boxes within boxes: discovering the atomic structure of … · boxes within boxes: discovering the...
TRANSCRIPT

Boxes within boxes: discovering the
atomic structure of an Al-Co-Ni
quasicrystal
Nan Gu, Marek Mihalkovic, and C.L. Henley,
Cornell University
LASSP Pizza talk, Tues July 11, 2006
1

1. Quasicrystals
Fourier transform (diffraction) has Bragg peaks
Rotational symmetry (e.g. 5-fold) excludes periodicity
Images of Al72Ni20Co8:
(L) electron microscope; (R) electron diffraction.
But this talk on how microscopic interactions relate to the atomic
structure; won’t focus on long range order/diffraction.
2

2. Effective pair potentials
Derived using Moriarty’s ‘Generalized Potential Theory” [I.
Al-Lehyani et al Phys. Rev. B 64, 075109 (2001)]
Depend implicitly on electron density.
Friedel oscillations [cos(2kF r + δ)/r3] from F.T. of sharp Fermi
surface.
Calibrated from LDA results for (comparatively) simple crystals.
3
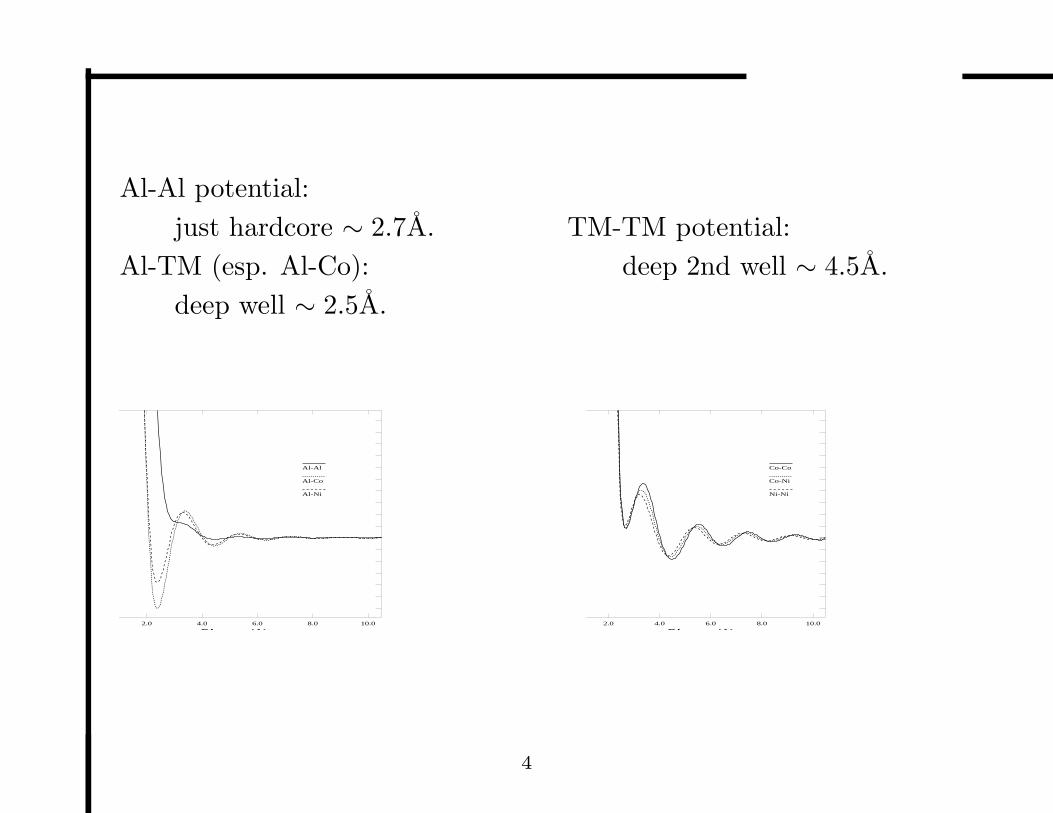
Al-Al potential:
just hardcore ∼ 2.7A.
Al-TM (esp. Al-Co):
deep well ∼ 2.5A.
TM-TM potential:
deep 2nd well ∼ 4.5A.
Al-Al
Al-Co
Al-Ni
Pair
poten
tial (e
V)
Distance (A)
-0.3
-0.2
-0.1
0.0
0.1
0.2
0.3
0.4
0.5
0.0 2.0 4.0 6.0 8.0 10.0
Co-Co
Co-Ni
Ni-Ni
Pair
poten
tial (e
V)
Distance (A)
-0.3
-0.2
-0.1
0.0
0.1
0.2
0.3
0.4
0.5
0.0 2.0 4.0 6.0 8.0 10.0
4

