anti-glycoprotein h antibody impairs the pathogenicity of varicella
TRANSCRIPT

JOURNAL OF VIROLOGY, Jan. 2010, p. 141–152 Vol. 84, No. 10022-538X/10/$12.00 doi:10.1128/JVI.01338-09Copyright © 2010, American Society for Microbiology. All Rights Reserved.
Anti-Glycoprotein H Antibody Impairs the Pathogenicity ofVaricella-Zoster Virus in Skin Xenografts in the
SCID Mouse Model�
Susan E. Vleck,1* Stefan L. Oliver,1 Mike Reichelt,1 Jaya Rajamani,1 Leigh Zerboni,1 Carol Jones,2James Zehnder,2 Charles Grose,3 and Ann M. Arvin1
Departments of Pediatrics and Microbiology & Immunology1 and Pathology,2 Stanford University School of Medicine, Stanford,California 94305, and Department of Pediatrics, University of Iowa, Iowa City, Iowa 522423
Received 29 June 2009/Accepted 6 October 2009
Varicella-zoster virus (VZV) infection is usually mild in healthy individuals but can cause severe disease inimmunocompromised patients. Prophylaxis with varicella-zoster immunoglobulin can reduce the severity ofVZV if given shortly after exposure. Glycoprotein H (gH) is a highly conserved herpesvirus protein withfunctions in virus entry and cell-cell spread and is a target of neutralizing antibodies. The anti-gH monoclonalantibody (MAb) 206 neutralizes VZV in vitro. To determine the requirement for gH in VZV pathogenesis invivo, MAb 206 was administered to SCID mice with human skin xenografts inoculated with VZV. Anti-gHantibody given at 6 h postinfection significantly reduced the frequency of skin xenograft infection by 42%. Virustiters, genome copies, and lesion size were decreased in xenografts that became infected. In contrast, admin-istering anti-gH antibody at 4 days postinfection suppressed VZV replication but did not reduce the frequencyof infection. The neutralizing anti-gH MAb 206 blocked virus entry, cell fusion, or both in skin in vivo. In vitro,MAb 206 bound to plasma membranes and to surface virus particles. Antibody was internalized into vacuoleswithin infected cells, associated with intracellular virus particles, and colocalized with markers for earlyendosomes and multivesicular bodies but not the trans-Golgi network. MAb 206 blocked spread, alteredintracellular trafficking of gH, and bound to surface VZV particles, which might facilitate their uptake andtargeting for degradation. As a consequence, antibody interference with gH function would likely prevent orsignificantly reduce VZV replication in skin during primary or recurrent infection.
Varicella-zoster virus (VZV) causes chicken pox (varicella)upon primary infection. Lifelong latency is established in neu-rons of the sensory ganglia, and reactivation leads to shingles(herpes zoster) (1). Disease is usually inconsequential in im-munocompetent people but can be severe in immunocompro-mised patients. The current prophylaxis for these high-riskindividuals exposed to VZV is high-titer immunoglobulin toVZV administered within 96 h of exposure. This prophylaxisdoes not always prevent disease, but the severity of symptomsand mortality rates are usually reduced (32).
Glycoprotein H (gH) is a type 1 transmembrane protein thatis required for virus-cell and cell-cell spread in all herpesvi-ruses studied (12, 15, 24, 26). gH is an important target of thehost immune system. Individuals who have had primary infec-tion with VZV or herpes simplex virus (HSV), the most closelyrelated human alphaherpesvirus, have humoral and cellularimmunity against gH (1, 56). Immunization of mice with arecombinant vaccinia virus expressing VZV gH and its chap-erone, glycoprotein L (gL), induced specific antibodies capableof neutralizing VZV in vitro (28, 37). Immunization of micewith purified HSV gH/gL protein resulted in the production ofneutralizing antibodies and protected mice from HSV chal-lenge (5, 44), and administration of an anti-HSV gH monoclo-
nal antibody (MAb) protected mice from HSV challenge (16).Antibodies to HSV and Epstein-Barr virus gH effectively neu-tralize during virus penetration but not during adsorption invitro, indicating an essential role for gH in the fusion of viraland cellular membranes but not in initial attachment of thevirus to the cell (18, 33).
Anti-gH MAb 206, an immunoglobulin G1 (IgG1) antibodywhich recognizes a conformation-dependent epitope on themature glycosylated form of gH, neutralizes VZV infection invitro in the absence of complement (35). MAb 206 inhibitscell-cell fusion in vitro, based on reductions in the number ofinfected cells and the number of infected nuclei within syncy-tia, and appears to inhibit the ability of virus particles to passfrom the surface of an infected epithelial cell to a neighboringcell via cell extensions (8, 35, 43). When infected cells weretreated with MAb 206 for 48 h postinfection (hpi), virus egressand syncytium formation were not apparent, but they wereevident within 48 h after removal of the antibody, suggestingthat the effect of the antibody was reversible and that there wasa requirement for new gH synthesis and trafficking to producecell-cell fusion. Conversely, nonneutralizing antibodies to gly-coproteins E (gE) and I (gI), as well as an antibody to imme-diate-early protein 62 (IE62), had no effect on VZV spread(46).
Like that of other herpesviruses, VZV entry into cells ispresumed to require fusion of the virion envelope with the cellmembrane or endocytosis followed by fusion. One of the hall-marks of VZV infection is cell fusion and formation of syncytia(8). Cell fusion can be detected as early as 9 hpi in vitro,
* Corresponding author. Mailing address: Stanford University, 300Pasteur Drive, Grant Building Room S366, Stanford, CA 94305.Phone: (650) 725-6555. Fax: (650) 725-9828. E-mail: [email protected].
� Published ahead of print on 14 October 2009.
141
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 30
Dec
embe
r 20
21 b
y 41
.157
.41.
148.

although VZV spread from infected to uninfected cells is ev-ident within 60 min (45). In vivo, VZV forms syncytia throughits capacity to cause fusion of epidermal cells. Syncytia areevident in biopsies of varicella and herpes zoster skin lesionsduring natural infection and in SCIDhu skin xenografts (34).VZV gH is produced, processed in the Golgi apparatus, andtrafficked to the cell membrane, where it might be involved incell-cell fusion (11, 29, 35). gH then undergoes endocytosis andis trafficked back to the trans-Golgi network (TGN) for incor-poration into the virion envelope (20, 31, 42). Since VZV ishighly cell associated in vitro, little is known about the glyco-proteins required for entry, but VZV gH is present in abun-dance in the skin vesicles during human chickenpox and zoster(55).
Investigating the functions of gH in the pathogenesis ofVZV infection in vivo is challenging because it is an essentialprotein and VZV is species specific for the human host. Theobjective of this study was to investigate the role of gH in VZVpathogenesis by establishing whether antibody-mediated inter-ference with gH function could prevent or modulate VZVinfection of differentiated human tissue in vivo, using theSCIDhu mouse model. The effects of antibody administrationat early and later times after infection were determined bycomparing infectious virus titers, VZV genome copies, andlesion formation in anti-gH antibody-treated xenografts. Invitro experiments were performed to determine the potentialmechanism(s) of MAb 206 interference with gH during VZVreplication, virion assembly, and cell-cell spread. The presentstudy has implications for understanding the contributions ofgH to VZV replication in vitro and in vivo, the mechanisms bywhich production of antibodies to gH by the host might restrictVZV infection, and the use of passive antibody prophylaxis inpatients at high risk of serious illness caused by VZV.
MATERIALS AND METHODS
Cells and virus. Human melanoma cells and human embryonic lung fibroblasts(HELFs) were grown at 37°C in Dulbecco’s modified Eagle’s medium supple-mented with 10% fetal bovine serum (Gemini Bio-Products, Woodland, CA),nonessential amino acids (melanoma cells only) (100 �M; Omega Scientific, Inc.,Tarzana, CA), and antibiotics (penicillin at 100 U/ml and streptomycin at 100�g/ml; Invitrogen, Carlsbad, CA). Wild-type recombinant VZV pOka was gen-erated in melanoma cells by use of a cosmid system (39) and then propagated inHELF cultures.
