atr atrip kinase complex triggers activation of the...
TRANSCRIPT

Molecular and Cellular Pathobiology
ATR–ATRIP Kinase Complex Triggers Activation ofthe Fanconi Anemia DNA Repair Pathway
Tomoko Shigechi1,2, Junya Tomida1, Koichi Sato3, Masahiko Kobayashi4, John K. Eykelenboom5,Fabio Pessina5, Yanbin Zhang6, Emi Uchida1, Masamichi Ishiai1, Noel F. Lowndes5, Kenichi Yamamoto4,Hitoshi Kurumizaka3, Yoshihiko Maehara2, and Minoru Takata1
AbstractATR kinase activates the S-phase checkpoint when replication forks stall at sites of DNA damage. This event
also causes phosphorylation of the Fanconi anemia (FA) protein FANCI, triggering its monoubiquitination of thekey DNA repair factor FANCD2 by the FA core E3 ligase complex, thereby promoting this central pathway of DNArepair which permits replication to be restarted. However, the interplay between ATR and the FA pathway hasbeen unclear. In this study, we present evidence that their action is directly linked, gaining insights into thisrelationship in a DT40 mutant cell line that is conditionally deficient in the critical ATR-binding partner proteinATRIP. Using this system, we showed that ATRIP was crucial for DNA damage–induced FANCD2 monoubiqui-tination and FANCI phosphorylation. ATR kinase phosphorylated recombinant FANCI protein in vitro, whichwasfacilitated by the presence of FANCD2. Mechanistic investigations revealed that the RPA region but not theTopBP1 region of ATRIP was required for FANCD2 monoubiquitination, whereas Chk1 phosphorylation reliedupon both domains. Together, ourfindings identify ATR as the kinase responsible for activating the FA pathway ofDNA repair. Cancer Res; 72(5); 1149–56. �2012 AACR.
IntroductionFanconi anemia (FA) is a genome instability disorder char-
acterized by increased incidence of cancer, progressive bonemarrow failure, developmental abnormalities, and hypersen-sitivity to DNA interstrand cross-links (ICL; refs. 1–3). FA iscaused by a mutational defect in the common FA pathway,which is composed of at least 15 FA proteins (3). The centralcomponent of the pathway is the FA core complex, whichcontains at least 8 FA (FANCA/B/C/E/F/G/L/M) proteins and4 FA-related factors (FAAP24, FAAP100, MHF1, and MHF2;refs. 3–5). In response to replication stress and during S phase,
the FA core complex functions as an E3 ligase to monoubi-quitinate the FANCD2/FANCI (ID) complex, which is thenloaded onto damaged chromatin to facilitate DNA repair (6, 7).In ICL repair, FANCD2 has a role in making incisions atboth sides of the damage and promoting translesion DNAsynthesis across the incised ICL (8, 9). Recent studiesshowed that monoubiquitinated FANCD2 recruits criticaleffector molecules such as the FAN1 nuclease or SLX4 viatheir ubiquitin-binding domains (2, 3, 10). Thus, monoubiqui-tination of FANCD2 is critical not only for its chromatinlocalization but also for acting as a scaffold for specific DNArepair factors.
Our previous study indicated that DNA damage–inducedFANCI phosphorylation at the S/TQ cluster domain serves as amolecular switch to activate FANCD2 monoubiquitination(11). Three S/TQ-directed phosphoinositide 3-kinase (PI3K)-related kinase family members ATM, DNA-PK, and ATR playpartially overlapping but distinct roles in the DNA damageresponse (12). ATR is activated by single-stranded (ss) DNAexposed during replication stress, whereas ATM and DNA-PKrespond primarily to DNA double strand breaks. A criticalinteracting partner of ATR, ATRIP protein, mediates theaccumulation of ATR on damaged chromatin via an interac-tion with the RPA complex, which recognizes and coatsssDNA (13). In parallel, the Rad9-Rad1-Hus1 (9-1-1) checkpointclamp, loaded onto DNA by the RAD17-replication factor Cclamp loader complex, recruits TopBP1 (14). These serialactions result in the catalytic activation of ATR through directbinding of ATR–ATRIP with the activation domain of TopBP1.The ATR–TopBP1 interaction is further stabilized by ATRautophosphorylation on S1989 (15). Active ATR results in
Authors' Affiliations: 1Laboratory of DNA Damage Signaling, Departmentof Late Effects Studies, Radiation Biology Center, Kyoto University, Kyoto;2Department of Surgery and Science, Graduate School of MedicalSciences, Kyushu University, Fukuoka; 3Graduate School of AdvancedScience and Engineering, Waseda University, Tokyo; 4Cancer ResearchInstitute, Kanazawa University, Kanazawa, Ishikawa, Japan; 5GenomeStability Laboratory, Center for Chromosome Biology, School of NaturalSciences, National University of Ireland Galway, Galway, Ireland; and6Department of Biochemistry & Molecular Biology, University of MiamiMiller School of Medicine, Miami, Florida
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
Current address for J. Tomida: Department of Molecular Carcinogenesis,The University of Texas MD Anderson Cancer Center, Smithville, Texas.
Corresponding Author: Minoru Takata, Radiation Biology Center, KyotoUniversity, Yoshidakonoe-cho, Sakyo-ku, Kyoto, Japan 606-8501. Phone:81-75-753-7563; Fax: 81-75-753-7564; E-mail: [email protected])
doi: 10.1158/0008-5472.CAN-11-2904
�2012 American Association for Cancer Research.
CancerResearch
www.aacrjournals.org 1149
on May 3, 2019. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 18, 2012; DOI: 10.1158/0008-5472.CAN-11-2904

phosphorylation and activation of Chk1, which in turn furtherorchestrates the biological responses to DNA damage.
