body water homeostasis: clinical disorders of urinary...
TRANSCRIPT

Review
Body Water Homeostasis: Clinical Disorders of UrinaryDilution and Concentration
Robert W. SchrierDepartment of Medicine, University of Colorado School of Medicine, Denver, Colorado
J Am Soc Nephrol 17: 1820–1832, 2006. doi: 10.1681/ASN.2006030240
T he discovery of the aquaporin-1 (AQP1) water channelby Agre and colleagues (1,2), which led to the NobelPrize in 2003, has revolutionized the understanding of
body fluid water regulation by the kidney. Moreover, the iden-tification of other water channels in the kidney, namely AQP2,3, and 4, along with urea and ion transporters, has allowed amuch improved understanding of urinary dilution and concen-tration in health and disease at the cellular and molecular levels(3–8).
The AQP have provided a pathway for water movementacross cellular membranes that could not be explained by sim-ple diffusion through the lipid bilayers of cell membranes.AQP1 has been found to be expressed constitutively on boththe apical and the basolateral membranes of the proximal tu-bule and descending limb of Henle’s loop. This water channelis not under control of vasopressin but is important in urinaryconcentration. Water efflux through these channels in the de-scending limb is an important factor in the countercurrentconcentrating mechanism, and diminished maximal urinaryosmolality has been shown in AQP1 knockout mice (9) andhumans without the AQP1 gene (10).
AQP2, 3, and 4 are expressed in the cortical and medullarycollecting duct (Figure 1) (11). AQP2 is found exclusively in theprincipal cells of the collecting tubule and collecting duct and isknown to be regulated by arginine vasopressin (AVP). AQP3and 4 are located on the basolateral membrane of the principalcells in the collecting duct. AQP3 knockout mice exhibit sub-stantial polyuria secondary to vasopressin-resistant nephro-genic diabetes insipidus (NDI) (12). AQP3 is regulated by AVP.AQP4 predominates on the basolateral membrane of the innermedulla and is not regulated by AVP. AQP4 knockout micealso exhibit an NDI that is less severe than that observed in theAQP3 knockout mice (13). Whereas AQP3 and AQP4 constitutethe exit channels for water movement across the basolateralmembrane of the collecting duct, AQP2 is the water channel forwater reabsorption across the apical membrane of the principalcells of the collecting duct. These transgenic mouse models of
AQP deletion/mutation are of importance not only for kidneyfunction but also for other epithelia (14).
AVP regulates AQP2 in both an acute (short term) (15) andchronic (long term) manner (16,17). The short-term regulationby AVP involves trafficking of vesicles that contain AQP2 to theapical membrane with resultant increased water permeability.Suppression of plasma AVP reverses this exocytosis of AQP2,and the vesicles are retrieved from the membrane (endocytosis)into the cytoplasm. Both the short- and long-term regulation ofAQP2 by AVP is initiated by activation of the V2 receptor onthe basolateral membrane of the collecting duct. The binding ofAVP to the V2 receptor, which is coupled to a guanine-nucle-otide–binding protein Gs, results in activation of adenylyl cy-clase. This results in an increase in intracellular cAMP concen-tration that mediates the activation of protein kinase A (PKA).Activated PKA phosphorylates the serine 256 residue at theC-terminus of AQP2 protein (18,19). The phosphorylated AQP2then is translocated to the collecting duct apical membrane,thus constituting short-term regulation of AQP2. The long-termregulation of AQP2 is mediated by the cAMP response elementin the 5� flanking region of the AQP2 gene in response to AVPstimulation (20,21). The AQP2 transcription and protein expres-sion involves the phosphorylation of the cAMP response el-ement–binding protein. In contrast to the involvement of PKAactivation in the short-term regulation of AQP2, there is somein vivo evidence for PKA-independent long-term regulation ofAQP2 expression by AVP (22). There also is recent in vitro (23)and in vivo (24) evidence for AQP2 regulation by hyperosmo-lality independent of AVP. The in vivo experiments were per-formed in Brattleboro rats, which have no detectable circulatoryAVP.
Disorders of water balance can lead to either clinically rele-vant hyponatremia or hypernatremia. Hyponatremia is muchmore common and is the most frequent fluid and electrolytedisturbance in hospitalized patients. When defined as plasmasodium concentration �135 mEq/L, the prevalence of hypona-tremia in hospitalized patients may be as high as 15 to 30%.Studies indicate that 40 to 75% of these cases are hospitalacquired (25,26). It is clear that hyponatremia may occur in thepresence of an increase in total body sodium, a decrease in totalbody sodium, or a near-normal total body sodium (Figure 2). Inany of these three circumstances, hyponatremia has occurredbecause of a relatively greater amount of total body water ascompared with total body solute. Recent advances of the mech-
Published online ahead of print. Publication date available at www.jasn.org.
Address correspondence to: Dr. Robert W. Schrier, Department of Medicine,University of Colorado School of Medicine, 4200 East Ninth Avenue B-173,Denver, CO 80262. Phone: 303-315-8059; Fax: 303-315-2685; E-mail:[email protected]
Copyright © 2006 by the American Society of Nephrology ISSN: 1046-6673/1707-1820

anisms of impaired urinary diluting capacity that lead to thesehyponatremic disorders are discussed.
HypothyroidismAdvanced primary hypothyroidism can lead to hyponatre-
mia, most frequently associated with myxedema. Milder formsof hypothyroidism can lead to a decrease in renal blood flow,glomerular filtration, and a decrease in maximal solute-free
water excretion. However, these defects generally are not ofsufficient magnitude to lead to hyponatremia in associationwith a normal fluid intake. With normal fluid intake, hypona-tremia generally results from the inability to decrease urineosmolality below plasma rather than a defect in maximal sol-ute-free water excretion. The failure to dilute the urine belowplasma generally is due to a failure to suppress maximally theantidiuretic hormone AVP (27). The failure of some hypothy-roid patients to suppress plasma AVP during an acute waterload was reported initially by Skowsky et al. (28).
Recently, experimental studies in rats with advanced hypo-thyroidism (serum total T4 0.6 � 0.02 ng/dl) further examinedurinary dilution and maximal solute-free water excretion (29).There was a significant decrease in cardiac index and heart ratein these hypothyroid animals that was normalized with l-thyroxine replacement. During an acute water load, these hy-pothyroid animals exhibited a significantly lower urine flowand higher urinary osmolality than either control animals orhypothyroid animals that were treated with l-thyroxine. Thehypothyroid animals also had significantly higher plasma AVPconcentrations despite a lower plasma osmolality that normal-ized with l-thyroxine treatment. The impaired water excretionalso was associated with an upregulation of AVP-mediatedAQP2 expression and membrane trafficking in the inner me-dulla (Figure 3). Taken together, these results suggested a majorrole of nonosmotic AVP release in the hyponatremia associatedwith advanced hypothyroidism. Studies therefore were under-taken with a vasopressin V2 receptor antagonist (OPC-31260) inthese animals with advanced hypothyroidism. Minimal urinaryosmolality in hypothyroid rats that were treated with the V2
Figure 1. Nephron sites of aquaporin (AQP) water channels(blue), ion transporters (black), and urea transporters (green).ROMK, renal outer medullary potassium channel; UT, ureatransporter.
Figure 2. Diagnostic and therapeutic approach to the hypovolemic, euvolemic, and hypervolemic hyponatremic patient.
J Am Soc Nephrol 17: 1820–1832, 2006 Body Water Homeostasis 1821

receptor antagonist was normalized as compared with un-treated hypothyroid rats (97 versus 430 mOsm/kg H2O; P �
0.0001). Maximal solute-free water excretion in the hypothyroidrats significantly increased but did not reach euthyroid values.These experimental results therefore support the pivotal role ofthe baroreceptor-mediated nonosmotic AVP release in the hy-ponatremia associated with advanced hypothyroidism. Thesubmaximal solute-free water excretion would not be of clinicalsignificance unless other factors, such as diuretics or large fluidintakes, intervene.
