, at 10 /LM-L-glutamate, the amplitude of the quisqualatereceptor current is negligible compared to that activated by NMDA receptors in thepresence of 3 aim-glycine with no added magnesium. For activation of NMDA
A0.2 mM-Ca2+ 2.0 mM-Ca2+ 0.2 mM-Ca2+
2 nA
is
B2 mm-Ca22
2S 35 1 6 s 0.84 ~~~~~ ~~~~~~~peakCAQ
: 2.5 o<> Peak - c0)4UB 1.50,;16s0
*~1.5 - V(D~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~0
pt 0.2 c0.50
200 350 500 650 800 Z
Time (s)
Fig. 2. Calcium-sensitive desensitization at NMDA receptors. A, the traces showmembrane current responses to rapid applications of 100 /Lm-NMDA, with 3 /um-glycinepresent continuously. Initially, NMIDA was applied with 0-2 mm-calcium in theextracellular solution, and produced responses with only a small amount of de-sensitization. XVith 2 mM-calcium, the amplitude of the NMDA-evoked current wasreduced, and the amount of desensitization markedly increased. On returning to 0-2 mm-calcium this effect was largely reversible. B, graphical analysis of this experiment showsthe amplitude of the peak response (0) and the response at the end of the 1-6 s applicationof NMDA (-) versus time following the start of whole-cell recording. The ratio of thesevalues (A) was used as a measure of desensitization.
receptors the potency of L-glutamate is 15 times higher than that of NMDA itself(M. L. Mayer & L. Vyklicky Jr, unpublished observations). However NMDA is a veryselective agonist, with virtually no agonist action at quisqualate receptors (M. L.Mayer & L. Vyklicky Jr, unpublished observations), and thus produces pure NMDAreceptor responses even at high concentrations of agonist. The similar action ofglycine on responses to both L-glutamate and NMDA, shown in Fig. 3, suggests thatglycine-sensitive desensitization at NMDA receptors is similar for agonists ofsubstantially different potency, and is in good agreement with a highly selectiveaction of 10 /M-L-glutamate at NMDA receptors.

MODUL.ATION OFNi.M1DA RE(CEPTOR DESEX'SITIZATIOJ)N 319
A N-Methyl-D-aspartate
0.1 pM-glycine 3 pM-glycine
L-Glutamate
0.1 pM-glycine 3 pM-glycine
12 nA
750 ms
B10
* Peak
Steady state
0NMDA NMDA L-Glu L-Glu
3,um-Gly 0-1 ,um-Gly 3 pm-Gly 0-1 pm-GlyFig. '3. GIveine-ev-okedl potentiation of'responises to -N..I)DA anid glutanmate is accompanliedbN- a redluction of cleseiisitizatioii. A. respoiises of a sliigle hippocamlpal nieuroiie to 1()( ,uam-NMAI). or l0O,uNI-L-glultamate. recorded in the presence of 0-1 or 3,lvm-glycine. asinidicated. There is stronig de;senisitizatioii NN-ith 0 1 but niot 3 p-m^-gly cine. B. clata fromi fouriieuronies, inoriiialized NN-ith respect to the steadys-state responise to N,-.IDA recorded U-ithi() 1 ,ulm-gl-\ in.ad plotted as means + S.D.
Expewrimeiits utilizing ani agonilst-stimulated iincrease in binding of the openichannel blockers -NIK-801 and its anialogues (Reynolds et al. 1987:-, nell et al. 1988)have been used to explore structure-activity relationships at the glvcine modulatorvsite on N\AIDA receptors. Such experimnents suggest that the D-isomers of serine and

L. VYKLICKY JR, M. BENVEATISTE ANVD M. L. MA YER
alanine also exhibit a potent glycine-like activity. Under our experimental conditionsthere was potentiation of responses to NMDA by both D-serine, 3 1m, and D-alanine,3 ,Im. This was also associated with a marked reduction of desensitization (Fig. 4),similar to responses recorded with 3 ,IM-glycine. The ratio steady state/peak currentwith 3 /tM-D-serine was 0-81+0-09 (nineteen observations on eight cells) and with3 /tM-D-alanine it was 0-87 +0-08 (fourteen observations on five cells). For responsesrecorded in the same cells, with 30 nm-glycine, the ratio was 017+ 0X04 (twentyobservations on eight cells). This, together with the high affinity of D-serine andD-alanine for NMDA receptors, demonstrated using displacement of radiolabelledglycine from brain membrane preparations (Snell et al. 1988), suggests thatoccupation of the glycine binding site on NMDA receptors by D-serine, D-alanine orglycine both potentiates responses to NMDA and blocks desensitization.
