article - igf structural basis for mor activation, here we report a 2.1 a˚ x-ray crystal structure...
TRANSCRIPT

ARTICLEdoi:10.1038/nature14886
Structural insights into m-opioidreceptor activationWeijiao Huang1*, Aashish Manglik1*, A. J. Venkatakrishnan1,2,3, Toon Laeremans4,5, Evan N. Feinberg1,2,3, Adrian L. Sanborn1,2,3,Hideaki E. Kato1, Kathryn E. Livingston6, Thor S. Thorsen1, Ralf C. Kling7, Sebastien Granier8, Peter Gmeiner7,Stephen M. Husbands9, John R. Traynor6, William I. Weis1,10, Jan Steyaert4,5, Ron O. Dror1,2,3 & Brian K. Kobilka1
Activation of the m-opioid receptor (mOR) is responsible for the efficacy of the most effective analgesics. To shed light onthe structural basis for mOR activation, here we report a 2.1 A X-ray crystal structure of the murine mOR bound to themorphinan agonist BU72 and a G protein mimetic camelid antibody fragment. The BU72-stabilized changes in the mORbinding pocket are subtle and differ from those observed for agonist-bound structures of the b2-adrenergic receptor(b2AR) and the M2 muscarinic receptor. Comparison with active b2AR reveals a common rearrangement in the packingof three conserved amino acids in the core of the mOR, and molecular dynamics simulations illustrate how theligand-binding pocket is conformationally linked to this conserved triad. Additionally, an extensive polar networkbetween the ligand-binding pocket and the cytoplasmic domains appears to play a similar role in signal propagationfor all three G-protein-coupled receptors.
The most powerful analgesic and addictive properties of opiate alka-loids are mediated by the mOR1. As the receptor primarily responsiblefor the effects of opium, the mOR is one of the oldest drug targetswithin the pharmacopeia2. Opioid receptors are highly versatile sig-nalling molecules. Activation of the mOR results in signalling throughthe heterotrimeric G protein Gi, resulting in analgesia and sedation aswell as euphoria and physical dependence3. The mOR can also signalthrough arrestin, and this pathway has been attributed to adverseeffects of opioid analgesics including tolerance, respiratory suppres-sion, and constipation4–6.
The mOR has been the subject of intense focus for drug-discoveryefforts over the past century, with the identification of numerousligands of varying efficacy. These drugs occupy a wide chemical spec-trum, from small organic molecules to a variety of endogenous andsynthetic peptides7. Structure–activity studies have revealed thatsubtle changes in ligand structure can convert an agonist into anantagonist7. These studies have yielded a general hypothesis for theinformation encoded within G-protein-coupled receptor (GPCR)ligands where distinct pharmacophores within a drug are responsiblefor efficacy (message) or selectivity (address)8 (Fig. 1a). For the mor-phinan ligands, our previous structural examination of the inactivestates of the mOR and the dOR revealed molecular insights into ligandselectivity9,10. To understand the structural basis for mOR activation,we obtained a structure of this receptor in the active state using acombination of a high-affinity agonist and a G protein mimetic camelidantibody fragment. A comparison of this structure with the inactive-state structures of the mOR9 and dOR10,11, as well as the inactive- andactive-state structures of the b2AR12–15, M2 muscarinic receptor(M2R)16,17, and rhodopsin18,19, provide insights into shared mechan-isms of GPCR activation.
Nanobody-stabilized structure of the mORThe active states of ligand-activated GPCRs are probably unstable,even when bound to full agonists20–23. However, the active conforma-tion can be stabilized by interactions between a receptor and its cog-nate G protein. This stabilization is reflected in a higher affinity foragonists when GPCRs are in complex with their cognate G protein24.In the case of the mOR, the affinity for the morphinan agonist BU72 isenhanced by 47-fold when coupled to the G protein Gi (Fig. 1b, c).Efforts to obtain a structure of activated mOR in complex with Gi havethus far not been successful. As an alternative, we have previouslyused camelid single-domain antibody fragments (nanobodies) as Gprotein mimetics to stabilize the active conformation of the b2AR andM2R for structural study12,13,17. For the b2AR, the conformation ofthe receptor obtained in complex with the Gs mimetic nanobody 80(Nb80) was nearly identical to that in the b2AR–Gs complex25 (rootmean squared deviation, 0.61 A).
To generate G protein mimetic nanobodies for the mOR, llamaswere immunized with purified mOR bound to the peptide agonist[Dmt1]DALDA (ref. 26) and reconstituted into phospholipid vesi-cles12. We examined the ability of selected nanobodies to stabilizethe high-affinity state for mOR agonists. Purified mOR was reconsti-tuted into high-density lipoprotein (HDL) particles and agonist com-petition assays were performed in the presence or absence ofnanobodies (Fig. 1b). In the presence of 5 mM nanobody 39 (Nb39),the affinity of the potent morphinan agonist BU7227 increases from470 pM to 16 pM (Fig. 1b). BU72 has a dissociation half-life of140 min in the presence of Nb39 (Extended Data Fig. 1b). Nb39 alsoenhances the affinity of mOR agonists DAMGO and endomorphin-2,indicating that the effect is not limited to morphinan agonists(Extended Data Fig. 1a).
*These authors contributed equally to this work.
1Departmentof Molecular and Cellular Physiology, Stanford UniversitySchool of Medicine, 279 CampusDrive, Stanford, California 94305,USA. 2Departmentof Computer Science, Stanford University, 318Campus Drive, Stanford, California 94305, USA. 3Institute for Computational and Mathematical Engineering, Stanford University, 475 Via Ortega, Stanford, California 94305, USA. 4Structural BiologyBrussels, Vrije Universiteit Brussel, Pleinlaan 2, B-1050 Brussels, Belgium. 5Structural Biology Research Center, VIB, Pleinlaan 2, B-1050 Brussels, Belgium. 6Department of Pharmacology, Universityof Michigan, Ann Arbor, Michigan 48109, USA. 7Department of Chemistry and Pharmacy, Friedrich Alexander University, Schuhstrasse 19, 91052 Erlangen, Germany. 8Institut de GenomiqueFonctionnelle, CNRS UMR-5203 INSERMU1191, University of Montpellier, F-34000Montpellier, France. 9Departmentof Pharmacy and Pharmacology, University of Bath, Bath BA2 7AY, UK. 10Departmentof Structural Biology, Stanford University School of Medicine, 299 Campus Drive, Stanford, California 94305, USA.
G2015 Macmillan Publishers Limited. All rights reserved
0 0 M O N T H 2 0 1 5 | V O L 0 0 0 | N A T U R E | 1

Crystals of the mOR bound to BU72 and Nb39 were obtained in amonoolein lipidic mesophase28 and a complete data set to 2.1 A wasobtained by merging diffraction data from four crystals (ExtendedData Table 1). Nb39 binds to the intracellular surface of mOR(Fig. 1d and Extended Data Fig. 2) and mediates the majority ofpacking interactions between lipidic layers and adjacent Nb39–mORcomplexes in the crystal lattice. There are no packing interactionsinvolving the extracellular surface of the receptor (Extended DataFig. 1c). We observe a limited parallel dimeric packing interactionbetween mOR molecules involving the extracellular end of transmem-brane helix 1 (TM1), TM2, the first extracellular loop (ECL1), andhelix 8 with a buried surface area of 460 A2 (Fig. 1e). A similar inter-face between TM1, TM2, and helix 8 was also observed in the inactivestructure of mOR with a slightly larger buried surface area of 615 A2
(Fig. 1e). The inactive structure also identified a more extensive par-allel dimer interaction involving TM5 and TM6 with a buried surfacearea of 1,460 A2 (Fig. 1f). This interaction involving TM5 and TM6would not be compatible with the conformational changes we observein the active state (Fig. 1f). It is important to note that the phys-iological relevance of these interfaces remains unclear.
Structural differences in the extracellular surface between inactiveand active mOR are relatively small (Fig. 2a) with the exception of theproximal N terminus, as discussed below. Conformational changes atthe cytoplasmic surface of the mOR observed upon activation aresimilar to those observed for the b2AR, M2R and rhodopsin, with
large outward movement of TM6 and a smaller inward movementof TM5 and TM7 (Fig. 2a and Extended Data Fig. 3). The conservedE/DRY motif at the intracellular end of TM3 plays a role in main-taining GPCRs in the inactive state. In rhodopsin, an ionic interactionbetween R1353.50 and E2476.30 in TM6 stabilizes TM6 in an inactiveconformation (superscript numbers follow the Ballesteros–Weinsteinnumbering method for GPCRs29). While there is no acidic amino acidat the end of TM6 of mOR that can form a similar salt bridge withR1653.50, R1653.50 can form a hydrogen bond with T2796.34 (Fig. 2b).In the active states of the mOR and metarhodopsin II, R3.50 forms ahydrogen bond with Y5.58, stabilizing the inward movement of TM5(Fig. 2b). R3.50 and Y5.58 assume a similar orientation in the M2R andthe b2AR–Gs complex; however, they are not close enough to form ahydrogen bond (Extended Data Fig. 3a).
Agonist binding pocketThe morphinan scaffold of BU72 binds to the activated mOR in asimilar orientation to that observed previously for the irreversibleantagonist b-funaltrexamine (b-FNA) at the inactive mOR (Fig. 3aand Extended Data Fig. 4). Fig. 3b shows that the overall structuraldifferences in the orthosteric binding pockets of active and inactivemOR are relatively subtle. The majority of interactions between BU72and active mOR are hydrophobic or aromatic in nature, with theexception of two conserved polar interactions (Fig. 3c and ExtendedData Fig. 4). As observed previously in the inactive structures of the
Message
Address
Nb39
O
HO
OH
NH+
HN
O
HO
OH
NH+
CH3
O
NH+
CH3
O
HO
OH
NH+
OOO
HO
NH+
CH3
O
HN
ActiveμOR
InactiveμOR
ActiveμOR
Ki = 16 pM
a b c
d
e f
Oxymorphone
(agonist)
Naltrexone
(antagonist)
Naltrindole
(δ selective antagonist)
No
rmaliz
ed
3H
-DP
N b
ind
ing
BU72
TM1–TM2–H8 TM5–TM6
Steric clash
Extracellular
Intracellular
log BU72 (M)
μOR–HDL
μOR–HDL+ 5 μM Nb39
+ 500 nM Gi
Khigh= 10 pM
Klow= 470 pM
0
20
40
60
80
100–14 –13 –12 –11 –10 –9 –8 –7 –6
–14 –13 –12 –11 –10 –9 –8 –7 –6
0
20
40
60
80
100
TM6
TM7
TM2
TM3
TM4
TM6
TM5
TM7TM1
TM2
TM3
TM4
90°
90°
90°
90°
Figure 1 | Activated structure of mOR bound to BU72 and Nb39.a, Structures of prototypical opioid ligands highlighting regions involved inencoding efficacy (message) and selectivity (address). b, 3H-diprenorphine(3H-DPN) radioligand competition binding of mOR in HDL particles. In thepresence of Gi, the affinity of the morphinan agonist BU72 increases 47-fold.The two observed binding sites indicate the affinity of BU72 for receptorcoupled to Gi (Khigh) and uncoupled to Gi (Klow). A similar 29-fold increase inaffinity is observed in presence of Nb39. The binding curves are representative
of at least three experiments performed in triplicate, and the data and error barsrepresent the mean 6 s.e.m. c, Structure of the high affinity agonist BU72.d, Overall structure of the mOR–BU72–Nb39 complex. e, An interface betweenTM1–TM2 and helix 8 (H8) is observed in both inactive and active structuresof the mOR. The residues comprising the interface are highlighted in darkcolours on the surface view. f, The TM5–TM6 interface observed for inactivemOR is not compatible with the active state due to clashing residues in TM5 andTM6 (highlighted in red).
RESEARCH ARTICLE
G2015 Macmillan Publishers Limited. All rights reserved
2 | N A T U R E | V O L 0 0 0 | 0 0 M O N T H 2 0 1 5