It is of interest to determine which kinase is actuallyresponsible for the phosphorylation of FANCI. Although itseems reasonable to assume that the ATR kinase plays animportant role in the FA pathway, the issue has not beensettled. Here, we clarify the role of theATR–ATRIP kinase in theFA pathway with a genetically defined system.
Materials and MethodsGene targeting in DT40 cells
Culture and transfections in DT40Cre1 were carried out asdescribed (11). DT40Cre1, a subline of DT40-expressing Mer-CreMer (16), was obtained from Drs. Jean-Marie Buersteddeand Hiroshi Arakawa and has been maintained by M. Takata.This cell line has been repeatedly verified in our laboratory byseveral criterion including morphology, karyotype, drug sen-sitivity, and gene targeting efficiency. To make conditionalATRIPflox-GFP/– cells (Fig. 1A and Supplementary Fig. S1), one ofthe ATRIP alleles was disrupted by gene targeting by replacingexons encoding chicken ATRIP amino acids 364 to 376 withthe Blastocidin resistance gene (bsr) cassette, which was thenremoved by TAM-activated Cre. Afterward, a pair of the loxP
sequence was knocked into the other allele so that the exonsencoding the C-terminal ATR-binding domain (amino acids614 to 822) could be floxed. The GFP sequence was alsoknocked-in at the termination codon to monitor the expres-sion of endogenous ATRIP protein. Removal of the bsr cassetteby transient expression of the Flp recombinase (Flp expressionplasmid was provided by Dr. Kyoji Horie, Osaka University)resulted in establishment ofATRIPflox-GFP/– cells. The cells werecharacterized after 20 nmol/L TAM treatment for 48 hoursunless otherwise stated.
Expression vectorsFull-length chicken ATRIP cDNA was amplified by reverse
transcriptase PCR from DT40 RNA and cloned into pDONRvector (Invitrogen). The sequence data were deposited at theDDBJ database (accession number AB684452). Mutations wereintroduced by the QuickChange Kit (Stratagene). Aftersequencing, the gateway system (Invitrogen) was used totransfer the cDNAs to the GFP expression vector containinga nuclear localization signal (NLS). GFP-chicken FANCIexpression vector has been described elsewhere (11).
Antibodies and reagentsAnti-chicken FANCD2 (17) and anti-Rad51 (18) serum have
been described. Anti-chicken FANCI serum was raised byimmunizing rabbits with GST fusion protein containing chick-en FANCI (1-251 amino acid region). Other antibodies werepurchased from Clontech (polyclonal anti-GFP), Santa CruzBiotechnology (polyclonal anti-ATR, monoclonal anti-Chk1),Sigma (monoclonal anti-FLAG M2 antibody), or Cell SignalingTechnology (monoclonal anti-phospho-Chk1-Ser345). ATMinhibitor Ku55933 and Chk1 inhibitor UCN-01 was purchasedfrom CalbioChem and Sigma, respectively.
Western blottingCells were treated with the indicated dose of TAM and/or
MMC (Kyowa-hakkou-Kirin) and lysed in SDS sample buff-er. Samples were separated by polyacrylamide gel electro-phoresis, transferred to a membrane, and detected withindicated antibodies and ECL reagents (GE Healthcare).Phostag-Western blotting was carried out as described(11).
Analysis of growth and cell sensitivity toward cisplatinCell cycle and cell proliferation rate with plastic microbeads
was assessed as described (19). Cell viability in liquid culturecontaining cisplatin (Nippon Kayaku) was assessed after 48hours using FACSCalibur (BD) and propidium iodide (PI)staining (11).
Subnuclear focus formation assayAfter MMC exposure, cytospin slides were fixed with 4%
paraformaldehyde/PBS and stained with antibodies againstchicken FANCD2 or FANCI followed by Alexa Fluor 594-conjugated secondary antibody (Invitrogen) with DAPI coun-terstaining. Images were captured by fluorescent microscopy(DM5500B; Leica) or confocal laser scanning microscopy (TCSSP5; Leica).
Figure 1. Generation of ATRIPflox-GFP/� mutant DT40 cells. A, schematicrepresentation of the gene targeting procedure. B, loss of ATRIP-GFPexpression following 4-OH TAM treatment (20 nmol/L).
Shigechi et al.
Cancer Res; 72(5) March 1, 2012 Cancer Research1150
on May 3, 2019. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 18, 2012; DOI: 10.1158/0008-5472.CAN-11-2904
Purification of recombinant chicken FANCD2 and FANCIBriefly, the DNA fragment encoding chicken FANCD2, wild-
type FANCI, or phospho-mimic mutant FANCI (Dx6) wasligated into the pET-15b vector, and was overexpressed in theEscherichia coli BL21(DE3) strain (Codon(þ)RIL; Stratagene).The cells were resuspended in buffer A containing 50 mmol/LTris-HCl (pH 8.0), 10% glycerol, 0.5 mol/L NaCl, 1 mmol/LPMSF, 12 mmol/L imidazole, and 5 mmol/L 2-mercaptoetha-nol, and disrupted by sonication. His6-tagged FANCD2 orFANCI was purified with nickel-nitrilotriacetic acid agaroseresin, a Heparin Sepharose CL-6B column (GEHealthcare), andSuperdex 200 gel filtration column (GE Healthcare). The His6tag was removed by digestion with thrombin protease (GEHealthcare). The ID complex was prepared by mixing FANCD2and FANCI preparations at 1:1 stoichiometry.