Of interest, a defect in maximal urinary concentration wasobserved at a somewhat less prolonged stage of experimentalhypothyroidism (30). Maximal urinary osmolality with fluiddeprivation was associated with decreased Na-K-2Cl expres-sion and diminished medullary osmolality. Even though AQP2expression was decreased, the failure to exhibit a differencebetween the diminished urinary and medullary osmolality sug-gested that the primary mediator of the concentrating defectwas a decrease in the Na-K-2Cl co-transporter expression andtherefore impaired countercurrent concentration rather thanthe decreased AQP2 expression. This urine-concentrating de-
fect in hypothyroidism would be of clinical relevance only inpatients who were undergoing excessive extrarenal fluid losses(e.g., diarrhea).
Addison’s Disease and HypopituitarismPrimary adrenal insufficiency (Addison’s disease) involves
deficiency of glucocorticoid and mineralocorticoid hormones,both of which can be associated with hyponatremia. Hypopi-tuitarism is associated with only glucocorticoid deficiency, be-cause the renin-angiotensin-aldosterone system is intact. Al-though secondary hypothyroidism is associated withhypopituitarism, in contrast to primary hypothyroidism, theseverity generally is not sufficient to be associated with hypo-natremia. Therefore, the hyponatremia that is related to hypop-ituitarism generally is due to glucocorticoid deficiency ratherthan thyroid deficiency.
Mineralocorticoid deficiency that is associated with primaryadrenal insufficiency exhibits a different pathophysiology thanglucocorticoid deficiency in causing hyponatremia. Because al-dosterone increases potassium and hydrogen ion secretion,hyperkalemia and nonanion gap metabolic acidosis is charac-
Figure 3. Effects in control (CTL) and hypothyroid (HT) and thyroid (T) replaced rats on urine flow (A), urine osmolality (B),plasma vasopressin (AVP; C), and inner medullary AQP2 plasma membrane (PM)/intracellular vesical (ICV) densitometry ratio(D). Reprinted from reference (27), with permission.
1822 Journal of the American Society of Nephrology J Am Soc Nephrol 17: 1820–1832, 2006

teristic of primary but not secondary adrenal insufficiency as aresult of hypopituitarism. The mineralocorticoid deficiency alsois responsible for the sodium chloride wasting and extracellularfluid volume depletion in primary adrenal insufficiency. Selec-tive mineralocorticoid deficiency has been studied in adrena-lectomized animals that received replacement glucocorticoidhormone (31–33). With isolated mineralocorticoid hormone de-ficiency and hyponatremia, plasma AVP concentrations werenot suppressed and collecting duct AQP2 and 3 expressionswere upregulated. Outer medullary Na-K-2Cl co-transporterand Na-K-ATPase were decreased in mineralocorticoid animals(32). Administration of AVP V2 antagonist in these animalssignificantly improved urinary dilution, but a modest defect inmaximal solute-free water excretion remained (33). This latterdefect relates to effects of extracellular fluid volume depletion,including decreased GFR and increased proximal tubular so-dium reabsorption with resultant diminished fluid delivery tothe distal diluting segment of the nephron. Avoidance of neg-ative sodium balance in mineralocorticoid-deficient rats nor-malized Na-K-2Cl co-transporter, Na-K-ATPase, and collectingduct AQP2 and 3 (32). It also is of interest that high sodiumchloride intake may correct not only the hyponatremia but alsothe hyperkalemia and metabolic acidosis in Addison’s disease,thereby supporting an important role for mineralocorticoiddeficiency.
If hyponatremia persists in primary adrenal insufficiencydespite avoiding negative sodium balance, then the hyponatre-mia is due to glucocorticoid deficiency. The mechanisms ofhyponatremia with glucocorticoid deficiency are different thanwith mineralocorticoid deficiency. Glucocorticoid deficiencydoes not cause a negative sodium balance, and in fact a positivesodium balance may occur. The absence of glucocorticoid hor-mone, however, has major effects on systemic hemodynamics,including a decrease in cardiac index with an inadequate re-sponse of systemic vascular resistance to maintain mean arte-rial pressure (34). These systemic effects result in several con-sequences. The resultant decrease in stretch on the arterialbaroreceptors in the carotid sinus and aortic arch removes thetonic vagal and glossopharyngeal inhibition on the central re-lease of AVP. Elevated plasma AVP concentrations have beendemonstrated in glucocorticoid-deficient animals (33,35) andpatients with hypopituitarism (36) despite a degree of hypos-molality that would maximally suppress AVP in normal indi-viduals. The messenger RNA for AVP in the hypothalamus alsohas been shown to be increased during glucocorticoid defi-ciency (37). AVP-synthesizing neurons terminate in the medianeminence of the hypothalamus; therefore, a role for ACTH alsocould be involved in the nonosmotic AVP stimulation that isassociated with glucocorticoid deficiency. There also is evi-dence of a central effect of glucocorticoid hormone in the hy-pothalamus whereby hypo-osmolality does not maximally sup-press AVP synthesis during glucocorticoid deficiency (38). Ineither case, the importance of this nonosmotic stimulation ofAVP in glucocorticoid deficiency was documented using pep-tide and nonpeptide vasopressin V2 receptor antagonists,which profoundly reversed the water retention (33,35).
A recent molecular analysis of the impaired diluting capacity
with glucocorticoid deficiency was undertaken in adrenalecto-mized rats that received replacement physiologic concentra-tions of mineralocorticoid hormone (35). As compared withadrenalectomized rats that received replacement of both min-eralocorticoid and glucocorticoid hormone, during an acutewater load, the glucocorticoid-deficient animals exhibitedhigher plasma AVP concentrations, diminished solute-free wa-ter excretion, and increased protein expression in the innermedulla of AQP2, phosphorylated AQP2, and apical mem-brane trafficking of AQP2. The administration of a nonpeptidevasopressin V2 receptor antagonist reversed these events (Fig-ure 4).
There also was insight into AVP-independent effects on wa-ter excretion during glucocorticoid deficiency. Pair feeding dur-ing glucocorticoid deficiency was associated with an upregula-tion of the Na�-K�-2Cl� co-transporter, Na�/H� exchangerisoform 3, and cortical � and � subunits of the epithelial sodiumchannel and sodium retention (35). These events, along with theeffect of a decrease in renal perfusion pressure, would lead todecreased fluid delivery to the distal nephron diluting seg-ments and attenuate maximal solute-free water excretion. ThisAVP-independent effect on maximal water excretion probablywould not be of clinical significance except in the circumstanceof large increases in water intake.
Of interest, maximal urinary concentration also was dimin-ished in glucocorticoid-deficient rats after 36 h of fluid depri-vation (34). The concentrating defect involved primarily animpairment of the countercurrent concentrating mechanismwith comparable diminutions in medullary and urinary osmo-lalities. With the fluid deprivation, lower expression of the ureatransporter and ion transporters (e.g., Na-K-2Cl) in the outermedulla seemed to account for the diminished medullary os-motic gradient.