mOR, dOR and kOR30, the phenolic hydroxyl of BU72 engages in awater-mediated interaction with H2976.52. In the active state, thisnetwork is more extended, and involves Y1483.33 and the backbonecarbonyl of K2335.39. An ionic interaction between the morphinantertiary amine and D1473.32 is seen in both the active mOR bound toBU72 and in inactive mOR bound to b-FNA (Fig. 3c).
We also observe an unexpected interaction between BU72 and theamino terminus of themOR, which forms a lid over the ligand-bindingpocket (Fig. 3d and Extended Data Fig. 5). In particular, the amino-terminal residue H54 is positioned 2.6 A from secondary amine ofBU72. It is of interest that binding of BU72, as well as other structur-ally unrelated peptide agonists like DAMGO and [Dmt1]DALDA,lead to conformational changes in the truncated amino terminus thatcan be detected by nuclear magnetic resonance (NMR) spectroscopy(Sounier et al., ref. 31). While interactions between the amino ter-minus and the transmembrane core may indeed be important formOR function32, the specific interaction observed here is unlikely tobe physiologically relevant as H54 is not highly conserved and theH54A mutation does not alter the affinity of BU72 in mOR with thefull-length amino terminus (mOR wild-type Ki 5 21 pM and mORH54A Ki 5 30 pM in HEK293 membranes). Additionally, using abioluminescence resonance energy transfer assay33, we observe nosignificant difference in the EC50 of Gi activation by BU72 between
the wild-type receptor and the H54A mutant (EC50 for wild type of79 6 17 pM and H54A mutant of 67 6 20 pM).
While the morphinan core of BU72 is unambiguously placedwithin the electron density, there are two unexplained features ofthe ligand. We observe a strong positive electron density (Fo 2 Fc
signal) between the H54 side chain and the BU72 amine (ExtendedData Fig. 5c). Attempts to identify the source of this density, includingmass spectrometry for an alternative ligand structure and thepresence of an anomalous signal for a coordinated metal were unsuc-cessful (Extended Data Fig. 4). Another unexpected finding is thenear-planar geometry of the sp3 hybridized carbon of BU72 to whichthe pendant phenyl group is attached (Extended Data Fig. 4e–h). Thisgeometry could be explained by a double bond between thiscarbon and the adjacent nitrogen. We observed a minor fraction ofsuch a compound by mass spectrometry in our preparation ofBU72. However, this compound was not observed in mass spectro-metry of the mOR–ligand–Nb39 complex used for crystallography.Ultimately, we modelled a higher-energy conformation of BU72within the observed electron density (see Extended Data Fig. 4 forfurther discussion).
In the active mOR, there is a water-filled cavity lined by polar andaromatic side chains that extends off of the intracellular end of theligand-binding pocket (Fig. 3e). While a similar cavity is observed in
Morphinan
scaffold
H54H54V1433.28V1433.28
W3187.35W3187.35
V3006.55V3006.55
I2966.51I2966.51
I3227.39I3227.39
H2976.52H2976.52Y3267.43Y3267.43
M1513.36M1513.36
W2936.48W2936.48
Y1483.33Y1483.33
D1473.32D1473.32
V2365.42V2365.42
D1473.32D1473.32
Y1483.33Y1483.33
V1433.28V1433.28
Y3267.43Y3267.43
W2936.48W2936.48
W3187.35W3187.35 I3227.39I3227.39
H2976.52H2976.52V3006.55V3006.55 M1513.36M1513.36
TM3TM3
TM5TM5
TM6TM6
N terminus
TM5TM5TM3TM3
TM2TM2
TM6TM6
TM7TM7
a b
d
H2976.52H2976.52
K2335.39K2335.39
Y1483.33Y1483.33
D1473.32D1473.32
TM2TM2
TM4TM4
TM3TM3
TM5TM5
TM6TM6
TM6
TM5
TM3
c
e f
Y3267.43Y3267.43
TM7TM7
V2365.42V2365.42
CyclopropylmethylMethyl
Active μORInactive μOR
BU74(docked)
BU72
Polar cavity
TM7TM7
BU72
β-FNA
Figure 3 | The mOR agonist-binding pocket. a, BU72 and b-funaltrexamine(b-FNA) occupy a similar pose in the mOR binding pocket. The commonmorphinan scaffold shared by both ligands is highlighted. b, Binding-pocketresidues of inactive (blue) and active (green) mOR viewed from the extracellularside. c, Polar interactions between BU72 and active mOR. d, BU72 andbinding-pocket residues shown with the proximal amino terminus (grey).e, View of the polar cavity extending towards the intracellular side ofmOR in theactive state. f, The cyclopropylmethyl group of the antagonist BU74 can bedocked to fit within the polar cavity of the active state. However, moleculardynamics simulations show that this pose is unstable (Extended Data Fig. 6).
90°
90°
TM5TM5
T6.34
TM2TM2
TM3TM3
TM1TM1
TM6TM6
TM7TM7
ECL2
TM5
H8
TM1
TM6TM5
TM3
ICL2
TM4
TM2
TM1
TM7
a
Intracellular
view
Extracellular
view
DRY motif
Y5.58
D(E)3.49R3.50
Y3.51
Y5.58
E6.30
R3.50
D(E)3.49
Y3.51
Active μORInactive μOR Active RhoInactive Rho
TM5
TM6
TM7H8
TM1
TM4
TM2
10 Å
10 Å TM6
TM7
Extracellular
IntracellularTM3
b
TM3
TM3
TM5 TM5
TM6
TM6
T6.34
Figure 2 | Structural comparison of inactive and active mOR. a, Active mORundergoes a 10 A outward displacement of TM6 on activation. Theextracellular domain of the receptor shows minimal changes upon activation.H8, helix 8. b, Left, comparison of the conserved E/DRY motif in the inactivestructures ofmOR (blue) and rhodopsin (Rho, brown) shows a polar interactionbetween R3.50 and T6.34 in mOR, analogous to the ionic lock between R3.50
and E6.30 observed for rhodopsin. Right, comparison of the same region in theactive state of mOR (green) and Rho (purple) shows a conserved interactionbetween R3.50 and Y5.58.
ARTICLE RESEARCH
G2015 Macmillan Publishers Limited. All rights reserved
0 0 M O N T H 2 0 1 5 | V O L 0 0 0 | N A T U R E | 3