In vitro kinase reactionCloning and expression of chicken ATR that has been N-
terminally HFSC tagged in DT40 cells lacking endogenous ATRwill be described elsewhere (JE, FP, and NFL; unpublishedresults). Cells were lyzed in lysis buffer (0.5% Triton X100, 50mmol/L Tris-HCl pH 7.5, 150 mmol/L NaCl, 2 mmol/L EDTA, 2mmol/L EGTA, 25 mmol/L NaF, 25 mmol/L b-glyceropho-sphate, 10% glycerol, 1 mmol/L Na3VO4, 1 mmol/L DTT,1mmol/L PMSF) containing proteinase inhibitor tablet(Roche), and HFSC-ATR was immunoprecipitated usinganti-FLAG-beads (Sigma). Following washes, the immunopre-
cipitates were incubated in kinase buffer [10 mmol/L HEPES,pH 7.4, 50 mmol/L b-glycerophosphate, 50 mmol/L NaCl, 10%glycerol, 10 mmol/L MgCl2, 10 mmol/L MnCl2, 1 mmol/L DTT,6 mmol/L Na3VO4, 1 mmol/L PMSF, 5 mmol/L ATP, g-32P-ATP(10 mCi per sample)] containing proteinase inhibitor tablet for30 minutes at 30�C. Recombinant protein (1 mg) of chickenFANCD2, FANCI, FANCD2/FANCI complex, or GST-p53 (kind-ly provided by Dr Junya Kobayashi) purified from E. coli wasadded as exogenous substrate. Samples were separated using5% to 20% gradient SDS-PAGE gel.
Results and DiscussionTo test the function of ATR-ATRIP, we have constructed a
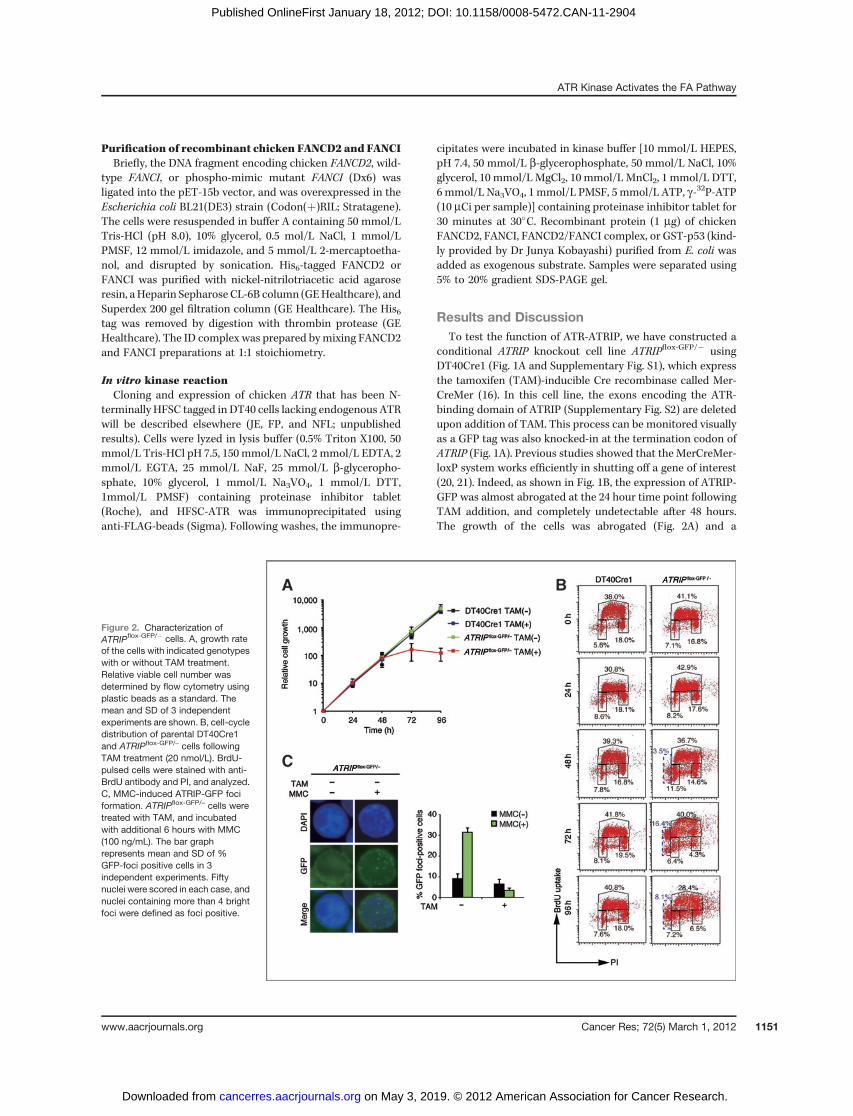
conditional ATRIP knockout cell line ATRIPflox-GFP/� usingDT40Cre1 (Fig. 1A and Supplementary Fig. S1), which expressthe tamoxifen (TAM)-inducible Cre recombinase called Mer-CreMer (16). In this cell line, the exons encoding the ATR-binding domain of ATRIP (Supplementary Fig. S2) are deletedupon addition of TAM. This process can be monitored visuallyas a GFP tag was also knocked-in at the termination codon ofATRIP (Fig. 1A). Previous studies showed that the MerCreMer-loxP system works efficiently in shutting off a gene of interest(20, 21). Indeed, as shown in Fig. 1B, the expression of ATRIP-GFP was almost abrogated at the 24 hour time point followingTAM addition, and completely undetectable after 48 hours.The growth of the cells was abrogated (Fig. 2A) and a
Figure 2. Characterization ofATRIPflox-GFP/� cells. A, growth rateof the cells with indicated genotypeswith or without TAM treatment.Relative viable cell number wasdetermined by flow cytometry usingplastic beads as a standard. Themean and SD of 3 independentexperiments are shown. B, cell-cycledistribution of parental DT40Cre1and ATRIPflox-GFP/– cells followingTAM treatment (20 nmol/L). BrdU-pulsed cells were stained with anti-BrdU antibody and PI, and analyzed.C, MMC-induced ATRIP-GFP fociformation. ATRIPflox-GFP/– cells weretreated with TAM, and incubatedwith additional 6 hours with MMC(100 ng/mL). The bar graphrepresents mean and SD of %GFP-foci positive cells in 3independent experiments. Fiftynuclei were scored in each case, andnuclei containing more than 4 brightfoci were defined as foci positive.
