bpog response to annex 2 - biophorum · bpog response to annex 2 ... to ensure that the messages it...
TRANSCRIPT

10.5731/pdajpst.2016.006502Access the most recent version at doi: 300-31170, 2016 PDA J Pharm Sci and Tech
Jeff Blake, David Estapé, Ken Green, et al. BPOG Response to Annex 2
on June 10, 2016journal.pda.orgDownloaded from on June 10, 2016journal.pda.orgDownloaded from

COMMENTARY
BPOG Response to Annex 2JEFF BLAKE (NOVAVAX), DAVID ESTAPE (M�W GROUP), KEN GREEN (SHIRE),KAVITA RAMALINGAM IYER (MERCK), JEFF JOHNSON (MERCK), PHIL MCDUFF (BIOGEN),MARC PELLETIER (CRB), ALAN POWELL (DME ENGINEERING),SCOTT PROBST (BAYER TECHNOLOGY SERVICES),PAUL SMOCK (MERIDIAN BIOGROUP LLC AND WAS EMPLOYED BY ASTRAZENECA AT THE TIMETHE WORK WAS COMPLETED), ROBIN PAYNE (BPOG FACILITATOR), TIM CORBIDGE* (BPOG FACILITATOR)
Introduction
This paper is an interpretive response to Annex 2 ofEudralex—Volume 4; the European Union (EU)guidelines for good manufacturing practice (GMP) ofmedicinal products for human and veterinary use (1).It was written collaboratively, by a team of experts inclosed system processing, from 26 companies in thebiopharmaceutical industry facilitated by BioPhorumOperations Group (BPOG), who have a history ofassessing the potential for new technologies to benefitthe industry (2, 3). The authors listed on the title pagewere lead contributors to the content of this document,writing sections, editing, and liaising with colleaguesto ensure that the messages it contains are represen-tative of current thinking across the biopharmaceuticalindustry. This paper is a consensus view of a responseto Annex 2, but it does not represent fully the internalpolicies, views, or opinions of the authors’ respectivecompanies.
The purpose of this paper is to share an interpretationof key areas of Annex 2, providing enhanced clarityfor the industry. This paper supports a scientific andrisk-based approach that identifies the biological ac-tive substance manufacturing requirements, and thetypes of control that meet those requirements.
Volume 4 of the EU GMP guidelines was originallydeveloped to complement EU Commission Directive91/356/EEC (subsequently amended by 91/412/EECand 2003/94/EC). It contains two annexes (Annex 1
and Annex 2) that are highly influential and provideguidance on the design, building, and regulatory in-spections of biopharmaceutical facilities. Their titlesare:
Annex 1: Manufacture of Sterile Medicinal Products.
Annex 2: Manufacture of Biological Active Substancesand Medicinal Products for Human Use.
Annex 1, pertaining to sterile manufacturing opera-tions and sterile products, will be revised through2015 and into 2016 (4).
Annex 2 focuses on the manufacture of biologicalactive substances and biological medicinal productsfor human use.
Annex 2 states the importance of quality risk manage-ment (QRM) principles in the development of controlstrategy [such as those principles described in ICH Q9(5)]. However, Annex 2 references Annex 1 in anumber of places, leading to potential misinterpreta-tion, that is, that the environmental classification cas-cades and associated controls described in Annex 1(sterile products) are required for facilities and man-ufacturing systems in scope for Annex 2 as well. Onthe other hand, QRM enables selection of controls thatare proportionate to the risk identified, as opposed tonon-risk-based conventional approaches. The authorsof this paper value the QRM approach, and it is theiropinion that QRM should drive the design of processand control strategies for the mitigation of contami-nation and cross-contamination risks.
A goal for QRM, in terms of facility design, is to applyrisk-based approaches to the management of contam-ination and cross-contamination that add the highestvalue regarding product quality and patient safety.It is not always necessary to employ conventionalmitigation methods like environmental controls. For
* Corresponding Author: Tim Corbidge, BiophorumOperations Group, 5 Westbrook Court, Sharrow ValeRoad, Sheffield, S11 8YZ, United Kingdom; E-mail:[email protected]; Telephone: � 44 (0)7970340073.
doi: 10.5731/pdajpst.2016.006502
DISCLAIMER: The following article is a special editorial contribution from the BioPhorum Operations Group (BPOG).Please note that it did not go through the PDA Journal of Pharmaceutical Science and Technology regular peer reviewprocess.
300 PDA Journal of Pharmaceutical Science and Technology
on June 10, 2016journal.pda.orgDownloaded from

example, if one uses a completely closed system ofoperation, QRM evaluation may determine that envi-ronmental controls are not required for the closedprocess steps. In this paper, the authors focus on theapplication of QRM to the following topics:
1. References to Annex 1 in Annex 2, which could beinterpreted to imply nonsterile facilities, shouldapply sterile facility controls.
The authors recognize Annex 1 will be revised (4) andsee this as an opportunity to clarify guidance in linewith some of the reasons for commenting in this paper.
2. Potential alternatives to specific text in Annex 2,pertaining to “Premises and Equipment”, “Startingand Raw Material” and “Operating Principles”.These are considered to be open to a range ofinterpretations and are frequently the subject oflengthy debate when biopharmaceutical facilitiesare designed and built.
1. References To Annex 1 In Annex 2
Annex 2 applies to the manufacture of biologicalactive substances and medicinal products for humanuse. The two instances in which Annex 2 refers toAnnex 1, which applies to the manufacture of sterilemedicinal products, sometimes lead the reader to theinterpretation that Annex 1 also applies verbatim tothe manufacture of biological active substances. Thatinterpretation is contrary to the QRM principles es-poused in Annex 2.
Annex 1 is referenced only to provide principles andguidance for establishing classified environments tomitigate contamination risks as deemed necessary.Annex 2 is clear that the environmental controlsshould be conceived and established through the exe-cution of a QRM process. The following sections showthe instances where Annex 2 references Annex 1 andhow these references instead could be applied to bio-pharmaceutical processes using the QRM framework.