inactive mOR, the active-state cavity is larger and completely contigu-ous with the morphinan-binding site. Substitution of morphinans witha cyclopropylmethyl at the tertiary amine generally results in a ligandwith antagonist activity7. As an example, BU74, which only differs fromBU72 in having a cyclopropylmethyl substituent on the tertiary amine(Fig. 3f), is an antagonist at the mOR34. While BU74 can be docked intothe active state mOR structure with the cyclopropylmethyl within thepolar cavity, there are potential clashes with Y3267.43 in TM7 andW2936.48 in TM6. Moreover, the cyclopropylmethyl substituent woulddisplace one or more water molecules that form part of a polar networkdescribed in more detail below. Consistent with these observations,molecular dynamics simulations reveal that the antagonist BU74 isunstable in the position occupied by the agonist BU72 and rapidlyshifts away from this initial pose (Extended Data Fig. 6).
Propagation of conformational changesThe structural difference in the orthosteric ligand-binding pocketbetween active and inactive mOR is relatively subtle (Fig. 3b). It isdifficult to identify specific interactions between the structurally rigidagonist and the receptor as being key for mOR activation. This standsin contrast to activation of the b2AR and M2R, where specific polarinteractions between the receptor and the smaller, more flexible ago-nists contribute to structural changes associated with activation.These polar interactions stabilize a 2 A inward movement of TM5in the b2AR and a 2 A inward movement of TM6 in the M2R.
While agonist-stabilized changes in the binding pocket differ forthe mOR, b2AR, and M2R, the overall structural changes observed atthe G-protein interface are very similar (Extended Data Fig. 3). Themechanism by which agonist-stabilized changes propagate to thecytoplasmic surface appears to be more similar between mOR andthe b2AR compared to the M2R (Fig. 4a). For both the b2AR and
the mOR, there is a rearrangement of the packing of a triad of con-served amino acids F6.44, P5.50 and I3.40 (which we term the conservedcore triad) that lay just below the binding pocket (Fig. 4a). Thisrearrangement is associated with a counter-clockwise rotation (whenviewed from the extracellular surface) and outward movement ofthe cytoplasmic end of TM6. Thus, the subtle agonist-stabilizedrearrangement in the conserved core triad may initiate the cascadeof structural changes involved in activation of the mOR and b2AR. Therole of the conserved core triad appears to differ in the M2R, wherethe smaller V1113.40 forms a weaker packing interaction with F3966.44
and P1985.50. As a result, the triad in both inactive and active states ofthe M2R appear similar to the active states of the b2AR (Fig. 4a).
Given the similarity in allosteric propagation between mOR andb2AR, we sought to identify how BU72 stabilizes the active conforma-tion of the conserved core triad. In the b2AR, the rearrangement of theconserved core triad can be attributed to an agonist-stabilized inwardmovement of TM512,13. For the mOR, however, there are no specificinteractions between the receptor and BU72 that stabilize the inwardmovement of TM5. Instead, we identify a set of interactions thattogether appear to stabilize the rearrangement of the conserved coretriad in the mOR.
Both b-FNA and BU72 share a common morphinan scaffold. Thiscommon scaffold, however, is positioned differently in the inactiveand active structures. In both states, residues I2966.51 and V3006.55 inTM6 and W3187.35 and I3227.39 in TM7 form a common hydrophobicsurface for binding the morphinan ligands (Fig. 4b, d, spheres). Whilethis surface is similar in both inactive and active states, differences inthe position of the rigid morphinan agonist and antagonist result indifferences in the positions of key ligand substituents relative to spe-cific residues in TM3 and TM6 that are coupled to the conserved coretriad of F2896.44, P2445.50 and I1553.40 (Fig. 4b, d).
D147D1473.323.32
N150N1503.353.35
TM3TM3
TM7TM7
W318W3187.357.35
I322I3227.397.39
V300V3006.556.55
M151M1513.363.36
TM3TM3
Na+
D1473.32
N1503.35
TM3
TM6TM6
W2936.48W2936.48
F2896.44F2896.44
H2976.52H2976.52
TM7
TM6TM6a b
W3187.35
I3227.39
V3006.55
I1553.40I1553.40
d
D1473.32D1473.32
BU72
β-FNA
M1513.36
F2896.44
I1553.40
P2445.50P2445.50
V3006.55V3006.55
W3187.35W3187.35
(e)(e)Active μORInactive μOR
P5.50P5.50
F6.44F6.44I3.40I3.40
P5.50P5.50
F6.44F6.44 I3.40I3.40
Active β2ARInactive β2AR
P5.50P5.50
F6.44F6.44 I3.40I3.40
P5.50P5.50
F6.44F6.44 I3.40I3.40
Active M2RInactive M2R
P5.50P5.50
F6.44F6.44
V3.40V3.40 V3.40V3.40
P5.50P5.50
F6.44F6.44
0 100 200 300 400 500 600 700
–180
–120
–60
0
60
120
180
Simulation time (ns)
c eActive μOR simulated without agonist
W293
6.4
8 χ
2 d
ihed
ral (d
eg
rees)
Active μOR simulated with co-crystallized BU72Inactive μOR simulated with co-crystallized β-FNA
Active μOR simulated without agonist
D1473.32:CαN1503.35:CγI1553.40:Cα Active μOR
Inactive μOR
Mo
vem
en
t to
ward
s in
active (Å
)
0 1,000 2,000 3,000 4,000
0
1
2
3
4
5
6
7
Simulation time (ns)
Figure 4 | Mechanisms of allosteric coupling in mOR. a, Comparison of thestructural rearrangements in the conserved core triad of mOR, b2AR, and M2R.b, The morphinan ligands BU72 and b-FNA bind to the mOR with a sharedhydrophobic surface shown in spheres. BU72 binding results in a 1.5 Adisplacement of TM3 towards TM2 and a rotameric change in the sodiumcoordinating residue N1503.35. Red arrows highlight displacement of the ligandor TM3 upon activation. c, In a molecular dynamics simulation initiatedfrom the active mOR structure but with the agonist BU72 removed from thebinding site, residues in TM3, including the conserved core triad residueI1553.40, adopt an inactive-like conformation. The motions of I1553.40 and
ligand-contacting residue D1473.32 are tightly coupled throughout thesimulation. Atom positions during simulation are plotted relative to the activestructure, with positive values representing displacement towards the positionin the inactive structure (see Methods). Dashed horizontal lines representthe positions of the indicated atoms in the inactive structure. d, W2936.48 isslightly closer to the phenolic aromatic of the morphinan in the active state ofmOR. e, Molecular dynamics simulations show that removal of the agonistBU72 from the active structure results in a change in the preferred rotamer ofW2936.48. Molecular dynamics results were consistent across multiplesimulations; see Supplementary Information.
RESEARCH ARTICLE
G2015 Macmillan Publishers Limited. All rights reserved
4 | N A T U R E | V O L 0 0 0 | 0 0 M O N T H 2 0 1 5

In the active mOR structure, the morphinan scaffold of BU72adopts a pose that is sterically incompatible with the inactive positionof TM3. As a result, the residues of TM3 that interact with the tertiaryamine (D1473.32) and methyl substituent (M1513.36) of the agonistshift 1.5 A towards TM2 relative to its position in the inactive struc-ture (Fig. 4b). We used molecular dynamics simulations to assesswhether the agonist favours this displacement in TM3. In simulationsof activated mOR with the agonist removed, the previously ligand-contacting residues on TM3 quickly relax towards their inactive posi-tions (Fig. 4c), often without global structural change of the receptor.These motions are tightly coupled to motion of the conserved coretriad residue I1553.40 towards its inactive position, causing it to repo-sition relative to F2896.44 (Fig. 4b, c). Notably, the position of D1473.32
is also coupled to the rotameric state of N1503.35, which coordinates anallosteric sodium ion in the inactive state and forms a hydrogen bondwith a backbone carbonyl of I1463.31 in the active state (Fig. 4b, c).
Another link between the ligand-binding pocket and the conservedcore triad may be mediated via TM6 through W2936.48 (Fig. 4d, e). Inthe active mOR structure, the aromatic group of BU72 is positionedonly slightly closer (0.6 A) to W2936.48 as compared to the samearomatic group of b-FNA in the inactive state. However, moleculardynamics simulations suggest that agonists stabilize W2936.48 in therotamer observed in the active-state crystal structure. In simulationsof the active state, removal of BU72 results in a change in the favouredrotamer of W2936.48 (Fig. 4e). Conversely, we assessed whether anagonist bound to the inactive state would stabilize the side chain ofW2936.48 in the rotamer associated with the active state. Here, wesimulated the inactive-state structure with the antagonist b-FNAreplaced with the agonist b-fuoxymorphamine (b-FOA), which dif-fers from b-FNA solely in a methyl substituent at the morphinantertiary amine35 (Extended Data Fig. 7a). In these simulations, thepose of b-FOA tends to shift towards that observed for BU72 andthe TM3 residues D1473.32 and N1503.35 shift towards their active-state positions; in concert, the side chain of W2936.48 shifts towards itsactive position (Extended Data Fig. 7b, c). W2936.48 is spatially juxta-posed to the conserved core triad residue F2896.44, and the conforma-
tion of W2936.48 may therefore serve as an important link between theligand-binding pocket and the triad (Fig. 4d).
Role of polar network in GPCR activationIn addition to rearrangement of the conserved core triad, comparisonof the inactive and active structures of mOR reveals an extensivenetwork of polar interactions between the orthosteric binding pocketand the G-protein-coupling interface that must rearrange uponactivation (Fig. 5a). The high-resolution electron density maps ofthe active mOR allow us to detect more ordered water moleculeswithin this polar network than have been observed in other active-state GPCR structures reported to date. To provide a better compar-ison of this polar network, we examined the high-resolution inactivestructure of the highly homologous dOR11. The inactive structure ofdOR at a resolution of 1.8 A identifies more water molecules withinthis polar network than the 2.8 A inactive structure of mOR and there-fore highlights the contribution of many hydrogen bonds in stabil-izing both the inactive and active states of opioid receptors (Fig. 5b).These hydrogen bonds represent many low-energy molecularswitches that have to be broken and reformed in a concerted mannerto achieve the active conformation. The polar network interactions forinactive-state mOR are likely to be identical to the inactive state of dORbecause the specific amino acids involved in this network and theirside chain conformations are identical between the two homologousreceptors.
Many of the residues in the polar network are conserved (ExtendedData Fig. 8a, b, e, f), suggesting that the polar network may also beconserved and play a similar role in activation of other family AGPCRs. While there are fewer water molecules observed in theactive-state structure of the b2AR, the ones that are resolved are inthe same positions as those observed in the mOR. Even though theresolution in the active b2AR and M2R structures is not sufficient toobserve as many ordered water molecules, the positions of the con-served side chains lining the polar core of the b2AR and M2R arenearly identical to those of the mOR (Extended Data Fig. 8f), suggest-ing that they are stabilized by a similar hydrogen-bonding network.
TM6TM3
TM7
TM2
NPxxY motif
Na+
H6.52I7.39
D3.32
W6.48
G7.42
N7.45
S3.39
S7.46
D2.50
N1.50
I2.43
N7.49
L3.43
V6.40
Y7.53Y5.58
H6.52
A5.46
W6.48
F6.44
V6.40
Y7.53
N7.49
N7.45
N1.50
D2.50
S3.39
S7.46
N3.35
D3.32
G7.42
Active μOR
Inactive δOR
a
b
Inactive δOR Active μOR
A5.46
Y5.58
L3.43
Y7.53 N7.49
P7.50
V6.40
TM5
TM6
TM7
TM3
Active μOR
Y5.58
L3.43
Y7.53
N7.49
P7.50
M6.40
Active Rho
c
d
N3.35
S3.39
W6.48
G7.42
N7.45
S7.46 D2.50
TM6TM3
TM7TM2
W6.48
G7.42
N3.35
I3.31
S3.39
D2.50
S7.46
N7.45
TM5
TM6
TM7
TM3
Figure 5 | Rearrangement of a conserved polar network. a, b, Comparison ofthe water-mediated polar network in the active mOR (a) and high-resolutioninactive dOR (b) (PDB ID: 4N6H). To simplify comparisons between differentreceptors, only Ballesteros–Weinstein numbers are used to label amino acidside chains. The network extends from the orthosteric ligand-binding site to the
G-protein-coupling domain of the receptor. c, The active structure of mORreveals the basis for sodium ion allosteric regulation of GPCR function.Rearrangement of S3.39 and N3.35 eliminates the sodium ion coordination site inthe active state. d, Conserved hydrogen-bonding network in the NPxxY regionbetween active mOR and rhodopsin.
ARTICLE RESEARCH
G2015 Macmillan Publishers Limited. All rights reserved
0 0 M O N T H 2 0 1 5 | V O L 0 0 0 | N A T U R E | 5