Comparison of the effect of glycine concentration on potentiation and desensitizationof responses to NMDAOur subsequent experiments were designed to explore mechanisms underlying the
reduction of desensitization and the potentiation of NMDA receptor responses byglycine, and to determine if these were related processes. Dose-response curve.analysis was performed by measuring glycine-evoked potentiation of responses to1'5 s applications of 100 1aM-NMDA at peak, and at steady state, following the onsetof desensitization, and then fitting the results with the logistic equation
11 + (EC50/[Gly])' (1)
allowing the maximum response (Imax), concentration for half-maximal activation(EC50) and Hill coefficient (n) to vary. Our first set of experiments was performedusing nominally glycine-free solutions which are likely to contain 20-50 nM-glycine(e.g. Kleckner & Dingledine, 1988). As a result, substantial NMDA-evoked responseswere recorded in solutions to which no glycine had been added, and the foot of thedose-response curve is distorted at glycine concentrations below 100 nm. This wasespecially true for the peak response to NMDA which is very sensitive to lowconcentrations of glycine. The deionized water used to prepare our recordingsolutions contained a background glycine concentration of 19 nm, assayed by pre-derivation high-performance liquid chromatography. During analysis of our datawe attempted to correct for this by assuming a background concentration of 20 nM-glycine in the calculation of glycine doses used for dose-response curve analysis, e.g.for 30 nm experimentally added glycine, we assumed a total concentration of 50 nM(Fig. 5). Analysis of such corrected dose-response curves with the logistic equationsuggests approximately a 3-fold higher potency of glycine for the peak response to100 1,M-NMDA (EC50 = 134 nM, n = 1-01) than for the steady-state response recordedafter the onset of desensitization (EC50 = 382 nM, n = 1-44). As noted in the Methods,the use of control solutions containing 1 mM-Mg2+, resulted in an underestimate ofthe peak response to NMDA. This contributes to the lower slope of the peak versusequilibrium glycine dose-response curve.The above analysis of glycine dose-response curves suggests that the NMDA
receptor has multiple binding sites for glycine. When data from the above
320

MODULATION OF NVMDA RECEPTOR DESENSITIZATION 321
100 nM-D-serine
3 pM-D-alanine
0 20
> 15'z
0
5
0Gly D-Ser D-Ser D-AIa
(30 nM) (100 nM) (3pM) (3,pM)
Fig. 4. D-Serine and D-alanine are potent glycine analogues. A, responses of a singlehippocampal neurone to applications of 100 /M-NNIDA, recorded in the presence of30 nM-glycine, 100 nM-D-serine, 3 JtM-D-serine or 3,uM-D-alanine, as indicated. There isstrong desensitization with 30 nM-glycine and 100 nM-D-serine, but not with 3 /tM-D-serineor 3 YuM-D-alanine. B, data from five to eight neurones, normalized with respect tothe steady-state response to NMDA recorded with 30 nM-glycine, and plotted as
means+ standard error of the mean (S.E.M.).
experiment was analysed using an independent two-site model, assuming that non-
co-operative binding of two molecules of glycine is required for NMDA receptoractivation (e.g. Colquhoun & Ogden, 1988), fits were nearly indistinguishable fromthose shown in Fig. 5. However, the microscopic equilibrium constants obtainedfrom the independent binding site model gave values of 47 and 170 nm for the peakand steady-state response to glycine.
11 PH Y 428
A30 nM-glycine
3 pM-D-serine
2 nA
750 ms
B30
I Peak
25 - [ Steady state-I
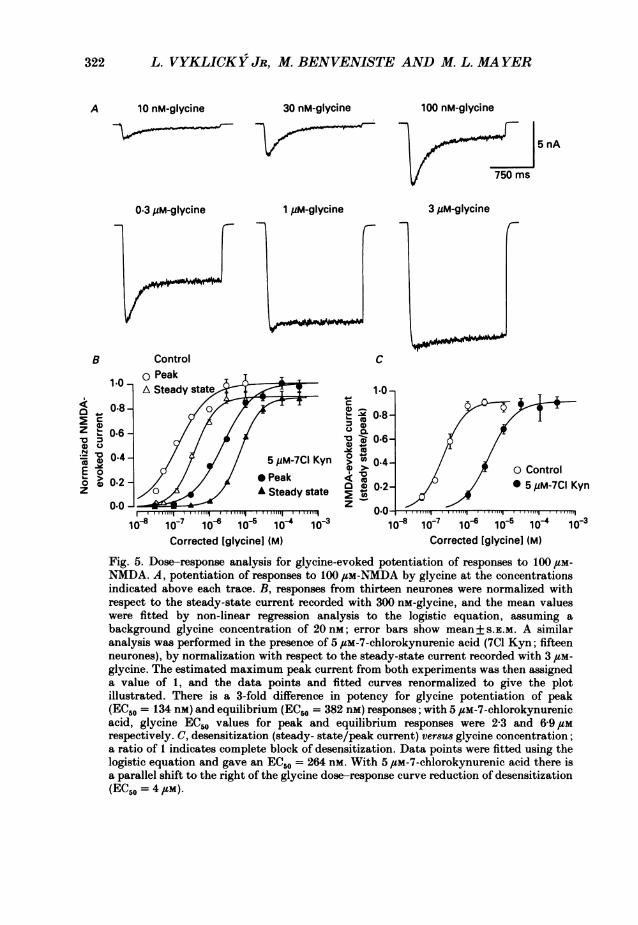
322 L. VYKLICKY JR, M. BENVENISTE AND M. L. MAYER
A 10 nM-glycine 30 nM-glycine 100 nM-glycine
5 nA
750 ms
0.3 jiM-glycine 1 ,M-glycine 3 4M-glycine
B Control c
10 .A Steady sitate I 1.0r
0.8-~~~~~~~~~~~~~.Z~~~~~~~~0.6~~ ~ ~c08
0.6-
indc0t4 5 eM-7C Kynfrm 0.4tEo g c of Control
anaysi was 0efo2e in th prsnea of 5,M7choro e aci (7C17Kyn;ite
~~~~0.2~~ ~ ~ ~ ~ ~ 002neurones), bynomaiztonwASteady state c 0.2ord5dwithC3 Kyn0.0 _ _ _ _ _ _ _ _ _ _ _ _ z _ _ _ _ _ _ _ _ _ _ _ _
Corrected [glycinel (M) Corrected [glycinel (M)
Fig. 5. Dose-response analysis for glycine-evoked potentiation of responses to 100pem-N(MDA. A, potentiation of respones to 100,um-NMDA by glycine at the concentrationsindicated above each trace. B, responses from thirteen neurones were normalized withrespect to the steady-state current recorded with 300 nm-glycine, and the mean valueswere fitted by non-linear regression analysis to the logistic equation, assuming abackground glycine concentration of 20 nm; error bars show mean+ S.E.m. A similaranalysis was performed in the presence of 5 /SM-7-chlorokynurenic acid (701 Kyn; fifteenneurones), by normalization with respect to the steady-state current recorded with3itm-glycine. The estimated maximum peak current from both experiments was then assigneda value of 1, and the data points and fitted curves renormalized to give the plotillustrated. There is a 3-fold difference in potency for glycine potentiation of peak(ECISO = 134 nm) and equilibrium (EC50 = 382 nm) responses; with 5 /Sm-7-chlorokynurenicacid, glycine EC,,, values for peak and equilibrium responses were 2-3 and 6-9 /Smrespectively. C, desensitization (steady- state/peak current) ver8u8 glycine concentration;a ratio of 1 indicates complete block of desensitization. Data points were fitted using thelogistic equation and gave an EC.50 = 264 nm~. With 5 /Lm-7-chlorokynurenic acid there isa parallel shift to the right of the glycine dose-response curve reduction of desensitization(EC50 = 4,sm).