1.1. Annex 2, Paragraph 6
Paragraph 6, in the “Premises and Equipment” section,states:
“6. Manufacturing and storage facilities, processes andenvironmental classifications should be designed toprevent the extraneous contamination of products. Pre-
vention of contamination is more appropriate thandetection and removal, although contamination islikely to become evident during processes such asfermentation and cell culture. Where processes are notclosed and there is therefore exposure of the product tothe immediate room environment (e.g., during addi-tions of supplements, media, buffers, gasses, manipu-lations during the manufacture of ATMPs) controlmeasures should be put in place, including engineeringand environmental controls on the basis of QRM prin-ciples. These QRM principles should take into accountthe principles and guidance from the appropriate sec-tions of Annex 118 to EudraLex, Volume 4, whenselecting environmental classification cascades and as-sociated controls.”
Footnote 18 provides clarification regarding the appli-cation of Annex 1 for active pharmaceutical ingredient(API) manufacturing:
“18 Although the title of Annex 1 refers to the manu-facture of sterile medicinal products it is not the in-tention to force the manufacture of sterile product at astage when a low bioburden is appropriate and autho-rized. Its use is because it is the only EU GMP sourceof guidance on all of the classified manufacturingareas including the lower grades D and C.”
Reason for Commenting
The application of QRM principles to the manufactureof biological products often indicates the use of de-signs that differ from the environmental control mea-sures dictated by Annex 1. The statement that controlmeasures should be based on QRM principles, fol-lowed by the statement that these should take intoaccount guidance from relevant sections in Annex 1,could lead to an interpretation that Annex 1 should beapplied directly to the manufacture of biological activesubstances. The statement in footnote 18, that it is notthe intention of Annex 1 to force sterile manufacturingwhen “. . . a low bioburden is appropriate and autho-rized”, provides some clarification but is open to num-ber of different interpretations.
Interpretation
It is proposed that decisions to assign area classifica-tion and implement other controls to prevent contam-ination should consider the nature of the operation,stage of the manufacturing process, and QRM out-come.
301Vol. 70, No. 3, May–June 2016
on June 10, 2016journal.pda.orgDownloaded from

Clearly, the ingress of contaminants into process fluidsor materials should be minimized to prevent adverseimpact on the product. The risk-based approach re-quires development of justified control measures topotential sources of environmental contamination.This requires justification beyond simply assumingthat filtration or other downstream purification stepsremove environmental contaminants that enter duringprocessing, or that contamination will be detected.
The following case illustrates the tenet that controlmeasures should be commensurate with the risks (5)and the consequences of simple adherence to Annex 1for biopharmaceutical processes:
One case where the risk profile of biological activesubstances manufacturing differs from sterile productmanufacturing is an open aseptic process for inoculumpreparation for cell culture. The operations often in-volve open manipulations of cell culture using asepticprocessing techniques. Across the industry, Grade Aconditions are achieved in the unidirectional air flowhood often used for such operations. The objective isto avoid microbial or viral contamination of the cul-ture during these operations.
In the above example, adherence to all Annex 1 re-quirements for Grade A environments is not necessar-ily required to avoid contamination of the culture.However, since Annex 2 refers the reader to Annex 1for principles and requirements of classified areas, onemight conclude that an open operation, like inoculumpreparation, where microbial contamination will resultin failure of the batch, requires adherence to all Annex1 background and in-process monitoring requirementsfor a Grade A environment.
Specifically, paragraph 33 from Annex 1 in the section“Aseptic Preparation” states:
“33. Handling and filling of aseptically prepared prod-ucts should be done in a grade A environment with agrade B background.”
Hence, adherence to Annex 1 aseptic requirementswould require a Grade B background environment forthe Grade A environment for aseptic operations inbiological active substances manufacturing. However,this interpretation may result in conditions that impedereliable manufacture of cell culture products. Appli-cation of Annex 1 requirements should be challengedand the design justified using QRM, to ensure thepatient is being protected in the most appropriate way.
In the case of open operations that fill sterile medicinalproducts, Annex 1 applies and requires a Grade Aenvironment with a Grade B background. This makessense, as there are no additional steps to remove orinactivate microbial contaminants and limited oppor-tunity to detect contamination before the product isgiven to the patient. In such a context, the Annex 1requirement provides appropriate controls.
However, the Annex 1 requirements are less appropri-ate for inoculum preparation. Additional layers ofprotection are provided in the process and controls andmitigate the impacts of potential contamination:
● A microbial load or viral contamination wouldlikely be detected through cell culture testing. An-nex 2 acknowledges this point in paragraph 6where it states “. . . contamination is likely tobecome evident during processes such as fermen-tation and cell culture.”
● Inoculum preparation is at the very beginning ofmany API manufacturing processes, with addi-tional detection points and downstream processsteps that remove contaminants.
Therefore, the risk profile of inoculum preparationdiffers from that of filling of sterile medicinal prod-ucts. QRM evaluation will often indicate differentrequirements for this process step.
In current practice, industry has significant experienceperforming cell culture expansions in a Grade A hoodwith Grade C or Grade D background environmentswith very good success and exceedingly low contam-ination rates.
Regulatory authorities have supported this practice.For example, the Irish Medicines Board, Reference 6states: “Typically, Grade C background with Grade Asupply BSC is considered acceptable”. An Aide Me-moire (071210BN) issued by the German Health Au-thority (ZLG) indicates that a Grade A unidirectionalflow hood in a Grade C background environment isappropriate for establishing a cell bank (7).
1.2. Annex 2, Paragraph 33
Paragraph 33 of Annex 2 in the “Starting and RawMaterials” section also refers to Annex 1:
33. Given that the risks from the introduction of con-tamination and the consequences to the finished prod-
302 PDA Journal of Pharmaceutical Science and Technology
on June 10, 2016journal.pda.orgDownloaded from

uct is the same irrespective of the stage of manufac-ture, establishment of a control strategy to protect theproduct and the preparation of solutions, buffers andother additions should be based on the principles andguidance contained in the appropriate sections of An-nex 1. The controls required for the quality of startingand raw materials and on the aseptic manufacturingprocess, particularly for cell-based products, wherefinal sterilization is generally not possible and theability to remove microbial by-products is limited,assume greater importance. Where an MA [MarketingAuthorization] or CTA [Clinical Trial Authorization]provides for an allowable type and level of bioburden,for example at active substance stage, the controlstrategy should address the means by which this ismaintained within the specified limits.”
