biodegradable and biocompatible inorganic–organic hybrid materials. i. synthesis and...
TRANSCRIPT

Biodegradable and Biocompatible Inorganic–OrganicHybrid Materials. I. Synthesis and Characterization
D. TIAN, PH. DUBOIS*, R. JEROME
Center for Education and Research on Macromolecules (CERM), University of Liege, Sart-Tilman, B6,4000 Liege, Belgium
Received 30 October 1996; accepted 30 January 1997
ABSTRACT: The hydroxyl or vinyl end-groups of linear or three-arm star-shaped poly(1-caprolactone) (PCL) chains have been derivatized into triethoxysilane groups reactivein the sol-gel process. New transparent hybrid materials that combine tetraethylor-thosilicate (TEOS) and PCL known for biodegradability and biocompatibility haveaccordingly been prepared. The sol-gel process is, however, limited by the early vitrifi-cation of the reactive system. However, thermal posttreatment can overcome thesediffusional and/or kinetic limitations as assessed by a set of analytical methods. Thethermal stability of PCL is improved by incorporation into the silica network. Con-versely, the thermal stability of the ceramer depends on the effective PCL content. Theextent of PCL incorporation into the silica network depends on PCL molecular weight,number, and reactivity of the PCL functional groups. IR spectroscopy has shown thathydrogen bonding occurs between the ester groups of PCL and residual OH groups ofthe silicate component. q 1997 John Wiley & Sons, Inc. J Polym Sci A: Polym Chem 35: 2295–2309, 1997Keywords: sol-gel process; poly(1-caprolactone); bioglass; ceramer; hybrid material
INTRODUCTION though originally devised for the production of ho-mogeneous ceramics and glasses, this techniquehas proved to be flexible enough for incorporatingDuring the last decade, a steadily increasing at-organic reagents and particularly oligomers ortention has been paid to inorganic–organic hybridpolymers bearing at least two groups reactive inmaterials due to a large range of potential appli-the hydrolysis-condensation process and prefera-cations.1 The sol-gel process is known today as ably attached as chain end-groups. The interest forvery straightforward way to prepare hybrid mate-inorganic–organic hybrid materials has to berials that advantageously combine inorganic ox-found in the possible tailoring of the solid-stateides and organic polymers. The sol-gel process isproperties in relation to the nature and relativebasically a two-step hydrolysis-condensation reac-content of the constitutive organic and inorganiction of metal alkoxides [M(OR)4] , most often tet-components.raethoxysilane [Si(OEt)4)] . In a first step, the
The validity of this concept has been assessedalkoxides are hydrolyzed with formation ofby using successfully polymeric reagents, such as
GM{OH groups, which are then condensed intotriethoxysilane-terminated polydimethylsiloxanea metal oxide network (GM{O{MG) . Al-(PDMS),1c–4 triethoxysilane end-capped poly(tet-ramethylene oxide) (PTMO),1c,3,5–8 triethoxysi-lane end-capped polyoxazoline (POZO),9 triethox-* Research Associate by the Belgian National Found for
the Scientific Research ysilane end-capped poly(oxypropylene) (PPO),3
Correspondence to: R. Jeromea,v-alkoxysilane-terminated hydrogenated poly-Contract grant sponsor: Services Federaux des Affaires Sci-butadiene (H-PBD),4 triethoxysilane end-cappedentifiques, Techniques et Culturelles
q 1997 John Wiley & Sons, Inc. CCC 0887-624X/97/112295-15 poly(arylene ether) ketone (PEK),10 polyisocya-
2295
8G48 96-162T/ 8g48$$162t 06-16-97 18:44:12 polca W: Poly Chem

2296 TIAN, DUBOIS, AND JEROME
nate bearing trialkoxysilyl side chains, 11 tri- of PCL chains bearing pendent alkoxysilanegroups as well. In a second step, PCL containingmethoxysilane end-capped aromatic polyamide,
for example, poly(phenyleneterephthal-amide).12 ceramers will be prepared by the sol-gel processfrom both these functional PCL chains and theThese inorganic–organic hybrid materials are
commonly designated as ‘‘ceramers,’’ because they nonfunctional precursors. The main characteris-tic features of the new hybrid materials will fi-combine the usual process of ceramics and poly-
mers. nally be investigated. The biodegradability andbiocompatibility will be reported in another arti-It is worth noting that organic polymers de-
prived of groups reactive in the sol-gel process but cle of this series.28
prone to hydrogen bonding with protic compoundshave been successfully incorporated into hybrid
EXPERIMENTALmaterials. As representative examples, poly (N-vinylpyrrolidone) (PVPr), poly(N,N-dimethylac-
Materialsrylamide) (PDMA), poly(2-methyl-2-oxazoline)(PMOx), and poly(vinylacetate) (PVAc) have High purity tetraethoxysilane (TEOS) (Janssen),been used to modify silicon oxide networks.9,13–16 3-isocyanatopropyltriethoxysilane (RNCO) (Lan-It has been shown that hydrogen bonds between caster), hydrochloric acid (12 N) (Lab Chemis-these organic polymers and silanol functions try ) , tetrahydrofuran (THF) (Janssen) , etha-formed by hydrolysis of tetraalkoxysilane have a nol (Riedel-de Haen ) , triethoxysilane (Fluka) ,decisive effect on the formation and final proper- hydrogen hexachloroplatinate ( IV) hydrateties of these hybrids. (Pt) (Aldrich), 4-dimethylaminopyridine (DMAP)
