astrocytes in brain aging and neurodegeneration
TRANSCRIPT

Hyman M. Schipper
Astrocytes in Brain Agingand Neurodegeneration
R.G. LANDESC O M P A N Y
NEUROSCIENCE I N T E L L I G E N C E U N I T 3

Hyman M. SchipperDepartment of Neurology and Neurosurgery
Department of Medicine (Geriatrics)and Centre for Studies in Aging
McGill Universityand
Bloomfield Centre for Research in AgingLady Davis Institute for Medical Research
Sir Mortimer B. Davis-Jewish General HospitalMontreal, Quebec, Canada
Astrocytesin Brain Aging andNeurodegeneration
NEUROSCIENCEINTELLIGENCEUNIT
AUSTIN, TEXAS
U.S.A.
R.G. LANDESCOMPANY
AUSTIN, TEXAS
U.S.A.

Astrocytes in brain aging and neurodegeneration / [edited by] Hyman M. Schipper.p. cm. -- (Neuroscience intelligence unit)
ISBN 1-57059-489-9 (alk. paper)1. Nervous system--Degeneration. 2. Nervous system--Aging. 3. Astrocytes.I. Schipper, Hyman M., 1954- . II. Series.
[DNLM: 1. Neurodegenerative Diseases--physiopathology. 2. Astrocytes--physiology. 3. Brain Diseases--physiopathology. 4. Brain--physiology. 5. Aging--physiology. WL 300A859 1998]RC365.A88 1998616.8'047--dc21DNLM/DLC 98-26335for Library of Congress CIP
Astrocytes in Brain Aging and Neurodegeneration
ISBN: 1-57059-489-9
Library of Congress Cataloging-in-Publication Data
NEUROSCIENCE INTELLIGENCE UNIT
R.G. LANDES COMPANYAustin, Texas, U.S.A.
Copyright © 1998 R.G. Landes Company
All rights reserved.No part of this book may be reproduced or transmitted in any form or by any means,electronic or mechanical, including photocopy, recording, or any information storage andretrieval system, without permission in writing from the publisher.Printed in the U.S.A.
Please address all inquiries to the Publishers:R.G. Landes Company, 810 South Church Street, Georgetown, Texas, U.S.A. 78626Phone: 512/ 863 7762; FAX: 512/ 863 0081
While the authors, editors and publisher believe that drug selection and dosage and the specifica-tions and usage of equipment and devices, as set forth in this book, are in accord with currentrecommendations and practice at the time of publication, they make no warranty, expressed orimplied, with respect to material described in this book. In view of the ongoing research, equipmentdevelopment, changes in governmental regulations and the rapid accumulation of informationrelating to the biomedical sciences, the reader is urged to carefully review and evaluate the informa-tion provided herein.

Hyman M. SchipperDepartment of Neurology and Neurosurgery
Department of Medicine (Geriatrics)and Centre for Studies in Aging
McGill Universityand
Bloomfield Centre for Research in AgingLady Davis Institute for Medical Research
Sir Mortimer B. Davis-Jewish General HospitalMontreal, Quebec, Canada
Astrocytesin Brain Aging andNeurodegeneration
NEUROSCIENCEINTELLIGENCEUNIT
AUSTIN, TEXAS
U.S.A.
R.G. LANDESCOMPANY
PUBLISHER’S NOTE
Landes Bioscience produces books in six Intelligence Unit series:Medical, Molecular Biology, Neuroscience, Tissue Engineering,Biotechnology and Environmental. The authors of our books areacknowledged leaders in their fields. Topics are unique; almostwithout exception, no similar books exist on these topics.
Our goal is to publish books in important and rapidly changingareas of bioscience for sophisticated researchers and clinicians. Toachieve this goal, we have accelerated our publishing program toconform to the fast pace at which information grows in bioscience.Most of our books are published within 90 to 120 days of receipt ofthe manuscript. We would like to thank our readers for theircontinuing interest and welcome any comments or suggestions theymay have for future books.
Judith KemperProduction Manager
R.G. Landes Company

DEDICATION
To my parents, Freda and Mendel, for their unflaggingdevotion.

CONTENTS
Part I: Biology of Astrocytes
1. Astrocyte Ontogenesis and Classification ................................................ 3James E. Goldman
Genesis of Radial Glia and Their Transformation into Astrocytes ....... 4Genesis of Astrocytes from SVZ Cells .................................................... 5Control of Astrocyte Differentiation ...................................................... 6Genesis of Astrocyte Heterogeneity ........................................................ 7Generation of Astrocytes in the Adult CNS ........................................... 8
2. Functions of Astrocytes ........................................................................... 15Harold K. Kimelberg and Michael Aschner
Introduction........................................................................................... 15Functions of Astrocytes ......................................................................... 16Homeostasis of the Extracellular Space ................................................ 17Transmitter Uptake Systems ................................................................. 21Receptors for Transmitters ................................................................... 22Astrocytes and the Blood-Brain Barrier (BBB) .................................... 26Astrocytes and Immune and Inflammatory Responses in the CNS ... 28
3. Astrocyte Pathophysiology in Disordersof the Central Nervous System ............................................................... 41Michael D. Norenberg
Introduction........................................................................................... 41Normal Functions ................................................................................. 41General Response to Injury ................................................................... 42Injury to Astrocytes in CNS Disorders (Passive Role) ........................ 43Active Role of Astrocytes in CNS Disorders ........................................ 44Clinical Considerations ......................................................................... 47Perspectives and Conclusions ............................................................... 53
Part II: Astrocytes in Human Brain Senescenceand Neurodegenerative Disorders
4. Glial Responses to Injury, Disease, and Aging ...................................... 71Lawrence F. Eng and Yuen Ling Lee
Introduction........................................................................................... 71Astrocyte Intermediate Filament, Glial Fibrillary Acidic Protein ....... 71Astrocytes in Experimental Gliosis ....................................................... 73Astrocytes in Disease ............................................................................. 73Astrocyte Activation of GFAP in Astrogliosis ...................................... 74Microglial Activation ............................................................................. 74Monocyte/Macrophage Activation ....................................................... 75Endothelial Cell Activation ................................................................... 75Astrocytes in Normal Aging .................................................................. 75Astrocyte Inclusions in Normal Aging ................................................. 77Astrocyte Inclusions in Disease ............................................................. 78

5. Astrocyte Pathology in Alzheimer Disease ............................................ 91Jerzy Wegiel and Henryk M. Wisniewski
Neuropathological Changes in Alzheimer Disease .............................. 91Relationships Between Amyloid-β, Neurons,
and Glial Cells in AD ......................................................................... 91Astrogliosis in Aging and AD ................................................................ 93Astrocyte Degeneration in AD .............................................................. 99
6. Parkinson’s Disease ............................................................................... 111Donato A. Di Monte
Introduction......................................................................................... 111Idiopathic Parkinson’s Disease ........................................................... 111MPTP-Induced Parkinsonism ............................................................ 113Neuronal-Astrocyte Interactions in Nigrostriatal Degeneration ...... 115Conclusion ........................................................................................... 121
7. Astrocytes in Transmissible Spongiform Encephalopathies(Prion Diseases) ..................................................................................... 127Pawel P. Liberski, Radzislaw Kordek, Paul Brown
and D. Carleton GajdusekIntroduction......................................................................................... 127KURU ................................................................................................... 130Creutzfeldt-Jakob Disease (CJD)
and Gerstmann-Straussler-Scheinker Disease (GSS) .................... 130GSS ....................................................................................................... 135The Involvement of Astrocytes in Formation
of Amyloid Plaques ......................................................................... 137Scrapie, Bovine Spongiform Encephalopathy (BSE),
and Chronic Wasting Disease (CWD) ........................................... 137BSE and CWD...................................................................................... 143Interaction Between Astrocytes and Oligodendrocytes ..................... 143A Particular Form of Astrocytic Reaction in TSES ............................ 145Expression of Glial Fibrillary Acidic Protein (GFAP)
and Its mRNA .................................................................................. 145Astrocytes and the Expression of Cytokines ...................................... 149Conclusions ......................................................................................... 153
8. Astrocytes in Other Neurodegenerative Diseases ............................... 165Dennis W. Dickson
Introduction......................................................................................... 165Neurofibrillary Tangles as an Archetype
of Cytoskeletal Inclusions ............................................................... 167Neurodegenerative Disorders with Filamentous Glial
Inclusion Bodies .............................................................................. 169Progressive Supranuclear Palsy (PSP) ................................................ 171Pick’s Disease ....................................................................................... 175

Corticobasal Degeneration (CBD) ..................................................... 176Argyrophilic Grain Dementia (AGD) ................................................ 179Familial Frontotemporal Dementia and Parkinsonism
Linked to Chromosome 17 (FTDP-17) ......................................... 180Multiple System Atrophy (MSA) ........................................................ 180Familial Amyotrophic Lateral Sclerosis (FALS) ................................. 181
Part III: Experimental Models of Astrocyte Senescence:Implications for Neurodegenerative Disease
9. The Peroxidase-Positive Subcortical Glial System .............................. 191Marc B. Mydlarski, James R. Brawer and Hyman M. Schipper
Introduction......................................................................................... 191Tinctorial and Histochemical Features .............................................. 191Topography of the Peroxidase-Positive Astroglia ............................. 192Modulation of the Peroxidase-Positive Glial System ........................ 193Peroxidase-Positive Astrocytes in Primary Culture ........................... 196Subcellular Precursors of Peroxidase-Positive
Astroglial Inclusions ........................................................................ 197Summary and Conclusions ................................................................. 202
10. Astrocyte Granulogenesis and the Cellular Stress Response .............. 207Marc B. Mydlarski and Hyman M. Schipper
HSP Expression in Acutely-stressed Neural Tissues:Effects of Aging ................................................................................ 208
Stress Protein Expression in the Agingand Degenerating Human Brain .................................................... 209
A Cellular Stress Model for the Biogenesisof Astroglial Inclusions ................................................................... 210
Astrocyte Senescence and the Origin of Corpora Amylacea ............. 221
11. Glial Iron Sequestration and Neurodegeneration ............................... 235Hyman M. Schipper
The Free Radical Hypothesis of Parkinson’s Disease ........................ 235The Redox Neurobiology of Alzheimer’s Disease .............................. 235Iron Deposition and Neurodegenerative Disease .............................. 236Iron Sequestration in Aging Astroglia ................................................ 237The Role of HO-1 in Brain Iron Deposition ...................................... 239Pro-toxin Bioactivation by Astrocytes in Primary Culture ............... 242Pathological Glial-Neuronal Interaction in Parkinson’s Disease ..... 243Conclusion ........................................................................................... 246
Index ................................................................................................................ 253

Hyman M. SchipperDepartment of Neurology and Neurosurgery
Department of Medicine (Geriatrics) and Centre for Studies in AgingMcGill University and
Bloomfield Centre for Research in AgingLady Davis Institute for Medical Research
Sir Mortimer B. Davis-Jewish General Hospital,Montreal, Quebec, Canada
Chapters 9, 10, 11
EDITOR
CONTRIBUTORSMichael AschnerDepartment of Physiology
and PharmacologyBowman Gray School of MedicineWinston-Salem, North Carolina, U.S.A.Chapter 2
James R. BrawerDepartment of Anatomy
and Cell BiologyMcGill UniversityMontreal, Quebec, CanadaChapter 9
Paul BrownLaboratory of Central Nervous
System Studies, National Instituteof Neurological Disorders and Stroke
National Institutes of HealthBethesda, Maryland, U.S.A.Chapter 7
Donato A. Di MonteThe Parkinson’s InstituteSunnyvale, California, U.S.A.Chapter 6
Dennis W. DicksonResearch DepartmentMayo Clinic JacksonvilleJacksonville, Florida, U.S.A.Chapter 8
Lawrence F. EngPathology ResearchVAPA Health Care SystemPalo Alto, California andStanford University School of MedicineStanford, California, U.S.A.Chapter 4
James E. GoldmanDepartment of Pathology
and The Center for Neurobiologyand Behavior
Columbia University College of P&SNew York, New York, U.S.A.Chapter 1
D. Carleton GajdusekLaboratory of Central Nervous
System Studies, National Instituteof Neurological Disorders and Stroke
National Institutes of HealthBethesda, Maryland, U.S.A.Chapter 7
Harold K. KimelbergDepartment of Pharmacology
and NeuroscienceDivision of NeurosurgeryAlbany Medical CollegeAlbany, New York, U.S.A.Chapter 2

Radzislaw KordekLaboratory of Central Nervous
System StudiesNational Institute of Neurological
Disorders and StrokeNational Institutes of HealthBethesda, Maryland, U.S.A. andLaboratories of Tumor BiologyLaboratory of Electron Microscopy
and NeuropathologyMedical Academy LodzLodz, PolandChapter 7
Yuen Ling LeePathology ResearchVAPA Health Care SystemPalo Alto, California andStanford University School of MedicineStanford, California, U.S.A.Chapter 4
Pawel P. LiberskiLaboratory of Central Nervous
System StudiesNational Institute of Neurological
Disorders and StrokeNational Institutes of HealthBethesda, Maryland, U.S.A. andLaboratories of Tumor BiologyLaboratory of Electron Microscopy
and NeuropathologyMedical Academy Lodz andLaboratory of Electron MicroscopyDepartment of PathologyPolish Mother Memorial HospitalLodz, PolandChapter 7
Marc B. MydlarskiDepartment of Neurology
and NeurosurgeryMcGill University andBloomfield Centre for Research in AgingLady Davis Institute for Medical ResearchSir Mortimer B. Davis-Jewish
General HospitalMontreal, Quebec, CanadaChapters 9, 10
Michael D. NorenbergLaboratory of NeuropathologyVeterans Administration Medical
Center andDepartments of Pathology, and
Biochemistry and Molecular BiologyUniversity of Miami School of MedicineMiami, Florida, U.S.A.Chapter 3
Jerzy WegielDepartment of Pathological
NeurobiologyNew York State Institute for Basic
Research in DevelopmentalDisabilities
Staten Island, New York, U.S.A.Chapter 5
Henryk M. WisniewskiDepartment of Pathological
NeurobiologyNew York State Institute for Basic
Research in DevelopmentalDisabilities
Staten Island, New York, U.S.A.Chapter 5

PREFACE
The last decade or so has witnessed a remarkable proliferation of originalscientific papers, review articles and books devoted to the neuroglia and their
involvement in health and disease. In the prefaces to the many excellent com-pendia currently available on this topic, the editors almost invariably take painsto point out that for almost 150 years the study of neuroglia in general, andastrocytes in particular, has been largely eclipsed by the effort to decipher theproperties of what has traditionally been regarded as the “business” end of thenervous system, the neurons and their connections. To be sure, no one woulddeny the paramount importance of neurons to the workings of the brain andits ailments. Yet, there is a rapidly-growing awareness, fueled by a biotechno-logical prowess permitting exquisitely refined analyses of cellular behavior, thatthe astroglia engage in intimate, mutually-dependent interactions with virtu-ally all neural cell types, including neurons, and subserve a multitude of adap-tive functions vital to the maintenance of normal brain structure and activity.To cite but a few examples, astrocytes are known to assume pivotal roles in theestablishment of the blood-brain barrier and the regulation of ion homeosta-sis, the elaboration of a scaffolding for neuronal migration during embryogen-esis, the sequestration and metabolism of various neurotransmitters and otherneuroactive substances, and the production of immunomodulatory and pro-inflammatory cytokines and neuropeptides. In this regard, it should come asno surprise that astrocyte dysfunction resulting from injury or disease may me-diate a host of dystrophic effects within the CNS and thereby contribute to adecline in neurological status. The formation of epileptogenic scar tissue inresponse to CNS trauma, the release of excitotoxic amino acids following tissuehypoxia, metal exposure or oxidative stress, neoplastic transformation andmalignant behavior, and the bioactivation of pro-toxins (such as MPTP) topotent neurotoxins (MPP+) are illustrative of some clinically-relevant patho-physiologic processes which directly implicate the astroglial compartment.
Astrocyte hypertrophy and hyperplasia, the biosynthesis of GFAP-associ-ated intermediate filaments (reactive gliosis) and the accumulation of discretecytoplasmic inclusions are characteristic pathological features of the major ag-ing-related neurodegenerative disorders, including Alzheimer’s disease,Parkinson’s disease, and amyotrophic lateral sclerosis. Gliosis and inclusion bodyformation also figure prominently in the relatively uncommon humanneurodegenerative conditions, such as Pick’s disease and corticobasal gangli-onic degeneration, and occur to a lesser extent in the course of normal brainaging. The raison d’être of this monograph was to consolidate information con-cerning the established and putative roles of astroglia in brain aging andneurodegeneration gleaned from vast and often disparate literatures on the bi-ology and pathology of these cells. To achieve this objective, I invited theparticipation of respected investigators from a mix of basic and clinical de-partments whose interests in the neuroglia are diverse and long-standing. Inaddition to providing thorough reviews of their respective fields, each team of

contributors was requested to speculate freely on the question “In the condi-tion under consideration, do the astrocytic changes actively contribute to thedegenerative process or do they merely represent passive responses to primaryneuronal injury?” Given the divergence of opinion on this question, a certaindegree of overlap of material covered by the authors (e.g., the role of astrogliain Alzheimer’s disease) was not only tolerated but encouraged.
The chapters in this monograph are grouped in three sections: I. Biologyof Astrocytes. Collectively, the chapters in this section constitute a comprehen-sive discussion of the origin and known functions of astroglia in the mamma-lian CNS and the roles these cells may play in the pathophysiology of neuro-logical disorders. II. Astrocytes in Human Brain Senescence and NeurodegenerativeDisorders. In this section, detailed accounts of the pathology of astrocytes andtheir involvement in human brain aging and various neurodegenerative condi-tions are presented. III. Experimental Models of Astrocyte Senescence: Implica-tions for Neurodegenerative Disease. In this final part, experimental approachesto the delineation of the role of astroglia in brain aging and degeneration aredescribed.
We hope that this compendium will appeal to basic neuroscientists inter-ested in various aspects of neuroglial biology, as well as to clinically-orientedinvestigators concerned with the pathogenesis of the major humanneurodegenerative disorders. I am deeply grateful to the many mentors, col-leagues and students at home and abroad who have helped shape my interestand refine my knowledge of the neuroglia and their place in clinical medicine.
Hyman M. Schipper

Part I
Biology of Astrocytes

CHAPTER 1
Astrocytes in Brain Aging and Neurodegeneration, edited by Hyman M. Schipper.©1998 R.G. Landes Company.
Astrocyte Ontogenesisand ClassificationJames E. Goldman
Astrocytes, first named for their star-shaped appearance as visualized with heavy metalimpregnations,1 in fact display a extensive variety of morphologies. All are united in
their astrocyte nature, however, by common features, including multiple, thin processes,close interactions with both the neuronal and mesenchymal elements of the CNS, the pres-ence of intermediate filaments of several types (vimentin, GFAP, nestin), and the expressionof a variety of other molecules, such as S-100β and glutamine synthetase.
Besides their complex, multiprocess shapes the other salient histological characteristicof astrocytes is their interactions with specific sets of other cells. First, the basal lamina thatsurrounds blood vessels in the brain and that lines the pial surface of the brain is coveredwith astrocyte end feet (the ends of astrocyte processes).2 This is indeed a very large surfacearea, and thus requires an exceedingly large number of astrocyte processes. Second, astro-cytes intimately associate with neurons, wrapping neuronal perikarya and dendrites, con-tacting neurons in zones between synaptic contacts.2-4 Thus, astrocytes serve to isolate indi-vidual synapses or groups of synapses, perhaps those that share functional connections orcharacteristics. Such isolation of synapses makes sense in view of the astrocytes’ abilities totake up neurotransmitters with high affinities and to buffer potassium (see chapter 2). Theseinteractions may well serve to condition and maintain astrocyte shape (see below).
Astrocytes are not distributed randomly in the brain, but rather lie in separate do-mains with some peripheral overlap. For example, the “domain” of a neocortical astrocyte isroughly spherical with a diameter of about 100 microns.5 Similarly, “domains” of retinalastrocytes are spatially separate at 100-150 microns, with a modest degree of overlap in theperipheral processes.6 Subpial astrocytes are not spherical, but look like truncated spheresor columns.7 Thus, astrocyte development must somehow produce a matrix of astrocytespheres which intersect only at their peripheries. It is at the periphery, incidentally, thatastrocytes are connected by gap junctions, allowing movement of ions and small moleculesthrough an astrocyte “syncytium” over many hundreds of cubic microns.8 How this regularspacing is accomplished is not known. Since glia continue to divide as they migrate throughthe brain (see below), sibling astrocytes begin life next to each other after a mitotic division.Do they migrate away from each other, or does the growth of the brain continue to separaterelated glial cells?
Astrocytes display a wonderful variety of sizes and shapes. In most gray matter regions,where astrocytes have been traditionally termed “protoplasmic,” the cell body and domainsof all processes roughly describe a sphere or ellipsoid. Processes branch into ever-finer twigs,more like the boughs of a tree than the rays of a star, eventually reaching tremendous numbers

Astrocytes in Brain Aging and Neurodegeneration4
and microscopic size.3 In the cerebellar granule cell layer, “velate” astrocytes wrap thin sheet-like extensions about the mossy fiber glomeruli.9,10
Astrocytes in white matter (classically termed “fibrous”) display fewer processes and aless complex branching pattern than their gray matter relatives. Processes separate fasciclesof axons, a characteristic particularly easily observed in spinal cord tracts and optic nerve.11,12
Astrocyte processes also contact nodes of Ranvier,13 where they may play a role in spatial ionbuffering.
Some astrocytes display processes oriented “radially,” perpendicular to the pial surface.These include the Bergmann glia of the cerebellar molecular layer, which retain their radi-ally-oriented nature first established for granule cell migration.9,14 Muller glia of the retinaare also first established as radially oriented cells, coursing through all retinal layers, andremain so for life. Radial type glia in periventricular regions, particularly around the thirdand fourth ventricles and aqueduct of Sylvius, display long processes beginning at the ven-tricular surface and extending for hundreds of microns into the parenchyma of the hypo-thalamus and brain stem.15-17
Genesis of Radial Glia and their Transformation into AstrocytesThe term “radial glia” is used to describe elongated, bipolar glial cells that arise during
early histogenesis of the CNS. Heavy metal impregnations and more recently, immunocy-tochemistry, have produced a detailed view of the radial glial scaffolding in the developingbrain.18-20 Oriented “radially” (perpendicular to the pial surface), these cells extend from theventricular zone to the pia and develop concurrently with the first sets of neurons.21-23 Notonly are radial glia generated contemporaneously with some neuronal populations, but alsothey share a lineage with neurons, since early progenitors give rise to both radial glia and tothe neurons that migrate along them.22-24 Thus, there is a glial-neuronal fate decision for asubpopulation of cells in the early ventricular zone, although how this decision is accom-plished is not known.
Radial glia have long been considered a form of astrocyte, based upon the expression ofthe intermediate filaments vimentin and nestin, and in primates, GFAP, as well as the stor-age of glycogen and the interactions with the pial surface, all characteristics of astrocytes.Furthermore, radial glia have been considered the source of many of the astrocytes in themature CNS. This transformation of radial glia into astrocytes has been inferred from sev-eral observations. Radial glia disappear in cortex concurrently with the emergence of themultiprocess forms of mature astrocytes. During this time several studies have noted forms“transitional” between radial glia and astrocytes: cells with both long, radially-oriented pro-cesses and smaller branches emerging from the cell body.18,19,25,26 While undoubtedly someof these “transitional” forms reflect passing stages from radial glia to astrocytes, similarforms are produced by subventricular zone (SVZ) cells that migrate into the cortex afterneurogenesis.27 Cells cultured from embryonic rodent CNS and expressing antigenic mark-ers for radial glia begin to express GFAP in culture and assume the morphologies of cul-tured astrocytes.28 One dynamic study provides direct evidence for such a transformation,however.29 In work with postnatal ferret brain, the application of the lipophilic fluorescenttracer dye, diI, to the cortex initially labeled radial glia. After maturation of the brain andthe disappearance of radial glia, the dye was found in astrocytes.
What controls this transformation of radial glia to astrocytes and why does such trans-formation apparently take place in some regions (cortex, for example), but not, or to a lesserextent, in others (periventricular zones in diencephalon and brain stem)? Studies in cellculture suggest a role of extrinsic factors in promoting the change in shape from elongatedto branched with many processes. Such a transformation takes place in primary culturesfrom embryonic forebrain,28 and can be reversibly promoted by soluble signals from the

5Astrocyte Ontogenesis and Classification
embryonic CNS.30 Cerebellar astrocytes cocultured with granule neurons assume elongatedshapes, suggesting that interactions with immature neurons helps determine astrocyteshape.31
A critical, and necessary, change in the transformation of radial glia is the loss of sub-pial connections. This process, which has not been examined, requires a loss of adhesionbetween the end of the glial process and the pial surface. Breaking such adhesion in turnmay require local extracellular protease activity or redistribution of surface adhesion mol-ecules such as integrins that may interact with mesenchymal tissue matrix, or contractionof the microfilament network in the process. Loss of adhesion to the pia does not representa lack of adhesive properties of the cell in general, since radial glia that transform intoastrocytes presumably contact blood vessels as they are detaching from the pia, or shortlythereafter.
Genesis of Astrocytes from SVZ CellsIn addition to the generation of astrocytes from radial glia, astrocytes are also derived
from immature cells of the subventricular zone, without apparently going through a radialintermediate stage. The genesis of astrocytes from immature cells in the forebrain SVZ wasoriginally suggested from thymidine labeling in the postnatal rodent brain.32-34 These clas-sic studies showed that the SVZ population is a highly proliferative one and that the thymi-dine label could be “chased” into mature glial cells in white matter and gray matter. Morerecent antigen expression studies35 and Golgi impregnations of the developing CNS7 havealso supported a nonradial glial derivation of some astrocytes. Through the use of recombi-nant retroviruses, a direct demonstration of SVZ cell migration and differentiation intomature glia has illuminated many of the details of this developmental process.5,36-39 In theseexperiments, immature, cycling cells of the postnatal SVZ were labeled in vivo by stereotac-tic injection of retroviruses directly into the SVZ. The fates of labeled cells and their routesof migration into the striatum, overlying white matter, and neocortex could then be traced.
How do astrocytes derived from the SVZ colonize the CNS? Glial colonization, to pro-duce the distributions described above, is not a random process, but takes place in definablespatial and temporal patterns. Migration of progenitors from SVZ into white matter andcortex occurs in a coronal plane.36 Perhaps the migratory pathways are defined in part bythe radial glial scaffolding. The idea that SVZ cells migrate along radial glia is supported byseveral observations. First, radial glia persist in the rodent neocortex through the first 1-2postnatal weeks.20,25,26 During this period, SVZ cells distribute into white matter and cor-tex.5,37 By postnatal day 14 (P14), however, progenitors that migrate out of the SVZ remainin white matter and do not enter cortex.36 Thus, a restriction in migration coincides exactlywith the loss of the cortical radial glial tracks. Second, we have observed progenitors fromthe SVZ aligning along radial glia in the cortex during early postnatal development.38 Third,progenitors from the SVZ migrate along “radial glial”-like cables in culture (Newman et al,in preparation).
In contrast to the laminar colonization of neurons of the neocortex, however, astro-cytes do not differentiate in a layered pattern. In fact, astrocytes derived from SVZ cellsappear to differentiate at all depths of the cortex, from the pial surface to deep layers, at thesame time. It is common to see radially oriented clusters of young astrocytes derived fromSVZ cells, clusters we believe are clonal. At present we favor a model in which progenitorsmigrate into cortex, and continue to divide therein. Some of the progeny cease migration,while others continue toward the pial surface, thus leaving progeny behind at a number ofcortical levels. What induces a particular progenitor to stop migrating and begin to differ-entiate into an astrocyte will be considered below.

Astrocytes in Brain Aging and Neurodegeneration6
In other regions of the CNS that do not have an SVZ, the genesis of astrocytes may bedifferent. For example, in the optic nerve, astrocytes arise prenatally, while those progeni-tors that migrate into and along the nerve in postnatal life do not differentiate into astro-cytes, but only into oligodendrocytes.40,41 Thus, there appear to be separate lineages forastrocytes and oligodendrodcytes in this tract. Astrocytes in optic nerve likely arise fromradial glial cells that formed during earlier telencephalic development and were carried intothe nerve when the optic outpouching occurred. And the oligodendrocytic fate of progeni-tors that migrate into the nerve postnatally may be analogous to the oligodendrocytic fateof forebrain SVZ cells that settle in subcortical white matter. In culture, the postnatal pro-genitors (O-2A cells) can differentiate into oligodendrocytes or into astrocytes,42 showingtheir bipotential nature, but there is apparently an oligodendrocyte fate restriction in vivo.
Astrocyte development in the spinal cord may be similar, with many of the astrocytesdeveloping from radial glia, as suggested from antigen and morphology studies.43,44 In thecord, oligodendrocytes arise from proliferating, immature cells in the centro-ventral re-gion,45,46 but whether astrocytes also arise from this proliferative population is not known.
Control of Astrocyte DifferentiationMuch recent work has utilized cell culture systems to examine the control of astrocyte
differentiation, and has led to the general conclusion that cell-extrinsic factors contributesubstantially to the determination of astrocyte cell fate. Oligodendrocyte progenitors iso-lated from the optic nerve are induced to express astrocyte genes and cease oligodendrocytedevelopment by exposure to serum,42 serum fractions,47 and ciliary neurotrophic factor(CNTF).48 Although the nature of the serum stimulus(i) is not known, extracellular mol-ecules isolated from endothelial and meningeal cells will also induce astrocyte genes,48,49 incombination with CNTF. This induction by matrix may well be an in vitro counterpart toastrocyte induction by cues from blood vessels and pia in vivo (see below). More recentstudies identify CNTF as an attractive candidate for an important inducer of astrocyticdifferentiation in immature CNS cells.50-52 CNTF induces GFAP expression and a flat, as-trocytic morphology in immature cortical cells via a JAK-STAT signaling pathway52 andalso upregulates GFAP transcription in the CG-4 glial cell line.53 The 5' upstream region ofthe GFAP gene contains a consensus STAT binding site,52,54 which in transfection assaysappears to be essential for the CNTF regulation of GFAP expression.52 The GFAP gene alsocontains consensus sequences for CREB, AP-2, and AP-1 binding,54,55 the former two possi-bly used for cyclic-AMP increases in GFAP transcription,54,56 the latter possibly utilized instress-regulated increases in GFAP.
Another candidate class of signaling molecules that can induce astrocyte differentia-tion are members of the transforming growth factor-β (TGF-β) family, in particular, thebone morphogenic proteins (BMP) 2 and 7, which cause astrocytic development and sup-press oligodendrocytic development in bipotential progenitors from neonatal rat forebrainand immature cells expanded from embryonic CNS by epidermal growth factor (EGF).57,58
Notably, serum and BMPs can induce GFAP expression in progenitors that have alreadybegun to express the early oligodendrocyte marker, O4, giving rise to a hybrid glial cell type.It is not known whether glial progenitors begin to express O4 and then become astrocytes invivo, during normal glial development, but the possibility seems unlikely. However, underpathological conditions (such as the development of brain tumors composed of progeni-tor-like cells) the acquisition of astrocyte gene expression in oligodendrocyte lineage cellsmight occur.
In contrast to BMPs, such growth factors as EGF, platelet-derived growth factor (PDGF),basic fibroblast growth factor (bFGF), thyroid hormone and insulin-like growth factor 1(IGF1) do not promote astrocyte differentiation of O-2A progenitors. Rather, they either

7Astrocyte Ontogenesis and Classification
promote division of progenitors without differentiation, as in the case of PDGF and bFGF,59
or are permissive for oligodendrocyte differentiation and/or survival.60,61
In most of the experiments cited above, astrocytic “differentiation” was measured bythe induction of GFAP expression. While this intermediate filament protein is characteris-tic of astrocytes and therefore denotes the acquisition of at least one astrocyte feature, it isnot clear whether there is a group of genes expressed coordinately during astrocyte devel-opment and whether in vitro systems fully capture that differentiated state. That other genesare both necessary and sufficient for astrocytic differentiation is clear from the several GFAPknockout transgenic mice, in which astrocytes do develop.62-64 In the future, control of specificreceptors, transporters, and astrocyte enzymes will be required to characterize the develop-mental pathway. As discussed below, perhaps a progenitor makes several decisions duringastrocyte development—the first to differentiate into an astrocyte and the second to acquirespecific characteristics required for specific functions in the local CNS environment.
Determinants of astrocyte fate in vivo has not been examined in as much detail asdeterminants in culture. Many of the factors suggested from the culture studies to play arole in fate determination exist in the developing brain. However, when a given progenitorbecomes responsive to those signals and even whether such signals play a role in vivo is notyet known.
Clues as to the nature of developmental signals may come from considering the ana-tomic changes that take place during astrocyte development. The peak period of astrocytegenesis coincides with the rapid growth of blood vessels65-67 and pial surface, the elabora-tion of dendritic arbors, and the establishment of synapses (both from cortical afferentsand from intracortical circuits). For example, in the rat forebrain, thalamocortical afferentsenter the cortex around P2-4 and cortico-cortical fibers around P6-8.68,69 Thus, the differ-entiation of astrocytes takes place during the establishment of synaptic connections and ofthe vascular supply. How is the development of glia coordinated with vascular and synapticgrowth to assure the appropriate glial-vascular and glial-neuronal interactions? Further-more, does the development of astrocytes and/or oligodendrocytes play a role in vasculargrowth or synapse formation?
There is evidence for mutual interactions between astrocytes and endothelial cells. As-trocytes may participate in the formation of endothelial tight junctions, the anatomic sub-strate of the blood-brain barrier, and in inducing specific endothelial cell properties, such aspolarization of transporters, increases in γ-glutamyl transaminase.70-72 Furthermore, thepresence of astrocytes in the mammalian retina correlates with the presence of blood ves-sels.73 In examining the fates of progenitors from the SVZ after migration into the cortex,we have noted a close concordance between the early stages of astrocyte differentiation, asjudged by an increase in intermediate filament expression and the beginnings of a complex,multiprocess cell shape and contact with blood vessels or the pial surface.39 These observa-tions do not prove a causal relationship between astrocyte differentiation and vessel con-tact, but the model suggests a way in which astrocyte development can be coordinated withthe tremendous growth of blood vessels and the pial surface in late gestational and postna-tal CNS development.
Genesis of Astrocyte HeterogeneityAstrocytes vary both in morphology and in the expression of certain antigens from
region to region. One example is the well known morphological distinctions between the“fibrous” astrocytes of white matter and the “protoplasmic” astrocytes of gray matter, theformer expressing a much higher level of GFAP than the latter.74 A number of studies haveclearly shown functional heterogeneity among astrocytes, although most of these experi-ments have been performed in vitro. Thus, astrocytes cultured from different regions of the

Astrocytes in Brain Aging and Neurodegeneration8
CNS differ in their abilities to support process growth of neurons, in their responses toneurotransmitters, and in their expressions of proteoglycans.75-77 Astrocytes from one re-gion appear to be matched functionally to support neurons of the same region; mesen-cephalic neurons grow better on mesencephalic astrocytes than on astrocytes from otherregions, for example.77 In cultures from neonatal forebrain, which includes all cortical areasand white matter and some subcortical gray matter nuclei, there is a heterogeneity in theuptake of and responses to neurotransmitters within the astrocyte population.78,79 Whetherthis heterogeneity was determined in vivo before the cultures were established or in vitro isnot clear, but the observations dramatically illustrate that astrocytes are able to acquire im-portant functional differences. In another study, the clonal progeny of single spinal cordastrocytes in culture were examined, and both homogeneous and heterogeneous clones wereobserved,80 showing clearly that an individual proliferating astrocyte, or an individual pro-genitor, is able to generate a mixture of astrocytic forms.
Less is known about heterogeneity in vivo, however, but techniques exist to study as-trocyte physiology in slices, where responses to transmitters or uptake mechanisms couldbe studied in real time. In vivo retroviral labeling studies suggest (although do not yet prove)that different astrocyte forms can arise from a single progenitor. For example, the proximityof retrovirally labeled Bergmann glia and velate astrocytes in the cerebellar cortex suggests aclonal heterogeneity (Fig. 1.1).81,82 And, as noted above, the astrocytic progeny of a singleprogenitor in the neocortex probably span the entire cortical depth, and would therefore beexposed to different microenvironments.
How is the heterogeneity of astrocytes determined? One model would suggest thatprogenitors first are induced to differentiate into astrocytes and then signals peculiar to thelocal environment dictate specific morphological and functional patterns. This model makessense if an astrocyte’s functional properties must match those of the neurons in the imme-diate proximity. Thus, the heterogeneity of astrocytes may not be lineage related, in thesense that such heterogeneity has little to do with the astrocyte fate decision. Astrocytes canchange morphology and expression of many molecules, including surface gangliosides, in-termediate filaments, enzymes, and stress proteins, in response to pathological conditions(see for examples refs. 83, 84). So, even in the mature CNS, astrocytes maintain a remark-able malleability.
Generation of Astrocytes in the Adult CNSThymidine labeling studies in the adult mammalian CNS show a low level of cell divi-
sion in the mature CNS85-87 and several investigators have inferred a slow turnover of astro-cytes. Genesis must be balanced by cell death, since numbers of astrocytes in the cortex donot appear to increase during adult life.88 The nature of the dividing cells is not clear; that is,astrocytes might be generated from dividing astrocytes or from dividing, immature cellsthat then differentiate into astrocytes.
Under pathological conditions, such as trauma, astrocytes in the region of the lesiondivide, although the capacity for proliferation appears limited.84 Whether new astrocytesare generated from immature cells in pathological circumstances is not known. Cyclingcells in adult rat white matter, labeled with recombinant retroviruses, do not differentiateinto astrocytes, either under normal conditions, demyelination, or trauma (refs. 89, 90 andour unpublished observations). This finding contrasts with studies that find a populationof immature cells isolated from adult optic nerve, cord, or forebrain that can differentiateinto either oligodendrocytes or astrocytes in culture (“adult O-2A progenitors”91,92). Again,there may be fate restrictions in vivo, or perhaps appropriate pathological conditions haveyet to be found in vivo to induce astrocyte differentiation in cycling immature cells.

9Astrocyte Ontogenesis and Classification
AcknowledgmentsThe work from the author’s lab has been supported by NIH grant NS-17125. Many
thanks to Bernetta Abramson, Cathy Chuang, JoAnn Gensert, Steven Levison, SharonNewman, Marielba Zerlin, and Lei Zhang for all of their many major contributions to ourstudies.
Fig. 1.1. Morphological transformations in the development of astrocytes as revealed by aLac-Z encoding retrovirus. Newborn rat pups were injected into the forebrain SVZ or cer-ebellar white matter as described.5,82 Labeled cells were visualized by X-gal staining.(a) two unipolar cells in the SVZ, 1 day after injection.(b) a bipolar cell oriented radially in the cortex, 3 days after injection; such cells do not expressastrocyte markers and presumably represent progenitors.(c) an early astrocyte in the cortex, 3 days after injection; one process has wrapped around ablood vessel (arrowhead); cells at this stage are expressing intermediate filament proteins.(d) two velate astrocytes in the cerebellar granule cell layer, 2 weeks after injection, displayingmature forms.(e) a Bergmann glial cell (top), with a cell body in the Purkinje cell layer and processes extend-ing into the molecular layer, adjacent to a velate astrocyte (bottom) in the granule cell layer, 2weeks after injection.

Astrocytes in Brain Aging and Neurodegeneration10
References1. Andriezen WL. The neuroglia elements in the human brain. Br J Med 2:227-230.2. Peters A, Palay SL, Webster H deF. The Fine Structure of the Nervous System, 3rd Ed.
Oxford: Oxford University Press, 1991.3. Hama K, Arii T, Kosaka T. Three-dimensional organization of neuronal and glial pro-
cesses: high voltage electron microscopy. Microsc Res Tech 1993; 29:357-367.4. Kosaka T, Hama K. Three-dimensional structure of astrocytes in the rat dentate gyrus. J
Comp Neurol 1986; 249:242-260.5. Levison SW, Goldman JE. Both oligodendrocytes and astrocytes develop from progenitors
in the subventricular zone of postnatal rat forebrain. Neuron 1993; 10:201-212.6. Chan-Ling T, Stone J. Factors determining the morphology and distribution of astrocytes
in the cat retina: a ‘contact-spacing’ model of astrocyte interaction. J Comp Neurol 1991;303:387-399.
7. Marin-Padilla M. Prenatal development of fibrous (white matter), protoplasmic (gray mat-ter), and layer I astrocytes in the human cerebral cortex: a Golgi study. J Comp Neurol1995; 357:554-572.
8. Dani JW, Chernjavsky A, Smith SJ. Neuronal activity triggers calcium waves in hippocam-pal astrocyte networks. Neuron 1992; 8:429-440.
9. Palay SL, Chan-Palay V. Cerebellar Cortex, Cytology, and Organization. New York: Springer-Verlag, 1974.
10. Chan-Palay V, Palay SL. The form of velate astrocytes in the cerebellar cortex of monkeyand rat: high voltage electron microscopy of rapid Golgi preparations. Z Anat Entwick-lungsgesch 1972; 138:1-19.
11. Bovolenta P, Liem RHK, Mason CA. Glial filament protein expression in astroglia in themouse visual pathway. Brain Res 1987; 430:113-126.
12. Butt AM, Ransom BR. Visualization of oligodendrocytes and astrocytes in the intact ratoptic nerve by intracellular injection of Lucifer yellow and horseradish peroxidase. Glia1989; 2:470-475.
13. Sims TJ, Gilmore SA, Waxman SG. Radial glia give rise to perinodal processes. Brain Res1991; 549:25-35.
14. Bovolenta P, Liem RKH, Mason CA. Development of cerebellar astroglia: transitions inform and cytoskeletal content. Dev Biol 1984; 102:248-259.
15. Seress L. Development and structure of the radial glia in the postnatal rat brain. AnatEmbryol (Berl) 1980; 160:213-226.
16. Mori K, Ikeda J, Hayaishi O. Monoclonal antibody R2D5 reveals midsagittal radial glialsystem in postnatally developing and adult brainstem. Proc Natl Acad Sci USA 1990;87:5489-5493.
17. Edwards MA, Yamamoto M, Caviness VS Jr. Organization of radial glia and related cells inthe developing murine CNS. An analysis based upon a new monoclonal antibody marker.Neuroscience 1990; 36:121-144.
18. Choi BH, Lapham LW. Radial glia in the human fetal cerebrum: A combined Golgi, im-munofluorescent, and electron microscopic study. Brain Res 1978; 148:295-311.
19. Schmechel DE, Rakic P. A Golgi study of radial glial cells in developing monkey telen-cephalon: morphogenesis and transformation into astrocytes. Anat Embryol (Berl.) 1979;156:115-152.
20. Misson J-P, Edwards ME, Yamamoto M et al. Identification of radial glial cells within thedeveloping murine central nervous system: studies based upon a new immunohistochemi-cal marker. Dev Brain Res 1988; 44:95-108.
21. Levitt P, Cooper ML, Rakic P. Coexistence of neuronal and glial precursor cells in thecerebral ventricular zone of the fetal monkey: An ultrastructural immunoperoxidase analy-sis. J Neurosci 1981; 1:27-39.
22. Halliday AL, Cepko CL. Generation and migration of cells in the developing striatum. 1992;Neuron 9:384-396.
23. Gray G, Sanes J. Lineage of radial glia in the chicken optic tectum. Development 1992;114:271-283.

11Astrocyte Ontogenesis and Classification
24. Galileo DS, Gray GC, Owens G et al. Neurons and glia arise from a common progenitor inchicken optic tectum: demonstration with two retroviruses and cell-type-specific antibod-ies. Proc Natl Acad Sci USA 1990; 87:458-462.
25. Misson J-P, Takahashi T, Caviness VS Jr. Ontogeny of radial and other astroglial cells inmurine cerebral cortex. Glia 1991; 4:138-148.
26. LeVine SM, Goldman JE. Embryonic divergence of oligodendrocyte and astrocyte lineagesin developing rat cerebrum. J Neurosci 1988; 8:3992-4006.
27. Zerlin M, Levison SW, Goldman JE. Early patterns of migration, morphogenesis, and in-termediate filament expression of subventricular zone cells in the postnatal rat forebrain. JNeuroscience 1995; 15:7238-7249.
28. Culican SM, Baumrind NL, Yamamoto M et al. Cortical radial glia: Identification in tissueculture and evidence for their transformation to astrocytes. J Neurosci 1990; 10:684-692.
29. Voigt T. Development of glial cells in the cerebral wall of ferrets: Direct tracing of theirtransformation from radial glia into astrocytes. J Comp Neurol 1989; 289:74-88.
30. Hunter KE, Hatten ME. Radial glial cell transformation to astrocytes is bidirectional: regu-lation by a diffusible factor in embryonic forebrain. Proc Natl Acad Sci USA 1995;92:2061-2065.
31. Hatten ME, Liem RKH, Mason CA. Two forms of cerebellar glial cells interact differentlywith neurons in vitro. J Cell Biol 1984; 98:193-204.
32. Altman J. Proliferation and migration of undifferentiated precursor cells in the rat duringpostnatal gliogenesis. Exp Neurol 1966; 16:263-278.
33. Imamoto K, Paterson JA, Leblond CP. Radioautographic investigation of gliogenesis in thecorpus callosum of young rats. I. Sequential changes in oligodendrocytes. J Comp Neurol1978; 180:115-138.
34. Paterson JA, Privat A, Ling EA et al. Investigation of glial cells in semithin sections III.Transformation of subependymal cells into glial cells as shown by radioautography after3H-thymidine injection into the lateral ventricle of the brain of young rats. J Comp Neurol1973; 149:83-102.
35. Gressens P, Richelme C, Kadhim HJ et al. The germinative zone produces the most corti-cal astrocytes after neuronal migration in the developing mammalian brain. Biol Neonate1992; 61:4-24.
36. Levison, SW, Chuang C, Abramson B et al. The migrational patterns and developmentalfates of glial precursors in the rat subventricular zone are temporally regulated. Develop-ment 1993; 119:611-622.
37. Luskin MB, McDermott K. Divergent lineages for oliogodendrocytes and astrocytes origi-nating in the neonatal forebrain subventricular zone. Glia 1994; 11:211-226.
38. Zerlin M, Levison SW, Goldman JE. Early patterns of migration, morphogenesis, and in-termediate filament expression of subventricular zone cells in the postnatal rat forebrain. JNeurosci 1995; 15:7238-7249.
39. Zerlin M, Goldman JE. Interactions between glial progenitors and blood vessels duringearly postnatal corticogenesis: blood vessel contact represents an early stage of astrocytedifferentiation. J Comp Neurol 1997; 387:537-546.
40. Skoff RP. Gliogenesis in rat optic nerve: Astrocytes are generated in a single wave beforeoligodendrocytes. Dev Biol 1990; 139:149-168.
41. Fulton BP, Burne JF, Raff MC. Visualization of O-2A progenitor cells in developing andadult rat optic nerve by quisqualate-stimulated cobalt uptake. J Neurosci 1995; 12:4816-4833.
42. Raff MC, Miller RH, Noble M. A glial progenitor cell that develops in vitro into an astro-cyte or an oligodendrocyte depending on culture medium. Nature 1983; 303:390-396.
43. Hirano M, Goldman JE. Gliogenesis in rat spinal cord: Evidence for origin of astrocytesand oligodendrocytes from radial precursors. J Neurosci Res 1988; 21:155-167.
44. Maier CE, Miller RH Development of glial architecture in the frog spinal cord. Dev Neurosci1995; 178:149-159.
45. Warf BC, Fok-Seang J, Miller RH. Evidence for the ventral origin of oligodendrocyte pre-cursors in the rat spinal cord. J Neurosci 1991; 11:2477-2488.

Astrocytes in Brain Aging and Neurodegeneration12
46. Richardson WD, Pringle NP, Yu W-P et al. Origins and early development of oligoden-drocytes. In: Jessen KR, Richardson WD, eds. Glial Cell Development, Basic Principles andClinical Relevance. Oxford, UK: Bios Scientific Publishers, 1996:53-70.
47. Levison SW, McCarthy KD. Characterization and partial purification of AIM: a plasmaprotein that induces rat cerebral type 2 astroglia from bipotential glial progenitors. JNeurochem 1991; 57:782-794.
48. Hughes SM, Lillien LE, Raff MC et al. Ciliary neurotrophic factor induces type-2 astrocytedifferentiation in culture. Nature 1988; 335:70-72.
49. Lillien LE, Sendtner M, Raff MC. Extracellular matrix-associated molecules collaborate withciliary neurotrophic factor to induce type-2 astrocyte development. J Cell Biol 1990;111:635-644.
50. Gard AL, Williams WC, Burrell MR. Oligodendroblasts distinguished from O-2A glial pro-genitors by surface phenotype (O4+/GalC-) and response to cytokines using signal trans-ducer LIFRβ. Dev Biol 1995; 167:596-608.
51. Johe KK, Hazel TG, Muller T et al. Single factors direct the differentiation of stem cellsfrom fetal and adult central nervous system Genes Dev 1996; 10:3129-3140.
52. Bonni A, Sun Y, Nadal-Vicens M et al. Regulation of gliogenesis in the central nervoussystem by the JAK-STAT signaling pathway. Science 1997; 278:477-483.
53. Kahn MA, Huang CJ, Caruso A et al. Ciliary neurotrophic factor activates JAK/Stat signaltransduction cascade and induces transcriptional expression of glial fibrillary acidic pro-tein in glial cells. J Neurochem 1997; 68:1413-1423.
54. Besnard F, Brenner M, Nakatani Y et al. Multiple interacting sites regulate astrocyte-spe-cific transcription of the human gene for tglial fibrillary acidic protein. J Biol Chem 1991;266:18877-18883.
55. Masood K, Besnard F, Su Y et al. Analysis of a segment of the human glial fibrillary acidicprotein gene that directs astrocyte-specific transcription. J Neurochem 1993; 61:160-166.
56. Shafit-Zagardo B, Iwaki AK, Goldman, JE. Astrocytes regulate GFAP mRNA levels by cAMPand protein kinase C dependent mechanisms. Glia 1988; 1:346-354
57. Gross RE, Mehler MF, Mabie PC et al. Bone morphogenetic proteins promote astrogliallineage commitment by mammalian subventricular zone progenitor cells. Neuron 1996;17:595-606.
58. Mabie P, Mehler MF, Marmur R et al. Bone morphogenetic proteins induce astroglial dif-ferentiation of oligodendroglial-astroglial progenitor cells. J Neurosci 1997; 117:4112-4120.
59. Noble M, Murray K, Stroobant P et al. Platelet-derived growth factor promotes divisionand inhibits premature differentiation of the oligodendrocyte/type 2 astrocyte progenitorcell. Nature 1988; 333:560-562.
60. Behar T, McMorris FA, Novotny EA, Barker JL, Dubois-Dalcq M. Growth and differentia-tion properties of O-2A progenitors purified from rat cerebral hemispheres. J NeurosciRes 1988; 21:168-180.
61. Barres BA, Raff M. Axonal control of oligodendrocyte development. In: Jessen KR,Richardson WD, eds. Glial Cell Development, Basic Principles and Clinical Relevance.Oxford, UK: Bios Scientific Publishers, 1996:71-83.
62. Pekny M, Leveen P, Pekna M et al. Mice lacking glial fibrillary acidic protein display as-trocytes devoid of intermediate filaments but develop and reproduce normally. EMBO J1995; 14:1590-1598.
63. Shibuki K, Gomi H, Chen L et al. Deficient cerebellar long term depression, impairedeyeblink conditioning, and normal motor coordination in glial fibrillary acidic proteinmutant mice. Neuron 1996; 16:587-599.
64. Liedtke W, Edelmann W, Bieri PL et al. GFAP is necessary for the integrity of CNS whitematter architecture and long-term maintenance of myelination. Neuron 1996; 17:607-615.
65. Caley DW, Maxwell DS. Development of the blood vessels and extracellular spaces duringpostnatal maturation of rat cerebral cortex. J Comp Neurol 1970; 138:31-48.
66. Phelps CH. The development of glio-vascular relationships in the rat spinal cord. ZZellforsch 1972; 128:555-563.

13Astrocyte Ontogenesis and Classification
67. Robertson PL, Du Bois M, Bowman PD et al. Angiogenesis in developing rat brain: an invivo and in vitro study. Dev Brain Res 1985; 23:219-223.
68. Wise SP, Jones ED. Organization and postnatal development of the commissural projec-tion of the rat somatic sensory cortex. J Comp Neurol 1976; 168:313-343.
69. Wise SP, Jones ED. Developmental studies of thalamocortical and commissural connec-tions in the rat somatic sensory cortex. J Comp Neurol 1978; 178:187-208.
70. DeBault LE, Cancilla PA. g-Glutamyl transpeptidase in isolated brain endothelial cells: in-duction by glial cells in vitro. Science 1980; 207:653-655.
71. Janzer, RC, Raff MC. Astrocytes induce blood-brain barrier properties in endothelial cells.Nature 1987; 325:253-257.
72. Beck DW, Roberts RL, Olson JJ. Glial cells influence membrane-associated enzyme activityat the blood-brain barrier. Brain Res 1986; 381:131-137.
73. Schnitzer J. Astrocytes in the guinea pig, horse, and monkey retina: their occurrence coin-cides with the presence of blood vessels. Glia 1988; 1:74-89.
74. Kitamura T, Nakanishi K, Watanabe S et al. GFA-protein gene expression on the astrogliain cow and rat brains. Brain Res 1987; 423:189-195.
75. Wilkin GP, Marriott DR, Cholewinski AJ. Astrocyte heterogeneity. TINS 1990; 13:43-46.76. Garcia-Abreu J, Neto VM, Carvelho SL et al. Regionally specific properties of midbrain
glia: I. Interactions with midbrain neurons. J Neurosci Res 1995; 40:471-477.77. Denis-Donini S, Glowinski J, Prochaintz A. Glial heterogeneity may define the three-di-
mensional shape of mouse mesencephalic dopaminergic neurons. Nature 1984; 307:641-643.78. Amundson RH, Goderie SK, Kimelberg HK. Uptake of [3H] serotonin and [3H] glutamate
by primary astrocyte cultures. II. Differences in cultures prepared from different brain re-gions. Glia 1992; 6:9-18.
79. McCarthy KD, Salm AK. Pharmacologically distinct subsets of astroglia can be identifiedby their calcium response to neuroligands. Neuroscience 1991; 41:325-333.
80. Miller RH, Szigeti V. Clonal analysis of astrocyte diversity in neonatal rat spinal cord cul-tures. Development 1991; 113:353-362.
81. Miyake T, Fujiwara T, Fukunaga T et al. Glial cell lineage in vivo in the mouse cerebellum.Develop Growth Differ 1995; 37:273-285.
82. Zhang L, Goldman JE. Developmental fates and migratory pathways of dividing progeni-tors in the postnatal rat cerebellum. J Comp Neurol 1996; 370:536-550.
83. Eddleston M, Mucke L. Molecular profile of reactive astrocytes—implications for their rolein neurologic disease. Neuroscience 1993; 54:15-36.
84. Norton WT, Aquino DA, Hozumi I et al. Quantitative aspects of reactive gliosis: a review.Neurochem Res 1992; 17:877-885.
85. Altman J Autoradiographic investigation of cell proliferation in the brains of rats and cats.Anat Rec 1963; 145:573-591.
86. Kaplan MS, Hinds JW. Gliogenesis of astrocytes and oligodendrocytes in the neocorticalgray and white matter of the adult rat: electron microscopic analysis of light radioauto-graphs. J Comp Neurol 1980; 193:711-727.
87. McCarthy GF, Leblond CP. Radoautographic evidence for slow astrocyte turnover andmodest oligodendrocyte production in the corpus callosum of adult mice perfused with3H-thymidine. J Comp Neurol 1988; 2761:589-603.
88. Ling EA, Leblond CP. Investigation of glial cells in semithin sections. II. Variation withage in the numbers of the various glial cell types in rat cortex and corpus callosum. JComp Neurol 1973; 149:73-82.
89. Gensert JM, Goldman JE. In vivo characterization of proliferating cells in adult rat subcor-tical white matter. Glia 1996; 17:39-51.
90. Gensert JM, Goldman JE. Remyelination by endogenous progenitors in the adult rat CNS.Neuron 1997; 19:197-203.
91. ffrench-Constant C, Raff MC. Proliferating bipotential glial progenitor cells in adult opticnerve. Nature 1986; 319:499-502.
92. Wren D, Wolswijk G, Noble M. In vitro analysis of origin and maintenance of O-2A adultprogenitor cells. J Cell Biol 1992; 116:167-176.


CHAPTER 2
Astrocytes in Brain Aging and Neurodegeneration, edited by Hyman M. Schipper.©1998 R.G. Landes Company.
Functions of AstrocytesHarold K. Kimelberg and Michael Aschner
Introduction
In the previous chapter, Goldman covered the structure and development of astrocytes,and that chapter should be read before reading this chapter to better understand the func-
tional properties we will discuss. Thus, an appreciation of the complex morphologies of alltypes of astrocytes, their interrelationships with other cells and brain structures such asblood vessels, and the complexity of astrocyte development must surely reasonably lead,based on the principle that form reflects function, to the conclusion that astrocytes arelikely to have many complex properties closely associated with many aspects of brain func-tion. Indeed, there has been no dearth of hypotheses regarding astrocyte function emergingsimply from contemplation of the complexities of astrocyte morphology and interrelation-ships dating from the work of Golgi, Cajal and others,1 which first showed their structuresin precise detail in the late nineteenth century. For example, per Lugaro2 in 1907; “the neu-ronal articulation* would be the center of the chemical exchange, and this would comprisetherefore in all the most proximal, vacant interstitial spaces, a region for infiltration of theprotoplasmic prolongations or feathery extensions of the neuroglia, perhaps with the pur-pose of collecting and instantly processing the smallest amount of waste product.” Golgiand Cajal among others speculated that the roles of glia included neuronal nutrition, struc-tural and metabolic support and involvement involved in nervous system development.1
However, these and other hypotheses could not then be tested. Experimental studies on glialfunction began with the work of Kuffler and his colleagues in the mid 1960s. They focusedon the electrical and ion transport properties of glia in simple invertebrate nervous systemsand the relatively simple preparation of the amphibian optic nerve.3 Beginning in the 1970sprimary astrocyte cultures from neonatal rodent CNS began to be used extensively to studythe properties of astroglia.4,5
The primary cultures prepared from neonatal rodents consist predominately of GFAP-positive astrocytes and provide preparations of cells in sufficient numbers to allow for avariety of biochemical, electrophysiological, molecular and general cell biological studies. Itis still unclear as to why all the cells in these astrocyte cultures, which consist primarily offlat cells which have been proposed to be analogous to protoplasmic astrocytes, stain forGFAP whereas protoplasmic astrocytes in situ in many regions, such as the cerebral cortex,stain variably for GFAP.6 This has led to the view that the cultures may consist predomi-nantly of reactive astrocytes, which in situ are characterized by prominent GFAP staining.7
* synapse

Astrocytes in Brain Aging and Neurodegeneration16
It needs to be emphasized that the bulk of the current information on the properties ofmammalian astrocytes has come from these preparations, as they are relatively easily stud-ied. However, their properties are often imprecisely referred to as astrocytic properties, with-out any qualifications. Studies on primary astrocyte cultures have always had the implicitcaveat that one is uncertain as to how their properties are altered by growth in vitro.8 In ourview, such cultures have had two major and critical advantages. One, they led to a majorexpansion of studies on astrocytes, albeit mainly in these culture systems, which otherwisewould probably not have been done in any other system. Second, the results of such studiessuggested to neuroscientists that astrocytes and other glial cells could have a number ofproperties such as receptors and uptake systems for transmitters and gated and rectifyingchannels which, based on a few negative studies on glial cells in situ had, rather prematurely,been considered specific to neurons in the CNS. The primary cultures have now provided along list of putative functions which need to be tested in systems more representative of thein vivo state, for the numerous differences that have now been reported between the prop-erties of the primary astrocyte cultures and the properties of astrocytes expressed in prepa-rations closer to the in situ state, such as brain slices, makes it impossible to use primarycultures by themselves to define astrocyte function. Thus there is currently less emphasis onsuch cultures and a reemphasis on in situ preparations that appear to more closely corre-spond to in vivo situations.
The relative paucity of reliable information is presumably why astrocytes so rarely fig-ure in discussions of brain function. In contrast, the characteristic properties of neuronshave been studied in great detail and were found to lend themselves relatively easily to hy-potheses of information processing by the postulation of electrically active loops and net-works. Also, experimental interference with neuronal function led to clear effects on neuralfunction, so that discussions of how the nervous system functions at the most complexlevels are currently almost exclusively based on the properties of neurons.9 Thus a certaincircularity of reasoning is apparent that can only be broken by sufficient rigorous study ofthe properties that astrocytes and other glia have in the CNS. In many respects we are stillsearching for experimental systems in which hypotheses advanced at the turn of this cen-tury can be rigorously tested.
Functions of AstrocytesWe will arrange this section based first on the properties of astrocytes established both
in cultured and acutely isolated cells or in slices of the intact brain, taking care to note theexperimental systems in which they were obtained. We will also discuss potential functionsthat these properties indicate, and then mention the very limited number of cases in whichfunctions have actually been demonstrated. It must be born in mind, however, that theproperties studied are not only limited by the experimental systems, but have been concep-tually restricted, notably by concepts based on what has been developed for neurons whichhave led to success in understanding nervous function. Thus, it is not surprising that manystudies of astrocytes have involved investigations of their membrane electrical propertiesand ion channels, even though this approach has led a well-respected worker in the field ofglial electrophysiology to conclude that “generation of glial electric signals is not amongtheir (i.e., astrocytes) functions.”10 This then begs the question: What is (are) the role(s) ofthe predominant K+ channels seen in astrocytes and what is (are) the role(s) of the -70 to-80 mV membrane potentials that are a consequence of them?

17Functions of Astrocytes
Homeostasis of the Extracellular Space
Regulation of Extracellular K+
One answer to the question just posed was proposed by Kuffler et al.3 Namely, that theselective K+ permeability implied a role in control of extracellular potassium levels ([K+]o).The original reason for Kuffler and his colleagues embarking on their pioneering studieswas that electron microscopic studies of mammalian CNS had shown, at this time, thatastrocytes generally formed enlarged watery compartments seemingly obliterating the ex-tracellular space (ECS). This led to the proposal that astrocytes actually formed the extra-cellular space of the brain. This would require that they would uniquely be high Na+ cells,and it was to examine this question that Kuffler and his associates studied the easily acces-sible glial cells in the leech nervous system and the amphibian optic nerve. In both cases,glial cells were found to have a membrane potential of -80 to -90 mV, and showed a close toNernstian response to varying [K+]o. Thus these glial cells had to have high intracellular K+
rather than Na+ concentrations, and thus could not form the ECS.The finding of an essentially selective K+-dependent membrane potential implied that
the cell membranes were operationally impermeable to sodium, and possibly chloride, andled to a mechanism for uptake of K+ released by active neurons.3 The mechanism would bethat a localized release of K+ from neurons during excitation would depolarize the astrocyteat this point with a 60 mV depolarization for a 10-fold increase in [K+]o. This would set upa current loop with other nondepolarized parts of the cell and, since the membrane waspermeable only to K+, there would be an inward current at the depolarized point carried byextracellular K+ crossing the membrane. Since K+ is the major electrolyte inside the cell, itwould also be the major current carrier inside the cell, and the current loop would be con-nected by efflux of K+ at some distant point. The return part of the loop would be carried bymajor extracellular ions such as Na+, or Cl– in the opposite direction. This led to the conceptof “K+ spatial buffering” in which K+ is transferred from a region of localized release tosome distant point, traveling through the astrocyte or the astrocytic syncytium.
Work since the studies of Kuffler and his colleagues has concentrated on identifyingthe types and location of K+ channels in different glial preparations using modern patchclamp methods. K+ channels are the most diverse ionic channel type11 and, as reviewed overthe past several years, a wide variety of K+ channels have been found, predominantly usingcultured astrocytes.12-15 These K+ channels include an inward rectifying K+ channel (K+
in),Ca2+-dependent K+ channels (K+
Ca), delayed rectifying channels (K+D) and an inactivating
potassium channel (K+a). K+ channels sensitive to ATP have also been found in astrocytes,
such as an ATP-regulated, strongly inward rectifying K+ channel that has been observed onBergmann glia in situ.16 Some of these channels may be related to the K+ spatial bufferingphenomenon just discussed. When there is also a significant chloride permeability (see later),net KCl uptake leading to swelling will occur when [K+]o rises. Some of these K+ channelsshould also be responsible for the large negative K+ diffusion potentials characteristic ofastrocytes. The work of Newman17 using acutely isolated astrocytes has indicated inward K+
rectifying channels at very high densities in areas of the astrocyte where it seems to be adaptedto K+ spatial buffering, namely at the capillary-facing astrocytic end-feet. If the membranepotential is very close to the K+ equilibrium potential, then the net outward leak of K+ willalways be very low, but this may be increased when there is depolarization of the astrocytecaused by other than an increased [K+]o, such as by receptor activation. In this case, therewill be an outward flux of potassium which would be later replenished by reuptake on theNa+/K+ pump or repolarization and reestablishment of Em ≅ Ek (see later).
If K+ channels are important in astrocyte function, then it is likely that alterations intheir functioning would affect astrocyte properties and they are likely to be targets of the

Astrocytes in Brain Aging and Neurodegeneration18
activation of astrocyte receptors. This is currently an active and fruitful area of investiga-tion. Thus, it has been shown that β-receptor activation modulates K+
IR currents in culturedrat spinal cord astrocytes,18 as well as altering astrocyte proliferation in vitro.15 AMPA/kainatereceptor activation blocks outward K+ currents in cultured stellate mouse cortical astro-cytes.19 This was suggested as a mechanism whereby astrocytes do not lose too much K+
when they become depolarized in pathological states. The K+ currents of glial cells in situ inhippocampal brain slices have been studied from different aged animals in both nonexcitable,GFAP-negative “complex cells” from younger animals and in GFAP-positive cells from olderanimals (>P20). The complex cells exhibited more types of ion currents. They showed adelayed outward K+ rectifier (K+
D) and a transient outward A-type K+ current. They alsoshowed a TTX-sensitive Na+ current. In the older cells, the voltage-gated Na+ and K+ out-ward currents downregulated and were replaced by passive and inward rectifier K+ conduc-tances.20 These changes are consistent with a precursor glial cell with a more complex arrayof ion channels changing into a mature astrocyte which exhibits K+ channels that have pre-dominantly [K+]o regulating properties.
It has also been shown that a variety of K+ channel blockers inhibit cell proliferation incultured astrocytes.15 Recent work has also shown that application of cesium for >2 min tohippocampal slices blocks long term depression (LTD) and synchronous, interictal-likebursting in the CA1 region.21 Studies using patch-clamp electrophysiology showed this tobe due to a direct blockade of the K+
IR currents of astrocytes. The increase in [K+]o wasconsidered to block the pyramidal cell activity since there was no change in the pyramidalcell conductance. This experiment is reminiscent of the 30 year old study of Krnjevic andSchwartz22 wherein they attempted to detect transmitter-induced conductance changes inglial cells from the cerebral cortex using sharp electrodes. They found no such changes,possibly due to the insensitivity of their techniques, and concluded that depolarization ofthe glial cells was due to a rise in [K+]o rather than a transmitter-mediated conductancechange in the astrocyte
Na+ ChannelsThis is a more controversial area because if astrocytes are nonexcitable there would
appear to be no need for Na+ channels, or at least voltage-sensitive ones. Na+ currents in gliawere first described in astrocytes in primary cultures.23,24 Like neuronal channels, these weresensitive to tetrodotoxin (TTX), but it was found that there were both TTX-sensitive andrelatively insensitive Na+ channels which had different characteristics in terms of the depo-larization required to activate them.25 The depolarizations needed to open these channelswere always thought to be greater than would ever be seen in astrocytes “clamped” at ahighly negative membrane potential by their large K+ conductances.5 However, recent workby Sontheimer et al26 has found that astrocytes cultured from certain regions of the brain,such as the spinal cord, have a very high density of Na+ channels which would have someopen probability at the resting membrane potential of these cells. It was hypothesized thatthe Na+ channels may function in regulating entry of Na+ in order to activate the Na+/K+
pump when active uptake K+ is required, such as when [K+]o rises from its normal level of3 mM to 5-10 mM during periods of sustained neuronal activity. This thus represents aself-regulating mechanism for active K+ clearance by astrocytes that does not require anyspecial properties of the Na+/K+ pump, and such special properties have not been clearlyshown (see below).
The major question in regard to the Na+ channels, as with other astrocytic propertiesmainly described in astrocytes in culture, is whether, when and in what cells Na+ channelsare expressed in situ. The type II sodium channel has been seen in astrocytes in situ in thedorsal and ventral columns of the spinal cord of the adult rat using immunocytochemistry,

19Functions of Astrocytes
but can only assumed to be functional.27 The function(s) of these channels must at presentbe purely speculative. Do they provide a voltage-dependent path for Na+ entry? There are,however, several other routes for Na+ entry into astrocytes. There are cotransporters forglutamate and aspartate and other substances which utilize the energy of the Na+ gradientto actively accumulate these substances. Are the channels a source of Na+ channels for theaxolemmal membrane of the node, as suggested by Ritchie?28 Do they in some way add tothe ability of astrocytes to control the ionic composition of the ECS? However, these chan-nels are only activated when the cell membrane is depolarized to at least -40 mV and thequestion still remains as to what extent such depolarizations do occur in astrocytes in situ.
Ca2+ ChannelsVoltage-gated L-type Ca2+ channels were also first identified in primary astrocyte cul-
tures.29 This was again a surprising finding because it was thought that such channels in theCNS were specific to neurons and were responsible for such properties as Ca2+ action po-tentials, depolarization-induced Ca2+ influx required for exocytosis at nerve terminals andmodulation of neuronal firing rates by hyperpolarization of the membrane potential viaCa2+-activated K+ channels. The question raised in regard to voltage-sensitive Na+ channelscan also be raised in regard to voltage-sensitive Ca2+ channels; namely, are the astrocytesever depolarized enough to activate these channels? Thus the function of the Ca2+ channelsin astrocytes has not yet been satisfactorily explained, but the occurrence of large changes in[Ca2+]i levels in astrocytes when stimulated by receptors such as glutamate, mechanical stimu-lation or swelling raise the possibility that part of the intracellular Ca2+ may be entering viasuch channels.30 Ca2+ channels will also be necessary for the entry of Ca2+ to replenish intra-cellular Ca2+ stores after their depletion.30 Intracellular Ca2+ is a pleiotropic intracellularsecond messenger affecting processes from gene regulation to ion channel activities. Thereader is referred to several reviews on this topic in astrocytes.30-35 In this context, the changesin [Ca2+]i levels can be viewed as a ubiquitous intracellular signaling mechanism present inastrocytes, as in other cells, rather than subserving a specific CNS function such as regulatedrelease of neurotransmitters at CNS synapses.
Anion ChannelsAs in other cells, anion channels have been less studied in astrocytes than cation chan-
nels, but it is now becoming clear that anion channels do have fundamental functions inmany, if not all, cells. A number of anion channels have now been identified in culturedastrocytes, including small conductance chloride channels (5-25 pS) and a high conduc-tance chloride channel (250-400 pS).14,15 These channels also transport Cl– and HCO3
–, butthe high conductance channel in astrocytes may also transport organic anions such as aminoacids,35 as may some of the other channels.36
The roles of anion channels may also include the uptake of HCO3– or chloride to ac-
company uptake of K+; a mechanism additional to K+ spatial buffering (see above) to con-trol [K+]o. However, this will lead to cell swelling, and many pathological states involve ex-aggerated astrocyte swelling.37 Cell swelling causes activation of a number of ion channelsand Strange et al38 and Okada39 have recently reviewed the different anion channels thatmay be involved in anion or amino acid efflux in swollen cells, including astrocytes. It isimportant to define the types of different anion channels normally present on astrocytes,and which ones are activated during swelling or are responsible for K+-dependent Donnanswelling, because, if release of excitatory amino acids in ischemia and other pathologicalstates occurs through a particular type of anion channel, then the identification of suchchannels would be of considerable practical benefit to either inhibit the swelling-inducedrelease or prevent the swelling in the first place. The inhibitory neurotransmitters GABA

Astrocytes in Brain Aging and Neurodegeneration20
and glycine also activate anion channels associated with some of their receptors, and suchreceptors appear to be present on astrocytes40-43 (see later).
Ion CarriersCarriers are distinct from channels in that a concerted movement of several ions usu-
ally occurs, rather than the independent diffusional movement of ions down their electro-chemical gradients as in channels.11 A number of carriers have been identified in astrocytes.
Na++K++2Cl– cotransporterOne important ion carrier is the Na++K++2Cl– uptake system utilized by cells for ac-
tive uptake of Cl–. This carrier has been found in astrocytes in primary culture, and intrac-ellular Cl– in astrocyte cultures has been found to be several-fold greater than expectedfrom electrochemical equilibrium.14 This carrier has also been localized by immunocy-tochemistry to Bergmann glia in situ.44 High [Cl–] in astrocytes may serve as a source tomaintain extracellular Cl–, based on the finding of GABAA receptors on astrocytes both invitro and in situ whose activation leads to efflux of Cl–.45 This was proposed as a mechanismto maintain extracellular concentrations at the same time as GABA causes influx of Cl– intoneurons, which contain low Cl–. In addition, the high Cl– in astrocytes may also be requiredfor the efflux of KCl in the process of volume regulation, as proposed by Kimelberg andFrangakis.46 Measurements with ion-specific microelectrodes in guinea-pig brain slices,however, have shown intracellular glial Cl– levels that were in equilibrium with the mem-brane potential and therefore <10 mM.47 However, a recent microprobe analysis has shownthat the intracellular Cl– levels in CNS glia are around 25 mM.48
(Na+/K+) pumpAs do all animal cells, astrocytes possess an active Na+/K+ pump responsible for accumu-
lating K+ and pumping out Na+. Na+/K+ pumps consist of isoforms of α and β subunits. Thework of Sweadner and colleagues49,50 has shown that the α1, α2, and α3 forms are distributedin a complex manner among different cells of the CNS. It appears that neurons can exhibitall three isoforms, either individually or in various combinations, and astrocytes and otherglia cells express α1, α2 or both, but not α3. The α1 mRNA has a broad distribution in brain,whereas the α2 mRNA is much more localized. While the β subunit is the same as an adhe-sion molecule on glia (AMOG), the β isoform is not specific to glia.51 These findings raiseinteresting questions regarding the relationship between ion transport and cell adhesion.
In terms of the kinetics of the different isoforms, there is evidence both for and againsta specialized role of glial Na+/K+ ATPase in uptake of K+ by astrocytes.50 As with other Na+
pumps this system seems to be driven mainly by intracellular [Na+]i. It has a high affinityfor K+ on the outside (Km ~ 1 mM), and a mid-activation level for Na+ of about 10 mM onthe inside. Thus, with a [K+]o of around 3 mM there will only be a small amount of activa-tion between 3 and 10 mM K+, the saturation level for K+ activation of the Na+/K+ pump,while with a Km for Na+
i of ~ 10 mM and [Na+]i ≅ 10 mM14 the pump is poised to be maxi-mally activated by increases or decreases in [Na+]i from its normal levels. Sontheimer etal52,25 found that some astrocytes have a high density of TTX-sensitive Na+ channels, andsuggested that these channels are responsible for maintaining the intracellular Na+ levelsrequired for the functioning of the Na+/K+ pump. Since these Na+ channels are voltage-activated, increased [K+]o would depolarize the astrocyte membrane potential and regulatethe influx of Na+ and thereby the Na+/K+ pump via increases in [Na+]i. This is an interestingsuggestion, since it would control active K+ uptake by astrocytes without requiring any spe-cialization of the astrocytic Na+/K+ pump.

21Functions of Astrocytes
pH carriersOther carrier systems for Cl– or Na+ involve co- or exchange transport with the pH
equivalents H+, HCO3– or OH–. These systems are the Na+/H+ and Cl–/HCO3
– or OH– ex-changers, and a variety of electrogenic or nonelectrogenic cotransport systems for Na+ plusnHCO3
–, where n can be two to three.53-55 It has been suggested, based on the possession ofthese transport systems and the fact that astrocytes in situ can undergo very large pH changesin ischemia (often in the opposite direction to the extracellular pH), that astrocytes arecritically important in maintaining pH homeostasis in the brain.56-58 In ischemia, the astro-cytes become very acidic, whereas in spreading depression the astrocytes undergo large in-tracellular alkaline shifts.56 The operation of such pH transporting systems could also leadto volume changes. For example, the simultaneous operation of the Na+/H+ and Cl–/HCO3
–
exchangers driven by intracellular hydration of CO2 to H+ and HCO3– could lead to a net
uptake of Na+ and Cl– with astrocyte swelling. Such swelling is seen, for example, in traumaor ischemia, when it is likely that the Na+/K+ pump will be compromised due to fallingenergy levels and therefore pump out the Na+ more slowly.59,38 Such swelling has been re-produced in vitro under such conditions using primary astrocyte cultures or C6 glioma celllines.60
Transmitter Uptake Systems
Amino Acid Carriers and Cellular Metabolic CompartmentationIt has long been known that there are very powerful uptake systems on astrocytes for a
number of amino acid transmitters, particularly the excitatory amino acids (EAA) glutamateand aspartate. It was proposed 25 years ago that glutamate released from terminals wastaken up into astrocytes where it is converted to glutamine by the astrocyte-specific enzymeglutamine synthetase.61 This was perhaps the first example of compartmentation of meta-bolic functions between neurons and glia. The general concept of metabolic compartment-ation between astrocytes and neurons has gained increasing experimental support with thefinding that key metabolic enzymes such as pyruvate carboxylase, specific isoforms of lacticdehydrogenase and malic enzyme are specific to astrocytes.62,63 Such compartmentation hasprofound implications for brain chemistry.
It is not clear whether the uptake of glutamate, seen in both cultured and acutely iso-lated astrocytes and by autoradiography and immunocytochemistry on astrocytes in situ,64,65
influences synaptic transmission. Uptake of the EAAs is likely to be slower than needed toalter synaptic transmission and therefore is more likely to serve for long term control. How-ever, it is now thought that binding to the high density of both neuronal and astrocytictransporters contributes to the decay of the EPSP, as diffusion of glutamate is considered tobe too slow.66 Since glutamine synthetase is astrocyte-specific, it is also intriguing for thefunctional implications of the transporters that administration of a specific inhibitor of thisenzyme just prior to a learning task in day-old chicks caused significant retention loss.67
Recent work has shown that the EAA transporters also transport K+ and OH–,68 andreversal of this transporter by ischemia-induced changes in cellular ion gradients has beensuggested to be responsible for some of the increased EAAs seen in ischemia.69,70 Increasedglutamate content in astrocytes and decreased neuronal content during ischemia have beenshown by quantitative immunocytochemistry at the electron microscope level.71 This in-crease in steady state glutamate levels could be due to a decreased conversion to glutaminewithin astrocytes, leading to a large increase in glutamate which more than offsets the in-creased release.
Recent molecular biology studies have identified at least three members of a family ofglutamate transporters of which two (GLT-1 and GLAST in the rat) are found exclusively or

Astrocytes in Brain Aging and Neurodegeneration22
predominantly on astrocytes.72-75 The GLT-1 is the major form in rat brain. GLT-1 andGLAST coexist on the same astrocyte membranes, but do not form complexes with eachother. Rather they form homo-oligomers.76 In an interesting study, Rothstein et al77 haveshown that separate intraventricular administration of antisense oligonucleotides to thethree different transporters led after 7-10 days to a marked reduction in all the transporterproteins. However, only antisense to the astrocyte-specific transporters GLT-1 and GLASTled to an increase in [Glu]o, as measured by microdialysis. The same group has also impli-cated a failure of the astrocyte transporters in the etiology of amyotrophic lateral sclerosis.78
There is also good evidence for GABA transporters on astrocytes. As with the EAAtransporters, different isoforms have been found, termed GAT-1, GAT-2 and GAT-3. UsingcDNA probes for in situ hybridization, it was found that GAT-1 was predominantly neu-ronal, while GAT-3 was also seen over glial profiles. GAT-2 was mainly seen in the meninges,and is also found outside the CNS.79 However, in the cerebellum GAT-1 is found as much inthe Bergmann glia surrounding the Purkinje cells as in the Purkinje cell bodies themselves.80
It was suggested that the GAT-1 localized to the basal pole and axon hillock of the Purkinjecells are primarily involved in terminating the action of GABA at basket synapses on thePurkinje axon hillocks, a role also presumably subserved by the ensheathing GAT-1-IR posi-tive glial processes as GABAergic cerebellar neurons did not show GAT-1-IR.
An amino acid transporter very active on astrocytes is that for the amino sulphonicacid taurine. Taurine occurs at high levels in many brain regions, and is considered to be aninhibitory transmitter and to also play a role in limiting cell swelling, especially in astro-cytes, by its release via swelling-activated organic anion channels.37,81
Monoamine TransportersUptake of a number of monoamine transmitters has been reported in primary astro-
cyte cultures.14,82 These systems resemble the high-affinity systems found in nerve termi-nals, being both Na+ -dependent and inhibitable by a variety of clinically relevant antide-pressants, such as fluoxetine (Prozac) for serotonin. Uptake systems for adenosine83 andhistamine84 have also been described in cultured astrocytes. Unlike the amino acid trans-port systems, the relevance of the monoamine uptake systems has not yet been establishedand may play a minor role, if it plays any role at all or exists on astrocytes in vivo, comparedto the very active monoamine transporters in nerve terminals. Recently, a direct compari-son of pure monolayer astrocyte cultures and explant cultures from different brain regionshas shown that while the high affinity norepinephrine and serotonin transporters were in-deed present in the pure astrocyte cultures, such uptake into astrocytes could not be de-tected in the explant cultures, which also contain neurons, under identical conditions.85 Asan appropriate control, the uptake of radiolabeled glutamate or GABA into astrocytes wasfound to be comparable in the pure astrocyte and the mixed explant cultures. This is an-other example of how neurons can influence astrocyte properties and how homogeneousastrocyte cultures can give idiosyncratic results. In this case it appears that neurons suppressan important function that astrocytes might otherwise express as a default property. It wouldbe of considerable interest to see whether this effect has any correlate in vivo, such as inreactive astrogliosis which occurs as a response to neuronal damage and death.
Receptors for Transmitters
Receptors in Primary Astrocyte CulturesA large number of neurotransmitter receptors have been found in monolayer primary
astrocyte cultures prepared from different brain regions of neonatal rats. These includeadrenergic (α1, α2, β1, β2), aminergic (5HT1, 5HT2, M1, M2, H1, H2), amino acid (mGluR,

23Functions of Astrocytes
KA/AMPA, GABAA, GABAB), peptide (AT II, somatostatin, endothelins, bradykinins, sub-stance P, ANP, neuropeptide Y, VIP, opioid), and purinergic (P2x, P2y, P2u, A1, A2) receptors.86
They have been identified by a variety of techniques, including stimulation of second mes-sengers (cAMP, IP3 or [Ca2+]i), electrophysiological studies, antibody staining, radio-ligandbinding studies, and most recently in situ hybridization and RT-PCR for receptor mRNA.In terms of regional specialization the receptors found so far in primary astrocyte culturesfrom the hippocampus which could be relevant to function in this region include glutamate(mGluR, KA/AMPA),34,87,88 5HT2A
89 and adenosine90 receptors. Adrenergic (α1, β) receptorshave also been found in mixed neuron-astroglial hippocampal cultures.91
Most of the work on receptor expression in astrocytes so far has been done in vitro.However, it is now clear from numerous studies that many of the properties of these cul-tures, including receptor expression, change with different culture conditions.55,92-99 Forexample, Shao and McCarthy99 demonstrated that cortical astroglia tended to lose theirresponsiveness to carbachol and histamine and to develop responsiveness to NE with growthin serum (FBS)-containing medium, even though they were cultured from single cells. Milleret al97 showed an upregulation of a phosphoinositide-coupled mGluR in cortical astrocytesin chemically-defined medium as compared to serum (FCS)-containing medium. This ef-fect was found to be due to the growth factors bFGF and EGF in chemically-defined me-dium. They also found that exposure to thrombin reduced mGluR5 level in astrocyte cul-tures,100 which might be the reason for the decrease in mGluR-mediated Ca2+ responsesseen in astrocytes cultured in horse serum-containing medium.101 One of our laboratoriesalso recently showed that the proportion of cells responding to 5-HT increased when acutelyisolated astrocytes were cultured in horse serum-containing medium,101,102 but did not in-crease in serum-free, chemically defined medium.95 These findings continue to raise ques-tions regarding which receptors seen in primary culture exist in astrocytes in vivo, or areupregulated or selected for in culture.
Receptor Studies in Astrocytes in Brain SlicesWithin the past few years, attempts have been made to study receptors on astrocytes in
situ. Studies in brain slices (mainly from the hippocampus) have shown that astrocytes insitu do respond to applied neurotransmitters. Techniques used to measure these responseshave been mainly electrophysiological, or calcium imaging with confocal microscopy. Thereceptors found so far in astrocytes have been GABAA receptors in hippocampal slices fromP21-42 rats,103,104 glutamate (mGluR, KA/AMPA, NMDA) receptors in hippocampal slicesfrom P9-13 rats105 and P9-12 mice,43 and P1 and less frequently P2 purinergic receptors inastrocytes in hippocampal slices from P9-13 rats.106 P2 receptors have also been found inBergmann glia in cerebellar slices from P6-45 day old mice107 and α1-adrenoreceptors andH1 histamine receptors in Bergmann glial cells in cerebellar slices from P20-25 mice.108
However, slice studies do present several difficulties. An important one is secondaryeffects on astrocytes due to the release of neurotransmitters or K+ from neurons stimulatedby the applied transmitters. TTX used in slice studies can only block action potential-in-duced terminal release of neurotransmitters and not transmitter release induced by TTX-insensitive action potentials or extrasynaptic release from dendrites or axons. The calciumresponses to iGluR agonists (KA, AMPA, NMDA) found in astrocytes in hippocampal slices,but not in our acutely isolated hippocampal astrocytes, could be due to the depolarizationof astrocytes by K+ released from excited neurons, which then activate voltage-activatedCa2+ channels leading to [Ca2+]i increases in astrocytes. For activation of the AMPA/KAreceptors to directly lead to an increase in [Ca2+]i they need to lack the GluR2 subunit thatresults in Ca2+ permeability of the AMPA receptors. This has been reported in Bergmannglia in mouse cerebellar slices.109

Astrocytes in Brain Aging and Neurodegeneration24
Porter and McCarthy105 showed that GFAP-positive astrocytes in rat hippocampal slicesresponded to application of NMDA-induced with an increase in [Ca2+]i. However, this mightbe due to a direct activation of NMDA receptors on neurons, leading to release of glutamate,K+ or some other substance which then activates the astrocytes via some different mecha-nism, and not indicative that the astrocytes themselves have NMDA receptors. Porter andMcCarthy106 also showed that most astrocytes in hippocampal slices responded to ATP, butthis was mediated by adenosine receptors (P1) and not ATP receptors. The P1 receptorswere presumably activated by adenosine produced by hydrolysis of the added ATP to ad-enosine by extracellular ectonucleotidases within the slice. This is another kind of indirecteffect which could have led to the erroneous conclusion that most hippocampal astrocytesexpress ATP receptors. However, a P2-induced Ca2+ transient was found in Bergmann gliain cerebellar slices from P6-45 mice,107 and this may imply a regional or species variation ofP2 receptor expression in astrocytes.
Another problem with slices is slowed access to applied transmitters due to long diffu-sion pathways and/or uptake, which can result in an insufficiently high concentration ofperfused transmitters at receptor sites within the slices. Slowed access is a particular prob-lem for rapidly desensitizing receptors such as the GluR receptors.110,111 Thus, cells close tothe center of slices are less likely to respond to glutamate than those close to the edge of theslice, and the glutamate uptake inhibitor THBA did increase the percentage of cells respondingto glutamate.105
Immunohistochemical Studies on Astrocytes In VivoImmunohistochemistry in brain sections shows the localization of receptors to astro-
cytes with perhaps the least involvement of confounding variables. However, such studiesdo not indicate whether the receptors are functional and what functions they subserve. Aokiand colleagues112,113 showed astrocytic localization of α2A and β-adrenergic and NMDA1
receptors to astrocytic profiles in visual cortex using electron microscopy with immunocy-tochemistry. 5HT1A,114 mGluR5,115 mGluR3,116 AMPA receptor subunits GluR1 and GluR4117
and muscarinic receptor118 immunoreactivities have also been localized to astrocytes in rathippocampus. Paspalas and Papadopoulos119 have recently reported that fine norepineph-rine-containing nerve terminals ended on astrocytes around capillaries in rat visual cortex,as well as directly on the basal lamina. However, no plasma membrane differentiation atthese sites on the astrocyte membranes was detected.
In situ hybridization is now beginning to be used to localize receptor mRNA in astro-cytes in intact brain. mRNAs for kainate receptors have been detected in astrocytes in vari-ous brain regions120 and mRNA for the NMDA2B subunit has been localized over Bergmannglia cells.121 Recent studies have also shown mGluR subtype mRNA expression in neuronsin the hippocampus, but strong labeling of astrocytes was only shown for mGluR3, pre-dominantly in the CA1 region.122,123 Light antibody staining for mGluR5 has been observedin astrocytes in hippocampal sections.115
Receptor Studies in Acutely Isolated AstrocytesAlternative preparations that also seem to more faithfully reflect in situ properties are
astrocytes studied as soon as practical after isolation from the CNS, as has been done forneurons.124 This approach, although subjecting cells to some degree of rough handling, hasclear experimental advantages, principally in avoiding the problem of indirect effects andslow access, as well as generally easier experimental techniques. More controlled experi-ments are possible but there is, of course, no possibility of studying interactions with neu-rons and other cells in the brain. Also, in cells from older animals, cell processes seem to be

25Functions of Astrocytes
lost (unpublished observations); this is a serious problem if there are differences in receptordistribution within the cell.
Acutely isolated astrocytes were first used to study voltage-gated ion channels in astro-cytes,12,13,125 and there has been less work on receptors. Fraser et al103 identified GABAA/ben-zodiazepine receptors in acutely isolated hippocampal astrocytes from 2-6 week old rats bya combination of whole-cell patch clamp, calcium imaging, immunocytochemistry and fluo-rescent ligand binding techniques. Duffy and MacVicar126 found that <5% of hippocampalastrocytes acutely isolated from P21-42 rats showed α1 adrenergic-mediated calcium re-sponses, although almost all the astrocytes in slices responded to norepinephrine. Glutamatedid not increase [Ca2+]i in both acutely isolated astrocytes and astrocytes in slices. Seifertand Steinhauser111 applied patch-clamp technique and single-cell RT-PCR to glial precursorcells acutely isolated from the juvenile mouse hippocampal CA1 stratum radiatum subre-gion and found responses due to activation of GluR2 and GluR4 AMPA receptor subunits.
Kimelberg et al101 studied receptor mediated Ca2+ responses of astrocytes acutely iso-lated from cerebral cortex to glutamate, 5-HT and ATP, and compared these with primaryastrocyte cultures from cortex. It was found that GFAP-positive astrocytes acutely isolatedfrom the cerebral cortices of postnatal 3-10 day old rats frequently showed increased intra-cellular [Ca2+] responses to L-glutamate. In contrast, responses to 10 µM ATP or 10 µM5-HT were much less frequent or absent, respectively. The same cells that failed to respondto ATP or 5-HT often responded to glutamate. Culturing acutely isolated cells in mediacontaining 10% horse serum decreased the percentage of GFAP-positive cells responsive toglutamate, but greatly increased the percentage that were responsive to ATP and 5-HT. Inprimary cultures prepared from the cerebral cortices of 1 day old rats and cultured in se-rum-containing medium for 2-4 weeks, fewer cells responded to glutamate than in acutelyisolated cells, but almost all cells responded to ATP and 5-HT. The lack of response to ATPand 5-HT in the acutely isolated cells seemed unlikely to be due to selective damage to therespective receptors during enzymatic dissociation because acutely isolated GFAP-negativecells in the same preparations showed responses to ATP, several different proteases andmechanical dissociation yielded cells which also responded to Glu but not ATP, andexposure of primary cultures to papain, the enzyme used during isolation, did not abolishCa2+ responses to several transmitters. We have now observed a very similar profile ofresponses for GFAP-positive astrocytes acutely isolated from the hippocampus.102 Thus,some of the receptor responses seen in primary astrocyte cultures may not reflect receptorspresent in astrocytes in vivo, but are rather upregulated or selected for in response to cultureconditions.
Of course, acutely isolated cells as models to study astrocytes have problems. First is thelow yield of cells compared to primary cultures, so that the responses of individual or smallgroups of cells only can be measured. Thus, some aspects of astroglial functions cannot beeasily studied (e.g., transmitter uptake and release). Second, and more seriously, there is thepotential possibility of selective proteolytic damage to receptors, or the shearing off of pro-cesses. The latter appears to be more of a problem in older animals of >15 days. Thus,damage during isolation always needs to be ruled out when we find that these cells respondto one transmitter but not another.101
Functional ImplicationsIt is of course of great interest to ask what effects receptor activation might have on
astrocyte properties. These will, of course, initially be the activation of second messengerswhich can then lead to a variety of functional effects, which in the case of astrocytes are stilllargely unknown. The activation of the KA/AMPA glutamate receptor in cultured astro-cytes elicits membrane potential depolarization and Na+ and K+ inward currents.127-129 In

Astrocytes in Brain Aging and Neurodegeneration26
cerebellar slices, KA has been shown to activate an AMPA receptor containing the GluR2subunit on Bergmann glia which allows Ca2+ entry.109 AMPA receptor activation has beenreported to reduce cell-cell junctional conductance between Bergmann glia in mouse cer-ebellar slices.130 Addition of glutamate to astrocytes has also been shown to engender self-propagating Ca2+ waves through the syncytium in primary astrocyte cultures,31-33,131,132 andin hippocampal slices from 7-12 day old rats.133 This will at a minimum allow astrocytes tosignal changes over a wide region of the brain. Because this superficially resembles how aneuronal network might function, there have been suggestions that these astrocytic Ca2+
waves are involved in some way in information processing, but it could simply be a mecha-nism whereby the astrocytes coordinate a homeostatic function, such as K+ transport withinthe entire astrocytic syncytium.
A recent study in hippocampal slices134 showed that, in the negative feedback byglutamate of its own release at Schaffer collateral-CA1 pyramidal cell synapses mediated bypresynaptic adenosine receptors, the adenosine seems to derive from perisynaptic astro-cytes and inhibited synaptic transmission on the msec time scale. The effect requires simul-taneous activation on the astrocytes of β adrenergic receptors by separate norepinephrineterminals and activation of an mGluR3 receptor on astrocytes by the synaptically releasedGlu, to raise astrocytic cAMP, which is the source of the adenosine. Thus, this system func-tions as a coincidence detector.
The effects of activation of β-adrenergic receptors on the shape of astrocytes in vitro135,136
implied that astrocytes may have the property of changing shape during neuronal activities,including learning. There have also been a number of studies that have shown an increase inastrocyte number in the brains of rats taught tasks as compared to nonlearning rats,137 andGFAP positive processes have been shown to increase in animals which have gone throughodor preference training.138 These changes, including the recent finding that inhibition ofthe astrocyte-specific enzyme glutamine synthetase by administration of methioninesulfoximine prior to a learning task inhibits retention of the task in day old chicks,67 implyroles for astrocytes in behavior.139
Astrocytes and the Blood-Brain Barrier (BBB)The BBB is a specialized structure responsible for the maintenance of the neuronal
microenvironment. It plays a pivotal role in tissue homeostasis, fibrinolysis and coagula-tion, vasotonus regulation, the vascularization of normal and neoplastic tissues, and bloodcell activation and migration during physiological and pathological processes, among otherfunctions.140-143 Such regulation of blood-tissue exchange is first accomplished by individualendothelial cells being continuously linked by occluding tight junctions (zonulaeoccludentes). This isolates the brain from the blood and also negates the oncotic and os-motic forces that govern blood-tissue exchange elsewhere in peripheral tissues. A numberof factors determine transport across the BBB.140 In the absence of specific carriers asubstance’s permeability is largely dependent upon its lipophilicity. Certain molecules neededfor brain metabolism, however, penetrate the BBB more readily than one would predictbased on their lipid solubility alone, and such substances cross the barrier on specific carri-ers. Some of these carriers are symmetrically distributed both on the luminal and abluminalmembranes of the endothelial cells, while others have an asymmetric distribution.143 Forexample, the carriers for the essential neutral amino acids, which are required in the brainfor neurotransmitter synthesis, are localized on both luminal and abluminal membranes. Incontrast, the carrier for the amino acid glycine appears to be located only on the abluminalmembrane. This asymmetric distribution functions to remove glycine from the CNS and tokeep its concentration in the brain low. Similarly, the abluminal membrane contains moreof the (Na++K+) ATPase than does the luminal membrane. This enzyme forms the basis of

27Functions of Astrocytes
a pump that simultaneously transports Na+ out of the endothelium into the brain, and K+
out of the brain into the endothelial cell. Like glycine, K+ has a potent effect on the trans-mission of nerve impulses and neuron firing, and this asymmetric distribution functions tomaintain low K+ concentrations in the extracellular fluid.
Astrocytes and BBB-InductionIn the mammalian CNS, brain capillaries develop from solid cords of endothelial cells.
These cords develop a slit-like lumen, which progressively increases its caliber.144 The newlyformed cords are separated from juxtaposed neurons by a basement membrane, and theyprogressively become ensheathed by resident astrocytes. The most rapid capillary sproutingcorresponds to the period of glial cell proliferation and neuronal dendritic developmentand arborization.142 It was thought at one time that the astrocytic foot-processes actuallyformed the restrictive barrier, since this was the most obvious distinguishing feature be-tween brain capillaries and all other capillaries in the periphery. However, electron micro-scope studies in the 1950s using electron-dense markers showed that the barrier to the dif-fusion of these markers was due to zonulae occludentes between the endothelial cells, andthat there was free passage of such markers between the astrocytic end-feet.140,143 Recentwork has indicated that the astrocytic end-feet processes may, however, play an importantrole in the induction of the BBB. Transplantation experiments have shown that the forma-tion of the BBB depends largely on the CNS environment, since it did not form in capillar-ies growing into systemic tissue transplanted into the CNS, whereas the converse was true.145
Janzer and Raff146 demonstrated that astrocytes might be responsible for this phenomenon.They showed that injection of primary astrocyte cultures into the anterior eye chamber orchorioallantoic membrane of the chick induced a permeability barrier in the endothelialcells of the capillaries of these tissues that would otherwise lack such a barrier. Another lineof evidence in support of the role of astrocytes in BBB induction derives from studies byTao-Cheng et al.147 When endothelial cells were cultured alone, their tight junctions ap-peared fragmentary. When cocultured with astrocytes, the length, breadth and complexityof the tight junctions between the endothelial cells was increased, more closely resemblingthe structures seen in vivo. Interestingly, when other cell types such as fibroblasts were sub-stituted for astrocytes the tight junctions remained fragmentary.
There are also several lines of evidence for astrocytic induction of functional proper-ties of CNS capillaries. These include gamma glutamyl transpeptidase (γ-GT) activity, aspecific marker of endothelial cells of the CNS endothelium, which was abolished by theabsence of astrocytes in a coculture system.148 Addition of astrocytes to endothelial cell cul-tures also increased the incorporation of neutral amino acids by the endothelial cells.149 Theexpression of the barrier-specific GLUT-1 isoform of the glucose transporter was markedlydownregulated in cultured bovine brain capillary endothelial cells in the absence of brain-derived or astrocyte trophic factors in the tissue culture medium.150 Astrocyte involvementin the differentiation and angiogenesis of the endothelial cells of the BBB is indirectly sup-ported by the observation that vascular endothelial cell growth factor (VEGF) expression isinduced and strongly upregulated in human malignant glioblastoma cells.141 VEGF is anangiogenic growth factor whose expression appears to parallel embryonic brain angiogen-esis. Also, morphological differentiation and induction of specific BBB proteins can be in-duced by primary astrocyte cultures in endothelial cells in vitro.151-153
Despite the above evidence, it still remains unsettled whether astrocytes have a generalrole in the induction and maintenance of the BBB in vivo. Brightman154 concluded that “theprecise role of perivascular astrocytes in the induction and maintenance of brain endo-thelium as a structural and functional barrier has yet to be fully elaborated.” Reasons fordoubting a general inductive effect of astrocytes on the BBB are that the cerebral capillaries