MIODULATIO.N OF NMDA RECEPTOR DESENSITIZATION 3
The reduction of desensitization (plotted as 'steady state/Ipeak) which occurs withincreasing glycine concentration has a steep dose-response relationship, and a curvefitted with the logistic equation rises from 10-90% over the concentration range100-700 nm (Fig. 5). At low concentrations of glycine desensitization of NMDAreceptors is intense, but never complete, and even for applications of agonist lastinggreater than 30 s sustained NMDA receptor activity is recorded, with no evidence ofslow desensitization. With 30 nM-glycine added to the extracellular solution the ratioof steady-state current measured at the end of 5 or 30 s applications of 100 /tm-NMDA (130s/I5s) had a value of 1<02+00095 (seven observations in three neurones).
Since we were unable to prepare solutions with a sufficiently low concentration ofglycine to examine the foot of the dose-response curve for glycine-evokedpotentiation of responses to NMDA, and because we were not certain that theambient concentration of glycine did not vary slightly from experiment toexperiment, we performed an additional set of measurements in which a 5,/tmconcentration of the competitive glycine antagonist 7-chlorokynurenic acid wasadded to the extracellular solution (Kemp et al. 1988). This was expected to producea rightward shift of the dose-response curve for glycine, and hence negate thedistorting effect due to contamination by low concentration of ambient glycine. Thisprocedure produced a large and similar displacement of the dose-response curves forboth the peak and steady-state response to glycine (Fig. 5). Thus, in the presence of5 pm-7-chlorokynurenic acid the 3-fold higher potency of glycine for the peak versussteady-state response to NMDA was maintained, but with EC50 values 17- to 18-foldgreater than in the absence of 7-chlorokynurenic acid (peak = 2-29 ,UM, 17 1 timescontrol; steady state = 6 89 /tM, 18-0 times control). Analysis of this experiment,using the EC50 values for glycine calculated from the control steady-statedose-response curve and assuming competitive inhibition of glycine binding by 7-chlorokynurenic acid (Kemp et al. 1988; Kleckner & Dingledine, 1989), can be usedto calculate the affinity of 7-chlorokynurenic (Ki) for its binding site on the NMDAreceptor from the equation
EC50, test - [7C] (2)EC50, control Ki
in which EC50 test and EC50 control are the concentrations of glycine required for 50%of the maximum response to NMDA in the presence and absence of 7-chlorokynurenicacid. The equilibrium dissociation constant of 7-chlorokynurenic acid is similar whencalculated at the peak of the response to NMDA (310 nm) or at equilibrium (294 nM),suggesting that the binding of glycine antagonists is not greatly influenced bydesensitization. This is supported by analysis of the shift of the desensitizationdose-response curve. plotted as the ratio Isteadystate/peak versus glycine concentrationin the presence of 5 flM-7-chlorokynurenic acid, since this is displaced parallel to thecontrol curve (EC50 = 40 I.m, 15'2 times greater than control) suggesting a Ki of353 nm for the action of 7-chlorokvnurenic acid on NMDA receptor desensitization(Fig. 5).Comparison of the peak and steady-state dose-response curves for potentiation of
NMDA receptor responses by glycine recorded in control experiments, and in thepresence of 7-chlorokynurenic acid suggests that, at sufficiently low concentrations
11-2
323

L. VYKLICKY JR, M. BENVVE.XVISTE AN..D M. L. AIA YER
of glycine, responses to NMDA should be eliminated. This was essentially true for thesteady-state response to NMDA, since responses recorded with ambient glycine wereon average only 0-7 % as large as those recorded with 10 /aM-glycine. However, for thepeak response to NMDA, more substantial responses were recorded with ambientconcentrations of glycine (4% of those with 10,uM-glycine). Our analysis of the
Control Recovery10 gM-7-chlorokynurenic acid
01 pM-Gly + 01 pM-Gly + 1 .0 pM-GIy + 0.1 pM-GIy + 0.1 pM-Gly +0.3 mM-NMDA 0.3 mM-NMDA 0.3 mM-NMDA 3.0 mM-NMDA 0.3 mM-NMDA
1.5 nA
is
Fig. 6. Glycine is required for expression of NKINDA receptor responses. The traces showsequential records from a single hippocampal neurone. during applications of 0-3 mm-N'MDA in the presence of 100 nM-glycine. 7-Chlorokynurenic acid (10,uM) almost com-pletely, but reversibly, blocks the response to NMIDA. Increasing the concentration ofglycine to 1 /,M partially overcomes the antagonist action of 7-chlorokynurenic acid, whileincreasing the concentration of NMDA from 0 3 to 3 mm has no effect.