ATR Kinase Activates the FA Pathway
www.aacrjournals.org Cancer Res; 72(5) March 1, 2012 1151
on May 3, 2019. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 18, 2012; DOI: 10.1158/0008-5472.CAN-11-2904
significant amount of dead cells (with sub-G1 DNA content)appeared (Fig. 2B) at 72 hours. TAM treatment also resulted infewer MMC-induced ATRIP-GFP foci (Fig. 2C). For all furtherexperiments, we decided to use cells treatedwith TAM for 24 or48 hours to examine activation of the FA pathway.
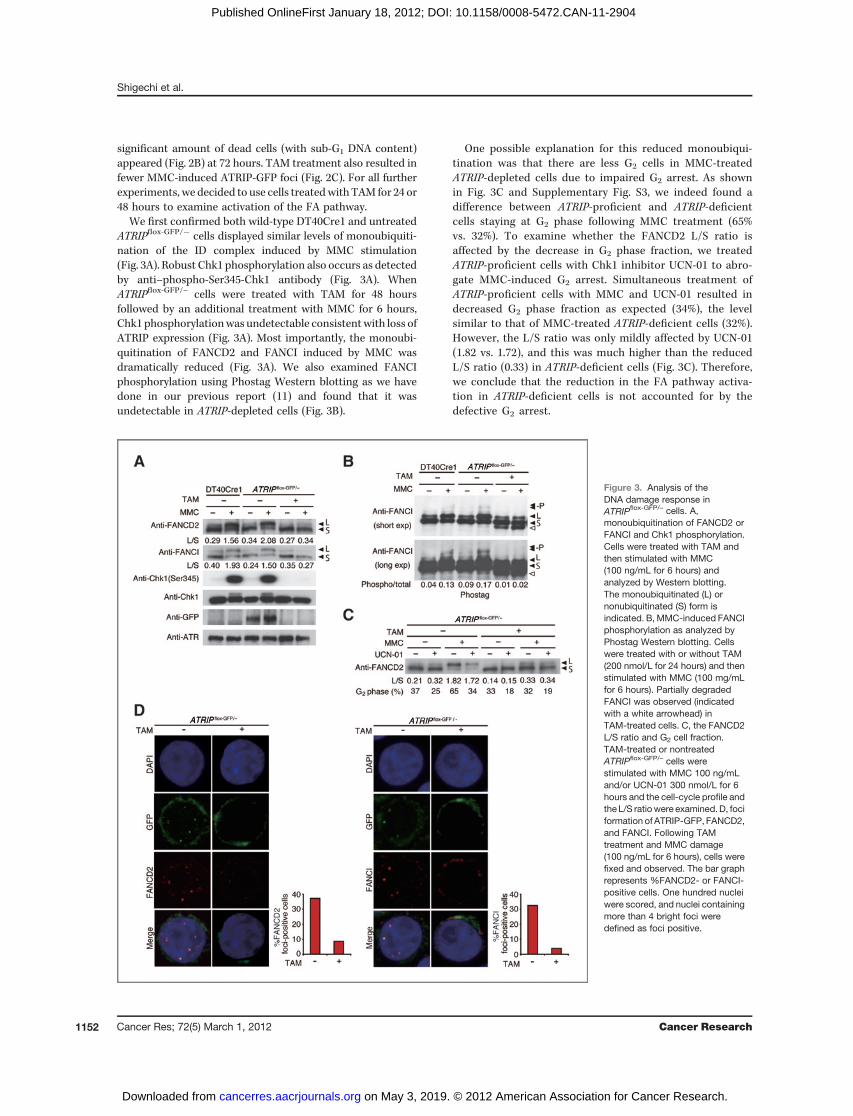
We first confirmed both wild-type DT40Cre1 and untreatedATRIPflox-GFP/� cells displayed similar levels of monoubiquiti-nation of the ID complex induced by MMC stimulation(Fig. 3A). Robust Chk1 phosphorylation also occurs as detectedby anti–phospho-Ser345-Chk1 antibody (Fig. 3A). WhenATRIPflox-GFP/– cells were treated with TAM for 48 hoursfollowed by an additional treatment with MMC for 6 hours,Chk1 phosphorylationwas undetectable consistentwith loss ofATRIP expression (Fig. 3A). Most importantly, the monoubi-quitination of FANCD2 and FANCI induced by MMC wasdramatically reduced (Fig. 3A). We also examined FANCIphosphorylation using Phostag Western blotting as we havedone in our previous report (11) and found that it wasundetectable in ATRIP-depleted cells (Fig. 3B).
One possible explanation for this reduced monoubiqui-tination was that there are less G2 cells in MMC-treatedATRIP-depleted cells due to impaired G2 arrest. As shownin Fig. 3C and Supplementary Fig. S3, we indeed found adifference between ATRIP-proficient and ATRIP-deficientcells staying at G2 phase following MMC treatment (65%vs. 32%). To examine whether the FANCD2 L/S ratio isaffected by the decrease in G2 phase fraction, we treatedATRIP-proficient cells with Chk1 inhibitor UCN-01 to abro-gate MMC-induced G2 arrest. Simultaneous treatment ofATRIP-proficient cells with MMC and UCN-01 resulted indecreased G2 phase fraction as expected (34%), the levelsimilar to that of MMC-treated ATRIP-deficient cells (32%).However, the L/S ratio was only mildly affected by UCN-01(1.82 vs. 1.72), and this was much higher than the reducedL/S ratio (0.33) in ATRIP-deficient cells (Fig. 3C). Therefore,we conclude that the reduction in the FA pathway activa-tion in ATRIP-deficient cells is not accounted for by thedefective G2 arrest.
