aromatizitaet benzol...
TRANSCRIPT

1
Aromatizität: Benzol und Naphthalin
Burkhard Kirste
Im folgenden Artikel soll ein Problem auf dem Gebiet der Aromatizität beleuchtet werden,
nämlich die Frage, inwieweit sich kleine Strukturänderungen auf die Stabilität auswirken.
Nach einem kurzen historischen Abriss werden – wegen ihrer relativen Anschaulichkeit und
Nachvollziehbarkeit – Hückel(HMO)-Rechnungen vorgestellt und schließlich
Dichtefunktional(DFT)-Rechnungen. Mit Hilfe der DFT werden reale und hypothetische
Strukturen untersucht. Es ist ein Ziel des Artikels, der nicht in die Tiefen der Quantenchemie
eindringt, zu eigenständigen Rechnungen zu ermuntern.
Kurzer historischer Abriss
Benzol wurde 1825 von Michael Faraday im Leuchtgaskondensat entdeckt. Der erste brauchbare Strukturvorschlag wurde erst 40 Jahre später, nämlich 1865 von August Kekulé von Stradonitz formuliert, und zwar als Sechsring mit alternierenden Einfach- und Doppelbindungen [1]. Dieser Strukturvorschlag war allerdings so nicht haltbar, weil bei ortho-disubstituierten Benzolen experimentell immer nur ein Isomer nachgewiesen werden konnte. Nach der Kekulé-Formel müsste es aber beispielsweise zwei verschiedene ortho-Xylole (1,2-Dimethylbenzole) geben, da es einen Unterschied macht, ob zwischen den Substituenten eine Einfach- oder eine Doppelbindung steht. Um diesem Dilemma zu entkommen, brachte 1872 Kekulé seine Oszillationshypothese ins Spiel, wobei Benzol zwischen den beiden möglichen Kekulé-Strukturen hin- und heroszilliert [2] (siehe auch [3, 4]). In moderner Diktion war wohl ein dynamisches Gleichgewicht gemeint. Damit ließ sich jedoch noch nicht die außergewöhnliche Stabilität des Benzols im Vergleich zu einem Polyen ("1,3,5-Cyclohexatrien") erklären.
Im 19. Jahrhundert hielt man ja das Atom für unteilbar, das Elektron war noch nicht entdeckt. Erst die Entwicklung der auf der Quantentheorie basierenden Theorien der chemischen Bindung versprach Erklärungsmöglichkeiten. Herausragende Beiträge lieferte Linus Pauling mit seiner Resonanztheorie. So formulierte er im Valence-Bond-Modell quantenchemische Darstellungen für die beiden Kekulé-Formeln des Benzols und zeigte, dass eine (quantentheoretische) "Resonanz" zwischen den Grenzformeln zu einer erheblichen Stabilisierung führt. Rechnerisch ergab sich eine noch etwas größere Stabilisierungsenergie, als er außerdem die drei Dewar-Formeln mit einbezog. Nach dieser Theorie liegt im Falle des Benzols kein dynamisches Gleichgewicht zwischen den beiden Kekulé-Strukturen vor (mit einem energiereicheren Übergangszustand), sondern der symmetrische Zwischenstand (D6h-Symmetrie) stellt die stabile Struktur des Benzols als Energieminimum der Potentialhyperfläche dar. An dieser Stelle sei auf ein Problem der Resonanztheorie hingewiesen, das häufig zu Missverständnissen geführt hat. Und zwar beziehen sich die fünf genannten Grenzformeln des Benzols auf die tatsächliche Geometrie des planaren regelmäßigen Sechsecks. Davon zu unterscheiden wäre das mittlerweile bekannte "Dewar-Benzol", ein nichtplanares bicyclisches Valenzisomer des Benzols, Bicyclo[2.2.0]hexa-2,5-dien.

2
2 Kekulé-Grenzformeln 3 Dewar-Grenzformeln
E
0 kJ/mol
150 kJ/mol
Resonanzhybrid
Abb. 1. Oben: Kekulé- und Dewar-Grenzformeln des Benzols. Unten: Stabilisierung des Benzolmoleküls durch Resonanz.
Es sei erwähnt, dass die Resonanztheorie von Linus Pauling heutzutage eher in einem qualitativen Sinn weiterhin Verwendung findet, man spricht von Mesomerie.
Hückel-Regel und HMO-Theorie
In den Jahren 1931/1932 entwickelte der theoretische Physiker Erich Hückel seine alternative quantenchemische Theorie der chemischen Bindung, eine Molekülorbitaltheorie (MO-Theorie) [5, 6]. Er beschränkte sich allerdings auf die Betrachtung von π-Elektronen in ungesättigten Kohlenwasserstoffen. Diese Theorie ist heute als HMO-Modell bekannt (Hückel Molekül-Orbital-Modell). Seiner Theorie entstammt auch die bekannte Hückel-Regel der Aromatizität, nach der 4n + 2 π-Elektronen zu einer Stabilisierung führen.
Hückel führte in seine Theorie die beiden Energie-Parameter α ("Coulomb-Integral") und β ("Resonanz-Integral") ein. Wie man zeigen kann, lassen sich die Orbitalenergien für einen monocyclischen Kohlenwasserstoff mit einem konjugierten Bindungssystem nach dem folgenden einfachen geometrischen Verfahren konstruieren. Man zeichnet einen Kreis mit dem Radius 2|β| und stellt das betreffende regelmäßige Polygon als einbeschriebenes Vieleck mit einer Spitze nach unten stehend hinein (s. Abb. 2, oben). Die Eckpunkte geben dann die energetische Lage der Orbitale an. Dieses Verfahren lässt sich sogar auf offenkettige Polyene mit n Kohlenstoffatomen erweitern, indem man ein Polygon mit 2n + 2 Ecken in den Kreis einbeschreibt und nur die linke (oder rechte) Hälfte ohne untere und obere Spitze verwendet (s. Abb. 2, Mitte).

