blocking brain-derived neurotrophic factor inhibits injury-induced hyperexcitability of hippocampal...
TRANSCRIPT

Blocking brain-derived neurotrophic factor inhibits injury-induced hyperexcitability of hippocampal CA3 neurons
Raminder Gill,1 Philip K.-Y. Chang,1 George A. Prenosil,1 Emily C. Deane2 and Rebecca A. McKinney1,21Department of Pharmacology & Therapeutics, McGill University, Bellini Life Sciences Complex, 3649 Promenade Sir WilliamOsler,Montreal, QC, Canada H3G 0B12Department of Neurology and Neurosurgery, McGill University, Montreal, QC, Canada
Keywords: axonal sprouting, brain-derived neurotrophic factor, GAP43, organotypic hippocampal slices, post-traumaticepilepsy
Abstract
Brain trauma can disrupt synaptic connections, and this in turn can prompt axons to sprout and form new connections. If these newaxonal connections are aberrant, hyperexcitability can result. It has been shown that ablating tropomyosin-related kinase B (TrkB), areceptor for brain-derived neurotrophic factor (BDNF), can reduce axonal sprouting after hippocampal injury. However, it is unknownwhether inhibiting BDNF-mediated axonal sprouting will reduce hyperexcitability. Given this, our purpose here was to determinewhether pharmacologically blocking BDNF inhibits hyperexcitability after injury-induced axonal sprouting in the hippocampus. Toinduce injury, we made Schaffer collateral lesions in organotypic hippocampal slice cultures. As reported by others, we observed a50% reduction in axonal sprouting in cultures treated with a BDNF blocker (TrkB-Fc) 14 days after injury. Furthermore, lesionedcultures treated with TrkB-Fc were less hyperexcitable than lesioned untreated cultures. Using electrophysiology, we observed atwo-fold decrease in the number of CA3 neurons that showed bursting responses after lesion with TrkB-Fc treatment, whereas wefound no change in intrinsic neuronal firing properties. Finally, evoked field excitatory postsynaptic potential recordings indicated anincrease in network activity within area CA3 after lesion, which was prevented with chronic TrkB-Fc treatment. Taken together, ourresults demonstrate that blocking BDNF attenuates injury-induced hyperexcitability of hippocampal CA3 neurons. Axonal sproutinghas been found in patients with post-traumatic epilepsy. Therefore, our data suggest that blocking the BDNF–TrkB signaling cascadeshortly after injury may be a potential therapeutic target for the treatment of post-traumatic epilepsy.
Introduction
Brain injury can physically disrupt axons and synapses within neu-ronal networks. When these connections are disrupted, axons mayrespond by sprouting their collaterals. Often, this process is aberrant,and can lead to the functional disturbance of an existing neuronalnetwork. For example, there is considerable evidence for mossyfiber sprouting from hippocampal dentate granule cells followingexcitotoxic injury with kainic acid (Davenport et al., 1990; Routbortet al., 1999), lesion of existing mossy fibers (Laurberg & Zimmer,1981; Hannesson et al., 1997), or injury to the cortex (Salin et al.,1995). However, the underlying cellular events that cause axonalgrowth following injury are poorly understood.Neurotrophic factors, in particular brain-derived neurotrophic fac-
tor (BDNF) and its receptor tropomyosin-related kinase B (TrkB),are important for axonal growth during development (Huang &Reichardt, 2001). Davies et al. were the first to show that applica-tion of BDNF to dissociated chick sensory neurons leads to profuseneurite outgrowth (Davies et al., 1986). More recent studies have
demonstrated that BDNF can lead to neurite elongation or branchingthrough distinct signaling cascades that are dependent on the con-centration of BDNF in the extracellular space (Ji et al., 2010).Moreover, the application of extracellular BDNF promotes neuritedifferentiation to axons (Shelly et al., 2007, 2011), and BDNF isrequired and sufficient for the formation of axons in cultured hippo-campal neurons (Cheng et al., 2011). We have also reported thatexogenous TrkB ligands can induce axon sprouting in unlesionedorganotypic hippocampal cultures, increasing the activity of CA3neurons (Schwyzer et al., 2002).Previously, we have shown that transecting the Schaffer collateral
pathway, which connects CA3 pyramidal neurons to those in areaCA1, can lead to axonal sprouting in area CA3 (McKinney et al.,1997). This resulted in more reciprocal excitatory synaptic connec-tions between CA3 cells, and led to hyperexcitability of CA3 neu-rons 14 days following transection (McKinney et al., 1997). Recentevidence has demonstrated that BDNF expression and TrkB expres-sion are upregulated 72 h after Schaffer collateral injury (Dinocourtet al., 2006; Aungst et al., 2013). Moreover, conditional partialknockdown of the BDNF receptor, TrkB, can reduce CA3 axonalsprouting after Schaffer collateral lesion (Dinocourt et al., 2006).However, it remains to be determined whether inhibiting thisBDNF-mediated axonal sprouting can attenuate the hyperexcitability
Correspondence: Dr Rebecca A. McKinney, 1Department of Pharmacology & Thera-peutics, as above.E-mail: [email protected]
Received 21 October 2011, revised 16 August 2013, accepted 28 August 2013
© 2013 Federation of European Neuroscience Societies and John Wiley & Sons Ltd
European Journal of Neuroscience, pp. 1–13, 2013 doi:10.1111/ejn.12367
European Journal of Neuroscience

of CA3 neurons. It is also unknown how this axonal sprouting func-tionally affects the hippocampal network within area CA3. More-over, the hippocampal localisation of BDNF and the time course ofits expression after Schaffer collateral lesion have not been estab-lished.Therefore, we hypothesised that BDNF initiates injury-induced
axonal remodeling and hyperexcitability, leading to changes innetwork activity. To test our hypothesis, we transected the Schaffercollateral pathway of organotypic hippocampal slice preparations.Following this, we immunostained for growth-associated protein of43 kDa (GAP43), a known marker for axonal growth, to determinethe extent of axonal sprouting. Expression levels of BDNF andTrkB were measured with quantitative polymerase chain reaction(qPCR) and western blot analysis. Finally, we used electrophysiol-ogy to determine the functional effect of the aberrant sprouting inarea CA3.Here, we demonstrate that transection of the Schaffer collateral
pathway can upregulate BDNF to induce axonal sprouting and hype-rexcitability of hippocampal CA3 neurons, leading to an increase innetwork activity within area CA3.
Materials and methods
Ethics statement
All animal handling procedures were carried out in accordance withguidelines set by the Canadian Council on Animal Care and theNational Institutes of Health in the USA. All procedures wereapproved by the Animal Resource Committee of the School ofMedicine at McGill University, and are outlined in McGill Univer-sity Animal Handling Protocol #5057.
Hippocampal slice cultures and Schaffer collateral lesions
We chose to study the hippocampus because it possesses a uniqueunidirectional network that is preserved within the organotypicculture system (G€ahwiler et al., 1997), making it an ideal candidatefor the study of microcircuitry remodeling. Organotypic hippocam-pal slices were prepared with the roller-tube method, as previouslydescribed (G€ahwiler, 1981; G€ahwiler et al., 1997). Briefly, 7-day-old C57B6 mice were decapitated, and a bilateral hippocampaldissection was performed, from which 400-lm-thick transverse hip-pocampal slices were made and plated on glass coverslips. Sliceswere fastened in place with a clot of chicken plasma (Cocalico Bio-logicals, Reamstown, PA, USA) coagulated with thrombin (Invitro-gen Gibco). Coverslips were placed in flat-sided culture tubeswith antibiotic-free serum-containing medium, and maintained in adry-air roller drum incubator at 36 °C for 3 weeks prior to experi-mentation. To prevent excitotoxic injury during lesioning, slices[> 21 days in vitro (DIV)] were placed into a cutting solution con-taining 0.5 lM tetrodotoxin (Alomone Labs, Jerusalem, Israel) and10 mM MgCl2. Lesions were made to the Schaffer collateralsbetween area CA3 and area CA1 with a razor blade shard; cutsextended from the alveus layer to the stratum radiatum. Control sis-ter slices were also exposed to the cutting medium for the same per-iod of time, but not lesioned. Slices were then returned to normalculture medium and processed simultaneously in all experiments.
Visualisation of CA3 pyramidal neurons
Farnesylated mCherry protein was expressed specifically in CA3neurons by infecting control and 14 days post-lesion (DPL) organo-
typic hippocampal slices with Semliki Forest virus strain PD, whichselectively targets neurons (Lundstrom & Ehrengruber, 2003; Lund-strom et al., 2003). The infection was achieved by microinjectionwith a picospritzer (Picospritzer III; Parker, Cleveland, OH, USA) of1–2 lL of virus via a glass pipette into area CA3 with 5–10 lb/in2
and 25–50-ms pulses at 0.1 Hz for 15–20 min. Approximately 16–20 h after infection, the slices were fixed in 4% paraformaldehyde for1 h at room temperature, and mounted onto microscope slides forconfocal imaging.