Poly(1-caprolactone) (PCL; {[C(O)O(CH2)5]n (Aldrich), PCL-diol (Mn Å 1250), and PCL-triol{) is a biocompatible and biodegradable aliphatic (Mn Å 900) (Aldrich) were used as received. Tolu-polyester, well known for a valuable set of proper- ene (Janssen) was dried by refluxing over calciumties, such as nontoxicity for living organisms, re- hydride and distilled under a nitrogen atmo-sorption after an appropriate period of implanta- sphere. Triethylamine (NEt3) (Aldrich) and 1,4-tion time, and good ultimate mechanical proper- diazabicyclo [2,2,2] octane (DABCO) (Aldrich)ties.17 Similarly to polylactides and polyglycolide, were dried by repeated (three times) azeotropicPCL releases nontoxic by-products upon hy- distillation of toluene just before use. THF wasdrolytic in vivo degradation.18 These aliphatic dried by refluxing over a benzophenone–sodiumpolyesters display a large range of biodegradabil- complex and distilled under nitrogen atmosphere.ity, because their half-life time varies from several Synthesis of hydroxyl end-capped linear PCL (Mn
weeks (polylactide) to several years (PCL), Å 2000, 4000), vinyl end-capped three-arm star-whereas copolymerization of the related mono- shaped PCL (MnÅ 12,000), and hydroxyl pendentmers is an easy way to adjust more precisely the groups containing PCL (Mn Å 14,500, 5.0 mol %biodegradation rate.17 Because of a unique combi- OH groups) was detailed elsewhere.22–27
nation of biocompatibility, permeability and bio-degradability, polymers and copolymers of 1-ca- Esterification of Hydroxyl End-Capped PCLprolactone, lactides, and glycolide have now wide-
A solution of hydroxyl end-capped PCL (Mnspread applications in medicine, as biodegradableÅ 4000) in dry THF was added with a fivefoldsuture, artificial skin, resorbable prostheses, andmolar excess of acetic anhydride, then with trieth-containers for sustained drug release.17–21
ylamine, and finally with a catalytic amount of 4-Depending on the polymerization mechanism,dimethylaminopyridine (DMAP, 0.2 equiv). ThePCL can be end-capped with functional groups,solution was kept under stirring for 3 days atsuch as hydroxyl groups or vinyl groups,22–25 reac-507C. The polymer was precipitated in cold metha-tive towards alkoxysilane. Moreover, we have re-nol and dried under reduced pressure.cently reported on the synthesis of PCL bearing
hydroxyl pendent groups.26,27 Therefore, thesePCL chains are polymeric reagents suited to the Synthesis of Triethoxysilane End-Capped PCLsol-gel process, which have potential of imparting
Reaction of Hydroxyl End-Groups withoriginal properties to novel ceramers.3-IsocyanatopropyltriethoxysilaneThe purpose of this article is to report on the
synthesis of linear and star-branched PCL chains a,v-Hydroxyl PCL was dried by repeated (threetimes) azeotropic distillation of toluene just beforeend-capped with alkoxysilane and the synthesis
8G48 96-162T/ 8g48$$162t 06-16-97 18:44:12 polca W: Poly Chem

INORGANIC–ORGANIC HYBRID MATERIALS. I 2297
use. In a carefully dried pyrex flask equipped with recorded in CDCl3 at 400 MHz in the FT modewith a Bruker AM 400 superconducting magneta rubber septum, PCL and 3-isocyanatopropyl-
triethoxysilane (RNCO) were dissolved in dry system. Glass transition temperature (Tg ) , melt-ing temperature (Tm ) , and melting enthalpyTHF or in dry toluene in a 1 : 1.2 hydroxyl group:
RNCO molar ratio and added with NEt3 or (DHm ) were measured by differential scanningcalorimetry (DSC) with a DuPont 9000 DSC ther-DABCO (1 equiv.) . This reaction proceeded under
stirring at 507C. After reaction, the a,v-triethoxy- mal analyzer. Thermogravimetric analysis wascarried out with a TA 51 Thermogravimetric Ana-silane PCL was recovered by precipitation into
cold methanol. lyzer over the 25–8507C temperature range at aheating rate of 207C/min. in air (purge rate 100mL/min). Dynamic Mechanical Analysis (DMA)Addition of Trialkoxysilane to Vinyl End-Groupswas performed with a DMA model 983 Du Pont
Vinyl end-capped three-arm star-shaped PCL was Instrument. Samples were tested from 0120 todried by repeated (three times) azeotropic distilla- 1507C at a heating rate of 37C/min. A frequencytion of dry toluene just before use. In a carefully of 0.5 Hz was used for all the dynamic mechanicaldried pyrex flask equipped with a rubber septum, experiments. PCL was extracted from hybrid ma-PCL and triethoxysilane were dissolved in dry terials in a standard Soxhlet apparatus with THF,THF in a 1 : 1.2 vinyl : silane molar ratio and for at least 24 h until no further weight loss wasadded with Pt as a catalyst. This reaction pro- observed. The sol fraction was calculated from theceeded under stirring at 257C for 4 h. The triethox- initial and the final weight of the sample.ysilane end-capped three-arm star-shaped PCLwas recovered by precipitation into cold methanol.
RESULTS AND DISCUSSION
Preparation of Tetraethylorthosilicate- Synthesis and Characterization of Triethoxysilanepoly(1-caprolactone) Hybrid Materials Containing PCL
For PCL to be covalently bonded to the silicatePCL/TEOS mixtures of various compositionsnetwork, the chain end-groups must be reactivewere dissolved in THF (20 wt %) and hydrolyzedtoward the silicon alkoxide precursor used in thewith a stoichiometric amount of water with re-
spect to the alkoxide functions. HCl was used asa catalyst in a 5/100 HCl/TEOS molar ratio. Arepresentative synthesis was as follows: 1.5 gTEOS was added to the PCL (0.5 g) solution inTHF (10.0 mL) and thoroughly mixed until a ho-mogeneous solution was formed. Then deionizedwater (0.52 mL), ethanol (0.80 mL), and HCl(0.01 mL) were added under rapid stirring at am-bient temperature for ca. 10 min. The clear solu-tion was then cast into a plastic Petri dish andcovered with a Parafilm. Based on a preliminaryseries of gelation experiments it was shown that,after several days, depending on the PCL end-groups (hydroxyl or triethoxysilane) the Parafilmhad to be removed.29 The gelified material wasthen dried under ambient conditions for 1 weekprior to testing.