Figure 3. Analysis of theDNA damage response inATRIPflox-GFP/– cells. A,monoubiquitination of FANCD2 orFANCI and Chk1 phosphorylation.Cells were treated with TAM andthen stimulated with MMC(100 ng/mL for 6 hours) andanalyzed by Western blotting.The monoubiquitinated (L) ornonubiquitinated (S) form isindicated. B, MMC-induced FANCIphosphorylation as analyzed byPhostag Western blotting. Cellswere treated with or without TAM(200 nmol/L for 24 hours) and thenstimulated with MMC (100 mg/mLfor 6 hours). Partially degradedFANCI was observed (indicatedwith a white arrowhead) inTAM-treated cells. C, the FANCD2L/S ratio and G2 cell fraction.TAM-treated or nontreatedATRIPflox-GFP/– cells werestimulated with MMC 100 ng/mLand/or UCN-01 300 nmol/L for 6hours and the cell-cycle profile andthe L/S ratiowere examined.D, fociformation of ATRIP-GFP, FANCD2,and FANCI. Following TAMtreatment and MMC damage(100 ng/mL for 6 hours), cells werefixed and observed. The bar graphrepresents %FANCD2- or FANCI-positive cells. One hundred nucleiwere scored, and nuclei containingmore than 4 bright foci weredefined as foci positive.
Shigechi et al.
Cancer Res; 72(5) March 1, 2012 Cancer Research1152
on May 3, 2019. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 18, 2012; DOI: 10.1158/0008-5472.CAN-11-2904

We noticed that small amounts of the L-form of the IDcomplex could still be observed after TAM treatment (Fig. 3Aand B). It was possible that the remaining I-D2 monoubiqui-tination could be due to ATM, whichmay substitute for ATR inMMC-induced activation of the FA pathway. However, specificpharmacologic inhibition (Ku55933) of ATM did not furtherdownregulate the levels of FANCD2 monoubiquitination inTAM-treated ATRIPflox-GFP/� cells (Supplementary Fig. S4). Wespeculate that deubiquitination of the ubiquitinated ID com-plex by USP1 may be inefficient and a recent structural studysuggests this might be the case, as the lysine-ubiquitin iso-peptide bond is protected from USP1 by the sequestration atthe ID interface (22). Alternatively, the efficient deubiquitina-tion by USP1 may require ATR–ATRIP kinase because thephosphorylation-regulated interaction between USP1/UAF1complex and FANCI is suggested to be important for thedeubiquitination (23).We further observed that MMC-induced ATRIP foci colo-
calized verywell with FANCD2or FANCI foci, and loss of ATRIPresulted in abrogation of the ID foci formation (Fig. 3D). Wealso observed that stable expression of a FANCI phospho-mimetic mutant (FANCI Dx6) in which 6 potential phosphor-ylation sites in the S/TQ cluster domain have been mutated toaspartic acid (11) partially rescued the loss of FANCD2 mono-ubiquitination caused by ATRIP deletion (SupplementaryFig. S5). This observation further supports our reasoningthat FANCI phosphorylation is required to trigger FANCD2monoubiquitination.We next examined whether ATR can directly phosphorylate
FANCI protein in vitro. We used chicken ATR immunopreci-pitated with an anti-FLAG antibody from DT40 cells stablyexpressing chickenATR amino terminally taggedwithmultipleepitopes (HA, FLAG, Strep TagII and Calmodulin-bindingprotein, termed HFSC-ATR) and recombinant chicken FANCI,FANCD2, or the FANCI–FANCD2 complex. Of note, the 2-step(FLAG-Strep tag) purified ATR preparation contained signif-icant amount of ATRIP as shown by a silver-stained gel(Supplementary Fig. S6) and mass spectrometric analysis(J.E. and N.L., unpublished results), similarly to the humanATR–ATRIP complex (24). We were able to detect autopho-sphorylation of ATR and the phosphorylated protein speciescorresponding to the phospho-ATRIP, phospho-FANCD2, andphospho-FANCI in these in vitro kinase assays (Fig. 4A). Asexpected, the mutant FANCI protein (Dx6 protein; ref. 11)could not be phosphorylated (Fig. 4B). Phosphorylation ofFANCD2 and FANCI was much less efficient compared withGST-p53 (data not shown). However, the phosphorylation onwild-type FANCI, but not on the Dx6-mutant FANCI, wassignificantly enhanced by the addition of FANCD2 (Fig. 4Aand B). This observation is consistent with our recent findingsthat efficient in vivo phosphorylation of FANCI requiresFANCD2 as well as the intact FA core complex (Tomida andcolleagues, submitted). It would be interesting to test whetherthe addition of the purified FA core complex could enhance theefficiency of the in vitro kinase reaction.To further elucidate how ATRIP participates in activation of