3
E
r = 2|β|
α
α - 2βα - β
α + βα + 2β
π*
π
Benzol EthenEπ = 6α + 8β Eπ = 2α + 2β
ψ6
ψ1
ψ2, ψ3
ψ4, ψ5
Abb. 2. Oben: geometrische Konstruktion zur Bestimmung der HMO-Energien. Mitte: HMO-Energien von Benzol und Ethen. Unten: Schematische Darstellung der sechs π-Molekülorbitale des Benzolmoleküls.
Die energetische Stabilisierung des Benzols lässt sich nach der HMO-Theorie abschätzen, indem man die gesamte π-Elektronen-Energie des Benzols mit derjenigen von drei Ethen-Molekülen vergleicht, also die Differenz bildet. Hierzu geht man jeweils nach dem Aufbau-Prinzip vor, d.h., man besetzt die Molekülorbitale beginnend mit dem energetisch am tiefsten liegenden unter Beachtung von Pauli-Prinzip und Hundscher Regel. (Pauli-Prinzip: Jedes Orbital kann nur zwei Elektronen aufnehmen, und zwar mit entgegengesetztem Spin. Hundsche Regel: Entartete, d.h. energiegleiche Orbitale werden zunächst einfach besetzt, wobei die Elektronen parallele Spins haben.)
Eπ(Ethen) = 2 α + 2 β
Eπ(Benzol) = 6 α + 8 β
∆E = Eπ(Benzol) - 3 Eπ(Ethen)
∆E = 2β
Vergleicht man mit der experimentell ermittelten Stabilisierungsenergie von etwa 150 kJ/mol für Benzol, so lässt sich β zu etwa -75 kJ/mol abschätzen.
In Abb. 2 (unten) sind die sechs π -Molekülorbitale des Benzolmoleküls schematisch dargestellt. In allen Fällen ist die Molekülebene eine Knotenebene der Wellenfunktion. Abgesehen von dem energetisch am tiefsten liegenden Orbital π1 treten weitere Knotenebenen senkrecht zur Molekülebene auf, wobei die Anzahl der Knotenebenen ein Maß für die Orbitalenergie ist.

4
HMO-Ergebnisse für Naphthalin
Tab. 1. HMO-Energien von Naphthalin
Orbital Energie, β Orbital Energie, β 1 2.3028 6 (LUMO) -0.6180 2 1.6180 7 -1.0000 3 1.3028 8 -1.3028 4 1.0000 9 -1.6180 5 (HOMO) 0.6180 10 -2.3028
Berechnung der Stabilisierungsenergie relativ zu fünf Ethen-Molekülen:
Eπ(Ethen) = 2 α + 2 β
Eπ(Naphthalin) = 10 α + 13.6832 β
∆E = Eπ(Naphthalin) - 5 Eπ(Ethen)
∆E = 3.6832 β
Unter Verwendung der Umrechnung β = -75 kJ/mol ergibt sich die Stabilisierungsenergie des Naphthalins zu -276 kJ/mol (Benzol: -150 kJ/mol). Hydriert man nur einen Ring, so gehen folglich nur 126 kJ/mol an Stabilisierungsenergie verloren, weshalb die Teilhydrierung des Naphthalins zu Tetralin in der Praxis leichter geht als die Hydrierung von Benzol zu Cyclohexan. Pro π -Zentrum gerechnet ist allerdings die Stabilisierungsenergie im Naphthalin (27.6 kJ/mol) sogar ein wenig größer als im Benzol (25.0 kJ/mol) (REPE, resonance energy per π -electron).
Im Gegensatz zum Benzol gibt es im Naphthalin keine entarteten Orbitale. Der HOMO-LUMO-Abstand (Benzol: 2 β) ist auf 1.236 β verkleinert (HOMO: highest occupied molecular orbital, LUMO: lowest unoccupied molecular orbital). Die Reduktion zum Radikalanion oder die Oxidation zum Radikalkation ist daher erleichtert. Im HMO-Modell ist die Spindichteverteilung in diesen beiden Spezies gleich. (Spindichten können mit Hilfe der EPR-Spektroskopie gemessen werden.) Sowohl die Spindichten (α: 0.180902, β: 0.069098) als auch die freien Valenzen (α: 0.4527, β: 0.4043) sind in den α -Positionen (1,4,5,8) größer als in den β -Positionen (2,3,6,7), im Einklang mit den beobachteten Reaktivitäten.
Dichtefunktionaltheorie, DFT-Rechnungen
Nach allen bis heute (2015) durchgeführten Experimenten gleicht die Struktur des Benzols einem regelmäßigen Sechseck. Unter Quantenchemikern (Janoschek [7], Shaik [8]) wird allerdings die Frage kontrovers diskutiert, ob hauptsächlich die π- oder die σ-Elektronen für die Stabilität und die Struktur des Benzols verantwortlich sind. Wie schon die Kekulé-Formel des Benzols suggeriert, sollten die π-Bindungen eventuell zu einer Bindungsalternanz Anlass geben. In der Tat findet man experimentell beim Naphthalin alternierende Bindungslängen, wenngleich sie näher bei der Aromatenbindung (140 pm) als bei Einfach- (154 pm) oder Doppelbindung (133 pm) liegen.
In diesem Rahmen wurden für Benzol und für Naphthalin Rechnungen nach der Dichtefunktionaltheorie [9] durchgeführt, und zwar mit den Programmen deMon2k (Kohn-Sham,

5
KS/DZVP-GGA) [10] und Gaussian 09 (B3LYP/6-311(d,p)) [11]. Zur Bearbeitung der Ein- und Ausgabe und zur Visualisierung z.B. der Orbitale wurde das Programm molden [12] verwendet.