In the high-resolution inactive-state structure of the dOR, there isan ordered sodium ion that is adjacent to the conserved core triad andis coordinated by the side chains of D2.50, N3.35 and S3.39, as well as byW6.48 through a water molecule (Fig. 5b, c). A sodium ion at a similarcoordination site has been observed in inactive structures of theprotease-activated receptor subtype 1 (PAR1)36, the adenosine A2A
receptor (A2AR)37 and the b1 adrenergic receptor (b1AR)38. For manyGPCRs, including themOR, agonist-binding affinity and/or G-proteinactivation is allosterically inhibited by sodium39. Consistent withallosteric stabilization of the inactive state by sodium, we do notobserve a sodium ion in the active-state structure of the mOR, andthe residues that formed the sodium-binding site rearrange to pre-clude sodium binding in the active state (Fig. 5c).
The polar network ends at the cytoplasmic surface with a web ofinteractions involving the conserved NPxxY sequence (Fig. 5d). Uponreceptor activation, the NPxxY motif in TM7 moves inward towardsTM5 where N3327.49 and Y3367.53 participate in a hydrogen-bondnetwork involving Y2525.58 in TM5, the backbone carbonyls ofL1583.43 in TM3 and V2856.40 in TM6, and three ordered water mole-cules (Fig. 5d). A similar hydrogen-bond network was previouslyobserved in the structure of metarhodopsin II19. Similar water-mediated hydrogen bonds are probably present in active b2AR andM2R as the side chains of key residues in this region occupy similarpositions (Extended Data Fig. 3b).
It is interesting to speculate on the role of this polar network in theenergetics of mOR activation. NMR studies reveal that allosteric coup-ling of the agonist-binding pocket and the G-protein-binding inter-face in the mOR is relatively weak, and structural changes in TM6 areobserved only in the presence of a G protein mimetic nanobody(Sounier et al., ref. 31). A similar observation has been made forthe b2AR20–22,40. Using both double electron–electron resonance(DEER) spectroscopy and NMR spectroscopy, we observed that inthe presence of a saturating concentration of the catecholamine ago-nist isoproterenol only 20% of b2AR has TM6 in an active-like con-formation24. This stands in contrast to the more efficient couplingbetween the orthosteric binding pocket and TM6 in rhodopsin, asreflected in the ability to crystallize rhodopsin in an active state with-out a G protein or a G protein mimetic nanobody41, and biophysicalstudies that reveal a stronger allosteric coupling between the orthos-teric binding pocket and TM6 in rhodopsin42. Comparison of theinactive-state structures of rhodopsin and the dOR reveals that thedOR has a more extensive polar network on the cytoplasmic side ofthe ligand-binding pocket (Extended Data Fig. 9). This is particularlynotable when considering the network of hydrogen bonds that main-tain TM6 in the inactive conformation (Extended Data Fig. 9). Asnoted above, a similar polar network probably stabilizes the inactivestates of the mOR and b2AR (Extended Data Fig. 8). The less extensivepolar network stabilizing the inactive state of rhodopsin is compen-sated for by the covalent inverse agonist, 11-cis-retinal. This balanceof non-covalent polar interactions and a covalent ligand imparts rho-dopsin with virtually no basal activity, but the ability to rapidly andefficiently respond to photoisomerization of retinal. In contrast, thepolar network in the mOR and b2AR help to maintain unligandedreceptors in an inactive state, but the need to disrupt this networkmakes activation of these receptors energetically less efficient.
ConclusionThe structure of activated mOR presented here provides a model forhow efficacy is encoded within small chemical differences in otherwisestructurally similar morphinan ligands. Comparison of active mORwith other active-state structures offers insights into shared andsubtype-specific mechanisms for the activation of family A GPCRs.Additionally, the extensive reorganization of the polar networkrequired to achieve the fully active state may explain the inefficientallosteric coupling of the orthosteric pocket and the G-protein-coupling interface observed in NMR studies for both the mOR and
the b2AR. It is possible that subtle ligand-specific differences in thepolar network connections may contribute to preferential activation ofdifferent signalling proteins.
Online Content Methods, along with any additional Extended Data display itemsandSourceData, are available in the online version of the paper; references uniqueto these sections appear only in the online paper.
Received 15 March; accepted 30 June 2015.
Published online 5 August 2015.
1. Matthes, H. W. et al. Loss of morphine-induced analgesia, reward effect andwithdrawal symptoms in mice lacking the m-opioid-receptor gene. Nature 383,819–823 (1996).
2. Brownstein, M. J. A brief history of opiates, opioid peptides, and opioid receptors.Proc. Natl Acad. Sci. USA 90, 5391–5393 (1993).
3. Schumacher, M. A., Basbaum, A. I. & Naidu, R. K., (McGraw-Hill Medical, 2015).4. Raehal, K. M., Walker, J. K. & Bohn, L. M. Morphine side effects in b-arrestin 2
knockout mice. J. Pharmacol. Exp. Ther. 314, 1195–1201 (2005).5. Bohn, L. M., Gainetdinov, R. R., Lin, F.-T., Lefkowitz, R. J. & Caron, M. G. m-Opioid
receptor desensitization by b-arrestin-2 determines morphine tolerance but notdependence. Nature 408, 720–723 (2000).
6. Bohn, L. M. et al. Enhanced morphine analgesia in mice lacking b-arrestin 2.Science 286, 2495–2498 (1999).
7. Pasternak,G.W.&Pan,Y.-X.Muopioids and their receptors: evolution ofa concept.Pharmacol. Rev. 65, 1257–1317 (2013).
8. Chavkin, C. & Goldstein, A. Specific receptor for the opioid peptide dynorphin:structure–activity relationships. Proc. Natl Acad. Sci. USA 78, 6543–6547 (1981).
9. Manglik, A. et al. Crystal structure of the m-opioid receptor bound to a morphinanantagonist. Nature 485, 321–326 (2012).
10. Granier, S. et al. Structure of the d-opioid receptor bound to naltrindole. Nature485, 400–404 (2012).
11. Fenalti, G. et al. Molecular control of d-opioid receptor signalling. Nature 506,191–196 (2014).
12. Rasmussen, S. G. F. et al. Structure of a nanobody-stabilized active state of the b2adrenoceptor. Nature 469, 175–180 (2011).
13. Ring, A.M.et al. Adrenaline-activatedstructureofb2-adrenoceptor stabilized byanengineered nanobody. Nature 502, 575–579 (2013).
14. Cherezov, V. et al. High-resolution crystal structure of an engineered human b2-adrenergic G protein-coupled receptor. Science 318, 1258–1265 (2007).
15. Rosenbaum, D. M. et al. GPCR engineering yields high-resolution structuralinsights into b2-adrenergic receptor function. Science 318, 1266–1273 (2007).
16. Haga,K.et al. Structureof thehuman M2muscarinicacetylcholine receptorboundto an antagonist. Nature 482, 547–551 (2012).
17. Kruse, A. C.et al. Activation and allosteric modulationof a muscarinic acetylcholinereceptor. Nature 504, 101–106 (2013).
18. Palczewski, K. et al. Crystal structure of rhodopsin: a G protein-coupled receptor.Science 289, 739–745 (2000).
19. Choe, H.-W. et al. Crystal structure of metarhodopsin II. Nature 471, 651–655(2011).
20. Rosenbaum, D. M. et al. Structure and function of an irreversible agonist-b2adrenoceptor complex. Nature 469, 236–240 (2011).
21. Nygaard, R. et al. The dynamic process of b2-adrenergic receptor activation. Cell152, 532–542 (2013).
22. Manglik, A. & Kobilka, B. The role of protein dynamics in GPCR function: insightsfrom the b2AR and rhodopsin. Curr. Opin. Cell Biol. 27, 136–143 (2014).
23. Manglik, A. et al. Structural insights into the dynamic process of b2-adrenergicreceptor signaling. Cell 161, 1101–1111 (2015).
24. De Lean, A., Stadel, J. M. & Lefkowitz, R. J. A ternary complex model explains theagonist-specific binding properties of the adenylate cyclase-coupled b-adrenergicreceptor. J. Biol. Chem. 255, 7108–7117 (1980).
25. Rasmussen, S. G. F.et al. Crystal structure of theb2 adrenergic receptor-Gsproteincomplex. Nature 477, 549–555 (2011).
26. Schiller, P. W. et al. Synthesis and in vitro opioid activity profiles of DALDAanalogues. Eur. J. Med. Chem. 35, 895–901 (2000).
27. Neilan, C. L. et al. Characterization of the complex morphinan derivative BU72 as ahigh efficacy, long-lasting mu-opioid receptor agonist. Eur. J. Pharmacol. 499,107–116 (2004).
28. Caffrey, M. Crystallizing membrane proteins for structure determination: use oflipidic mesophases. Annu. Rev. Biophys. 38, 29–51 (2009).
29. Ballesteros, J. A. & Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations inG protein-coupled receptors. Methods Neurosci. 25, 366–428 (1995).
30. Wu, H. et al. Structure of the human k-opioid receptor in complex with JDTic.Nature 485, 327–332 (2012).
31. Sounier, R. et al. Propagation of conformational changes during m-opioid receptoractivation. Nature http://dx.doi.org/10.1038/nature14680 (2015).
32. Chaturvedi, K., Shahrestanifar, M. & Howells, R. D. m Opioid receptor: role for theamino terminus as a determinant of ligand binding affinity. Brain Res. Mol. BrainRes. 76, 64–72 (2000).
33. Gales, C. et al. Probing the activation-promoted structural rearrangements inpreassembled receptor-G proteincomplexes.NatureStruct. Mol. Biol. 13, 778–786(2006).
RESEARCH ARTICLE
G2015 Macmillan Publishers Limited. All rights reserved
6 | N A T U R E | V O L 0 0 0 | 0 0 M O N T H 2 0 1 5