Preparation, inoculation, and harvest of skin xenografts in SCID mice. Skinxenografts were prepared in homozygous CB-17scid/scid mice, using human fetalskin tissue obtained according to federal and state regulations (34). Animalprotocols complied with the Animal Welfare Act and were approved by theStanford University Administrative Panel on Laboratory Animal Care. Humantissues were obtained, in accordance with state and federal regulations, fromAdvanced Bioscience Resources (Alameda, CA). VZV pOka-infected HELFcultures were used to inoculate the xenografts. Infectious virus titer was deter-mined at the time of inoculation by 10-fold serial dilution on melanoma cells.Skin xenografts were harvested at 7, 14, 21, 28, 35, and 42 days postinfection(dpi). Half of each xenograft was stored in 4% paraformaldehyde for histology,and half was homogenized and resuspended in 1 ml phosphate-buffered saline(PBS) for virus titration and DNA extraction. Serum was prepared from bloodcollected from mice at the time of tissue harvest, allowed to coagulate at roomtemperature (RT), and then centrifuged at 6,000 � g for 20 min.
Treatment of SCIDhu mice with anti-gH antibody. Anti-gH MAb 206 is anIgG1 complement-independent neutralizing antibody that recognizes a confor-mational epitope on mature glycosylated gH (35). Either 100 �l PBS containing25 �g MAb 206 or 100 �l PBS alone was administered to mice intraperitoneallyevery 4 days starting at 6 hpi. Mice were treated with antibody beginning at 6 hpior 4 dpi, and repeated doses were given every 4 days through 12 dpi. The twoantibody-treated groups consisted of mice treated with antibody at 6 hpi, 4 dpi,
8 dpi, and 12 dpi (Ab-0-12 group) and mice treated at 4 dpi, 8 dpi, and 12 dpi(Ab-4-12 group). PBS was administered at time points when antibody was notgiven out to 42 dpi. A control group (PBS group) was given PBS at all compa-rable time points. The number of xenografts evaluated at each time point was asfollows: 7 to 21 dpi, n � 11 or 12; 28 dpi, Ab-0-12 group, n � 5; 28 dpi, Ab-4-12and PBS groups, n � 11 or 12; and 35 to 42 dpi, n � 5 or 6.
Infectious plaque assay. Melanoma cells were seeded in a 24-well plate andinoculated in triplicate with 0.1 ml of a 10-fold serial dilution of xenografthomogenate or the inoculum virus to be titrated. For the titration of virus fromhomogenates, the medium was changed 24 h after inoculation. Cells were cul-tured for 5 days, and plaques were stained with anti-VZV polyclonal serum.Titers were analyzed using Student’s t test to determine if a statistically signifi-cant difference (P � 0.05) in titer existed. The number of xenografts positive forvirus was analyzed using Fisher’s exact test to determine if a statistically signif-icant difference (P � 0.05) existed.
Plaque neutralization assay. Melanoma cells were seeded in a 24-well plateand inoculated in triplicate with 0.1 ml of 10-PFU/ml pOka in the absence orpresence of 0.1 ml of a 10-fold dilution of xenograft homogenate. The mediumwas changed after 24 h, and plates were incubated for 5 days. Plates were stainedas described above, and titers were analyzed using Student’s t test to determineif anti-gH antibody within the homogenate neutralized the 10-PFU/ml inoculum.
Enzyme-linked immunosorbent assay. The IgG1 MAb 206 in mouse serumwas measured using a mouse IgG1 enzyme-linked immunosorbent assay quan-titation kit from Bethyl Laboratories, Inc. (Montgomery, TX), following themanufacturer’s recommended protocol. Briefly, plates were coated with captureantibody and blocked with postcoat solution. Serum samples were diluted 1:100and 1:1,000 in duplicate and incubated for 60 min at RT. Horseradish peroxidaseconjugate and tetramethylbenzidine (TMB) with an acid stop were used to detectthe presence of IgG1 antibody. Plates were read in a SpectraMax 190 instrument(Molecular Devices, Sunnyvale, CA). The IgG1 concentration was determinedfrom a standard curve with a range of 250 to 3.9 ng/ml, analyzed using afour-parameter logistic curve fit, as recommended by the manufacturer.
Immunohistochemistry of skin xenograft sections. Mouse anti-gE antibody(MAb 8612; Millipore, Temecula, CA) was used at 1:2,000 to detect VZV lesionsin sectioned xenografts. Slides were developed using an alkaline phosphatase-based enzyme detection method (Millipore, Temecula, CA) with PermaRedsubstrate (VWR, West Chester, PA). Slides were counterstained with hematox-ylin. Xenografts were examined using an Axiovert 200 microscope (Zeiss).
Quantitative PCR. DNA was isolated from xenograft homogenates by use ofDNAzol (Gibco-BRL, Grand Island, NY) following the manufacturer’s protocol.VZV genome copy number was assessed using primers/probes to detect ORF31(encoding gB), ORF62 (encoding IE62), and ORF63 (encoding IE63), as pre-viously reported (58). Each gene target was measured in duplicate, and the meanof each was used to determine the number of genome copies.
VZV DNA in situ hybridization. A VZV-specific DNA probe was prepared aspreviously described (45). Paraffin-embedded sections were deparaffinized, in-cubated with proteinase K (Roche, Indianapolis, IN) in proteinase K buffer (0.1M Tris-HCl, pH 7.5, 150 mM NaCl, 12.5 mM EDTA) for 10 min at 37°C, andthen dried completely. Hybridization mix (15 �l) was added to each section,covered with a glass coverslip, denatured for 10 min at 95°C, and then hybridizedovernight at 60°C. Sections were washed twice in 2� SSC (1� SSC is 0.15 MNaCl plus 0.015 M sodium citrate) and once in 0.2� SSC for 10 min at 50°C andthen blocked for 30 min in digoxigenin (DIG) blocking solution (Roche, India-napolis, IN). Sections were incubated with anti-DIG MAb (Roche, Indianapolis,IN) (1:50 in blocking solution) for 1 h at 37°C and then with secondary anti-mouse–DIG antibody (1:100 in blocking solution) for 1 h at 37°C to amplify theDIG signal. Finally, sections were incubated with anti-DIG–alkaline phosphatase(1:200 in blocking solution) for 1 h at 37°C, followed by nitroblue tetrazolium–5-bromo-4-chloro-3-indolylphosphate (NBT-BCIP) staining to detect the VZVDNA-specific signal. Sections were counterstained in methyl green and imagedusing an Axiovert 200 microscope (Zeiss).
Antibody treatment of pOka-infected HELF cultures in vitro. Following aprotocol adapted from the work of Rodriguez et al. (46), 1 � 106 HELFs/wellwere seeded in six-well plates 24 h prior to inoculation. Monolayers were inoc-ulated with 3 log10 PFU pOka for 90 min at 37°C. The medium was changed, 25�g of anti-gH MAb 206 was added, and this was repeated at 24-h intervals. Mockcells received only medium. HELF cultures were harvested for titration andDNA extraction at 24-h intervals.
EM. HELF monolayers were seeded in 10-cm dishes containing glass cover-slips, infected with pOka, and treated with antibody, as detailed above. After48 h, the cells on the coverslips were washed in PBS, fixed, and processed fortransmission electron microscopy (EM) as previously described (7), althoughwithout the use of propylene oxide in the dehydration steps. The remaining cells
142 VLECK ET AL. J. VIROL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 30
Dec
embe
r 20
21 b
y 41
.157
.41.
148.

were washed in PBS and collected for cryo-EM. For cryo-EM, treated anduntreated samples were fixed in 4% paraformaldehyde with 0.1% glutaraldehydein phosphate buffer (0.1 M, pH 7.2), washed in PBS, and infiltrated with 2.3 Msucrose overnight at 4°C. The samples were then mounted on pins for cryo-ultramicrotomy and frozen in liquid nitrogen. Ultrathin cryosections (80 nm)were prepared with a diamond knife (Diatome, Hatfield, PA) at �130°C, usingan ultramicrotome (Ultracut; Leica) equipped with a cryosectioning chamber.Thawed cryosections were transferred to Formvar- and carbon-coated EM grids(nickel) within a drop of 2.3 M sucrose, washed in PBS, labeled with immuno-gold, and counterstained with 0.5% uranyl acetate in 2% methylcellulose for 10min on ice. For immunogold labeling, thawed cryosections were blocked with 1�DIG blocking solution. The mouse monoclonal antibody MAb 206, used fortreating the samples, was detected by incubation with 1:100-diluted polyclonalrabbit anti-mouse antibody (Cappel Laboratories) for 1 h at RT, followed byprotein A-gold (15 nm) incubation for 30 min at RT.