Primary Polydipsia or Compulsive WaterDrinking
The renal capacity to excrete solute-free water is substantial(27), yet the average daily fluid intake for hyponatremic pa-tients is only 2.4 L (39). The delivery of tubular fluid to thedistal diluting segment of the nephron is estimated to be ap-proximately 20% of glomerular filtrate. The diluting segmentbegins with the water-impermeable ascending limb of Henle’sloop and in the absence of vasopressin includes virtually theremainder of the nephron, i.e., connecting tubule, collectingtubule, and cortical and medullary collecting ducts. With a GFRof 100 ml/min, 140 L of filtrate occurs daily. If 20% of thisfiltrate reaches the distal diluting segment and plasma AVP ismaximally suppressed, then 28 L of solute-free water theoreti-cally could be excreted. Therefore, the normal capacity to ex-crete solute-free water may approximate 1 L/h if administeredconsistently over 24 h. Patients with primary polydipsia, how-ever, ingest their water intake mostly during their awake hoursof the day. Even so, individuals who have primary polydipsiaand normal renal, endocrine, cardiac, and hepatic function andare not volume depleted or taking diuretics do not becomehyponatremic from drinking up to 10 to 12 L of water per day.Their plasma osmolalities, however, may be in the lower nor-
J Am Soc Nephrol 17: 1820–1832, 2006 Body Water Homeostasis 1823

mal range (275 to 285 mOsm/kg H2O). If any of the abovecircumstances intervene, then patients with primary polydipsiaare in danger of developing “water intoxication” with severecentral nervous system symptoms, including headache, nausea,vomiting, decreased mentation, confusion, obtundation, sei-zures, and even cerebral hernia, cardiopulmonary arrest, anddeath. Pure water intoxication rarely can occur without anintervening event with massive water intake, e.g., drinkingdirectly from a faucet or with an open hose in the stomach.
Psychiatric patients in hospital frequently have polydipsia,because of dry mouth from medications with anticholinergicproperties and/or delusions (e.g., “water cleanses the soul”). Infact, another term that has been used for primary polydipsiahas been psychogenic water drinking. In 1973, acute psychoseswith hyponatremia mimicking the syndrome of antidiureticsyndrome first was described (40). Subsequently, hyponatremiaand water intoxication with failure to suppress plasma AVPhave been described with psychotic disorders, such as schizo-phrenia (41). Psychiatric patients also have a high incidence ofsmoking, and nicotine is a very potent acute stimulus for AVPrelease (42,43).
Recently, molecular and cellular events were analyzed in a
unique rat model of primary polydipsia in which the samedaily food intake was ingested in control animals with adlibitum water intake and polyuric rats that drank 100 ml/d (44).This model therefore allowed assessment of the renal effects ofpolyuria at comparable electrolyte, caloric, and protein intakes.As compared with controls, serum osmolality was lower in thepolydipsic rats (293 versus 277 mOsm/kg H2O; P � 0.04) as wasurine osmolality (1365 versus 139 mOsm/kg H2O; P � 0.001).As occurs in humans after 10 d of increased water intake (45),a form of AVP-resistant NDI emerged after 10 d in the poly-dipsic rats. With 36 h of fluid deprivation, the polydipsic ani-mals exhibited significantly higher urine output associatedwith a lower urine osmolality despite significant higher plasmaAVP concentrations (Figure 5). The ion and urea transporterswith potential to alter the integrity of the countercurrent con-centrating mechanism were unaltered, including outer medul-lary Na-K-2Cl, Na-K-ATPase, and inner medullary urea trans-porter, in polydipsic as compared with control rats. The AQP1water channel expression, which if mutated can cause a con-centrating defect (9,10), also was no different in the polydipsicanimals. What was remarkably different was a highly signifi-cant suppression of AQP2 protein expression in the outer and
Figure 4. Effect of V2 receptor antagonist (OPC) in glucocorticoid-deficient rats to reverse the impaired water excretion (A) andurinary dilution (B) as well as the increased AQP2 (C) and phosphorylated AQP2 expression (D). Reprinted from reference (33),with permission.
1824 Journal of the American Society of Nephrology J Am Soc Nephrol 17: 1820–1832, 2006

inner medulla. The AQP2 expression in the membrane fraction,as an index of trafficking to the collecting duct membrane, wasdecreased. The AQP3 protein abundance in the outer medullaalso was significantly diminished in the polydipsic rats. Afterthe 36-h fluid deprivation, the medullary osmolalities in thecontrol and polydipsic rats were no different, even though thepolydipsic rats demonstrated a significant decrease in maximalurinary osmolality. This failure of osmotic equilibration be-tween the medullary interstitium and urine in the polydipsicrats supported a critical role of the downregulation of AQP2and, possibly, AQP3 in the impaired urine-concentrating capac-ity associated with polydipsia.
PregnancyA decrease in plasma osmolality of 8 to 10 mOsm/kg H2O is
an early occurrence in normal pregnancy. Sodium and waterretention occurs during pregnancy with an expansion of extra-cellular fluid ranging from 30 to 50% (46). Water retentionexceeds the sodium retention in normal pregnancy and thus thefall in plasma osmolality and sodium concentration. Recentstudies have shown that systemic arterial vasodilation occursearly in the first trimester of pregnancy (47). The mediator(s) ofthis decrease in vascular resistance is(are) not well defined, butsome experimental results indicate a role of estrogen-mediatednitric oxide (48) and relaxin (49). In any case, the standardresponses to arterial underfilling secondary to arterial vasodi-
lation also occur in pregnancy. There is a compensatory rise incardiac output and stimulation of the renin-angiotensin-aldo-sterone system that attenuates the vasodilation-mediated fall inBP during the first trimester (47). In other circumstances ofarterial vasodilation, such as cirrhosis or a large arteriovenousfistula, the activation of arterial baroreceptors is accompaniedby the nonosmotic release of AVP (50,51). In pregnancy, plasmaAVP concentrations are still detectable in the presence of adegree of hypo-osmolality that normally would suppress AVPrelease, i.e., 1 to 2% decrease in plasma osmolality. The hypo-osmolality of pregnancy has been termed a “resetting” of thehypothalamic osmoreceptor threshold to a lower level (52) butmay be due merely to the nonosmotic release of AVP thatoccurs with arterial vasodilation (50,51). It also is of interest thatearly in pregnancy, there is an increase in thirst and fluid intakedespite a fall in plasma osmolality, which normally suppressesthirst. This also is compatible with a nonosmotic stimulation ofthirst as a result of arterial underfilling.
Arterial underfilling as a result of arterial vasodilation, asoccurs in cirrhosis and pregnancy, is associated with compen-satory increases in total blood volume and cardiac output.Moreover, arterial vasodilation stimulates the nonosmotic AVPrelease and activates the renin-angiotensin-aldosterone axis, asoccurs in cirrhosis and pregnancy. These hormonal responsesalso compensate for arterial underfilling, at the expense ofhyponatremia and edema (50,51,53).
Figure 5. During 36 h of water deprivation, rats with polydipsia (POLY) demonstrated increased urine output (A), decreased urineosmolality (B), increased plasma AVP (C), and decreased AQP2 expression as compared with control (CTL) rats (D). Reprintedfrom reference (44), with permission.
J Am Soc Nephrol 17: 1820–1832, 2006 Body Water Homeostasis 1825

With the discovery of the renal water channels, i.e., AQP,body water regulation in pregnancy could be investigated inmore depth. Pregnancy in the rat exhibits most of the charac-teristics of human pregnancy and therefore has been the stan-dard experimental model. Studies therefore were undertaken inpregnant rats to examine the effect on renal AQP (54). If thenonosmotic release of AVP secondary to arterial vasodilation isinvolved in the water retention in pregnancy (55), then vaso-pressin-mediated upregulation and trafficking of AQP2 shouldbe demonstrable. Alternatively, if pregnancy “resets” the os-motic threshold for AVP release, then the regulation of theAQP2 around this reset osmostat should mimic the nonpreg-nant state. As compared with nonpregnant littermates, preg-nant rats were found to have in the first trimester a profoundupregulation of inner medullary AQP2 mRNA and proteinexpression that persisted throughout the pregnancy (Figure 6).In the pregnant rats, there also was an increase in AQP2 in themembrane fraction, indicating increased AQP2 trafficking tothe apical membrane of the collecting duct. It is known that theeffect of AVP on AQP2 protein expression and trafficking ismediated by vasopressin V2 receptor. Studies therefore wereundertaken with a nonpeptide V2 receptor antagonist in preg-nant and nonpregnant animals. The AQP2 protein expressionand apical membrane location by immunofluorescence werereturned to the nonpregnant state during the V2 receptor an-tagonist administration to pregnant rats (53). These experimen-tal results therefore strongly support a role of the nonosmoticAVP release in regulation AQP2 expression and trafficking inpregnancy. This was supported further by the observation thatincreased urinary AQP2 occurs during pregnancy (56). It must
be remembered, however, that the antidiuretic effect of oxy-toxin also is mediated via the V2 receptor on the basolateralmembrane of the collecting duct and therefore could be acontributing factor in the water retention of pregnancy (57). Italso has been shown that circulating vasopressinase from theplacenta, which increases AVP degradation, can uncover orcause diabetes insipidus in pregnancy (58).