Benzol
H
H
H
H
H
H
H
H
H
H
H
H
138.969 138.969
138.637
138.637
139.343
139.343
140.418
110.278
105.980
105.980107.162
107.162
108.433
108.433
exp [pm] ber [pm]
Abb. 3. Bindungslängen im Benzol-Molekül (in pm); links: experimentell (Neutronenbeugung, BENZEN aus der Cambridge Structural Database); rechts: berechnet (DFT: KS/DZVP-GGA, deMon2k).
Für Benzol wurde bei der Geometrieoptimierung (erwartungsgemäß) die D6h-Struktur, also das regelmäßige Sechseck, als energetisch günstigste Struktur erhalten (Abb. 3). In der Abbildung 4 wird ein Ausschnitt aus dem Orbitalschema des Benzols (in der optimierten Struktur) mit den Darstellungen der π- und π*-Orbitale gezeigt.
Abb. 4. Die sechs π - bzw. π*-Orbitale des Benzols und die Kohn-Sham-Orbitalenergien in atomaren Einheiten (Hartree).
Der HOMO-LUMO-Abstand im Benzol beträgt 0.18776 au = 492.96 kJ/mol, entsprechend einer Wellenlänge von 242.7 nm im Elektronenspektrum, berechnet nach

6
λ = c/ν = hc/∆E
λ [nm] = 119630/∆E [kJ/mol]
Experimentell findet man eine intensitätsschwache UV-Bande bei λmax = 254 nm (siehe dazu jedoch den Exkurs).
Weitere Rechnungen wurden für schwach alternierende Bindungslängen (wie im Naphthalin) sowie für eine ausgeprägte Bindungsalternanz, also die tatsächliche Kekulé-Struktur durchgeführt.
Tab. 2. DFT-Energien von Benzol und verzerrtem Benzol
totale Energie [au] relative Energie [kJ/mol] Bindungslängen [pm] -232.308550228 0.0 140.4 (alle) -232.305451857 8.1 138.5, 142.5 (wie Naphthalin) -232.300055609 22.3 135, 145 -232.274818137 88.6 133, 154 (wie Ethen, Ethan)
Die Energiewerte in der voranstehenden Tabelle stammen aus DFT-Rechnungen, B3LYP/6-311G(d,p) (Gaussian 09). Eine schwache Alternanz der Bindungslängen wie im Naphthalin führt zu einer Destabilisierung um etwa 8 kJ/mol. Verstärkt man die Alternanz so weit, dass die Bindungslängen abwechselnd typischen Doppel- bzw. Einfachbindungen entsprechen, so beträgt die Destabilisierung etwa 89 kJ/mol, d.h., der größte Teil der aromatischen Stabilisierungsenergie (ca. 150 kJ/mol) geht verloren.
Naphthalin
H
H
H
HH
H
H
H H
H
H
HH
H
H
H
137.4
141.6
141.7
142.1
138.6
142.2
142.6
144.1
108.4 110.4
0.7246
0.6032
0.5547
0.5182
HMO-π-Bindungsordnungen (Benzol: 0.6667) [10]Annulen
exp [pm] ber [pm]
Abb. 5. Oben: Bindungslängen im Naphthalin-Molekül (in pm); links: experimentell (Röntgenbeugung, NAPHTA15 aus der Cambridge Structural Database); rechts: berechnet (KS/DZVP-GGA, deMon2k). Unten links: nach dem HMO-Modell berechnete π-Bindungsordnungen; sie verhalten sich umgekehrt proportional zu den Bindungslängen. Unten rechts: [10]Annulen, als Modell für ein Naphthalin-Molekül mit sehr langer zentraler Bindung.

7
Das HMO-Modell erlaubt einen Vergleich der π-Energien der beiden 10-π-Elektronensysteme Naphthalin (13.6832 β) und [10]Annulen (12.9443 β); demnach ist das π-Elektronensystem des Naphthalins um etwa 55 kJ/mol stabiler.
Die DFT-Rechnungen für das Naphthalin liefern erwartungsgemäß eine optimierte Struktur mit schwach alternierenden Bindungslängen, siehe Abbildung 5. Weiterhin wurden Rechnungen für eine Struktur mit gleichen Bindungslängen sowie für die beiden ungleichartigen Kekulé-Strukturen und eine"pericyclische Struktur" (mit gleichen Bindungslängen im Pericyclus und längerer zentraler Bindung) durchgeführt.
Abb. 6. Strukturen und berechnete Energien (B3LYP/6-311(d,p), Gaussian 09) für verschiedene Naphthalin-Modelle. a-c: drei mesomere Grenzformeln des Naphthalins (in Klammern: unter der Annahme berechnete Energien, dass die eingezeichneten Doppelbindungen 133 pm und die Einfachbindungen 154 pm lang sind); d: Modell, in dem alle Bindungen gleich lang sind (140.4 pm); e: Modell, in dem alle pericyclischen Bindungen gleich lang sind (140.4 pm), während sich im Zentrum eine Einfachbindung befindet (154 nm); f: steht für die optimierte, energieärmste Struktur ("Resonanzhybrid").
Nach den DFT-Rechnungen (Abb. 6) wäre eine Naphthalin-Struktur, in der alle C-C-Bindungen die gleiche "aromatische" Länge hätten wie die C-C-Bindungen im Benzol (140.4 pm) um 17 kJ/mol instabiler als die optimale Struktur. (An der D2h-Symmetrie würde sich nichts ändern.) In qualitativer Übereinstimmung mit dem HMO-Modell (s.o.) ergibt auch die DFT-Rechnung, dass eine [10]Annulen-artige Struktur (e) etwa 64 kJ/mol instabiler wäre als die optimale (f). Von den drei "Kekulé-Strukturen" (mit Bindungslängen von 133 bzw. 154 pm) wäre a (siehe Abb. 6) etwas günstiger (138 kJ/mol) als die beiden gleichwertigen Strukturen b und c (151 kJ/mol); sie ist auch aus Symmetriegründen vorzuziehen und gibt (mit Ausnahme der zentralen Bindung) die Bindungslängen besser wieder.