Reason for Commenting
The statement that the consequences of contaminationare “. . . the same irrespective of the stage of manu-facture”, and that a control strategy should be based on“. . . appropriate sections of Annex 1”, though “appro-priate sections” are not specified, indicates that verystrong contamination controls should be in placethroughout every process. However, a QRM approachwould indicate that the risk of contamination is lowerupstream in the process than downstream, as down-stream filtration and purification steps remove andprovide opportunities to detect contamination. Stron-ger controls may be required further downstream inthe process, where there are fewer subsequent purifi-cation steps and opportunities for detection.
While the initial statement of the paragraph is not inaccordance with QRM, the statement at the end of theparagraph that “the Marketing Authorization (MA) orthe Clinical Trial Authorization (CTA) may providefor an allowable bioburden level and that controlsneed to be in place to ensure those specified bioburdenlevels” are consistent with the risk-based approachthat is prescribed throughout Annex 2. Apparentlycontradictory statements in paragraph 33 may lead toconfusion and misinterpretation for an end user.
Interpretation
As stated in the previous section, paragraph 6 statesthat prevention of contamination is more appropriatethan detection and removal. The authors agree withthis approach so long as it is interpreted within a QRMframework. Some operations, such as solution prepa-
ration, are typically open operations, and microbialcontamination is successfully removed by filtration.However, applying Grade C conditions to solutionpreparation in biological active substance manufactur-ing operations (as specified under paragraph 17 ofAnnex 1) may not be an appropriate control for arisk-based approach where many factors are consid-ered.
In the following example for solution preparation, it isobvious that raw material is the primary contaminationsource as opposed to influences from the environment.Hence, controls developed to address bioburden fromraw materials will also address bioburden from theenvironment.
Consider a solution preparation operation in which1000 L of solution is prepared with a total solidsconcentration in the solution of 50 g/L. The solidsused in the solution are manufactured under con-trolled, nonclassified conditions (at best). None of thesolids are tested for viruses, but all are tested forbioburden and have a maximum bioburden specifica-tion of 100 CFU/g. This means that the final solutioncould contain 5,000,000 CFU of bioburden from theraw materials alone. This does not include microbialpropagation in the solution during preparation.
The solids are added through an open port on thesolution preparation vessel in an open manner. Envi-ronmental conditions should be set so that the poten-tial additional contamination is negligible in compar-ison to the inherent microbial load in the startingmaterials. Even if the air contained 1000 CFU/m3, fivetimes higher than the recommended values for GradeD specification in operation, 50 m3 of air (50,000 L)would have to enter the tank during the addition pro-cess in order to introduce 50,000 CFUs of bioburden,which is 1% of the maximum allowable amount ofbioburden based on the raw material specifications.For a briefly exposed operation, exchange of 50 m3 ofair between the vessel and surroundings is not credi-ble, because both must be at the same pressure for theopen operation. No driving force exists for rapid airexchange between the tank and the room environment.In this context, QRM should drive the appropriatemanufacturing controls.
However, the tables in paragraph 17 and paragraph 32of Annex 1 both mandate a Grade C environment forthe preparation of solutions that are subsequently fil-tered when those solutions are used in a process in
303Vol. 70, No. 3, May–June 2016
on June 10, 2016journal.pda.orgDownloaded from

which the product is not terminally sterilized. Thiscould lead to the interpretation that solution prepara-tion for biological active substances, when the productis not terminally sterilized, should also be conductedin a Grade C environment. This should not be the case.Due to the inherently greater capability to detect andremove contaminants, the risk profile of biologicalactive substance manufacturing is very different frommanufacturing sterile medicinal products. Therefore, arisk-based approach should be taken when setting en-vironmental conditions for such operations. See Ap-pendix A for an example of the application of riskanalysis to solution preparation operations. As amodel, this example is intended to help when deter-mining appropriate area classifications for such oper-ations.
In summary, the two instances in which Annex 2refers to Annex 1 may lead the reader to believe thatAnnex 1 also applies to the manufacture of biologicalactive substance. Annex 1 is only referenced becauseit provides considerations and guidance for establish-ing classified environments that may be used to miti-gate contamination risks if deemed necessary. In sce-narios where Annex 2 is applicable, appropriateenvironmental controls should be implemented whenthe requirement is determined through the executionof a QRM process.
2. Potential Alternatives To Specific Text InAnnex 2
The second part of this paper describes interpretationsof statements in Annex 2 that the authors consideroverly prescriptive and/or that may be interpreted inways that are inconsistent with a risk-based approach.
2.1. Paragraph 5
“As part of the control strategy, the degree of envi-ronmental control of particulate and microbial con-tamination of the production premises should beadapted to the active substance, intermediate or fin-ished product and the production step, bearing in mindthe potential level of contamination of the startingmaterials and the risks to the product. The environ-mental monitoring program should be supplementedby the inclusion of methods to detect the presence ofspecific microorganisms (i.e., host organism, yeast,molds, anaerobes, etc.) where indicated by the QRMprocess.”
Reason for Commenting
Paragraph 5 highlights the need for an environmentalmonitoring program and supplemental requirements asindicated by the QRM process. This might be inter-preted to mandate a strong and conservative environ-mental monitoring program of a type that might berequired only in some instances for open processing.
Also, at the end of the paragraph, very specific rec-ommendations for an environmental monitoring pro-gram are given. However, the QRM process for aclosed system likely would show minimal risk ofparticulate or microbial contamination when located ina controlled nonclassified (CNC) space where require-ments for environmental monitoring are minimal ornonexistent. Usage of the abbreviation “i.e.” also in-dicates an expectation that tests for the listed organ-isms will be included. Therefore, the team believesthat the recommendations in this paragraph relating toan environmental monitoring program are overly pre-scriptive.
Interpretation
The degree of environmental control for contamina-tion should be risk based and not driven by conven-tional practices or preset expectation. Elements to beconsidered when determining the risk of contamina-tion from the environment are:
● Properties of the process materials, for example,growth promoting characteristics could affect holdtime.
● The degree of closure of equipment.
For closed and functionally closed systems, the QRMprocess may determine that the risk of particulate ormicrobial contamination is minimal and that a CNCenvironment is appropriate, in which case the environ-mental monitoring program can be minimized or elim-inated as required and appropriate for CNC environ-ments. In the example below, the risk from roomenvironment during cell expansion steps is renderednegligible by an approach to closing the operations.
