birinapant (tl32711), a bivalent smac mimetic, …...chi, [email protected]; and...
TRANSCRIPT

Small Molecule Therapeutics
Birinapant (TL32711), a Bivalent SMAC Mimetic, TargetsTRAF2-Associated cIAPs, Abrogates TNF-Induced NF-kBActivation, and Is Active in Patient-Derived XenograftModels
Christopher A. Benetatos1, Yasuhiro Mitsuuchi1, Jennifer M. Burns1, Eric M. Neiman1, Stephen M. Condon1,Guangyao Yu1, Martin E. Seipel1, Gurpreet S. Kapoor1, Matthew G. LaPorte1, Susan R. Rippin1, Yijun Deng1,Mukta S. Hendi1, Pavan K. Tirunahari1, Yu-Hua Lee1, Thomas Haimowitz1, Matthew D. Alexander1,Martin A. Graham1, David Weng1, Yigong Shi2, Mark A. McKinlay1, and Srinivas K. Chunduru1
AbstractThe acquisition of apoptosis resistance is a fundamental event in cancer development. Among the
mechanisms used by cancer cells to evade apoptosis is the dysregulation of inhibitor of apoptosis (IAP)
proteins. The activity of the IAPs is regulated by endogenous IAP antagonists such as SMAC (also termed
DIABLO). Antagonism of IAP proteins by SMAC occurs via binding of the N-terminal tetrapeptide (AVPI) of
SMAC to selected BIR domains of the IAPs. Small molecule compounds that mimic the AVPI motif of SMAC
have been designed to overcome IAP-mediated apoptosis resistance of cancer cells. Here, we report the
preclinical characterization of birinapant (TL32711), a bivalent SMAC-mimetic compound currently in clinical
trials for the treatment of cancer. Birinapant bound to the BIR3 domains of cIAP1, cIAP2, XIAP, and the BIR
domain of ML-IAP in vitro and induced the autoubiquitylation and proteasomal degradation of cIAP1 and
cIAP2 in intact cells, which resulted in formation of a RIPK1:caspase-8 complex, caspase-8 activation, and
induction of tumor cell death. Birinapant preferentially targeted the TRAF2-associated cIAP1 and cIAP2 with
subsequent inhibition of TNF-induced NF-kB activation. The activity of a variety of chemotherapeutic cancer
drugswas potentiated by birinapant both in a TNF-dependent or TNF-independentmanner. Tumor growth in
multiple primary patient–derived xenotransplant models was inhibited by birinapant at well-tolerated doses.
These results support the therapeutic combination of birinapant with multiple chemotherapies, in particular,
those therapies that can induce TNF secretion. Mol Cancer Ther; 13(4); 867–79. �2014 AACR.
IntroductionApoptosis or programmed cell death is a genetically
encoded process that results in the clearance of damagedcells without inducing an inflammatory response and canbe initiated by either intrinsic or extrinsic cellular cues.Apoptotic signaling culminates in the activation of afamily of cysteine proteases with aspartate specificity
(termed caspases), which cleaves key protein substratesensuring that the cell is dismantled in a controlled man-ner. Resistance to apoptosis is a fundamental property ofcancer (1). The acquisition of genetic lesions, which dys-regulate the execution of apoptosis, is known to cooperatewith other cellular defects such as oncogene activationleading to tumor initiation and progression (1, 2). Inaddition, the emergence of drug resistance has also beenattributed, in part, to defects in apoptosis (3, 4). Thedevelopment ofdrugs aimedat regulation of the apoptoticsignaling of cancer cells represents a new paradigm incancer treatment and several of these agents have enteredclinical trials.
The inhibitor of apoptosis (IAP) proteins are a highlyconserved family of signaling regulators that play keyroles in cell death as well as immunity, inflammation, andcell division (5, 6). Originally discovered for their cyto-protective effect in baculovirus-infected insect cells, theIAP proteins have been conserved throughout evolution.The Baculovirus IAP Repeat (BIR) domain is the definingfeature shared by all IAP proteins and facilitates specificinteractions with other proteins. Some family members
Authors' Affiliations: 1TetraLogic Pharmaceuticals, 343Phoenixville Pike,Malvern, Pennsylvania; and 2Tsinghua University School of Medicine,Beijing, China
Note: Supplementary data for this article are available at Molecular CancerTherapeutics Online (http://mct.aacrjournals.org/).
C.A. Benetatos and Y. Mitsuuchi contributed equally to this work.
Corresponding Authors:Christopher A. Benetatos, 343 Phoenixville Pike,Malvern, PA 19355. Phone: 610-889-9900; Fax: 610-889-9994;E-mail: [email protected]; Yasuhiro Mitsuu-chi, [email protected]; and Srinivas K. Chunduru,[email protected]
doi: 10.1158/1535-7163.MCT-13-0798
�2014 American Association for Cancer Research.
MolecularCancer
Therapeutics
www.aacrjournals.org 867
on June 13, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 21, 2014; DOI: 10.1158/1535-7163.MCT-13-0798
on June 13, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 21, 2014; DOI: 10.1158/1535-7163.MCT-13-0798
on June 13, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 21, 2014; DOI: 10.1158/1535-7163.MCT-13-0798
on June 13, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 21, 2014; DOI: 10.1158/1535-7163.MCT-13-0798

also possess a C-terminal really interesting new gene(RING) domain with E3 ubiquitin ligase activity. Thereare 8 human IAPs with XIAP being the only direct inhib-itor of the proteolytic activity of caspases (7). The cellularIAPs, cIAP1, and cIAP2 (referred collectively as cIAP1/2)play key roles in the promotion of survival signalinginduced by members of the TNF superfamily of cellsurface receptors. Upon ligand binding, cIAP1/2 arerecruited to the membrane-bound TNF receptor 1(TNFR1) complex via the TNF receptor-associated factor(TRAF) family of proteins. In this context, cIAP1/2 func-tion as ubiquitin E3 ligases,which catalyze the attachmentof ubiquitin or polyubiquitin chains through the action oftheir C-terminal RING domains. These ubiquitylationevents serve to either provide a scaffold for the furtherassembly of protein complexes and subsequent signaltransduction or target-specific proteins for proteasome-mediated degradation (8).
The second mitochondria-derived activator of caspases(SMAC) also referred to as the direct IAP binding proteinwith low pI (DIABLO) is an endogenous antagonist of IAPproteins. SMAC is released from the mitochondrial com-partment in response to apoptotic stimuli and neutralizesIAP proteins thus allowing sustained caspase activity andexecution of the apoptotic process. The contact betweenIAPs and SMAC ismediated through theN-terminal AVPItetrapeptide of SMAC, which binds within a groove on theIAP BIR domain. The design of small molecules that mimicthe IAP binding motif of SMAC and pharmacologicallyinhibit IAP protein function has been described (9).
The IAP proteins are frequently dysregulated in manycancers and thus have been suggested as contributingto chemoresistance and treatment failure. DNA amplifi-cation of the cIAP1 and cIAP2 genes (BIRC2 and BIRC3,respectively), located on chromosome 11q22-q23, hasbeen observed in human lung cancers (10), liver carcino-mas (11), oral squamous cell carcinomas (12, 13), medul-loblastomas (14), glioblastomas (15), and pancreaticcancers (16). Frequent dysregulation of IAP proteins hasalso been observed at the protein level in multiple cancercell lines and tumor samples (17, 18). Consequently, theIAP proteins have been pursued as anticancer drug tar-gets. Various approaches have been utilized to target IAPproteins, including antisense oligonucleotides (19) andsmall molecules with specificity toward BIR domains ofXIAP, survivin, ML-IAP, and cIAP1/2 (9).
Here we report the preclinical activity of birinapant(TL32711), a biindole-based bivalent SMAC mimetic.We show that birinapant binds with high affinity tothe isolated BIR3 domains of cIAP1, cIAP2, and XIAPand the single BIR domain of ML-IAP and rapidlydegrades TRAF2-bound cIAP1 and cIAP2 thereby inhibit-ing TNF-mediated NF-kB activation. In addition, birina-pant promotes caspase-8:RIPK1 complex formation inresponse to TNF stimulation, resulting in the activationof downstream caspases. Moreover, birinapant treatmentafforded synergistic cytotoxicity of several widely usedchemotherapeutic agents. In murine xenograft models of
ovarian and colorectal cancers and melanoma patient–derived tumors, birinapant displayed antitumor activityas a single agent at well-tolerated doses. These datasuggest that birinapant targets IAP proteins to trigger anapoptotic response both in vitro and in vivo and haspotential application in multiple tumor malignancies.
Materials and MethodsReagents, plasmids, and tissue culture
pGEX-2T plasmids with N-terminal glutathione-S-trans-ferase (GST)-tagged XIAP-BIR3 (amino acids 238–358) andcIAP1 BIR3 (amino acids 255–364) were obtained fromYigong Shi (Tsinghua University, China). TheML-IAP BIRdomain was cloned into a pGEX-6P3 vector and the cIAP2BIR3 domain was cloned into a pET28a vector. The NF-kBluciferase reporter plasmid was from Stratagene. ThecDNA encoding hemagglutinin (HA)-tagged enhancedgreen fluorescence protein (EGFP) was cloned into theN-termini of human cIAP1 and cIAP2 in the mammalianexpression plasmid pcDNA3 and designated as HA2x-EGFP-cIAP1 and HA2xEGFP-cIAP2, respectively. Celllines were obtained from various sources (American TypeCultureCollection; FoxChaseCancer Center, Leibniz-Insti-tutDSMZ-Deutsche SammlungvonMikroorganismenundZellkulturen GmbH) andmaintained in appropriate medi-um. No authentication was performed. Defined FBS waspurchased from HyClone and all cells were grown in ahumidified 37�C, 5% CO2 incubator. Human recombinantTNF, TRAIL, Lipofectamin2000, G418, restriction enzymes,protein A/G agarose, and a NuPAGE system werepurchased from Invitrogen. Birinapant (TL32711) andbiotin-labeled birinapant were synthesized at TetraLogicPharmaceuticals. Other reagents included AbuRPFK(5-Fam)-NH2 peptide (Biomer Technologies), anti-cIAP1,-cIAP2, and -ML-IAP antibodies were from R&D, Anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH)and -PARP antibodies were from Santa Cruz Biotechnolo-gy, anti-XIAPantibody (Stressgen), anti-IkBa antibodywasfrom Cell Signaling, anti-RIPK1 antibody was from BD,anti–caspase-8 andTRAF2antibodieswere fromboth SantaCruz Biotechnology and Cell Signaling.