Electrophysiological recordings and analysis
Slices were placed in a temperature-controlled chamber (30 °C)mounted on an upright microscope (DM LFSA; Leica Microsys-tems), and continuously perfused with Tyrode solution containing:137 mM NaCl, 2.7 mM KCl, 2.5 mM CaCl2, 2 mM MgCl2, 11.6 mM
NaHCO3, 0.4 mM NaH2PO4, and 5.6 mM glucose (pH 7.4). Toassess excitability, traces were recorded from CA3 neurons (twocells per slice) in current-clamp mode. Whole-cell recording elec-trodes were pulled from borosilicate glass (resistance of 4–6 MΩ;GC150TC; Clark Instruments, UK) with a P-97 electrode puller(Stutter Instruments, Novato, CA, USA). All electrophysiologicalrecordings were made with an Axopatch 200A amplifier (MolecularDevices, Sunnyvale, CA, USA). Signals were recorded at 20 kHz,and then filtered through a Bessel low-pass filter at 2 kHz with theCLAMPEX 9.2 acquisition program (Axon Instruments). Recordingpipettes were filled with an intracellular solution containing:120 mM potassium gluconate, 1 mM EGTA, 10 mM HEPES, 5 mM
Mg-ATP, 0.5 mM Na-GTP, 5 mM NaCl, 5 mM KCl, and 10 mM
phosphocreatine (pH adjusted to 7.3 with KOH; 285–295 mOsm).CA3 neurons were chosen at ~ 300 lm from the site of lesion inthe case of transected slices. Once whole-cell configuration wasestablished, cells were monitored for 5 min for equilibration of theinternal solution and to ensure that the seal and opening were main-tained; cells were then recorded for 20 min. Input resistance andmembrane potential were monitored at regular intervals throughoutthe experiment, and no differences were noted between conditions.Data were discarded if recordings in cells showed a seal resistanceof < 5 GΩ or a holding current of more than –100 pA, or if resis-tance deviated by more than � 20% through the course of record-ing. Also, cells that did not survive the entire 20 min of recordingwere excluded from further analysis. The digitised data were codedprior to analysis, so that analysis could be carried out blinded.Upward deflecting excitatory postsynaptic potentials (EPSPs)> 40 mV in amplitude were considered to be somatic action poten-tials (APs), and were detected offline. The AP threshold was identi-fied as the membrane potential at the start point of the first AP.Three or more APs were grouped into bursts if the inter-spike inter-val was < 600 ms. For spontaneous EPSP frequency analysis, tracesshowing typical EPSP upward deflections were considered and ana-lysed as EPSPs if they were > 3 mV in size. APs were not takeninto account in EPSP analysis.To assess network activity, evoked field EPSPs (fEPSPs) were
recorded in area CA3 with the recording electrode placed in the stra-tum oriens at 30 °C. Field recording pipettes were pulled to0.5 MΩ, and filled with Tyrode’s solution. The extracellular solutionalso consisted of Tyrode’s solution without the addition of any phar-macological agents. CA3 axonal stimulation was achieved with aninsulated platinum–iridium bipolar electrode [diameter, 50 lm; 25-lm layer of Teflon insulation (A-M Systems, Carlsborg, WA,USA)]. Stimulus strength ranged from 20 to 200 lA to elicit a mini-mum response, and a single stimulus was delivered every 20 s.
© 2013 Federation of European Neuroscience Societies and John Wiley & Sons LtdEuropean Journal of Neuroscience, 1–13
2 R. Gill et al.

Stimulus strength was increased every five stimuli by 10% to amaximum of 300% of the minimum response. The signal was low-pass filtered at 2 kHz and recorded at 20 kHz with PCLAMP 9.2 soft-ware (Axon Instruments). The digitised data were coded prior toanalysis, so that analysis could be carried out blinded. fEPSPs weredetected offline, and analysed for compound responses. Com-pounded responses were considered to be any fEPSP responses thatincluded two or more downward deflections following the deliveryof a stimulus before reaching maximum amplitude. The number ofpeaks during the rise phase were calculated and compared with cor-responding stimulation strengths.
Immunohistochemistry
Slices were fixed at 4 °C overnight in 4% paraformaldehyde in0.1 M phosphate buffer (PB) (pH 7.4). Following fixation, sliceswere washed in 0.1 M PB, permeabilised in 0.4% Triton X-100, andblocked with 1.5% heat-inactivated horse serum overnight at 4 °C.Primary antibodies were incubated for 5 days at 4 °C in permeabi-lising buffer with a 1 : 20 dilution of GAP43 mouse antiserum(clone 10E8/E7, provided by K. Meiri, Tufts University, Boston,MA, USA; Meiri et al., 1986), or a 1 : 250 dilution of anti-phos-phorylated TrkB (pTrkB; Y515; Abcam) or a 1 : 250 dilution ofanti-postsynaptic density protein 95 (PSD95; Millipore). Followingseveral washes in 0.1 M PB, slices were incubated overnight at 4 °Cin a 1 : 250 dilution of secondary Alexa Fluor 594-conjugated anti-mouse IgG1 (Invitrogen Molecular Probes) diluted in 0.1 M PB con-taining 1.5% heat-inactivated horse serum. Finally, slices weremounted with Dako Fluorescent Mounting medium (Dako Canada,Mississauga, Canada) onto microscope slides. Following mounting,slices were imaged in area CA3 with a Leica TCS SP2 scanhead(Leica Microsystems) on a Leica DM6000 B upright microscope,equipped with an HCX PL APO 9 40 NA 1.4 oil immersion objec-tive, with a 543-nm HeNe laser line. Image stacks were collected atZ = 0.4 lm, and averaged four times to improve the signal-to-noiseratio. For quantification of the presence of GAP43 fibers, imagestacks were obtained with identical parameters (laser intensity,filters, pinhole size, and photomultiplier tube gain and offset). Rep-resentative images are maximum intensity projections of five sec-tions from each stack.
Reverse Transcription-qPCR
Slices were lesioned in cutting solution, and areas CA1 and CA3 weremicrodissected, snap frozen on dry ice, and preserved in RNAlater(Ambion, Austin, TX, USA) until experimentation. Control sliceswere exposed to cutting medium, and were maintained for 6 h prior tomicrodissection. Five or six slices were used for each experimentalgroup. Each condition was tested with three biological samples in trip-licate. Total RNA was extracted with TRIzol reagent (Invitrogen), and1 lg of RNA was reverse transcribed to cDNA with 20 U of avianmyeloblastosis virus reverse transcriptase (Roche Diagnostics),according to the manufacturer’s recommendation. Subsequently,qPCRs were performed with 2 lL of cDNA in a 20-lL reaction mix-ture containing SYBR green mastermix (Roche Diagnostics), and0.5 lM of forward and reverse primers (all primers were obtainedfrom Invitrogen). Reactions were performed with a Roche LightCy-cler LC480 under the following conditions: denaturation for 10 minat 95 °C, 45 steps of denaturation at 95 °C for 10 s, annealing for10 s, and extension at 72 °C for 10 s, and finally extension at 72 °Cfor 10 min. The following primers and annealing temperatures wereused for experimentation: BDNF targeting all mRNA transcript vari-
ants, sense 5′-GAAGAGCTGCTGGATGAGGAC-3′ and antisense5′-TTCAG TTGGCCTTTTGATACC-3′, annealing temperature59 °C; TrkB targeting the transcript for the full-length receptor, sense5′-TGATGGC AGAGGGTAACC-3′ and antisense 5′-CTAC-TATCGGGTCGGTG GC-3′, annealing temperature 57 °C; glyceral-dehyde-3-phosphate dehydrogenase, sense 5′-AAATGGTGAAGGTCGGTGTG-3′ and antisense 5′-TGAAGGGGTCGTTGATGG-3′,annealing temperature 59 °C; and b-actin, sense 5′-TGGTGGGTATGGGTCAGAAGGAC TC-3′ and antisense 5′-CATGGCTGGGGTGTTGAAGGTCTCA-3′, annealing temperature 59 °C. Reactionefficiencies for each assay were calculated from serial dilutions ofcDNA. All qPCR data were quantified and normalised with RocheLightCycler 480 software.
Western blotting
Ten control or 10 lesioned slices were grouped together for eachparadigm (defined as one sample group). Slices were freed from theplasma clot, pooled, and lysed in ice-cold RIPA buffer [1% NonidetP-40, 0.5% sodium deoxycholate, 0.1% sodium dodecylsulfate,50 mM Tris–HCl (pH 8.0), 150 mM NaCl] and completeMini prote-ase inhibitors (Roche). The protein concentration was determinedwith a bicinchoninic acid dye-binding assay (Thompson Scientific),according to the manufacturer’s protocol, with bovine serum albu-min (BSA) as a standard. Twenty-five micrograms of total proteinwas separated by sodium dodecylsulfate polyacrylamide gel electro-phoresis (10 or 15% resolving) under reducing conditions, andwet-transferred onto 0.22-lm-pore poly(vinylidene difluoride) mem-branes (Millipore). Membranes were blocked for 1 h at roomtemperature with 5% BSA in Tris-buffered saline with 0.05%Tween-20 (TBST). Membranes were then incubated at 4 °C over-night with anti-BDNF (Santa Cruz Biotechnology; 1 : 500), anti-TrkB (BD Falcon; 1 : 1000) or anti-b-tubulin (Sigma-Aldrich;1 : 3000; internal loading control) diluted in 5% BSA in TBST. Pri-mary antibodies were revealed with horseradish peroxidase-conju-gated secondary antibodies (1 : 50 000; Bio-Rad Laboratories)diluted in 5% BSA in TBST for 1 h at room temperature. Immuno-reactive bands were detected with enhanced chemiluminescence (GEHealthcare), according to the manufacturer’s protocol, and revealedwith Kodak BioMax Light Film (Sigma-Aldrich). Blots were ana-lysed with ADOBE PHOTOSHOP software for mean pixel intensity.
Immunoprecipitation
To detect the phosphorylation of TrkB, immunoprecipitation wascarried out on 10 pooled control or 3 hours post-lesion (HPL) sliceswith 500 lL of ice-cold RIPA buffer containing completeMini prote-ase and phosphatase inhibitors (Roche). Lysates were incubated over-night at 4 °C with 1 lg of primary anti-pTrkB/tropomyosin-relatedkinase A antibody (pY515; Cell Signaling, Beverly, MA, USA). Fiftymicroliters of Protein G–agarose beads (Thermo Scientific) was addedto the lysates and incubated for 90 min at 4 °C. After three washeswith 0.1 M PB, the beads were spun down and boiled at 95 °C for5 min in 2 9 Laemmli buffer (Bio-Rad Laboratories) containing b-mercaptoethanol. Proteins were separated by sodium dodecylsulfatepolyacrylamide gel electrophoresis and analysed by western blotting.TrkB expression was assessed with anti-TrkB (BD Falcon; 1 : 1000).
Pharmacological treatments
To scavenge BDNF, slices were treated with TrkB-Fc (R&DSystems, Minneapolis, MN, USA), a fusion protein comprising the
© 2013 Federation of European Neuroscience Societies and John Wiley & Sons LtdEuropean Journal of Neuroscience, 1–13
BDNF and injury-induced hyperexcitability 3

Fc domain of human IgG1 and the ectodomain of TrkB. As a con-trol, we treated sister lesioned slices with IgG-Fc. We found thatTrkB-Fc treatment of hippocampal slices for 24 h downregulatedTrkB receptor phosphorylation (data not shown). TrkB-Fc or IgG-Fcwas diluted in culture medium at a final concentration of 10 lg/mL,and chronic treatment began 12 h following Schaffer collateraltransection of slices maintained in vitro for > 21 days. Control cul-ture medium or drug-containing medium was replaced three times aweek for 1–3 weeks following transection.