Cardiac FailureAdvanced heart failure frequently is associated with hypo-
natremia. In fact, hyponatremia has been found to be a riskfactor for poor survival for patients with congestive heart fail-ure (59). The pathogenesis of the hyponatremia of cardiac fail-ure initially seemed not to involve AVP, primarily because thebioassay for antidiuretic hormone was relatively insensitive.With the development of the RIA to measure plasma AVP,studies were undertaken to examine the cause of hyponatremiain patients with heart failure. In the first study, the hypo-osmolality in patients with heart failure was of a degree thatwould maximally suppress plasma AVP in normal individuals,yet 30 (81%) of 37 patients had detectable plasma AVP by RIA(60). Therefore, the term nonosmotic AVP release emerged todescribe hyponatremic patients with heart failure and otheredematous disorders. There also is evidence for increased AVPsynthesis in the hypothalamus in experimental heart failure(61). Hyponatremia may occur both in low-output cardiac fail-ure (e.g., ischemic or nonischemic cardiomyopathy) and inhigh-output cardiac failure (e.g., thyrotoxicosis, beriberi, largearteriovenous fistula) (62). This apparent dilemma is under-standable because arterial underfilling with baroreceptor-me-diated AVP stimulation can occur with either a decrease incardiac output or systemic arterial vasodilation as occurs withhigh-output cardiac failure (50,51,53).
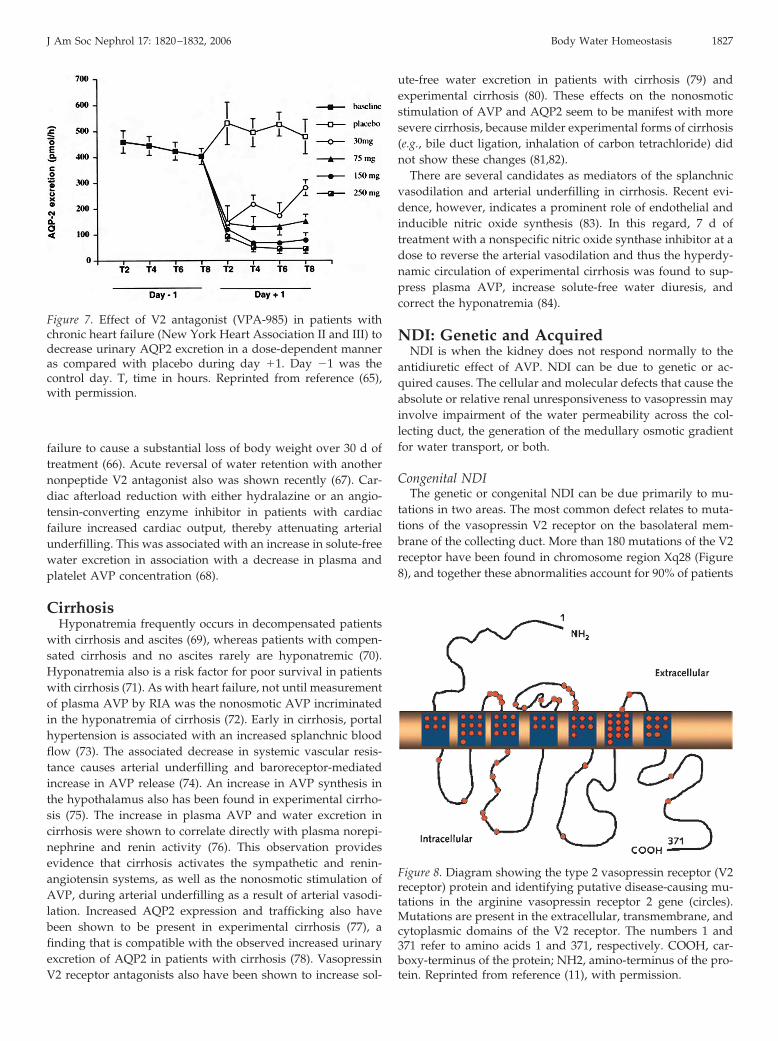
With the discovery of water channels, investigations wereundertaken to examine whether increased plasma AVP in car-diac failure would be associated with increased renal AQP2protein expression and trafficking to the apical membrane ofthe collecting duct (63,64). As in humans, plasma AVP in-creased in advanced cardiac failure in rats despite hypo-osmo-lality (63). In these animals with heart failure secondary tocoronary artery ligation, AQP2 expression and membrane traf-ficking were increased in the inner medulla of the kidney(63,64). Administration of a nonpeptide V2 receptor antagonistto these animals with heart failure reversed the increased ex-pression and trafficking of AQP2 (63). Support for this relation-ship between nonosmotic AVP and AQP2 expression has beendemonstrated by studies in patients with heart failure by mea-surement of urinary AQP2 (65). Approximately 3 to 6% ofAQP2 can be measured by RIA or Western immunoblotting inthe urine. In patients with New York Heart Association class IIor III heart failure, a V2 receptor antagonist caused a solute-freewater diuresis and increased plasma sodium concentration.Urinary AQP2 decreased in these patients during the V2 recep-tor antagonist administration (Figure 7) (65), thus indicatingthat less of this water channel reached the apical membrane ofthe collecting duct. An orally active, nonpeptide vasopressin V2receptor antagonist was shown recently in patients with cardiac
Figure 6. Inner medulla AQP2 protein expression (densitometryabove and immunoblots below) in nonpregnant (NP) rats andfirst-trimester (P7), second-trimester (P14), and third-trimester(P20) pregnant rats. Glycosylated (36 to 45 kD) and nonglyco-sylated (29 kD) expression is shown. Reprinted from reference(54), with permission.
1826 Journal of the American Society of Nephrology J Am Soc Nephrol 17: 1820–1832, 2006
failure to cause a substantial loss of body weight over 30 d oftreatment (66). Acute reversal of water retention with anothernonpeptide V2 antagonist also was shown recently (67). Car-diac afterload reduction with either hydralazine or an angio-tensin-converting enzyme inhibitor in patients with cardiacfailure increased cardiac output, thereby attenuating arterialunderfilling. This was associated with an increase in solute-freewater excretion in association with a decrease in plasma andplatelet AVP concentration (68).
CirrhosisHyponatremia frequently occurs in decompensated patients
with cirrhosis and ascites (69), whereas patients with compen-sated cirrhosis and no ascites rarely are hyponatremic (70).Hyponatremia also is a risk factor for poor survival in patientswith cirrhosis (71). As with heart failure, not until measurementof plasma AVP by RIA was the nonosmotic AVP incriminatedin the hyponatremia of cirrhosis (72). Early in cirrhosis, portalhypertension is associated with an increased splanchnic bloodflow (73). The associated decrease in systemic vascular resis-tance causes arterial underfilling and baroreceptor-mediatedincrease in AVP release (74). An increase in AVP synthesis inthe hypothalamus also has been found in experimental cirrho-sis (75). The increase in plasma AVP and water excretion incirrhosis were shown to correlate directly with plasma norepi-nephrine and renin activity (76). This observation providesevidence that cirrhosis activates the sympathetic and renin-angiotensin systems, as well as the nonosmotic stimulation ofAVP, during arterial underfilling as a result of arterial vasodi-lation. Increased AQP2 expression and trafficking also havebeen shown to be present in experimental cirrhosis (77), afinding that is compatible with the observed increased urinaryexcretion of AQP2 in patients with cirrhosis (78). VasopressinV2 receptor antagonists also have been shown to increase sol-
ute-free water excretion in patients with cirrhosis (79) andexperimental cirrhosis (80). These effects on the nonosmoticstimulation of AVP and AQP2 seem to be manifest with moresevere cirrhosis, because milder experimental forms of cirrhosis(e.g., bile duct ligation, inhalation of carbon tetrachloride) didnot show these changes (81,82).