Confocal microscopy. HELF cultures were infected with pOka and treatedwith antibody or mock treated as detailed above. At 48 hpi, cells were fixed in 4%paraformaldehyde and blocked with PBS containing 10% donkey serum and0.1% Triton X-100. Cellular localization of VZV proteins was performed usingprimary antibodies to VZV proteins gH (SG3 monoclonal mouse anti-gH;Biodesign, Saco, ME), ORF23 (rabbit polyclonal) (7), and gE (rabbit polyclonal)(25) and to cellular proteins TGN46 (AHP500 polyclonal sheep anti-TGN46;AbD Serotec, Oxford, United Kingdom), early endosome antigen 1 (EEA1)(NB300-502 rabbit anti-EEA1; Novus Biologicals, Littleton, CO), and vacuolarprotein sorting 4 (Vps4) (sc-32922 rabbit anti-Vps4; Santa Cruz Biotechnology,Inc., Santa Cruz, CA). Secondary antibodies used were fluorescein isothiocya-nate-coupled donkey anti-sheep (Jackson ImmunoResearch, West Grove, PA),Alexa Fluor 555-coupled donkey anti-mouse (Molecular Probes, Carlsbad, CA),Alexa Fluor 647-coupled donkey anti-rabbit (Molecular Probes, Carlsbad, CA),and Hoechst 33342 (Molecular Probes, Carlsbad, CA). Confocal microscopy wasperformed using a Zeiss LSM510 confocal microscope equipped with two-pho-ton excitation.
RESULTS
Treatment with MAb 206 at 6 hpi significantly reduced thenumber of skin xenografts infected with VZV. To investigatethe effect of anti-gH neutralizing antibody on VZV pathogen-esis in vivo, skin xenografts were inoculated with pOka at atiter of 5.3 log10 PFU/ml, and mice were administered intra-peritoneally either MAb 206 or PBS. Of 56 xenografts frommock-treated mice (PBS group), 70% were positive for VZV
(Fig. 1). Only 28% of the xenografts from the mice treated withthe anti-gH MAb 206 at 6 hpi (Ab-0-12 group) were positivefor virus, significantly less than the positive number in the PBSgroup (P � 0.01; Fisher’s exact test). In contrast, anti-gH MAb206 treatment at 4 dpi (Ab-4-12 group) did not significantlyreduce the number of infected skin xenografts compared to thePBS group (P � 0.33; Fisher’s exact test), as 60% of thexenografts were positive for infectious virus. The number ofvirus-positive xenografts in the Ab-0-12 group was also signif-icantly reduced compared to that in the Ab-4-12 group (P �0.01; Fisher’s exact test), indicating that early (6 hpi) treatmentwith MAb 206 was capable of preventing VZV infection in42% of skin xenografts but that delaying treatment until 4 dpifailed to prevent infection in any skin xenografts.
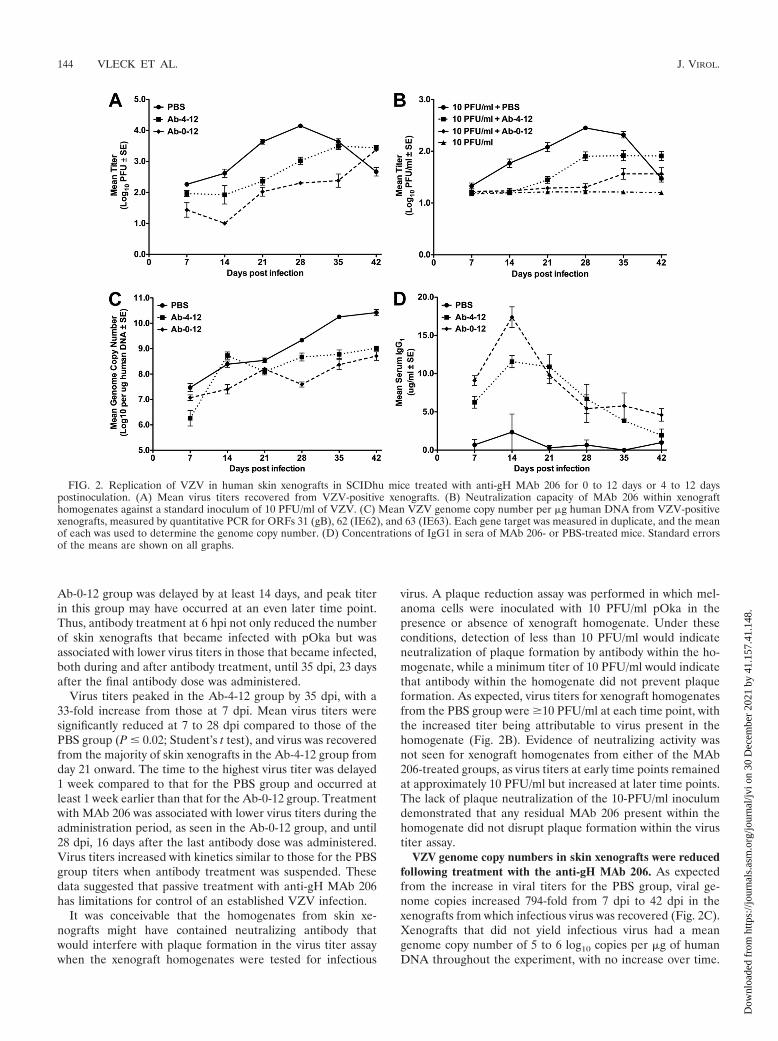
VZV titer was significantly reduced following treatment withthe anti-gH MAb 206. For the PBS group, the mean virus titerpeaked by 28 dpi, with a 77-fold increase in titer compared tothat at day 7 (Fig. 2A). The mean virus titer was unchanged by35 dpi but fell by 42 dpi. The 30-fold decrease from 28 to 42 dpiwas consistent with the depletion of cells permissive for infec-tion in the skin xenograft as a consequence of VZV replication.
Administration of MAb 206 starting at 6 hpi (Ab-0-12group) significantly reduced the titers of recoverable virus at 7to 35 dpi compared to those for the PBS group (P � 0.01;Student’s t test), with 7- to 70-fold differences between the twogroups. By 35 dpi, 67% of the Ab-0-12 xenografts were positivefor virus, the maximum percentage for this group. This was theonly time point at which �50% of the xenografts were positive,compared with �50% of xenografts being positive for virus atfour and five of the time points for the Ab-4-12 and PBSgroups, respectively (Fig. 1). By 42 dpi, the maximum titerreached in the Ab-0-12 group was 88-fold higher than the titerat day 7. The Ab-0-12 titer at 42 dpi was equivalent to the meantiter in the PBS group at 21 dpi, showing a considerable delayin reaching this titer in the Ab-0-12 group. The peak titer in thePBS group occurred at 28 dpi, whereas the maximal titer in the
FIG. 1. VZV infection of human skin xenografts in SCIDhu mice treated with anti-gH MAb 206 for 0 to 12 days or 4 to 12 days postinoculation.The timeline of treatment is shown above the percentage of xenografts from which infectious virus was recovered. Skin xenografts were inoculatedwith pOka-infected HELFs at a titer of 5.3 log10 PFU/ml, and mice were administered anti-gH MAb 206 (solid line; administration time pointsindicated with “A”) or PBS (dashed line; administration time points indicated with “P”). PBS, control group treated with PBS; Ab-0-12, grouptreated with MAb 206 at 6 hpi, 4 dpi, 8 dpi, and 12 dpi; Ab-4-12, group treated with MAb 206 at 4 dpi, 8 dpi, and 12 dpi. Xenografts were collectedat 7-day intervals up to 42 dpi. The total percentage of infected xenografts over the 42-day interval is shown on the right.
VOL. 84, 2010 ANTI-gH ANTIBODY NEUTRALIZATION OF VZV 143
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 30
Dec
embe
r 20
21 b
y 41
.157
.41.
148.
Ab-0-12 group was delayed by at least 14 days, and peak titerin this group may have occurred at an even later time point.Thus, antibody treatment at 6 hpi not only reduced the numberof skin xenografts that became infected with pOka but wasassociated with lower virus titers in those that became infected,both during and after antibody treatment, until 35 dpi, 23 daysafter the final antibody dose was administered.