Statistical analysis
Student’s t-test (with Bonferroni correction when necessary), Fish-er’s exact test or one-way ANOVA with a post hoc Tukey test wasused when appropriate. In cases where data were not normally dis-tributed (tested with Lilliefors), a Kruskal–Wallis test with a posthoc Mann–Whitney U-test was utilised, unless otherwise specified.Results are expressed as mean � standard error of the mean. ForFigs 2B and 6B, linear regression was performed for each slice, andslopes were then compared by use of one-way ANOVA with a posthoc Tukey test.
Results
Schaffer collateral transection in mouse organotypic slicesleads to axonal sprouting and hyperexcitability 14 days post-lesion
We wished to determine the role of BDNF and TrkB in injury-induced hyperexcitability. Previously, we studied axonal sproutingby transecting the Schaffer collateral pathway in mature rat hippo-campal slice cultures (McKinney et al., 1997). Here, we sought toconsolidate our findings in a mouse organotypic hippocampalsystem, and re-establish the timeframe for axonal sprouting.Initially, we tested whether a similar outcome and timeframe
could be observed following lesion to Schaffer collaterals in mouseorganotypic hippocampal slices. Three days after transection of theSchaffer collateral pathway, we found a small gap between areaCA3 and area CA1 (Fig. S1A), isolating the two areas. Similar toour previous observations in rat slices (McKinney et al., 1997,1999), we detected a few propidium iodide-positive cells close to
the lesion site in area CA3 and area CA1 (data not shown), indicat-ing selective cell death close to the lesion site.However, what effect did this lesion have on axonal morphology
and reorganisation of axonal fibers? 14 days after transection, wemicroinjected a virus expressing an mCherry plasmid into area CA3,which transfected the mCherry plasmid into a pyramidal cell situatedclose to the lesion site (> 500 lm). We found that the typical
A B
C
F
E
G
H I
D
Fig. 1. Schaffer collateral lesion induced axonal sprouting and hyperexcit-ability at 14 DPL in area CA3. (A) A CA3 neuron, visualised with virustransfection of an mCherry plasmid in a 14 DPL preparation. (B) Expandedinset of A, showing axons regrowing towards the lesion site. (C) The numberof GAP43-immunopositive fibers was reduced in mature slices (> 21 DIV).(D) When mature hippocampal cultures were lesioned, GAP43-immunoposi-tive fibers were observed in area CA3 adjacent to the lesion site at 14 DPL.C and D are representative images of maximum intensity projectionsrendered from confocal stacks of slices immunohistochemically stained forGAP43, and all images were taken in area CA3 (n = 10 slices for each para-digm). Note: dotted lines indicate the sites of lesion; the scale bar for A, Cand D (shown in C) is 50 lm, and the scale bar in B is 20 lm. (E) Repre-sentative traces of whole-cell current-clamp electrophysiological recordingsfrom CA3 pyramidal neurons. (F) Quantification of AP frequency. We founda significant increase in AP firing at 14 DPL as compared with control slices.(G) Analysis of burst frequency. We detected a significant increase in burstfrequency in 14 DPL slices as compared with control slices. Note: for F andG, *P < 0.05, two-tailed Student’s t-test; data are depicted as mean � stan-dard error of the mean. (H) Percentage of CA3 neurons showing spontaneousAPs. (I) Percentage of cells showing spontaneous bursting. Note: for H andI, **P < 0.01, two-tailed Fisher’s exact test.
© 2013 Federation of European Neuroscience Societies and John Wiley & Sons LtdEuropean Journal of Neuroscience, 1–13
4 R. Gill et al.

Schaffer collateral morphology, a parallel bundle of axonal fibers,was disrupted, and the axonal processes were undulating and twist-ing, seeming to lack a primary direction of growth (Fig. 1A and B).To determine whether this disrupted morphology was caused bynewly sprouted axons, we performed immunofluorescence stainingfor GAP43, an established marker for axonal growth (Meiri et al.,1986). GAP43 is upregulated during development, when there is ahigh level of axonal growth, but it is downregulated when an animalmatures to adulthood; we found that, in hippocampal slices, GAP43was downregulated at 21 DIV (Fig. S1B and Fig. 1C), the timepoint at which we made Schaffer collateral lesions. We found that,14 days following lesion of Schaffer collaterals, at a time pointwhen GAP43 is normally downregulated (Fig. 1C), a number offibers were GAP43-immunoreactive immediately adjacent to thelesion site in area CA3 (Fig. 1D) but not in area CA1. The GAP43-immunopositive fibers were visible for 327.2 � 12.8 lm (n = 24slices) away from the lesion site into area CA3. Hence, lesion of theSchaffer collateral pathway leads to the upregulation of GAP43 andaxonal sprouting of CA3 pyramidal neurons. However, does theobserved axonal sprouting in mouse hippocampal slices result inhyperexcitability of CA3 neurons, as seen after rat hippocampalculture transections?To determine whether a functional change accompanied the
morphological changes, we measured spontaneous activity withwhole-cell current-clamp recordings from CA3 pyramidal neurons ineither control or 14 DPL slices. We found that lesioning markedlyincreased excitation, with increased AP firing (Fig. 1E and F;3.95 � 1.17 APs/min; n = 20 cells) as compared with control slices(0.40 � 0.16 APs/min; n = 14 cells). Moreover, there was a signifi-cant increase in the average number of bursts (defined as three ormore consecutive spikes with inter-spike intervals of < 600 ms) in14 DPL slices (Fig. 1G; 0.56 � 0.16 bursts/min; n = 21 cells) ascompared with control slices (0.094 � 0.034 bursts/min; n = 14cells). In addition, we found that 90% of 14 DPL slices (n = 18 of20 cells) showed spontaneous APs, whereas 42.9% of control slicesfired spontaneously (Fig. 1H; n = 6 of 14 cells). Furthermore, 75%of CA3 neurons in 14 DPL slices (n = 21 of 28 cells) showed burst-ing, as compared with only 31.6% of control CA3 neurons (Fig. 1I;n = 6 of 19 cells). Therefore, at 14 DPL, CA3 neurons are morelikely to have spontaneous hyperexcitability than CA3 neurons fromcontrol cultures.Consistent with our published data in rats, these results show that
at 14 days after Schaffer collateral transection in mouse organotypichippocampal cultures, we consistently observe GAP43 labeling ofde novo axons and an increase in excitation, which confirmed thatthis is a suitable model to address the mechanisms of reactive axo-nal sprouting-induced hyperexcitability. Since we observed anincrease in hyperexcitability, we then sought to determine why theCA3 neurons were more excitable.
Intrinsic electrical properties of CA3 neurons remainunchanged after Schaffer collateral transection
Next, we evaluated the intrinsic electrical properties of CA3 pyrami-dal cells, as we had observed an increase in the firing and burstingof these neurons. We found that the input resistance of CA3 pyrami-dal cells was unchanged in 14 DPL slices (124.9 � 20.9 MΩ;n = 16) as compared with control slices (126 � 28.8 MΩ; n = 12).We then investigated the resting membrane potentials of these neu-rons, and we found they were also unchanged in 14 DPL slices(–71.8 � 0.48 mV; n = 16) as compared with control cultures(�69.7 � 1.4 mV; n = 12), and were also within the normal range
of CA3 pyramidal neurons (Anderson et al., 2007). Finally, wetested the input–output response of CA3 pyramidal cells by perform-ing current step experiments. We found no change in the number ofAPs fired in response to various injected currents; some examplesare shown in Table S1. Therefore, we concluded that Schaffer col-lateral lesion did not affect the intrinsic electrical properties of CA3pyramidal neurons at 14 DPL.Our next objective was to determine whether there were any
changes in GABA production, as an increase in hyperexcitabilitycould be related to changes in inhibitory GABAergic transmission.Therefore, we immunostained for glutamate decarboxylase 67(GAD67) to determine whether GABA production was increased at14 DPL, the time when we had seen the most robust axonal sprout-ing. We found that GAD67 staining was similar to that in controlslices (Fig. S2), indicating that there was no change in the produc-tion of GABA post-lesion.
Hyperexcitability of CA3 neurons after Schaffer collateraltransection can be attributed to increased excitatory inputs
As we did not observe any changes in the intrinsic electrical proper-ties of CA3 pyramidal neurons or in GAD67 staining at 14 DPL,we performed evoked fEPSP recordings to further characterise net-work activity in area CA3. We stimulated axons in area CA3 closerto the dentate gyrus, and recorded potentials in area CA3 near thelesion site. We observed that there was an increase in compoundedfEPSPs (defined as responses that included two or more downwarddeflections following stimulus before reaching maximum amplitude)in relation to the stimulus strength in area CA3 of 14 DPL slices ascompared with control slices (Fig. 2A). We found that, with thesame stimulus intensity as used in control slices, we were able torecruit more fibers in 14 DPL slices (Fig. 2A and B). Moreover,overall, we found an increase in the percentage of slices that showedcompounded fEPSPs at any stimulus intensity in 14 DPL slices(100%; n = 10; Fig. 2C) as compared with control slices (50%;n = 8; Fig. 2C). This indicates that there are more connections,probably because of the formation of a recurrent network.Our next objective was to determine what cellular signal could be
causing these axons to sprout and form new connections. As recentevidence has suggested that BDNF is important for injury-inducedaxonal sprouting following Schaffer collateral transection (Dinocourtet al., 2006; Aungst et al., 2013), we wished to determine whenBDNF was upregulated following injury.