Establishment of stable cellsA375 cellswere transfectedwithHA2xEGFP-cIAP1and
HA2xEGFP-cIAP2 linearized with Pvu I by using Lipo-fectamine 2000 as described by the manufacturer andstable transfectant cells were selected by 800 mg/mLG418. The expression of GFP-cIAP1 and GFP-cIAP2 pro-teins in the G418-resistant cells was verified by Westernblot analysis and fluorescence-activated cell sorting(FACS) analysis.
Coimmunoprecipitation and Western blot analysisFor immunoprecipitation of TRAF2 protein complex,
cells were collected and lysed with buffer-A consisting of20 mmol/L Tris-HCl (pH 7.5), 150 mmol/L sodium chlo-ride, 10% glycerol, 2 mmol/L EDTA, 50 mmol/L sodiumfluoride, 25 mmol/L b-glycerophosphate, 0.2 mmol/L
Benetatos et al.
Mol Cancer Ther; 13(4) April 2014 Molecular Cancer Therapeutics868
on June 13, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 21, 2014; DOI: 10.1158/1535-7163.MCT-13-0798

sodium vanadate, 10 mmol/L sodium pyrophosphate, 2mmol/L DTT, 0.5% Triton X-100, 1 mmol/L phenyl-methylsulfonyl fluoride, and protease inhibitor cocktail(Roche). Cleared total cell lysate (2 mg) was incubatedwith anti-TRAF2 antibody overnight at 4�C. The immuno-protein complex was purified by using Protein A/Gagarose beads and subjected toWestern blot analysiswithindicated antibodies. Immunoprecipitation of ripopto-some, complex-I, and complex-II were essentially carriedout as described (20, 21). Protein samples were applied toNuPAGE gel, followed by the electrotransfer to Immobi-lon-FLmembranes (Millipore).Membraneswere incubat-ed in blocking buffer (LI-COR Bioscience, Inc.). Detectionof proteins on the membrane was performed with astandard procedure using primary antibodies indicatedin the figures and their appropriate secondary antibodiesconjugated to Alexa Fluor 680 or 800 IRDye. Membraneswere scanned and analyzed byOdyssey infrared imagingsystem (LI-COR Biosciences, Inc.).
Fluorescence polarization assayThe binding affinities of compounds to the purified
BIR3 domains of cIAP1, cIAP2, and XIAP, and the singleML-IAP BIR domain were determined as described pre-viously (22) and are reported as dissociation constant (Kd)values. The Kd values of competitive inhibitors werecalculated using the equation described in ref. 22 usingIC50 values, the Kd value of the protein, and the concen-trations of the protein and fluorescence polarization (FP)peptide in the competition assay.
Measurement of caspase-3 activity in cell lysatesMDA-MB-231 breast carcinoma cellswere harvested by
trypsinization and collected by centrifugation at 1,000� gfor 10 minutes at room temperature. Cell pellet waswashed one time by resuspending in 5 mL hypotoniclysis buffer (20 mmol/L HEPES, pH 7.5, 10 mmol/L KCl,1.5mmol/LMgCl2, 1.0mmol/L EDTA, 1.0mmol/LDTT)and recollected by centrifugation. Pellet was next resus-pended in 1 volume of hypotonic lysis buffer supplemen-ted with a complete protease inhibitor tablet and allowedto swell on ice for 30 minutes. Cells were disrupted byapproximately 50 passages through a 27-gauge needle.Lysis was monitored by light microscopy. Lysate wascentrifuged at 12,000 � g for 10 minutes at 4�C to removemembrane fraction, unlysed cells, and debris. The solublefraction was collected for protein concentration determi-nation and subsequent assay analysis.The hypotonic lysate (25 mg protein), 50 mg/mL cyto-
chrome c and 10 mmol/L dATP were combined in amicrocentrifuge tube to afinal volumeof 9mL inhypotoniclysis buffer followed by addition of birinapant and incu-bated for 30 minutes at room temperature. Followingincubation, 50 mL of hypotonic lysis buffer containing5 mmol/L of profluorescent rhodamine-110(2)–based cas-pase-3 substrate zDEVD-R110r was added and fluores-cence intensity was monitored using a Perkin-ElmerVictor2V multilabel plate reader.
Measurement of caspase activity in intact cellsMDA-MB-231 cells were seeded in 96-well plates at an
approximate density of 20,000 cells per well and allowedto adhere overnight. The next day, birinapant was addedat various concentrations, and the plates were incubatedat 37�C and 5% CO2 for an additional 24 hours. Followingthe 24 hours incubation, Caspase-Glo assay (Promega)was performed according to manufacturer’s instructions.Luminescense was read on a Perkin-Elmer Victor2V mul-tiplate reader.
GFP-cIAP degradation assayA375 GFP-cIAP1 and GFP-cIAP2 cells plated in a 96-
well plate were treated with various concentrations ofbirinapant as indicated in the figures for either 2 hours or24 hours. After the incubation, cells were collected bytrypsinization and suspended in PBS. A total of 104 cellswere analyzed using a FACScan (Becton Dickinson). GFPfluorescence was detected using 488 nm excitation and530 nm emission wavelengths.
Cell viability assayViabilitywasdetermined in 96-well plates byMTTassay.
Briefly, cells were seeded in 96-well plates at an approxi-mate density of 5,000 to 10,000 cells perwell and allowed toadhereovernight. Thenextday, test agentswere addedandthe plates were incubated at 37�C and 5% CO2 for anadditional 72 hours. Following the 72 hours incubation,50 mL of 5 mg/mL MTT reagent was added to each well(final concentration, 1 mg/mL). The IC50 was derived bynonlinear regression analysis using GraphPad Prism 4.
Xenograft methodsFemale athymic nude (nu/nu)mice (Harlan) between 5
to 8 weeks of age were housed on irradiated corncobbedding (Teklad) in individual HEPA-ventilated cages(Sealsafe Plus, Techniplast) on a 12 hours light–dark cycleat 21�C to 23�C and 40% to 60% humidity. Animals werefed water ad libitum (reverse osmosis, 2 ppm Cl2) and anirradiated standard rodent diet (Teklad2919) consisting of18% protein, 9% fat, and 34% fiber. Low-passage patient-derived xenograft models representing human ovariancancer, melanoma, and colorectal cancer were implantedunilaterally on the right flank with tumor fragmentsharvested from 2 to 4 host animals. Prestudy tumorvolumes were recorded for each experiment beginningapproximately 1 week before its estimated start date.When tumors reached approximately 122 to 208 mm3,animals were matched by tumor volume into treatmentand control groups and dosing initiated (Day 0); animalswere tagged and followed individually throughout theexperiment. Birinapant was formulated in 12.5% Captisolin sterile water and adjusted to pH 4.0 using concentratedhydrochloric acid. Dosing for vehicle and birinapantbegan on Day 0; animals in all groups were dosed byweight (0.01 mL per gram; 10 mL/kg). Control animals incolorectal studies are untreated. Animals were dosed byintraperitoneal injection 3 times a day, as indicated.
Preclinical Activity of Birinapant, a Bivalent SMAC Mimetic
www.aacrjournals.org Mol Cancer Ther; 13(4) April 2014 869
on June 13, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 21, 2014; DOI: 10.1158/1535-7163.MCT-13-0798
Beginning on Day 0, animals were observed daily andweighed twice weekly (data not shown) using a digitalscale. Beginning on Day 0, tumor dimensions were mea-sured twice weekly by digital caliper (Fowler Ultra-CalIV) and datawere recorded for each group; tumor volumewas calculated using the formula: TV¼width2� length�0.52.