glycine dose-response curve for the peak response to NMDA suggests that, for anNMDA response 0 1 % of that recorded with 10 /tM-glycine, the glycine concentrationwould need to be reduced to 150 pM. Although it is unlikely that it will be possibleto achieve this experimentally, as an alternative we performed further experimentswith 7-chlorokynurenic acid. At 10 JIM this glycine antagonist produced total blockof the equilibrium response to 300 ,tm-NMDA recorded with 100 nM-glycine added tothe extracellular solution (Fig. 6). The peak response to NMDA was also essentiallyabolished by 10 /Lm-7-chlorokynurenic acid (< 2% of the control response recordedwith 300 ,tM-NMDA and 100 nM-glycine). We suggest that this result is identical tothat which would be recorded if the glycine concentration could be lowered topicomolar concentrations. Because control experiments with 100 nm-glycine show!that peak responses to NAIDA are on average 52% of the maximum which can berecorded with a saturating concentration of glycine (Fig. 5), the above experimentsuggests that if 7-chlorokynurenic acid acts as a pure competitive glycine antagonist.then in the absence of glycine there would be virtually no response to NMDA.Antagonism of the NMDA response recorded with 10 /aM-7-chlorokynurenic acid
could be reduced on raising the glycine concentration from 01 to 1 Jum. but not byraising the NMDA concentration from 0-3 to 3 mait (Fig. 6). consistent with aselective, competitive action of 7-chlorokynurenic acid at the glycine recognition siteon the NMDA receptor. Analysis of ligand binding experiments, which show affinitiesof 7-chlorokynurenic acid for the glycine and NMDA binding sites of 0-56 and 169 JIMrespectively (Kemp et al. 1988). also suggest that with 300 JIM-NMDA and 100 nm-
324

MODUlTLATION OF NMDA RECEPTOR DESENSITIZATION 3
glycine, 10 Ium-7-chlorokynurenic acid should selectively block NMDA responses bybinding to the glycine recognition site. Together with our analysis of thedose-response curve for the action of glycine on the peak response to NMDA (Fig.5), the above result suggests that for both the peak and steady-state responses toNMDA, glycine is absolutely required for the activation of ion channel activity. Thisis in accord with the results of Kleckner & Dingledine (1988) who studied equilibriumresponses to NMDA using a slow perfusion system. Because of the contamination ofexperimental solutions by nanomolar concentrations of glycine, our results suggestit is unnecessary to postulate an inverse agonist action for 7-chlorokynurenic acid(Kemp et al. 1988).
The glycine antagonist 7-chlorokynurenic acid promotes desensitization of responses toNVMDA
The 7-chlorokynurenic acid-induced rightward shift of the dose-response curve forglycine-evoked potentiation of responses to NMDA (Fig. 5) suggests that, at a fixedconcentration of glycine, increasing doses of this antagonist should produce similareffects to those recorded on lowering the concentration of glycine in controlexperiments such as those shown in Figs 3-5. For responses to 300 /IM-NMDA,recorded with 3 /Im-glycine, we found that doses of 7-chlorokynurenic acid from 0-1to 300 /,M produced a progressive block of both the peak and steady-state responseto NMDA, with greater sensitivity of the steady-state response (Fig. 7). Thus, atintermediate doses of 7-chlorokynurenic acid, the response to NMDA showed strongdesensitization, similar to responses recorded in control experiments with 30-300 nM-glycine.