34. Husbands, S. M. et al. BU74, a complex oripavine derivative with potent kappaopioid receptor agonism and delayed opioid antagonism. Eur. J. Pharmacol. 509,117–125 (2005).
35. Takemori, A. E., Larson, D. L. & Portoghese, P. S. The irreversible narcoticantagonistic and reversible agonistic properties of the fumaramate methyl esterderivative of naltrexone. Eur. J. Pharmacol. 70, 445–451 (1981).
36. Zhang, C. et al. High-resolution crystal structure of human protease-activatedreceptor 1. Nature 492, 387–392 (2012).
37. Liu, W. et al. Structural basis for allosteric regulation of GPCRs by sodium ions.Science 337, 232–236 (2012).
38. Miller-Gallacher, J. L. et al. The 2.1 A resolution structure of cyanopindolol-boundb1-adrenoceptor identifies an intramembrane Na1 ion that stabilises the ligand-free receptor. PLoS ONE 9, e92727 (2014).
39. Pert, C. B., Pasternak, G. & Snyder, S. H. Opiate agonists and antagonistsdiscriminated by receptor binding in brain. Science 182, 1359–1361 (1973).
40. Manglik, A. et al. Structural insights into the dynamic process of b2-adrenergicreceptor signaling. Cell 161, 1101–1111 (2015).
41. Park, J.H., Scheerer, P.,Hofmann,K.P., Choe,H.W.&Ernst,O.P.Crystal structureofthe ligand-free G-protein-coupled receptor opsin. Nature 454, 183–187 (2008).
42. Knierim, B., Hofmann, K. P., Gartner, W., Hubbell,W. L. & Ernst, O. P. Rhodopsin and9-demethyl-retinal analog: effect of a partial agonist on displacement oftransmembrane helix 6 in class A G protein-coupled receptors. J. Biol. Chem. 283,4967–4974 (2008).
Supplementary Information is available in the online version of the paper.
Acknowledgements We acknowledge support from the Stanford Medical ScientistTraining Program and the American Heart Association (A.M.), National Institutes ofHealth grants R37DA036246 (B.K.K. and S.G.) and R01GM083118 (B.K.K.), a TermanFaculty Fellowship (R.O.D.), Eli Lilly and Company through the Lilly Research Program(R.O.D.), and the Mathers Foundation (B.K.K. and W.I.W). We also acknowledge the
National Institute of Drug Abuse Drug Supply Program for providing [Dmt1]DALDA. Wethank D. Maurel and S. Agnel from the ARPEGE facility (Institut de GenomiqueFonctionnelle) for assistance with cell-based Gi coupling assays, H. El Hassan for experttechnical assistance, and S. Hertig, N. Latorraca and K. Cavalotti for assistance withmolecular dynamics simulations and analysis.
Author Contributions W.H. developed functional purification protocols, expressed andpurified mOR, characterized the effect of nanobodies and Gi on mOR ligand affinity,identified Nb39 for crystallography of the mOR–Nb complex, performed crystallizationtrials, data collection, structure determination and refinement. A.M. established theproject with biochemistry of active mOR, prepared samples for llama immunization,validated nanobody activity, performed crystallization trials, and identified initialcrystals of the mOR–BU72–Nb complex suitable for diffraction studies. A.J.V. analysedthe polar network. A.J.V., E.F. and A.S. performed and analysed molecular dynamicssimulations with supervision from R.O.D. T.L. identified mOR-binding nanobodieswith supervision from J.S. S.G. established the biochemistry for purification ofagonist-bound mOR and prepared samples for mOR immunization. H.E.K. helpedwith data collection and processing. T.S.T helped with the characterization of theamino-terminal region. R.K. and P.G. analysed BU72 and assessed alternative ligandstructures. S.M.H. synthesized BU72. K.E.L. and J.R.T. helped with selection of opioidligands including BU72 and performed dissociation kinetics experiments. W.I.W.supervised structure refinement. A.M. and B.K.K. provided overall project supervisionand wrote the manuscript with W.H. and R.O.D.
Author Information Coordinates and structure factors for the mOR–BU72–Nb39complex have been deposited in the Protein Data Bank under accession code 5C1M.Reprints and permissions information is available at www.nature.com/reprints. Theauthors declare competing financial interests: details are available in the online versionof the paper. Readers are welcome to comment on the online version of the paper.Correspondence and requests for materials should be addressed to A.M.([email protected]) or B.K.K. ([email protected]).
ARTICLE RESEARCH
G2015 Macmillan Publishers Limited. All rights reserved
0 0 M O N T H 2 0 1 5 | V O L 0 0 0 | N A T U R E | 7