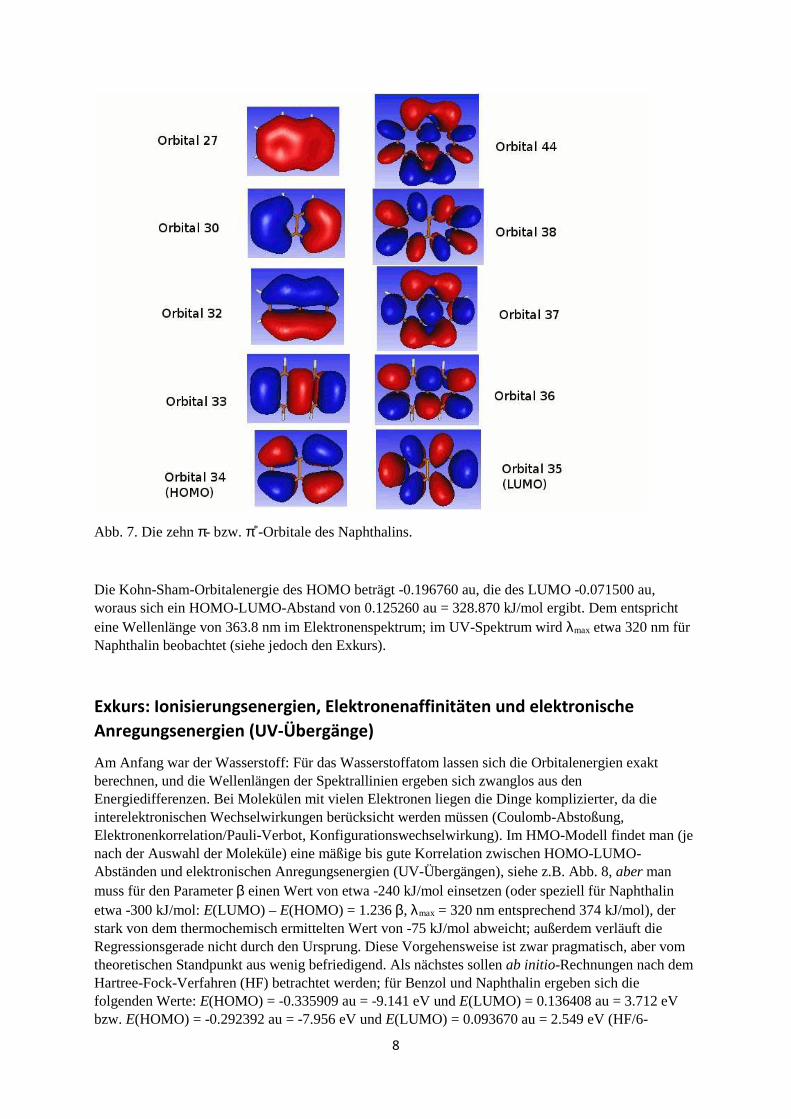
8
Abb. 7. Die zehn π- bzw. π*-Orbitale des Naphthalins.
Die Kohn-Sham-Orbitalenergie des HOMO beträgt -0.196760 au, die des LUMO -0.071500 au, woraus sich ein HOMO-LUMO-Abstand von 0.125260 au = 328.870 kJ/mol ergibt. Dem entspricht eine Wellenlänge von 363.8 nm im Elektronenspektrum; im UV-Spektrum wird λmax etwa 320 nm für Naphthalin beobachtet (siehe jedoch den Exkurs).
Exkurs: Ionisierungsenergien, Elektronenaffinitäten und elektronische
Anregungsenergien (UV-Übergänge)
Am Anfang war der Wasserstoff: Für das Wasserstoffatom lassen sich die Orbitalenergien exakt berechnen, und die Wellenlängen der Spektrallinien ergeben sich zwanglos aus den Energiedifferenzen. Bei Molekülen mit vielen Elektronen liegen die Dinge komplizierter, da die interelektronischen Wechselwirkungen berücksicht werden müssen (Coulomb-Abstoßung, Elektronenkorrelation/Pauli-Verbot, Konfigurationswechselwirkung). Im HMO-Modell findet man (je nach der Auswahl der Moleküle) eine mäßige bis gute Korrelation zwischen HOMO-LUMO-Abständen und elektronischen Anregungsenergien (UV-Übergängen), siehe z.B. Abb. 8, aber man muss für den Parameter β einen Wert von etwa -240 kJ/mol einsetzen (oder speziell für Naphthalin etwa -300 kJ/mol: E(LUMO) – E(HOMO) = 1.236 β, λmax = 320 nm entsprechend 374 kJ/mol), der stark von dem thermochemisch ermittelten Wert von -75 kJ/mol abweicht; außerdem verläuft die Regressionsgerade nicht durch den Ursprung. Diese Vorgehensweise ist zwar pragmatisch, aber vom theoretischen Standpunkt aus