Characterization
IR analysis was performed with a Perkin–Elmer106 FT–IR spectrometer. A minimum of 64 scans Figure 1. IR spectra of the reaction product of a,v-of a resolution of 2 cm01 was used. Samples were hydroxyl PCL with 3-isocyanatopropyltriethoxysilaneanalyzed as thin films cast on NaCl windows or in the presence of DABCO as a catalyst in toluene at
507C for different reaction times.in the form of KBr pellets. 1H-NMR spectra were
8G48 96-162T/ 8g48$$162t 06-16-97 18:44:12 polca W: Poly Chem

2298 TIAN, DUBOIS, AND JEROME
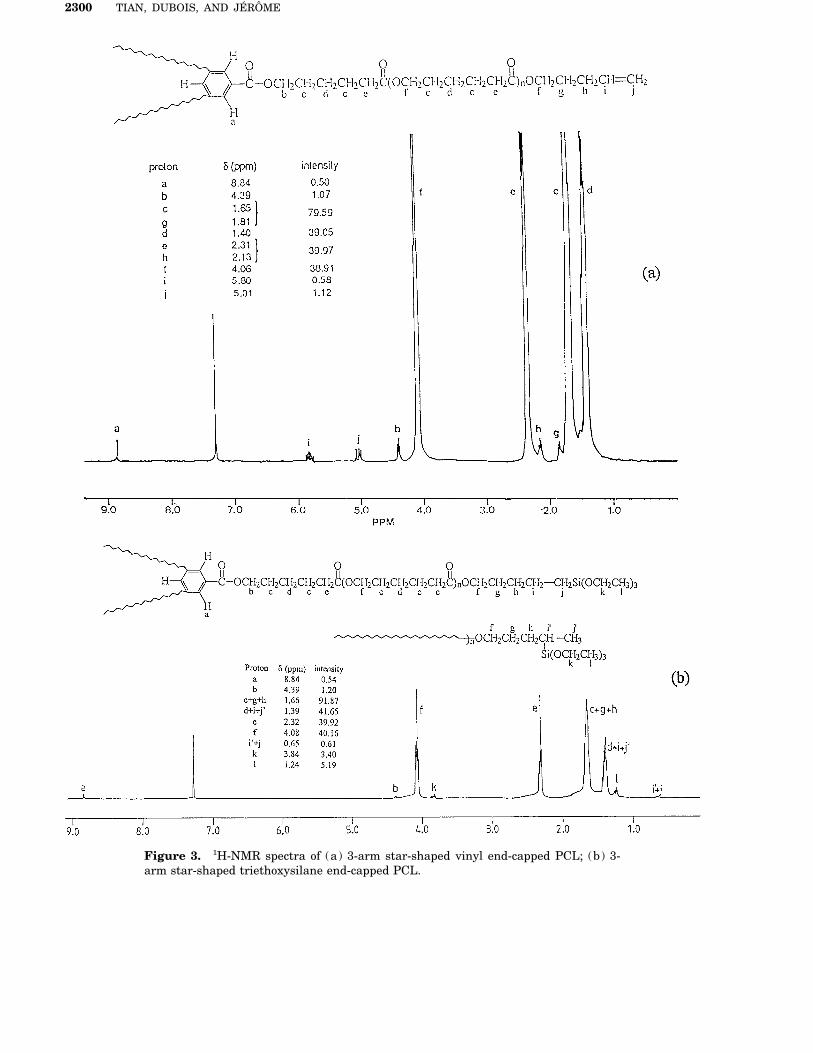
Figure 2. 1H-NMR spectra of (a) a,v-hydroxyl PCL; (b) a,v-triethoxysilane end-capped PCL.
sol-gel process. Depending on the polymerization Equation (1) relies upon the nucleophilic addi-tion of the amine onto the carbonyl of the isocya-mechanism, PCL can be end-capped by a hydroxyl
group or by a vinyl group,22–25 whereas PCL nate.32–35
Equation (2) takes the possible hydrogen bond-chains can be modified by hydroxyl pendentgroups.26,27 Accordingly, triethoxysilane groups ing of amines with OH containing compounds into
account.36,37 Although the reactivity of hydroxyl-can be attached to PCL chains, either by reactionof the hydroxyl groups with 3-isocyanatopropyl- ated reagents may be increased by hydrogen
bonding, no direct correlation between the aminetriethoxysilane or by addition of trialkoxysilaneto vinyl containing PCL as detailed hereafter. basicity and the catalytic activity has been ob-
served, which does not give credit to eq. (2).Reaction of 3-Isocyanatopropyltriethoxysilane withHydroxyl Containing PCL
R3N / R *OH e R3Nr r r rHd/
r r r rOR *d0
Reaction of isocyanate with alcohol is a traditionalreaction of polyurethane chemistry catalyzed, forexample, by amines.30,31 The amine can activate R3Nr r r rH
d/
r r r rOR *d0
/ C6H5NCO rboth the isocyanate and the hydroxyl com-pound,32,33 which explains why two mechanismshave been proposed [eqs. (1) and (2)] :
Ox
C6H5{N{C{OR *w
H
/ NR3 (2)
IR spectra show that the intensity of the hy-droxyl absorption band of a,v-hydroxyl PCL (Mn
Å 3.61 103) changes with time when reacted with3-isocyanatopropyltriethoxysilane in the presenceof triethylamine as a catalyst in THF at 507C.Even after 9 days, however, the absorption of thehydroxyl groups at ca. 3500 cm01 persists, inagreement with the lower reactivity of aliphaticisocyanates compared to aromatic ones.38,39 Be-
CflHfi©N®C®O 1 R‹N
(X) 1 RπOH CflHfi©N©C©ORπ 1 NR‹
CflHfi©N®C©O
NR‹
(X)
CflHfi©N©C®O)h h
g
(
(1)
H
O
NR‹g
8G48 96-162T/ 8g48$$162t 06-16-97 18:44:12 polca W: Poly Chem

INORGANIC–ORGANIC HYBRID MATERIALS. I 2299
cause no aromatic isocyanate substituted by an Hd/d * and He protons] and for the final polymer[Fig. 2(b): Hd/d * and Hi protons), respectively,alkyltriethoxysilane group is commercially avail-
able, experimental conditions, such as catalyst are in agreement within the limits of experimen-tal errors (2.0 1 103 (10%). PCL bearing trieth-and solvent, have to be optimized in order to im-
prove the reaction rate and completeness. In this oxysilane pendent groups has also been success-fully prepared by the same technique from therespect, Ephrain reported on the effect of solvent
on the reaction of phenyl isocyanate with metha- hydroxyl containing precursor:nol at 207C.40 The reaction rate increases as fol-lows when the solvent is changed, i.e., benzene,toluene ú nitrobenzene ú di-n -butyl ether ú n -butyl acetate ú methyl ethyl ketone ú dioxaneú acetonitrile, in dependence on the dielectricconstant of the solvent and to some extent on itspropensity to hydrogen bond formation. Burkushas also studied the catalytic activity of tertiaryamines on the reaction of phenyl isocyanate with1-butanol in toluene.41 Twenty-three tertiaryamines have been tested, and 1,4-diazabicyclo(2,2,2) octane (N(CH2CH2)3N: DABCO) hasproved to be the most efficient catalyst, morelikely because no steric hindrance prevents theaccess to the electron doublet of the nitrogenatoms. Although organometallic compounds suchas dibutyltin diacetate42 and lithium alkoxide,43
exhibit a catalytic activity higher than tertiaryamines by two to four orders of magnitude, theiruse is not suited to aliphatic polyesters becauseof possible scission of the ester linkage. For allthese reasons, toluene and DABCO have been
©C©CH¤CH¤CH¤CH¤CH¤©O©
O
n
m
( )
)(©C©CH¤CH¤©CH©CH¤CH¤©O©
©C©CH¤CH¤CH¤CH¤CH¤©O©
O
n( )
O
OH
OCN©(CH¤)‹©Si(OEt)‹ DABCO, in toluene
(3)m)(©C©CH¤CH¤©CH©CH¤CH¤©O©
O
O
O®C©NH©(CH¤)‹©Si(OEt)‹chosen as solvent and catalyst, respectively. Un-der these experimental conditions, the absorption Addition of Triethoxysilane to Vinyl Groupsof the hydroxyl group at ca. 3500 cm01 completelydisappears in favor of the urethane linkage (3384 Triethoxysilane has been added to the vinyl end-
groups of star-shaped PCL by hydrosilylation incm01) (Fig. 1). The reaction is complete after 24h and the polymer is then recovered and purified the presence of Speier’s catalyst [i.e., hydrogen
hexachloroplatinate (IV) hydrate] in THF at 257Cby precipitation into cold methanol, in which thecatalyst and the excess of isocyanate (even after for 4 h. After reaction, PCL was recovered by pre-
cipitation into cold methanol and washed withreaction with methanol) are soluble. Moreover, nocarboxylic acid end-group can be detected by FT– that solvent. 1H-NMR spectra show that the hy-