3. Where are (or should be) the atoms?
Fundamental problem of crystal chemistry: Given we can perfectly
compute total energy of a complex compound, what’s the atomic
arrangement?
Why?
(i) Diffraction is insufficient in quasicrystal case. Most
structure fits include some impossible distances; if you pluged into
an ab-initio code, the high energies from these rare (and spurious)
environments would swamp the small differences that decide the
equilibrium phase diagram.
Also, usually partial occupations – which combinations are
simultaneously occupied?
ternary, but X-rays (or electron diffraction) won’t separate
transition metals with similar atomic numbers [Co (Z=27) vs Ni
(Z=28)]
5

(ii) Check agreement with experiment to see how far we can
trust the pair potentials.
(iii) Do interactions favor an essentially unique ground state
(matching-rule scenario) or does order emerge from an
entropy-dominated state (random tiling scenario)? Modeling may
be the fastest way to answer this old question.
6

why is it so hard?
Implicitly demands comparing with infinity of possible structures!
What people do:
i. compare a few known candidate structures
Usually, too few!
ii. Brute-force Monte Carlo / Molecular Dynamics?
Typically gets stuck in glassy states.
7

4. The solution......
An empirical fact: quasicrystal atomic structure is well described
by tilings. (That is, atoms are close to tile vertices.) The same tile
appears with different surroundings.
Input information:
Quasilattice constant a0 = 2.45A, period (2 layers) c = 4.08A
Experimental density and composition.
8

Here is the Penrose tiling of rhombi.
9

Another important tiling – Hexagon-Boat-Star.
Basis for structure model of Al70Ni20Co10 by Mihalkovic, Widom,
Henley, and collaborators, 2002. (Figure: Al=gray, Ni=black,
Co=blue; size is top or bottom layer.)
Note: you can describe the same configuration by tiles of different
size scales. (Note supertiling here which is also an H-B-S tiling.)
10

11

4. Our recipe – Multiscale Procedure
Represent structures as decorations of Penrose rhombi
1 MC simulations, atoms as lattice gas on fixed list of ideal sites.
Allow atom swaps and ‘tile-flips’ (a0 = 2.45A rhombi). View
low E configuration from each run, identify common motifs.
2 Promote observations [1] to rules for larger tiles: fewer degrees
of freedom.
3 New MC simulations (fixed sites) on larger-scale tiling; iterate.
4 Relaxation/MD simulations to find true equilibrium positions.
Thus “boxes within boxes.”
It worked for “best case” Al70Ni20Co10, [M. Mihalkovic et al, PRB
65, 104205 (2002)]. Generally? Try on Al70Co20Ni10, Nan Gu et al,
2005: it works, but trickier than we thought!
12

5. Simulations:
First stage
rhombus edge a0 = 2.45A
Two independent layers
Tile flips and atom swaps.
Second stage
rhombus edge τa0 = 4.0 A
One layer tiling //
two-layer decoration
of candidate sites
2 flavors of fat rhombus;
Atom swaps only
13

Simulation image (first stage result)
This breaks up into Hexagon-Boat-Star like the Ni-rich composition
did, but there are also 8A Decagons with the same edge – in fact
5-fold symmetry extends farther out. But many defects present.
Idealizing clusters lowers the energy (so we make it a rule in the
next stage).
14