Virus titers peaked in the Ab-4-12 group by 35 dpi, with a33-fold increase from those at 7 dpi. Mean virus titers weresignificantly reduced at 7 to 28 dpi compared to those of thePBS group (P � 0.02; Student’s t test), and virus was recoveredfrom the majority of skin xenografts in the Ab-4-12 group fromday 21 onward. The time to the highest virus titer was delayed1 week compared to that for the PBS group and occurred atleast 1 week earlier than that for the Ab-0-12 group. Treatmentwith MAb 206 was associated with lower virus titers during theadministration period, as seen in the Ab-0-12 group, and until28 dpi, 16 days after the last antibody dose was administered.Virus titers increased with kinetics similar to those for the PBSgroup titers when antibody treatment was suspended. Thesedata suggested that passive treatment with anti-gH MAb 206has limitations for control of an established VZV infection.
It was conceivable that the homogenates from skin xe-nografts might have contained neutralizing antibody thatwould interfere with plaque formation in the virus titer assaywhen the xenograft homogenates were tested for infectious
virus. A plaque reduction assay was performed in which mel-anoma cells were inoculated with 10 PFU/ml pOka in thepresence or absence of xenograft homogenate. Under theseconditions, detection of less than 10 PFU/ml would indicateneutralization of plaque formation by antibody within the ho-mogenate, while a minimum titer of 10 PFU/ml would indicatethat antibody within the homogenate did not prevent plaqueformation. As expected, virus titers for xenograft homogenatesfrom the PBS group were �10 PFU/ml at each time point, withthe increased titer being attributable to virus present in thehomogenate (Fig. 2B). Evidence of neutralizing activity wasnot seen for xenograft homogenates from either of the MAb206-treated groups, as virus titers at early time points remainedat approximately 10 PFU/ml but increased at later time points.The lack of plaque neutralization of the 10-PFU/ml inoculumdemonstrated that any residual MAb 206 present within thehomogenate did not disrupt plaque formation within the virustiter assay.
VZV genome copy numbers in skin xenografts were reducedfollowing treatment with the anti-gH MAb 206. As expectedfrom the increase in viral titers for the PBS group, viral ge-nome copies increased 794-fold from 7 dpi to 42 dpi in thexenografts from which infectious virus was recovered (Fig. 2C).Xenografts that did not yield infectious virus had a meangenome copy number of 5 to 6 log10 copies per �g of humanDNA throughout the experiment, with no increase over time.
FIG. 2. Replication of VZV in human skin xenografts in SCIDhu mice treated with anti-gH MAb 206 for 0 to 12 days or 4 to 12 dayspostinoculation. (A) Mean virus titers recovered from VZV-positive xenografts. (B) Neutralization capacity of MAb 206 within xenografthomogenates against a standard inoculum of 10 PFU/ml of VZV. (C) Mean VZV genome copy number per �g human DNA from VZV-positivexenografts, measured by quantitative PCR for ORFs 31 (gB), 62 (IE62), and 63 (IE63). Each gene target was measured in duplicate, and the meanof each was used to determine the genome copy number. (D) Concentrations of IgG1 in sera of MAb 206- or PBS-treated mice. Standard errorsof the means are shown on all graphs.
144 VLECK ET AL. J. VIROL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 30
Dec
embe
r 20
21 b
y 41
.157
.41.
148.
Xenografts that did not become infected from either of theanti-gH MAb 206-treated groups had similar levels of DNA.This was likely to be residual VZV DNA from the inoculum,and these data were not included in the average copy numberfor each group.
The mean genome copy number in the infected xenograftsfrom mice treated with MAb 206 at 6 hpi (Ab-0-12 group)followed the same trend as virus titer. The number of genomecopies increased 40-fold from 7 dpi to 42 dpi (Fig. 2C). Meangenome copy numbers were significantly lower at 14 to 42 dpithan those in infected xenografts from the PBS group (P �0.05; Student’s t test). Similar to the Ab-0-12 group, treatmentof mice with MAb 206 starting at 4 dpi (Ab-4-12 group) sig-nificantly reduced the mean genome copy number at each timepoint, except at 14 dpi, compared to that for the PBS group(P � 0.01; Student’s t test). There was a 501-fold increase from7 dpi to 42 dpi (Fig. 2C). The significantly lower numbers ofgenome copies in both MAb 206 treatment groups than in thePBS group were highly likely to be caused by the reduced virusspread, as determined by virus titer.
Kinetics of anti-gH MAb 206 accumulation and clearance.As expected, the PBS group had extremely low levels of serumIgG1 throughout the duration of the 42-day experiment. SCIDmice are known to spontaneously develop partial immune re-activity as they age, resulting in low levels of serum IgG inapproximately 87% of 2- to 3-month-old mice (40). The high-est concentration of IgG1 in the PBS group at 14 dpi was notsignificantly higher than those at other times points (P � 0.3;Student’s t test) (Fig. 2D). The low levels of IgG1 were con-sidered to be background.
The serum levels of IgG1 in mice administered MAb 206starting at 6 hpi (Ab-0-12 group) were significantly higher thanlevels in mock-treated mice (P � 0.02; Student’s t test), andtherefore above background, at all time points (Fig. 2D). IgG1levels in both MAb 206-treated groups peaked by 14 dpi andthen decreased over time to 42 dpi. The Ab-0-12 group had
significantly higher IgG1 levels than the Ab-4-12 group at 7, 14,and 42 dpi (P � 0.02; Student’s t test). IgG1 levels in theAb-4-12 group were significantly higher than those in the PBSgroup at 7 to 35 dpi (P � 0.01; Student’s t test). The decreasein serum antibody level correlated with the increases in viraltiter and genome copy number for both antibody-treatedgroups. The additional dose of MAb 206 for the Ab-0-12 groupat 6 hpi resulted in higher levels of serum IgG1, likely leadingto greater reductions of viral titers and genome copy numbers.
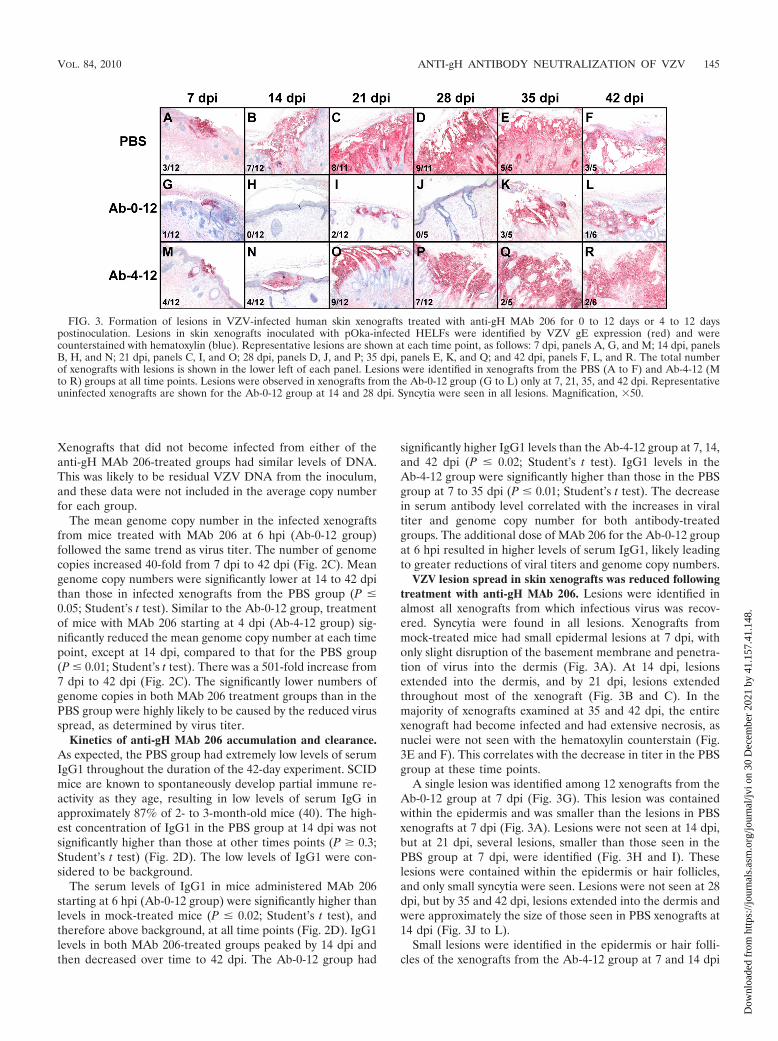
VZV lesion spread in skin xenografts was reduced followingtreatment with anti-gH MAb 206. Lesions were identified inalmost all xenografts from which infectious virus was recov-ered. Syncytia were found in all lesions. Xenografts frommock-treated mice had small epidermal lesions at 7 dpi, withonly slight disruption of the basement membrane and penetra-tion of virus into the dermis (Fig. 3A). At 14 dpi, lesionsextended into the dermis, and by 21 dpi, lesions extendedthroughout most of the xenograft (Fig. 3B and C). In themajority of xenografts examined at 35 and 42 dpi, the entirexenograft had become infected and had extensive necrosis, asnuclei were not seen with the hematoxylin counterstain (Fig.3E and F). This correlates with the decrease in titer in the PBSgroup at these time points.