drosilylation is complete (Fig. 4). The signalsIR, which supports that no chain degradation hasoccurred during the end-capping reaction. 1H- characteristic of the unsaturated end-group (pro-
tons Hi at 5.80 ppm and protons Hj at 5.01 ppm)NMR analysis confirms these results because thesignal characteristic of the a-hydroxymethylene have completely disappeared, and a new tetrad
peak at d Å 3.84 ppm is observed, which is as-protons at 3.66 ppm (He ) has disappeared in favorof a new peak at 3.16ppm (Hf ) (Fig. 2), which has signed to the methylene protons of the ethoxysi-
lane group: {Si{ ({OCH2{CH3)3 (protonsto be assigned to the N-methylene amide protons(Hf ) . A new tetrad peak at d Å 3.83 ppm can be Hk ) . Moreover, 1H-NMR analysis indicates that
PCL chains remain unchanged during this addi-assigned to the methylene protons of the ethoxysi-lane group: {Si{ ({OCH2-CH3)3 (Hi ) . Fur- tion reaction. Indeed, molecular weight of PCL
has been calculated form the relative intensity ofthermore, 1H-NMR analysis also indicates thatPCL chains have not been degraded during the the Hf and Hb or Ha or Hk protons after hydrosily-
lation [Fig. 3(b)] , and found to be in agreementchain end-capping reaction. For example, the mo-lecular weight calculated from 1H-NMR spectra with the precursor molecular weight [Fig. 3(a),
Hf and Hb or Ha , or Hi , or Hj protons) within thefor the a,v-hydroxyl PCL precursor [Fig. 2(a):
8G48 96-162T/ 8g48$$162t 06-16-97 18:44:12 polca W: Poly Chem
2300 TIAN, DUBOIS, AND JEROME
Figure 3. 1H-NMR spectra of (a) 3-arm star-shaped vinyl end-capped PCL; (b) 3-arm star-shaped triethoxysilane end-capped PCL.
8G48 96-162T/ 8g48$$162t 06-16-97 18:44:12 polca W: Poly Chem

INORGANIC–ORGANIC HYBRID MATERIALS. I 2301
limits of experimental errors (12.0 1 103 { 10%). into triethoxysilane groups. The polycondensationreaction can then be schematized as follows:This value also agrees with the molecular weight
measured form vapor pressure osmometry (Mn
Å 13.0 1 103).25
Si(OEt)4 / HOCH2{PCL{CH2OH rH/
Preparation of Tetraethylorthosilicate-poly(1-caprolactone) Hybrid Materials
The eqs. (4) – (6) [eq. (6) is unbalanced] are an
F F
O Ow w
RO{Si{O{SiOCH2{PCLw w
O Of f
oversimplified view of the sol-gel reaction path-way envisioned for the synthesis of PCL con-taining ceramers. Indeed, hydrolysis of the eth-oxysilane groups whatever their origin (TEOS orPCL end-groups) is assumed to be complete beforepolycondensation occurs. Undoubtedly, the actualnetwork forming process is much more complex
F F
O Ow w
{CH2OSi{O{Si{Or
w w
O Of f
/ H2O / EtOH (7)due to the interplay of hydrolysis and condensa-tion. Even though the actual stepwise reactionmechanism cannot be detailed, it is clear that PCLchains must be end-capped by ethoxysilanegroups for being most probably incorporated into
This polycondensation reaction, however, oc-the final network.curs at a much slower rate compared to the casewhere the PCL chains have previously been
Si(OEt)4 / 4H2O rH/
capped by triethoxysilane groups. The extent ofthe reaction depends on the solvent used. Al-Si(OH)4 / 4 CH3CH2OH (4)though ethanol is found as a reaction by-product,it is often used as solvent, more likely because it
(EtO)3SiCH2{PCL{CH2Si(OEt)3 / 6H2O rH/
has been observed to provide the final materialwith transparency.29 Ethanol seems to perturb(HO)3SiCH2{PCL{CH2Si(OH)3
the hydrolysis-condensation balance in such a/ 6C2H5OH (5) way that the incorporation of the PCL chains into
the silica network is favored. This observation isSi(OH)4 / (HO)3SiCH2{PCL{CH2Si(OH)3 r
H/
consistent with a hydrolysis reaction faster thanthe polycondensation reaction, so that the proba-bility for silica to be formed independently of thePCL component is decreased.
Warping, shrinking, and cracking problems
F F
O Ow w
RO{Si{O{SiCH2{PCLw w
O Of f
have often been reported for materials preparedby the sol-gel process. Warping is expected to re-sult from a difference in the evaporation rate ofthe volatiles from the top to the bottom of thesample. Shrinking and cracking result from theexceedingly large weight loss during the dryingstep. These problems are as more severe as the
F F
O Ow w
{CH2Si{O{Si{Or
w w
O Of f
/ H2O (6) PCL content becomes smaller. They can, however,be alleviated by casting the film onto a plasticPetri dish instead of a glass one and reducing theevaporaton rate of the volatiles. The sample adhe-sion to the plastic surface prevents warping fromoccurring during the first few days of drying.However, we reported in a previous communi-
cation29 that a,v-hydroxyl PCL can also be used When the sample is dry enough, it is detachedfrom the substrate and let to dry further slowlyas such in the sol-gel process, i.e., without the
previous conversion of the hydroxyl end-groups enough so that cracking and warping do not occur.
8G48 96-162T/ 8g48$$162t 06-16-97 18:44:12 polca W: Poly Chem

2302 TIAN, DUBOIS, AND JEROME
Figure 4. Specimen of biocompatible and biodegradable ceramers, (A) 47.5 wt %SiO2/52.5 wt % PCL (a,vhydroxyl PCL, Mn Å 2000); (B) 46.5 wt % SiO2/53.5 wt %PCL (a,v-triethoxysilae PCL, Mn Å 2000); (C) 53.5 wt % SiO2/46.5 wt % PCL (noreactive end-groups on PCL, Mn Å 4000); (D) 47.5 wt % SiO2/52.5 wt % PCL (PCL-triol, Mn Å 900); (E) pure silica.