Analysis of the block of the equilibrium response to NMDA as a function of7-chlorokynurenic acid concentration (Fig. 7) was used to calculate the dose ofantagonist required for 50% block (IC50) of the response to NMDA from therelationship
Test 1Control 1 + ([Antag]/lC50)' (3)
the IC50 value was then used to calculate an equilibrium dissociation constant (Ki),using the Cheng-Prusoff equation (Cheng & Prusoff, 1973):
1 + ([Gly]/EC50)' (4)
and EC50 values for glycine obtained as illustrated in Fig. 5. This experimentrevealed a 4-fold greater sensitivity for block of the steady-state current by 7-chlorokynurenic acid (IC50 = 2-25 ,UM) than for block of the peak current (IC50 =8-96 /tM), and gave similar Ki values for the steady-state (240 nm) and peak (330 nm)responses respectively. These estimates are in good agreement with the values of 294and 310 nm calculated from shift of the dose-response curve for glycine by 7-chlorokynurenic acid (Fig. 5), and provide further evidence for our suggestion thatglycine is absolutely required for activation of NMDA receptors. Analysis of thedegree of desensitization of responses to 300 ,aM-NIDA and 3 ,uM-glycine (plottedas Isteadystate/1peak versus antagonist concentration), and evoked by several doses of
325
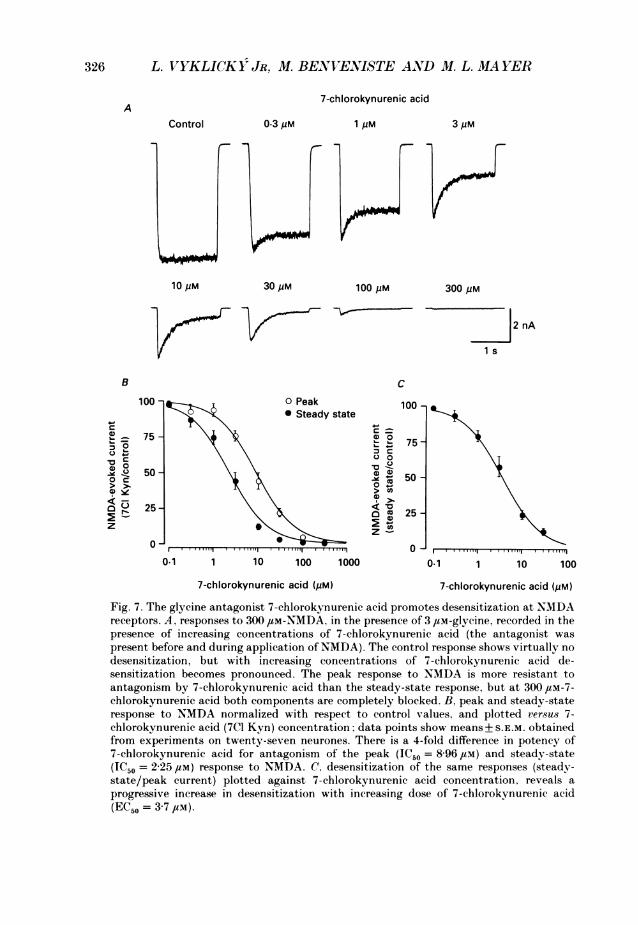
326 L. VYKLICOKY JR, M. BENVENISTE AND M. L. MA YER
7-chlorokynurenic acidA
Control 0.3 pM 1 pM 3 pM
10pM 30uM 100PM 300pM
2 nA
i s
B C
100 100Peak 100 1* Steady state
7575 75-c ~~~~~00
0~~~~~~~~~~~~~50 -~-
0 ~~~~~~~~~~~~~~50-
25 o ~~~~~~~~~~ ~25-0)
z -
0 ~~~~~~~~~~~~00.1 1 10 100 1000 0.1 1 10 100
7-chlorokynurenic acid (pM) 7-chlorokynurenic acid (pM)
Fig. 7. The glycine antagonist 7-chlorokynurenic acid promotes desensitization at NMDAreceptors. A, responses to 300 ,#M-NMDA, in the presence of 3 /LM-glycine, recorded in thepresence of increasing concentrations of 7-chlorokynurenic acid (the antagonist waspresent before and during application of NMDA). The control response shows virtually nodesensitization, but with increasing concentrations of 7-chlorokynurenic acid de-sensitization becomes pronounced. The peak response to NMDA is more resistant toantagonism by 7-chlorokynurenic acid than the steady-state response, but at 300 4uM-7-chlorokynurenic acid both components are completely blocked. B, peak and steady-stateresponse to NMDA normalized with respect to control values, and plotted versus 7-chlorokynurenic acid (7C1 Kyn) concentration; data points show means+ S.E.M. obtainedfrom experiments on twenty-seven neurones. There is a 4-fold difference in potency of7-chlorokynurenic acid for antagonism of the peak (IC50 = 8-96 yM) and steady-state(IC50 = 2-25 /LM) response to NMDA. C, desensitization of the same responses (steady-state/peak current) plotted against 7-chlorokynurenic acid concentration, reveals aprogressive increase in desensitization with increasing dose of 7-chlorokynurenic acid(EC50 = 3-7 /LM).

MODULATION OF NMDA RECEPTOR DESENSITIZATION 3
7-chlorokynurenic acid, gave an estimate of 240 nfm for the Ki of 7-chloro-kynurenic acid, calculated from an IC50 of 3 68 JIM (Fig. 7), and the control datashown in Fig. 5. This is similar to the estimate of 275 nm calculated from shift of thedesensitization dose-response curve, assuming a competitive interaction betweenglycine and 7-chlorokynurenic acid.
In summary, our results suggest that binding of glycine to the NMDA receptorboth promotes activation of the receptor-channel complex, and reduces desen-sitization. The competitive glycine antagonist 7-chlorokynurenic acid reducesactivation, and promotes desensitization, with similar potency for both effects. Infurther attempts to determine if these processes were closely related, we performeda series of experiments in which the kinetics of desensitization and the rate constantsfor binding and dissociation of glycine were examined directly, as described in thecompanion paper (Benveniste, Clements, Vyklicky & Mayer, 1990).
DISCUSSION
The modulation of NMDA receptor activity by glycine is an unusual example ofion channel regulation, in that the potentiating action of glycine is very strong.Indeed, our results support the suggestion made by Kleckner & Dingledine (1988)that glycine is absolutely required for the receptor-channel complex to enter theopen state. At equilibrium, our results with extracellular solutions to which noglycine was added are similar to those reported by Kleckner & Dingledine (1988), inthat we obtained essentially no response to NMDA. We propose that the generationof more substantial initial responses to NMDA applied by fast perfusion, andrecorded from neurones bathed in solutions to which no glycine has been addedexperimentally, is due to the inevitable background contamination of experimentalsolutions by nanomolar concentrations of endogenous glycine.