METHODSNo statistical methods were used to predetermine sample size. The experimentswere not randomized. The investigators were not blinded to allocation duringexperiments and outcome assessment.Expression and purification of mOR. Full length Mus musculus mOR bearing anamino-terminal Flag epitope tag and a carboxy-terminal 6 3 His tag wasexpressed in Sf9 insect cells using the BestBac baculovirus system (ExpressionSystems). To facilitate removal of flexible amino- and carboxy-terminal regions, atobacco etch virus protease recognition sequence was inserted after residue 51and a rhinovirus 3C protease recognition sequence was inserted before residue359. Insect cells were infected with baculovirus encoding mOR at a density of4 3 106 cells ml21 for 48–60 h at 27 uC. Receptor was solubilized and purified in afinal buffer comprised of 25 mM HEPES pH 7.4, 100 mM NaCl, 0.01% MNG(Anatrace), and 0.001% cholesterol hemisuccinate (CHS), as previouslydescribed9.Llama immunization and selection of mOR-binding nanobodies. Purified mORbound to the antagonist naloxone and with the amino and carboxy termini cleavedwas incubated with an excess of the agonist [Dmt1]DALDA (NIDA Drug SupplyProgram), and further purified by size-exclusion chromatography in a buffer com-prised of 20 mM HEPES pH 7.5, 100 mM NaCl, 0.1% dodecylmaltoside (DDM,Anatrace), 0.03% CHAPS, 0.01% CHS, and 1mM [Dmt1]DALDA. The resultingagonist-bound receptor was reconstituted into phospholipid vesicles composed ofDOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine, Avanti Polar Lipids) and LipidA in a 10:1 (w:w) ratio at a final receptor concentration of 1.3 mg ml21. Theresulting reconstituted receptor was flash frozen in liquid nitrogen in 100mg ali-quots for llama immunization.
One llama was immunized over a period of 6 weeks with 0.3 mg of liposome-reconstituted mOR purified as described above and bound to the agonist[Dmt1]DALDA. A phage display library of nanobodies was prepared from peri-pheral blood lymphocytes as previously described43. mOR-binding nanobodieswere identified by selecting phages that bound liposome-reconstitutedmOR in thepresence or absence of agonist. Ten clones from three families were enrichedduring rounds of selection. One family, which includes Nb39, bind the intracel-lular surface and function as G-protein mimetics. Another family of nanobodies,including clone Nb35, was identified that competes directly with orthostericantagonists of the mOR and binds at the extracellular surface.Expression and purification of nanobodies. Nanobodies bearing a carboxy-terminal 6 3 His tag were expressed in the periplasm of Escherichia coli strainWK6. Cultures were grown to an OD600 of 1.0 at 37 uC in Terrific Broth mediumcontaining 0.1% glucose, 2 mM MgCl2, and 50 mg ml21 ampicillin and inducedwith 0.5 mM isopropyl-b-D-thiogalactoside (IPTG). Cells were harvested afterovernight growth at 25 uC and incubated in a buffer containing 200 mM TrispH 8.0, 0.5 mM EDTA, 500 mM sucrose and 0.5 mg ml21 lysozyme for 1 h at25 uC. Bacteria were osmotically lysed by rapid dilution in a 43 volume of water.The periplasmic fraction was isolated by centrifugation of cell debris, and wassupplemented with NaCl to a final concentration of 150 mM as well as imidazoleto a final concentration of 25 mM. Nanobodies were purified from the periplas-mic fraction by nickel affinity chromatography, and were subsequently purifiedby size-exclusion chromatography in a buffer comprised of 25 mM HEPES pH 7.5and 100 mM NaCl. Peak fractions were pooled and concentrated to approxi-mately 5 mM.Radioligand characterization of BU72 and Nb39. Radioligand competitionassays were performed using purified mOR reconstituted into high-density lipo-protein (HDL) particles comprised of the lipids POPC and POPG (Avanti PolarLipids) in a 3:2 molar ratio as previously described44,45. Heterotrimeric Gi wasprepared by co-expressing human Gia1, human Gb1 and Gc2 subunits in Hi5insect cells using baculoviruses encoding the individual subunits. The G proteinwas purified as previously described25.
For competition binding experiments, a mixture of 0.02 nM mOR and 2.3 nM3H-diprenorphine (3H-DPN, Perkin Elmer) was incubated with varying concen-trations of agonist in a binding buffer comprised of 25 mM HEPES pH 7.4,100 mM NaCl, and 0.1% BSA. Experiments were also performed with either5 mM Nb39 or 500 nM Gi. Binding reactions were incubated for 4 h at 25 uC.Free radioligand was separated from bound radioligand by rapid filtration ontoa Whatman GF/B filter pretreated with 0.1% polyethylenimine with the aid of a48-well harvester (Brandel). Radioligand activity was measured by liquid scintil-lation counting. Competition binding data were fit to a one-site model for mORalone and mOR with Nb39 and a two-site model for mOR incubated with Gi usingGraphPad Prism 6.0.
Dissociation studies for BU72 were performed using the method of Motulskyand Mahan46. 3H-DPN was diluted in an assay buffer comprised of 20 mM Tris,pH 7.4, 150 mM NaCl, and 0.05% BSA containing mOR in HDL particles with
either vehicle or different concentrations of BU72 alone or in the presence ofNb39. Binding reactions were incubated at 25 uC in the dark and nonspecificbinding was determined in the presence of 10mM naloxone. Aliquots of thisbinding reaction were removed at specified time points over the course of 2 or8 h and filtered through Whatman GF/C filters with the aid of a Brandel harvester.As above, radioligand activity was measured by liquid scintillation counting.Dissociation rates for BU72 were determined by fitting data in the ‘kinetics ofcompetitive binding’ program in GraphPad Prism 6.02. For K1 and K2, rates of3H-DPN association and dissociation were determined through independentstudies following the same method as above.Purification and crystallization of the mOR–BU72–Nb39 complex. Initial crys-tals of a mOR–BU72–Nb complex diffracted to low resolution. In an effort toimprove the specific activity of purified mOR, we used an extracellular bindingnanobody, Nb35 during the purification of the receptor. Nb35 binds in theorthosteric pocket of mOR with a Ki of 12 nM. Purified ligand-free mOR, withthe amino and carboxy termini cleaved was mixed with a 2.53 excess of Nb35,and the complex was isolated by Ni-NTA chromatography. Functional mOR incomplex with Nb35 was eluted in a buffer containing 25 mM HEPES pH 7.5,100 mM NaCl, 0.01% MNG, 0.001% CHS, and 250 mM imidazole. The agonistBU72 and Nb39 were then added in excess. In the presence of Nb39, BU72 hasexceptionally high affinity for the mOR and displaces Nb35, resulting in thedesired mOR–BU72–Nb39 complex. The mOR–BU72–Nb39 complex was iso-lated by size-exclusion chromatography and concentrated to approximately50 mg ml21 for crystallization trials.
For crystallization, the purified mOR–BU72–Nb39 complex was reconstitutedinto a 10:1 (w:w) mixture of monoolein and cholesterol (Sigma). The proteinsolution and lipid were mixed in a 1:1.5 ratio (w:w), and the lipidic cubic phasewas attained using the two-syringe method47. Thirty nanolitres of the resultingmesophase was dispensed onto 96-well glass plates and overlaid with 500 nl ofprecipitant solution using a Gryphon LCP robot (Art Robbins Instruments).Crystals grew in a precipitant comprised of 15–25% PEG300, 100 mM HEPESpH 7.0–7.5, 1% 1,2,3-heptanetriol, 0.5–1.0% polypropylene glycol P 400(Hampton Research) and 100–300 mM (NH4)2HPO4. Crystals were observedafter 2 days and reached full size in 1 week. Crystals were harvested with meshgrid loops (MiTeGen) and flash frozen in liquid nitrogen. Loops werescreened at the Advanced Photon Source GM/CA beamlines 23ID-B and23ID-D. Diffraction data were collected at a wavelength of 1.033 A using a10 mm beam with fivefold attenuation and exposed for 0.5–1 s. An oscillationwidth of 0.1–0.5u was used and diffraction images from four crystals were mergedto create the final data set.Structure determination and refinement. Diffraction images were indexed,scaled, and merged by XDS48 with statistics summarized in Extended DataTable 1. The structure was determined by molecular replacement in Phaser49
with initial search models including inactive mOR (PBD ID: 4DKL) and Nb80(PDB ID: 3P0G). The structural model was iteratively rebuilt in Coot50 andrefined using Phenix51. Final refinement statistics are summarized in ExtendedData Table 1. Molprobity52 was used for structure analysis and indicated that96.1% of residues were within favoured Ramachandran regions with 3.9% inallowed regions. No residues were identified as Ramachandran outliers. Figureswere prepared using MacPyMOL (Schrodinger, Inc.).System setup for molecular dynamics simulations. Simulations of themOR werebased on both the antagonist-bound inactive-state crystal structure (PDB ID:4DKL) and the agonist-bound active-state crystal structure described in thismanuscript. Coordinates were prepared by first removing all non-ligand andnon-receptor molecules except for the cholesterol neighbouring TM7 and forcrystallographic water molecules near the receptor. For inactive mOR simulations,the T4 lysozyme was removed and acetyl and methylamide capping groups wereplaced on R263ICL3 and E270ICL3. For active mOR simulations, the nanobody wasremoved. In both cases, Prime (Schrodinger, Inc.) was used to model missingside-chains, and capping groups were then added to the N and C termini of thereceptor. Histidine residues were simulated as the neutral Ne tautomer. Othertitratable residues were simulated in their dominant protonation state at pH 7except for D1142.50, which was charged in inactive simulations and neutral inactive simulations. A sodium ion was placed adjacent to D1142.50 in inactivesimulations.
The mOR was simulated in seven distinct conditions. These include: (1) theunliganded, inactive mOR, prepared by deleting the covalently bound, co-crystal-lized ligand, b-FNA, and adding a proton in its place to K2335.39; (2) the inactivemOR with the co-crystallized ligand b-FNA; (3) the inactive mOR with agonistb-FOA (which does not bind covalently); (4) the unliganded, active mOR, pre-pared by deleting the co-crystallized ligand, BU72; (5) the active mOR with theco-crystallized ligand BU72; (6) the active mOR with the co-crystallized ligandBU72, with the N-terminal peptide deleted; and (7) the active mOR with the
RESEARCH ARTICLE
G2015 Macmillan Publishers Limited. All rights reserved