BDNF transcript and protein levels increase after Schaffercollateral lesion
In order to see area-specific changes in BDNF expression, weused qPCR to quantify bdnf transcripts shortly after injury. Weobserved a two-fold increase in bdnf mRNA expression, whichwas observed in area CA3 but not in area CA1 (Fig. 3A). Fur-thermore, this two-fold increase was sustained until 4 HPL, andexpression was then downregulated to control levels by 6 HPL(Fig. 3A). Conversely, full-length trkb transcript expression didnot vary in either area CA3 or area CA1 up to 6 HPL (Fig. 3B).As we detected a transient change in expression of bdnf mRNA,we also wanted to know whether this resulted in changes inBDNF levels post-lesion. Using western blot analysis, we foundan increase in BDNF expression at 24 HPL, but no change inTrkB expression (Fig. 3C). As we did not observe a change inTrkB expression, we next determined whether BDNF was activat-ing TrkB receptors by probing for autophosphorylation of TrkB at
© 2013 Federation of European Neuroscience Societies and John Wiley & Sons LtdEuropean Journal of Neuroscience, 1–13
BDNF and injury-induced hyperexcitability 5

tyrosine 515 (Y515), a known site of TrkB autophosphorylationfollowing BDNF binding to TrkB. By performing a western blot,we found an increase in pTrkB(Y515) as compared with controlslices at 3 HPL (Fig. 3D). Immunofluorescence revealed a two-fold increase in pTrkB(Y515; Fig. 3E and F), which was mostlylocated adjacent to the lesion site (Fig. 3E), similar to the distribu-tion of GAP43 immunoreactivity at 14 DPL.
Taken together, our data show that lesioning of Schaffer collater-als results in elevated BDNF and hyperexcitability. Next, we investi-gated whether BDNF is necessary for injury-induced sprouting.
BDNF inhibition downregulates axonal sprouting followinginjury
To determine whether BDNF is necessary for sprouting after Schaf-fer collateral lesion, we blocked BDNF signaling by chronicallytreating lesioned slices with TrkB-Fc, a BDNF scavenger, for 14DPL, and assessed axonal sprouting with GAP43 immunofluores-cence. We found that 31.3% (n = 5 of 16) of TrkB-Fc-treated 14DPL slices had GAP43-immunoreactive fibers in area CA3(Fig. 4Dii and E), whereas 88.9% (n = 24 of 27) of untreated 14DPL slices had GAP43-immunoreactive fibers in area CA3(Fig. 4Cii and E). Moreover, TrkB-Fc-treated lesioned slices weresimilar to control sister slices (Fig. 4Aii and E). Our results showthat blocking BDNF reduced axonal sprouting following transectionof Schaffer collaterals.However, it is possible that the TrkB-Fc treatment either acceler-
ated or simply delayed the axonal sprouting. Therefore, we investi-gated GAP43 expression at 7 and 21 DPL. When we treatedlesioned slices for 7 days with TrkB-Fc, we found that no slicesshowed GAP43 immunoreactivity (n = 5), in comparison with37.5% of untreated 7 DPL slices (n = 8; Fig. 4Ci, Di, and E).TrkB-Fc-treated 7 DPL slices were similar to control sister slices(Fig. 4Ai, Di, and E). Moreover, lesioned slices that were treatedwith TrkB-Fc for 21 days also did not show any GAP43 immunore-activity (0%; n = 6; Fig. 4Diii and E), and were similar to controlslices (0%; n = 5; Fig. 4Aiii and E). However, 50.3% of 21 DPLslices showed GAP43-immunopositive fibers (n = 8; Fig. 4Ciii andE) when GAP43 expression was downregulated, indicating that theconnections were maturing. Finally, when we returned 14 DPLslices treated with TrkB-Fc-containing medium to control culturemedium for an additional week, we did not detect GAP43-immuno-positive sprouting (data not shown). This indicates that there is anearly time window during which BDNF can induce axonal sprout-ing, and that, after this window is closed, axonal sprouting does notoccur.From these findings, we conclude that BDNF is necessary for
lesion-induced sprouting, but is BDNF-induced axonal sproutingrequired for injury-induced hyperexcitability of CA3 pyramidalneurons?
BDNF inhibition blocks the development of hyperexcitability at14 DPL
To determine whether BDNF-induced axonal sprouting is necessaryfor hyperexcitability, we recorded spontaneous AP firing from CA3cells. We found that 75% of lesioned slices (Fig. 5A and B; n = 21of 28 cells) showed spontaneous APs at 14 DPL, which led to anincrease in firing rate (Fig. 5C; 4.11 � 1.07 APs/min; n = 28 cells).This was attenuated in TrkB-Fc-treated lesioned slices (Fig. 5A andB; 44%; n = 11 of 25 cells), which showed significantly fewer APs(Fig. 5A and C; 1.22 � 0.48 APs/min; n = 25 cells). Treatedlesioned slices were similar to control slices (Fig. 5A and B; 40%;n = 8 of 20 cells), with a spontaneous firing frequency of0.46 � 0.16 APs/min (Fig. 5C; n = 20 cells). TrkB-Fc treatmentdid not affect control slices, as only 40.9% (Fig. 5A and B; n = 9of 22 cells) of TrkB-Fc-treated control slices showed spontaneousfiring (Fig. 5C; 0.68 � 0.28 APs/min; n = 22 cells). To ensure thatthe Fc fragment was not itself affecting the functionality of the neu-
A
B
C
Fig. 2. Axonal sprouting leads to an increase in area CA3 network activity.(A) Representative traces of evoked fEPSP recordings within area CA3 for100, 150 and 200% of minimal stimulation. Note: the arrows indicate addi-tional peaks in compounded fEPSP responses. (B) Quantification of the num-ber of peaks as a function of stimulus strength. There is a significantincrease in the number of compounded fEPSPs with an incremental increasein stimulus strength in 14 DPL slices as compared with control slices(*P < 0.05; linear regression was performed for each slice, and the twoslopes were then compared by use of an independent Student’s t-test; wethen performed pairwise comparisons for individual data points with anunprotected independent Student’s t-test). (C) Quantification of percentage ofslices showing compounded fEPSPs (*P < 0.05; Fisher’s exact probabilitytest, two-tailed).
© 2013 Federation of European Neuroscience Societies and John Wiley & Sons LtdEuropean Journal of Neuroscience, 1–13
6 R. Gill et al.

rons, we treated lesioned sister slices with IgG-Fc, and found thatthose slices were similar to untreated lesioned slices, as 87.5%(Fig. 5A and B; n = 14 of 16 cells) fired spontaneously (Fig. 5C;5.44 � 2.64 APs/min; n = 16 cells).When we quantified the burst frequency (three or more spikes
were grouped into bursts if the inter-spike interval was ≤ 600 ms),we found that 67.9% (Fig. 5A and D; n = 19 of 28 cells) of 14DPL slices showed spontaneous bursting (Fig. 5E; 0.51 � 0.13bursts/min; n = 28 cells). This increase in excitability was signifi-cantly attenuated by TrkB-Fc treatment, when 28% of treatedlesioned slices (Fig. 5A and D; n = 7 of 25 cells) burst at afrequency of 0.10 � 0.04 bursts/min (Fig. 5E; n = 25 cells). Theseslices were similar to control slices; 20% (Fig. 5A and D; n = 4 of20 cells) of sister control slices showed spontaneous bursting(Fig. 5E; 0.06 � 0.02 bursts/min; n = 20 cells). We observed spon-taneous bursting in 31.8% (Fig. 5A and D; n = 7 of 22 cells) ofTrkB-Fc-treated control slices (Fig. 5E; 0.10 � 0.04 bursts/min;n = 22 cells). Finally, IgG-Fc-treated lesioned slices were similar tountreated lesioned slices, as 50% (Fig. 5A and D; n = 8 of 16 cells)of cells showed spontaneous bursting at a frequency of 0.24 � 0.07bursts/min (Fig. 5E; n = 16 cells).When the inter-spike intervals were plotted as a cumulative proba-
bility distribution, we found a leftward shift in the distribution of 14DPL and IgG-Fc-treated lesioned slices (Fig. 5F), whereas TrkB-Fc-treated lesioned slices were similar to control and TrkB-Fc-treated
control slices (Fig. 5F). This indicated that the inter-spike intervalswere much shorter in 14 DPL and IgG-Fc-treated lesioned slicesthan in other conditions.This evidence strongly indicates that BDNF blockade attenuates
the Schaffer collateral injury-induced hyperexcitability of individualCA3 neurons. Next, we wished to determine whether BDNF block-ade could inhibit the formation of a recurrent network.
Chronic treatment with TrkB-Fc for 14 DPL inhibits lesion-induced hyperexcitable network activity in area CA3
To determine whether BDNF was important for sprouting-inducednetwork activity, we carried out evoked fEPSP recordings. Wefound that there was an increase in the number of compoundedfEPSPs in relation to the stimulus strength in area CA3 of 14 DPLslices as compared with control slices, and that this was attenuatedby TrkB-Fc treatment (Fig. 6A and B). To control for non-specificeffects of the Fc fragment, we treated lesioned sister slices withIgG-Fc, and found no difference from untreated lesioned slices(Fig. 6A and B). Moreover, unlesioned TrkB-Fc-treated slicesbehaved like control slices (Fig. 6A and B). Control slices typicallyshowed non-compounded fEPSPs at any given moment (Fig. 6A),with only 50% of slices showing compounded fEPSPs (Fig. 6C;n = 8); this was similar to what was found in TrkB-Fc-treated con-trol slices (Fig. 6B and C; 62.5%; n = 8). However, we detected
A
E F
B C D
Fig. 3. BDNF expression was upregulated at 2 HPL. (A) Region-specific mRNA expression of BDNF at 2, 4 and 6 HPL. (*P < 0.05, two-tailed Student’s t-test with Bonferroni correction). (B) Full-length TrkB transcript expression at 2, 4 and 6 HPL. Note: for A and B, three separate experiments with five or sixpooled slices were performed for each group; b-actin was used as a reference gene; error bars are � standard error of the mean; Ctrl, control. (C) Western blotof TrkB and BDNF protein expression at 24 HPL. (D) Western blot of TrkB after immunoprecipitation with a pTrk antibody (Y515; an autophosphorylation sitefor dimerisation) at 3 HPL. Note: b-tubulin was used as a loading control for C and D, and all western blots were performed in three separate experiments with10 pooled slices for each group. (E) The increase in pTrkB(Y515) was especially prominent in area CA3 at 3 HPL, and many pTrkB puncta co-localised withPSD95, a marker for excitatory postsynaptic terminals (scale bar: 5 lm). Note: representative images are maximum intensity projections rendered from confocalstacks immunostained for pTrkB and PSD95. In the merge panel, pTrkB is represented by red, PSD95 by green, and co-localisation by yellow. (F) Quantifica-tion of fluorescence intensity normalised to control (**P < 0.01, two-tailed Student’s t-test). A.U., arbitrary units.