Cluster motifsDominant motif “13A Decagon”
(really 12.8A):
Secondary motif “Star cluster”
(fills spaces):
Al Co Ni
�����
�����
��� ���
��� ������ ���
� �
��� ���
� � � �� � � �� � � �� � �� � �� � �� � �� � �� � �
� � �� � �� � �� � �� � �� � �
� � � �� � � �� � � �� � � �� � � �� � � �� � � �� � � �� � � �� � � �� � � �
� � � �� � � �� � � �� � � �� � � �� � � �� � � �� � � �� � � �� � � �� � � �
� � � �� � � �� � � �� � � �� � � �
� � � �� � � �� � � �� � � �� � � �
� � �� � �
� � � �� � � �� � � �� � � �
� � � �� � � �� � � �� � � �ring 3
ring 2
ring 1
ring 2.5
Ni/AlCo/Ni
���
���
�� ��
!
"�"
#�#
$�$
%�%
Concentric rings... some
irregularities on rings 2.5/3.
Circled: are puckering channels
when relaxation allowed
Ring of five “TM” sites: only
∼30% TM (context dep.), oth-
erwise Al.
15

6. Decoration on 10.4A Binary tiling
At this composition, Decagon motif always appears. How are the
clusters placed relative to each other? We identified a tiling on a
larger scale – the “binary rhombus tiling”– with large/small cluster
centers on the vertices. The edge length is τ 3 times larger than the
original small rhombi – 10.4A.
Ni(s )Co(1)
Co(3)
Ni(3)
Ni(s)
“+” “-” mark orientations σi of TM’s in centers. Other mark-
ings refer to a rule for TM decoration, which depends on the
orientations. Composition around Al70Co21Ni9.
16

Another version of idealized decoration
This image emphasizes a decomposition into 2.45A Hexagon,
Boat, and Star tiles around 2.45A Decagon tiles. The different
possible tilings of the HBS tiles correspond to options for the Al
in rings 2.5/3, all of which maximize the number of Al-Co bonds.
17

Orientational order
How did we know the relative orientation of Decagons? (Since
each actually has 5-fold symmetry.)
Laborious to answer. Turns out best if the transition-metal atoms
(from the central Co ring) are in the same layers; this creates a
deeper modulation of the potential energy of an Al atom (at
second-neighbor distances), allowing certain variable Al atoms to
lower their energies. This happens if all Decagons oriented the
same – quasicrystal is pentagonal, not decagonal!
Note there are competing contributions: answer is affected by
composition. Get wrong if we stick with the discrete-site
approximation – need MD/relaxation.
18

7. Relaxation and puckeringUnder relaxation, a subset of atoms “puckers” (deviates from
layer plane). Also, the period doubled to c′ = 2c = 8A, the real
period for most d(AlCoNi) subphases
Why? There are “channels” of potential minima, running verti-
cally, with just room to place three Al in every four layers, hence
the period doubles to c′ = 2c.
A4c
aτ 0 A4
idealposition
� �� �� �� �� �� �� �� �� �� �� �� �
� �� �� �� �� �� �
� �� �� �� �� �� �� �� �� �
U(z)
(b).
Alz
(a).
TM Co
channel
19

Puckering pattern...
An obvious result (related to the orientation order of clusters) is
that every other layer becomes singled out as a mirror layer (flat by
symmetry and richer in transition metal atoms), the other layers
being puckered alternately. Nearby channels interacts: adjacent
ones want to have opposite puckering sense, and more globally the
interaction is frustrated. Again, we have several important length
scales – boxes within boxes again!
20

Conclusion
Our approach “works” in that prominent features match
experiment (NiNi pairs and decagons with triangle feature in
center, in previous Ni-rich case; decagons and overall 5-fold
symmetry, in present Co-rich case; puckering). The inherent bias in
starting with fixed-site simulations doesn’t necessarily prevent us
from finding the correct answers for relaxed structures.
But it’s an art, not a science. We missed some things by not trying
a broad enough range of densities, compositions, and system sizes
in the initial stages, and by taking too big a step in devising a
constrained model. To do better on some of these points requires
technical improvements on the code which are underway. Further
simulations are being done by undergrad Sejoon Lim, which (so
far) explored toy models in which matching rules do emerge from
potentials, also the Al-Ni-Fe decagonal phase.
21

Oops!
One run of the more constrained simulation, using a somewhat
enlarged site list, showed emergence of a larger decagon unit. The
local order is very similar, but at large scales it has a different set
of supertiles. Quite possibly, both structures are “correct” in that
they occur for some nearby composition – the Al-Co-Ni decagonal
phase is known to be fragmented into modifications with various
larger scale modulations.
22

23