antagonist BU74, with the N-terminal peptide deleted. Simulations of the activemOR without an N-terminal peptide were prepared by deleting residues 52through 64 of the receptor. Simulations with b-FOA were prepared by dockingb-FOA to the crystallographic pose of b-FNA. Simulations with BU74 wereprepared by docking BU74 to the crystallographic pose of BU72 and rotatingthe torsion angle of the methylcyclopropyl group to agree with that of b-FNA’smethylcyclopropyl group.
We performed three to six simulations per condition (SupplementaryInformation). Simulations in a given condition were initiated from identicalstructures, but with initial atom velocities assigned independently and randomly.
It should be noted that in all liganded simulations, including those withb-FNA,b-FOA, BU72, and BU74, the ligand’s tertiary amine nitrogen was protonatedand therefore the ligand was simulated as a cation. This is necessary for the ligandto form the conserved salt bridge with neighbouring D1473.32.
Each of the resulting prepared mOR structures was then aligned to the orienta-tions of proteins in membranes (OPM)53 entry for the inactive mOR usingMacPyMOL (Schrodinger, Inc.). The mOR was modified with disulphide bridgesand inserted into a hydrated, equilibrated palmitoyloleoylphosphatidylcholine(POPC) bilayer using the CHARMM-GUI interface54–57. Sodium and chlorideions were added to neutralize the system, reaching a final concentration ofapproximately 150 mM. All simulations contained one mOR embedded in a lipidbilayer with 160 POPC molecules.Simulation protocol and analysis. Each simulation was performed on two GPUsusing the CUDA version of PMEMD (Particle Mesh Ewald Molecular Dynamics)in Amber1458–60 with 2.5-fs time steps. Simulations were heated from 0 K to 100 Kin the NVT ensemble and then from 100 K to 310 K in the NPT ensemble, bothwith 10.0 kcal mol21 A22 harmonic restraints applied to the protein and to thelipids. Initial velocities were assigned randomly at the first heating step withLangevin dynamics. Subsequently, simulations were equilibrated in the NPTensemble at 310 K (controlled with a Langevin thermostat) with pressure of1 bar (controlled with anisotropic Berendsen weak-coupling barostat), with har-monic restraints on all protein atoms tapered off 1.0 kcal mol21 A22 in a stepwisefashion every 2.0 ns starting at 5.0 kcal mol21 A22 to 0.0 kcal mol21 A22, for atotal of 12.0 ns of equilibration. Bond lengths to hydrogen atoms were con-strained using SHAKE. Production simulations were performed in the NPTensemble at 310 K and 1 bar, using a Langevin thermostat for temperature coup-ling and a Monte Carlo barostat for pressure coupling, and were initiated from thefinal snapshot of the corresponding equilibration simulation. Non-bonded inter-actions were cut off at 9.0 A, and long-range electrostatic interactions were com-puted using the particle mesh Ewald (PME) method, with an Ewald coefficient bof approximately 0.31 A21 and B-spline interpolation of order 4. The FFT gridsize was 84 3 84 3 96 for all simulations. We performed a total of 27 simulations,which are summarized in the Supplementary Information.
We used the CHARMM36 parameter set for protein molecules, lipid mole-cules, and salt ions, and the CHARMM TIP3P model for water; protein para-meters incorporated CMAP terms61–64. These parameters were assigned using theParmEd implementation of Chamber, provided with AmberTools14. Ligandparameters were based on the results from the CHARMM ParamChem webserver, version 0.9.7.165, and incorporated specific modifications to partialcharges. When ParamChem reported large penalties (errors) for estimated partialcharges, better estimates were determined by submitting appropriately chosenfragments to ParamChem (see below).
Parameterization of b-FNA, the co-crystallized ligand in the inactive mORstructure, required additional steps due to the added complexity of its covalentbond with K2335.39 on the receptor. To assign parameters to the rest of b-FNA,the molecule was fragmented as shown in Supplementary Information section‘‘Parameterization’’ and then re-assembled. In addition, a molecule consisting ofK2335.39 bonded to the portion of b-FNA on the extracellular side of its morphi-nan scaffold was submitted to ParamChem, and the results were used to modifycharges near the bond between the b-FNA carbon and Lys side-chain nitrogen.A modified version of the ParmEd program was used to form a bond betweenb-FNA and K2335.39, to parameterize that and neighbouring bonds, and to mod-ify neighbouring partial charges according to the derived parameters. The relatedligand b-FOA (which does not bind covalently to the mOR) was divided into thesame three fragments as b-FNA for parameterization, with the following mod-ifications: the bond between the ligand and K2335.39 was cleaved; a C5C doublebond was formed between the two carbons adjacent to this bond; and the methyl-cyclopropyl group was replaced with a methyl group. The co-crystallized ligand
of the active mOR, BU72, was similarly parameterized by splitting it into frag-ments, individually uploading the fragments to the ParamChem web server, andassembling the results together. The fragments can be found in theSupplementary Information. The ligand, BU74, was parameterized identicallyto BU72, except that its tertiary amine methyl group was replaced with a methyl-cyclopropyl group.
Quantitative analysis of trajectories was conducted in both cpptraj, anAmber14 software package, and VMD66. Plots were smoothed with a movingaverage, with a symmetric triangular smoothing window. The window length was20 ns, except during the first 10 ns of each simulation, when the smoothingwindow extended from time zero to double the time at which the data wasrecorded. Plots were rendered in R with the ggplot2 package as well as inPython with the Matplotlib package.
To compute movement of specified atoms from the active position towards theinactive position, as plotted in Fig. 4c, the active and inactive crystal structuresand each simulation frame were aligned using the Ca atoms of residues 80–95,102–13, and 181–205 (TMs 1, 2 and 4). For each specified atom, the atom positionin simulation was projected onto the line connecting the atom positions in theactive and inactive crystal structures, and the distance of the projected point fromthe position in the active crystal structure position was determined.
43. Pardon, E. et al. A general protocol for the generation of nanobodies for structuralbiology. Nature Protocols 9, 674–693 (2014).
44. Whorton, M. R. et al. A monomeric G protein-coupled receptor isolated in a high-density lipoprotein particle efficiently activates its G protein. Proc. Natl Acad. Sci.USA 104, 7682–7687 (2007).
45. Kuszak, A. J. et al. Purification and functional reconstitution of monomeric mu-opioid receptors: allosteric modulation of agonist binding by Gi2. J. Biol. Chem.284, 26732–26741 (2009).
46. Motulsky, H. J. & Mahan, L. C. The kinetics of competitive radioligand bindingpredicted by the law of mass action. Mol. Pharmacol. 25, 1–9 (1984).
47. Caffrey, M. & Cherezov, V. Crystallizing membrane proteins using lipidicmesophases. Nature Protocols 4, 706–731 (2009).
48. Kabsch, W. XDS. Acta Crystallogr. D 66, 125–132 (2010).49. McCoy, A. J. et al. Phaser crystallographic software. J. Appl. Cryst. 40, 658–674
(2007).50. Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta
Crystallogr. D 60, 2126–2132 (2004).51. Afonine, P. V. et al. Towards automated crystallographic structure refinement with
phenix.refine. Acta Crystallogr. D 68, 352–367 (2012).52. Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular
crystallography. Acta Crystallogr. D 66, 12–21 (2010).53. Lomize, M. A., Lomize, A. L., Pogozheva, I. D. & Mosberg, H. I. OPM: orientations of
proteins in membranes database. Bioinformatics 22, 623–625 (2006).54. Brooks, B. R. et al. CHARMM: The biomolecular simulation program. J. Comput.
Chem. 30, 1545–1614 (2009).55. Jo, S., Kim, T. & Im, W. Automated builder and database of protein/membrane
complexes for molecular dynamics simulations. PLoS ONE 2, e880 (2007).56. Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user
interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008).57. Wu, E. L. et al. CHARMM-GUI Membrane Builder toward realistic biological
membrane simulations. J. Comput. Chem. 35, 1997–2004 (2014).58. Case, D. A. et al. AMBER 14. (University of California, San Francisco, 2014).59. Le Grand, S., Gotz, A. W. & Walker, R. C. SPFP: Speed without compromise—A
mixed precision model for GPU accelerated molecular dynamics simulations.Comput. Phys. Commun. 184, 374–380 (2013).
60. Salomon-Ferrer, R., Gotz, A. W., Poole, D., Le Grand, S. & Walker, R. C. Routinemicrosecond molecular dynamics simulations with Amber on GPUs. 2. Explicitsolvent particle mesh Ewald. J. Chem. Theory Comput. 9, 3878–3888 (2013).
61. Best, R. B. et al. Optimization of the additive CHARMM all-atom protein force fieldtargeting improved sampling of the backbone w, y and side-chain x1 and x2dihedral angles. J. Chem. Theory Comput. 8, 3257–3273 (2012).
62. Klauda, J. B. et al. Update of the CHARMM all-atom additive force field for lipids:validation on six lipid types. J. Phys. Chem. B 114, 7830–7843 (2010).
63. MacKerell, A. D. et al. All-atom empirical potential for molecular modeling anddynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 (1998).
64. Mackerell, A. D., Feig, M. & Brooks, C. L. Extending the treatment of backboneenergetics in protein force fields: limitations of gas-phase quantum mechanics inreproducing protein conformational distributions in molecular dynamicssimulations. J. Comput. Chem. 25, 1400–1415 (2004).
65. Vanommeslaeghe, K. et al. CHARMM general force field: a force field for drug-likemolecules compatible with the CHARMM all-atom additive biological force fields.J. Comput. Chem. 31, 671–690 (2010).
66. Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol.Graph. 14, 33–38 (1996).
ARTICLE RESEARCH
G2015 Macmillan Publishers Limited. All rights reserved

Extended Data Figure 1 | Characterization of Nb39 and lattice interactionsin mOR–Nb39 crystals. a, 3H-diprenorphine (3H-DPN) competitionbinding shows increased affinity for mOR-selective agonists DAMGO andendomorphin-2 in the presence of Nb39. b, The dissociation half-life (t1/2)of BU72 was determined by measuring the association rate of the antagonist3H-DPN in the presence of the indicated concentrations of BU72. The
dissociation t1/2 of BU72 is 43 min and increases to 140 min in presence ofNb39. Panels a and b are representative of at least three experiments performedin triplicate, and the data and error bars represent the mean 6 s.e.m. c, Crystallattice packing of the mOR–Nb39 complex shows that most of the contactsare mediated by Nb39. The mOR extracellular domain is not involved in anycontacts.
RESEARCH ARTICLE
G2015 Macmillan Publishers Limited. All rights reserved

Extended Data Figure 2 | mOR–Nb39 interface. a, Nb39 does not penetrateas deeply into the core of the mOR when compared with the b2AR–Nb80complex and the M2R–Nb9-8 complex. In the b2AR–Nb80 and M2R–Nb9-8complexes, nanobody CDR3 residues bind within the core of the receptortransmembrane bundle. In comparison, Nb39 binding involves more
framework residues. Notably, seven residues of CDR3 remained unresolved inthe final model of themOR–Nb39 complex. b, Nb39 interacts primarily throughhydrogen bonds with residues from ICL2, ICL3 and helix 8 of the mOR.c, Schematic representation of the interactions between mOR and Nb39highlighting the numerous Nb39 framework interactions.
ARTICLE RESEARCH
G2015 Macmillan Publishers Limited. All rights reserved

RESEARCH ARTICLE
G2015 Macmillan Publishers Limited. All rights reserved

Extended Data Figure 3 | Cytoplasmic domain rearrangements inconserved regions. a, The E/DRY sequence is a highly conserved motif withinfamily A GPCRs responsible for constraining receptors in an inactiveconformation. Comparisons of inactive- and active-state structures around theconserved E/DRY residues at the cytoplasmic surface of the mOR, the M2muscarinic receptor (M2R), the b2 adrenergic receptor (b2AR) and rhodopsin(Rho) are shown here. Hydrogen bonds are shown as dotted lines. b, The
NPxxY motif is a highly conserved sequence in TM7 among family A GPCRs.In the active state mOR, Y7.53 and N7.49 in TM7 interact with Y5.58 in TM5 andthe backbone carbonyl of L3.43 in TM3 through a water-mediated polarnetwork. A similar network is observed in the active state of rhodopsin. Whilewaters are not observed in the lower-resolution structures of the b2AR andM2R, the positions of the side chains of Y7.53, N7.49 and Y5.58 suggest a similarwater-mediated network with putative waters represented by red circles.
ARTICLE RESEARCH
G2015 Macmillan Publishers Limited. All rights reserved