Astrocytes in Brain Aging and Neurodegeneration28
of a number of elasmobranchs are ensheathed by astrocytes, but their endothelial cells donot express tight junctions. Rather, they exhibit open pores which are permeable to largemolecules, including horseradish peroxidase (HRP).155,156 As further suggested byBrightman,154 “either these particular astrocytes ensure that the endothelial junctions re-main open or the junctional configuration is an inherent one that is not determined by theastrocytic investment.” Astrocytes are also found in close association with pituicytes, yet theendothelium in the neural lobe is of the fenestrated phenotype and it is largely permeable todyes such as HRP. Cloned endothelial cells in vitro can establish a barrier with a relativelyhigh resistance of 700-800 ohm⋅cm2.157 In further studies of injection of astrocytes into theanterior chamber and chorioallantoic membrane of the eye, they failed to form richly vas-cularized grafts.158 Rather, grafting of the astrocytes to the chorioallantoic membrane led toan extensive inflammatory response which, in turn, led to poor delivery of tracers to thegraft vasculature. The iridial vessels associated with astrocyte grafts did not change theirultrastructure to resemble brain capillaries, and the astrocyte graft vasculature also failed toexpress high levels of the GLUT-1 isoform of the glucose transporter, even after treatmentwith anti-inflammatory agents.158 Hence, the authors questioned the general utility of theanterior chamber and chorioallantoic membrane for studying BBB induction, as used byJanzer and Raff.146
As pointed out by Abbott,159 in evolutionarily lower animals such as the cephalopodmollusks, the blood-brain barrier is formed between the glial cells and not between theendothelial cells. Abbott159 suggested that during evolution the barrier in vertebrates haslikely shifted from glial cells to endothelial cells, “perhaps to allow greater complexity andcontrol of the CNS interstitial environment by the glial cells, superimposed upon a barrierwhich prevented interference by large and rapid changes in the blood.”
Astrocytes and Immune and Inflammatory Responses in the CNSThe CNS was regarded for decades as an “immunologically privileged” organ. This
long-standing view that the brain is isolated from the effects of the immune system hasbeen challenged with convincing experimental evidence that in response to invasion bymicroorganisms the CNS can mount its own defense by resident cells, such as the microgliaand astrocytes.160-162 As summarized by Benveniste,163 “cells of the CNS constitutively ex-press low levels of antigens encoded for by major histocompatibility complex (MHC) geneswhose products play a fundamental role in the induction and regulation of immune re-sponses.” However, both activated microglia and astrocytes can secrete a number of cytokineswhich can modulate the function of lymphoid-mononuclear cells, thus establishing an in-tegrative communication pathway between resident cells of the CNS and those of the im-mune system. For a detailed discussion on the function of microglia in CNS immune me-diation, the reader is referred to reviews in Graeber et al164 (also see chapter 4 of this volume).For a comprehensive review on CNS cytokines and their respective origins (i.e., astrocytes,microglia, macrophages) the reader is directed to a review chapter by Benveniste.163 As anexample of how astrocytes are potentially involved in immune responses in the CNS, wewill focus on astrocyte-specific cytokine elaboration and their potential role in initiationand suppression of immune responses, as well as the role of astrocyte-derived cytokines insustaining and propagating CNS-induced damage
Do Astrocytes Modulate CNS Immune Responses?Evidence has accumulated for a role for cytokines in the CNS. For example, when di-
rectly injected into the brain, IL-1 promotes glial scarring or astrogliosis, suggesting thatIL-1 may be important in mediating astrocytic hypertrophy upon neuronal injury.165 Whenprimary astrocyte cultures derived from newborn mice are treated with lipopolysaccharide

29Functions of Astrocytes
(LPS, E. coli) the astrocytes secrete interleukin-1 (IL-1).166 Intracerebral synthesis of IL-1has been implicated as a prerequisite to intracerebral T cell activation, primarily becauseIL-1 enhances the production of IL-2 and expression of IL-2 receptors on T cells.167 It wouldappear, therefore, that astrocytes may be both responsive to IL-1 and capable of synthesiz-ing it, providing for autocrine regulation of IL-1 levels within the CNS.
The signals leading to the recruitment of circulating blood monocytes, and possiblyresident CNS macrophages, are poorly understood. Astrocytes have been implicated as ac-tive participants in this process in view of their ability in primary culture to secrete an IL-3-like factor which induces growth of cultured mouse peritoneal exudate cells (PEC) andbrain tissue macrophages.168 Astrocytes also secrete granulocyte-macrophage colony-stimu-lating factor (GM-CSF), as evidenced by induction of colony formation in bone marrowcells and growth of FDC-P1 cells.169 GM-CSF is a cytokine necessary for growth and differ-entiation of macrophages and has been found to lead to an accumulation of macrophagesat the site of inflammatory lesions. GM-CSF enhances a number of functional activities ofmature macrophages such as their phagocytic, cytotoxic, and microbicidal activities.
Migration of activated T cells across the compromised BBB in the course of CNS dis-ease is associated with parenchymal production of interferon-γ (IFN-γ). IFN-γ interacts withastrocytes, as well as microglia, in the CNS, where it has been shown to modulate MHCgene expression and increase class I antigen expression.170 Expression of class I antigens onthe astrocytic membranes increases their susceptibility to lysis by class 1-restricted cyto-toxic T cells. Like IL-1, IFN-γ can lead to increased expression of adhesion molecules onastrocytes.171 Although IFN-γ does not appear to directly stimulate astrocytic cytokine pro-duction, it appears to “prime” the astrocytes to respond to other cytokines, such as TNF-α.172
The latter induces cytokine production by astrocytes, and leads to secretion of IL-6.173 Theresponse of cultured astrocytes to IFN-γ results in increased expression of MHC antigensand costimulatory molecules (intercellular adhesion molecule-1, LFA-1 alpha) which me-diate astrocyte-T cell interactions.174 IFN-γ can induce the production of the cytotoxic aminoacid quinolinic acid, an NMDA agonist, and in conjunction with IL-1β it can increase NOsynthetase (NOS) expression in astrocytes.175 Inducible NOS (iNOS) induction in astro-cytes, as well as macrophages, has recently been postulated to contribute to HIV-relatedneurotoxicity.176
Recent studies also suggest that astrocyte-derived cytokines may be detrimental. Asnoted in the preceding paragraph, after an initial penetration of T cells into the CNS, astro-cytes can further support the intracerebral T cell activation process. GM-CSF would there-fore be expected to increase granulocyte and macrophage survival within the CNS and aug-ment their activity against invading microbes.177 However, because viral replication incultured HIV-infected monocytes is increased by GM-CSF, the potential for these cytokinesto augment viral production in monocytes and microglia in HIV encephalitis exists, poten-tially worsening the spread of the infection within the CNS.178,179 Another cytokine, trans-forming growth factor (TGF) β1, was also recently implicated in facilitating the recruitmentof peripheral infected monocytic cells, and thus it may contribute to HIV-1-related inflam-mation and the spread of the virus into the CNS.180 However, in view of the fact that manyof the in vitro studies await in vivo confirmation, it is essential that future studies be di-rected toward determining the role of cytokines in inflammatory invasion of the CNS byblood-borne factors in vivo.
There is circumstantial evidence that implicates astrocytes in mediating the neurotoxiceffects of the HIV-1 soluble protein gp120. The latter has been reported to modulate severalastrocyte functions, inducing intracellular signaling, ion transport, release of endogenousamino acids, and protein phosphorylation.181-185 A prominent effect of gp120 on astrocytefunction includes increased efflux of K+. Since the glutamate carrier is both voltage- and ion

Astrocytes in Brain Aging and Neurodegeneration30
gradient-dependent,68 increased [K+]o should increase [glutamate]o both because of reducedeffectiveness of astrocytic glutamate uptake due to depolarization as well as swelling-in-duced glutamate release.
A well-reproduced laboratory model for the CNS autoimmune disease, multiple scle-rosis, is experimental allergic encephalomyelitis (EAE). As early as 1933, Rivers et al186 notedthat when monkeys were injected with a rabbit brain extract they developed encephalitisthat was characterized by destruction of the white matter. During the course of EAE, mono-nuclear leukocytes preferentially accumulate in the CNS. Ransohoff et al187 have recentlymonitored the factors that govern leukocyte trafficking in EAE. Using in situ hybridization,Ransohoff et al187 noted that astrocytes were the major source of mRNAs encoding for IP-10 and JE/MCP-1, members of a family of chemoattractant cytokines. This suggested thatastrocyte-derived cytokines may function as chemoattractants for inflammatory cells dur-ing EAE. For additional information on the role of cytokines in multiple sclerosis/autoim-mune encephalitis the reader should consult the review by Benveniste.188
Infection of mice with the neurotropic JHM strain of mouse hepatitis virus (MHV-JHM) leads within several weeks of infection to a demyelinating encephalomyelitis diseaseassociated with prominent astrogliosis and infiltration of inflammatory cells. Analysis ofinfected spinal cords derived from these mice have recently revealed that three pleiotropiccytokines, TNF-α, IL-1β, and IL-6, as well as type 2 nitric oxide synthase (iNOS) are ex-pressed by activated astrocytes localized to areas of virus infection and demyelination.189
These data also show that, by analogy to the human demyelinating disease multiple sclero-sis, astrocytes are a major cellular source for these cytokines in mice with chronic MHV-JHM infection and the findings are consistent with a role of astrocyte-derived cytokinesand nitric oxide in the demyelinating process.
It is now appreciated that, “different cytokines activate distinct functional programs inastrocytes, which may play a specific role in different brain diseases or at different stages ofthe same disease.”174 In addition, it appears that astrocytic responses to various cytokineslargely depends on the presence or absence of neurons in the culture. Accordingly, neu-ronal-glial interactions may be of utmost importance in determining the activation thresh-old of astrocytes to inflammatory cytokines.174
Astrocytes as CNS Antigen Presenting CellsAstrocytes have been proposed to function as antigen-presenting cells (APCs), i.e., those
cells with the ability to present antigens to lymphocytes.167,190 When astrocytes from Lewisrats were cocultured with a syngenic, MBP-specific, Ia-restricted T cell line of Lewis ratorigin, they stimulated T cell proliferation. This process is antigen-specific and restricted tothe MHC.191 During such cocultivation of T cells and astrocytes, the latter are induced bythe preactivated T cells to express MHC type II molecules.160,191 Furthermore, supernatantsof lectin-stimulated spleen cells containing IFN can induce murine astrocytes in culture toexpress Ia antigens,192 underscoring the point that astrocytes depend on the presence of Ia-inducing signals, such as IFN-γ, to function as APCs. However, the validity of these studiesdepends on the complete absence of microglia in the cultures,193 because, in both rat194 andhuman,195 microglia potently express MHC type II antigens in situ. Microglia constitute5-10% of the total glial cell population and are considered to be the macrophages of thebrain.164 Their major function is as a scavenger cell, ingesting cellular debris, a process whichmay be important for tissue modeling in the developing CNS. Microglia may also be in-volved in inflammation and repair in the adult CNS due to their phagocytic ability, releaseof neutral proteinases, and production of oxidative radicals. Microglia have been shown toexpress MHC antigens upon activation, and they are known to secrete a number ofimmunoregulatory cytokines and to respond to cytokine stimulation. At this stage, the evi-

31Functions of Astrocytes
dence favors the microglia, and not the astrocytes, functioning as the brain’s APCs. Micro-glia are the more likely source of IL-1 during acute-phase brain injury because microglia arethe first brain cells to appear in increased numbers at sites of trauma or infection. In addi-tion, IL-1 appears to be produced more efficiently in microglia than in astrocytes.196 Otherevidence favoring microglia as the principle APCs of the CNS includes observations onmixed glial cultures from adult human brain where only a limited number of astrocytesexpress MHC class II molecules, whereas the majority of the microglial cells were MHC II-positive.197 In addition, microglia were readily induced by IFN-γ to express MHC II, whereasastrocytes were nonresponsive to the same treatment.198,197 Although earlier studies sug-gested that astrocytes can be induced by the preactivated T cells to express MHC type IImolecules160,191 more recent studies challenge this view. Microglia treated with IFN-γ werecapable of presenting MBP to MBP-specific T cells, whereas astrocytes could not fulfill sucha role even in the presence of high concentrations of IFN-γ.197
AcknowledgmentsWe would like to acknowledge support from the NIH to HKK (NS 19492 and NS 35205)
and to MA (NIEHS 7331) for the period during which this chapter was written and for theexperimental work cited from our laboratories. We thank Erin Grasek and TinaGiannakopoulos for help in preparing the manuscript.
References1. Somjen GG. Nervenkitt: Notes on the history of the concept of neuroglia. Glia 1988;1:2-9.2. Lugaro E. Sulle funzioni della nevroglia. Riv D Pat Nerv Ment 1907; 12:225-233.3. Kuffler SW, Nicholls JG, Orkand RK. Physiological properties of glial cells in the central
nervous system of amphibia. J Neurophysiol 1966; 29:768-787.4. Fedoroff S, Vernadakis A. Astrocytes—Development, Morphology, and Regional Special-
ization of Astrocytes, Volumes 1-3. Orlando: Academic Press, 1986.5. Kettenmann H, Ransom BR, eds. Neuroglia. New York: Oxford University Press, 1995.6. Dahl D, Bjorklund H, Bignami A. Immunological markers in astrocytes. In: Fedoroff S,
Vernadakis A, eds. Astrocytes: Cell Biology and Pathology of Astrocytes. Orlando: Aca-demic Press, 1986:1-25.
7. Kimelberg HK, Norenberg MD. Astrocytic responses to central nervous system trauma. In:Salzman SK, Faden AI, eds. The Neurobiology of Central Nervous System Trauma. NewYork, Oxford: Oxford University Press, 1994:193-208.
8. Kimelberg HK. Primary astrocyte cultures—A key to astrocyte function. Cell and MolecNeurobiol 1983; 3:1-16.
9. Crick F. The Astonishing Hypothesis: The Scientific Search for the Soul. New York: CharlesScribner’s Sons, 1994.
10. Somjen, G. Electrophysiology of mammalian glial cells in situ. In: Kettenmann H, RansomBR, eds. Neuroglia. New York, Oxford: Oxford University Press, 1995:319-334.
11. Hille B. Ionic Channels of Excitable Membranes. Sunderland, MA: Sinauer Assoc, Inc.,1992:1-20,115-139.
12. Barres BA, Chun LLY, Corey DP. Ion channels in vertebrate glia. Annual Rev Neurosci1990; 13:441-474.
13. Duffy S, MacVicar BA. Voltage-dependent ionic channels in astrocytes. In: Murphy S, ed.Astrocytes, Pharmacology and Function. San Diego: Academic Press, 1993:137.
14. Kimelberg HK, Jalonen T, Walz W. Regulation of the brain microenvironment: Transmit-ters and ions. In: Murphy S, ed. Astrocytes, Pharmacology and Function. San Diego: Aca-demic Press, Inc., 1993:193-228.
15. Sontheimer H. Voltage-dependent ion channels in glial cells. Glia 1994; 11:156-172.16. Takumi T, Ishii T, Horio Y et al. A novel ATP-dependent inward rectifier potassium chan-
nel expressed predominantly in glial cells. J Biol Chem 1995; 270:16339-16346.

Astrocytes in Brain Aging and Neurodegeneration32
17. Newman EA. High potassium conductance in astrocyte endfeet. Science 1986; 233:453-454.18. Roy ML, Sontheimer H. β-adrenergic modulation of glial inwardly rectifying potassium
channels. J Neurochem 1995; 64:1576-1584.19. Robert A, Magistretti PJ. AMPA/kainate receptor activation blocks K+ currents via internal
Na+ increase in mouse cultured stellate astrocytes. Glia 1997; 20:38-50.20. Kressin K, Kuprijanova E, Jabs R et al. Developmental regulation of Na+ and K+ conduc-
tances in glial cells of mouse hippocampal brain slices. Glia 1995; 15:173-187.21. Janigro D, Gasparini S, D’Ambrosio R et al. Reduction of K+ uptake in glia prevents long-
term depression maintenance and causes epileptiform activity. J Neurosci 1997;17:2813-2824.
22. Krnjevic K, Schwartz S. Some properties of unresponsive cells in the cerebral cortex. ExpBrain Res 1967; 3:306-319.
23. Bevan S, Chiu SY, Gray PTA et al. Presence of voltage-gated sodium, potassium and chlo-ride channels in rat cultured astrocytes. Phil Trans Roy Soc of London: Biol 1985;225:299-313.
24. Bowman CL, Kimelberg HK, Frangakis MV et al. Astrocytes in primary culture have chemi-cally activated sodium channels. J Neurosci 1984; 4:1527-1534.
25. Sontheimer H, Ransom BR, Waxman SG. Different Na+ currents in P0- and P7-derivedhippocampal astrocytes in vitro: Evidence for a switch in Na+ channel expression in vivo.Brain Res 1992; 597:24-29.
26. Sontheimer H, Waxman SG. Ion channels in spinal cord astrocytes in vitro. II. Biophysicaland pharmacological analysis of two Na+ current types. J Neurophysiol 1992; 68:1001-1011.
27. Black JA, Westenbroek R, Ransom BR et al. Type II sodium channels in spinal cord astro-cytes in situ: Immunocytochemical observations. Glia 1994; 12:219-227.
28. Ritchie JM. Current perspectives in glial electrophysiology. Ann N Y Acad Sci 1991;633:331-342.
29. MacVicar BA. Voltage-dependent calcium channels in glial cells. Science 1984; 2265:1345-1347.
30. Verkhratsky A, Kettenmann H. Calcium signaling in glial cells. Trends in NeurosciencesSpecial Issue: glial signaling 1996; 19:346-352.
31. Cornell-Bell AH, Finkbeiner S. Ca2+ waves in astrocytes. Cell Calcium 1991; 12:185-204.32. Finkbeiner S. Calcium waves in astrocytes—Filling in the gaps. Neuron 1992; 8:1101-1108.33. Finkbeiner SM. Glia calcium. Glia 1993; 9:83-104.34. Kim WT, Rioult MG, Cornell-Bell AH. Glutamate-induced calcium signaling in astrocytes.
Glia 1994; 11:173-184.35. Jalonen T. Single-channel characteristics of the large-conductance anion channel in rat
cortical astrocytes in primary culture. Glia 1993; 9:227-237.36. Roy G. Amino acid current through anion channels in cultured human glial cells. J Membr
Biol 1995; 147:35-44.37. Kimelberg HK. Current concepts of brain edema. J Neurosurg 1995; 83:1051-1059.38. Strange K, Emma F, Jackson PS. Cellular and molecular physiology of volume-sensitive
anion channels. Am J Physiol Cell Physiol 1996; 270:C711-C730.39. Okada, Y. Volume expansion-sensing outward-rectifier Cl- channel: Fresh start to the
molecular identity and volume sensor. Am J Physiol 1997; 273:C755-C789.40. Berger T, Walz W, Schnitzer J et al. GABA- and glutamate-activated currents in glial cells
of the mouse corpus callosum slice. J Neurosci Res 1992; 31:21-27.41. Kirchhoff F, Mülhardt C, Pastor A et al. Expression of glycine receptor subunits in glial
cells of the rat spinal cord. J Neurochem 1996; 66:1383-1390.42. Pastor A, Chvátal A, Syková E et al. Glycine- and GABA-activated currents in identified
glial cells of the developing rat spinal cord slice. Eur J Neurosci 1995; 7:1188-1198.43. Steinhäuser C, Jabs R, Kettenmann H. Properties of GABA and glutamate responses in
identified glial cells of the mouse hippocampal slice. Hippocampus 1994; 4:19-36.44. Zalc B, Collet A, Monge M et al. Tamm-Horsfall protein, a kidney marker is expressed on
brain sulfogalactosylceramide-positive astroglial structures. Brain Res 1984; 291:182-187.

33Functions of Astrocytes
45. Kettenmann H, Bormann J. Patch-clamp study of gamma-aminobutyric acid receptor Cl–
channels in cultured astrocytes. Proc Natl Acad Sci U S A 1988; 85:9336-9340.46. Kimelberg HK, Frangakis MV. Furosemide- and bumetanide-sensitive ion transport and
volume control in primary astrocyte cultures from rat brain. Brain Res 1985; 361:125-134.47. Ballanyi K., Grafe P, Bruggencate GT. Ion activities and potassium uptake mechanisms of
glial cells in guinea-pig olfactory cortex slices. J Physiol 1987; 382:159-174.48. LoPachin RM Jr, Stys PK. Elemental composition and water content of rat optic nerve
myelinated axons and glial cells: Effects of in vitro anoxia and reoxygenation. J Neurosci1995; 15:6735-6746.
49. Sweadner KJ. Overlapping and diverse distribution of Na-K ATPase isozymes in neuronsand glia. Can J Physiol Pharmacol 1992; 70:S255-S259.
50. Sweadner KJ. Na,-K-ATPase and its isoforms. In: Kettenmann H, Ransom BR, eds. Neuro-glia. New York, Oxford: Oxford University Press, 1995:259-272.
51. Gloor S, Antonicek H, Sweadner KJ et al. The adhesion molecule on glia (AMOG) is ahomologue of the B subunit of the Na, K-ATPase. J Cell Biology 1990; 110:165-174.
52. Sontheimer H, Black JA, Ransom BR et al. Ion channels in spinal cord astrocytes in vitro.I. Transient expression of high levels of Na+ and K+ channels. J Neurophysiol 1992;68:985-1000.
53. Boyarsky G, Ganz MB, Sterzel RB et al. pH regulation in single glomerular mesangial cellsI. Acid extrusion in absence and presence of HCO3. Am J Physiol: Cell Physiol 1993;255:C844-C856.
54. Newman EA, Astion ML. Localization and stoichiometry of electrogenic sodium bicarbon-ate co-transport in retinal glial cells. Glia 1991; 4:424-428.
55. Pappas CA, Ransom BR. Depolarization-induced alkalinization (DIA) in rat hippocampalastrocytes. J Neurophysiol 1994; 72:2816-2826.
56. Chesler M. Regulation and modulation of pH in the nervous system. Prog Neurobiol 1990;34:401-427.
57. Chesler M, Kraig RP. Intracellular pH transients of mammalian astrocytes. J Neurosci 1989;9:2011-2019.
58. Grichtchenko II, Chesler M. Depolarization-induced alkalinization of astrocytes in gliotichippocampal slices. Neurosci 1994; 62:1071-1078.
59. Kimelberg HK. Astrocytic edema in CNS trauma. J Neurotrauma 1992; 9:Suppl.1:S71-S81.60. Kempski O, Zimmer M, Neu A et al. Control of glial cell volume in anoxia in vitro studies
on ischemic cell swelling. Stroke 1987; 18:623-628.61. Balazs R, Patel AJ, Richter D. Metabolic Compartmentation in the Brain. London:
MacMillan Press, 1973:167-184.62. Farinelli SE, Nicklas WJ. Glutamate metabolism in rat cortical astrocyte cultures. J
Neurochem 1992; 58:1905-1915.63. Shank RP, Leo GC, Zielke HR. Cerebral metabolic compartmentation as revealed by nuclear
magnetic resonance analysis of D-[1-13C]glucose metabolism. J Neurochem 1993;61:315-323.
64. Aas J, Berg-Johnson J, Hegstead E et al. Redistribution of glutamate and glutamine in slicesof human neocortex exposed to combined hypoxia and glucose deprivation in vitro. J CerebBlood Flow Metab 1993; 13:503-515.
65. Torp R, Andine P, Hagberg H et al. Cellular and subcellular redistribution of glutamate-,glutamine- and taurine-like immunoreactivities during forebrain ischemia: A semiquanti-tative electron microscopic study in rat hippocampus. Neurosci 1991; 41(2/3):433-447.
66. Diamond JS, Jahr CE. Transporters buffer synaptically released glutamate on asubmillisecond time scale. J Neurosci 1997; 17:4672-4687.
67. Gibbs ME, O’Dowd BS, Hertz L et al. Inhibition of glutamine synthetase activity preventsmemory consolidation. Cognit Brain Res 1996; 4:57-64.
68. Bouvier M, Szatkowski M, Amato A et al. Glial cell glutamate uptake carrier countertrans-ports pH-changing anions. Nature 1992; 360:471-474.
69. Attwell D, Barbour B, Szatkowski M. Nonvesicular release of neurotransmitter. Neuron1993; 11:403-407.

Astrocytes in Brain Aging and Neurodegeneration34
70. Rutledge EM, Kimelberg HK. Release of [3H]-D-aspartate from primary astrocyte culturesin response to raised potassium. J Neurosci 1996; 16:7803-7811.
71. Ottersen OP, Laake JH, Reichelt W et al. Ischemic disruption of glutamate homeostasis inbrain: Quantitative immunocytochemical analyses. J Chem Neuroanat 1996; 12:1-14.
72. Lehre KP, Levy LM, Ottersen OP et al. Differential expression of two glial glutamate trans-porters in the rat brain: Quantitative and immunocytochemical observations. J Neurosci1995; 15:1835-1853.
73. Kanner BI. Glutamate transporters from brain: A novel neurotransmitter transporter fam-ily. FEBS Letters 1993; 325:95-99.
74. furuta A, Rothstein JD, Martin LJ. Glutamate transporter protein subtypes are expresseddifferentially during rat CNS development. J Neurosci 1997; 17:8363-8375.
75. Rothstein JD, Martin L, Levey AI et al. Localization of neuronal and glial glutamate trans-porters. Neuron 1994; 13:713-725.
76. Haugeto O, Ullensvang K, Levy LM et al. Brain glutamate transporter proteins formhomomultimers. J Biol Chem 1996; 271:27715-27722.
77. Rothstein JD, Dykes-Hoberg M, Pardo CA et al. Knockout of glutamate transporters re-veals a major role for astroglial transport in excitotoxicity and clearance of glutamate.Neuron 1996; 16:675-686.
78. Bristol LA, Rothstein JD. Glutamate transporter gene expression in amyotrophic lateralsclerosis motor cortex. Ann Neurol 1996; 39:676-679.
79. Durkin MM, Smith KE, Borden LA et al. Localization of messenger RNAs encoding threeGABA transporters in rat brain: An in situ hybridization study. Mol Brain Res 1995; 33:7-21.
80. Morara S, Brecha NC, Marcotti W et al. Neuronal and glial localization of the GABA trans-porter GAT-1 in the cerebellar cortex. NeuroReport 1996; 7:2993-2996.
81. Sanchez-Olea R, Morales M, Garcia O et al. C1 channel blockers inhibit the volume-acti-vated efflux of C1 and taurine in cultured neurons. J Amer Physiol Soc 1996; 270:C1703-C1708.
82. Kimelberg HK, Katz DM. High-affinity uptake of serotonin into immunocytochemicallyidentified astrocytes. Science 1985; 228:889-891.
83. Matz H, Hertz L. Effects of adenosine deaminase inhibition on active uptake and metabo-lism of adenosine in astrocytes in primary cultures. Brain Res 1990; 515:168-172.
84. Huszti Z, Imrik P, Madarász E. [3H]histamine uptake and release by astrocytes from ratbrain: Effects of sodium deprivation, high potassium, and potassium channel blockers.Neurochem Res 1994; 19:1249-1256.
85. Hosli L, Hosli E. Autoradiographic studies on the uptake of 3H-noradrenaline and 3H-serotonin by neurones and astrocytes in explant and primary cultures of rat CNS: Effectsof antidepressants. Int J Devl Neuroscience 1995; 13:897-908.
86. Kimelberg HK. Receptors on astrocytes—What possible functions. Neurochem Int 1995;26:27-40.
87. Cornell-Bell AH, Finkbeiner SM, Cooper MS et al. Glutamate induces calcium in culturedastrocytes: Long-range glial signaling. Science 1990; 247:470-473.
88. Glaum SR, Miller RJ. Acute regulation of synaptic transmission by metabotropic glutamatereceptors. In: Conn PJ, Patel J eds. The Metabotropic Glutamate Receptors. Totowa, NJ:Humana Press, 1994:147-172.
89. Deecher DC, Wilcox BD, Dave V et al. Detection of 5-hydroxytryptamine2 receptors byradioligand binding, Northern blot analysis, and Ca2+ responses in rat primary astrocytecultures. J Neurosci Res 1993; 35:246-256.
90. Ogata T, Nakamura Y, Tsuji K et al. Adenosine enhances intracellular Ca2+ mobilization inconjunction with metabotropic glutamate receptor activation by t-ACPD in cultured hip-pocampal astrocytes. Neurosci Letts 1994; 170:5-8.
91. Lerea LS, McCarthy KD. Neuron-associated astroglial cells express α1 and β adrenergicreceptors in vitro. Brain Res 1990; 521:7-14.
92. Barres BA, Chun LLY, Corey DP. Calcium current in cortical astrocytes: Induction by cAMPand neurotransmitters and permissive effect of serum factors. J Neurosci 1989; 9:3169-3175.

35Functions of Astrocytes
93. Kimelberg HK, Goderie SK, Conley PA et al. Uptake of [3H]serotonin and [3H]glutamateby primary astrocyte cultures I. Effects of different sera and time in culture. Glia 1992;6:1-8.
94. Landis DMD, Weinstein LA, Skordeles CJ. Serum influences the differentiation of mem-brane structure in cultured astrocytes. Glia 1990; 3:212-221.
95. Jalonen TO, Margraf RR, Wielt DB. et al. Serotonin induces inward potassium and cal-cium currents in rat cortical astrocytes. Brain Res 1997; 758:69-82.
96. Michler-Stuke A, Wolff JR, Bottenstein JE. Factors influencing astrocyte growth and devel-opment in defined media. Int J Devl Neuroscience 1984; 2:575-584.
97. Miller S, Romano C, Cotman CW. Growth factor upregulation of a phosphoinositide-coupled metabotropic glutamate receptor in cortical astrocytes. J Neurosci 1995;15:6103-6109.
98. Raff MC, Miller RH, Noble M. A glial progenitor cell that develops in vitro into an astro-cyte or an oligodendrocyte depending on culture medium. Nature 1983; 303:390-396.
99. Shao Y, McCarthy KD. Regulation of astroglial responsiveness to neuroligands in primaryculture. Neurosci 1993; 55:991-1001.
100. Miller S, Sehati N, Romano C et al. Exposure of astrocytes to thrombin reduces levels ofthe metabotropic glutamate receptor mGluR5. J Neurochem 1996; 67:1435-1447.
101. Kimelberg HK, Cai Z, Rastogi P et al. Transmitter-induced calcium responses differ inastrocytes acutely isolated from rat brain and in culture. J Neurochem 1997; 68:1088-1098.
102. Cai Z, Kimelberg HK. Glutamate receptor-mediated calcium responses in acutely isolatedhippocampal astrocytes. Glia 1997; 21:380-389.
103. Fraser DD, Duffy S, Angelides KJ et al. GABAa/Benzodiazepine receptors in acutely iso-lated hippocampal astrocytes. J Neurosci 1995; 15:2720-2732.
104. MacVicar BA, Tse FWY, Crichton SA et al. GABA-activated Cl– channels in astrocytes inhippocampal slices. J Neurosci 1989; 9(10):3577-3583.
105. Porter JT, McCarthy KD. GFAP-positive hippocampal astrocytes in situ respond toglutamatergic neuroligands with increases in [Ca2+]i.. Glia 1995; 13:101-112.
106. Porter JT, McCarthy KD. Adenosine receptors modulate [Ca2+]i in hippocampal astrocytesin situ. J Neurochem 1995; 65:1515-1523.
107. Kirischuk S, Möller T, Voitenko N et al. ATP-induced cytoplasmic calcium mobilizationin Bergmann glial cells. J Neurosci 1995; 15:7861-7871.
108. Kirischuk S, Tuschick S, Verkhratsky A et al. Calcium signaling in mouse Bergmann glialcells mediated by a1-adrenoreceptors and H1 histamine receptors. Eur J Neurosci 1996;8:1198-1208.
109. Muller T, Moller T, Berger T et al. Calcium entry through kainate receptors and resultingpotassium-channel blockade in Bergmann glial cells. Science 1992; 256:1563-1566.
110. Gallo V, Patneau DK, Mayer ML et al. Excitatory amino acid receptors in glial progenitorcells: Molecular and functional properties. Glia 1994; 11:94-101.
111. Seifert G, Steinhäuser C. Glial cells in the mouse hippocampus express AMPA receptorswith an intermediate Ca2+ permeability. Eur J Neurosci 1995; 7:1872-1881.
112. Aoki C, Go C-G, Venkatesan C et al. Perikaryal and synaptic localization of α2A-adrenergicreceptor immunoreactivity in brain as revealed by light and electron microscopic immu-nocytochemistry. Brain Res 1994; 650:181-204.
113. Aoki C, Venkatesan C, Go C-G et al. Cellular and subcellular localization of NMDA-R1subunit immunoreactivity in the visual cortex of adult and neonatal rats. J Neurosci 1994;14:5202-5222.
114. Whitaker-Azmitia PM, Clarke C, Azmitia EC. Localization of 5-HT1A receptors to astroglialcells in adult rats: Implications for neuronal-glial interactions and psychoactive drug mecha-nism of action. Synapse 1993; 14:201-205.
115. Van den Pol AN, Romano C, Ghosh P. Metabotropic glutamate receptor mGluR5 subcel-lular distribution and developmental expression in hypothalamus. J Comp Neurol 1995;362:134-150.

Astrocytes in Brain Aging and Neurodegeneration36
116. Jeffery G, Sharp C, Malitschek B et al. Cellular localization of metabotropic glutamate re-ceptors in the mammalian optic nerve: A mechanism for axon-glia communication. BrainRes 1996; 741:75-81.
117. Petralia RS, Wang YX, Zhao HM et al. Ionotropic and metabotropic glutamate receptorsshow unique postsynaptic, presynaptic, and glial localizations in the dorsal cochlear nucleus.J Comp Neurol 1996; 372:356-383.
118. Van der Zee EA, De Jong GI, Strosberg AD et al. Muscarinic acetylcholine receptor-ex-pression in astrocytes in the cortex of young and aged rats. Glia 1993; 8:42-50.
119. Paspalas CD, Papadopoulos GC. Ultrastructural relationships between noradrenergic nervefibers and non-neuronal elements in the rat cerebral cortex. Glia 1996; 17:133-146.
120. Petralia RS, Wang YX, Niedzielski AS et al. The metabotropic glutamate receptors, mGluR2and mGluR3, show unique postsynaptic, presynaptic and glial localizations. Neurosci 1996;71:949-976.
121. Luque JM, Richards JG. Expression of NMDA 2B receptor subunit mRNA in Bergmannglia. Glia 1995; 13:228-232.
122. Shigemoto R, Nakanishi S, Mizuno N. Distribution of the mRNA for a metabotropicglutamate receptor (mGluR1) in the central nervous system: An in situ hybridization studyin adult and developing rat. J Comp Neurol 1992; 322:121-135.
123. Yokoi M, Kobayashi K, Manabe T et al. Impairment of hippocampal mossy fiber LTD inmice lacking mGluR2. Science 1996; 273:645-647.
124. Kay AR, Wong RKS. Calcium current activation kinetics in isolated pyramidal neurones ofthe CA1 region of the mature guinea-pig hippocampus. J Physiol (Lond) 1987; 392:603-616.
125. Tse FW, Fraser DD, Duffy S et al. Voltage-activated K+ currents in acutely isolated hip-pocampal astrocytes. J Neurosci 1992; 12(5):1781-1788.
126. Duffy S, MacVicar BA. Adrenergic calcium signaling in astrocyte networks within the hip-pocampal slice. J Neurosci 1995; 15:5535-5550.
127. Bowman CL, Kimelberg HK. Excitatory amino acids directly depolarize rat brain astro-cytes in primary culture. Nature 1984; 311:656-659.
128. Kettenmann H, Schachner M. Pharmacological properties of gamma-aminobutyric acid-,glutamate- and aspartate-induced depolarizations in cultured astrocytes. J Neurosci 1985;5:3295-3301.
129. Sontheimer H, Kettenmann H, Backus KH et al. Glutamate opens Na+/K+ channels in cul-tured astrocytes. Glia 1988; 1:328-336.
130. Muller T, Moller T, Neuhaus J et al. Electrical coupling among Bergmann glial cells andits modulation by glutamate receptor activation. Glia 1996; 17:274-284.
131. Van den Pol AN, Finkbeiner S, Cornell-Bell AH. Calcium excitability and oscillations insuprachiasmatic nucleus neurons and glia in vitro. J Neurosci 1992; 12:2648-2664.
132. Cornell-Bell AH, Finkbeiner SM., Cooper MS. Glutamate induces calcium in cultured as-trocytes: Long-range glial signaling. Science 1990; 247:470-473.
133. Pasti L, Volterra A, Pozzan T et al. Intracellular calcium oscillations in astrocytes: A highlyplastic, bidirectional form of communication between astrocytes and neurons in situ.J.Neurosci 1997; 17:7817-7830.
134. Winder DG, Ritch PS, Gereau RW et al. Novel glial-neuronal signaling by coactivation ofmetabotropic glutamate and β-adrenergic receptors in rat hippocampus. J Physiol (Lond)1996; 494:743-755.
135. Narumi S, Kimelberg HK, Bourke RS. Effects of norepinephrine on the morphology andsome enzyme activities of primary monolayer cultures from rat brain. J Neurochem 1978;31:1479-1490.
136. Shain W, Forman DS, Madelian V et al. Morphology of astroglial cells is controlled bybeta-adrenergic receptors. J Cell Biol 1987; 105:2307-2314.
137. Diamond MC, Law F, Rhodes H et al. Increases in cortical depth and glia numbers in ratssubjected to enriched environment. J Comp Neurol 1966; 128:117-126.
138. Matsutani S, Leon M. Elaboration of glial cell processes in the rat olfactory bulb associatedwith early learning. Brain Res 1993; 613:317-320.

37Functions of Astrocytes
139. Kimelberg HK, Jalonen TO, Aoki C, McCarthy K. Transmitter receptor and uptake sys-tems in astrocytes and their relation to behaviour. In: Laming PR, Sykova E, ReichenbachA, Hatton G, Bauer H. Glial Cells: Their Role in Behaviour. Cambridge: Cambridge Uni-versity Press 1998; 6:107-129.
140. Rapoport SI. Blood-Brain Barrier in Physiology and Medicine. New York: Raven Press,1976.
141. Risau W. Molecular biology of blood-brain barrier ontogenesis and function. Acta Neurochir1994; 60:109-112.
142. Risau W. Differentiation of endothelium. FASEB J 1995; 9:926-933.143. Goldstein GW, Betz AL. Blood-brain barrier. Scientific Amer 1986; 255:74-83.144. Jacobson M. Histogenesis and morphogenesis of the central nervous system. In: Jacobson
M, ed. Developmental Neurobiology. New York: Plenum Press, 1978:57-114.145. Stewart PA, Wiley MJ. Developing nervous tissue induces formation of blood-brain barrier
characteristics in invading endothelial cells: A study using quail-chick transplantation chi-meras. Develop Biology 1981; 84:183-192.
146. Janzer RC, Raff MC. Astrocytes induce blood-brain barrier properties in endothelial cells.Nature 1987; 325:253-257.
147. Tao-Cheng J, Nagy Z, Brightman MW. Tight junction of brain endothelium in vitro areenhanced by astroglia. J Neurosci 1987; 7:3293-3299.
148. DeBault LE, Cancilla PA. γ-Glutamyl transpepitidase in isolated brain endothelial cells:Induction by glial cells in vitro. Science 1980; 207:653-655.
149. Cancilla PA, DeBault LE. Neutral amino acid transport properties of cerebral endothelialcells in vitro. J Neuropathol Exp Neurol 1997; 191-199.
150. Boado RJ, Wang L, Pardridge WM. Enhanced expression of the blood-brain barrier GLUT1glucose transporter gene by brain-derived factors. Mol Brain Res 1994; 22:259-267.
151. Dehouck M-P, Meresse S, Delorme P et al. An easier, reproducible, and mass-productionmethod to study the blood-brain barrier in vitro. J Neurochem 1990; 54:1798-1801.
152. Lobrinus JA, Juillerat-Jeanneret L, Darekar P et al. Induction of the blood-brain barrierspecific HT7 and neurothelin epitopes in endothelial cells of the chick chorioallantoic ves-sels by a soluble factor derived from astrocytes. Dev Brain Res 1992; 70:207-211.
153. Tagami M, Yamagata K, Fujino H et al. Morphological differentiation of endothelial cellsco-cultured with astrocytes on type-I or type-IV collagen. Cell and Tissue Res 1992;268:225-232.
154. Brightman MW. Implications of astroglia in the blood-brain barrier. Ann NY Acad Sci1991; 633:343-347.
155. Brightman MW, Reese TS, Olson Y et al. Morphological aspects of the blood-brain barrierto peroxidase in elasmobranchs. Progr Neuropathol 1971; 1:146-161.
156. Bundgaard M, Cserr HF. A glial blood-brain barrier in elasmobranchs. Brain Res 1981;226:61-73.
157. Rutten MJ, Hoover RL, Karnovsky MJ. Electrical resistance and macromolecular perme-ability of brain endothelial monolayer cultures. Brain Res 1987; 425:301-310.
158. Holash JA, Noden DM, Stewart PA. Re-evaluating the role of astrocytes in blood-brainbarrier induction. Dev Dynamics 1993; 197:14-25.
159. Abbott NJ, Raff MC. Glial-neuronal interaction. Preface. Ann N Y Acad Sci 1991; 633:xiii-xv.160. Fierz W, Endler B, Reske K et al. Astrocytes as antigen presenting cells: I. Induction of Ia
antigen expression on astrocytes by T cells via immune interferon and its effect on antigenpresentation. J Immunol 1990; 134:3785-3793.
161. Fontana A, Erb P, Pircher H et al. Astrocytes as antigen-presenting cells. Part II: UnlikeH-2K-dependent cytotoxic T cells H-2Ia-restricted T cells are only stimulated in the pres-ence of interferon-gamma. J Neuroimmunol 1986; 12:15-28.
162. Schnyder B, Weber E, Fierz W et al. On the role of astrocytes in polyclonal T cell activa-tion. J Neuroimmunol 1986; 10:209-218.
163. Benveniste EN. Astrocyte-microglia interactions. In: Murphy S, ed. Astrocytes: Pharmacol-ogy and Function. San Diego: Academic Press, 1993:355-382.
164. Graeber MB, Kreutzberg GW, Streit WJ. Microglia. Glia 1993; 7:2-118.

Astrocytes in Brain Aging and Neurodegeneration38
165. Giulian D. Interleukin-1 injected into mammalian brain stimulates astrogliosis andneovascularization. J Neurochem 1988; 8:2485-2490.
166. Fontana A, Kristensen F, Dubs R et al. Production of prostaglandin E and interleukin 1-likefactors by cultured astrocytes and C-6 glioma cells. J Immunol 1982; 129:2413-2419.
167. Fontana A, Frei K, Bodmer S et al. Immune-mediated encephalitis: On the role of antigenpresenting cells in brain tissue. Immunol Rev 1987; 100:185-201.
168. Frei K, Bodmer S, Schwerdel C et al. Astrocytes of the brain synthesize interleukin 3-likefactors. J Immunol 1985; 135:4044-4047.
169. Malpiero UV, Frei K, Fontana A. Production of hemopoietic colony-stimulating factors byastrocytes. J Immunol 1990; 144:3816-3821.
170. Wong GHW, Barlett PF, Clark-Lewis I et al. Inducible expression of H-2 and Ia antigenson brain cells. Nature 1984; 310:688-691.
171. Frohman EM, Frohman TC, Dustin ML et al. The induction of intracellular adhesionmolecule 1 (ICAM-1) expression on human fetal astrocytes by interferon-γ tumor necrosisfactor-α, lymphotoxin, and interleukin-1: Relevance to intracerebral antigen presentation.Neuroimmunol 1989; 23:117-124.
172. Chung IY, Benveniste EN. Tumor necrosis factor-α production by astrocytes: Induction bylipopolysaccharide, IFN-gamma, and IL-1β. J Immunol 1990; 144:2999-3007.
173. Benveniste EN, Sparacio SM, Morris JG et al. Induction and regulation of interleukin-6gene expression in rat astrocytes. J Neuroimmunol 1990; 30:201-212.
174. Aloisi F, Borsellino G, Caré A et al. Cytokine regulation of astrocyte function: In-vitrostudies using cells from the human brain. Int J Dev Neurosci 1995; 13:265-274.
175. Nathan C. Nitric oxide as a secretory product of mammalian cells. FASEB 1992; 6:3051-3064.176. Nottet HSLM, Gendelman HE. Unraveling the neuroimmune mechanisms for the HIV-1
associated cognitive/motor complex. Immunol Today 1995; 16:441-448.177. Garland JM. Colony stimulating factors. In: Thomson A, ed. The Cytokine Handbook. San
Diego: Academic Press, 1991:269-300.178. Mucke L, Eddleston M. Astrocytes in infectious and immune-mediated diseases of the cen-
tral nervous system. FASEB 1993; 7:1226-1232.179. Tweardy DJ, Mott PL, Glazer EW. Monokine modulation of human astroglial cell produc-
tion of granulocyte colony-stimulation factor and granulocyte-macrophage colony-stimu-lating factor. 1. Effects of IL-1 alpha and IL-1 beta. J Immunol 1990; 144:2233-2241.
180. Wahl SM, Allen JB, McCartney-Francis N et al. Macrophage- and astrocyte-derived trans-forming growth factor beta as a mediator of central nervous system dysfunction in ac-quired immune deficiency syndrome. J Exp Med 1991; 173:981-991.
181. Benos DJ, Hahn BH, Bubien JK et al. Envelope glycoprotein gp120 of human immunode-ficiency virus type 1 alters ion transport in astrocytes: Implications for AIDS dementiacomplex. Proc Natl Acad Sci USA 1994; 91:494-498.
182. Ciardo A, Meldolesi J. Effects of the HIV-1 envelope glycoprotein gp 120 in cerebellarcultures. [Ca2+] increases in glial cell subpopulation. Euro J Neurosci 1993; 5:1711-1718.
183. Levi G, Patrizio M, Bernardo A et al. Human immunodeficiency virus coat protein gp120inhibits the β-adrenergic regulation of astroglial and microglial functions. Proc Natl AcadSci U S A 1993; 90:1541-1545.
184. Pulliam L, West D, Haigwood N et al. HIV-1 envelope gp 120 alters astrocytes in humanbrain cultures. AIDS Res Hum Retrovirus 1993; 9:439-444.
185. Schneider-Schaulies J, Schneider-Schaulies S, Brinkman R et al. HIV-1 and gp 120 recep-tor on CD-4-negative brain cells activates a tyrosine kinase. Virology 1992; 191:765-772.
186. Rivers TM, Sprunt DH, Berry GP. Observations on attempts to produce acute dissemi-nated encephalomyelitis in monkeys. J Exp Med 1933; 58:39-53.
187. Ransohoff RM, Hamilton TA, Tani M et al. Astrocyte expression of mRNA encodingcytokines IP-10 and JE/MCP-1 in experimental autoimmune encephalomyelitis. FASEB J1993; 7:592-600.
188. Benveniste EN. The role of cytokines in multiple sclerosis/autoimmune encephalitis andother neurological disorders. In: Agrawal B, Puri R, eds. Human Cytokines, Their Role inResearch and Therapy. Boston: Blackwell Science Publications, 1995:195-216.

39Functions of Astrocytes
189. Sun N, Grzybicki D, Castro RF et al. Activation of astrocytes in the spinal cord of micechronically infected with a neurotropic coronavirus. Virology 1995; 213:482-493.
190. Erb P, Kennedy M, Hagmann I et al. Accessory cells and the activation and expression ofdifferent T cell functions. In: Feldmann M, McMichael A, eds. Regulation of Immune GeneExpression. Clifton: The Humana Press, 1986:187
191. Fontana A, Fierz W, Wekerle H. Astrocytes present myelin basic protein to encephalitoge-nic T-cell lines. Nature 1984; 307:273-276.
192. Hirsch M-R, Wietzerbin J, Pierres M et al. Expression of Ia antigens by cultured astrocytestreated with gamma-interferon. Neurosci Lett 1988; 41:199-204.
193. Giulian D, Baker TJ. Characterization of ameboid microglia isolated from developing mam-malian brain. Neurosci 1986; 6(8):2163-2178.
194. Vass K, Lassmann H. Intrathecal application of interferon gamma. Progressive appearanceof MHC antigens within the rat nervous system. Am J Pathol 1990; 137:789-800.
195. Lampson LA, Hickey WF. Monoclonal antibody analysis of MHC expression human brainbiopsies: Tissue ranging from “histologically normal” to that showing different levels ofglial tumor involvement. J Immunol 1986; 136:4054-4062.
196. Giulian DJ, Baker TJ, Shih L-CN et al. Interleukin 1 of the central nervous system is pro-duced by ameboid microglia. J Exp Med 1986; 164:594-604.
197. Williams K, Bar-Or A, Ulvestad E et al. Biology of adult human microglia in culture: Com-parisons with peripheral blood monocytes and astrocytes. J Neuropath Exp Neurol 1992;51:538-549.
198. Matsumoto Y, Ohmori K, Fujiwara M. Immune regulation by brain cells in the centralnervous system: Microglia but not astrocytes present myelin basic protein to encephalito-genic T cells under in vivo-mimicking conditions. Immunol 1992; 76:209-216.


CHAPTER 3
Astrocytes in Brain Aging and Neurodegeneration, edited by Hyman M. Schipper.©1998 R.G. Landes Company.
Astrocyte Pathophysiologyin Disorders of the CentralNervous SystemMichael D. Norenberg
Introduction
Astrocytes play a prominent role in the central nervous system (CNS) response to injury.These responses may be useful in restoring the integrity of the CNS microenvironment
as well as in promoting reparative and regenerative events. On the other hand, some astro-cytic actions may harm the CNS and possibly impede regeneration. This article will con-sider the contributions of astrocytes to neurologic disease in the context of passive andactive glial responses: Astrocytes may be injured and become incapable of carrying out theirnormal function, resulting in a gliopathy (i.e., passive response), or ostensibly “healthy”astrocytes may secrete potentially harmful molecules and thus play an active role in CNSdisorders.
Normal FunctionsA growing body of evidence indicates that astrocytes are involved in many functions
that are critical to the CNS. Their activities involve important interactions with neurons,1
oligodendrocytes,2 microglia,3 and endothelial cells.4 A particularly important function isthe maintenance and regulation of the extracellular environment. Such actions include buff-ering of K+, H+ and Ca2+ and osmoregulation.5 While most studies have employed cell cul-ture methods to examine glial function, recent in vivo investigations utilizing the selectivegliotoxin fluoroacetate have given added support for the critical role of astrocytes in themaintenance of the extracellular environment.6,7 Other putative astroglial functions includeneurotransmitter and neuromodulator uptake and release;8,9 regulation of synaptic trans-mission and neuronal excitability;10 provision of nutrients, energy substrates and neurotrans-mitter precursors;11,12 neurotrophism;13 metabolism and detoxification of ammonia, drugsand hormones;14,15 free radical scavenging;16 metal sequestration;17 development and main-tenance of the blood-brain barrier;18 guidance of neuronal migration during development;19
and immune/inflammatory functions.20
The astrocyte uptake of the excitatory neurotransmitter glutamate is a particularly criti-cal astrocytic function.21 It not only serves to recycle glutamate, regulate glutamatergic neu-rotransmission and detoxify ammonia, but it is also necessary to avoid excitotoxic injury.22

Astrocytes in Brain Aging and Neurodegeneration42
Glial uptake of glutamate is achieved by powerful transporters. Three glutamate transport-ers have been cloned: GLT-1,23 GLAST,24 EAAC1.25 In situ hybridization26 and immunohis-tochemical studies27-29 indicate that GLT-1 is strictly astrocytic, GLAST is mostly glial butalso found in neurons, while EAAC1 is chiefly neuronal. Astrocyte cultures express chieflythe GLAST transporter.30 A defect in astrocyte glutamate transport contributes to the patho-genesis of several neurological disorders (see below).
General Response to Injury
Reactive AstrocytosisGlial transformation to reactive astrocytes (gemistocytes) represents one of the earliest
and dramatic responses of the CNS to tissue injury. Reactive astrocytosis (gliosis, astrogliosis)occurs following physical, chemical, ischemic, infectious, immunologic and neuro-degen-erative disorders.This response is characterized by cellular hypertrophy and a profusion ofnew, thicker and longer cytoplasmic processes. The end product of gliosis is often referredto as a “glial scar”.
Electron microscopic findings show changes consistent with enhanced metabolic ac-tivity, i.e., increased numbers of mitochondria and ribosomes, enlarged Golgi complexesand increased amounts of glycogen.31 A most striking change is the accumulation of bundlesof 10 nm intermediate glial filaments, composed chiefly of glial fibrillary acidic protein(GFAP) and vimentin.32 Overexpression of GFAP is currently the most commonly usedindicator of reactive astrocytosis.
The reactive astrocyte produces a wide array of molecules including growth factors,extracellular matrix molecules (glycoproteins and proteoglycans), adhesion molecules,β-amyloid precursor protein (APP), proteases, protease inhibitors, and immune/inflam-matory molecules (MHC molecules, cytokines, chemokines). Additionally, many enzymesare upregulated; those particularly related to CNS disorders include the inducible form ofnitric oxide synthase, monoamine oxidase-B, superoxide dismutase, catalase, gluta-thione-S-transferase (an enzyme involved in the detoxification of various xenobiotics),kynurenine aminotransferase, 3-hydroxyanthranilic acid oxygenase, and quinolinic acidphosphoribosyltransferase. For reviews on factors produced by reactive astrocytes seerefs. 33, 34.
The morphologic changes of reactive astrocytes are those of metabolically activatedcells. The precise significance of this activation is uncertain and there is considerable con-troversy regarding its beneficial or deleterious effect on the CNS. One might predict thatearly stages of reactive astrocytosis may be involved in the restoration of the ionic milieu,provision of nutrients, removal of toxins (including excitotoxins such as glutamate), freeradical scavenging, and metal sequestration. Later, astroglial responses are perhaps gearedtowards promoting repair and regeneration. Nevertheless, the dominant view until recentlywas that gliosis created an inhospitable, non-permissive environment for neurite outgrowth,interfered with remyelination, and possibly disturbed neuronal circuitry leading to seizures.These issues will be covered below.
Astrocyte SwellingAstrocyte swelling represents one of the earliest pathological features of most CNS
injuries, and at times may be the only abnormality found. It occurs following ischemia,trauma, hypoglycemia, status epilepticus, hypotonicity, and fulminant hepatic failure. It isusually best seen in the perivascular astrocytic endings, possibly because of the greater numberof transport systems at that site. Various factors have been implicated in the mechanism ofswelling, including osmotic changes, abnormalities in ion transport, and excessive concen-

43Astrocyte Pathophysiology in Disorders of the Central Nervous System
trations of glutamate, lactic acid, arachidonic acid, potassium, free radicals, and ammonia.For reviews on glial swelling see ref. 35.
Astrocyte swelling may lead to increased intracranial pressure and its associated compli-cations. Swelling may also impair astrocyte integrity and function. Swollen glia depolarizeand thus lose their ability to maintain the necessary ionic gradients for the uptake of glutamateand other amino acids. Moreover, swollen astrocytes release K+ and glutamate, which mayresult in changes in the level of excitability and contribute to excitotoxicity.36,37 The reduc-tion in the size of the extracellular space following astrocyte swelling may also elevate extra-cellular ionic concentrations, which could affect neuronal excitability.38 Astrocyte swellingmay also compress capillaries, contributing to a reduction in cerebral blood flow.39 Ulti-mately, when swelling is severe, the cell membrane may rupture, resulting in cell death.
Astrocyte swelling also causes the release of large amounts of taurine.36 While suchrelease is likely to help restore cell volume (due to loss of an osmolyte), taurine hasneuroinhibitory effects which can affect the state of neuronal excitability.40 Whether thisreduction in excitability is useful or not is difficult to predict. Taurine may also exert aneuroprotective effect,41 possibly through its antioxidant properties.42
Alzheimer Type II ResponseAlzheimer type II astrocytes are seen in a variety of metabolic encephalopathies in-
cluding hepatic encephalopathy (HE), uremia, hypercapnia and the early stages of anoxiaand hypoglycemia, especially in infants (for review see ref. 43). This change often occurs inthe setting of elevated brain or blood ammonia. The process is occasionally referred to asprotoplasmic astrocytosis or metabolic gliosis. Alzheimer type II astrocytes have enlarged,pale nuclei with peripheral margination of chromatin and often prominent nucleoli. Inexperimental models of HE, Alzheimer type II astrocytes contain increases in mitochon-dria, smooth and rough endoplasmic reticulum, and cytoplasmic glycogen. Eventually, de-generative changes characterized by hydropic changes, cytoplasmic vacuoles and degener-ated mitochondria are observed.44 The initial change in the Alzheimer type II astrocytesuggests that it is a metabolically active cell responding to a perturbation in the extracellularmilieu (presumably to excessive levels of ammonia). The later changes are indicative of cellinjury. Culture studies, and more recently in vivo studies, have identified various functionaldeficits that may contribute to the encephalopathy of HE and related conditions (see below).
Injury to Astrocytes in CNS Disorders (Passive Role)Although astrocytes are more resistant to various CNS insults than neurons and oligo-
dendrocytes, they are nevertheless vulnerable to many injurious processes and may evenundergo necrosis. Injury or death of astrocytes can cause severe impairment in the regula-tion of extracellular potassium concentrations, amino acid levels, and extracellular pH.7
Astrocytes may occasionally be the primary target of injury (primary gliopathy). Hepaticencephalopathy, associated with the Alzheimer type II response, is probably the best ex-ample of such a process.
Many factors are released in injured brain, including lactic acid, potassium, arachi-donic acid, ammonia, free radicals, glutamate, nitric oxide and cytokines.45 All of these fac-tors negatively impact on glial function.
Elevation of extracellular K+ occurring in tissue injury leads to astrocyte depolariza-tion,46 intracellular alkalinization,47 and extracellular acidification.48 Astrocytic depolariza-tion diminishes the ability of astrocytes to take up glutamate.49 Additional effects of potas-sium on astrocytes include increased glycogenolysis,50,51 which may contribute to thegeneration of lactic acid. Elevated K+ may also be a factor in glial swelling.52

Astrocytes in Brain Aging and Neurodegeneration44
The release of glutamate and associated excitotoxicity contributes to the pathogenesisof ischemia, trauma, hypoglycemia and various neurodegenerative conditions.53,54 Glutamatemay also have profound effects on astrocytes, including depolarization, swelling, activationof phosphoinositide hydrolysis, increase in intracellular Ca2+, generation of calcium waves,morphologic changes, stimulation of glucose utilization and lactate release, enhanced gly-cogen synthesis, decreased cell proliferation, production of growth factors, protooncogeneand transcription factor expression, inhibition of MHC class II expression, and the releaseof GABA, glutamate, aspartate, glycine, taurine, alanine, and serine.55,56
Lactic acid causes injury to astrocytes,57,58 contributes to glial swelling,59,60 and impairsthe astrocyte’s capacity to take up glutamate.61,62 Arachidonic acid is a potent inhibitor ofglial glutamate uptake and a cause of glial swelling,63,64 and free radicals are have been shownto inhibit glutamate uptake by astrocytes65,66 and to cause glial swelling.63,64
Active Role of Astrocytes in CNS DisordersThis section reviews the possibility that in some circumstances astrocytes actively pro-
duce compounds that are potentially harmful to the CNS.
ExcitotoxinsAstrocytes have been shown to release glutamate and/or aspartate in the following con-
ditions: treatment with kainic acid and depolarization with high concentrations of K+;67 ina culture model of hypoxia/ischemia;68 following inhibition of glycolysis;69,70 and after treat-ment with the HIV-1 coat protein gp120,71 mercuric chloride,72 trimethyltin,73 and alumi-num.74 Astrocyte swelling can also cause glutamate to be released into the extracellular space.36
Such increases in extracellular glutamate may contribute to excitotoxic injury.Quinolinic acid, a tryptophan metabolite with excitotoxic properties, is synthesized in
the CNS by glial cells. While the majority of studies indicate that astrocytes are the cellsinvolved in this process,75 evidence for a microglial source is also available.76 The syntheticenzyme 3-hydroxyanthranilic acid oxygenase is upregulated following tissue injury.77 Inap-propriate release of quinolinic acid may contribute to excitotoxic injury. However, the samemetabolic pathway that generates quinolinic acid also generates kynurenic acid, which hasinhibitory effects on glutamate receptors.78 The net effect of this dual release is unknown.Quinolinic acid may play a role in AIDS encephalopathy (see below), seizures and variousneurodegenerative diseases.54,75
Glycine, a known activator of the NMDA receptor, can be synthesized and released byastrocytes after treatment with kainic acid, quisqualate and high concentrations of K+.67,79,80
GlutamineAstrocytes are well known to synthesize and release glutamine.81,82 Simantov showed
that in mixed CNS cultures, high concentrations of glutamine are toxic.83 The potentialtoxicity of glutamine is likely due to its being the principal precursor of glutamate.84
Glutamine may additionally contribute to excitotoxicity through its inhibition of kynure-nine aminotransferase,85 thereby preventing the synthesis of kynuretic acid, a glutamatereceptor antagonist. The neuroprotective effect of methionine sulfoximine, an inhibitor ofglutamine synthetase which is mainly found in astrocytes,86 could be due to its ability toabrogate the potential excitotoxic properties of glutamine.87
Lactic AcidLactic acid can be released from astrocytes as a result of stimulation of anaerobic gly-
colysis. The almost exclusive localization of glycogen to astrocytes may contribute to thisrelease.88 Lactic acid may result in extracellular acidosis and contribute to tissue injury.89

45Astrocyte Pathophysiology in Disorders of the Central Nervous System
Recent studies have additionally shown that it reduces astrocytic glutamate uptake,61,62 pos-sibly contributing to excitotoxicity. It also causes glial swelling.90 The deleterious effects oflactic acid, however, may be partially offset by the fact that it is a source of fuel for neighbor-ing neurons,11,91 and has additionally been found to reduce NMDA receptor activation.92
Nitric OxideNitric oxide (NO) contributes to normal physiological processes,93 including those in
brain.94 While NO plays a role in host defense,95,96 it may also contribute to tissue injury andmay contribute to the pathogenesis of various neurological diseases.93,97 NO is synthesizedfrom L-arginine by nitric oxide synthase (NOS). Astrocytes possess both the constitutiveand inducible forms of NOS.98
The inducible form of NOS has been shown to be elevated in reactive astrocytes fol-lowing CNS injury.99 The significance of this increase is uncertain. NO possesses cytotoxicproperties that could contribute to neuronal death.93,97 However, because of its vasodilatingeffect, NO may also improve blood flow.100 While microglia are the major producers of NOfollowing injury, astrocytes are the principal source of NO in humans.99
Free RadicalsIn general, it appears that astrocytes serve a protective role in mitigating the actions of
free radicals.16 In certain sites, such as the hypothalamus, there is evidence that astrocytesmay be a source of free radicals.101
Extracellular Matrix Molecules (ECM)Proteoglycans are complex molecules consisting of a protein core to which chains of
glycosaminoglycans are covalently bound. Sulfate groups confer a high anionic charge tothese molecules. Astrocytes have been shown to synthesize chondroitin sulfate proteoglycans,dermatan sulfate proteoglycan, and heparan sulfate proteoglycans.102,103 Proteoglycans ap-pear to stimulate neurite outgrowth, guidance and remodeling during development.104,105
Despite these beneficial actions of ECMs, astrocyte-derived proteoglycans have been shownto inhibit neurite outgrowth, to contribute to the development of neuritic plaques inAlzheimer’s disease, and to play a role in establishing the epileptic focus (see below).
Various astrocyte-derived glycoproteins such as tenascin,102,106,107 hyaluronate-bindingprotein,108 Thy-1 glycoprotein,109 and other as yet unidentified proteins110 have been shownto exert a repulsive action on neurite outgrowth and may contribute to aberrantsynaptogenesis associated with the epileptic lesion (see below).
Inflammatory/Immune MoleculesThe inflammatory response contributes to the destruction of microorganisms and re-
moval of necrotic debris (phagocytosis), which sets the scene for appropriate reparativeresponses. CNS inflammation is associated with the activation of microglia, recruitment ofblood monocytes and neutrophils and increased vascular permeability. However, anoverreactive inflammatory response may also be deleterious. Indeed, a fine balance is atwork and the outcome of the inflammatory response may be difficult to predict.111
It has become clear that astrocytes contribute to immune/inflammatory phenomena.20
Various cytokines have been identified as products of astrocytes including IL-1β, IL-3, IL-6and TNF-α (for reviews see refs. 112, 113). Some cytokines may contribute to tissue injuryin AIDS, multiple sclerosis and experimental allergic encephalomyelitis (see below).
Chemokines are recently described molecules that potently recruit inflammatory cellsfollowing injury and play a key role in wound healing.114 In the CNS they are made mostlyby astrocytes and are upregulated in astrocytes following treatment with various

Astrocytes in Brain Aging and Neurodegeneration46
cytokines.115-117 Among these chemokines include monocyte chemoattractant protein-1α(MCP-1), a chemoattractant and stimulator of monocytes and macrophage inflammatoryprotein 1 (MIP-1), which is strongly chemotactic for neutrophils and other leukocytes.
Class II major histocompatability (MHC) antigens are critical molecules involved inantigen presentation and are vital in the initiation of immune responses. These moleculeshave been identified on astrocytes,118-121 although this matter is controversial.122,123
The protease cathepsin G is found in reactive astrocytes after trauma.124 Proteases maybe beneficial in the destruction of toxic cytokines, neurite extension, remodeling of extra-cellular matrix, chemotaxis and hemostasis.125-127 They may also contribute to tissue dam-age and to the accumulation of β-amyloid.128 Matrix metalloproteinases can also be pro-duced by astrocytes;129,130 these proteases break down connective tissue and have beenimplicated in ischemia, multiple sclerosis, and amyotrophic lateral sclerosis.131
α2-Macroglobulin, a broad spectrum protease inhibitor that can be synthesized by as-trocytes,132,133 is an acute phase protein that acts to eliminate proteases and cytokines frominflammatory processes. It appears to play a role in development through an interactionwith plasminogen activator and other proteases. Another protease inhibitor, α1-antichymotrypsin, appears to be involved in the genesis of neuritic plaques in Alzheimer’sdisease (see below).
Adhesion molecules are involved in cell-cell interaction, cell migration, neurite out-growth and guidance, synaptogenesis, synaptic reorganization, and myelination.134 Adhe-sion molecules also possess potent chemotactic properties for microglia and/or leukocytes.Excessive recruitment of these inflammatory cells may produce further tissue injury. Theintracellular adhesion molecule, ICAM-1, has been found on astrocytes and can beupregulated in these cells by IFN-γ and TNF-α.121,135,136 A preliminary study has also identi-fied vascular adhesion molecule-1 in astrocytes.137 Adhesion molecules may play a role inregeneration, inflammatory disorders, Alzheimer’s disease and in the production of seizurefoci (see below).
Astrocyte-Microglial InteractionsMicroglia are the resident histiocytes of the CNS and are the principal cells involved in
inflammatory and immunological responses as well as in phagocytosis. Microglia releasecertain factors that are potentially harmful to the CNS including nitric oxide, superoxideanions, reactive oxygen radicals, and excitotoxins. Microglia also synthesize and releasecytokines as well as various proteases. Interestingly, astrocytes appear capable of counter-acting the harmful effects of microglia138,139 (see refs. 140-142 for review).
Important interactions occur between astrocytes and microglia/macrophages. Micro-glia are a prime source of gliotic mediators (IL-1β, TNF-α, IL-6). Inhibiting microglia withcolchicine decreases the extent of astrogliosis.143 Microglial-derived IL-1 stimulates nervegrowth factor production by astrocytes,144,145 possibly contributing to regeneration. In turn,astrocytes are the principal producers of granulocyte-macrophage colony stimulating fac-tor in brain (GM-CSF),146,147 which serves as a growth factor for microglia, and inducesgranulocytes and macrophages to migrate into inflammatory foci, thereby increasing theirretention and survival.148 Since GM-CSF increases viral replication in cultured HIV infectedmonocytes, it has been postulated that this cytokine increases the viral load in HIV en-cephalitis.149 Astrocytes can also stimulate microglial proliferation through their produc-tion of IL-3150 and laminin.151
While abundant amounts of extracellular matrix are deposited in gliotic tissue, not allof it necessarily comes from astrocytes. Microglia/monocytes are capable of making chon-droitin sulfate proteoglycan.152 Furthermore, macrophage/microglial release of IL-1β153 mayincrease the production of chondroitin sulfate proteoglycan by astrocytes in certain brain

47Astrocyte Pathophysiology in Disorders of the Central Nervous System
lesions. Ness and David154 have additionally shown that astrocytes cocultured with meningealcells or in the presence of conditioned media from meningeal cells inhibited neurite growth.These cocultured astrocytes had more tenascin-C and chondroitin sulfate proteoglycan andhad less laminin, a factor that supports neurite outgrowth.
Clinical Considerations
RegenerationMost axons in the vertebrate CNS fail to regenerate following injury. This failure has
been largely attributed to a “hostile terrain” due to the presence of inhibitory molecules,155
or inadequate supply of growth factors or growth promoting molecules.156 One of the im-pediments to repair and regeneration has been the deposition of a glial scar, a notion origi-nally expressed by Ramon y Cajal.157 By acting as a mechanical barrier, the glial scar couldinterfere with the process of axonal regeneration. Reactive astrocytes grown on nitrocellu-lose filters,158 as well as membranes isolated from gliotic lesions,159 have been shown to bepoor substrates for neurite outgrowth. Interestingly, this inhibition was only observed inmembranes derived from isomorphic gliosis, whereas no such inhibitory effect was ob-served in membranes derived from anisomorphic gliosis.159 As reviewed above, some of theinhibitory factors are proteoglycans and glycoproteins.
Gliosis at the root entry zone blocks regeneration of dorsal roots,160,161 possibly due tothe development of synaptic-like contacts with reactive astrocytes.162,163 Astrocytes may thusactivate a physiologic stop pathway as opposed to inhibiting neurite growth.
Before assigning too sinister a role to the astrocyte, it may be useful to remember thatastrocytes can exert a beneficial role, at least in the young or immature brain.164,165 Thegeneration of a glial scar forms a glia limitans which may isolate the lesion from the remain-ing viable tissue. Astrocytes have been shown to promote neuronal survival in culture andin vivo, protect neurons against excitotoxic and anoxic injury, and aid in neuronal repairand functional recovery. Astrocytes produce various neurotrophic factors, and provide anexcellent substrate for neurite outgrowth, mediated by a number of adhesion moleculesand extracellular matrix molecules. They are also involved in the removal of degeneratingsynapses and the provision of axonal guidance (see refs. 13, 166 for review).
While it is clear that astrocytes, and in particular young or immature astrocytes, aresupportive of neurite outgrowth, older astrocytes have a diminished capacity to do so.109,167,168
Perhaps mature cells do not provide sufficient surface molecules, extracellular matrix mol-ecules or other substances for regeneration to occur.169
It should be recalled that meninges, fibroblasts, microglia/macrophages, and oligoden-drocytes also contribute to the formation of the glial scar. Consequently, any or all of thesecells may adversely affect the outcome of regeneration. Certainly, the fibroblastic/glioticscar is impermeable to axonal growth.32,170,171 Furthermore, as noted above, microglia/mac-rophages play a major role in the deposition of proteoglycans and glycoproteins followingCNS lesions. There is also growing evidence that oligodendrocytes/myelin contribute to theinhospitable environment following CNS injury.172,173
In summary, the issue remains unresolved whether astrocytes, or particularly reactiveastrocytes, play a beneficial or detrimental role in CNS regeneration. Overall, it appears thatwhile immature astrocytes have a positive influence on neurite outgrowth, reactive astro-cytes in adults may be injurious. Although reactive astrocytes may serve to wall off an areaof injury, generate various growth promoting substances and mediate a number of homeo-static functions, excessive production of certain proteoglycans and glycoproteins appear toimpede the regeneration process.

Astrocytes in Brain Aging and Neurodegeneration48
Acquired Immunodeficiency Syndrome (AIDS)The majority of people with AIDS eventually develop neurological symptoms (cogni-
tive, motor, behavioral). Pathological changes in the CNS include inflammation, microgliosis,multinucleated giant cells, pallor and vacuolization of the white matter, cerebral atrophy,and a vacuolar myelopathy. Astrocytic gliosis is a common and early finding in HIV-1infection.174,175
The mechanism for neuronal injury is not known, as neurons appear not to be directlyinvolved by HIV, suggesting that the viral effects on neurons are indirect.176 A commonlyheld view focuses on the deleterious effects of HIV-1-infected microglia/macrophages. Thesecells are capable of releasing cytokines, nitric oxide, arachidonic acid and its metabolites,unknown neurotoxins, free radicals, and platelet-activating factor (PAF).
Important interactions between astrocytes and microglia occur in AIDS. It appearsthat astroglial cells are required to activate HIV-1-infected monocytes to produce the vari-ous neurotoxic factors.177,178 Conditioned media of lipopolysaccharide-treated astrocytesincrease HIV-1 gene expression in monocytes,179 and astrocyte-derived IL-6 promotes HIV-1replication.180 Conversely, supernatants from HIV-treated macrophages induce cultured as-trocytes to release TGF-β.181
There is evidence that astrocytes may be also be infected with HIV-1, particularly inthe pediatric age group.182-184 Astrocyte dysfunction induced by direct HIV-1 infection couldthus possibly affect CNS development.
An indirect mechanism for neuronal injury has been the involvement of the HIV-1envelope glycoprotein gp120. Pulliam and colleagues185 were the first to suggest that AIDSdementia may partially involve a perturbation of astrocyte function by gp120. These work-ers showed that treatment of human brain tissue with gp120 caused astrocyte alterationsand death. Studies on astrocyte cultures showed decreased expression of glial fibrillary acidicprotein (GFAP), as well as the diminution of a major 66 kDa phosphoprotein. Levi et alsubsequently reported that gp120 inhibits β-adrenergic regulation of astroglial and micro-glial functions.186 Other studies have also shown that gp120 interacts with microglia and/orastrocytes to release neurotoxic compounds, some of which act synergistically with glutamateto activate NMDA receptors.187
Several studies have demonstrated that gp120 also produces abnormalities in astro-cytic glutamate transport. Benos and coworkers71,188 showed that gp120 stimulated a Na+/H+
antiporter, resulting in loss of the Na+ gradient, intracellular alkalinization, activation ofoutward K+ conductance, membrane depolarization and increased glutamate efflux. Dreyerand Lipton189 showed that gp120 impairs astrocyte uptake of excitatory amino acids, andthe resulting excess glutamate may lead to neuronal damage. They suggested that the effectof gp120 on astroglial glutamate uptake may be indirect, as a consequence of a direct effectof gp120 on macrophages, which in turn releases arachidonic acid, a known inhibitor ofglutamate uptake.190-192
Quinolinic acid is a tryptophan derivative that has excitotoxic properties. A potentialrole for quinolinic acid in AIDS has been described.193 Since enzymes involved in the syn-thesis of quinolinic acid have been identified in astrocytes,75 excessive quinolinate produc-tion by astrocytes could contribute to excitotoxic damage. It should be noted, however, thatmicroglia may also be a source of quinolinic acid76 (for general reviews on astrocytes andAIDS, see refs. 194-196).
Multiple SclerosisMultiple sclerosis (MS) is an inflammatory demyelinating disease of unknown etiol-
ogy that is widely considered to have an immune pathogenesis.197 MS is associated withprominent astrocytosis and indeed the term sclerosis refers to this astrocytic response. Re-

49Astrocyte Pathophysiology in Disorders of the Central Nervous System
active astrocytes in the MS lesion (plaque) can be extremely large, are often binucleated andmay even show atypical nuclei.197 The precise role of astrocytes in MS is not clear. Someinvestigators support the view that astrocytes are capable of actively promoting demyelina-tion and suppressing remyelination,198,199 while others believe that astrocytes supportremyelination.200,201 Astrocytes may also play a role in phagocytosis of myelin debris.120,202
As discussed above, reactive astrocytes contribute to the process of inflammation byvirtue of their production of cytokines, proteases,203 MHC class II antigens, adhesion mol-ecules, and nitric oxide. Reactive astrocytes associated with MS plaques also contain sub-stance P,204 a potent mediator of vasodilatation and local immune responses.205
Experimental allergic encephalomyelitis (EAE) is an autoimmune disorder that is widelyused as an experimental model of MS.197,206 Reactive astrocytosis is a prominent his-topathological feature in EAE.207,208 As in MS, astrocytes have been shown to producecytokines, chemokines and adhesion molecules,121,209-212 all of which could contribute todemyelination.
There is controversy regarding the presence or absence of MHC class II antigens inastrocytes in EAE. As reviewed by Benveniste,113 such failure may be due to the time duringthe disease process in which they were looked for, and the fact that class II antigens are morereadily downregulated in astrocytes as compared to microglia.213,214 Furthermore, astro-cytes may be destroyed via an MHC class II restricted cytotoxicity.119
On the other hand, Massa and colleagues215 have shown that astrocytes from a suscep-tible rat strain (Lewis) express higher amounts of class II antigens in vitro after IFN or virustreatment compared to a less susceptible strain (Brown-Norway). In keeping with this ob-servation are studies showing that EAE resistant and susceptible strains of rats also differ intheir ability to express TNF.209
Despite these detrimental factors potentially contributing to demyelination, it shouldbe noted that astrocytes can promote process outgrowth by adult human oligodendrocytesin vitro through the interaction between astrocyte derived bFGF and extracellular matrixmolecules (vitronectin, fibronectin, laminin, heparan sulfate proteoglycans) (see ref. 216and references cited therein). Additionally, insulin-like growth factor is present in astro-cytes,217 is upregulated following injury218,219 and has trophic actions on oligodendrocytes.220
Alzheimer’s DiseaseReactive gliosis is a prominent finding in Alzheimer’s disease (AD),221,222 which may
actively contribute to its pathogenesis as opposed to merely representing a nonspecific reac-tion to tissue injury. Such gliosis is associated with an increase in GFAP223,224 and GFAPmRNA.225 Reactive astrogliosis is also a prominent feature of the neuritic plaque.226
Neuritic plaques represent a major component of the pathology of AD and consist ofextracellular masses of amyloid intimately associated with dystrophic neurites, activatedmicroglia and reactive astrocytes. Important advances have been made in recent years sothat a clearer picture of the evolution of plaques is beginning to emerge. Fundamental tothe initiation of the plaque is the formation of β-amyloid1-42 derived from the abnormalproteolytic processing of β-amyloid precursor protein (APP).227 The deposition of β-amy-loid (diffuse plaque) initiates a cascade of events that culminate in the formation of theneuritic plaque. An excellent review on the genesis of neuritic plaques has recently beenpublished228 and a detailed account of the role of astroglia in plaque formation is presentedin chapter 5 of this volume.
The neurotoxic β-amyloid1-42 is a 4 kDa, 42 amino acid peptide that contains a comple-ment activation domain (initiating inflammation), glycation binding sites (which contrib-ute to the recruitment of microglia) as well as binding sites for apolipoprotein E (Apo E),α1-antichymotrypsin and proteoglycans. Through the interaction with microglia and

Astrocytes in Brain Aging and Neurodegeneration50
astrocytes (see below), the soluble β-amyloid is converted to an insoluble, fibrillary proteinwith a folded, β-pleated sheet configuration which is resistant to protease digestion andstains positive with thioflavin S and Congo red (congophilia) and is neurotoxic.229,230
An important effect of β-amyloid is the activation of microglia. This activation is as-sociated with the release of proinflammatory cytokines, reactive oxygen species, proteases,acute phase proteins and other poorly defined toxic molecules,231-233 and represents a keypathogenetic step in the progression of AD. Activated microglia and β-amyloid are alsoresponsible for the reactive gliosis associated with the neuritic plaque.234,235 While reactiveastrocytes in the plaque may act to wall off the amyloid from the surrounding neuropil,226 itmay have a more sinister role. Reactive astrocytes in this setting appear to secrete a numberof molecules that are critical to the formation of neuritic plaques (see below) as well asvarious toxic compounds (cytokines, free radicals, nitric oxide, proteases).
Reactive astrocytes in AD express elevated levels of α1-antichymotrypsin (ACT),236,237
probably secondary to microglial-derived IL-1β and TNF-α. ACT is an acute phase protein,serine protease inhibitor, capable of inhibiting cathepsin G and chymotrypsin that nor-mally prevent excessive proteolysis during inflammation. ACT is intimately associated withβ-amyloid;238,239 this association appears to promote the assembly of β-amyloid into fila-ments and contributes to the resistance of β-amyloid to inflammatory protease damage.240-242
Similarly, astrocytes are a source of proteoglycans (see above) which are found in neuriticplaques.243,244 Proteoglycans induce β-amyloid to form insoluble β-pleated structures (fibrils),and protect β-amyloid from proteolytic degradation.245-247
Apolipoprotein E (Apo E) is normally involved in the transport (recycling) of triglyc-erides and cholesterol.248 The identification of the Apo E type 4 allele is of importance, as itinfluences the risk of acquiring AD.249 Apo E in brain is primarily a product of astrocytes250,251
and its increased deposition has been described in AD.250,252 Apo E (presumably a mutantform) binds to β-amyloid, thereby leading to the transformation of β-amyloid to fibrillaryamyloid.241,253,254
Abundant neurite sprouting and formation of dystrophic neurites is characteristic ofneuritic plaques.255 Pike et al256 showed that appearance of reactive astrocytes occur earlierthan dystrophic neurites in mild AD suggesting that reactive astrocytes are the cause of thedystrophic neurites. It has been suggested that ACT,238 bFGF,257 adhesion molecules258 andS-100β, all derived from astrocytes, may contribute to the formation of abnormal sproutsand dystrophic neurites found in plaques.
While astrocytes play a key role in the formation of fibrillary amyloid from β-amyloid,the latter also has potent affects on astrocytes. β-amyloid may be responsible for astrocyteactivation in AD.234,235,259 It also stimulates the production of matrix metalloproteinases byastrocytes,130 which may break down myelin260 and impair the BBB.131 It also stimulates theproduction of bFGF by astrocytes,259 possibly contributing to increased production of APPby reactive astrocytes.261,262 It additionally causes a loss of astroglial calcium homeostasis,impairment of ion transport, generation of free radicals, and a decrease in glutamate uptake(for review, see ref. 263).
EpilepsyThe potential involvement of astrocytes in epilepsy dates back to the work of Penfield264
who emphasized the role of the glio-mesenchymal scar as a factor in seizure production.Gliosis is arguably the most common and consistent neuropathologic feature in the seizurefocus,265-267 and the extent of gliosis often parallels the severity of seizure activity.268,269 Asexpected, astrocytes show an increase in GFAP, GFAP mRNA and vimentin.270-273 Otherastrocytic changes have included an elevated activity of various oxidative enzymes,274 in-

51Astrocyte Pathophysiology in Disorders of the Central Nervous System
creased number of gap junctions,275 and alterations in Ca2+ homeostasis.276 Interestingly,astrocytic alterations may precede the onset of seizures.277
The mechanism by which astrocytes contribute to the epileptic state remains specula-tive. An older view suggested that gliosis resulted in mechanical deformation and height-ened excitability.278 It is likely, however, that perturbations of glial functions are the factorsthat lead to altered states of excitability. Any imbalance between excitatory and inhibitoryprocesses is likely to result in seizure activity.279,280 In view of the key role of astrocytes inglutamate, GABA and taurine uptake and release, abnormalities in these functions maycontribute to the epileptic state.281 Consistent with this possibility is the finding that a glial-selective inhibitor of GABA uptake, THPO,282 can protect against experimental seizures.283
Other mechanisms by which glial dysfunction may contribute to the epileptic state includeabnormalities in the metabolism of K+,284,285 H+,47,286 ammonia,287,288 and quinolinicacid.289,290
Experimental models of epilepsy are associated with aberrant synaptic sprouting thatmay lead to abnormal recurrent excitatory circuits that result in seizures.291-294 Many ofthese models are associated with the production of various astrocyte-derived moleculesincluding growth factors (bFGF),295 fibronectin,296 vitronectin,297 tenascin,107,294 neural celladhesion molecules,298,299 and S-100.300 It has been suggested that such molecules may beresponsible for the production of the aberrant axonal sprouting.301,302
Parkinson’s Disease and MPTP NeurotoxicityThe involvement of astrocytes in Parkinson’s disease is presented in detail in chapter 6
of this volume. Here we describe a recent observation on the potential involvement ofastroglial glutamate uptake in the pathogenesis of 1-methyl-4-phenyl-1,2,3,6-tetrahydro-pyridine (MPTP) neurotoxicity. There is evidence to suggest that excessive glutamatergicactivity may play a role in Parkinson’s disease.303-305 We have recently shown that MPTPsignificantly diminished astroglial glutamate uptake.306 Thus, in addition to the oxidationof MPTP to the toxin MPP+ by astroglial MAO B, interference in glutamate uptake may beanother means by which astrocytes contribute to MPTP neurotoxicity and parkinsonism.
Amyotrophic Lateral Sclerosis (ALS)ALS is a progressive disorder of motor neurons of the cortex, brainstem and spinal
cord leading to muscle atrophy and weakness. As in other degenerative diseases, it is associ-ated with a prominent gliosis.307
Abnormalities in glutamate metabolism and excitotoxicity have been proposed as patho-genetic mechanisms for ALS.308 Patients show a marked decrease in glutamate uptake insynaptosomes from spinal cord, motor cortex, and somatosensory cortex.309 Subsequentstudies by the same research group have shown that the astrocyte-specific transporter GLT-1was markedly decreased in ALS, in motor cortex and spinal cord.310 Additionally, loss ofeither GLAST or GLT-1 transporter by the use of antisense oligonucleotides to the glialglutamate transporters resulted in a progressive motor deficit in rats.311 All of these findingssupport the concept that defective clearing of glutamate from the extracellular space maylead to neurotoxic levels of extracellular glutamate and contribute to the neuronal damagein ALS.
The mechanism for defects in glutamate uptake in ALS is not known. A mutation inCu/Zn superoxide dismutase (SOD-1) has been identified in about 20% of patients withfamilial ALS.312 Eosinophilic inclusions containing SOD-1 and ubiquitin have been identi-fied in astrocytes of patients with familial ALS.313 Several SOD-1 mutant mice have beendescribed that show clinical and pathological features of ALS.314,315 As in cases of familialALS, astrocytic SOD-1 and ubiquitin-containing cytoplasmic inclusions are also found.

Astrocytes in Brain Aging and Neurodegeneration52
Importantly, these inclusions appear before the clinical onset of disease.315 This model alsoshows reduced expression of the GLT-1 glutamate transporter. Recent studies have also de-scribed a reduction or absence of SOD-1 in reactive astrocytes in cases of sporadic ALS.316
In view of the important role of SOD in protection against oxidative stress,317,318 a defect inscavenging free radicals may partially explain the abnormality in glial glutamate uptake.
StrokeA detailed account of the role of astrocytes in stroke is given in a recent review.319
Astrocyte swelling and cellular hypertrophy is seen as early as 1-3 hours following reversibleischemia.320 These astrocytes show increased numbers of mitochondria and rough endo-plasmic reticulum in keeping with evidence of increased protein synthesis.321 The nuclei areenlarged and pale and these cells resemble Alzheimer type II astrocytes that have been de-scribed in hyperammonemia and hepatic encephalopathy.43 Whether these astroglial changesare secondary to elevated levels of ammonia that have been documented in ischemia,322,323
or are mediated through some other mechanism, is not known. In the early transitionalphase of the Alzheimer type II change, astrocytes appear metabolically activated, consistentwith data suggesting an increased glucose utilization by glial cells.324
Cell culture studies have shown that anoxic-ischemic injury diminishes the capacity ofastrocytes for glutamate uptake,321,325,326 which can exacerbate excitotoxicity.327,328 A tran-sient decrease in the glutamate transporter GLT-1 has been identified in the CA1 region ofthe hippocampus following global ischemia.329 Glutamate uptake may also be compromisedbecause of energy failure, leading to depolarization and the inability of astrocytes to main-tain ionic gradients that are necessary for glutamate transport.21,49 Alternatively, variousfactors are generated in the ischemic process that are known to interfere with glutamateuptake including arachidonic acid, free radicals, lactic acid, and possibly nitric oxide.330
Hepatic Encephalopathy (HE)HE is a common neurological complication of severe liver disease which occurs in
acute and chronic forms. Acute HE presents with the abrupt onset of delirium, seizures, andcoma. The principal cause of death in acute HE is brain edema associated with increasedintracranial pressure. Chronic HE, sometimes referred to as portal-systemic encephalopa-thy, is characterized by altered mental state, change in personality, diminished intellectualcapacity, abnormal muscle tone and tremor.
The pathogenesis of HE remains poorly understood. The dominant view over manydecades has been the generation of gut-derived neurotoxins, with the greatest emphasis onthe role of ammonia.331 In more recent years, the involvement of heightened GABAergicneurotransmission, possibly through the action of endogenous benzodiazepines, has beenstressed.332
The pathology of HE suggests that astrocytes play a crucial role in this condition. As-trocyte swelling represents the principal component of acute HE and likely contributes tothe brain edema found in this condition, while the Alzheimer type II astrocytic change isthe histological hallmark of chronic HE. No significant or consistent neuronal changes havebeen identified (for reviews of astrocyte changes in HE, see refs. 43, 333).
Astrocytes are the cells in brain where ammonia is metabolized, as glutamine synthetase(GS) is predominantly located in astrocytes.86 Astrocytes are also involved in glutamateuptake, and failure of astrocytes to do so may contribute to abnormal glutamatergic neu-rotransmission.334 Additionally, astrocytes may be sites of action of benzodiazepines, puta-tive factors in the pathogenesis of HE.
The view we have espoused is that astrocytes are primarily injured in HE14,333 and thatastrocytic dysfunction contributes to neuronal derangements, leading to encephalopathy.14

53Astrocyte Pathophysiology in Disorders of the Central Nervous System
Administration of ammonium chloride to cultured astrocytes reproduces the pathologicalchanges observed in HE.335,336 Such treatment also decreases glial fibrillary acidic protein(GFAP) and GFAP mRNA, consistent with the loss of GFAP observed in humans with HE.337
Additional effects of ammonia on astrocytes have included a decreased cAMP response toβ-adrenergic agonists, decreased Ca2+ influx, altered protein phosphorylation, diminishedglycogen levels, and reduced glutamate and GABA uptake. Ammonia also causes astrocyteswelling, alterations in energy and amino acid metabolism, and upregulation of the periph-eral-type benzodiazepine receptor (for reviews, see refs. 333, 338).
There is growing evidence that HE is associated with major derangements of glutamateneurotransmission resulting from an ammonia-induced alteration in glutamate metabo-lism.334,339 We have recently carried out studies on the effect of ammonia on glutamate up-take in cultured astrocytes.340 Chronic treatment (days) resulted in inhibition of glutamateuptake that was associated with a fall in the Vmax, suggesting that the number of glutamatetransporters was decreased. Consistent with this finding is a fall in mRNA for the GLASTglutamate transporter in ammonia-treated astrocyte cultures.341 Treatment of mice withthioacetamide (a hepatotoxin) or with ammonia for 3 days caused a decrease in GLT-1mRNA steady state levels in cerebral cortex and striatum.342
The mechanism for enhancement of GABAergic tone in HE is unclear. Our laboratoryhas been investigating the potential involvement of the “peripheral-type” benzodiazepinereceptor (PBR). In contrast to the “central” benzodiazepine receptor that is present on theplasma membrane as part of the neuronal GABAA receptor complex, the PBR in the CNS isconfined to glial cells343-345 where it is primarily located on the outer mitochondrialmembrane.346
The best studied function of the PBR is the regulation of steroid biosynthesis.347
Neurosteroids, particularly tetrahydroprogesterone (THP, allopregnanolone), andtetrahydrodeoxycorticosterone (THDOC) have potent CNS depressant effects that are me-diated through their actions on the GABAA receptor.348-350 Recent findings from our labora-tory have shown that: the number of PBR binding sites is increased in ammonia-treatedcultured astrocytes using PK 11195 as the PBR ligand; the PBR is upregulated in mice withacute HE caused by thioacetamide (TAA) as well as in hyperammonemic mice; treatmentwith PK 11195, a putative antagonist of the PBR, significantly attenuates ammonia toxicityin mice; pregnenolone levels are increased in TAA- and ammonia-treated animals; and brainlevels of THP and THDOC are elevated in hyperammonemic mice and mice with acuteliver failure produced by TAA.338,351 Upregulation of the astrocytic PBR by ammonia canpotentially result in increased production of neurosteroids which have positive modulatoryeffects on the GABAA receptor, that in turn may lead to neuroinhibition and neurologicdysfunction.
Perspectives and ConclusionsAstrocytes are active and dynamic cells involved in many aspects of CNS function and
are early responders to CNS injury. They also contribute to the pathogenesis of many neu-rological conditions, although it is not always clear whether their effects are beneficial ordetrimental. Such issues as the concentrations of various factors and the clinical setting maymaterially determine whether the astrocyte response is appropriate or not. Additionally,astrocytes have important interactions with microglia and other mesenchyme-derived cellsthat strongly influence the outcome of disease. It should also be stressed that astrocytes maybe injured in disease states and thus astroglial functional failure may contribute to the patho-genesis of some CNS disorders.
While astrocytes may occasionally exert deleterious actions, on balance their activityappears more geared towards generating homeostatic responses and promoting repair and

Astrocytes in Brain Aging and Neurodegeneration54
regeneration. Indeed, the regulation of the astrocyte response may in future years provide akey strategy in influencing the outcome of CNS injury.
AcknowledgmentsThis work was supported by research grants from the Department of Veterans Affairs
and the National Institutes of Health (NS30291 and NS34951).
References1. Abbott NJ, ed. Glial-Neuronal Interactions. New York: New York Acad Sci, 1991.2. Gard AL. Astrocyte-oligodendrocyte interactions. In: Murphy S, ed. Astrocytes: Pharma-
cology and Function. San Diego: Academic Press, 1993:331-354.3. Benveniste EN. Astrocyte-microglia interactions. In: Murphy S, ed. Astrocytes, Pharmacol-
ogy and Function. New York: Academic Press, 1993:355-382.4. Cancilla PA, Bready J, Berliner J. Astrocyte-endothelial interactions. In: Murphy S, ed.
Astrocytes: Pharmacology and Function. San Diego: Academic Press, 1993:383-397.5. Walz W. Role of glial cells in the regulation of the brain ion microenvironment. Prog
Neurobiol 1989; 33:309-333.6. Largo C, Cuevas P, Herreras O. Is glia dysfunction the initial cause of neuronal death in
ischemic penumbra. Neurol.Res. 1996; 18:445-448.7. Largo C, Cuevas P, Somjen GG et al. The effect of depressing glial function in rat brain in
situ on ion homeostasis, synaptic transmission, and neuron survival. J Neurosci 1996;16:1219-1229.
8. Schousboe A. Transport and metabolism of glutamate and GABA in neurons and glialcells. Int Rev Neurobiol 1981; 22:1-45.
9. Martin DL. Synthesis and release of neuroactive substances by glial cells. Glia 1992; 5:81-94.10. Keyser DO, Pellmar TC. Synaptic transmission in the hippocampus: Critical role for glial
cells. Glia 1994; 10:237-243.11. Dringen R, Gebhardt R, Hamprecht B. Glycogen in astrocytes: Possible function as lactate
supply for neighboring cells. Brain Res 1993; 623:208-214.12. Westergaard N, Sonnewald U, Schousboe A. Metabolic trafficking between neurons and
astrocytes: The glutamate glutamine cycle revisited. Dev Neurosci 1995; 17:203-211.13. Muller HW, Junghans U, Kappler J. Astroglial neurotrophic and neurite promoting fac-
tors. Pharmacol Ther 1995; 65:1-18.14. Norenberg MD. The role of astrocytes in hepatic encephalopathy. Neurochem Pathol 1987;
6:13-33.15. Abramovitz M, Homma H, Ishigaki S et al. Characterization and localization of
glutathione-S-transferases in rat brain and binding of hormones, neurotransmitters, anddrugs. J Neurochem 1988; 50:50-57.
16. Makar TK, Nedergaard M, Preuss A et al. Vitamin E, ascorbate, glutathione, glutathionedisulfide, and enzymes of glutathione metabolism in cultures of chick astrocytes and neu-rons: Evidence that astrocytes play an important role in antioxidative processes in the brain.J Neurochem 1994; 62:45-53.
17. Sawada J, Kikuchi Y, Shibutani M et al. Induction of metallothionein in astrocytes bycytokines and heavy metals. Biol Signals 1994; 3:157-168.
18. Janzer RC, Raff MC. Astrocytes induce blood-brain barrier properties in endothelial cells.Nature 1987; 325:253-257.
19. Stitt TN, Gasser UE, Hatten ME. Molecular mechanisms of glial-guided neuronal migra-tion. Ann NY Acad Sci 1991; 633:113-121.
20. Frei K, Fontana A. Immune regulatory functions of astrocytes and microglial cells withinthe central nervous system. In: Goetzl EJ, Spector NH eds. Neuroimmune Networks: Physi-ology and Diseases. New York: Alan R. Liss, 1989:127-136.
21. Nicholls D, Attwell D. The release and uptake of excitatory amino acids. Trends PharmacolSci 1990; 11:462-468.