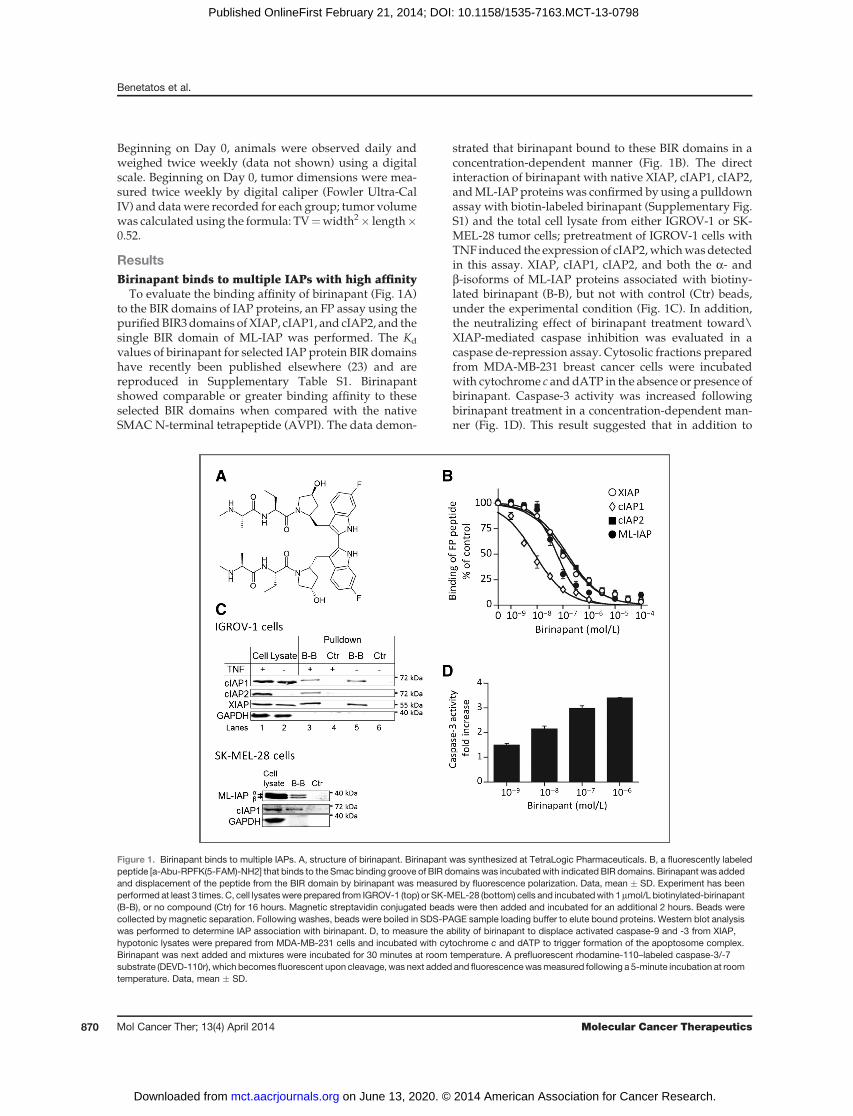
ResultsBirinapant binds to multiple IAPs with high affinity
To evaluate the binding affinity of birinapant (Fig. 1A)to the BIR domains of IAP proteins, an FP assay using thepurifiedBIR3domains of XIAP, cIAP1, and cIAP2, and thesingle BIR domain of ML-IAP was performed. The Kd
values of birinapant for selected IAP protein BIR domainshave recently been published elsewhere (23) and arereproduced in Supplementary Table S1. Birinapantshowed comparable or greater binding affinity to theseselected BIR domains when compared with the nativeSMAC N-terminal tetrapeptide (AVPI). The data demon-
strated that birinapant bound to these BIR domains in aconcentration-dependent manner (Fig. 1B). The directinteraction of birinapant with native XIAP, cIAP1, cIAP2,andML-IAP proteinswas confirmed by using a pulldownassay with biotin-labeled birinapant (Supplementary Fig.S1) and the total cell lysate from either IGROV-1 or SK-MEL-28 tumor cells; pretreatment of IGROV-1 cells withTNF induced the expression of cIAP2,whichwasdetectedin this assay. XIAP, cIAP1, cIAP2, and both the a- andb-isoforms of ML-IAP proteins associated with biotiny-lated birinapant (B-B), but not with control (Ctr) beads,under the experimental condition (Fig. 1C). In addition,the neutralizing effect of birinapant treatment toward\XIAP-mediated caspase inhibition was evaluated in acaspase de-repression assay. Cytosolic fractions preparedfrom MDA-MB-231 breast cancer cells were incubatedwith cytochrome c anddATP in the absence or presence ofbirinapant. Caspase-3 activity was increased followingbirinapant treatment in a concentration-dependent man-ner (Fig. 1D). This result suggested that in addition to
Figure 1. Birinapant binds to multiple IAPs. A, structure of birinapant. Birinapant was synthesized at TetraLogic Pharmaceuticals. B, a fluorescently labeledpeptide [a-Abu-RPFK(5-FAM)-NH2] that binds to the Smac binding groove of BIR domains was incubatedwith indicated BIR domains. Birinapant was addedand displacement of the peptide from the BIR domain by birinapant was measured by fluorescence polarization. Data, mean � SD. Experiment has beenperformed at least 3 times. C, cell lysateswere prepared from IGROV-1 (top) or SK-MEL-28 (bottom) cells and incubatedwith 1mmol/L biotinylated-birinapant(B-B), or no compound (Ctr) for 16 hours. Magnetic streptavidin conjugated beads were then added and incubated for an additional 2 hours. Beads werecollected by magnetic separation. Following washes, beads were boiled in SDS-PAGE sample loading buffer to elute bound proteins. Western blot analysiswas performed to determine IAP association with birinapant. D, to measure the ability of birinapant to displace activated caspase-9 and -3 from XIAP,hypotonic lysates were prepared from MDA-MB-231 cells and incubated with cytochrome c and dATP to trigger formation of the apoptosome complex.Birinapant was next added and mixtures were incubated for 30 minutes at room temperature. A prefluorescent rhodamine-110–labeled caspase-3/-7substrate (DEVD-110r), which becomes fluorescent upon cleavage, was next added and fluorescencewasmeasured following a 5-minute incubation at roomtemperature. Data, mean � SD.
Benetatos et al.
Mol Cancer Ther; 13(4) April 2014 Molecular Cancer Therapeutics870
on June 13, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 21, 2014; DOI: 10.1158/1535-7163.MCT-13-0798

binding to XIAP, birinapantwas able to displace activatedcaspases from XIAP sequestration.
Birinapant induces degradation of cIAP1 and cIAP2in cellsTo evaluate the ability of birinapant to degrade cIAP1
and cIAP2 in intact cells, we established cell lines thatstably expressed GFP-tagged cIAP1 (GFP-cIAP1) or GFP-cIAP2. This allowed for the quantitative determination ofIC50 values for the degradation of each cIAP protein bymonitoring GFP fluorescence by flow cytometry. Follow-ing 2-hour birinapant treatment, IC50 values for GFP-cIAP1 and GFP-cIAP2 were 17 � 11 nmol/L and 108 �46 nmol/L, respectively (Supplementary Fig. S2A). Weconfirmed that both endogenous cIAP1 and cIAP2 weredegraded synchronously with their GFP-tagged counter-parts by western blot analysis (Supplementary Fig. S2B).As described previously, pretreatment with TNF wasused to induce the expression of endogenous cIAP2 inthe A375 cell line to facilitate this analysis.
Birinapant was active in a variety of cancer cell linesThe activity of birinapant was tested as a single agent
and in combinationwith TNF and TRAIL in a panel of 111cell lines representing multiple tumor types (Supplemen-tary Table S2). Eighteen of the 111 cell lines (18/111) weresensitive to birinapant as a single agent, with IC50 values<1 mmol/L. For evaluation of the activity of birinapant incombinationwith TNF or TRAIL, cell lineswere treated ina matrix pattern consisting of a range of concentrations ofboth birinapant and TNF or TRAIL (Supplementary Fig.S3).Datawere analyzedusing theMacSynergy II program(24). Combinations resulting in synergy volumes >100mmol/L2% were considered synergistic and are also indi-cated in Supplementary Table S2. The addition of TNF orTRAIL resulted in the sensitization to birinapant ofapproximately 45% of the single agent birinapant-resis-tant cell lines. Several of these cell lines were also resistantto both TNF and/or TRAIL as single agents, highlightingthe synergistic nature of these combinations. In addition,no normal cells tested (HUVEC,MRC-5) were sensitive tobirinapant alone or in combination with either TNF orTRAIL (data not shown). Taken together, these resultsillustrate that approximately 60% of cell lines were sen-sitive to birinapant as a single agent or in combinationwith TNF or TRAIL.It has been shown that the single-agent activity of
SMAC mimetics is dependent upon autocrine secretionof TNF (25–30). Consistent with these findings, birina-pant-sensitive tumor cell lines secreted TNF followingbirinapant treatment whereas no TNF secretion wasobserved in birinapant-resistant tumor cell lines (Supple-mentary Fig. S4). To confirm that the autocrine productionof TNF was responsible for the cytotoxic effect of birina-pant, pretreatment of the birinapant-sensitive cell linesSK-OV-3 and EVSA-T with a TNF neutralizing antibodyreduced birinapant-mediated cytotoxicity (Supplementa-ry Fig. S5).
The activation of caspases and induction of cell deathfollowing birinapant treatment occurred below thedrug concentration required for completedegradation of total cIAP2
The rapid loss of both cIAP1 and cIAP2 in tumor cellsfollowing treatment with certain SMAC mimetics hasbeen associated with tumor cell death in sensitive celllines (28–30). However, in some birinapant-sensitive can-cer cell lines, we observed that the induction of cell deathfollowing birinapant treatment occurred below the drugconcentration required for a detectable decrease in totalcIAP2 (Fig. 2). For example, in the MDA-MB-231 breastcancer cell line, 1 nmol/L birinapant treatment resulted in>50% cIAP1 loss, without any observed effect on totalcIAP2 levels (Fig. 2A), togetherwith activation of caspasesand cell death induction (Fig. 2C and D). At 10 nmol/Lbirinapant treatment, cIAP1 and cIAP2 loss was approx-imately 90% and 40%, respectively (Fig. 2B). Moreover,Western blot analysis of birinapant-treatedMDA-MB-231cells indicated a dose-dependent loss of cIAP1 whereascIAP2 levels remained relatively stable until caspase-3–mediated proteolysis of PARP—a measure of apoptosisinduction—became evident at 10 nmol/L birinapant(Fig. 2E).