wenig befriedigend. Als nächstes sollen ab initio-Rechnungen nach dem Hartree-Fock-Verfahren (HF) betrachtet werden; für Benzol und Naphthalin ergeben sich die folgenden Werte: E(HOMO) = -0.335909 au = -9.141 eV und E(LUMO) = 0.136408 au = 3.712 eV bzw. E(HOMO) = -0.292392 au = -7.956 eV und E(LUMO) = 0.093670 au = 2.549 eV (HF/6-
9
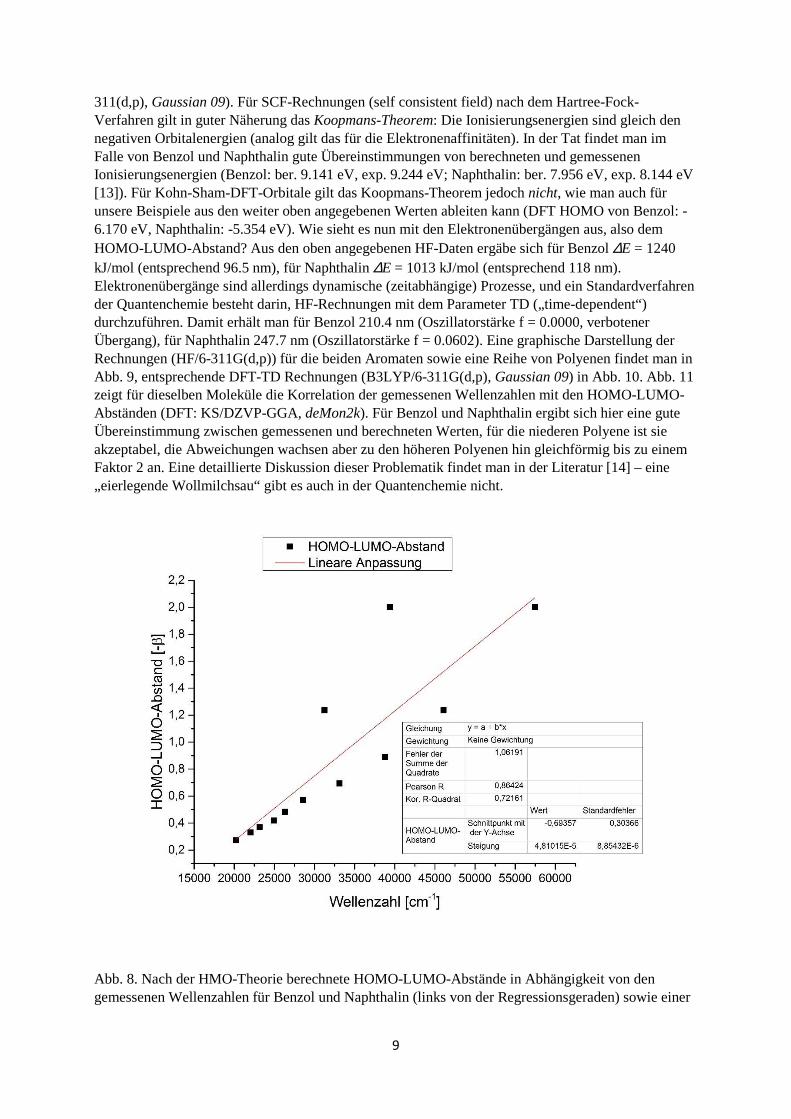
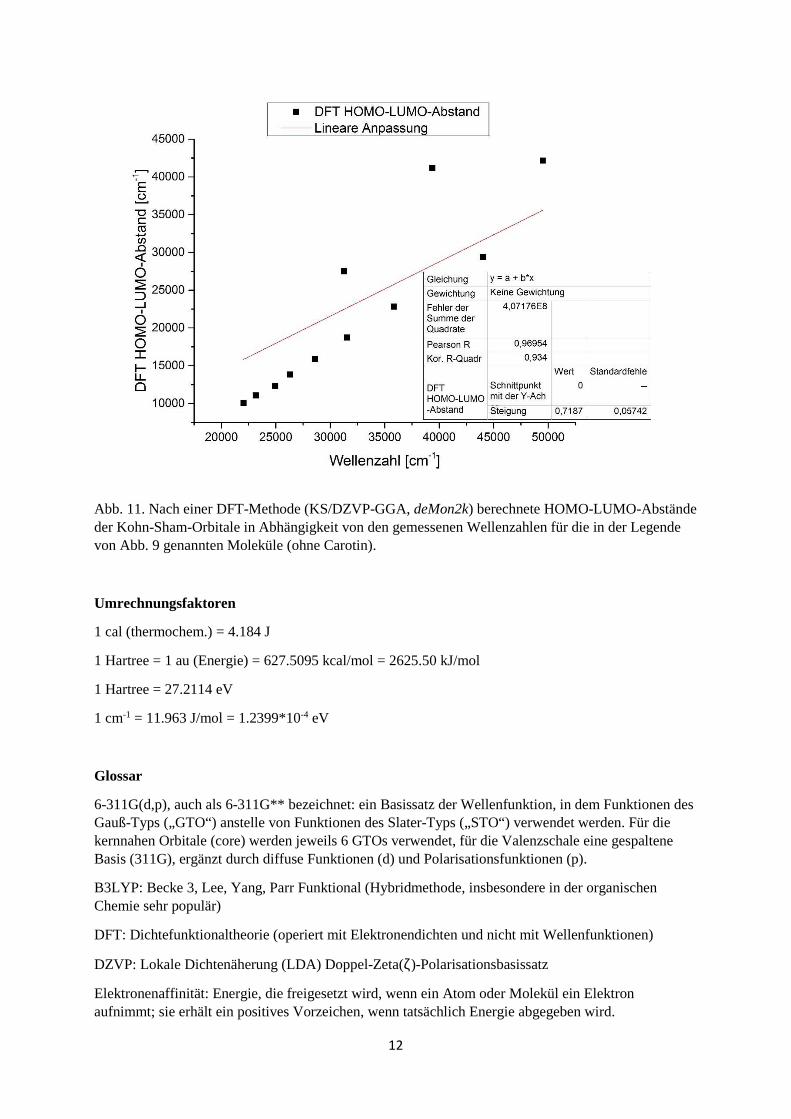
311(d,p), Gaussian 09). Für SCF-Rechnungen (self consistent field) nach dem Hartree-Fock-Verfahren gilt in guter Näherung das Koopmans-Theorem: Die Ionisierungsenergien sind gleich den negativen Orbitalenergien (analog gilt das für die Elektronenaffinitäten). In der Tat findet man im Falle von Benzol und Naphthalin gute Übereinstimmungen von berechneten und gemessenen Ionisierungsenergien (Benzol: ber. 9.141 eV, exp. 9.244 eV; Naphthalin: ber. 7.956 eV, exp. 8.144 eV [13]). Für Kohn-Sham-DFT-Orbitale gilt das Koopmans-Theorem jedoch nicht, wie man auch für unsere Beispiele aus den weiter oben angegebenen Werten ableiten kann (DFT HOMO von Benzol: -6.170 eV, Naphthalin: -5.354 eV). Wie sieht es nun mit den Elektronenübergängen aus, also dem HOMO-LUMO-Abstand? Aus den oben angegebenen HF-Daten ergäbe sich für Benzol ∆E = 1240 kJ/mol (entsprechend 96.5 nm), für Naphthalin ∆E = 1013 kJ/mol (entsprechend 118 nm). Elektronenübergänge sind allerdings dynamische (zeitabhängige) Prozesse, und ein Standardverfahren der Quantenchemie besteht darin, HF-Rechnungen mit dem Parameter TD („time-dependent“) durchzuführen. Damit erhält