55Astrocyte Pathophysiology in Disorders of the Central Nervous System
22. Rosenberg PA, Amin S, Leitner M. Glutamate uptake disguises neurotoxic potency ofglutamate agonists in cerebral cortex in dissociated culture. J Neurosci 1992; 12:56-61.
23. Pines G, Danbolt NC, Bjoras M et al. Cloning and expression of a rat brain L-glutamatetransporter. Nature 1992; 360:464-467.
24. Stork T, Schulte S, Hofmann K et al. Structure, expression and functional analysis of aNa+ dependent glutamate/aspartate transporter from rat brain. Proc Natl Acad Sci USA1992; 89:10955-10959.
25. Kanai Y, Hediger MA. Primary structure and functional characterization of a high affinityglutamate transporter. Nature 1992; 360:467-471.
26. Torp R, Danbolt NC, Babaie E et al. Differential expression of two glial glutamate trans-porters in the rat brain: An in situ hybridization study. Eur J Neurosci 1994; 6:936-942.
27. Danbolt NC, Storm-Mathisen J, Kanner BI. An [Na+ + K+] coupled L-glutamate trans-porter purified from brain is located in glial cell processes. Neuroscience 1992; 51:295-310.
28. Levy LM, Lehre KP, Rolstad B et al. A monoclonal antibody raised against an [Na+ +K+]coupled L-glutamate transporter purified from rat brain confirms glial cell localization.FEBS Lett 1993; 317:79-84.
29. Rothstein JD, Martin L, Levey AI et al. Localization of neuronal and glial glutamate trans-porters. Neuron 1994; 13:713-725.
30. Kondo K, Sashimoto H, Kitanaka J et al. Expression of glutamate transporters in culturedglial cells. Neurosci Lett 1995; 188:140-142.
31. Nathaniel EJH, Nathaniel DR. The reactive astrocyte. Adv Cell Neurobiol 1981; 2:249-301.32. Eng LF, Reier PJ, Houle JD. Astrocyte activation and fibrous gliosis: Glial fibrillary acidic
protein in CNS tissue. Prog Brain Res 1987; 71:439-455.33. Eddleston M, Mucke L. Molecular profile of reactive astrocytes - Implications for their
role in neurologic disease. Neuroscience 1993; 54:15-36.34. Norenberg MD. Reactive astrocytosis. In: Aschner M, Kimelberg HK, eds. The Role of Glia
in Neurotoxicity. Boca Raton: CRC Press, 1996:93-107.35. Kimelberg HK. Current concepts of brain edema. J Neurosurg 1995; 83:1051-1059.36. Kimelberg HK, Goderie SK, Higman S et al. Swelling-induced release of glutamate, aspar-
tate, and taurine from astrocyte cultures. J Neurosci 1990; 10:1583-1591.37. Bender AS, Norenberg MD. Calcium dependence of hypoosmotically induced potassium
release in cultured astrocytes. J Neurosci 1994; 14:4237-4243.38. Smith SJ. Do astrocytes process neural information? In: Yu ACH, Hertz L, Norenberg MD,
Sykova E, Waxman S, eds. Neuronal-Astrocytic Interactions: Implications for Normal andPathological CNS Function. Amsterdam: Elsevier, 1992:119-136.
39. Garcia JH, Liu KF, Yoshida Y et al. The brain microvessels: Factors altering their patencyafter the occlusion of a middle cerebral artery (Wistar rat). Am J Pathol 1994; 145:1-13.
40. Kuriyama K. Taurine as an neuromodulator. Fed Proc 1980; 39:2680-2684.41. Schurr A, Tseng MT, West CA et al. Taurine improves the recovery of neuronal function
following cerebral hypoxia: An in vitro study. Life Sci 1987; 40:2059-2066.42. Aruoma OI, Halliwell B, Hoey BY et al. The antioxidant action of taurine, hypotaurine
and their metabolic precursors. Biochem J 1988; 256:251-255.43. Norenberg MD. The astrocyte in liver disease. In: Fedoroff S, Hertz L, eds. Advances in
Cellular Neurobiology, Vol. 2. New York: Academic Press, 1981:303-352.44. Norenberg MD. A light and electron microscopic study of experimental portal-systemic
(ammonia) encephalopathy. Lab Invest 1977; 36:618-627.45. Faden AI, Salzman S. Pharmacological strategies in CNS trauma. Trends Neurosci 1992;
13:29-35.46. Orkand RK, Nicholls JG, Kuffler SW. Effects of nerve impulses on the membrane potential
of glial cells in the central nervous system of amphibia. J Neurophysiol 1966; 29:788-806.47. Chesler M, Kraig RP. Intracellular pH transients of mammalian astrocytes. J Neurosci 1989;
9:2011-2019.48. Kraig RP, Pulsinelli WA, Plum F. Hydrogen ion buffering during complete brain ischemia.
Brain Res 1985; 342:281-290.

Astrocytes in Brain Aging and Neurodegeneration56
49. Barbour B, Brew H, Attwell D. Electrogenic uptake of glutamate and aspartate into glialcells isolated from the salamander (Ambystoma) retina. J Physiol (Lond) 1991; 436:169-193.
50. Cambray-Deakin M, Pearce B, Morrow C et al. Effects of extracellular potassium on glyco-gen stores of astrocytes in vitro. J Neurochem 1988; 51:1846-1851.
51. Subbarao KV, Stolzenburg JU, Hertz L. Pharmacological characteristics of potassium-in-duced, glycogenolysis in astrocytes. Neurosci Lett 1995; 196:45-48.
52. Kimelberg HK, Ransom BR. Physiological aspects of astrocyte swelling. In: Fedoroff S,Vernadakis A, eds. Astrocytes, Vol. 3. Orlando: Academic Press, 1986:129-166.
53. Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron 1988;1:623-634.
54. Whetsell WO, Shapira NA. Biology of disease: Neuroexcitation, excitotoxicity and humanneurological disease. Lab Invest 1993; 68:372-387.
55. Teichberg VI. Glial glutamate recepors: likely actors in brain signaling. FASEB J 1991;5:3086-3091.
56. Hansson E, Rönnbäck L. Astrocytes in glutamate neurotransmission. FASEB J 1995;9:343-350.
57. Norenberg MD, Mozes LW, Gregorios JB et al. Effects of lactic acid on astrocytes in pri-mary culture. J Neuropathol Exp Neurol 1987; 46:154-166.
58. Giffard RG, Monyer H, Choi DW. Selective vulnerability of cultured glia to injury by ex-tracellular acidosis. Brain Res 1990; 530:138-141.
59. Jakubovicz D, Klip A. Lactic acid-induced swelling in C6 glial cells via Na+/H+ exchange.Brain Res 1989; 485:215-224.
60. Staub F, Peters J, Kempski O et al. Swelling of glial cells in lactacidosis and by glutamate:Significance of Cl--transport. Brain Res 1993; 610:69-74.
61. Swanson RA, Farrell K, Simon RP. Acidosis causes failure of astrocyte glutamate uptakeduring hypoxia. J Cereb Blood Flow Metab 1995; 15:417-424.
62. Bender AS, Young LP, Norenberg MD. Effect of lactic acid on L-glutamate uptake in cul-tured astrocytes: Mechanistic considerations. Brain Res 1997; 750:59-66.
63. Chan PH, Longar S, Chen S et al. The role of arachidonic acid and oxygen radical metabo-lites in the pathogenesis of vasogenic brain edema and astrocytic swelling. Ann NY AcadSci 1989; 559:237-247.
64. Staub F, Winkler A, Peters J et al. Swelling, acidosis, and irreversible damage of glial cellsfrom exposure to arachidonic acid in vitro. J Cereb Blood Flow Metab 1994; 14:1030-1039.
65. Piani D, Frei K, Pfister H-W et al. Glutamate uptake by astrocytes is inhibited by reactiveoxygen intermediates but not by other macrophage-derived molecules including cytokines,leukotrienes or platelet-activating factor. J Neuroimmunol 1993; 48:99-104.
66. Volterra A, Trotti D, Tromba C et al. Glutamate uptake inhibition by oxygen free radicalsin rat cortical astrocytes. J Neurosci 1994; 14:2924-2932.
67. Levi G, Patrizio M. Astrocyte heterogeneity: Endogenous amino acid levels and releaseevoked by non-N-methyl-D-aspartate receptor agonists and by potassium-induced swell-ing in type-1 and type-2 astrocytes. J Neurochem 1992; 58:1943-1952.
68. Ogata T, Nakamura Y, Shibata T et al. Release of excitatory amino acids from culturedhippocampal astrocytes induced by a hypoxic-hypoglycemic stimulation. J Neurochem 1992;58:1957-1959.
69. Gemba T, Oshima T, Ninomiya M. Glutamate efflux via the reversal of the sodium-depen-dent glutamate transporter caused by glycolytic inhibition in rat cultured astrocytes. Neu-roscience 1994; 63:789-795.
70. Longuemare MC, Swanson RA. Excitatory amino acid release from astrocytes during en-ergy failure by reversal of sodium-dependent uptake. J Neurosci Res 1995; 40:379-386.
71. Benos DJ, Hahn BH, Bubien JK et al. Envelope glycoprotein gp120 of human immunode-ficiency virus type 1 alters ion transport in astrocytes: Implications for AIDS dementiacomplex. Proc Natl Acad Sci USA 1994; 91:494-498.
72. Mullaney KJ, Vitarella D, Albrecht J et al. Stimulation of D-aspartate efflux by mercuricchloride from rat primary astrocyte cultures. Devl Brain Res 1993; 75:261-268.

57Astrocyte Pathophysiology in Disorders of the Central Nervous System
73. Dawson R, Jr., Patterson TA, Eppler B. Endogenous excitatory amino acid release frombrain slices and astrocyte cultures evoked by trimethyltin and other neurotoxic agents.Neurochem Res 1995; 20:847-858.
74. Albrecht J, Simmons M, Dutton GR et al. Aluminum chloride stimulates the release ofendogenous glutamate, taurine and adenosine from cultured rat cortical astrocytes. NeurosciLett 1991; 127:105-107.
75. Schwarcz R, Guidetti P, Roberts RC. Quinolinic acid and kynurenic acid: Glia-derivedmodulators of excitotoxic brain injury. In: Aschner M, Kimelberg HK, eds. The Role ofGlia in Neurotoxicity. Boca Raton: CRC Press, 1996:245-262.
76. Heyes MP, Achim CL, Wiley CA et al. Human microglia convert L-tryptophan into theneurotoxin quinolinic acid. Biochem J 1996; 320:595-597.
77. Speciale C, Okuno E, Schwarcz R. Increased quinolinic acid metabolism following neu-ronal degeneration in the rat hippocampus. Brain Res 1987; 436:18-24.
78. Perkins MN, Stone TW. An iontophoretic investigation of the actions of convulsantkynurenines and their interaction with the endogenous excitant quinolinic acid. Brain Res1982; 247:183-187.
79. Holopainen I, Kontro P. Uptake and release of glycine in cerebellar granule cells and as-trocytes in primary culture: Potassium-stimulated release from granule cells is calcium-dependent. J Neurosci Res 1989; 24:374-383.
80. Sato K, Yoshida S, Fujiwara K et al. Glycine cleavage system in astrocytes. Brain Res 1991;567:64-70.
81. Waniewski RA, Martin DL. Exogenous glutamate is metabolized to glutamine and exportedby rat primary astrocyte cultures. J Neurochem 1986; 47:304-313.
82. Farinelli SE, Nicklas WJ. Glutamate metabolism in rat cortical astrocyte cultures. JNeurochem 1992; 58:1905-1915.
83. Simantov R. Glutamate neurotoxicity in culture depends on the presence of glutamine:Implications for the role of glial cells in normal and pathological brain development. JNeurochem 1989; 52:1694-1699.
84. Bradford HF, Ward HK. Glutamine--a major substrate for nerve endings. J Neurochem1978; 30:1453-1459.
85. Okuno E, Nakamura M, Schwarcz R. Two kynurenine aminotransferases in human brain.Brain Res 1991; 542:307-312.
86. Norenberg MD, Martinez-Hernandez A. Fine structural localization of glutamine synthetasein astrocytes of rat brain. Brain Res 1979; 161:303-310.
87. Swanson RA, Shiraishi K, Morton MT et al. Methionine sulfoximine reduces cortical inf-arct size in rats after middle cerebral artery occlusion. Stroke 1990; 21:322-327.
88. Cataldo AM, Broadwell RD. Cytochemical identification of cerebral glycogen andglucose-6-phosphatase activity under normal and experimental conditions. Neurons andglia. Electron Microscop Tech 1986; 3:413-437.
89. Siesjö BK. Acidosis and ischemic brain damage. Neurochem Pathol 1988; 9:31.90. Staub F, Baethmann A, Peters J et al. Efects of lactacidosis on glial cell volume and viabil-
ity. J Cereb Blood Flow Metab 1990; 10:866-876.91. Poitry-Yamate CL, Poitry S, Tsacopoulos M. Lactate released by Müller glial cells is me-
tabolized by photoreceptors from mammalian retina. J Neurosci 1995; 15:5179-5191.92. Giffard RG, Monyer H, Christine CW et al. Acidosis reduces NMDA receptor activation,
glutamate neurotoxicity, and oxygen-glucose deprivation neuronal injury in cortical cul-tures. Brain Res 1990; 506:339-342.
93. Moncada S, Palmer RM, Higgs EA. Nitric oxide: Physiology, pathophysiology, and phar-macology. Pharmacol Rev 1991; 43:109-142.
94. Dawson TM, Dawson VL, Snyder SH. A novel neuronal messenger molecule in brain: Thefree radical, nitric oxide. Ann Neurol 1992; 32:297-311.
95. Lee SC, Dickson DW, Brosnan CF et al. Human astrocytes inhibit Cryptococcus neoformansgrowth by a nitric oxide-mediated mechanism. J Exp Med 1994; 180:365-369.
96. Peterson PK, Gekker G, Hu S et al. Human astrocytes inhibit intracellular multiplicationof Toxoplasma gondii by a nitric oxide-mediated mechanism. J Infect Dis 1995; 171:516-518.

Astrocytes in Brain Aging and Neurodegeneration58
97. Beckman JS. The double-edged role of nitric oxide in brain function and superoxide-me-diated injury. J Dev Physiol 1991; 15:53-59.
98. Murphy S, Simmons ML, Agullo L et al. Synthesis of nitric oxide in CNS glial cells. TrendsNeurosci 1993; 16:323-328.
99. Brosnan CF, Battistini L, Raine CS et al. Reactive nitrogen intermediates in human neuro-pathology: An overview. Dev Neurosci 1994; 16:152-161.
100. Goadsby P, Kaube H, Hoskin KL. Nitric oxide synthesis couples cerebral blood flow andmetabolism. Brain Res 1992; 595:167-170.
101. Schipper HM. Astrocytes, brain aging, and neurodegeneration. Neurobiol Aging 1996;17:467-480.
102. Pindzola RR, Doller C, Silver J. Putative inhibitory extracellular matrix molecules at thedorsal root entry zone of the spinal cord during development and after root and sciaticnerve lesions. Dev Biol 1993; 156:34-48.
103. DeWitt DA, Richey PL, Praprotnik D et al. Chondroitin sulfate proteoglycans are a com-mon component of neuronal inclusions and astrocytic reaction in neurodegenerative dis-eases. Brain Res 1994; 656:205-209.
104. Letourneau PC, Condic ML, Snow DM. Extracellular matrix and neurite outgrowth. CurrOpin Genet Devel 1992; 2:625-634.
105. Faissner A, Steindler DA. Boundries and inhibitory molecules in developing neural tissue.Glia 1995; 13:233-254.
106. Laywell ED, Dörries U, Bartsch U et al. Enhanced expression of the developmentally regu-lated extracellular matrix molecule tenascin following adult brain injury. Proc Natl AcadSci USA 1992; 89:2634-2638.
107. Scheffler B, Faissner A, Beck H et al. Hippocampal loss of tenascin boundaries in Ammon’shorn sclerosis. Glia 1997; 19:35-46.
108. Mansour H, Asher R, Dahl D et al. Permissive and non-permissive reactive astrocytes:Immunofluorescence study with antibodies to the glial hyaluronate-binding protein. JNeurosci Res 1990; 25:300-311.
109. Tiveron M-C, Barboni E, Pliego Rivero FB et al. Selective inhibition of neurite outgrowthon mature astrocytes by Thy-1 glycoprotein. Nature 1992; 355:745-748.
110. Liuzzi FJ. Proteolysis is a critical step in the physiological stop pathway: Mechanisms in-volved in the blockade of axonal regeneration by mammalian astrocytes. Brain Res 1990;512:277-283.
111. Perry VH, Andersson P-B, Gordon S. Macrophages and inflammation in the central ner-vous system. Trends Neurosci 1993; 16:268.
112. Morganti-Kossmann MC, Kossmann T, Wahl SM. Cytokines and neuropathology. TrendsPharmacol Sci 1992; 13:286-291.
113. Benveniste EN. Inflammatory cytokines within the central nervous system: Sources, func-tion, and mechanism of action. Am J Physiol Cell Physiol 1992; 263:C1-C16.
114. Furie MB, Randolph GJ. Chemokines and tissue injury. Am J Pathol 1995; 146:1287-1301.115. Goldman JE, Chiu FC. DbcAMP causes intermediate filament accumulation and actin
re-organization in astrocytes. Brain Res 1984; 306:85-95.116. Faden AI et al. Experimental spinal cord injury: Effects of trauma or ischemia on TRH
and muscarinic receptors. Neurology 1986; 36:723-726.117. Ghirnikar RS, Lee YL, He TR et al. Chemokine expression in rat stab wound brain injury.
J Neurosci Res 1996; 46:727-733.118. Hickey WF, Osborn JP, Kirby WM. Expression of Ia molecules by astrocytes during acute
experimental allergic encephalomyelitis in the Lewis rat. Cell Immunol 1985; 91:528-535.119. Sun D, Wekerle H. Ia-restricted encephalitogenic T lymphocytes mediating EAE lyse
autoantigen-presenting astrocytes. Nature 1986; 320:70-72.120. Lee SC, Moore GR, Golenwsky G et al. Multiple sclerosis: A role for astroglia in active
demyelination suggested by class II MHC expression and ultrastructural study. J Neuro-path Exp Neurol 1990; 49:122-136.

59Astrocyte Pathophysiology in Disorders of the Central Nervous System
121. Cannella B, Cross AH, Raine CS. Adhesion-related molecules in the central nervous sys-tem: Upregulation correlates with inflammatory cell influx during relapsing experimentalautoimmune encephalomyelitis. Lab Invest 1991; 65:23-31.
122. Vass K, Lassmann H, Wekerle H et al. The distribution of Ia antigen in the lesions of ratacute experimental allergic encephalomyelitis. Acta Neuropathol 1986; 70:149-160.
123. Matsumoto Y, Kawai K, Fujiwara M. In situ Ia expression on brain cells in the rat: Au-toimmune encephalomyelitis-resistant strain (BN) and susceptible strain (Lewis) compared.Immunology 1989; 66:621-627.
124. Abraham CR, Kanemaru K, Mucke L. Expression of cathepsin G-like and alpha1-antichymotrypsin-like proteins in reactive astrocytes. Brain Res 1993; 621:222-232.
125. Monard D. Cell-derived proteases and protease inhibitors as regulators of neurite outgrowth.Trends Neurosci 1988; 11:541-544.
126. Neveu I, Jehan F, Jandrot Perrus M et al. Enhancement of the synthesis and secretion ofnerve growth factor in primary cultures of glial cells by proteases: A possible involvementof thrombin. J Neurochem 1993; 60:858-867.
127. Eddleston M, De la Torre JC, Oldstone MB et al. Astrocytes are the primary source oftissue factor in the murine central nervous system. A role for astrocytes in cerebral hemo-stasis. J Clin Invest 1993; 92:349-358.
128. Abraham CR, Driscoll J, Potter H et al. A calcium-activated protease from Alzheimer’sdisease brain cleaves at the N-terminus of the amyloid beta-protein. Biochem Biophys ResCommun 1991; 174:790-796.
129. Wells GMA, Catlin G, Cossins JA et al. Quantitation of matrix metalloproteinases in cul-tured rat astrocytes using the polymerase chain reaction with a multi-competitor cDNAstandard. Glia 1996; 18:332-340.
130. Deb S, Gottschall PE. Increased production of matrix metalloproteinases in enriched as-trocyte and mixed hippocampal cultures treated with β-amyloid peptides. J Neurochem1996; 66:1641-1647.
131. Rosenberg GA. Matrix metalloproteinases in brain injury. J Neurotrauma 1995; 12:833-842.132. Gebicke-Haerter PJ, Bauer J, Brenner A et al. Alpha2-Macroglobulin synthesis in an astro-
cyte subpopulation. J Neurochem 1987; 49:1139-1145.133. Higuchi M, Ito T, Imai Y et al. Expression of the α2-macroglobulin-encoding gene in rat
brain and cultured astrocytes. Gene 1994; 141:155-162.134. Reichardt LF, Tomaselli KJ. Extracellular matrix molecules and their receptors: Functions
in neural development. Annu Rev Neurosci 1991; 14:531-570.135. Frohman EM, Frohman TC, Dustin ML et al. The induction of intracellular adhesion
molecule 1 (ICAM-1) expression on human fetal astrocytes by interferon-gamma, tumornecrosis factor-alpha, lymphotoxin, and interleukin-1: Relevance to intracerebral antigenpresentation. J Neuroimmunol 1989; 23:117-124.
136. Sobel RA, Mitchell ME, Fondren G. Intracellular adhesion molecule-1 (ICAM-1) in cellu-lar immune reactions in the human central nervous system. Am J Pathol 1990;136:1309-1316.
137. Hurwitz AA, Lyman WD, Guida MP et al. Tumor necrosis factor alpha induces adhesionmolecule expression on human fetal astrocytes. J Exp Med 1992; 176:1631-1636.
138. Vaca K, Wendt E. Divergent effects of astroglial and microglial secretions on neuron growthand survival. Exp Neurol 1992; 118:62-72.
139. Giulian D, Vaca K, Corpuz M. Brain glia release factors with opposing actions upon neu-ronal survival. J Neurosci 1993; 13:29-37.
140. Graeber MB, Streit WJ. Microglia: Immune network in the CNS. Brain Pathol 1990; 1:2-5.141. Kreutzberg GW. Microglia, the first line of defence in brain pathologies. Arzneimittel-
forschung 1995; 45:357-360.142. Thomas WE. Brain macrophages: Evaluation of microglia and their functions. Brain Res
Rev 1992; 17:61-74.143. Giulian D, Chen J, Ingeman JE et al. The role of mononuclear phagocytes in wound heal-
ing after traumatic injury to adult mammalian brain. J Neurosci 1989; 9:4416-4429.

Astrocytes in Brain Aging and Neurodegeneration60
144. Gadient RA, Cron KVC, Otten U. Interleukin-1 and tumor necrosis factor-alpha synergis-tically stimulate nerve growth factor (NGF) release from cultured astrocytes. Neurosci Lett1990; 117:335-340.
145. Gage FH, Olejniczak P, Armstrong DM. Astrocytes are important for sprouting in theseptohippocampal circuit. Exp Neurol 1988; 102:2-13.
146. Hao C, Guilbert LJ, Fedoroff S. Production of colony-stimulating factor-1 (CSF-1) by mouseastroglia in vitro. J Neurosci Res 1990; 27:314.
147. Aloisi F, Carè A, Borsellino G et al. Production of hemolymphopoietic cytokines (IL-6,IL-8, colony-stimulating factors) by normal human astrocytes in response to IL-1beta andtumor necrosis factor-alpha. J Immunol 1992; 149:2358-2366.
148. Garland JM. Colony stimulating factors. In: Thomson A, ed. The Cytokine Handbook. SanDiego: Academic Press, 1991:269-300.
149. Tweardy DJ, Mott PL, Glazer EW. Monokine modulation of human astroglial cell produc-tion of granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimu-lating factor. I. Effects of IL-1alpha and IL-beta. J Immunol 1990; 144:2233-2241.
150. Frei K, Bodmer S, Schwerdel C et al. Astrocytes of the brain synthesize interleukin 3-likefactors. J Immunol 1985; 135:4044-4047.
151. Chamak B, Mallat M. Fibronectin and laminin regulate the in vitro differentiation of mi-croglial cells. Neuroscience 1991; 45:513-527.
152. Levitt D, Ho PL. Induction of chondroitin sulfate proteoglycan synthesis and secretion inlymphocytes and monocytes. J Cell Biol 1983; 97:351-358.
153. Johnson-Green PC, Dow KE, Riopelle RJ. Neurite growth modulation associated with as-trocyte proteoglycans: Influence of activators of inflammation. Glia 1992; 5:33.
154. Ness R, David S. Leptomeningeal cells modulate the neurite growth promoting propertiesof astrocytes in vitro. Glia 1997; 19:47-57.
155. Schwab ME, Bartholdi D. Degeneration and regeneration of axons in the lesioned spinalcord. Physiol Rev 1996; 76:319-370.
156. Silos-Santiago I, Greenlund LJ, Johnson EM Jr et al. Molecular genetics of neuronal sur-vival. Curr Opin Neurobiol 1995; 5:42-49.
157. Ramon y Cajal S. Degeneration and Regeneration of the Nervous System. Oxford, OxfordUniversity Press, 1928.
158. Rudge JS, Silver J. Inhibition of neurite outgrowth on astroglial scars in vitro. J Neurosci1990; 10:3595-3603.
159. Bovolenta P, Wandosell F, Nieto-Sampedro M. CNS glial scar tissue: A source of mol-ecules which inhibit central neurite outgrowth. In: Yu ACH, Hertz L, Norenberg MD,Sykova E, Waxman SG, eds. Neuronal-Astrocytic Interactions: Pathological Implications.Amsterdam: Elsevier, 1992:367-379.
160. Stensaas LJ, Partlow LM, Burgess PR et al. Inhibition of regeneration: the ultrastructure ofreactive astrocytes and abortive axon terminals in the transition zone of the dorsal root.Prog Brain Res 1987; 71:457-468.
161. Carlstedt T, Cullheim S, Risling M et al. Nerve fiber regeneration across the PNS-CNSinterface at the root-spinal cord junction. Brain Res Bull 1989; 22:93-102.
162. Carlstedt T. Regenerating axons form nerve terminals at astrocytes. Brain Res 1985;347:188-191.
163. Liuzzi FJ, Lasek RJ. Astrocytes block axonal regeneration in mammals by activating thephysiological stop pathway. Science 1987; 237:642-645.
164. Noble M, Fok-Seang J, Cohen J. Glia are a unique substrate for the in vitro growth ofcentral nervous system neurons. J Neurosci 1984; 4:1892-1903.
165. Fallon JR. Preferential outgrowth of central nervous system neurites on astrocytes andSchwann cells as compared with nonglial cells in vitro. J Cell Biol 1985; 100:198-207.
166. Rudge JS. Astrocyte-derived neurotrophic factors. In: Murphy S, ed. Astrocytes. Pharma-cology and Function. San Diego: Academic Press, 1993:267-305.
167. Fawcett JW, Housden E, Smith-Thomas L et al. The growth of axons in three-dimensionalastrocyte cultures. Dev Biol 1989; 135:449-458.

61Astrocyte Pathophysiology in Disorders of the Central Nervous System
168. Smith GM, Rutishauser U, Silver J et al. Maturation of astrocytes in vitro alters the extentand molecular basis of neurite outgrowth. Dev Biol 1990; 138:377-390.
169. Schwartz M, Cohen A, Stein-Izsak C et al. Dichotomy of the glial cell response to axonalinjury and regeneration. FASEB J 1989; 3:2371-2378.
170. Kruger S, Sievers J, Hansen C et al. Three morphologically distinct types of interface de-velop between adult host and fetal brain transplants: Implications for scar formation inthe adult central nervous system. J Comp Neurol 1986; 249:103-116.
171. Berry M, Maxwell WL, Logan A et al. Deposition of scar tissue in the central nervoussystem. Acta Neurochirurg Suppl 1983; 32:31-53.
172. Caroni P, Schwab ME. Antibody against myelin-associated inhibitor of neurite outgrowthneutralizes non-permissive substrate properties of CNS white matter. Neuron 1988; 1:85-96.
173. Savio T, Schwab ME. Lesioned corticospinal tract axons regenerate in myelin-free rat spi-nal cord. Proc Natl Acad Sci USA 1990; 87:4130-4133.
174. Petito CK, Cho E-L, Lemann W et al. Neuropathology of acquired immunodeficiency syn-drome (AIDS): an autopsy review. J Neuropathol Exp Neurol 1986; 45:635-646.
175. Gray F, Gherardi R, Scaravilli F. The neuropathology of the acquired immune deficiencysyndrome (AIDS). A review. Brain 1988; 111:245-266.
176. Epstein LG, Gendelman HE. Human immunodeficiency virus type-1 infection of the ner-vous system: Pathogenetic mechanisms. Ann Neurol 1993; 33:429-436.
177. Genis P, Jett M, Bernton EW et al. Cytokines and arachidonic metabolites produced dur-ing human immunodeficiency virus (HIV)-infected macrophage-astroglia interactions:Implications for the neuropathogenesis of HIV disease. J Exp Med 1992; 176:1703-1718.
178. Nottet HSLM, Jett M, Flanagan CR et al. A regulatory role for astrocytes in HIV-1 en-cephalitis: An overexpression of eicosanoids, platelet-activating factor, and tumor necrosisfactor-α by activated HIV-1-infected monocytes is attenuated by primary human astro-cytes. J Immunol 1995; 154:3567-3581.
179. Vitkovic L, Kalebic T, de Cunha A et al. Astrocyte-conditioned media stimulates HIV-1expression in chronically infected promonocyte clone. J Neuroimmunol 1990; 30:153.
180. Vitkovic L, Wood GP, Major EO et al. Human astrocytes stimulate HIV-1 expression in achronically infected promonocyte clone via interleukin-6. AIDS Res Human Retroviruses1991; 7:723-727.
181. Wahl SM, Allen JB, McCartney-Francis N et al. Macrophage- and astrocyte-derived trans-forming growth factor beta as a mediator of central nervous system dysfunction in ac-quired immune deficiency syndrome. J Exp Med 1991; 173:981-991.
182. Saito Y, Sharer LR, Epstein LG et al. Overexpression of nef as a marker for restricted HIV-1infection of astrocytes in postmortem pediatric central nervous tissues. Neurology 1994;44:474-481.
183. Tornatore C, Chandra R, Berger JR et al. HIV-1 infection of subcortical astrocytes in thepediatric central nervous system. Neurology 1994; 44:481-487.
184. Nath A, Hartloper V, Furer M et al. Infection of human fetal astrocytes with HIV-1: Viraltropism and the role of cell to cell contact in viral transmission. J Neuropath Exp Neurol1995; 54:320-329.
185. Pulliam L, West D, Haigwood N et al. HIV-1 envelope gp120 alters astrocytes in humanbrain cultures. AIDS Res Hum Retroviruses 1993; 9:439-444.
186. Levi G, Patrizio M, Bernardo A et al. Human immunodeficiency virus coat protein gp120inhibits the beta-adrenergic regulation of astroglial and microglial functions. Proc Natl AcadSci USA 1993; 90:1541-1545.
187. Lipton SA. Models of neuronal injury in AIDS: Another role for the NMDA receptor?Trends Neurosci 1992; 15:75-79.
188. Benos DJ, McPherson S, Hahn BH et al. Cytokines and HIV envelope glycoprotein gp120stimulate Na+/H+ exchange in astrocytes. J Biol Chem 1994; 269:13811-13816.
189. Dreyer EB, Lipton SA. The coat protein gp120 of HIV-1 inhibits astrocyte uptake of exci-tatory amino acids via macrophage arachidonic acid. Eur J Neurosci 1995; 7:2502-2507.

Astrocytes in Brain Aging and Neurodegeneration62
190. Yu ACH, Chan PH, Fishman RA. Arachidonic acid inhibits uptake of glutamate andglutamine but not of GABA in cultured cerebellar granule cells. J Neurosci Res 1987;17:424-427.
191. Barbour B, Szatkowski M, Ingledew N et al. Arachidonic acid induces a prolonged inhibi-tion of glutamate uptake into glial cells. Nature 1989; 342:918-920.
192. Volterra A, Trotti D, Cassutti P et al. High sensitivity of glutamate uptake to extracellularfree arachidonic acid levels in rat cortical synaptosomes and astrocytes. J Neurochem 1992;59:600-606.
193. Heyes MP, Brew B, Martin A et al. Cerebrospinal fluid quinolinic acid concentrations areincreased in acquired immune deficiency syndrome. Adv Exp Med Biol 1991; 294:687-690.
194. Merrill JE, Chen ISY. HIV-1, macrophages, glial cells, and cytokines in AIDS nervous sys-tem disease. FASEB J 1991; 5:2391-2397.
195. Blumberg BM, Gelbard HA, Epstein LG. HIV-1 infection of the developing nervous sys-tem: Central role of astrocytes in pathogenesis. Virus Res 1994; 32:253-267.
196. Patton HK, Benveniste EN, Benos DJ. Astrocytes and the AIDS dementia complex. JNeuro-AIDS 1996; 1:111-131.
197. Raine CS. Demyelinating diseases. In: Davis RL, Robertson DM, eds. Textbook of Neuro-pathology. Baltimore: Williams and Wilkins, 1997:627-714.
198. Raine CS, Bornstein MB. EAE: A light and electron microscope study of remyelinationand “sclerosis” in vitro. J Neuropathol Exp Neurol 1970; 29:552-574.
199. Vick RS, Neuberger TJ, DeVries GH. Role of adult oligodendrocytes in remyelination afterneural injury. J Neurotrauma 1992; 9 Suppl 1:S93-103.
200. Blakemore WF. Remyelination by Schwann cells of axons demylinated by intraspinal injec-tion of 6-aminonicotinamide in the rat. J Neurocytol 1975; 4:745-757.
201. Franklin RJM, Crang AJ, Blakemore WF. The role of astrocytes in the remyelination ofglia-free areas of demyelination. Adv Neurol 1993; 59:125-133.
202. Toshniwal P, Tiku ML. Astrocytes can degrade myelin basic protein. Neurology 1986;36:146-147.
203. Maeda A, Sobel RA. Matrix metalloproteinases in the normal human central nervous sys-tem, microglial nodules, and multiple sclerosis lesions. J Neuropathol Exp Neurol 1996;55:300-309.
204. Kostyk SK, Kowall NW, Hause SL. Substance P immunoreactive astrocytes are present inmultiple sclerosis plaques. Brain Res 1989; 504:284-288.
205. May PC, Lampert-Etchells M, Johnson SA et al. Dynamics of gene expression for a hip-pocampal glycoprotein elevated in Alzheimer’s disease and in response to experimentallesions in rat. Neuron 1990; 5:831-839.
206. Wekerle H, Linington C, Lassmann H. Cellular immune reactivity within the CNS. TrendsNeurosci 1986; 9:271-277.
207. Goldmuntz EA, Brosnan CF, Chiu FC et al. Astrocytic reactivity and intermediate filamentmetabolism in experimental autoimmune encephalomyelitis: The effect of suppression withprazosin. Brain Res 1986; 397:16-26.
208. Cammer W, Tansey FA, Brosnan CF. Reactive gliosis in the brains of Lewis rats with ex-perimental allergic encephalomyelitis. J Neuroimmunol 1990; 27:111-120.
209. Chung IY, Norris JG, Benveniste EN. Differential tumor necrosis factor alpha expressionby astrocytes from experimental allergic encephalomyelitis-susceptible and -resistant ratstrains. J Exp Med 1991; 173:801-811.
210. Merrill JE, Kono DH, Clayton J et al. Inflammatory leukocytes and cytokines in thepeptide-induced disease of experimental allergic encephalomyelitis in SJL and B10.PL mice.Proc Natl Acad Sci USA 1992; 89:574-578.
211. Ransohoff RM, Hamilton TA, Tani M et al. Astrocyte expression of mRNA encodingcytokines IP-10 and JE/MCP-1 in experimental autoimmune encephalomyelitis. FASEB J1993; 7:592-600.
212. Glabinski AR, Tani M, Aras S et al. Regulation and function of central nervous systemchemokines. Int J Dev Neurosci 1995; 13:153-165.

63Astrocyte Pathophysiology in Disorders of the Central Nervous System
213. Frohman EM, Vayuvegula B, Gupta S et al. Norepinephrine inhibits gamma interferon-induced major histocompatability class II (Ia) antigen expression on cultured astrocytesvia beta2-adrenergic signal transduction mechanisms. Proc Natl Acad Sci USA 1988;85:1292-1296.
214. Frohman E, Frohman T, Vayuvegula B et al. Vasoactive intestinal polypeptide inhibits theexpression of the MHC class II antigens on astrocytes. J Neurol Sci 1988; 88:339-346.
215. Massa PT, ter Meulen V, Fontana A. Hyperinductibility of Ia antigen on astrocytes corre-lates with strain-specific susceptibility to experimental autoimmune encephalomyelitis. ProcNatl Acad Sci USA 1987; 84:4219-4223.
216. Oh LYS, Yong VW. Astrocytes promote process outgrowth by adult human oligodendro-cytes in vitro through interaction between bFGF and astrocyte extracellular matrix. Glia1996; 17:237-253.
217. García-Segura LM, Pérez J, Pons S et al. Localization of insulin-like growth factor I(IGF-I)-like immunoreactivity in the developing and adult rat brain. Brain Res 1991;560:167-174.
218. Garcia-Estrada J, Garcia-Segura LM, Torres-Aleman I. Expression of insulin-like growthfactor I by astrocytes in response to injury. Brain Res 1992; 592:343-347.
219. Gluckman P, Klempt N, Guan J et al. A role for IGF-1 in the rescue of CNS neuronsfollowing hypoxic-ischemic injury. Biochem Biophys Res Commun 1992; 182:593-599.
220. Mozell RL, McMorris FA. Insulin-like growth factor I stimulates oligodendrocyte develop-ment and myelination in rat brain aggregate cultures. J Neurosci Res 1991; 30:382-390.
221. Schecter R, Yen S-HC, Terry RD. Fibrous astrocytes in senile dementia of the Alzheimertype. J Neuropathol Exp Neurol 1981; 40:95-101.
222. Mancardi GL, Liwnicz BH, Mandybur TI. Fibrous astrocytes in Alzheimer’s disease andsenile dementia of Alzheimer type. Acta Neuropathol 1983; 61:76-80.
223. Delacourte A. General and dramatic glial reaction in Alzheimer brains. Neurology 1990;40:33-37.
224. Jorgensen OS, Brooksbank BWL, Balazs R. Neuronal plasticity and astrocytic reaction inDown syndrome and Alzheimer’s disease. J Neurol Sci 1990; 98:63-79.
225. Le Prince G, Delaere P, Fages C et al. Alterations in glial fibrillary acidic protein mRNAlevel in the aging brain and in senile dementia of the Alzheimer type. Neurosci Lett 1993;151:71-73.
226. Mandybur TI, Chuirazzi CC. Astrocytes and the plaques of Alzheimer’s disease. Neurology1990; 40:635-639.
227. LeBlanc AC. The role of β-amyloid peptide in Alzheimer’s disease. Metab Brain Dis 1994;9:3-31.
228. Dickson DW. The pathogenesis of senile plaques. J Neuropathol Exp Neurol 1997;56:321-339.
229. Price DL, Borchelt DR, Walker LC et al. Toxicity of synthetic A beta peptides and model-ing of Alzheimer’s disease. Neurobiol Aging 1992; 13:623-625.
230. Pike CJ, Burdick D, Walencewicz AJ et al. Neurodegeneration induced by β-amyloid pep-tides in vitro: The role of peptide assembly state. J Neurosci 1993; 13:1676-1687.
231. Meda L, Cassatella MA, Szendrei GI et al. Activation of microglial cells by β-amyloid pro-tein and interferon-gamma. Nature 1995; 374:647-650.
232. Lorton D, Kocsis JM, King L et al. β-Amyloid induces increased release of interleukin-1beta from lipopolysaccharide-activated human monocytes. J Neuroimmunol 1996; 67:21-29.
233. Giulian D, Haverkamp LJ, Yu JH et al. Specific domains of β-amyloid from Alzheimerplaque elicit neuron killing in human microglia. J Neurosci 1996; 16:6021-6037.
234. Pike CJ, Cummings BJ, Monzavi R et al. β-amyloid-induced changes in cultured astrocytesparallel reactive astrocytosis associated with senile plaques in Alzheimer’s disease. Neuro-science 1994; 63:517-531.
235. Canning DR, McKeon RJ, DeWitt DA et al. β-Amyloid of Alzheimer’s disease induces re-active gliosis that inhibits axonal outgrowth. Exp Neurol 1993; 124:289-298.
236. Pasternack JM, Abraham CR, Van Dyke BJ et al. Astrocytes in Alzheimer’s disease graymatter express α1-antichymotrypsin mRNA. Am J Pathol 1989; 135:827-834.

Astrocytes in Brain Aging and Neurodegeneration64
237. Gollin PA, Kalaria RN, Eikelenboom P et al. Alpha1-antitrypsin and alpha1-antichymo-trypsin are in the lesions of Alzheimer’s disease. NeuroReport 1992; 3:201-203.
238. Abraham CR, Shirahama T, Potter H. α1-Antichymotrypsin is associated solely with amy-loid deposits containing the beta-protein. Amyloid and cell localization of 1-antichymo-trypsin. Neurobiol Aging 1990; 11:123-129.
239. Rozemuller JM, Abbink JJ, Kamp AM et al. Distribution pattern and functional state ofα1-antichymotrypsin in plaques and vascular amyloid in Alzheimer’s disease. An immuno-histochemical study with monoclonal antibodies against native and inactivated α1-anti-chymotrypsin. Acta Neuropathol 1991; 82:200-207.
240. Fraser PE, Nguyen JT, McLachlan DR et al. α1-Antichymotrypsin binding to Alzheimer Aβpeptides is sequence specific and induces fibril disaggregation in vitro. J Neurochem 1993;61:298-305.
241. Ma J, Yee A, Brewer BJ et al. Amyloid-associated proteins α1-antichymotrypsin andapolipoprotein E promote assembly of Alzheimer β-protein into filaments. Nature 1994;372:92-94.
242. Kanemaru K, Meckelein B, Marshall DCL et al. Synthesis and secretion of activeα1-antichymotrypsin by murine primary astrocytes. Neurobiol Aging 1996; 17:767-771.
243. Su JH, Cummings BJ, Cotman CW. Localization of heparan sulfate glycosaminoglycan andproteoglycan core protein in aged brain and Alzheimer’s disease. Neuroscience 1992;51:801-813.
244. Snow AD, Nochlin D, Sekiguchi R et al. Identification and immunolocalization of a newclass of proteoglycan (keratan sulfate) to the neuritic plaques of Alzheimer’s disease. ExpNeurol 1996; 138:305-317.
245. Brunden KR, Richtercook NJ, Chaturveti N et al. pH-Dependent binding of syntheticβ-amyloid peptides to glycosaminoglycans. J Neurochem 1993; 61:2147-2154.
246. Crutcher KA, Anderton BH, Barger SW et al. Cellular and molecular pathology inAlzheimer’s disease. Hippocampus 1993; 3 Spec No:271-287.
247. Snow AD, Malouf AT. In vitro and in vivo models to unravel the potential roles of beta/A4 in the pathogenesis of Alzheimer’s disease. Hippocampus 1993; 3 Spec No:257-267.
248. Mahley RW. Apolipoprotein E: Cholesterol transport protein with expanding role in cellbiology. Science 1988; 240:622-630.
249. Roses AD. On the metabolism of apolipoprotein E and the Alzheimer diseases. Exp Neurol1995; 132:149-156.
250. Diedrich JF, Minnigan H, Carp RI et al. Neuropathological changes in scrapie andAlzheimer’s disease are associated with increased expression of apolipoprotein E and cathe-psin D in astrocytes. J Virol 1991; 65:4759-4768.
251. Namba Y, Tomonaga M, Kawasaki H et al. Apolipoprotein E immunoreactivity in cerebralamyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amy-loid in Creutzfeldt-Jakob disease. Brain Res 1991; 541:163-166.
252. Coria F, Castano E, Prelli F et al. Isolation and characterization of amyloid P componentfrom Alzheimer’s disease and other types of cerebral amyloidosis. Lab Invest 1988;58:454-458.
253. Strittmatter WJ, Saunders A, Schmechel D et al. Apolipoprotein E: High avidity binding toβ-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease.Proc Natl Acad Sci USA 1993; 90:1977-1981.
254. Wisniewski T, Golabek A, Matsubara E et al. Apolipoprotein E: Binding to a solubleAlzheimer’s beta-amyloid. Biochem Biophys Res Commun 1993; 192:359-365.
255. Geddes JW, Monaghan DT, Cotman CW et al. Plasticity of hippocampal circuitry inAlzheimer’s disease. Science 1985; 230:1179-1181.
256. Pike CJ, Cummings BJ, Cotman CW. Early association of reactive astrocytes with senileplaques in Alzheimer’s disease. Exp Neurol 1995; 132:172-179.
257. Gray CW, Patel AJ. Characterization of a neurotrophic factor produced by cultured astro-cytes involved in the regulation of subcortical cholinergic neurons. Brain Res 1992;574:257-265.

65Astrocyte Pathophysiology in Disorders of the Central Nervous System
258. Akiyama H, Kawamata T, Yamada T et al. Expression of intercellular adhesion molecule(ICAM)-1 by a subset of astrocytes in Alzheimer disease and some other neurodegenerativedisorders. Acta Neuropathol 1993; 85:628-634.
259. Araujo DM, Cotman CW. Beta-amyloid stimulates glial cells in vitro to produce growthfactors that accumulate in senile plaques in Alzheimer’s disease. Brain Res 1992; 569:141-145.
260. Gijbels K, Proost P, Masure S et al. Gelatinase B is present in the cerebrospinal fluid dur-ing experimental autoimmune encephalomyelitis and cleaves myelin basic protein. J NeurosciRes 1993; 36:432-440.
261. Siman R, Card JP, Nelson RB et al. Expression of beta-amyloid precursor protein in reac-tive astrocytes following neuronal damage. Neuron 1989; 3:275-285.
262. Berkenbosch F, Refolo LM, Friedrich VL Jr et al. The Alzheimer’s amyloid precursor pro-tein is produced by type I astrocytes in primary cultures of rat neuroglia. J Neurosci Res1990; 25:431-440.
263. Mark RJ, Blanc EM, Mattson MP. Amyloid β-peptide and oxidative cellular injury inAlzheimer’s disease. Mol Neurobiol 1996; 12:211-224.
264. Penfield W, Humphreys S. Epileptogenic lesions of the brain. A histologic study. ArchNeurol Psychiat 1940; 43:240-261.
265. Harris AB. Cortical neuroglia in experimental epilepsy. Exp Neurol 1975; 49:691-715.266. Westrum LE, White LEJ, Ward AAJ. Morphology of the experimental epileptic focus. J
Neurosurg 1964; 21:1033-1044.267. Armstrong DD. The neuropathology of of temporal lobe epilepsy. J Neuropathol Exp Neurol
1993; 52:433-443.268. Fischer J, Holubar J, Malik V. Neurohistochemical study of the development of the experi-
mental epileptogenic cortical cobalt-gelatine focus in rats and its correlation with the on-set of epileptic electrical activity. Acta Neuropathol 1968; 11:45-54.
269. Payan H, Toga M, Berard-Badier M. The pathology of post-traumatic epilepsies. Epilepsia1970; 11:81-94.
270. Orzi F, Zoli M, Passarelli F et al. Repeated electroconvulsive shock increases glial fibrillaryacidic protein, ornithine decarboxylase, somatostatin and cholecystokinin immunoreactivi-ties in the hippocampal formation of the rat. Brain Res 1990; 533:223-231.
271. Steward O. Electroconvulsive seizures upregulate astroglial gene expression selectively inthe dentate gyrus. Mol Brain Res 1994; 25:217-224.
272. Stringer JL. Repeated seizures increase GFAP and vimentin in the hippocampus. Brain Res1996; 717:147-153.
273. Khurgel M, Ivy GO. Astrocytes in kindling: Relevance to epileptogenesis. Epilepsy Res 1996;26:163-175.
274. Brotchi J. The activated astrocyte - a histochemical approach to the epileptic focus. In:Schoffeniels E, Franck G, Towers DB, Hertz L, eds. Dynamic Properties of Glial Cells.Oxford: Pergamon, 1978:429-434.
275. Lee SH, Magge S, Spencer DD et al. Human epileptic astrocytes exhibit increased gap junc-tion coupling. Glia 1995; 15:195-202.
276. Cornell-Bell AH, Williamson A. Hyperexcitability of neurons and astrocytes in epileptichuman cortex. In: Fedoroff S, Juurlink BHJ, Doucette R, eds. Biology and Pathology ofAstrocyte-Neuron Interactions. New York: Plenum Press, 1993:51-65.
277. Norenberg MD, Chu N-S. Aminophylline-induced preictal alterations in cortical astrocytes.Exp Neurol 1977; 54:340-351.
278. Ward A. Glia and epilepsy. In: Schoffeniels E, Franck G, Hertz L, Tower DB, eds. DynamicProperties of Glial Cells. Oxford: Pergamon Press, 1978:413-427.
279. Meldrum BS. Pharmacology of GABA. Clin Neuropharmacol 1982; 5:293-316.280. Bradford HF. Glutamate, GABA and epilepsy. Prog Neurobiol 1995; 47:477-511.281. Schousboe A, Larsson OM, Frandsen A et al. Neuromodulatory actions of glutamate, GABA
and taurine: Regulatory role of astrocytes. Adv Exp Med Biol 1991; 296:165-180.282. Schousboe A, Thorbek P, Hertz L et al. Effects of GABA analogues of restricted conforma-
tion on GABA transport in astrocytes and brain cortex slices and on GABA receptor bind-ing. J Neurochem 1979; 33:181-189.

Astrocytes in Brain Aging and Neurodegeneration66
283. Schousboe A, Hjeds H, Engler J et al. Tissue distribution, metabolism, anticonvulsant effi-cacy and effect on brain amino acid levels of the glia-selective gamma-aminobutyric acidtransport inhibitor 4,5,6,7-tetrahydroisoxazolo (4,5-c) pyridin-3-ol in mice and chicks. JNeurochem 1986; 47:758-63.
284. Pollen DA, Trachtenberg MC. Neuroglia: Gliosis and focal epilepsy. Science 1970;167:1252-1253.
285. Lewis DV, Mutsuga N, Schuette WH et al. Potassium clearance and reactive gliosis in thealumina gel lesion. Epilepsia 1977; 18:132-136.
286. Somjen GG. Acidification of interstitial fluid in hippocampal formation caused by seizuresand by spreading depression. Brain Res 1984; 311:186.
287. Benjamin AM. Ammonia. In: Lajtha A, ed. Handbook of Neurochemistry, Vol 1. New York:Plenum Press, 1982:117-137.
288. Cooper AJL, Plum F. Biochemistry and physiology of brain ammonia. Physiol Rev 1987;67:440-519.
289. Lapin IP. Convulsant action of intracerebroventricularly administered l-kynurenine sul-fate, quinolinic acid and other derivatives of succinic acid, and effects of amino acids:Structure-activity relationships. Neuropharmacology 1982; 21:1227-1233.
290. Du F, Williamson J, Bertram E et al. Kynurenine pathway enzymes in a rat model of chronicepilepsy: Immunohistochemical study of activated glial cells. Neuroscience 1993; 55:975-989.
291. Sutula TP, Cascino G, Cavazos JIP et al. Mossy fiber synaptic reorganization in the epilep-tic human temporal lobe. Ann Neurol 1989; 26:321-330.
292. Babb TL, Kupfer WR, Pretorius JK et al. Synaptic reorganization by mossy fibers in hu-man epileptic fascia dentata. Neuroscience 1991; 42:351-363.
293. Represa A, Jorquera I, Le Gal La Salle G et al. Epilepsy induced collateral sprouting ofhippocampal mossy fibers: Does it induce the development of ectopic synapses with gran-ule cell dendrites? Hippocampus 1993; 3:257-268.
294. Niquet J, Jorquera I, Faissner A et al. Gliosis and axonal sprouting in the hippocampus ofepileptic rats are associated with an increase of tenascin-C immunoreactivity. J Neurocytol1995; 24:611-624.
295. Gall CM, Berschauer R, Isackson PJ. Seizures increase basic fibroblast growth factor mRNAin adult rat forebrain neurons and glia. Mol Brain Res 1994; 21:190-205.
296. Niquet J, Jorquera I, Ben-Ari Y et al. Proliferative astrocytes may express fibronectin-likeprotein in the hippocampus of epileptic rats. Neurosci Lett 1994; 180:13-16.
297. Niquet J, Gillian A, Ben-Ari Y et al. Reactive glial cells express a vitronectin-like protein inthe hippocampus of epileptic rats. Glia 1996; 16:359-367.
298. Le Gal La Salle G, Rougon G, Valin A. The embryonic form of neural cell surface molecule(E-NCAM) in the rat hippocampus and its reexpression on glial cells following kainicacid-induced status epilepticus. J Neurosci 1992; 12:872-882.
299. Niquet J, Jorquera I, Ben-Ari Y et al. NCAM immunoreactivity on mossy fibers and reac-tive astrocytes in the hippocampus of epileptic rats. Brain Res 1993; 626:106-116.
300. Griffin WST, Yeralan O, Sheng JG et al. Overexpression of the neurotrophic cytokine S100in human temporal lobe epilepsy. J Neurochem 1995; 65:228-233.
301. Tauck DL, Nadler JV. Evidence for functional mossy fiber sprouting in hippocampal for-mation of kaininc acid treated rats. J Neurosci 1985; 5:1016-1022.
302. Ben-Ari Y, Represa A. Brief seizure episodes induce long-term potentiation and mossy fi-bre sprouting in the hippocampus. Trends Neurosci 1990; 13:312-318.
303. DeLong MR. Primate models of movement disorders of basal ganglia origin. TrendsNeurosci 1990; 13:281-285.
304. Turski L, Bressler K, Rettig KJ et al. Protection of substantia nigra from MPP+ neurotoxic-ity by N-methyl-D-aspartate antagonists. Nature 1991; 349:414-418.
305. Greenamyre JT. Glutamate-dopamine interactions in the basal ganglia: Relationship toParkinson’s disease. J Neural Trans 1993; 91:255-269.
306. Hazell AS, Itzhak Y, Liu H et al. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)decreases glutamate uptake in cultured astrocytes. J Neurochem 1997; 68:2216-2219.

67Astrocyte Pathophysiology in Disorders of the Central Nervous System
307. Schiffer D, Cordera S, Cavalla P et al. Reactive astrogliosis of the spinal cord in amyo-trophic lateral sclerosis. J Neurol Sci 1996; 139 Suppl.27-33.
308. Rothstein JD. Excitotoxic mechanisms in the pathogenesis of amyotrophic lateral sclerosis.Adv Neurol 1995; 68:7-20.
309. Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by brain and spinalcord in amyotrophic lateral sclerosis. New Engl J Med 1992; 326:1464-1468.
310. Rothstein JD, Van Kammen M, Levey AI et al. Selective loss of glial glutamate transporterGLT-1 in amyotrophic lateral sclerosis. Ann Neurol 1995; 38:73-84.
311. Rothstein JD, Dykes-Hoberg M, Pardo CA et al. Knockout of glutamate transporters re-veals a major role for astroglial transport in excitotoxicity and clearance of glutamate.Neuron 1996; 16:675-686.
312. Rosen DR, Siddique T, Patterson D et al. Mutations in Cu/Zn superoxide dismutase geneare associated with familial amyotrophic lateral sclerosis. Nature 1993; 362:59-62.
313. Kato S, Shimoda M, Watanabe Y et al. Familial amyotrophic lateral sclerosis with a twobase pair deletion in superoxide dismutase 1 gene: Multisystem degeneration with intracy-toplasmic hyaline inclusions in astrocytes. J Neuropathol Exp Neurol 1996; 55:1089-1101.
314. Gurney ME, Pu H, Chiu AY et al. Motor neuron degeneration in mice that express a hu-man Cu,Zn superoxide dismutase mutation. Science 1994; 264:1772-1775.
315. Bruijn LI, Becher MW, Lee MK et al. ALS-linked SOD1 mutant G85R mediates damage toastrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neu-ron 1997; 18:327-338.
316. O’Reilly SA, Roedica J, Nagy D et al. Motor neuron-astrocyte interactions and levels ofCu,Zn superoxide dismutase in sporadic amyotrophic lateral sclerosis. Exp Neurol 1995;131:203-210.
317. Yu BP. Cellular defenses against damage from reactive oxygen species. Physiol Rev 1994;74:139-161.
318. Halliwell B. Free radicals, antioxidants and human disease: Curiosity, cause, or consequence?Lancet 1994; 344:721-724.
319. Norenberg MD. Astrocytes in ischemic injury. In: Ginsberg MD, Bogousslavsky J, eds.Cerebrovascular Disease: Pathophysiology, Diagnosis, and Management. Cambridge:Blackwell, 1997.
320. Petito CK, Babiak T. Early proliferative changes in astrocytes in postischemic noninfarctedrat brain. Ann Neurol 1982; 11:510-518.
321. Hori O, Matsumoto M, Maeda Y et al. Metabolic and biosynthetic alterations in culturedastrocytes exposed to hypoxia/reoxygenation. J Neurochem 1994; 62:1489-1495.
322. Richter D, Dawson RMC. The ammonia and glutamine content of the brain. J Biol Chem1948; 176:1119-1210.
323. Goldberg RN, Cabal LA, Sinatra FR et al. Hyperammonemia associated with perinatal as-phyxia. Pediatrics 1979; 64:336-341.
324. Rischke R, Krieglstein J. Postischemic neuronal damage causes astroglial activation andincrease in local cerebral glucose utilization of rat hippocampus. J Cereb Blood Flow Metab1991; 11:106-113.
325. Yu ACH, Gregory GH, Chan PH. Hypoxia-induced dysfunction and injury of astrocytes inprimary cell cultures. J Cereb Blood Flow Metab 1989; 9:20-28.
326. Swanson RA. Astrocyte glutamate uptake during chemical hypoxia in vitro. Neurosci Lett1992; 147:143-146.
327. Rothstein JD, Jin L, Dykes-Hoberg M et al. Chronic inhibition of glutamate uptake pro-duces a model of slow neurotoxicity. Proc Natl Acad Sci USA 1993; 90:6591-6595.
328. Robinson MB, Djali S, Buchhalter JR. Inhibition of glutamate uptake with L-trans-pyrrolidine-2,4-dicarboxylate potentiates glutamate neurotoxicity in primary hippocampalcultures. J Neurochem 1993; 61:2099-2103.
329. Torp R, Lekieffre D, Levy LM et al. Reduced postischemic expression of a glial glutamatetransporter, GLT1, in the rat hippocampus. Exp Brain Res 1995; 103:51-58.
330. Pogun S, Dawson V, Kuhar MJ. Nitric oxide inhibits 3H-glutamate transport in synapto-somes. Synapse 1994; 18:21-26.

Astrocytes in Brain Aging and Neurodegeneration68
331. Butterworth RF. Pathophysiology of hepatic encephalopathy; the ammonia hypothesis re-visited. In: Bengtsson F, Jeppsson B, Almdal T, Vilstrup H, eds. Hepatic Encephalopathyand Metabolic Nitrogen Exchange. Boca Raton: CRC Press, 1993:9-24.
332. Basile AS, Jones EA, Skolnick P. The pathogenesis and treatment of hepatic encephalopa-thy: Evidence for the involvement of benzodiazepine receptor ligands. Pharmacol Rev 1991;43:27-71.
333. Norenberg MD. Hepatic encephalopathy. In: Kettenmann H, Ransom BR, eds. Neuroglia.New York: Oxford, 1995:950-963.
334. Rao VLR, Murthy CRK, Butterworth RF. Glutamatergic synaptic dysfunction inhyperammonemic syndromes. Metab Brain Dis 1992; 7:1-20.
335. Gregorios JB, Mozes LW, Norenberg LOB et al. Morphologic effects of ammonia on pri-mary astrocyte cultures. I. Light microscopic studies. J Neuropathol Exp Neurol 1985;44:397-403.
336. Gregorios JB, Mozes LW, Norenberg MD. Morphologic effects of ammonia on primaryastrocyte cultures. II. Electron microscopic studies. J Neuropathol Exp Neurol 1985;44:404-414.
337. Sobel RA, DeArmond SJ, Forno LS et al. Glial fibrillary acidic protein in hepatic encepha-lopathy. An immunohistochemical study. J Neuropathol Exp Neurol 1981; 40:625-632.
338. Norenberg MD. Astrocytic-ammonia interactions in hepatic encephalopathy. Semin LiverDis 1996; 16:245-253.
339. Szerb JC, Butterworth RF. Effect of ammonium ions on synaptic transmission in the mam-malian central nervous system. Prog Neurobiol 1992; 39:135-153.
340. Bender AS, Norenberg MD. Effects of ammonia on L-glutamate uptake in cultured astro-cytes. Neurochem Res 1996; 21:567-573.
341. Zhou B, Norenberg MD. Ammonia downregulates GLAST mRNA glutamate transporterin cultured astrocytes. Soc Neurosci Abstr 1997; 23:1461.
342. Norenberg MD, Huo Z, Neary JT et al. The glial glutamate transporter in hyperammonemiaand hepatic encephalopathy: Relation to energy metabolism and glutamatergic neurotrans-mission. Glia 1997; 21:124-133.
343. McCarthy KD, Harden TK. Identification of two benzodiazepine binding sites on cells cul-tured from rat cerebral cortex. J Pharmacol Exp Ther 1981; 216:183-191.
344. Bender AS, Hertz L. Binding of (3H) R05-4864 in primary cultures of astrocytes. Brain Res1985; 341:41-9.
345. Itzhak Y, Baker L, Norenberg MD. Characterization of the peripheral-type benzodiazepinereceptor in cultured astrocytes: evidence for multiplicity. Glia 1993; 9:211-218.
346. Anholt RRH, Pedersen PL, DeSouza EB et al. The peripheral-type benzodiazepine recep-tor: Localization to the mitochondrial outer membrane. J Biol Chem 1986; 261:576-583.
347. Papadopoulos V. Peripheral-type benzodiazepine/diazepam binding inhibitor receptor: Bio-logical role in steroidogenic cell function. Endocr Rev 1993; 14:222-240.
348. Mok WM, Herschkowitz S, Krieger NR. In vivo studies identify 5alpha-pregnan-3alpha-ol-20-one as an active anesthetic agent. J Neurochem 1991; 57:1296-1301.
349. Bitran D, Hilvers RJ, Kellog CK. Anxiolytic effects of 3alpha-hydroxy-5alpha[beta]-pregnan-20-one: Endogenous metabolites of progesterone that are active at the GABA-A receptor.Brain Res 1991; 561:157-161.
350. Wieland S, Lan NC, Mirasedeghi S et al. Anxiolytic activity of the progesterone metabolite5alpha-pregnan-3alpha-ol-20-one. Brain Res 1991; 565:263-268.
351. Norenberg MD, Itzhak Y, Bender AS. The peripheral benzodiazepine receptor andneurosteroids in hepatic encephalopathy. Adv Exp Biol Med 1997; 420:95-111.

Part II
Astrocytes in Human Brain Senescenceand Neurodegenerative Disorders


CHAPTER 4
Astrocytes in Brain Aging and Neurodegeneration, edited by Hyman M. Schipper.©1998 R.G. Landes Company.
Glial Responses to Injury,Disease, and AgingLawrence F. Eng and Yuen Ling Lee
Introduction
Astrocytes comprise 25% of the cells and 35% of the mass in the central nervous system(CNS). They have intimate contact with the pia of the brain, neurons, endothelial cells,
pericytes, myelin membrane internodes, synapses, and microglia. Astrocytes have special-ized functions depending on their location in the CNS and participate in a variety of impor-tant physiologic and pathologic processes. Any kind of stress or injury to the CNS induces astereotypic response in the astrocytes termed reactive gliosis and, in the extreme case,astrogliosis and plaque or scar formation. The prominence of astroglial reactions in variousinjuries/diseases, the rapidity of the astroglial response and the evolutionary conservationof reactive astrogliosis indicate that reactive astrocytes fulfill important functions in theCNS.1,2 Astrogliosis is best characterized by rapid synthesis of glial fibrillary acidic protein(GFAP), a cytoskeletal intermediate filament. Numerous in vitro and in vivo studies on themolecular profiles of substances which are upregulated during astrocyte activation docu-ment the complex and varied responses of astrocytes to injury. Besides morphologicalchanges, reactive astrocytes have also been shown to upregulate a number of different mol-ecules including other glial markers (S-100β), cytokines (IL-1, IL-6, IFN and TNF), growthfactors (FGF, NGF, NT-3, CNTF) and heat shock proteins.3 Reactive astrocytes are thoughtto play a role in the healing phase following CNS injury by actively monitoring and control-ling the molecular and ionic contents of the extracellular space of the CNS. It has beenhypothesized that activated astroglia may benefit the damaged neurons by participating inseveral important biological processes such as regulation of neurotransmitter levels, repairof the extracellular matrix, control of the blood-CNS interface, control of transport pro-cesses, and trophic support to the damaged cells. On the other hand, gliosis which occursduring normal aging and after injury may result in detrimental pathological effects by in-terfering with the residual neuronal circuits through inhibiting regeneration or preventingremyelination.
Astrocyte Intermediate Filament, Glial Fibrillary Acidic ProteinAs a member of the cytoskeletal protein family, GFAP is thought to be important in
modulating astrocyte motility and shape by providing structural stability to extensions ofastrocytic processes. It was first isolated from a multiple sclerosis (MS) plaque in 1969(Fig. 4.1). GFAP is the principal 8-9 nm intermediate filament in mature astrocytes.2 Ge-nomic clones have been obtained from human, mouse and rat GFAP genes. Each gene is

Astrocytes in Brain Aging and Neurodegeneration72
composed of nine exons distributed over about 10 kb of DNA and yields a mature mRNAof about 3 kb. The coding sequences for the three genes are highly homologous. Stronghomology also extends upstream of the RNA start site for about 200 bp, recurs betweenabout -1300 and 1700 (RNA startpoint = +1) and is present in some intronic regions. Theprimary sites for the initiation of RNA and protein synthesis are essentially identical for thethree genes, and each contains a TAT-like sequence (CATAAA or AATAA) in the expected5'-flanking position. In addition to GFAP-α, two additional mRNAs that start at differentsites have been identified, GFAP-β and GFAP-γ. The different tissue distributions of GFAP-α,-β, and -γ mRNAs suggest that the synthesis of each is subject to unique control. All tran-scriptional studies to date either have explicitly measured GFAP-α or have not distinguishedamong the possible mRNA isotypes. GFAP transgenes have been used extensively to studysignaling pathways that operate during development, disease, and injury—all states thatincrease GFAP gene activity.4,5
Fig. 4.1. Diagram illustrates the isolation of GFAP from a MS plaque for amino acid analysis.

73Glial Responses to Injury, Disease, and Aging
Astrocytes in Experimental GliosisIncreased protein content or immunostaining of GFAP has been found in experimen-
tal models involving gliosis, such as cryogenic lesions,6 stab wounds,7-15 toxic lesions16-18
and experimental allergic encephalomyelitis (EAE).19-22 Increased levels of GFAP mRNAhave been found in the 6-hydroxydopamine lesion-bearing substantia nigra in the rat,17,23
mechanical lesions of rat cerebral cortex,24-26 entorhinal cortex lesions,27 corticospinalaxotomy,28 EAE29 and lesions of the dentate gyrus.30
Astrocytes in DiseaseWhile reactive gliosis occurs with any type of insult to the CNS, the anatomical region,
severity of gliosis, and developmental time sequence vary in amyotrophic lateral sclerosis(ALS), Gerstmann-Straussler syndrome (GSS), Huntington’s, Wilson’s, Pick’s, Parkinson’s,Alzheimer’s, and Down’s diseases.31 Astrocytic gliosis is a prominent neuropathologic changein Alzheimer’s disease (AD). Numerous reports have shown reactive astrocytes in AD brains,most frequently in association with neuritic plaques.32-38 It is not known whether gliosisprecedes the appearance of β-amyloid peptide (βAP) or whether it might be induced byβAP. Moreover, cases have been reported where diffuse βAP plaques in advanced AD are notassociated with reactive astrocytes.39-41 Diffuse plaques in advanced AD cases may not beidentical to the early lesions which may characterize preclinical AD cases. Our approach toidentifying early pathological changes in AD has been to study young cases of Down’s syn-drome (DS), because individuals with DS develop the neuropathological changes of ADprematurely.42 DS cases show a very mild form of AD pathology from the second and thirddecades of life; however, DS cases from the fifth decade and older show fully developedAD.43 Since the amygdala is a site of neuropathologic change, including extensive gliosis, inboth AD and DS,44-46 we examined the amygdala for evidence of astrocytic gliosis in youngand old cases of DS and AD. We also compared the distribution of astrocytes with the distri-bution of βAP deposits in the amygdala to determine whether the βAP deposits were spa-tially related to astrocytes. Our results demonstrated that astrocytic hypertrophy is not anearly change in the AD-like process of DS and that astrocyte morphology did not differ inyoung DS cases from that of controls. Furthermore, there was no consistent spatial relation-ship between the numerous βAP deposits observed in the young DS cases and astrocytes.47
In agreement with our study, Michetti et al48 did not observe a difference in the morphologyof S-100 labeled astrocytes in the cerebellum of DS cases (ranging from newborn to 26months) relative to controls. Griffin et al,49 however, have reported that S-100 labeled astro-cytes were increased in size in DS cases aged 2 days, 3.5 months, and 34 years. Our datademonstrate that the βAP deposits in young DS brain, which may be similar to those inpreclinical AD, are not associated with reactive gliosis.47 In a recent study of DS brains,GFAP was found to be expressed at levels significantly below those of controls, suggestingthat trisomy 21 exerts a suppressive effect on GFAP gene expression.50
Moossy et al51 have described two cases of “primary dementia” not distinguishable fromAD but devoid of neurofibrillary tangles. Astrocytosis was observed in several subcorticalnuclei and mainly in the thalamus. Astrogliosis has been demonstrated by chemical analysisand immunocytochemistry for GFAP in Huntington’s disease.52,53 Huntington disease brainswere used as positive controls for gliosis in a study of schizophrenic patient brains.53 Em-ploying quantitative image analysis of brains immunostained for GFAP, Roberts et al54 didnot find significant differences in 20 different brain areas between schizophrenic andHuntington’s disease groups, in contrast to previous reports of gliosis in schizophrenic brains.Holzer’s histological stain for glial fibrils showed increased fibrillary gliosis that affectedmainly the periventricular structures of the diencephalon, the periventricular structures ofthe periaqueductal region of the mesencephalon, and the basal forebrain of schizophrenic

Astrocytes in Brain Aging and Neurodegeneration74
subjects. The hypothalamus, midbrain tegmentum, and innominata were also involved.55
Astrocytes in human brain immunostain less intensely when the death-autopsy interval isprolonged, because GFAP is very sensitive to proteolysis.56,57 In contrast, Holzer’s stain promi-nently labels mildly gliotic tissue which may not be evident by GFAP immunostaining(L. Forno, personal communication). Significant astrogliosis is also characteristic of ALS.In a study of 13 ALS brains, gliosis was present in six different control areas and was differ-ent from that seen in AD, Pick’s disease, and Parkinson’s disease. GFAP staining within thesubcortical white matter of ALS was unlike that of any other disease examined with theexception of cerebral infarction.58 The prion diseases, which include CJD, GSS, and kuru inhumans and scrapie in animals, have been characterized as being “hypergliotic” because thegliosis often appears to be out of proportion to the degree of nerve cell loss or injury.59,60
Astrocyte hypertrophy and gliosis were concentrated in the cerebellum of the ataxic form ofCJD.61,62 The brains from hamsters infected with the human CJD agent showed a gradualincrease of GFAP and GFAP mRNA during the course of the disease.63 Molecular studiessuggest that a single abnormal nerve cell protein PrPsc may cause both the neuronal degen-eration and reactive gliosis.64 Finally, in Wilson’s disease and hepatic encephalopathy, gliosisoccurs in demyelinated foci of the brain; however, there is a population of protoplasmicastrocytes in the gray matter (Alzheimer’s type 2 cells) that show a decrease in GFAPcontent.65-68
Astrocyte Activation of GFAP in AstrogliosisWhat are the signals that induce the upregulation of GFAP in normal aging and dis-
ease? Astrocytes can be activated by molecules expressed by microglia, monocyte/macroph-ages, endothelial cells, lymphocytes, by various blood proteins, ions, free radicals, neurotrans-mitters, enzymes, and their degradation products resulting from damaged neurons and glialcells. Whatever the factors may be, they probably induce mild or transient activation whichis less intense than that seen in injury or disease.
Microglial ActivationThe broad distribution of microglia in the CNS is similar to that of astrocytes. Micro-
glial cells are intimately associated with astrocytes, endothelial cells, oligodendrocytes andneurons. Microglial activation appears to be independent of the form of pathological stimulussince uniform changes occur in all models, including proliferation, transformation intophagocytic cells with macrophage morphology, and upregulation of cell surface moleculessuch as the MHC antigens. Microglia responds quickly to a variety of signaling molecules ata very early stage of injury.69-74 Activation often precedes reactions of any other cell type inthe CNS, even before the reactive astrocytic response. They respond to changes in the brain’sstructural integrity, and also to very subtle alterations in their microenvironment, such asan imbalance in homeostasis that precedes histologically detectable pathological changes.75
This may be due to the unique collection of microglial membrane channels which includesan inward-rectifying K+ channel (see below).76,77
Although little is known about the in vivo regulation of microglial proliferation andactivation, both in vivo and in vitro experiments suggest an involvement of cytokines in thisprocess.78,79 T cell derived IFN-γ is the cytokine most widely implicated in the activation ofmicroglia. The long list of events that occur after IFN-γ treatment includes the productionof reactive oxygen intermediates, increase in MHC class II expression and synthesis of comple-ment components. Colony stimulating factors (CSF) have been identified as mediating po-tent activation of microglial cells. The source of these molecules in the brain in vivo is stillcontroversial. Astrocytes have been shown to express macrophage (M)-CSF, granulocyte(G)-CSF, and granulocyte-macrophage (GM)-CSF. Neurons have been shown to secrete

75Glial Responses to Injury, Disease, and Aging
M-CSF in vitro.80 Chemotactic effects on resident microglia and blood monocytes are ex-erted by TGF-β and the β-chemokines, a group of 8-10 kDa proteins with highly conservedcysteines linked by disulfide bonds. Other β-chemokines like monocyte chemoattractantprotein-1 (MCP-1/JE) or macrophage inflammatory protein-1α (MIP-1α) are produced byIL-1β- and TNF-α-activated astrocytes and microglia.81 Early metabolic and ultrastructuralalterations in the neurons, including disintegration of cytoskeletal proteins, synaptic mem-brane changes, decrease in protein synthesis, altered polyamine metabolism or elevated K+
in the intracellular space, may activate the microglia. Potassium can depolarize microgliavia an inward potassium channel; however, they lack a rectifying outward current so thateven a small inward current leads to membrane depolarization with unknown metabolicconsequences. Activated microglia have been reported to release the following compounds:IL-1, IL-6, IL-10, TGF-β and TNF-α. Activated microglia increase with normal aging andexhibit increased expression of MHC II, leukocyte common antigen CD4, and ED1, thelatter a marker of the lysosomal apparatus whose upregulation indicates activation of theendosomal-lysosomal system in aging microglia.82,83
Monocyte/Macrophage ActivationBone marrow-derived monocytes and macrophages infiltrate the CNS following in-
jury or disease. Because of the variety of effects they mediate, monocytes and macrophagesmay play a crucial role in neuroimmunologic disorders such as MS and EAE. Their activa-tion and recruitment into the CNS occur in response to chemokines and cytokines secretedby endogenous cells of the CNS (astrocytes, microglia, and endothelial cells) as well as acti-vated lymphocytes. Activated macrophages secrete inflammatory mediators (IL-1, IL-4, IL-6,IL-8, TNF-α, TGF-β, MIP-1, MIP-2, M-CSF, MCP-1, γIP-10, GRO, RANTES), nitric oxideand proteases which serve to enhance the inflammatory response, promote vascular perme-ability and initiate myelin destruction.84,85 In addition, their role as antigen presenting cellshas been well documented.86
Endothelial Cell ActivationInteraction between various subclasses of inflammatory cells and endothelial cells (ECs)
comprising the mammalian blood-brain barrier (BBB) is an early, important event in thecourse of CNS inflammation, as exemplified by EAE. Breakdown of the BBB through theaction of vasoactive amines and other soluble inflammatory factors (cytokines) and en-hanced endothelial transcytotic activity facilitates the migration of inflammatory cells inthe CNS.87-89 However, studies by Knobler et al90 have suggested that ECs at the intact BBBmay also actively participate in the trafficking of lymphocytes into the CNS. Vascular adhe-sion molecules (selectins) are elaborated in acute brain inflammation and appear to fosterlymphocyte entry into the CNS.91-93 In addition, activated/injured ECs have been shown toexpress factors such as PDGF, plasminogen activator, prostacylin, TNF-β, IL-1 and IL-8.Several of these cytokines play a role in the adhesion of leukocytes to vascular endotheliumand thereby contribute to the inflammatory response.94
Astrocytes in Normal AgingSenescent astrocytes increase in size, become fibrous and exhibit a gradual increase in
GFAP and GFAP mRNA. The increase of GFAP during senescence can be seen in speciesfrom several long-separated evolutionary orders of mammals.95 The augmented GFAP mRNAlargely corresponds to enhanced astrocyte hypertrophy rather than to increases in total num-bers of these cells.96,97 In humans, the increase in GFAP mRNA appears to be negligiblebefore 60 years, but increases occur in the hippocampus and in frontal and temporal cortexduring an average life-span in the absence of specific neurological disease.70 GFAP mRNA

Astrocytes in Brain Aging and Neurodegeneration76
and protein increase 2-fold after midlife in mice and rats.98-102 In a study of aging femalemouse brains employing combined immunocytochemical and in situ hybridization tech-niques, GFAP mRNA and protein exhibited significant age-related increases in the majorwhite matter tracts, including the corpus callosum, fimbria, stria terminalis, and optic tract.Gray matter showed large increases in GFAP mRNA with age in the thalamus and hypo-thalamus, areas typically expressing little GFAP in the young.103 A parallel, age-related in-crease in GFAP intron RNA in the hippocampus, internal capsule, and corpus callosum ofmale rats indicates that the regulation of GFAP expression during aging occurs at the trans-lational level.104
In mice, rats, and humans, increases in GFAP expression with aging occur graduallyand there may be significant inter-individual variability in the degree of hypertrophy.70 Thus,minor degrees of reactive gliosis accompanying early degeneration or inflammatory CNSconditions may be easily overlooked. In other disorders, such as MS or adrenoleukodystrophy,reactive gliosis is generally intense and readily distinguishable from normal backgroundlevels of GFAP expression.105-107
There are two recent studies suggesting that low grade or transient stress might inducea gradual increase in GFAP expression in the course of normal aging. The first is the spread-ing depression (SD) experiments in which resident microglia were shown to release factorsthat activate the GFAP gene without altering astrocyte morphology. Cortical SD (CSD) waselicited in rat brain for one hour by topical application of 4 M potassium chloride solution.This treatment was sufficient to induce a microglial reaction throughout the cortex at 24hours. Activated microglial cells exhibited apparent cellular hypertrophy, increasedimmunostaining with macrophage/microglial antibodies (MUC 100, 102, and OX-42), andde novo expression of MHC class II antigens.75 No neuronal damage or increase in GFAPimmunoreactivity was detected three days after treatment. In a second study, Kraig et al108
reported that enhanced GFAP immunostaining could be demonstrated in rat brain astro-cytes two days after induction of SD (21 DC shift) by application of KCl to the parietalcortex for 3 hr. The increased GFAP staining, however, returned to normal after two weeks.Both of these studies suggest that SD may be a useful technique to delineate the cellularmechanisms subserving GFAP upregulation in reactive astrocytes. For example, SD studiesmay help determine which factors released by transiently activated microglia are respon-sible for subsequent induction of GFAP in these astrocytes.
Finch and co-workers have published a series of timely papers10,98,99,103 which demon-strate that GFAP and GFAP mRNA increase with normal aging in the rodent and humanbrain. Moreover, they showed that this increase in GFAP mRNA could be attenuated inaging rat if they were maintained on a calorie-restricted diet109,110 and hypothesized that theaging-related increase in GFAP is due to oxidative stress. One possible source of oxidativestress in the aging brain could be reactive microglia. A 2-fold increase in activated microgliawas found in the aging rat hippocampus based on immunostaining with OX6, a marker formicroglia/macrophages. Both the increase in reactive microgliosis and the parallel increasesin GFAP expression could be delayed by dietary restriction.111 Such results are consistentwith the effects of food restriction in attenuating oxidative damage to brain membranesduring aging.112,113 To test this hypothesis, glial cell cultures were treated with H202, cys-teamine or bacterial lipopolysaccharide to directly activate microglia. Treatment with H2O2
and cysteamine, but not bacterial lipopolysaccharide, induced GFAP mRNA in mixed cul-tures containing astrocytes, oligodendrocytes, and microglia as well as purified astrocytesalone. Furthermore, the GFAP response to oxidative stress was shown to be regulated at thetranscriptional level.104,114 It was suggested that this control could be mediated by transcrip-tional response elements in the 5' upstream promoter region of the GFAP gene that respond

77Glial Responses to Injury, Disease, and Aging
to oxidative stress, including sites for Fos, Jun and NFκB.114 However, various soluble factorsin addition to reactive oxygen species are released from activated microglia which may regu-late Fos, Jun or NFκB and, therefore, GFAP expression in nearby astrocytes.
Astrocyte Inclusions in Normal AgingCorpora amylacea (CA) arise in the human CNS in the course of normal aging and in
several disease states. They are well-circumscribed, rounded inclusions ranging from 5 to20 µm in diameter, and are well demonstrated by a wide variety of stains, including hema-toxylin and eosin, iodine, Nile blue sulfate, methyl violet, PAS, and Best’s carmine. Most CAare comprised of a densely staining central round zone of amorphous material surroundedby a lighter peripheral rim. Chemical analysis of purified CA showed that they consist of upto 80% glycogen-like substance bound to approximately 1% sulfate and phosphate. In onestudy, protein content was estimated at around 5% whereas reactions for lipids, nucleicacids and sialic acid were negative.115 In another study, purified CA were found to containabout 4% total protein. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis showed protein bands with molecular weights ranging from 24 to 133 kDa.Four bands with molecular weights of 24, 92, 94, and 133 kDa represented the major pro-teins present.116 Immunocytochemical analysis of human brain and spinal cord revealedsmall, rounded bodies expressing epitopes of the 72 kDa heat shock protein (HSP) in bothnormal and neurologically-abnormal individuals. These bodies were interpreted as theprecorpora amylacea (pre-CA) which gradually enlarged into mature CA. Mature CA ex-pressed HSP epitopes chiefly in their peripheral rims while the smaller, immature pre-CAstained intensely for HSP throughout the entire structure.117 In still another SDS-PAGEstudy, ubiquitin and HSP27 and 70 were demonstrated in CA by immunocytochemistry.118
In a combined light and electron microscopic study, CA in control normal aging brainswere found to contain tau, tubulin, ubiquitin, and amyloid serum component P protein.Energy dispersive X-ray microanalysis data on isolated CA and those found in tissue sec-tions corroborate findings obtained by wavelength dispersive analysis.119 CA characteristi-cally contained high levels of iron, some copper and minimal aluminum. Elemental com-position analysis also revealed a high content of phosphorus.120 In a second study from thesame laboratory, CA were positively immunostained with antibodies directed against my-elin basic protein, proteolipid protein, galactocerebroside, ferritin, and myelin-associatedglycoprotein (MAG).121 A more recent X-ray microprobe analysis of CA revealed significantconcentrations of sodium, phosphorus, sulfur, and chloride.122
Ultrastructurally, Ramsay123 demonstrated that CA were intracytoplasmic bodies witha smooth outline, lacking surrounding membranes. They consist of many dense linear struc-tures of approximately 10 nm diameter; in those with more dense central cores, fibrils ap-peared to intermingle with granular deposits, and glycogen particles were often present atthe periphery of the inclusions. Ramsay123 identified the structures as lying within the pro-cesses of fibrous astrocytes and there is general agreement that this is their usual, albeit notexclusive, location. Identical bodies have been identified in small numbers within myeli-nated axons,124 in the ventral and lateral horns of the human spinal cord, and within neuritesof the human orbital cortex.125
The origin of corpora amylacea is not known. They occur particularly in the subpialand subependymal zones of the cerebral hemispheres, in the cerebral white matter particu-larly around small blood vessels, and in the hippocampus and long tracts of the spinal cordin elderly subjects, with or without neurological disorders. They are present in great num-bers in degenerating white matter tracts and are reportedly increased in many sites in nu-merous pathological states. In accord with their largely intra-astrocytic localization, they

Astrocytes in Brain Aging and Neurodegeneration78
tend vary greatly in number in areas of reactive gliosis and are occasionally observed infibrillary astrocytomas. A functional role for CA has been proposed based on the presenceof classical complement pathway components, the activation product C3d, the terminalcomplement complex (TCC), the C3 convertase regulator cofactor protein (MCP), the fluidphase regulator S-protein, and clusterin. It has been proposed that the formation of CAconfers the CNS with a degree of protection from the consequence of complement activa-tion, and that the functional role of CA formation is to provide an efficient mechanism forisolating potentially dangerous proteins following cell death in the aging and diseased CNS.126
The presence of ubiquitin and a number of HSPs strongly suggest that they are the result ofsome type of low-level chronic or transient stress.127 While the astrocyte is the cell that mostcommonly contain CA, proteins derived from other neural cells such as neurons and oligo-dendrocytes have occasionally been demonstrated in CA. An ultrastructural study of thevestibular nerve in patients suffering from Meniere’s disease and vascular cross-compres-sion syndrome of the root entry zone revealed that CA produced in astrocytes can be trans-ported to pial connective tissue across the glial-limiting lamina. The authors of that studysuggested that CA are components of a glio-pial system devoted to the clearance of sub-stances from the CNS.128 As in the case of augmented GFAP expression in normal aging,oxidative stress has also been implicated as an important mechanism mediating CA forma-tion129-132 (see chapter 10). A review of the structural and biochemical changes which occurin subpopulation of astroglia in the normal aging brain recently has been published.133
Astrocyte Inclusions in DiseaseChin and Goldman134 have recently compiled a detailed survey of the morphologic
and histochemical properties of a variety of glial cytoplasmic inclusion bodies (GCIs) whicharise in astrocytes and oligodendroglia under pathological conditions. The diseases in thisreview include multiple system atrophy, progressive supranuclear palsy, corticobasal gangli-onic degeneration,. Pick disease, and Alexander disease. GCIs stain intensely with antibod-ies to ubiquitin and αB-crystallin and less intensely against α- and β-tubulin.135-138 Immuno-staining with various antibodies to tau and paired helical filaments tended to be weak ornegative.135-137 Ari et al139 and Abe et al140 have reported strong staining with monoclonalantibodies against microtubule-associated protein (MAP).5 Staining with antibodies to ac-tin, vimentin, desmin, cytokeratin and GFAP has been consistently negative.
Among the most prominent of the stress protein-rich neural inclusions are the Rosenthalfibers (RFs). RFs are eosinophilic, cytoplasmic inclusions of astrocytes present in pilocyticastrocytomas, in astrocytic scars, in MS plaques, chronic infarcts, and most prominently inAlexander’s disease.141-144 The RFs appear to be similar among the different disorders. RFsvary from round, focal deposits of a few microns in diameter to elongated, cigar-shapedfibers one hundred microns or more in length, the latter usually residing within astrocyteprocesses. At the ultrastructural level, RFs appear as dense, osmiophilic masses lying on ameshwork of intermediate filaments.43 The inclusion is composed of two small molecularweight heat shock proteins, αB-crystallin and HSP27.145,146 Some of the αB-crystallin is con-jugated to ubiquitin.147 Levels of αB-crystallin and HSP27 mRNA are elevated in Alexander’sdisease.148,149 It has been suggested that a variety of “stressors” might induce the accumula-tion of RFs in astrocytes (reviewed in ref. 134).
To study the behavior of astrocytes overexpressing GFAP, six lines of transgenic micewere generated which carry multiple copies of the human GFAP gene.150,151 Mice that ex-pressed high levels of the human GFAP gene (Tg73.1, Tg73.7, and Tg73.8) died by postnatalday 8-24 days while mice that expressed lower levels of the transgene (Tg73.2, Tg73.3, and

79Glial Responses to Injury, Disease, and Aging
Tg73.5) survived and attained adulthood. At the light microscopic level, astrocytes in thehigh-expressing lines were distended by aggregates of globular eosinophilic material. Ultra-structural examination of optic nerve from a 13 day old Tg73.7 mouse showed astrocytesthat contained abundant cytoplasmic filaments in association with irregular osmiophilicdeposits resembling Rosenthal fibers (RFs) characteristic of Alexander’s disease.141,142
Astrocyte cultures were prepared from a transgenic mouse (Tg73.2) that carries mul-tiple copies of the human GFAP gene and from its wild type littermate. Astrocytes in theTg73.2 cultures appear irregularly shaped and enlarged, expressed increased human GFAPand its mRNA, exhibited both human and mouse GFAP, and expressed αB-crystallin pro-tein and mRNA, HSP27 protein, and vimentin protein. At the light microscopic level, theTg73.2 astrocytes appeared filled with eosinophilic deposits surrounded by GFAP positiveimmunostaining. Many, but not all, astrocytes in 20 day Tg73.2 cultures exhibited large,oddly shaped cells that immunostained with antibody specific for human GFAP (SMI-21),while most astrocytes in Tg73.2 cultures immunostained with a polyclonal anti-bovine GFAPantiserum (R-68) that reacts with human and mouse GFAP. This demonstrated that not allastrocytes in the Tg73.2 mouse contained the human GFAP gene. Tg73.2 astrocytes in cul-ture for 14 days immunostained for αB-crystallin, but the wild type astrocyte cultures grownfor equivalent lengths of time did not. Tg73.2 cultures at 20 days contain elevated αB-crys-tallin message when compared to time-matched wild type cells. At 14 days in culture, boththe transgenic and wild type cells immunostained sparsely for HSP27.
Conventional ultrastructural examination of Tg73.2 astrocytes showed numerous os-miophilic deposits in a bed of intermediate filaments (Fig. 4.2A) identical to that seen in acase of Alexander’s disease (Fig. 4.2B). Tg73.2 astrocyte cultures exhibited double labelingwith antibovine GFAP antiserum (R-68) and anti-human GFAP (SMI-21) using animmunogold technique (Fig. 4.3B). Wild type astrocytes (Fig. 4.3A) exhibited staining withR-68 but not SMI-21.
R-68 immunostaining of a paraffin-embedded brain section from an infant diagnosedwith Alexander’s disease152 revealed numerous astrocytic processes surrounding the bloodvessels which were replete with RFs. A RF exhibiting GFAP staining in its periphery is de-picted in Figure 4.4A. A sample of white matter from an infant with Alexander’s disease washomogenized with 0.05 M phosphate buffer (pH 8) and centrifuged to give a clear superna-tant and an insoluble pellet. This pellet was fixed in paraformaldehyde, embedded in paraf-fin, and sections were immunostained with R-68. The Rosenthal fibers in the pellet weresurrounded by GFAP immunoreactivity (Fig. 4.4B).
Evidence has been provided to show that the abnormal deposits present in Tg73.2 as-trocytes are very similar to RFs found in Alexander’s disease. We conclude that Tg73.2 mouseastrocytes in culture do not require exposure to additional stressors from external sourcesor contact with other neural cells to produce RFs. This suggests that the human GFAPtransgene is sufficient to induce the formation of RFs and that an excess of GFAP in astro-cytes may be detrimental to normal neural development. In contrast, other studies havedemonstrated that a lack of GFAP in astrocytes is not detrimental to normal breeding anddevelopment.153-155 We have previously shown that the metabolic turnover of GFAP is slow.2
Based on our present findings, we hypothesize that the normal mechanism for GFAP turn-over may be insufficient to handle the excess GFAP produced by transgenic expression orunder pathological conditions, thus resulting in the induction and accumulation of variousstress proteins. Together, the aberrant intracellular deposition of GFAP and its attendantHSP chaperones constitute the astroglial RFs which accumulate in Alexander’s disease andother neuropathological conditions.

Astrocytes in Brain Aging and Neurodegeneration80
Fig. 4.2. (A) Astrocytes in culture for 20 days from a Tg73.2 mouse were analyzed at the ultra-structural level. Note the dense Rosenthal fibers among the glial filaments. (B) Astrocytes froma 17 month old infant brain with Alexander’s disease were examined at the ultrastructurallevel. Note the dense deposits in the astrocyte which are identical to those seen in the Tg73.2astrocyte cultures.

81Glial Responses to Injury, Disease, and Aging
Fig. 4.3. Ultrastructural analysis of wild type (A) and Tg73.2 (B) astrocytes double-labeled withR-68 (12 nm gold particle) antiserum and SMI-21 (18 nm gold particle). The wild type astro-cytes exhibit R-68 immunoreactivity only, whereas the Tg73.2 astrocytes exhibit immunostainingfor both R-68 and SMI-21. The astrocyte cultures were fixed in 0.3 M NaCl in 70% aqueousethanol.

Astrocytes in Brain Aging and Neurodegeneration82
Fig. 4.4. (A) R-68 immunostain of a paraffin-embedded brain section from an infant diagnosedwith Alexander’s disease. (B) R-68 immunostain of an insoluble pellet prepared from anAlexander’s disease brain.
AcknowledgmentsThis chapter is supported by the Office of Research and Development, Medical Re-
search Service, Department of Veterans Affairs and by NIH grant NS-11632. Dr. L.F. Eng isthe Chief of the Chemistry Section, Pathology and Laboratory Service, Department of Vet-erans Affairs Medical Center, Palo Alto, CA 94304 and Professor of Pathology, StanfordUniversity School of Medicine, Stanford, CA 94305.
References1. Eng LF, Ghirnikar RS. GFAP and gliosis. Brain Pathol 1994; 4:229-237.2. Eng LF, Lee YL, Intermediate filaments in astrocytes. In: Kettermann H, Ransom BR, eds.
Neuroglia. Oxford: Oxford University Press, 1995; 650-667.3. Eddleston M, Mucke L. Molecular profile of reactive astrocytes: Implications for their role
in neurologic disease. Neuroscience 1993; 54:15-36.4. Brenner M. Structure and transcriptional regulation of the GFAP gene. Brain Pathol 1994;
4:245-257.5. Brenner M, Messing A. GFAP transgenic mice. Methods: A companion to methods in en-
zymology. 1996; 10:352-364.6. Amaducci L, Forno KI, Eng LF. Glial fibrillary acidic protein in cryogenic lesions of the rat
brain. Neurosci. Lett 1981; 21:27-32.7. Jeneczko K. The proliferative response of astrocytes to injury in neonatal rat brain. A com-
bined immunocytochemical and autoradiographic study. Brain Res 1988; 456:280-285.8. Latov N, Nilaver G, Zimmerman EA et al. Fibrillary astrocytes proliferate in response to
brain injury, a study combining immunoperoxidase technique for glial fibrillary acidic pro-tein and radioautography of tritiated thymidine. Develop Biol 1979; 72:381-384.

83Glial Responses to Injury, Disease, and Aging
9. Mathewson AJ, Berry M Observations on the astrocyte response to a cerebral stab woundin adult rats. Brain Res 1985; 327:61-69.
10. Miyake T, Hattori T, Fukuda M et al. Quantitative studies on proliferative changes of re-active astrocytes in mouse cerebral cortex. Brain Res 1988; 451:133-138.
11. Takamiya Y, Kohsaka S, Toya S et al. Possible association of platelet-derived growth factor(PDGF) with the appearance of reactive astrocytes following brain injury in situ. Brain Res1986; 383:305-309.
12. Takamiya Y, Kohsaka S, Otani M et al. Immunohistochemical studies on the proliferationof reactive astrocytes and the expression of cytoskeletal proteins following brain injury inrats. Develop Brain Res 1988; 38:201-210.
13. Topp KS, Faddis BT, Vijayan VK. Trauma-induced proliferation of astrocytes in the brainsof young and aged rats.Glia 1989; 2:201-211.
14. Hozumi I, Chiu FC, Norton WT. Biochemical and immunocytochemical changes in glialfibrillary acidic protein after stab wounds. Brain Res 1990; 524:64-71.
15. Vijayan VK, Lee Y, Eng F. Increase in glial fibrillary acidic protein following neural trauma.Mol Chem Neuropathol 1990; 13:107-118.
16. Brock TO, O’Callaghan JP. Quantitative changes in the synaptic vesicle proteins synapsin1 and P38 and the astrocyte-specific protein glial fibrillary acidic protein are associatedwith chemical-induced injury to the rat central nervous system. J Neurosci 1987;7:931-942.
17. Rataboul P, Vernier P, Biguet NF et al. Modulation of GFAP mRNA levels following toxiclesions in the basal ganglia of the rat. Brain Res Bull 1989; 22:155-161.
18. Reinhard JF Jr, Miller DB, O’Callaghan JP. The neurotoxicant MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) increases glial fibrillary acidic protein and decreases dopaminelevels of the mouse striatum: Evidence for glial response to injury. Neurosci Lett 1988;95:246-251.
19. Aquino DA, Chiu FC, Brosnan CF et al. Glial fibrillary acidic protein increases in the spi-nal cord of Lewis rats with acute experimental autoimmune encephalitis. J Neurochem1988; 51:1085-1096.
20. Goldmuntz EA. Brosnan CF, Chiu FC et al. Astrocytic reactivity and intermediate filamentmetabolism in experimental autoimmune encephalomyelitis: The effect of suppression withprazosin. Brain Res 1986; 397:16-26.
21. Smith ME, Somera FP, Eng LF. Immunocytochemical staining for glial fibrillary acidic pro-tein and the metabolism of cytoskeletal proteins in experimental allergic encephalomyeli-tis. Brain Res 1983; 264:241-253.
22. Smith ME, Somera FP, Swanson, K. et al. Glial fibrillary acidic protein in acute and chronicrelapsing experimental allergic encephalomyelitis (EAE). Prog Clin Biol Res 1984;146:139-144.
23. Rataboul P, Biguet NF, Vernier P et al. Identification of a human glial fibrillary acidicprotein cDNA: A tool for the molecular analysis of reactive gliosis in the mammalian cen-tral nervous system. J Neurosci Res 1988; 20:165-175.
24. Condorelli DF, Dell’Albani P, Kaczmarek L et al. Glial fibrillary acidic protein messengerRNA and glutamine synthetase activity after nervous system injury. J Neurosci Res 1990;26:251-257.
25. Hozumi I, Aquino DA, Norton, WT. GFAP mRNA levels following stab wounds in ratbrain. Brain Res 1990; 534:291-294.
26. Landry CF, Ivy GO, Brown IR. Effect of a discrete dorsal forebrain lesion in the rat on theexpression of neuronal and glial-specific genes: Induction of calmodulin, NF-L, SC1, andGFAP mRNA. J Neurosci Res 1992; 32:280-289.
27. Poirier J, Hess M, May PC et al. Astrocytic apolipoprotein E mRNA and GFAP mRNA inhippocampus after entorhinal cortex lesioning. Mol Brain Res 1991; 11:97-106.
28. Kost-Mikucki SA, Oblinger MM. Changes in glial fibrillary acidic protein mRNA expres-sion after corticospinal axotomy in the adult hamster. J Neurosci Res 1991; 28:182-191.
29. Aquino DA, Shafit-Zagardo B, Brosnan CF et al. Expression of glial fibrillary acidic pro-tein and neurofilament mRNA in gliosis induced by experimental autoimmune encephalo-myelitis. J Neurochem 1990; 54:1398-1404.