Birinapant preferentially induces the degradation ofthe TRAF2-associated pool of cIAPs
Cellular IAP2 levels are controlled, at least in part, by theE3 ligaseactivityof cIAP1 (31). The apparentdissociationofcIAP2 loss and tumor cell death induction suggested thateither cIAP1 alone or a specific subset of cIAP proteinsmight instead be the physiologic targets of birinapant. Wetherefore investigated the action of birinapant on thosecIAP proteins, which are specifically associated withTNFR1 signaling. The TNF receptor–associated factor 2(TRAF2) is a recognized cIAP binding partner that recruitsboth cIAP1andcIAP2 toTNFR1 followingTNFstimulation(32–34). A375 cells stably expressing GFP-cIAP1 or GFP-cIAP2 were utilized for coimmunoprecipitation with ananti-TRAF2 antibody owing to the high expression levelof both GFP-cIAP constructs (infra). Following treatmentwith 100 nmol/L birinapant, endogenous TRAF2-boundcIAP1 and TRAF2-bound GFP-cIAP1 were synchronouslydegraded with similar kinetics, which accurately reflectedtotal cIAP1 loss (Fig. 3A, top left). Birinapant also inducedthe degradation of TRAF2-boundGFP-cIAP2 in 2 indepen-dent experiments (Fig. 3A, top right and middle). Interest-ingly, the majority of non–TRAF2-bound GFP-cIAP2remained intact. Quantitation of the second Western blotanalysis confirmed that the TRAF2-bound pool of cIAP2was, in fact, degraded preferentially to the non–TRAF2-bound pool (Fig. 3A, bottom). Notably, endogenous cIAP2was poorly detected owing to its low expression level. Toconfirm these observations using endogenous cIAP2, wepretreated HeLa cells with TNF (10 ng/mL) to induce theexpression of endogenous cIAP2 followed by treatmentwith a range of birinapant doses (Supplementary Fig. S6A).Consistent with the previous results, TRAF2-bound cIAP2
Preclinical Activity of Birinapant, a Bivalent SMAC Mimetic
www.aacrjournals.org Mol Cancer Ther; 13(4) April 2014 871
on June 13, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 21, 2014; DOI: 10.1158/1535-7163.MCT-13-0798

was reduced following birinapant treatment. By analyzingthe TRAF2 immunoprecipitation Western blots, we esti-mated that the TRAF2-bound cIAP1 in HeLa cells wasapproximately 25% of the total cIAP1 (data not shown).
In a similar fashion, treatment of HeLa cells with 10nmol/L of birinapant induced the degradation of TRAF2-bound cIAP1with slightly faster kinetics than total cIAP1,including non–TRAF2-bound cIAP1, and quantitation ofthis Western blot analysis reflected these observations(Fig. 3A, leftmiddle andbottom). Like cIAP2,weanalyzedthe effects on cIAP1 levels inHeLa cells using adose rangeof birinapant (Supplementary Fig. S6B). Owing to theobservable levels of endogenous cIAP1, the HeLa cellswere not pretreated with TNF. Consistently, the TRAF2-bound pool of cIAP1 was degraded with slightly greaterefficiency than the total cIAP1 following birinapanttreatment.
These results suggested that birinapant was preferen-tially targeting theTRAF2-bound cIAPs to elicit tumor celldeath. In addition, cIAP1 was more sensitive to birina-pant-induced degradation than cIAP2, which may reflectan inherent difference between these 2 proteins towardSMAC-mimetic treatment (35). To exclude the possibilitythat birinapant was displacing cIAP1 or cIAP2 fromTRAF2, similar TRAF2 pulldown experiments were per-
formed in the presence of the proteasome inhibitor borte-zomib. Following treatment with birinapant plus bortezo-mib, both cIAP1 and cIAP2 remained associated withTRAF2 upon pulldown analysis (data not shown). Thisdemonstrated that birinapant was not disrupting the asso-ciationofTRAF2witheither cIAP1orcIAP2butwas insteadinducing their proteasome-mediated degradation.
TRAF2-deficient or cIAP1/cIAP2 double knockoutmouse fibroblast cells are unable to activate NF-kB inresponse to TNF stimulation (34, 36). To further confirmthat birinapant induced the preferential degradation ofTRAF2-bound cIAPs,we used anNF-kBpromoter-drivenluciferase assay to evaluate birinapant treatment on NF-kB activity. Pretreatment of HeLa cells that stably harborthe NF-kB-luciferase reporter gene with birinapantblocked TNF-mediated NF-kB activation in a concentra-tion-dependent manner (Fig. 3B). TNF treatment of thesecells caused a rapid (10 minutes) loss of the endogenousinhibitor of NF-kB a (IkBa), which could be abrogated bybirinapant pretreatment (Fig. 3C). In A375 cells stablyoverexpressing GFP-cIAP1 or GFP-cIAP2, the TNF-medi-ated loss of IkBa was also blocked by pretreatment withbirinapant (Supplementary Fig. S7). Taken together, theseresults demonstrated that birinapant treatment resultedin the loss of TRAF2-bound cIAP1/2 and the subsequent
Figure 2. The activation ofcaspases and induction of celldeath following birinapanttreatment occurred below the drugconcentration required forcomplete degradation of totalcIAP2. A and B, time-courseanalysis of birinapant-mediateddegradation of cIAP1 and cIAP2 inMDA-MB-231, birinapant-sensitive breast cancer cells.MDA-MB-231 cells were treatedwith 1 nmol/L (A) or 10 nmol/L (B) ofbirinapant and the total cell lysateat each time pointwas subjected toWestern blot analysis. The intensityof cIAP1 andcIAP2detected on theWestern blot analysis wasquantified. C, dose–responseanalysis of birinapant-inducedcaspase activity in MDA-MB-231cells. D, dose–response analysis ofbirinapant in MDA-MB-231 cellsmeasured by MTT assay. Data,mean � SD and representative ofmultiple experiments. E, Westernblot analysis of cIAP1, cIAP2, andPARP cleavage in MDA-MB-231cells treated with variousconcentrations of birinapant.
Benetatos et al.
Mol Cancer Ther; 13(4) April 2014 Molecular Cancer Therapeutics872
on June 13, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 21, 2014; DOI: 10.1158/1535-7163.MCT-13-0798
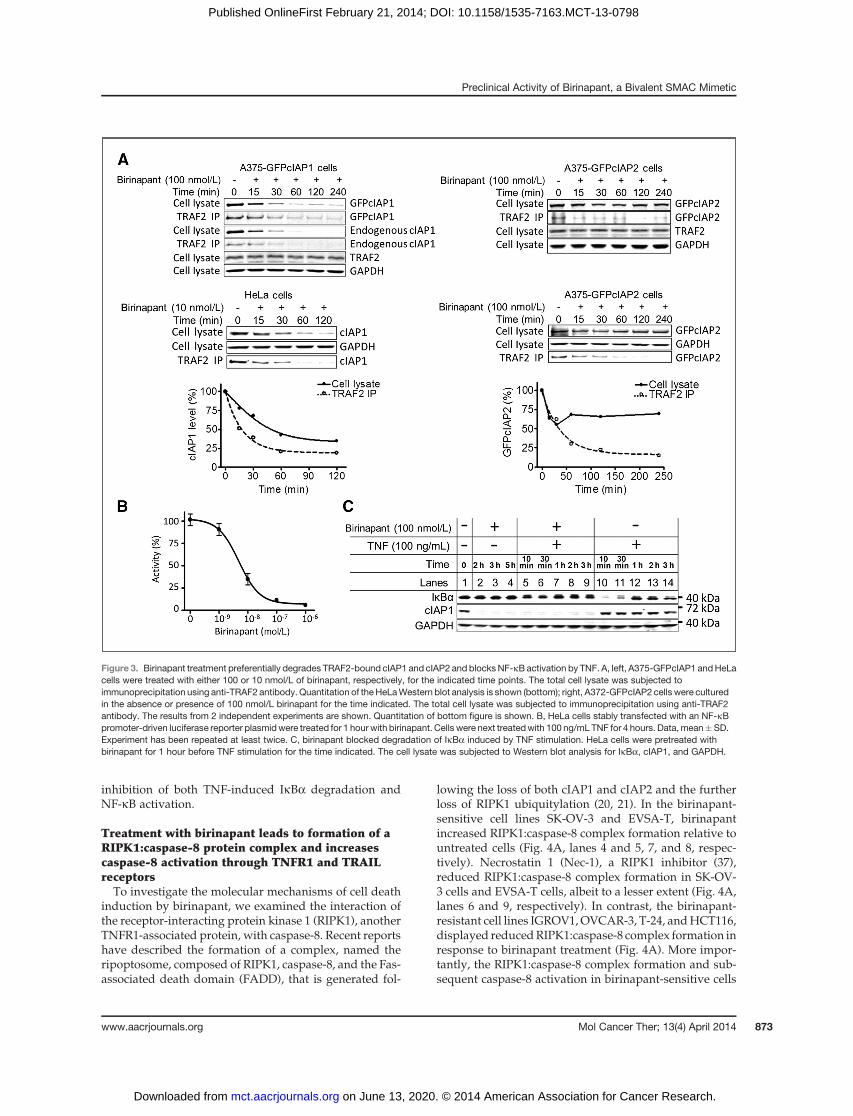
inhibition of both TNF-induced IkBa degradation andNF-kB activation.
Treatment with birinapant leads to formation of aRIPK1:caspase-8 protein complex and increasescaspase-8 activation through TNFR1 and TRAILreceptorsTo investigate the molecular mechanisms of cell death
induction by birinapant, we examined the interaction ofthe receptor-interacting protein kinase 1 (RIPK1), anotherTNFR1-associated protein, with caspase-8. Recent reportshave described the formation of a complex, named theripoptosome, composed of RIPK1, caspase-8, and the Fas-associated death domain (FADD), that is generated fol-
lowing the loss of both cIAP1 and cIAP2 and the furtherloss of RIPK1 ubiquitylation (20, 21). In the birinapant-sensitive cell lines SK-OV-3 and EVSA-T, birinapantincreased RIPK1:caspase-8 complex formation relative tountreated cells (Fig. 4A, lanes 4 and 5, 7, and 8, respec-tively). Necrostatin 1 (Nec-1), a RIPK1 inhibitor (37),reduced RIPK1:caspase-8 complex formation in SK-OV-3 cells and EVSA-T cells, albeit to a lesser extent (Fig. 4A,lanes 6 and 9, respectively). In contrast, the birinapant-resistant cell lines IGROV1,OVCAR-3, T-24, andHCT116,displayed reducedRIPK1:caspase-8 complex formation inresponse to birinapant treatment (Fig. 4A). More impor-tantly, the RIPK1:caspase-8 complex formation and sub-sequent caspase-8 activation in birinapant-sensitive cells
Figure 3. Birinapant treatment preferentially degrades TRAF2-bound cIAP1 and cIAP2 and blocksNF-kB activation by TNF. A, left, A375-GFPcIAP1 andHeLacells were treated with either 100 or 10 nmol/L of birinapant, respectively, for the indicated time points. The total cell lysate was subjected toimmunoprecipitation using anti-TRAF2 antibody. Quantitation of the HeLaWestern blot analysis is shown (bottom); right, A372-GFPcIAP2 cells were culturedin the absence or presence of 100 nmol/L birinapant for the time indicated. The total cell lysate was subjected to immunoprecipitation using anti-TRAF2antibody. The results from 2 independent experiments are shown. Quantitation of bottom figure is shown. B, HeLa cells stably transfected with an NF-kBpromoter-driven luciferase reporter plasmidwere treated for 1 hour with birinapant. Cells were next treatedwith 100 ng/mL TNF for 4 hours. Data, mean�SD.Experiment has been repeated at least twice. C, birinapant blocked degradation of IkBa induced by TNF stimulation. HeLa cells were pretreated withbirinapant for 1 hour before TNF stimulation for the time indicated. The cell lysate was subjected to Western blot analysis for IkBa, cIAP1, and GAPDH.