There are several candidates as mediators of the splanchnicvasodilation and arterial underfilling in cirrhosis. Recent evi-dence, however, indicates a prominent role of endothelial andinducible nitric oxide synthesis (83). In this regard, 7 d oftreatment with a nonspecific nitric oxide synthase inhibitor at adose to reverse the arterial vasodilation and thus the hyperdy-namic circulation of experimental cirrhosis was found to sup-press plasma AVP, increase solute-free water diuresis, andcorrect the hyponatremia (84).
NDI: Genetic and AcquiredNDI is when the kidney does not respond normally to the
antidiuretic effect of AVP. NDI can be due to genetic or ac-quired causes. The cellular and molecular defects that cause theabsolute or relative renal unresponsiveness to vasopressin mayinvolve impairment of the water permeability across the col-lecting duct, the generation of the medullary osmotic gradientfor water transport, or both.
Congenital NDIThe genetic or congenital NDI can be due primarily to mu-
tations in two areas. The most common defect relates to muta-tions of the vasopressin V2 receptor on the basolateral mem-brane of the collecting duct. More than 180 mutations of the V2receptor have been found in chromosome region Xq28 (Figure8), and together these abnormalities account for 90% of patients
Figure 7. Effect of V2 antagonist (VPA-985) in patients withchronic heart failure (New York Heart Association II and III) todecrease urinary AQP2 excretion in a dose-dependent manneras compared with placebo during day �1. Day �1 was thecontrol day. T, time in hours. Reprinted from reference (65),with permission.
Figure 8. Diagram showing the type 2 vasopressin receptor (V2receptor) protein and identifying putative disease-causing mu-tations in the arginine vasopressin receptor 2 gene (circles).Mutations are present in the extracellular, transmembrane, andcytoplasmic domains of the V2 receptor. The numbers 1 and371 refer to amino acids 1 and 371, respectively. COOH, car-boxy-terminus of the protein; NH2, amino-terminus of the pro-tein. Reprinted from reference (11), with permission.
J Am Soc Nephrol 17: 1820–1832, 2006 Body Water Homeostasis 1827

with congenital NDI (85). Studies suggest that most mutationsinvolve protein misfolding, which traps the V2 receptor intra-cellularly, thereby not allowing the receptor to translocate tothe basolateral membrane of the collecting duct (86). Therefore,the adenylate cyclase-cAMP signaling pathway, which medi-ates AQP2 expression and trafficking to the apical membrane,is not activated. Recent in vivo studies indicate that nonpeptideV2, V1, and V1/V2 receptor antagonists may allow some of themutant V2 receptors to be transported to the basolateral mem-brane and to function normally (87). These antagonists act aschaperones for the misfolded V2 receptors to reach the mem-brane. A recent study in patients with NDI demonstrated anapproximate decrease in daily urine flow from 12 to 8 L and anincrease in urinary osmolality from 98 to 170 mOsm/kg usingsuch a receptor antagonist (88). This variety of congenital NDIhas an X-linked recessive mode of inheritance, and affectedmale individuals cannot concentrate their urine in response tovasopressin. Heterozygous female individuals generally areasymptomatic but may have a modest degree of polyuria andpolydipsia. In contrast, male individuals may have up to 20 L ofurine every day. The need to drink sufficient water to maintainwater balance, therefore, is challenging and certainly affects thequality of life of the individual. Before genetic screening ofnewborns in affected families, dehydration and mental retar-dation frequently developed. Now affected individuals whoreceive adequate water intake progress to normal adulthoodwithout any mental or physical retardation.
Another cause of congenital NDI involves mutations of theAQP2 gene in chromosome region 12q13 (89). This accounts for10% of families with congenital NDI. Currently, more than 30such mutations have been identified (Figure 9). In the autoso-mal recessive variety, the mutant AQP2 proteins are trapped inthe endoplasmic reticulum, whereas the autosomal dominantvarieties are mutations of the carboxy terminus.
Acquired NDIHypokalemia and hypercalcemia are electrolyte disorders
that may be associated with polyuria and polydipsia as a resultof a defect in the renal response of vasopressin to concentratethe urine maximally. Experimental studies in rats have beenundertaken to study the mechanisms involved. A potassium-deficient diet for 11 d caused hypokalemic-related NDI (90),and 7 d of vitamin D (dihydrotachysterol) caused hypercalce-mic-related NDI (91). In both experimental models, there was adownregulation of AQP2 expression in the inner medulla. Inthe hypercalcemic model, there also was a decrease in theNa-K-2Cl co-transporter in the outer medulla. As the initiatorof the countercurrent concentrating mechanism for generationof the corticomedullary osmotic gradient, this decrease in theNa-K-2Cl co-transporter no doubt also contributed to the ac-quired NDI.
Polyuria secondary to NDI also may accompany bilateralurinary tract obstruction, in part due to the resultant volumeexpansion and urea retention. Experimental studies, however,have shown that unilateral ureteral obstruction, which avoidsthese sequelae of bilateral ureteral obstruction, also causes NDI(92). As with the above electrolyte disorders, AQP2 downregu-lation has been demonstrated to be associated with acquiredNDI related to urinary tract obstruction. Lithium also has beendemonstrated in experimental animals to be associated withdownregulation of AQP2 expression and trafficking (93). Lith-ium therapy has been a worldwide, effective treatment forbipolar affective disorder for many years. Therefore, under-standing the mechanism for the NDI that is associated with thismedication is important, particularly because it occurs in ap-proximately 20% of treated patients. In this regard, recent invitro and in vivo experiments suggest that the effect of lithium todownregulate AQP2 may occur independent of adenylyl cy-clase activity (94,95).
A vasopressin-resistant urine-concentrating defect is one ofthe earliest abnormalities associated with acute renal failure.This variety of acquired NDI also seems to involve an inabilityto establish a high medullary solute content, i.e., the osmoticdriving force for water reabsorption. Moreover, AQP1, 2, and 3expression has been shown to be reduced in rats with botholiguric and nonoliguric ischemic acute renal failure (96). Sim-ilar results have been found in the 5/6 nephrectomy–inducedmodel of chronic renal failure (97). In patients with advancedchronic renal failure, vasopressin-resistant hypotonic urine hasbeen described (98). This finding suggests a defect in watertransport across the medullary collecting duct, because abso-lute impairment of the countercurrent concentrating mecha-nism still would be associated with an isotonic, not hypotonic,medullary interstitium. In contrast to congenital NDI, the poly-uria of acquired NDI generally is of a moderate degree (e.g., 3to 4 L/24 h). The thirst mechanism generally protects againsthypernatremia in NDI states unless age (newborn, elderly) orillness restricts availability of fluid intake.
ConclusionThe ability to analyze the renal water channels and ion and
urea transporters has allowed for the better understanding of
Figure 9. Diagram showing the AQP2 protein and identifyingputative disease-causing mutations in AQP2 (circles). Muta-tions are present in the extracellular, transmembrane, and cy-toplasmic domains of AQP2. The numbers 1 and 271 refer toamino acids 1 and 271, respectively. Reprinted from reference(11), with permission.
1828 Journal of the American Society of Nephrology J Am Soc Nephrol 17: 1820–1832, 2006

several clinical disorders with impaired urinary dilutionand/or concentration.
AcknowledgmentsThis work was supported by National Institutes of Health grant DK
19928.I appreciate the assistance of Jan Darling in the preparation of the
manuscript. Most important, I acknowledge Peter Agre, MD, and hisassociates, whose discovery of the first mammalian water channel hasallowed much of this work to be undertaken.