A typical cell culture seed train process progressesthrough multiple cell expansion steps and increasesbatch volume in a process that requires multiple steriletransfers. In each transfer, connections are requiredthat increase the process contamination risk by poten-
304 PDA Journal of Pharmaceutical Science and Technology
on June 10, 2016journal.pda.orgDownloaded from

tially exposing process contact surfaces to the outsideenvironment. To minimize this risk it is critical tokeep process vessel connections closed until ready touse and then to join them together while minimizingexposure to the outside environment.
The availability and successful use of sterile closedsystems like presterilized disposable media bags andbioreactor bags that can be fitted with transfer, addi-tion, and sample lines significantly reduce or eliminatethe process contamination risks that otherwise can bea concern. The closed systems can be integratedthrough the use of techniques such as tube weldingand/or disposable aseptic connectors, therefore ensur-ing that the process remains closed even when theseconnections are made. Because the entire seed traincan now be performed as a closed system, it is notexposed to the room environment and the operationhas been successfully performed without contamina-tion in CNC areas.
The nature of the process step being executed can, ofcourse, have an impact on how best to protect the drugsubstance, upstream versus downstream of viral clear-ance. The following example illustrates this for man-ufacture of a drug substance intermediate:
● Operations like fermentation are a classic exampleof an upstream drug substance manufacturing pro-cess. Fermentation processes are conducted in aclosed system to prevent contamination (i.e., main-tain pure culture) and QRM evaluation would sup-port conducting such operations in CNC space.
● Downstream processes like purification of a drugsubstance have typically been located in classifiedareas. The higher room classification would oftenhave been selected following a QRM evaluation,because purification operations typically havefewer layers of protection and often include openoperations.
Implementation of QRM presents the opportunity toperform downstream operations in CNC or lower clas-sification areas. Modern downstream processes oftenachieve a high degree of closure, such as utilizingfunctionally closed systems and single-use technolo-gies (e.g., tube welding and sterile connections). Witheffectively closed systems coupled with other layers ofprotection, QRM evaluation may support locating theprocess in a CNC space, with minimal or no environ-
mental monitoring requirements as determined byQRM evaluation.
For most systems, an effective way to minimize therisk of contamination is not to control the environmentbut to close the system so that the risk associated withthe environment is minimized or eliminated.
Recommendation
Currently paragraph 5 describes environmental moni-toring requirements that might be applicable for opensystems. It will be beneficial if Annex 2 is revised toindicate that QRM evaluation should determine if EMis required and to what extent for a process step.
2.2. Paragraph 50
“A control strategy for the entry of articles and mate-rials into production areas should be based on QRMprinciples. For aseptic processes, heat stable articlesand materials entering a clean area or clean/con-tained area should preferably do so through a dou-bled-ended autoclave or oven. Heat labile articlesand materials should enter through an air lock withinterlocked doors where they are subject to effectivesurface sanitization procedures. Sterilization of ar-ticles and materials elsewhere is acceptable pro-vided that they are multiple wrappings, as appropri-ate to the number of stages of entry to the cleanarea, and enter through an airlock with the appro-priate surface sanitization precautions.”
Reason for Commenting
While paragraph 50 supports the implementation of aQRM approach and the authors fully agree with thefirst sentence, the remainder of the text describes insome detail the expectations for aseptic processingareas. While this direction may be appropriate forareas where open aseptic processing is conducted, thedetailed attention to aseptic processes might lead areader to conclude that similar controls are expectedfor a broader spectrum of processes, that is, typicalbulk biopharmaceutical manufacturing processes thatare not open aseptic processes. The amount of textdevoted to aseptic processes and the failure to distin-guish closed processes from open processes may in-fluence the end-user to implement designs that do notalign with QRM principles, providing an overburdenof controls.
305Vol. 70, No. 3, May–June 2016
on June 10, 2016journal.pda.orgDownloaded from

Interpretation
Paragraph 50 focuses on the requirements appropriatefor aseptic processing with no mention of the typicalbulk biopharmaceutical nonaseptic requirements. TheQRM process for entry into nonaseptic areas willindicate lower risk than for aseptic areas and minimalrisk for entry into nonaseptic areas with functionallyclosed systems with no need for multiple wrappingsand/or multiple stages of entry to the production area.Most of the discussion in paragraph 50 applies for thehigh-risk aseptic processes, but there is no discussionof lower risk options. Paragraph 50 could be inter-preted as requiring multiple wrapping and/or stages ofentry and surface sanitization requirements even forlow contamination risk situations, such as for nona-septic operations in closed systems. It is importantto have the understanding that QRM evaluation andoutcome should determine and inform the controlstrategy instead of incorporating process controlsbased on the nature of the process whether it isaseptic or nonaseptic.
Recommendation
Suggested wording: “A control strategy for the entryof articles and materials into production areas shouldbe determined based on QRM principles as applicableto the stage and nature of the process.”
2.3. Paragraph 54
“Centrifugation and blending of products can lead toaerosol formation and containment of such activitiesto minimize cross-contamination is necessary.”
Reason for Commenting
Centrifuges are often used in manufacturing, often forupstream cell and cellular debris separations. The for-mation of aerosols may transfer live microorganismsor other contaminants into the environment.
The requirement for “containment of such activities”in paragraph 54 is interpreted as an unconditional needto isolate centrifuges without a rational assessment ofthe likelihood that aerosols will contaminate the envi-ronment and present a significant risk for cross-con-tamination. As a result, there is a clear expectation thata centrifuge must be located in a separate room, inde-pendent of the type, scale, operation, operating time,etc.
Most of the centrifuges now used in biopharmaceuticaloperations operate as functionally closed systems thatdo not release aerosols into the environment. Forexample, while windage and mechanical action mayresult in considerable aerosol formation inside a discstack centrifuge, the design of the casing and connec-tions for ventilation, solid and liquid discharges renderthe system closed and effectively prevent release ofaerosols. While other requirements may dictate theisolation of centrifuges, for example, noise abatement,vibration control, proper maintenance, and othersafety requirements, QRM may show that a centrifugemay be located and operated in the same room withother unit operations in the same space without a riskof cross-contamination due to aerosols.