Several observations suggest that the action of glycine occurs via substantiallydifferent mechanisms than those determined previously in studies on other ligand-gated receptor channel complexes subject to modulation. First, the absoluterequirement of glycine for activation of NMDA receptors has no precedent. In thecase of another well-characterized ligand-gated channel, the GABAA receptor,activation by GABA is sufficient to generate large responses in the absence of anyadded modulatory substances (e.g. Bormann, Hamill & Sakmann, 1987), despite thefact that the activity of GABAA receptors can be very strongly modulated by drugsand endogenous ligands. Second, stabilization of the activated state of the GABAAreceptor-channel complex by barbiturates, and to a lesser extent by benzodiazepines,which is recorded as a prolongation of single-channel open times, provides anexample for a molecular mechanism for modulation of ligand-gated channels (Barker& McBurney, 1979; Study & Barker, 1981). In contrast, the potentiation of NMDAreceptor responses by glycine must occur via a different mechanism, since changes inglycine concentration evoke no marked changes in single-channel open time orconductance (Johnson & Ascher, 1987a). These observations, and the results of thepresent experiments, suggest alternative mechanisms for the potentiating action ofglycine on NMDA receptors, which may include regulation of transitions of theNMDA receptor channel complex to the active state, or a reduction of desensitization.
327

L. VYKLICKYJJR, M. BENVENISTE AND M. L. MAYER
We suggest that both mechanisms are likely to be important. The kinetic basis forthis dual action of glycine is presented in the accompanying paper (Benveniste et al.1990).
Desensitization at NMDA receptors compared to desensitization at nicotinic receptorsThe relief of NMDA receptor desensitization by glycine appears to have no
precedent from studies on other ligand-gated ion channels, although modulation ofdesensitization at neuronal nicotinic acetylcholine receptors has been observedpreviously. At low concentrations, substance P enhances desensitization at nicotinicreceptors, but does not reduce the initial amplitude of responses to acetylcholine(Clapham & Neher, 1984; Boyd & Leeman, 1987). In contrast, glycine potentiatesboth the initial and steady-state response to NMDA. In the present experiments thisappears to be due to two effects: at low concentrations of NMDA, between 3 and10/ M, responses to fast application of agonist recorded with 10 or 100 nM-glycineadded to the extracellular solution show little desensitization, but are stronglypotentiated by further increases in glycine concentration. When NMDA is applied athigher concentrations, glycine-sensitive desensitization is recorded, the amount ofwhich becomes progressively larger with increasing dose of NMDA. This suggeststhat glycine could promote activation of NMDA receptors by two mechanisms, andthat the relief of desensitization by glycine is important only at higher concentrationsof NMDA. However, kinetic experiments described in a companion paper suggest acommon mechanism which accounts for both actions of glycine (Benveniste et al.1990).In the above studies on nicotinic receptors, substance P applied together with
acetylcholine markedly increased the rate of onset of desensitization of the responseto agonist. Although by analogy potentiation of equilibrium responses to NMDA byglycine might be expected to occur via a slowing of the rate of onset ofdesensitization,the action of glycine actually appears to result from an increase in the rate ofrecovery from desensitization (see Mayer et al. 1989b and Benveniste et al. 1990). Itseems likely that the mechanisms underlying desensitization at NMDA and nicotinicreceptors are substantially different. For nicotinic receptors, pre-incubation withacetylcholine, or with high concentrations of substance P in the absence of agonist,produces strong desensitization of responses to subsequent application of acetyl-choline; recovery from substance P-evoked desensitization occurs with similarkinetics to that observed following desensitization evoked by application ofacetylcholine applied alone (Boyd & Leeman, 1987). In contrast, when neurones areincubated with 100 ,SM-NMDA in solutions with no added glycine, for periodsexceeding one minute, the subsequent application of glycine produces rapid ionchannel activation of similar amplitude to that recorded in the absence of pre-incubation with NMDA (see Benveniste et al. 1990), suggesting that in the absenceof glycine NMDA receptors do not enter a desensitized state of long duration.
Implications for other glutamate-activated channelsBecause modulation of the NMDA receptor complex by glycine has several unique
features, it is probable the mechanisms underlying desensitization at other types ofglutamate receptor channel complex are likely to be appreciably different. Aninteresting comparison can be made with the quisqualate subtype of L-glutamate
328

AMODUtLATION OF NJIDA RECEPTOR DESENSITIZATION
receptor, desensitization of which is reduced by the lectin concanavalin A (Mathers& Usherwood, 1976; Mayer & Vyklick', 1989). In preliminary experiments we havefound that the potentiation of responses to quisqualate by concanavalin A isassociated with a slowing of the onset of desensitization, with less effect on the rateof recovery from desensitization. This is in marked contrast to the increase in the rateof recovery from desensitization that occurs for the interaction of glycine withNMDA receptors, and provides further evidence that NMDA and quisqualatereceptors are likely to be different proteins.