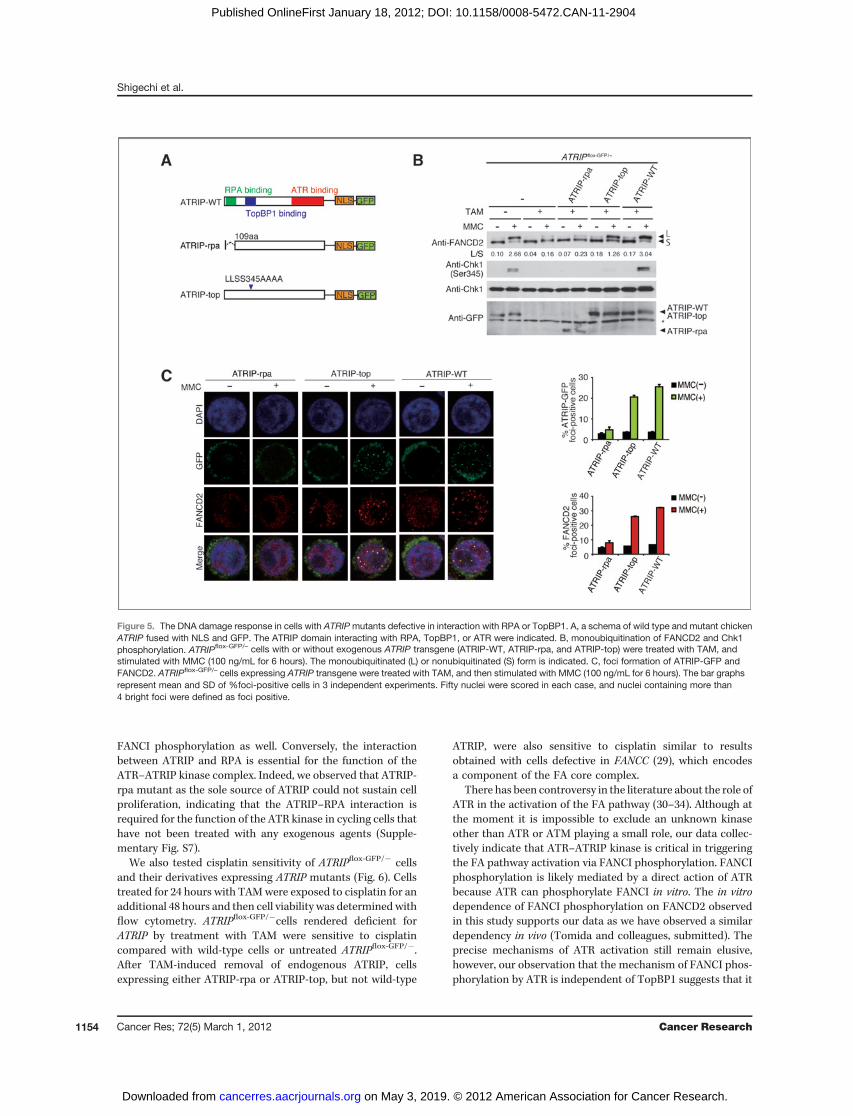
the FA pathway, we expressed wild-type or mutant chickenATRIP (Fig. 5A) in ATRIPflox-GFP/� cells. The N-terminal dele-
tion mutant lacking amino acids 1-108 (hereafter designatedATRIP-rpa) is expected to abrogate RPA binding becausethis region contains the conserved acidic amino acids (seeSupplementary Fig. S2) known to interact with RPA70 (25–27). The LLSS345AAAA mutation (designated ATRIP-top)should disrupt interaction with TopBP1, as these 4 residues(LLSS) are completely conserved between human and chickenATRIP (Supplementary Fig. S2) and have been reported tobe required for the ATRIP–TopBP1 interaction in human cells(28). Note that the ATRIP expression constructs included anNLS and GFP at their C-terminus, and transfectants wereselected by flow cytometry on the basis of increased GFPfluorescence.
Upon switching off expression of integrated ATRIP-GFP bytreatment of cells with TAM, the wild-type ATRIP transgenecould complement the cells with respect to both Chk1 phos-phorylation and FANCD2monoubiquitination following treat-ment with MMC (Fig. 5B). In contrast, Chk1 phosphorylationcould not be induced in cells expressing ATRIP-rpa or ATRIP-top transgenes (Fig. 5B). Interestingly, robust FANCD2 mono-ubiquitination was not supported by the ATRIP-rpa transgene,whereas cells expressing ATRIP-top were fully proficient forthis modification (Fig. 5B). The latter finding is consistentwith our recent observation that chicken cells lacking RAD17or human cells treated with siRNA targeting TOPBP1 werestill able to induce FANCD2 monoubiquitination (Tomida andcolleagues, submitted). Furthermore, colocalization of ATRIP-FANCD2 foci was abrogated in cells expressing ATRIP-rpa,but not the ATRIP-top mutant (Fig. 5C). These results indi-cated that the association between ATRIP and TopBP1 isdispensable for FANCD2 monoubiquitination and hence for
Figure 4. FANCI phosphorylation in in vitro ATR kinase reaction. ChickenHFSC-tagged ATR was immunoprecipitated with anti-FLAG andsubjected to in vitro kinase reaction. Anti-FLAG immunoprecipitates fromwild-typeDT40 cells was used as negative control. Recombinant chickenFANCI andFANCD2 (A) or eitherwild-typeFANCI (WT) or phospho-mimicFANCI (Dx6) and FANCD2 (B; 1 mg per reaction) were added as indicated.An asterisk indicates a nonspecific phosphorylation band. CBB,Coomasie brilliant blue staining.
ATR Kinase Activates the FA Pathway
www.aacrjournals.org Cancer Res; 72(5) March 1, 2012 1153
on May 3, 2019. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 18, 2012; DOI: 10.1158/0008-5472.CAN-11-2904
FANCI phosphorylation as well. Conversely, the interactionbetween ATRIP and RPA is essential for the function of theATR–ATRIP kinase complex. Indeed, we observed that ATRIP-rpa mutant as the sole source of ATRIP could not sustain cellproliferation, indicating that the ATRIP–RPA interaction isrequired for the function of the ATR kinase in cycling cells thathave not been treated with any exogenous agents (Supple-mentary Fig. S7).
We also tested cisplatin sensitivity of ATRIPflox-GFP/� cellsand their derivatives expressing ATRIP mutants (Fig. 6). Cellstreated for 24 hours with TAMwere exposed to cisplatin for anadditional 48 hours and then cell viability was determined withflow cytometry. ATRIPflox-GFP/�cells rendered deficient forATRIP by treatment with TAM were sensitive to cisplatincompared with wild-type cells or untreated ATRIPflox-GFP/�.After TAM-induced removal of endogenous ATRIP, cellsexpressing either ATRIP-rpa or ATRIP-top, but not wild-type
ATRIP, were also sensitive to cisplatin similar to resultsobtained with cells defective in FANCC (29), which encodesa component of the FA core complex.