© 2013 Federation of European Neuroscience Societies and John Wiley & Sons LtdEuropean Journal of Neuroscience, 1–13
BDNF and injury-induced hyperexcitability 7

compounded fEPSPs in 100% of 14 DPL slices (Fig. 6B and C;n = 11) and in 100% of IgG-Fc-treated lesioned slices (Fig. 6B andC; n = 9), indicating that there were increased excitatory inputs
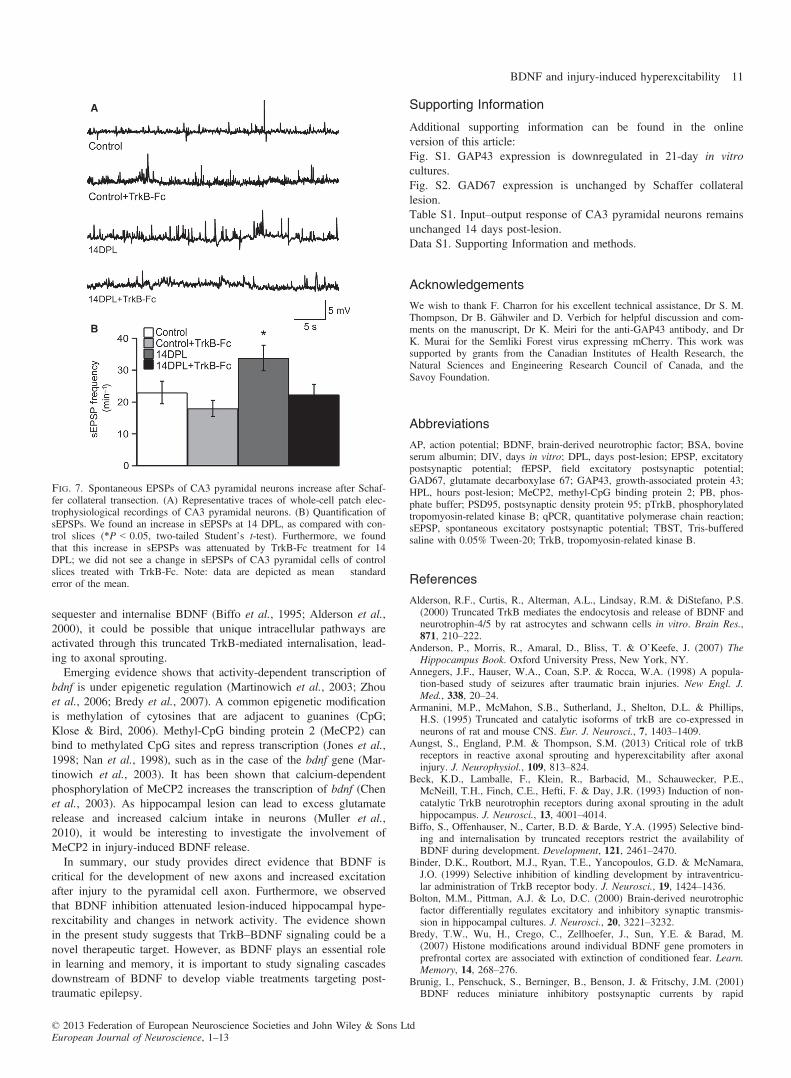
within area CA3 after lesioning. Interestingly, TrkB-Fc-treatedlesioned slices were similar to control slices (Fig. 6B and C; 30%;n = 10). In addition, we quantified individual spontaneous EPSPs(sEPSPs), and found an increase in the number of sEPSPs in 14DPL slices (Fig. 7A and B; 33.84 � 4.02; n = 26) as comparedwith control slices (Fig. 7A and B; 22.98 � 3.56; n = 20); thisincrease was blocked by TrkB-Fc treatment (Fig. 7A and B;22.33 � 3.23; n = 24). We did not see a change in sEPSPs of CA3pyramidal cells of control slices treated with TrkB-Fc (Fig. 7A andB; 18.0 � 2.53; n = 22). Therefore, our data demonstrate thatlesion-induced hyperexcitability and increased network activity areattributable to BDNF-induced axonal sprouting.
Discussion
Here, we demonstrate that following Schaffer collateral transectionin mouse organotypic hippocampal slices, there is an increase in thenumber of GAP43-immunopositive axon collaterals specific to areaCA3 close to the lesion site, which coincides with an increase in theextrinsic excitability of CA3 pyramidal neurons coupled with anincrease in network activity. Pharmacological inhibition of BDNFattenuated the observed morphological and functional changes.Furthermore, we show that BDNF is increased in a tight therapeutictime window and activates TrkB receptors, which we measured viaphosphorylation. Therefore, we show that BDNF is necessary forlesion-mediated axonal sprouting and the subsequent increase inexcitation.
Time course of BDNF expression following injury
Our results also reveal the timeframe for bdnf mRNA upregulationfollowing Schaffer collateral transection in area CA3. We found thattransection of Schaffer collaterals led to a two-fold change in bdnfmRNA transcripts at 2 and 4 HPL, which was specific to area CA3.Interestingly, no changes were observed in area CA1, consistentwith the observation that GAP43 immunoreactivity was notobserved in area CA1 at any of the time points tested. Moreover,the post-lesion change in bdnf mRNA expression in area CA3 led toan increase in BDNF expression at 24 HPL, consistent with the pre-vious finding that BDNF expression was increased at 24 and 48 hafter Schaffer collateral transection (Dinocourt et al., 2006). Interest-ingly, the BDNF increase immediately preceded GAP43 upregula-tion and axonal sprouting. Moreover, upregulation of BDNF in thehippocampus over a similar timeframe has been reported with invivo animal models of brain injury (Mudo et al., 1993; Hicks et al.,1997, 1998; Grundy et al., 2000; Griesbach et al., 2002), in chil-dren following severe head trauma (Chiaretti et al., 2003), and within vivo kainic acid injury (Zafra et al., 1990; Dugich-Djordjevicet al., 1992a,b). As bdnf mRNA and BDNF expression wasincreased after injury, we tested for TrkB expression at 24 HPL, anddid not observe any change in expression. However, a previousstudy reported increased TrkB expression between 24 and 48 h afterSchaffer collateral lesion (Dinocourt et al., 2006). As intracellularphosphorylation of TrkB receptors occurs after brain injury inrodents (Binder et al., 1999; Hu et al., 2004), we tested for TrkBreceptor activation by phosphorylation of Y515, a residue that medi-ates Shc binding following activation of the TrkB receptor (Huang& Reichardt, 2001). Trk receptor activation of the Shc pathway isresponsible for the local axon outgrowth effects of neurotrophins viaRas and extracellular signal-related kinase second messengers (Hu-ang & Reichardt, 2003). We found a two-fold increase in pTrkB(Y515) at 3 HPL, which was specifically localised in area CA3.
A
B
C
D
E
Fig. 4. Chronic BDNF blockade downregulates GAP43 expression at 7, 14and 21 days following lesion to Schaffer collaterals. (A) We detected littleGAP43 immunoreactivity in control slices at 7 days (i) 14 days (ii) and21 days (iii) after exposure to cutting solution. (B) GAP43-immunopositivefibers were also not seen in TrkB-Fc-treated control slices at 7 days (i)14 days (ii) and 21 days (iii) following the start of the experiment. (C) At 7DPL, a few GAP43-immunopositive fibers were observed (i), which disap-peared by 21 DPL (iii); as seen earlier, GAP43 expression reached its peakat 14 DPL (ii). (D) TrkB-Fc significantly decreased the expression of GAP43at 14 DPL (ii), and completely abolished GAP43-labeled axonal sprouting at7 DPL (i) and 21 DPL (ii). B–D are representative images of maximumintensity projections rendered from confocal stacks of slices immunohisto-chemically stained for GAP43, and all images were taken in area CA3. Scalebar: 50 lm. The dotted line indicates the site of lesion. (E) Percentage ofslices expressing GAP43. We found a significant increase in the percentageof slices with GAP43-immunopositive fibers at 14 DPL, and this was signifi-cantly decreased by TrkB-Fc treatment (***P < 0.001; two-tailed Fisher’sexact test; control data were pooled from different time points).
© 2013 Federation of European Neuroscience Societies and John Wiley & Sons LtdEuropean Journal of Neuroscience, 1–13
8 R. Gill et al.

Therefore, our finding that there is an increase in pTrkB(Y515) 3 hfollowing Schaffer transection indicates that the Shc pathway hasbeen activated and may underlie the observed axon outgrowth.Taken together with the temporally restricted increase in BDNFexpression, our results suggest that BDNF induces axonal sproutingfollowing Schaffer collateral lesion.