The thickness of the cast films is usually in the ent, even when PCL does not bear end-functionalgroups. This observation indicates that the tworange of 0.1 to 1 mm, and the diameter in the
range of 20 to 55 mm, although thicker and larger constitutive components (PCL and silica) interactenough for preventing any macrophase separa-films can be prepared.
Figure 4 shows some specimen of biocompatible tion from occurring at least at that composition.However, when the initial PCL/TEOS wt ratio isand biodegradable ceramers. All these samples
that finally contain ca. 50 wt % SiO2 are transpar- higher than 30 : 70, i.e., ceramers with more than
Figure 5. DSC traces of a,v-triethoxysilane PCL/TEOS (25 : 75, wt/wt) hybrid mate-rial for different curing conditions. (a) at 257C; (b) at 1007C for 6 h; (c) at 1007C for 1day; (d) at 1007C for 2 days.
8G48 96-162T/ 8g48$$162t 06-16-97 18:44:12 polca W: Poly Chem

INORGANIC–ORGANIC HYBRID MATERIALS. I 2303
Figure 6. Dynamic mechanical spectra of ceramers prepared from 80 wt % TEOSand 20 wt % a,v-hydroxyl PCL (Mn Å 4000) for different curing conditions. (a) at 257C( — - — - —); (b) at 1007C for 6 h (------) ; (c) at 1007C for 1 day ( — -- — -- —); (d)at 1007C for 2 days ( — — —).
ca. 60 wt % PCL, the final hybrid material be- served for a,v-hydroxyl PCL/TEOS (25 : 75, wt/wt) hybrid materials. All these observations sup-comes translucent if not opaque.port that the sol-gel reaction is not complete aslong as it is carried out at 257C, whatever the
Thermal Analysis functional end-groups of PCL : triethoxysilane orhydroxyl. Vitrification of the material clearlyDifferential scanning calorimetry (DSC) has been
used to probe the possible crystallization of PCL places limitation on the diffusion of the not yetreacted species. Curing at high temperature, forin the hybrid materials. In agreement with the
sample transparency, no melting endotherm is ob- example, 1007C, allows overcoming, at leastpartly, this drawback. Annealing above 2007C is,served when the initial PCL/TEOS wt ratio is
smaller than 30 : 70, whether the PCL chains be however, prohibited due to the poor thermal sta-bility of PCL.45end-capped by triethoxysilane or hydroxyl groups.
Typical DSC curves are shown in Figure 5. Curve As a rule, Tg of PCL is observed to increasewith the SiO2 content. Indeed, Tg increasea shows an endotherm at ca. 807C, which is cur-
rently attributed to the vaporization of volatile from 0617C for pure PCL up to to 0557C in thecase of 50 wt % PCL containing the hybrid mate-compounds, including water, ethanol, and sol-
vent.44 The very broad endotherm originally ob- rial.Dynamic mechanical analysis (DMA) confirmsserved at ca. 1507C completely disappears when
the sample is cured at 1007C for more than 6 h the DSC data. Annealing at 1007C of the samplesprepared at 257C, results in a shift of the glass(Fig. 5; curves b, c, and d). The glass transition
of PCL at0557C (Fig. 5) becomes less visible upon transition of PCL toward higher temperatures.However, an annealing time longer than 1 daycuring at 1007C. The same behavior is also ob-
8G48 96-162T/ 8g48$$162t 06-16-97 18:44:12 polca W: Poly Chem
2304 TIAN, DUBOIS, AND JEROME
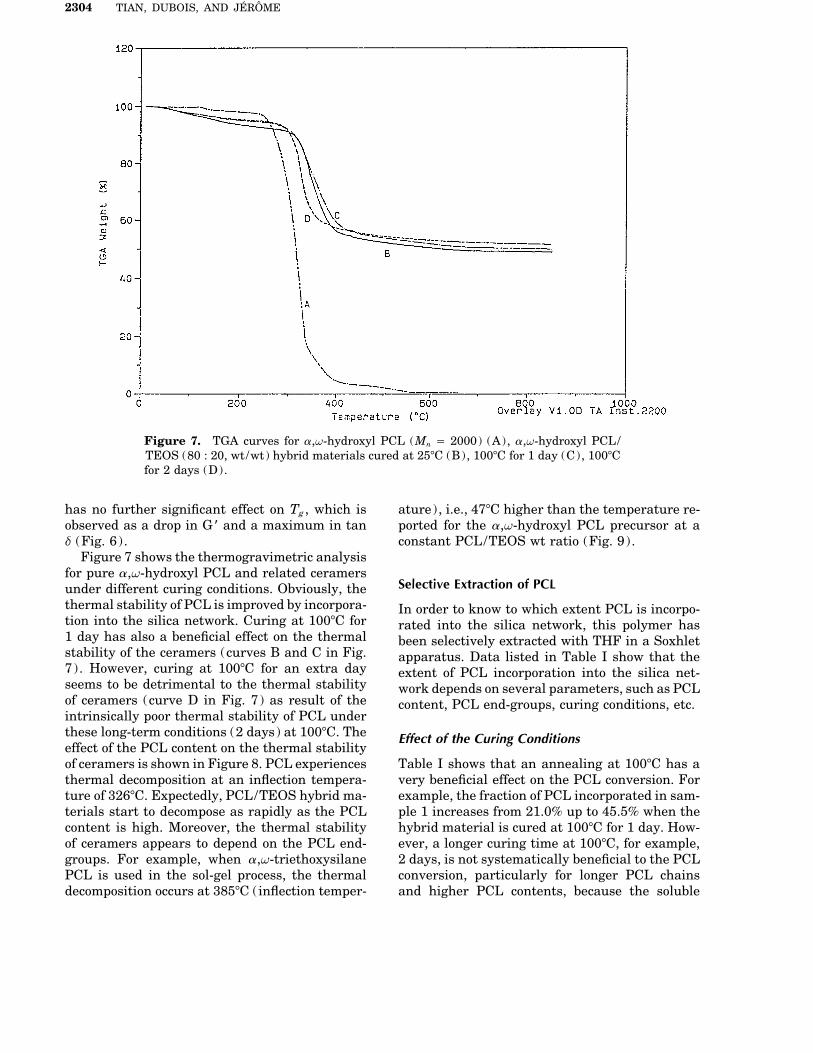
Figure 7. TGA curves for a,v-hydroxyl PCL (Mn Å 2000) (A), a,v-hydroxyl PCL/TEOS (80 : 20, wt/wt) hybrid materials cured at 257C (B), 1007C for 1 day (C), 1007Cfor 2 days (D).