Astrocytes in Brain Aging and Neurodegeneration84
30. Steward O, Torre ER, Phillips LL et al. The process of reinnervation in the dentate gyrusof adult rats: Time course of increases in mRNA for glial fibrillary acidic protein. J Neurosci1990; 10:2373-2384.
31. Calne DB. Neurodegenerative Diseases. Philadelphia: WB Saunders, 1994.32. Duffy PE, Rapoport M, Graf L. Glial fibrillary acidic protein and Alzheimer-type senile
dementia. Neurology 1980; 30:778-782.33. Schechter R, Yen S-HC, Terry RD. Fibrous astrocytes in senile dementia of the Alzheimer
type. J Neuropathol Exp Neurol 1981; 40:95-101.34. Mancardi GL, Liwnicz BH, Mandybur TI. Fibrous astrocytes in Alzheimer’s disease and
senile dementia of Alzheimer’s type. An immunohistochemical and ultrastructural study.Acta Neuropathol (Berl) 1983; 61:76-80.
35. Beach TB, McGeer EG. Lamina-specific arrangement of astrocytic gliosis and senile plaquesin Alzheimer disease visual cortex. Brain Res 1988; 463:357-361.
36. Beach TG, Walker R, McGeer EG. Patterns of gliosis in Alzheimer’s disease and aging ce-rebrum. Glia 1989; 2:420-436.
37. Mandybur TI. Cerebral amyloid angiopathy and astrocytic gliosis in Alzheimer’s disease.Acta Neuropathol (Berlin) 1989; 78:329-331.
38. Vijayan V, Geddes JW, Anderson KJ et al. Astrocyte hypertrophy in the Alzheimer’s dis-ease hippocampal formation. Exp Neurol 1991; 112:72-78.
39. Joachim CL, Morris JH, Selkoe DJ. Diffuse senile plaques occur commonly in the cerebel-lum in Alzheimer’s disease. Am J Pathol 1989; 135:309-319.
40. Rozemuller JM, Eikelenboom P, Stam FC, Beyreuther K, Masters CL. A4 protein inAlzheimer’s disease: Primary and secondary cellular events in extracellular amyloid deposi-tion. J Neuropathol Exp Neurol 1989; 48:674-691.
41. Suenaga T, Hirano A, Llena JF et al. Modified immunocytochemical studies in cerebellarplaques in Alzheimer’s disease. J Neuropathol Exp Neurol 1990; 49:31-40.
42. Malamud N. Neuropathology of organic brain syndromes associated with aging. In: GaitzC, ed. Aging and the Brain. New York: Plenum, 1972:63-87.
43. Burger PC, Vogel S. The development of pathologic changes of Alzheimer’s disease andsenile dementia in patients with Down’s syndrome. Am J Pathol 1973; 73: 457-476.
44. Mann DMA. The pathological association between Down’s syndrome and Alzheimer’s dis-ease. Mech Aging Dev 1988; 43:99-136.
45. Murphy GM, Eng LF, Ellis WG et al. Antigenic profile of plaques and neurofibrillary tanglesin the amygdala in Down’s syndrome: A comparison with Alzheimer’s disease. Brain Res1990; 537:02-108.
46. Murphy GM Jr, Murphy E, Greenberg BD et al. Alzheimer’s disease: β-amyloid precursorprotein expression in plaques varies among cytoarchitectonic areas of the medial temporallobe. Neurosci Lett 1991; 131:100-104.
47. Murphy GM Jr, Ellis WG, Lee YL et al. Astrocytic gliosis in the amygdala in Down’s syn-drome and Alzheimer’s disease. In: Yu ACH, Hertz L, Norenberg MD, Sykova E, WaxmanSG, eds. Progress in Brain Research, Vol. 94. Amsterdam: Elsevier Science Publishers B.V.,1992:475-483.
48. Michetti F, Larocca LM, Rinelli A et al. Immunocytochemical distribution of S-100 pro-tein in patients with Down’s syndrome. Acta Neuropathol (Berlin) 1990; 80:475-478.
49. Griffin WST, Stanley LC, Ling C et al. Brain interleukin 1 and S-100 immunoreactivity areelevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA 1989;86:7611-7615.
50. Goodison KL, Parhad IM, White CL III et al. Neuronal and glial gene expression in neo-cortex of Down’s syndrome and Alzheimer’s disease. J Neuropathol Exp Neurol 1993;52:192-198.
51. Moossy J, Martinez AJ, Hanin I et al. Thalamic and subcortical gliosis with dementia. ArchNeurol 1987; 44:510-513.
52. Selkoe DJ, Salazar FJ, Abraham C et al. Huntington’s disease: Changes in striatal proteinsreflect astrocytic gliosis. Brain Res 1982; 245:117-125.

85Glial Responses to Injury, Disease, and Aging
53. Stevens CD, Altshuler LL, Bogerts B et al. Quantitative study of gliosis in schizophreniaand Huntington’s chorea. Biol Psychiat 1988; 24:697-700.
54. Roberts GW, Colter N, Lofthouse R et al. Gliosis in schizophrenia: A survey. Biol Psychiat1986; 2:1043-1050.
55. Stevens JR. Neuropathology of schizophrenia. Arch Gen Psychiatry 1982; 39:1131-1139.56. Bigbee JW, Bigner DD, Pegram C et al. Study of glial fibrillary acidic protein in a human
glioma cell line grown in culture and as a solid tumor. J Neurochem 1983; 40:460-467.57. DeArmond SJ, Fajardo M, Naughton SA et al. Degradation of glial fibrillary acidic protein
by a calcium dependent proteinase: An electroblot study. Brain Res 1983; 262:275-282.58. Kushner PD, Stephenson DT, Wright S. Reactive astrogliosis is widespread in the subcor-
tical white matter of amyotrophic lateral sclerosis brain. J Neuropathol Exp Neurol 1991;50:263-277.
59. Dormont D, Delpech A, Courcel M-N et al. Hyperproduction de proteine glio-fibrillaireacide (GFA) au cours de l’évolution de la tremblante experimentale de la souris. C R AcadSci (Paris) 1981; 293:53-56.
60. Mackenzie A. Immunohistochemical demonstration of glial fibrillary acidic protein inscrapie. J Comp Path 1983; 3:251-259.
61. Gomori AF, Partnow MJ, Horoupian DS et al. The ataxic form of Creutzfeldt-Jakob dis-ease. Arch Neurol 1973; 29:318-323.
62. Lafarga M, Berciano MT, Suarez I et al. Reactive astroglia-neuron relationships in the hu-man cerebellar cortex: A quantitative, morphological and immunocytochemical study inCreutzfeldt-Jakob disease. Int J Devl Neurosci 1993; 11:199-213.
63. Manuelidis L, Tesin DM, Sklaviadis T et al. Astrocyte gene expression in Creutzfeldt-Jakobdisease. Proc Natl Acad Sci USA 1987; 84:5937-5941.
64. DeArmond SJ, Kristensson K, Bowler RP. In: Yu ACH, Hertz L, Norenberg MD, Sykova E,Waxman SG, eds. Progress in Brain Research, Vol. 94. Amsterdam: Elsevier Science Pub-lishers 1992:437-446.
65. Sobel RA, DeArmond SJ, Forno LS et al. Glial firillary acidic protein in hepatic encephal-opathy. An immunohistochemical study. J Neuropathol Exp Neurol 1981;40: 625-632.
66. Kretzschmar HA, DeArmond SJ, Forno LS. Measurement of GFAP in hepatic encephal-opathy by ELISA and transblots. J Neuropathol Exp Neurol 1985; 44:459-471.
67. Norenberg MD. The role of astrocytes in hepatic encephalopathy. Neurochem Pathol 1987;6:13-33.
68. Ma KC, Ye ZR, Fang J et al. Glial fibrillary acidic protein immunohistochemical study ofAlzheimer I & II astrogliosis in Wilson’s disease. Acta Neurol Scand 1988; 78:290-296.
69. Perry VH, Andersson PB, Gordon S. Macrophages and inflammation in the central ner-vous system. Trends Neurosci 1993; 16:268-273.
70. Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: Intrinsic immunoeffector cells ofthe brain. Brain Res Rev 1995; 20:269-287.
71. Streit WJ, Graeber MB, Kreutzberg GW. Functional plasticity of microglia: A review. Glia1988; 1:301-307.
72. Dickson DW, Mattiace LA, Kure K et al. Microglia in human disease, with an emphasis onacquired immunodeficiency syndrome. Lab Invest 1991; 64:135-156.
73. Giulian D, Chen J, Ingwman JE et al. The role of mononuclear phagocytes in wound heal-ing after traumatic injury to adult mammalian brain. J Neurosci 1989; 9:4416-4429.
74. McGeer PL, Itagaki S, McGeer EG. Expression of the histocompatibility glycoprotein HLA-DR in neurological disease. Acta Neuropathol 1988; 76:550-557
75. Gehrmann J, Mies G, Bonnekoh P et al. Microglial reaction in the rat cerebral cortex in-duced by cortical spreading depression. Brain Pathol 1993; 3:11-17.
76. Kettenmann H, Hoppe D, Gottman K et al. Cultured microglial cells have a distinct pat-tern of membrane channels different from peritoneal macrophages. J Neurosci Res 1990;26:278-287.
77. Kettenmann H, Banati R, Walz W. Electrophysiological behavior of microglia. Glia 1993;7:93-101.

Astrocytes in Brain Aging and Neurodegeneration86
78. Imamura K, Suzumura A, Sawada M et al. Induction of MHC class II antigen expressionon murine microglia by interleukin 3. J Neuroimmunol 1994; 55:119-125.
79. Eng LF, Ghirnikar RS, Lee YL. Inflammation in EAE: Role of chemokine/cytokine expres-sion by resident and infiltrating cells. Neurochem Res 1966; 21:511-525.
80. Nohava K, Malipiero U, Frei K et al. Neurons and neuroblastoma as a source of macroph-age colony-stimulating factor. Eur J Immunol 1992; 22:2539-2545.
81. Hayashi M, Luo Y, Laning JH et al. Production and function of monocyte chemoattractantprotein-1 nd other β-chemokines in murine glial cells. J Neuroimminol 1995; 60:143-150
82. Perry VH, Matyszak MK, Fearn S. Altered antigen expression in the aged rodent CNS. Glia1993; 7:60-59.
83. Ogura K, Ogawa M, Yoshida M. Effect of aging on microglia in the normal rat brain:Immunohistochemical observations. Neuroreport 1994; 5:1224-1226.
84. Dijkstra CD, DeGroot CJ, Huitinga I. The role of macrophages in demyelination. JNeuroimmunol 1992; 40:183-188.
85. Hulkower K, Brosnan C F, Aquino DA et al. Expression of CSF-1, c-fms, and MCP-1 inthe central nervous system of rats with experimental allergic encephalomyelitis. J Immunol1993; 150:2525-2533.
86. McCombe PA, de Jersey J, Pender MP. Inflammatory cells, microglia and MHC class IIantigen-positive cells in the spinal cord of Lewis rats with acute and chronic relapsingexperimental autoimmune encephalomyelitis. J Neuroimmunol 1994; 51:153-167.
87. Norton WT, Brosnan CF, Cammer W et al. Mechanisms and suppression of inflammatorydemyelination. Acta Neurobiol Exp 190; 50:225-235.
88. Claudio L, Kress Y, Factor J et al. Mechanisms of edema formation in experimental au-toimmune encephalomyelitis. The contribution of inflammatory cells. Am J Pathol 1990;137:1033-1045.
89. Claudio L, Martiney J A, Brosnan CF. Ultrastructural studies of the blood-retina barrierafter exposure to interleukin-1 beta or tumor necrosis factor-alpha. Lab Invest 1994;70:850-861.
90. Knobler RL, Marini JC, Goldowitz D et al. Distribution of the blood brain barrier in het-erotopic brain transplants and its relationship to the lesions of EAE. J Neuropathol ExpNeurol 1992; 51:36-39.
91. Cannella B, Cross AH, Raine CS. Relapsing autoimmune demyelination: A role for vascu-lar addressins. J Neuroimmunol 1991; 35:295-300.
92. Cannella B, Cross AH, Raine CS. Adhesion related molecules in the central nervous sys-tem. Upregulation correlates with inflammatory cell influx during relapsing experimentalautoimmune encephalomyelitis. Lab Invest 1991; 65:23-31.
93. Raine CS, Cannella B, Duijvestijn A M et al. Homing to central nervous system vascula-ture by antigen-specific lymphocytes. II. Lymphocyte/endothelial cell adhesion during theinitial stages of autoimmune demyelination. Lab Invest 1990; 63:476-489.
94. Seki T, Joh K, Oh-ishi T. Augmented production of interleukin-8 in cerebrospinal fluid inbacterial menengitis. Immunology 1993; 80:333-335.
95. Nichols NR, Day JR, Laping NJ et. al. GFAP mRNA increases with age in rat and humanbrain. Neurobiol Aging 1993; 14:421-429.
96. Landfield PW, Waymire JL, Lynch GS. Hippocampal aging and adrenocorticoids: Quanti-tative correlations. Science 1978; 202:1098-1102.
97. Gordon MN, Morgan DG. Increased GFAP expression in the aged brain does not resultfrom increased astrocyte density. Soc Neurosci Abstr.1993;19:1042.
98. Goss JR, Finch CE, Morgan DG. GFAP RNA prevalence is increased in aging and in wast-ing mice. Exp Neurol 1990; 108:266-268.
99. Goss JR, Finch CE, Morgan DG. Age-related changes in glial fibrillary acidic protein mRNAin mouse brain. Neurobiol Aging 1991; 12:165-170.
100. O’Callaghan JP, Miller DB. The concentration of glial fibrillary acidic protein increaseswith age in the mouse and rat brain. Neurobiol Aging 1991; 12:171-174.
101. Bronson RT, Lipman RD, Harrison DE. Age-related gliosis in the white matter of mice.Brain Res 1993; 609:124-128.

87Glial Responses to Injury, Disease, and Aging
102. Laping NJ, Morgan TE, Nichols NR et al. Transforming growth factor-1 induces neuronaland astrocyte genes; Tubulin alpha-1, glial fibrillary acidic protein, and clusterin. Neuro-science 1994; 58:563-572.
103. Kohama SG, Goss JR, Finch CE et al. Increases of glial fibrillary acidic protein in the agedfemale mouse brain. Neurobiol Aging 1995; 16:59-67
104. Yoshida T, Goldsmith SK, Morgan TE et al. Transcription supports age-related increasesof GFAP gene expression in the male rat brain. Neurosci Ltt 1966; 215:107-110.
105. Hallpike JF, Adams CWM, Tourtellotte WW. Multiple Sclerosis. Pathology, Diagnosis andManagement. Baltimore: Williams & Wilkins, 1983.
106. Schaumburg HH, Powers JM, Raine CS et al. Adrenoleukodystrophy: A clinical and patho-logical study of 17 cases. Arch Neurol 1975; 33: 577-591.
107. Goldman JE, Schaumburg HH, Norton WT. Isolation and characterization of glial fila-ments from human brain. J Cell Biol 1978; 78:426-440.
108. Kraig RP, Dong L, Thisted R. Spreading depression increases immunohistochemical stain-ing of glial fibrillary acidic protein J Neurosci 1991; 11:2187-2198.
109. Major DE, Kessiak JP, Cotman CW, et.al. Life-long dietary restriction attenuates age-re-lated increases in glial fibrillary acidic protein. GFAP mRNA in the rat hippocampus. SocNeurosci abstr 1994; 20:50
110. Nichols RN, Caleb EF, Nelson JF. Food restriction delays the age-related increase in GFAPmRNA in rat hypothalamus. Neurobiol Aging 1995; 16:105-110.
111. Morgan TE, Rozovshy I, Goldsmith SK et al. Increased transcription of the astrocyte geneGFAP during middle-age is attenuated by food restriction: Implications for the role ofoxidative stress. Free Rad Biol Med 1997; 23:524-528.
112. Tacconi MT, Lligona L, Salmona M et al. Aging and food restriction: Effects on lipids ofthe cerebral cortex. Neurobiol Aging 1991; 12:55-59.
113. Choi JH, Yu BP. Brain synaposomal aging: Free radicals and membrane fluidity. Free RadBiol Med 1995; 18:133-139.
114. Laping NJ, Teter B, Nichols NR et al. Glial fibrillary acidic protein: Regulation by hor-mones, cytokines, and growth factors. Brain Pathol 1994b; 4:259-275.
115. Stam FC, Roukema PA. Histochemical and biochemical aspects of corpora amylacea. ActaNeuropathol 1973; 25:95-102.
116. Steyaert A, Cissé S, Merhi Y et al. Purification and polypeptide composition of corporaamylacea from aged human brain. J Neurosci Meth 1990; 31:59-64.
117. Martin JE, Mather K, Swash M et al. Heat-shock protein expression in corpora amylaceain the central nervous system—clues to their origin. Neuropathol Appl Neurobiol 1991;17:113-119.
118. Cissé S, Perry G, Lacoste-Royal G et al. Immunochemical identification of ubiquitin andheat-shock proteins in corpora amylacea from normal aged and Alzheimer’s disease brains.Acta Neuropathol 1993; 85:233-240.
119. Sakai M, Austin J, Witmer F et al. Studies of corpora amylacea. Arch Neurol 1969;21:526-544.
120. Singhrao SK, Neal JW, Newman GR. Corpora amylacea could be an indicatior ofneurodegeneration. Neuropathol Appl Neurbiol 1993; 19:269-276.
121. Singhrao SK, Neal JW, Piddlesdent SJ et al. New immunocytochemical evidence for neu-ronal/oligodendroglial origin for corpora amylacea. Neuropathol Appl Neurol 1994;20:66-73.
122. Tokutake H, Nagase TH, Morisaki S et al. X-ray microprobe analysis of corpora amylacea.Neuropathol Appl Neurobiol 1995; 21:269-273.
123. Ramsay HJ. Ultrastructure of corpora amylacea. J Neuropath Exp Neurol 1956; 24:25-39.124. Takahashi K, Agari M, Nakamura H. Intra-axonal corpora amylacea in ventral and lateral
horns of the human spinal cord. Acta Neuropathol (Berlin) 1975; 31:151-158.125. Anzil AP, Herrlinger H, Blinzinger Ket al. Intraneuritic corpora amylacea. Demonstration
in orbital cortex of elderly subjects by means of early post-mortem brain sampling andelectron microscopy. Virchows Archives of Pathological Anatomy and Histology 1974;364:297-302.

Astrocytes in Brain Aging and Neurodegeneration88
126. Singhrao SK, Morgan BP, Neal JW et al. A functional role for corpora amylacea based onevidence from complement studies. Neurodegeneration 1995; 4:335-345.
127. Cissé S, Lacoste-Royal G, Laperriere G et al. Ubiquitin is a component of polypeptidespurified from corpora amylacea of aged human brain. Neurochemical Res 1991; 16:429-433
128. Sbarbati A, Carner M, Colletti V et al. Extrustion of corpora amylacea from the marginalglia at the vestibular root entry zone. J Neuropath Exp Neurol 1996; 55:196-201.
129. Schipper HM, Cissé S. Mitochondrial constituents of corpora amylacea and autofluorescentastrocytic inclusions in senescent human brain. Glia 1995; 14:55-64
130. Schipper HM, Cissé S, Stopa EG. Expression of heme oxygenase-1 in the senescent andAlzheimer-diseased brain. Ann Neurol 1995; 37:758-768.
131. Chopra VS, Chalifour LE, Schipper HM. Differential effects of cysteamine on heat shockprotein induction and cytoplasmic granulation in astrocytes and glioma cells. MolecularBrain Res 1995; 31:173-18.
132. Cissé S, Schipper HM. Experimental induction of corpora amylacea-like inclusions in ratastroglia. Neuropathol Appl Neurobiol 1995; 21:423-431.
133. Schipper HM. Astrocytes, brain aging, and neurodegeneration. Neurobiol Aging 1996;17:467-480.
134. Chin SM, Goldman JE. Glia inclusions in CNS degenerative diseases. J Neuropathol ExpNeurol 1996; 55:499-508.
135. Papp MI, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients withmultiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J Neurol Sci 1989; 94:79-100.
136. Nakazato Y, Yamazaki H, Hirato J et al. Oligodendroglial microtubular tangles inolivopontocerebellar atrophy. J Neuropath Exp Neurol 1990; 49:521-530.
137. Kato S, Nakamura H, Hirano A et al. Argyrophilic ubiquitinated cytoplasmic inclusions ofLeu-7-positive glial cells in olivopontocerebellar atrophy (multiple system atrophy). ActaNeuropath 1991; 82:488-493.
138. Murayama S, Arima K, Nakazato Y et al. Immunocytochemical and ultrastructural studiesof neuronal and oligodendroglial cytoplasmic inclusions in multiple system atrophy. 2.Oligodendroglial cytoplasmic inclusions. Acta Neuropath 1992; 84:32-38.
139. Arai N, Nishimura M, Oda M et al. Immunohistochemical expression of microtubule-as-sociated protein 5 (MAP5) in glial cells in multiple system atrophy. J Neurol 1992;109;102-106.
140. Abe H, Yagishita S, Amano N et al. Argyrophilic glial intracytoplasmic inclusions in mul-tiple system atrophy: Immunocytochemical and ultrastructural study. Acta Neuropathol1992; 273-277.
141. Alexander WS. Progressive fibrinoid degeneration of fibrillary astrocytes associated withmental retardation in a hydrocephalic infant. Brain 1949; 72:373-381.
142. Herndon RM, Rubinstein LV, Freeman JM, Mathieson G. Light and electron microscopicobservations on Rosenthal fibres in Alexander’s disease and in multiple sclerosis. JNeuropathol Exp Neurol 1970; 29:524-550.
143. Grcevic N, Yates PO. Rosenthal fibres in tumours of the central nervous system. J PatholBact 1957; 73:467-472.
144. Borrett G, Becker LE. Alexander’s disease: A disease of astrocytes. Brain 1985; 108:367-385.145. Iwaki T, Kume-Iwaki A, Liem RKH, Goldman J. αB-crystallin is expressed in non-lenticu-
lar tissues and accumulates in Alexander’s disease. Cell 1989; 57:71-78.146. Tomokane N, Iwaki T, Tateishi J et al. Rosenthal fibers share epitopes with αB-crystallin,
glial fibrillary acidic protein and ubiquitin, but not with vimentin: Immunoelectron mi-croscopy with colloidal gold. Am J Pathol 1991; 138:875-885.
147. Goldman JE, Corbin E. Rosenthal fibers contain ubiquitinated αB-crystallin. Am J Pathol1991; 139:933-938.
148. Head M, Corbin E, Goldman JE. Overexpression and abnormal modification of the stressproteins αB-crystallin and Hsp27 in Alexander’s disease. Am J Pathol 1993; 143:1743-1753.
149. Head MW, Corbin E, Goldman JE. Coordinate and independent expression of αB-crystal-lin and hsp27. J Cell Physiol 1994; 159:41-50.

89Glial Responses to Injury, Disease, and Aging
150. Messing A., Galbreath EJ, Sijapati KK, Brenner M. Overexpression of GFAP in transgenicmice. J Neuropath Exp Neurol 1996; 55:620 (abstract).
151. Messing A, Head MW, Galles K, Galbreath EJ, Goldman UE, Brenner M. Fatalencephalopothy with astrocyte inclusions in FGAP transgenic mice. Am J Pathol 1998;152:391-398.
152. Ramsay P, Norman M, Eng LF. Chemical study of an Alexander brain. Trans Am SocNeurochem 10:125, 1979, Abstract.
153. Gomi H., Yokoyama T, Fujimoto K et al. Properties of astrocytes from mice devoid ofGFAP. Mice devoid of the glial fibrillary acidic protein develop normally and are suscep-tible to scrapie prions. Neuron 1995; 14:29-41.
154. Pekny M, Leveen P, Pekna M et al. Mice lacking glial fibrillary acidic protein display as-trocytes devoid of intermediate filaments but develop and reproduce normally. EMBO J1995; 14:1590-1598.
155. McCall. MA, Gregg. RG, Behringer RR et al. Targeted deletion in astrocyte intermediatefilament (GFAP) alters neuronal physiology. Proc Natl Acad Sci USA 1996; 93:6361-6366.


CHAPTER 5
Astrocytes in Brain Aging and Neurodegeneration, edited by Hyman M. Schipper.©1998 R.G. Landes Company.
Astrocyte Pathologyin Alzheimer DiseaseJerzy Wegiel and Henryk M. Wisniewski
Neuropathological Changes in Alzheimer Disease
Alzheimer disease (AD) is a degenerative cerebral disorder with progressive dementia. Inthe brain of subjects with Alzheimer-type dementia, several percent of the volume of the
gray matter is infiltrated with 39- to 43-amino-acid amyloid-β (Aβ), which forms parenchy-mal diffuse nonfibrillar deposits and fibrillar neuritic plaques. In AD, microglial cells changemorphology, immunophenotype, function, and distribution. The subpopulation of micro-glial cells produces fibrillar amyloid deposits in classical and primitive plaques.1-3 Aβ depos-its in the wall of capillaries are the product of perivascular cells and perivascular microglialcells,4-5 both of which are of monocyte/microglial cell lineage.4,6-8 Neurons are the source ofparenchymal nonfibrillar amyloid in diffuse plaques.9-11 Nonfibrillar and fibrillar amyloiddeposits in the tunica media of leptomeningeal and parenchymal arteries and veins are theproduct of smooth muscle cells.12-13 Fibrillar parenchymal amyloidosis is associated withsuch secondary changes as neuronal degeneration and astrocytosis.14 The effect of vascularamyloidosis is local impairment in blood circulation, neuronal degeneration and loss, andastrocytosis.4
The second hallmark of AD is neurofibrillary degeneration with cytoplasmic accumu-lation of abnormally phosphorylated tau-protein and paired helical filaments (PHFs), whichcause severe, structure-specific neuronal loss reaching up to 80-90% in the hippocampalformation15 and up to 50% in the neocortex.16
During the course of AD, the number of astrocytes increases by several times.17 In part,these changes are related to the hallmarks of AD—amyloidosis-β and neurofibrillary de-generation of neurons. Activated astrocytes are engaged in the dispersion, degradation, andremoval of Aβ14 as well as in the removal of ghost tangles and dark neurons. Astrocyte pa-thology in AD comprises not only their proliferation and activation but also several formsof degeneration, such as cytoplasmic accumulation of PHFs, Rosenthal fibers, anchoragedensities with desmosome-like structures, eosinophilic inclusions, and corpora amylacea.18
This complex Alzheimer-type pathology is the result of numerous interactions betweenamyloid-β, neurons affected and not affected by neurofibrillary changes, microglial cells,and astrocytes.
Relationships Between Amyloid-βββββ, Neurons, and Glial Cells in ADNeurons, astrocytes, microglial cells, and oligodendrocytes maintain a complex paren-
chymal milieu in the normal brain. Amyloidosis-β and neurofibrillary pathology modify

Astrocytes in Brain Aging and Neurodegeneration92
the brain environment and initiate several reinforcing feedbacks loops, which change theinterneuronal and interglial balance.
Astrocytes produce apolipoprotein E (Apo E),19-20 interleukin-1,21-22 endothelin-1,23
prostaglandin E, and a subset of these cells expresses intercellular adhesion molecule-1.24 Inpart, their function depends on microglial cells, which produce growth factors includinginterleukin-1,25-27 which in turn induces astrogliosis and activates astrocytes.28-29 The acti-vation of astrocytes is associated indirectly with another form of microglial cell function,namely, with the production of several cytotoxins—glutamate, tumor necrosis factor α(TNF~α), nitric oxide, hydrogen peroxide, and oxygen-containing free radicals—which af-fect neurons.30-31 Neuronal degeneration and death cause both activation of astrocytes andastrocytosis.
Interleukin 1Microglial cells of the neuritic plaques are activated, and they overexpress acute-phase
cytokine interleukin-1. Interleukin-1 beta has a strong mitogenic impact on cultured astro-cytes.32 In tissue, this cytokine activates astrocytes and induces expression of the astrocyte-derived cytokine, S-100β, which increases the intraneuronal free calcium level and may causeneuronal injury and death. Interleukin-1 upregulates expression and processing of βAPP,favoring amyloid-β deposition,33 and induces expression of protease inhibitor alpha-1-antichymotrypsin, thromboplastin, complement protein C, and Apo E, all of which arepresent in neuritic plaques. Interleukin-1 induces increased synthesis of alpha-1-antichymotrypsin, which acts as a pathological chaperone,34-35 binding to the beta proteinand strongly promoting its polymerization into amyloid filaments in vitro.36
Apolipoprotein EBoth astrocytes and microglia produce Apo E.19-20 The synthesis of Apo E increases
after neuronal injury in the central37 and peripheral nervous system.38 Apo E interacts withboth normal soluble Aβ and fibrillar amyloid in plaques.39 More than 60% of persons withone allelic form of Apo E4 suffer from AD by the time they reach 75 years of age, and morethan 90% of the subjects with two copies of the Apo E4 gene have the disease by 75 years ofage.40 Apo E4 is an important pathological chaperon protein in soluble amyloid-beta pro-tein fibrillization and tau phosphorylation.39,41 In vitro studies show that Apo E4 binds Aβfaster and with a different pH dependence than Apo E3.42 Persons who inherit two Apo E4genes bind more Aβ to form plaques.41
Amyloid-βββββThe amyloidogenic processing of APP might be upregulated by extracellular Aβ43-44
through the cellular receptor for Aβ.45-46 Astrocytes recognize extracellular amyloid and re-spond to this antigen.14 In vitro studies indicate that Aβ enhances the secretion ofinterleukin-147-48 and basic fibroblast growth factor (bFGF) from cultured microglia andastrocytes as well as proliferation of microglial cells. That interleukin-1 and bFGF elevatethe synthesis of βAPP suggests that this cascade effect contributes to plaque development.47
In cultured astrocytes, all three major transcripts of βAPP are expressed, with the ratio forAPP 695, APP 751, and APP 770 isoform mRNAs being 1:4:2. Treatment with transforminggrowth factor beta 1 (TGF-beta 1) produces about a 6-fold increase in total APP mRNA.32
EndothelinEndothelin-1 immunoreactive astrocytes are very rare in non-AD cases but are very
numerous in AD brains in the periphery of plaques, in the molecular layer of the cerebralcortex, in the subcortical white matter, and the folia of cerebellum.23 Endothelin-1 expres-

93Astrocyte Pathology in Alzheimer Disease
sion increases not only in AD but also in infarcts and traumatic injuries.49 Astrocytes grownin vitro also release endothelin-1 and -3 into the culture medium.50-53 In vitro endothelinacts as a mitogen for astrocytes and tumor cells.51,54 Receptors to endothelin-1 were identi-fied in the plasma membrane of cultured astrocytes of rats and mice51 and to endothelin-3in rat astrocytes.55-56 It has been suggested that endothelins may modulate neuronal activ-ity.57-59 Endothelin-1 is considered the most potent and long-lasting vasoconstrictor pep-tide known to date.60-64 Endothelins discharged from reactive astrocytes in AD brains mayinduce constriction of arterioles and contribute to the local reduction of blood flow.65-66
Simultaneous expression of endothelin and endothelin receptors in primary astrocytes im-plies the presence of an autocrine control mechanism in astrocytes.52
These cytokines and the molecular and cellular processes that they support form acomplex of interactions that may be capable of self-propagation facilitated by means ofseveral reinforcing feedback loops (Fig. 5.1).
Astrogliosis in Aging and ADAstrocytic gliosis, characterized by cellular hypertrophy and augmented GFAP expres-
sion, is a morphological marker of cerebral aging.67 These changes occur not only in thebrains of aged, nondemented people and in individuals with Alzheimer disease,68 but alsoin the brains of aged monkeys,69 rats,70-74 and mice.67,75 Astrogliosis in brains of species freeof plaques, such as mice, the great topographical variability of astrogliosis, and the presenceof astrocytosis in many other pathological processes indicate that astrocyte proliferation isa regional reaction elicited by many different factors.
In the brain of subjects with Alzheimer disease, focal and diffuse astrocytosis devel-ops.17,76-84 In advanced stages of AD, the number of astrocytes increases approximately4-fold.17 In AD brains, the astrocytes appear in plaques, around ghost tangles, dark neurons,capillaries obliterated by Aβ, and in areas of ischemic changes. Astrocytes reveal specificmorphological properties in each of the above-listed pathological interactions, which indi-cates that their role in each of these events may be different. Astrocytes surrounding plaquesare usually so rich in glial fibrillary acidic protein-positive fibers17,80 that the number ofplaques demonstrated by GFAP is usually higher than the number obtained with other tech-niques.83 Astrocytic accumulation in plaque is considered the reaction to focal extracellularaccumulation of Aβ.1,67,79,83,85 Because almost all fibrillar plaques develop in gray matter,Aβ-associated astrocytosis is restricted to the cerebral cortex and subcortical gray matter.
Neuritic PlaquesLight and electron microscopic studies show that neuritic plaques consist of amyloid;
dystrophic, degenerating and regenerating neuronal processes; astrocytes; and microglialcells. Plaques with a central amyloid core, called also amyloid star, surrounded by six toeight microglial cells, degenerated neurites, and a ring of astrocytic processes, are calledclassical plaques. Primitive plaques do not have amyloid star and are composed of wisps ofamyloid associated with one to four microglial cells, and chaotically distributed bundles ofdegenerated neurites and astrocytic processes. Two or three astrocytes are involved directlyin classical plaque formation.14,17,86 Rozemuller and coworkers87 hypothesized that the pro-teolytic cleavage of amyloidogenic proteins and formation of amyloid fibrils are related toastroglia. However, ultrastructural studies indicate that microglial cells are the cells engagedin fibrillar Aβ deposition in classical and primitive plaques.1-4
The spatial distribution of proliferating astrocytic processes in the plaque indicatesthat astrocytic reaction in the classical plaque relates mainly to the amyloid deposits. Astro-cyte receptors recognize many molecules,88-89 and extracellular amyloid appears also to bedetectable by astrocytes. Astrocytic processes proliferate in the plaque periphery, isolate Aβ

Astrocytes in Brain Aging and Neurodegeneration94Fi
g. 5
.1. S
chem
atic
dia
-gr
am s
how
ing
the
re-
lati
on
ship
s b
etw
een
astr
ocy
tes,
mic
rogl
ial
cell
s, a
nd
neu
ron
s in
AD
.

95Astrocyte Pathology in Alzheimer Disease
aggregates, and divide the latter into smaller clusters (Fig. 5.2). Transformation of fibrillaramyloid in floccular and amorphous material in areas of proliferating astrocytic processessuggests that astrocyte ectoenzymes degrade Aβ. The proportion 1:9.2 between the volumeof amyloid and the volume of proliferating astrocytic processes appears to be the measureof astrocytic response to Aβ in classical plaques.
Diffuse PlaquesAstrocytic reaction is undetectable in diffuse plaques, which are nonfibrillar and
thioflavin S and Congo red negative. Their formation is associated with neuronal release ofAβ peptide. Nonfibrillar amyloid is present in relatively low concentrations, is probablybound to chaperon proteins thereby preventing fibrillization, and appears not to be able toactivate surrounding astrocytes. As a result, in diffuse plaques, there is no sign of local in-creases in the numbers of astrocytes or astrocyte activation.
Astrogliosis in the Area of Amyloid AngiopathyDeposition of amyloid in the wall of the capillary vessels by perivascular cells and perivas-
cular microglial cells is the cause of vessel obliteration. Amyloid deposits and remnants ofthe capillary wall are surrounded by astrocytic processes and degraded. The presence ofonly amyloid deposits indicates that this material is more resistant to degradation thanendothelial cell residues.4
Astrocyte Response to Neurofibrillary ChangesNeurofibrillary degeneration appears to be the major cause of neuronal loss in such
brain structures as the hippocampal formation,15 but much less so in the isocortex.16 Lightand electron microscopic studies indicate that in the end stage of neurofibrillary changesneurons are degraded by astrocytic processes that penetrate ghost tangles,84,90 separate ag-gregates of cellular debris and bundles of PHF. The pathological fibrillar component is themost resistant residue in neurons showing neurofibrillary changes. Scars formed by astro-cytes and residues of NFTs are often encountered in cortical biopsy specimens derived fromAD subjects (Fig. 5.3). In the end stage of ghost tangle resolution, the characteristic featuresof PHF disappear, and fibrils are not detectable with antibodies to abnormally phosphory-lated tau. They are detectable with antibody (mAb 3-39) to ubiquitin bound to PHF (50-65 aaof ubiquitin). Degradation of the ghost tangles is probably a very slow process. The differ-ences in the number of ghost tangles—very few in the isocortex16 and very numerous in thecornu Ammonis or subiculum15—suggest that the rate of neuronal death associated withneurofibrillary changes and the local reaction of astrocytes vary and are structure specific.
Astrocytes Response to Ischemia-Related Neuronal DegenerationThe second factor contributing to neuronal loss in AD is amyloid angiopathy. The depo-
sition of amyloid-β in the wall of capillary vessels causes their obliteration and regionalischemic changes, with neuronal degeneration and death.4-5 These changes also are associ-ated with astrocyte proliferation and activation. Interaction between astrocytes and darkneurons similar to the interaction between astrocytes and ghost tangles suggests that astro-cytes are involved in removal of bodies and processes of necrotic neurons with and withoutneurofibrillary changes (Fig. 5.4). The number and size of astrocytic processes surroundingdark neuronal perikarya and neurites increases. The volume of cytoplasm in the neuronalperikarya is reduced and the hyaloplasm is condensed, whereas astrocytic processes becomedistended.

Astrocytes in Brain Aging and Neurodegeneration96
Fig. 5.2. Periphery of a classical plaque with aggregates of fibrillar amyloid-β (asterisks), whichare separated from the neuronal processes by a dense network of astrocytic processes (As). Neu-ronal processes (np), which are in direct contact (arrows) with amyloid deposits (asterisks), re-veal degenerative changes, with an accumulation of abnormal mitochondria, osmophilic bodies,and vacuoles and an increase in the diameter of the processes.

97Astrocyte Pathology in Alzheimer Disease
Fig. 5.3. Tangential (arrows) and longitudinal (double arrows) sections of bundles of fibrils ofthe remnants of a ghost tangle between astrocytic processes (As). In this stage of ghost tangledegradation, the twisted structure of the PHFs is indistinguishable.

Astrocytes in Brain Aging and Neurodegeneration98
Fig. 5.4. Neuronal pathology in cerebral cortex affected by severe amyloid angiopathy and oblit-eration of capillary vessels. There is proliferation of edematous astrocytic processes (As) aroundthe body of a dark neuron (N) exhibiting condensed nucleoplasm and aggregated chromatin(arrow) in the deformed cell nucleus, vacuolation of endoplasmic reticulum (er), and degen-erative changes in mitochondria (m).

99Astrocyte Pathology in Alzheimer Disease
Astrocyte Degeneration in ADIn brains of patients with severe Alzheimer-type pathology, some astrocytes degener-
ate with accumulation of abnormally phosphorylated tau, Rosenthal fibers, eosinophilicinclusions, anchorage densities, or corpora amylacea. Interindividual differences in the type,distribution, and severity of these changes suggest that they may represent responses toseveral different factors. They might be a nonspecific expression of chronic oversaturationof the cell environment with extracellular proteins. The nature of accumulated proteinsvaries, depending on the pathological conditions to which astrocytes are exposed, and as aresult, different morphological forms of degeneration are observed. Abnormal tau proteinphosphorylation with deposition of fibrillar tau-positive inclusions might be the effect of acellular imbalance between the process of phosphorylation, dependent upon specific ki-nases, and dephosphorylation, dependent upon specific phosphatases. As a result,hyperphosphorylated tau accumulates in the neurons, and in some cases in astrocytes and/oroligodendrocytes of individuals with AD.
Tau-Positive InclusionsThe abnormal fibrils, which resemble PHFs in neurons, were described also in astro-
cytes,91-93 where they appear as fibrils with periodical constrictions (twisted tubules) orstraight tubules. The presence of tau-positive twisted and nontwisted tubules in astrocytesand oligodendrocytes94 in AD and other progressive disorders, including progressive supra-nuclear palsy95-98 and Pick’s disease,99 suggests that glial elements also are affected by insultssimilar to those that affect neurons.
The presence of tau protein in astrocytes and oligodendrocytes100-101 indicates that taucan no longer be considered a neuron-specific protein. Tau-positive inclusions in neuronsand glial cells have been recently recognized in many neurodegenerative diseases includingAlzheimer disease, Pick disease, progressive supranuclear palsy, subacute sclerosingpanencephalitis, and corticobasal degeneration.92,96-98,102-104 In corticobasal degeneration,they are even more numerous in astrocytes and oligodendroglia than in neurons.104 Tau-positive inclusions share common phosphorylation characteristics irrespective of the un-derlying disease or cell type in which they occur.103
In comparison with neuronal tangles, glial inclusions show some morphological andimmunocytochemical differences. In corticobasal degeneration, they are tau- and Gallyas-positive but Bielschowsky-negative.104 NFTs in neurons and tau-positive inclusions in as-trocytes in progressive supranuclear palsy are composed mainly of straight tubules103 andhave less phosphorylated tau than in AD.105
Rosenthal FibersIn three of the six examined cortical biopsies from patients with AD, Rosenthal fibers
were found in the astrocyte body and cytoplasmic processes.18 In some cells, Rosenthal fi-bers are associated with glial filaments (Fig. 5.5), but in many astrocytes, there is no spatialrelationship between intermediate filaments and Rosenthal fibers. The proportion of corti-cal astrocytes affected by Rosenthal fibers ranges from 5 to 40%. This form of astrocytedegeneration is associated with condensation of cell cytoplasm, deformation of the cellnucleus, and aggregation of the chromatin and nucleoplasm. This form of astrocyte degen-eration is detectable not only in the area of AD pathology but also in the surrounding,morphologically unchanged neuropil. Astrocytes with Rosenthal fibers involved in ghosttangle removal are seen sporadically (Fig. 5.6).
Rosenthal fibers develop in human astrocytes, in astrocytic tumors, in Alexander’s dis-ease, and in astrocytes where reactive gliosis has been present for a long time.106-113 Theywere described also in astrocytes of sheep.114-116 Rosenthal fibers have a heterogenous chemical

Astrocytes in Brain Aging and Neurodegeneration100
Fig. 5.5. Astrocyte degeneration with accumulation of Rosenthal fibers (RF) between cytoplasmicintermediate filaments (if), condensation of nucleoplasm, aggregation of chromatin (ch), anddeformation of cell nucleus. Neuronal degeneration with PHF accumulation in cell processes isalso depicted.

101Astrocyte Pathology in Alzheimer Disease
structure and share epitopes with αB-crystallin, GFAP, and ubiquitin.111,117-118 Because ofspatial relationships to intermediate filaments, overproduction or incomplete degradationof glial filaments was considered the cause of Rosenthal fiber formation.119 In AD, in manyastrocytes this contact is undetectable, which may indicate that these changes are not re-lated to GFAP (but see chapter 4). Rosenthal fibers are ubiquitinated from the earliest stepsof their formation111 but ubiquitination is considered a secondary reaction.111,118 Ubiquitinbinds to abnormal proteins destined for ATP-dependent proteolysis.120-121 In experimentalstudies, Rosenthal fiber formation has been attributed to the intake of foreign proteins byastrocytes and their cytoplasmic pathology in degradation and disposal of these proteins.116,122
Eosinophilic InclusionsThis form of degeneration manifests with the accumulation of eosinophilic material in
the cytoplasm of astrocytes in AD. The inclusions contain two morphologically distinguish-able components:
Fig. 5.6. Remnants of ghost tangle (arrows) in deep cytoplasmic invagination of an astrocytewith numerous Rosenthal fibers (RF) in the cytoplasm and aggregated chromatin (ch) in thenucleus (Nu).

Astrocytes in Brain Aging and Neurodegeneration102
Fig. 5.7. Numerous eosinophilic inclusions (ei) in the cytoplasm of an astrocyte. There is con-densed nucleoplasm and aggregated chromatin (ch) in the deformed cell nucleus (Nu).

103Astrocyte Pathology in Alzheimer Disease
1. electron-dense granular or floccular material, which is very condensed in some as-trocytes (Fig. 5.7) and more loosely arranged in other cells, especially in those withcytoplasm that is watery and poor in organelles; and
2. the remnants of cytoplasmic organelles in different stages of disintegration. Thepresence of fragments of rough endoplasmic reticulum, clusters of ribosomes, andsmall vesicles embedded in electron-dense granular or floccular material suggestsnonlysosomal degradation of cell components in this form of astrocyte degeneration.
The distribution, size, and shape of the inclusions in astrocytes vary. They prevail inthe cell body, but in cells with numerous inclusions, they are also present in cell processes.In severely affected astrocytes, the cytoplasm is occupied by numerous inclusions 6-12 µmin diameter. In other cells, tiny aggregates of granular or floccular osmophilic material prevail.
This form of astrocyte degeneration was observed originally in the brain of a 5 year oldchild with Aicardi’s syndrome123 and in a 20 year old man with cerebral palsy, mental retar-dation and brain malformation.124 However, development of eosinophilic inclusions in the
Fig. 5.8. Anchorage densities (arrows) with hemidesmosome-like structures (double arrow) inend-feet of perivascular-reactive astrocyte. There is pathological reinforcement of the astrocyticinterface between the vessel and neuropil. bm = basement membrane; E = endothelial cell.

Astrocytes in Brain Aging and Neurodegeneration104
brain of aged persons with AD indicates that eosinophilic degeneration of astrocytes is notrestricted to brain developmental malformations or congenital astrocytic dysfunction.
Anchorage DensitiesAnchorage densities associated with hemidesmosome-like structures develop in perivas-
cular reactive astrocytes (Fig. 5.8). The fusion of hundreds of hemidesmosomes producescontinuous hemidesmosomes, which are coated by thickened basal lamina. Anchorage den-sities appear about 200-300 nm away from hemidesmosomes. Hemidesmosomes are con-nected to anchorage densities by numerous fibrils. The anchorage densities, in turn, areoften contiguous with intermediate cytoplasmic filaments. This complex structure appearsto reinforce the cell membrane facing the perivascular space.
Astrocytic anchorage densities associated with hemidesmosome-like structures weredescribed in two cases of brain atrophy.92 The presence of the same pathological changes intwo cases of AD18 and in one case of Gerstmann-Sträussler-Scheinker disease (unpublishedobservation in material obtained from Dr. Budka, Vienna), all affected by severe brain atro-phy, indicates that astrocytic anchorage densities associated with hemidesmosome-like struc-tures may develop in many pathological conditions associated with brain atrophy andastrogliosis.
Corpora AmylaceaCorpora amylacea (CA) are cytoplasmic inclusions of human astrocytes and neurons
and are often deposited in the extracellular space following disintegration of the host cell.They are associated with normal aging and neurodegenerative diseases such as Alzheimer’sdisease, Lafora disease, and progressive supranuclear palsy. CA are discussed in consider-able detail in chapters 4 and 10 of this volume.
AcknowledgmentThe authors wish to thank Dr. B. Lach and Dr. A.P. Anzil for biopsy material, Dr. K.C.
Wang for assistance in preparing the material for electron microscopic studies, and Ms. M.Stoddard Marlow for copy-editing the manuscript. The study was supported by funds fromthe New York State Office of Mental Retardation and Developmental Disabilities and a grantfrom the National Institutes of Health, National Institute of Aging No. PO1-AG11531.
References1. Wisniewski HM, Wegiel J, Wang KC et al. Ultrastructural studies of the cells forming
amyloid fibers in classical plaques. Can J Neurol Sci 1989; 16:535-542.2. Wisniewski HM, Vorbrodt AW, Wegiel J et al. Ultrastructure of the cells forming amyloid
fibers in Alzheimer disease and scrapie. Am J Med Genet, Suppl 1990; 7:287-297.3. Wegiel J, Wisniewski HM. The complex of microglial cells and amyloid star in three- di-
mensional reconstruction. Acta Neuropathol 1990; 81:116-124.4. Wisniewski HM, Wegiel J, Wang K-C et al. Ultrastructural studies of the cells forming
amyloid in the vessel wall in Alzheimer disease. Acta Neuropathol 1992; 84:117-127.5. Wisniewski HM, Wegiel J. Migration of perivascular cells into the neuropil and their in-
volvement in β-amyloid plaque formation. Acta Neuropathol 1993; 85:586-595.6. Hickey WF, Kimura H. Perivascular microglial cells of the CNS are bone marrow-derived
and present antigen in vivo. Science 1988; 239:290-292.7. Perry VH, Gordon S. Modulation of CD4 antigen on macrophages and microglia in rat
brain. J Exp Med 1987; 166:1138-1143.8. Perry VH, Crocker RP, Gordon S. The blood-brain barrier regulates the expression of a
macrophage sialic acid-binding receptor on microglia. J Cell Sci 1992; 101:201-207.

105Astrocyte Pathology in Alzheimer Disease
9. Probst A, Langui D, Ipsen S et al. Deposition of beta/A4 protein along neuronal plasmamembranes in diffuse senile plaques. Acta Neuropathol 1991; 83:21-29.
10. Pappolla MA, Omar RA, Vinters HV. Image analysis microspectroscopy shows that neu-rons participate in the genesis of early primitive (diffuse) senile plaques. Am J Pathol 1991;139:599-607.
11. Wisniewski HM, Wegiel J, Kotula L. Some neuropathological aspects of Alzheimer’s dis-ease and its relevance to other disciplines. Neuropath Appl Neurobiol 1996; 22:3-11.
12. Wisniewski HM, Wegiel J. β-amyloid formation by myocytes of leptomeningeal vessels.Acta Neuropathol 1994; 87:233-241.
13. Wegiel J, Wisniewski HM, Dziewiatkowski J et al. The origin of amyloid in cerebral vesselsof aged dogs. Brain Res 1995; 705:225-234.
14. Wisniewski HM, Wegiel J. Spatial relationships between astrocytes and classical plaquecomponents. Neurobiol Aging 1991; 12:593-600.
15. Bobinski M, Wegiel J, Wisniewski HM et al. Neurofibrillary pathology—correlation withhippocampal formation atrophy in Alzheimer disease. Neurobiol Aging 1996; 17:909-919.
16. Gomez-Isla T, Hollister R, West H et al. Neuronal loss correlates with but exceeds neu-rofibrillary tangles in Alzheimer’s disease. Ann Neurol 1997; 41:17-24.
17. Schechter R, Yen SHC, Terry RD. Fibrous astrocytes in senile dementia of Alzheimer type.J Neuropath Exp Neurol 1981; 40:95-101.
18. Wegiel J, Wisniewski HM. Rosenthal fibers, eosinophilic inclusions, and anchorage densi-ties with desmosome-like structures in astrocytes in Alzheimer disease. Acta Neuropathol1993; 87:355-361.
19. Pitas RE, Boyle JK, Lee et al. Astrocytes synthesize apolipoprotein E and metabolizeapolipoprotein E-containing lipoproteins. Biochem Biophys Acta 1987; 917:148-161.
20. Poirier J, Hess M, May PC et al. Astrocytic apolipoprotein E mRNA and GFAP mRNA inhippocampus after entorhinal cortex lesioning. Mol Brain Res 1991; 11:97-106.
21. Fontana A, Kristensen F, Dubs R et al. Production of prostaglandin E and an interleukin-1 like factor by cultured astrocytes and C6 glioma cells. J Immunol 1982; 129:2413-2419.
22. Bakhit C, Armanini M, Bennett GL et al. Increase in glia-derived nerve growth factor fol-lowing destruction of hippocampal neurons. Brain Res 1991; 560:76-83.
23. Zhang WW, Badonic T, Höög A et al. Astrocytes in Alzheimer’s disease express immu-noreactivity to the vaso-constrictor endothelin-1. J Neurol Sci 1994; 122:90-96.
24. Akiyama H, Kawamata T, Yamada T et al. Expression of intercellular adhesion molecule(ICAM)-1 by a subset of astrocytes in Alzheimer disease and some other degenerative neu-rological disorders. Acta Neuropathol 1993; 85:628-634.
25. Giulian D, Baker TJ. Characterization of ameboid microglia isolated from developing mam-malian brain. J Neurosci 1986; 6:2163-2178.
26. Giulian D, Baker TJ, Shih LN et al. Interleukin-1 of the central nervous system is pro-duced by ameboid microglia. J Exp Biol Med 1986; 164:594-604.
27. Giulian D, Chen J, Ingeman JE et al. The role of mononuclear phagocytes in wound heal-ing after traumatic injury to adult mammalian brain. J Neurosci 1989; 9:4416-4429.
28. Giulian D, Lachman LB. Interleukin-1 stimulation of astroglial proliferation after braininjury. Science 1985; 228:497-499.
29. Giulian D, Woodward J, Young DG et al. Interleukin-1 injected into mammalian brainstimulates astrogliosis and neovascularization. J Neurosci 1988; 8:2485-2490.
30. Boje KM, Arora PK. Microglial-produced nitric oxide and reactive nitrogen oxides medi-ate neuronal cell death. Brain Res 1992; 587:250-256.
31. Chao CC, Hu S, Molitor TW et al. Activated microglia mediate neuronal cell injury via anitric oxide mechanism. J Immunol 1992; 149:2736-2741.
32. Gray CW, Patel AJ. Regulation of beta-amyloid precursor protein isoform mRNAs by trans-forming growth factor-beta 1 and interleukin-1 beta in astrocytes. Brain Res Mol BrainRes 1993; 19:251-256.
33. Sheng JG, Ito K, Skinner RD et al. In vivo and in vitro evidence supporting a role for theinflammatory cytokine interleukin-1 as a driving force in Alzheimer pathogenesis. NeurobiolAging 1996; 17:761-766.

Astrocytes in Brain Aging and Neurodegeneration106
34. Picken MM, Larrondo-Lillo M, Coria F et al. Distribution of the protease inhibitor α1-antichymotrypsin in cerebral and systemic amyloid. J Neuropathol Exp Neurol 1990;49:41-48.
35. Potter H, Abraham CR, Dressler D. The Alzheimer amyloid components α1-antichymo-trypsin and β-protein form a stable complex in vitro. In: Iqbal K, McLachlan DRC, WinbladB et al, eds. Alzheimer’s Disease. Chichester: John Wiley and Sons Ltd, 1991:275-279.
36. Das S, Potter H. Expression of the Alzheimer amyloid-promoting factor antichymotrypsinis induced in human astrocytes by IL-1. Neuron 1995; 14:447-456.
37. Snipes GJ, McGuire CB, Norden JJ et al. Nerve injury stimulates the secretion ofapolipoprotein E by nonneuronal cells. Proc Natl Acad Sci U S A 1986; 83:1130-1134.
38. Boyles JK, Zoellner CD, Anderson LJ et al. A role for Apolipoprotein E, Apolipoprotein A-1, low density lipoprotein receptors in cholesterol transport during regeneration andremyelination of the rat sciatic nerve. J Clin Invest 1989; 83:1015-1031.
39. Wisniewski T, Golabek A, Matsubara E et al. Apolipoprotein E: Binding to solubleAlzheimer’s β-amyloid. Biochem Biophys Res Commun 1993; 192:359-365.
40. Schmechel DE, Saunders AM, Strittmatter WJ et al. Increased amyloid β-deposition as aconsequence of apolipoprotein E genotype in late-onset Alzheimer’s disease. Proc Natl AcadSci U S A 1993; 90:9649-9653.
41. Strittmatter WJ, Weisgraber KH, Goedert M et al. Hypothesis: Microtubule instability andpaired helical filament formation in the Alzheimer disease brain are related to apolipoproteinE genotype. Exp Neurol 1994; 125:163-171.
42. Strittmatter WJ, Weisgraber KH, Huang et al. Binding of human apolipoprotein E to βA4peptide: Isoform-specific effects and implications for late-onset Alzheimer disease. ProcNatl Acad Sci U S A 1993; 90:8098-8102.
43. Yang AJ, Knauer M, Burdick DA et al. Intracellular Aβ1-42 aggregates stimulate the accu-mulation of stable, insoluble amyloidogenic fragments of the amyloid precursor protein intransfected cells. J Biol Chem 1995; 270:14786-14792.
44. Davis-Salinas J, Saporito-Irvin SM, Cotman CW et al. Amyloid β-protein induces its ownproduction in cultured degenerating cerebrovascular smooth muscle cells. J Neurochem1995; 65:931-934.
45. Yan SD, Chen X, Fu J et al. RAGE and amyloid-β peptide neurotoxicity in Alzheimer’sdisease. Nature 1996; 382:685-691.
46. Khoury EJ, Hickman SE, Thomas CA et al. Scavenger receptor-mediated adhesion of mi-croglia to β-amyloid fibrils. Nature 1996; 382:716-719.
47. Araujo DM, Cotman CW. Beta-amyloid stimulates glial cells in vitro to produce growthfactors that accumulate in senile plaques in Alzheimer’s disease. Brain Res 1992; 569:141-145.
48. Del Bo R, Angeretti N, Lucca E. Reciprocal control of inflammatory cytokines, IL-1 andIL-6, and beta-amyloid production in cultures. Neurosci Lett 1995; 188:70-74.
49. Jiang MH, Höög A, Ma K et al. Endothelin-1-like immune-reactivity is expressed in reac-tive astrocytes of the human brain. Neuroreport 1993; 4:935-937.
50. Miller RC, Pelton JT, Huggins JP. Endothelins—from receptors to medicine. TrendsPharmacol Sci 1993; 14:54-60.
51. MacCumber M, Ross CA, Snyder H. Endothelin in brain: Receptors, mitogenesis, and bio-synthesis in glial cells. Proc Natl Acad Sci U S A 1990; 87:2359-2363.
52. Ehrenreich H, Anderson RW, Fox CH et al. Endothelins, peptides with potent vasoactiveproperties are produced by human macrophages. J Exp Med 1990; 172:1741-1748.
53. Ehrenreich H, Kehrl JH, Anderson RW et al. A vasoactive peptide, endothelin-3, is pro-duced by and specifically binds to primary astrocytes. Brain Res 1991; 538:54-58.
54. Supattapone S, Simpson AWM, Ashley CC. Free calcium rise and mitogenesis in glial cellscaused by endothelin. Biochem Biophys Res Commun 1989; 165:1115-1122.
55. Hösli E, Hösli L. Autoradiographic evidence for endothelin receptors on astrocytes in cul-tures of rat cerebellum, brainstem and spinal cord. Neurosci Lett 1991; 129:55-58.
56. Couraud P-O, Durieu-Trautmann O, Mahe E et al. Comparison of binding characteristicsof endothelin receptors on subpopulations of astrocytes. Life Sci 1991; 49:1471-1476.

107Astrocyte Pathology in Alzheimer Disease
57. Giaid A, Gibson S, Ibrahim N et al. Endothelin 1 and endothelium-derived peptide is ex-pressed in neurons of the human spinal cord and dorsal root ganglia. Proc Natl Acad SciU S A 1989; 86:7634-7638.
58. Giaid A, Gibson SJ, Herrero MT et al. Topographical localisation of endothelin mRNAand peptide immunoreactivity in neurones of the human brain. Histochemistry 1991;95:303-314.
59. Yamaji T, Johshita H, Ishibashi M et al. Endothelin family in human plasma and cere-brospinal fluid. J Clin Endocrinol Metab 1990; 71:1611-1615.
60. Shigeno T, Mima T. A new vasoconstrictor peptide, endothelin: Profiles as vasoconstrictorand neuropeptide. Cerebrovasc Brain Metab Rev 1990; 2:227-239.
61. Yanagisawa M, Kurihara H, Kimura S et al. A novel potent vasoconstrictor peptide pro-duced by vascular endothelial cells. Nature 1988; 332:411-415.
62. Ide K, Yamakawa K, Nakagomi T et al. The role of endothelin in the pathogenesis of va-sospasm following subarachnoid hemorrhage. Neurol Res 1989; 11:101-104.
63. Kauser K, Rubanyi G, Harder D. Endothelium dependent modulation of endothelin-in-duced vasoconstriction and membrane depolarization in cat cerebral arteries. J PharmacolExp Ther 1989; 252:93-97.
64. Lee M-E, Monte SMDL, Ng S-C et al. Expression of the potent vasoconstrictor endothelinin the human central nervous system. J Clin Invest 1990; 86:141-147.
65. O’Brien JT, Eagger S, Syed GMS et al. A study of regional cerebral blood flow and cogni-tive performance in Alzheimer’s disease. J Neurol Neurosurg Psychiatry 1992; 55:1182-1187.
66. Bonte FJ, Tintner R, Weiner MF et al. Brain blood flow in the dementias: SPECT withhistopathologic correlation. Radiology 1993; 186:361-365.
67. Mandybur TI, Ormsby I, Zemlan FP. Cerebral aging: A quantitative study of gliosis in oldnude mice. Acta Neuropathol 1989; 77:507-513.
68. Delacourte A. General and dramatic glial reaction in Alzheimer brains. Neurology 1990;40:33-37.
69. O’Kusky J, Colonier M. Postnatal changes in the number of astrocytes, oligodendrocytes,and microglia in the visual cortex (area 17) of the macaque monkey: A stereological analy-sis in normal and monocularly deprived animals. J Comp Neurol 1982; 210:307-315.
70. Adams I, Jones DG. Synaptic remodelling and astrocytic hypertrophy in rat cerebral cortexfrom early to late adulthood. Neurobiol Aging 1983; 3:179-186.
71. De la Roza C, Cano J, Reinoso-Suarez F. An electron microscopic study of astroglia andoligodendroglia in the lateral geniculate nucleus of aged rats. Mech Ageing Dev 1985;29:267-281.
72. Geinisman Y, Bondareff W, Dodge JT. Hypertrophy of astroglial processes in the dentategyrus of the senescent rat. Am J Anat 1978; 153:537-544.
73. Landfield PW, Rose G, Sandles L et al. Patterns of astroglial hypertrophy and neuronaldegeneration in the hippocampus of aged, memory-deficient rats. J Gerontol 1977; 32:3-12.
74. Lindsay JD, Landfield PW, Lynch G. Early onset and topographical distribution of hyper-trophied astrocytes in hippocampus of aging rats: A quantitative study. J Gerontol 1979;34:661-671.
75. Lamar CH, Hinsman EJ, Henrickson CK. Alterations in the hippocampus of aged mice.Acta Neuropathol 1976; 36:387-391.
76. von Braunmuhl A. Alterserkrankungen des Zentralnervensystems. Senile Involution. SenileDemenz. Alzheimerische Krankheit. In: Lubarsch O, Henke F, Rossle R, eds. Handbuchder Speziellen Pathologischen Anatomie und Histologie XIII/1. Berlin: Springer Verlag, 1957;13:337-539.
77. Senitz D, Goertchen R. Über Astrozytenveränderungen in der orbitofrontalen Hirnrindebei seniler Demenz. Zentral Allg Pathol 1978; 122:515-521.
78. Hansen LA, Armstrong DM, Terry RD. An immunohistochemical quantification of fibrousastrocytes in the aging human cerebral cortex. Neurobiol Aging 1987; 8:1-6.
79. Dickson DW, Farlo J, Davies P. Alzheimer’s disease. A double-labeling immunohistochemicalstudy of senile plaques. Am J Pathol 1988; 132:86-101.

Astrocytes in Brain Aging and Neurodegeneration108
80. Duffy PE, Rapoport M, Graf L. Glial fibrillary acidic protein and Alzheimer-type seniledementia. Neurology 1980; 30:778-780.
81. Fischer O. Die presbyophrene Demenz, deren anatomische Grundlage und klinischeAbgrenzung. Z Gesamte Neurol Psychiat 1910; 3:371-471.
82. Mancardi GL, Liwnicz BH, Mandybur TI. Fibrous astrocytes in Alzheimer’s disease andsenile dementia of Alzheimer’s type. An immunohistochemical and ultrastructural study.Acta Neuropathol 1983; 61:76-80.
83. Mandybur TI. Cerebral amyloid angiopathy and astrocytic gliosis in Alzheimer’s disease.Acta Neuropathol 1989; 78:329-331.
84. Probst A, Ulrich J, Heitz PU. Senile dementia of Alzheimer type: Astroglial reaction toextracellular neurofibrillary tangles in the hippocampus. Acta Neuropathol 1982; 57:75-79.
85. Wisniewski HM, Sinatra RS, Iqbal K et al. Neurofibrillary and synaptic pathology in theaged brain. In: Johnson JE, ed. Aging and Cell Structure. New York: Plenum Publishing,1981:105-142.
86. Itagaki S, McGeer PL, Akiyama H et al. Relationship of microglia and astrocytes to amy-loid deposits of Alzheimer disease. J Neuroimmunol 1989; 24:173-182.
87. Rozemuller JM, Eikelenboom P, Stam FC et al. A4 protein in Alzheimer’s disease: Primaryand secondary cellular events in extracellular amyloid deposition. J Neuropathol Exp Neurol1989; 48:674-691.
88. Ebersolt C, Perez M, Bockaernt J. Neuronal, glial, and monoglial localization of neurotrans-mitter: Sensitive adenylate cyclases in cerebral cortex of mice. Brain Res 1981; 213: 139-150.
89. Whitaker-Azmitia PM, Azmitia EC. Autoregulation of fetal serotonergic neuronal develop-ment: role of high affinity serotonin receptors. Neurosci Lett 1986; 67:307-312.
90. Yamaguchi H, Morimatsu M, Hirai S et al. Alzheimer’s neurofibrillary tangles are pen-etrated by astroglial processes and appear eosinophilic in their final stages. Acta Neuropathol1987; 72:214-217.
91. Ikeda K, Haga C, Akiyama H et al. Coexistence of paired helical filaments and glial fila-ments in astrocytic processes within ghost tangles. Neurosci Lett 1992; 148:126-128.
92. Nakano I, Iwatsubo T, Otsuka N et al. Paired helical filaments in astrocytes: electron mi-croscopy and immunocytochemistry in a case of atypical Alzheimer’s disease. ActaNeuropathol 1992; 83:228-232.
93. Yamazaki M, Nakano I, Imazu O et al. Paired helical filaments and straight tubules inastrocytes: An electron microscopic study in dementia of the Alzheimer type. ActaNeuropathol 1995; 90:31-36.
94. Nishimura M, Tomimoto H, Suenaga T et al. Immunocytochemical characterization ofglial fibrillary tangles in Alzheimer’s disease brain. Am J Pathol 1995; 146:1052-1058.
95. Nishimura M, Namba Y, Ikeda K et al. Glial fibrillary tangles with straight tubules in thebrains of patients with progressive supranuclear palsy. Neurosci Lett 1992; 143:35-38.
96. Yamada T, McGeer PL. Oligodendroglial microtubular masses: An abnormality observedin some human neurodegenerative diseases. Neurosci Lett 1990; 120:163-166.
97. Yamada T, McGeer PL, McGeer EG. Appearance of paired nucleated, tau-positive glia inpatients with progressive supranuclear palsy brain tissue. Neurosci Lett 1992; 135:99-102.
98. Yamada T, Calne DB, Akiyama H et al. Further observations on tau-positive glia in thebrains with progressive supranuclear palsy. Acta Neuropathol 1993; 85:308-315.
99. Yamazaki M, Nakano I, Imazu O et al. Astrocytic straight tubules in the brain of patientwith Pick’s disease. Acta Neuropathol 1994; 88:587-591.
100. Migheli A, Butler M, Brown M et al. Light and electron microscope localization of themicrotubule-associated tau protein in rat brain. J Neurosci 1988; 8:1846-1851.
101. Papasozomenos SC, Binder LI. Phosphorylation determines two distinct species of tau inthe central nervous system. Cell Motil Cytoskeleton 1987; 8:210-226.
102. Wakabayashi K, Oyanagi K, Makifuchi T et al. Corticobasal degeneration: Etiopathologicalsignificance of the cytoskeletal alterations. Acta Neuropathol 1994; 87:545-553.
103. Iwatsubo T, Hasegawa M, Ihara Y. Neuronal and glial tau-positive inclusions in diverseneurologic diseases share common phosphorylation characteristics. Acta Neuropathol 1994;88:129-136.

109Astrocyte Pathology in Alzheimer Disease
104. Horoupian DS, Chu PL. Unusual case of corticobasal degeneration with tau /Gallyas-posi-tive neuronal and glial cells. Acta Neuropathol 1994; 88:592-598.
105. Flament S, Delacourte A, Verny M et al. Abnormal tau proteins in progressive supranuclearpalsy. Similarities and differences with the neurofibrillary degeneration of the Alzheimertype. Acta Neuropathol 1991; 81:591-596.
106. Alexander WS. Progressive fibrinoid degeneration of fibrillary astrocytes associated withmental retardation in a hydrocephalic infant. Brain 1949; 71:373-381.
107. Borrett D, Becker LE. Alexander’s disease: A disease of astrocytes. Brain 1985; 108:367-385.108. Crome L. Megaloencephaly associated with hyaline pan-neuropathy. Brain 1953; 76:215-228.109. Grcevic N, Yates PO. Rosenthal fibers in tumors of the central nervous system. J Pathol
Bacteriol 1957; 73:467-472.110. Herndon RM, Rubenstein LJ, Freeman JM et al. Light and electron microscopic observa-
tions on Rosenthal fibers in Alexander’s disease and in multiple sclerosis. J Neuropath ExpNeurol 1970; 29:524-551.
111. Lach B, Sikorska M, Rippstein P et al. Immunoelectron microscopy of Rosenthal fibers.Acta Neuropathol 1991; 81:503-509.
112. Ogasawara N. Multiple Sclerose mit Rosenthalschen Fasern. Acta Neuropathol 1956; 5:61-68.113. Russell DS, Rubinstein LJ. In: Pathology of Tumors of the CNS. Baltimore: Williams and
Wilkins 1989; 977:155-167.114. Frankhauser R, Fatzer R, Bestetti JP et al. Encephalopathy with Rosenthal fiber formation
in a sheep. Acta Neuropathol 1980; 50:57-60.115. McGrath JT. Spontaneous animal models of human disease. In: Andrews EJ, Ward C,
Altman NH, eds. Spontaneous Animal Models of Human Disease. New York: AcademicPress, 1979; 2:147-148.
116. Horoupian DS, Kress Y, Yen SH et al. Nickel induced changes and reappraisal of Rosenthalfibers in focal CNS lesions. J Neuropathol Exp Neurol 1982; 41:664-675.
117. Iwaki T, Kume-Iwaki A, Corbin E et al. Expression of the B-chain of α-crystallin in CNSglia. J Neuropathol Exp Neurol 1990; 49:344.
118. Tomokane N, Iwaki T, Tateishi J et al. Rosenthal fibers share epitopes with αB-crystallin,glial fibrillary acidic protein, and ubiquitin, but not with vimentin. Immunoelectron mi-croscopy with colloidal gold. Am J Pathol 1991; 138:875-885.
119. Goldman JE, Corbin E. Isolation of major protein component of Rosenthal fibers. Am JPathol 1988; 130:569-578.
120. Hershko A, Ciechanover A, Heller H et al. Proposed role of ATP in protein breakdown:Conjugation of proteins with multiple chains of the early peptide of ATP-dependent pro-teolysis. Proc Natl Acad Sci U S A 1980; 77:1783-1786.
121. Rechsteiner M. Ubiquitin-mediated pathways for intracellular proteolysis. Ann Rev CellBiol 1987; 3:1-30.
122. Kress Y, Gaskin F, Horoupian DS et al. Nickel induction of Rosenthal fibers in rat brain.Brain Res 1981; 210:419-425.
123. Abe H, Yagashita S, Itoh K et al. Novel eosinophilic inclusion in astrocytes. ActaNeuropathol 1992; 83:659-663.
124. Minagawa M, Shioda K, Shimizu Y et al. Inclusion bodies in cerebral cortical astrocytes: Anew change of astrocytes. Acta Neuropathol 1992; 84:113-116.


CHAPTER 6
Astrocytes in Brain Aging and Neurodegeneration, edited by Hyman M. Schipper.©1998 R.G. Landes Company.
Parkinson’s DiseaseDonato A. Di Monte
Introduction
Astrocytes have often been seen as mere “supportive” elements of the brain architectureand have generally received little consideration in discussions on the pathogenesis of
neurodegeneration. This view is slowly but inevitably being reconsidered. The more weknow about biochemical, pharmacologic and pathological interactions between neuronalcells and astrocytes, the more the concept of a “hierarchic supremacy” of neurons versusastrocytes becomes outdated. Neuron-astrocyte interaction is an essential component ofbrain function and, as such, would also be expected to play a role in pathological processes.Indeed, evidence pointing to astrocytes as “active” participants in the toxic events leading toneurodegeneration is quite convincing, starting with the simple observation of gliosis as acommon astrocyte response to injury. In the case of Parkinson’s disease, astrocytes formglial scars in the areas of neurodegeneration.1 It would be a mistake, however, to view for-mation of these scars as the only neuron-astrocyte interaction, secondary to nerve cell loss.Most likely, glial scars are the ultimate manifestation of an ongoing interaction which hasbeen active during the entire course of the neurodegenerative process in the Parkinsonianbrain.
Both the pathogenesis of nigrostriatal degeneration and the role of astrocytes inParkinson’s disease are still poorly understood. Hypothetical scenarios can be visualizedbased on the current knowledge of mechanisms of nigrostriatal injury. For example, impor-tant clues can be derived from studies of models of toxicant-induced parkinsonism. Inparticular, the mechanisms of neuronal loss caused by the parkinsonism-inducing agent 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) have confirmed an important role ofastrocytes and directed our research toward specific aspects of neuron-astrocyte interac-tions that are most likely involved in dopaminergic degeneration. In this paper, I will firstpresent a general overview on idiopathic Parkinson’s disease and the MPTP model of par-kinsonism. Then, in the second part of the review, specific examples of known or suggestedneuron-astrocyte interactions will be discussed in relation to nigrostriatal dopaminergicdegeneration.
Idiopathic Parkinson’s DiseaseIdiopathic Parkinson’s disease is one of the most common neurodegenerative disor-
ders of aging. The age-specific prevalence is estimated to be 41/100,000 in the population atage 50, but it increases dramatically to 1,518/100,000 by age 80.2 From the clinical point ofview, this age-related neurodegenerative disorder is characterized by the classic symptom-atic triad of tremor, rigidity and bradykinesia as well as a number of other symptoms and

Astrocytes in Brain Aging and Neurodegeneration112
signs such as postural instability and loss of mimicry. The primary pathological feature ofParkinson’s disease is the loss of pigmented neurons in the substantia nigra pars compacta.3
The degenerative process, however, also involves neurons in other anatomical regions and,in particular, in the locus ceruleus and nucleus basalis of Meynert. Reactive microglia andglial scars, often made up of astrocytes with delicate glial fibrils, are usually observed in theareas most severely affected by neurodegeneration.1 Since nigral neurons project their axonsto the striatum and use dopamine as their neurotransmitter, depletion of dopamine in thenigrostriatal pathway represents the most relevant neurochemical alteration in the Parkin-sonian brain.4 It is also the basis for the primary treatment of the disease with L-dopa which,after crossing the blood-brain barrier, is decarboxylated to and replenishes the lackingdopamine.
As already mentioned, the toxic mechanisms underlying nigrostriatal degeneration re-main to be identified. However, at least two hypotheses concerning dopaminergic cell in-jury are often debated and will be discussed here because of their implications for possibleastrocyte involvement (see section below on Neuron-Astrocyte Interactions in NigrostriatalDegeneration). It has been suggested that dopamine, in addition to being the neurotrans-mitter used by nigrostriatal neurons, may also act as an endogenous toxin. As a corollary ofthis hypothesis, oxygen radical-induced oxidative stress could be responsible for or contrib-ute to neurodegeneration.5 The scheme in Figure 6.1 summarizes the relationship betweendopamine, oxidative stress and neuronal injury. The enzymatic catabolism of dopamine iscatalyzed by monoamine oxidase (MAO) and generates the 3,4-dihydroxyphenyl-acetalde-hyde metabolite, as well as hydrogen peroxide (H2O2). H2O2 is an oxidizing agent whichcould damage cells both directly and after its further reduction to the hydroxyl radical, areaction catalyzed by transition metals (Fenton reaction). Nonenzymatic conversion ofdopamine could also lead to oxidative stress via the formation of 6-hydroxydopamine and/or toxic quinone metabolites.6 Several lines of evidence support a role of dopamine-in-duced oxidative stress in nigrostriatal degeneration. This evidence includes a finding of in-creased oxidized glutathione (GSSG) levels as a consequence of higher dopamine turnoverin animals treated with reserpine or haloperidol.7-8 GSSG production is likely to reflect oxi-dative stress since GSSG is formed from the reaction between reduced glutathione (GSH)and H2O2, catalyzed by glutathione peroxidase. Additional evidence derives from studies inhumans indicating, for example, an increased total iron content in nigral tissue of patientsdying of Parkinson’s disease.9-10 As a transition metal, iron could be a catalyst in the Fentonreaction, generating the highly toxic hydroxyl radical (OH•) from H2O2.
The other hypothesis that is often put forward to explain neuronal loss in Parkinson’sdisease is that abnormalities in energy metabolism and, in particular, in mitochondrial oxi-dative phosphorylation may make the nigrostriatal tissue vulnerable to neurodegeneration.Findings in vitro support this hypothesis, showing that dopaminergic neurons are relativelymore susceptible than other neuronal cell types to the toxic effects of energy deprivation.11
Convincing evidence also derives from studies in humans revealing a decrease in mitochon-drial complex I activity in the brain of patients with idiopathic Parkinsonism.12-13 This de-crease appears to be selective for the substantia nigra, as it was not found in the caudatenucleus, medial and lateral globus pallidus, cerebral cortex or cerebellum.14 More recently,Swerdlow and colleagues performed an experiment in which a clonal line of human neuro-blastoma cells containing no mitochondrial DNA was repopulated with mitochondria fromcontrol subjects or patients with Parkinson’s disease.15 Cell lines from patients showed de-creased complex I activity, increased oxygen radical production and a greater susceptibilityto MPP+-induced cell death as compared to control cell lines. These results seem to supportthe hypothesis that abnormalities in mitochondrial DNA underlie the complex I impair-ment in Parkinson’s disease.

113Parkinson’s Disease
It is important to emphasize that the oxidative stress and mitochondrial hypotheses ofnigrostriatal degeneration should not be seen as antithetical. In fact, it is most likely that alink exists between impairment of mitochondrial activity and oxidative stress since mito-chondrial damage may ultimately cause an increased generation of oxygen radicals, and,vice versa, mitochondria are critical targets for oxidative stress.16 Thus, both toxic mecha-nisms may contribute to neuronal damage, perhaps in different phases in the course ofParkinson’s disease and/or to various extents in different patient populations.
MPTP-Induced ParkinsonismThe description 15 years ago of the clinical syndrome that abruptly developed in young
drug addicts who injected themselves with MPTP-contaminated illicit drugs represents anhistorical hallmark for scientists working in the field of neurodegenerative disorders.17 MPTPpoisoning mimicked as closely as anyone could have anticipated the clinical features ofParkinson’s disease18 and has since become a valuable model for in vitro and animal studieson nigrostriatal degeneration. The intense basic science work that followed the MPTP dis-covery has confirmed a striking number of similarities between MPTP-induced parkin-sonism and the idiopathic disease. In particular, neurochemical measurements in the mon-key and rodent brain have demonstrated dramatic MPTP-induced depletion of nigrostriataldopamine.19-20 From a pathological point of view, examination of the monkey brain afterMPTP exposure has revealed a rather selective action of the neurotoxicant toward thenigrostriatal system.19,21 Furthermore, similar to observations in the Parkinsonian brain,dopaminergic cell loss in MPTP-treated monkeys is accompanied by definite glial scars.1
The mechanisms of MPTP neurotoxicity have also been considerably, though notcompletely, clarified by studies in the last 15 years. A seemingly concerted sequence ofmetabolic, biochemical and toxic events appears to be triggered by MPTP exposure. A brief
Fig. 6.1. Mechanisms of dopaminergic degeneration inferred from the MPTP and dopaminemodels of neurotoxicity.

Astrocytes in Brain Aging and Neurodegeneration114
review of these events represents a necessary background for our discussion on neuronal-astrocyte interactions in nigrostriatal degeneration. Shortly after its discovery, MPTP wasfound to be metabolized by MAO,22 an enzyme localized to the outer membrane of mito-chondria. Of the two forms of this enzyme, MAO type B appears to be much more efficientthan type A in catalyzing MPTP conversion.23 This conversion occurs via a two-step pro-cess: First, MPTP is oxidized to the 1-methyl-4-phenyl-2,3-dihydropyridinium (MPDP+)intermediate, and then MPDP+ is further oxidized to the final product, the 1-methyl-4-phenylpyridinium (MPP+) metabolite. MAO B is responsible for the conversion of MPTPto MPDP+, while the rapid oxidation of MPDP+ to MPP+ is unlikely to be mediated byeither MAO or other enzymatic activities.24 The biotransformation of MPTP to MPP+ is anessential step leading to neurotoxicity, since MPP+ is thought to be the ultimate mediatorof the biologic effects of MPTP. Indeed, MPTP toxicity can be prevented by MAO Binhibitors25-26 and MPP+ itself causes toxic effects similar to those seen after MPTPadministration.27-28
The next critical step following MPTP bioactivation and leading to neurotoxicity is theaccumulation of MPP+ by nigrostriatal dopaminergic neurons. This is an active processoccurring via the catecholamine uptake system and is thought to play an important role inthe selective action of MPTP.29 Dopaminergic neurons are significantly more vulnerable toMPTP toxicity because they are exposed to higher levels of MPP+ for a prolonged period oftime. The mechanism of MPP+-induced cell death has also been extensively studied. In1985, a report by Nicklas and colleagues,30 showing that MPP+ was an inhibitor of mito-chondrial complex I, provided the first experimental evidence linking MPP+ cytotoxicity toan impairment of mitochondrial energy metabolism. A number of studies have since sup-ported the now widely accepted view that, upon reaching the intracellular space, MPP+:
1. accumulates in mitochondria;31
2. blocks mitochondrial respiratory chain activity at the level of complex I;30 and3. causes a dramatic depletion of the cellular energy substrate, ATP.24,32-34
Oxygen radicals could also be generated as a consequence of mitochondrial electronflow inhibition.35 Although the occurrence of these sequential events remains to be directlydemonstrated in nigrostriatal dopaminergic neurons, MPP+-induced ATP depletion hasbeen linked to cytotoxicity in a variety of in vitro models24,32-33 and an ATP decrease hasbeen observed in the striatum and ventral mesencephalon of mice injected with MPTP.34
The impairment of energy supplies caused by MPP+ has prompted scientists to sug-gest that excitatory amino acids (EAAs) may contribute to its neurotoxicity. ATP depletionand excitotoxicity could be linked by one or more of the following biochemical events:
1. the reduction of ATP levels could affect the normal uptake and inactivation of EAAs,resulting in their accumulation in the synaptic cleft;36
2. the membrane depolarization resulting from ATP depletion could relieve the volt-age-dependent Mg2+ block of NMDA channels.37 This would facilitate the activa-tion of EAA receptors and thus increase the sensitivity of neuronal cells to EAA-mediated damage;
3. overstimulation of the EAA receptors could itself cause membrane depolarizationleading to a further increase in energy consumption;38-39
4. one of the consequences of NMDA receptor activation is an influx of calcium andthus a rise in cytosolic calcium. In order to counteract this potentially cytotoxiccondition, calcium is:a. actively taken up by both mitochondria and the endoplasmic reticulum,40 andb. pumped out of the cells via the plasma membrane calcium translocase.41 Mito-
chondrial uptake and membrane translocation of calcium are energy-dependentprocesses and would significantly contribute to ATP consumption.

115Parkinson’s Disease
Thus, impaired mitochondrial activity and excitotoxicity might trigger a toxic cyclewith failing energy metabolism, leading to EAA receptor activation which, in turn, causesfurther depletion of energy supplies. That excitotoxicity plays a role in MPTP-inducednigrostriatal injury remains to be convincingly demonstrated. While some studies have shownprotection against MPTP neurotoxicity by antagonists of the N-methyl-D-aspartate (NMDA)excitotoxic receptor,42-43 other investigators have failed to achieve such protective effects.44-45
The energy impairment and ATP loss caused by MPP+ could lead to an increase incytosolic Ca2+ either directly or indirectly via NMDA receptor activation (Fig. 6.1). Thecytotoxic consequences of this Ca2+ perturbation have been related to the stimulation ofenzyme activities and, in particular, attention has recently been focused on the activity ofCa2+/calmodulin-dependent nitric oxide synthase (NOS) in the CNS. Stimulation of NOSwould generate excessive amounts of nitric oxide (NO), a neurotransmitter with potentialcytotoxic properties.46 The possibility that NOS stimulation may play a role in MPTP-in-duced nigrostriatal injury is suggested by studies in which the NOS inhibitor, 7-nitroindazole(7-NI), prevented neurotoxicity.47-49 This inhibitor has been found to counteract dopaminedepletion and neuronal cell loss in the striatum and substantia nigra of mice47-48 and toprotect against the behavioral, neurochemical and pathological effects of MPTP in the pri-mate model.49
Neuronal-Astrocyte Interactions in Nigrostriatal DegenerationThe scheme in Figure 6.1 summarizes the pathways leading to nigrostriatal degenera-
tion as suggested by evidence from studies on idiopathic and MPTP-induced Parkinsonismreviewed in the previous paragraphs. Next, the involvement of astrocytes in these toxic path-ways will be evaluated. Experimental findings will be discussed together with more hypo-thetical mechanisms in order to provide a general view of current knowledge as well aspossible directions for future research.
Astrocytes, MAO and Oxidative StressReactions catalyzed by MAO seem to be involved in different toxic pathways leading to
nigrostriatal damage. As shown in Figure 6.1, MAO has been implicated in the oxidativestress hypothesis of neurodegeneration because of the formation of H2O2, a product ofdopamine catabolism. This involvement of MAO in dopamine metabolism suggests thatastrocytes play an important role in neurodegeneration in light of the fact that these cellsmay comprise a major compartment for the extraneuronal metabolism of dopamine. Arecent study in monkeys has revealed that, after administration of the dopamine precursorL-DOPA, a significant proportion (approximately 50%) of dopamine deamination is medi-ated through MAO B.50 This reaction does not occur within dopaminergic neurons, whichexpress only MAO A,51 but could involve astrocytes because:
1. these cells contain both MAO A and MAO B;52-53 and2. they possess uptake sites for dopamine on their plasma membranes.54
Thus, MAO-mediated catabolism of dopamine within astrocytes could represent a sig-nificant source of H2O2. Hydrogen peroxide readily traverses cell membranes55 and maypromote neuronal degeneration.
Oxidative stress as a mechanism of cell injury only occurs when the production ofoxidizing species (e.g., H2O2) overwhelms anti-oxidant defense mechanisms. In the case ofH2O2, a primary anti-oxidant effect is achieved by the activity of glutathione peroxidase.This enzyme reduces H2O2 to H2O at the expense of glutathione, which is converted fromits reduced state (GSH) to its oxidized form (GSSG). Interestingly, astrocytes are known tocontain relatively higher levels of GSH than neuronal cells,56-57 raising the possibility thatH2O2 generated either intra- or extraneuronally may be scavenged within astrocytes.

Astrocytes in Brain Aging and Neurodegeneration116
Decreased levels of glutathione have been reported in the substantia nigra of patients withParkinson’s disease as compared to control subjects.9,58 This decrease may reflect specificneuron-astrocyte interactions, with intraglial glutathione playing an important role againstH2O2 accumulation and oxidative stress. An imbalance between pro-oxidant generating re-actions and anti-oxidant defenses may ultimately contribute to neurodegeneration in theParkinsonian brain.
Astrocytes, MAO and MPTPAnother pathway by which MAO-catalyzed reactions could lead to nigrostriatal dam-
age is through the bioactivation of MPTP-like endogenous and/or exogenous toxins (Fig. 6.2).The MPTP model of parkinsonism also provides a clear example of a relationship betweenMAO, astrocytes and dopaminergic cell injury. As already mentioned, immunocytochemi-cal studies have shown that brain MAO B is localized to serotonergic neurons and astro-cytes, but not to dopamine-containing neuronal groups.51-52 Therefore, although dopamin-ergic neurons of the nigrostriatal system are the main targets for MPTP neurotoxicity, theymay be incapable of mediating the bioactivation of MPTP to its toxic metabolite, MPP+.59
The possibility that serotonergic neurons may play a significant role in the production ofMPP+ in the brain in vivo has been ruled out by studies indicating that lesions of theseneurons do not attenuate MPTP neurotoxicity.60 Thus, indirect evidence points to astro-cytes as a primary locus for the MAO B-catalyzed conversion of MPTP to MPP+. The roleof astrocytes in MPTP biotransformation has been extensively documented by in vitro studiesshowing that:
1. glial cells in culture are indeed able to oxidize MPTP, first to MPDP+ via MAO andthen to MPP+;61 and
2. neuronal cells which are killed by MPP+ but not by MPTP, become sensitive to thetoxic effects of MPTP when cocultured with glial cells.62
If astrocytes represent the major source of MPP+, one might expect them to be par-ticularly vulnerable to its cytotoxic properties. However, an astrocytic reaction, rather thanovert glial damage, is a predominant feature of MPTP exposure in vivo. Changes in theexpression of glial fibrillary acidic protein (GFAP) have been observed in the rodent brainafter systemic administration of MPTP.63 Furthermore, neuropathological examination ofthe brain of monkeys with relatively long survival times after MPTP exposure (1 to 4 years)has revealed proliferation of cell processes and glial filaments and focal glial scars localizedto the ventral and lateral cell groups of the substantia nigra.1
Recent studies in vitro using primary cultures of astrocytes have provided clues thatmay explain the apparent resistance of glial cells to MPTP toxicity.64-65 In order to causecytotoxicity, relatively high intracellular levels of MPP+ have to be generated and main-tained within astrocytes. Two mechanisms may prevent this from happening. First, MPP+may be produced extracellularly rather than within astrocytes as a consequence of the fol-lowing events:
1. MPTP is oxidized to MPDP+ by intraglial MAO;2. MPDP+ crosses the plasma membranes of astrocytes in its lipophilic form of 1,2-
MPDP; and3. MPDP+ generates MPP+ in the extracellular space, possibly via autoxidation.Second, even if produced within glial cells and despite its charged chemical structure,
MPP+ appears capable of crossing astrocyte membranes and gaining access to the extracel-lular compartment;65 thus, MPP+ may not reach concentrations great enough and/or maynot persist long enough to cause irreversible injury to astrocytes. In contrast, the activeaccumulation of MPP+ via the catecholamine uptake system29 (see section on MPTP-in-

117Parkinson’s Disease
duced Parkinsonism, above) explains its selective toxic effects on dopaminergic neurons. Aschematic view of the interaction between astrocytes and dopaminergic neurons leading toMPTP bioactivation and MPP+ neurotoxicity is presented in Figure 6.2.
Astrocytes, MAO and AgingOnset of Parkinson’s disease before the age of 50 is quite rare, while the incidence of the
disease increases with age in the over-50 population. Therefore, aging represents the mostevident risk factor for Parkinson’s disease. The age-related mechanisms underlyingneurodegeneration, though still unknown, may involve astrocytes and MAO. This possibil-ity is supported by studies with the MPTP model of parkinsonism. It was noted that, in themouse, the sensitivity to MPTP-induced dopamine depletion and degeneration of dopam-inergic neurons increased with age.66-68 However, while MPTP-induced striatal dopaminedepletion was more pronounced in older mice, the effects of direct exposure to MPP+ didnot seem to be age-related.69 These findings suggested that differences in MPTP neurotox-icity with age involved changes in its conversion to the toxic MPP+ metabolite, catalyzed byMAO B. Subsequent studies proved a direct correlation between susceptibility to MPTP-induced nigrostriatal damage and MAO B activity; both increase in mice between 2 and 10and between 10 and 16 months of age.20
The involvement of MAO B in the age-related toxicity of MPTP is unlikely to be pecu-liar to the mouse model. In fact, an increase in MAO B activity is thought to be a commonfeature of the aging mammalian brain and has also been described in humans.70-72 Thisincrease is generally attributed to a greater proportion of MAO B-containing astrocytes inthe aging brain.70-71 Thus, if MAO B catalyzes the metabolic activation of MPTP-like neuro-toxins, astrocytes are likely to play an important role in rendering the aging brain increas-ingly susceptible to neurodegeneration.
The age-related increase in MAO B activity within astrocytes may also lead to enhancedgeneration of H2O2, a product of any reaction catalyzed by MAO B. As previously discussed,dopamine itself may be a substrate for astrocytic MAO B. The contribution of this
Fig. 6.2. Bioactivation of MPTP by astrocytic MAO B and accumulation of MPP+ indopaminergic neurons.

Astrocytes in Brain Aging and Neurodegeneration118
extraneuronal pathway of dopamine deamination could therefore increase with age, andH2O2 generated within astrocytes may become a relatively more prominent factor forneurodegeneration in the aging striatum and substantia nigra.
Astrocytes, Iron and AgingAnother possible link between astrocytes, aging and neurodegeneration arises from
the following observations:1. increased total iron content has been found in nigral tissue of patients dying with
Parkinson’s disease;9-10
2. a significant proportion of the excess iron reported in the Parkinsonian brain ap-pears to be localized within astrocytes;73 and
3. an age-dependent increase in iron-containing astrocytic inclusions has been de-scribed in different areas of the brain, including the striatum.74 The role of iron as acatalyst of cytotoxic reactions may be attributed to its ability to reduce H2O2 to OH•
in the Fenton reaction (Fig. 6.1). However, it is improbable that the generation ofOH• within astrocytes would damage adjacent neuronal cells; OH• is a highly reac-tive oxygen species which, unlike H2O2, would not be expected to cross cell mem-branes. Thus, the mechanisms by which astrocytic iron may contribute toneurodegeneration must involve the formation of toxic metabolites other than OH•.
An important requirement for iron to be involved in toxic reactions is its presence in aredox-active form. In this form, iron can act like a nonenzymatic peroxidase, oxidizing sub-strates and transferring electrons to H2O2.75 It is noteworthy therefore that inclusions ob-served within striatal astrocytes of the aging rodent and human brain exhibit peroxidaseactivity, most likely mediated by ferrous iron.74 This suggests that an increase in astrocyticiron content with age and possibly in pathological conditions may lead to thepseudoperoxidase-dependent formation of reactive metabolites and thus contribute toneurodegeneration (see chapter 11).
A potential substrate for iron-catalyzed oxidation is dopamine itself. Indeed, experi-mental evidence indicates that catecholamines like dopamine are substrates for peroxidaseactivity.76-78 Furthermore, addition of iron to a solution of dopamine has been shown tocause increased dopamine autoxidation as measured by aminochrome (a dopamine-de-rived quinone) formation.79 Products of dopamine oxidation such as quinone derivativescould cross astrocyte membranes and inflict damage to neuronal cells via oxidative stressand/or binding to cellular macromolecules.6
Iron-catalyzed reactions could also play a role in the bioactivation of MPTP-like neu-rotoxins. Although the conversion of MPTP to MPP+ is mostly dependent on the activity ofMAO B (see section on MPTP-induced Parkinsonism, above), recent results have shownthat a small proportion of MPP+ formation still occurs in astrocyte cultures in the presenceof MAO inhibitors and appears to require the catalytic activity of transition metals such asiron.80 A possible mechanism for this MAO-independent MPP+ generation involves thefollowing reactions:
1. formation of the superoxide radical from oxidation of ferrous iron in the presenceof oxygen;81
2. reaction of the superoxide radical with MPTP to produce a reactive intermediatewhich in turn would generate MPDP+;81 and
3. rapid nonenzymatic oxidation of MPDP+ to MPP+.24 The contribution of iron-mediated activation to the overall conversion of MPTP, although minor under nor-mal conditions, may become more significant with aging and in the Parkinsonianbrain due to iron accumulation within neuronal and astrocytic cells.

119Parkinson’s Disease
Astrocytes, Energy Metabolism and ExcitotoxicityPrompted at least in part by the finding that MPTP is a mitochondrial poison, research
on the pathogenesis of nigrostriatal damage has focused on the role of energy metabolism.A deficiency in mitochondrial oxidative phosphorylation and, more generally, in the abilityof neurons to utilize glucose and to convert it into energy-rich pyrophosphate bonds in ATPwould certainly be expected to place neurons at serious risk for degenerative processes. Ifthis is the case for Parkinson’s disease, then an important aspect of the relationship betweenenergy deficiency and neurodegeneration would be the interaction of neuronal cells withastrocytes. There is little doubt that the metabolic requirements of neuronal cells are depen-dent upon reactions that occur within astrocytes.82 These glial cells have been shown toprotect neurons in culture against anoxia,83 and glycogen stores within astrocytes appear toenable neighboring neurons to survive glucose deprivation.84 Therefore, any future studyaimed at elucidating the role of energy deficiency in nigrostriatal degeneration should ap-proach the issue from the point of view of neuron-astrocyte interactions.
We have recently investigated a specific mechanism by which a perturbation of energymetabolism in astrocytes may ultimately cause neuronal damage. This involves the abilityof astrocytes to take up glutamate through a high-affinity glutamate-aspartate carrier.85
Maintenance of low extracellular concentrations of glutamate is a critical function for pro-tecting neurons against the cytotoxic effects that result from a sustained activation of exci-tatory amino acid receptors (e.g., the NMDA receptor). Indeed, as illustrated in Figure 6.1,excitotoxicity has been proposed as a mechanism of nigrostriatal degeneration in the MPTPmodel of neurotoxicity as well as in idiopathic Parkinsonism. A series of experiments wereconducted in our laboratory in order to test the consequences of energy impairment onglutamate uptake from the extracellular space in primary cultures of astrocytes.
In one of these experiments, astrocytes were preincubated for 5 hours in the absence orpresence of MPP+. Then, glutamate (500 µM) was added and its extracellular levels weremonitored at 45, 90 and 150 min. As reported in Table 6.1, glutamate was efficiently re-moved by control astrocytes (without MPP+) and only 8% of its initial concentration waspresent extracellularly at the 150 min time point. In contrast, the ability of astrocytes toremove glutamate was significantly impaired after preincubation with MPP+ and, with 50 µMMPP+, 65% of the initial concentration of glutamate was still measured at 150 min in theextracellular compartment (Table 6.1). Since:
1. no sign of cytotoxicity was observed in MPP+-treated cultures during the time ofthe experiment; and
2. results similar to those seen with MPP+ could be obtained in cultures pretreatedwith rotenone (an inhibitor of mitochondrial complex I activity), it can be impliedthat any perturbation of energy metabolism in astrocytes significantly impairs theircritical function of removing extracellular glutamate.
The following sequence of events may be hypothesized that would link neurodegen-eration to specific neuron/astrocyte interactions. Changes in energy metabolism take placein the astrocyte population because of:
1. aging (a decline in mitochondrial oxidative phosphorylation appears to occur inthe aging brain of primates86);
2. pathological processes (a decrease in mitochondrial complex I activity has beenreported in the Parkinsonian substantia nigra12-14); and/or
3. toxic exposure (MPTP is metabolically activated to the mitochondrial poison MPP+within astrocytes).
Astrocytes would then become unable to take up efficiently glutamate from the extra-cellular space, leading to a sustained activation of NMDA receptors and ultimately toexcitotoxic injury.
Astrocytes in Brain Aging and Neurodegeneration120
Astrocytes and Nitric OxideIf the neurodegenerative process underlying Parkinson’s disease involves excitotoxicity,
then it is possible that neuronal injury is mediated at least in part through the generation ofnitric oxide. As already mentioned, when NMDA receptors are activated, the NMDA chan-nel allows an influx of calcium into the cells. Several lines of evidence indicate that, aftersustained receptor activation, an increase in cytosolic calcium concentrations stimulatescalmodulin-regulated NOS activity and may lead to neuronal damage via excessive NOproduction. Indeed, NMDA-induced toxicity in neuronal cultures can be prevented by in-hibitors of NOS.87 NOS inhibitors are also effective in protecting against ischemic cerebraldamage, which is thought to be NMDA receptor-mediated in animal models.88
The involvement of NO in the pathogenesis of nigrostriatal damage in experimentaland idiopathic Parkinsonism is suggested not only by findings concerning the MPTP andmethamphetamine models of dopaminergic injury47-49,89 (see section on MPTP-inducedParkinsonism, above), but also by observations in the Parkinsonian brain. Hunot and col-leagues have recently reported that immunoreactivity for the inducible form of NOS (iNOS)is increased in dopaminergic brain regions (i.e., the substantia nigra pars compacta, theventral tegmental area and the A8 catecholaminergic cell group) of Parkinsonian patients ascompared to control subjects.90 Interestingly, iNOS appeared to be localized to nonneuronalelements, possibly activated macrophages and/or astrocytes.
Whether astrocyte-generated NO plays a causal role in dopaminergic degeneration orwhether iNOS stimulation in glial cells occurs as a consequence of neuronal damage re-mains to be ascertained. It is already known, however, that astrocytes express iNOS whenexposed to specific inducers, such as lipopolysaccharide or tumor necrosis factor-α (TNF-α).91
The effects of TNF-α may be of particular importance, since TNF-immunoreactive glialcells have been described in the substantia nigra of patients with Parkinson’s disease but notin control subjects.92 Thus, it is possible that induction of astrocytic NOS by cytokines likeTNF-α may contribute to nigrostriatal damage, providing another mechanism by whichastrocyte/neuronal interactions may lead to neurodegeneration in Parkinson’s disease.
Table 6.1. Glutamate uptake by primary cultures of astrocytes in the presenceand absence of MPP+
Pretreatment Time (min)
0 45 90 150
None 100 ± 2.8 81.3 ± 3.9 41.7 ± 0.9 7.9 ± 0.8MPP+ (5 µM) 100 ± 6.8 101.3 ± 4.8* 64.7 ± 2.0** 32.4 ± 2.1**MPP+ (50 µM) 100 ± 6.3 95.1 ± 9.4* 81.0 ± 4.2** 65.9 ± 6.0**
Astrocytes were preincubated for 5 hours in the absence or presence of MPP+. Then glutamate(500 µM) was added, and its extracellular levels were monitored at 45, 90 and 150 min.Data (± SEM) are expressed as per cent of glutamate concentration measured at the 0 time point.* Statistically different (p<0.05) from the control group preincubated in the absence of MPP+.** Statistically different (p<0.05) from the other two experimental groups.