References1. Agre P, Saboori AM, Asimos A, Smith BL: Purification and
partial characterization of the M 30,000 integral membraneprotein associated with the erythrocyte Rh (D) antigen.J Biol Chem 262: 17497–17503, 1987
2. Agre P, Preston GM, Smith BL, Jung JS, Raina S, Moon C,Guggino WB, Nielsen S: Aquaporin CHIP: The archetypalmolecular water channel. Am J Physiol Renal Fluid Electro-lyte Physiol 265: F463–F476, 1993
3. Nielsen S, Frokiaer J, Marples D, Kwon T-H, Agre P, Knep-per M: Aquaporins in the kidney: From molecules to med-icine. Physiol Rev 82: 205–244, 2002
4. Schrier RW, Cadnapaphornchai MA: Renal aquaporin wa-ter channels: From molecules to human disease. Prog Bio-phys Mol Biol 81: 117–131, 2003
5. Schrier RW, Chen Y-C, Cadnapaphornchai MA: From finchto fish to man: Role of aquaporins in body fluid and brainwater regulation. Neuroscience 129: 897–904, 2004
6. Chen Y-C, Cadnapaphornchai MA, Schrier RW: Clinicalupdate on renal aquaporins. Biol Cell 97: 357–371, 2005
7. Ishikawa S, Schrier RW: Pathophysiological roles of argi-nine vasopressin and aquaporin-2 in impaired water ex-cretion. Clin Endocrinol 58: 1–17, 2003
8. Sands J: Mammalian urea transporters. Annu Rev Physiol65: 543–566, 2003
9. Ma T, Yang B, Gillespie A, Carlson E, Epstein GC, Verk-man A: Severely impaired urinary concentrating ability intransgenic mice lacking aquaporin-1 water channels. J BiolChem 273: 4296–4299, 1998
10. King LS, Choi M, Ferenandez PC, Cartron J-P, Agre P:Defective urinary concentrating ability due to a completedeficiency of aquaporin-1. N Engl J Med 345: 175–179, 2001
11. Sands J, Bichet D: Nephrogenic diabetes insipidus. AnnIntern Med 144: 186–194, 2006
12. Ma T, Song Y, Yang B, Gillespie A, Carlson E, Epstein C,Verkman A: Nephrogenic diabetes insipidus in mice lack-ing aquaporin-3 water channels. Proc Natl Acad Sci U S A97: 4386–4391, 2000
13. Ma T, Yang B, Gillespie A, Carlson E, Epstein C, VerkmanA: Generation and phenotype of a transgenic knockoutmouse lacking the mercurial-insensitive water channelaquaporin-4. J Clin Invest 100: 957–962, 1997
14. Verkman AS: Novel roles of aquaporins revealed by phe-notype analysis of knockout mice. Rev Physiol BiochemPharmacol 155: 31–55, 2005
15. Nielsen S, Chou C, Marples D, Christensen E, Knepper M,Harris H: Cellular and subcellular immunolocalization ofvasopressin-regulated water channel in rat kidney. ProcNatl Acad Sci U S A 90: 11663–11667, 1993
16. DiGiovanni S, Nielsen S, Christensen E, Knepper M: Reg-ulation of collecting duct water channel expression byvasopressin in Brattleboro rats. Proc Natl Acad Sci U S A 91:8984–8988, 1994
17. Wade J, Nielsen S, Coleman R, Knepper M: Long-termregulation of collecting duct water permeability: Freeze-fracture analysis of isolated perfused tubules. Am J PhysiolRenal Fluid Electrolyte Physiol 166: F723–F730, 1994
18. Fushimi K, Sasaki S, Marumo F: Phosphorylation of serine256 is required for cAMP-dependent regulatory exocytosisof the aquaporin-2 water channel. J Biol Chem 272: 14800–14804, 1997
19. Katsura T, Gustafson C, Ausiello A, Brown D: Proteinkinase A phosphorylation is involved in regulated exocy-tosis of aquaporin-2 transfected LLC-PK1 cells. Am JPhysiol 272: F817–F822, 1997
20. Hozawa S, Holtzman E, Ausiello D: cAMP motifs regulat-ing transcription in the aquaporin-2 gene. Am J Physiol 270:C1695–C1702, 1996
21. Matsumura Y, Uchida S, Rai T, Sasaki S, Marumo F: Tran-scriptional regulation of aquaporin-2 water channel geneby cAMP. J Am Soc Nephrol 8: 861–867, 1997
22. Umenishi F, Narikiyo T, Vandewalle A, Schrier RW: cAMPregulates vasopressin-induced AQP-2 expression via pro-tein kinase A-independent pathway. BBA Biomembranes2006, in press
23. Umenishi F, Narikiyo T, Schrier RW: Effect on stability,degradation, expression, and targeting of aquaporin-2 wa-ter channel by hyperosmolality in renal epithelial cells.Biochm Biophys Res Commun 338: 1593–1599, 2005
24. Li C, Wang W, Summer S, Cadnapaphornchai MA, Falk S,Umenishi F, Schrier RW: Hyperosmolality in vivo upregu-lates aquaporin 2 water channel and Na-K-2Cl cotrans-porter in Brattleboro rats. J Am Soc Nephrol 17: 1657–1664,2006
25. Anderson R, Chung H, Kluge R, Schrier RW: Hyponatre-mia: A prospective analysis of its epidemiology and thepathogenetic role of vasopressin. Ann Intern Med 102: 164–168, 1985
26. Hoorn EJ, Lindemans J, Zietse R: Development of severehyponatraemia in hospitalized patients: Treatment-relatedrisk factors and inadequate management. Nephrol DialTransplant 21: 70–76, 2006
27. Berl T, Schrier RW: Disorders of water metabolism. In:Renal and Electrolyte Disorders, 6th Ed., edited by SchrierRW, Philadelphia, Lippincott Williams & Wilkins, 2002, pp1–63
28. Skowsky W, Kikuchi T: The role of vasopressin in theimpaired water excretion of myxedema. Am J Med 64:613–621, 1978
29. Chen Y-C, Cadnapaphornchai MA, Yang J, Summer S, FalkS, Li C, Wang W, Schrier RW: Nonosmotic release of va-sopressin and renal aquaporins in impaired urinary dilu-tion in hypothyroidism. Am J Physiol Renal Physiol 289:F672–F678, 2005
30. Cadnapaphornchai MA, Kim Y-W, Gurevich A, Summer S,Falk S, Thurman J, Schrier RW: Urinary concentrating de-fect in hypothyroid rats: Role of sodium, potassium,2-chloride co-transporter and aquaporins. J Am Soc Nephrol14: 566–574, 2003
31. Ufferman R, Schrier RW: Importance of sodium intake andmineralocorticoid hormone in the impaired water excre-
J Am Soc Nephrol 17: 1820–1832, 2006 Body Water Homeostasis 1829

tion in adrenal insufficiency. J Clin Invest 51: 1639–1646,1972
32. Ohara M, Cadnapaphornchai MA, Summer S, Falk S, YangJ, Togawa T, Schrier RW: Effect of mineralocorticoid defi-ciency on ion and urea transporters and aquaporin waterchannels in the rat. Biochem Biophys Res Commun 299: 285–290, 2002
33. Ishikawa S, Schrier RW: Effect of arginine vasopressinantagonist on renal water excretion in glucocorticoid andmineralocorticoid deficient rats. Kidney Int 22: 587–593,1982
34. Chen Y-C, Cadnapaphornchai MA, Summer S, Falk S, Li C,Wang W, Schrier RW: Molecular mechanisms of impairedurinary concentrating ability in glucocorticoid-deficientrats. J Am Soc Nephrol 16: 2864–2871, 2005
35. Wang W, Li C, Summer S, Falk S, Cadnapaphornchai MA,Chen Y-C, Schrier RW: Molecular analysis of impairedurinary diluting capacity in glucocorticoid deficiency. Am JPhysiol Renal Physiol 290: F1135–F1142, 2006