A single lesion was identified among 12 xenografts from theAb-0-12 group at 7 dpi (Fig. 3G). This lesion was containedwithin the epidermis and was smaller than the lesions in PBSxenografts at 7 dpi (Fig. 3A). Lesions were not seen at 14 dpi,but at 21 dpi, several lesions, smaller than those seen in thePBS group at 7 dpi, were identified (Fig. 3H and I). Theselesions were contained within the epidermis or hair follicles,and only small syncytia were seen. Lesions were not seen at 28dpi, but by 35 and 42 dpi, lesions extended into the dermis andwere approximately the size of those seen in PBS xenografts at14 dpi (Fig. 3J to L).
Small lesions were identified in the epidermis or hair folli-cles of the xenografts from the Ab-4-12 group at 7 and 14 dpi
FIG. 3. Formation of lesions in VZV-infected human skin xenografts treated with anti-gH MAb 206 for 0 to 12 days or 4 to 12 dayspostinoculation. Lesions in skin xenografts inoculated with pOka-infected HELFs were identified by VZV gE expression (red) and werecounterstained with hematoxylin (blue). Representative lesions are shown at each time point, as follows: 7 dpi, panels A, G, and M; 14 dpi, panelsB, H, and N; 21 dpi, panels C, I, and O; 28 dpi, panels D, J, and P; 35 dpi, panels E, K, and Q; and 42 dpi, panels F, L, and R. The total numberof xenografts with lesions is shown in the lower left of each panel. Lesions were identified in xenografts from the PBS (A to F) and Ab-4-12 (Mto R) groups at all time points. Lesions were observed in xenografts from the Ab-0-12 group (G to L) only at 7, 21, 35, and 42 dpi. Representativeuninfected xenografts are shown for the Ab-0-12 group at 14 and 28 dpi. Syncytia were seen in all lesions. Magnification, �50.
VOL. 84, 2010 ANTI-gH ANTIBODY NEUTRALIZATION OF VZV 145
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 30
Dec
embe
r 20
21 b
y 41
.157
.41.
148.

(Fig. 3M and N). At 21 dpi, lesions extended into the dermis(Fig. 3O). Lesion size continued to increase until the majorityof the tissue was infected, by 35 and 42 dpi, (Fig. 3P to R).Genome-positive cells, identified by in situ hybridization todetect VZV genomic DNA, correlated with protein-positivecells in both Ab-4-12 and PBS xenografts at 14 dpi (data notshown). Overall, lesion size increased in infected xenografts inall groups over time, but the increase was delayed in bothgroups administered anti-gH MAb 206, with a more pro-nounced delay in the Ab-0-12 group.
Spread and replication of VZV were decreased in vitro aftertreatment with anti-gH MAb 206. MAb 206 was previouslyshown to neutralize cell-cell spread (8, 35, 43, 46), but itseffects on VZV titer and genome copy number have not beendetermined. To confirm the neutralizing activity of this MAb,HELFs were inoculated with pOka and cultured in the pres-ence or absence of antibody. Small plaques were seen inHELFs infected with pOka in mock-treated cultures at 24hpi and increased in size until 96 hpi, when nearly all cellswere infected (Fig. 4A to D). Treatment of HELFs with
MAb 206 (Ab-96 cells) reduced infection to single cells inthe monolayer at 24 hpi, with only a few small plaquesvisible at 48 hpi. A small increase was seen in plaque size at72 hpi, but no further increase was observed at 96 hpi (Fig.4E to H). The plaques seen at 72 and 96 hpi were not muchlarger than the plaques at 24 hpi in the mock-treated cells,and the overall increase between 24 hpi and 72 to 96 hpi wasmuch less than that seen from 24 to 48 hpi in the mock-treated cells, indicating extremely inefficient spread of thevirus in the presence of MAb 206.
In the absence of MAb 206 (mock-treated cells), the meanvirus titer peaked at 72 hpi, and the titer increased 73-foldfrom that at 24 hpi (Fig. 5A). The number of genome copiesincreased 251-fold during the 96-hour period (Fig. 5B). Virustiter and genome copy number did not increase significantlyover the first 48 h in HELFs continuously treated with MAb206 (Ab-96 cells) but increased significantly (P � 0.01; Stu-dent’s t test) between 48 and 72 hpi (Fig. 4A). No furtherincrease was seen between 72 and 96 hpi (Fig. 5A and B). Thepeak titer in the Ab-96 wells at 72 to 96 hpi was 12-fold higher
FIG. 4. Cell-cell spread of VZV in vitro in the presence of anti-gH MAb 206. HELFs were mock treated (A to D) or treated with antibody (Eto H) and then fixed and stained with polyclonal sera against VZV at 24-h intervals. (A and E) 24 hpi; (B and F) 48 hpi; (C and G) 72 hpi; (Dand H) 96 hpi. Magnification, �50.
FIG. 5. VZV replication in vitro in the presence of anti-gH MAb 206. HELFs were inoculated with pOka and grown in medium withoutantibody (mock) or medium supplemented with MAb 206 for 24, 48, or 96 h (Ab-24, Ab-48, or Ab-96, respectively). Cells were harvested at 24-hintervals for 96 h. (A) Mean titer in HELF cultures. (B) Mean genome copy number based on quantitative PCR for ORFs 31 (gB), 62 (IE62), and63 (IE63) per ng human DNA. Each gene target was measured in duplicate, and the mean of each was used to determine the number of genomecopies. Standard errors of the means are shown for both graphs.
146 VLECK ET AL. J. VIROL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 30
Dec
embe
r 20
21 b
y 41
.157
.41.
148.

than that at 24 hpi. At all time points, titers in Ab-96 wells weresignificantly lower than the titers in mock-treated wells (P �0.01; Student’s t test), and genome copy numbers remainedstatistically equivalent to those in the mock-treated cells at24 hpi. The slight increases in titer and genome copy numberoccurred at the same time as the small increases in plaque size.
Virus titers were significantly lower than those in mock-treated HELFs at all time points (P � 0.02; Student’s t test) forcells treated with MAb 206 for only 24 h (Ab-24) or 48 h(Ab-48) following inoculation (Fig. 5A). The genome copynumber in Ab-24 wells was significantly lower than that formock-treated cells at 48 hpi (P � 0.01; Student’s t test) (Fig.5B). The virus titer increased 440-fold and genome copy num-ber increased 122-fold over the 96 h in HELFs treated for 24 h.At 48 and 72 hpi, the titer was statistically equivalent to that inmock-treated cells at 24 hpi and 48 hpi (P � 0.13; Student’s ttest), but by 96 hpi, it was significantly lower than that inmock-treated cells at 72 hpi (P � 0.04; Student’s t test). Asimilar trend was seen for HELFs treated with MAb 206 for 48hpi. Virus titers increased 199-fold and genome copy numberincreased 89-fold between 24 and 96 hpi. The virus titer at 96hpi was statistically equivalent to that in mock-treated cells at48 hpi, and the numbers of genome copies were statisticallyequivalent at 48 to 96 hpi to the mock values 24 h prior. The invitro data reflected what was seen in vivo, with neutralizingactivity against pOka causing decreased virus spread, titers,and genome copies, but this effect persisted only while anti-body was present.
Anti-gH MAb 206 localized to intracellular vacuoles, virusparticles on the cell surface, and virus particles within infectedcells. Normal nucleocapsids and enveloped virus particles werefound on mock-treated HELFs infected with pOka (Fig. 6A).Some virus particles were observed in aggregates on the sur-
faces of cells. Nucleocapsids and enveloped virus particleswere also identified in HELFs infected with pOka and treatedwith MAb 206 and were identical to those from mock-treatedcells (Fig. 6B). Thus, MAb 206 did not appear to disrupt virionassembly.