man für Benzol 210.4 nm (Oszillatorstärke f = 0.0000, verbotener Übergang), für Naphthalin 247.7 nm (Oszillatorstärke f = 0.0602). Eine graphische Darstellung der Rechnungen (HF/6-311G(d,p)) für die beiden Aromaten sowie eine Reihe von Polyenen findet man in Abb. 9, entsprechende DFT-TD Rechnungen (B3LYP/6-311G(d,p), Gaussian 09) in Abb. 10. Abb. 11 zeigt für dieselben Moleküle die Korrelation der gemessenen Wellenzahlen mit den HOMO-LUMO-Abständen (DFT: KS/DZVP-GGA, deMon2k). Für Benzol und Naphthalin ergibt sich hier eine gute Übereinstimmung zwischen gemessenen und berechneten Werten, für die niederen Polyene ist sie akzeptabel, die Abweichungen wachsen aber zu den höheren Polyenen hin gleichförmig bis zu einem Faktor 2 an. Eine detaillierte Diskussion dieser Problematik findet man in der Literatur [14] – eine „eierlegende Wollmilchsau“ gibt es auch in der Quantenchemie nicht.
Abb. 8. Nach der HMO-Theorie berechnete HOMO-LUMO-Abstände in Abhängigkeit von den gemessenen Wellenzahlen für Benzol und Naphthalin (links von der Regressionsgeraden) sowie einer
10
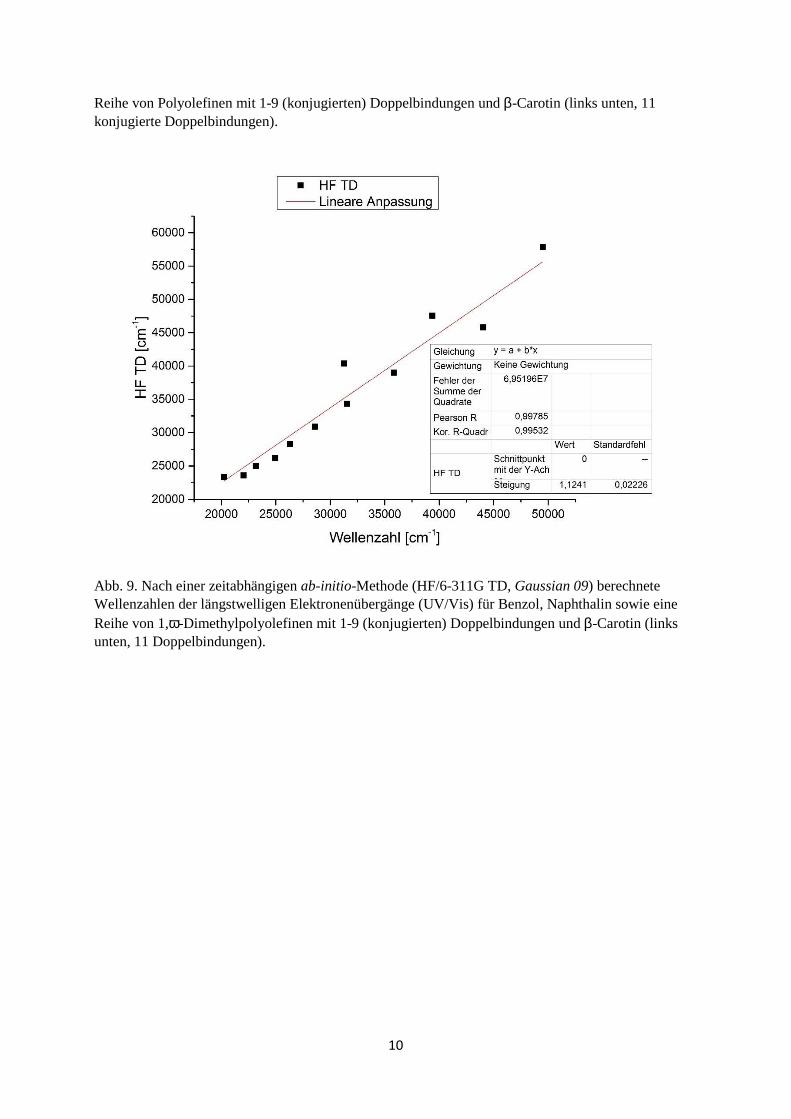
Reihe von Polyolefinen mit 1-9 (konjugierten) Doppelbindungen und β-Carotin (links unten, 11 konjugierte Doppelbindungen).
Abb. 9. Nach einer zeitabhängigen ab-initio-Methode (HF/6-311G TD, Gaussian 09) berechnete Wellenzahlen der längstwelligen Elektronenübergänge (UV/Vis) für Benzol, Naphthalin sowie eine Reihe von 1,ω-Dimethylpolyolefinen mit 1-9 (konjugierten) Doppelbindungen und β-Carotin (links unten, 11 Doppelbindungen).