RESEARCH ARTICLE
G2015 Macmillan Publishers Limited. All rights reserved

Extended Data Figure 4 | Conformation of the binding pocket and BU72.The 2Fo 2 Fc electron density contoured at 2.0s and within 1.8 A of residuescomprising the active mOR ligand-binding pocket is shown as grey mesh ina and b. The same views are shown in c and d with the omit Fo 2 Fc density forBU72 displayed as an orange mesh. Displayed Fo 2 Fc electron density iscontoured at 3.0s. e, Placement of an energetically minimized conformation ofBU72 within the Fo 2 Fc electron density shows a poor fit for the pendantphenyl ring. The conformation of BU72 was minimized using quantummechanical Hartree–Fock methods. f, An alternative possible ligand structurewith sp2 geometry at the carbon adjacent to the phenyl (highlighted in reddashed circle) was initially considered due to a better fit within the electron
density. This alternative ligand is predicted to be 2 Da smaller than BU72.g, In order to resolve potential ambiguity in the co-crystallized ligand, weperformed mass spectrometry on the same protein sample used to generatecrystals of the active mOR. The protein was precipitated in methanol and thesupernatant was subjected to MALDI–MS which revealed a strong peak atm/z 5 429.226, consistent with the expected mass of BU72. h, Shown is ourfinal crystallographic model for BU72 within the Fo 2 Fc electron density. Thismodel probably represents a high-energy conformation of BU72. Notably, theposition of the morphinan scaffold is invariant between these alternativemodels for the crystallized ligand.
ARTICLE RESEARCH
G2015 Macmillan Publishers Limited. All rights reserved

Extended Data Figure 5 | The N terminus of the mOR interacts with BU72.a, Surface cut-away view showing that the N terminus forms a lid over theligand-binding pocket. Shown in the lower panel is the ligand-binding pocket inthe absence of the N terminus. b, Blue mesh shows the 2Fo 2 Fc omit map
contoured at 1.0s for the N terminus. c, Shown in green mesh is the Fo 2 Fc
omit map contoured at 4.0s of an unidentified density between BU72 andHis54.
RESEARCH ARTICLE
G2015 Macmillan Publishers Limited. All rights reserved

Extended Data Figure 6 | Molecular dynamics simulation of active mORbound to antagonist BU74. a, Structures of agonist BU72, and antagonistsBU74 and b-funaltrexamine (b-FNA). The inactive-state structure of mOR wasco-crystallized with b-FNA. b, BU74 was docked into the active-state structureof the mOR based on the crystallographic pose of BU72, but in a moleculardynamics simulation it rapidly moves away from this initial pose. The middlepanel highlights the movements of BU74 after 560 ns of simulation and the
rightmost panel shows the comparison of the BU74 pose as compared to thecrystal structure of b-FNA bound to inactive mOR. c, Molecular dynamicstrajectory measuring the distance between the phenolic hydroxyl of Y3267.43
and the tertiary amine of BU74. Dotted lines show the distance betweenY3267.43 and the same amine of BU72 in the crystal structure of active mOR andb-FNA in the structure of inactive mOR.
ARTICLE RESEARCH
G2015 Macmillan Publishers Limited. All rights reserved
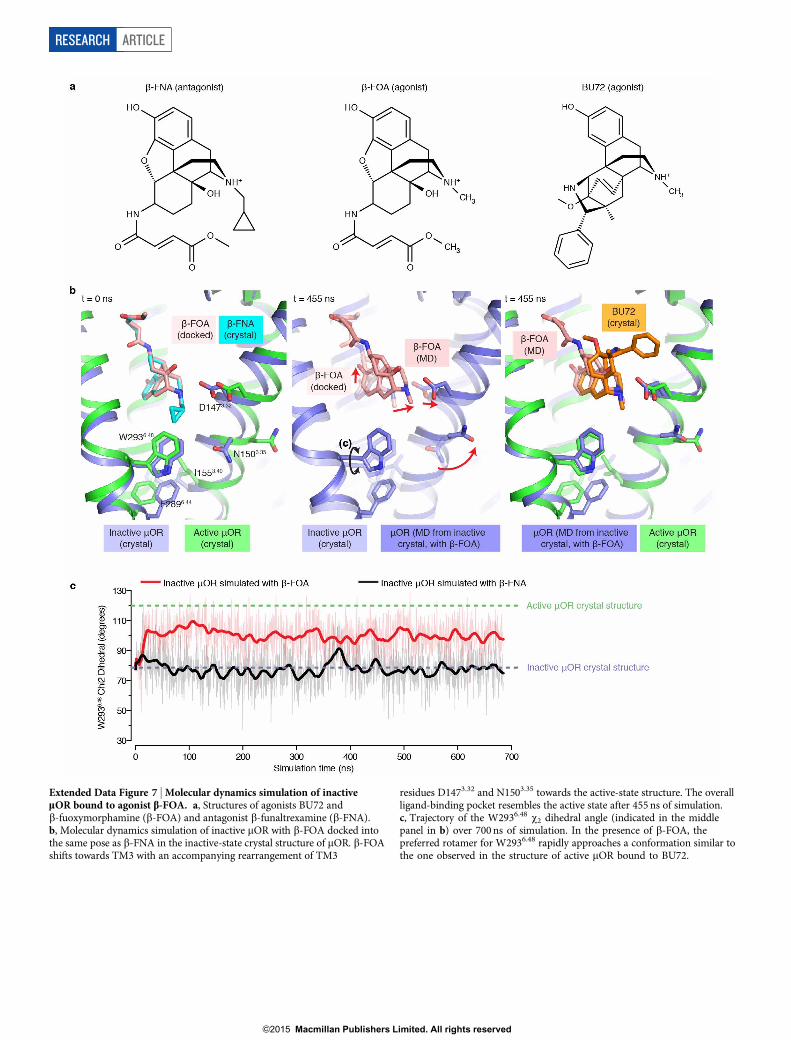
Extended Data Figure 7 | Molecular dynamics simulation of inactivemOR bound to agonist b-FOA. a, Structures of agonists BU72 andb-fuoxymorphamine (b-FOA) and antagonist b-funaltrexamine (b-FNA).b, Molecular dynamics simulation of inactive mOR with b-FOA docked intothe same pose as b-FNA in the inactive-state crystal structure of mOR. b-FOAshifts towards TM3 with an accompanying rearrangement of TM3
residues D1473.32 and N1503.35 towards the active-state structure. The overallligand-binding pocket resembles the active state after 455 ns of simulation.c, Trajectory of the W2936.48 x2 dihedral angle (indicated in the middlepanel in b) over 700 ns of simulation. In the presence of b-FOA, thepreferred rotamer for W2936.48 rapidly approaches a conformation similar tothe one observed in the structure of active mOR bound to BU72.
RESEARCH ARTICLE
G2015 Macmillan Publishers Limited. All rights reserved

Extended Data Figure 8 | Comparison of polar networks involved in GPCRactivation. a, Residues involved in the polar network in the inactive state ofthe dOR (PDB ID: 4N6H) and conservation of those residues in b2AR, M2R,and rhodopsin. b, Residues involved in the polar network in active statemOR and conservation in b2AR, M2R, and rhodopsin. c, Water-mediated polarnetwork in the inactive structure of the dOR involves residues from TM1, TM2,
TM3, TM5, TM6 and TM7. d, An identical view as in c of the polar networkin the active mOR. e, Residues involved in the polar network in inactivestructures of dOR, b2AR and M2R are conserved both in sequence andconformation. f, In active mOR, b2AR and M2R, the residues within the polarnetwork are again conserved in sequence and conformation.
ARTICLE RESEARCH
G2015 Macmillan Publishers Limited. All rights reserved

Extended Data Figure 9 | Differences in TM6 polar network in opioidreceptors and rhodopsin. a, The entire set of contacts within the polar networkthat include a residue within TM6 is displayed for the inactive dOR, activemOR,and inactive and active rhodopsin (Rho). b, Helix wheel representation of
TM6 showing polar contacts. Notably, the inactive dOR engages in many morepolar contacts with neighbouring residues as compared to inactive rhodopsin.Additionally, the active states of both mOR and rhodopsin have fewer polarcontacts than the inactive state.
RESEARCH ARTICLE
G2015 Macmillan Publishers Limited. All rights reserved

Extended Data Table 1 | Data collection and refinement statistics (molecular replacement) µOR-BU72-Nb39a Data collectionb Space group I212121 Cell dimensions a, b, c (Å) 44.4, 144.0, 209.9 , , (°) 90.0, 90.0, 90.0 Resolution (Å) 50.0-2.07 (2.12-2.07) Rmerge (%) 11.2 (153.6) <I/ I> 11.0 (1.7) CC1/2 (%) 99.2 (64.7) Completeness (%) 99.8 (100.0) Redundancy 8.6 (8.8) Refinement Resolution (Å) 42.5-2.1 (2.15-2.10) Number of reflections 39,948 Rwork/Rfree (%) 18.53/22.15 (25.83/28.71) Number of atoms Protein 3,278 Ligand (BU72) 32 Lipid,water and others 208 B-factors (Å2) Protein 56.89 Ligand (BU72) 39.50 Lipid,water and others 69.91 R.M.S. deviation from ideality Bond lengths (Å) 0.008 Bond angles (º) Ramachandran statisticsc (%) Favored Allowed Outliers
1.193 96.1 3.9 0
a Diffraction data from 4 crystals were merged into a complete data set b Highest resolution shell statistics are shown in parentheses c As calculated by Molprobity
ARTICLE RESEARCH
G2015 Macmillan Publishers Limited. All rights reserved