121Parkinson’s Disease
Astrocytes and Neurotrophic FactorsIt is not surprising that neurotrophic factors have increasingly become the subject of
intense studies concerning neurodegenerative disorders. Their important role in neuronaldevelopment and subsistence has raised great expectations for their potential therapeuticefficacy against progressive cell loss and/or in support of neuronal regeneration. Nerve growthfactor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin 3 and 5 (NT-3 andNT-5) and basic fibroblast growth factor (bFGF) have all been tested as potentialneuroprotective agents against dopaminergic degeneration; in the MPP+ model of neuro-toxicity, they have shown various degrees of effectiveness.93 However, the identification of aglial cell line-derived neurotrophic factor (GDNF) in 1993 has significantly redirected thefocus of scientists in the field of Parkinson’s disease to this new member of the transforminggrowth factor-β superfamily.94 This is because GDNF appears to be both more specific andmore potent than other factors toward dopaminergic neurons.
GDNF is expressed in the developing striatum and promotes the survival of mesen-cephalic dopaminergic neurons in culture.94-95 When used in animal models of nigrostriatalinjury, it has been reported not only to protect against dopaminergic damage but also toinduce repair mechanisms. In C57Bl/6 mice exposed to MPTP, GDNF injected over thesubstantia nigra or in the striatum prior to MPTP protected dopaminergic cell bodies andterminals.96 Interestingly, GDNF was partially effective against dopamine depletion evenwhen injected 1 week after MPTP. This ability of GDNF to improve the recovery of dopam-inergic neurons after toxic injury has also been documented in vitro. Short-term exposureof mesencephalic cultures to MPP+ for 1 hour resulted in a continuous loss of cells over theensuing 5 days.97 However, if GDNF was added after MPP+ removal, it significantly pre-vented neuronal depletion and stimulated the regrowth of dopaminergic fibers.
The therapeutic potential of GDNF in the treatment of Parkinson’s disease is alreadybeing explored.98 One major challenge is the need for a delivery strategy that would allowGDNF to reach target areas of the brain, bypassing the blood-brain barrier. Possible solu-tions to this problem may emerge from studies on the mechanisms of induction, produc-tion and secretion of neurotrophic factors by astrocytes during neuronal development andin the course of neurotoxic injury. These studies may lead to treatment of nigrostriatal de-generation with agents that stimulate GDNF production in situ, rendering its direct admin-istration unnecessary.
ConclusionSecretion of neurotrophic factors by astrocytes as a mechanism of neuronal pruning
and differentiation during development and as a possible signal for neuronal sprouting andregeneration during pathological/toxic events provides a clear illustration of astrocyte/neu-ron interactions. In this review article, we have discussed a number of other mechanisms bywhich such interactions may contribute to the pathogenesis of idiopathic and toxicant-induced Parkinsonism. In some cases, astrocytes could play a protective role against thedevelopment and progression of nigrostriatal degeneration. For example, they may coun-teract oxidative stress by virtue of their relatively high content of GSH and support tissuerecovery via the production of neurotrophic factors. On the other hand, astrocytes may bedirectly involved in the biochemical/pathological changes leading to neurodegeneration. Aspreviously seen, astrocytes may bioactivate MPTP-like neurotoxins and promote, for ex-ample, the generation of NO. It is also possible that neuron/astrocyte interactions underliethe increased vulnerability to nigrostriatal damage with aging. Changes in astrocyte levelsof MAO and/or iron may contribute to this effect.
Neurodegeneration is likely to result from an imbalance between toxic events and de-fense capabilities within the nigrostriatal tissue. Both toxic and defense mechanisms would

Astrocytes in Brain Aging and Neurodegeneration122
therefore be expected to involve astrocyte/neuron interactions, accounting for the role ofastrocytes in both counteracting and promoting neuronal injury. It is also important toreemphasize that the precise sequence of events leading to death of nigrostriatal dopam-inergic neurons remains to be unraveled. For this reason, discussion on the role of astro-cytes (and neurons, for that matter) remains somewhat speculative in nature. Perhaps thesingle most important goal of future research on Parkinson’s disease is to evaluate all of thehypothesized mechanisms of neurodegeneration (e.g., oxidative stress, mitochondrial fail-ure and excitotoxicity) in order to identify causative factors. Astrocyte/neuron interactionsshould be an important component of these future studies, because there is little doubt thatastrocytes are indeed active participants in both physiologic and pathological processes ofthe CNS.
AcknowledgmentsThe author wishes to thank Dr. S.A. Jewell for her comments on the manuscript. This
work was supported by The Parkinson’s Institute.
References1. Forno LS, DeLanney LE, Irwin I et al. Astrocytes and Parkinson’s disease. In: Yu ACH,
Hertz L, Norenberg MD, Sykova E, Waxman SG, eds. Progress in Brain Research. Vol 94.Amsterdam: Elsevier Science Publishers, 1992:429-436.
2. Tanner CM, Thelen JA, Offord KP et al. Parkinson’s disease (PD) incidence in OlmstedCounty, MN: 1935-1988. Neurology 1992; 42 (suppl 3): 194.
3. Forno LS. Neuropathology of Parkinson’s disease. J Neuropathol Exp Neurol 1996;55:259-272.
4. Bernheimer H, Birkmeyer W, Hornykiewicz O. Brain dopamine and the syndromes ofParkinson and Huntington. J Neurol Sci 1973; 20:925-964.
5. Fahn S, Cohen G. The oxidant stress hypothesis in Parkinson’s disease: Evidence support-ing it. Ann Neurol 1992; 32:804-812.
6. Graham DG. Oxidative pathways for catecholamines in the genesis of neuromelanin andcytotoxic quinones. Mol Pharmacol 1978; 14:633-643.
7. Spina MB, Cohen G. Dopamine turnover and glutathione oxidation: Implications forParkinson’s disease. Proc Natl Acad Sci USA 1989; 86:1398-1400.
8. Cohen G, Spina MB. Deprenyl suppresses the oxidant stress associated with increaseddopamine turnover. Ann Neurol 1989; 26:689-690.
9. Riederer P, Sofic E, Rausch WD et al. Transition metals, ferritin, glutathione, and ascorbicacid in Parkinsonian brains. J Neurochem 1989; 52:515-520.
10. Dexter DT, Weils FR, Lees AJ et al. Increased nigral iron content and alterations in othermetal ions occurring in brain in Parkinson’s disease. J Neurochem 1989; 52:1830-1836.
11. Marey-Semper I, Gelman M, Levi-Strauss M. A selective toxicity toward cultured mesen-cephalic dopaminergic neurons is induced by the synergistic effects of energetic metabo-lism impairment and NMDA receptor activation. J Neurosci 1995; 15:5912-5918.
12. Schapira AHV, Cooper JM, Dexter DT et al. Mitochondrial complex I deficiency inParkinson’s disease. J Neurochem 1990; 54:823-827.
13. Mizuno Y, Otha S, Tanaka M et al. Deficiencies in complex I subunits of the respiratorychain in Parkinson’s disease. Biochem Biophys Res Commun 1989; 163:1450-1455.
14. Schapira AHV, Mann VM, Cooper JM et al. Anatomic and disease specificity of NADHCoQ1 reductase (complex I) deficiency in Parkinson’s disease. J Neurochem 1990;55:2142-2145.
15. Swerllow RH, Parks JK, Miller SW et al. Origin and functional consequences of the com-plex I defect in Parkinson’s disease. Ann Neurol 1996; 40:663-671.
16. Di Monte DA, Chan P, Sandy MS. Glutathione in Parkinson’s disease: A link betweenoxidative stress and mitochondrial damage? Ann Neurol 1992; 32: S111-S115.

123Parkinson’s Disease
17. Langston JW, Ballard PA, Tetrud JW et al. Chronic Parkinsonism in humans due to aproduct of meperidine analog synthesis. Science 1983; 219:979-980.
18. Langston JW. MPTP and Parkinson’s disease. TINS 1985; 8:79-83.19. Burns RS, Chiueh CC, Markey SP et al. A primate model of Parkinsonism: Selective de-
struction of dopaminergic neurons in the pars compacta of the substantia nigra byN-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proc Natl Acad Sci USA 1983; 80:4546-4550.
20. Irwin I, Finnegan KT, DeLanney LE et al. The relationships between aging, monoamineoxidase, striatal dopamine and the effects of MPTP in C57BL/6 mice: A critical reassess-ment. Brain Res 1992; 572:224-231.
21. Langston JW, Forno LS, Rebert CS et al. Selective nigral toxicity after systemic administra-tion of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in the squirrel monkey. BrainRes 1984; 292:390-394.
22. Chiba K, Trevor AJ, Castagnoli N Jr. Metabolism of the neurotoxic tertiary amine, MPTP,by brain monoamine oxidase. Biochem Biophys Res Commun 1984; 120:574-578.
23. Salach JI, Singer TP, Castagnoli N Jr et al. Oxidation of the neurotoxic amine 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) by monoamine oxidases A and B and suicideinactivation of the enzymes by MPTP. Biochem Biophys Res Commun 1984; 125:831-835.
24. Wu EY, Langston JW, Di Monte DA. Toxicity of the 1-methyl-4-phenyl-2,3-dihydropy-ridinium and 1-methyl-4-phenylpyridinium species in primary cultures of mouse astro-cytes. J Pharmacol Exp Ther 1992; 262:225-230.
25. Langston JW, Irwin I, Langston EB et al. Pargyline prevents MPTP-induced Parkinsonismin primates. Science 1984; 225:1480-1482.
26. Heikkila RE, Manzino L, Cabbat FS et al. Protection against the dopaminergic neurotoxic-ity of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine by monoamine oxidase inhibitors.Nature 1984; 311:467-469.
27. Markey SP, Johannessen JN, Chiueh CC et al. Intraneuronal generation of a pyridiniummetabolite may cause drug-induced Parkinsonism. Nature 1984; 311:464-467.
28. Bradbury AJ, Costall B, Domeney AM et al. 1-Methyl-4-phenylpyridine is neurotoxic tothe nigrostriatal dopamine pathway. Nature 1986; 319:56-57.
29. Javitch JA, D’Amato RJ, Strittmatter SM et al. Parkinsonism-inducing neurotoxin, N-me-thyl-4-phenyl-1,2,3,6-tetrahydropyridine: Uptake of the metabolite N-methyl-4-phenyl-pyridine by dopamine neurons explains selective toxicity. Proc Natl Acad Sci USA 1985;82:2173-2177.
30. Nicklas WJ, Vyas I, Heikkila RE. Inhibition of NADH-linked oxidation in brain mitochon-dria by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Life Sci 1985; 36:2503-2508.
31. Ramsay RR., Singer TP. Energy-dependent uptake of N-methyl-4-phenylpyridinium, theneurotoxic metabolite of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, by mitochondria.J Biol Chem 1986; 261:7585-7587.
32. Di Monte DA, Jewell SA, Ekstrom G et al. 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine(MPTP) and 1-methyl-4-phenylpyridine (MPP+) cause rapid ATP depletion in isolatedhepatocytes. Biochem Biophys Res Commun 1986; 137:310-315.
33. Denton T, Howard BD. A dopaminergic cell line variant resistant to the neurotoxin 1-me-thyl-4-phenyl-1,2,3,6-tetrahydropyridine. J Neurochem 1987; 49:622-629.
34. Chan P, Langston JW, Irwin I et al. 2-Deoxyglucose enhances 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced ATP loss in the mouse brain. J Neurochem 1993;61:610-616.
35. Cleeter MWJ, Cooper JM, Schapira AHV. Irreversible inhibition of complex I by 1-me-thyl-4-phenylpyridinium: Evidence for free radical involvement. J Neurochem 1992;58:786-789.
36. Nicholls D, Attwell D. The release and uptake of excitatory amino acids. TIPS 1990;11:462-468.
37. Henneberry RC, Novelli A, Vigano MA et al. Energy-related neurotoxicity at the NMDAreceptor: A possible role in Alzheimer’s disease and related disorders. Prog Clin Biol Res1989; 317:143-156.

Astrocytes in Brain Aging and Neurodegeneration124
38. Tanaka S, Sako K, Tanaka T et al. Uncoupling of local blood flow and metabolism in thehippocampal CA3 in kainic acid-induced limbic seizure status. Neuroscience 1990;36:339-48.
39. Tuong MDT, Brion F, Schwartz JC. Stimulation of deoxy[3H]glucose uptake into slice fromcerebral cortex elicited by excitatory amino acids. Neuroscience 1984; 12:385-393.
40. Carafoli E, Crompton M. The regulation of intracellular calcium by mitochondria. AnnNY Acad Sci 1978; 307:269-284.
41. Kraus-Friedmann N, Biber J, Murer H et al. Calcium uptake in isolated hepatic plasma-membrane vesicles. Eur J Biochem 1982; 129:7-12.
42. Turski L, Bressler K, Rettig K et al. Protection of substantia nigra from MPP+ neurotoxic-ity by N-methyl-D-aspartate antagonists. Nature 1991; 349:414-418.
43. Zuddas A, Oberto G, Vaglini F et al. MK-801 prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism in primates. J Neurochem 1992; 59:733-739.
44. Sonsalla PK, Zeevalk GD, Manzino L et al. MK-801 fails to protect against the dopaminer-gic neuropathology produced by systemic 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine inmice or intranigral 1-methyl-4-phenylpyridinium in rats. J Neurochem 1992; 58:1979-1982.
45. Chan P, Langston JW, Di Monte DA. MK-801 temporarily prevents MPTP-induced acutedopamine depletion and MPP+ elimination in the mouse striatum. J Pharmacol Exp Ther1993; 267:1515-1520.
46. Varner PD, Beckman JS. Nitric oxide toxicity in neuronal injury and degeneration. In:Vincent S, ed. Nitric Oxide in the Nervous System. San Diego: Academic Press,1995:191-206.
47. Schultz JB, Matthews RT, Muqit MMK et al. Inhibition of neuronal nitric oxide synthaseby 7-nitroindazole protects against MPTP-induced neurotoxicity in mice. J Neurochem1995; 64:936-939.
48. Przedborski S, Jackson-Lewis V, Yokoyama R et al. Role of neuronal nitric oxide in MPTP(1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine)-induced dopaminergic neurotoxicity. ProcNatl Acad Sci USA 1996; 93:4565-4571.
49. Hantraye P, Brouillet E, Ferrante R et al. Inhibition of neuronal nitric oxide synthase pre-vents MPTP-induced Parkinsonism in baboons. Nature Med 1996; 2:1017-1022.
50. Di Monte DA, DeLanney LE, Irwin I et al. Monoamine oxidase-dependent metabolism ofdopamine in the striatum and substantia nigra of L-dopa-treated monkeys. Brain Res 1996;738:53-59.
51. Westlund KN, Denney RM, Kochersperger LM et al. Distinct monoamine oxidase A and Bpopulations in primate brain. Science 1985; 230:181-183.
52. Levitt P, Pintar JE, Breakefield XO. Immunocytochemical demonstration of monoamineoxidase B in brain astrocytes and serotonergic neurons. Proc Natl Acad Sci USA 1982;79:6385-6389.
53. Yu PH, Hertz L. Differential expression of type A and type B monoamine oxidase of mouseastrocytes in primary cultures. J Neurochem 1982; 39:1492-1495.
54. Semenoff D, Kimelberg HK. Autoradiography of high affinity uptake of catecholamine byprimary astrocyte cultures. Brain Res 1985; 348:125-136.
55. Halliwell B, Gutteridge JMC. Oxygen toxicity, oxygen radicals, transition metals and dis-ease. Biochem J 1984; 219:1-14.
56. Slivka A, Mytilineou C, Cohen G. Histochemical evaluation of glutathione in brain. BrainRes 1987; 409:275-284.
57. Raps SP, Lai JCK, Hertz L et al. Glutathione is present in high concentrations in culturedastrocytes but not in cultured neurons. Brain Res 1989; 493:398-401.
58. Perry TL, Godin DV, Hansen S. Parkinson’s disease: A disorder due to nigral glutathionedeficiency? Neurosci Lett 1982; 33:305-310.
59. Vincent SR. Histochemical localization of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridineoxidation in the mouse brain. Neuroscience 1989; 28:189-199.
60. Melamed E, Pikarski E, Goldberg A et al. Effect of serotonergic, corticostriatal and kainicacid lesions on the dopaminergic neurotoxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydro-pyridine (MPTP) in mice. Brain Res 1986; 399:178-180.

125Parkinson’s Disease
61. Di Monte DA, Wu EY, Irwin I et al. Biotransformation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in primary cultures of mouse astrocytes. J Pharmacol Exp Ther 1991;258:594-600.
62. Notter MSD, Irwin I, Langston JW et al. Neurotoxicity of MPTP and MPP+ in vitro: Char-acterization using specific cell lines. Brain Res 1988; 456:254-262.
63. Reinhard JF Jr, Miller D, O’Callaghan JP. The neurotoxicant MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) increases glial fibrillary acidic protein and decreases dopaminelevels of the mouse striatum: Evidence for glial response to injury. Neurosci Lett 1988;95:246-251.
64. Di Monte DA, Wu EY, DeLanney LE et al. Toxicity of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in primary cultures of mouse astrocytes. J Pharmacol Exp Ther 1992;261:44-49.
65. Di Monte DA, Wu EY, Irwin I et al. Production and disposition of 1-methyl-4-phenylpyridinium in primary cultures of mouse astrocytes. Glia 1992; 5:48-55.
66. Jarvis MF, Wagner GC. Age-dependent effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydro-pyridine (MPTP). Neuropharmacology 1985; 24:581-583.
67. Gupta M, Gupta BK, Thomas R et al. Aged mice are more sensitive to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine than young adults. Neurosci Lett 1986; 70:326-331.
68. Ricaurte GA, Irwin I, Forno LS et al. Aging and MPTP-induced degeneration of dopamin-ergic neurons in the substantia nigra. Brain Res 1987; 403:43-51.
69. Irwin I, Ricaurte GA, DeLanney LE et al. The sensitivity of nigrostriatal dopamine neuronsto MPP+ does not increase with age. Neurosci Lett 1988; 87:51-56.
70. Benedetti MS, Keane PE. Differential changes in monoamine oxidase A and B activity inthe aging rat brain. J Neurochem 1980; 35:1026-1032.
71. Fowler J, Wilberg A, Oreland L et al. The effect of age on the activity and molecular prop-erties of human brain monoamine oxidase. J Neural Transm 1980; 49:1-20.
72. Robinson DS, Davis JM, Nies A et al. Relation of sex and aging to monoamine oxidaseactivity of human brain, plasma, and platelets. Arch Gen Psychiatry 1971; 24:536-539.
73. Jellinger K, Paulus W, Grundke-Iqbal I et al. Brain iron and ferritin in Parkinson’s andAlzheimer diseases. J Neural Transm 1990; 2:327-340.
74. Schipper HM. Gomori-positive astrocytes: Biological properties and implications for neu-rologic and neuroendocrine disorders. Glia 1991; 4:365-377.
75. Miller DM, Buettner GR, Aust SD. Transition metals as catalysts of “autoxidation” reac-tions. Free Radical Biol Med 1990; 8:95-108.
76. Kalyanaraman B, Felix CC, Sealy RC. Peroxidatic oxidation of catecholamines: A kineticelectron spin resonance investigation using the spin stabilization approach. J Biol Chem1984; 259:7584-7589.
77. Metodiewa D., Dunford HB. The role of myeloperoxidase in the oxidation of biologicallyactive polyhydroxyphenols (substitutes catechols). Eur J Biochem 1990; 193:445-448.
78. Schipper HM, Kotake Y, Janzen EG. Catechol oxidation by peroxidase-positive astrocytesin primary culture: An electron spin resonance study. J Neurosci 1991; 11:2170-2176.
79. Poirier J, Donaldson J, Barbeau A. The specific vulnerability of the substantia nigra toMPTP is related to the presence of transition metals. Biochem Biophys Res Commun 1985;128:25-33.
80. Di Monte DA, Schipper HM, Hetts S et al. Iron-mediated bioactivation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in glial cultures. Glia 1995; 15:203-206.
81. Poirier J, Barbeau A. A catalyst function for MPTP in superoxide formation. BiochemBiophys Res Commun 1985; 131:1284-1289.
82. Hamprecht B, Dringen R. Energy metabolism. In: Kettenman H, Ransom BR, eds. Neuro-glia. New York: Oxford University Press, 1995:473-487.
83. Vibulsreth S, Hefti F, Ginsberg MD et al. Astrocytes protect cultured neurons from degen-eration induced by anoxia. Brain Res 1987; 422:303-311.
84. Swanson RA, Choi DW. Glial glycogen stores affect neuronal survival during glucose dep-rivation in vitro. J Cereb Blood Flow Metab 1993; 13:162-169.

Astrocytes in Brain Aging and Neurodegeneration126
85. Schousboe A, Westergaard N. Transport of neuroactive amino acids in astrocytes. In:Kettenman H, Ransom BR, eds. Neuroglia. New York: Oxford University Press,1995:246-258.
86. Di Monte DA, Sandy MS, DeLanney LE et al. Age-dependent changes in mitochondrialenergy production in striatum and cerebellum of the monkey brain. Neurodegeneration1993; 2:93-99.
87. Dawson VL, Dawson TM, Bartley DA et al. Mechanisms of nitric oxide-mediated neuro-toxicity in primary brain cultures. J Neurosci 1993; 13:2651-2661.
88. Nowicki JP, Duval D, Poignet H et al. Nitric oxide mediates neuronal death after focalcerebral ischemia in the mouse. Eur J Pharmacol 1991; 204:339-340.
89. Di Monte DA, Royland J, Jakowec MW et al. Role of nitric oxide in methamphetamineneurotoxicity: Protection by 7-nitroindazole, an inhibitor of neuronal nitric oxide syn-thase. J Neurochem 1996; 67:2443-2450.
90. Hunot S, Boissiere F, Faucheux B et al. Nitric oxide synthase and neuronal vulnerability inParkinson’s disease. Neuroscience 1996; 72:355-363.
91. Vigne P, Damais C, Frelin C. IL1 and TNFα induce cGMP formation in C6 astrocytomacells via the nitridergic pathway. Brain Res 1993; 606:332-336.
92. Boka G, Anglade P, Wallach D et al. Immunocytochemical analysis of tumor necrosis fac-tor and its receptors in Parkinson’s disease. Neurosci Lett 1994; 172:151-154.
93. Kirschner PB, Jenkins BG, Schulz JB et al. NGF, BDNF and NT-5, but not NT-3 protectagainst MPP+ toxicity and oxidative stress in neonatal animals. Brain Res 1996; 713:178-185.
94. Lin L-FH, Doherty DH, Lile JD et al. GDNF: A glial cell line-derived neurotrophic factorfor midbrain dopaminergic neurons. Science 1993; 260:1130-1132.
95. Schaar DG, Sieber B-A, Dreyfus CF et al. Regional and cell-specific expression of GDNF inrat brain. Exp Neurol 1993; 124:368-371.
96. Tomac A, Lindqvist E, Lin L-FH et al. Protection and repair of the nigrostriatal dopamin-ergic system by GDNF in vivo. Nature 1995; 373:335-339.
97. Hou J-GG, Lin L-FH, Mytilineou C. Glial cell line-derived neurotrophic factor exerts neu-rotrophic effects on dopaminergic neurons in vitro and promotes their survival and re-growth after damage by 1-methyl-4-phenylpyridinium. J Neurochem 1996; 66:74-82.
98. Gash DM, Zhang Z, Ovadla A et al. Functional recovery in Parkinsonian monkeys treatedwith GDNF. Nature 1996; 380:252-255.

CHAPTER 7
Astrocytes in Brain Aging and Neurodegeneration, edited by Hyman M. Schipper.©1998 R.G. Landes Company.
Astrocytes in TransmissibleSpongiform Encephalopathies(Prion Diseases)Pawel P. Liberski, Radzislaw Kordek, Paul Brown and D. Carleton Gajdusek
Introduction
The transmissible spongiform encephalopathies (TSE) or prion diseases are a group ofneurodegenerative disorders which include kuru (Fig. 7.1), Creutzfeldt-Jakob disease
(CJD), Gerstmann-Straussler-Scheinker (GSS) disease, and fatal familial insomnia (FFI) inman; natural scrapie in sheep, goats and mufflons; transmissible mink encephalopathy (TME)in ranch-reared mink; chronic wasting disease (CWD) of captive and free living mule deerand elk in the USA; bovine spongiform encephalopathy (BSE) or “mad cow disease” and itsanalogues in several exotic species of ungulates, a puma and several cheetahs from Britishzoological gardens; and feline spongiform encephalopathy in domestic cats.1-5 The status ofspongiform encephalopathy in ostrich is still unclear but it is probably unrelated to TSEs.6
Recently, a new variant of CJD (vCJD) was described among young people in the UK.7 ThisvCJD is characterized by florid PrP plaques surrounded by a corona of vacuoles (Fig. 7.2).As virtually identical plaques were reproduced in macaques inoculated with BSE material8
and the same glycosylation9 pattern is observed in BSE and vCJD, the transmission fromBSE to humans seems increasingly likely.
TSEs are caused by a still incompletely understood pathogen variously referred to as avirus (usually with the adjectives slow, unconventional or atypical), agent, “prion” or “virino”.These names reflect, in part, different views on the molecular structure of the pathogen and,by the same token, our ignorance of its nature.10 Those who prefer to view this pathogen ascomposed “predominantly or entirely” of one protein, PrP, use the term “prion” hence theterm “prion disorders”.11-12 The last term, however, implies more than a semantic prefer-ence: It suggests that neuropathologically confirmed cases of TSE are only the “tip of theiceberg” of poorly delineated conditions (“dementias without a characteristic pathology”)which share abnormalities in the PRNP gene (the gene which encodes for PrP).13 To evalu-ate the validity of this claim, one of us searched for PrP in 46 cases of “nonspongiform”dementias; none was positive.14 It was concluded that “for all intents and purposes ‘priondementia’ and ‘spongiform encephalopathy’ are one and the same”.
Irrespective of the nature of the agent, it is widely accepted that the abnormal isoform(probably also an abnormal conformer) PrPsc plays a crucial role in the pathogenesis of thewhole group of TSEs.15-16 This protein accumulates in all TSE-affected brains, either as

Astrocytes in Brain Aging and Neurodegeneration128
Fig. 7.1. A preadolescent child, totally incapacitated by kuru in 1957. The child hadsuch severe dysarthria that he could no longer communicate by word, but was stillintelligent and alert. He had spastic strabismus. He could not stand, sit without sup-port, or even roll over; he had been ill for less than six months and died within a fewmonths of the time of photography.

129Astrocytes in Transmissible Spongiform Encephalopathies (Prion Diseases)
amyloid plaques or plaque-like deposits or in the so-called synaptic forms.17 Furthermore,alterations (point mutations, insertions and deletions) in the gene encoding PrP (PRNP inhumans) cosegregate with various phenotypic presentations of CJD, GSS or FFI.18
The “virino” hypothesis suggests that the pathogen is an unprecedented molecular chi-mera composed of a still to be discovered nucleic acid and a shell protein which is host-encoded (possibly even PrP).19 The virus hypothesis simply suggests that the pathogen is yetto be purified, as there is no conclusive evidence to prove that it is outside the spectrum ofconventional viruses.20 The “unified theory” of Weissmann suggests that, not unlike thevirino theory,19 the agent is a molecular chimera,21-22 in which PrPsc confers infectivity, whilea still undetected oligonucleotide specifies strain characteristics; in other words, the agenthas a genome that is unnecessary for infectivity.
Astrocytosis or reactive gliosis is a prominent feature of naturally occurring and ex-perimentally produced TSEs.23-25 It is also a feature of a new entity “familial progressivesubcortical gliosis” in which PrP accumulates in the brain.26 In contrast to familial CJD andGSS cases, which are linked to chromosome 20, familial progressive gliosis is linked to chro-mosome 17.
In this chapter we shall review diverse aspects of astrocytic gliosis in naturally occur-ring and experimentally induced TSE.
Fig. 7.2. Astrocytes within the perimeter of florid plaques in a case of the new variant of CJD.Inset, a typical florid plaque surrounded by a corona of spongiform change. GFAP immunohis-tochemistry, original magnification x 1000; inset, H & E. By courtesy of Dr. James Ironside,Edinburgh, Scotland.

Astrocytes in Brain Aging and Neurodegeneration130
KURU
Natural DiseaseHistorically, astrocytic hypertrophy and proliferation have been stressed as a hallmark
of kuru27-28 and supported by more recent systematic immunohistochemical studies.29 As-trocytic proliferation was widespread and more abundant in gray than in white matter.Usually, astrocytosis paralleled neuronal destruction, but it has also been observed in re-gions with only minimal brain pathology. In the pons, severe gliosis was observed in thetegmental and basal portions, with a conspicuous sparing of the pyramidal tracts and me-dial lemnisci. Gliosis was severe in the midbrain, basal ganglia, thalamus, subcortical whitematter and in the cerebellum where the vermis was mostly affected. Conspicuously, Fañanáscell proliferation has been noticed. In the cerebral cortex, proliferation of astrocytes was inexcess of other pathological changes. Furthermore, astrocytosis was diffusely present in theanterior horns of the spinal cord. Some astrocytes showed clasmatodendrosis. In contrast,Neuman et al30 did not observe severe astrocytosis in three kuru patients, while Scrimgeourat al31 found only mild astrocytic changes with the presence of rare binucleated forms in thecerebral cortex.
Experimental StudiesExperimental kuru in chimpanzees is characterized by widespread astrocytosis, and
both hypertrophy and proliferation of astrocytes have been observed.32-34 Gliosis seems toparallel the severity of spongiform change and neuronal loss, being most abundant in mark-edly vacuolated sensory cortex and less so in better preserved motor areas. Striatum, dien-cephalon, cerebral white matter and cerebellum show severe gliosis. Ultrastructurally, as-trocytes show focal and, in our opinion, artifactual, attenuation of the cytoplasm andaccumulations of glycogen granules.34 In a separate unpublished study of early changes inNew World monkeys infected with kuru, Liberski, Brown and Gajdusek found, using GFAP-immunohistochemistry and electron microscopy, only moderate astrocytosis. Interestingly,astrocytes were observed adjacent to cerebellar granule cells undergoing faulty myelination.The biological significance of this phenomenon is unknown.
Creutzfeldt-Jakob Disease (CJD) andGerstmann-Straussler-Scheinker Disease (GSS)
CJD
Natural diseaseVariably severe astrocytosis is observed among almost all neurodegenerative condi-
tions, and CJD is no exception. Hypertrophic astrocytes, detected by means of metal tech-niques (Holzer, Kanzler or Cajal’s stains) or more recently by immunostaining against glialfibrillary acid protein (GFAP), are seen in all vacuolated areas (Figs. 7.3, 7.4). In cerebralcortex they are particularly prominent in deeper cortical layers. Gemistocytic forms arefrequently observed (Fig. 7.5). When destruction is so severe as to lead to the collapse ofvacuolated neuropil, proliferating astrocytes may virtually replace all other cellular elements.In such a situation the spongiform changes may no longer be recognizable. In the cerebel-lum, the proliferation of Bergman glia is frequently observed.
Marin and Vial35 were the first to report on the ultrastructure of human CJD. Theydescribed hypertrophic astrocytes, a proportion of which suffered from suboptimal fixa-tion and produced areas of “watery” cytoplasm. Astrocytic cytoplasm contained numerouslipofuscin granules. A similar distinction between hypertrophic astrocytes containing glial

131Astrocytes in Transmissible Spongiform Encephalopathies (Prion Diseases)
fibrils and those of “watery” cytoplasm was made by Gonatas et al36 Torrack,37-38 Brion etal,39 Bubis et al40 and Ribadeau-Dumas and Escourolle.41-42 Astrocytic nuclei frequently con-tain inclusion bodies.43 The first type consists of granular and filamentous profiles, fre-quently forming paracrystalline arrays, and most probably represents deformed chromatin.The other type corresponds to the IVth type of “nuclear bodies” (Fig. 7.6) according to theclassification of Bouteille et al.44 The first type of inclusion results most probably from sub-optimal fixation, but the type IV nuclear body has been frequently found in infectious andneoplastic disorders and is regarded as a nonspecific reaction of cells to noxious stimuli.45
Still another type of intranuclear inclusion reported by Jellinger46 corresponds to type Anuclear inclusions. As these inclusions have been reported in numerous viral and degenera-tive conditions, as well as a result of abnormal mitoses, they also most probably representnonspecific changes.
There are only two overlapping morphometric studies of astrogliosis in the cerebella(both Bergmann and velate astrocytes) in two cases of the ataxic form of CJD.47-48 Astro-cytes increased from 192.76 + 117.98 cells per mm2 in controls to 278.08 + 137.73 per mm2
in CJD. An increase in the cross-sectioned nuclear area of Bergmann glia (32.72 + 6.8 mm2
vs. 42.75 + 9.61 mm2) and of velate astrocytes (34.86 + 7.29 mm2 vs. 39.37 + 7.10 mm2) wasseen when control values were compared with those of CJD values. Of note, the basic three-dimensional geometry of the astrocytic scaffold of the cerebellum was maintained despitesevere loss of granule cells. Electron microscopy revealed several subcellular organelles, rarebut otherwise typical for reactive astrocytes, single cilia consisting of ciliary shafts (Fig. 7.7),
Fig. 7.3. Whole mount section of kuru brain stained with Kanzler stain to detect astrocytosis.The distribution of fibrillary gliosis is similar in the middle and deep layers of parasagittal andinterhemispheric neocortex and cingulate cortex but more diffuse in the thalamus and under-neath the insular and temporal cortices. Reprinted with permission from Hainfellner JA et al,Brain Pathol 1997; 7:547-553.

Astrocytes in Brain Aging and Neurodegeneration132
Fig. 7.4. Natural CJD. Severe astrocytosis detected by Cajal gold sublimate impregnation method(a) and GFAP immunohistochemistry (b). Note binucleated astrocytes (arrows) in (b). Origi-nal magnification, x 1000.
a
b

133Astrocytes in Transmissible Spongiform Encephalopathies (Prion Diseases)
clusters of interchromatin and perichromatin granules, various adhesive plaque junctions(Fig. 7.8) and simple and granular nuclear bodies. Of particular interest is the presence ofinfoldings of plasma membranes in the perivascular regions of astrocytic end-feet. Theseinfoldings were covered by an interrupted or continuous electron-dense undercoat of30-60 nm in diameter. The latter observation is in agreement with the earlier freeze-etchingstudy of Dubois-Dalcq et al49 who showed an increased number of astrocyte-specific par-ticles, as opposed to their depletion on membranes, forming vacuoles.
Experimental studiesAstrocytosis represents a substantial component of the neuropathological profile of
experimental CJD. In the first reported transmission experiment, Beck et al50 found moder-ate to severe astrocytosis in both biopsy and necropsy specimens of CJD virus-infected chim-panzees. In the cerebral cortex, the hypertrophic astrocytes completely distorted the neu-ronal architecture. Many astrocytes were of the gemistocytic type, similar to those in humanCJD. Severe glial reaction was also seen in the striatum, diencephalon and cerebellar cortex.Beck et al50 raised the problem of astrocytes as a primary target for CJD agent, in otherwords, the location of CJD within a vague spectrum of so-called “glial dystrophies”. Thisnotion was based primarily on observed discrepancies between the severity of astrocytosisand neuronal damage. While such differences have been unequivocally noted, it must bestressed that in most situations the most severely vacuolated brain regions also presentedthe highest level of astrocytosis. Manuelidis and colleagues51-54 found particularly severeastrocytosis in experimental CJD in guinea pigs, hamsters and mice. In CJD affected-ham-sters “clusters of these cells appear almost as pure astrocytic cultures; this collection is far inexcess of what classically in human and experimental neuropathology is known as reactiveastrocytosis”. To further substantiate the notion of a primary involvement of astrocytes inCJD, Manuelidis and Manuelidis reported that astrocytes from CJD-affected brains couldbe maintained in vivo (immortalized) for a long time.54 In contrast, those established from
Fig. 7.5. Numerous GFAP-immunopositive gemistocytic astrocytes in the cerebral cortex of CJD-affected brain. x 400.

Astrocytes in Brain Aging and Neurodegeneration134
a
b
Fig. 7.6. (a, b). Two examples of nuclear bodies. Lead citrate and uranyl acetate,original magnification, x 30,000.

135Astrocytes in Transmissible Spongiform Encephalopathies (Prion Diseases)
uninfected brains all died after a short period of time. This problem was further addressedin a serial killing experiment, in which we found by means of electron microscopy thatastrocytosis paralleled spongiform change in parietal cortex and adjacent corpus callosumof mice infected with the Fujisaki strain of CJD agent.55 As dilated and swollen astrocyticprocesses were found occasionally in both CJD-infected and sham-inoculated animals, thesewere regarded as a result of local suboptimal fixation and not as part of CJD neuropathol-ogy as reported by others.3,4,51-52
Recently, the neuropathology of TSE has been partially reproduced in transgenic micecreated by microinjection of the chimeric murine cosmid containing a codon 101 Pro toLeu substitution in the ORF of mouse PrP gene.56 The 101 codon substitution is regarded asan equivalent to that found in GSS (a deletion of codon 55 in the mouse PrP gene).57-58
Transgenic mice presented severe spongiform change but rather mild or moderateastrocytosis, except in the cerebellum where severe Bergmann radial gliosis was observed.Again, spongiform change rather than astrocytosis seems to be the primary neuropatho-logical phenomenon in TSEs.
GSSWhile astrocytic gliosis is a prominent and ubiquitous finding in CJD, kuru, scrapie
and bovine spongiform encephalopathy, it still remains a controversial issue in GSS. Hudsonet al59 found mild astrocytosis associated mostly with amyloid plaques in 3 cases of GSS. Ina case reported by Kuzuhara et al,60 moderate astrocytosis of the cerebellar white matter wasfound, while a severe astrocytic reaction was seen in the inferior colliculus. Vinters et al61
reported astrocytic gliosis throughout the neocortex while, in contrast, Tateishi et al62 foundastrocytosis only in an area of concomitant infarct. Similarly, Ghetti et al,63 Nochlin et al64
and Pearlman et al65 reported severe gliosis only in areas where numerous plaques and neu-ronal loss were also seen. In 3 cases of GSS studied by us at the Laboratory of Central NervousSystem Studies (LCNSS), National Institutes of Health in Bethesda, and in the NeurologicalInstitute of the University of Vienna, astrocytosis was found throughout the cerebral and
Fig. 7.7. An astrocyte containing a cilium (arrow). Lead citrate and uranyl acetate,original magnification, x 12,000.

Astrocytes in Brain Aging and Neurodegeneration136
cerebellar cortex, but the severity of this change never approached that found in CJD cases.In particular, gemistocytic astrocytes were never seen in these cases; rather, the astrocyteswere characteristically elongated and slender, reminiscent of pilocytic astrocytes. However,in a recent case from the original Austrian GSS family, astrocytosis in the cerebral cortexapproached that of CJD brains and innumerable gemistocytic astrocytes were seen.66-68 Thus,the diversity of neuropathology of GSS is perhaps of the same magnitude as that of CJD.
Fig. 7.8. (a) Symmetric desmosome-like adhesive plaque junction (arrows); (b) longtortuous adhesive plaque junctions (arrows) in an astrocyte in the subependymalspace. Note that one of these junctions opens to the extracellular space. Lead citrateand uranyl acetate, original magnification, x 50,000.
a
b

137Astrocytes in Transmissible Spongiform Encephalopathies (Prion Diseases)
The Involvement of Astrocytes in Formation of Amyloid PlaquesAmyloid plaques are a neuropathological feature of TSEs.69 There is an “unnecessarily
complex”69 classification of amyloid deposits in humans into several partially overlappingcategories.70 Kuru plaques and multicentric plaques are characteristic features of kuru (orCJD) and GSS, respectively (Fig. 7.9). Cortical kuru (unicentric) plaques of GSS consist ofamyloid fibrils within a narrow extracellular space between distended astrocytic processes(Fig. 7.10).71-73
Amyloid fibrils invaginated “deeply the surrounding profiles of astrocytes so that thefilaments sometimes seemed to be intracellular”. Such peripheral accumulations of astro-cytic processes in close connection with the amyloid fibrils was noted even in the earliestamyloid plaques.71-73 This intimate association of amyloid and astrocytes in GSS led Boellaardet al72 to coin the term glial plaques. Glial plaques are plaques of TSEs and contrast withneuritic plaques of Alzheimer’s disease,74 but, like the latter, are invaded by microglial cells.75-76
A systematic immunohistochemical approach disclosed that 30% to 50% of unicentricplaques contain microglia, while astrocytes are located around these plaques with long pro-cesses penetrating them.75 In contrast, only some of the multicentric plaques contain mi-croglial cells, but the pattern of astrocytic involvement is practically the same. These au-thors also discriminate “cores with satellite deposits” (a variant of multicentric plaques).Seventy to 80% of the latter contain microglia cells; the pattern of astrocyte involvementremains unchanged.
In contrast, diffuse (primitive) plaques in mice exhibit neither amyloid cores nor amy-loid filaments.77-80 They are infiltrated by neither astrocytes nor microglial cells. However,as plaques mature and the PrP within them fibrilizes, the number of both microglial cellsand astrocytes tends to increase. Thus, it seems that both categories of glial cells may merelybe reactive cells, and elegant immunogold studies have shown that PrP is indeed localized tolysosomal compartments of these cells.
Scrapie, Bovine Spongiform Encephalopathy (BSE),and Chronic Wasting Disease (CWD)
Scrapie
Natural diseaseGenerally, the degenerative brain pathology of natural sheep and goat scrapie consists
of spongiform change and astrocytosis. The latter change is highly variable; many cases ofnatural scrapie in sheep show inconspicuous or undetectable astrocytosis. In the majorityof textbook descriptions “neuronal loss” is also mentioned. However, the only referencesupporting this statement is that of Beck et al81 who studied brain areas which are charac-terized by highly variable numbers of neurons. In contrast, neuronal loss clearly does occurin BSE.82
The true nature of the astrocytic changes in scrapie remains poorly understood. It stillremains to be resolved, for example, whether astrocytic proliferation (hyperplasia), astro-cytic hypertrophy or both are responsible for the apparent increases in numbers of astro-cytes observed in scrapie brain under light microscopy.83 Hadlow,84 studying scrapie-in-fected dairy goats, reported that both hypertrophy and, to a lesser extent, hyperplasia broughtabout an overall increase in the apparent number of astrocytes seen in sections. The estima-tion of the number of astrocytes was difficult, however, as the Cajal method used also stainsdifferent proportions of astrocytes in normal goat brains. The “scrapie” astrocytes were notalways easily discriminated from pleomorphic “normal” astrocytes, but typically they mea-sured up to 14 mm in diameter and contained a few chromatin granules. In particular, the

Astrocytes in Brain Aging and Neurodegeneration138
presence of kidney-shaped, elongated and irregularly lobulated nuclei, frequently noted inclusters of 3 to 4 cells reminiscent of those in Alzheimer II cells or the “naked nuclei” ofAlzheimer, have been reported. Hypertrophy and proliferation of astrocytes were confinedto the affected (vacuolated) gray matter. The adjacent white matter was involved only occa-sionally. However, in the midbrain and several thalamic nuclei, astrocytosis was more severethan vacuolation and cerebral cortex characterized by minimal spongiform change occa-sionally presented disproportionately spectacular astrocytosis.
Topographically, various brain regions were involved to different degrees. The lesionstended to be bilaterally symmetrical and the boundaries between affected and unaffected
Fig. 7.9. General view of GSS pathology. Note a multicentric plaque (circle)and three kuru (unicentric) plaques (squares). A microglial cell (arrow) isvisible at the periphery. Lead citrate and uranyl acetate, original magnifica-tion, x 4400.

139Astrocytes in Transmissible Spongiform Encephalopathies (Prion Diseases)
regions were remarkably sharp. Dense astrocytosis was observed in the pallidum, septalnuclei, and diencephalon. Moderate astrocytosis was seen in the striatum and the brainstem, where hypertrophy prevailed above hyperplasia. Minimal astrocytic hypertrophy withslight proliferation was seen in deeper layers of cerebral and cerebellar cortex which other-wise remained largely unaffected by the disease. In the cerebellar cortex, radially arrangedglial processes and clusters of Bergmann glial cells constituted distinctive features. The hip-pocampus formation was unaffected, but diffuse astrocytosis was evident, mostly betweenpyramidal cells and the alveus. Hypertrophy and “undoubted” proliferation has been alsodetected in natural scrapie in goats.85 Topographically, astrocytosis of both experimentaland natural caprine scrapie were alike, except that the striatum, pallidum and septal nucleiwere only slightly affected in the latter. A similar increase in the number of hypertrophicastrocytes in sheep with natural scrapie was reported recently.86 Of note, the number ofastrocytes diminished with age both in controls and in scrapie-affected sheep. In the lattergroup, however, this decrease was not as pronounced. No association was found betweenthe degree of astrocytosis and duration of clinical disease or severity of spongiform change.
Experimental studiesPattison and Jones stated that astrocyte hypertrophy, but not proliferation, was a fea-
ture of rats infected with the Chandler strain of scrapie agent.87 Astrocytosis mostly paral-leled spongiform change and was greater after intracerebral inoculation than after intrap-eritoneal inoculation. Astrocytosis preceded the vacuolation by 14 days. Astrocytic end-plateswere hypertrophic, and in the later stages of disease “capillaries appeared to be embedded inswollen, darkly staining astrocytic cytoplasm.”
The problem of hypertrophy versus hyperplasia has been studied by Fraser and col-leagues88-89 in several models of murine scrapie and by Liberski and colleagues90-93 in ham-sters infected with the 263K strain of scrapie agent. Fraser88 coined the term gliocytosis todenote proliferation of astrocytes accompanied by some changes in their morphology andsubstantial proliferation of rod-like microglial cells. In murine scrapie, gliocytosis, encoun-tered in the hippocampus and the thalamus, is an extremely rare phenomenon found inapproximately 3% of ten thousand murine scrapie-affected brains. Gliocytosis occurs in awide range of scrapie isolates passaged in different strains of mice, but almost exclusivelyafter intracerebral inoculation (256 examples of 260 studied brains with gliocytosis88). Inmore detailed studies of gliocytosis (sclerosis) of the hippocampus formation, Scott andFraser89 found that its presence paralleled that of severe vacuolation.
Liberski and colleagues found “gliocytosis” in hamsters infected with the 263K strainof scrapie in much higher proportion than that of murine scrapie models.90-93 Both astro-
Fig. 7.10. Light microscopy of kuru(unicentric) plaques of GSS. GFAP-immunoreactive astrocytes are seenaround the unstained (star) plaque;,GFAP-immunohistochemistry; x 400.By courtesy of Dr. Maria Barcikowska,Medical Research Centre, the PolishAcademy of Sciences, Warsaw.

Astrocytes in Brain Aging and Neurodegeneration140
cytic hypertrophy and proliferation were observed. Astrocytosis apparently correlated withspongiform change but not with neuronal loss. In the hippocampus, astrocytic changeswere seen in both the pyramidal cell layer and the granular cell layer of fascia dentata. Astro-cytic hyperplasia was evident and different stages of mitosis were recognized (Fig. 7.11).Many astrocytes were similar to “naked nuclei” of Alzheimer II cells (Fig. 7.12). Others con-tained lobulated and bizarre nuclei more reminiscent of those which characterize hyper-trophic reactive astrocytes or astrocytes encountered in multifocal leukoencephalopathy.The presence of glial fibers and Rosenthal fibers, regarded as products of gliofilament con-densation and degeneration, were frequently noted. Proliferation of astrocytes was accom-panied by the presence of rod-like microglial cells.
To clarify the proliferative potential of astrocytes in TSEs, we have studiedimmunohistochemically the immunoreactivity of proliferating cell nuclear antigen (PCNA)(Fig. 7.13),94 an auxillary protein of polymerase-δδδδδ which is active in DNA leading-strandsynthesis, (an established marker for cell proliferation95-99 in experimental scrapie and CJDand in human cases of kuru, CJD and GSS). In CJD-infected mouse and scrapie-infectedhamster brains, astrocytes expressing PCNA exhibited homogeneously stained, intenselyblack nuclei. Astrocyte PCNA-specific immunostaining was confined entirely to the cellnuclei. Faint cytoplasmic staining detected in many hypertrophic astrocytes was regarded asnonspecific in the absence of nuclear staining but proved useful for identification of cellmorphology. During the early stages of experimental CJD with minimal spongiform change,PCNA-immunopositive nuclei were occasionally observed in the subependymal zone (PCNAlabeling index, PCNA LI, 0% to 1.0%). From 18 weeks postinoculation, PCNA-immunopositive astrocytes were most frequent in the corpus callosum (PCNA LI, 0% to3.6%) and cerebellar white matter (0% to 3.7%), regions which characteristically exhibitrobust vacuolation. The gray matter lesions were practically devoid of PCNA-immunopositive
Fig. 7.11. Experimental scrapie in hamsters. A dividing astrocyte. Note chromatin (arrows) anda part of apparatus (arrowheads) including a centriole (open arrow). Lead citrate and uranylacetate, original magnification, x 7000.

141Astrocytes in Transmissible Spongiform Encephalopathies (Prion Diseases)
Fig. 7.12. “Naked nuclei” in scrapie-affected hamster brain. Original magnification, x 1000.
Fig. 7.13. PCNA-immunoreactive astrocytic nuclei (arrows) in CJD-affected mouse brains. PCNAimmunohistochemistry. Original magnification, x 1000.

Astrocytes in Brain Aging and Neurodegeneration142
astrocytes except in the deep cortical layers of the parietal cortex adjacent to the corpuscallosum (PCNA LI, 0% to 4.5%). In the latter region, PCNA LIs even exceeded those of thecorpus callosum. No other cells, particularly no ferritin-immunopositive cells with themorphology of ramified microglia,76,100 expressed PCNA, and no PCNA expression wasobserved in the brains of control animals.
In CJD-affected mouse brains, PCNA LIs correlate significantly with the grade ofastrocytosis in both deep layers of the cerebral cortex and the corpus callosum (r = 0.78 and0.5; p<0.01 and p<0.05, respectively) but not in the subependymal zone or in the cerebellarwhite matter. The correlation of PCNA LI and incubation time (measured in weeks) wasstatistically significant only in the subependymal zone (r = 0.41; p<0.05) while the grade ofastrocytosis correlates significantly with incubation period only in the deep layers of cere-bral cortex and in the subependymal zone (r = 0.47 and 0.51; p<0.05 and p<0.01,respectively).
In CJD-affected mice of all stages, the number of PCNA-immunopositive astrocyteswas low, less than 5% of the visible population of astrocytes (the highest PCNA LI, 4.5%).By contrast, in brain tissues from human patients with kuru, CJD and GSS, in which abun-dant PrP-immunopostive plaques were seen,76,100 no PCNA-immunopositive cells were de-tected despite the presence of numerous microglial cells and reactive astrocytes, which wereclearly identified on adjacent sections following immunostaining with antibodies againstferritin and glial fibrillary acidic protein (GFAP), respectively. Ultrastructural studies are ingeneral agreement concerning the glial changes.101-103 Astrocytes did not show any featureswhich discriminated them from reactive astrocytes found in a plethora of neurodegenerativedisorders.
A few “serial killing” experiments performed so far provide conflicting data on whetherastrocytosis appears before or after vacuolation. Marsh and Kimberlin104 found hypertrophicastrocytes in scrapie-infected hamsters 9 weeks after intracerebral inoculation and preced-ing vacuolation by 2 weeks. This initial astrocytic hypertrophy was first observed at the pia-arachnoid surfaces and adjacent to the ventricles. In contrast, Liberski and Alwasiak dem-onstrated that astrocytosis actually followed vacuolation in hamsters infected with the 263Kstrain of scrapie agent.105 Scrapie-specific vacuoles appeared 8 weeks postinoculation, whileat that time astrocytosis unequivocally surpassed vacuolation. Masters et al106 foundastrocytosis detectable at week 7 or 5 by means of routine neuropathological staining orindirect immunofluorescence. Unequivocal spongiform change appeared in this model 7-8weeks after inoculation. While the spongiform change stabilized in intensity at week 9-10postinoculation, the number of astrocytes increased steadily until the clinical phase of dis-ease and thus paralleled the progressive increase in the infectivity titers. This correlationmay implicate astrocytes as a target for the replicating agent rather than merely constitutingpassively reactive cells. Unfortunately, both experimental studies suffered from the obviousweakness of the use of “poorly vacuolated” models. Thus, the problem of whether astrocytosisis merely a reaction toward the destruction of neuronal elements, or whether astrocytesundergo primary proliferation and hypertrophy, cannot be settled.
Scrapie passaged to cattle produce prominent but moderate astrocytosis and only mini-mal or no spongiform change in numerous brain structures.107-108 The latter discriminatescrapie in cattle from BSE in this species. Astrocytes frequently appeared in clusters and thetopographic distribution of astrocytosis was reminiscent of sheep scrapies, i.e., septal nucleiwere prominently affected while the other forebrain structures were not. The other affectedstructures included the thalamus, midbrain tegmentum (especially the periaqueductal graymatter), pontine nuclei, and the nucleus of the solitary tract. In the cerebellum, Bergmannglia increased in number. The moderate degree of astrocytosis was confirmed by GFAP-immunohistochemistry. Analogous findings were reported for cattle infected with TME.109

143Astrocytes in Transmissible Spongiform Encephalopathies (Prion Diseases)
BSE and CWDThe data on astrocytic reaction in bovine spongiform encephalopathy (BSE) and chronic
wasting disease (CWD) are very limited. In the first report of BSE by Wells et al,110 only mildgliosis was noted in BSE-affected cattle brains and this finding has been subsequently con-firmed.111-112 Liberski et al113-114 found that numerous hypertrophic astrocytes, not infre-quently binucleated ones containing abundant glial filaments, accompanied the neuronaldegeneration. Jeffrey et al82 reported on astrocytosis in BSE-infected mice, but as the astro-cytic response is highly variable among different experimental models, this study has littlerelevance to the problem of astrocytosis in natural BSE. In a captive puma (Felis concolor)infected with BSE, both astro- and microgliosis was readily apparent; the latter formed typi-cal microglial nodules.115
Analogously, in chronic wasting disease (CWD) in mule deer, hybrids of mule deer andwhite tailed deer and Rocky Mountain elk, numerous hypertrophic astrocytes werenoted.116-120
Interaction Between Astrocytes and OligodendrocytesInteractions between astrocytes and oligodendrocytes121 have been previously reported
only in early lesions of multiple sclerosis (MS) and a few other conditions.122-123 Its presencein both naturally occurring and experimentally induced TSE suggests an early cellular eventthat may trigger further tissue destruction (see section: Astrocytes and expression of cytokines,this chapter). In the brain biopsy of a patient with CJD,124 low-power electron microscopyrevealed numerous examples of astrocytes and oligodendroglial cells in close apposition toone another; cellular membranes of one cell type were often molded on those of another(Fig. 7.14). Occasionally two oligodendroglial cells were seen in close contact with the sameastrocyte. At higher magnification, both types of cells were connected by rare adhesive plaquejunctions (Fig. 7.15). These subcellular organelles were composed of two symmetric or asym-metric subplasmalemmal densities (attachment plaques) collectively forming “attenuateddesmosomes” or “desmosome-like” structures. In both the 263K and 22C-H hamster mod-els, similar phenomena were observed. A narrow intercellular space between these attach-ment plaques was visible, containing one or two intermediate lines. More complex struc-tures were also seen in both hamster models. Astrocytic cytoplasm was penetrated by a fewoligodendroglial processes, or oligodendroglial cells were completely surrounded by astro-cytic processes which formed multilayered onion-like “collars” around the former. Suchinteractions were previously reported in early lesions of MS122 and at that time they wereregarded as unusual and possibly specific for this demyelinating process. In a subsequentdetailed study, however, Wu and Raine123 showed that such interactions, while frequentlyencountered in MS lesions, are nonspecific, being observed in other neurological disordersincluding Krabbe’s disease, Toxoplasma encephalitis and brain infarcts. The common de-nominator in all these processes is the presence of inflammatory lymphocytic infiltrates,which tend to be minimal or totally absent in TSEs.125,126 It is of further interest that astro-cytes and oligodendrocytes show weak electric coupling in vitro, which has been interpretedas evidence that these cells are physically connected.127,128
The significance of the interactions between astrocytes and oligodendrocytes is un-clear at the present time. As in MS and other brain lesions in which it has been studied, thisinteraction is not associated with a response to any infectious pathogen. Rather, it may bean event that triggers brain tissue destruction mediated by pro-inflammatory cytokinessecreted from astrocytes, lymphocytes and macrophages.

Astrocytes in Brain Aging and Neurodegeneration144
Fig. 7.14. A general view of complex structure formed by astrocytes (circles) and oligodendro-cytes (squares). Lead citrate and uranyl acetate, original magnification, x 12,000.

145Astrocytes in Transmissible Spongiform Encephalopathies (Prion Diseases)
A Particular Form of Astrocytic Reaction in TSESThe majority of TSEs are polioencephalopathies (diseases of the gray matter) and cor-
responding fine structural changes are relatively well described. However, thepanencephalopathic type of CJD characterized by the predominant involvement of the whitematter has also been reported129 and axonal and myelin pathology at the ultrastructurallevel has been described.55,130-132
Myelinated axons presented various pathological lesions. While these changes wereobserved simultaneously in different areas of the same sample, the following description isorganized as if it followed a sequence of events. Initially, the myelin sheath was separated bycytoplasmic tongues into several concentric bands. Cellular processes penetrated betweenlayers of myelin and lifted away the outermost lamellae. Then a complicated labyrinth ofconcentric cellular processes, clearly belonging to either astrocytes or macrophages, investedmyelinated axons (Fig. 7.16). In terminal stages, axons completely denuded of myelin wereseen in the center of a concentric network of cellular processes, or spirals of myelin wereseen surrounded by such processes. The myelin fragments penetrated into astrocytes ormacrophages, where they underwent final digestion (Fig. 7.17). Macrophages containingfragments of axons, paracrystalline lamellar bodies and myelin debris were abundant in thismodel.
Expression of Glial Fibrillary Acidic Protein (GFAP) and Its MRNAFirst isolated from mature multiple sclerosis plaques,133-134 glial fibrillary acidic pro-
tein (GFAP) is a major protein component of glial filaments, a class of intermediate fila-ments specific for astrocytes. On polyacrylamide gel electrophoresis (PAGE), GFAP gener-ally appears as a 49 kDa protein accompanied by proteolytic cleavage products down to a
Fig. 7.15. An astrocyte (star) and oligodendrocyte (square) connected by well-formed adhesiveplaque junction (arrows). Lead citrate and uranyl acetate, original magnification, x 50,000.
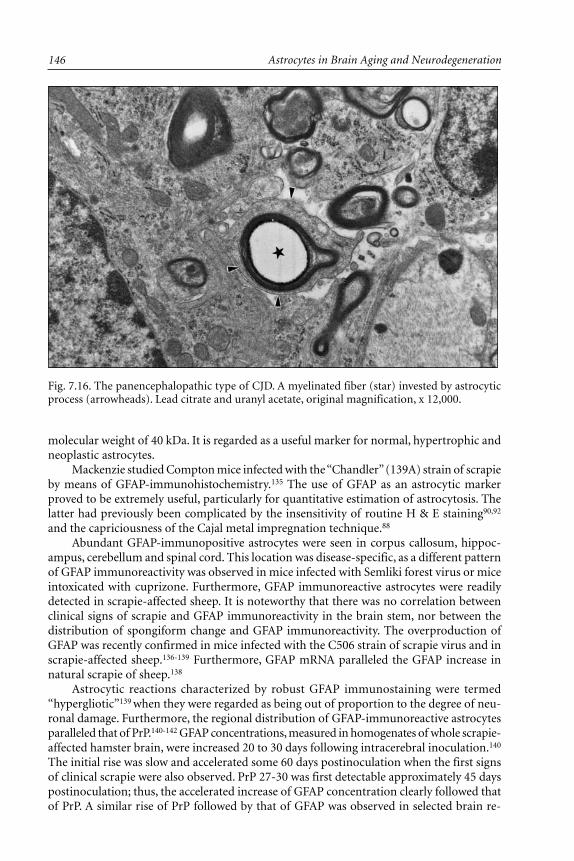
Astrocytes in Brain Aging and Neurodegeneration146
molecular weight of 40 kDa. It is regarded as a useful marker for normal, hypertrophic andneoplastic astrocytes.
Mackenzie studied Compton mice infected with the “Chandler” (139A) strain of scrapieby means of GFAP-immunohistochemistry.135 The use of GFAP as an astrocytic markerproved to be extremely useful, particularly for quantitative estimation of astrocytosis. Thelatter had previously been complicated by the insensitivity of routine H & E staining90,92
and the capriciousness of the Cajal metal impregnation technique.88
Abundant GFAP-immunopositive astrocytes were seen in corpus callosum, hippoc-ampus, cerebellum and spinal cord. This location was disease-specific, as a different patternof GFAP immunoreactivity was observed in mice infected with Semliki forest virus or miceintoxicated with cuprizone. Furthermore, GFAP immunoreactive astrocytes were readilydetected in scrapie-affected sheep. It is noteworthy that there was no correlation betweenclinical signs of scrapie and GFAP immunoreactivity in the brain stem, nor between thedistribution of spongiform change and GFAP immunoreactivity. The overproduction ofGFAP was recently confirmed in mice infected with the C506 strain of scrapie virus and inscrapie-affected sheep.136-139 Furthermore, GFAP mRNA paralleled the GFAP increase innatural scrapie of sheep.138
Astrocytic reactions characterized by robust GFAP immunostaining were termed“hypergliotic”139 when they were regarded as being out of proportion to the degree of neu-ronal damage. Furthermore, the regional distribution of GFAP-immunoreactive astrocytesparalleled that of PrP.140-142 GFAP concentrations, measured in homogenates of whole scrapie-affected hamster brain, were increased 20 to 30 days following intracerebral inoculation.140
The initial rise was slow and accelerated some 60 days postinoculation when the first signsof clinical scrapie were also observed. PrP 27-30 was first detectable approximately 45 dayspostinoculation; thus, the accelerated increase of GFAP concentration clearly followed thatof PrP. A similar rise of PrP followed by that of GFAP was observed in selected brain re-
Fig. 7.16. The panencephalopathic type of CJD. A myelinated fiber (star) invested by astrocyticprocess (arrowheads). Lead citrate and uranyl acetate, original magnification, x 12,000.

147Astrocytes in Transmissible Spongiform Encephalopathies (Prion Diseases)
gions. For instance, in the thalamus, GFAP concentrations were increased at 50 to 55 dayspostinoculation and were preceded by increased concentrations of PrP 33-35sc
(at 5-10 dayspostincubation). It thus seemed that PrP induces reactive gliosis. To test this hypothesisdirectly, the influence of PrP on astrocytic growth in vitro has been studied.140 Primaryastrocytic cultures (more than 90% astrocytes) exhibited a significant increase in cell num-bers after 3 days of PrP exposure. Furthermore, a dramatic increase in GFAP-immunopositiveglial filaments was observed following PrP supplementation of the culture medium. Suchan increase in GFAP concentration was paralleled by an increase in GFAP mRNA. In con-clusion, PrP was found to be a potent stimulant for astrocytes. It has been suggested thatPrP released into extracellular spaces induces reactive astrocytic gliosis. PrPsc has also beenlocalized to astrocytes.143 In serial experiments, PrPsc was detected in astrocytes 8 weeksfollowing inoculation, then increased to the stage where it was detectable diffusely through-out the neuropil. The accumulation of PrPsc within astrocytes preceded both astrocytosis,which was first observed 12 weeks following inoculation and the appearance of scrapie amy-loid 16 weeks postinoculation. From the above discussion it would appear that PrP, GFAP,and astrocytosis are functionally related and that PrP is a growth factor for astrocytes.
Molecular studies of GFAP have had an interesting twist. Weitgrefe et al144 constructeda cDNA from purified poly(A+)RNA from scrapie-infected mouse brain. For differentialhybridization, this cDNA library was screened by [32P]-labeled cDNA reverse-transcribedfrom poly(A+)RNA of scrapie-infected and control brains. One clone (Scr-1) hybridizedpreferentially to scrapie-infected brains. However, in dot-blot experiments, Scr-1 was shownto also hybridize to control material, although the extent was 20-fold less. On Northernblots, Scr-1 hybridized to the 3.3 kb RNA species. In in situ hybridization experiments, Scr-1was located to neurons, mostly in scrapie-affected brains, and to dystrophic neurites withinneuritic plaques in human brains with Alzheimer’s disease and rare senile plaques of multi-infarct dementia brains.145 While the significance of the Scr-1 gene was unknown at thetime of its discovery, it has subsequently been established that the Scr-1 clone merely repre-sented the 3' noncoding region of Gfap.145,146 The Scr-1 cDNA sequence is 98% homologousto the 3' untranslated region of the mouse Gfap cDNA. Indeed, Scr-1 was further used as aprobe to examine the expression of GFAP mRNA in CJD-infected hamsters.146 The mRNAfor GFAP was studied in regions that show no spongiform change and compared with thoseexhibiting severe vacuolation. An increased amount of GFAP mRNA was found in the cere-bral cortex toward the end stage of disease, and its increase preceded the appearance ofspongiform change. However, in some cerebral areas which had prominent vacuolation, itsincrease was not readily apparent. Conversely, a large increase of GFAP mRNA was noticedin the cerebellum, in which spongiform changes were absent. Analogous data were pro-vided for scrapie-infected newborn mice.147
Andreas-Barquin et al138 found upregulation at the protein and mRNA level of bothGFAP and glutamine synthetase (GS). The latter finding may suggest that the traffic ofglutamate and glutamine is distorted in scrapie, but it was not confirmed in a subsequentstudy. GFAP is not, however, necessary for scrapie infection.148-149 Mice in which the firstexon of the Gfap gene was disrupted by replacing it with lacZ gene (Gfap–/–) are susceptibleto scrapie infection, develop typical pathology (including astrocytosis), and exhibit the samelevel and distribution (by histoblots) of PrPsc. Mutated astrocytes showed subtle differencesin immunostaining with antibodies against vimentin and S-100 protein. Both vimentin andS-100 protein signals tend to be granular and limited in the perinuclear space, as opposed towild type astrocytes where both signals are rather filamentous and fill the whole cytoplasm.Indeed, in a recently described Brazilian family with spongiform encephalopathy and a novelmutation at codon 183 of the PRNP gene, gliosis was minimal or even absent throughoutthe brain.149a

Astrocytes in Brain Aging and Neurodegeneration148
Fig. 7.17a.
Moreover, it was demonstrated that not only GFAP but several biologically active sub-stances localized to astrocytes (methallothionein, crystallins, apolipoprotein E, cathepsin Dand various lymphokines; see below) are upregulated in scrapie150-156 and some of thesecompounds are also upregulated in another neurodegenerative disorder, Alzheimer’s dis-ease, suggesting a convergent pathological mechanism.145,147,148 Finally, tissue factor, the tis-sue activator of the coagulation protease cascade, was also upregulated in astrocytes of scrapie-infected brains.157 In experimental scrapie, the upregulation of astrocytic enzymes precedesthe development of neuropathological lesions but follows the rise of PrP.148 ApolipoproteinE4, which is a risk factor for Alzheimer’s disease,158 may increase when astrocytes assume
a

149Astrocytes in Transmissible Spongiform Encephalopathies (Prion Diseases)
the role of macrophages, as demonstrated in experimental CJD,55,130-132 and crystallin, beinga heat shock protein, may participate in the early response to CNS damage.151-152 Collec-tively, these data suggest that astrocytes induced by PrP may play a substantially more im-portant role in the pathogenesis of TSE than merely reacting passively to other brain tissuelesions.
Astrocytes and the Expression of CytokinesIn the brain, a potential role of cytokines in the pathogenesis of neurological disorders
was first considered in multiple sclerosis (MS) and related demyelinating disorders.159 Incontrast to TSEs, the neuropathology of MS is characterized by focal perivascular lympho-cytic infiltrates and macrophages associated with areas of demyelination, disappearance ofoligodendroglial cells and proliferation of astrocytes. Similar features are observed in ex-perimental allergic encephalomyelitis (EAE)—a disorder induced by active immunizationwith myelin basic protein (MBP), which may be passively transferred by T cells.160 Numer-ous data indicate a pivotal role for tumor necrosis factor-α (TNF-α) in the pathogenesis ofEAE,161-166 and several studies suggest a similar pathomechanism for MS.167-174 Myelin bal-looning accompanied by oligodendrocyte degeneration and astrocytic hypertrophy has beenproduced in mouse spinal cord cultures treated with recombinant human TNF-α,175 andcytotoxic activity of TNF-α and lymphotoxin on oligodendroglial cells was reported. Simi-lar findings in EAE have prompted the hypothesis that TNF-α, a cytokine released fromactivated microglia/macrophages176 and astrocytes,177,178 is directly involved in myelin break-down in several demyelinating disorders, presumably by interacting with sodium channelson the axolemma.179 This hypothesis has been further substantiated by immunohistochemicaldetection of TNF-α expression in astrocytes in brain tissues of patients with MS168,174 as wellas by blocking of passive transfer of EAE by anti-TNF-α neutralizing antibodies164 or by thepotent TNF-α inhibitor pentoxyfilline.180 Thus, at least in demyelinating disorders, TNF-α
Fig. 7.17. (a, opposite) General view of brain lesions of the panencephalopathictype of CJD. Note several macrophages (stars) and an astrocyte (circle) digesting amyelinated fiber (arrow); (b, above) remnants of myelinated fiber (arrow) withinthe cytoplasm of an astrocyte. Lead citrate and uranyl acetate, original magnifica-tion: (a) x 7000; (b) x 12,000.
b

Astrocytes in Brain Aging and Neurodegeneration150
may play a crucial role in the activation of immune cells. Although immunocyte activationhas not been directly implicated in the pathogenesis of the TSEs,181 the data accruing fromthe aforementioned MS and EAE studies raise the possibility that TNF-α and other pro-inflammatory cytokines may play important roles in the development of the former condi-tions, in particular those panencephalopathic types of TSE with severe white matterinvolvement.
Tumor Necrosis Factor-ααααα (TNF-ααααα)TNF-α was first described in 1975 when Carswell and colleagues182 found, that in se-
rum of mice infected with bacillus Calmette-Guerin (BCG) and treated with endotoxin(lipopolysaccharide W Escherichia coli), there was an additional toxin capable of inducingnecrosis of certain neoplasms. This toxin is also cytostatic or cytotoxic to a variety of hu-man cell lines in vitro.183 Discovered earlier, cachectin (a toxin causing cachexia by blockinglipoprotein lipase) was shown to be identical to TNF-α.184,185 In contrast, lymphotoxin—abiologically active compound released by T cells in immunized rats and causing cytolysis ofmice L929 fibroblasts—is a different molecule designated TNF-β.
In the central nervous system (CNS), TNF-α is produced by both microglial cells andastrocytes.178,186 Reactive astrocytes also produce prostaglandins and IL-1.187 These cells mayalso express class II major histocompatibility complex (MHC) antigens following exposureto interferon-γ (IFN-γ), probably by increasing expression of a receptor for TNF-α.188,189
Astrocytes are postulated to act as antigen presenting cells in the CNS, presenting antigensto T cell active clones, in an MHC-restricted fashion, upon expression of class II molecules.190
TNF-α damages oligodendroglial cells in vitro and is a mitogen for astrocytes but not foroligodendrocytes.191,192
TNF-α is merely the violin section of a large “orchestra” of cytokine molecules whichmay act synergically or in opposition to one another. TNF-α induces production of IL-1,IL-6, TGF-α and other cytokines, and may thus activate multiple cell types in the course ofan inflammatory reaction.193 In addition, IL-1 may induce production of TNF-α194 and fur-ther amplify the reaction. These cytokines are produced within the CNS by both astrocytesand microglial cells150 Transgenic mice with IL-6 overexpression show hippocampal lesionssimilar to those observed in scrapie,195 although IL-6 was not shown to be overexpressed inthis disease.150 Intracerebral inoculation with IL-1α or IF-γ causes extensive astrocytosis.196-198
Expression of these cytokines may increase after activation of IF-γ released from lympho-cytes172 but other cytokines, such as IL-10 or TGF-β, may act antagonistically and blocksecretion of TNF-α and IL-1α.199
An example of synergistically acting cytokines are TNF-α and IL-1α, which induce thereciprocal release of cytokines.150,194,200,201 IL-1α, one of the first cytokines described (lym-phocyte activating factor, endogenous pyrogen, mitogenic protein), exhibits multiple bio-logical activities, with the activation of T cells being the most important.202 Other activitiesof IL-1α include activation of natural killer (NK) cells, induction of IL-2 receptor expres-sion, induction of B cell proliferation and induction of colony stimulating factor (G-CSF,M-CSF, GM-CSF) secretion.203-205 Moreover, IL-1 may induce TNF-α production.194 Thereare two forms of IL-1: IL-1α and β which, although products of two distinct genes, arecharacterized by a similar molecular weight of 17 kDa. Both gene products independentlybind to two distinct classes of IL-1 receptor (I and II). A number of cell types produce IL-1:monocytes and macrophages, Langerhan’s and dendritic cells, NK cells, endothelial cellsand, within the CNS, astrocytes and microglial cells.203 IL-1α is mitogenic for astrocytes, butis similar to TNF-α in causing a decrease in the number of oligodendrocytes.170
TNF-α may be responsible for some of the neuronal cell loss which occurs in CJD,scrapie and BSE. TNF-α may play a role in the induction of apoptosis.206 In experimental

151Astrocytes in Transmissible Spongiform Encephalopathies (Prion Diseases)
CJD, in scrapie207 and in experimental BSE,82 neuronal autophagic vacuoles, similar to thoseobserved in cells undergoing apoptosis, were observed. Although Selmaj et al208 failed todemonstrate DNA fragmentation typical of apoptosis in cultured oligodendroglial cellstreated with TNF-α, DNA fragmentation was found after application of lymphotoxin.
TNF-ααααα in TSEsThe potential role of TNF-α in the development of neuropathological changes has been
studied in the TSEs.209 Recombinant murine TNF-α injected into the vitreous of the mouseeye produced myelin ballooning in the optic nerve (Fig. 7.18).210 This myelin dilatation wasultrastructurally indistinguishable from that observed in the panencephalopathic type ofCJD. Furthermore, TNF-α immunoreactivity in astrocytes of scrapie- and CJD-infectedmouse brain has been shown.209,211
Campbell et al150 in their sequential study of experimental scrapie demonstrated thatthe mRNA of TNF-α, IL-1α and IL-1β are overexpressed in scrapie-affected brains. On theother hand the expression of brain TNF-α mRNA and IL-4, IL-5, IFN-γ, IL-2, IL-6 and IL-3mRNAs were not altered during scrapie infection. The expression of TNF-α, IL-1α, IL-1β,and other cytokine mRNAs in the kidneys, spleen, and liver were not altered by scrapieinfection, indicating that overexpression of these cytokines is brain-specific. Detailed timecourse experiments showed that significant increase of TNF-α, IL-1α and IL-1β mRNAsoccurred by week 15 postinoculation and increased progressively until end-stage disease atweek 25. This study showed not only that there is pronounced activation of cerebral TNF-α,IL-1α and IL-1β gene product expression in scrapie but also that the increased expression of
Fig. 7.18. Large intramyelin vacuole (star) and numerous astrocytic processes(squares) within an optic nerve following intraocular injection of recombinantTNF-α. Lead citrate and uranyl acetate, original magnification, x 7000

Astrocytes in Brain Aging and Neurodegeneration152
these cytokines’ mRNA correlates well with the progression of the clinical disease and mo-lecular neuropathological changes. This correlation is suggestive of a causal relationship,but this remains unproven.
Similar results were obtained in another experimental TSE—the panencephalopathictype of CJD.211 Here, the brain tissues from CJD virus-infected mice were examined at 1week intervals postinoculation for TNF-α and IL-1α transcript expression using reversetranscriptase-directed polymerase chain reaction (RT-PCR). TNF-α expression was alsoexamined by Western and Northern blot analyses and by immunocytochemistry. Total RNAsamples from control brains were diluted to minimize banding intensity and this dilution
Fig. 7.19. RT-PCR products for TNF-α, IL-1α, GFAP, and β-actin. Lanes 1-11,controls, 2 weeks apart from 2nd to 22nd week; lanes 12-33, CJD-infected ani-mals, one week apart from 1st to 22nd week after inoculation; Lane 23, 100 bpladder.

153Astrocytes in Transmissible Spongiform Encephalopathies (Prion Diseases)
(0.4 mg/ml) was used throughout the study (comparative PCR). Similar to the results ob-tained by Campbell and colleagues,150 low intensity bands for TNF-α increased significantlyafter 15 week postinoculation, being unchanged in the brains of control animals (Figs. 7.19and 7.20). Analogous results were obtained for IL-1α. In contrast, bands for PrP and β-actinderived from brains of infected animals were similar to those of controls and did not showany correlation with disease progression. These results were subsequently confirmed byWestern and Northern blot analysis and by immunohistochemistry, implicating hypertrophicastrocytes as TNF-α-secreting cells.
Collectively, the aforementioned studies demonstrate that TNF-α and IL-1α areupregulated in CJD-affected brains and suggest that these cytokines may serve as molecularmediators of white matter degeneration in experimental CJD. On the other hand, theoverexpression of TNF-α in diseases as diverse as CJD, AIDS vacuolar myelopathy and MSmay suggest that pro-inflammatory lymphokines may merely act as final end-stage media-tors of axon and myelin damage, irrespective of its cause. Accumulation of PrP may initiatea cascade of events inducing the release of TNF-α, IL-1α and β which, in turn, amplify othercytokine responses, produce secondary astrocytosis and microglial infiltration, and culmi-nate in oligodendroglial injury and white matter damage. As mentioned above, overpro-duction of these cytokines correlates with the progression of the clinical features and mo-lecular neuropathological changes, but although this correlation is suggestive of a causalrelationship, this still remains to be proven.
ConclusionsAt present, it is very difficult, or even impossible, to answer the question, “What is the
role of astrocytes in TSEs?” In general, astrocytes undergo proliferative and hypertrophic
Fig. 7.20. Agarose gel with RT-PCR products for TNF-α, IL-1α, GFAP, PrP, and β-actin. Forcontrols (lanes 1, 3, 5, 7, 9) and CJD-infected animals, week 22 postinoculation (lanes 2, 4, 6, 8,10). Lane 11, 100 bp ladder.

Astrocytes in Brain Aging and Neurodegeneration154
changes in the course of TSEs. Also, and this is particularly true for the natural diseases, thepresence and extent of astrocyte hypertrophy are highly variable. In some cases there is afocal astrocytosis, in others the astrocytic reaction is diffuse, and occasionally it is nonexist-ent. The same appears to be true for numerous models of scrapie in mice and animal mod-els of CJD, GSS and BSE. Astrocytes participate in PrP plaque formation, but probably onlyits later stages. In an exceptional model of the panencephalopathic type of CJD, astrocytestake part in myelin stripping and, finally, in myelin digestion. Astrocytes secrete cytokines,in particular TNF-α, which may amplify further tissue damage. In all these activities, theydo not seem to differ from astrocytes in other brain lesions; the reader may find furtherexamples in any textbook of brain pathology, including this volume.
In the TSEs, astrocytes do not appear to be the primary targets of the infectious agent.Their responses are predominantly reactive in nature, although they may participate in theperpetuation of tissue injury.
AcknowledgmentsThe research performed by PPL and RD is supported by the Maria Sklodowska-Curie
Foundation in Poland and by Fogarty International Center and the Kosciuszko Foundationin the USA. It is a part of European Community Concerted Action “Biomed 1 and 2”—“Prion diseases: From neuropathology to pathobiology and molecular genetics” awarded toProfessor Herbert Budka, Vienna, Austria.
References1. Gajdusek DC. Unconventional viruses and the origin and disappearance of kuru. Science
1977; 197:943-960.2. Gajdusek DC. Transmissible amyloidoses of the brain. In: Liberski PP, ed.Light and Elec-
tron Microscopic Neuropathology of Slow Virus Disorders. Boca Raton: CRC Press,1993:1-31.
3. Liberski PP. The enigma of slow viruses: Facts and artifacts. Arch Virol 1993 (suppl 6):1-267.4. Liberski PP. Prions, β-sheets and transmissible dementias: Is there still something missing?
Acta Neuropathol (Berl) 1995; 90:113-125.5. Kretzschmar HA. Human prion diseases (spongiform encephalopathies). Arch Virol [suppl]
1993; 7:261-293.6. Schoon HA, Brunckhorst D, Pohlenz J. Spongiform encephalopathie beim Rothalsstrauss
(Struthio camelus). Ein kasuistischer Beitrag. Therarztl Prax 1991; 19:263-265.7. Will RG, Ironside JW, Zeidler M et al. A new variant of Creutzfeldt-Jakob disease in UK.
Lancet 1996; 347:921-925.8. Lasmezas CI, Deslys JP, Demaimay R et al. BSE transmission to macaques. Nature 1996;
381:743-744.9. Collinge J, Sidle KCL, Meads J et al. Molecular analysis of prion strain variation and the
aetiology of “new variant” CJD. Nature 1996; 383:685-688.10. Gajdusek DC. Infectious amyloids: Subacute spongiform encephalopathies as transmissible
cerebral amyloidoses. In: Fields BN, Knipe DM, Howley PM et al, eds, Fields Virology, 3rded. New York: Lippincott-Raven Publishers, 1997:2851-2900.
11. Prusiner SB. Prions. In: Fields BN, Knipe DM, Howley PM et al, eds. Fields Virology, 3rded. New York: Lippincott-Raven Publishers, 1997:2901-2950.
12. Prusiner SB. Novel proteinaceous infection particles cause scrapie. Science 1982;216:136-144.
13. Collinge J, Owen F, Poulter M et al. Prion dementia without characteristic pathology. Lan-cet 1990; 336:7-9.
14. Brown P, Kaur P, Sulima M et al. Real and imagined clinicopathological limits of “priondementia”. Lancet 193; 341:127-129.

155Astrocytes in Transmissible Spongiform Encephalopathies (Prion Diseases)
15. Kocisko DA, Come JH, Priola SA et al. Cell-free formation of protease-resistant prion pro-tein. Nature 1994; 370:471-4.
16. Lansbury PT, Caughey B. The chemistry of scrapie infection: Implications of the “ice 9”metaphor. Chem Biol 1995; 2:1-5.
17. Budka H, Aguzzi A, Brown P et al. Neuropathological diagnostic criteria for Creutzfeldt-Jakob disease (CJD) and other human spongiform encephalopathies. Brain Pathol 1995;4:459-466.
18. Brown P, Goldfarb LG, Gajdusek DC. The new biology of spongiform encephalopathy:Infectious amyloidoses with a genetic twist. Lancet 1991; 337:1019-1022.
19. Kimberlin RH. Scrapie and possible relationships with viroids. Sem Virol 1990; 1:153-162.20. Diringer H. Hidden amyloidoses. Exp Clin Immunogenet 1993; 9:212-229.21. Weissmann C. Sheep disease in human clothing. Nature 1989; 338:298-299.22. Weissmann C. A “unified theory” of prion propagation. Nature 1991; 352:679-683.23. Liberski PP, Budka H. An overview of neuropathology of the slow unconventional virus
infections. In: Liberski PP, ed. Light and Electron Microscopic Neuropathology of SlowVirus Disorders. Boca Raton: CRC Press, 1992:111-151.
24. Liberski PP, Guiroy DC, Yanagihara R et al. Astrocytic changes. In: Liberski PP, ed. Lightand Electron Microscopic Neuropathology of Slow Virus Disorders. Boca Raton: CRC Press,1992:187-232.
25. Liberski PP, Nerurkar V, Yanagihara R et al. Immunohistochemistry of astrocytic reaction.In: Liberski PP, ed. Light and Electron Microscopic Neuropathology of Slow Virus Disor-ders. Boca Raton: CRC Press, 1992:233-250.
26. Petersen RB, Tabaton M, Chen SG et al. Familial progressive subcortical gliosis: Presenceof prions and linkage to chromosome 17. Neurology 1995; 45:1062-1067.
27. Klatzo I, Gajusek DC. Pathology of kuru. Lab Invest 1959; 8:799-847.28. Klatzo I, Gajusek DC, Zigas V. Evaluation of pathological findings in twelve cases of kuru.
In: Van Boagert L, Radermecker J, Hozay J, Lowenthal A, eds. Encephalitides. Amsterdam:Elsevier Publ Comp, 1959:172-190.
29. Hainfellner JA, Liberski PP, Guiroy DC et al. Pathology and immunocytochemistry of akuru brain. Brain Pathol 1997; 7:547-553.
30. Neuman MA, Gajdusek DC, Zigas V. Neuropathologic findings in exotic neurologic disor-der among natives of the highlands of New Guinea. J Neuropathol Exp Neurol 1964;23:486-507.
31. Scrimgeour EM, Masters CL, Alpers MP et al. A clinico-pathological study of a case ofkuru. J Neurol Sci 1983; 59:265-275.
32. Beck E, Daniel PM, Asher DM et al. Experimental kuru in chimpanzee: A neuropathologi-cal study. Brain 1973; 96:31-38.
33. Beck E, Daniel PM, Alpers M et al. Experimental “kuru” in chimpanzees. A pathologicalreport. Lancet 1966; 2:1056-1059.
34. Lampert PW, Earle KM, Gibbs CJ Jr et al. Experimental kuru encephalopathy in chimpan-zees and spider monkeys. Electron microscopic studies. J Neuropathol Exp Neurol 1969;28:353-370.
35. Marin O, Vial JD. Neuropathological and ultrastructural findings in two cases of subacutespongiform encephalopathy. Acta Neuropathol (Berl) 1964; 4:218-229.
36. Gonatas NK, Terry RD, Weiss M. Electron microscopic study in two cases of Jakob-Creutzfeldt disease. J Neuropathol Exp Neurol 1965; 24:575-598.
37. Torack RM. Ultrastructure and histochemical studies in a cse of progressive dementia andits relationship to protein metabolism. Am J Pathol 1966; 49:77-97.
38. Torack RM. Ultrastructural and histochemical studies of cortical biopsies in subacute de-mentia. Acta Neuropathol (Berl) 1969; 13:43-55.
39. Brion S. La efermedad de Creutzfeldt-Jakob. Neurol Neurocir Psiq (Mexico) 1968; 9:103-115.40. Bubis JJ, Goldhammer Y, Braham J. Subacute spongiform encephalopathy. Electron mi-
croscopic studies. J Neurol Neurosurg Psychiatr 1965; 35:881-887.

Astrocytes in Brain Aging and Neurodegeneration156
41. Ribadeau-Dumas JL, Escourolle R. The Creutzfeldt-Jakob syndrome. A neuropathologicaland electron-microscopic study. In: Subirana A, Espadaler JM, Burrows EH, eds. Neurol-ogy. Proc of the Xth Int Cong Neurol. Amsterdam: Excerpta Medica, 1974:315-329.
42. Ribadeau-Dumas JL, Escourolle R, Castaigne P. Syndrome de Creutzfeldt-Jakob: etudeultrastructurale de trois observations. Rev Neurolog (Paris) 1969; 121:405-422.
43. Payne CM, Sibley WA. Intranuclear inclusions in a case of Creutzfeldt-Jakob disease. ActaNeuropathol (Berl) 1975; 31:353-361.
44. Bouteille M, Kalifat S, Delarue J. Ultrastructural variations of nuclear bodies in humandiseases J Ultrastruct Res 1967; 19:474-486.
45. Liberski PP, Papierz W, Alwasiak J et al. Diagnostic difficulties in a case of subacute scle-rosing encephalitis. Light and electron microscopic studies. Neuropatol Polska 1988;26:98-110.
46. Jellinger K. Nuclear inclusions in subacute spongiform encephalopathy. Acta Neuropathol(Berl) 1971; 17:283-286.
47. Lafarga M, Berciano MT, Suarez I et al. Cytology and organization of reactive astroglia inhuman cerebellar cortex with severe loss of granule cells: A study on the ataxic form ofCreutzfeldt-Jakob disease. Neuroscience 1991; 40:337-352.
48. Lafarga M, Berciano MT, Suarez I et al. Reactive astroglia-neuron relationships in the hu-man cerebellar cortex: A quantitative, morphological and immunohistochemical study inCreutzfeldt-Jakob disease. Int J Devl Neuroscience 1993; 11:199-213.
49. Dubois-Dalcq M, Rodriguez M, Reese TS et al. Search for a specific marker in the neuralmembranes of scrapie mice. Lab Invest 1977; 36:547-553.
50. Beck E, Daniel PM, Matthews WB et al. Creutzfeldt-Jakob disease. The neuropathology ofa transmission experiment. Brain 1969; 92:699-716.
51. Kim JH, Manuelidis EE. Serial ultrastructural study of experimental Creutzfeldt-Jakob dis-ease in guinea pigs. Acta Neuropathol (Berl) 1986; 69:81-90.
52. Kim JH, Manuelidis EE. Ultrastructural findings in experimental Creutzfeldt-Jakob diseasein guinea pigs. J Neuropathol Exp Neurol 1983; 42:29-43.
53. Manuelidis EE, Manuelidis L. Clinical and morphological aspects of of transmissibleCreutzfeldt-Jakob disease. In: Zimmerman HM, ed.Progress in Neuropathology. Vol 4. NewYork: Raven Press, 1979:1-26.
54. Manuelidis EE, Manuelidis L. Observations on Creutzfeldt-Jakob disease propagated in smallrodents. In: Prusiner SB, Hadlow WJ, eds.Slow Transmissible Diseases of the Nervous Sys-tem. Vol 2. New York: Academic Press, 1979:147-172.
55. Liberski PP, Yanagihara R, Asher DM et al. Reevaluation of the ultrastructural pathologyof experimental Creutzfeldt-Jakob disease. Brain 1990; 113:121-137.
56. Hsiao K, Scott M, Foster D et al. Spontaneous neurodegeneration in transgenic mice withmutant prion protein. Science 1990; 250:1587-1590.
57. Hsiao K, Baker HF, Crow TJ et al. Linkage of a prion protein missense variant toGerstmann-Straussler syndrome. Nature 1989; 338:342-345.
58. Hsiao K, Doh-Ura K, Kitamoto T et al. A prion protein amino acid substitution in ataxicGerstmann-Straussler syndrome. Ann Neurol 1989; 26:106.
59. Hudson AJ, Farrell MA, Kalnins R et al. Gerstmann-Straussler-Scheinker disease with co-incidental familial onset. Ann Neurol 1983; 14:670-678.
60. Kuzuhara S, Kanazawa I, Sasaki H et al.Gerstmann-Straussler-Scheinker’s disease. AnnNeurol 1983; 14:216-225.
61. Vinters HV, Hudson AJ, Kaufmann JCE. Gerstmann-Straussler-Scheinker disease: Autopsystudy of a familial case. Ann Neurol 1986; 20:540-543.
62. Tateishi J, Kitamoto T, Tashima T et al. Gerstmann-Straussler-Scheinker’s disease: Dis-similarities in pathology and transmission. In: Court LA, Dormont D, Brown P, KingsburyDT, eds.Unconventional Virus Diseases of the Central Nervous System. Paris: Commisariata’ l’Energie Atomique, 1989:151-157.
63. Ghetti B, Tagliavini F, Masters CL et al. Gerstmann-Straussler-Scheinker disease. II. Neu-rofibrillary tangles and plaques coexist in an affected family. Neurology 1989; 39:1453-1461.

157Astrocytes in Transmissible Spongiform Encephalopathies (Prion Diseases)
64. Nochlin D, Sumi SM, Bird TD et al. Familial dementia with PrP positive amyloid plaques:A variant of Gerstmann-Straussler syndrome. Neurology 1989; 39:910-918.
65. Pearlman RL, Towfighi J, Pezeshkpour GH et al. Clinical significance of types of cerebellaramyloid plaques in human spongiform encephalopathies. Neurology 1988; 38:1249-1254.
66. Liberski PP, Budka H. Ultrastructural pathology of Gerstmann-Straussler-Scheinker dis-ease. Ultrastr Pathol 1995; 19:23-36.
67. Hainfellner J, Brantner-Inhaler S, Cervenakova L et al. The original Gerstmann-Straussler-Scheinker family of Austria: Divergent clinicopathological phenotypes but constant PrPgenotype. Brain Pathol 1995; 5:201-213.
68. Liberski PP. Reappraisal of ultrastructural diversity of amyloid plaques in Gerstmann-Straussler-Scheinker syndrome. Pat Polska 1995; 46:33-41.
69. Jeffrey M, Goodbrand IA, Goodsir A. Pathology of the transmissible spongiform encepha-lopathies with special emphasis on ultrastructure. Micron 1995; 26:277-298.
70. Watanabe R, Duchen L. Cerebellar amyloid in human prion diseases. Neuropathol Applneurobiol 1993; 19:253-260.
71. Boellaard JW, Schlote W. Subakute spongiforme Encephalopathie mit multiformerPlaquebildung. Eigenartige familiar-hereditare Kranknheit des Zentralnervensystems [spino-cerebellare Atrophie mit Demenz, Plaques and plaqueahnlichen im Klein- and Grosshirn(Gerstmann, Straussler, Scheinker)]. Acta Neuropathol (Berl) 1980; 49:205-212.
72. Boellaard JW, Schlote W. Glial plaques: Amyloid deposits characteristic of slow transmis-sible encephalopathies. Virchows Arch [Cell pathol] 1981; 37:337-341.
73. Boellaard JW, Schlote W, Heldt N. The development of amyloid plaques in human trans-missible encephalopathy and the role of astrocytes in their formation. In: Court LA,Dormont D, Brown P, Kingsbury DT, eds. Unconventional Virus Diseases of the CentralNervous System. Paris: Commissariat a’ l’Energie Atomique, 1989:162-171.
74. Wisniewski HM, Terry RD. Re-examination of the pathogenesis of the senile plaques. In:Zimmerman HM, ed.Progress in Neuropathology, Vol. 2. New York: Grune & Stratton,1973:1-26.
75. Miyazono M, Iwaki T, Kitamoto T et al. A comparative immunohistochemical study ofkuru and senile plaques with a special reference to glial reactions at various stages of amy-loid plaque formation. Am J Pathol 1991; 589-598.
76. Barcikowska M, Liberski PP, Boellaard Jet al. Microglia is a component of the prion pro-tein amyloid plaque in the Gerstmann-Straussler-Scheinker syndrome. Acta Neuropathol(Berl.) 1993; 85:623-627.
77. Jeffrey M, Goodsir CM, Bruce ME et al. Infection specific prion protein (PrP) accumulateson neuronal plasmalemma in scrapie infected mice. Neurosci Lett 1992; 147:106-109.
78. Jeffrey M, Goodsir C, Bruce ME et al. Morphogenesis of amyloid plaque in 87V murinescrapie. Neuropathol Appl Neurobiol 1994; 20:535-542.
79. Jeffrey M, Goodsir CM, Bruce ME et al. Correlative light and electron microscopic studiesof PrP localization in 87V mice. Brain Res 1994; 656:329-343.
80. Jeffrey M, Goodsir CM, Fowler N et al. Ultrastructural immuno-localization of syntheticprion protein peptide antibodies in 87V murine scrapie. Neurodegeneration 1996; 5:101-109.
81. Beck E, Daniel PM, Parry HB. Degeneration of the cerebellar and hypothalamo-neurophysialsystems in sheep with scrapie; and its relationship to human system degenerations. Brain1964; 87:153-176.
82. Jeffrey M, Scott JR, Williams A et al. Ultrastructural features of spongiform encephalopa-thy transmitted to mice from three species of bovidae. Acta Neuropathol (Berl) 1992;84:559-569.
83. Field EJ. The significance of astroglial hypertrophy in scrapie, kuru, multiple sclerosis andold age, together with a note on the possible nature of the scrapie agent. Deutsch ZNervenheil 1967; 192:265-274.
84. Hadlow WJ. The pathology of experimental scrapie in dairy goats. Res Vet Sci 1961;2:289-315.
85. Hadlow WJ, Kennedy RC, Race RE et al. Virologic and histologic findings in dairy goatsaffected with natural scrapie. Vet Pathol 1980; 17:187-199.

Astrocytes in Brain Aging and Neurodegeneration158
86. Georgsson G, Gisladottir E, Arnadottir S. Quantitative assessment of the astrocytic responsein natural scrapie of sheep. J Comp Pathol 1994; 108:229-240.
87. Pattison IH, Jones KM. The astrocytic reaction in experimental scrapie in the rat. Res VetSci 1967; 8:160-165.
88. Fraser H. Neuropathology of scrapie: The precision of the lesions and their significance.In: Prusiner SB, Hadlow WJ, eds. Slow Transmissible Diseases of the Nervous System. Vol.1. New York: Academic Press, 1979:387-406.
89. Scott JR, Fraser H. Degenerative hippocampal pathology in mice infected with scrapie.Acta Neuropathol (Berl) 1984; 65:62-68.
90. Liberski PP. Astrocytic reaction in experimental scrapie. J Comp Pathol 1987; 97:73-78.91. Liberski PP. The brain fine structure in experimental scrapie. The 263K strain in golden
syrian hamsters. Neuropatol Polska 1987; 25:35-51.92. Liberski PP. Gliocytosis in experimental scrapie (263K strain of scrapie) in golden syrian
hamsters. Neuropatol. Polska 1986; 24:221-230.93. Liberski PP, Asher DM, Yanagihara R et al. Serial ultrastructural studies of scrapie in ham-
sters. J Comp Pathol 1989; 101:429-442.94. Biernat W, Liberski PP, Guiroy DC et al. Proliferating cell nuclear antigen immunohis-
tochemistry in astrocytes in experimental Creutzfeldt-Jakob disease and in human kuru,Creutzfeldt-Jakob disease and Gerstmann-Straussler-Scheinker syndrome. Neurodegenera-tion 1995; 4:195-201.
95. Bravo R, Frank R, Blundell PA et al. Cyclin/PCNA is the auxillary protein of DNA poly-merase-δ. Nature 1987; 326:515-517.
96. Bravo R, Macdonald-Bravo H. Changes in the nuclear distribution of cyclin (PCNA) butnot its synthesis depend on DNA replication. EMBO J 1985; 4:655-661.
97. Bravo R, Macdonald-Bravo H. Existence of two populations of cyclin/proliferating cellnuclear antigen during the cell cycle: association with DNA replication sites. J Cell Biol1987; 105:1549-1554.
98. Matthews MB, Benbstein RM, Franza BR et al. Identity of the proliferating cell nuclearantigen. Nature 1984; 309:374-376.
99. Prelich G, Cheng-Keat T, Kostura M et al. Functional identity of proliferating cell nuclearantigen and a DNA polymerase-δ auxillary protein. Nature 1987; 326:517-520.
100. Guiroy DC, Wakayama I, Liberski PP et al. Relationship of microglia and scrapie amyloid-immunoreactive plaques in kuru, Creutzfeldt-Jakob disease and Gerstmann-Straussler syn-drome. Acta Neuropathol. (Berl) 1994; 87:526-530.
101. Chandler RL Electron microscopic and other observations on the ependyma and small ce-rebral blood vessels in mice and rats infected with scrapie. Res Vet Sci 1967; 8:166-169.
102. Chandler RL Ultrastructural pathology of scrapie in the mouse: An electron microscopicstudy of spinal cord and cerebellar areas. Br J Exp Pathol 1968; 49:52-59.
103. Chandler RL Ultrastructural observations on scrapie in the gerbil. Res Vet Sci 1968;15:322-328.
104. Marsh RF, Kimberlin RH. Comparison of scrapie and transmissible mink encephalopathyin hamsters. II. Clinical signs, pathology and pathogenesis. J Infect Dis 1975; 131:104-110.
105. Liberski PP, Alwasiak J. Neuropathology of experimental transmissible spongiform encepha-lopathy. The 263K strain of scrapie in golden syrian hamsters. Neuropatol Polska 1983;21:378-392.
106. Masters CL, Rohwer RG, Franko MC et al. The sequential development of spongiformchange and gliosis of scrapie in the golden syrian hamsters. J Neuropathol Exp Neurol1984; 43:242-252.
107. Clark WW, Hourigan JL, Hadlow WJ. Encephalopathy in cattle experimentally infectedwith the scrapie agent. Am J vet Res 1995; 56:606-612.
108. Cutlip RC, Miller JM, Race RE et al. Intracerebral transmission of scrapie to cattle. J InfDis 1994; 169:814-820.
109. Robinson MM, Hadlow WJ, Knowles DP et al. Experimental infection of cattle with theagents of transmissible mink encephalopathy and scrapie. J Comp Pathol 1995; 113:241-251.