There has been controversy in the literature about the role ofATR in the activation of the FA pathway (30–34). Although atthe moment it is impossible to exclude an unknown kinaseother than ATR or ATM playing a small role, our data collec-tively indicate that ATR–ATRIP kinase is critical in triggeringthe FA pathway activation via FANCI phosphorylation. FANCIphosphorylation is likely mediated by a direct action of ATRbecause ATR can phosphorylate FANCI in vitro. The in vitrodependence of FANCI phosphorylation on FANCD2 observedin this study supports our data as we have observed a similardependency in vivo (Tomida and colleagues, submitted). Theprecise mechanisms of ATR activation still remain elusive,however, our observation that the mechanism of FANCI phos-phorylation by ATR is independent of TopBP1 suggests that it
Figure 5. The DNA damage response in cells with ATRIPmutants defective in interaction with RPA or TopBP1. A, a schema of wild type and mutant chickenATRIP fused with NLS and GFP. The ATRIP domain interacting with RPA, TopBP1, or ATR were indicated. B, monoubiquitination of FANCD2 and Chk1phosphorylation. ATRIPflox-GFP/– cells with or without exogenous ATRIP transgene (ATRIP-WT, ATRIP-rpa, and ATRIP-top) were treated with TAM, andstimulated with MMC (100 ng/mL for 6 hours). The monoubiquitinated (L) or nonubiquitinated (S) form is indicated. C, foci formation of ATRIP-GFP andFANCD2. ATRIPflox-GFP/– cells expressing ATRIP transgene were treated with TAM, and then stimulated with MMC (100 ng/mL for 6 hours). The bar graphsrepresent mean and SD of %foci-positive cells in 3 independent experiments. Fifty nuclei were scored in each case, and nuclei containing more than4 bright foci were defined as foci positive.
Shigechi et al.
Cancer Res; 72(5) March 1, 2012 Cancer Research1154
on May 3, 2019. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 18, 2012; DOI: 10.1158/0008-5472.CAN-11-2904

should be distinct from that of Chk1. This also suggests thatATR activation can occur without TopBP1 association, incontrast to the widely accepted model (13). Supporting this
notion, a recent study showed that ATR autophosphorylatesitself in a TopBP1-independent manner (15). Additional stud-ies on ATR signaling will be required to provide further novelinsights into the activation of the FA pathway as well ascheckpoint regulation.
Disclosure of Potential Conflic of InterestNo potential conflicts of interest were disclosed.
AcknowledgmentsThe authors thank Dr. Kyoji Horie (Osaka University) for Flp recombi-
nase expression vector, Dr. Junya Kobayashi (Radiation Biology Center,Kyoto University) for GST-p53 vector, Dr James Brown (NUI Galway) foradvice on in vitro kinase reaction, and Ms. Seiko Arai for secretarialassistance.
Grant SupportThis work was supported in part by grants-in-aid from the Ministry of
Education, Science, Sports, and Culture of Japan and also by The UeharaMemorial Foundation and Takeda foundation.
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicate thisfact.
Received August 28, 2011; revised December 14, 2011; accepted January 1, 2012;published OnlineFirst January 18, 2012.
References1. Wang W. Emergence of a DNA-damage response network consisting
of Fanconi anaemia and BRCA proteins. Nat Rev Genet 2007;8:735–48.
2. KeeY,D'AndreaAD. Expanded roles of the Fanconi anemia pathway inpreserving genomic stability. Genes Dev 2010;24:1680–94.
3. Kitao H, Takata M. Fanconi anemia: a disorder defective in the DNAdamage response. Int J Hematol 2011;93:417–24.
4. Yan Z, Delannoy M, Ling C, Daee D, Osman F, Muniandy PA, et al.A histone-fold complex and FANCM form a conserved DNA-remo-deling complex to maintain genome stability. Mol Cell 2010;37:865–78.
5. Singh TR, Saro D, Ali AM, Zheng XF, Du CH, Killen MW, et al. MHF1-MHF2, a histone-fold-containing protein complex, participates in theFanconi anemia pathway via FANCM. Mol Cell 2010;37:879–86.
6. Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C,Hejna J, et al. Interaction of the Fanconi anemia proteins andBRCA1 ina common pathway. Mol Cell 2001;7:249–62.
7. MatsushitaN, KitaoH, IshiaiM,NagashimaN,HiranoS,OkawaK, et al.A FancD2-monoubiquitin fusion reveals hidden functions of Fanconianemia core complex in DNA repair. Mol Cell 2005;19:841–7.
8. Raschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, Griffith JD, et al.Mechanism of replication-coupled DNA interstrand crosslink repair.Cell 2008;134:969–80.
9. Knipscheer P, Raschle M, Smogorzewska A, Enoiu M, Ho TV, ScharerOD, et al. The Fanconi anemia pathway promotes replication-depen-dent DNA interstrand cross-link repair. Science 2009;326:1698–701.
10. Garner E, Smogorzewska A. Ubiquitylation and the Fanconi anemiapathway. FEBS Lett 2011;585:2853–60.
11. Ishiai M, Kitao H, Smogorzewska A, Tomida J, Kinomura A, Uchida E,et al. FANCI phosphorylation functions as amolecular switch to turn onthe Fanconi anemia pathway. Nat Struct Mol Biol 2008;15:1138–46.
12. Shiloh Y. ATM and related protein kinases: safeguarding genomeintegrity. Nat Rev Cancer 2003;3:155–68.
13. Flynn RL, Zou L. ATR: amaster conductor of cellular responses toDNAreplication stress. Trends Biochem Sci 2011;36:133–40.