Paragraph 54 also refers to blending of products as apotential source of aerosols that present a risk ofcross-contamination. Similarly, a perceived risk ofblending operations like buffer and media preparationor additions to product vessels is the spread contami-nants. When these operations are open and may re-lease particles or aerosols, then QRM may well dictateuse of dedicated areas or rooms for such operations.However, QRM also considers the process and othercontrols, for example, the nature of the blending op-eration, material properties (e.g., chemical or raw ma-terial type), levels of protection (e.g., closed systems,temporal separation, air flow direction), time andlength of the operation, etc., to determine whetherseparate rooms are required.
In summary, dedicated rooms are often built to containoperations like centrifugation or solution preparationeven though other methods to prevent aerosol contam-ination may be more appropriate and may already beintrinsic to the design.
Isolation of other operations that can lead to potentialaerosol formation, like homogenization, is often notconsidered because these operations are not explicitlymentioned in Annex 2.
Interpretation
It is essential to understand the probability or level ofaerosol formation and spread and to evaluate the ac-tual risk and consequences for cross-contaminationposed by aerosols. Proportional containment measure-ments should be adopted according to the risk that hasbeen determined. This may not require segregation orisolation of such operations when more robust solu-tions (like closed processing) are used.
306 PDA Journal of Pharmaceutical Science and Technology
on June 10, 2016journal.pda.orgDownloaded from

This QRM approach is not limited to centrifuges andblending operations, the two examples mentioned inAnnex 2. It should include any unit operation that canlead to aerosol formation.
2.4. Paragraph 57
“57. In cases where a virus inactivation or removalprocess is performed during manufacture, measuresshould be taken to avoid the risk of recontamination oftreated products by non-treated products.”
Reason for Commenting
Although methods to prevent recontamination are notprescribed in Annex 2, a frequent subject of discussionwithin industry and regulatory agencies is where andhow to segregate pre- and post-viral product processstreams to comply with paragraph 57. These discus-sions and expectations result in inconsistant levels ofimplementation and strategies.
If a firm decides not to segregate, it is likely that atsome point the firm will engage in difficult discussionswith regulatory agencies. This is perceived as a busi-ness risk rather than a product quality issue. As aresult, separate rooms are frequently used for pre- andpost-virus removal processing even if a QRM ap-proach indicates that alternative techniques are appro-priate.
Interpretation
Achieving viral safety of biological active substancesrequires more attention than simply undertaking pre-and post-virus reduction steps into separate rooms.Based on a sound QRM process and appropriate jus-tification, use of physical barriers between pre- andpost-virus operations (e.g., separate rooms or closedprocessing) is just one component of a complete riskmitigation strategy for virus contamination control.Other components include: barriers to virus entry(e.g., characterization of cell banks), raw materialssourcing, robust virus clearance steps, and testingduring production to detect any contaminating virus(8).
Robust control strategies will only be achievedthrough a sound QRM process and not by solelyfollowing the current common practice. Where a virusinactivation or removal process is performed duringmanufacturing, the QRM approach should provide
measures to identify and mitigate the risk of recon-tamination of treated products by nontreated products,components, or equipment.
Conclusions
While Annex 2 of the EU GMP regulatory guidanceclearly embraces QRM, it refers to Annex 1 in waysthat could be misinterpreted to imply controls used forsterile product processes should be applied to low-bioburden processes. Recognizing that the ultimategoal of regulatory guidance documents is protection ofpatients, thus prior to final product being renderedsterile, the bioprocess manufacturing scheme mustprovide layers of protection or barriers to protect theprocess from environmental contamination. However,our interpretation of the guidance is that controls forsterile products (i.e., those referred to in Annex 1) arenot typically required for biological active substancesprior to them being rendered sterile.
Use of a QRM approach ensures the selection ofcontrol measures that are commensurate with the po-tential risks to product and ultimately to the patient. AQRM approach should define the control strategy inline with Annex 2. Protecting the drug substance andintermediates by closing the process is a consistent,reproducible, and robust approach—more so than at-tempting to control the environment around the man-ufacturing process. Closing the process yields a lowerrisk to the manufactured drug substance, and ulti-mately to the patient.
The authors are aware that there will be a revision ofAnnex 1 and believe this represents an opportunity toclarify guidance as suggested in this paper (4).
Acknowledgments
BPOG and the authors wish to acknowledge the workof the following people, who acted as reviewers of thispaper and who support and endorse the content andproposals made:
Jose Caraballo (Bayer), Liz Dooley (Janssen), PaulDriesprong (Janssen), Marcella Goodnight (AstraZeneca Biologics), Richard Gummer (Baxalta), LarsHovmand-Lyster (Novo Nordisk), Jorgen Magnus(Bayer), Benjamin Montano (Baxalta), Russ Moser(Janssen), Iv Neov (Novavax), Lawrence Pranzo(Merck), Stephanie Ramsey (Baxalta), and GeraldUitz (Baxalta).
307Vol. 70, No. 3, May–June 2016
on June 10, 2016journal.pda.orgDownloaded from

Conflict of Interest Declaration
The author(s) declare that they have no competinginterests.
References
1. EudraLex—Volume 4. Good Manufacturing Prac-tice (GMP) Guidelines: http://ec.europa.eu/health/documents/eudralex/vol-4/index_en.htm. Annex 1:http://ec.europa.eu/health/files/eudralex/vol-4/2008_11_25_gmp-an1_en.pdf. Annex 2: http://ec.europa.eu/health/files/eudralex/vol-4/vol4-an2__2012-06_en.pdf.
2. Gill, P.; Rogalewicz, J.; Chalk, S.; Probst, S.; Palberg,P.; Kennedy, M.; Johnson, J.; Green, K. Challenging theCleanroom Environment for Biopharmaceutical Manu-facture of Bulk Drug Substances. BioPharm. Int. 2011,24, (8), http://www.biopharminternational.com/challenging-cleanroom-paradigm-biopharmaceutical-manufacturing-bulk-drug-substances.
3. Probst, S.; Chalk, S.; Zicaro, M.; McDuff, P.;Pranzo, L.; Dooley, L.; Moser, R.; Urbanski, F.;Smock, P.; Green, K. New Challenges to theCleanroom Environment for Multi-Product Facil-ities. BioPharm. Int. 2013, 26 (5). http://www.biopharminternational .com/new-challenges-cleanroom-paradigm-multi-product-facilities.