Based on immunogold EM analysis, the anti-gH MAb wasenriched in numerous endosome-like structures within treatedcells (Fig. 6C and D). Gold particles within endosomes wereidentified more frequently within infected cells than in neigh-boring uninfected cells, where only one or two gold particlesper cell were found, compared to much more dense labeling ininfected cells (Fig. 6D). The limited detection of MAb 206 inuninfected but treated HELFs might have been due to passiveuptake of the anti-gH antibody from the medium or to minimalbackground labeling with unbound gold particles. The in-creased uptake within infected cells suggested that MAb 206bound to cell surface gH and was internalized. MAb 206 lo-calized to the surfaces of infected cells (Fig. 6E) and to virusparticles (Fig. 6E to G). These antibody-virus complexes wereobserved on the cell surface and within the cell near the plasmamembrane, indicative of endocytosis.
MAb 206 was not trafficked to the TGN but colocalized withthe endocytic pathway. gH localized to the cell surfaces ofinfected HELFs and to the TGN at 48 hpi, as determined byconfocal microscopy (Fig. 7A and C). The TGN is thought tobe the site of virus particle secondary envelopment (20). gHoften colocalized with gE both in the TGN and on the surfacesof infected HELFs (Fig. 7A). gH also colocalized with a majorcapsid protein, ORF23 (Fig. 7C). It has previously been dem-onstrated that ORF23 is found predominantly in the nuclei ofinfected cells (7), but ORF23 staining on the surfaces of cellsor near nuclear membranes has been associated with incomingvirions clustered on the plasma membrane or capsids along the
FIG. 6. Virus particle formation and localization of MAb 206 in infected cells in vitro. HELFs inoculated with pOka were mock treated (A) ortreated with antibody (B to G) and were fixed at 48 hpi. (A and B) Transmission EM of infected and untreated (A) or treated (B) cells. Arrowheadsand right insets show virion particles on the cell surface. Arrows and left insets show nucleocapsids. (C to F) Immunogold labeling of MAb 206in treated cells. Arrows indicate MAb 206 on the cell surface or within vacuoles. Arrowheads indicate MAb 206 on virus particles. (D) Infectedcell (top) with MAb 206 labeling next to uninfected cell (bottom) lacking dense MAb 206 labeling. Magnification, �10,000 (A and B). Bars, 0.2 �m.
VOL. 84, 2010 ANTI-gH ANTIBODY NEUTRALIZATION OF VZV 147
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 30
Dec
embe
r 20
21 b
y 41
.157
.41.
148.

nuclear envelope (45). Therefore, in this study, ORF23 stain-ing that did not colocalize with the nuclear marker but didcolocalize with gH was identified as representing envelopedvirions clustered on the surfaces of infected cells, in agreementwith the small aggregates of virion particles or individual viri-ons lining the surfaces of infected cells, as seen by electronmicroscopy.
Similar to the gH localization by confocal microscopy,MAb 206 localized to the cell surface and colocalized withgE and ORF23 on infected cell surfaces but did not localizeto the TGN (Fig. 7B and D). Colocalization between ORF23and gH or MAb 206 on the cell surface was confirmed innonpermeabilized cells (data not shown). The similaritiesbetween gH and MAb 206 localization demonstrated thatMAb 206 bound to gH on the surfaces of infected cells andon virions, but the lack of colocalization between MAb 206and the TGN was markedly different from the localization ofgH within the TGN. In treated uninfected cells, minimalMAb 206 was seen, mostly localizing to the surfaces of cells(Fig. 7B and D).
gH and MAb 206 often had a punctate distribution patternwithin the cytoplasm of infected cells. Some, but not all, gH
and MAb 206 colocalized with the early endosome markerEEA1 (Fig. 8A and B, arrowheads). This indicated that vesi-cles labeled with MAb 206 by immunogold EM were likely tobe endosomes. gH and MAb 206 also colocalized with Vps4, amarker for the multivesicular body (MVB) pathway, in some-what diffuse patches or punctae within cells (Fig. 8C and D,arrowheads). The colocalization with EEA1 and Vps4 indi-cated that the endocytic and MVB pathways were involved intrafficking of both gH and MAb 206-bound gH, albeit to dif-ferent destinations, as the MAb 206-gH complex did not reachthe TGN, while gH did.
DISCUSSION
VZV gH is predicted to play an essential role in VZVvirulence and is a known target of the humoral immune systemduring infection (1). The present study of the SCIDhu mousemodel showed for the first time that gH contributes to VZVinfection and cell-cell spread of virus in skin xenografts in vivoand confirmed this contribution in vitro. The low levels ofpersistent VZV replication and spread observed in some VZV-infected xenografts in vivo and in HELF cells in vitro in the
FIG. 7. Localization of gH and MAb 206 relative to VZV gE and ORF23 proteins in fibroblasts in vitro. HELFs inoculated with pOka weremock treated (A and C) or antibody treated (B and D) for 48 h, fixed, permeabilized, stained, and examined by confocal microscopy. UninfectedHELFs were also mock treated (A and C) or antibody treated (B and D) for 48 h, fixed, permeabilized, stained, and examined. (A) gH, red; gE,blue; TGN46, green; nuclei, gold. (B) MAb 206, red; gE, blue; TGN46, green; nuclei, gold. (C) gH, red; ORF23, blue; TGN46, green; nuclei, gold.(D) MAb 206, red; ORF23, blue; TGN46, green; nuclei, gold. Bar, 5 �m.
148 VLECK ET AL. J. VIROL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 30
Dec
embe
r 20
21 b
y 41
.157
.41.
148.

presence of anti-gH antibody indicate that passive antibodyinterference with gH functions has some limitations. This re-sidual spread might have resulted from incomplete binding ofantibody to all functional gH. Alternatively, other VZV glyco-proteins might be capable of mediating some virion-cell orcell-cell fusion in the absence of functional gH. It has beensuggested that VZV gH and gL or gB and gE might inducefusion (12, 30), although if so, VZV would be the only herpes-virus investigated to date that does not require both gB and gHfor this event.
This report also demonstrated for the first time that theadministration of anti-gH antibody was effective at preventingor reducing VZV pathogenicity in human skin. Since SCIDhuanimals are immunodeficient (4), this inhibitory effect could beassessed in the absence of a polyclonal B-cell response tomultiple viral proteins and without VZV-specific cell-mediatedimmunity. The prevention of infection might be attributed to ablock of virus attachment and entry into cells. The reducedpathogenicity in xenografts that became infected might be at-tributed to the block in spread of virus from cell to cell, inhi-bition of canonical gH trafficking, and potential virus particledegradation. MAb 206 binds to a conformation-dependent
epitope on mature glycosylated gH, and neutralization of VZVis complement independent (35). Two potential mechanismsfor MAb 206 neutralization of VZV are inhibition of receptorbinding and attachment and inhibition of fusion. The antibodymight also disrupt postentry events, such as interactions be-tween virus proteins and trafficking of proteins or virion par-ticles (Fig. 9).
Neutralizing antibodies to herpesviruses block attachmentby physically blocking interaction and preventing binding ofvirus proteins to cellular receptors (38, 41, 57). A VZV gHreceptor has not been identified, and the cell-associated natureof VZV prevents study of VZV attachment steps. If gH inter-action with a cellular protein is required for attachment, thena neutralizing antibody could disrupt binding and prevent in-fection (Fig. 9, step 1).
Epstein-Barr virus, human cytomegalovirus, and human her-pesvirus 6 and 7 gH/gL form a complex with additional glyco-proteins. Some of these complexes appear to determine cell-specific infectivity, receptor specificity, or the route of virusentry into the cell (36, 48, 53, 54). No interactions with cellularproteins have been identified for VZV gH, but antibody bind-ing to gH could disrupt or prevent formation of a protein
FIG. 8. Localization of gH and MAb 206 relative to EEA1 and Vps4 in fibroblasts in vitro. HELFs inoculated with pOka were mock treated(A to D, K to N) or antibody treated (F to I, P to S) for 48 h, fixed, permeabilized, stained, and examined by confocal microscopy. UninfectedHELFs were mock treated (E and O) or antibody treated (J and T) for 48 h, fixed, permeabilized, stained, and examined. Arrowheads highlightcolocalization. (A) gH, red; EEA1, blue; TGN46, green; nuclei, gold. (B) MAb 206, red; EEA1, blue; TGN46, green; nuclei, gold. (C) gH, red;Vps4, blue; TGN46, green; nuclei, gold. (D) MAb 206, red; Vps4, blue; TGN46, green; nuclei, gold. Bar, 5 �m.