36. Celkjers W: Hyponatremia and inappropriate secretion ofvasopressin (antidiuretic hormone) in patients with hy-popituitarism. N Engl J Med 321: 492–496, 1989
37. Pyo H, Summer S, Kim J, Schrier RW: Vasopressin geneexpression in glucocorticoid hormone-deficient rats. AnnN Y Acad Sci 689: 659–662, 1993
38. Kim J, Summer S, Wood W, Schrier RW: Role of glucocor-ticoid hormones in arginine vasopressin gene regulation.Biochem Biophys Res Commun 289: 1252–1256, 2001
39. Gross P, Pehrisch H, Rascher W, Schomig A, Hackenthal E,Ritz E: Pathogenesis of clinical hyponatremia: Observationof vasopressin and fluid intake in 100 hyponatremic med-ical patients. Eur J Clin Invest 17: 123–129, 1987
40. Dubovsky S, Grabon S, Berl T, Schrier RW: Syndrome ofinappropriate secretion of antidiuretic hormone with exac-erbated psychosis. Ann Intern Med 79: 551–554, 1973
41. deLeon J, Verghese C, Tracy J, Josiassen R, Simpson G:Polydipsia and water intoxication in psychiatric patients: Areview of the epidemiological literature. Biol Psychiatry 35:408–419, 1994
42. Blum A: The possible role of tobacco cigarette smoking inhyponatremia of long-term psychiatric patients. JAMA 252:2864–2865, 1984
43. Allon M, Allen H, Deck L, Clark M: Role of cigarette use inhyponatremia in schizophrenic patients. Am J Psychiatry147: 1075–1077, 1990
44. Cadnapaphornchai MA, Summer S, Falk S, Thurman J,Knepper M, Schrier RW: Effect of primary polydipsia onaquaporin and sodium transporter abundance. Am JPhysiol Renal Physiol 285: F965–F971, 2003
45. DeWardener H, Herxheimer A: The effect of a high waterintake on the kidney’s ability to concentrate urine in man.J Physiol 139: 42–52, 1957
46. Briner V, Cadnapaphornchai M, Schrier RW: Hypertensionand pregnancy. In: Diseases of the Kidney and Urinary Tract,8th Ed., edited by Schrier RW, Philadelphia, LippincottWilliams & Wilkins, 2006, in press
47. Chapman AB, Abraham WT, Zamudio S, Coffin C, Mer-ouani A, Young D, Johnson A, Osorio F, Goldberg C,Moore LG, Dahms T, Schrier RW: Temporal relationshipsbetween hormonal and hemodynamic changes in earlyhuman pregnancy. Kidney Int 54: 2056–2063, 1998
48. Cadnapaphornchai MA, Ohara M, Morris K, Knotek M,
Rogachev B, Ladtkow T, Carter E, Schrier RW: Chronicnitric oxide synthase inhibition reverses systemic vasodi-lation and glomerular hyperfiltration in pregnancy. Am JPhysiol Renal Physiol 280: F592–F598, 2001
49. Conrad K, Jeyabalen A, Danielson L, Kerchner L, Novak J:Role of relaxin in maternal renal vasodilation of preg-nancy. Ann N Y Acad Sci 1041: 147–154, 2005
50. Schrier RW: Pathogenesis of sodium and water retention inhigh and low output cardiac failure, cirrhosis, nephroticsyndrome, and pregnancy (1). N Engl J Med 319: 1065–1072,1988; published erratum appears in N Engl J Med 320: 676,1989
51. Schrier RW: Pathogenesis of sodium and water retention inhigh and low output cardiac failure, cirrhosis, nephroticsyndrome, and pregnancy (2). N Engl J Med 319: 319:1127–1134, 1988; published erratum appears in N EnglJ Med 320: 676, 1989
52. Durr J, Stamoutsos B, Lindheimer M: Osmoregulation dur-ing pregnancy in the rat. Evidence for resetting of thethreshold for vasopressin secretion during gestation. J ClinInvest 68: 337–346, 1981
53. Schrier RW: Body fluid volume regulation in health anddisease: A unifying hypothesis. Ann Intern Med 113: 155–159, 1990
54. Ohara M, Martin P-Y, Xu D-L, St. John J, Pattison T, Kim J,Schrier RW: Upregulation of aquaporin 2 water channelexpression in pregnant rats. J Clin Invest 101: 1076–1083,1998
55. Schrier RW, Briner VA: Peripheral arterial vasodilationhypothesis of sodium and water retention in pregnancy:Implications for pathogenesis of preeclampsia-eclampsiastate. Obstet Gynecol 77: 632–639, 1991
56. Buemi M, D’Anna R, DiPasquale G, Floccari F, Ruello A,Aloisi C, Leonardi I, Frisina N, Corica F: Urinary excretionof aquaporin-2 water channel during pregnancy. CellPhysiol Biochem 11: 203–208, 2001
57. Chou C, DiGiovanni S, Luther A, Knepper M: Oxytocin asan antidiuretic hormone. II. Role of V2 vasopressin recep-tor. Am J Physiol Renal Fluid Electrolyte Physiol 269: F78–F85,1995
58. Durr J, Hoggard J, Hunt J, Schrier RW: Diabetes insipidusdue to abnormally high circulating vasopressinase activityin a pregnancy. N Engl J Med 316: 1070–1074, 1987
59. Lee W, Packer M: Prognostic importance of serum concen-tration and its modification by converting-enzyme inhibi-tion in patients with severe chronic heart failure. Circula-tion 73: 257–267, 1986
60. Szatalowicz V, Arnold P, Chaimovitz C, Bichet D, SchrierRW: Radioimmunoassay of plasma arginine vasopressin inhyponatremic patients with congestive heart failure.N Engl J Med 305: 263–266, 1981
61. Kim J, Michel J, Soubrier F, Durr J, Corvol P, Schrier RW:Arginine vasopressin gene expression in chronic cardiacfailure in rats. Kidney Int 38: 818–822, 1990
62. Schrier RW, Abraham WT: Hormones and hemodynamicsin heart failure. N Engl J Med 341: 577–585, 1999
63. Xu D, Martin P-Y, Ohara M, St. John J, Pattison T, Meng X,Morris K, Kim J, Schrier RW: Upregulation of aquaporin-2water channel expression in chronic heart failure. J ClinInvest 99: 1500–1505, 1997
64. Nielsen S, Terris J, Andersen D, Ecelbarger C, Frokiaer J,Jonassen T, Marples D, Knepper M, Petersen J: Congestive
1830 Journal of the American Society of Nephrology J Am Soc Nephrol 17: 1820–1832, 2006

heart failure in rats is associated with increased expressionand targeting of aquaporin-2 water channel in collectingduct. Proc Natl Acad Sci U S A 94: 5450–5455, 1997
65. Martin P-Y, Abraham W, Lieming X, Olson B, Oren R,Ohara M, Schrier RW: Selective V2-receptor vasopressinantagonism decrease urinary aquaporin-2 excretion in pa-tients with chronic heart failure. J Am Soc Nephrol 10:2165–2170, 1999
66. Gheorghiade M, Niazi I, Ouyang J, Czerwiec F, Kamba-yashi J-I, Xampino M, Orlandi C: Vasopressin V2-receptorblockade with Tolvaptan in patients with chronic heartfailure. Results from a double-blind, randomized trial. Cir-culation 107: 2690–2696, 2003
67. Abraham W, Shamshirsaz A, McFann K, Oren R, SchrierRW: Aquaretic effect of lixivaptan, an oral, non-peptide,selective V2 receptor vasopressin antagonist, in New YorkHeart Association functional class II and III heart failurepatients. J Am Coll Cardiol 47: 1615–1621, 2006