4. Concept Paper on the Revision of Annex 1 of theGuidelines on Good Manufacturing Practice—Manufacture of Sterile Medicinal Products: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/02/WC500181863.pdf.
5. ICH Q9 —Quality Risk Management, November2005: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q9/Step4/Q9_Guideline.pdf.
6. Moody, P. Irish Medicines Board GMP Informa-tion Seminar, September 27, 2012.
7. Aide memoire Bio- und Gentechnologie071210BN, Zentralstelle der Länder fur Gesund-heitsschutz bei Arzneimitteln und Medizinproduk-ten, June 16, 2004.
8. Aranha, H. Virus Safety of Biopharmaceuticals.Contract Pharma 2011, 13.
9. ISPE Baseline® Guide. Risk-Based Manufacture ofPharmaceutical Products (Risk-MaPP), 1st ed.;2010; Volume 7.
Appendix: A
As previously stated in the discussion of Annex 2,paragraph 33, Grade C background for solution prep-aration activities is often accepted as a defacto regu-latory requirement.
The following risk assessment example demonstratesthat environmental classification controls are only oneconsideration, and probably not the most significantconsideration, when evaluating risks associated withthe preparation and storage of buffers used in drugsubstance purification. In this example, the risk ofmicrobial endotoxins entering the purification processwith buffers is evaluated. Application of the scoringcriteria indicate that the operation is low risk overallwith all the controls in place and that environmentalsources of bioburden-related endotoxin are bothhighly unlikely and highly detectable, rendering themthe lowest risk of all.
The example buffer preparation process shown inFigure 1 is conducted in a CNC environment. TableI shows a number of measures that are in place toavoid introduction and propagation of microorgan-isms that could produce endotoxins. The preparationvessel remains closed, and when solution compo-nents are added, they are added in a protected man-ner that minimizes exposure of the internal surfacesof the tank to the external environment. After prep-aration, the buffer is held for a short maximum holdtime (12 h) then filtered into a sterile storage bag toreduce bioburden that may be present in raw mate-rials. The buffer is stored under conditions in whichthere is no, or very low, bioburden and thereforelimited opportunity for endotoxins to form duringthe storage period.
Table I provides a risk assessment for the base casescenario shown in Figure 1. Severity, probability ofoccurrence, and detection point are all evaluated withrespect to the failure mode (microbial endotoxins enterprocess equipment from storage bag). Table I alsoprovides a risk assessment for a select set of alterna-tives to the base case (in red and blue text). Values forthese alternative cases are also assigned with respectto the failure mode. The analysis of the alternatives
308 PDA Journal of Pharmaceutical Science and Technology
on June 10, 2016journal.pda.orgDownloaded from

shows that increasing the classification of the back-ground environment does not reduce the risk associ-ated with the microbial load or endotoxins in thesolution; nonenvironmental factors have a greater im-pact. The potential contribution of microbial load tothe solution from the environment is already negligi-ble in the base case, with controls, in comparison toother potential sources of microbial load. However,other factors can influence the risk. For example,doubling the allowable preparation and filtration timeprovides more opportunity for microbial contaminantsto grow. Reduced preparation temperature lowers therate at which microbes will proliferate (with only asmall risk-reduction impact). Finally, if the addition
of solids to the tank is not as protected as describedin the base case, there is more opportunity formicrobial load to enter from the background envi-ronment or personnel.
Table II shows the scoring guidelines for severity,probability of occurrence, and detection point thathave been used in this example. This guidance hasbeen taken directly from the ISPE Baseline® Guide:Risk-Based Manufacture of Pharmaceutical Products(Risk-MaPP) (9).
An addendum is added to provide a context forinterpreting the resultant Risk Process Number[RPN].
Figure 1
Process for preparing and storing a purification buffer in a CNC environment. Solids are added so thatexposure of the interior of the vessel to the external environment is minimized. After preparation, thebuffer is 0.2 micron filtered into a gamma-irradiated storage bag. The solution remains in the storage baguntil it is ready for use. The buffer is dispensed to the target equipment when needed through gamma-irradiated tubing.
309Vol. 70, No. 3, May–June 2016
on June 10, 2016journal.pda.orgDownloaded from

Tab
leI.
Ass
essm
ent
ofth
eR
isk
Tha
tM
icro
bial
End
otox
ins
Cou
ldE
nter
the
Pur
ifica
tion
Pro
cess
Stre
amG
iven
the
Pre
para
tion
Equ
ipm
ent
and
Pro
cedu
res
Illu
stra
ted
inF
igur
e1.
Bas
eC
ase
Ana
lysi
sA
naly
sis
ofA
lter
nati
veC
ases
Fai
lure
Mod
eP
oten
tial
Eff
ects
ofF
ailu
reM
ode
Seve
rity
Pot
enti
alC
ause
sC
urre
ntC
ontr
ols
Occ
urre
nce
Det
ecti
onR
PN
Per
mut
atio
nSe
veri
tyO
ccur
renc
eD
etec
tion
RP
NC
omm
ent/
Exp
lana
tion
Mic
robi
alen
doto
xins
ente
rpu
rifi
cati
oneq
uipm
ent
from
buff
er.
End
otox
ins
may
ente
rth
edr
ugsu
bsta
nce
proc
ess
stre
aman
dm
ayno
tbe
rem
oved
.H
igh
endo
toxi
nco
ncen
trat
ions
inth
edr
ugpr
oduc
tw
ill
resu
ltin
adve
rse
pati
ent
reac
tion
s.
10[A
]B
iobu
rden
(tha
tca
nfo
rmen
doto
xins
*)is
pres
ent
insa
lts.
[B]
Bio
burd
enpr
esen
tin
CW
FI.
[C]
Bio
burd
enen
ters
from
the
envi
ronm
ent
(CN
Cba
ckgr
ound
)[D
]B
iobu
rden
tran
sfer
red
toin
teri
orof
tank
byop
erat
ors.
[E]
Bio
burd
enpr
esen
ton
exte
rior
surf
aces
ofra
wm
ater
ial
pack
agin
gan
dis
tran
sfer
red
toin
teri
orof
prep
arat
ion
vess
el.