159Astrocytes in Transmissible Spongiform Encephalopathies (Prion Diseases)
110. Wells GAH, Scott AC, Johnson CT et al. A novel progressive spongiform encephalopathyin cattle. Vet Rec 1987; 121:419-420.
111. Wells GAH, Wilesmith JW, McGill IS. Bovine spongiform encephalopathy. Brain Pathol1991; 1:69-78.
112. Wells GAH, Wilesmith JW. The neuropathology and epidemiology of bovine spongiformencephalopathy. Brain Pathol 1995; 5:91-103.
113. Liberski PP. Ultrastructural neuropathologic features of bovine spongiform encephalopa-thy. In: Bolis L, Gibbs CJ Jr. Proceedings of an International Roundtable on BovineSpongiform Encephalopathy. J AmVet Med Assoc 1990; 196:1682.
114. Liberski PP, Yanagihara R, Wells GAH et al.. Comparative ultrastructural studies of bovinespongiform encephalopathy, scrapie and Creutzfeldt-Jakob disease. J Comp Pathol 1992;106:361-38.
115. Willoughby K, Kelly DF, Lyon DG et al. Spongiform encephalopathy in a captive puma(Felis concolor). Vet Rec 1992; 131:431-434.
116. Williams ES, Young S. Chronic wasting disease of captive mule deer: A spongiform en-cephalopathy. J Wildlife Dis 1980; 16:89-98.
117. Williams ES, Young S. Spongiform encephalopathy of Rocky Mountain elk. J Wildlife Dis1982; 18:465-471.
118. Williams ES, Young S. Spongiform encepalopathies in Cervidae. Review of Scientific Tech-nology Office and International Epizootiology 1992; 11:551-567.
119. Guiroy DC, Williams ES, Liberski PP et al. Ultrastructural pathology of chronic wastingdisease in captive mule deer. Acta Neuropathol. (Berl) 1993; 85:437-444.
120. Guiroy DC, Liberski PP, Williams ES et al. Electron microscopic findings in brain of RockyMountain elk with chronic wasting disease. Folia Neuropathol. (Warsaw) 1994; 32:171-173.
121. Liberski PP, Brown P, Cervenakova L, Gajdusek DC. Interactions between astrocytes andoligodendroglia in human and experimental Creutzfeldt-Jakob disease and scrapie. ExpNeurol 1997; 144:227-234.
122. Prineas JW, Kwon EE, Goldenberg PZ et al. Interaction of astrocytes and newly formedoligodendrocytes in resolving multiple sclerosis lesions. Lab Invest 1990; 63:624-636.
123. Wu E, Raine CS. Interactions between oligodendrocytes and hypertrophic astrocytes andtheir occurrence in other, nondemyelinating conditions. Lab Invest 1992; 67:88-99.
124. Liberski PP, Sklodowski P, Klimek A. Creutzfeldt-Jakob disease: Ultrastructural study ofbrain biopsy: Unusual interaction between astrocytes and oligo- and microglia. FoliaNeuropathol (Warsaw) 1995; 33:85-92.
125. Museteanu C, Diringer H. Perivascular infiltrates of leukocytes in brains of scrapie-infectedmice. Nature 1991; 294:360-361.
126. Williams AE, Ryder S, Blakemore WF. Monocyte recruitment into the scrapie-affected brain.Acta Neuropathol (Berl) 1995; 90:164-169.
127. Ransom BR, Kettemann H Electrical coupling, without dye coupling, between mammalianastrocytes and oligodendrocytes in cell culture. Glia 1990; 3:258-266.
128. Couraud P-O. Interactions between lymphocytes, macrophages, and central nervous sys-tem. J Leukocyte Biol 1994; 56:407-415.
129. Tateishi J, Ohta M, Koga M, Sato Y, Kuroiwa Y. Transmission of chronic spongiform en-cephalopathy with kuru plaques from humans to rodents. Ann Neurol 1978; 24:35-40.
130. Liberski PP, Yanagihara R, Gibbs CJ Jr, Gajdusek DC. White matter ultrastructural pathol-ogy of experimental Creutzfeldt-Jakob disease in mice. Acta Neuropathol (Berl.) 1989b;79:1-9.
131. Liberski PP, Yanagihara R, Gibbs CJ Jr, Gajdusek DC. Mechanism of the damage to myeli-nated axons in experimental Creutzfeldt-Jakob disease in mice: An ultrastructural study.Eur J Epidemiol 1991; 7:545-550.
132. Liberski PP, Gajdusek DC. Myelinated axon undergoes complete demyelination in thepanencephalopathic but it is merely subjected to the Wallerian degeneration in thepolioencephalopathic-type of transmissible spongiform encephalopathies. Polish J Pathol1997; 48:163-171.

Astrocytes in Brain Aging and Neurodegeneration160
133. Eng LF, Vanderhaegen JJ, Bignami A et al. An acidic protein isolated from fibrous astro-cytes. Brain Res 1971; 28:351-354.
134. Eng LE, DeArmond SJ. Immunochemistry of the glial fibrillary acidic protein. In:Zimmerman HM, ed. Progress in Neuropathology. Vol 5. New York: Raven Press,1983:19-39.
135. Mackenzie A. Immunohistochemical demonstration of glial fibrillary acidic protein inscrapie. J Comp Pathol 1983; 93:251-259.
136. Lefrancois T, Tardy M. L’astrocyte, une cellule cle’ dans la pathogenie des encephalopa-thies spongiformes. Rec Med Vet 1995; 171:29-32.
137. Lefrancois T, Fages C, Brugere-Picoux et al. Astroglial reactivity in natural scrapie of sheep.Microbial Pathogenesis 1994; 17:283-289.
138. Andreas-Barquin PJ, le Prince G, Fages C et al. Expression of glial fibrillary acidic proteinand glutamine synthetase genes in the natural scrapie of sheep. Mole Chem Neuropathol1994; 22:57-65.
139. Dormont D, Delpech B, Delpech A et al. Hyperproduction de proteine gliofibrillaire acide(GFA) au cors de l’evolution de la tremblante experimentale de la Souris. C R Acad SciParis 1981; 293:53-56.
140. DeArmond SJ, Gonzales M, Mobley WC et al. PrPsc in scrapie-infected hamster brain isspatially and temporally related to histopathology and infectivity titer. In: Iqbal K,Wisniewski HM, Winblad B, eds. Alzheimer’s Disease and Related Disorders. New York:Alan R. Liss, Inc., 1989:601-618.
141. DeArmond SJ, Kretzschmar HA, McKInley MP et al. Molecular pathology of prion dis-ease. In: Prusiner SB, McKinley MP, eds. Prions. Novel Infectious Pathogens causing Scrapieand Creutzfeldt-Jakob Disease. New York: Academic Press, 1987:387-414.
142. DeArmond SJ, Mobley WC, DeMott DL et al. Changes in the localization of brain prionproteins during scrapie infection. Neurology 1987; 37:1721-1280.
143. Brown HR, Goller NL, Rudelli RD et al. The mRNA encoding the scrapie agent protein ispresent in a variety of non-neuronal cells. Acta Neuropathol (Berl) 1987; 80:1-6.
144. Weitgrefe S, Zupanic M, Haase A et al. Cloning of a gene whose expression is increased inscrapie and in senile plaques in human brain. Science 1985; 230:1177-1179.
145. Diedrich J, Wietgrafe S, Zupancic M et al. The molecular pathogenesis in scrapie andAlzheimer’s disease. Microb Pathogen 1987; 2:435-442.
146. Manuelidis L, Tesin DM, Sklaviadis T et al. Astrocyte gene expression in Creutzfeldt-Jakobdisease. Proc Natl Acad Sci USA 1987; 84:5937-5941.
147. Lazarini F, Deslys JP, Dormont D. Variations in prion protein and glial fibrillary acidicprotein mRNAs in the brain of scrapie-infected newborn mouse. J Gen Virol 1992;73:1645-1648.
148. Gomi H, Yokoyama T, Fujimoto K et al. Mice devoid of the glial fibrillary acidic proteindevelop normally and are susceptible to scrapie prions. Cell 1995; 14:29-41.
149. Tatzelt J, Maeda N, Pekny M et al. Scrapie in mice deficient in apolipoprotein E or glialfibrillary acidic protein. Neurology 1996; 47:449-453.
149a.Nitrini R, Rosemberg S, Passos-Bueo MR et al. Familial spongiform encephalopathy withdistinct clinico-pathological features associated with a novel prion protein gene mutationat codon 183. Ann Neurol 1997; 42:138-146.
150. Campbell IL, Eddleston M, Kemper P. Activation of cerebral cytokine gene expression andits correlation with onset of reactive astrocyte and acute-phase response gene expression inscrapie. J Virol 1994; 68:2383-2387.
151. Diedrich J, Duguid JR, Haase AT. The role of astrocytes in the neuropathology of scrapieand Alzheimer’s disease. Sem Virol 1991; 2:233-238.
152. Diedrich J, Minnigan H, Carp RI. Neuropathological changes in scrapie and Alzheimer’sdisease are associated with increased expression of apolipoprotein E and cathepsin D inastrocytes. J Virol 1991; 65:4759-4768.
153. Duguid JR, Bohmont CW, Liu N et al. Changes in gene expression shared by scrapie andAlzheimer disease. Proc Natl Acad Sci USA 1987; 86:7260-7264.

161Astrocytes in Transmissible Spongiform Encephalopathies (Prion Diseases)
154. Williams AE, van Dam AM, Man-A-Hing WK et al. Cytokines, prostaglandins andlipocortin-1 are present in the brains of scrapie-infected mice. Brain Res 1994; 654:200-206.
155. Renkawek K, de Jong WW, Merck KB et al. A,b-crystallin is present in reactive glia inCreutzfeldt-Jakob disease. Acta Neuropathol (berl) 1992; 83:324-327.
156. Kenward N, Hope J, Landon M et al. Expression of polyubiquitin and heat-shock protein70 genes increases in the latter stages of disease progression in scrapie-infected mouse brain.J Neurochem 1994; 62:1870-1877.
157. Eddleston M, de la Torre JC, Oldstone MBA et al. Astrocytes are the primary source oftissue factor in the murine central nervous system. A role of astrocytes in cerebral hemo-stasis. J Clin Invest 1993; 92:349-358.
158. Roses AD, Einstein G, Gilbert J et al. Morphological, biochemical and genetic support foran apolipoprotein E effect on microtubular metabolism. Ann New York Acad Sci 1996;777:146-57.
159. Brosnan CF, Selmaj K, Raine CS. Hypothesis: A role of tumor necrosis factor in immune-mediated demyelination and its relevance to multiple sclerosis. J Neuroimmunol 1988; 18:87-94.
160. Ben-nun A, Wekerle H, Cohen I. The rapid isolation of clonable antigen specific linescapable of mediating autoimmune encephalomyelitis. Eur J Immunol 1981; 11:195-199.
161. Chung IY, Norris J G, Benveniste EN. Differential TNF expression by astrocytes from EAE-susceptible and -resistant rat strains. J Exp Med 1991; 173:801-811.
162. Martin D, Near S L, Bendele A et al. Inhibition of tumor necrosis factor is protectiveagainst neurologic dysfunction after active immunization of Lewis rats with myelin basicprotein. Exp Neurol 1995; 131:221-228.
163. Powell M B, Mitchell D, Lederman J et al. Lymphotoxin and TNF production by myelinbasic protein-specific T cell clones correlates with encephalitogenicity. Int Immunol 1990;2:539-544.
164. Selmaj KW, Raine CS, Cross A H. Anti-tumor necrosis factor therapy abrogates autoim-mune demyelination. Ann Neurol 1991; 30:694-700.
165. Selmaj K, Papierz W, Glabinski A et al. Prevention of chronic relapsing experimental au-toimmune encephalomyelitis by soluble tumor necrosis factor receptor I. J Neuroimmunol1995; 56:135-141.
166. Selmaj KW, Raine CS. Experimental autoimmune encephalomyelitis: immunotherapy withanti-tumor necrosis factor antibodies and soluble tumor necrosis factor receptors. Neurol-ogy 1995; 45, Suppl. 6: S 44-49.
167. Benvenuto R, Paroli M, Buttinelli C et al. TNF synthesis by CSF-derived T cell clonesfrom patients with multiple sclerosis. Clin Exp Immunol 1991; 84:97-102.
168. Hoffman FM, Hinton DR, Johnson K et al. TNF-α identified in multiple sclerosis brain. JExp Med 1989; 170, 607-612.
169. Glabiñski A, Mirecka M, Pokoca L. Tumor necrosis factor α but not lymphotoxin is over-produced by blood mononuclear cells in multiple sclerosis. Acta Neurol Scand 1995;91:276-279.
170. Merrill JE. Proinflamatory and antiinflamatory cytokines in multiple sclerosis and centralnervous system acquired immunodeficiency syndrome. J Immunother 1992; 12:167-170.
171. Merrill JE, Strom SR, Ellison GW et al. In vitro study of mediators of inflammation inmultiple sclerosis. J Clin Immunol 1989; 9:84-96.
172. Rieckmann P, Albrecht M, Ehrenreich H, Weber T et al. Semi-quantitative analysis ofcytokine gene expression in blood and cerebrospinal fluid cells by reverse transcriptasepolymerase chain reaction. Res Exp MedBerl 1995; 195:17-29.
173. Rieckmann P, Albrecht M, Kitze B et al. Tumor necrosis factor α messenger RNA expres-sion in patients with relapsing-remitting multiple sclerosis is associated with disease activ-ity. Ann Neurol 1995; 37:82-88.
174. Selmaj KW, Raine CS, Cannella B et al. Identification of lymphotoxin and tumor necrosisfactor in multiple sclerosis lesions. J Clin Invest 1991; 87:949-954.
175. Selmaj K W, Raine CS. Tumor necrosis factor mediates myelin and oligodendrocyte dam-age in vitro. Ann Neurol 1988; 23:339-346.

Astrocytes in Brain Aging and Neurodegeneration162
176. Hetier E, AyalaJ, BousseauA et al. Ameboid microglial cells and not astrocytes synthesizeTNF-α in Swiss mouse brain cell cultures. Eur J Neurosci 1990; 2:762-768.
177. Chung IY, Benveniste EN. TNF-α production by astrocytes. Induction by polysaccharide,interferon-α and interleukin-1α. J Immunol 1990; 144:2999-3007.
178. Robbins D.S, Shirazi Y, Drysdale B-E et al. Production of cytotoxic factor for oligodendro-cytes by stimulated astrocytes. J Immunol 1987; 139:2593-2597.
179. Barna BP, Estes ML, Jacobs BS et al. Human astrocytes proliferate in response to tumornecrosis factor-α. J Neuroimmunol 1990; 30:239-243.
180. Nataf S, Louboutin JP, Feve JR et al. Pentoxifylline inhibits experimental allergic encepha-lomyelitis. Acta Neurol Scandinav 1993; 88:97-99.
181. Brown P. The phantasmagoric immunology of transmissible spongiform encephalopathy.In: Waksman BH, ed.Immunologic Mechanisms in Neurologic and Psychiatric Disease. NewYork: Raven Press, Ltd., 1990:305-313.
182. Carswell EA, Old LJ, Kassel RL et al. An endotoxin-induced serum factor that causes ne-crosis of tumors. Proc Natl Acad Sci USA 1975; 72:3666-3669.
183. Sugarman BJ, Aggarwal BB, Hass PE et al. Recombinant human tumor necrosis factor-alpha: Effects on proliferation of normal and transformed cells in vitro. Science 1985;230:943-945.
184. Kawakami M, Pekala PH, Lane MD et al. Lipoprotein lipase suppression in 3T3-L1 cellsby an endotoxin-induced mediator from exudate cells. Proc Natl Acad Sci 1982; 79:912-915.
185. Beutler B, Cerami A. Cachectin and tumor necrosis factor as two sides of the same bio-logical coin. Nature 1986; 320:584-588.
186. Lieberman AP, Pitha PM, Shin HS et al. Production of TNF-α and other cytokines byastrocytes stimulated with lipopolysaccharide or a neurotropic virus. Proc Natl Acad SciUSA 1989; 86:6348-6352.
187. Fontana A, Kristensen F, Dubs R et al. Production of prostaglandin E and an interleukin-1 like factor by cultured astrocytes and C6 glioma cells. J Immunol 1982; 129:2413-2419.
188. Fierz W, Endler B, Reske K. Astrocytes as antigen presenting cells. I. Induction of Ia anti-gen expression on astrocytes by T cells via immune interferon and its effect on antigenpresentation. J Immunol 1985; 134:3785-3793.
189. Benveniste EN, Sparacio SM, Bethea JR. Tumor necrosis factor-α enhances interferon-γ-mediated class II antigen expression on astrocytes. J Neuroimmunol 1989; 25:209-219.
190. Takiguchi M, Frelinger JA. Induction of antigen presentation ability in purified cultures ofastroglia by interferon-γ. J Mol Cell Immunol 1986; 2:269-280.
191. Lachman LB, Brown DC, Dinarello CA. Growth promoting effects of recombinant IL-1and TNF for a human astrocytoma cell line. J Immunol 1987; 138:2913-2916.
192. Selmaj K, Shafit-Zagardo B, Aquino DA et al. Tumor necrosis factor-induced proliferationof astrocytes from mature brain is associated with downregulation of glial fibrillary acidicprotein mRNA. J Neurochem 1991; 57:823-830.
193. Philip R, Epstein LB. TNF-α as immunomodulator and mediator of monocyte cytotoxicityinduced by itself, gamma interferon and interleukin 1. Nature 1986; 323:86-89.
194. Campbell IL, Abraham CR, Mazliah E et al. Neurologic disease induced in transgenic miceby cerebral overexpression of IL-6. Proc Natl Acad Sci USA 1993; 90:10061-10065.
195. Giulian D, Woodward J, Young DG et al. Interleukin-1 injected into mammalian brainstimulates astrogliosis and neovascularisation. J Neurosci 1988, 8:2485-2490.
196. Giulian D, Lachman LB. Interleukin-1 stimulation of astroglial proliferation after braininjury. Science 1985, 228:497-499.
197. Yong VW, Moumdjian R, Yong FP et al. Gamma-interferon promotes proliferation of adulthuman astrocytes in vitro and reactive gliosis in the adult mouse brain in vivo. Proc NatlAcad Sci USA 1991; 88:7016-7020.
198. Olsson T. Cytokine-producing cells in experimental autoimmune encephalomyelitis andmultiple sclerosis. Neurology 1995; 45:11-15.
199. Le J, Vilcek J. Tumor necrosis factor and interleukin 1: Cytokines with multiple overlap-ping biological activities. Lab Invest 1987; 56:234-248.

163Astrocytes in Transmissible Spongiform Encephalopathies (Prion Diseases)
200. Dinarello CA, Cannon JG, Wolff SM et al. Tumor necrosis factor (cachectin) is an endog-enous pyrogen and induces production of interleukin 1. J Exp Med 1986; 163:1433-1450.
201. Dinarello CA. Biology of interleukin-1. FASEB J 1988; 2:108-115.202. Dinarello CA. Biologic basis for interleukin in disease. Blood 1996; 87:2095-147.203. Lai Ch-F, Baumann H. Interleukin-1β induces production of granulocyte colony-stimulat-
ing factor in human hepatoma cells. Blood 1996; 87:4143-4148.204. Lee M, Segal GM, Bagby GC. Interleukin-1 induces human bone marrow-derived fibro-
blasts to produce multilineage hematopoietic growth factors. Exp Hematol 1987; 15:983-988.205. Jeffrey M, Fraser JR, Halliday WG et al. Early unsuspected neuron and axon terminal loss
in scrapie-infected mice revealed by morphometry and immunocytochemistry. NeuropatholAppl Neurobiol 1995; 21:41-49.
206. Robaye B, Mosselmans R, Fiers W et al. Tumor necrosis factor induces apoptosis (pro-grammed cell death) in normal endothelial cells in vivo. Am J Pathol 1991; 138:447-453.
207. Liberski PP, Yanagihara R, Gibbs CJ Jr, Gajdusek DC. Neuronal autophagic vacuoles inexperimental scrapie and Creutzfeldt-Jakob disease. Acta Neuropathol (Berl) 1992;83:134-139.
208. Selmaj K, Raine CS, Faroq M et al. Cytokine cytotoxicity against oligodendrocytes. Apoptosisinduced by lymphotoxin. J Immunol 1991; 147:1522-1529.
209. Liberski PP, Nerurkar VR, Yanagihara R et al. Tumor necrosis factor: Cytokine-mediatedmyelin vacuolation in experimental Creutzfeldt-Jakob disease. Abstract no P 68-15 in Ab-stracts of the VIIIth International Congress of Virology, Berlin, West Germany, August26-31, 1990, 421.
210. Liberski PP, Yanagihara R, Nerurkar VR et al. Tumor necrosis factor produces CJD-likelesions in vivo. Neurodegeneration 1933; 2:215-225.
211. Kordek R, Nerurkar VR, Liberski PP et al. Heightened expression of tumor necrosis-α,interleukin 1α, and glial fibrillary acidic protein in experimental Creutzfeldt-Jakob diseasein mice. Proc Natl Acad Sci USA 1996; 93:9754-9758.


CHAPTER 8
Astrocytes in Brain Aging and Neurodegeneration, edited by Hyman M. Schipper.©1998 R.G. Landes Company.
Astrocytes in OtherNeurodegenerative DiseasesDennis W. Dickson
Introduction
Glial pathology is increasingly recognized in several neurodegenerative diseases. The rela-tionship of the glial changes to neurodegeneration is uncertain, but the discovery of
glial inclusion bodies in select neurodegenerative diseases suggests that glial dysfunctionmay contribute to disease pathogenesis. While it has not been specifically studied in thedisorders under consideration, basic studies have provided evidence for cross talk betweenglia and neurons with production of mutual trophic factors and their receptors. It is thuspossible that glial pathology may contribute to, or be a direct consequence of, neurodegen-eration rather than a curious epiphenomenon. It would indeed be groundbreaking if someof the disorders that are currently considered to be neurodegenerative diseases were in factdue to primary abnormalities in glia—i.e., gliodegenerative diseases. Only further researchinto fundamental biology of glia and their interactions with neurons will produce answersto these questions.
All neurodegenerative diseases are associated with reactive gliosis that usually is topo-graphically coincident with neuronal degeneration and loss. Gliosis in this setting is un-doubtedly an important pathologic finding, and in the case of spongiform encephalopa-thies, such as Creutzfeldt-Jakob disease, a cardinal histopathologic feature of the disease.On the other hand, gliosis in the setting of neurodegeneration offers few clues to diseasepathogenesis since there is no way to know if it is anything other than a reactive or second-ary change due to the processes that lead to neuronal degeneration and loss. Increasingly,the genetic bases for many of the disorders are being discovered, but the pathogenesis ofeven the most common of the disorders, namely Alzheimer’s disease, is unresolved and thefocus of many current investigations.
Particular among neurodegenerative disorders are those in which glial cytoplasmic in-clusions are composed of filamentous aggregates; these disorders will be the focus of thepresent discussion. Other types of glial inclusions, such as Rosenthal fibers and autofluo-rescent inclusions that are seen in tumors, storage diseases and aging, are discussed in detailin other chapters of this book. A recent review covers many of these same topics.1 By con-vention, when we speak of neurodegenerative disorders we mean those disorders associatedwith progressive and selective loss of neurons, whose etiology in most cases remains uncer-tain. The particular subset of neurons that is vulnerable to cell loss defines the condition.For example, in idiopathic Parkinson’s disease, selective loss of neurons in the pars com-pacta of the substantia nigra in the midbrain leads to depletion of dopaminergic nerve

Astrocytes in Brain Aging and Neurodegeneration166
terminals in the basal ganglia, which manifests as a characteristic movement disorder. In-clusion bodies composed of neurofilament in the cytoplasm of vulnerable neurons, whichare referred to as Lewy bodies,2 define the pathology of idiopathic Parkinson’s disease, butglial inclusions are not widely recognized in Lewy body disease. Only recently has a reportappeared that Gallyas-positive glial inclusions can be detected in some cases of idiopathicParkinson’s disease.3 The inclusions were within astrocytes, since they were colabeled withantibodies to glial fibrillary acidic protein, but they were negative for tau and tubulin. Nosuch inclusions were observed in Alzheimer’s disease or aged controls. Further studies areneeded to confirm these observations and to determine if these argyrophilic astrocytic in-clusions are specific to Lewy body disease.
Evidence suggests that most of the filamentous aggregates within glia in manyneurodegenerative diseases are derived from cytoskeletal elements, in particular, the micro-tubule-associated protein tau. Tau protein is also the major structural protein of neurofibril-lary tangles (NFT), one of the major histopathological hallmarks of Alzheimer’s disease.4
NFT are numerous and widespread in the brain in Alzheimer’s disease; they are restricted indistribution in normal aging.5,6 NFT or NFT-like neuronal inclusions can also be found incertain other neurodegenerative diseases. In the great majority of the latter conditions, in-clusions are found within astrocytes or oligodendrocytes, or both, as well as in neurons. InAlzheimer’s disease, glial inclusions, though occasionally detected,7 are far less common.
NFT and glial tau inclusions are argyrophilic, which means that they are intensely stainedwith silver impregnation methods, such as the Bielschowsky, Bodian and Gallyas stains.This property has been traced to the presence of highly charged molecules, in particular,highly phosphorylated proteins.8 The inclusion bodies are also variably stained with his-tochemical methods for amyloid, such as thioflavin-S. This presumably reflects a highlyordered secondary structure that permits intercalation of the chromogens into the filaments.In contrast to NFT, which are intensely positive with amyloid stains, glial tau inclusions arefar more difficult to detect with amyloid stains. These and other results discussed subse-quently suggest that tau proteins in glial inclusions are not identical to tau proteins in NFT.Currently, the only means of directly studying the composition of glial inclusions in braintissue is with descriptive methods, such as histochemical and immunohistochemical stainsand electron microscopy. Methods to separately analyze tau protein in neurons and gliawith direct brain tissue biochemical methods are currently not possible. Unfortunately, allpublished biochemical studies of these disorders do not distinguish the cell of origin for theprotein in question.
Studies of cytoskeletal proteins in cultured cells may provide a clue to the nature ofproteinaceous inclusions in neurodegenerative diseases, but such results must be interpretedconservatively. The simple and defined environment in which cultured glial cells grow is fardifferent from the complex and highly ordered environment of brain tissue. There is noguarantee that biochemical features observed for glia in vitro are comparable to those invivo, especially in the diseased brain. Nevertheless, tau protein in neurodegenerative dis-eases and tau protein in cultured glial cells, especially oligodendroglia, have some intriguingsimilarities (see below).
A great deal of effort has been devoted to studying the biochemical composition ofNFT in Alzheimer’s disease, but much less is known about the biochemical basis of inclu-sions in other neurodegenerative disorders. One may infer by analogy that what is knownabout NFT may have some relevance to glial inclusions. It must, however, be acknowledgedthat even with all the advances in understanding the molecular biology of NFT, we do notknow what induces NFT formation. Depletion of trophic factors, aberrant expression ofdevelopmental antigens and excitotoxicity are a few of the hypotheses that are under cur-rent investigation. There being no animal model for NFT, research is guided largely by de-

167Astrocytes in Other Neurodegenerative Diseases
scriptive analyses. A brief discussion of NFT seems warranted as a background for under-standing the nature of glial inclusions. Subsequently, glial inclusions will be discussed withreference to the different diagnostic entities in which they have been described. Commonthemes will be emphasized where possible.
Neurofibrillary Tangles as an Archetype of Cytoskeletal InclusionsNFT are composed of aggregates of filaments that appear to be composed of pairs of
10 nm-diameter filaments with a helical arrangement.9 Although recent fine structural studieswith atomic force microscopy suggest that a more accurate model may be that of twistedribbon,10 the filamentous inclusions that make up NFT are commonly referred to as pairedhelical filaments (PHF).9 PHF have a diameter of about 22 nm with cross-over points in thePHF at about every 80 nm. In disorders with tau-positive glial inclusions the filaments areusually straight, rather than twisted, ranging in diameter from 13 nm to 18 nm. In somedisorders with neuronal and glial tau inclusions, twisted wider filaments (about 24 nm indiameter) are present with a distance between crossover points that is almost twice (160 nm)that of Alzheimer-type PHF.11,12 While precise cell type identification at the fine structurallevel is often difficult in autopsy tissue in the absence of double-immunolabeling with celltype-specific markers, most results suggest that the wider filaments with longer periodicityare neuronal rather than glial.12 Alzheimer-type PHF have not been described in glia. Inaddition to filaments, NFT invariably contain poorly characterized granular material. Com-parable granular material is also present in glial inclusions, where it has been described asan integral component of the filaments (“granule-coated filaments”13). There are currentlyno clues as to the composition of the granular material.
Given the great abundance of NFT in AD and the unusual solubility properties of PHF,PHF can be purified to near homogeneity and biochemically characterized.14,15 Biochemi-cal and immunochemical studies have demonstrated that PHF are primarily composed ofmicrotubule-associated protein tau.14-19 NFT are immunoreactive with antibodies to epitopesspanning the full length of tau,20 suggesting that full length tau is present in NFT. Similarstudies of tau-positive glial inclusions also suggest that full length tau protein is present;21
however, there is suggestive evidence in some of the disorders with abundant glial inclu-sions that certain splice forms of tau may preferentially accumulate.12,22
Tau protein is a phosphoprotein with multiple isoforms derived from alternative splic-ing of a single gene on chromosome 1723 and also from posttranslational modification (re-viewed in refs. 24 and 25). The best studied of the posttranslational modifications is phos-phorylation. The phosphorylation state of tau determines its ability to promotepolymerization of tubulin and to stabilize microtubules. More highly phosphorylated formsshow decreased ability to promote polymerization of tubulin and to stabilize microtubules.Tau protein in PHF has increased and abnormal phosphorylation based on indirect immu-nochemical and immunocytochemical methods16 and direct chemical analysis of phosphatecontent.26 Hyperphosphorylated tau protein is incompetent with respect to tubulin assem-bly, and it is this form that accumulates in brains of neurodegenerative diseases.27 The cur-rent theory is that tau protein which has dissociated from tubulin due to increased phos-phorylation undergoes self-assembly to form filaments.28 The tau protein in glial inclusionsis also hyperphosphorylated based on immunostaining with antibodies to multiple phos-phorylation sites in the tau molecule.21 Direct phosphate analysis of tau from glia in thesedisorders has not been reported.
Whether phosphorylation of tau has any impact on microtubules in glia has not beeninvestigated, but in neurons it is speculated that microtubule instability leads to impairedaxoplasmic and dendritic transport and may thereby contribute directly to neurode-generation.28 In contrast to neurons where microtubules are abundant in all cell domains

Astrocytes in Brain Aging and Neurodegeneration168
(especially axons) and where they are crucial to essential cellular functions such as axoplas-mic transport, the presence and function of microtubules in glia is far less clear. Microtu-bules are sparse in normal astrocytes,29 but more abundant in normal oligodendroglia.30
Furthermore, the major microtubule associated protein (MAP) in astrocytes is not tau, butrather a specific splice variant of MAP2.31-34 MAP2 expression is increased in astrocytessubjected to cell injury or stress.32,33 While a number of antibody based methods have indi-cated that inclusion bodies in astrocytes contain tau protein, direct proof for tau protein inastrocytes is lacking. All available evidence is based upon indirect immunocytochemicalanalyses. The fact that antibodies to multiple epitopes in tau stain the inclusions is fairlydefinitive evidence that tau is present in astrocytes, but not all studies have used more thanone antibody to tau. It must be realized that microtubule-associated proteins share certainstructural motifs and even conserved domains. For example, the microtubule-binding do-main of MAP2 and tau share significant sequence homology.24 Additional studies are war-ranted to determine if any of the immunoreactivity observed in astrocytes in these disor-ders is related to crossreactivity with MAP2.
Although tau was once felt to be restricted to neurons,35 more recent studies suggestthat tau can be detected in normal glial cells; however, only trace amounts of tau can bedetected at basal levels in normal human (unpublished data), bovine and rodent astro-cytes.31,36,37 Even in cultured human fetal astrocytes exposed to activating conditions (e.g.,interleukin-1β),38 or phosphatase inhibitors (e.g., okadaic acid),36 tau is difficult to detect(unpublished data). These results suggest that additional factors lead to production andaggregation of tau protein within astrocytes in neurodegenerative diseases or that adultastrocytes have properties that are distinct from fetal astrocytes.
Tau is far easier to detect in oligodendrocytes, especially in brains that have been sub-jected to injury or stress, such as ischemia.40,41 It is also readily detected in cultured oligo-dendrocytes,36,37 where, interestingly, it appears to be composed of restricted tau isoforms,similar to isoform restriction that occurs in neurodegenerative diseases with glial tau inclu-sions.36 Specifically, exon 3 of tau is an alternatively spliced exon that contributes to theheterogeneity in tau isoforms.24 The function of this domain is not known, but tau inneurodegenerative diseases with abundant glial tau-positive inclusions have accumulationof tau splice forms that preferentially lack exon 3 based on lack of immunostaining withexon 3 specific antibodies.42, 43 The same is true for cultured oligodendrocytes.36
Native tau protein is a soluble protein that does not readily form filaments. Structuralanalysis indicates that it is an elongated molecule, but one that does not have a regularshape, which is an unusual feature for a soluble protein.25 There are multiple discrete do-mains in tau, with 3 or 4 conserved repeats in a domain involved in binding of tau protein tomicrotubules.25 Alternative RNA splicing generates tau proteins with either 3 or 4 repeats inthe microtubule-binding domain.24 The microtubule-binding domain of tau appears to beessential for assembly into pathological filaments, since recombinant tau protein composedof little more than the microtubule-binding domain spontaneously assembles into PHF-like structures.44 Full length recombinant tau molecules do not spontaneously form fila-ments.45 This may indicate that proteolysis may play a role in producing tau fragments thatspontaneously form filaments. On the other hand, recent studies have shown that full lengthrecombinant tau protein forms filaments resembling the pathological filaments in braindiseases when it is mixed with acidic polymers such as heparan sulfate proteoglycan45 orlipids such as arachidonic acid.46 Additional factors, such as protein crosslinking,ubiquitination,47 glycation,48-50 or association with polymers or other proteins, appear to beessential in the formation and aggregation of PHF (reviewed in ref. 51). Whether or notthese processes are relevant to glial tau inclusions awaits further study.

169Astrocytes in Other Neurodegenerative Diseases
Clearly, the phosphorylation state is one way in which pathological tau differs fromnormal tau. Given this observation, there has been much interest in identifying the kinasesthat may be responsible for catalyzing tau phosphorylation. Alternatively, increased phos-phate content in tau may also be due to decreased activity or inhibition of specific phos-phatases.52 It is of interest that freshly isolated tau proteins undergo rapid dephosphoryla-tion,53 which suggests that phosphorylation of tau protein is under tight control. Both kinasesand phosphatases are likely to be important in maintaining this tight control. On the otherhand, experimental paradigms suggest that tau aggregates occur in the absence of abnormalphosphorylation and that phosphate-dependent epitopes may appear subsequently, con-tributing to stabilization of the aggregates.54 Regardless, phosphorylation remains an im-portant phenotypic difference between normal tau and abnormal tau in most cytoskeletalinclusions.
Among the several kinases that have been implicated in phosphorylation of tau protein(reviewed in ref. 55), proline-directed kinases have drawn a great deal of attention. This isbecause there are multiple such phosphorylation sites in tau and because phosphorylationat these sites affects the function of tau. Cyclin-dependent kinases are proline-directed ki-nases that can phosphorylate tau protein in vitro.56,57 While many different kinases can beshown to phosphorylate tau in a test tube, only the cyclin-dependent kinases have beenshown to consistently colocalize with NFT and even to copurify with PHF.56-58 Cyclin-de-pendent kinases are members of the family of kinases that are involved in cell cycle regula-tion, and their expression in differentiated cells has been considered to be aberrant.57,59
Aberrant expression of such kinases has been implicated in programmed cell death orapoptosis,59 which is the mode of neuronal (and glial?) loss in most neurodegenerative dis-eases in which it has been studied. Of known cyclin-dependent kinases, one particular spe-cies that may be relevant to tau phosphorylation in glia is similar to cdc2 and has beenreferred to as KKIALRE.60 The latter terminology refers to an amino acid sequence in thecarboxyl terminus of the kinase that is unique and distinguishes KKIALRE from authenticcdc2 kinase. Antibodies to the KKIALRE domain of this cdc2-like kinase label astrocytes ingray and white matter of human brain (Fig. 8.1). The expression of KKIARLE is increasedin diseased brains in regions with gliosis, as seen in Alzheimer’s disease and otherneurodegenerative diseases. Further studies are needed on the distribution of this and otherkinases in neurodegenerative disorders associated with glial tau pathology.
Neurodegenerative Disorderswith Filamentous Glial Inclusion Bodies
The most common disorders with glial tau pathology (Table 8.1) include progressivesupranuclear palsy (PSP), corticobasal degeneration (CBD) and Pick’s disease. Less frequently,tau-positive astrocytic inclusions have been detected in Guam Parkinson-dementia complex(GPDC), postencephalitic Parkinsonism and other neurodegenerative disorders with NFT.Rarely, tau-positive glial inclusions are found in AD. Tau-immunoreactive astrocytes arealso detected in aged human brains, but in this context the immunoreactivity is not associ-ated with inclusion bodies. Mixed and variable numbers of glial and neuronal tau-positiveinclusions are seen in familial frontotemporal dementia with Parkinsonism linked to chro-mosome 17 (FTDP-17). Predominantly oligodendroglial inclusions are seen in dementiawith argyrophilic grains (AGD). Inclusion bodies reminiscent of Lewy bodies have recentlybeen described in astrocytes in some families with amyotrophic lateral sclerosis (FALS) and,interestingly, also in transgenic animals bearing the same mutation as FALS. Finally, glialinclusions have come to be the defining histopathologic hallmark of multiple system atro-phy (MSA). These inclusions are found in oligodendroglia rather than astrocytes, and differ

Astrocytes in Brain Aging and Neurodegeneration170
Table 8.1. Classification of glial pathology
Glial Cell Type Name Composition AssociatedDisorders
Astrocytes Tufted astrocyte and thorn Hyperphosphorylated tau PSP, PD, PDC,shaped astrocytes PEP, SSPE
Astrocytes Astrocytic plaque Hyperphosphorylated tau CBD
Oligodendrocytes Coiled bodies; oligodendrocyte Hyperphosphorylated tau CBD, PSP,microtubule bodies; AGD, ADglial fibrillary tangles
Oligodendrocytes Glial cytoplasmic inclusion Normal tau; Ubiquitin MSA
Astrocytes Lewy body-like inclusion Superoxide dismutase FALS
Abbreviations: PSP = progressive supranuclear palsy; PD = Pick’s disease; PDC = Parkinson dementiacomplex of Guam; PEP = postencephalitic Parkinsonism; SSPE = subacute sclerosing panencephalitis;CBD = corticobasal degeneration; AGD; argyrophilic grain dementia; AD = Alzheimer’s disease;MSA = multiple system atrophy (Shy-Drager syndrome); FALS = familial amyotrophic lateral sclerosis.
Fig. 8.1. Astrocytes in cortical gray matter of Alzheimer’s disease are immunoreactive with acdc2-related (KKIARLE) antibody.

171Astrocytes in Other Neurodegenerative Diseases
from the other inclusions noted above in their inconsistent immunoreactivity with anti-bodies to phosphate-dependent tau epitopes.61 A scheme for classifying neurodegenerativedisorders based on presence and type of inclusion body has recently been proposed62 andmodified here to fit with new information (Fig. 8.2).
The major inclusion bodies in astrocytes in the aged brain are corpora amylacea, whichare composed of glycosidic polymers rather than cytoskeletal proteins.29 (Corpora amylaceaare discussed in chapters 4 and 10 of this volume.) A much less recognized change in glia inaging is tau immunoreactivity. This can be demonstrated with immunocytochemical meth-ods and specific tau antibodies.63,64 Initial studies of tau protein distribution and immu-noreactivity failed to recognize tau in glial cells. The fact that tau was considered to be aneuron-specific molecule restricted to the axoplasmic domain65 no doubt influenced inter-pretation of early studies. Redistribution of tau epitopes, especially phosphorylated tauepitopes, to the somatodendritic domain of neurons was considered a pathological trait.64
Presence of phospho-tau within the soma of small cells was more apt to be considered apathological process in neurons than in glia. Rigorous double-labeling immunocytochemi-cal studies with cell type-specific markers, such as glial fibrillary acidic protein, were neededbefore pathological tau was widely recognized in glia. While antibodies to tau protein donot stain astrocytes in brains of young adults and of other species, they do stain astrocytesin elderly individuals and especially in Alzheimer’s disease (Fig. 8.3). Certain astrocytes con-sistently show tau immunoreactivity in aged and Alzheimer’s disease brains. Tau-positiveastrocytes are located in subpial regions at the base of the brain (e.g., basal forebrain), in themedial temporal lobe (e.g., amygdala) and subependymal regions (e.g., temporal horn oflateral ventricle). That the immunoreactivity is within astrocytes is unambiguous since thereis staining of astrocytic end-feet at the glia limitans and around blood vessels (Fig. 8.4). Theastrocytic tau is cytoplasmic, without formation of discrete inclusion bodies. Tau-positiveastrocytes in aging and Alzheimer’s disease have not been well studied and no ultrastruc-tural studies have been reported. More recently, glial inclusion bodies have been describedin Alzheimer’s disease, but these are not a common finding compared to the other condi-tions under consideration.7 The tau-positive glia in Alzheimer’s disease are labeled by anti-bodies against transferrin and 2'3'-cyclic nucleotide 3'-phosphohydrolase, which are mark-ers for oligodendrocytes. Ultrastructurally, they were composed of bundles of straightfilaments about 16 nm in diameter.
Progressive Supranuclear Palsy (PSP)PSP is a sporadic degenerative disease associated with axial rigidity, vertical eye move-
ment abnormalities and subcortical dementia, first described by Steele, Richardson andOlszewski.66,67 The pathology of PSP is characterized by neuronal loss and gliosis in a num-ber of interrelated subcortical nuclei of the extrapyramidal system, including basal ganglia,motor nuclei of the thalamus, dopaminergic and other nuclei in the midbrain, noradrener-gic neurons and neurons in the pontine base, inferior olivary nucleus and cerebellar dentatenucleus. In most of these locations neurons have NFT, and tau aggregates are detected withincell processes.68 The NFT in PSP differ from those in Alzheimer’s disease by the presence of15 nm to 18 nm diameter straight, rather than twisted, filaments.69-71 Nevertheless, the neu-ronal inclusions in PSP contain tau protein that is very similar to PHF in Alzheimer’s dis-ease with immunocytochemical methods.72 On the other hand, biochemical studies of tauprotein in PSP reveal differences from those in Alzheimer’s disease73, 74 that may to someextent reflect the fact that pathological tau is derived from both neurons and glia in PSP.Specifically, the abnormal tau in PSP is composed predominantly of two isoforms while inAlzheimer’s disease PHF-tau is composed of three major isoforms. In the scheme proposedfor classifying neurodegenerative disorders in Figure 8.2 these two forms are referred to as

Astrocytes in Brain Aging and Neurodegeneration172
Fig. 8.2. Diagram illustrating a scheme for classifying neurodegenerative diseases depending onthe presence and type of inclusion body. Note that glial inclusions are found in several branchesof the tree, but that tau-positive glial inclusions are most frequent in those disorders with tauisoform restriction. Glial tau pathology is particularly prominent in those disorders with a patho-logical tau “doublet” isoform. Tau-positive glial inclusions are also seen in “triplet” tau disorders,but they are a minor or inconspicuous feature in most cases.

173Astrocytes in Other Neurodegenerative Diseases
“doublet” and “triplet” tau. In PSP pathological tau, some evidence suggests that this isoformpattern may reflect preferential accumulation of specific splice forms of tau. This observa-tion, along with information about tau in glia, might suggest that observed biochemicaldifferences in tau in PSP compared to Alzheimer’s disease may be related in part to thegreater contribution of glial tau in PSP.
Since tau was considered to be a neuron-specific protein, tau-positive inclusions inPSP were initially considered to be NFT in small neurons. Double labeling methods havenow demonstrated unequivocally that many of the tau-positive small cells in PSP, especiallyin the basal ganglia, are astrocytes.1,21,75-81 Immunocytochemical studies with antibodies totau protein also revealed abnormal filamentous profiles in cell processes in affected regionsof gray matter and also white matter (e.g., pencil fibers in the caudate and putamen) in PSP(Fig. 8.5). These so-called “neuropil threads”81,82 were initially interpreted to be within neu-ronal processes, but ultrastructural immunolabeling studies have now shown that many ofthese profiles are within the cytoplasm of oligodendrocytes and also within loops of myelinsheaths, which are extensions of the cytoplasm of oligodendroglia.83
Oligodendroglial tau-positive inclusions have become a recognized pathological fea-ture of PSP (Fig. 8.5). Similar oligodendroglial inclusions can be found in otherneurodegenerative disorders, where they are often more abundant than PSP. They have beenreferred to as “coiled bodies.”84 Coiled bodies were first described in argyrophilic grain de-mentia (AGD), an uncommon, or at least under-recognized, neurodegenerative disorderdescribed by the Braaks.84-86 AGD is named for grain-like lesions within the neuropil thatcan be detected with silver stains or tau immunostains. The grains correspond to filamen-tous aggregates within segmental domains of cell processes. The latter are mostly neuronal
Fig. 8.3. Astrocytes in cortical gray matter are immunoreactive with an anti-body to tau protein that recognizes an epitope near the carboxyl terminus oftau. Tissue was fixed in periodate-lysine paraformaldehyde and sectioned witha Vibratome.

Astrocytes in Brain Aging and Neurodegeneration174
Fig. 8.4. Subpial astrocytes at the base of the brain near the basal forebrain show tau immunore-activity. Note absence of immunostaining of corpora amylacea in some of the processes.
processes (i.e., dendrites) based upon immunoelectron microscopic studies, but some arealso clearly within glial cell processes.86 Like oligodendroglial coiled bodies, grains are notdisease-specific and can be found in several neurodegenerative disorders, most notablycorticobasal degeneration (see below). Oligodendroglial coiled bodies should be distinguishedfrom the glial cytoplasmic inclusions (GCI) within oligodendroglia that are the hallmark ofmultiple system atrophy (MSA).87-90 Oligodendroglial coiled bodies contain phosphory-lated tau epitopes and are actually best recognized with immunocytochemical methods. Incontrast, GCI are essentially limited to MSA and have a different morphology and antigenicmakeup. Phospho-tau antibodies either fail to stain GCI or they do so inconsistently. Recentevidence suggests that tau in GCI has properties closer to normal tau than to the abnormaltau that aggregates in neuronal and glial inclusions in other disorders.61 GCI are also in-tensely immunoreactive with antibodies to ubiquitin, which is a small heat shock moleculeinvolved in ATP-dependent proteolysis of abnormal or denatured proteins.87-90 In contrast,oligodendroglial coiled bodies and tau-positive argyrophilic inclusions in astrocytes in otherdisorders are very weakly immunoreactive for ubiquitin.
As mentioned previously, astrocytes are invariably affected in PSP. The tau-positiveinclusions in neurons and glia in PSP are composed of straight filaments at the ultrastruc-tural level. A variety of names have been attached to the abnormal astrocytes in PSP, such as“tufted astrocytes” or “thorn-shaped astocytes”76-79,91 (Fig. 8.5). This nomenclature is purelydescriptive and has not proven to be useful in discriminating astrocytic lesions in one disor-der from another. Within a given disorder there is morphologic diversity of abnormal astro-

175Astrocytes in Other Neurodegenerative Diseases
cytes. There is also variability in the appearance of the astrocytes depending upon the ana-tomical region in which they reside (Fig. 8.5).
Tufted astrocytes similar to those in PSP can be seen in Pick’s disease,78,79,92 GuamParkinson dementia complex,93 postencephalitic Parkinsonism94 and subacute sclerosingpanencephalitis.95 All of these disorders have both neuronal (i.e., NFT) and glial tau inclu-sions. The major means of differentiating the disorders is not by the appearance of theastrocyte inclusions, but rather by the clinical presentation and pathological findings, aswell as the distribution and nature of the glial and neuronal pathology. Biochemical analysisof the major tau isoforms that accumulate in brain tissue may offer another means of differ-entiating the disorders (see Fig. 8.2).
Pick’s DiseasePick’s disease is a rare late-life degenerative disorder presenting as dementia and
personality deterioration due to circumscribed (“lobar”) atrophy with marked neuronalloss and gliosis in the frontal and anterior temporal lobes.96 While there is no universalagreement as to the defining feature of Pick’s disease, the presence of argyrophilic, roundinclusion bodies within neurons is an increasingly accepted pathological hallmark. These
Fig. 8.5. Astrocytes and oligodendroglia have tau-positive inclusions in PSP. (A), (B) gray matterastrocytes [(A) cortex, (B) basal ganglia] and (C), (D) white matter glial inclusions [(C) cortex,(D) basal ganglia]. Note variability in tufted astrocyte morphology [(A) and (B)] dependingupon anatomical region. Coiled bodies are evident in the cerebral white matter and the pencilfibers of the basal ganglia.

Astrocytes in Brain Aging and Neurodegeneration176
inclusion bodies were originally described by Alzheimer97 and have come to be known asPick bodies. Pick bodies are filamentous inclusions composed of altered tau protein that ishighly phosphorylated, similar to tau proteins in Alzheimer’s disease.98 Few biochemicalstudies have been performed on cytoskeletal proteins in Pick’s disease, but those that havebeen reported suggest that the tau protein abnormalities in Pick’s disease are distinct fromAlzheimer’s disease and closer to those in PSP, with expression of two major pathologicaltau isoforms.99 Pick bodies differ most from NFT in their distinct anatomical distributionand characteristic microscopic appearance. At the ultrastructural level, Pick bodies containfilaments of variable morphology, including wide and long-period, twisted filaments orstraight filaments.100-102
Astrocytic inclusions in Pick’s disease have a distinctive appearance, but show mor-phologic overlap with lesions in the various disorders under consideration. In Pick’s diseaseand PSP the inclusions are most often of the tufted or thorn-shaped appearance (Fig. 8.6).In Pick’s disease, abnormal astrocytes are found in affected cortical regions, while in PSPthey are most abundant in the basal ganglia. In the cortex in PSP they are confined to motorand premotor cortex. Thus, glial pathology in these disorders parallels the distribution ofthe other more widely recognized neuronal pathology. The tufted astrocytes in PSP oftenhave filamentous aggregates that are often displaced into the cell processes with less stainingin the perinuclear region, which accounts for the tufted appearance. In contrast, in Pick’sdisease the filamentous aggregates are often in a more proximal perinuclear cellular domain(Fig. 8.6). Despite this general observation, the overlap in appearance precludes a morpho-logical basis for neuropathological diagnosis. Far more important is the distribution of thepathology.105
White matter pathology is a well-known feature of Pick’s disease, with loss of myeli-nated fibers in a distribution that parallels the areas with most marked cortical atrophy. Thedegree of myelin loss correlates with the severity of the cortical atrophy and is reflected inloss of myelin-related lipids.103 These observations strongly suggest that white matter pa-thology is a type of Wallerian degeneration secondary to cortical neuronal loss; however,the recent discovery of cytoskeletal inclusion bodies in oligodendroglia and astrocytes inwhite matter in Pick’s disease raises the possibility that the white matter pathology may bean integral part of the disease rather than a secondary change. An interesting phenotype ofoligodendroglial inclusions in Pick’s disease includes round inclusion bodies that are highlyreminiscent of neuronal Pick bodies.78 More often oligodendroglial lesions in Pick’s diseasehave the appearance of coiled bodies or oligodendroglial microtubular inclusions similar tothose of PSP and other disorders21,104 (Fig. 8.6).
Corticobasal Degeneration (CBD)CBD is a rare, sporadic disorder whose classical clinical picture is one of asymmetrical
rigidity, dystonia and apraxia, with mild or inapparent cognitive deterioration.106,107 It isbecoming clear that the clinical phenotype is broader than originally described. Many sub-jects have progressive dysphasia, reflecting the fact that the brunt of the pathology is oftenin the dominant cerebral hemisphere.100,108,109 The hallmark lesion of CBG is the achro-matic neuron.106 Achromatic neurons are swollen and weakly stained with routine his-tochemical methods, hence their name. They are also referred to as ballooned or swollenchromatolytic neurons.110 Ballooned neurons lack diagnostic specificity when found in limbicareas, but are highly characteristic of CBD when found in the convexity cerebral cortex,especially in the superior frontal and parietal lobes. They are intensely immunoreactivewith phosphorylated neurofilament antibodies and variably stained with tau antibodies.111
Tau antibodies also reveal a host of other pathologies in CBD, foremost among them beinginclusions in both oligodendroglia and astrocytes.12,21,42,43,78,79,112-117

177Astrocytes in Other Neurodegenerative Diseases
Glial inclusions are more numerous in CBD than in any of the other disorders dis-cussed, which may account for the many reports in recent years on this disor-der.12,21,42,43,78,79,112-117 While morphological distinctions may not differentiate the disorders,the relative abundance of neuronal versus glial inclusions, and the distribution in gray ver-sus white matter and forebrain vs. hindbrain, clearly differs in PSP, Pick’s disease and CBD.105
In particular, Pick’s disease is predominantly a neuronal disorder, CBD predominantly gliaand PSP both. PSP affects mostly deep gray matter, Picks’ disease mostly cortical and CBDboth. In CBD the predominant distribution of abnormal tau protein is within cell processesof glia and neurons with abundant white matter disease. Neither Picks’ disease nor PSP havewhite matter pathology as marked as that of CBD.
If any of the astrocytic lesions is diagnostically useful, perhaps the so-called “astrocyticplaque” of CBD comes closest to meeting this criterion (Fig. 8.7). The astrocytic plaque ismost apparent in the affected cortical regions, with fewer lesions in deep gray matter. In theastrocytic plaque abnormal tau accumulates in distal cellular processes of reactive astro-cytes, forming an annular arrangement of miliary structures in the neuropil.105,115 The ap-pearance is suggestive of a neuritic plaque in Alzheimer’s disease, but the central core doesnot contain amyloid. Instead, specific cellular markers demonstrate a central astrocyte withdilated distal processes containing tau immunoreactivity. Ultrastructural studies demon-strate that filaments within the cell body of astrocytes resemble glial filaments in fibrousastrocytes, while those in distal segmental cell processes are thicker. The filaments in the cellbodies are not immunostained with tau and PHF antibodies, while the ones in distal pro-cesses are positive (Fig. 8.8). In considering the predominant cellular domain of the astro-cyte affected by tau aggregates, Pick’s disease, PSP and CBD appear to differ. In Pick’s dis-ease aggregates are in or close to the cell body, in PSP proximal processes tend to be affectedand in CBD distal segmental domains are preferentially affected.
Cerebral white matter shows marked pathology in CBD characterized by numeroustau-positive thread-like processes as well as many tau-positive glial cells (i.e., coiled bodies)(Fig. 8.7). The abnormal white matter tau aggregates have been shown with double labelingmethods to reside in both neuronal axons and glial processes.115 The white matter tau
Fig. 8.6. Astrocyticand neuronal inclu-sions in Pick’s diseaseare immunoreactivewith a tau antibody.(A) Note neuronalPick body (arrowhead) and two tau-positive astrocytes(arrows). (B) Astro-cytic nature of theglial inclusions is ob-vious in that some ofthe cells form end-feet to blood vessels(arrow). (C) In whitematter some of theglial inclusions in ap-parent oligodendro-cytes appear similarto small Pick bodies. Reprinted with permission from Vincent I et al, J Cell Biol 1996; 132:413-425.

Astrocytes in Brain Aging and Neurodegeneration178
Fig. 8.7. Tau-positive glia are numerous in CBD. (A) Many tau-positive astrocytic plaques arevisible in this low power image of superior frontal cortex. (B) Double stained sections (mono-clonal antibodies to vimentin and to tau) show a central astrocyte in the middle of the clusters ofstubby tau-positive processes. (C) The cerebral white matter has many glial inclusions in oligo-dendrocytes (coiled bodies). (D) White matter processes are numerous in certain cortical andsubcortical fiber tracts. Other studies have demonstrated that these processes are within glial andneuronal processes. Ultrastructural studies suggest that predominantly oligodendrocytes pro-duce this lesion.
pathology in CBD is greater than in any of the other conditions. It is most marked in areasthat also show the greatest cortical pathology, but is also very severe in certain diencephalicfiber tracts where neuronal pathology is not marked, most notably the thalamic fasciculus.In some cases the pattern of white matter tau-positive pathology follows defined anatomicpathways, such as the corticospinal tract in the cerebral peduncle and medullary pyramid.Given the abundance of oligodendroglial and astrocytic tau pathology in CBD, if biochem-istry of brain samples reflects glial changes in any disorder it is most likely to be representa-tive of glial changes in CBD. In this disorder biochemical studies show an abnormal taupattern with two major tau isoforms, probably due to preferential alternative splicing of tauDNA.12,118 Exon 3 is also underrepresented in pathological tau in CBD.42,43 There is evi-dence to suggest that exon 10 is abnormally expressed in the abnormal tau protein of CBD.12,22
Exon 10 is another alternatively spliced exon in tau,24 which determines whether tau willhave three or four repeats in the microtubule-binding domain. It is suggested that preferen-tial tau splicing contributes to the restricted isoform pattern in CBD. What remains to be
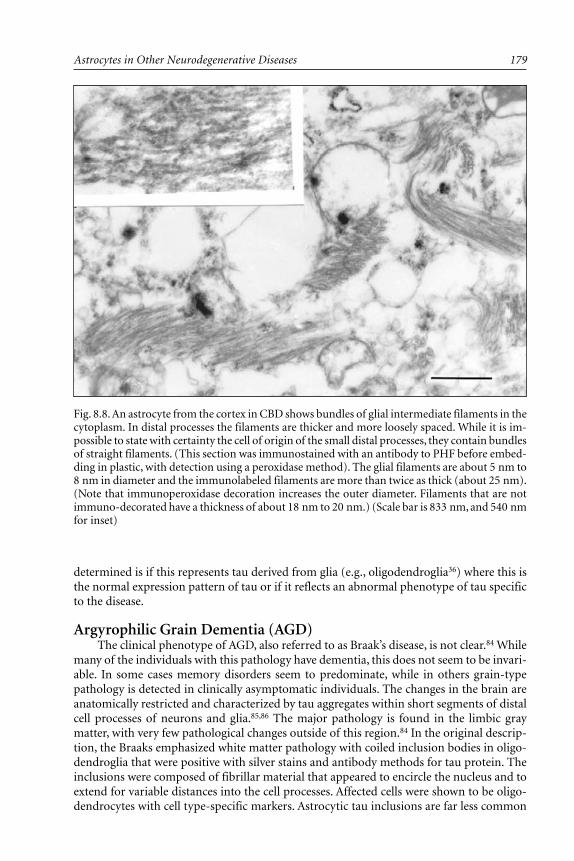
179Astrocytes in Other Neurodegenerative Diseases
determined is if this represents tau derived from glia (e.g., oligodendroglia36) where this isthe normal expression pattern of tau or if it reflects an abnormal phenotype of tau specificto the disease.
Argyrophilic Grain Dementia (AGD)The clinical phenotype of AGD, also referred to as Braak’s disease, is not clear.84 While
many of the individuals with this pathology have dementia, this does not seem to be invari-able. In some cases memory disorders seem to predominate, while in others grain-typepathology is detected in clinically asymptomatic individuals. The changes in the brain areanatomically restricted and characterized by tau aggregates within short segments of distalcell processes of neurons and glia.85,86 The major pathology is found in the limbic graymatter, with very few pathological changes outside of this region.84 In the original descrip-tion, the Braaks emphasized white matter pathology with coiled inclusion bodies in oligo-dendroglia that were positive with silver stains and antibody methods for tau protein. Theinclusions were composed of fibrillar material that appeared to encircle the nucleus and toextend for variable distances into the cell processes. Affected cells were shown to be oligo-dendrocytes with cell type-specific markers. Astrocytic tau inclusions are far less common
Fig. 8.8. An astrocyte from the cortex in CBD shows bundles of glial intermediate filaments in thecytoplasm. In distal processes the filaments are thicker and more loosely spaced. While it is im-possible to state with certainty the cell of origin of the small distal processes, they contain bundlesof straight filaments. (This section was immunostained with an antibody to PHF before embed-ding in plastic, with detection using a peroxidase method). The glial filaments are about 5 nm to8 nm in diameter and the immunolabeled filaments are more than twice as thick (about 25 nm).(Note that immunoperoxidase decoration increases the outer diameter. Filaments that are notimmuno-decorated have a thickness of about 18 nm to 20 nm.) (Scale bar is 833 nm, and 540 nmfor inset)

Astrocytes in Brain Aging and Neurodegeneration180
and have not been emphasized in the descriptions of AGD. Disorders that have been de-scribed as being an overlap between PSP and AGD,119 having many glial tau-positive inclu-sions, likely represent CBD on second consideration, given the fact that ballooned neuronsare consistently present in these cases (E. Masliah, personal communication).
Familial Frontotemporal Dementia and ParkinsonismLinked to Chromosome 17 (FTDP-17)
Frontotemporal dementia describes a clinical phenotype with variable pathology. Theclinical syndrome includes dementia with variable personality changes, frontal lobe signs,motor involvement and language disturbance. There is often asymmetry, with the left hemi-sphere more often affected than the right. Some cases of FTD have Pick’s disease, while mosthave dementia lacking distinctive histology.120,121 A variable degree of Parkinsonism is notuncommon in FTD and about 10% of cases are hereditary.121 A recent consensus confer-ence proposed the term FTDP as an umbrella term to describe this group of disorders thathad previously gone under a wide diversity of terms.122 All familial cases included in thereport were variably linked to a gene of chromosome 17.122 Interestingly, the tau gene islocated within the consensus region. Closer inspection of many of the cases of FTDP-17 hasshown that tau-positive inclusions in neurons and glia are common.22,123 In some cases taupathology is marked, with numerous inclusions in neurons, oligodendroglia and astrocytes.This has prompted the designation for the disease in one of the families as a “tau-opathy”.122
Such cases have a great deal of overlap with PSP and CBD.22 Furthermore, recent studiessuggest that PSP is also linked to a polymorphism in the tau gene on chromosome 17.124,125
The polymorphism is within an intron in tau (between exons 9 and 10) that is subject toalternative splicing.124 Glial pathology in FTDP-17 includes coiled bodies, white matterthreads, tufted astrocytes and in some cases astrocytic plaques similar to those in CBD.While CBD is usually a nonfamilial disorder, the great similarity in pathology between somecases of FTDP-17 and CBD suggests that some cases may be a familial form of CBD.
Multiple System Atrophy (MSA)Multiple system atrophy refers to a symptom complex that includes cerebellar ataxia,
Parkinsonism, orthostatic hypotension and autonomic dysfunction.126 The pathology isvariable and reflects the predominant clinical phenotype. The pathological diagnoses thatare subsumed under the rubric of MSA include sporadic olivopontocerebellar degenera-tion, striatonigral degeneration and Shy-Drager syndrome.87,126 The hallmark of MSA is theglial cytoplasmic inclusion (GCI) which is a round or crescent-shaped inclusion that is in-tensely argyrophilic and positive with ubiquitin antibodies, but variably stained or negativewith tau antibodies61,87-90 (Fig. 8.9). Recent studies suggest that GCI contain tau protein thatis not as highly phosphorylated as tau in PHF and more analogous to normal tau.61 Thisalone indicates that aggregation and polymerization of cytoskeletal elements, and tau inparticular, are not dependent on high phosphate content of the cytoskeletal components.
The discovery of glial inclusions in MSA was a breakthrough in the nosology and clas-sification of the various spinocerebellar degenerations. The presence of glial cytoplasmicinclusions has become a means of differentiating sporadic from hereditary forms of spinoc-erebellar degeneration.127 The latter, which are often trinucleotide repeat disorders,128 areonly rarely associated with glial cytoplasmic inclusions that are positive with tau immuno-cytochemistry.129 On the other hand, a different type of inclusion body has recently beenidentified in many (if not all) of the trinucleotide repeat disorders, including Huntington’sdisease, and that is the intranuclear inclusion.130 The latter are most often within nuclei ofneurons, but some of these disorders, as well as sporadic disorders, have similar hyalineinclusions with glial nuclei.131 In these disorders, evidence suggests that the protein that

181Astrocytes in Other Neurodegenerative Diseases
aggregates is also the mutated protein, but further investigation is clearly needed in thisemerging area. It is unknown how, or if, nuclear inclusions in neurons and glia affect cellu-lar function.
Familial Amyotrophic Lateral Sclerosis (FALS)ALS is usually a sporadic condition associated with progressive degeneration of motor
neurons in brainstem and spinal cord. The clinical syndrome is one of progressive weaknessand muscle atrophy with preservation of higher cognitive funcitons.132 The pathology isthat of selective neuronal loss and gliosis relatively confined to the upper and lower motorneurons and denervation atrophy of skeletal muscle.133 It is variably associated with degen-eration in other systems, such as the spinocerebellar and somatic sensory pathways, espe-cially in familial forms.133 There are few histological hallmarks that are specific to the disor-der. The exception is the Bunina body, an inclusion in affected neurons in ALS that appearsto be derived from membranous organelles, possibly related to the endoplasmic reticu-lum.108 In familial cases a number of investigators have reported hyaline cytoplasmic inclu-sions in affected neuronal populations in the motor cortex, brainstem and anterior horn ofthe spinal cord.134-136
The neuronal inclusions in familial ALS resemble Lewy bodies of idiopathic Parkinson’sdisease (for review see ref. 2). These spherical hyaline cytoplasmic inclusions containneurofilament protein and are highly ubiquitinated.134-136 More recently, Lewy body-likeinclusions have also been described in astrocytes13,137 in certain familial forms of amyo-trophic lateral sclerosis associated with mutations in Cu/Zn superoxide dismutase.138 Com-parable inclusion bodies are also detected in transgenic mice carrying the mutation in Cu/Znsuperoxide dismutase.139 Developmental studies in this animal model suggest that glial pa-thology may actually precede neuronal changes. This may be a valuable model for exploringthe role of glial pathology in neurodegeneration.
At the ultrastructural level the inclusions in both the transgenic animals and in astrocytesin humans with FALS are granule-coated filaments.13,138 Since they are immunoreactive
Fig. 8.9. Glial cytoplasmic inclusions are the hallmark lesions of multiple system atrophy. In thissection the white matter glia from cerebellum (A) and pontine base (B) have crescent-shapedinclusion bodies [arrows in (A)]. These inclusions were completely negative with phospho-tauantibodies. Occasional thread-like profiles are also visible with ubiquitin staining [arrowhead in(B)], which also probably represents inclusions in oligodendroglial cytoplasm.

Astrocytes in Brain Aging and Neurodegeneration182
with antibodies to SOD, they may be composed of polymers of this molecule, but this re-mains to be demonstrated by direct biochemical analysis. This would be unprecedented. Allother common filamentous inclusions in neurons and glia are derived from cytoskeletalproteins. On the other hand, the lesson from trinucleotide repeat disorders and from vari-ous amyloidoses is that pathological fibrils may be formed from proteins of diverse molecu-lar nature, particularly if they have an altered conformation that favors the low energy stateof stable filaments.
AcknowledgmentsImmunocytochemical studies of Drs. Mel Feany and Linda Mattiace were important in
defining glial pathology in non-AD disorders. Biochemical studies were performed byDr. Hanna Ksiezak-Reding. Tissue culture studies of human fetal astrocytes were performedby Dr. Deke He. Additional tissue culture studies were performed by Drs. Sunhee Lee andMeng-Liang Zhao. Discussion of various aspects of this work with Drs. Peter Davies, BridgetShafit-Zagardo and Shu-Hui Yen are acknowledged. Yvonne Kress assisted with ultrastruc-tural studies. The efforts of these individuals at Albert Einstein College of Medicine is grate-fully acknowledged. Support for this research was provided by NIA AG06803.
References1. Chin SS, Goldman JE. Glial inclusions in CNS degenerative diseases. J Neuropathol Exp
Neurol 1996; 55:499-508.2. Pollanen MS, Dickson DW, Bergeron C. Pathology and biology of the Lewy body. J
Neuropathol Exp Neurol 1993; 52:183-191.3. Wakabayashi K, Takahashi H. Gallyas-positive, tau-negative glial inclusions in Parkinson’s
disease midbrain. Neurosci Lett 1996; 217:133-136.4. Goedert M, Spillantini MG, Jakes R et al. Multiple isoforms of human microtubule-associ-
ated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s dis-ease. Neuron 1989; 3:519-526.
5. Morrison JH, Hof PR. Life and death of neurons in the aging brain. Science 1997;278:413-419.
6. Dickson DW, Crystal HA, Mattiace LA et al. Identification of normal and pathologicalaging in prospectively studied nondemented elderly humans. Neurobiol Aging 1992; 13:1-11.
7. Nishimura M, Tomimoto H, Suenaga T et al. Immunocytochemical characterization ofglial fibrillary tangles in Alzheimer’s disease brain. Am J Pathol 1995; 146:1052-1058.
8. Phillips LL, Autilio-Gambetti L, Lasek RJ. Bodian’s silver method reveals molecular varia-tion in the evolution of neurofilament proteins. Brain Res 1983; 278:219-223.
9. Wisniewski HM, Narang HK, Terry RD. Neurofibrillary tangles of paired helical filaments.J Neurol Sci 1976; 27:173-181.
10. Pollanen MS, Markiewicz P, Goh MC. Paired helical filaments are twisted ribbons com-posed of two parallel and aligned components: Image reconstruction and modeling of fila-ment structure using atomic force microscopy. J Neuropathol Exp Neurol 1997; 56:79-85.
11. Yen S-H, Dickson DW, Peterson C et al. Cytoskeletal abnormalities in neuropathology. In:Zimmerman HM, ed. Progress in Neuropathology, Vol 6. New York: Raven Press,1986:63-90.
12. Ksiezak-Reding H, Morgan K, Mattiace LA et al. Ultrastructure and biochemical composi-tion of paired helical filaments in corticobasal degeneration. Am J Pathol 1994;145:1496-1508.
13. Kato S, Shimoda M, Watanabe Y et al. Familial amyotrophic lateral sclerosis with a twobase pair deletion in superoxide dismutase 1: Gene multisystem degeneration with intracy-toplasmic hyaline inclusions in astrocytes. J Neuropathol Exp Neurol 1996; 55:1089-1101.
14. Greenberg SG, Davies P. A preparation of Alzheimer paired helical filaments that displaysdistinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA 1990;87:5827-5831.

183Astrocytes in Other Neurodegenerative Diseases
15. Lee VM, Balin BJ, Otvos L Jr et al. A68: A major subunit of paired helical filaments andderivatized forms of normal tau. Science 1991; 251:675-678.
16. Grundke-Iqbal I, Iqbal K, Tung YC et al. Abnormal phosphorylation of the microtubule-associated protein tau (τ) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA1986; 83:4913-1917.
17. Yen S-H, Dickson DW, Crowe A et al. Alzheimer neurofibrillary tangles contain uniqueepitopes and epitopes in common with heat-stable microtubule-associated proteins, MAP2and tau. Am J Pathol 1987; 126:63-73.
18. Goedert M, Wischik CM, Crowther RA et al. Cloning and sequencing of the cDNA encod-ing a core protein of the paired helical filament of Alzheimer disease: Identification as themicrotubule-associated protein tau. Proc Natl Acad Sci USA 1988; 85:4051-4055.
19. Goedert M, Spillantini MG, Cairns NJ et al. Tau proteins of Alzheimer paired helical fila-ments: Abnormal phosphorylation of all six brain isoforms. Neuron 1992; 8:159-168.
20. Kosik KS, Orecchio LD, Binder L et al. Epitopes that span the tau molecule are sharedwith paired helical filaments. Neuron 1988; 1:817-825.
21. Iwatsubo T, Hasegawa M, Ihara Y. Neuronal and glial tau-positive inclusions in diverseneurologic diseases share common phosphorylation characteristics. Acta Neuropathol 1994;88:129-136.
22. Spillantini MG, Goedert M, Crowther RA et al. Familial multiple system tauopathy withpresenile dementia: A disease with abundant neuronal and glial tau filaments. Proc NatlAcad Sci USA 1997; 94:4113-4118.
23. Neve RL, Harris P, Kosik KS et al. Identification of cDNA clones for the human microtu-bule-associated protein tau and chromosomal localization of the genes for tau and micro-tubule-associated protein 2. Brain Res 1986; 387:271-280.
24. Goedert M, Crowther RA, Garner CC. Molecular characterization of microtubule-associ-ated proteins tau and MAP2. TINS 1991; 14:193-199.
25. Mandelkow E-W, Schweers O, Drewes G et al. Structure, microtubule interactions andphosphorylation of tau protein. In: Wurtman RJ, Corkin S, Growdon JH, Nitsch RM, eds.The Neurobiology of Alzheimer’s Disease. New York: New York Academy of Science,1996:96-106.
26. Ksiezak-Reding H, Liu WK, Yen SH. Phosphate analysis and dephosphorylation of modi-fied tau associated with paired helical filaments. Brain Res 1992; 597:209-219.
27. Bramblett GT, Trojanowski JQ, Lee VM. Regions with abundant neurofibrillary pathologyin human brain exhibit a selective reduction in levels of binding-competent tau and accu-mulation of abnormal tau-isoforms (A68 proteins). Lab Invest 19992; 66:212-222.
28. Trojanowski JQ, Schmidt ML, Shin R-W et al. PHFτ (A68): From pathological marker topotential mediator of neuronal dysfunction and degeneration in Alzheimer’s disease. ClinNeuroscience 1993; 1:184-191.
29. Hirano A. Neurons and astrocytes. In: Davis RL, Robertson DM, eds. Textbook of Neuro-pathology. Baltimore: Williams & Wilkins, 1997:1-110.
30. Raine CS. Oligodendrocytes and central nervous system myelin. In: Davis RL, RobertsonDM, eds. Textbook of Neuropathology. Baltimore: Williams & Wilkins, 1997:111-164.
31. Zientek GM, Herman MM, Katsetos CD et al. Absence of neuron-associated microtubuleproteins in the rat C-6 glioma cell line. A comparative immunoblot and immunohistochemi-cal study. Neuropathol Appl Neurobiol 1993; 19:346-349.
32. Geisert EE Jr, Johnson HG, Binder LI. Expression of microtubule-associated protein 2 byreactive astrocytes. Proc Natl Acad Sci USA 1990; 87:3967-3971.
33. Lin RC, Matesic DF. Immunohistochemical demonstration of neuron-specific enolase andmicrotubule-associated protein 2 in reactive astrocytes after injury in the adult forebrain.Neuroscience 1994; 60:11-16.
34. Doll T, Meichsner M, Riederer BM et al. An isoform of microtubule-associated protein 2(MAP2) containing four repeats of the tubulin-binding motif. J Cell Sci 1993; 106:633-639.
35. Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian centralnervous system. J Cell Biol 1985; 101:1371-1378.

Astrocytes in Brain Aging and Neurodegeneration184
36. Ksiezak-Reding H, Farooq M, Norton W et al. A distinct subset of tau isoforms is ex-pressed in bovine brain oligodendrocytes. Mol Biol Cell 1997; 8:263a.
37. Muller R, Heinrich M, Heck S et al. Expression of microtubule-associated proteins MAP2and tau in cultured rat brain oligodendrocytes. Cell Tissue Res 1997; 288:239-249.
38. Liu W, Shafit-Zagardo B, Aquino D et al. Cytoskeletal alterations in human fetal astrocytesinduced by IL-1β. J Neurochem 1994; 63:1625-1634.
39. Arias C, Sharma N, Davies P et al. Okadaic acid induces early changes in microtubule-associated protein 2 and tau phosphorylation prior to neurodegeneration in cultured cor-tical neurons. J Neurochem 1993; 61:673-682.
40. Dewar D, Dawson D. Tau protein is altered by focal cerebral ischemia in the rat: An im-munohistochemical and immunoblotting study. Brain Research 1995; 684:70-78.
41. Irving EA, Yatsushiro K, McCulloch J et al. Rapid alteration of tau in oligodendrocytesafter focal ischemic injury in the rat: involvement of free radicals. J Cerebral Blood FlowMet 1997;17:612-622.
42. Feany MB, Ksiezak-Reding H, Liu WK et al. Epitope expression and hyperphosphorylationof tau protein in corticobasal degeneration: Differentiation from progressive supranuclearpalsy. Acta Neuropathol 1995; 90:37-43.
43. Nishimura T, Ikeda K, Akiyama H et al. Glial tau-positive structures lack the sequenceencoded by exon 3 of the tau protein gene. Neurosci Lett 1997; 224:169-172.
44. Wille H, Drewes G, Biernat J et al. Alzheimer-like paired helical filaments and antiparalleldimers formed from microtubule-associated protein tau in vitro. J Cell Biol 1992;118:573-584.
45. Goedert M, Jakes R, Spillantini MG et al. Assembly of microtubule-associated protein tauinto Alzheimer-like filaments induced by sulfated glycosaminoglycans. Nature 1996;383:550-553.
46. Wilson DM, Binder LI. Free fatty acids stimulate the polymerization of tau and amyloidbeta peptides. In vitro evidence for a common effector of pathogenesis in Alzheimer’s dis-ease. Am J Pathol 1997; 150:2181-2195.
47. Iqbal K, Grundke-Iqbal I. Ubiquitination and abnormal phosphorylation of paired helicalfilaments in Alzheimer’s disease. Molec Neurobiol 1991;5:399-410.
48. Smith MA, Taneda S, Richey PL et al. Advanced Maillard reaction end products are asso-ciated with Alzheimer disease pathology. Proc Natl Acad Sci USA 1994; 91:5710-5714.
49. Yan S-D, Chen X, Schmid A-M et al. Glycated tau protein in Alzheimer disease: A mecha-nism for induction of oxidant stress. Proc Natl Acad Sci USA 1994; 91:7787-7791.
50. Ledesma MD, Bonay P, Colaco C et al. Analysis of microtubule-associated protein tauglycation in paired helical filaments. J Biol Chem 1994; 269:21614-21619.
51. Yen S-H, Liu W-K, Hall FL et al. Alzheimer neurofibrillary lesions: Molecular nature andpotential roles of different components. Neurobiol Aging 1995; 16:381-387.
52. Goedert M, Jakes R, Qi Z et al. Protein phosphatase 2A is the major enzyme in brain thatdephosphorylates tau protein phosphorylated by proline-directed protein kinases or cyclicAMP-dependent protein kinase. J Neurochem 1995; 65:2804-2807.
53. Garver TD, Harris KA, Lehman RA et al. Tau phosphorylation in human, primate, and ratbrain: Evidence that a pool of tau is highly phosphorylated in vivo and is rapidly dephos-phorylated in vitro. J Neurochem 19994; 63:2279-2287.
54. Savory J, Huang Y, Herman MM et al. Quantitative image analysis of temporal changes intau and neurofilament proteins during the course of acute experimental neurofibrillarydegeneration; Non-phosphorylated epitopes precede phosphorylation. Brain Res 1996;707:272-281.
55. Goedert M. Tau protein and the neurofibrillary pathology of Alzheimer’s disease. In:Wurtman RJ, Corkin S, Growdon JH, Nitsch RM, eds. The Neurobiology of Alzheimer’sDisease. New York: Ann NY Acad Sci 1996; 777:121-131.
56. Liu W-K, Williams RT, Hall FL et al. Detection of a cdc2-related kinase associated withAlzheimer paired helical filaments. Am J Pathol 1995; 146:228-238.
57. Vincent I, Jicha G, Rosado M et al. Aberrant expression of mitotic cdc2/cyclin B1 kinasein degenerating neurons of Alzheimer’s disease brain. J Neuroscience 1997; 17:3588-3598.

185Astrocytes in Other Neurodegenerative Diseases
58. Vincent I, Rosado M, Davies P. Mitotic mechanisms in Alzheimer’s disease? J Cell Biol1996; 132:413-425.
59. Ross ME. Cell division and the nervous system: Regulating the cycle from neural differen-tiation to death. TINS 1996; 19:62-68.
60. Yen SH, Kenessey A, Lee SC et al. The distribution and biochemical properties of a Cdc2-related kinase, KKIALRE, in normal and Alzheimer brains. J Neurochem 1995; 65:2577-2584.
61. Cairns NJ, Atkinson PF, Hanger DP et al. Tau protein in the glial cytoplasmic inclusionsof multiple system atrophy can be distinguished from abnormal tau in Alzheimer’s disease.Neurosci Lett 1997; 230:49-52.
62. Dickson DW. Neurodegenerative diseases with cytoskeletal pathology: A biochemical clas-sification. Ann Neurol 1997; 42:541-544.
63. Dickson DW, Ksiezak-Reding H, Liu W-K et al. Immunocytochemistry of neurofibrillarytangles (NFT) with antibodies to subregions of tau protein: Identification of hidden andcleaved epitopes and a new phosphorylation site. Acta Neuropathol 1992; 84:596-605.
64. Papasozomenos SC, Binder LI. Phosphorylation determines two distinct species of Tau inthe central nervous system. Cell Motil Cytoskeleton 1987; 8:210-226.
65. Dotti CG, Banker GA, Binder LI. The expression and distribution of the microtubule-asso-ciated proteins tau and microtubule-associated protein 2 in hippocampal neurons in therat in situ and in cell culture. Neuroscience 1987; 23:121-130.
66. Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy: A heterogeneousdegeneration involving the brain stem, basal ganglia and cerebellum, with vertical gazeand pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol 1964; 10:333-339.
67. Litvan I, Agid Y. Progressive Supranuclear Palsy: Clinical and Research Approaches. NewYork: Oxford University Press, 1992.
68. Jellinger K, Riederer P, Tomonaga M. Progressive supranuclear palsy: Clinico-pathologicaland biochemical studies. J Neural Transm 1980; 16(Suppl):111-128.
69. Roy S, Datta CK, Hirano A et al. Electron microscopic study of neurofibrillary tangles inSteele-Richardson-Olszewski syndrome. Acta Neuropathol 1974; 29:175-179.
70. Tellez-Nagel I, Wisniewski HM. Ultrastructure of neurofibrillary tangles in Steele-Richardson-Olszewski syndrome. Arch Neurol 1973; 29:324-327.
71. Yagishita S, Itoh Y, Amano N et al. Ultrastructure of neurofibrillary tangles in progressivesupranuclear palsy. Acta Neuropathol 1979; 48:27-30.
72. Schmidt ML, Lee VM-Y, Hurtig H et al. Properties of antigenic determinants that distin-guish neurofibrillary tangles in progressive supranuclear palsy and Alzheimer’s disease. LabInvest 1988; 59:460-466.
73. Flament S, Delacourte A, Verny M et al. Abnormal tau proteins in progressive supranuclearpalsy. Similarities and differences with the neurofibrillary degeneration of the Alzheimertype. Acta Neuropathol 1991; 81:591-596.
74. Vermersch P, Robitaille Y, Berneir L et al. Biochemical mapping of neurofibrillary degen-eration in a case of progressive supranuclear palsy: Evidence for general cortical involve-ment. Acta Neuropathol 1994; 87:572-577.
75. Nishimura M, Namba Y, Ideda K et al. Glial fibrillary tangles with straight tubules in thebrains of patients with progressive supranuclear palsy. Neurosci Lett 1992; 143:35-38.
76. Yamada T, Calne DB, Akiyama H et al. Further observations on tau-positive glia in thebrains with progressive supranuclear palsy. Acta Neuropathol 1993; 85:308-315.
77. Nishimura T, Ikeda K, Akiyama H et al. Immunohistochemical investigation of tau-posi-tive structures in the cerebral cortex of patients with progressive supranuclear palsy.Neurosci Lett 1995; 201:123-126.
78. Feany MB, Mattiace LA, Dickson DW. Neuropathologic overlap of progressive supranuclearpalsy, Pick’s disease and corticobasal degeneration. J Neuropathol Exp Neurol 1996;55:53-67.
79. Dickson DW, Feany MB, Yen SH et al. Cytoskeletal pathology in non-Alzheimer degenera-tive dementia: New lesions in diffuse Lewy body disease, Pick’s disease, and corticobasaldegeneration. J Neural Transm. Suppl 1996;47:31-46.

Astrocytes in Brain Aging and Neurodegeneration186
80. Li F, Iseki E, Kosaka K et al. Progressive supranuclear palsy with fronto-temporal atrophyand various tau-positive abnormal structures. Clin Neuropathol 1996; 15:319-323.
81. Probst A, Langui D, Lautenschlager C et al. Progressive supranuclear palsy: Extensive neu-ropil threads in addition to neurofibrillary tangles. Acta Neuropathol 1988; 77:61-68.
82. Nelson SJ, Yen S-H, Davies P et al. Basal ganglia neuropil threads in progressive supra-nuclear palsy. J Neuropathol Exp Neurol 1989; 48:324.
83. Arima K, Nakamura M, Sunohara N et al. Ultrastructural characterization of the tau-im-munoreactive tubules in the oligodendroglial perikarya and their inner loop processes inprogressive supranuclear palsy. Acta Neuropathol 1997; 93:558-566.
84. Braak H, Braak E. Argyrophilic grains: Characteristic pathology of cerebral cortex in casesof adult onset dementia without Alzheimer changes. Neurosci Lett 1987; 76:124-127.
85. Tolnay M, Spillantini MG, Goedert M et al. Argyrophilic grain disease: Widespreadhyperphosphorylation of tau protein in limbic neurons. Acta Neuropathol 1997; 93:477-484.
86. Ikeda K, Akiyama H, Kondo H et al. A study of dementia with argyrophilic grains. Pos-sible cytoskeletal abnormality in dendrospinal portion of neurons and oligodendroglia. ActaNeuropathol 1995; 89:409-414.
87. Papp MI, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients withmultiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J Neurol Sci 1989; 94:79-100.
88. Horoupian DS, Dickson DW. Striatonigral degeneration, olivoponto-cerebellar atrophy and“atypical” Pick disease. Acta Neuropathol 1991; 81:287-295.
89. Kato S, Nakamura H. Cytoplasmic argyrophilic inclusions in neurons of pontine nuclei inpatients with olivopontocerebellar atrophy: Immunohistochemical and ultrastructural stud-ies. Acta Neuropathol 1990; 79:584-594.
90. Tamaoka A, Mizusawa H, Mori H et al. Ubiquitinated alpha B-crystallin in glial cytoplas-mic inclusions from the brain of a patient with multiple system atrophy. J Neurol Sci1995; 129:192-198.
91. Ikeda K, Akiyama H, Kondo H et al. Thorn-shaped astrocytes: Possibly secondarily in-duced tau-positive glial fibrillary tangles. Acta Neuropathol 1995; 90:620-625.
92. Yamazaki M, Nakano I, Imazu O et al. Astrocytic straight tubules in the brain of a patientwith Pick’s disease. Acta Neuropathol 1994; 88:587-591.
93. Oyanagi K, Makifuchi T, Ohtoh T et al. Distinct pathological features of the Gallyas- andtau-positive glia in the Parkinsonism-dementia complex and amyotrophic lateral sclerosisof Guam. J Neuropathol Exp Neurol 1997; 56:308-316.
94. Ikeda K, Akiyama H, Kondo H et al. Anti-tau-positive glial fibrillary tangles in the brainof postencephalitic Parkinsonism of Economo type. Neurosci Lett 1993; 162:176-178.
95. Ikeda K, Akiyama H, Kondo H et al. Numerous glial fibrillary tangles in oligodendrogliain cases of subacute sclerosing panencephalitis with neurofibrillary tangles. Neurosci Lett1995; 194:133-135.
96. Constantinidis J. Pick dementia: Anatomoclinical correlations and pathophysiological con-siderations. In: Rose FC, ed. Modern Approaches to the Dementias, Part I: Etiology andPathophysiology. Basel: Karger, 1985:72-97.
97. Alzheimer A. Über eigenartige Krankheitsfälle des späteren Alters. Z ges Neurol Psychiat1911; 4:356-385.
98. Love S, Saitoh T, Quijada S et al. Alz-50, ubiquitin and tau immunoreactivity of neu-rofibrillary tangles, Pick bodies and Lewy bodies. J Neuropathol Exp Neurol 1988;47:393-405.
99. Delacourte A, Robitaille Y, Sergeant N et al. Specific pathological tau protein variants char-acterize Pick’s disease. J Neuropathol Exp Neurol 1996; 55:159-168.
100. Murayama S, Mori H, Ihara Y et al. Immunocytochemical and ultrastructural studies ofPick’s disease. Ann Neurol 1990; 27:394-405.
101. Wisniewski HM, Coblentz JM, Terry RD. Pick’s disease: A clinical and ultrastructural study.Arch Neurol 1972; 26:97-108.
102. Takauchi S, Hosomi M, Marasigan S et al. An ultrastructural study of Pick bodies. ActaNeuropathol 1984; 64:344-348.

187Astrocytes in Other Neurodegenerative Diseases
103. Scicutella A, Davies P. Marked loss of cerebral galactolipids in Pick’s disease. Ann Neurol1987; 22:606-609.
104. Yamada T, McGeer PL. Oligodendroglial microtubular masses: An abnormality observedin some human neurodegenerative diseases. Neurosci Lett 1990; 120:163-166.
105. Feany MB, Dickson DW. Neurodegenerative disorders with extensive tau pathology: A com-parative study and review. Ann Neurol 1996; 40:139-148.
106. Rebeiz JJ, Kolodny EH, Richardson EP. Corticodentatonigral degeneration with neuronalachromasia. Arch Neurol 1968; 18:20-33.
107. Gibb WRG, Luthert PJ, Marsden CD. Corticobasal degeneration. Brain 1989; 112:1171-1192.108. Ikeda K, Akiyama H, Iritani S, Corticobasal degeneration with primary progressive aphasia
and accentuated cortical lesion in superior temporal gyrus: Case report and review. ActaNeuropathol 1996; 92:534-539.
109. Kertesz A, Hudson L, Mackenzie IR et al. The pathology and nosology of primary progres-sive aphasia. Neurology 1994; 44:2065-2072.
110. Clark AW, Manz HJ, White CL III et al. Cortical degeneration with swollen chromatolyticneurons: Its relationship to Pick’s disease. J Neuropathol Exp Neurol 1986; 45:268-284.
111. Dickson DW, Yen S-H, Suzuki KI et al. Ballooned neurons in select neurodegenerativediseases contain phosphorylated neurofilament epitopes. Acta Neuropathol 1986; 71:216-223.
112. Horoupian DS, Chu PL. Unusual case of corticobasal degeneration with tau/Gallyas-posi-tive neuronal and glial tangles. Acta Neuropathol 1994; 88:592-598.
113. Mori H, Nishimura M, Namba Y et al. Corticobasal degeneration: A disease with wide-spread appearance of abnormal tau and neurofibrillary tangles, and its relation to progres-sive supranuclear palsy. Acta Neuropathol 1994; 88:113-121.
114. Wakabayashi K, Oyanagi K, Makifuchi T et al. Corticobasal degeneration: Etiopathologicalsignificance of the cytoskeletal alterations. Acta Neuropathol 1994; 87:545-553.
115. Feany MB, Dickson DW. Widespread cytoskeletal pathology characterizes corticobasal de-generation. Am J Pathol 1995; 146:1388-1396.
116. Takahashi T, Amano N, Hanihara T et al. Corticobasal degeneration: Widespread argento-philic threads and glia in addition to neurofibrillary tangles. Similarities of cytoskeletalabnormalities in corticobasal degeneration and progressive supranuclear palsy. J NeurolSci 1996; 138:66-77.
117. Bergeron C, Pollanen MS, Weyer L et al. Cortical degeneration in progressive supranuclearpalsy. A comparison with cortical-basal ganglionic degeneration. J Neuropathol Exp Neurol1997; 56:726-734.
118. Buee Scherrer V, Hof PR, Buee L et al. Hyperphosphorylated tau proteins differentiatecorticobasal degeneration and Pick’s disease. Acta Neuropathol 1996; 91:351-359.
119. Masliah E, Hansen LA, Quijada S et al. Late onset dementia with argyrophilic grains andsubcortical tangles or atypical progressive supranuclear palsy? Ann Neurol 1992; 29:389-396.
120. Mann DMA, South PW, Snowden JS et al. Dementia of frontal lobe type: Neuropathologyand immunohistochemistry. J Neurol Neurosurg Psychiatry 1993; 56:605-614.
121. Knopman DS, Mastri AR, Frey F et al. Dementia lacking distinctive histologic features: Acommon non-Alzheimer degenerative dementia. Neurology 1990; 40:251-256.
122. Foster NL, Wilhelmsen K, Sima AAF et al. Frontotemporal dementia and Parkinsonismlinked to chromosome 17: A consensus conference. Ann Neurology 1997; 41:706-715.
123. Sima AA, Defendini R, Keohane C et al. The neuropathology of chromosome 17-linkeddementia. Ann Neurol 1996; 39:734-743.
124. Conrad C, Andreadis A, Trojanowski JQ et al. Genetic evidence for the involvement of τin progressive supranuclear palsy. Ann Neurol 1997; 41:277-281.
125. Lazzarini AM, Golbe LI, Dickson DW et al. Tau intronic polymorphism in Parkinson’sdisease and progressive supranuclear palsy. Neurology 1997; 48:A427.
126. Wenning GK, Tison F, Ben Shlomo Y et al. Multiple system atrophy: A review of 203pathologically proven cases. Movement Dis 1997; 12:133-147.
127. Harding AE. Inherited ataxias. Curr Opinion Neurol 1995; 8:306-309.128. Paulson HL, Fischbeck KH. Trinucleotide repeats in neurogenetic disorders. Ann Rev
Neurosci 1996; 19:79-107.

Astrocytes in Brain Aging and Neurodegeneration188
129. Gilman S, Sima AA, Junck L et al. Spinocerebellar ataxia type 1 with multiple system de-generation and glial cytoplasmic inclusions. Ann Neurol 1996; 39:241-255.
130. DiFiglia M, Sapp E, Chase KO et al. Aggregation of huntingtin in neuronal intranuclearinclusions and dystrophic neurons in brain. Science 1997; 277:1990-1993.
131. Weidenheim KM, Dickson DW. Intranuclear inclusion bodies in an elderly dementedwoman: A form of intranuclear inclusion body disease. Clin Neuropathol 1995; 14:93-99.
132. Bradley WG Overview of motor neuron disease: Classification and nomenclature. ClinNeurosci 1995; 3:323-326.
133. Hirano A. Neuropathology of ALS: An overview. Neurology 1996; 47(Suppl 2):S63-66.134. Mizusawa H, Matsumoto S, Yen SH et al. Focal accumulation of phosphorylated
neurofilaments within anterior horn cell in familial amyotrophic lateral sclerosis. ActaNeuropathol 1989; 79:37-43.
135. Murayama S, Ookawa Y, Mori H et al. Immunocytochemical and ultrastructural study ofLewy body-like hyaline inclusions in familial amyotrophic lateral sclerosis. Acta Neuropathol1989; 78:143-152.
136. Lowe J, Aldridge F, Lennox G et al. Inclusion bodies in motor cortex and brainstem ofpatients with motor neurone disease are detected by immunocytochemical localization ofubiquitin. Neurosci Lett 1989; 105:7-13.
137. Kato S, Hayashi H, Nakashima K et al. Pathological characterization of astrocytic hyalineinclusions in familial amyotrophic lateral sclerosis. Am J Pathol 1997; 151:611-620.
138. Rosen DR, Siddique T, Patterson D et al. Mutations in Cu/Zn superoxide dismutase geneare associated with familial amyotrophic lateral sclerosis. Nature 1993; 362:59–62.
139. Bruijn LI, Becher MW, Lee MK et al. ALS-linked SOD1 mutant G85R mediates damage toastrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neu-ron 1997; 18:327-338.