BDNF initiates axonal sprouting of CA3 pyramidal neuronsafter lesion
To determine whether the observed increase in BDNF expression isnecessary for axonal sprouting, we interfered with this increase bytreating lesioned cultures with TrkB-Fc. We found marked downre-gulation of GAP43-expressing axon fibers at 7, 14 and 21 DPL ascompared with untreated lesioned cultures. This finding builds onthe work of Dinocourt et al., who showed that partial knockdown ofTrkB receptors downregulated axonal sprouting 5 days after Schaf-fer collateral injury (Dinocourt et al., 2006). As we chronicallyblocked BDNF for up to 21 days after transection, our data indicatethat, at least for 3 weeks, no compensatory mechanism was initiatedto overcome the BDNF blockade to induce axonal sprouting.Exactly how BDNF regulates this aberrant sprouting is currentlyunknown. However, on the postsynaptic side, emerging evidenceshows that TrkB may regulate synapse number in early developmentby stimulating filopodial motility (Luikart et al., 2008) and increas-ing the probability that a dendritic filopodium will encounter anearby axon. This suggests that BDNF facilitates excitatory synapto-genesis, which could disrupt the subtle balance between excitationand inhibition in favor of excitation to yield hyperexcitability. It hasalso been shown that BDNF can enhance glutamatergic synaptictransmission (Lu, 2003; Gottmann et al., 2009), and BDNF maytherefore potentially exacerbate the hyperexcitability that we foundfollowing Schaffer collateral injury.Interestingly, BDNF levels can also enhance the formation of
GABAergic synapses (Vicario-Abejon et al., 1998; Marty et al.,
2000; Rico et al., 2002) and increase the expression of GAD67 incultured hippocampal neurons (Bolton et al., 2000; Yamada et al.,2002) and GAD65 in organotypic hippocampal slices (Marty et al.,2000). Here, we observed no change in the expression of GAD67,although we did not test for the expression of GAD65. However,whether there is an alteration of inhibitory transmission after Schaf-fer collateral injury to organotypic slices remains to be determined.BDNF can affect GABAergic transmission by regulating cell surfaceexpression of GABAA receptor subunits (Brunig et al., 2001; Jova-novic et al., 2004; Kanematsu et al., 2006); it is unknown whetherthere is a change in GABAA receptor subunit expression after Schaf-fer collateral injury. In addition, exogenous application of BDNF toprimary hippocampal neurons can increase the frequency (Boltonet al., 2000) and amplitude (Brunig et al., 2001) of miniature inhibi-
A B
C
D
E
F
Fig. 5. Chronic treatment with TrkB-Fc for 14 DPL prevents the developmentof hyperexcitability following transection of Schaffer collaterals. (A) Represen-tative whole-cell current-clamp recordings. (B) Percentage of cells showingspontaneous activity. The number of slices with spontaneous APs was signifi-cantly increased in 14 DPL slices, but was reduced by TrkB-Fc treatment, to alevel similar to that in control slices. We found no change in the 14 DPL IgG-Fc-treated group (**P < 0.01 between 14 DPL + IgG-Fc and all groupsexcluding 14 DPL; *P < 0.05 between 14 DPL and all groups excluding 14DPL + IgG-Fc; Fisher’s exact probability test, two-tailed). (C) Quantificationof AP frequency. The observed increase in firing rate of CA3 neurons in 14DPL slices was decreased in TrkB-Fc-treated lesioned slices, to a level similarto that in control sister slices; no change was seen in 14 DPL slices treated withIgG-Fc (***P < 0.005 between 14 DPL and all groups excluding 14DPL + IgG-Fc; *P < 0.05 between 14 DPL + IgG-Fc and all groups exclud-ing 14 DPL; Kruskal–Wallis test with post hoc Mann–Whitney U-test). (D)Percentage of cells with spontaneous bursting. The number of slices thatshowed spontaneous bursting after 14 DPL was increased; this was not seen in14 DPL slices treated with TrkB-Fc. No change was seen in the 14 DPL IgG-Fc-treated slices (*P < 0.05 between 14 DPL and all groups excluding 14DPL + IgG-Fc; Fisher’s exact probability test, two-tailed). (E) Quantificationof burst frequency. The observed increase in burst frequency of 14 DPL sliceswas attenuated by TrkB-Fc treatment; neurons in 14 DPL TrkB-Fc-treatedslices behaved like control CA3 neurons. No change was seen in 14 DPL slicestreated with IgG-Fc (**P < 0.01 between 14 DPL and all groups excluding 14DPL + IgG-Fc; *P < 0.05 between 14 DPL + IgG-Fc and all groups exclud-ing 14 DPL; Kruskal–Wallis test with post hoc Mann–Whitney U-test). Data inC and E are mean � standard error of the mean (bars). (F) Cumulative proba-bility distribution of inter-spike intervals demonstrated a leftward shift in the14 DPL and 14 DPL IgG-Fc-treated cultures (P < 0.001, Kolmogorov–Smir-nov test).
© 2013 Federation of European Neuroscience Societies and John Wiley & Sons LtdEuropean Journal of Neuroscience, 1–13
BDNF and injury-induced hyperexcitability 9

tory postsynaptic currents. Previously, we have shown that there isno change in miniature inhibitory events 14 days following Schaffercollateral lesion in rat organotypic hippocampal slices, indicatingthat inhibitory drive has not changed, at least in a rat culture system(McKinney et al., 1997).
Chronic TrkB-Fc treatment attenuates injury-inducedhyperexcitability and network activity
As axonal sprouting caused by Schaffer collateral transectionincreases the connectivity between CA3 pyramidal neurons(McKinney et al., 1997), it may facilitate the synchronous dischargeof the CA3 cell population. Therefore, we tested the hypothesis thatBDNF-mediated axonal sprouting can lead to hyperexcitability ofexcitatory CA3 pyramidal neurons 14 days after transection. Wefound that chronically treated TrkB-Fc slices had fewer spontaneousAPs than untreated lesioned slices. Moreover, Schaffer collaterallesion caused an increase in compound fEPSPs in area CA3, consis-tent with epileptiform activity and hyperexcitability, similarly toin vivo Schaffer collateral lesion (Aungst et al., 2013). This changein network activity was also attenuated with TrkB-Fc treatment.Interestingly, epileptogenesis in a kindling model is much more dif-ficult to induce in BDNF heterozygous knockout mice (Kokaiaet al., 1995) or with pretreatment with TrkB-Fc (Binder et al.,1999). Moreover, BDNF-overexpressing transgenic mice can havespontaneous seizures (Croll et al., 1999). Taken together, these find-ings indicate that BDNF-mediated local microcircuitry remodelingmay be important for epileptogenesis.This may have implications for post-traumatic epilepsy, as recur-
rent epileptic seizures are a common clinical consequence of trau-
matic brain injury (Annegers et al., 1998). Others have shown thatSchaffer collateral lesion can result in increased network activityafter the addition of a mildly proconvulsive amount of bicuculline(0.1 lM), a GABAA receptor antagonist (Aungst et al., 2013).Moreover, evidence from in vivo models of epilepsy has shownthat axonal sprouting can occur in the hippocampus after inductionof status epilepticus with convulsants. This is well characterised inhippocampal granule cells, where an insult leads to aberrant mossyfiber sprouting and a recurrent network with increased excitatorydrive (Tauck & Nadler, 1985; Cronin et al., 1992; Okazaki et al.,1999). This phenomenon has also been described in patients withtemporal lobe epilepsy (de Lanerolle et al., 1989; Sutula et al.,1989; Houser, 1990) and in rodent models of head trauma (Golaraiet al., 2001; Santhakumar et al., 2001; Dinocourt et al., 2011). Asthere is considerable evidence correlating axonal sprouting andhyperexcitability in epilepsy models, BDNF maybe a potentialtherapeutic target.
Trigger and target for BDNF release
Our evidence implicates BDNF in injury-related axonal sproutingand hyperexcitability. However, little is known about the cell typesresponsible for the observed increase in BDNF expression, and thelocation of the essential TrkB receptors remains unknown. BothBDNF expression and TrkB expression are widespread in the hippo-campus, and TrkB receptors are found on both neurons and astro-cytes (Ernfors et al., 1990; Wetmore et al., 1990; Armanini et al.,1995). Interestingly, there is an induction of non-catalytic truncatedtrkb mRNA in glial cells 6–14 days following lesion of the perfo-rant pathway (Beck et al., 1993). As truncated TrkB receptors
A
B C
Fig. 6. Chronic treatment with TrkB-Fc for 14 DPL prevents the lesioned-induced increase in network activity. (A) Representative traces of evoked fEPSPrecordings. Note: arrows indicate additional peaks in compounded fEPSP responses. (B) Quantification of the number of peaks as a function of stimulusstrength. We found that 14 DPL slices required less stimulation than control slices for induction of compound fEPSPs. This was not observed in 14 DPL slicestreated with TrkB-Fc, which were similar to control unlesioned TrkB-Fc-treated sister slices (*P < 0.05; linear regression was performed for each slice, and thenslopes were compared by use of one-way ANOVA with a post hoc Tukey’s test, following which we performed a two-tailed independent Student’s t-test for pair-wise comparisons between data points). Moreover, IgG-Fc-treated lesioned slices were similar to 14 DPL slices. (C) Quantification of percentage of culturesshowing compounded fEPSPs. We found that all 14 DPL slices showed compounded fEPSPs. However, TrkB-Fc-treated slices were similar to control slicesand TrkB-Fc-treated control slices. IgG-Fc-treated 14 DPL slices were similar to 14 DPL slices (*P < 0.05 between 14 DPL and all groups excluding 14DPL + IgG-Fc; also *P < 0.05, between 14 DPL + IgG-Fc and all groups excluding 14 DPL; Fisher’s exact probability test, two-tailed). Note: control and 14DPL data are repeated from Fig. 2B and C for the purpose of comparison.
© 2013 Federation of European Neuroscience Societies and John Wiley & Sons LtdEuropean Journal of Neuroscience, 1–13
10 R. Gill et al.
sequester and internalise BDNF (Biffo et al., 1995; Alderson et al.,2000), it could be possible that unique intracellular pathways areactivated through this truncated TrkB-mediated internalisation, lead-ing to axonal sprouting.Emerging evidence shows that activity-dependent transcription of
bdnf is under epigenetic regulation (Martinowich et al., 2003; Zhouet al., 2006; Bredy et al., 2007). A common epigenetic modificationis methylation of cytosines that are adjacent to guanines (CpG;Klose & Bird, 2006). Methyl-CpG binding protein 2 (MeCP2) canbind to methylated CpG sites and repress transcription (Jones et al.,1998; Nan et al., 1998), such as in the case of the bdnf gene (Mar-tinowich et al., 2003). It has been shown that calcium-dependentphosphorylation of MeCP2 increases the transcription of bdnf (Chenet al., 2003). As hippocampal lesion can lead to excess glutamaterelease and increased calcium intake in neurons (Muller et al.,2010), it would be interesting to investigate the involvement ofMeCP2 in injury-induced BDNF release.In summary, our study provides direct evidence that BDNF is
critical for the development of new axons and increased excitationafter injury to the pyramidal cell axon. Furthermore, we observedthat BDNF inhibition attenuated lesion-induced hippocampal hype-rexcitability and changes in network activity. The evidence shownin the present study suggests that TrkB–BDNF signaling could be anovel therapeutic target. However, as BDNF plays an essential rolein learning and memory, it is important to study signaling cascadesdownstream of BDNF to develop viable treatments targeting post-traumatic epilepsy.
Supporting Information
Additional supporting information can be found in the onlineversion of this article:Fig. S1. GAP43 expression is downregulated in 21-day in vitrocultures.Fig. S2. GAD67 expression is unchanged by Schaffer collaterallesion.Table S1. Input–output response of CA3 pyramidal neurons remainsunchanged 14 days post-lesion.Data S1. Supporting Information and methods.