has no further significant effect on Tg , which is ature), i.e., 477C higher than the temperature re-ported for the a,v-hydroxyl PCL precursor at aobserved as a drop in G * and a maximum in tan
d (Fig. 6). constant PCL/TEOS wt ratio (Fig. 9).Figure 7 shows the thermogravimetric analysis
for pure a,v-hydroxyl PCL and related ceramersSelective Extraction of PCLunder different curing conditions. Obviously, the
thermal stability of PCL is improved by incorpora- In order to know to which extent PCL is incorpo-tion into the silica network. Curing at 1007C for rated into the silica network, this polymer has1 day has also a beneficial effect on the thermal been selectively extracted with THF in a Soxhletstability of the ceramers (curves B and C in Fig. apparatus. Data listed in Table I show that the7). However, curing at 1007C for an extra day extent of PCL incorporation into the silica net-seems to be detrimental to the thermal stability work depends on several parameters, such as PCLof ceramers (curve D in Fig. 7) as result of the content, PCL end-groups, curing conditions, etc.intrinsically poor thermal stability of PCL underthese long-term conditions (2 days) at 1007C. The Effect of the Curing Conditionseffect of the PCL content on the thermal stabilityof ceramers is shown in Figure 8. PCL experiences Table I shows that an annealing at 1007C has a
very beneficial effect on the PCL conversion. Forthermal decomposition at an inflection tempera-ture of 3267C. Expectedly, PCL/TEOS hybrid ma- example, the fraction of PCL incorporated in sam-
ple 1 increases from 21.0% up to 45.5% when theterials start to decompose as rapidly as the PCLcontent is high. Moreover, the thermal stability hybrid material is cured at 1007C for 1 day. How-
ever, a longer curing time at 1007C, for example,of ceramers appears to depend on the PCL end-groups. For example, when a,v-triethoxysilane 2 days, is not systematically beneficial to the PCL
conversion, particularly for longer PCL chainsPCL is used in the sol-gel process, the thermaldecomposition occurs at 3857C (inflection temper- and higher PCL contents, because the soluble
8G48 96-162T/ 8g48$$162t 06-16-97 18:44:12 polca W: Poly Chem

INORGANIC–ORGANIC HYBRID MATERIALS. I 2305
Figure 8. TGA curves for a,v-hydroxyl PCL/TEOS hybrid materials (cured at 1007Cfor 2 days) of various PCL content. PCL/TEOS ratio is (A) 100 : 0, (B) 20 : 80, (C)15 : 85, (D) 10 : 90.
fraction tends to increase with curing time (e.g., Effect of the PCL Molecular Weightsamples 1, 2, 9, and 10 in Table I) . This effect
While keeping constant the number of functionalwould result from a more rapid degradation ofend-groups of, for example, a,v-hydroxyl PCL, aPCL with release of soluble fragments, as as-higher PCL molecular weight is favorable to thesessed by 1H-NMR analysis of the sol fraction,incorporation yield of PCL (Table I, samples 11,which shows small amounts of carboxylic acid3, and 6, and samples 7 and 10). This merelyend-groups.indicates that when the TEOS : PCL weight ratiois kept constant, an increasing PCL molecular
Effect of the PCL Content weight results in a decreasing number of reactivePCL end-groups, which means that fewer TEOS/
When the sol-gel conditions are kept constant ex- PCL crossreactions are required for incorporatingcept for the PCL content (Table I, samples 2–5), the same amount of PCL in the silica network.the PCL conversion is higher in case of smallerPCL contents. For instance, the fraction of PCL Effect of PCL Functional End-Groupsincorporated in the sample 5 compared to the the-oretical PCL content (15.5%) is increased from The PCL incorporation yield is 40% at 257C when
a,v-hydroxyl PCL has previously been reacted40.6% (curing at 257C) up to 100% upon curingat 1007C for 1 day. In the case of sample 2 (theo- with 3-isocyanatopropyltriethoxysilane (sample
1; Table I) compared to 21.5% when used as suchretical PCL content of 53.5%), this fraction is in-creased from 21.5% up to 79.5%. A longer curing (sample 2; Table I) . After annealing at 1007C, the
amount of extractable PCL remains higher whentime at 1007C does not change the PCL conver-sion, which indicates that the reaction of the PCL a,v-hydroxyl PCL has been used rather than the
triethoxysilane derivative. This observationend-groups is slower and more restricted whenthe PCL content is higher. agrees with the lower reactivity of the PCL hy-
8G48 96-162T/ 8g48$$162t 06-16-97 18:44:12 polca W: Poly Chem

2306 TIAN, DUBOIS, AND JEROME
Figure 9. TGA curves for ceramers of various PCL end-groups (PCL : TEOS Å 20 :80 wt/wt). (A) a,v-hydroxyl PCL (Mn Å 2000); (B) a,v-triethoxysilane PCL (Mn
Å 2000).