Part III
Experimental Models of AstrocyteSenescence: Implications forNeurodegenerative Disease


CHAPTER 9
Astrocytes in Brain Aging and Neurodegeneration, edited by Hyman M. Schipper.©1998 R.G. Landes Company.
The Peroxidase-PositiveSubcortical Glial SystemMark B. Mydlarski, James R. Brawer and Hyman M. Schipper
Introduction
A subpopulation of astrocytes bearing unique cytoplasmic inclusions which progressivelyaccumulate with advancing age has been described in limbic and periventricular brain
regions of all vertebrates thus far examined, including frogs,1 rats,2 mice,3,4 dogs,5 cats,5
monkeys6 and humans.7-10 These cells were initially identified as astrocytes by electron mi-croscopy on the basis of their attenuated cytoplasm, ellipsoidal, euchromatic nuclei andbundles of intermediate filaments.3,11-13 The cytoplasmic granules that distinguish thesecells are round to angular in shape, of varying dimensions and intensely osmiophilic (Fig. 9.1).The inclusions are often invested with limiting membranes and occasionally appear con-tiguous with short, tubular elements filled with material of similar electron density.3,12,14 Atthe light microscopic level, this glial subpopulation is shown to advantage by dual labelingfor endogenous peroxidase activity (see below) and the astrocyte marker, GFAP.
Tinctorial and Histochemical FeaturesThese cells are commonly referred to as Gomori-positive or peroxidase-positive astro-
cytes on the basis of their tinctorial and histochemical characteristics. The cytoplasmic in-clusions exhibit metachromasia in toluidine blue-stained sections6 and have an affinity forthe Gomori stains, aldehyde fuchsin and chrome alum hematoxylin.15 Gomori stains wereoriginally used to identify pancreatic β cells, and a high sulfur content of β cells (proinsulindisulfide bonds) was thought to account for their Gomoriphilia.16 Histochemical and mi-croprobe analyses have confirmed that the Gomori-positive astrocyte granules are indeedrich in sulfhydryl groups.17,18 However, the Gomori stains are relatively nonspecific and,under certain circumstances, will complex with sulfuric acid esters and with sulfonic, alde-hyde, carboxyl and phosphate groups in neuronal and other nonastrocytic substrates.2,6,19-21
Thus, in addition to the glial granules, aldehyde fuchsin stains oxidized neuronal lipofuscin,neuromelanin,19 neuronal dense bodies, neurosecretory material of the hypothalamo-hy-pophyseal system6 and corpora amylacea.19 On the basis of their tinctorial properties andpropensity to accumulate with aging (see below), the Gomoriphilic astrocyte granules wereinitially regarded as a form of the senescent pigment, lipofuscin,22 or as phagocytosed neu-rosecretion.21,23 However, both views were challenged in the face of studies demonstratingthat:
1. few Gomori-positive astrocyte granules are present in the supraoptic nucleus, whereneurons replete with sulfur-rich neurosecretory material (neurophysins) abound;24

Astrocytes in Brain Aging and Neurodegeneration192
2. in unstained sections viewed under light microscopy, the astrocytic inclusions re-veal no visible pigment characteristic of lipofuscin;25
3. under transmission EM, lipofuscin exhibits heterogeneous electron-lucent and denseregions in osmicated preparations,26 whereas osmicated Gomori-positive astrocytegranules tend to be uniformly electron-dense;3,12
4. unlike lipofuscin, the glial granules are not labeled with the conventional lipid mark-ers Sudan Black, Sudan III25 or oil red O;27 and
5. the glial inclusions emit an orange-red autofluorescence (610-640 nm) consistentwith the presence of porphyrins24,28 or oxidized flavoproteins29,30 and distinct fromthe green or yellow-orange autofluorescence (400-545 nm) typically emitted bylipofuscins in situ.28,31
Perhaps most importantly, the Gomori-positive gliosomes are rich in iron,32-34 maycontain other transition metals such as copper14 and chromium14 and express the metal-binding protein metallothionein.35 The gliosomes stain intensely with diaminobenzidine(DAB), a marker of endogenous peroxidase activity.36-38 In these cells, DAB staining persistsafter tissue preheating, at extremes of pH and in the presence of the catalase inhibitoraminotriazole.9,39 The peroxidase activity is therefore nonenzymatic in nature(pseudoperoxidase) and is most likely mediated by ferrous iron or other redox-active tran-sition metals.40
Topography of the Peroxidase-Positive AstrogliaPeroxidase-positive astrocytes are relatively abundant in the subependymal zone
throughout the neuraxis and in blood-brain barrier-deficient regions including all of thecircumventricular organs.6,17,25,36 In the rat telencephalon, relatively high concentrations ofthese cells are found in the olfactory bulb, the caudate nucleus adjacent to the lateral ven-tricle, the putamen-globus pallidus, and the hippocampus.36,41 A highly stratified distribu-tion of these cells was delineated within the dorsal hippocampus of adult rats in a studyemploying dual histochemical/immunohistochemical labeling to identify peroxidase (DAB)-positive cytoplasmic granules within GFAP-positive astrocytes. In this region, numerousDAB-positive astrocytes are confined to the hilus of the dentate gyrus and the lacunosummolecular layer and stratum oriens of subsectors CA1-3. Other hippocampal layers exhibitGFAP-positive astrocytes with little or no detectable DAB reaction product, such as thegranule cell and inner molecular layers of the dentate gyrus, and the pyramidal cell layerand stratum radiatum of CA1-3.41 In the diencephalon, peroxidase-positive astroglia areprominent in the arcuate nucleus and ventral premammillary area of the basal hypothala-mus, in the third ventricular subependymal zone and in the organum vasculosum of thelamina terminalis. In the mesencephalon, these cells are frequently observed in theperiaqueductal gray, dorsal to the raphe nuclear complex and in the superficial aspect of thesuperior colliculi. Although few DAB-positive astrocytes are seen in the substantia nigra of3 month old rats, this area becomes heavily populated with these cells by 15 months ofage.41a In the rhombencephalon, peroxidase-positive astrocytes are most numerous in thearea postrema and occur, to a lesser extent, in the nucleus gracilis, dorsal motor nucleus ofthe vagus, locus coeruleus, olivary nuclear complex and lateral cerebellum. In the spinalcord, small numbers of these cells have been identified in Rexed’s laminae 1 and 2 of thedorsal horn.36 Further details concerning the topography of these cells within the rat neuraxisare presented in Keefer and Christ,36 Schipper8 and Schipper and Mateescu-Cantuniari.41
In serial sections derived from adult human autopsy material, topographicallysuperimposable chrome alum hematoxylin-positive and DAB-positive astrocytes were foundthroughout the periventricular forebrain, in the optic tract and globus pallidus and withinthe diencephalon.7,8 In the latter, these cells appear to be concentrated in the organum

193The Peroxidase-Positive Subcortical Glial System
vasculosum of the lamina terminalis, infundibular region and capsule of the mammillarybody. As in other vertebrates, DAB staining in the human astrocytes occurred over a widerange of pH and was resistant to tissue preheating and aminotriazole, indicating that stain-ing was due to a nonenzymatic (likely iron-mediated) pseudoperoxidase reaction.
Modulation of the Peroxidase-Positive Glial System
AgingGomori-positive astroglia first appear at the end of the first week in the postnatal rab-
bit hypothalamus42 and in the fourth week in rats and mice.15,43 In the hypothalamus of ratsand mice, numbers of peroxidase-positive astrocytes and their granule content progres-sively increase between 6 and 14 months of age.44 In female rodents, early ovariectomy sig-nificantly attenuates the senescence-dependent proliferation of these gliosomes in the hy-pothalamic arcuate nucleus (Fig 9.2), indicating that, in this neuroendocrine locus, theaccumulation of peroxidase-positive astrocytic inclusions is influenced by exposure to cir-culating sex hormones (see below).44 Relative to other subcortical brain regions, few peroxi-dase-positive glial granules are present in the rat substantia nigra at 3 months of age. By 15months, however, many nigral astroglia are replete with large, DAB-positive inclusions whichincrease in abundance by a factor of four in comparison with 3 month old animals.41a Inhumans, numbers of peroxidase-positive astrocyte granules increase in the basal gangliaand throughout the periventricular forebrain between the ages of 3 and 69 years.7 Despitetheir consistent increase with aging, the results of various histochemical and morphologicalstudies (described above) indicate that the peroxidase-positive granules are a unique formof glial inclusion constitutively different from the aging pigment lipofuscin.
Fig. 9.1. (A) Ultrastructure of Gomori-positive astrocyte in the hypothalamic arcuate nucleus ofa normal adult female rat. Osmiophilic, Gomori-positive gliosomes (g) and a bundle of interme-diate filaments (arrows) are depicted. x17,220 magnification. (B) Gomori-positive astrocyte inthe hypothalamic arcuate nucleus of an adult female rat rendered anovulatory with estradiolvalerate. This treatment induces a marked accumulation of Gomori-positive cytoplasmic inclu-sions (g) in close proximity to degenerating dendritic profiles (arrow near bottom). N, astrocytenucleus; F, intermediate filaments: x17,220. Reprinted with permission from Brawer JR et al,Endocrinol 1978; 103:501-512.

Astrocytes in Brain Aging and Neurodegeneration194
Sex Hormones and Reproductive SenescenceIn adult female rodents, the persistent estrus (PE) state, an anovulatory syndrome char-
acterized by the development of polycystic ovaries and persistent vaginal cornification, spon-taneously develops as a function of advancing age.44,45 In young postpubertal female rats,ablation of the preoptic-suprachiasmatic region of the diencephalon or transection of itsprojections(s) into the medial basal hypothalamus abolishes sexual cyclicity and elicits thePE state.46 The PE state also arises in adult female rats:
1. exposed to continuous illumination;47
2. treated neonatally with systemic or intrahypothalamic testosterone;48,49 and3. following the intramuscular injection of estradiol valerate.11,50
Concomitant with the induction of premature reproductive failure, constant light treat-ment50 and multiple12 or single11,51 intramuscular injections of 2 mg EV greatly acceleratethe aging-related accumulation of peroxidase-positive astrocyte granules in the hypotha-lamic arcuate nucleus, a neuroendocrine locus rich in estrogen receptors.12,13,50,51 Further-more, EV treatment promotes the collapse of dendritic profiles, degeneration of axon ter-minals, synaptic loss and remodeling, depletion of neuronal b-endorphin, formation ofmyelin figures and the accumulation of phagocytic microglial cells within the arcuatenucleus.11,12,50,52-55 The degenerating neuronal processes often occur in close proximity tohypertrophic astrocytes exhibiting a massive proliferation of electron-dense (metal-rich)cytoplasmic inclusions (Fig. 9.1). Using numbers of reactive microglia and astrocytic gran-ules as quantitative indices of steroid-related neural damage, we demonstrated that the EVand light-induced pathological changes in the arcuate nucleus are completely abrogated byprior ovariectomy.50 Thus, an ovarian product, and not the EV or constant light treatmentper se, is responsible for the progressive development of degenerative changes in the arcuatenuclei of PE rats.50 Repeated monthly injections of EV in male rats50 and tonic, high-physi-ologic levels of unconjugated estradiol maintained by Silastic implants in gonadectomized
Fig. 9.2. Effects of aging and long-term gonadectomy on numbers of Gomori-positiveastrocytic granules in the arcuate nucleus of male and female rats. (A) Females: Numbersof astrocytic granules increase significantly with advancing age (fine crosshatching). Earlyovariectomy markedly attenuates this aging effect (heavy crosshatching). (B) Male rats:The age-related increase in numbers of astrocytic granules in male rats is less robust thanin females and early castration in the former does not significantly suppress this agingphenomenon. Reprinted with permission from Schipper HM et al, Biol Reprod 1981;25:413-419.

195The Peroxidase-Positive Subcortical Glial System
females56 produce identical histopathological changes in the arcuate nucleus. In contradis-tinction to estrogens, the administration of androgens56 or progestins57 fails to elicit similarglial reactions in this brain region (Fig. 9.3). In normal aging female rats and mice, theprogressive accumulation of peroxidase-positive astrocyte granules within the arcuate nucleuscan be blocked by early gonadectomy.44 Taken together, these observations indicate thataging of the neuroendocrine hypothalamus may be hastened by abnormal patterns of cir-culating ovarian estradiol resulting from EV or constant light exposure.44,58 Conceivably,estrogen-induced neurodegeneration within the arcuate nucleus compromises the integrityof gonadotropin-regulating neural circuitry in this brain region with ensuing anovulatorysterility.59 Our histomorphological observations are consistent with earlier physiologicalstudies demonstrating that E2 withdrawal by early ovariectomy enables female rats to cycle(young or old) ovarian grafts at very advanced ages relative to sham-operated littermates.45
Along similar lines, corticosterone administration enhances, whereas adrenalectomy attenu-ates, senescence-dependent gliosis in glucocorticoid receptor-rich regions of the rodent hip-pocampus.60,61 Thus, during aging, several classes of steroid hormones may render dys-functional the neural circuitry subserving their own regulation.
Metal-mediated peroxidase reactions within arcuate astroglia may play a pivotal role inthe development of estrogen-related hypothalamic injury during aging. In various estrogentarget tissues, the oxidative metabolism of estrogen incurs the formation of cytotoxicsemiquinones and other free radical intermediates.62-65 The mammalian hypothalamus con-tains estrogen 2/4-hydroxylase and peroxidases which promote the conversion of estradiolto 2- or 4-hydroxyestradiol (catecholestrogen).66-68 Highly reactive semiquinone radicalsare formed when catecholestrogens are oxidized further in peroxidase/H2O2-catalyzed re-actions. Catecholestrogens may also undergo spontaneous autoxidation, resulting in thegeneration of semiquinone radicals and reactive oxygen species including H2O2 and super-oxide anion.63,64 In peripheral sex steroid target tissues, estrogen-derived semiquinones andreactive oxygen species have been shown to facilitate membrane lipid peroxidation and DNAdamage, which may in part account for the teratogenic and carcinogenic effects of estrogenin these tissues.62,65,69,70 In an analogous fashion, glial peroxidase activity in the hypotha-lamic arcuate nucleus may promote the bioactivation of estrogens and catecholestrogens to
Fig. 9.3. Effects of sex hor-mones on numbers ofGomori-positive astrocyticgranules in the arcuatenucleus of castrated femalerats. All animals receivedSilastic implants designedto release high-physiologiclevels of steroid hormonesor vehicle only (control)for 3 months. (a) control,(b) estradiol-17β, (c) test-osterone, (d) dihydrotes-tosterone, (e) estradiol plustestosterone, (f) estradiolplus dihydrotestosterone.Estradiol induces a massive accumulation of Gomori-positive astrocytic granules.Dihydrotestosterone, and to a lesser extent testosterone, suppress this estradiol effect. Reprintedwith permission from Brawer JR et al, Endocrinol 1983; 112:194-199.

Astrocytes in Brain Aging and Neurodegeneration196
cytotoxic semiquinone radicals and reactive oxygen species. The latter, in turn, may be di-rectly responsible for the axodendritic pathology and loss of β-endorphin observed in thearcuate nucleus of PE rats. In support of this hypothesis, we demonstrated that dietarysupplementation with potent antioxidants such as α-tocopherol55 or the 21-aminosteroidU7438971 blocks the depletion of hypothalamic β-endorphin in EV-treated rats. Furtherevidence implicating peroxidase-positive astroglia in oxidative neural injury is derived fromstudies on the metabolism of catecholestrogens and catecholamines by these cells in pri-mary brain cell cultures (see chapter 11).
X-Irradiation and TraumaIncreased numbers of Gomori-positive glia have been documented in the rat arcuate
nucleus and third ventricular subependymal zone following exposure to cranial X-irradia-tion.38 The author of that study suggested that the accumulation of peroxidase-positivegranules in glia inhabiting periventricular brain regions may contribute to the protection ofblood-brain barrier-deficient regions by degrading cytotoxic, blood-borne substances.38
Recent studies have indicated, however, that the metal-rich glial inclusions, rather than con-ferring protection to the surrounding neuropil, may promote the generation of neurotoxicfree radical intermediates (see chapter 11). In 1990, Noble and coworkers72 reported promi-nent augmentation of GFAP staining and endogenous glial peroxidase activity in rat spinalcord at two weeks following contusion injury. They conjectured that heme-derived com-pounds ingested from the extracellular space may be the source of endogenous peroxidaseactivity in the cells. As described below and in chapter 11, nonheme iron sequestered within“stressed” astroglial mitochondria is responsible for nonenzymatic DAB oxidation in thesecells.
Peroxidase-Positive Astrocytes in Primary Culture
In Vitro “Aging”In the early 1970s, Srebro and Macinska reported that Gomori-positive cells with tinc-
torial and fluorescent attributes akin to those of periventricular astrocytes in situ are presentin cultures of rodent embryonic and human fetal brain tissue.10,34 Gomori-positive glia inhamster and mouse diencephalic explants were first observed on day 14 in vitro and pro-gressively accumulated over the ensuing 2-3 weeks.34 In periventricular brain explants de-rived from a 6 week old human embryo, Gomori-positive glia appeared after 5 weeks inculture and increased in number thereafter.10 Over the last ten years we have been investi-gating the structural, histochemical and functional properties of peroxidase-positive astro-cytes in dissociated embryonic and neonatal rat brain primary cell cultures.27,41 As in the rathypothalamus,51 peroxidase-positive inclusions were localized to GFAP-positive astrocytesin culture by combining DAB histochemistry with anti-GFAP immunohistochemistry. Thenumbers of peroxidase-positive astrocytes and their granule content progressively increasebetween days 10 and 46 in vitro, consistent with earlier observations in diencephalic ex-plants and in the intact rodent hypothalamus. In contrast with 10 day old cultures whereDAB-positive astrocytes represented fewer than 1% of all cells, older cultures exhibited nu-merous peroxidase-positive granules deposited within the cytoplasm of isolated flat andstellate astroglia and in astrocytes forming confluent monolayers. In unstained prepara-tions, the astrocytic inclusions are invisible and phase-dark under light and phase-contrastmicroscopy, respectively. As in situ, the astrocytic inclusions are Gomoriphilic, emit an or-ange-red autofluorescence, and exhibit nonenzymatic peroxidase activity resistant toaminotriazole, tissue preheating or broad pH modification.8,27,73

197The Peroxidase-Positive Subcortical Glial System
The Cysteamine ModelWe demonstrated that exposure to the sulfhydryl agent, 2-mercaptoethylamine or cys-
teamine (CSH; 880 µM in culture medium administered twice weekly from in vitro days6-18) induces a marked accumulation of peroxidase-positive astrocytes in primary culture.Cystamine, the oxidized disulfide of CSH, generated a similar glial response at relatively lowdoses (8.8-88 µM) but was pancytotoxic at 880 µM concentrations. Equimolar concentra-tions of L-cysteine or ethanolamine, which differ from CSH by single functional groupmodifications, did not stimulate the accumulation of Gomori-positive cytoplasmic inclu-sions in cultured astroglia.27 The CSH-treated astrocytes exhibit orange-red autofluorescentgranules and nonenzymatic peroxidase activity indistinguishable from that of Gomori-posi-tive astrocytes in unstimulated, older cultures and in senescent subcortical brain regions insitu (Fig. 9.4).25,27,28 At the ultrastructural level, 18 day old CSH-treated astrocytes containnumerous membrane-bound cytoplasmic inclusions which are variable in size and roundor ovoid in shape. The inclusions consistently exhibit an intensely osmiophilic matrix iden-tical to that observed in senescent subcortical astroglia in situ.11-33 Occasionally, concentricstacks of membrane reminiscent of myelin-like figures or fingerprint bodies are seen at theperiphery of larger inclusions (Fig. 9.5). As noted in situ, clusters of the dense inclusionsinfrequently appear contiguous with cisternal elements filled with a similar electron-opaquesubstance.3,11,12,33 In nonosmicated material, the DAB reaction product is visualized as amoderately dense, granular precipitate deposited within many, but not all, of the inclusions.Within strongly-labeled inclusions, the DAB reaction product is either dispersed homoge-neously throughout the granule matrix or is restricted to discrete intraorganellar compart-ments. Elemental iron is detected in the inclusions by energy dispersive X-ray microanaly-sis, and the presence and concentration of the metal correlates closely with the presence andintensity of DAB staining (Fig. 9.6).33 These astrocyte granules exhibit little or no affinityfor Prussian blue, a marker of ferric and hemosiderin iron, arguing that ferrous iron isresponsible for the nonenzymatic peroxidase activity in these cells.27
Subcellular Precursors of Peroxidase-Positive Astroglial InclusionsFine structural, cytochemical and X-ray microprobe analyses of cultured neonatal rat
astroglia were performed at various time points following CSH exposure in an effort todelineate the subcellular precursors of the peroxidase-positive, cytoplasmic inclusions.74 InCSH-treated astroglia, the earliest morphological changes appear restricted to the mito-chondrial compartment. Within six hours of treatment, many (but not all) of the astrocytescontained mitochondria with irregular swollen cristae which often assumed tubular or sac-cular morphologies. By 24-72 hours, numerous mitochondrial profiles were characterizedby double membranes completely devoid of organized cristae encompassing homogeneous,dense matrices (Fig. 9.7). In some cases, multiple concentric stacks of membrane surroundedthe mitochondrial matrix for variable distances along its perimeter. Occasional profiles ex-hibited narrow, tail-like extensions of the double membrane which tended to terminate assmall bulbs. Typically, the acristic mitochondrial profiles formed large clusters intermixedwith normal-appearing mitochondria.
The alterations in mitochondrial morphology at the different CSH exposure intervalswere paralleled by changes in elemental composition. Within 6 hours of CSH exposure,many mitochondria with distorted or absent cristae displayed X-ray emission peaks forchromium. These mitochondrial forms contained no detectable iron and were consistentlyDAB-negative. Within 24-72 hours many acristic mitochondria probed positively for bothchromium and iron and exhibited variable DAB reaction product (visualized innonosmicated preparations). The intensity of DAB staining correlated with the size of theiron peaks further indicating that (ferrous) iron is the likely source of nonenzymatic per-

Astrocytes in Brain Aging and Neurodegeneration198
Fig. 9.4. Embryonic day 17 rat brain cell cultures (18 days in vitro). (A) Untreated control. DABstain for endogenous peroxidase activity. Astrocytes devoid of DAB-positive granules are ob-served. Methyl green counterstain. x292. (B) Effects of cysteamine (880 µM twice weekly in me-dium from day 6). DAB stain. Astrocytes exhibit a massive accumulation of cytoplasmic peroxi-dase-positive inclusions. Methyl green counterstain. x292. (C) Untreated embryonic day 17 ratbrain cell culture photographed for autofluorescence. Astrocytes exhibit faint or no orange-redautofluorescence. x292. (D) Cysteamine-treated brain cell culture. Cysteamine-induced astro-cyte granules and cytoplasm emit intense orange-red autofluorescence. x292. Reprinted withpermission from Schipper HM et al, Dev Brain Res 1990; 54:71-79.
Fig. 9.5.(A) Transmission electron microscopy of astrocytes from control culture (unexposed tocysteamine). Segments of four cells are visible. Polysomes, short cisternae of rough endoplasmicreticulum and bundles of intermediate filaments are scattered throughout the cytoplasm. A smallGolgi apparatus (arrowheads) and a dense inclusion body (arrow) are depicted. Bar = 0.83 µm.(B) Astrocyte from cysteamine-treated culture. The cytoplasm is replete with large osmiophilicinclusions. Many of the gliosomes exhibit concentric stacks of membrane along segments oftheir periphery (arrows).Bar = 0.45 µm. Reprinted with permission from McLaren J et al, JHistochem Cytochem 1992; 40:1887-1897.

199The Peroxidase-Positive Subcortical Glial System
Fig. 9.6.(a) Peroxidase activ-ity in astrocyte from cys-teamine-treated culture. In-clusions within this nonos-micated cell exhibit varyingdegrees of peroxidase activ-ity indicated by the granu-lar DAB reaction product.Inclusions D and E showlittle DAB reaction product,whereas inclusions C and Fare strongly positive. TheDAB precipitate in the lat-ter appears localized to spe-cific intraorganellar com-partments. Bar = 0.4 µm.(b) X-ray emission spectraderived from cell depicted in(a). Emission peaks indicat-ing the various elementalconstituents are labeled. Theconcentration of a given el-ement is proportional to thearea under the peak(s) forthat element. The large peakat the right of each histo-gram indicates copper re-sulting from the use of cop-per grids. (A) This emissionspectrum was generated bya region of clear cytoplasm.Note the absence of a peakfor iron. (B) This spectrumwas generated by a euchro-matic region of nucleus.Note the absence of a peakfor iron. (C) This spectrumwas generated by inclusionC in (a). The two iron peaks(arrows) are indicative of ahigh iron concentrationwithin this inclusion. (D)Spectrum for inclusion D in(a). There is only a single small iron peak (arrow), indicative of a relatively low concentration ofiron. (E) Spectrum for inclusion E in (a) The single small iron peak (arrow) indicates a low ironconcentration. (F) Spectrum for inclusion F in (a). The twin peaks (arrows) indicate a high con-centration of iron. Reprinted with permission from McLaren J et al, J Histochem Cytochem1992; 40:1887-1897.

Astrocytes in Brain Aging and Neurodegeneration200
oxidase activity in these cells. X-ray microanalysis of peroxidase-positive astroglial granulesin several subcortical brain regions of adult rats have revealed the presence of elementalcopper rather than iron and chromium.14,14a Copper, a transition metal also capable of pro-moting pseudoperoxidase reactions,74 was not detected in control and CSH-treated astroglialcultures.75 These observations suggest that various redox-active metals may be sequesteredin senescent astroglial mitochondria, and that the relative abundance of a particular metalin these organelles reflects, at least in part, its bioavailability within specific brain regionsand neural cell cultures. In support of this notion, rat astroglial mitochondria have beenshown to accumulate exogenous iron,76 chromium,77 lead78 and manganese79 following ad-ministration of these metals to the culture media.
In young adult rats, subcutaneous CSH injections (150-300 mg/kg twice weekly for3 weeks) elicit striking astrocyte hypertrophy (gliosis) and 2- to 3-fold increases in numbersof peroxidase-positive astrocyte granules in hippocampus, striatum, and other subcorticalbrain regions related to vehicle-injected controls (Fig. 9.8).80 As in the case of CSH-treatedcultures, peroxidase-positive glial granules in the intact rat and human brain invariablyexhibit mitochondrial epitopes in immunohistochemical preparations.9,14 In rat brain, Youngand co-workers recently demonstrated that immunoreactive acyl-CoA binding protein81
and brain fatty acid binding protein (FABP)82 are most prominent in brain regions repletewith peroxidase-positive astrocytes, and that these proteins colocalize significantly to thisglial population. In rat hepatocytes, chemically-mediated inhibition of mitochondrial β-fattyacid oxidation results in enhanced expression of these lipid-binding proteins and increasedcytoplasmic lipid levels.83 Thus changes in mitochondrial β-oxidation may represent a bio-chemical lesion corresponding to the pathological alterations in mitochondrial structureobserved in senescent subcortical astroglia. Whether or not CSH exposure incurs similarchanges in β-oxidation and levels of fatty acid binding proteins in cultures of immatureastroglia remains to be determined.
Although mitochondria appear to be the fundamental subcellular precursors of theCSH-induced glial granules, other organelles participate in the biogenesis of these inclu-sions to varying degrees. Progressively over a period of 3-12 days of CSH exposure, manyaberrant astroglial mitochondria (incipient peroxidase-positive inclusions) become incor-
Fig. 9.7. Cysteamine-induced mitochondrial pathology in cultured astroglia. (A) Mitochondrialswelling and dissolution of cristae after 24 h of CSH exposure. Bar = 300 nm. (B) Mitochondrialmacroautophagy after 12 days of CSH exposure. Bar = 500 nm. Reprinted with permission fromBrawer JR et al, Brain Res 1994; 633:9-20.

201The Peroxidase-Positive Subcortical Glial System
porated along with other cytoplasmic elements in apparent autophagosomes (Fig. 9.7). Manyof the latter exhibit acid phosphatase activity indicative of the participation of lysosomes.75
Using a panel of FITC-labelled antibodies directed against organelle-specific proteins andlaser scanning confocal microscopy, we confirmed partial colocalization of lysosomes, andto a lesser extent early endosomes and rough endoplasmic reticulum, to the redautofluorescent (peroxidase-positive) granules induced in cultured astroglia by CSH expo-sure.84 We determined that CSH stimulates the differential expression of specific lysosomalhydrolases in cultured astroglia. CSH suppresses cathepsin B mRNA levels and immunore-active protein, whereas cathepsin D mRNA and protein levels are significantly augmentedin these cells. Moreover, cathepsin D but not cathepsin B exhibits robust colocalization tothe autofluorescent glial inclusions.85 Furthermore, concordant with our in vitro observa-tions, cathepsin B immunoreactivity is prominent in the hypothalamic ventromedial nucleus,which accumulates few peroxidase-positive glial inclusions during aging, and is relativelyinapparent in the heavily-granulated hypothalamic arcuate nucleus. Cathepsin D, on theother hand, is heavily expressed in the aging arcuate nucleus, where it colocalizes to theautofluorescent glial inclusions and exhibits scant immunoreactivity in the adjacent ven-tromedial nuclear complex.85 These findings further support our contention that the astroglial
Fig. 9.8. Effects of subcutaneous CSH administration (see text for treatment regimen)on numbers of DAB-positive astrocyte and ependymal granules in various brain re-gions. Columns and vertical bars represent means and standard errors of the means,respectively. Asterisks denote statistically significant increases in granule numbers rela-tive to untreated controls (p<0.05). Reprinted with permission from Schipper HM et al,J Neuropathol Exp Neurol 1993; 52:399-410.

Astrocytes in Brain Aging and Neurodegeneration202
inclusions induced by CSH in vitro are identical to those which naturally accumulate in theaging subcortical brain.
Summary and ConclusionsA subpopulation of astrocytes in hippocampus, striatum and other subcortical brain
regions accumulates cytoplasmic inclusions with advancing age that are histochemicallyand morphologically distinct from lipofuscin. The gliosomes exhibit an affinity for Gomoristains, orange-red autofluorescence, and intense nonenzymatic peroxidase activity medi-ated by iron and other redox-active transition metals.
In the hypothalamic arcuate nucleus of adult rats and mice, chronic estrogen exposureinduces the proliferation of peroxidase-positive astrocytic inclusions in close proximity todegenerating neuritic processes. The glial peroxidase activity may promote oxidative dam-age of adjacent neuropil constituents in this brain region by metabolizing catecholestrogensand catecholamines to potentially neurotoxic free radical derivatives. In support of this hy-pothesis, dietary supplementation with potent antioxidants (α-tocopherol, 21-aminosteroids)prevents estradiol-related depletion of hypothalamic β-endorphin, a marker of estrogentoxicity.
The sulfhydryl agent, cysteamine (CSH), induces the accumulation of peroxidase-posi-tive astrocytic inclusions in situ and in primary brain cell cultures. In the latter, electronmicroprobe analysis in conjunction with diaminobenzidine cytochemistry confirmed thatredox-active (likely ferrous) iron is largely responsible for the manifest nonenzymatic per-oxidase activity in these cells.
In CSH-treated glial cultures and in the aging subcortical brain, the peroxidase-posi-tive glial inclusions are derived from effete, metal-laden mitochondria which become incor-porated with cathepsin D-positive lysosomes in a complex autophagic process.
Taken together, the morphological and histochemical data indicate that CSH acceler-ates the appearance of a senescent phenotype in subpopulations of astroglia both in situand in primary culture. As such, the aminothiol compound may serve as a useful tool todelineate further the structural and biochemical correlates of astroglial aging and facilitateour understanding of inclusion biogenesis in the senescent and degenerating CNS. In thefollowing chapter, cellular mechanisms implicated in the formation of peroxidase-positiveastroglial inclusions and the relationship of the latter to human corpora amylacea are re-viewed.
AcknowledgmentsThe authors thank Mrs. Kay Berckmans and Mrs. Adrienne Liberman for assistance
with the preparation of this manuscript. This work is supported by grants from the MedicalResearch Council of Canada (HMS.JRB) and the Fonds de la Recherche en Santé du Québec(HMS).
References1. Srebro Z, Lach H, Krawczyk S et al. Observations on the presence of Gomori-positive cells
in the telencephalon of various forms of green frogs. Acta Biologica Cracoviensia 1975;18:203-212.
2. Wislocki GB, Leduc EH. The cytology of the subcommissural organ, Reissner’s fibre,periventricular glial cells and posterior collicular recess of the rat’s brain. J Comp Neurol1954; 101:283-309.
3. Srebro Z, Lach H. The ultrastructure of the periventricular glia in the brains of rats andmice. Folia Biologica (Krakow) 1987; 35:131-136.
4. Srebro Z, Cichocki T, Godula J. Peroxidase activity in glia and ependymocytes of the mousebrain. Acta Morphologica Academiae Scientiarum Hungaricae 1971; 19:389-395.

203The Peroxidase-Positive Subcortical Glial System
5. Diepen R, Engelhardt F, Smith-Agreda V. Uber Ort und Art der Entstehung desNeurosekrets im supraoptico-hypophysaren system bei Hund und Katze. Verh Anat GesAnat Anz (suppl) 1954; 101:276-286.
6. Creswell GF, Reis DJ, MacLean PD. Aldehyde-fuchsin positive material in brain of squirrelmonkey (Saimiri sciureus). Am J Anat 1964; 115:543-558.
7. Schipper HM, Stopa EG, Wheelock T. Gomori-positive periventricular glia in the humanbrain. Society for Neuroscience, Phoenix, AZ (Abstract 232.7), 1988.
8. Schipper HM. Gomori-positive astrocytes: Biological properties and implications for neu-rologic and neuroendocrine disorders. Glia 1991; 4:365-377.
9. Schipper HM, Cissé S. Mitochondrial constituents of corpora amylacea and autofluorescentastrocytic inclusions in senescent human brain. Glia 1995; 14:55-64.
10. Srebro Z, Macinska A. Gomori-positive glial cells in in vitro cultures of human fetal braintissue. Acta Biologica Cracoviensia 1973; 16:147-152.
11. Brawer JR, Naftolin F, Martin J et al. Effects of a single injection of estradiol valerate onthe hypothalamic arcuate nucleus and on reproductive function in the female rat. Endocrinol1978; 103:501-512.
12. Brawer JR, Sonnenschein C. Cytopathological effects of estradiol on the arcuate nucleus ofthe female rat. A possible mechanism for pituitary tumorigenesis. Am J Anat 1975;144:57-88.
13. Casanueva F, Cocci D, Locatelli V et al. Defective central nervous system dopaminergicfunction in rats with estrogen-induced pituitary tumors, as assessed by plasma prolactinconcentrations. Endocrinol 1982; 110:590-599.
14. Brawer JR, Stein R, Small L et al. Composition of Gomori-positive inclusions in astrocytesof the hypothalamic arcuate nucleus. Anat Rec 1994; 240:407-415.
15. Koritsanszky S, Vigh B, Aros B. Studies on the Gomori-positive glial cells. I. Changes inthe Gomori-positive glial cells in rats of various ages. Acta Biologica Hungaricae 1967;18:9-19.
16. Gomori F. A differential stain for cell types in the pancreatic islets. Am J Pathol 1939;15:497-499.
17. Srebro Z, Cichocki T. A system of periventricular glia in brain characterized by large per-oxisome-like organelles. Acta Histochem Bd 1971; 41:108-114.
18. Wagner HJ, Pilgrim C. A microprobe analysis of Gomori-positive glial cells in the rat ar-cuate nucleus. Histochem 1978; 55:147-157.
19. Barden H. Further histochemical studies characterizing the lipofuscin component of hu-man neuromelanin. J Neuropath Exp Neurol 1978; 37:437-451.
20. Barden H. The oxidative generation of sulfonic acid groups in neuro-melanin and lipofus-cin in the human brain. J Histochem Cytochem 1984; 32:329-336.
21. Noda H. On the Gomorophil findings other than the neurosecretory system in the obser-vation of the hypothalamo-hypophysial system. Gunma Journal of Medical Sciences 1959;8:223-232.
22. Röhlich P, Vigh B, Teichmann I et al. Electron microscopic examination of the medianeminence of the rat. Acta Biologica Hungaricae 1965; 15:431-457.
23. Löfgreen F. The glial-vascular apparatus in the floor of the infundibular cavity. Acta Uni-versitatis Lundensis 1961; 57:1-18.
24. Cottle MKW, Silver A. Fluorescent granules in the Guinea-pig hypothalamus and theirpossible relation to neurosecretory substance. Zeitschrist fur Zellforschung, Bd 1970;103:559-569.
25. Goldgefter L. Studies on the structure and function of Gomori-positive glial cells in the rathypothalamus. Acta Anat 1976; 95:545-557.
26. Sohal RS, Wolfe LS. Lipofuscin: Characteristics and significance. Progress in Brain Res 1986;70:171-183.
27. Schipper HM, Scarborough DE, Lechan RM et al. Gomori-positive astrocytes in primaryculture: Effects of in vitro age and cysteamine exposure. Dev Brain Res 1990; 54:71-79.
28. Goldgefter L, Schejter AS, Gill D. Structure and microspectrofluorometric studies on glialcells from the periventricular and arcuate nuclei of the rat hypothalamus. Cell Tissue Res1980; 211:503-510.

Astrocytes in Brain Aging and Neurodegeneration204
29. Duchen MR, Biscoe TJ. Mitochondrial function in type I cells isolated from arterialchemoreceptors. J Physiol 1992; 450:13-31.
30. Kohler M, Fromter E. Identification of mitochondrial-rich cells in unstained vital prepara-tion of epithelia by autofluorescence. Eur J Physiol 1985; 403:47-49.
31. Udenfriend S. Fluorescence assay in biology and medicine. New York and London: Aca-demic Press, 1962; 293-294.
32. Hill J, Switzer R. The regional distribution and cellular localization of iron in the rat brain.Neurosci 1984; 3:595-603.
33. McLaren J, Brawer JR, Schipper HM. Iron content correlates with peroxidase actvity incysteamine-induced astroglial organelles. J Histochem Cytochem 1992; 40:1887-1897.
34. Srebro Z, Macinska A. Cytochemical demonstration of ferric iron and fluorescence mi-croscopy observations on Gomori-positive glia grown in vitro. Brain Res 1972; 42:53-58.
35. Young JK, Garvey JS, Huang PC. Glial immunoreactivity for metallothionein in the ratbrain. Glia 1991; 4:602-610.
36. Keefer DA, Christ JF. Distribution of endogenous diaminobenzidine-staining cells in thenormal rat brain. Brain Res 1976; 116:312-316.
37. Sherlock DA, Field PM, Raisman G. Retrograde transport of horseradish peroxidase in themagnocellular neurosecretory system of the rat. Brain Res 1975; 88:403-414.
38. Srebro Z. Periventricular Gomori-positive glia in brains of X-irradiated rats. Brain Res1971; 35:463-468.
39. Kumamoto T. Histochemical study on endogenous diaminobenzidine-positive granules inthe glia cell of rat brain. Acta Histochem Cytochem 1981; 14:173-185.
40. Goldfischer S, Villaverde H, Forschirm R. The demonstration of acid hydrolase, thermo-stable-reduced diphosphopyridine nucleotide tetrazolium reductase and peroxidase activi-ties in human lipofuscin pigment granules. J Histochem Cytochem 1966; 14:641-652.
41. Schipper HM, Mateescu-Cantuniari A. Identification of peroxidase-positive astrocytes bycombined histochemical and immunolabeling techniques in situ and in cell culture. JHistochem Cytochem 1991; 39:1009-1016.
41a. Schipper HM, Vininsky R, Brull R, Small L, Brawer JR. Astrocyte mitochondria: A sub-strate for iron deposition in the aging rat substantia nigra. Exp Neurol 1998; in press.
42. Srebro Z, Slebodzinski A. Periventricular Gomori-positive glial cells in the hypothalamusof the rabbit. Folia Biologica (Krakow) 1966; 14:391-395.
43. Maksymowicz K, Srebro Z. Gormori-positive glia in the mouse: Post-natal developmentand topographical distribution. Folia Biologica (Krakow) 1972; 20:135-146.
44. Schipper HM, Brawer JR, Nelson JF et al. Role of the gonads in the histologic aging of thehypothalamic arcuate nucleus. Biol Reprod 1981; 25:413-419.
45. Aschheim P. Aging in the hypothalamic-hypophyseal ovarian axis in the rat. In: EverittAV, Burgess JA, eds. Hypothalamus, Pituitary and Aging. Springfield: Charles C. Thomas1976:376-418.
46. Blake C, Weiner R, Gorski R et al. Secretion of pituitary luteinizing hormone and folliclestimulating hormone in female rats made persistently estrous or diestrous by hypotha-lamic deafferentation. Endocrinol 1972:90:855-861.
47. Singh, KB, Greenwald GS. Effects of continuous light on the reproductive cycle of thefemale rat: Induction of ovulation and pituitary gonadotrophins during persistent oestrus.J. Endocrinol 1967; 38:389-394.
48. Hahn DH, McGuire JL. The androgen-sterilized rat: Induction of ovulation and implanta-tion by luteinizing hormone-releasing hormone. Endocrinol 1978; 102:1741-1747.
49. Nadler RD. Intrahypothalamic locus for induction of androgen sterilization in neonatalfemale rats. Neuroendocrinol 1972; 8:349-357.
50. Brawer JR, Schipper HM, Naftolin F. Ovary-dependent degeneration in the hypothalamicarcuate nucleus. Endocrinol 1980; 107:274-279.
51. Schipper HM, Lechan RM, Reichlin S. Glial peroxidase activity in the hypothalamic arcu-ate nucleus: Effects of estradiol valerate-induced persistent estrus. Brain Res 1990;507:200-207.

205The Peroxidase-Positive Subcortical Glial System
52. Olmos G, Naftolin F, Perez J et al. Synaptic remodeling in the rat arcuate nucleus duringthe estrous cycle. Neurosci 1989; 32:663-667.
53. Perez J, Naftolin F, Garcia S et al. Sexual differentiation of synaptic connectivity and neu-ronal plasma membrane in the arcuate nucleus of the rat hypothalamus. Brain Res 1990;527:116-122.
54. Naftolin F, Garcia-Segura LM, Keefe D et al. Estrogen effects on the synaptology and neu-ral membranes of the rat hypothalamic arcuate nucleus. Biol Reprod 1990; 42:21-28.
55. Desjardins GC, Beaudet A, Schipper HM et al. Vitamin E protects hypothalamic β-endor-phin neurons from estradiol neurotoxicity. Endocrinol 1992; 131:2482-2484.
56. Brawer JR, Schipper HM, Robaire B. Effects of long-term androgen and estradiol exposureon the hypothalamus. Endocrinol 1983; 112:194-199.
57. Schipper HM, Piotte M, Brawer JR. Effects of progestins on the estradiol-related accumu-lation of astrocytic granules in the hypothalamic arcuate nucleus. Brain Res 1990;527:176-179.
58. Schipper HM. Role of peroxidase-positive astrocytes in estradiol-related hypothalamic dam-age. In: Fedoroff S, Juurlink B, Doucette, R, eds. Biology and Pathology of Astrocyte-neu-ron Interactions. New York: Plenum Publishing Corp., 1993:125-139.
59. Brawer JR, Beaudet A, Desjardins GC et al. Pathologic effect of estradiol on the hypothala-mus. Biol Reprod 1993; 49:647-652.
60. Landfield PW. An endocrine hypothesis of brain aging and studies on brain-endocrinecorrelations and monosynaptic physiology during aging. In: Finch CE, Potter DE, KennyAD, eds. Parkinson’s Disease II. New York: Plenum Publishing Corp., 1978:179-199.
61. Landfield PW, Waymire JC, Lynch G. Hippocampal aging and adrenocorticoids: Quantita-tive correlations. Sci 1978; 202:1098-1102.
62. Horning EC, Thenot J-P, Helton E. Toxic agents resulting from the oxidative metabolismof steroid hormones and drugs. J Toxicol Environ Health 1978; 4:341-361.
63. Kalyanaraman B, Sealy R, Sivarajah K. An electron spin resonance study of o-semiquinonesformed during the enzymatic and autoxidation of catechol estrogens. J Biol Chem 1984;259:14018-14022.
64. Kalyanaraman B, Felix CC, Sealy RC. Semiquinone anion radicals of catechol(amine)s, cat-echol estrogens, and their metal ion complexes. Environ Health Perspect 1985; 64:185-198.
65. Liehr JG, Roy D. Free radical generation by redox cycling of estrogens. Free Radic Biol1990; 8:415-423.
66. Ball P, Knuppen R. Formation of 2- and 4-hydroxy-estrogens by brain, pituitary, and liverof the human fetus. J Clin Endocrinol Metab 1978; 47:732-737.
67. MacLusky NJ, Barnea ER, Clark CR et al. Catechol estrogens and estrogen receptors. In:Merriam GR, Lipsett MB, eds. Catechol Estrogens New York: Raven Press 1983:151-165.
68. Mondschein J, Hersey R, Weisz J. Purification and characterization of estrogen-2/4-hy-droxylase activity from rabbit hypothalamus: Peroxidase-mediated catechol estrogen for-mation. Endocrinol 1986; 119:1105-1112.
69. Jellinck PH, Fletcher R. Peroxidase-catalyzed conjugation of [4-14C]-estradiol with albu-min and thiols. Can J Biochem 1970; 48:1192-1198.
70. Metzler M, McLachlan JA. Peroxidase-mediated oxidation, a possible pathway for the meta-bolic activation of diethylstilbestrol. Biochem Biophys Res Commun 1978; 85:874-884.
71. Schipper HM, Desjardins GC, Beaudet A et al. The 21-aminosteroid antioxidant, U74389F,prevents estradiol-induced depletion of hypothalamic β-endorphin in adult female rats.Brain Res 1994; 652:161-163.
72. Noble LJ, Cortez SC, Ellison JA. Endogenous peroxidatic activity in astrocytes after spinalcord injury. J Comp Neurol 1990; 296:674-685.
73. Cissé S, Schipper HM. Isolation of pseudoperoxidase-positive astrocyte granules from in-tact rat brain and cysteamine-treated neuroglial cultures. Brain Res 1993; 615:141-146.
74. Stohs SJ, Bagchi D. Oxidative mechanisms in the toxicity of metal ions. Free Radical BiolMed 1995; 18:321-336.
75. Brawer JR, Reichard G, Small L et al. The origin and composition of peroxidase-positivegranules in cysteamine-treated astrocytes in culture. Brain Res 1994; 633:9-20.

Astrocytes in Brain Aging and Neurodegeneration206
76. Wang X, Manganaro F, Schipper HM. A cellular stress model for the sequestration of re-dox-active glial iron in the aging and degenerating nervous system. J Neurochem 1995;64:1868-1877.
77. Brawer JR, Small L, Wang X et al. Uptake and subcellular distribution of 15Cr in Gomori-positive astrocytes in primary culture. Neurotoxicol 1995; 16:327-336.
78. Holtzman D, Olson J, de Vries C et al. Lead toxicity in primary cultured cerebral astro-cytes and cerebellar granular neurons. Toxicol Appl Pharmacol 1987; 89:211-235.
79. Wedler FC, Vichnin MC, Ley BW et al. Effects of Ca(II) ions on Mn(II) dynamics in chickglia and rat astrocytes—Potential regulation of glutamine synthetase. Neurochem Res 1994;19:145-151.
80. Schipper HM, Mydlarski MB, Wang X. Cysteamine gliopathy in situ: A cellular stress modelfor the biogenesis of astrocytic inclusions. J Neuropathol Exp Neurol 1993; 52:399-410.
81. Young JK. Immunoreactivity for diazepam binding inhibitor in Gomori-positive astrocytes.Regulatory Peptides 1994; 50:159-165.
82. Young JK, Baker JH, Muller T. Immunoreactivity for brain-fatty acid binding protein inGomori-positive astrocytes. Glia 1996; 16:218-226.
83. Vanden Heuvel JP, Sterchele PF, Nesbit, DJ et al. Coordinate induction of acyl-CoA bind-ing protein, fatty acid binding protein and peroxisomal β-oxidation by peroxisomeproliferators. Biochem Biophys Acta 1993; 1177:183-190.
84. Schipper HM, Cissé S, Walton PA. Colocalization of organelle-specific proteins toautofluorescent astrocyte granules by laser scanning confocal microscopy. Exp Cell Res 1993;207:62-67.
85. Chopra VS, Moozar KL, Mehindate K, Schipper HM. A cellular stress model for the differ-ential expression of glial lysosomal cathepsins in the aging nervous system. Exp Neurol147:221-228.

CHAPTER 10
Astrocytes in Brain Aging and Neurodegeneration, edited by Hyman M. Schipper.©1998 R.G. Landes Company.
Astrocyte Granulogenesisand the Cellular Stress ResponseMark B. Mydlarski and Hyman M. Schipper
A host of cellular insults, including sublethal exposure to heat, reactive oxygen species,metal ions, amino acid analogues, denatured proteins and sulfhydryl agents, stimulate
both prokaryotic and eukaryotic cells to elaborate a number of highly-conserved stress pro-teins. The superfamily of stress proteins includes high molecular weight heat shock proteins(HSPs) such as HSP90 and 72, certain low molecular weight peptides (e.g., HSP27) and agroup of glucose-regulated proteins (e.g., GRP94). The latter appear to respond to a morerestricted range of stimuli such as glucose deprivation and calcium ionophores but not togeneralized intracellular oxidative stress. The transcription of heat shock genes is regulatedby cis-acting heat shock elements in the promoter regions and trans-acting heat shock fac-tors. In mammalian cells, heat shock factors undergo posttranslational modification afterheat shock or exposure to other stressors and thereby acquire DNA-binding capability. Inaddition to the classic HSPs, genes coding for heme oxygenase-1 (HO-1), ubiquitin andsome α-crystallins contain heat shock element consensus sequences and may be upregulatedwith the former in a concerted cellular stress response. HSPs are thought to protect cellsundergoing stress by preventing damage to the translational apparatus, by maintenance oflipid membrane integrity, by accelerating degradation of misfolded or denatured proteinsand by obviating deleterious protein aggregation by binding to exposed hydrophobic sur-faces. Ubiquitin binds to normal and abnormal short-lived proteins and targets them forATP-dependent proteolysis. In addition, ubiquitin may exhibit complex interactions withheat shock factors and thereby coordinate transcription of HSP genes. In response to acutecellular stress, induction of HO-1 may also protect cells by catabolizing pro-oxidantmetalloporphyrins, such as heme, to bile pigments with free radical-scavenging capabilities.
In this chapter, we shall consider the role of the cellular stress response in the biogen-esis of astrocytic inclusions and establishment of gliosis in aging and degenerating neuraltissues. The chapter is subdivided into four major sections: First, the behavior of neuralHSPs under conditions of acute stress is discussed, with emphasis on the participation ofastrocytes and the impact of aging on these processes. Next, we review literature implicatingvarious stress proteins in the formation of neural inclusions in the aging and degeneratinghuman CNS. In a penultimate section, the role of the cellular heat shock response in theformation of peroxidase-positive astroglial inclusions is explored in considerable detail. Weconclude by providing a cellular stress model for the formation of corpora amylacea (CA)in the aging human brain.

Astrocytes in Brain Aging and Neurodegeneration208
HSP Expression in Acutely-stressed Neural Tissues: Effects of AgingAn understanding of the behavior of neural HSPs under conditions of acute stress,
such as that incurred by ischemia or hyperthermia, may shed light on the role(s) of stressproteins in brain aging and neurodegeneration which, on account of their indolent nature,often defy direct, methodical analysis. In brain, elevated demands for oxygen and energyaccompany an enhanced sensitivity to metabolic stress compared with other tissues. Heatshock protein (HSP)70c is constitutively expressed to a high degree in the CNS and repre-sents approximately 1% of total axonal protein.1 Furthermore, numerous studies have dem-onstrated that the CNS responds to a diverse array of insults, including hyperthermia, is-chemia, and physical and chemical stressors, by transcriptionally upregulating various HSPs,especially members of the HSP70 family.2-4 In a rat model of global ischemia, althoughstrong immunoreactivity for HSP72 was evidenced in surviving hippocampal CA3 and CA4pyramidal neurons, the stress protein was also markedly expressed in CA1 neurons whichexhibit pronounced cytotoxicity under hypoxic conditions.5,6 Similarly, enhanced accumu-lation of Ub-protein conjugates in the ischemia-sensitive CA1 region was shown to occurwith increasing severity of ischemic insult.7 The authors conjectured that accumulation ofubiquitin (Ub)-protein conjugates may precede, and play a role in, the development of is-chemia-related neurotoxicity. Others have cited the role of Ub in developmental apoptosisand suggested that during pathogenic processes this function of Ub is reactivated.8 Hayashiet al7 nonetheless conceded that it was not clear whether the observed ubiquitination ofproteins promoted or resulted from neuronal cell death.
The enhanced resistance to forebrain ischemic cytotoxicity observed in rats precondi-tioned with mild, transient heat shock has been attributed to the heat-induced upregulationof stress protein expression.9 Additionally, widespread induction of neural HSP72 follow-ing mild ischemia in gerbils correlated with tolerance to subsequent severe ischemic chal-lenge.10 Following hyperthermia, HSP72 immunoreactivity is most prominent in astrocytesand vascular endothelia in situ, and is observed to a lesser extent in neurons.11 Similarly,relative to cultured astrocytes, neurons exhibit an attenuated heat shock response in vitro.11,12
Large numbers of astroglia and ependymal cells upregulate HO-1 expression in situ follow-ing thermal stress, whereas the neuronal HO-1 response is relatively muted.13 Furthermore,treatment of rats with chemical depletors of the antioxidant glutathione results in robustinduction of HO-1 in astrocytes, ependymal cells, Bergmann glia and leptomeninges, butnot in neurons.14 Cultured astrocytes, but not neurons, strongly overexpress HO-1 follow-ing exposure to oxidative stress.15 Induction of HO-1 may promote the accumulation offree radical-scavenging bile pigments,16 and thereby fortify the brain’s antioxidant poten-tial. The more pronounced heat shock response exhibited by astrocytes in comparison withneurons may underlie the former’s relative resistance to a host of noxious stimuli includinghyperthemia,12 oxidative stress15 and ischemia.17
The aforementioned studies have given rise to the concept of a “hierarchy of vulner-ability” of cells in an ischemic territory.17 Brief focal ischemia results in enhanced transcrip-tion and elaboration of the HSP70 gene product restricted to hippocampal pyramidal neu-rons residing in the ischemic region. In contrast, ischemia of longer duration causesupregulation of HSP70 within a rim of astrocytes surrounding the damaged neurons, andapparently results in translational inhibition of the latter. Finally, severe ischemia sufficientto cause hippocampal infarction results in HSP70 overexpression in capillary endothelialcells, but not in neurons or astroglia, within the ischemic core. Based on these observations,it has been suggested that translational blockade resulting from ischemic injury occurs firstin neurons, and then in astroglia. Capillary endothelial cells, on the other hand, appearrelatively resistant to similar levels of hypoxia, and may be the last cell type in an infarctedarea to cease expression of stress proteins. The induction of HSP70 in astrocytes and neu-

209Astrocyte Granulogenesis and the Cellular Stress Response
rons within the CNS has also been documented in various studies following stereotaxicinjection of kainic acid,18 flurothyl-induced status epilepticus,19 cortical stab wounds,20 andspinal cord trauma.21
Several studies have examined the relationship between aging and the ability to mounta cellular stress response in the brain. For example, relative to younger rats, older animalsexhibit an age-related deficiency in HSP70 mRNA induction in brain (and other tissues)following heat stress.22 However, the heat-induced increase in colonic temperature of theaged rats in this study was lower than that observed in younger controls. Based on thisobservation, the authors concluded that impaired upregulation of HSP70 in heat-stressed,older animals resulted from an age-associated decline in heat-generating capacity ratherthan an age-related failure to mount a heat shock response. Subsequently, Pardue et al23
performed a comparable study with young and old rats which controlled for potential dif-ferences in body temperature between the groups. Relative to younger thermal-stressed ani-mals, induction of HSP70 mRNA was again blunted in dentate gyrus granule cells and py-ramidal cells of the hippocampus in heat-stressed, older rats. Few studies have examinedthe normal distribution of HSP expression in the CNS of young versus old, unstressed ani-mals. In one such study, Tytell et al24 assayed retinal HSP70 expression in young and old ratsand could not detect any significant age-related differences. However, following hyperther-mia, levels of inducible HSP70 in older animals were significantly attenuated relative toyounger controls.24,25 There is a well documented reduction in the ability of older organ-isms to cope with stress and maintain vital homeostatic mechanisms under adverse condi-tions.26 Age-related impairments in the heat shock response system may contribute to thedecreased ability of senescent organisms to mount adaptive responses under stressful con-ditions.27 It has been suggested that inability of older cells to adequately promote the post-translational conversion of inactive heat shock factor (HSF) to its oligomeric DNA-bindingform underlies this functional deficit.27 A reduced capacity of senescent neural tissues tomount a cytoprotective heat shock response may be a factor predisposing the aging CNS toneurodegeneration.
Stress Protein Expression in the Agingand Degenerating Human Brain
A number of studies have examined the distribution of various stress proteins in nor-mal and diseased human neural tissues. Pappolla et al28 demonstrated punctate deposits ofUb-immunoreactivity distributed throughout the white matter of normal aged (≥70 years),but not normal young (≤33 years), human brain. Although ultrastructural evaluation of theUb deposits was not performed, Ub immunoreactivity appeared to localize to axonal ele-ments by light microscopy. In another study, immunoelectron microscopy detected granu-lar Ub-immunoreactivity within glia and myelin lamellae of white matter.29 Ub-positivedystrophic neurites in cortical areas and axonal spheroids in the substantia nigra and stria-tum were also found to accumulate with advancing age in normal subjects.29 More recently,an age-dependent accumulation of vacuole-laden, Ub-immunopositive astrocytes withinthe globus pallidus of normal human brain was demonstrated.30 The authors suggested thatthis was a normal, age-related effect because the numbers of these astrocytes were not fur-ther increased in basal ganglia derived from subjects with Alzheimer’s disease (AD),Parkinson’s disease (PD), multiple system atrophy, or multiple sclerosis.
The expression of Ub and various HSPs has been associated with a large number ofpathologic structures found in neurodegenerative states. Of these, the neurofibrillary pa-thology of AD has been most extensively studied. Ub levels in the AD brain are reportedlyhigher than in nondemented, age-matched controls.31 Several studies revealed that Ub is acomponent of the neurofibrillary tangles (NFT) and senile plaques characteristic of AD.32,33

Astrocytes in Brain Aging and Neurodegeneration210
Relative to age-matched controls, HSP72 is dramatically overexpressed in AD brain, and,like Ub, exhibits colocalization to neuritic plaques and NFT.34 HSP27 is similarlyoverexpressed in AD relative to nondemented controls.35 In AD, HSP27 was immunolocalizedto degenerating reactive astrocytes, particularly in areas rich in senile plaques, and exhibitedoccasional colocalization to NFT.35 In another study, HSP28 (HSP27) was increased relativeto controls in the temporal, frontal and parietal lobes of AD, and staining appeared local-ized to damaged neuronal elements and senile plaques.36 αB-crystallin-immunopositivehypertrophic astrocytes and microglia appeared more numerous in AD in comparison withcontrols and were concentrated in areas replete with plaques and NFT.37 In the AD brain,HO-1 immunostaining is dramatically increased in cortical and hippocampal neurons andastrocytes relative to age-matched, nondemented controls, and is found in association withneuritic plaques, NFT and corpora amylacea.38,39
Several other neuropathological states are characterized by the accumulation of aber-rant, cytoplasmic inclusions in neural cells (see chapter 8). Many of these inclusions, in-cluding Lewy bodies in PD, Lewy body-like inclusions in ALS, as well as Pick bodies andballooned neurons in Pick’s disease, are ubiquitinated and may be associated with otherstress proteins. In brains of patients with multiple system atrophy, glial cytoplasmic inclu-sions accumulate within oligodendroglia and appear to contain ubiquitinated forms of αB-crystallin.40 This finding is reminiscent of an earlier report in which ubiquitinated αB-crys-tallin was localized to Rosenthal fibers.41 The latter represent eosinophilic, GFAP-containing,HSP27-positive inclusions which accumulate within astrocytic processes in long-standinggliosis, certain cerebellar astrocytomas, and in Alexander’s disease (see chapter 4). Hypoxicencephalopathy during infancy is associated with the accretion of gliofibrillary inclusions,predominantly in white matter. This astrocytic inclusion body appears related to Rosenthalfibers and similarly contains GFAP, Ub and αB-crystallin, but diverges from the latter in theultrastructural pattern of intermediate filament deposition.42 In certain neurodegenerativestates, astrocytes may occasionally exhibit cytopathological changes reminiscent of thosetypically encountered in affected neuronal populations. For example, gliofibrillary tanglesassociated with PHF-tau have been reported in white matter astrocytes in subjects withcorticobasal ganglionic degeneration and progressive supranuclear palsy (see chapter 8).42
A summary of stress protein-containing neural inclusion bodies is presented in Table 10.1.It should be borne in mind that many of the neurodegenerative changes associated
with HSP expression represent end-stage or “graveyard” histopathology. In such cases, thereis considerable difficulty resolving cause and effect relationships between HSP inductionand the development of specific neuropathologic features. Regarding the ubiquitination ofabnormal structures in neurodegenerative states, Wilkinson43 stated that “It is not knownwhether ubiquitin is present at the time of deposition or only detected late in the process asa reflection of the cells’ attempt to deal with this pathological situation.” Due to the inherentdifficulties in studying inclusion biogenesis using human tissues, there is considerable valuein the generation of tissue culture and animal models in which the development of stress-related inclusions can be thoroughly investigated. The work described in the following sec-tion was performed with the view of elucidating, in prospective fashion, the role of thecellular stress (heat shock) response in the biogenesis of astroglial inclusions.
A Cellular Stress Model for the Biogenesis of Astroglial Inclusions
Cell Stress and Astrocyte Granulation in Primary CultureUsing the cysteamine (CSH) model for accelerated astrocyte granulation (chapter 9),
we determined that this aminothiol compound induces stress protein expression in astrogliaakin to its effects on rat liver.44 Astrocyte cultures exposed to CSH for 6 h exhibit a robust

211Astrocyte Granulogenesis and the Cellular Stress Response
cellular stress response characterized by the enhanced expression of HO-1 and HSPs 27, 72and 90, long before increases in numbers of DAB (peroxidase)-positive cytoplasmic gran-ules become apparent at the LM level.45 Furthermore, in comparison to control cultures,CSH-pretreated cells exhibited enhanced resistance to both trypsinization-related cell deathand killing by H2O2 exposure, providing physiologic evidence that CSH induces a cellularheat shock response in cultured astroglia. Thus, activation of a cellular stress response pre-cedes, and may be a prerequisite for, the formation of peroxidase-positive astrocyte gran-ules in CSH-treated glial cultures. In this respect, the astrocytic inclusions may represent atype of “stress granule” reminiscent of heat shock granules which have been shown to arisein other cell types following sustained stress.46,47
Cell Stress and Astrocyte Granulation In SituFurther studies were undertaken to determine whether CSH administration to young
adult rats accelerates the aging-related accumulation of peroxidase-positive astrocyte gran-ules in situ, and whether this also occurs in the context of a glial cellular stress response.48
We noted that subcutaneous CSH treatment (150-300 mg/kg body weight twice weekly x 3weeks) produced 2- to 3-fold increases in numbers of DAB-positive astrocyte granules withinthe dorsal hippocampus, corpus callosum, striatum and the third ventricular subependymalzone. Brain regions normally containing few or no peroxidase-positive glia, such as thecerebellum and cerebral cortex, exhibited little or no response to CSH treatment (see chap-ter 9, Fig. 9.8). Thus, systemic CSH administration appears to accelerate the appearance ofa normal “aging” phenotype in subpopulations of rat astroglia. Furthermore, using duallabel immunohistochemistry, we observed that both acute (24 h) and prolonged (3 weeks)CSH exposure induced the expression of HSPs 27, 72 and 90 and GRP94 by GFAP-positiveastrocytes residing precisely within those brain regions shown to be susceptible to CSH-induced glial granulation. The upregulation of GRP94 in situ probably represents an indi-rect effect of CSH (perhaps mediated by perturbations in glucose or calcium homeostasis)because, in contradistinction to its direct stimulatory action on other HSPs, this aminothioldoes not appreciably alter patterns of GRP94 expression in primary astrocyte cultures.45
Taken together, our findings indicate that, as in primary astroglial cultures, induction of thecellular heat shock response is a proximal event in the biogenesis of peroxidase-positiveastrocyte granules in the aging subcortical brain.
In addition to promoting glial stress protein biosynthesis and cytoplasmic granulation,prolonged exposure to CSH elicited robust astrocytic hypertrophy and GFAP expression(gliosis) in the corpus callosum, ventral hippocampal commissure and striatum of adultrats. Akin to the effects of CSH, multiple systemic injections of estradiol valerate (EV) in-duce overexpression of glial HSPs and the accumulation of peroxidase-positive astroglialgranules in estrogen receptor-rich brain regions (see below).49 However, unlike chronic CSHtreatment, long term EV exposure did not promote astrogliosis and enhanced GFAP ex-pression in situ. Taken together, these results indicate that the accumulation of peroxidase-positive astrocyte inclusions may occur within the context of, or entirely independently of,classical astrocyte hypertrophy (gliosis).
In contrast to the striking effects of chronic CSH exposure on glial morphology andhistochemistry in situ, this treatment regimen engendered no overt neuronal pathology atthe LM level. Specifically, there was no evidence of neuronal damage, demyelination orlipofuscin accumulation in preparations stained with cresyl violet, hematoxylin-eosin, modi-fied Bielschowsky’s silver method, Luxol fast blue, or periodic acid-Schiff.48 Prolonged CSHexposure did reduce somatostatin immunoreactivity in fiber tracts of the ventral striatum48
as had been reported in earlier short term CSH studies.50 However, these findings may notrepresent true depletion of somatostatin levels or loss of somatostatin-containing neurons.

Astrocytes in Brain Aging and Neurodegeneration212
Tabl
e 10
.1. S
tres
s pr
otei
n-co
ntai
ning
neu
ral i
nclu
sion
bod
ies
Cel
l Typ
eC
hara
cter
isti
cSt
ruct
ural
Stre
ssIn
clus
ion
Bod
yC
linic
al C
ondi
tion
Loca
tion
Prot
ein
Prot
ein
AST
RO
CY
TES
Cor
pora
am
ylac
eaA
ging
; AD
Subp
ial a
ndU
nkno
wn
Ubi
quiti
na ;pe
rive
ntri
cula
r re
gion
sH
SP27
a 72a ;
HO
-1b
Ros
enth
al fi
bers
Ale
xand
er’s
dis
ease
Subp
ial a
nd p
eriv
ascu
lar
GFA
PU
biqu
itin;
αB
-cry
stal
lin;
regi
ons;
whi
te m
atte
rH
SP27
Glio
fibri
llary
incl
usio
nIn
fant
ile h
ypox
icW
hite
mat
ter
GFA
PU
biqu
itin;
αB
-cry
stal
linen
ceph
alop
athi
es
Glio
fibri
llary
tang
les
CB
G; P
SPW
hite
mat
ter
PHF-
tau
Ubi
quiti
n
NEU
RO
NS
Gra
nulo
vacu
olar
bod
iesA
ging
; AD
Hip
poca
mpu
sTa
u, N
FU
biqu
itin;
HO
-1c
Mar
ines
co b
odie
sA
ging
Subs
tant
ia n
igra
Unk
now
nU
biqu
itin
Neu
roax
onal
dys
trop
hyA
ging
Dor
sal c
olum
n nu
clei
;U
nkno
wn
Ubi
quiti
nsu
bsta
ntia
nig
ra;
glob
us p
allid
us
Dys
trop
hic
neur
ites
Agi
ng A
D &
pri
on d
isea
ses
Ass
ocia
ted
with
sen
ile p
laqu
esU
nkno
wn
Ubi
quiti
n; H
O-1
b
Dys
trop
hic
neur
ites
DLB
DC
A2/
3; b
asal
fore
brai
n;N
FU
biqu
itin
brai
nste
m n
ucle
i
Cor
tical
Lew
y B
odie
sD
LBD
limbi
c &
par
a-lim
bic
NF
Ubi
quiti
n; α
B-c
ryst
allin
;co
rtic
es; a
myg
dala

213Astrocyte Granulogenesis and the Cellular Stress Response
Bal
loon
ed N
euro
nsC
BG
; Pic
k’s
dise
ase
limbi
c &
par
a-lim
bic
NF
Ubi
quiti
n; α
B-c
ryst
allin
;co
rtic
es; a
myg
dala
HSP
27b
Bun
ina
bodi
esA
LSA
nter
ior
horn
cel
lsoc
casi
onal
ly N
FU
biqu
itine
Lew
y B
odie
sPD
:DLB
DSu
bsta
ntia
nig
ra;
NF
Ubi
quiti
n; α
B-c
ryst
allin
brai
nste
m m
onoa
min
ergi
c;ba
sal f
oreb
rain
Lew
y bo
dy-l
ike
ALS
-spo
radi
cA
nter
ior
horn
cel
lsN
FU
biqu
itin;
αB
-cry
stal
linin
clus
ions
in A
LSan
d fa
mili
al
Neu
rofib
rilla
ryA
D;P
SPC
orte
x; h
ippo
cam
pus;
PHF-
tau
Ubi
quiti
n; H
O-1
b
tang
les
&ba
sal f
oreb
rain
;ne
urop
il th
read
sbr
ains
tem
nuc
lei
Pick
bod
ies
Pick
’s d
isea
seH
ippo
cam
pal d
enta
te fa
scia
;PH
F-ta
uU
biqu
itin
cere
bral
cor
tex
Neu
ropi
l gra
ins
CB
G; D
emen
tia w
ith g
rain
sC
orte
x; h
ippo
cam
pus
PHF-
tau
Ubi
quiti
n
Ubi
quiti
n re
activ
eD
emen
tia la
ckin
g di
stin
ctiv
eC
orte
x (s
mal
l neu
rons
inU
nkno
wn
Ubi
quiti
nin
clus
ions
hist
opat
holo
gyla
yer2
); de
ntat
e fa
scia
OLI
GO
DEN
DR
OG
LIA
L
Gra
nula
rA
ging
Bra
in a
nd s
pina
l cor
dU
nkno
wn
Ubi
quiti
nde
gene
ratio
n of
mye
linm
yelin
Glia
l cyt
opla
smic
Mul
tisys
tem
atr
ophy
Whi
te m
atte
rPH
F-ta
uU
biqu
itin;
αB
-cry
stal
linf
incl
usio
ns
Mod
ified
afte
r D
icks
on a
nd Y
en, 1
994.
Add
ition
al d
ata
are
refe
renc
ed. A
bbre
viat
ions
: AD
, Alz
heim
er’s
dis
ease
; ALS
, am
yotr
ophi
c la
tera
l scl
eros
is;
CB
G, c
ortic
obas
al g
angl
ioni
c de
gene
ratio
n; D
LBD
, diff
use
Lew
y bo
dy d
isea
se; G
FAP,
glia
l fib
rilla
ry a
cidi
c pr
otei
n; N
F, n
euro
filam
ent;
PD, P
arki
nson
’s d
isea
se;
PHF,
pai
red
helic
al fi
lam
ents
; PSP
, pro
gres
sive
sup
ranu
clea
r pa
lsy.
Ref
eren
ces:
a, C
issé
et a
l, 19
93; b
, Sch
ippe
r et
al,
1995
; c, S
mith
et a
l, 19
94; d
, Kat
o et
al,
1992
; e, M
ighe
li et
al,
1994
; f, T
amao
ka e
t al,
1995

Astrocytes in Brain Aging and Neurodegeneration214
Rather, by virtue of disulfide bond interactions, CSH may chemically alter somatostatinmolecules, rendering them undetectable by conventional immunohistochemistry.51 Theabsence of overt neuronal pathology, in conjunction with observations that CSH-inducedHSP expression and cytoplasmic granulation are restricted to astrocytes (and certain thirdventricular ependymal cells) in situ and in pure neuroglial cultures,45 strongly suggest thatthis aminothiol compound elicits a primary “gliopathy” independent of any antecedentneuronal injury.
The development of neuropathological features, including basal ganglia necrosis andpatchy demyelination, has been documented in young patients chronically receiving CSHfor the treatment of nephropathic cystinosis.52 However, these degenerative changes appearto derive from the disease process itself, rather than from iatrogenic CSH toxicity, becausesimilar neuropathologic profiles have been observed in rare individuals with nephropathiccystinosis who have survived into the second or third decades without CSH treatment.Whether or not sustained CSH treatment induces astrocyte hypertrophy, HSP elaborationand cytoplasmic granulation in the brains of these patients (as it does in rats) remains to bedetermined.
Stress Protein Content of Peroxidase-Positive Astrocyte GranulesExperiments were performed to ascertain whether HSPs are actual constituents of the
peroxidase-positive astrocyte granules in adult rat brain sections and in CSH-treated astro-cyte cultures.53 Using fluorescein-conjugated antibodies directed against a host of stressproteins in conjunction with laser scanning confocal microscopy, we determined that HSP27exhibits intense colocalization to the red autofluorescent (peroxidase-positive) astrocyteinclusions which derive from degenerative mitochondria both in situ and in vitro(Figs. 10.1-10.3). GRP94 exhibited partial colocalization to peripheral regions of the astro-cytic inclusions in both preparations. Colocalization of GRP94, an endoplasmic reticulum(ER)-derived stress protein, suggests that elements of the ER may participate in the biogen-esis of peroxidase-positive astrocyte granules. Using a panel of antibodies directed againstorganelle-specific proteins, it was shown that the ER exhibits occasional colocalization tothe astrocytic inclusions in CSH-treated cultures.48 Furthermore, ultrastructural studies ofthese gliosomes in periventricular brain regions of aging and estrogen-treated rodents54,55
and in CSH-treated glial cultures56 revealed infrequent contiguity of the inclusions withelectron-dense cisternal elements reminiscent of ER. HO-1, another ER-derived stress pro-tein, also exhibits partial immunolocalization to the autofluorescent astrocyte granules inCSH-treated cultures and in the intact rat brain (Mydlarski and Schipper, unpublished re-sults). In contrast to the aforementioned stress proteins, HSP72 was a minor constituent ofthe astrocytic inclusions in situ and in culture, and the glial granules appeared consistentlydevoid of HSP90 and αB-crystallin53 (Fig. 10.4). Thus, patterns of stress proteinimmunolocalization to astroglial inclusions in subcortical regions of the adult rat brain andin CSH-treated glial cultures are virtually indistinguishable from each other (Table 10.2).These findings greatly extend previous histochemical and morphological data underscor-ing the identical origin of the CSH-induced astroglial inclusions and those which spontane-ously accumulate in the aging subcortical brain.
Role of Ubiquitin in the Biogenesis of Astroglial InclusionsAfter 6 days of primary culture, control (untreated) neonatal rat astroglia exhibited
weak, diffuse Ub-immunostaining. In contrast, short term (6 h) CSH treatment induced ashift to intense, granular deposition of Ub53 suggestive of the formation of Ub-protein con-jugates.57 The induction of Ub and the formation of high molecular weight conjugates isknown to occur in a variety of tissues following heat shock or oxidative stress.58-60 Activa-

215Astrocyte Granulogenesis and the Cellular Stress Response
tion of the Ub system in our model represents an early event in the biogenesis of the glialinclusions. These results contest the “common view of ubiquitin being involved in [neuro-pathological] reactions that are both secondary and late”.61 Furthermore, our results dem-onstrate that the Ub system is activated shortly after CSH exposure (6 h), concomitant withthe upregulation of HSPs 27, 72, 90 and HO-1.45 This finding argues against the view thatactivation of the Ub system is contingent on prior failure of the HSPs in their attempt torenature damaged polypeptides and reconfer normal protein homeostasis.43
As in the case of HSP27, ubiquitin exhibits intense colocalization to the autofluorescentastrocytic inclusions both in CSH-treated glial cultures and in situ (Fig. 10.5).53 Peroxidase-positive astrocyte granules often appear to be delimited by membranes under transmissionEM,56 and the larger inclusions are heavily labeled with lysosome-specific markers.48,62
Ubiquitination of mature, autofluorescent inclusions53 is therefore consistent with earlierstudies demonstrating the accumulation of free Ub and Ub-protein conjugates within lyso-somes,63-65 and contradicts the view that Ub-immunoreactivity in neural and other tissuesis restricted to nonmembrane-bound, nonlysosomal inclusions.66 Additional studies will berequired to determine whether Ub or other specific HSPs colocalize to aberrant mitochon-dria prior to autophagy, or if incorporation of the stress proteins into astroglial inclusionsoccurs during or after lysosomal fusion (see chapter 9).
Ub has been associated with a number of senescence and disease-related neural inclu-sions, including granulovacuolar bodies, Marinesco bodies, granular degeneration of my-elin, dystrophic axons and neurites, and corpora amylacea.42,67 Of these inclusions, onlycorpora amylacea have previously been shown to predominate in astrocytes of the normalaging human brain.68-70 The aforementioned results, along with those of a study71 depictingthe presence of Gomori-positive astroglial granules in aging human neural tissues, establishthe ubiquitinated, peroxidase-positive glial inclusions as a second, highly consistentbiomarker of astrocyte senescence in the mammalian brain.
The presence of Ub in these inclusions may provide important clues concerning theirmode of formation and chemical constituents. The accumulation of aberrant proteins andprotein aggregates within a variety of cytoplasmic inclusions is characteristic of many se-nescence and disease-related neurodegenerative changes.42 Shang and Taylor72 demonstratedthat H2O2-related oxidative stress compromises the Ub conjugation activity of culturedmammalian cells with a resultant reduction in proteolytic activity. Moreover, their resultsindicated that activation of the Ub system only occurs following removal of the stress andcell recovery. The activities of the Ub-activating and conjugating enzymes, E1 and E2,
Table 10.2. Gomori-positive astrocyte granules: Stress protein expression patterns
Stress Proteins Astrocyte Granules
12-Week Rat Brain 12-Day CSH-Treated Cultures
HSP27 intense, larger granules intense, larger granulesHSP72 occasional infrequentHSP90 no no; nuclear translocationGRP94 intense, granule periphery intense, granule peripheryUbiquitin intense, larger granules intense, larger granulesαB-crystallin no no
GRP = glucose-regulated protein; HSP = heat shock protein

Astrocytes in Brain Aging and Neurodegeneration216
Fig. 10.1. (See color plate 1 for color representation of these figures.) Identification of Gomori-positive astrocytes by DAB-GFAP double label immunohistochemistry. Hypothalamic arcuatenucleus. Long arrow: brown reaction product (endogenous peroxidase activity) within pink(GFAP-positive) astrocyte. Short arrow: astrocytic process replete with endogenous peroxidaseactivity. Arrowhead: astrocyte largely devoid of DAB-positive inclusions. 40 micron section; x630.Reprinted with permission from Schipper HM et al, Brain Res 1990; 507:200-207.
Fig. 10.2. Laser scanning confocal micrograph of adult arcuate nucleus stained with the mito-chondrial marker CLSO. Consistent colocalization of the mitochondrial marker (green) to theautofluorescent glial granules (red) produces yellow fluorescence. Bar = 10 µM. Reprinted withpermission from Brawer JR et al, Anat Rec 1994; 240:407-415.

217Astrocyte Granulogenesis and the Cellular Stress Response
respectively, are dependent on their constituent free thiol groups.73,74 The authors suggestedthat oxidative modification of these thiol groups may inactivate Ub-dependent proteolysisand contribute to the intracellular accrual of damaged proteins. In a similar fashion, unre-mitting oxidative stress in the aging and diseased nervous system, and in primary astrocytecultures repeatedly exposed to CSH (see below), may prevent Ub-dependent degradationof aberrant proteins and promote their accumulation within astrocytic inclusions. Further-more, Ub-dependent proteolysis and the release of renatured target proteins from mostHSPs require ATP hydrolysis.75,76 Although we have not measured ATP concentrations inCSH-treated cells, others have shown that ATP levels in mammalian cells decline rapidlyfollowing heat shock.77-79 Thus, ATP depletion, as a direct consequence of cellular stress orresulting from a progressive, aging-related decline in mitochondrial function, could con-tribute to the accrual of HSP-bound proteins and compromise the availability of free HSPsnecessary for the chaperoning of newly-damaged polypeptides. The role of oxidative stressand the Ub system in the accumulation of aberrant proteins and protein aggregates in theaging and CNS is currently an active area of research with important ramifications for theelucidation (and possible treatment) of various human neurodegenerative disorders. Theuse of CSH-exposed astroglia and other well-characterized models of neural cell senescenceshould facilitate this line of inquiry by providing opportunities to test salient hypotheses insimplified, but biologically-relevant, contexts.
Estrogen-Related Astrocyte GranulationAs described in chapter 9, administration of estrogen to adult female rats accelerates
the accretion of peroxidase-positive astrocyte granules in periventricular brain regions ex-pressing sex steroid receptors.54,80-83 We subsequently determined that three monthly intra-muscular injections of estradiol valerate (EV; 0.2 or 2.0 mg) elicit the overexpression ofHSPs 27, 72, and 90 and augment cytoplasmic granulation in GFAP-positive astrocytes ofthe arcuate nucleus and third ventricular subependymal zone. In contrast, long term EVtreatment induced little or no HSP upregulation or astrocyte granulation in estrogen recep-tor-deficient brain regions such as the caudate-putamen and corpus callosum (Table 10.3).49
Olazábal and coworkers have previously reported the induction of HSPs 70 and 90 in ro-dent hypothalamic neurons following estrogen treatment.84-88 Our study, on the other hand,demonstrated estrogen-related upregulation of HSPs in astrocytes. Of significance, shortterm administration of EV (48 h) induced similar HSP expression, but no concomitantcytoplasmic granulation, in arcuate astrocytes.49 These findings, in conjunction with resultsderived from CSH- and H2O2-treated astrocyte cultures45,53 and CSH-exposed rats,48 further
Figs. 10.3-10.5 (opposite). Immunolocalization of stress proteins to autofluorescent astrocytegranules in rat brain sections and CSH-treated glial cultures. Fig. 10.3: HSP27 shows intensecolocalization (yellow fluorescence) to astrocyte granules in situ (10.3A; empty arrows) and inculture (10.3B; empty arrows). Solid arrows indicate smaller granules devoid of HSP27-immu-noreactivity [bars = 100 µm for (A); 10 µm for (B)].Fig. 10.4. In brain sections (A), strong αB-crystallin staining can be seen in cells along the thirdventricular wall (V). Both in situ (A) and in vivo (B), αB-crystallin manifests strongimmunolabeling of astroglia but no colocalization to the autofluorescent inclusions (arrows)[bars = 10 µm for (A) and (B)].Fig. 10.5. Within the subependymal zone of the third ventricle (V), ubiquitin exhibits strongcolocalization (yellow fluorescence) to the autofluorescent granules (A), arrows. [Bar = 25 µm].In vivo, the larger autofluorescent granules are ubiquitinated (B), empty arrows, whereas manysmaller granules are not ubiquitin-immunoreactive (solid arrow). Occasional ubiquitin stainingof granule-free cytoplasm is shown in (B) (arrowhead) [bar = 10 µm]. Reprinted with permis-sion from Mydlarski MB et al, Brain Res 1993; 627:113-121.

Astrocytes in Brain Aging and Neurodegeneration218
Tabl
e 10
.3.P
erce
nt o
f GFA
P-po
siti
ve a
stro
cyte
s (m
ean
±SD
) pe
r re
gion
exp
ress
ing
HSP
27, 7
2, o
r 90
or
GR
P94
afte
r lo
ng t
erm
EV
trea
tmen
t
AR
C P
eri-
III
Con
trol
Ev 0
.2 m
gEV
2.0
mg
Con
trol
EV 0
.2 m
gEV
2.0
mg
HSP
2716
.7 ±
6.9
68.2
±7.
1*56
.2 ±
27.2
28.5
±4.
458
.6 ±
14.2
*55
.9 ±
29.3
HSP
72 7
.6 ±
7.8
17.2
±18
.123
.8 ±
11.7
4.4
±3.
416
.8 ±
10.9
30.6
±5.
9*H
SP90
2.7
±1.
937
.8 ±
15.5
*37
.1 ±
6.4*
6.1
±7.
935
.5 ±
9.0*
32.8
±6.
0*G
RP9
416
.0 ±
14.4
23.5
±24
.415
.6 ±
18.6
12.0
±13
.924
.5 ±
17.0
25.7
±25
.2
CC
CP
Con
trol
EV 0
.2 m
gEV
2.0
mg
Con
trol
EV 0
.2 m
gEV
2.0
mg
HSP
2726
.3 ±
26.0
38.2
±24
.946
.8 ±
22.3
33.0
±4.
536
.6 ±
12.7
43.3
±8.
4H
SP72
13.2
±16
.6 9
.3 ±
9.4
15.2
±4.
8 7
.4 ±
10.2
4.6
±4.
8 2
.7 ±
4.0
HSP
9019
.5 ±
15.4
12.2
±8.
114
.9 ±
9.1
3.7
±3.
1 4
.8 ±
6.0
12.9
±5.
3*G
RP9
416
.1 ±
22.4
13.1
±11
.0 5
.8 ±
2.9
8.3
±4.
1 9
.8 ±
5.6
7.6
±4.
0
Abb
revi
atio
ns: A
RC
, arc
uate
nuc
leus
; EV
, est
radi
ol v
aler
ate;
CC
, cor
pus
callo
sum
; CP,
cau
date
-put
amen
; HSP
, hea
t sho
ck p
rote
in;
Peri
-III,
thir
d pe
rive
ntri
cula
r re
gion
.*
Sign
ifica
ntly
incr
ease
d fr
om c
ontr
ol v
alue
s (p
<0.
05).

219Astrocyte Granulogenesis and the Cellular Stress Response
indicate that induction of a cellular stress (heat shock) response precedes, and is not a con-sequence of, the development of iron-laden astroglial inclusions.
Intracellular Oxidative Stress: A “Final Common Pathway”for the Biogenesis of Astrocytic Inclusions
Oxidation of CSH in the presence of transition metals generates several pro-oxidantspecies including H2O2, and the superoxide, hydroxyl and thiyl radicals.87 HO-1, Ub, andthe various HSPs induced by CSH in cultured astroglia45,53 are commonly upregulated inresponse to hyperthermic challenge as well as oxidative stress. In contrast, addition of CSHto the glial monolayers did not enhance the expression of GRP94, a stress protein known torespond to glucose deprivation and calcium ionophores, but not to heat shock or oxidativestress.88,89 Systemic administration of CSH also resulted in overexpression of these redox-sensitive HSPs in GFAP-positive astroglia in situ48 providing further, albeit indirect, evi-dence of a free radical mechanism of CSH action.
The gene coding for manganese superoxide dismutase (MnSOD) is modulated in bac-teria and in mammalian cells by oxidative stress.90,91 This mitochondrial enzyme, whichcatalyzes the dismutation of superoxide anion to H2O2, protects mitochondria from inordi-nate or inadvertent superoxide radical generation during the normal electron transportprocess and following exposure to mitochondrial toxins. MnSOD gene expression and ac-tivity are significantly enhanced in CSH-treated glial cultures and in the intact diencepha-lon of rats given subcutaneous injections of CSH relative to vehicle-injected controls.92 In-creased MnSOD activity in liver mitochondria derived from aged humans has been proposedas a mechanism whereby senescent tissues cope with an increased oxidative burden.93 El-evated MnSOD levels reported in the substantia nigra of subjects with Parkinson’s disease94
have similarly been interpreted as a response to excessive oxidative challenge in this condi-tion.95 Oxidative stress is thus a likely mediator of increased MnSOD gene transcriptionand enzymatic activity observed in CSH-exposed astroglia.92
As described above, EV-related astrocyte granulation in the hypothalamic arcuatenucleus occurs in the context of an antecedent cellular stress response.52 We conjecturedthat, analogously to the action of CSH, estradiol may promote a concerted HSP response inARC astroglia via the generation of pro-oxidant intermediates. The mammalian hypothala-mus contains estrogen 2/4-hydroxylases which catalyze the conversion of estradiol to 2- and4-hydroxyestradiol (catecholestrogens).96,97 Subsequent peroxidase-catalyzed reactions trans-form catecholestrogens to highly reactive semiquinone radicals.98 Spontaneous autoxida-tion of catechol groups may, additionally, generate pro-oxidant species including H2O2 andsuperoxide anion.98 Thus, although native estradiol-17β has been shown under certain cir-cumstances to possess antioxidant properties,99 catecholestrogen-derived free radical spe-cies may mediate a cellular stress response and the accumulation of iron-rich cytoplasmicgranules in subpopulations of hypothalamic astroglia. A vicious cycle may then ensuewhereby iron-mediated oxidation of catechol moieties within astroglia produces oxyradicalswhich further stimulate HSP overexpression and the formation of redox-active cytoplasmicinclusions. In summary, prolonged or repeated exposure to oxidative stress may be the “fi-nal common pathway” responsible for activation of the cellular stress (heat shock) responseand subsequent biogenesis of peroxidase-positive astroglial inclusions in vitro and in theintact aging nervous system. This hypothesis is supported by the fact that X-irradiation, aknown generator of intracellular free radical intermediates, increases numbers of peroxi-dase-positive glial granules in the rat hypothalamus in a dose-dependent manner.100 Directevidence implicating oxidative stress in the generation of peroxidase-positive astroglial in-clusions is presented in the following section.

Astrocytes in Brain Aging and Neurodegeneration220
Pro-oxidant Effects of CSH on Astroglial MitochondriaEffete, iron-laden mitochondria are the primary subcellular precursors of peroxidase-
positive cytoplasmic inclusions in CSH-exposed astroglial cultures, and these glial inclu-sions invariably exhibit mitochondrial epitopes in the intact aging rat and human brain(chapter 9). We determined that mitochondrial distension and disorganization of cristaeare the earliest morphological changes visible in cultured rat astroglia by transmission elec-tron microscopy following CSH exposure (chapter 9). Mitochondrial swelling is acknowl-edged to be an important bio-marker of intracellular oxidative stress101 and it occurs withaging,102 following heat shock,103 and under conditions of increased osmotic pressure dueto compromised membrane integrity.104 Direct evidence of oxidative damage in biologicalsystems can be obtained by measuring the accumulation of oxidized proteins, lipids, andnucleic acids in whole cells and in various subcellular compartments.105 Significant increasesin lipid peroxide levels and oxidative DNA lesions have been amply documented in mam-malian mitochondria as a function of advancing age.93,106,107 Of all subcellular compart-ments, mitochondria normally represent the most abundant source of endogenous pro-oxidants.108 In young tissues, free radicals generated in the inner mitochondrial membraneby step-wise reduction of molecular oxygen during oxidative phosphorylation are normallytightly bound to the cytochromes of the electron transport chain and produce relativelylittle oxidative damage. In aging cells, on the other hand, fidelity of electron transport isprogressively compromised, resulting in increased oxidative damage to the mitochondrialmembranes, to mitochondrial DNA and other cellular constituents. The mutated mito-chondrial genome, in turn, codes for aberrant electron transport chain proteins, resultingin a vicious spiral of further free radical “leakage” and oxidative injury.108 In addition, vari-ous hemoproteins constituting the mitochondrial electron transport system sustain thioloxidation reactions with concomitant generation of cytotoxic pro-oxidant species.86
In light of the above, we determined whether oxidative stress is an important mecha-nism mediating CSH-related injury to isolated astroglial mitochondria (granule precur-sors).92 Administration of CSH (600-1000 µM) to purified mitochondrial suspensions de-rived from cultured rat astroglia resulted in significant mitochondrial lipid peroxidationrelative to non-CSH-treated (control) preparations. Conceivably, abundant mitochondrialheme ferrous iron sustains the autoxidation of CSH (to cystamine) with concurrent genera-tion of reactive oxygen species.87 The latter, in turn, may initiate or exacerbate oxidativedamage to the mitochondrial compartment. These observations are consistent with earlierdata indicating that inhibition of CSH autoxidation prevents CSH-induced lipid peroxidationof rat liver mitochondria.109 Addition of catalase significantly attenuates CSH-related mito-chondrial lipid peroxidation, suggesting that H2O2 plays an important role in the transfor-mation of normal astroglial mitochondria to metal-laden cytoplasmic inclusions in vitroand in astrocytes of the aging subcortical brain. Indeed, prolonged H2O2 exposure pro-motes the accumulation of (mitochondria-derived) peroxidase-positive inclusions in cul-tured rat astroglia akin to the effects of CSH.45
CSH (Paradoxically) Confers Cytoprotection to Astroglia Concomitantwith Mitochondrial Injury
Although purified astroglial mitochondria exhibited enhanced sensitivity to lipidperoxidation in the presence of CSH,92 whole cell lysates derived from CSH-treated astroglialcultures consistently manifested lower levels of lipid peroxidation relative to untreated con-trols.83 This latter observation is consistent with the observation that CSH reduces lipidperoxidation in rat liver microsomes in vitro.110 The apparent discrepancy in the behaviorof CSH is reconciled by the fact that aminothiols may act either as oxidizing or reducingagents depending on the redox status of their microenvironments.87,111 Redox potentials of

221Astrocyte Granulogenesis and the Cellular Stress Response
different subcellular compartments have been shown to vary within a given cell.112 In cellu-lar compartments containing relatively low amounts of redox-active transition metals, theantioxidant properties of CSH may prevail over its oxyradical-generating capacity. Indeed,CSH has been used for many years in experimental oncology to protect against excessiveradiation-induced tissue damage.113 CSH may confer cytoprotection under these circum-stances by direct scavenging of reactive oxygen species111,114 or by chelation of catalytic tran-sition metals.109,115 We observed that short term (6 hr) exposure of cultured astroglia toCSH confers enhanced resistance to mechanoenzymatic stress (trypsinization) in compari-son with untreated controls. Prolonged (12 day) CSH treatment additionally protects cul-tured astrocytes from subsequent H2O2 toxicity relative to control preparations (Fig. 10.6).45
Since glial CSH is no longer detectable by HPLC at the end of a 24 h washout period preced-ing the H2O2 challenge,45 it would seem that the aminothiol does not serve as a directprotectant in the cytotoxicity assays. It is far more likely that the upregulation of MnSODgene expression and the elaboration of various stress proteins in cultured astroglia follow-ing CSH exposure (vide supra) are important mediators of this glioprotective effect. Theaugmented MnSOD activity may curtail the accumulation of cytotoxic superoxide anion,while the HSPs may function to prevent deleterious aggregation of unfolded or aberrantproteins and protect lipid membranes and the translational apparatus from stress-induceddamage.116-119 Furthermore, rapid activation of Ub in CSH-exposed astroglia may facilitateglial recovery and survival by assisting in the degradation of denatured protein complexesand the re-establishment of normal protein homeostasis. Finally, the enhanced HO-1 activ-ity may confer some degree of cytoprotection by augmenting the degradation of pro-oxi-dant metalloporphyrins (heme) to antioxidant bile pigments,16 thereby promoting the res-toration of a more favorable redox microenvironment. That similar glioprotective responsesmay be manifest in situ is supported by the observations that:
1. glial cultures are easier to establish when the cells are harvested from postmortemAlzheimer brain than from age-matched, nondemented controls (A. LeBlanc, per-sonal communication); and
2. astrocytes derived from rodent hippocampi previously lesioned (“stressed”) withibotenic acid exhibit enhanced survival and proliferation in vitro relative to thoseprocured from nonlesioned controls.120
As alluded to throughout this volume, astrocyte hypertrophy, GFAP biosynthesis, andpossibly astroglial hyperplasia (reactive gliosis) are fundamental pathological features ofvirtually all major human neurodegenerative disorders and occur, albeit to a lesser extent,in the course of normal brain senescence. By promoting glial survival under these condi-tions, stress-induced upregulation of cytoprotective mechanisms in astrocytes (simulatedby CSH exposure) may facilitate the establishment of gliosis and the accumulation of intra-cellular inclusions in the face of concomitant neuronal depletion.
Astrocyte Senescence and the Origin of Corpora AmylaceaCorpora amylacea (CA) are glycoproteinacious, cytoplasmic inclusions that accumu-
late in subpial and periventricular regions of human brain in the course of normal aging.Numbers of CA are reportedly increased in AD121,122 and other neurodegenerative condi-tions123-126 relative to age-matched, normal controls. CA are most frequently encounteredwithin astroglia or as extracellular deposits.68,70,127 CA may occasionally arise within neu-ritic processes68,128-131 and they have also been reported in a variety of nonneural tissues.132-134
Many of the tinctorial and histochemical properties of CA have been delineated (see chap-ter 4). Yet, their subcellular origin and the mechanism(s) responsible for their biogenesisremain enigmatic. Human CA share many topographical, histochemical and antigenic fea-tures with the peroxidase-positive astrocytic granules considered in this and the preceding

Astrocytes in Brain Aging and Neurodegeneration222
chapter. Both types of inclusion predominate in periventricular brain regions,101,135,136 pro-gressively accumulate with aging,81,120,137 and exhibit affinities for periodic acid-Schiff (PAS)and Gomori’s chrome alum hematoxylin stains,138 metachromasia with toluidine blue,138,139
and nonenzymatic peroxidase activity.82,135,138 In addition, both CA and the peroxidase-positive glial granules are ubiquitinated and contain several heat shock proteins.53,71,67,140
We recently demonstrated consistent immunolocalization of two mitochondrial proteins,sulfite oxidase141 and HSP60,142,143 to the red autofluorescent (peroxidase-positive) astro-cyte granules and CA in subependymal regions of senescent and Alzheimer-diseased hu-man brain, as well as in smears of purified human CA.71 In addition, CA in situ and in thepurified fractions were noted to contain nucleic acids on the basis of anti-DNA staining.
Fig. 10.6. Automated MTT cell viability as-say depicts cytotoxic effects of trypsiniza-tion and H2O2 exposure on fixed numbers(40,000 cells per well) of control and CSH-pretreated astrocytes. Optical density cor-relates directly with cell viability. (A) Longterm CSH exposure (880 µM DIV 6-18).CSH-pretreated cells (♦) exhibit increasedresistance to mechanical trauma and H2O2
exposure relative to controls ( ). (B) Shortterm CSH exposure (880 µM x 6 h). As in(A), CSH-pretreated cells (♦) exhibit robustresistance to mechanical trauma relative tocontrols ( ). However, normalizing forthe effects of mechanical stress, both groupsshow similar declines in cell viability withincreasing H2O2 concentrations. An aster-isk denotes a significant difference fromcontrol values (p<0.050 by Student-Newman-Keuls post hoc test). An open star denotes firstsignificant decline in cell viability relative to respective conditions at 0 µM H2O2 (p<0.050 byStudent-Newman-Keuls). Reprinted with permission from Mydlarski MB et al, J Neurochem1993; 61:1755-1765.

223Astrocyte Granulogenesis and the Cellular Stress Response
That these nucleic acids are of mitochondrial origin is strongly suggested by the robustcolocalization of sulfite oxidase and DNA within CA in dual-labeled preparations. Thesedata are consistent with earlier electron micrographs depicting damaged mitochondrialcomponents within CA of the optic nerve144 and within “granular glycogen bodies” in se-nescent human astrocytes.145 Thus far, the only major divergent histochemical feature be-tween peroxidase-positive astrocyte granules and CA is the presence of orange-redautofluorescence in the former and the absence of endogenous fluorescence in the latter.On the basis of the evidence available to date, we submit that:
1. the peroxidase-positive astrocyte granules may be structural precursors of CA insenescent human brain; and
2. degenerate mitochondria within periventricular astrocytes are a major source ofautofluorescent cytoplasmic inclusions and CA in the aging human brain.
Conceivably, during the putative maturation of peroxidase-positive granules to CA,progressive glycation of autofluorescent mitochondrial substrates may be responsible forthe quenching of endogenous fluorescence in the larger inclusions.71 The histochemistryand morphology of CA and Gomori (peroxidase)-positive astrocyte granules are summa-rized in Table 10.4.
As described above, CSH-derived free radical intermediates stimulate the transforma-tion of normal mitochondria to peroxidase-positive, autofluorescent inclusions in primaryastrocyte cultures62,146 and in the intact rat brain.48,147 More recently, we observed that longterm (90 day) exposure of neonatal rat glial cultures to CSH148 and subcutaneous adminis-tration of CSH to adult albino rats148a results in the formation of large spherical, PAS-posi-tive astrocytic inclusions which are highly reminiscent of, if not identical to, human CA. Asin the case of human CA, the CSH-induced CA-like inclusions lack endogenous fluores-cence and exhibit nonenzymatic peroxidase activity and consistent immunostaining for themitochondrial protein sulfite oxidase (Fig. 10.7).148,148a These findings further support ourcontention that mitochondrial damage and autophagy play an important role in the bio-genesis of CA (and peroxidase-positive granules) in astrocytes of the aging periventricularbrain. Taken together with observations reported in chapter 9, the data reviewed hereinsuggest a mechanism for the biogenesis of CA in aging astrocytes whereby sustained orrepeated intracellular oxidative stress serves as a “final common pathway” mediating thefollowing sequence of events (Fig. 10.8).148
Stage 1: Mitochondria swell, become autofluorescent, and sequester redox-active iron(nonenzymatic peroxidase activity).56,62 The astrocytes undergo a cellular stress reaction asevidenced by upregulation of HSP27, 72 and 90, ubiquitin, and HO-1.45,53,135
Stage 2: The abnormal mitochondria fuse with lysosomes, undergo macroautophagyand incorporate HSP27 and ubiquitin (formation of Gomori-positive “stress granules”).Other stress proteins exhibit partial or no colocalization to the autofluorescent inclusionsand remain largely confined to granule-free cytoplasm (HSP72, HO-1) or undergo translo-cation to the nucleus (HSP90).48,53
Stage 3: Granule constituents (proteins) become glycosylated,149,150 with quenching ofautofluorescence and displacement of mitochondrial components to the inclusion periph-ery (nascent CA). It is conceivable that during the formation of CA, progressive glycosylationof damaged astrocyte mitochondria may abrogate free radical generation by iron-contain-ing mitochondrial proteins. The glycosylation of redox-active mitochondria may thus rep-resent a protective mechanism which serves to limit oxidative injury within the aging ner-vous system.
Stage 4: Many stressed astroglia eventually degenerate and mature, residual CA aredeposited in the extracellular space. In accord with this model, excessive oxidative stressreported in the brains of Alzheimer subjects39,151-154 may exacerbate senescence-related injury

Astrocytes in Brain Aging and Neurodegeneration224
Tabl
e 10
.4. H
isto
chem
istr
y an
d m
orph
olog
y of
CA
and
Gom
ori-
posi
tive
ast
rocy
te in
clus
ions
in h
uman
and
rat
bra
in t
issu
esa
Rat
Bra
inH
uman
Bra
in
Sect
ions
Cul
ture
sSe
ctio
nsSe
ctio
nsC
ell F
ract
ions
GA
IG
AI
GA
IC
AC
A
Cel
l ori
gin
Ast
rocy
tes
Ast
rocy
tes
Ast
rocy
tes
Ast
rocy
tes
Ast
rocy
tes
and
(epe
ndym
a)(e
pend
yma)
(epe
ndym
a)an
d ne
uron
s(?e
pend
yma)
neur
ons
(?ep
endy
ma)
Dis
trib
utio
nPV
, lim
bic
—PV
, lim
bic
PV, l
imbi
c, s
ubpi
al—
Acc
umul
atio
n w
ith a
geY
esY
esY
esY
es—
Size
0.5-
10 µ
m0.
5-10
µm
0.5-
10 µ
m10
-50
µm
10-5
0 µm
Shap
ePl
eom
, rou
ndPl
eom
, rou
ndPl
eom
, rou
ndR
ound
Rou
nd
Vis
ible
pig
men
tN
oN
oN
oN
oN
o
Indu
ctio
nsC
yste
amin
eY
esY
es?
??
Estr
ogen
Yes
??
??
X-i
rrad
iatio
nY
esY
es?
??
Aut
oflu
ores
cenc
eY
es (r
ed)
Yes
(red
)Y
es (r
ed)
No
No
Pero
xida
se a
ctiv
ityY
esY
esY
esY
esY
espH
ran
ge3-
10.5
3-11
3-11
3-10
.53-
10.5
AT
inhi
bitio
nN
oN
oN
oN
oN
oH
eat i
nact
ivat
ion
No
No
No
No
No

225Astrocyte Granulogenesis and the Cellular Stress Response
PAS
Yes
(lar
ger)
Yes
(lar
ger)
Yes
(lar
ger)
Yes
Yes
CA
HY
esY
esY
esY
esY
es
DN
AY
esY
esY
esY
esY
es
Arg
yrop
hilia
Yes
Yes
?Y
es?
Tolu
idin
e bl
ueY
esY
esY
esY
es?
met
achr
omas
ia
Iron
-ric
hY
esY
es?
??
Sulfu
rY
esY
es?
Yes
Yes
Hea
t sho
ck p
rote
ins
Yes
Yes
Yes
Yes
Yes
Ubi
quiti
nY
esY
esY
esY
esY
es
Mito
chon
dria
lY
esY
esY
esY
esY
esA
ntig
ens
a AT,
am
inot
riaz
ole;
CA
H, c
hrom
e al
um h
emat
oxyl
in; G
AI,
Gom
ori-
poiti
ve a
stro
cyte
incl
usio
ns; P
AS,
per
iodi
c ac
id S
chiff
; ple
om, p
leom
orph
ic; P
V,
peri
vent
ricu
lar.