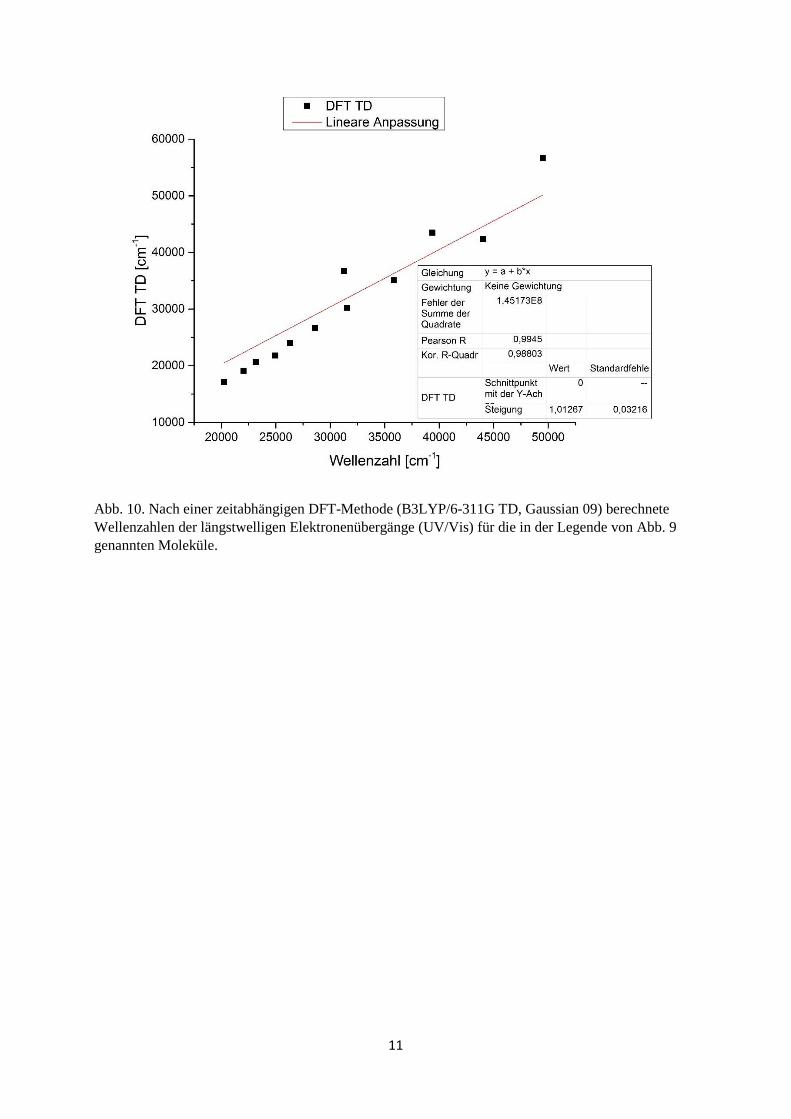
11
Abb. 10. Nach einer zeitabhängigen DFT-Methode (B3LYP/6-311G TD, Gaussian 09) berechnete Wellenzahlen der längstwelligen Elektronenübergänge (UV/Vis) für die in der Legende von Abb. 9 genannten Moleküle.
12
Abb. 11. Nach einer DFT-Methode (KS/DZVP-GGA, deMon2k) berechnete HOMO-LUMO-Abstände der Kohn-Sham-Orbitale in Abhängigkeit von den gemessenen Wellenzahlen für die in der Legende von Abb. 9 genannten Moleküle (ohne Carotin).
Umrechnungsfaktoren
1 cal (thermochem.) = 4.184 J
1 Hartree = 1 au (Energie) = 627.5095 kcal/mol = 2625.50 kJ/mol
1 Hartree = 27.2114 eV
1 cm-1 = 11.963 J/mol = 1.2399*10-4 eV
Glossar
6-311G(d,p), auch als 6-311G** bezeichnet: ein Basissatz der Wellenfunktion, in dem Funktionen des Gauß-Typs („GTO“) anstelle von Funktionen des Slater-Typs („STO“) verwendet werden. Für die kernnahen Orbitale (core) werden jeweils 6 GTOs verwendet, für die Valenzschale eine gespaltene Basis (311G), ergänzt durch diffuse Funktionen (d) und Polarisationsfunktionen (p).
B3LYP: Becke 3, Lee, Yang, Parr Funktional (Hybridmethode, insbesondere in der organischen Chemie sehr populär)
DFT: Dichtefunktionaltheorie (operiert mit Elektronendichten und nicht mit Wellenfunktionen)
DZVP: Lokale Dichtenäherung (LDA) Doppel-Zeta(ζ)-Polarisationsbasissatz
Elektronenaffinität: Energie, die freigesetzt wird, wenn ein Atom oder Molekül ein Elektron aufnimmt; sie erhält ein positives Vorzeichen, wenn tatsächlich Energie abgegeben wird.

13
GGA: „Generalized Gradient Approximation“, verallgemeinerte Gradientennäherung
HF: Hartree-Fock-Verfahren, eine quantenchemische ab-initio-Methode, die auf dem SCF-Prinzip basiert (s.d.)
HMO: Hückel-Molekülorbital
HOMO: „highest occupied molecular orbital“, höchstes besetztes Molekülorbital
Ionisierungsenergie: Energie, die aufgewendet werden muss, um ein Elektron aus einem Atom oder Molekül zu entfernen
KS: Kohn-Sham, ein Dichtefunktional
LUMO: „lowest unoccupied molecular orbital“, tiefstes unbesetztes Molekülorbital (ein virtuelles Orbital)
REPE: „resonance energy per π-electron”, Resonanzenergie pro π-Elektron
SCF: „self consistent field“ – Methode des selbstkonsistenten Feldes, bei der Ein-Elektronen-Wellenfunktionen im effektiven Potential der übrigen Elektronen sukzessive optimiert werden, bis sich praktisch nichts mehr ändert
Zusammenfassung
DFT-Rechnungen an Benzol und Naphthalin stehen im Einklang mit den experimentellen Befunden, wonach im Benzolmolekül alle sechs Bindungen exakt gleich lang sind, während man im Naphthalin-Molekül eine schwach ausgeprägte Alternanz findet. Die Abweichungen von der "aromatischen" Bindungslänge (140 pm) liegen aber bei maximal etwa 25 % der Werte, die man für reine Einfach- oder Doppelbindungen erwarten würde. Ein hypothetisches Naphthalin mit gleich langen Bindungen (wie im Benzol) wäre lediglich um 17 kJ/mol energiereicher als die optimale Struktur, ein hypothetisches Benzol mit schwach alternierenden Bindungslängen (wie im Naphthalin) sogar nur um 8 kJ/mol energiereicher als die tatsächlich vorliegende D6h-Struktur. Bei einer echten „Kekulé-Struktur“ würde allerdings der größte Teil der Stabilisierungsenergie verloren gehen.