Do XMDA receptors desensitize in vivo?The relief of desensitization of NMDA receptors by high concentrations of glycine
provides an important mechanism for the generation of sustained responses toNMDA in vitro. Whether this is important for the function of NMDA receptors invivo will depend on both the range of extracellular glycine concentrations that canoccur in the brain, and the concentration-time profile for extracellular L-glutamateconcentration following release from nerve terminals. Our results suggest that evenfollowing intense activation of excitatory amino acid synapses, substantialdesensitization of responses to NMDA is unlikely to occur via a glycine-sensitiveroute, if the extracellular glycine concentration in vivo is much greater than 500 nm.In experiments with prolonged applications of low concentrations of NMDA (<10/M) we have found that substantial, non-desensitizing responses can be recordedover a wide range of glycine concentrations, during NMDA applications lastingseveral minutes, even with physiological concentrations of calcium (e.g. Mayer et al.1988). It seems probable that similar sustained NMDA receptor activation could beprovoked by L-glutamate in vivo, consistent with recent observations in brain slicepreparations of hippocampus, which suggest tonic activation of NMDA receptors inthe absence of added agonists, most likely due to accumulation of L-glutamate in theextracellular fluid (Sah, Hestrin & Nicoll, 1989). Thus, sustained NMDA receptoractivity seems quite possible in vivo, and is likely to play an important role inregulating neuronal excitability. An unfortunate consequence of sustained NMDAreceptor activity occurs during cerebral anoxia, since there is considerable evidencethat the activation of NMDA receptors participates in triggering neuronal deathduring ischaemia (Choi, 1988).
We thank Sandy Fitzgerald and Christine Winters for preparing and maintaining the culturesused in our experiments, Dr B. Smith, Dr S. Hsiao, W. Holsinger, J. Ries and N. Simmons forbuilding the rapid perfusion apparatus, and Dr V. Nadler for HPLC measurements. L. V. was aFogarty visiting fellow. M. B. is an NRC research fellow.
REFERENCES
ASCHER, P., HENDERSON, G. & JOHNSON, J. (1988). Dual inhibitory actions of kynurenate on theN-methyl-D-aspartate (NMDA)-activated response of cultured mouse cortical neurones. Journalof Physiology 406, 141P.
BARKER, J. L. & McBURNEY, R. N. (1979). Phenobarbitone modulation of post-synaptic GABAreceptor function on cultured mammalian neurones. Proceedings of the Royal Society B 206,318-326.
BENVENISTE, M., CLEMENTS, J., VYKLICKY, L. JR & MAYER, M. L. (1990). A kinetic analysis of the
329

30L. VYKLICKY JR, M. BENVEXVISTE AN..D M. L. MAYER
modulation of N-methyl-D-aspartic acid receptors by glycine in mouse cultured hippocampalneurones. Journal of Physiology 428. 333-357.
BORMANN, J., HAMILL. 0. P. & SAKMANN, B. (1987). Mechanism of anion permeation through
channels gated by glycine and y-aminobutyric acid in mouse cultured spinal neurones. Journalof Physiology 385. 243-286.
BoYD, N. D. & LEEMAN. S. E. (1987). Multiple actions of substance P that regulate the functional
properties of acetylcholine receptors. Journal of Physiology 389, 69-97.BRISTOW, D. R., BOWERY, N. & WOODRUFF, G. N. (1986). Light microscopic autoradiographic
localization of [3H]glycine and [3H]strychnine binding sites in rat brain. European Journal ofPharmacology 126, 303-307.
CHENG. Y. C. & PRUSOFF. WV. H. (1973). Relationship between the inhibition constant (K1) and theconcentration of inhibitor which causes 50 per cent inhibition(I50) of an enzymatic reaction.Biochemical Pharmacology 22, 3099-3108.
CHOI, D. W. (1988). Glutamate neurotoxicity and diseases of the nervous system. Neuron 1,623-634.
CLAPHAM. D. E. & NEHER, E. (1984). Substance P reduces acetylcholine-induced currents inisolated bovine chromaffin cells. Journal of Physiology 347, 255-277.
COLQUHOUN. D. & OGDEN, D. C. (1988). Activation of ion channels in the frog end-plate by highconcentrations of acetylcholine. Journal of Physiology 395, 131-159.
FOSTER, A. C. & KEMP, J. A. (1989). HA-966 antagonizes N-methyl-D-aspartate receptors througha selective interaction with the glycine modulatory site. Journal of Neuroscience 9, 2191-2196.
GUTHRIE, P. B., BRENNEMAN, D. E. & NEALE, E. A. (1987). Morphological and biochemicaldifferences expressed in separate dissociated cell cultures of dorsal and ventral halves of themouse spinal cord. Brain Research 420, 313-323.
HUETTNER, J. E. (1989).Indole-2-carboxylic acid: a competitive antagonist of potentiation by
glycine at the NMDA receptor. Science 243. 1611-1613.JOHNSON, J. W. & ASCHER, P. (1986). Response of neurones to fast application of N-methyl-D-
aspartate. Society for Neuroscience Abstracts 12, 58.JOHNSON, J.WV. & ASCHER, P. (1987 a). Glycine potentiates the NMDA response of mouse central
neurones. Nature 325, 529-531.JOHNSON, J. W. & ASCHER, P. (1987b). Interaction of glycine with the N-methyl-D-aspartate
receptor. Society for Neuroscience Abstracts 13, 383.KEMP, J. A., FOSTER, A. C.. LEESON, P. D., PRIESTLEY, T.. TRIDGETT, R., IVERSEN, L. L. &WOODRUFF. G. N. (1988). 7-Chlorokynurenic acid is a selective antagonist at the glycinemodulatory site of the N-methyl-D-aspartate receptor complex. Proceedings of the NationalAcademy of Sciences of the USA 85, 6547-6550.