Preclinical Activity of Birinapant, a Bivalent SMAC Mimetic
www.aacrjournals.org Mol Cancer Ther; 13(4) April 2014 873
on June 13, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 21, 2014; DOI: 10.1158/1535-7163.MCT-13-0798

was attenuated by a neutralizing anti-TNF antibody,suggesting that autocrine TNF secretion played a rolein promoting RIPK1:caspase-8 formation in these birina-pant-sensitive cells (Fig. 4B). Consistentwith a role of TNFin ripoptosome formation, the addition of exogenous TNFto the birinapant-resistant A375 or HeLa cells enhancedthe formation of the RIPK1:caspase-8 complex in thepresence of birinapant (Fig. 4C, top and bottom, lane 7).The RIPK1:caspase-8 complex formation was transient.This is consistent with reports that RIPK1 is a target ofactive caspase-8 (38) and thus as caspase-8 is activated,RIPK1 is cleaved, and the RIPK1:caspase-8 complex isdestabilized (Fig. 4C, top and bottom, lanes 8 and 9). Ofnote, the upregulation of cIAP2 by TNF was reduced inthe presence of birinapant (Fig. 4C, lanes 5 and 9), which
may contribute to the enhanced apoptosis induction bybirinapant/TNF cotreatment. In addition, birinapantenhanced (ca. 10-fold) the activation of caspase-8 byTRAIL in A375 cells (Fig. 4D). These results suggestedthat birinapant promoted the intracellular cell death com-plex formation, which activated caspase-8 and sensitizedtumor cells to TNF and TRAIL receptor–mediated cellkilling.
Birinapant can potentiate the cytotoxicity of certainchemotherapeutic drugs in vitro
Birinapant increased the potency of several chemother-apeutic agents, including SN-38 (the active metabolite ofirinotecan), gemcitabine, and 5-azacytidine (Fig. 5A). Thisproperty of birinapant seemed to be independent of p53
Figure 4. Birinapant treatment leads to the formation of a RIPK1 and caspase-8 protein complex in a RIPK1 kinase activity-dependent manner and enhancescaspase-8 activation by TRAIL. A, birinapant promoted caspase-8 and RIPK1 protein complex formation in birinapant-sensitive cancer cells, which wasattenuated by Nec-1, a RIPK1 kinase inhibitor. Indicated cell lines were treated with 1 mmol/L birinapant in the absence or presence of 5 mmol/L Nec-1. Celllysates were subjected to immunoprecipitation with anti–caspase-8 antibody as described in the Materials and Methods section. B, RIPK1 and caspase-8complex formation inducedbybirinapant is augmentedby autocrine TNF. EVSA-T andSK-OV-3cellswere pretreatedwith anti-TNFantibody for 1 hour beforebirinapant treatment for 4 hours. Cell lysates were subjected to immunoprecipitation with anti–caspase-8 antibody and Western blot analysis. C, incombination with TNF, birinapant induces RIPK1 and caspase-8 interaction before activation of caspases in birinapant-resistant cells. A375 and HeLa cellswere pretreated with birinapant for 1 hour before TNF treatment. The immunoprecipitations and Western blot analyses were carried out as describedabove. D, birinapant enhances caspase-8 activation by TRAIL. A375 cells were pretreated with birinapant for 1 hour before TRAIL treatment followed byWestern blot analysis. �, immunoglobulin heavy chain.
Benetatos et al.
Mol Cancer Ther; 13(4) April 2014 Molecular Cancer Therapeutics874
on June 13, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 21, 2014; DOI: 10.1158/1535-7163.MCT-13-0798
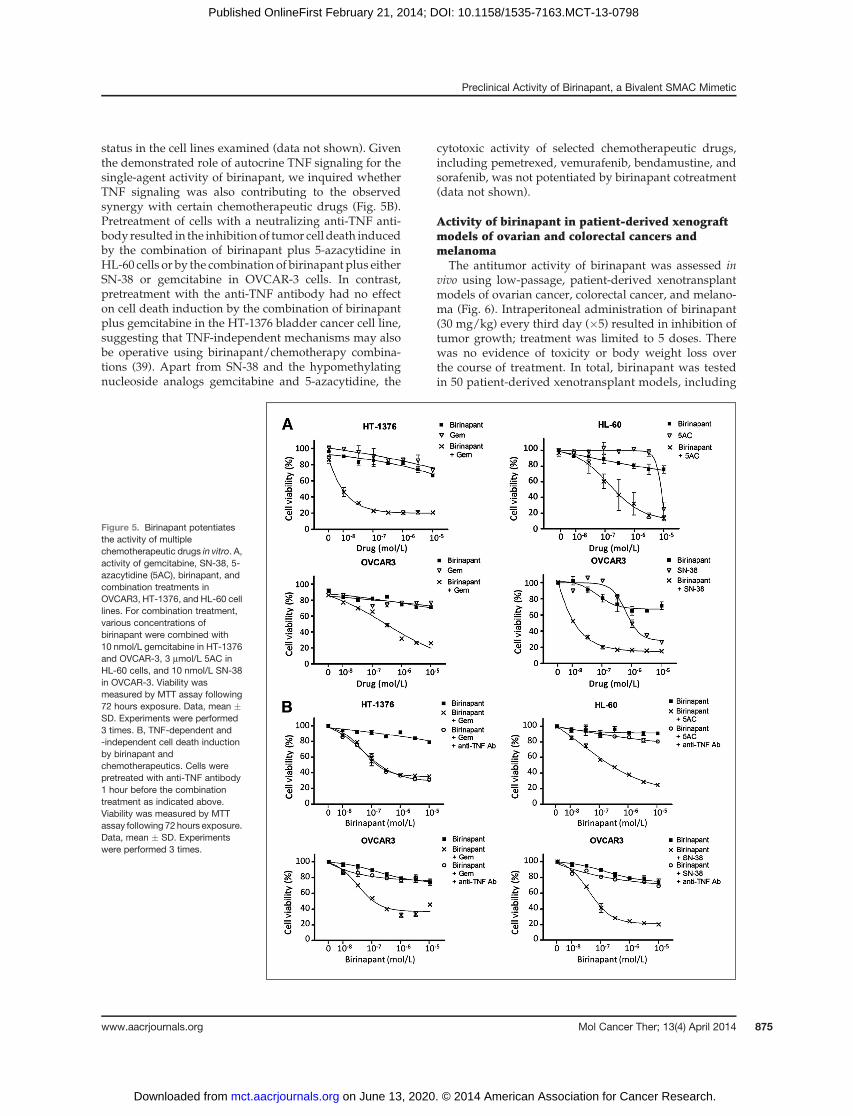
status in the cell lines examined (data not shown). Giventhe demonstrated role of autocrine TNF signaling for thesingle-agent activity of birinapant, we inquired whetherTNF signaling was also contributing to the observedsynergy with certain chemotherapeutic drugs (Fig. 5B).Pretreatment of cells with a neutralizing anti-TNF anti-body resulted in the inhibition of tumor cell death inducedby the combination of birinapant plus 5-azacytidine inHL-60 cells or by the combination of birinapant plus eitherSN-38 or gemcitabine in OVCAR-3 cells. In contrast,pretreatment with the anti-TNF antibody had no effecton cell death induction by the combination of birinapantplus gemcitabine in the HT-1376 bladder cancer cell line,suggesting that TNF-independent mechanisms may alsobe operative using birinapant/chemotherapy combina-tions (39). Apart from SN-38 and the hypomethylatingnucleoside analogs gemcitabine and 5-azacytidine, the
cytotoxic activity of selected chemotherapeutic drugs,including pemetrexed, vemurafenib, bendamustine, andsorafenib, was not potentiated by birinapant cotreatment(data not shown).
Activity of birinapant in patient-derived xenograftmodels of ovarian and colorectal cancers andmelanoma
The antitumor activity of birinapant was assessed invivo using low-passage, patient-derived xenotransplantmodels of ovarian cancer, colorectal cancer, and melano-ma (Fig. 6). Intraperitoneal administration of birinapant(30 mg/kg) every third day (�5) resulted in inhibition oftumor growth; treatment was limited to 5 doses. Therewas no evidence of toxicity or body weight loss overthe course of treatment. In total, birinapant was testedin 50 patient-derived xenotransplant models, including
Figure 5. Birinapant potentiatesthe activity of multiplechemotherapeutic drugs in vitro. A,activity of gemcitabine, SN-38, 5-azacytidine (5AC), birinapant, andcombination treatments inOVCAR3, HT-1376, and HL-60 celllines. For combination treatment,various concentrations ofbirinapant were combined with10 nmol/L gemcitabine in HT-1376and OVCAR-3, 3 mmol/L 5AC inHL-60 cells, and 10 nmol/L SN-38in OVCAR-3. Viability wasmeasured by MTT assay following72 hours exposure. Data, mean �SD. Experiments were performed3 times. B, TNF-dependent and-independent cell death inductionby birinapant andchemotherapeutics. Cells werepretreated with anti-TNF antibody1 hour before the combinationtreatment as indicated above.Viability was measured by MTTassay following 72 hours exposure.Data, mean � SD. Experimentswere performed 3 times.