droxyl end-groups compared to the silanol (or sila- PCL chain decreases the probability of bindingPCL to the silica network. It is noteworthy thatnolate) ones, as previously reported.31 wt % PCL deprived of any reactive group isincorporated into the silica network after a 1-dayEffect of the Number of Functional (End-) Groups
per PCL Chain curing at 1007C (sample 8 in Table I) . This obser-vation can only be accounted for by specific inter-As shown in Table I, the PCL incorporation yield actions between PCL and silica as it will be dis-also depends on the number of functional groups cussed hereafter.per chain (Table I, samples 11 and 12, or 6, 7
and 8, or 9 and 13). The incorporation yield isexpectedly increasing with the number of func- FT–IR Investigationtional groups per chain at constant molecularweight. For instance, comparison of samples 11 PCL ceramers have been investigated by IR spec-
troscopy in relation to the curing time and tem-and 12 (Table I) of the same theoretical composi-tion shows that using a PCL-triol rather than a perature. Figure 10 shows typical IR spectra for
sample 2 in Table I. The relative intensity (R) ofdiol of a comparable molecular weight increasesthe PCL incorporation yield whatever the curing the absorption of Si{O{Si and Si{O{C
groups at 1110–1000 cm01 with respect to theconditions, for example, the incorporation of thePCL-triol is close to completion upon annealing methylene absorption of PCL at 2943 cm01 has
been measured. From Figure 10, it appears thatat 1007C for 2 days compared to 85% for the parentdiol. The same conclusion holds when samples 6 R increases from 3.75 to 5.30 and finally to 5.80
when sample 2 is cured at 257C, 1007C for 6 h,and 7 are compared; thus, when a,v-hydroxylPCL is used rather then v-hydroxyl PCL of the and 1007C for 24 h, respectively. This observation
confirms that increasing curing time and temper-same molecular weight. It is not surprising thatdecreasing the number of functional groups per ature improve the molecular mobility so that the
8G48 96-162T/ 8g48$$162t 06-16-97 18:44:12 polca W: Poly Chem

INORGANIC–ORGANIC HYBRID MATERIALS. I 2307
Table I. Main Characteristics of the Prepared PCL Ceramers
PCL Wt %b
Curing ConditionsPCL
End-Groups of Mn TEOS : PCL Wt %a at 1007C at 1007CSamples PCL Chains PCL (Wt. Ratio) (Theor.) at 257C for 1 day for 2 days
1 a,v-triethoxysilane 2000 75 : 25 52.7% 21.0% (39.8%) 45.5% (86.3%) 42.5% (80.6%)2 a,v-hydroxyl 2000 75 : 25 53.6% 11.5% (21.5%) 42.5% (79.3%) 41.0% (76.5%)3 a,v-hydroxyl 2000 80 : 20 46.5% 9.0% (19.4%) 34.0% (73.1%) 35.0% (75.3%)4 a,v-hydroxyl 2000 90 : 10 27.8% 9.2% (33.1%) 15.5% (55.8%) 26.5% (95.3%)5 a,v-hydroxyl 2000 95 : 5 15.5% 6.3% (40.6%) 15.5% (100%) 15.5% (100%)6 a,v-hydroxyl 4000 80 : 20 46.5% 11.5% (24.7%) 38.5% (82.8%) 42.0% (90.3%)7 v-hydroxyl 4000 80 : 20 46.5% 9.5% (20.4%) 31.5% (67.7%) 31.5% (67.7%)8 no reactive end- 4000 80 : 20 46.5% 8.5% (18.3%) 31.0% (67.1%) 31.5% (67.7%)
group9 triethoxysilane 12,000 80 : 20 46.5% 10.5% (22.6%) 37.0% (79.6%) 36.5% (78.5%)
end-capped 3-arm star-shapedPCL
10 v-hydroxyl 80,000 80 : 20 46.5% 12.5% (26.9%) 40.0% (86.0%) 37.5% (80.6%)11 PCL-diol 1250 80 : 20 46.5% 7.5% (16.1%) 37.0% (79.6%) 39.5% (84.9%)12 PCL-triol 900 80 : 20 46.5% 13.5% (29.0%) 44.5% (95.7%) 46.0% (98.9%)13 pendent 16,000 80 : 20 44.4% 12.5% (28.2%) 39.0% (87.8%) 42.5% (95.7%)
triethoxysilanegroups (5.0 mol%)
a Theoretical PCL content of ceramers calcualted for a complete sol-gel reaction.b PCL content of ceramers after Soxhlet extraction (see Experimental Section) and PCL incorporation yield (noted in parenthe-
ses).
crossreaction between TEOS and triethoxysilane wavenumber. Similar observation has been re-ported for the carbonyl absorption of PVPr,end-capped PCL occurs to a larger extent.
Figure 11 shows the infrared absorption region PDMA, PMOx, PVAc, and PMMA when used aspart of hybrid materials with silica.9,13–16 Rela-for the carbonyl stretching vibration in case of
the PCL/TEOS (80 : 20, wt/wt) hybrid material, tively strong hydrogen bonding of the ester groupswith residual OH groups on silica is the most rea-containing unreactive, i.e., PCL, whose hydroxyl
end-groups have been esterified prior to addition sonable explanation. Furthermore, the relativeintensity of the absorption at 1733 cm01 and 1715of TEOS. The frequency of the carbonyl stretching
vibration of PCL depends on whether PCL is cm01 [Fig. 11(c)] , respectively, allows calculationthat ca. 20% ester groups of PCL are involved inamorphous or crystalline. Indeed, the absorption
is reported at 1725 cm01 for crystalline PCL com- hydrogen bonding. This conclusion might explainwhy ethanol has a favorable effect on the trans-pared to 1733 cm01 for the amorphous polymer
[Fig. 11(a) and (b)] . A new absorption band at parency of the final material, because a networkof hydrogen bonds is able to prevent PCL from1715 cm01 is observed for hybrid materials [Fig.
11(c) and (d)] . In case of a 46.5% PCL content, crystallizing.the carbonyl absorption of PCL persists at a wave-number that might be assigned to amorphousPCL (1733 cm01) , in agreement with the trans- CONCLUSIONparency of the sample. The relative intensity ofthis original PCL absorption decreases with re- PCL has been successfully end-capped with trie-
thoxysilane by derivatization of hydroxyl or vinylspect to the new absorption when the PCL contentof the hybrid material is decreased [Fig. 11(c) end-capped PCL. Transparent hybrid materials
incorporating PCL into TEOS-based silica net-and (d)] . Thus, the carbonyl groups of PCL inter-act with the hydrolyzed TEOS, which results in a work have then been prepared by the sol-gel pro-
cess. PCL is so intimately incorporated into theshift of the carbonyl absorption toward smaller
8G48 96-162T/ 8g48$$162t 06-16-97 18:44:12 polca W: Poly Chem

2308 TIAN, DUBOIS, AND JEROME
Figure 10. IR spectra of a,v-hydroxyl PCL/TEOS (25 : 75, wt/wt) hybrid materialfor different curing conditions. (A) 257C; (B) 1007C for 6 h; (C) 1007C for 24 h.
polymer network that it remains completely reactive system. DSC, DMA, and PCL extraction,and IR analysis have shown that this drawbackamorphous. The progress of the sol-gel process is,
however, limited by the early vitrification of the could be alleviated by curing the sample at hightemperature (1007C). The thermal stability ofPCL is improved by incorporation into the silicanetwork. The thermal stability of the final ceram-ers increases when the PCL content decreases andwhen PCL is end-capped with triethoxysilanegroups rather than hydroxyl ones. The extent ofthe PCL incorporation into the silica network de-pends on the PCL content and molecular weight,on the reactivity of the PCL end-groups, and theirnumber per chain. Lower PCL content, higherPCL molecular weight, more functional groupsper chain, and triethoxysilane end-groups are fa-vorable to the PCL incorporation. The IR analysishas shown that the carbonyl groups of PCL partic-ipate to hydrogen bonding with residual OHgroups on silica, and that ca. 20% of the estergroups would be involved in these interactions.Thus, the organic (PCL) and the inorganic (SiO2)components are associated not only by covalentbond (in case of end-reactive PCL) but also byhydrogen bonding. The analysis of phase morphol-ogy and structure of the PCL ceramers by TEM,SAXS, DMA, AFM, SEM–EDAX, and solid-stateNMR will be reported in a forthcoming article.