KESSLER. M., TERRAMANI, T., LYNCH, G. & BAUDRY, M. (1989). A glycine site associated with
N-methyl-D-aspartic acid receptors: characterization and identification of a new class of
antagonists. Journal of Neurochemistry 52, 1319-1328.KLECKNER, N. WV. & DINGLEDINE, R. (1988). Requirement for glycine in activation of N-methyl-
D-aspartic acid receptors expressed in Xenopus oocytes. Science 241, 835-837.KLECKNER, N. WV. & DINGLEDINE, R. (1989). Selectivity of quinoxalines and kynurenines as
antagonists of the glycine site onN-methyl-D-aspartate receptors.Mlolecular Pharmacology 36,430-436.
KUSHNER, L., LERMA, J.. ZUKIN, R. S. & BENNETT, M. V. L. (1988). Coexpression of N-methyl-D-aspartate and phencyclidine receptors in Xenopus oocytes injected with rat brain mRNA.Proceedings of the National Academy of Sciences of the USA 85, 3250-3254.
LARSON, A. L. & BETZ, A. J. (1988). Glycine potentiates strychnine-induced convulsions: role ofNMDA receptors. Journal ofNeuroscience 8, 3822-3826.
LODGE, D. (1988). Excitatory Amino Acids in Health and Disease. John Wiley & Sons, New York.MATHERS, D. A. & USHERWOOD, P. N. R. (1976). Concanavalin A blocks desensitization of
glutamate receptors in insect muscle fibers. Nature 259, 409-411.MAYER. M. L. (1989). Activation and desensitization of glutamate receptors in mammalian CNS.
In Ion Transport, ed. BENHAM, C. D. & KEELING, D. J., pp. 183-194. Academic Press, London.MAYER, M. L., MAcDERMOTT, A. B., WESTBROOK, G. L., SMITH, S. J. & BARKER, J. L. (1987).
Agonist- and voltage-gated calcium entry in cultured mouse spinal cord neurons under voltageclamp measured using arsenazo III. Journal of Neuroscience 7, 3230-3244.
3330

MODULATIO1N OF NMDA RECEPTOR DESENSITIZATION 3
MAYER, M. L. & VYKLICKY, L. JR (1989). Concanavalin A selectively reduces desensitization ofmammalian neuronal quisqualate receptors. Proceedings of the National Academy of Sciences of theUSA 86, 1411-1415.
MAYER, M. L., VYKLICKY, L. JR & CLEMENTS, J. D. (1989b). Regulation of NMDA receptordesensitization in mouse hippocampal neurones by glycine. Nature 338, 425-427.
MAYER, M. L., VYKLICKY. L. JR & SERNAGOR. E. (1989a). A physiologist's view of the N-methyl-D-aspartate receptor: an allosteric ion channel with multiple regulatory sites. Drug DevelopmentResearch 17, 263-280.
MAYER, M. L., VYKLICKY, L. JR & WVESTBROOK, G. L. (1989c). Modulation of excitatory aminoacid receptors by group II B metal cations in cultured mouse hippocampal neurones. Journal ofPhysiology 415, 329-350.
MAYER, M. L. & WESTBROOK, G. L. (1984). Mixed-agonist action of excitatory amino acids onmouse spinal neurones under voltage clamp. Journal of Physiology 354, 29-53.
MAYER, M. L. & WESTBROOK, G. L. (1985). The action ofN-methyl-D-aspartic acid on mouse spinalneurones in culture. Journal of Physiology 361. 65-90.
MAYER, M. L. & WESTBROOK, G. L. (1987). The physiology of excitatory amino acids in thevertebrate central nervous system. Progress in Neurobiology 28, 197-276.
MAYER, M. L., WESTBROOK. G. L. & VYKLICKY, L. JR (1988). Sites of antagonist action on N-methyl-D-aspartic acid receptors studied using fluctuation analysis and a rapid perfusiontechnique. Journal of Neurophysiology 60. 645-663.
MODY, I., SALTER, M. W. & MACDONALD, J. F. (1988). Requirement of NMDA receptor/channelsfor intracellular high-energy phosphates and the extent of intraneuronal calcium buffering incultured mouse hippocampal neurones. Neuroscience Letters 93, 73-78.
REYNOLDS, I. J., MURPHY, S. N. & MILLER, R. J. (1987). 3H-labelled MK-802 binding to theexcitatory amino acid receptor complex from rat brain is enhanced by glycine. Proceedings of theNational Academy of Sciences of the USA 84, 7744-7748.
SAH, P., HESTRIN, S. & NICOLL, R. A. (1989). Tonic activation of NMDA receptors by ambientglutamate enhances excitability of hippocampal neurons. Science 246, 815-818.
SNELL, L. D., MORTER, R. S. & JOHNSON, K. M. (1988). Structural requirements for activation ofthe glycine receptor that modulates the N-methyl-D-aspartate operated ion channel. EuropeanJournal of Pharmacology 156, 105-1 10.
STUDY, R. E. & BARKER, J. L. (1981). Diazepam and (-)pentobarbitone: fluctuation analysisreveals different mechanisms for potentiation of GABA responses in cultured central neurones.Proceedings of the NVational Academy of Sciences of the USA 78, 7180-7184.
THOMSON, A. M., WALKER, V. E. & FLYNN, D. M. (1989). Glycine enhances NMDA-receptormediated synaptic potentials in neocortical slices. NVature 338, 422-424.
YELLEN, G. (1982). Single Ca2l-activated nonselective cation channels in neuroblastoma. Nature296, 357-359.
ZORUMSKI, C. F., YANG, J. & FISCHBACH, G. D. (1989). Calcium-dependent, slow desensitizationdistinguishes different types of glutamate receptors. Cellular and .Molecular Nveurobiology 9,95-104.
331