Astrocytes in Brain Aging and Neurodegeneration226
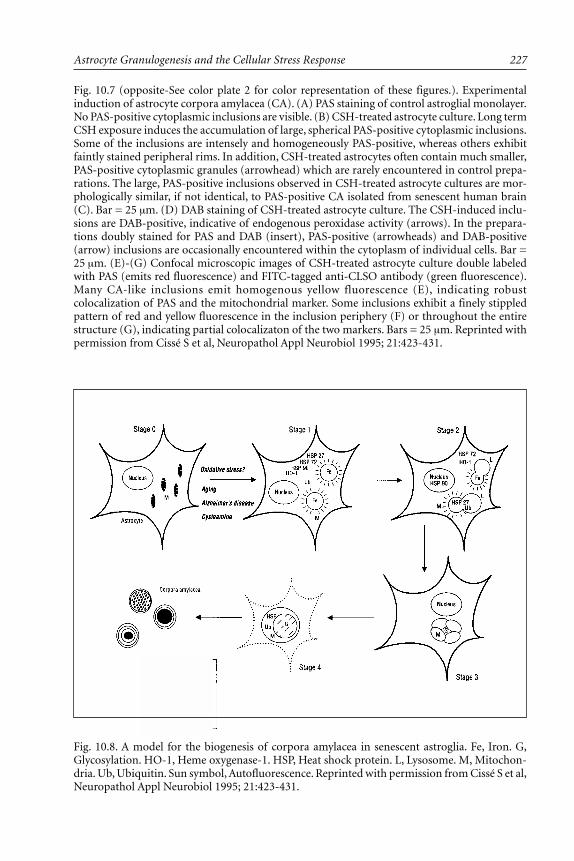
227Astrocyte Granulogenesis and the Cellular Stress Response
Fig. 10.7 (opposite-See color plate 2 for color representation of these figures.). Experimentalinduction of astrocyte corpora amylacea (CA). (A) PAS staining of control astroglial monolayer.No PAS-positive cytoplasmic inclusions are visible. (B) CSH-treated astrocyte culture. Long termCSH exposure induces the accumulation of large, spherical PAS-positive cytoplasmic inclusions.Some of the inclusions are intensely and homogeneously PAS-positive, whereas others exhibitfaintly stained peripheral rims. In addition, CSH-treated astrocytes often contain much smaller,PAS-positive cytoplasmic granules (arrowhead) which are rarely encountered in control prepa-rations. The large, PAS-positive inclusions observed in CSH-treated astrocyte cultures are mor-phologically similar, if not identical, to PAS-positive CA isolated from senescent human brain(C). Bar = 25 µm. (D) DAB staining of CSH-treated astrocyte culture. The CSH-induced inclu-sions are DAB-positive, indicative of endogenous peroxidase activity (arrows). In the prepara-tions doubly stained for PAS and DAB (insert), PAS-positive (arrowheads) and DAB-positive(arrow) inclusions are occasionally encountered within the cytoplasm of individual cells. Bar =25 µm. (E)-(G) Confocal microscopic images of CSH-treated astrocyte culture double labeledwith PAS (emits red fluorescence) and FITC-tagged anti-CLSO antibody (green fluorescence).Many CA-like inclusions emit homogenous yellow fluorescence (E), indicating robustcolocalization of PAS and the mitochondrial marker. Some inclusions exhibit a finely stippledpattern of red and yellow fluorescence in the inclusion periphery (F) or throughout the entirestructure (G), indicating partial colocalizaton of the two markers. Bars = 25 µm. Reprinted withpermission from Cissé S et al, Neuropathol Appl Neurobiol 1995; 21:423-431.
Fig. 10.8. A model for the biogenesis of corpora amylacea in senescent astroglia. Fe, Iron. G,Glycosylation. HO-1, Heme oxygenase-1. HSP, Heat shock protein. L, Lysosome. M, Mitochon-dria. Ub, Ubiquitin. Sun symbol, Autofluorescence. Reprinted with permission from Cissé S et al,Neuropathol Appl Neurobiol 1995; 21:423-431.

Astrocytes in Brain Aging and Neurodegeneration228
to astrocyte mitochondria which, in turn, gives rise to the plethora of CA reported in thiscondition.
Our mitochondrial hypothesis of CA biogenesis does not preclude the formation ofthese inclusions within nonastrocytic substrates. The relative preponderance of CA in se-nescent astroglia may be due to the sheer abundance of these cells as well as to their uniquemetabolic properties. In contradistinction to neurons, astroglia exposed to mitochondrialtoxins exhibit long term survival by converting to robust anaerobic metabolism.155 Thiscould allow sufficient time for the gradual transformation of damaged mitochondria to CAin these cells. Conversely, CA may be less often encountered in neuronal processes and othernonastrocytic substrates, because in these tissues the toxicity “window” permitting bothsufficient mitochondrial injury and sustained cell viability may be relatively narrow.148
The data reviewed in chapters 9 and 10 of this volume provide, in our estimation,compelling evidence that exposure to the simple aminothiol compound CSH fully reca-pitulates many of the morphological and biochemical changes incurred by populations ofsubcortical astroglia as these cells naturally age. As such, CSH-treated astroglia should con-tinue to serve as a useful model to delineate further the role of the cellular stress (heatshock) response in the biogenesis of glial inclusions and the establishment of reactive gliosisin the aging and degenerating CNS.
AcknowledgmentsThe authors thank Mrs. Kay Berckmans and Mrs. Adrienne Liberman for assistance
with the preparation of this manuscript. This work is supported by grants from the MedicalResearch Council of Canada (HMS.JRB) and the Fonds de la Recherche en Santé du Québec(HMS).
References1. De Waegh S, Brady ST. Axonal transport of a clathrin uncoating ATPase (HSC70): A role
for HSC70 in the modulation of coated vesicle assembly in vivo. J Neurosci Res 1989;23:433-440.
2. Brown IR. Induction of heat shock (stress) genes in the mammalian brain by hyperther-mia and other traumatic events: A current perspective. J Neurosci Res 1990; 27:247-255.
3. Dienel GA, Kiessling M, Jacewicz M, Pulsinelli W. Synthesis of heat shock proteins in ratbrain cortex after transient ischemia. J Cereb Blood Flow Metab 1986; 6:505-510.
4. Tytell M, Barbe MF, Brown IR. Stress (heat shock) protein accumulation in the centralnervous system. Adv Neurol 1993; 59:293-303.
5. Chopp M, Li Y, Dereski MO et al. Neuronal injury and expression of 72-kDa heat-shockprotein after forebrain ischemia in the rat. Acta Neuropathol 1991; 83:66-71.
6. Simon RP, Cho H, Gwinn R, Lowenstein DH. The temporal profile of 72-kDa heat-shockprotein expression following global ischemia. J Neurosci 1991; 11:881-889.
7. Hayashi T, Takada K, Matsuda M. Subcellular distribution of ubiquitin-protein conjugatesin the hippocampus following transient ischemia. J Neurosci Res 1992; 31:561-564.
8. Mayer RJ, Arnold J, Laszlo L et al. Ubiquitin in health and disease. Biochim Biophys Acta1991; 1089:141-157.
9. Chopp M, Chen H, Ho KL et al. Transient hyperthermia protects against subsequent fore-brain ischemic cell damage in the rat. Neurol 1989; 39:1396-1398.
10. Kitagawa K, Matsumoto M, Kuwabara K et al. Ischemia tolerance phenomenon detectedin various brain regions. Brain Res 1991; 561:203-211.
11. Marini AM, Kozuka M, Lipsky RL, Nowak TS Jr. 70-Kilodalton heat shock protein induc-tion in cerebellar astrocytes and cerebellar granule cells in vitro: Comparison with immu-nocytochemical localization after hyperthermia in vivo. J Neurochem 1990; 54:1509-1516.
12. Nishimura RN, Dwyer BE, Clegg K et al. Comparison of the heat shock response in cul-tured cortical neurons and astrocytes. Mol Brain Res 1991; 9:39-45.

229Astrocyte Granulogenesis and the Cellular Stress Response
13. Ewing JF, Maines MD. Rapid induction of heme oxygenase 1 mRNA and protein by hy-perthermia in rat brain: Heme oxygenase 2 is not a heat shock protein. Proc Natl Acad SciUSA 1991; 88:5364-5368.
14. Ewing JF, Maines MD. Glutathione depletion induces heme oxygenase-1 (HSP32) mRNAand protein in rat brain. J. Neurochem 1993; 60:1512-1519.
15. Dwyer BE, Nishimura RN, Lu SY. Differential expression of heme oxygenase-1 in culturedcortical neurons and astrocytes determined by the aid of a new heme oxygenase antibody.Response to oxidative stress. Mol Brain Res 1995; 30:37-47.
16. Stocker R. Induction of haem oxygenase as a defense against oxidative stress. Free RadicRes Comm 1990; 9:101-112.
17. Sharp FR, Sagar SM. Alterations in gene expression as an index of neuronal injury: Heatshock and the immediate early gene response. Neurotoxicol 1994; 15:51-60.
18. Uney JB, Leigh PN, Marsden CD et al. Stereotaxic injection of kainic acid into the stria-tum of rats induces synthesis of mRNA for heat shock protein 70. FEBS Lett 1988;235:215-218.
19. Lowenstein DH, Simon RP, Sharp FR. The pattern of 72-kDa heat shock protein-like im-munoreactivity in the rat brain following flurothyl-induced status epilepticus. Brain Res1990; 531:173-182.
20. Brown IR, Rush S, Ivy GO. Induction of a heat shock gene at the site of tissue injury inthe rat brain. Neuron 1989; 2:1559-1564.
21. Gower DJ, Hollman C, Lee S, Tytell M. Spinal cord injury and the stress protein response.J Neurosurg 1989; 70:605-611.
22. Blake MJ, Fargnoli J, Gershon D, Holbrook NJ. Concomitant decline in heat-induced hy-perthermia and hsp70 mRNA expression in aged rats. Am J Physiol 1991; 260:R663-667.
23. Pardue S, Groshan K, Raese JD, Morrison-Bogorad M. Hsp70 mRNA induction is reducedin neurons of aged rat hippocampus after thermal stress. Neurobiol Aging 1992; 13:661-672.
24. Tytell M, Yamaguchi K, Yamaguchi K. Role of heat shock protein 70 (hsp70) in photore-ceptor cell survival in the aged rat. In: Hollyfield JG, LaVail MM, Anderson RE, eds. Reti-nal Degeneration: Clinical and Laboratory Applications. New York: Plenum PublishingCorp., 1993.
25. Tytell M. Heat shock proteins in the retina and optic nerve. In: Mayor RJ, Brown IR, eds.Heat Shock Proteins in the Nervous System. San Diego: Academic Press, 1994:83-100.
26. Shock NW. Systems integration. In: Finch CE, Hayflick L, eds. Handbook of the Biologyof Aging. New York: Van Nostrand Reinhold Co., 1977:639-665.
27. Heydari AR, Takahashi R, Gutsmann A et al. Hsp70 and aging. Experientia 1994;50:1092-1098.
28. Pappolla MA, Omar R, Saran B. Abnormal ubiquitinilated deposits highlight an age-re-lated protein change. Am J Pathol 1989; 135:585-591.
29. Dickson DW, Wertkin A, Kress Y et al. Ubiquitin immunoreactive structures in normalhuman brains. Lab Invest 1990; 63:87-99.
30. Abe H, Mehraein P, Weis S. Unusual ubiquitin-positive glial cells in the globus pallidus ofnormal elderly human brains. Neuropathol Appl Neurobiol 1994;20:609-613.
31. Wang GP, Khatoon S, Iqbal K, Grundke-Iqbal I. Brain ubiquitin is markedly elevated inAlzheimer disease. Brain Res 1991; 566:146-151.
32. Mori H, Kondo J, Ihara Y. Ibiquitin is a component of paired helical filaments inAlzheimer’s disease. Science 1987; 235:1641-1644.
33. Perry G, Friedman R, Shaw G, Chau V. Ubiquitin is detected in neurofibrillary tangles andsenile plaque neurites of Alzheimer disease brains. Proc Natl Acad Sci USA 1987;84:3033-3036.
34. Hamos JE, Oblas B, Pulaski-Salo D et al. Expression of heat shock proteins in Alzheimer’sdisease. Neurol 1991; 41:345-350.
35. Renkawek K, Bosman GJCGM, de Jong WW. Expression of small heat-shock protein hsp27in reactive gliosis in Alzheimer disease and other types of dementia. Acta Neuropathol1994; 87:511-519.

Astrocytes in Brain Aging and Neurodegeneration230
36. Shinohara H, Inaguma Y, Goto S et al. αB-crystallin and HSP28 are enhanced in the cere-bral cortex of patients with Alzheimer’s disease. J Neurol Sci 1993; 119:203-208.
37. Renkawek K, Voorter CEM, Bosman GJCGM et al. Expression of αB-crystallin inAlzheimer’s disease. Acta Neuropathol 1994; 87:155-160.
38. Smith MA, Kutty RK, Richey PL et al. Heme oxygenase-1 is associated with the neurofibril-lary pathology of Alzheimer’s disease. Am J Pathol 1994; 145:42-47.
39. Schipper HM, Cissé S, Stopa EG. Expression of heme oxygenase-1 in the senescent andAlzheimer-diseased brain. Ann Neurol 1995; 37:758-768.
40. Tamaoka A, Mizusawa H, Mori H, Shoji S. Ubiquinated αB-crystallin in glial cytoplasmicinclusions from the brain of a patient with multiple system atrophy. J Neurol Sci 1995;129:192-198.
41. Goldman JE, Corbin E. Rosenthal fibers contain ubiquitinated αB-crystallin. Am J Pathol1991; 139:933-938.
42. Dickson DW, Yen SHC. Ubiquitin, the cytoskeleton and neurodegenerative diseases. In:Mayer J, Brown I, eds. Heat Shock Proteins in the Nervous System. San Diego: AcademicPress, 1994:235-262.
43. Wilkinson KD. Cellular roles of ubiquitin. In: Mayor RJ, Brown IR, eds. Heat Shock Pro-teins in the Nervous System. San Diego: Academic Press, 1994:191-234.
44. Peterson TC, Peterson MR, Williams CN. The role of heme-oxygenase and aryl hydrocar-bon hydroxylase in the protection by cysteamine from acetaminophen hepatotoxicity.Toxicol Appl Pharmacol 1989; 97:430-439.
45. Mydlarski MB, Liang J-J, Schipper HM. Role of the cellular stress response in the biogen-esis of cysteamine-induced astrocytic inclusions in primary culture. J Neurochem 1993;61:1755-1765.
46. Collier NC, Heuser J, Levy MA, Schlesinger MJ. Ultrastructural and biochemical analysisof the stress granula in chicken embryo fibroblasts. J Cell Biol 1988; 106:1131-1139.
47. Nover L, Scharf KD, Neumann D. Cytoplasmic heat shock granules are formed from pre-cursor particles and are associated with a specific set of mRNAs. Mol Cell Biol 1989;9:1298-1308.
48. Schipper HM, Mydlarski MB, Wang X. Cysteamine gliopathy in situ: A celular stress modelfor the biogenesis of astrocytic inclusions. J Neuropathol Exp Neurol 1993; 52:399-410.
49. Mydlarski MB, Liberman A, Schipper HM. Estrogen induction of glial heat shock proteins:Implications for hypothalamic aging. Neurobiol Aging 1995; 16:977-981.
50. Ceccatelli S, Hokfelt T, Hallman H et al. Immunohistochemical analysis of the effects ofcysteamine on somatostatin-like immunoreactivity in the rat central nervous system. Pep-tides 1987; 8:371-384.
51. Kwok RPS, Cameron JL, Faller DV, Fernstrom JD. Effects of cysteamine administration onsomatostatin biosynthesis and levels in rat hypothalamus. Endocrinol 1992; 131:2999-3009.
52. Vogel DG, Malekzadeh MH, Cornford ME et al. Central nervous system involvement innephropathic cystinosis. J Neuropathol Exp Neurol 1990; 49:591-599.
53. Mydlarski MB, Schipper HM. Stress protein co-localization to autofluorescent astrocyticinclusion in situ and in cysteamine-treated glial cultures. Brain Res 1993; 627:113-121.
54. Brawer JR, Sonnenschein C. Cytopathological effects of estradiol on the arcuate nucleus ofthe female rat. A possible mechanism for pituitary tumorigenesis. Am J Anatomy 1975;144:57-88.
55. Srebro Z, Lach H. The ultrastructure of the periventricular glia in the brains of rats andmice. Folia Biol (Krakow) 1987; 35:131-136.
56. McLaren J, Brawer JR, Schipper HM. Iron content correlates with peroxidase activity incysteamine-induced astroglial organelles. J Histochem Cytochem 1992; 40:1887-1897.
57. Chronopoulos S, Lembo P, Alizadeh-Khiavi K, Ali-Khan Z. Ubiquitin: Its potential signifi-cance in murine AA amyloidogenesis. J Pathol 1992; 167:249-259.
58. Carlson N, Rogers S, Rechsteiner M. Microinjection of ubiquitin: Changes in protein deg-radation in HeLa cells subjected to heat-shock. J Cell Biol 1987; 104:547-555.
59. Finley D, Ozkaynak E, Varshavsky A. The yeast polyubiquitin gene is essential for resis-tance to high temperature, starvation and other stresses. Cell 1987; 49:1035-1046.

231Astrocyte Granulogenesis and the Cellular Stress Response
60. Haas AL, Bright PM. The immunochemical detection and quantitation of intracellularubiquitin-protein conjugates. J Biol Chem 1985; 260:12464-12473.
61. Landon M, Lowe J, Mayer RJ. Ubiquitin, endosomes-lysosomes and neurodegenerative dis-eases. In: Mayer J, Brown I, eds. Heat Shock Proteins in the Nervous System. San Diego:Academic Press, 1994:263-287.
62. Brawer JR, Reichard G, Small L, Schipper HM. The origin and composition of peroxidase-positive granules in cysteamine-treated astrocytes in culture. Brain Res 1994; 633:9-20.
63. Doherty FJ, Osborn NU, Wassell JA et al. Ubiquitin-protein conjugates accumulate in thelysosomal system of fibroblasts treated with cysteine proteinase inhibitors. Biochem J 1989;263:47-55.
64. Laszlo L, Doherty FJ, Osborn NU, Mayer RJ. Ubiquitinated-protein conjugates are specifi-cally enriched in the lysosomal system of fibroblasts. FEBS Lett 261:365-368.
65. Schwartz AL, Ciechanover A, Brandt RA, Geuze HJ. Immunoelectron microscopic local-ization of ubiquitin in hepatoma cells. EMBO J 1988; 7:2961-2966.
66. Manetto V, Abdul-Karim FW, Perry G et al. Selective presence of ubiquitin in intracellularinclusions. Am J Pathol 1989; 134:505-513.
67. Cissé S, Perry G, Lacoste-Royal G et al. Immunochemical identification of ubiquitin andheat-shock proteins in corpora amylacea from normal aged and Alzheimer’s disease brains.Acta Neuropathol 1993; 85:233-240.
68. Anzil AP, Herrlinger H, Blinzinger K, Kronski D. Intraneuritic corpora amylacea. VirchowsArch [A] 1974; 364:297-304.
69. Palmucci L, Anzil AP, Luh S. Intra-astrocytic glycogen granules and corpora amylacea stainpositively for polyglucosan: A cytochemical contribution on the fine structural polymor-phism of particulate polysaccharides. Acta Neuropathol (Berl) 1982; 57:99-102.
70. Ramsay HJ. Ultrastructure of corpora amylacea. J Neuropathol Exp Neurol 1965; 24:25-39.71. Schipper HM, Cissé S. Mitochondrial constituents of corpora amylacea and autofluorescent
astrocytic inclusions in senescent human brain. Glia 1995; 14:55-64.72. Shang F, Taylor A. Oxidative stress and recovery from oxidative stress are associated with
altered ubiquitin conjugating and proteolytic activities in bovine lens epithelial cells.Biochem J 1995; 307:297-303.
73. Haas AL, Warms JV, Hershko A, Rose IA. Ubiquitin-activating enzyme. Mechanism androle in protein-ubiquitin conjugation. J Biol Chem 1982; 257:2543-2548.
74. Herskho A, Heller H, Elias S, Ciechanover A. Components of ubiquitin-protein ligase sys-tem. Resolution, affinity purification, and role in protein breakdown. J Biol Chem 1983;258:8206-8214.
75. Becker J, Craig EA. Heat-shock proteins as molecular chaperones. Eur J Biochem 1994;219:11-23.
76. Welch WJ. Mammalian stress response: cell physiology, structure/function of stress pro-teins, and implications for medicine and disease. Physiol Rev 1992; 72:1063-1081.
77. Findly RC, Gillies RJ, Shulman RG. In vivo phosphorous-31 nuclear magnetic resonancereveals lowered ATP during heat shock of Tetrahymena. Science 1983; 219:1223-1225.
78. Stevenson MA, Calderwoud SK, Hahn GM. Rapid increases in inositol trisphosphate andintracellular Ca++ after heat shock. Biochem Biophys Res Commun 1981; 137:826-833.
79. Weitzel G, Pilatus U, Rensing L. Similar dose response of heat shock protein synthesis andintracellular pH change in yeast. Exp Cell Res 1985; 159:252-256.
80. Brawer J, Schipper HM, Robaire B. Effects of long-term androgen and estradiol exposureon the hypothalamus. Endocrinology 1983; 112:194-199.
81. Schipper HM, Brawer JR, Nelson JF, Felicio LS, Finch CE. Role of the gonads in the histo-logic aging of the hypothalamic arcuate nucleus. Biol Reprod 1981; 25:413-419.
82. Schipper HM, Lechan RM, Reichlin S. Glial peroxidase activity in the hypothalamic arcu-ate nucleus: Effects of estradiol valerate-induced persistent estrus. Brain Res 1990;507:200-207.
83. Schipper HM. Role of peroxidase-positive astrocytes in estradiol-related hypothalmic dam-age. In: Fedoroff S, Juurlink B, Doucette R. eds. Biology and Pathology of Astrocyte-neu-ron Interactions. New York: Plenum Publishing Corporation, 1993:125-139.

Astrocytes in Brain Aging and Neurodegeneration232
84. Kleopoulos SP, Olazabal UE, Lauber AH et al. Heat-shock proteins 90 Kd and 70Kd in ratbrain and uterus: Cellular localization by immunocytochemistry and in situ hybridization.Society for Neuroscience 1991; 17:432 (abstr).
85. Olazabal UE, Pfaff DW, Mobbs CV. Estrogenic regulation of heat shock protein 90 kDa inthe rat ventromedial hypothalamus and uterus. Mol Cell Endocrinol 1991; 84:174-183.
86. Olazabal UE, Pfaff DW, Mobbs CV. Sex differences in the regulation of heat shock protein70 kDa and 90 kDa in the rat ventromedial hypothalamus by estrogen. Brain Res 1992;596; 311-314.
87. Munday R. Toxicity of thiols and disulphides: Involvement of free-radical species. FreeRad Biol Med 1989; 7:659-673.
88. Lee AS. Coordinated regulation of a set of genes by glucose and calcium ionophores inmammalian cells. TIBS 1987; 12:20-23.
89. Lee AS. Mammalian stress response: Induction of the glucose-regulated protein family. CurrOp Cell Biol 1992; 4:267-273.
90. Liochev SI, Fridovich I. Superoxide radical in Escherichia coli. In: Scandalios JG, ed. Cur-rent Communications in Cell and Molecular Biology. Molecular Biology of Free RadicalScavenging Systems. New York: Cold Spring Harbor Laboratory Press, 1992:213-229.
91. Wong GHW, Kamb A, Tartaglia LA, Goeddel DV. Possible protective mechanisms of tu-mour necrosis factors against oxidative stress. In: Scandalios JG, ed. Current Communica-tions in Cell and Molecular Biology. Molecular Biology of Free Radical Scavenging Sys-tems. New York: Cold Spring Harbor Laboratory Press, 1992:69-96.
92. Manganaro F, Chopra VS, Mydlarski MB et al. Redox perturbations in cysteamine-stressedastroglia: Implications for inclusion formation and gliosis in the aging brain. Free Rad BiolMed 1995; 19:823-835.
93. Yen T-C, King K-L, Lee H-C et al. Age-dependent increase of mitochondrial DNA dele-tions together with lipid peroxides and superoxide dismutase in human liver mitochon-dria. Free Rad Biol Med 1994; 16:207-214.
94. Saggu H, Cooksey J, Dexter D et al. A selective increase in particulate superoxide dismutaseactivity in parkinsonian substantia nigra. J Neurochem 1989; 53:692-697.
95. Jenner P. What process causes nigral cell death in Parkinson’s disease? In: Cedarbaum JM,Gancher ST, eds. Neurological Clinics; Parkinson’s Disease. Montreal: WB Saunders Co.,1992:387-404.
96. Ball P, Knuppen R. Formation of 2- and 4-hydroxyestrogens by brain, pituitary, and liverof the human fetus. J Clin Endocrinol Metab 1978; 47:732-737.
97. Inoue K, Yoshizawa I. Immunocytochemical localization of catecholestrogens in the ratpituitary gland. II. Median eminence. Acta Histochem Cytochem 1989; 22:65-76.
98. Kalyanaraman B, Felix CC, Sealy RC. Semiquinone anion radicals of catechol(amine)s, cat-echol estrogens, and their metal ion complexes. Environ Health Perspect 1985; 64:185-198.
99. Goodman Y, Bruce AJ, Cheng B, Mattson MP. Estrogens attenuate and corticosterone ex-acerbates excitotoxicity, oxidative injury and amyloid β-peptide toxicity in hippocampalneurons. J Neurochem 1996; 66:1836-1844.
100. Srebro Z. Periventricular Gomori-positive glia in brains of X-irradiated rats. Brain Res1971; 35:463.
101. Castilho RF, Kowaltowski AJ, Meinicke AR et al. Permeabilization of the inner mitochon-drial membrane by Ca2+ ions is stimulated by t-butylhydroperoxide and mediated by reac-tive oxygen species generated by mitochondria. Free Rad Biol Med 1995; 18:479-486.
102. Wilson PD, Franks LM. The effect of age on mitochondrial ultrastructure and enzymes.Adv Exp Med Biol 1975; 53:171-183.
103. Welch WJ, Suhan JP. Morphological study of the mammalian stress response: Character-ization of changes in cytoplasmic organelles, cytoskeleton, and nucleoli, and appearance ofintranuclear actin filaments in rat fibroblasts after heat-shock treatment. J Cell Biol 1985;101:1198-1211.
104. Skrede S. Effects of cystamine and cysteamine on the adenosine-triphosphatase activity andoxidative phosphorylation of rat-liver mitochondria. Biochem J 1966; 98:702-710.
105. Gutteridge JMC, Halliwell B. The measurement and mechanism of lipid peroxidation inbiological systems. TIBS 1990; 15:129-135.

233Astrocyte Granulogenesis and the Cellular Stress Response
106. Laganiere S, Yu VP. Modulation of membrane phospholipid fatty acid composition by ageand food restriction. Gerontology 1993; 39:7-18.
107. Yu BP, Suescum EA, Yang SY. Effect of age-related lipid peroxidation on membrane fluid-ity and phospholipase A2: Modulation by dietary restriction. Mech Aging Dev 1992;65:17-33.
108. Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in ag-ing. Proc Natl Acad Sci USA 1994; 91:10771-10778.
109. Skrede S, Christophersen BO. Effects of cystamine and cysteamine on the peroxidation oflipids and the release of proteins from mitochondria. Biochem J 1966; 101:37-41.
110. Haenen GRMM, Vermeulen NPE, Timmerman H, Bast A. Effect of thiols on lipidperoxidation in rat liver microsomes. Chem Biol Interaction 1989; 71:201-212.
111. Weiss KF, Kumar KS. Antioxidant mechanisms in radiation injury and radioprotection.In: Chow CK, ed. Cellular Antioxidant Defense Mechanisms. Vol. II. Boca Raton: CRCPress Inc., 1994:163-189.
112. Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmicreticulum. Science 1992; 257:1496-1502.
113. Bacq ZM. Chemical protection against X- and γ-ray. In: Bacq ZM, Alexander P, eds. Fun-damentals of Radiobiology. London: Butterworths Scientific Publications, 1955:290-327.
114. Halliwell B, Gutteridge JMC, Cross CE. Review article: Free radicals, antioxidants, andhuman disease: Where are we now? J Lab Clin Med 1992; 119:598-620.
115. Knoblock EC, Purdy EC. Apparent instability of 2-mercaptoethylamine complexes. RadiatRes 1961; 15:94-96.
116. Ananthan J, Boldberg AL, Voellmy R. Abnormal proteins serve as eukaryotic stress signalsand trigger the activation of heat shock genes. Science 1986; 232:522-524.
117. Burdon RH, Gill VM, Rice-Evans C. Oxidative stress and heatshock protein induction inhuman cells. Free Rad Res Commun 1987; 3:129-139.
118. Finley D, Ciechanover A, Varshavsky A. Thermolability of ubiquitin-activating enzyme fromthe mammalian cell cycle mutant ts85. Cell 1984; 37:43-55.
119. Liu R, Li X, Li GC. Expression of human hsp70 in rat fibroblasts enhances cell survivaland facilitates recovery from translational and transcriptional inhibition following heatshock. Cancer Rec 1992; 52:3667-3673.
120. Condorelli DF, Bellurado N, Avola R et al. Effect of trophic factors, released after hippoc-ampal injury, on astroglial cell proliferation. Metab Brain Dis 1989; 4:41-46.
121. Fleming PD, Rogers J. Neuropathology of olfactory system in Alzheimer’s disease and nor-mal aging. Soc Neurosci Abstract 1986; 12:1314.
122. Fleming PD, Cordoza ME, Woods SG et al. Corpora amylacea increased in Alzheimer’sdisease. Neurology (Suppl 1) 1987; 37:157.
123. Behrman S, Carrol JD, Janota I, Matthews WB. Progressive supranuclear palsy. Clinico-pathological study of four cases. Brain 1969; 92:663-678.
124. Gambetti P, DiMauro S, Hirt L, Blume RP. Myoclonic epilepsy with Lafora bodies: Someultrastructural, histochemical and biochemical aspects. Arch Neurol 1971; 25:483-493.
125. Janeway R, Ravens JR, Pearce LA et al. Progressive myoclonic epilepsy with Lafora inclu-sion bodies. I. Clinical, genetic, histopathologic and biochemical aspects. Arch Neurol 1967;16:565-582.
126. Robitaille Y, Carpenter S, Karpati G, DiMauro S. A distinct form of adult polyglucosanbody disease with massive involvement of central and peripheral neuronal processes andastrocytes. Brain 1980; 103:315-336.
127. Palmucci L, Anzil AP, Luh S. Intra-astrocytic glycogen granules and corpora amylacea stainpositively for polyglucosan: A cytochemical contribution on the fine structural polymor-phism of particulate polysaccharides. Acta Neuropathol (Berl) 1982; 57:99-102.
128. Takahashi K, Agari M, Nakamura H. Intra-axonal corpora amylacea in ventral and lateralhorns of the spinal cord. Acta Neuropathol (Berl) 1975; 31:151-158.
129. Wolozin BL, Prucnki A, Dickson DW, Davies P. Neuronal antigen in the brains of Alzheimerpatients. Science 1986; 232:648-650.
130. Yagashita S. Ultrastructural observations on axonal swelling in the human gracile nucleus.Virch Arch Path and Hist 1979; 382:217-226.

Astrocytes in Brain Aging and Neurodegeneration234
131. Yagashita S, Itoh Y. Corpora amylacea in the peripheral nerve axon. Acta Neuropathol(Berl) 1977; 37:73-76.
132. Hollander DM, Hutchins GM. Central spherules in pulmonary corpora amylacea. ArchPathol Lab Med 1978; 102:629-630.
133. Seman G. Fibrillar bodies in human prostate. The Prostate 1983; 54:179.134. Sun CN. Ultrastructural study of corpora amylacea in human thyroid gland. Exp Path 1983;
23:219-225.135. Schipper HM, Mateescu-Cantuniari A. Identification of peroxidase-positive astrocytes by
combined histochemical and immunolabeling techniques in situ and in cell culture. JHistochem Cytochem 1991; 39:1009-1016.
136. Wislocki GB, Leduc EH. The cytology of the subcommissural organ, Reissner’s fiber,periventricular glial cells and posterior collicular recess of the rat’s brain. J Comp Neurol1954; 101:283-309.
137. Koritsanszky S, Vigh B, Aros B. Studies on the Gomori-positive glial cells. I. Changes inthe Gomori-positive glial cells in rats of various ages. Acta Biol Hung 1967; 18:9-19.
138. Schipper HM. Gomori-positive astrocytes: Biological properties and implications for neu-rologic and neuroendocrine disorders. Glia 1991; 4:365-377.
139. Stam FC, Roukema PA. Histochemical and biochemical aspects of corpora amylacea. ActaNeuropathol (Berlin) 1973; 25:95-102.
140. Cissé S, Lacoste-Royal G, Laperriere J et al. Ubiquitin is a component of polypeptides pu-rified from corpora amylacea of aged human brain. Neurochem Res 1991; 16:429-433.
141. Rajagopalan KV. Sulfite oxidase. In: Coughlan MP, ed. Molybdenum and Molybdenum-containing Enzymes. New York: Pergamon Press, 1980:241-272.
142. Ellis RJ, van der Vies SM. Molecular chaperones. Ann Rev Biochem 1991; 60:321-347.143. Lawrence EH. Heat shock, stress proteins chaperones, and proteotoxicity. Cell 1991;
66:191-197.144. Woodford B, Tso MOM. An ultrastructural study of the corpora amylacea of the optic
nerve head and retina. Am J Ophthalmol 1980; 90:492-502.145. Gertz HJ, Cervos-Navarros J, Frydl V, Shultz F. Glycogen accumulation of the aging hu-
man brain. Mech Ageing Dev 1985; 35:25-35.146. Schipper HM, Scarborough DE, Lechan RM, Reichlin S. Gomori-positive astrocytes in pri-
mary culture: Effects of in vitro age and cysteamine exposure. Dev Brain Res 1990; 54:71-79.147. Brawer JR, Stein R, Small L et al. Composition of Gomori-positive inclusions in astrocytes
of the hypothalamic arcuate nucleus. Anat Rec 1994; 240:407-415.148. Cissé S, Schipper HM. Experimental induction of corpora amylacea-like inclusions in rat
astroglia. Neuropathol Appl Neurobiol 1995; 21:423-431.148a.Schipper HM. Experimental indcution of corpora amylacea in adult rat brain. Microsc Res
Techniq, 1998; (in press).149. Cerami A. Aging of proteins and nucleic acids: What is the role of glucose? Trends Biochem
Sci 1986; 11:311-314.150. Yan S-D, Chen X, Schmidt A-M, Brett J. Non-enzymatic glycation of Tau in neurofibril-
lary tangles of Alzheimer’s disease: a mechanism for aggregation and neurotoxicity. Neu-rology 1994; 44 (Supple 2):A371, Abstract 960S.
151. Balazs L, Leon M. Evidence of an oxidative challenge in the Alzheimer’s brain. NeurochemRes 1994; 19:1131-1137.
152. Butterfield AD, Hensley K, Harris M et al. β-amyloid peptide free radical fragments ini-tiate synaptosomal lipoperoxidation in a sequence-specific fashion: Implications toAlzheimer’s disease. Biochem Biophys Res Comm 1994; 200:710-715.
153. Palmer AM, Burns MA. Selective increase in lipid peroxidation in the inferior temporalcortex in Alzheimer’s disease. Brain Res 1994; 645:338-342.
154. Subbarao KV, Richardson JS, Ang LC. Autopsy samples of Alzheimer’s cortex show in-creased peroxidation in vitro. J Neurochem 1990; 55:342-345.
155. Bolanos JP, Peuchen S, Heales SJR et al. Nitric oxide-mediated inhibition of the mito-chondrial respiratory chain in cultured astrocytes. J Neurochem 1994; 63:910-916.

CHAPTER 11
Astrocytes in Brain Aging and Neurodegeneration, edited by Hyman M. Schipper.©1998 R.G. Landes Company.
Glial Iron Sequestrationand NeurodegenerationHyman M. Schipper
The Free Radical Hypothesis of Parkinson’s Disease
Idiopathic Parkinson’s disease (PD) is a movement disorder of uncertain etiology charac-terized by the accelerated loss of dopaminergic (DA) neurons in the pars compacta of the
substantia nigra.1 Although dissenting opinions exist,2 there is currently a broad consensusimplicating oxidative stress as a major factor in the pathogenesis of PD.3-6 The free radicalhypothesis of PD draws support from the following observations:
1. The accelerated oxidative deamination of DA by monoamine oxidase B (MAO B)in idiopathic and experimental Parkinsonism subjects the nigrostriatal projectionsto excessive concentrations of hydrogen peroxide (H2O2).3,7,8
2. The neurotoxins, 6-hydroxydopamine, manganese and, to some extent, MPTP, in-duce parkinsonism in animals via the generation of free radicals.8-12
3. Basal lipid peroxidation in the substantia nigra of postmortem human PD brainwas found to be significantly elevated relative to non-Parkinsonian controls matchedfor age and postmortem interval.13
4. Free radical scavenger enzymes (such as catalase) and intracellular reducing sub-stances (such as reduced glutathione) are reportedly deficient in the basal ganglia ofpatients with Parkinson’s disease.14,15 In contrast, the mitochondrial antioxidantmanganese superoxide dismutase (MnSOD) appears to be augmented in the basalganglia of PD subjects and may represent an adaptive response to oxidative stressand mitochondrial injury in this condition.16,17
5. Although still controversial, results of a large multicenter clinical trial (DATATOP)and a more recent study suggest that treatment of early PD with the MAO B inhibi-tor 1-deprenyl may slow the progression of this neurodegenerative disorder by cur-tailing the production of dopamine-derived H2O2.18-20
The Redox Neurobiology of Alzheimer’s DiseaseAlzheimer’s disease (AD) is characterized by progressive neuronal degeneration, glio-
sis, and the accumulation of intracellular inclusions (neurofibrillary tangles [NFTs]) andextracellular deposits of amyloid (senile plaques [SPs]) in discrete regions of the basal fore-brain, hippocampus, and association cortices.21 Although the etiology of sporadic AD re-mains unknown, evidence amassed over the last 5 years has implicated free radicals andoxidative stress in the pathogenesis of this dementing illness. For example, end products oflipid peroxidation are elevated in the brains of AD subjects22,23 and various antioxidant

Astrocytes in Brain Aging and Neurodegeneration236
defenses are reportedly deranged in AD brain and peripheral tissues.24-26 The excessive gen-eration of free radicals may promote both paired helical filament formation and amyloiddeposition in the AD brain.26,27 Moreover, the results of recent biochemical studies suggestthat the neurotoxic effects of certain amyloid fragments may, in part, be mediated by freeradical intermediates.28,29 A growing body of evidence suggests that brain cell mitochondriamay be prime targets of chronic oxidative injury in Alzheimer-affected tissues and that bioen-ergetic failure (mitochondrial insufficiency) may play an important role in the pathogen-esis of this disease.30,31 In support of the latter, cytochrome C oxidase and complex V activi-ties,32 the pyruvate dehydrogenase complex, and various Krebs cycle intermediates arepurportedly deficient in AD brain,30,32,33 and excessive mutations in mitochondrial genesencoding subunits of complex I and IV have been reported in the CNS and blood of ADsubjects.34,35 It has been suggested that further increases in free radical generation resultingfrom infidelity of electron transport within the inner membranes of damaged mitochon-dria may perpetuate oxidative neuropil injury in the AD brain long after initiating neuro-toxic insults have dissipated.31,36 As in the case of idiopathic PD, epigenetic factors contrib-uting to the excessive oxidative stress and mitochondrial electron transport chain deficits inthe brains of AD subjects remain poorly understood.
Iron Deposition and Neurodegenerative DiseaseThe pathological sequestration of redox-active brain iron has been implicated as a major
generator of reactive oxygen species in PD, AD, and other aging-related neurodegenerativedisorders.
Parkinson’s DiseaseAbnormally high levels of tissue iron have been consistently reported in the substantia
nigra and basal ganglia of PD subjects.5,6,17,37 In PD, the excessive iron deposition primarilyaffects the zona compacta of the substantia nigra and correlates with loss of dopaminergicneurons in this brain region.6,38,39 Using conventional histochemical stains, the excessivenigral iron appears to be predominantly deposited within astrocytes, microglia, macroph-ages and microvessels within areas depleted of neuromelanin-containing (dopaminergic)neurons. Although minor concentrations of iron have been detected in neuronalneuromelanin using micro-analytical techniques,6,40 histochemical evidence for substantialiron deposition in PD-affected nigral or striatal neurons is scant or nonexistent.6,41,42 Thus,glia and other nonneuronal cells may represent the chief substrates of excessive iron seques-tration in the basal ganglia of PD subjects. In the latter, the augmented tissue iron levels areaccompanied by alterations in the expression of several important iron-binding proteinsand their receptors. In general, increased expression of tissue ferritin, the major intracellu-lar sequestrant of ferric iron, parallels the distribution of the excess iron and largely impli-cates nonneuronal (glial) cellular compartments.6 The iron-binding protein transferrin isresponsible for the extracellular transport of ferric iron and its delivery to virtually all mam-malian tissues. After binding to transferrin receptors within the plasma membrane, the trans-ferrin-transferrin receptor complex is internalized via endocytosis, free iron is liberated fromthe complex by a temperature and energy-dependent process involving endosomal acidifi-cation, the iron translocates to the cytosol and is sequestered in ferritin, and theapotransferrin-transferrin receptor complex is recycled to the cell surface, where it dissoci-ates.43-45 To maintain tissue iron homeostasis, plasma membrane transferrin receptor den-sities and intracellular ferritin concentrations are tightly regulated (at transcriptional andposttranscriptional levels) by iron bioavailability and intracellular iron stores.43-45 In nor-mal rat and human brain tissues, there appears to be an overt mismatch between local brainiron concentrations and the densities in cell surface transferrin binding sites.46-48 Moreover,

237Glial Iron Sequestration and Neurodegeneration
in glaring contrast to the ferritin data, the density of transferrin binding sites remains un-changed or varies inversely with augmented iron stores in the substantia nigra and striatumof PD subjects.42,46,49,50 An important interpretation of these findings is that, in contradis-tinction to most peripheral tissues, transferrin and its receptor play a limited role, if any, inthe sequestration of iron by aging and degenerating CNS tissues.42,46,49,50 Indeed, attentionis shifting to alternative iron transport mechanisms such as that mediated by lactoferrinand the lactoferrin receptor, which are reportedly augmented in neurons, astrocytes, andblood vessels of PD-affected, iron-laden neural tissues.51,52
Alzheimer’s DiseaseAs in the case of PD, abnormalities of iron homeostasis and excessive deposition of this
transition metal are characteristic of Alzheimer-affected brain tissues. In AD, increases inbulk brain iron have been reported in both gray and white matter regions.53 Interestingly,although a significant proportion of ferritin iron in normal human brain is stored withinoligodendroglia, in AD white matter there appears to be a shift towards pathological irontrapping within the astrocytic compartment.53 In the AD hippocampus, augmented depo-sition of nonheme iron has been shown to occur in NFT-bearing neurons, astrocytes, mi-croglia, and in the vicinity of neuritic plaques.37,42,53,54 Although transferrin immunoreac-tivity has been noted in AD astrocytes and senile plaques, there appears to be an overalldecrease in levels of immunoreactive transferrin in AD-affected cortical and subcorticalbrain tissue relative to that in age-matched, nondemented controls.53 Moreover, transferrinreceptor densities (determined by [125I]-transferrin binding) are significantly reduced inpostmortem hippocampus and temporal cortex derived from AD subjects relative to con-trols.50 This apparent mismatch of brain iron and transferrin/transferrin receptor is similarto that observed in the substantia nigra of PD subjects (vide supra) and further suggeststhat the transferrin pathway of iron mobilization may contribute little to the pathologicalsequestration of brain iron observed in the major aging-related neurodegenerative disor-ders. As in the case of the PD nigra, increased lactoferrin and/or lactoferrin receptor immu-noreactivity has been reported in neurons, glia and extracellular amyloid plaques withinbrain regions undergoing degeneration in AD, Down’s syndrome, Pick’s disease, and ALS-Parkinsonism/dementia complex of Guam.52,55-58 Unlike transferrin, lactoferrin binding toits receptor is not affected by degrees of tissue iron saturation and could theoretically per-mit toxic levels of this metal to accumulate in these degenerating neural tissues.52 By par-ticipating in Fenton reactions, the aberrantly-sequestered brain iron could promote oxida-tive stress and lipid peroxidation and thereby directly contribute to the neurodegenerativeprocess. Furthermore, the amyloid precursor protein gene contains iron response element-like consensus sequences, raising the possibility that brain amyloid deposition in AD andother human neurodegenerative disorders may be iron-sensitive.42
Iron Sequestration in Aging AstrogliaEfforts to ameliorate iron-mediated neuronal injury in AD and PD presupposes some
understanding of the regulatory mechanisms subserving iron metabolism and sequestra-tion in the aging and degenerating CNS. The following important, but as yet unanswered,questions in this regard arise from the pathological studies considered in the previous section:
1. What is the role of heme vs. nonheme iron in aging-related neurodegenerative con-ditions?
2. Which cell type(s) and subcellular compartments are responsible for the abnormalsequestration of brain iron in these degenerative disorders?
3. Does induction of a cellular stress (heat shock) response facilitate trapping of re-dox-active iron in neural tissues? and

Astrocytes in Brain Aging and Neurodegeneration238
4. What is the role of the iron-transport protein, transferrin, in this process?We have begun to explore these and related issues by focusing on the mechanisms
responsible for the accumulation of iron-rich cytoplasmic inclusions in aging subcorticalastrocytes and in astroglial cultures subjected to oxidative stress. As described in chapter 9,the sulfhydryl agent cysteamine (CSH) accelerates the aging-related accumulation of iron-rich cytoplasmic inclusions in hippocampal, striatal and other subcortical astroglia in situand in primary neuroglial cultures. Evidence was also provided that these iron-laden glialinclusions and related corpora amylacea are derived from oxidatively-damaged mitochon-dria in the context of a cellular stress (heat shock) response (chapters 9 and 10). Severallaboratories including our own59,60 have previously concluded on the basis of histochemicaland spectrofluorometric data that porphyrins and heme ferrous iron are responsible, re-spectively, for the orange-red autofluorescence and nonenzymatic peroxidase activity inthese glial inclusions. However, we subsequently determined that CSH suppresses the in-corporation of the heme precursors ∆-amino[14C]-levulinic acid (ALA) and [14C]-glycineinto astroglial porphyrin and heme in primary culture, prior to and during the time whenincreased iron content is detectable in swollen astrocyte mitochondria by microprobe analysis(Fig.11.1).61,62 Thus, contrary to hypothesis, de novo biosynthesis of porphyrins and hemeis not responsible for the increased mitochondrial iron content, autofluorescence, and per-oxidase activity observed in cultured astroglia following CSH exposure. Because the CSH-induced astroglial inclusions are morphologically and histochemically identical to the iron-laden astrocyte granules that normally accumulate in the aging periventricular brain, it wouldseem highly unlikely that augmentation of porphyrin-heme biosynthesis plays a role in thebiogenesis of the latter as well. Oxidized mitochondrial flavoproteins exhibit fluorescenceemission spectra that may be difficult to distinguish from porphyrins63,64 and are likelymediators of orange-red autofluorescence in these astrocytic inclusions.
Following inhibition of porphyrin-heme biosynthesis, CSH augments the incorpora-tion of 59Fe (or 55Fe) into astroglial mitochondria without significantly affecting transfer ofthe metal into whole-cell and lysosomal compartments (Fig. 11.2).62 This CSH effect wasclearly demonstrable when inorganic [59Fe]Cl3, but not [59Fe]-diferric transferrin (Fig. 11.3),served as the metal donor. These findings are consistent with previous reports that intracel-lular transport of low molecular weight, inorganic iron may be 5- to 10-fold more efficientthan that of transferrin-bound iron in various tissues, including melanoma cells,65-67 Chi-nese hamster ovary cells,68 and K562 cells.69 Our observations support the conclusion ofAdams and coworkers70 that inhibition of heme biosynthesis stimulates the selective trans-port of low molecular weight iron from the cytoplasm to the mitochondrial compartment.
Recent work from our laboratory suggests that dopamine may be an important endog-enous stressor mediating nigrostriatal glial iron trapping in PD and, to a lesser extent, in thecourse of normal aging. Akin to the effects of CSH, physiologically-relevant concentrationsof dopamine (1 µM) stimulate the sequestration of nontransferrin-bound 55Fe in the mito-chondrial compartment of cultured astroglia without affecting the disposition of transfer-rin-derived 55Fe. L-DOPA (25 µM) weakly recapitulated the effects of dopamine on glialiron sequestration, whereas equimolar concentrations of norepinephrine were entirely in-ert in this regard.71 The effects of dopamine on glial iron trapping were abrogated bycoadministration of ascorbate (200 µM) but not by the D1 and D2 antagonists, SCH 23390and sulpiride, respectively, suggesting that, analogous to the CSH mechanism of action,dopamine-derived free radicals promote the sequestration of nontransferrin-derived ironwithin astroglial mitochondria. That such dopamine-astrocyte interactions may be opera-tional in vivo is supported by:
1. a recent nuclear microscopy study demonstrating increased elemental iron in thesubstantia nigra of 6-hydroxydopamine-lesioned rats;72 in conjunction with

239Glial Iron Sequestration and Neurodegeneration
2. immunocytochemical evidence of direct contact between tyrosine hydroxylase-posi-tive (dopaminergic) processes and Gomori-positive (metal-laden) astrocytes in therat arcuate nucleus73 and basal ganglia (Schipper, unpublished results).
The Role of HO-1 in Brain Iron DepositionAs described in chapter 10, HO-1 is a 32 kDa member of the stress protein superfamily
that catalyses the rapid conversion of heme to biliverdin in brain and other tissues. In re-sponse to oxidative stress, induction of HO-1 may protect cells by catabolizing prooxidantmetalloporphyrins such as heme to bile pigments (biliverdin, bilirubin) with free radical-scavenging capabilities.74 On the other hand, HO-1-catalyzed heme degradation liberatesfree iron and carbon monoxide (CO) which exacerbate intracellular oxidative stress by stimu-lating oxyradical generation within the mitochondrial compartment.75 We74 and others76
have recently shown that HO-1 is massively upregulated in neurons and astrocytes ofAlzheimer-diseased human temporal cortex and hippocampus (but not in unaffected sub-stantia nigra) relative to age-matched, nondemented controls. Conversely, the percentage ofGFAP-positive astrocytes expressing HO-1 in substantia nigra (but not in other brain re-gions) of PD subjects is significantly increased in comparison with age-matched controls76a
(Fig. 11.4). Although HO-1 upregulation in these conditions may confer some degree of
Fig. 11.1. Incorporation of [14C]-ALA into (A) uroporphyrin, (B) coproporphyrin, (C) proto-porphyrin, and (D) hemin in control untreated (O) and CSH-treated (● ) astrocytes. Data arepresented as mean ± SD (bars) of three to six observations. *p<0.05, **p<0.01 for significance ofdifference relative to untreated controls. CSH suppresses porphyrin-heme biosynthesis in cul-tured astroglia. Reprinted with permission from Wang X et al, J Neurochem 1995; 64:1868-1877.

Astrocytes in Brain Aging and Neurodegeneration240
Fig. 11.2. Iron-59 uptake in control (O)and CSH-treated (● ) astrocytes exposedto [59Fe]Cl3: (A) total cell, (B) lysosomal,and (C) mitochondrial fractions. Data arepresented as mean ± SD (bars) of three tosix observations. *p<0.05 for significanceof difference relative to untreated controls.CSH promotes the sequestration ofnontransferrin derived iron within the mi-tochondrial compartment. Reprinted withpermission from Wang X et al, J Neuro-chem 1995; 64:1868-1877.
Fig. 11.3. Iron-59 uptake in cultured control un-treated (O) and CSH-treated (● ) astrocytes ex-posed to [59Fe]-transferrin: (A) total cell, (B)lysosomal, and (C) mitochondrial fractions.Data are mean ± SD (bars) values of three to sixobservations. CSH has no significant effect onthe disposition of transferrin-derived iron inthese cells. Reprinted with permission fromWang X et al, J Neurochem 1995; 64:1868-1877.

241Glial Iron Sequestration and Neurodegeneration
cytoprotection by degrading pro-oxidant heme to anti-oxidant bile pigments, heme-de-rived free iron and CO may contribute, at least in part, to the development of mitochon-drial electron transport chain deficiencies and excess mitochondrial DNA mutations re-ported in the brains of AD and PD subjects.77,78
The upregulation of HO-1 may have important implications for the biogenesis of mi-tochondria-derived astrocytic inclusions in senescent and oxidatively challenged astroglia.Within 6 h of CSH exposure, cultured astroglia exhibit 4- to 10-fold increases in HO-1mRNA and protein levels, robust HO-1 immunofluorescent staining, and a 3-fold increasein HO enzymatic activity.79-81 As in the case of CSH, H2O2, menadione and dopamine (butnot norepinephrine) consistently upregulate HO-1 in cultured astroglia prior to promotingthe sequestration of nontransferrin-bound 55Fe by the mitochondrial compartment.71,82 Indopamine-exposed glial cultures and in senescent subcortical astroglia in situ, the libera-tion of free iron and CO resulting from HO-1-catalyzed heme degradation may promoteearly oxidative injury to mitochondrial membranes and thereby facilitate the transforma-tion of normal astrocyte mitochondria to iron-rich cytoplasmic inclusions. In support ofthis contention, we observed that dopamine-induced sequestration of mitochondrial iron
Fig. 11.4. Percentage of glial fibrillary acid protein (GFAP)-positive astrocytes concomitantlyexpressing HO-1 in various brain regions of control and PD subjects. Vertical lines denote stan-dard errors of the mean and asterisks denote statistical significance (p<0.05). ( ) = number ofspecimens per group. Reprinted with permission from Schipper HM et al. Exp Neurol 1998;150:60-68.

Astrocytes in Brain Aging and Neurodegeneration242
in cultured rat astroglia is prevented by coadministration of the competitive heme oxyge-nase inhibitor tin-mesoporphyrin, or the HO-1 transcriptional suppressor dexamethasone(Schipper and Bernier, manuscript in preparation).
Pro-toxin Bioactivation by Astrocytes in Primary CultureElectron spin resonance spectroscopy (ESR) with magnesium spin stabilization was
used to determine whether CSH-induced peroxidase activity (mitochondrial iron deposi-tion) in cultured astroglia is capable of oxidizing catecholestrogens and catecholamines totheir respective orthosemiquinone radicals.83 Incubation of 2-hydroxyestradiol withhomogenates derived from untreated (control) astroglial monolayers in the presence ofH2O2 and NADPH (pH 7.0) yielded no or barely detectable o-semiquinone spectra. In con-trast, intense o-semiquinone spectra indicative of robust catechol oxidation were consis-tently observed following incubation of equimolar concentrations of 2-hydroxyestradiolwith homogenates obtained from CSH-pretreated (iron-enriched) astrocyte monolayers inthe presence of appropriate cofactors (Fig. 11.5). In the absence of H2O2 substrate, there wasa marked reduction in signal amplitude, attesting to the important role of glial peroxidaseactivity in the augmentation of catecholestrogen metabolism in our system.83 The results ofthe ESR experiments, in conjunction with the protective effects of α-tocopherol and21-aminosteroids on estradiol-induced depletion of hypothalamic β-endorphin (chapter 9),support the notion that free radical generation by iron-laden hypothalamic astrocytes maymediate, at least in part, the dystrophic effects of estradiol in this brain region.
As in the case of 2-hydroxyestradiol, we demonstrated that the iron-dependent peroxi-dase activity induced in cultured astroglia by CSH exposure significantly enhances the oxi-dation of the catecholamine, dopamine, to its dopamine-o-semiquinone derivative in thepresence of H2O2.83 This observation is consistent with previous reports that dopamine andnorepinephrine are readily oxidized to semiquinones with proven neurotoxic activity invitro via peroxidase-mediated reactions.84 Because aging subcortical astrocytes may exhibitboth enhanced MAO B activity (see chapter 6) and abundant mitochondrial iron, it is con-ceivable that H2O2 produced by MAO B oxidation of dopamine serves as a cofactor forfurther dopamine oxidation (to potentially neurotoxic ortho-semiquinones) by peroxidase-mediated reactions. In addition to dopamine, redox-active glial iron may also facilitate thenonenzymatic oxidation of:
1. the pro-toxin MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) to the dopam-inergic toxin MPP+ in the presence of MAO inhibitors;85 and
2. the dopamine precursor DOPA to 2,4,5-trihydroxyphenylalanine (TOPA) and thenon-NMDA excitotoxin TOPA-quinone.86
The high stress protein content of peroxidase-positive astrocytes and a compensatoryupregulation of manganese superoxide dismutase (MnSOD) observed in these cells couldserve to limit the extent of oxidative injury within the glia themselves (see chapter 10).However, the dystrophic effects of reactive oxygen species need not be confined to the cellu-lar compartment in which they are generated. For example, H2O2 is lipid soluble and caneasily traverse plasma membranes to reach the intercellular space, while superoxide (poten-tially generated in our model by semiquinone-quinone redox cycling or by infidelity ofelectron transport in damaged inner mitochondrial membranes) can be extruded from cellsvia anion channels.87 In support of this formulation, we recently observed that catechola-mine-secreting PC12 cells grown atop monolayers of CSH-pretreated (iron-enriched) as-trocytes are far more susceptible to dopamine/H2O2-related killing than PC12 cells coculturedwith nonpretreated (DAB-negative) astroglia. In both coculture paradigms, astroglial death,determined by ethidium monoazide bromide nuclear staining and GFAP immunofluores-cence, was not significantly augmented by dopamine-H2O2 exposure.88 On the basis of these

243Glial Iron Sequestration and Neurodegeneration
in vitro findings, we hypothesize that, in the intact basal ganglia, leakage of free radicalsfrom peroxidase-positive astrocytes into the surrounding neuropil may promote lipidperoxidation and degeneration of nearby dopaminergic terminals and other vulnerableneuronal constituents. In this regard, the progressive increase in numbers of peroxidase-positive astrocytes which have been documented in the basal ganglia and other subcorticalregions of the aging rodent and human brain may render the latter particularly prone toParkinsonism and other free radical-related neurodegenerations.
Pathological Glial-Neuronal Interaction in Parkinson’s DiseaseOur observations on CSH-stressed astroglia suggest a model for inclusion formation,
iron sequestration, and the perpetuation of oxidative injury in the aging and degeneratingnervous system (Fig. 11.7):
Fig. 11.5. ESR spectra of mag-nesium-complexed semiquin-ones from the peroxidase-H2O2
oxidation of 2-hy-droxy(catechol)estradiol .(A) Top: autoxidation of 2-hydroxyestradiol in serum-freemedium in the absence of cellsfollowing alkalinization to pH10.0 with NaOH. Medium con-tained 2-hydroxyestradiol (10–2
M), MgCl2 (0.5 M) and NaOHin DMEM. The characteristic o-semiquinone spectrum of oxi-dized 2-hydroxyestradiol isshown. Bottom: computer-simulated spectrum of the 2-hydroxyestradiol o-semi-quinone derived from measuredhyperfine coupling constants. (B) Incubation of 2-hydroxy-estradiol (10–2 M), MgCl2
(0.5 M), NADPH (0.3 M), andH2O2 (0.1 mM) with tissue ho-mogenate derived from un-treated (control) brain cell cul-ture (pH 7.0). The gain settingsin (B) and (C) are identical, per-mitting direct amplitude com-parisons. (C) Incubation as in(B) with tissue homogenate de-rived from cysteamine pre-treated (peroxidase-enriched)brain cell culture. An intense o-semiquinone signal is observedwith hyperfine structure identi-cal to the pattern obtained in thecell-free 2-hydroxyestradiol au-toxidation experiment (A). Theperoxidase activity induced in astrocytes by cysteamine catalyses catechol oxidation to o-semiquinoneradicals. Reprinted with permission from Schipper HM et al, J Neurosci 1991; 11:2170.

Astrocytes in Brain Aging and Neurodegeneration244
1. In the senescent basal ganglia and other subcortical brain regions, dopamine and/or other unidentified oxidative stressors (simulated by CSH exposure) induce a cel-lular stress response in subpopulations of astroglia, characterized by upregulationof various HSP and HO-1. Free iron and CO derived from HO-1-mediated hemedegradation may initiate or potentiate injury to the mitochondrial compartment.
2. Stress-related inhibition of porphyrin-heme biosynthesis and/or direct oxidativedamage to mitochondrial membranes promotes the selective transport ofnontransferrin-derived, nonheme iron into the mitochondrial compartment. Com-
Fig. 11.6. ESR spectra of magnesium-complexed semiquinones derived fromthe peroxidase-H2O2 oxidation ofdopamine. (A) Top: Autoxidation ofdopamine in serum-free medium inthe absence of cells at pH 10.0. Me-dium contained dopamine (1 mM),MgCl2 (0.2 M), and NaOH in DMEM.The characteristic dopamine-o-semi-quinone spectrum is observed. Bot-tom: Computer-simulated spectrum ofthe dopamine-o-semiquinone derivedfrom the measured hyperfine couplingconstants. (B) Incubation of dopam-ine (1 mM), MgCl2 (0.2 M), NADPH(0.3 M), and H2O2 (0.1 mM) with tis-sue homogenate derived from an un-treated (control) astrocyte culture (pH7.0). The gain settings in (B) and (C)are identical. (C) Incubation as in (B)with tissue derived from cysteamine-pretreated (peroxidase-enriched) as-trocyte culture. ESR spectra ampli-tudes are approximately 2.5-foldgreater than those observed in (B). Thecysteamine-induced peroxidase activ-ity catalyzes catechol oxidation to o-semiquinone radicals. Reprinted withpermission from Schipper HM et al, JNeurosci 1991; 11:2170.

245Glial Iron Sequestration and Neurodegeneration
pensatory upregulation of MnSOD may provide some degree of protection to themitochondrial compartment by limiting the accumulation of superoxide.
3. By promoting further oxidative stress, the redox-active mitochondrial iron partici-pates in a vicious cycle of pathologic events whereby damage to glial mitochondriaas well as to the surrounding neuropil is perpetuated. This model of astrocyte se-nescence is consistent with the Mitochondrial Hypothesis of Aging, which statesthat oxidative damage to mitochondria results in bioenergetic failure, a vicious spi-ral of augmented mitochondrial free radical generation and injury and progressive
Fig. 11.7. A model for glial inclusion formation, iron sequestration, and oxidative injury in theaging and degenerating nervous system.

Astrocytes in Brain Aging and Neurodegeneration246
tissue aging.19,89-91 Our model also accounts for the observation that mosaicism forspecific mitochondrial DNA mutations in the normal aging human brain is moststriking in regions particularly rich in intracellular iron such as the caudate, puta-men and substantia nigra.92 Our findings recapitulate the discordant pattern of iron/transferrin receptor localization observed in the PD nigra (see above) and raise thepossibility that exacerbation of stress-related trapping of nontransferrin-derivediron by astroglial mitochondria may be an important mechanism underlying thepathological accumulation of this redox-active metal in the basal ganglia of PDsubjects. Such iron could conceivably originate from degenerating neurons, glia, ormyelin or from the cerebrospinal fluid (CSF). Micromolar quantities of chelatable,low-molecular-weight iron are present in normal CSF, and the concentration ofthis metal in CSF has been shown to increase under neuropathological conditions.93
As described above, a portion of this chelatable iron may be derived from HO-1-mediated degradation of cellular heme within oxidatively-challenged neural tissues.Consistent with our model are reports that a significant proportion of the excessiron in PD brain may indeed be localized to astroglial mitochondria,37,94,95 and thatdeficiencies of mitochondrial electron transport are prevalent in the brains of PDsubjects.77,78 By oxidizing dopamine and environmentally-derived xenobiotics toneurotoxic intermediates, the redox-active glial iron could serve as a “final commonpathway” perpetuating nigrostriatal degeneration initiated by as yet undeterminedgenetic and epigenetic factors in patients with PD.
ConclusionThere is considerable evidence implicating excessive basal ganglia iron and catechola-
mine-derived free radicals in the pathogenesis of idiopathic PD. Yet, the mechanisms re-sponsible for the pathological sequestration of brain iron in this and other debilitatingneurodegenerative conditions remain enigmatic. The progressive accumulation of iron-rich(peroxidase-positive) astrocytic granules represents a fundamental and highly consistentbiomarker of aging in the vertebrate CNS. Although these glial inclusions were first identi-fied almost half a century ago on the basis of their affinity for Gomori stains, it is only inrecent years, and largely through the advent of in vitro toxicologic modeling of inclusionbiogenesis, that we have begun to elucidate the subcellular origin of these inclusions, themechanism(s) governing their formation, and their potential role in brain aging andneurodegeneration. The current state of our knowledge indicates that these gliosomes are“stress granules” which ultimately derive from effete, metal-laden mitochondria engaged ina complex autophagic process. Determination of the topography of these glial inclusionsmay permit “mapping” of CNS regions at increased risk for chronic oxidative injury duringnormal aging and under pathological conditions. More importantly, the ability to experi-mentally recapitulate the development of this senescent glial phenotype in primary cultureprovides a powerful model to investigate:
1. the role of HO-1 and other heat shock proteins in the biogenesis of potentiallydeleterious neural inclusions;
2. stress-related (dys)regulation of MnSOD and other antioxidant enzymes in the ag-ing and degenerating nervous system; and
3. mechanisms of pathological brain iron sequestration and mitochondrial insuffi-ciency characteristic of aging and degenerating neural tissues.
Our findings support the notion that stress-related trapping of nonheme,nontransferrin-bound iron by astroglial mitochondria is a primary mechanism underlyingthe pathological accumulation of redox-active iron in the basal ganglia of PD subjects. Wehave provided evidence that the nonenzymatic peroxidase activity manifest in senescent,

247Glial Iron Sequestration and Neurodegeneration
iron-laden astroglia promotes the bioactivation of endogenous catechols and environmen-tally-derived xenobiotics to potential neurotoxins which, in turn, may perpetuate “second-ary” neural damage long after initiating neurotoxic insults have dissipated. If the latter istrue, attempts to pharmacologically inhibit metal sequestration by “stressed” astroglial mi-tochondria (e.g., using HO-1 inhibitors and centrally-active iron chelators) may constitutea rational and effective strategy in the management of Parkinson’s disease and other aging-related neurodegenerative afflictions.
AcknowledgmentsThe authors thank Mrs. Kay Berckmans and Mrs. Adrienne Liberman for assistance
with the preparation of this manuscript. This work is supported by grants from the MedicalResearch Council of Canada and the Fonds de la Recherche en Santé du Québec.
References1. Hornykiewicz O. Neurochemical pathology and etiology of Parkinson’s disease: Basic facts
and hypothetical possibilities. Mt Sinai J Med 1988; 45:35-43.2. Calne DB. Initiating treatment for idiopathic Parkinsonism (Review). Neurology 1994; 44(7
Suppl 6):S19-22.3. Cohen G, Werner P. Free radicals, oxidative stress, and neurodegeneration. In: Calne DB,
ed. Neurodegenerative Diseases. Philadelphia: WB Saunders Co., 1994:139-161.4. Fahn S, Cohen G. The oxidant stress hypothesis in Parkinson’s disease; evidence support-
ing it (Review). Ann Neurol 1992; 32:804-812.5. Jenner P. What process causes nigral cell death in Parkinson’s disease. In: Cederbaum JM,
Gancher ST, eds. Neurological Clinics, Part 2: Parkinson’s Disease. Philadelphia: SaundersCo., 1992:387-403.
6. Youdim MBH. Inorganic neurotoxins in neurodegenerative disorders without primary de-mentia. In: Calne DB, ed. Neurodegenerative Diseases. Philadelphia: Saunders WB Co.,1994:251-276.
7. Hefti F, Melamed E, Wurtman R. Partial lesions of the dopanergic nigrostriatal system inthe rat brain: Biochemical characterization. Brain Res 1980; 195:123-137.
8. Zigmond M, Berger TW, Grace AA, Stricker BM. Compensatory responses to nigrostriatalbundle injury. Studies with 6-hydroxydopamine in an animal model of Parkinsonism (Re-view). Mol Chem Neuropath 1989; 10:185-200.
9. Barbeau A. Etiology of Parkinson’s disease: A research strategy. Can J Neurol Sci 1984;11:24-28.
10. Cadet JL, Katz M, Jackson-Lewis V, Fahn S. Vitamin E attenuates the toxic effects ofintrastriatal injection of 6-hydroxydopamine (6-OHDA) in rats: Behavioral and biochemi-cal evidence. Brain Res 1989; 476:10-15.
11. Cohen G. Oxygen radicals and Parkinson’s disease. Upjohn symposium on Oxygen Radi-cals, Augusta, MI 1987; 130-135.
12. Parenti M, Rusconi L, Cappabianca V et al. Role of dopamine in manganese neurotoxicity.Brain Res 1988; 473:236-240.
13. Dexter DT, Carter CJ, Wells FR et al. Basal lipid peroxidation in substantia nigra is in-creased in Parkinson’s disease. J Neurochem 1989; 52:381-389.
14. Ambani LM, Van Woert MH, Murphy S. Brain peroxidase and catalase in Parkinson dis-ease. Arch Neurol 1975; 32:114-118.
15. Perry TL, Godin DV, Hansen S. Parkinson’s disease: A disorder due to nigral glutathionedeficiency? Neurosci Lett 1982; 33:305-310.
16. Kalra J, Rajput AH, Mantha SV et al. Oxygen free radical producing activity of polymor-phonuclear leukocytes in patients with Parkinson’s disease. Mol Cell Biochem 1992;112:181-186.
17. Riederer P, Sofic E, Rausch WD et al. Transition metals, ferritin, glutathione, and ascorbicacid in Parkinsonian brains. J Neurochem 1989; 52:515-520.

Astrocytes in Brain Aging and Neurodegeneration248
18. Olanow CW, Hauser RA, Gauger L et al. The effects of deprenyl and levodopa on theprogression of Parkinson’s disease. Ann Neurol 1995; 38:771-777.
19. Parkinson Study Group. Effects of deprenyl on the progression of disability in earlyParkinson’s disease. N Engl J Med 1989; 321:1364-1371.
20. Parkinson Study Group. Effects of tocopherol and deprenyl on the progression of disabil-ity in early Parkinson’s disease. N Engl J Med 1993; 328:176-183.
21. Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron 1991; 6:487-498.22. Subbarao KV, Richardson JS, Ang LC. Autopsy samples of Alzheimer’s cortex show in-
creased peroxidation in vitro. J Neurochem 1990; 55:342-345.23. Palmer AM, Burns MA. Selective increase in lipid peroxidation in the inferior temporal
cortex in Alzheimer’s disease. Brain Res 1994; 645:338-342.24. Perrin R, Briancon S, Jeandel C et al. Blood activity of Cu/Zn superoxide dismutase, glu-
tathione peroxidase and catalase in Alzheimer’s disease. A case-control study. Gerontology1990; 36:306-313.
25. Jeandel C, Nicolas MB, Dubois F et al. Lipid peroxidation and free radical scavengers inAlzheimer’s disease. Gerontology 1989; 35:275-282.
26. Zemlan FP, Thienhaus OJ, Bosmann HB. Superoxide dismutase activity in Alzheimer’s dis-ease: Possible mechanism for paired helical filament formation. Brain Res 1989; 476:160-162.
27. Dyrks T, Weidemann A, Multhaup G et al. Identification, transmembrane orientation andbiogenesis of the amyloid A4 precursor of Alzheimer’s disease. EMBO J 1988; 7:949-957.
28. Butterfield DA, Hensley K, Harris M et al. β-Amyloid peptide free radical fragments ini-tiate synaptosomal lipoperoxidation in a sequence-specific fashion: Implications toAlzheimer’s disease. Biochem Biophys Res Commun 1994; 200:710-715.
29. Hensley K, Carney JM, Mattson MP et al. A model for β-amyloid aggregation and neuro-toxicity based on free radical generation by the peptide: Relevance to Alzheimer disease.Proc Natl Acad Sci USA 1994; 91:3270-3274.
30. Leblanc A. Increased production of 4 kDa amyloid beta peptide in serum deprived humanprimary neuron cultures: possible involvement of apoptosis. J Neurosci 1995; 15:7837-7846.
31. Beal MF. Does impairment of energy metabolism result in excitotoxic neuronal death inneurodegenerative illnesses? (Review) Ann Neurol 1992; 31:119-130.
32. Schagger H, Diekmann S, Ohm TG. Oxphos protein defects in Alzheimer’s disease, inParkinson’s disease and in mitochondrial encephalomyopathies revealed by blue native elec-trophoresis. Soc Neurosci Abstract 1995; 21:1492.
33. Gonzalez-Lma F, Matos-Collazo S, Garrosa M. Cytochrome oxidase activity in the humaninferior calliculus: Quantification at cellular and tissue levels and comparison to Alzheimer’sbrains. Soc Neurosci. Abstract 1995; 21:739.
34. Fukuyama R, Hatanpaa K, Rapoport SI, Chandrasekaran K. Gene expression of ND4, asubunit of complex I of oxidative phosphorylation in mitochondria, is decreased in tem-poral cortex of brain of Alzheimer’s disease patients. Brain Res 1996; 713:290-293.
35. Davis RE, Miller S, Herrnstadt C et al. Mutations in mitochondrial cytochrome-c oxidasegenes segregate with late-onset Alzheimer disease. Proc Natl Acad Sci USA 1997;94:4526-4531.
36. Benzi G, Moretti A. Are reactive oxygen species involved in Alzheimer’s disease? NeurobiolAging 1995; 16:661-674.
37. Jellinger P, Paulus W, Grundke-Iqbal I et al. Brain iron and ferritin in Parkinson’s andAlzheimer’s diseases. J Neural Transm Park Dis Dement Sect 1990; 2:327-340.
38. Youdim MBH, Ben-Shachar D, Riederer P. The role of iron in etiopathology of Parkinson’sdisease. Mov Disord 1993; 8:1-12.
39. Janetzky B, Reichman H, Youdim MBN et al. Iron and oxydative damage in neurodegen-erative diseases. In: Beal MF, Howell N, Bodis-Walker I, eds. Mitochondria and Free Radi-cals in Neurodegenerative Diseases. New York: Wiley-Liss 1997; 20:407-421.
40. Good PF, Olanow CW, Perl DP. Neuromelanin-containing neurons of the substantia nigraaccumulate iron and aluminum in Parkinson’s disease: A LAMMA study. Brain Res 1992;593:343-346.

249Glial Iron Sequestration and Neurodegeneration
41. Morris CM, Edwardson JA. Iron histochemistry of the substantia nigra in Parkinson’s dis-ease. Neurodegen 1994; 3:277-282.
42. Gelman BB. Iron in CNS disease. J Neuropath Exp Neurol 1995; 54:477-486.43. Thiel EC. Regulation of ferritin and transferrin receptor mRNAs. J Biol Chem 1990;
265:4771-4774.44. Klausner RD, Rouault TA, Harford JB. Regulating the fate of mRNA: The control of cellu-
lar iron metabolism. Cell 1993; 72:19-28.45. Ponka, P. Physiology and pathophysiology of iron metabolism: Implications for iron che-
lation therapy in iron overload. In: Bergeron RJ, Brittehham GM, eds. The Developmentof Iron Chelators for Clinical Use. Boca Raton: CRC Press, 1994.
46. Faucheux BA, Mirch EC, Villares T et al. Distribution of 125I-ferrotransferrin binding sitesin the mesencephalon of control subjects and patients with Parkinson’s disease. J Neurochem1993; 60:2338-2341.
47. Dwork AJ, Schon EA, Herbert J. Nonidentical distribution of transferrin and ferric iron inhuman brain. Neurosci 1988; 27:333-345.
48. Mash DC, Sanchez-Ramos J, Weiner WJ. Transferrin receptor regulation in Parkinson’sdisease and MPTP-treated mice. In: Narabayashi H, Nagatsu T, Xanagisawa N, Maurino Y,eds. Advances in Neurology. New York: Raven Press Ltd., 1993; 60:133-139.
49. Mash DC, Pablo J, Buck BE et al. Distribution and number of transferrin receptors inParkinson’s disease and in MPTP-treated mice. Exp Neurol 1991; 114:73-81.
50. Kalaria RN, Sromek SM, Grahovac I, Harik SI. Transferrin receptors of rat and humanbrain and cerebral microvessels and their status in Alzheimer’s disease. Brain Res 1992;87-93.
51. Faucheux BA, Nillesse N, Damier P et al. Expression of lactoferrin receptors is increasedin the mesencephalon of patients with Parkinson disease. Proc Natl Acad Sci USA 1995;92:9603-9607.
52. Leveugle B, Fauxheux BA, Bouras C et al. Cellular distribution of the iron-binding proteinlactotransferrin in the mesencephalon of Parkinson’s disease cases. Acta Neuropath 1996;91:566-572.
53. Connor JR, Menzies SL, St. Martin SM, Mufson EJ. A histochemical study of iron, trans-ferrin, and ferritin in Alzheimer’s diseased brains. J Neurosci Res 1992; 31:75-83.
54. Morris CM, Kerwin JM, Edwarsan JA. Non-haem iron histochemistry of the normal andAlzheimer’s disease hippocampus. Neurodegen 1994; 3:267-275.
55. Kawamata T, Tooyama I, Yamada T et al. Lactotransferrin immunocytochemistry inAlzheimer and normal human brains. Am J Pathol 1993; 142:1574-1585.
56. Leveugle B, Spik G, Perl DB et al. The iron binding protein lactotransferrin is present inpathologic lesions in a variety of neurodegenerative disorders: A comparative immunohis-tochemical analysis. Brain Res 1994; 650:20-31.
57. Osmand AP, Switzer RC III. Differential distribution of lactoferrin and Alz-50 immunore-activities in neuritic plaques and neurofibrillary tangles in Alzheimer’s disease. In: Iqbal K,McLachlan RC, Winblad B, Wisniewski HM, eds. Alzheimer’s Disease: Basic Mechanisms,Diagnosis and Therapeutic Strategies. New York: Wiley 1991:219-227.
58. Rebeck GW, Harr SD, Strickland KD, Hyman BT. Multiple, diverse senile plaque-associ-ated proteins are ligands of an apolipoprotein E receptor, the alpha-macroglobulin recep-tor/low density lipoprotein receptor-related protein. Ann Neurol 1995; 37:211-217.
59. Goldgefter L, Schejter AS, Gill D. Structural and microspectrofluorometric studies on glialcells from the periventricular and arcuate nuclei of the rat hypothalamus. Cell Tissue Res1980; 211:503.
60. Schipper HM. Gomori-positive astrocytes: Biological properties and implications for neu-rologic and neuroendocrine disorders. Glia 1991; 4:365.
61. Brawer JR, Reichard G, Small L, Schipper HM. The origin and composition of peroxidase-positive granules in cysteamine-treated astrocytes in culture. Brain Res 1994; 633:9-20.
62. Wang X, Manganaro F, Schipper HM. A cellular stress model for the sequestration of re-dox-active glial iron in the aging and degenerating nervous system. J Neurochem 1995;64:1868-1877.

Astrocytes in Brain Aging and Neurodegeneration250
63. Duchen MR, Biscoe TJ. Mitochondrial function in type I cells isolated from arterialchemoreceptors. J Physiol 1992:450:13-31.
64. Kohler M, Fromter E. Identification of mitochondrial-rich cells in unstained preparationof epithelia by autofluorescence. Eur J Physiol 1985; 403:47-49.
65. Richardson DR, Baker E. The uptake of inorganic iron complexes by human melanomacells. Biochim Biophys Acta 1991; 1093:20-28.
66. Richardson DR, Baker E. The uptake of iron and transferrin by human malignant malanomacells. Biochim Biophys Acta 1990; 1053:1-12.
67. Richardson DR, Baker E. Two mechanisms of iron uptake from transferrin by melanomacells. The effects of desferrioxamine and ferric ammonium citrate. J Biol Chem 1992;267:13972-13979.
68. Chan RYY, Ponka P, Schulman HM. Transferrin-receptor-independent but iron-depen-dent proliferation of variant Chinese hamster ovary cells. Exp Cell Res 1992; 202:326-336.
69. Inman RS, Wessling-Resnick M. Characterization of transferrin-independent iron trans-port in K562 cells. Unique properties provide evidence of multiple pathways of iron up-take. J Biol Chem 1993; 268:8521-8528.
70. Adams ML, Ostapiuk I, Grasso JA. The effects of inhibition of heme synthesis on the in-tracellular localization of iron in rat reticulocytes. Biochim Biophys Acta 1989; 1012:243-253.
71. Schipper HM, Bernier L, Bernatchez G. Pathological glial-neuronal interaction in Parkinson’sdisease. Soc Neuro Abst 1996; 22:219.
72. He Y, Thong PSP, Lee T et al. Increased iron in the substantia nigra of 6-OHDA inducedParkinsonian rats: A nuclear microscopy study. Brain Res 1996; 735:149-153.
73. Young JK, McKenzie JC, Baker JH. Association of iron-containing astrocytes with dopam-inergic neurons of the arcuate nucleus. J Neurosci Res 1990; 25:204-213.
74. Schipper HM, Cissé S, Stopa EG. Expression of heme oxygenase-1 in senescent andAlzheimer-diseased brain. Ann Neurol 1995; 37:758-768.
75. Zhang J, Piantadosi CA. Mitochondrial oxydative stress after carbon monoxide hypoxia inthe rat brain. J Clin Invest 1992; 90:1193-1199.
76. Premkumar DRD, Smith MA, Pichey PL et al. Induction of heme oxygenase-1 mRNA andprotein in neocortex and cerebral vessels in Alzheimer’s disease. J Neurochem 1995;65:1399-1402.
76a. Schipper HM, Liberman A, Stopa EG. Neural heme oxygenase-1 expression in idiopathicParkinson's disease. Exp Neurol 1998; 150:60-68.
77. Beal MF. Mitochondrial Dysfunction and Oxidative Damage in Neurodegenerative Dis-eases. Austin: RG Landes, 1995:1-128.
78. Reichmann H, Riederer P. Mitochondrial disturbances in neurodegeneration. In: Calne DB,ed. Neurodegenerative Diseases. Philadelphia: Saunders 1994:195-204.
79. Chopra VS, Chalifour LE, Schipper HM. Differential effects of cysteamine on heat shockprotein induction and cytoplasmic granulation in astrocytes and glioma cells. Mol BrainRes 1995; 31:173-184.
80. Manganaro F, Chopra VS, Mydlarski MB et al. Redox perturbations in cysteamine-stressedastroglia: Implications for inclusion formation and gliosis in the aging brain. Free Rad BiolMed 1995; 19:823-835.
81. Mydlarski MB, Liberman A, Schipper HM. Estrogen induction of glial heat shock proteins:Implications for hypothalamic aging. Neurobiol Aging 1995:16:977-981.
82. Bernier L, Bernachez G, Schipper HM. Oxidative stress promotes mitochondrial iron se-questration in cultured astroglia. Soc Neuroscience Abst 1996; 22:1495.
83. Schipper HM, Kotake Y, Janzen EG. Catechol oxidation by peroxidase-positive astrocytesin primary culture: An electron spin resonance study. J Neurosci 1991; 11:2170.
84. Metodiewa D, Reszka K, Dunford H. Evidence for a peroxidatic oxidation of norepineph-rine, a catecholamine, by lactoperoxidase. Biochem Biophys Res Commun 1989; 160:1183.
85. DiMonte DA, Schipper HM, Hetts S, Langston JW. Iron-mediated bioactivation of 1-me-thyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in glial cultures. Glia 1995; 15:203-206.
86. Newcomer TA, Rosenberg PA, Aizenman E. Iron-mediated oxidation of 3,4-dihydrophenyl-alanine to an excitotoxin. J Neurochem 1995; 64:1742-1748.

251Glial Iron Sequestration and Neurodegeneration
87. Kontos H, Wei W, Ellis E et al. Appearance of superoxide anion radical in cerebral extra-cellular space during increased prostaglandin synthesis in cats. Cir Res 1985; 57:142.
88. Frankel D, Schipper HM. Does astroglial senescence facilitate oxidative neuronal injury?Soc Neurosci Abstr 1997; 23:1371.
89. Bowling AC, Mutisya EM, Walker LC et al. Age-dependent impairment of mitochondrialfunction in primate brain. J Neurochem 1993; 60:1964-1967.
90. Linnane AW, Zhang C, Baumer A, Nagley P. Mitochondrial DNA mutation and the agingprocess: Bioenergy and pharmacological intervention. Mutat Res 1992; 275:195-208.
91. Miquel J, Fleming J. Theoretical and experimental support for an “oxygen radical-mito-chondrial injury” hypothesis of cell aging. In: Johnson JE Jr, ed. Free Radicals, Aging, andDegenerative Diseases. New York: Alan R. Liss, 1986:51-74.
92. Shigenaga MK, Hagen TM, Ames BN. Oxidative damage and mitochondrial decay in ag-ing. Proc Natl Acad Sci USA 1994; 91:1077-10778.
93. Soong NW, Hinton DR, Cortopassi G, Arnheim M. Mosaicism for specific somatic mito-chondrial DNA mutation in adult human brain. Nat Genet 1992; 2:318-323.
94. Gutteridge JM. Iron and oxygen radicals in brain. Ann Neurol (Suppl.) 1992; 32:S16-S21.95. Connor JR, Menzles S, St. Martin SM, Mufson EJ. Cellular distribution of transferrin, fer-
ritin and iron in normal and aged human brains. J Neurosci 1990; 27:595-611.96. Olanow CW. Magnetic resonance imaging in Parkinsonism. In: Cederbaum JM, Gancher
ST, eds. Neurological Clinics Par 2, Vol. 10. Philadelphia: Saunders, 1992; 405-420.


Index
A
αB-crystallin 78, 79, 210, 212-215, 217Achromatic (ballooned) neuron 176, 180,
210, 213Acyl-CoA binding protein 200Adenosine triphosphate (ATP) 17, 24, 25,
101, 114, 115, 119, 174, 207, 217Adhesion molecule 5, 20, 29, 42, 46, 47, 49,
50, 51, 75, 92Aging 71, 74-78, 93, 104, 111, 117-119, 121,
165, 166, 171, 191, 193-196, 201, 202,207-209, 211-215, 217, 219-223, 228,236-238, 242, 243, 245-247
Aicardi’s syndrome 103Alexander’s disease 78-80, 82, 99, 210Alpha-1-antichymotrypsin 46, 49, 50, 92Alzheimer type II astrocytes/cells 43, 52Alzheimer’s disease (AD) 45, 46, 49, 50, 73,
74, 91-95, 99, 101, 104, 137, 147, 148,165-167, 169-171, 173, 176, 177, 182,209, 210, 212, 213, 235-237, 241
Ammonia 41, 43, 51-53AMPA/kainate receptor 18Amyloid (Aβ) 42, 46, 49, 50, 73, 77, 91-93,
95, 96, 98, 129, 135, 137, 147, 166, 177,182, 235-237
Amyotrophic lateral sclerosis (ALS) 22, 46,51, 52, 73, 74, 169, 170, 181, 210, 213,237
Anchorage densities 91, 99, 103, 104Antioxidant 43, 196, 202, 208, 219, 221, 235,
246Apolipoprotein E (Apo E) 49, 50, 92, 148Arcuate nucleus 192-196, 201, 202, 216, 217,
219, 239Argyrophilic inclusions 174Ascorbate 238Aspartate 19, 21, 44, 115, 119Astrocyte 3-9, 15-31, 41, 43-56, 71, 73-79, 81,
82, 91-93, 95, 97, 99, 101-104, 111, 112,114-122, 137, 139, 140, 142, 143,145-158, 168, 169, 174, 177, 180-187,197-209, 212, 213, 215, 217, 219-223,228-231, 233, 234, 242, 245, 249-251
Astrocytoma 78, 210Astrogliosis (gliosis) 22, 28, 30, 42, 43, 46-51,
71, 73, 74, 76, 78, 92, 93, 95, 99, 104, 111,129-131, 135, 143, 147, 165, 169, 171,175, 181, 195, 200, 207, 210, 211, 221,228, 235
Autofluorescence 192, 196, 198, 202, 223,224, 227, 238
Autoxidation 116, 118, 195, 219, 220, 243,244
B
Bergmann glia 4, 8, 9, 17, 20, 22-24, 26, 131,139, 142, 208
Blood-brain barrier (BBB) 7, 28, 29, 41, 50,75, 112, 121, 192, 196
Bone morphogenic proteins (BMP) 6Bovine spongiform encephalopathy (BSE)
127, 135, 137, 142, 143, 150, 151, 154
C
Carbon monoxide (CO) 239, 241, 244Catalase 42, 192, 220, 235Catecholamine 114, 116, 118, 120, 196, 202,
242, 246Catecholestrogen 195, 196, 202, 219, 242Cathepsin 46, 50, 148, 201, 202Chemokines 42, 45, 46, 49, 75Chronic wasting disease (CWD) 127, 137,
143Ciliary neurotrophic factor (CNTF) 6, 71Coiled bodies 170, 173-178, 180Copper 77, 199, 200Corpora amylacea (CA) 77, 78, 91, 99, 104,
171, 174, 191, 202, 207, 210, 215,221-224, 227, 228, 238
Corticobasal degeneration (CBD) 99, 169,170, 174, 176-180
Creutzfeldt-Jakob disease (CJD) 74, 127,129-133, 135-137, 140-143, 145-147,149-154, 165
Cysteamine 76, 197-200, 202, 210, 224, 238,243, 244
Cytochrome C oxidase 236Cytokine 28-30, 42, 43, 45, 46, 48-50, 71, 74,
75, 92, 93, 120, 143, 149-154Cytoprotection 220, 221, 241
D
Dopamine 112, 113, 115-118, 121, 235, 238,241, 242, 244, 246
Down’s syndrome (DS) 73, 237

Astrocytes in Brain Aging and Neurodegeneration254
E
Electron spin resonance (ESR) spectroscopy242-244
Endothelin 23, 92, 93Eosinophilic inclusions 51, 91, 99, 101-103Epidermal growth factor (EGF) 6, 23Epilepsy 50, 51Estrogen 194, 195, 202, 211, 214, 217, 219,
224Experimental allergic encephalomyelitis
(EAE) 30, 45, 49, 73, 75, 149, 150Extracellular matrix molecules (ECM) 45,
47, 49
F
Fatal familial insomnia (FFI) 127, 129Fatty acid binding protein (FABP) 200Ferritin 77, 142, 236, 237Fibrils 50, 73, 77, 93, 95, 97, 99, 104, 112, 131,
137, 182Fibrous astrocyte 77, 177Frontotemporal dementia (FTDP-17) 169,
180
G
Gemistocytic astrocytes 133, 136Gerstmann-Straussler-Scheinker disease
(GSS) 73, 74, 127, 129, 130, 135-140,142, 154
Glial cytoplasmic inclusion (GCI) 78, 165,170, 174, 180, 181, 210
Glial derived neurotrophic factor (GDNF)121
Glial fibrillary acidic protein (GFAP) 3, 4, 6,7, 15, 18, 24-26, 42, 48-50, 53, 71-79, 93,101, 116, 129, 130, 132, 133, 139, 142,145-148, 152, 153, 166, 171, 191, 192,196, 210-213, 216, 217, 219, 221, 239,241, 242
Glucose-regulated protein (GRP) 207, 211,214, 215, 219
Glutamate 19, 21-26, 29, 30, 41-45, 48, 50-53,92, 119, 120, 147
Glutamine 3, 21, 26, 44, 52, 147Glutathione (GSH) 42, 112, 115, 116, 121,
208, 235Glycogen 4, 42, 43, 44, 53, 77, 119, 130, 223Glycosylation 127, 223, 227Gray matter 3, 4, 5, 7, 8, 74, 76, 91, 93, 138,
140, 142, 145, 170, 173, 175, 177, 179Guam Parkinson-dementia complex (GPDC)
169, 170, 175, 237
H
Heat shock factor (HSF) 207, 209Heat shock protein (HSP) 71, 77-79, 149,
207-215, 217, 219, 221-223, 225, 227,244, 246
Heme 196, 207, 220, 221, 227, 237-239, 241,242, 244, 246
Heme oxygenase-1 (HO-1) 207, 208,210-212, 214, 215, 219, 221, 223, 227,239, 241, 242, 244, 246, 247
Hepatic encephalopathy (HE) 43, 52, 53, 74Hippocampus 23-25, 52, 75-77, 139, 140,
146, 192, 195, 200, 202, 209, 211-213,235, 237, 239
HIV-1 29, 44, 48Huntington’s disease 73, 180Hydrogen peroxide (H
2O
2) 76, 92, 112,
115-118, 195, 211, 215, 217, 219-222,235, 241-244
Hypothalamus 4, 45, 74, 76, 192-196, 219
I
Interferon-γ (IFN-γ) 29-31, 46, 74, 150, 151Interleukin-1 (IL-1) 28-31, 45, 46, 50, 71, 75,
92, 150-153, 168Ion channel 16, 18, 19, 25Iron (Fe) 77, 112, 118, 121, 192, 193, 196,
197, 199, 200, 202, 219, 220, 223, 225,227, 236-247
Ischemia 19, 21, 42, 44, 46, 52, 95, 168, 208
K
Kuru 74, 127, 128, 130, 131, 135, 137-140,142
L
L-Dopa 112, 115, 238Lactic acid 43, 44, 45, 52Lactoferrin 237Lafora disease 104Lipid peroxidation 195, 220, 235, 237, 243Lipofuscin 130, 191, 192, 202, 211Lymphocyte 30, 74, 75, 143, 150Lysosome 201, 202, 215, 223, 227

255Index
M
Manganese superoxide dismutase (MnSOD)219, 221, 235, 242, 245, 246
Metalloproteinase 46, 501-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
(MPTP) 51, 111, 113-121, 235, 242Microglia 28-31, 41, 44-50, 53, 71, 74-77,
91-95, 112, 137-140, 142, 143, 149, 150,153, 194, 210, 236, 237
Microtubule associated protein (MAP) 78,168
Mitochondria 42, 43, 52, 53, 96, 98, 112-115,119, 122, 196, 197, 200, 202, 214-217,219, 220, 222, 223, 225, 227, 228, 235,236, 238-242, 244-247
Monoamine oxidase (MAO) 42, 51, 112, 114-118, 121, 235, 242
Monocyte/macrophage 74, 75Muller glia 4Multiple sclerosis (MS) 30, 45, 46, 48, 49, 71,
72, 75, 76, 78, 143, 145, 149, 150, 153, 209Multiple system atrophy (MSA) 78, 169, 170,
174, 180, 181, 209, 210
N
Nerve growth factor (NGF) 46, 71, 121Neuritic plaque 45, 46, 49, 50, 73, 91-93, 137,
147, 177, 210, 237Neurofibrillary tangles (NFT) 73, 95, 99, 166,
167, 169, 171, 173, 175, 176, 209, 210,235, 237
Neuropil threads 173Neurotoxin 48, 52, 117, 118, 121, 235, 247Nitric oxide (NO) 29, 30, 42, 43, 45, 46,
48-50, 52, 75, 92, 115, 120, 121NMDA receptor 24, 44, 45, 48, 114, 115, 119,
120Nuclear inclusions 131, 181
O
Oligodendrocyte 6-8, 41, 43, 47, 49, 74, 76,78, 91, 99, 143-145, 149, 150, 166, 168,170, 171, 173, 177-179
Oxidative stress 52, 76-78, 112, 113, 115, 116,118, 121, 122, 207, 208, 214, 215, 217,219, 220, 223, 235-239, 244, 245
P
Paired helical filaments (PHF) 78, 91, 95, 97,99, 100, 167-169, 171, 177, 179, 180, 210,213, 236
Parkinson’s disease (PD) 51, 73, 74, 111-113,116-122, 165, 166, 181, 209, 210, 213,219, 235-239, 241, 243, 246, 247
PC12 cells 242Peroxidase-positive astrocyte 191-197, 200,
211, 214, 215, 217, 223, 242, 243Phosphorylation 29, 53, 92, 99, 112, 119, 167,
169, 220Pick bodies 176, 177, 210, 213Pick’s disease 74, 99, 169, 170, 175-177, 180,
210, 213, 237Pilocytic astrocytes 136Porphyrin 238, 239, 244Potassium (K+) 3, 16-21, 23-27, 29, 30, 41,
43, 44, 48, 51, 74-76Prion protein (PrP) 74, 127, 129, 135, 137,
142, 146-149, 153, 154Progressive supranuclear palsy (PSP) 78, 99,
104, 169-171, 173-177, 180, 210, 212, 213Protoplasmic astrocyte 15, 74Pyruvate dehydrogenase 236
Q
Quinolinic acid 29, 42, 44, 48, 51
R
Radial glia 4-6Reactive oxygen species 50, 77, 118, 195, 196,
207, 220, 221, 236, 242Rosenthal fibers (RFs) 78-80, 91, 99, 100,
101, 140, 165, 210
S
S-100b 3, 50, 71, 92Scrapie 74, 127, 135, 137, 139-142, 146-148,
150, 151, 154Semiquinone 195, 196, 219, 242-244Stress granule 211, 223, 246Substantia nigra 73, 112, 115, 116, 118-121,
165, 192, 193, 209, 212, 213, 219,235-239, 246
Subventricular zone (SVZ) 4-7, 9

Astrocytes in Brain Aging and Neurodegeneration256
T
Tau 77, 78, 91, 92, 95, 99, 166-181, 210, 212,213
Tau-positive inclusions 99, 168, 169,173-175, 180
Taurine 22, 43, 44, 51Thorn shaped astrocytes 170Transferrin 171, 236-238, 240, 246Transforming growth factor-β (TGF-β) 6, 48,
75, 92, 121, 150Transmissible mink encephalopathy (TME)
127, 142Transmissible spongiform encephalopathies
(TSE) 127, 129, 135, 137, 140, 143, 145,149-154
Trauma 8, 21, 31, 42, 44, 46, 196, 209, 222Tufted astrocyte 170, 174-176, 180Tumor necrosis factor α (TNF α) 29, 30, 45,
46, 92, 120, 149-154
U
Ubiquitin (Ub) 51, 77, 78, 95, 101, 170, 174,180, 181, 207-210, 212, 214, 215, 217,219, 221-223, 225, 227
V
Velate astrocyte 8, 9, 131Vimentin 3, 4, 42, 50, 78, 79, 147, 178
W
Wilson’s disease 73, 74
X
X-irradiation 196, 219, 224