Acknowledgements
We wish to thank F. Charron for his excellent technical assistance, Dr S. M.Thompson, Dr B. G€ahwiler and D. Verbich for helpful discussion and com-ments on the manuscript, Dr K. Meiri for the anti-GAP43 antibody, and DrK. Murai for the Semliki Forest virus expressing mCherry. This work wassupported by grants from the Canadian Institutes of Health Research, theNatural Sciences and Engineering Research Council of Canada, and theSavoy Foundation.
Abbreviations
AP, action potential; BDNF, brain-derived neurotrophic factor; BSA, bovineserum albumin; DIV, days in vitro; DPL, days post-lesion; EPSP, excitatorypostsynaptic potential; fEPSP, field excitatory postsynaptic potential;GAD67, glutamate decarboxylase 67; GAP43, growth-associated protein 43;HPL, hours post-lesion; MeCP2, methyl-CpG binding protein 2; PB, phos-phate buffer; PSD95, postsynaptic density protein 95; pTrkB, phosphorylatedtropomyosin-related kinase B; qPCR, quantitative polymerase chain reaction;sEPSP, spontaneous excitatory postsynaptic potential; TBST, Tris-bufferedsaline with 0.05% Tween-20; TrkB, tropomyosin-related kinase B.
References
Alderson, R.F., Curtis, R., Alterman, A.L., Lindsay, R.M. & DiStefano, P.S.(2000) Truncated TrkB mediates the endocytosis and release of BDNF andneurotrophin-4/5 by rat astrocytes and schwann cells in vitro. Brain Res.,871, 210–222.
Anderson, P., Morris, R., Amaral, D., Bliss, T. & O’Keefe, J. (2007) TheHippocampus Book. Oxford University Press, New York, NY.
Annegers, J.F., Hauser, W.A., Coan, S.P. & Rocca, W.A. (1998) A popula-tion-based study of seizures after traumatic brain injuries. New Engl. J.Med., 338, 20–24.
Armanini, M.P., McMahon, S.B., Sutherland, J., Shelton, D.L. & Phillips,H.S. (1995) Truncated and catalytic isoforms of trkB are co-expressed inneurons of rat and mouse CNS. Eur. J. Neurosci., 7, 1403–1409.
Aungst, S., England, P.M. & Thompson, S.M. (2013) Critical role of trkBreceptors in reactive axonal sprouting and hyperexcitability after axonalinjury. J. Neurophysiol., 109, 813–824.
Beck, K.D., Lamballe, F., Klein, R., Barbacid, M., Schauwecker, P.E.,McNeill, T.H., Finch, C.E., Hefti, F. & Day, J.R. (1993) Induction of non-catalytic TrkB neurotrophin receptors during axonal sprouting in the adulthippocampus. J. Neurosci., 13, 4001–4014.
Biffo, S., Offenhauser, N., Carter, B.D. & Barde, Y.A. (1995) Selective bind-ing and internalisation by truncated receptors restrict the availability ofBDNF during development. Development, 121, 2461–2470.
Binder, D.K., Routbort, M.J., Ryan, T.E., Yancopoulos, G.D. & McNamara,J.O. (1999) Selective inhibition of kindling development by intraventricu-lar administration of TrkB receptor body. J. Neurosci., 19, 1424–1436.
Bolton, M.M., Pittman, A.J. & Lo, D.C. (2000) Brain-derived neurotrophicfactor differentially regulates excitatory and inhibitory synaptic transmis-sion in hippocampal cultures. J. Neurosci., 20, 3221–3232.
Bredy, T.W., Wu, H., Crego, C., Zellhoefer, J., Sun, Y.E. & Barad, M.(2007) Histone modifications around individual BDNF gene promoters inprefrontal cortex are associated with extinction of conditioned fear. Learn.Memory, 14, 268–276.
Brunig, I., Penschuck, S., Berninger, B., Benson, J. & Fritschy, J.M. (2001)BDNF reduces miniature inhibitory postsynaptic currents by rapid
A
B
Fig. 7. Spontaneous EPSPs of CA3 pyramidal neurons increase after Schaf-fer collateral transection. (A) Representative traces of whole-cell patch elec-trophysiological recordings of CA3 pyramidal neurons. (B) Quantification ofsEPSPs. We found an increase in sEPSPs at 14 DPL, as compared with con-trol slices (*P < 0.05, two-tailed Student’s t-test). Furthermore, we foundthat this increase in sEPSPs was attenuated by TrkB-Fc treatment for 14DPL; we did not see a change in sEPSPs of CA3 pyramidal cells of controlslices treated with TrkB-Fc. Note: data are depicted as mean � standarderror of the mean.
© 2013 Federation of European Neuroscience Societies and John Wiley & Sons LtdEuropean Journal of Neuroscience, 1–13
BDNF and injury-induced hyperexcitability 11

downregulation of GABA(A) receptor surface expression. Eur. J. Neuro-sci., 13, 1320–1328.
Chen, W.G., Chang, Q., Lin, Y., Meissner, A., West, A.E., Griffith, E.C.,Jaenisch, R. & Greenberg, M.E. (2003) Derepression of BDNF transcrip-tion involves calcium-dependent phosphorylation of MeCP2. Science, 302,885–889.
Cheng, P.L., Song, A.H., Wong, Y.H., Wang, S., Zhang, X. & Poo, M.M.(2011) Self-amplifying autocrine actions of BDNF in axon development.Proc. Natl. Acad. Sci. USA, 108, 18430–18435.
Chiaretti, A., Piastra, M., Polidori, G., Di Rocco, C., Caresta, E., Antonelli,A., Amendola, T. & Aloe, L. (2003) Correlation between neurotrophic fac-tor expression and outcome of children with severe traumatic brain injury.Intens. Care Med., 29, 1329–1338.
Croll, S.D., Suri, C., Compton, D.L., Simmons, M.V., Yancopoulos, G.D.,Lindsay, R.M., Wiegand, S.J., Rudge, J.S. & Scharfman, H.E. (1999)Brain-derived neurotrophic factor transgenic mice exhibit passive avoid-ance deficits, increased seizure severity and in vitro hyperexcitability inthe hippocampus and entorhinal cortex. Neuroscience, 93, 1491–1506.
Cronin, J., Obenaus, A., Houser, C.R. & Dudek, F.E. (1992) Electrophysiol-ogy of dentate granule cells after kainate-induced synaptic reorganizationof the mossy fibers. Brain Res., 573, 305–310.
Davenport, C.J., Brown, W.J. & Babb, T.L. (1990) Sprouting of GABAergicand mossy fiber axons in dentate gyrus following intrahippocampal kainatein the rat. Exp. Neurol., 109, 180–190.
Davies, A.M., Thoenen, H. & Barde, Y.A. (1986) The response of chick sen-sory neurons to brain-derived neurotrophic factor. J. Neurosci., 6,1897–1904.
Dinocourt, C., Gallagher, S.E. & Thompson, S.M. (2006) Injury-inducedaxonal sprouting in the hippocampus is initiated by activation of trkBreceptors. Eur. J. Neurosci., 24, 1857–1866.
Dinocourt, C., Aungst, S., Yang, K. & Thompson, S.M. (2011) Homeostaticincrease in excitability in area CA1 after Schaffer collateral transectionin vivo. Epilepsia, 52, 1656–1665.
Dugich-Djordjevic, M.M., Tocco, G., Lapchak, P.A., Pasinetti, G.M., Najm,I., Baudry, M. & Hefti, F. (1992a) Regionally specific and rapid increasesin brain-derived neurotrophic factor messenger RNA in the adult rat brainfollowing seizures induced by systemic administration of kainic acid. Neu-roscience, 47, 303–315.
Dugich-Djordjevic, M.M., Tocco, G., Willoughby, D.A., Najm, I., Pasinetti,G., Thompson, R.F., Baudry, M., Lapchak, P.A. & Hefti, F. (1992b)BDNF mRNA expression in the developing rat brain following kainicacid-induced seizure activity. Neuron, 8, 1127–1138.
Ernfors, P., Wetmore, C., Olson, L. & Persson, H. (1990) Identification ofcells in rat brain and peripheral tissues expressing mRNA for members ofthe nerve growth factor family. Neuron, 5, 511–526.
G€ahwiler, B.H. (1981) Organotypic monolayer cultures of nervous tissue. J.Neurosci. Meth., 4, 329–342.
G€ahwiler, B.H., Capogna, M., Debanne, D., McKinney, R.A. & Thompson,S.M. (1997) Organotypic slice cultures: a technique has come of age.Trends Neurosci., 20, 471–477.
Golarai, G., Greenwood, A.C., Feeney, D.M. & Connor, J.A. (2001) Physio-logical and structural evidence for hippocampal involvement in persistentseizure susceptibility after traumatic brain injury. J. Neurosci., 21,8523–8537.
Gottmann, K., Mittmann, T. & Lessmann, V. (2009) BDNF signaling in theformation, maturation and plasticity of glutamatergic and GABAergicsynapses. Exp. Brain Res., 199, 203–234.
Griesbach, G.S., Hovda, D.A., Molteni, R. & Gomez-Pinilla, F. (2002) Alter-ations in BDNF and synapsin I within the occipital cortex and hippocam-pus after mild traumatic brain injury in the developing rat: reflections ofinjury-induced neuroplasticity. J. Neurotraum., 19, 803–814.
Grundy, P.L., Patel, N., Harbuz, M.S., Lightman, S.L. & Sharples, P.M.(2000) Glucocorticoids modulate BDNF mRNA expression in the rathippocampus after traumatic brain injury. NeuroReport, 11, 3381–3384.
Hannesson, D.K., Armitage, L.L., Mohapel, P. & Corcoran, M.E. (1997)Time course of mossy fiber sprouting following bilateral transection of thefimbria/fornix. NeuroReport, 8, 2299–2303.
Hicks, R.R., Numan, S., Dhillon, H.S., Prasad, M.R. & Seroogy, K.B.(1997) Alterations in BDNF and NT-3 mRNAs in rat hippocampus afterexperimental brain trauma. Brain Res. Mol. Brain Res., 48, 401–406.
Hicks, R.R., Zhang, L., Dhillon, H.S., Prasad, M.R. & Seroogy, K.B. (1998)Expression of trkB mRNA is altered in rat hippocampus after experimentalbrain trauma. Brain Res. Mol. Brain Res., 59, 264–268.
Houser, C.R. (1990) Granule cell dispersion in the dentate gyrus of humanswith temporal lobe epilepsy. Brain Res., 535, 195–204.