Preclinical Activity of Birinapant, a Bivalent SMAC Mimetic
www.aacrjournals.org Mol Cancer Ther; 13(4) April 2014 875
on June 13, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 21, 2014; DOI: 10.1158/1535-7163.MCT-13-0798

ovarian and colorectal cancers and melanoma, andactivity was observed in approximately one third ofthe models tested (data not shown). These studies high-light the efficacy and tolerability of birinapant in pre-clinical models.
DiscussionOne hallmark of cancer cells is a compromised ability to
undergo apoptosis. Therefore, strategies targeting celldeath regulators such as the IAP family of proteins withthe goal of overcoming resistance to apoptosis have ther-apeutic potential for the development of new classes ofanticancer drugs.
Unlike XIAP, the cIAPs are not direct inhibitors ofcaspases; however, cIAPs play an important role in inhi-biting the TNF-mediated extrinsic cell death pathwayand enabling the activation of TNF-induced NF-kB pro-survival signaling (6, 25, 40). In this study, we showthat birinapant, a biindole-based bivalent SMACmimetic,is able to induce the degradation of specific pools of cIAPs
while leaving others intact. In addition, birinapant inhi-bits TNF-mediated NF-kB activity, induces cell death as asingle agent and in combination with TNF or TRAIL in avariety of cancer cell lines, and potentiates the activity ofchemotherapeutic drugs in both TNF-dependent andindependent manners. Birinapant was also active in pri-mary patient-derived xenotransplant models.
Western blot analysis of birinapant-induced cIAP1 andcIAP2 degradation in total protein lysates as well as theanalysis of the degradation of GFP-tagged cIAP1 andcIAP2 in intact cells suggested that birinapantwas capableof inducing apoptosis at concentrations that resulted inthe complete degradation of cIAP1 but only partial deg-radation of cIAP2. This resultwas in conflictwithmultiplereports describing the redundant functions of cIAP1 andcIAP2 within the TNFR1 complex as well as the require-ment of antagonizing both cIAP1 and cIAP2 for inducingTNF-dependent cell death by SMAC mimetics (41). Rea-soning that distinct pools of cIAP1 and cIAP2 may bepreferentially targeted by birinapant, we examined theeffects of birinapant on TRAF2-associated cIAPs in order
A
0 10 20 30 40 50 60
100
1,000
Time (days)
Tu
mo
r vo
lum
e (
mm
3)
Ovarian PDX 88
0 10 20 30 40 50–20
–10
0
10
20
Time (days)B
od
y w
eig
ht ch
an
ge
(%
) Ovarian PDX 88
0 10 20 30 40 50–20
–10
0
10
20
Time (days)
Bo
dy w
eig
ht ch
an
ge
(%
)
Ovarian PDX 38
0 10 20 30 40 50
100
1,000
Time (days)
Tu
mo
r vo
lum
e (
mm
3)
Ovarian PDX 38Vehicle control Birinapant (30 mg/kg; IP)
0 10 20 30 40
100
1,000
Time (days)
Tu
mo
r vo
lum
e (
mm
3)
Colon PDX 42
0 10 20 30–20
–10
0
10
20
Time (days)
Bo
dy w
eig
ht ch
an
ge
(%
)
Colon PDX 42
0 10 20–20
–10
0
10
20
Time (days)
Bo
dy w
eig
ht ch
an
ge
(%
)
Colon PDX 125
0 10 20 30
100
1,000
Time (days)
Tu
mo
r vo
lum
e (
mm
3)
Colon PDX 125
B
C
0 10 20 30–20
–10
0
10
20
Time (days)
Bo
dy w
eig
ht ch
an
ge
(%
)
Melanoma PDX 150
0 10 20 30 40 50
100
1,000
Time (days)
Tu
mo
r vo
lum
e (
mm
3)
Melanoma PDX 150
0 10 20 30 40 50
100
1,000
Time (days)
Tu
mo
r vo
lum
e (
mm
3)
Melanoma PDX 54
0 10 20 30 40 50–20
–10
0
10
20
Time (days)B
od
y w
eig
ht ch
an
ge
(%
)
Melanoma PDX 54
Vehicle control Birinapant (30 mg/kg; IP)
Vehicle control Birinapant (30 mg/kg; IP)
Figure 6. Birinapant exhibits single-agent activity in patient-derived xenograft models of ovarian and colorectal cancer andmelanoma at awell-tolerated dose.Birinapant activity in low-passage, patient-derived ovarian cancer (A), colorectal cancer (B), andmelanoma (C). Vehicle control (*) or birinapant (30mg/kg;*)dosed intraperitoneally 3 times a day as indicated (").
Benetatos et al.
Mol Cancer Ther; 13(4) April 2014 Molecular Cancer Therapeutics876
on June 13, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 21, 2014; DOI: 10.1158/1535-7163.MCT-13-0798

to understand these seemingly contradictory results. Wefound that a characteristic feature of birinapant was thepreferential degradation of TRAF2-bound cIAP1 andTRAF2-bound cIAP2.Recruitment of cIAP1 or cIAP2 to the TNFR1 complex
by TRAF2 is necessary for TNF-mediated canonical NF-kB activation (34). Both cIAP1 and cIAP2, via their RINGdomain-mediatedE3 ligase activity, can transfer ubiquitinor poly-ubiquitin chains (i.e., ubiquitylation) to RIPK1,which allows for the recruitment of the IKK complex(IKKa/IKKb/IKKg) to TNFR1 thus enabling the initiationof TNF-induced NF-kB activation. In the absence ofTRAF2 or cIAPs, this ubiquitylated RIPK1-dependentcomplex fails to assemble and, hence, TNF-mediatedNF-kB signaling is blocked and apoptosis is initiated.The specificmechanismsunderlying the regulationof the
TRAF2-boundandnon–TRAF2-boundcIAP1andcIAP2bybirinapant are unknown. It was recently demonstrated thata bivalent SMACmimetic, BV6, induced the autoubiquity-lation anddegradationof the single BIRdomain-containingprotein, ML-IAP, whereas a related monovalent SMACmimetic had no effect. This result suggested that BIRdomain crosslinking may have a mechanistic role in biva-lent SMAC-mimetic–induced E3 ligase activity (42). It istherefore possible that a similar cIAP BIR3 domain cross-linking may be responsible for the selective degradation ofTRAF2-bound cIAPs by birinapant.It has also been reported that cIAP2 is a substrate for
cIAP1-mediated ubiquitylation and proteasomal degra-dation and that cIAP1 is necessary for the efficient SMAC-mimetic–induced degradation of cIAP2 (43, 44). In addi-tion, following SMAC-mimetic treatment, cIAP2 levelsare stabilized possibly because of prolonged suppressionof cIAP1 E3 ligase activity (44, 45). We found that birina-pant degrades both TRAF2-bound and non–TRAF2-bound cIAP1.Although TRAF2-bound cIAP2 is subject to birinapant-
mediated degradation, non–TRAF2-bound cIAP2 is part-ly spared from birinapant-induced degradation. In con-trast, treatment with the pan-IAP antagonist, CompoundA, induced the degradation of TRAF2-bound and non–TRAF2-bound pools of both cIAP1 and cIAP2, indicatingthat different SMAC mimetics can elicit selective effectson these 2 proteins (46). To affect the ubiquitylation/degradation of TRAF2-bound cIAP2 while sparing non–TRAF2-bound cIAP2, birinapant may selectively stabilizethe homodimeric TRAF2-bound cIAP2 E3 ligase complexor, alternatively, a heterodimeric TRAF2-bound cIAP1:cIAP2 E3 complex might be engaged. Heterodimeric E3ligases such as BRCA:BARD and MDM2:MDMX havebeen reported (47, 48). Alternatively, different SMAC-mimetic–ligated homodimeric cIAP1 E3 ligases may offerunique ubiquitin transfer kinetics, which allows non–TRAF2-bound cIAP2 to partially escape birinapant-medi-ated ubiquitylation whereas a Compound A–derivedcIAP1 E3 ligase would offer no selectivity.Following TNF binding, the cIAPs ubiquitylate RIPK1
allowing the assembly of a TNFR1 signaling complex
composed of the IKK (IKKa/IKKb/IKKg) and TAK/TABcomplexes. Recent reports have described the spontane-ous formation of the ripoptosome, composed of RIPK1,caspase-8, and the FADD, generated via loss of cIAP1 andcIAP2 (20, 21). In the tumor cell lines tested, birinapanttreatment induced cell death despite sparing of non–TRAF2-bound cIAP2. This suggested that RIPK1 was nota substrate for non–TRAF2-bound cIAP2 and thus non–TRAF2-bound cIAP2 does not seem to contribute to apo-ptosis resistance.
The ability to potentiate the cytotoxicity of mechanis-tically distinct chemotherapeutic drugs is relevant to theclinical development of birinapant. Drugs such as irino-tecan and gemcitabine are important components of clin-ical anticancer regimens (49). We observed that TNFcontributes to the activity of birinapant in combinationwith SN-38 (the active metabolite of irinotecan), gemcita-bine, and5-azacytidine in certain tumor cell lines,which isconsistent with other reports of SMAC-mimetic use incombination with chemotherapeutic drugs (50). Theseobservations, as well as the ability of TNF to broadlysensitize cell lines to birinapant, support the clinical par-adigm of combining birinapant with agents capable ofincreasing TNF levels within the tumor microenviron-ment. TNF-independent mechanisms of SMAC-mimetic–mediated synergywith chemotherapeutics have also beenreported in lung cancer cell lines (39). Consistent withthese observations, we have observed that TNF neutral-ization does not block birinapant-mediated potentiationof all chemotherapeutic agents, suggestingmultiple proa-poptotic mechanisms of interaction.