Summary (Abstract)
DFT calculations of benzene and naphthalene are in accordance with experimental observations that all six bonds in benzene have the same length, whereas bond lengths in naphthalene alternate moderately. The energy of a hypothetical naphthalene with equal bond lengths would be only 17 kJ/mol higher than that of the optimum structure, and the energy of a hypothetical benzene with weakly alternating bond lengths (as in naphthalene) would exceed that of the regular structure by merely 8 kJ/mol. In a true “Kekulé structure”, however, most of the stabilization energy would be lost.
Schlagworte: Aromatizität Hückel-Theorie HMO Dichtefunktionaltheorie DFT
Literatur
Ergänzendes elektronisches Material (Ausgabedateien der Rechnungen, Excel-Tabelle zum Exkurs, HMO-Programm): http://www.chemie.fu-berlin.de/benzol2015.

14
Weiterführende Literatur:
Thomas Heine, Jan-Ole Joswig und Achim Gelessus, Computational Chemistry Workbook (Learning Through Examples), Wiley-VCH, Weinheim, 2009; ISBN: 978-3-527-32442-2 (mit CD-ROM).
E. Heilbronner und H. Bock, Das HMO-Modell und seine Anwendung, 3 Bände, Verlag Chemie, Weinheim, 1968-1970.
Linus Pauling, Die Natur der chemischen Bindung, Verlag Chemie, Weinheim, 2. Aufl., 1964.
Peter J. Garratt und Peter Vollhardt, Aromatizität, Georg Thieme Verlag, Stuttgart, 1987.
Aromaticity, Chem. Rev. 2001, 101, Heft 5 (Sonderheft zum Thema Aromatizität), http://pubs.acs.org/toc/chreay/101/5.
Zitierte Literatur:
[1] Kekulé, "Sur la constitution des substances aromatiques," Bull. Soc. Chim. de Paris 1865, 3, 98-110 (englische Übersetzung: D. Wilcox und F. Greenbaum, J. Chem. Ed. 1965, 42, 266-267); DOI: 10.1021/ed042p266); "Untersuchungen über aromatische Verbindungen," Liebigs Ann. Chem. 1866, 137, 129-136; DOI: 10.1002/jlac.18661370202.
[2] Kekulé, "Ueber einige Condensationsproducte des Aldehyds," Liebigs Ann. Chem. 1872, 162, 77-124; DOI: 10.1002/jlac.18721620110
[3] Kekulé, "Benzolfest: Rede," Ber. Dt. Chem. Ges. 1890, 23, 1302-1311.
[4] H. Hartmann, "Die Benzolformel. Eine kurze Problemgeschichte," Angew. Chem. 1965, 77, 750-752; DOI: 10.1002/ange.19650771703.
[5] Erich Hückel, "Quantentheoretische Beiträge zum Benzolproblem. I. Die Elektronenkonfiguration des Benzols und verwandter Verbindungen," Z. Phys. 1931, 70, 204-286; DOI: 10.1007/BF01339530.
[6] Erich Hückel, "Quantentheoretische Beiträge zum Benzolproblem. II. Quantentheorie der induzierten Polaritäten," Z. Phys. 1931, 72, 310-337; DOI: 10.1007/BF01341953.
[7] Rudolf Janoschek, "Has the benzene molecule an extra stability?" J. Mol. Struct. (Theochem) 1991, 229, 197-203; DOI: 10.1016/0166-1280(91)90146-B.
[8] Sason Shaik, Avital Shurki, David Danovich und Philippe C. Hiberty: "A different story of benzene," J. Mol. Struct. (Theochem) 1997, 398-399, 155-167; DOI: 10.1016/S0166-1280(96)04934-2.
[9] Aron J. Cohen, Paula Mori-Sánchez und Weitao Yang, Chem. Rev. 2012, 112, 289–320; DOI: 10.1021/cr200107z.
[10] A.M. Koster, P. Calaminici, M.E. Casida, V.D. Dominguez, Flores-Moreno, G. Geudtner, A. Goursot, T. Heine, A. Ipatov, F. Janetzko, J.M. del Campo, J.U. Reveles, A. Vela, B. Zuniga und D.R. Salahub, deMon2k, Version 2, The deMon developers, Cinvestav, Mexico City, 2006.
[11] Gaussian 09, Revision D.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross,

15
V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski und D. J. Fox, Gaussian, Inc., Wallingford CT, 2013
[12] G. Schaftenaar, CAOS/CAMM Center, Faculty of Science, Nijmegen University, Nijmegen, The Netherlands, http://www.cmbi.ru.nl/molden/; G. Schaftenaar, J. H. Noordik, J. Comp.-Aided Mol. Des. 2000, 14, 123; DOI: 10.1023/A:1008193805436.
[13] NIST Chemistry WebBook, http://webbook.nist.gov/chemistry/
[14] Gang Zhang und Charles B. Musgrave, J. Phys. Chem. A 2007, 111, 1554-1561; DOI: 10.1021/jp061633o
© Priv.-Doz. Dr. Burkhard Kirste, 2015, 2016
E-Mail: [email protected]
WWW: http://www.chemie.fu-berlin.de/kirste
Institut für Chemie und Biochemie der Freien Universität Berlin, Takustr. 3, 14195 Berlin
Letzte Bearbeitung: 2016-08-21