[F]
Pre
para
tion
vess
elan
d/or
tran
sfer
line
isin
adeq
uate
lycl
eane
dan
dsa
niti
zed
befo
reus
e.T
here
isbi
obur
den
pres
ent
inth
eve
ssel
ortr
ansf
erli
nepr
ior
toth
est
art
ofth
eba
tch.
[G]
Sol
utio
nsi
tsin
vess
ela
long
tim
eat
room
tem
pera
ture
befo
refi
ltra
tion
into
stor
age
bag
and
biob
urde
ngr
ows
wit
hin
the
tank
.[H
]In
itia
lle
vel
ofbi
obur
den
wit
hin
the
solu
tion
isto
ohi
ghan
dfil
ter
isun
able
tore
tain
the
biob
urde
n.[I
]0.
2m
icro
nfi
lter
isno
tin
tegr
al.
Bio
burd
enca
npa
ssth
roug
hdu
ring
filt
rati
on.
[J]
Sin
gle-
use
tubi
ngor
stor
age
bag
isno
tin
tegr
al.
Bio
burd
enca
nen
ter
thro
ugh
inte
grit
ybr
each
.[K
]S
ingl
e-us
etu
bing
orst
orag
eba
gin
adeq
uate
lysa
niti
zed
byth
eve
ndor
and
cont
ains
biob
urde
n.[L
]B
iobu
rden
accu
mul
ates
wit
hin
the
bag
duri
ngst
orag
e.O
rgan
ism
sdi
ean
dre
leas
een
doto
xins
into
the
form
ulat
ion
buff
er.
[A]
Bio
burd
ensp
ecifi
cati
onli
mit
sfo
rsa
lts.
[B]
Man
yde
sign
and
engi
neer
ing
cont
rols
inpl
ace
toav
oid
biob
urde
nin
CW
FI.
[B]
Per
iodi
csa
niti
zati
onof
CW
FI
dist
ribu
tion
syst
em.
[B]
Reg
ular
envi
ronm
enta
lm
onit
orin
gof
CW
FI.
[C,
D,
E]
Soli
dsad
ded
soth
atex
posu
reof
the
inte
rior
toth
eve
ssel
toth
een
viro
nmen
tis
min
imiz
ed.
[F]
Tan
kan
dtr
ansf
erli
near
ede
sign
edfo
rpr
oper
clea
ning
and
drai
ning
.A
vali
date
dcl
eani
ngan
dsa
niti
zati
onse
quen
ceis
exec
uted
for
the
vess
elan
dth
etr
ansf
erli
nepr
ior
toea
chlo
t.P
erio
dic
envi
ronm
enta
lm
onit
orin
g(i
.e.,
rins
esa
mpl
ing)
tove
rify
cont
inue
def
fect
iven
ess
ofcl
eani
ngpr
oced
ure.
[G,
H]
Val
idat
edti
me
esta
blis
hed
from
begi
nnin
gof
batc
hpr
epar
atio
nto
end
offi
ltra
tion
.T
ime
stam
psar
ech
ecke
din
the
batc
hre
cord
toen
sure
batc
hw
aspr
epar
edan
dfi
lter
edw
ithi
nth
eal
low
edti
me.
[I]
Fil
ter
vend
orqu
alit
ysy
stem
.[I
]P
ost-
use
inte
grit
yte
stpe
rfor
med
onea
chfi
lter
.[J
,L
]S
ingl
e-us
eve
ndor
qual
ity
prog
ram
.[J
]O
pera
tor
trai
ning
for
insp
ecti
ngsi
ngle
-use
elem
ents
prio
rto
use
and
toav
oid
dam
agin
gsi
ngle
-use
elem
ents
duri
ngha
ndli
ng.
[J]
Inte
grit
ybr
each
may
beco
me
evid
ent
duri
ngfi
ltra
tion
and
tran
sfer
.[K
]R
adia
tion
indi
cato
rson
gam
ma
ster
iliz
edel
emen
tsth
atar
ere
ceiv
ed.
[L]
Sol
utio
nis
0.2
mic
ron
filt
ered
prio
rto
stor
age.
Mea
sure
sJ
and
Kar
ein
plac
eto
prev
ent
the
buff
erfr
ombe
ing
cont
amin
ated
duri
ngth
est
orag
epe
riod
afte
rfi
ltra
tion
.N
OT
E:
Dru
gsu
bsta
nce
iste
sted
for
mic
robi
alen
doto
xins
.T
hem
ater
ial
isno
tre
leas
edfo
rfi
nal
drug
prod
uct
man
ufac
turi
ngif
the
spec
ified
lim
itis
exce
eded
.
31
30
[C’1
]So
luti
onpr
epar
atio
nan
dst
orag
eis
perf
orm
edw
ithi
na
Gra
deA
back
grou
nden
viro
nmen
tra
ther
than
ina
CN
Cen
viro
nmen
t.[D
’1,
E’1
)]So
lid
com
pone
nts
are
adde
dth
roug
han
open
man
way
rath
erth
anvi
aa
prot
ecte
dco
nnec
tion
.[G
’1,
H’1
]P
repa
rati
onan
dfi
ltra
tion
tim
eis
incr
ease
dfr
om12
to24
h.[G
’2]
Dec
reas
epr
epar
atio
nte
mpe
ratu
refr
om20
°Cto
5°C
.
10 10 10 10
3 5 7 3
1 1 1 1
30 50 70 30
[C’1
]P
roba
bili
tyof
occu
rren
cedo
esno
tch
ange
ifop
erat
ion
ispe
rfor
med
ina
Gra
deA
back
grou
nden
viro
nmen
t.T
hein
gres
sof
mic
robi
allo
adfr
omth
een
viro
nmen
tis
alre
ady
negl
igib
lein
the
base
case
inco
mpa
riso
nto
othe
rpo
tent
ial
sour
ces
(e.g
.,fr
omra
wm
ater
ials
).[D
’1,E
’1]
The
open
man
way
prov
ides
agr
eate
rop
port
unit
yfo
rbi
obur
den
from
the
envi
ronm
ent
orth
eop
erat
orto
ente
rth
eve
ssel
duri
ngpr
epar
atio
n.[G
’1,H
’1]
Incr
easi
ngth
eal
low
able
prep
arat
ion
and
filt
rati
onti
me
coul
din
crea
seth
epo
tent
ial
mic
roor
gani
sms
pres
ent
inth
era
wm
ater
ials
anop
port
unit
yto
grow
and
prod
uce
endo
toxi
ns.