The primary patient-derived xenotransplant studieshighlight the activity and tolerability of birinapant in vivo.These models, which were developed from patient-derived tumor explants and passaged in vivo, are consid-ered tomore accurately reflect the original tumor than cellline–derived xenograft models. As described in this arti-cle, significant antitumor efficacy was observed at con-centrations of birinapant that are devoid of overt toxicityto the animals, which highlight the safety and efficacy ofbirinapant in preclinical models. Furthermore, the resultsdescribed in this article support the clinical investigationof birinapant both as a single agent as well as in combi-nation with multiple chemotherapies.
Disclosure of Potential Conflicts of InterestM.A. Graham has ownership interest (including patents) in TetraLogic
Pharmaceuticals. D. Weng has ownership interest (including patents) inTetraLogic Pharmaceuticals Corp. Y. Shi is a scientific advisory boardmember. M.A. McKinlay has ownership interest (including patents) inTetraLogic Pharma. S.K. Chunduru has ownership interest (includingpatents) in TetraLogic. No potential conflicts of interest were disclosedby the other authors.
Authors' ContributionsConception anddesign:C.A.Benetatos, Y.Mitsuuchi, J.M.Burns,Y.Deng,Y.-H. Lee, M.A. Graham, Y. Shi, M.A. McKinlay, S.K. ChunduruDevelopment of methodology: C.A. Benetatos, Y. Mitsuuchi, S.M. Con-don, G. Yu, M.G. LaPorte, S.R. Rippin, M.S. Hendi, P.K. Tirunahari, Y.-H.Lee, M.A. Graham, M.A. McKinlay, S.K. Chunduru
Preclinical Activity of Birinapant, a Bivalent SMAC Mimetic
www.aacrjournals.org Mol Cancer Ther; 13(4) April 2014 877
on June 13, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 21, 2014; DOI: 10.1158/1535-7163.MCT-13-0798

Acquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): Y. Mitsuuchi, E.M. Neiman, G. Yu, M.E. Seipel,M.S. Hendi, M.D. Alexander, M.A. GrahamAnalysis and interpretation of data (e.g., statistical analysis, biostatis-tics, computational analysis):C.A.Benetatos, Y.Mitsuuchi, J.M. Burns,M.S. Hendi, P.K. Tirunahari, T. Haimowitz, M.A. Graham, D. Weng, Y. Shi,M.A. McKinlayWriting, review, and/or revision of the manuscript: C.A. Benetatos, Y.Mitsuuchi, J.M. Burns, S.M. Condon, G.S. Kapoor, M.G. LaPorte, M.S.Hendi, T. Haimowitz, M.D. Alexander, M.A. GrahamAdministrative, technical, or material support (i.e., reporting or orga-nizing data, constructing databases):M.E. Seipel, Y. Deng, T. Haimowitz,D. Weng, M.A. McKinlay, S.K. ChunduruStudy supervision: Y. Mitsuuchi, J.M. Burns, D.Weng, M.A. McKinlay, S.K. Chunduru
AcknowledgmentsThe authors thank Staci Heise (Ph.D.) and Katie Gersh (Ph.D.) of
MedErgy for writing and editorial assistance.
Grant SupportThis work is supported by TetraLogic Pharmaceuticals.The costs of publication of this article were defrayed in part by the
payment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received September 23, 2013; revised January 28, 2014; accepted January30, 2014; published OnlineFirst February 21, 2014.
References1. HanahanD,WeinbergRA.Thehallmarksof cancer.Cell 2000;100:57–70.2. Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell
survival and cooperates with c-myc to immortalize pre-B cells. Nature1988;335:440–2.
3. Schmitt CA, Lowe SW. Apoptosis and therapy. J Pathol 1999;187:127–37.
4. Lowe SW, Lin AW. Apoptosis in cancer. Carcinogenesis 2000;21:485–95.
5. Srinivasula SM, Ashwell JD. IAPs: what's in a name? Mol Cell2008;30:123–35.
6. Gyrd-HansenM, Meier P. IAPs: from caspase inhibitors to modulatorsof NF-kB, inflammation and cancer. Nat Rev Cancer 2010;10:561–74.
7. Eckelman BP, Salvesen GS, Scott FL. Human inhibitor of apoptosisproteins: why XIAP is the black sheep of the family. EMBO Rep2006;7:988–94.
8. WalczakH. TNF and ubiquitin at the crossroads of gene activation, celldeath, inflammation, and cancer. Immunol Rev 2011;244:9–28.
9. Fulda S, Vucic D. Targeting IAP proteins for therapeutic intervention incancer. Nat Rev Drug Disc 2012;11:109–24.
10. Dai Z, ZhuWG,Morrison CD, Brena RM, Smiraglia DJ, Raval A, et al. Acomprehensive search for DNA amplification in lung cancer identifiesinhibitorsof apoptosis cIAP1andcIAP2ascandidateoncogenes.HumMol Genet 2003;12:791–801.
11. Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J,et al. Identification and validation of oncogenes in liver cancer using anintegrative oncogenomic approach. Cell 2006;125:1253–67.
12. Imoto I, Yang ZQ, Pimkhaokham A, Tsuda H, Shimada Y, Imamura M,et al. Identification of cIAP1 as a candidate target gene within anamplicon at 11q22 in esophageal squamous cell carcinomas. CancerRes 2001;61:6629–34.
13. Snijders AM, Schmidt BL, Fridlyand J, Dekker N, Pinkel D, Jordan RC,et al. Rare amplicons implicate frequent deregulation of cell fatespecification pathways in oral squamous cell carcinoma. Oncogene2005;24:4232–42.
14. ReardonDA,Michalkiewicz E, Boyett JM, Sublett JE, Entrekin RE, et al.Extensive genomic abnormalities in childhood medulloblastoma bycomparative genomic hybridization. Cancer Res 1997;57:4042–7.
15. Weber RG, Sommer C, Albert FK, Kiessling M, Cremer T. Clinicallydistinct subgroups of glioblastomamultiforme studied by comparativegenomic hybridization. Lab Invest 1996;74:108–19.
16. Bashyam MD, Bair R, Kim YH, Wang P, Hernandez-Boussard T,Karikari CA, et al. Array-based comparative genomic hybridizationidentifies localized DNA amplifications and homozygous deletions inpancreatic cancer. Neoplasia 2005;7:556–62.
17. Tamm I, Kornblau SM, Segall H, Krajewski S, Welsh K, Kitada S, et al.Expression and prognostic significance of IAP-family genes in humancancers and myeloid leukemias. Clin Cancer Res 2000;6:1796–803.
18. Vucic D, Stennicke HR, PisabarroMT, Salvesen GS, Dixit VM.ML-IAP,a novel inhibitor of apoptosis that is preferentially expressed in humanmelanomas. Curr Biol 2000;10:1359–66.
19. LaCasse EC, Cherton-Horvat GG, Hewitt KE, Jerome LJ, Morris SJ,Kandimalla ER, et al. Preclinical characterization of AEG35156/GEM
640, a second-generation antisense oligonucleotide targeting X-linkedinhibitor of apoptosis. Clin Cancer Res 2006;12:5231–41.
20. Feoktistova M, Geserick P, Kellert B, Dimitrova DP, Langlais C, HupeM, et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8containing intracellular cell death complex differentially regulated bycFLIP isoforms. Mol Cell 2011;43:449–63.
21. TenevT,BianchiK,DardingM,BroemerM,LanglaisC,WallbergF, et al.The Ripoptosome, a signaling platform that assembles in response togenotoxic stress and loss of IAPs. Mol Cell 2011;43:432–48.
22. Nikolovska-Coleska Z, Wang R, Fang X, Pan H, Tomita Y, Li P, et al.Development and optimization of a binding assay for the XIAP BIR3domain using fluorescence polarization. Anal Biochem 2004;332:261–73.
23. Allensworth JL, Sauer SJ, Lyerly HK,MorseMA, DeviGR. SmacmimeticBirinapant induces apoptosis and enhances TRAIL potency in inflam-matorybreast cancer cells in an IAP-dependent andTNF-a-independentmechanism. Breast Cancer Res Treat 2013;137:359–71.
24. Prichard MN, Shipman C Jr. A three-dimensional model to analyzedrug-drug interactions. Antiviral Res 1990;14:181–205.
25. Petersen SL, Wang L, Yalcin-Chin A, Li L, Peyton M, Minna J, et al.Autocrine TNF-a signaling renders human cancer cells susceptible toSmac-mimetic-induced apoptosis. Cancer Cell 2007;12:445–56.
26. Vince JE,WongWW,KhanN, FelthamR,ChauD, AhmedAU, et al. IAPantagonists target cIAP1 to induce TNF-a-dependent apoptosis. Cell2007;131:682–93.
27. Gaither A, Porter D, Yao Y, Borawski J, Yang G, Donovan J, et al. ASmac mimetic rescue screen reveals roles for inhibitor of apoptosisproteins in tumor necrosis factor-a signaling. Cancer Res 2007;67:11493–8.
28. Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, KayagakiN, Garg P, et al. IAP antagonists induce autoubiquitination of c-IAPs,NF-kB activation, and TNF-a-dependent apoptosis. Cell 2007;131:669–81.
29. Lu J, McEachern D, Sun H, Bai L, Peng Y, Qiu S, et al. Therapeuticpotential and molecular mechanism of a novel, potent, nonpeptide,Smac mimetic SM-164 in combination with TRAIL for cancer treat-ment. Mol Cancer Ther 2011;10:902–14.
30. BertrandMJ, Milutinovic S, Dickson KM, HoWC, Boudreault A, DurkinJ, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning asE3 ligases that promote RIP1 ubiquitination. Mol Cell 2008;30:689–700.
31. Cheung HH, Plenchette S, Kern CJ, Mahoney DJ, Korneluk RG. TheRING domain of cIAP1 mediates the degradation of RING-bearinginhibitor of apoptosis proteins by distinct pathways. Mol Biol Cell2008;19:2729–40.