VOL. 84, 2010 ANTI-gH ANTIBODY NEUTRALIZATION OF VZV 149
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 30
Dec
embe
r 20
21 b
y 41
.157
.41.
148.

complex required for receptor binding, thereby preventing in-fection (Fig. 9, step 1).
Neutralizing anti-gH MAb might mask functional domainsof VZV gH, preventing fusion and entry. VZV gH and gL canmediate fusion between cell membranes when expressed invitro in a vaccinia virus vector (12). Many anti-HSV gH neu-tralizing antibodies block virus penetration and prevent entry(18). HSV gH is required for hemifusion, and one MAb spe-cifically blocks this fusion step (49). Deletion or mutation ofpredicted HSV gH heptad repeats and �-helical coiled coilsdisrupts virus infectivity and cell-cell fusion (21, 23). Mimeticpeptides of the �-helices interact with lipid membranes (19,21). When the HSV �-helix 1 was replaced with the position-ally conserved �-helix from VZV, the resulting chimeric gHwas capable of promoting cell-cell fusion and rescuing infec-tivity of an HSV gH-negative virus, indicating that VZV gHcontains a functional �-helix capable of mediating fusion (22).Thus, the reduced number of infected skin xenografts andcell-cell spread during MAb 206 treatment could potentiallyhave resulted from inhibition of virus entry into cells (Fig. 9,steps 1 and 2).
Glycoprotein trafficking from the plasma membrane to theGolgi apparatus can occur via clathrin-coated vesicles (3).VZV gH endocytosis is antibody independent but clathrin de-pendent (42), and this report has demonstrated that anti-gHMAb 206 binds gH and is internalized with gH (Fig. 9, step 2).MAb 206 and gH colocalized with EEA1, a marker for earlyendosomes, and with Vps4, a marker for MVB, indicating thatMAb 206 and gH underwent endocytosis and sorting via theMVB pathway. Vps4 is required for transport of endocytosedproteins between prevacuolar endosomes and vacuoles (2).Vps4 is also required for autophagy, presumably for autophago-some-endolysosome fusion (47). Endocytosed proteins can besorted to the Golgi apparatus via the endocytic recycling path-way or late endosomes. The colocalization between gH andVps4 might occur during one of the sorting steps, as gH istargeted to the TGN. Alternatively, the association of gH andVps4 might indicate that VZV uses the MVB pathway forassembly and egress of virus particles, as has been suggestedfollowing studies of HSV gB colocalization with Vps4 duringenvelopment and egress (6). The MAb 206-gH complexes were
not targeted to the TGN. The lack of these complexes in theTGN may have resulted from the experimental system, butthey may also demonstrate that the antibody disrupts gH traf-ficking, although the complexes did travel through endosomesand the MVB pathway. VZV gH interacts with gE on theplasma membrane and in virions, and it has been suggestedthat this interaction results in the targeting of gH to the TGNfor secondary envelopment via the TGN-targeting motif of gE(31, 43). Binding of MAb 206 to gH particles expressed on thesurfaces of infected cells does not disrupt endocytosis of gH,but the resulting MAb-gH complex might not interact withother virion proteins, resulting in the altered trafficking of gHto sites other than the TGN (Fig. 9, step 3). These complexescolocalize with Vps4, similar to gH, but rather than sorting tothe TGN, they could be sorted through late endosomes tolysosomes or autophagosomes, which are induced during VZVinfection (50).
MAb 206 might also direct gH complexes to be degraded byinteracting with Fc receptors on the surfaces of infected cells.Antigen-antibody complexes bound to Fc receptors on humanfibroblasts undergo endocytosis and are targeted for degrada-tion, and these fibroblast Fc receptors mainly interact withmonomeric immunoglobulins, especially IgG1 (9, 17). Skinkeratinocytes have also been shown to express functional Fcreceptors that interact with IgG (51). Antibodies against pseu-dorabies virus gD and gB expressed on monocytes induce an-tibody-dependent, clathrin-dependent endocytosis of the gly-coproteins, mediated by tyrosine-based endocytosis motifs (13,14, 52). VZV gH contains a functional tyrosine-based endocy-tosis motif (42). Thus, MAb 206 would not have to disrupt gHprotein interactions, but instead could interact with cellular Fcreceptors in order to alter trafficking of gH and target it fordegradation via the Vps4/MVB pathway and sorting to eitherlysosomes or autophagosomes (Fig. 9, step 3).
Immunogold EM analysis of MAb 206 localization withintreated infected cells demonstrated that MAb 206 bound to gHon virus particles on the cell surface, which resulted in neu-tralization of these particles and prevention of virus spreadfrom cell to cell (Fig. 9, step 4). The MAb 206-labeled virionswere found within cells, suggesting that labeled particles on thesurfaces of infected cells were internalized. A MAb 206 inter-
FIG. 9. Schematic of mechanisms for antibody disruption of gH function and trafficking and potential effects on the pathogenesis of VZV skininfection in vivo. (1) Anti-gH antibody present soon after inoculation can bind and neutralize virus particles on the inoculum cells, preventing VZVattachment or entry into skin epidermal cells. (2) Antibody present after infection has been established can bind gH expressed on the infected cellsurface, preventing cell-cell fusion. (3) These antibody-gH complexes are internalized, and antibody-bound gH is not targeted for secondaryenvelopment and incorporation into virions, but instead is targeted for degradation. (4) Antibody binds to gH on the envelopes of surface virusparticles, blocking envelope fusion with neighboring cell membranes and inhibiting cell-cell spread of the virus. (5) Antibody-virion complexes areinternalized and targeted for degradation.
150 VLECK ET AL. J. VIROL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 30
Dec
embe
r 20
21 b
y 41
.157
.41.
148.

action with Fc receptors could direct the internalization anddegradation not only of gH but also of virions containing gH intheir envelope (Fig. 9, step 5).
Interferon (IFN) production is activated in human epider-mal cells in VZV-infected skin xenografts and modulates theprogression of lesion formation (27). The experiments re-ported here with anti-gH antibody showed that infection wassuppressed during treatment but progressed to complete de-struction of the skin tissue when anti-gH antibody was clearedfrom the circulation. Together, these findings indicate that thecombination of the innate IFN response of epidermal cells andpassively administered antibody is not sufficient to eliminateVZV when replication has been initiated in skin. This suggeststhat an effective cell-mediated immune response to VZV islikely to be necessary to resolve primary VZV infection. Her-pes zoster as a result of VZV reactivation has been suggestedto result in part from a decreased cellular immune response aspatients age, even though the humoral immune response re-mains high (1). The observations presented here suggest thatpreexisting anti-gH antibodies might reinforce the innate IFNresponse and slow the progression of cell-cell spread for aninterval that allows for clonal expansion of memory VZV-specific T cells that make IFN-� and stimulate B cells andcytotoxic T lymphocytes to respond to VZV reactivation.
Anti-gH antibody administration to SCIDhu mice withpOka-infected skin xenografts prevented infection or reducedpathogenicity if infection had been established. This mirrorsthe prophylaxis of immunocompromised patients, who are ad-ministered varicella-zoster immunoglobulin (VZIG) as soon aspossible after exposure, up to 96 h postexposure to the virus(32). Neutralization assays demonstrated that humanized MAb206 had a biological activity that was 2,400-fold that of thestandard VZIG preparation (10). Administration of VZIGdoes not consistently prevent varicella following exposure butcan ameliorate the severity of the disease, although severedisease and death can still occur. The data presented heredemonstrated that administration of anti-gH antibody imme-diately after inoculation prevented infection, but delayingtreatment by 4 days resulted only in suppression of infection,emphasizing the need to give prophylaxis to exposed patientsas soon after exposure as possible. This has potential clinicalrelevance because maintaining the supply of VZIG has beenchallenging, and an anti-gH MAb might be developed as analternative for prophylaxis of patients at risk of severe primaryVZV infection.
ACKNOWLEDGMENTS
This work was supported by training grants from the National In-stitutes of Health (R01 AI 020459 and P01CA49605). S. Vleck re-ceived support from grants T32 GM007279 and T32 AI07328.
We thank Nafisa Ghori, Department for Microbiology & Immunol-ogy, Stanford University, for assistance with transmission electron mi-croscopy and Barbara Berarducci for technical help and scientific dis-cussion.