68. Bichet D, Kortas C, Mettauer B, Manzini C, Marc-Aurele J,Rouleau J, Schrier RW: Modulation of plasma and plateletvasopressin by cardiac function in patients with heart fail-ure. Kidney Int 29: 1188–1196, 1986
69. Gines P, Berl T, Bernardi M, Bichet D, Hamon G, JimenezW, Liard J, Martin PY, Schrier RW: Hyponatremia in cir-rhosis: From pathogenesis to treatment. Hepatology 28: 851–864, 1988
70. Madsen M, Pedersen E, Danielsen H, Jensen L, Sorensen S:Impaired renal water excretion in early hepatic cirrhosis.Lack of relationship between renal water excretion andplasma levels of arginine vasopressin, angiotensin II, andaldosterone after water loading. Scand J Gastroenterol 21:749–755, 1986
71. Gines A, Escorsell A, Gines P, Salo J, Jimenez W, Inglada L,Navasa M, Claria J, Rimola A, Arroyo V, et al.: Incidence,predictive factors, and prognosis of the hepatorenal syn-drome in cirrhosis with ascites. Gastroenterology 105: 229–236, 1993
72. Bichet D, Szatalowicz V, Chaimovitz C, Schrier RW: Role ofvasopressin in abnormal water excretion in cirrhotic pa-tients. Ann Intern Med 96: 413–417, 1982
73. Bosch J, Garcia-Pagan J: The splanchnic circulation in cir-rhosis. In: Ascites and Renal Dysfunction in Liver Disease, 2ndEd., edited by Gines P, Arroyo V, Rodes J, Schrier RW,Oxford, Blackwell Publishing, 2005, pp 156–163
74. Schrier RW, Arroyo V, Bernardi M, Epstein M, HenricksenJ, Rodes J: Peripheral arterial vasodilation hypothesis: Aproposal for the initiation of renal sodium and water re-tention in cirrhosis. Hepatology 8: 1151–1157, 1988
75. Kim J, Summer S, Howard R, Schrier RW: Vasopressingene expression in rats with experimental cirrhosis. Hepa-tology 17: 143–147, 1993
76. Bichet D, Van Putten V, Schrier RW: Potential role ofincreased sympathetic activity in impaired sodium andwater excretion in cirrhosis. N Engl J Med 397: 1552, 1982
77. Fujita N, Ishikawa S, Sasaki S, Fujisawa G, Fushimi K,Marumo F, Saito T: Role of water channel AQP-CD inwater retention in SIADH and cirrhotic rats. Am J Physiol269: F926–F931, 1995
78. Ivarsen P, Frokiaer J, Aagaard N, Hansen E, Bendtsen F,Nielsen S, Vilstrup H: Increased urinary excretion of aqua-porin 2 in patients with liver cirrhosis. Gut 52: 1194–1199,2003
79. Inoue T, Ohnishi A, Matsuo A, Kawai B, Kunihiro N, TadaY, Koizumi F, Chau T, Okada Y, Yamamura Y, Tanaka T:Therapeutic and diagnostic potential of a vasopressin-2antagonist for impaired water handling in cirrhosis. ClinPharmacol Ther 63: 561–570, 1998
80. Claria J, Jiminez W, Arroyo V, Guarner F, Lopez C, La VillaG, Asbert M, Rivera F, Rodes J: Blockade of the hydroos-motic effect of vasopressin normalizes water excretion incirrhotic rats. Gastroenterology 97: 1294–1299, 1989
81. Fernandez-Llama P, Turner R, Dibona G, Knepper M: Re-nal expression of aquaporins in liver cirrhosis induced bychronic common bile duct ligation in rats. J Am Soc Nephrol10: 195–1957, 1999
82. Fernandez-Llama P, Jimenez W, Bosch-Marce M, ArroyoV, Nielsen S, Knepper M: Dysregulation of renal aquapor-ins and Na-Cl cotransporter in CC14-induced cirrhosis.Kidney Int 58: 216–228, 2000
83. Martin PY, Gines P, Schrier RW: Nitric oxide as a mediatorof hemodynamic abnormalities and sodium and water re-tention in cirrhosis. N Engl J Med 33: 533–541, 1998
84. Martin PY, Ohara M, Gines P, Xu D, St. John J, Nieder-berger M, Schrier RW: Nitric oxide synthase (NOS) inhibi-tion for one week improves renal sodium and water excre-tion in cirrhotic rats with ascites. J Clin Invest 101: 235–242,1998
85. Morello J, Bichet D: Nephrogenic diabetes insipidus. AnnuRev Physiol 63: 607–630, 2001
86. Arid M, Balch W: Integration of endoplasmic reticulumsignaling in health and disease. Nat Med 5: 745–751, 1999
87. Fujiwara T, Bichet D: Molecular biology of hereditary di-abetes insipidus. J Am Soc Nephrol 16: 2836–2846, 2005
88. Bernier V, Morello J-P, Zarruk A, Debrand N, Salahpour A,Lonergan M, Arthus M-F, Laperriere A, Brouard R, Bou-vier M, Bichet D: Pharmacologic chaperones as a potentialtreatment for x-linked nephrogenic diabetes insipidus.J Am Soc Nephrol 17: 232–243, 2006
89. Deen P, Croes H, van Aubel R, Ginsel L, van Os C: Waterchannels encoded by mutant aquaporin-2 genes in neph-rogenic diabetes insipidus are impaired in their cellularrouting. J Clin Invest 95: 2291–2296, 1995
90. Marples D, Frokiaer J, Dorup J, Knepper M, Nielsen S:Hypokalemia-induced downregulation of aquaporin-2 wa-ter channel expression in rat kidney medulla and cortex.J Clin Invest 97: 1960–1968, 1996
91. Earm J, Christensen B, Frokiaer J, Marples D, Han J, Knep-per M, Nielsen S: Decreased aquaporin-2 expression andapical plasma membrane delivery in kidney collectingducts of polyuric hypercalcemic rats. J Am Soc Nephrol 9:2181–2193, 2998
92. Frokiaer J, Christensen B, Marples D, Djurhuus J, Jensen U,Knepper M, Nielsen S: Downregulation of aquaporin-2parallels changes in renal water excretion in unilateralureteral obstruction. Am J Physiol Renal Physiol 273: F213–F223, 1997
93. Marples D, Christensen S, Christensen E, Ottosen P,Nielsen S: Lithium-induced downregulation of aqua-porin-2 water channel expression in rat kidney medulla.J Clin Invest 95: 1838–1845, 1995
94. Bichet DG: Lithium, cyclic AMP signaling, A-kinase an-choring proteins, and aquaporin-2. J Am Soc Nephrol 17:920–922, 2006
95. Li Y, Shaw S, Kamsteeg E-J, Vandewalle A, Deen P: De-
J Am Soc Nephrol 17: 1820–1832, 2006 Body Water Homeostasis 1831

velopment of lithium-induced nephrogenic diabetes insip-idus is dissociated from adenylyl cyclase activity. J Am SocNephrol 17: 1063–1072, 2006
96. Fernandez-Llama P, Andrews P, Turner R, Saggi S, DimariJ, Kwon T, Nielsen S, Safirstein R, Knepper M: Decreasedabundance of collecting duct aquaporins in post-ischemicrenal failure in rats. J Am Soc Nephrol 10: 1658–1668, 1999
97. Kwon T, Frokiaer J, Knepper M, Nielsen S: Reduced AQP1,-2, and -3 levels in kidneys of rats with CRF induced bysurgical reduction in renal mass. Am J Physiol Renal Physiol275: F724–F741, 1998
98. Tannen RL, Regal EM, Dunn MJ, Schrier RW: Vasopressin-resistant hyposthenuria in advanced chronic renal disease.N Engl J Med 280: 1135–1141, 1969
Access to UpToDate on-line is available for additional clinical informationat http://www.jasn.org/
1832 Journal of the American Society of Nephrology J Am Soc Nephrol 17: 1820–1832, 2006