14. Burrows AE, Elledge SJ. How ATR turns on: TopBP1 goes on ATRIPwith ATR. Genes Dev 2008;22:1416–21.
15. Liu S, Shiotani B, Lahiri M, Marechal A, Tse A, Leung CC, et al. ATRautophosphorylation as a molecular switch for checkpoint activation.Mol Cell 2011;43:192–202.
16. Arakawa H, Hauschild J, Buerstedde JM. Requirement of the activa-tion-induced deaminase (AID) gene for immunoglobulin gene conver-sion. Science 2002;295:1301–6.
17. Yoshikiyo K, Kratz K, Hirota K, Nishihara K, Takata M, Kurumizaka H,et al. KIAA1018/FAN1 nuclease protects cells against genomic insta-bility induced by interstrand cross-linking agents. Proc Natl Acad SciU S A 2010;107:21553–7.
18. Tachiwana H, Shimura M, Nakai-Murakami C, Tokunaga K, TakizawaY, Sata T, et al. HIV-1 Vpr induces DNA double-strand breaks. CancerRes 2006;66:627–31.
19. Yamamoto K, Ishiai M, Matsushita N, Arakawa H, Lamerdin JE,Buerstedde JM, et al. Fanconi anemia FANCG protein in mitigatingradiation- and enzyme-induced DNA double-strand breaks by homol-ogous recombination in vertebrate cells. Mol Cell Biol 2003;23:5421–30.
20. Fujimori A, Tachiiri S, Sonoda E, Thompson LH, Dhar PK, Hiraoka M,et al. Rad52 partially substitutes for the Rad51 paralog XRCC3 inmaintaining chromosomal integrity in vertebrate cells. EMBO J2001;20:5513–20.
21. Arakawa H. Excision of floxed-DNA sequences by transient inductionof Mer-Cre-Mer. Subcell Biochem 2006;40:347–9.
22. Joo W, Xu G, Persky NS, Smogorzewska A, Rudge DG, BuzovetskyO, et al. Structure of the FANCI-FANCD2 complex: insights intothe Fanconi anemia DNA repair pathway. Science 2011;333:312–6.
23. Yang K,MoldovanGL, Vinciguerra P,Murai J, Takeda S, D'Andrea AD.Regulation of the Fanconi anemia pathway by a SUMO-like deliverynetwork. Genes Dev 2011;25:1847–58.
24. Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners incheckpoint signaling. Science 2001;294:1713–6.
Figure 6. Cisplatin sensitivity caused by loss ormutation ofATRIP in DT40cells. Indicated cells were cultured for 48 hours in the medium containingcisplatin. Cell viability was assessed with flow cytometry and normalizedto cells not treated with cisplatin.
ATR Kinase Activates the FA Pathway
www.aacrjournals.org Cancer Res; 72(5) March 1, 2012 1155
st
on May 3, 2019. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 18, 2012; DOI: 10.1158/0008-5472.CAN-11-2904

25. Itakura E, Takai KK, Umeda K, Kimura M, Ohsumi M, Tamai K, et al.Amino-terminal domain of ATRIP contributes to intranuclear relocationof the ATR-ATRIP complex following DNA damage. FEBS Lett2004;577:289–93.
26. Namiki Y, Zou L. ATRIP associates with replication protein A-coatedssDNA through multiple interactions. Proc Natl Acad Sci U S A2006;103:580–5.
27. Ball HL, Ehrhardt MR, Mordes DA, Glick GG, Chazin WJ, Cortez D.Function of a conserved checkpoint recruitment domain in ATRIPproteins. Mol Cell Biol 2007;27:3367–77.
28. Mordes DA,GlickGG, ZhaoR,CortezD. TopBP1 activatesATR throughATRIP and a PIKK regulatory domain. Genes Dev 2008;22:1478–89.
29. Hirano S, Yamamoto K, Ishiai M, Yamazoe M, Seki M, Matsushita N,et al. Functional relationships of FANCC to homologous recombina-tion, translesion synthesis, and BLM. EMBO J 2005;24:418–27.
30. Andreassen PR, D'Andrea AD, Taniguchi T. ATR couples FANCD2monoubiquitination to the DNA-damage response. Genes Dev 2004;18:1958–63.
31. Sobeck A, Stone S, Hoatlin ME. DNA structure-induced recruitmentand activation of the Fanconi anemia pathway protein FANCD2. MolCell Biol 2007;27:4283–92.
32. Sobeck A, Stone S, Landais I, de Graaf B, Hoatlin ME. The Fanconianemia protein FANCM is controlled by FANCD2 and the ATR/ATMpathways. J Biol Chem 2009;284:25560–8.
33. Chan KL, Palmai-Pallag T, Ying S, Hickson ID. Replication stressinduces sister-chromatid bridging at fragile site loci in mitosis. NatCell Biol 2009;11:753–60.
34. Toledo LI, Murga M, Gutierrez-Martinez P, Soria R, Fernandez-Cape-tillo O. ATR signaling can drive cells into senescence in the absence ofDNA breaks. Genes Dev 2008;22:297–302.
Shigechi et al.
Cancer Res; 72(5) March 1, 2012 Cancer Research1156
on May 3, 2019. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 18, 2012; DOI: 10.1158/0008-5472.CAN-11-2904

2012;72:1149-1156. Published OnlineFirst January 18, 2012.Cancer Res Tomoko Shigechi, Junya Tomida, Koichi Sato, et al. Anemia DNA Repair Pathway
ATRIP Kinase Complex Triggers Activation of the Fanconi−ATR
Updated version
10.1158/0008-5472.CAN-11-2904doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2012/01/18/0008-5472.CAN-11-2904.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/72/5/1149.full#ref-list-1
This article cites 34 articles, 18 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/72/5/1149.full#related-urls
This article has been cited by 3 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/72/5/1149To request permission to re-use all or part of this article, use this link
on May 3, 2019. © 2012 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst January 18, 2012; DOI: 10.1158/0008-5472.CAN-11-2904