Hu, B., Liu, C., Bramlett, H., Sick, T.J., Alonso, O.F., Chen, S. & Dietrich,W.D. (2004) Changes in trkB–ERK1/2–CREB/Elk-1 pathways in hippo-campal mossy fiber organization after traumatic brain injury. J. Cerebr.Blood F. Met., 24, 934–943.
Huang, E.J. & Reichardt, L.F. (2001) Neurotrophins: roles in neuronal devel-opment and function. Annu. Rev. Neurosci., 24, 677–736.
Huang, E.J. & Reichardt, L.F. (2003) Trk receptors: roles in neuronal signaltransduction. Annu. Rev. Biochem., 72, 609–642.
Ji, Y., Lu, Y., Yang, F., Shen, W., Tang, T.T., Feng, L., Duan, S. & Lu, B.(2010) Acute and gradual increases in BDNF concentration elicit distinctsignaling and functions in neurons. Nat. Neurosci., 13, 302–309.
Jones, P.L., Veenstra, G.J., Wade, P.A., Vermaak, D., Kass, S.U., Landsber-ger, N., Strouboulis, J. & Wolffe, A.P. (1998) Methylated DNA andMeCP2 recruit histone deacetylase to repress transcription. Nat. Genet.,19, 187–191.
Jovanovic, J.N., Thomas, P., Kittler, J.T., Smart, T.G. & Moss, S.J. (2004)Brain-derived neurotrophic factor modulates fast synaptic inhibition byregulating GABA(A) receptor phosphorylation, activity, and cell-surfacestability. J. Neurosci., 24, 522–530.
Kanematsu, T., Yasunaga, A., Mizoguchi, Y., Kuratani, A., Kittler, J.T.,Jovanovic, J.N., Takenaka, K., Nakayama, K.I., Fukami, K., Takenawa,T., Moss, S.J., Nabekura, J. & Hirata, M. (2006) Modulation of GABA(A)receptor phosphorylation and membrane trafficking by phospholipase C-related inactive protein/protein phosphatase 1 and 2A signaling complexunderlying brain-derived neurotrophic factor-dependent regulation of GAB-Aergic inhibition. J. Biol. Chem., 281, 22180–22189.
Klose, R.J. & Bird, A.P. (2006) Genomic DNA methylation: the mark andits mediators. Trends Biochem. Sci., 31, 89–97.
Kokaia, M., Ernfors, P., Kokaia, Z., Elmer, E., Jaenisch, R. & Lindvall, O.(1995) Suppressed epileptogenesis in BDNF mutant mice. Exp. Neurol.,133, 215–224.
de Lanerolle, N.C., Kim, J.H., Robbins, R.J. & Spencer, D.D. (1989) Hippo-campal interneuron loss and plasticity in human temporal lobe epilepsy.Brain Res., 495, 387–395.
Laurberg, S. & Zimmer, J. (1981) Lesion-induced sprouting of hippocampalmossy fiber collaterals to the fascia dentata in developing and adult rats. J.Comp. Neurol., 200, 433–459.
Lu, B. (2003) BDNF and activity-dependent synaptic modulation. Learn.Memory, 10, 86–98.
Luikart, B.W., Zhang, W., Wayman, G.A., Kwon, C.H., Westbrook, G.L. &Parada, L.F. (2008) Neurotrophin-dependent dendritic filopodial motility: aconvergence on PI3K signaling. J. Neurosci., 28, 7006–7012.
Lundstrom, K. & Ehrengruber, M.U. (2003) Semliki Forest virus (SFV) vec-tors in neurobiology and gene therapy. Methods Mol. Med., 76, 503–523.
Lundstrom, K., Abenavoli, A., Malgaroli, A. & Ehrengruber, M.U. (2003)Novel Semliki Forest virus vectors with reduced cytotoxicity and tempera-ture sensitivity for long-term enhancement of transgene expression. Mol.Ther., 7, 202–209.
Martinowich, K., Hattori, D., Wu, H., Fouse, S., He, F., Hu, Y., Fan, G. &Sun, Y.E. (2003) DNA methylation-related chromatin remodeling in activ-ity-dependent BDNF gene regulation. Science, 302, 890–893.
Marty, S., Wehrle, R. & Sotelo, C. (2000) Neuronal activity and brain-derived neurotrophic factor regulate the density of inhibitory synapses inorganotypic slice cultures of postnatal hippocampus. J. Neurosci., 20,8087–8095.
McKinney, R.A., Debanne, D., G€ahwiler, B.H. & Thompson, S.M. (1997)Lesion-induced axonal sprouting and hyperexcitability in the hippocampusin vitro: implications for the genesis of posttraumatic epilepsy. Nat. Med.,3, 990–996.
McKinney, R.A., Luthi, A., Bandtlow, C.E., G€ahwiler, B.H. & Thompson,S.M. (1999) Selective glutamate receptor antagonists can induce or preventaxonal sprouting in rat hippocampal slice cultures. Proc. Natl. Acad. Sci.USA, 96, 11631–11636.
Meiri, K.F., Pfenninger, K.H. & Willard, M.B. (1986) Growth-associatedprotein, GAP-43, a polypeptide that is induced when neurons extendaxons, is a component of growth cones and corresponds to pp 46, a majorpolypeptide of a subcellular fraction enriched in growth cones. Proc. Natl.Acad. Sci. USA, 83, 3537–3541.
Mudo, G., Persson, H., Timmusk, T., Funakoshi, H., Bindoni, M. & Bellu-ardo, N. (1993) Increased expression of trkB and trkC messenger RNAsin the rat forebrain after focal mechanical injury. Neuroscience, 57,901–912.
Muller, C.M., Vlachos, A. & Deller, T. (2010) Calcium homeostasis ofacutely denervated and lesioned dentate gyrus in organotypic entorhino-hippocampal co-cultures. Cell Calcium, 47, 242–252.
© 2013 Federation of European Neuroscience Societies and John Wiley & Sons LtdEuropean Journal of Neuroscience, 1–13
12 R. Gill et al.

Nan, X., Ng, H.H., Johnson, C.A., Laherty, C.D., Turner, B.M., Eisenman,R.N. & Bird, A. (1998) Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature,393, 386–389.
Okazaki, M.M., Molnar, P. & Nadler, J.V. (1999) Recurrent mossy fiberpathway in rat dentate gyrus: synaptic currents evoked in presence andabsence of seizure-induced growth. J. Neurophysiol., 81, 1645–1660.
Rico, B., Xu, B.J. & Reichardt, L.F. (2002) TrkB receptor signaling isrequired for establishment of GABAergic synapses in the cerebellum. Nat.Neurosci., 5, 225–233.
Routbort, M.J., Bausch, S.B. & McNamara, J.O. (1999) Seizures, cell death,and mossy fiber sprouting in kainic acid-treated organotypic hippocampalcultures. Neuroscience, 94, 755–765.
Salin, P., Tseng, G.F., Hoffman, S., Parada, I. & Prince, D.A. (1995) Axonalsprouting in layer V pyramidal neurons of chronically injured cerebralcortex. J. Neurosci., 15, 8234–8245.
Santhakumar, V., Ratzliff, A.D., Jeng, J., Toth, Z. & Soltesz, I. (2001)Long-term hyperexcitability in the hippocampus after experimental headtrauma. Ann. Neurol., 50, 708–717.
Schwyzer, L., Mateos, J.M., Abegg, M., Rietschin, L., Heeb, L., Thompson,S.M., Luthi, A., G€ahwiler, B.H. & McKinney, R.A. (2002) Physiologicaland morphological plasticity induced by chronic treatment with NT-3 orNT-4/5 in hippocampal slice cultures. Eur. J. Neurosci., 16, 1939–1948.
Shelly, M., Cancedda, L., Heilshorn, S., Sumbre, G. & Poo, M.M. (2007)LKB1/STRAD promotes axon initiation during neuronal polarization. Cell,129, 565–577.
Shelly, M., Cancedda, L., Lim, B.K., Popescu, A.T., Cheng, P.L., Gao, H. &Poo, M.M. (2011) Semaphorin3A regulates neuronal polarization by
suppressing axon formation and promoting dendrite growth. Neuron, 71,433–446.
Sutula, T., Cascino, G., Cavazos, J., Parada, I. & Ramirez, L. (1989) Mossyfiber synaptic reorganization in the epileptic human temporal lobe. Ann.Neurol., 26, 321–330.
Tauck, D.L. & Nadler, J.V. (1985) Evidence of functional mossy fibersprouting in hippocampal formation of kainic acid-treated rats. J. Neuro-sci., 5, 1016–1022.
Vicario-Abejon, C., Collin, C., McKay, R.D. & Segal, M. (1998) Neuro-trophins induce formation of functional excitatory and inhibitory syn-apses between cultured hippocampal neurons. J. Neurosci., 18, 7256–7271.
Wetmore, C., Ernfors, P., Persson, H. & Olson, L. (1990) Localization ofbrain-derived neurotrophic factor mRNA to neurons in the brain by in situhybridization. Exp. Neurol., 109, 141–152.
Yamada, M.K., Nakanishi, K., Ohba, S., Nakamura, T., Ikegaya, Y., Nishiy-ama, N. & Matsuki, N. (2002) Brain-derived neurotrophic factor promotesthe maturation of GABAergic mechanisms in cultured hippocampal neu-rons. J. Neurosci., 22, 7580–7585.
Zafra, F., Hengerer, B., Leibrock, J., Thoenen, H. & Lindholm, D. (1990)Activity dependent regulation of BDNF and NGF mRNAs in the rat hip-pocampus is mediated by non-NMDA glutamate receptors. EMBO J., 9,3545–3550.
Zhou, Z., Hong, E.J., Cohen, S., Zhao, W.N., Ho, H.Y., Schmidt, L., Chen,W.G., Lin, Y., Savner, E., Griffith, E.C., Hu, L., Steen, J.A., Weitz, C.J.& Greenberg, M.E. (2006) Brain-specific phosphorylation of MeCP2 regu-lates activity-dependent Bdnf transcription, dendritic growth, and spinematuration. Neuron, 52, 255–269.
© 2013 Federation of European Neuroscience Societies and John Wiley & Sons LtdEuropean Journal of Neuroscience, 1–13
BDNF and injury-induced hyperexcitability 13