32. Rothe M, Pan MG, Henzel WJ, Ayres TM, Goeddel DV. The TNFR2-TRAF signaling complex contains two novel proteins related to bacu-loviral inhibitor of apoptosis proteins. Cell 1995;83:1243–52.
33. Shu HB, Takeuchi M, Goeddel DV. The tumor necrosis factor receptor2 signal transducers TRAF2 and c-IAP1 are components of the tumornecrosis factor receptor 1 signaling complex. Proc Natl Acad Sci U SA1996;93:13973–8.
Benetatos et al.
Mol Cancer Ther; 13(4) April 2014 Molecular Cancer Therapeutics878
on June 13, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 21, 2014; DOI: 10.1158/1535-7163.MCT-13-0798

34. Vince JE, Pantaki D, Feltham R, Mace PD, Cordier SM, Schmukle AC,et al. TRAF2 must bind to cellular inhibitors of apoptosis for tumornecrosis factor to efficiently activate NF-kBb and to prevent TNF-induced apoptosis. J Biol Chem 2009;284:35906–15.
35. Feltham R, Bettjeman B, Budhidarmo R, Mace PD, Shirley S, CondonSM, et al. Smacmimetics activate theE3 ligase activity of cIAP1proteinby promoting RING domain dimerization. J Biol Chem 2011;286:17015–28.
36. YehWC, Shahinian A, Speiser D, Kraunus J, Billia F, Wakeham A, et al.Early lethality, functional NF-kB activation, and increased sensitivityto TNF-induced cell death in TRAF2-deficient mice. Immunity 1997;7:715–25.
37. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al.Chemical inhibitor of nonapoptotic cell deathwith therapeutic potentialfor ischemic brain injury. Nat Chem Biol 2005;1:112–9.
38. Lin Y, Devin A, Rodriguez Y, Liu ZG. Cleavage of the death domainkinase RIP by caspase-8 prompts TNF-induced apoptosis. GenesDev1991;13:2514–26.
39. GreerRM,PeytonM, Larsen JE,Girard L, XieY,GazdarAF, et al. SMACmimetic (JP1201) sensitizes non-small cell lung cancers to multiplechemotherapy agents in an IAP-dependent but TNF-a-independentmanner. Cancer Res 2011;71:7640–8.
40. Wang L, Du F, Wang X. TNF-a induces two distinct caspase-8 acti-vation pathways. Cell 2008;133:693–703.
41. MahoneyDJ,CheungHH,MradRL, Plenchette S, SimardC, Enwere E,et al. Both cIAP1 and cIAP2 regulate TNF-a-mediated NF-kB activa-tion. Proc Natl Acad Sci U S A 2008;105:11778–83.
42. Varfolomeev E, Moradi E, Dynek JN, Zha J, Fedorova AV, Deshayes K,et al. Characterization ofML-IAPprotein stability andphysiological rolein vivo. Biochem J 2012;447:427–36.
43. Conze DB, Albert L, Ferrick DA, Goeddel DV, Yeh WC, Mak T, et al.Posttranscriptional downregulation of c-IAP2 by the ubiquitin proteinligase c-IAP1 in vivo. Mol Cell Biol 2005;25:3348–56.
44. Darding M, Feltham R, Tenev T, Bianchi K, Benetatos C, Silke J, et al.Molecular determinants of Smac mimetic induced degradation ofcIAP1 and cIAP2. Cell Death Differ 2011;18:1376–86.
45. Petersen SL, Peyton M, Minna JD, Wang X. Overcoming cancer cellresistance to Smac mimetic induced apoptosis by modulating cIAP-2expression. Proc Natl Acad Sci U S A 2010;107:11936–41.
46. Geserick P, Hupe M, Moulin M, Wong WW, Feoktistova M, Kellert B,et al. Cellular IAPs inhibit a cryptic CD95-induced cell death by limitingRIP1 kinase recruitment. J Cell Biol 2009;187:1037–54.
47. Nishikawa H, Wu W, Koike A, Kojima R, Gomi H, Fukuda M, et al.BRCA1-associated protein 1 interferes with BRCA1/BARD1 RINGheterodimer activity. Cancer Res 2009;69:111–9.
48. Linke K,Mace PD, SmithCA, Vaux DL, Silke J, DayCL. Structure of theMDM2/MDMX RING domain heterodimer reveals dimerization isrequired for their ubiquitylation in trans. Cell Death Differ 2008;15:841–8.
49. Diaz-Rubio E. New chemotherapeutic advances in pancreatic, colo-rectal, and gastric cancers. Oncologist 2004;9:282–94.
50. ProbstBL, Liu L,RameshV, Li L, SunH,MinnaJD, et al. Smacmimeticsincrease cancer cell response to chemotherapeutics in a TNF-a-dependent manner. Cell Death Differ 2010;17:1645–54.
www.aacrjournals.org Mol Cancer Ther; 13(4) April 2014 879
Preclinical Activity of Birinapant, a Bivalent SMAC Mimetic
on June 13, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 21, 2014; DOI: 10.1158/1535-7163.MCT-13-0798

Correction
Correction: Birinapant (TL32711), a BivalentSMAC Mimetic, Targets TRAF2-AssociatedcIAPs, Abrogates TNF-Induced NF-kBActivation, and Is Active in Patient-DerivedXenograft Models
In this article (Mol Can Ther 2014 April; 13:867–79), which was published intheApril 2014 issue ofMolecular Cancer Therapeutics (1), the authors regret thatdosing is incorrectly stated as "3 times a day" in the legend for Figure 6 and inthe xenograft section of the Materials and Methods. The correct dosing is"every three days." In addition, the authors acknowledge that the titles of thexenograft models in Fig. 6 incorrectly read as "PDX" followed by a number.They should instead be titled "ST" followed by the number.
The authors would like to acknowledge South Texas Accelerated Research
Therapeutics (START) for performing the in vivo studies.
A
0 10 20 30 40 50 60
100
1,000
Time (days)
Tu
mo
r vo
lum
e (
mm
3)
Ovarian ST088
0 10 20 30 40 50−20
−10
0
10
20
Time (days)
Body w
eig
ht change (
%) Ovarian ST088
0 10 20 30 40 50−20
−10
0
10
20
Time (days)
Bo
dy w
eig
ht
ch
an
ge
(%
) Ovarian ST038
0 10 20 30 40 50
100
1,000
Time (days)
Tu
mo
r vo
lum
e (
mm
3)
Ovarian ST038
Vehicle control Birinapant (30 mg/kg; IP)
0 10 20 30 40
100
1,000
Time (days)
Tu
mo
r vo
lum
e (
mm
3)
Colon ST042
0 10 20 30−20
−10
0
10
20
Time (days)
Body w
eig
ht change (
%) Colon ST042
0 10 20−20
−10
0
10
20
Time (days)
Body w
eig
ht change (
%) Colon ST125
0 10 20 30
100
1,000
Time (days)
Tu
mo
r vo
lum
e (
mm
3)
Colon ST125B
C
0 10 20 30−20
−10
0
10
20
Time (days)
Body w
eig
ht
change (
%)
Melanoma ST150
0 10 20 30 40 50
100
1,000
Time (days)
Tum
or
volu
me (
mm
3)
Melanoma ST150
0 10 20 30 40 50
100
1,000
Time (days)
Tu
mo
r vo
lum
e (
mm
3)
Melanoma ST054
0 10 20 30 40 50−20
−10
0
10
20
Time (days)
Body w
eig
ht
change (
%)
Melanoma ST054
Vehicle control Birinapant (30 mg/kg; IP)
Vehicle control Birinapant (30 mg/kg; IP)
Figure6. Birinapant exhibits single-agent activity in patient-derived xenograftmodels of ovarian andcolorectal cancer, andmelanomaat awell-tolerated dose.Birinapant activitywas shown in lowpassage, patient-derived xenograftmodels of ovarian cancer (A), colorectal cancer (B), andmelanoma (C). Vehicle control(*) or birinapant (30 mg/kg; *) was dosed intraperitoneally once every 3 days as indicated (").
MolecularCancer
Therapeutics
Mol Cancer Ther; 13(9) September 20142246

Reference1. BenetatosCA,Mitsuuchi Y, Burns JM,NeimanEM,CondonSM,YuG, et al. Birinapant (TL32711),
a bivalent SMAC mimetic, targets TRAF2-associated cIAPs, abrogates TNF-induced NF-kBactivation, and is active in patient-derived xenograft models. Mol Can Ther 2014;13:867–79.
Published online August 20, 2014.doi: 10.1158/1535-7163.MCT-14-0497�2014 American Association for Cancer Research.
Correction
www.aacrjournals.org Mol Cancer Ther; 13(9) September 2014 2247

2014;13:867-879. Published OnlineFirst February 21, 2014.Mol Cancer Ther Christopher A. Benetatos, Yasuhiro Mitsuuchi, Jennifer M. Burns, et al. Activation, and Is Active in Patient-Derived Xenograft Models
BκTRAF2-Associated cIAPs, Abrogates TNF-Induced NF-Birinapant (TL32711), a Bivalent SMAC Mimetic, Targets
Updated version
10.1158/1535-7163.MCT-13-0798doi:
Access the most recent version of this article at:
Material
Supplementary
http://mct.aacrjournals.org/content/suppl/2014/02/21/1535-7163.MCT-13-0798.DC1
Access the most recent supplemental material at:
Cited articles
http://mct.aacrjournals.org/content/13/4/867.full#ref-list-1
This article cites 50 articles, 19 of which you can access for free at:
Citing articles
http://mct.aacrjournals.org/content/13/4/867.full#related-urls
This article has been cited by 14 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://mct.aacrjournals.org/content/13/4/867To request permission to re-use all or part of this article, use this link
on June 13, 2020. © 2014 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst February 21, 2014; DOI: 10.1158/1535-7163.MCT-13-0798