[G’2
]D
ecre
asin
gth
epr
epar
atio
nte
mpe
ratu
rede
crea
ses
the
grow
thra
teof
pote
ntia
lm
icro
bial
cont
amin
ants
pres
ent
inra
wm
ater
ials
.
*T
his
isas
sum
edw
hene
ver
biob
urde
nis
men
tion
edw
ithi
nth
isex
ampl
e.
310 PDA Journal of Pharmaceutical Science and Technology
on June 10, 2016journal.pda.orgDownloaded from
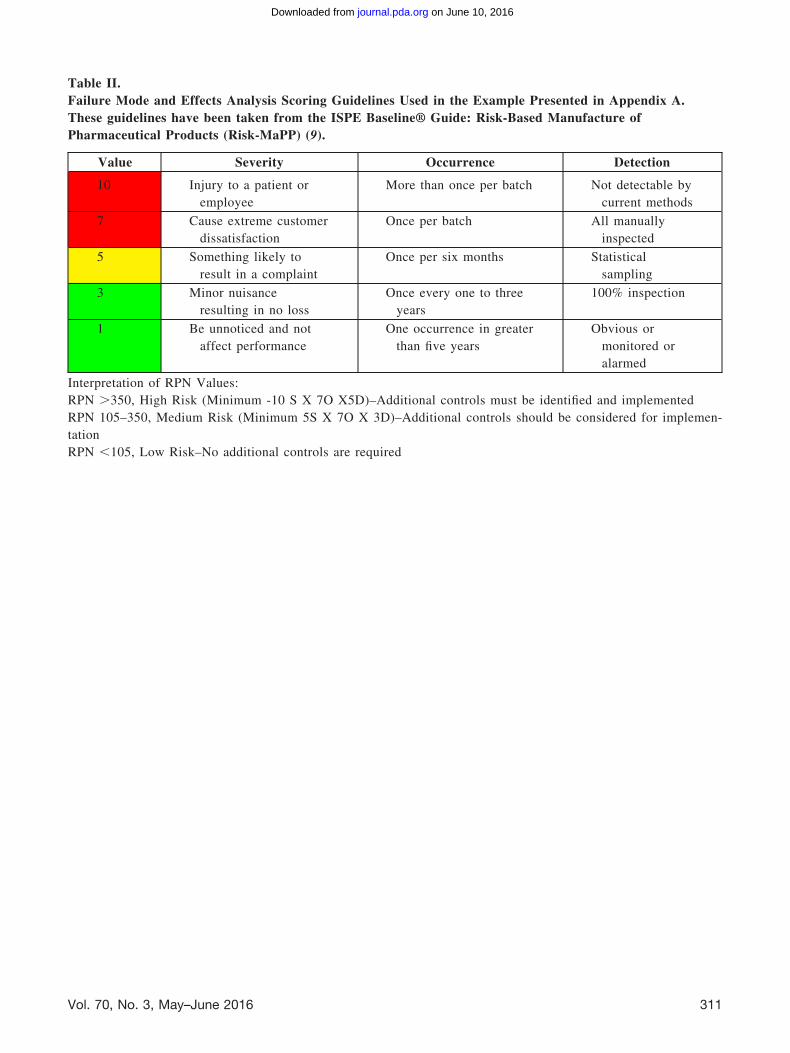
Table II.Failure Mode and Effects Analysis Scoring Guidelines Used in the Example Presented in Appendix A.These guidelines have been taken from the ISPE Baseline® Guide: Risk-Based Manufacture ofPharmaceutical Products (Risk-MaPP) (9).
Value Severity Occurrence Detection
10 Injury to a patient oremployee
More than once per batch Not detectable bycurrent methods
7 Cause extreme customerdissatisfaction
Once per batch All manuallyinspected
5 Something likely toresult in a complaint
Once per six months Statisticalsampling
3 Minor nuisanceresulting in no loss
Once every one to threeyears
100% inspection
1 Be unnoticed and notaffect performance
One occurrence in greaterthan five years
Obvious ormonitored oralarmed
Interpretation of RPN Values:RPN �350, High Risk (Minimum -10 S X 7O X5D)–Additional controls must be identified and implementedRPN 105–350, Medium Risk (Minimum 5S X 7O X 3D)–Additional controls should be considered for implemen-tationRPN �105, Low Risk–No additional controls are required
311Vol. 70, No. 3, May–June 2016
on June 10, 2016journal.pda.orgDownloaded from

Authorized User or for the use by or distribution to other Authorized Users·Make a reasonable number of photocopies of a printed article for the individual use of an·Print individual articles from the PDA Journal for the individual use of an Authorized User ·Assemble and distribute links that point to the PDA Journal·Download a single article for the individual use of an Authorized User·Search and view the content of the PDA Journal permitted to do the following:Technology (the PDA Journal) is a PDA Member in good standing. Authorized Users are An Authorized User of the electronic PDA Journal of Pharmaceutical Science and
copyright information or notice contained in the PDA Journal·Delete or remove in any form or format, including on a printed article or photocopy, anytext or graphics·Make any edits or derivative works with respect to any portion of the PDA Journal including any·Alter, modify, repackage or adapt any portion of the PDA Journaldistribution of materials in any form, or any substantially similar commercial purpose·Use or copy the PDA Journal for document delivery, fee-for-service use, or bulk reproduction orJournal or its content·Sell, re-sell, rent, lease, license, sublicense, assign or otherwise transfer the use of the PDAof the PDA Journal ·Use robots or intelligent agents to access, search and/or systematically download any portion·Create a searchable archive of any portion of the PDA JournalJournal·Transmit electronically, via e-mail or any other file transfer protocols, any portion of the PDAor in any form of online publications·Post articles from the PDA Journal on Web sites, either available on the Internet or an Intranet,than an Authorized User· Display or otherwise make any information from the PDA Journal available to anyone otherPDA Journal·Except as mentioned above, allow anyone other than an Authorized User to use or access the Authorized Users are not permitted to do the following:
on June 10, 2016journal.pda.orgDownloaded from