Figure 11. IR spectra in the carbonyl stretching re-gion for (A) crystalline PCL, (B) amorphous PCL, (C)46.5 wt % PCL hybrid materials, (D) 15.5 wt % PCL The authors are very much indebted to the ‘‘Services
Federaux des Affaires Scientifiques, Techniques et Cul-containing hybrid materials. PCL is deprived of anyfunctional group (i.e., hydroxyl or triethoxysilane). turelles’’ for general support to CERM and a fellowship
8G48 96-162T/ 8g48$$162t 06-16-97 18:44:12 polca W: Poly Chem

INORGANIC–ORGANIC HYBRID MATERIALS. I 2309
to T.D. in the frame of the ‘‘Poles d’Attraction Interuni- E. M. Schaefger, Eds., Plenum, New York, 1977, p.251.versitaires: Polymeres.’’
19. M. Vert, Makromol. Chem., Macromol. Symp., 6,109 (1986).
20. S. Gogolewski and A. Pennings, Macromol. Chem.,REFERENCES AND NOTES Rapid. Commun., 3, 839 (1982).
21. J. Pak, J. L. Ford, C. Rostron, and V. Walters,Pharm. Acta Helv., 60, 160 (1985).1. For most recent reviews, see (a) B. M. Novak, Adv.
22. Ph. Dubois, R. Jerome, and Ph. Teyssie, Polym.Mater., 5, 422 (1993); (b) D. R. Ulrich, J. Non-Bull., 22, 475 (1989).Cry. Solids, 121, 465 (1990); (c) G. L. Wilkes, H. H.
23. Ph. Dubois, Ph. Degee, R. Jerome, and Ph. Teyssie,Huang, and R. H. Glaser, ACS Symp. Silicon-Macromolecules, 26, 2730 (1993).Based Polym. Sci., 224, 207 (1990).
24. A. Duda, Macromolecules, 27, 576 (1994).2. H. H. Huang, B. Orler, and G. L. Wilkes, Macro-25. D. Tian, Ph. Dubois, R. Jerome, and Ph. Teyssie,molecules, 20, 1322 (1987).
Macromolecules, 27, 4134 (1994).3. S. Kohjiya, K. Ochiai, and S. Yamashita, J. Non-26. D. Tian, Ph. Dubois, Ch. Grandfils, and R. Jerome,cryst. Solids, 119, 132 (1990).
Macromolecules, 30, 406 (1997).4. F. Surivet, T. M. Lam, J. P. Pascault, and C. Nai,27. D. Tian, Ph. Dubois, and R. Jerome, Macromole-Macromolecules, 25, 5748 (1992).
cules, 30, 1947 (1997).5. H. H. Huang, R. H. Glaser, and G. L. Wilkes, ACS28. D. Tian, Ph. Dubois, Ch. Grandfils, R. Jerome, P.Symp. Inorg. Org. Polym., 360, 354 (1987).
Viville, R. Lazzaroni, J.-L, Bredas, and P. Leprince,6. H. H. Huang and G. L. Wilkes, Polym. Bull., 18,Chem. Mater., 9, 871 (1997).455 (1987).
29. D. Tian, Ph. Dubois, and R. Jerome, Polymer, 37,7. A. B. Brennar, B. Wang, D. E. Rodrigues, and G. L. 3983 (1996).Wilkes, J. Inorg. Org. Polym., 1, 167 (1991). 30. S. G. Entelis and O. V. Nesterov, Russ. Chem. Rev.,
8. A. B. Brennan and G. L. Wilkes, Polymer, 32, 733 35, 917 (1966).(1991). 31. S. Ozaki, Chem. Rev., 72, 457 (1971).
9. T. Saegusa, J. Macromol. Sci.-Chem., A28, 817 32. J. W. Baker and J. B. Holdsworth, J. Chem. Soc.,(1991). 713 (1947).
10. J. L. W. Noell, G. L. Wilkes, D. K. Mohanty, and 33. J. W. Baker and J. Gaunt, J. Chem. Soc., 9, 19, 27J. E. McGrath, J. Appl. Polym. Sci., 40, 1177 (1949).(1990). 34. J. Brukus, J. Am. Chem. Soc., 26, 779 (1961).
11. B. M. Novak, S. M. Hoff, and Y. He, Polym. Pre- 35. A. Farkas and P. F. Strohm, Ind. Eng. Chem. (Fun-prints, 34, 258 (1993). damentals) , 4, 32 (1965).
12. J. E. Mark, S. Wang, and Z. Ahmad, Macromol. 36. S. Nagakura and M. Goeterman, J. Chem. Phys.,Symp., 98, 731 (1995). 26, 881 (1957).
13. Y. Chujo and T. Saegusa, Adv. Polym. Sci., 100, 11 37. M. D. Joesten and R. S. Drago, J. Am. Chem. Soc.,(1992). 84, 3817 (1962).
14. C. J. T. Landry, B. K. Coltrain, and B. K. Brady, 38. L. L. Ferstanding and R. A. Scherrer, J. Am. Chem.Polymer, 33, 1486 (1992). Soc., 81, 4838 (1959).
15. C. J. T. Landry, B. K. Coltrain, J. A. Wesson, N. 39. M. Sato, J. Org. Chem., 27, 819 (1962).Zumbulyadis, and J. L. Lippert, Polymer, 33, 1496 40. S. Ephraim, A. E. Woodward, and R. B. Mesrobian,(1992). J. Am. Chem. Soc., 80, 1326 (1958).
16. J. J. Fitzgerald, C. J. T. Landry, and J. M. Pochan, 41. J. Burkus, J. Org. Chem., 26, 779 (1961).Macromolecules, 25, 3715 (1992). 42. J. W. Brittain and P. G. Gemeinhardt, J. Appl.
17. C. G. Pitt and T. A Marks, Schindler, in Controlled Polym. Sci., 4, 207 (1960).Release of Bioactive Materials, R. Baker, Ed., Aca- 43. W. J. Bailey, J. Org. Chem., 43, 2690 (1978).demic Press, New York, 1980. 44. C. Lemoine, B. Gilbert, B. Michaux, J. P. Pirard,
18. A. Schindler, R. Jeffcoat, G. L. Kimmel, C. G. Pitt, and A. J. Lecloux, Non-cryst. Solids, 175, 1 (1994).M. E. Wall, and R. Zweidinger, in Contemporary 45. Ph. Dubois, I. Barakat, R. Jerome, and Ph. Teyssie,
Macromolecules, 26, 4407 (1993).Topics in Polymer Science, Vol. 2, J. R. Pearce and
8G48 96-162T/ 8g48$$162t 06-16-97 18:44:12 polca W: Poly Chem