REFERENCES
1. Arvin, A. M. 2001. Varicella-zoster virus, p. 2731–2768. In D. M. Knipe et al.(ed.), Fields virology. Lippincott-Williams & Wilkins, Philadelphia, PA.
2. Babst, M., T. K. Sato, L. M. Banta, and S. D. Emr. 1997. Endosomaltransport function in yeast requires a novel AAA-type ATPase, Vps4p.EMBO J. 16:1820–1831.
3. Bos, C. R., S. L. Shank, and M. D. Snider. 1995. Role of clathrin-coated
vesicles in glycoprotein transport from the cell surface to the Golgi complex.J. Biol. Chem. 270:665–671.
4. Bosma, G. C., R. P. Custer, and M. J. Bosma. 1983. A severe combinedimmunodeficiency mutation in the mouse. Nature 301:527–530.
5. Browne, H., V. Baxter, and T. Minson. 1993. Analysis of protective immuneresponses to the glycoprotein H-glycoprotein L complex of herpes simplexvirus type 1. J. Gen. Virol. 74:2813–2817.
6. Calistri, A., P. Sette, C. Salata, E. Cancellotti, C. Forghieri, A. Comin, H.Gottlinger, G. Campadelli-Fiume, G. Palu, and C. Parolin. 2007. Intracel-lular trafficking and maturation of herpes simplex virus type 1 gB and virusegress require functional biogenesis of multivesicular bodies. J. Virol. 81:11468–11478.
7. Chaudhuri, V., M. Sommer, J. Rajamani, L. Zerboni, and A. M. Arvin. 2008.Functions of varicella-zoster virus ORF23 capsid protein in viral replicationand the pathogenesis of skin infection. J. Virol. 82:10231–10246.
8. Cole, N. L., and C. Grose. 2003. Membrane fusion mediated by herpesvirusglycoproteins: the paradigm of varicella-zoster virus. Rev. Med. Virol. 13:207–222.
9. Danilova, T. A., L. M. Bartova, R. L. Panurina, and I. M. Lyampert. 1981.Studies of Fc receptors of heart valve and joint fibroblasts. Clin. Exp. Im-munol. 46:575–580.
10. Drew, P. D., M. T. Moss, T. J. Pasieka, C. Grose, W. J. Harris, and A. J.Porter. 2001. Multimeric humanized varicella-zoster virus antibody frag-ments to gH neutralize virus while monomeric fragments do not. J. Gen.Virol. 82:1959–1963.
11. Duus, K. M., and C. Grose. 1996. Multiple regulatory effects of varicella-zoster virus (VZV) gL on trafficking patterns and fusogenic properties ofVZV gH. J. Virol. 70:8961–8971.
12. Duus, K. M., C. Hatfield, and C. Grose. 1995. Cell surface expression andfusion by the varicella-zoster virus gH:gL glycoprotein complex: analysis bylaser scanning confocal microscopy. Virology 210:429–440.
13. Favoreel, H. W., G. Van Minnebruggen, H. J. Nauwynck, L. W. Enquist, andM. B. Pensaert. 2002. A tyrosine-based motif in the cytoplasmic tail ofpseudorabies virus glycoprotein B is important for both antibody-inducedinternalization of viral glycoproteins and efficient cell-to-cell spread. J. Virol.76:6845–6851.
14. Ficinska, J., G. Van Minnebruggen, H. J. Nauwynck, K. Bienkowska-Szewc-zyk, and H. W. Favoreel. 2005. Pseudorabies virus glycoprotein gD containsa functional endocytosis motif that acts in concert with an endocytosis motifin gB to drive internalization of antibody-antigen complexes from the surfaceof infected monocytes. J. Virol. 79:7248–7254.
15. Forrester, A., H. Farrell, G. Wilkinson, J. Kaye, N. Davis-Poynter, and T.Minson. 1992. Construction and properties of a mutant of herpes simplexvirus type 1 with glycoprotein H coding sequences deleted. J. Virol. 66:341–348.
16. Forrester, A. J., V. Sullivan, A. Simmons, B. A. Blacklaws, G. L. Smith, A. A.Nash, and A. C. Minson. 1991. Induction of protective immunity with anti-body to herpes simplex virus type 1 glycoprotein H (gH) and analysis of theimmune response to gH expressed in recombinant vaccinia virus. J. Gen.Virol. 72:369–375.
17. Frey, J., M. Janes, W. Engelhardt, E. G. Afting, C. Geerds, and B. Moller.1986. Fc gamma-receptor-mediated changes in the plasma membrane po-tential induce prostaglandin release from human fibroblasts. Eur. J. Bio-chem. 158:85–89.
18. Fuller, A. O., R. E. Santos, and P. G. Spear. 1989. Neutralizing antibodiesspecific for glycoprotein H of herpes simplex virus permit viral attachment tocells but prevent penetration. J. Virol. 63:3435–3443.
19. Galdiero, S., A. Falanga, M. Vitiello, H. Browne, C. Pedone, and M.Galdiero. 2005. Fusogenic domains in herpes simplex virus type 1 glycopro-tein H. J. Biol. Chem. 280:28632–28643.
20. Gershon, A. A., D. L. Sherman, Z. Zhu, C. A. Gabel, R. T. Ambron, andM. D. Gershon. 1994. Intracellular transport of newly synthesized varicella-zoster virus: final envelopment in the trans-Golgi network. J. Virol. 68:6372–6390.
21. Gianni, T., R. Fato, C. Bergamini, G. Lenaz, and G. Campadelli-Fiume.2006. Hydrophobic �-helices 1 and 2 of herpes simplex virus gH interact withlipids, and their mimetic peptides enhance virus infection and fusion. J. Vi-rol. 80:8190–8198.
22. Gianni, T., P. L. Martelli, R. Casadio, and G. Campadelli-Fiume. 2005. Theectodomain of herpes simplex virus glycoprotein H contains a membranealpha-helix with attributes of an internal fusion peptide, positionally con-served in the Herpesviridae family. J. Virol. 79:2931–2940.
23. Gianni, T., A. Piccoli, C. Bertucci, and G. Campadelli-Fiume. 2006. Heptadrepeat 2 in herpes simplex virus 1 gH interacts with heptad repeat 1 and iscritical for virus entry and fusion. J. Virol. 80:2216–2224.
24. Haddad, R. S., and L. M. Hutt-Fletcher. 1989. Depletion of glycoproteingp85 from virosomes made with Epstein-Barr virus proteins abolishes theirability to fuse with virus receptor-bearing cells. J. Virol. 63:4998–5005.
25. Ito, H., M. H. Sommer, L. Zerboni, A. Baiker, B. Sato, R. Liang, J. Hay, W.Ruyechan, and A. M. Arvin. 2005. Role of the varicella-zoster virus geneproduct encoded by open reading frame 35 in viral replication in vitro and indifferentiated human skin and T cells in vivo. J. Virol. 79:4819–4827.
VOL. 84, 2010 ANTI-gH ANTIBODY NEUTRALIZATION OF VZV 151
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 30
Dec
embe
r 20
21 b
y 41
.157
.41.
148.

26. Klupp, B. G., R. Nixdorf, and T. C. Mettenleiter. 2000. Pseudorabies virusglycoprotein M inhibits membrane fusion. J. Virol. 74:6760–6768.
27. Ku, C. C., L. Zerboni, H. Ito, B. S. Graham, M. Wallace, and A. M. Arvin.2004. Varicella-zoster virus transfer to skin by T cells and modulation of viralreplication by epidermal cell interferon-alpha. J. Exp. Med. 200:917–925.
28. Kutinova, L., P. Hainz, V. Ludvikova, L. Maresova, and S. Nemeckova. 2001.Immune response to vaccinia virus recombinants expressing glycoproteinsgE, gB, gH, and gL of varicella-zoster virus. Virology 280:211–220.
29. Maresova, L., L. Kutinova, V. Ludvikova, R. Zak, M. Mares, and S. Nem-eckova. 2000. Characterization of interaction of gH and gL glycoproteins ofvaricella-zoster virus: their processing and trafficking. J. Gen. Virol. 81:1545–1552.
30. Maresova, L., T. J. Pasieka, and C. Grose. 2001. Varicella-zoster virus gBand gE coexpression, but not gB or gE alone, leads to abundant fusion andsyncytium formation equivalent to those from gH and gL coexpression.J. Virol. 75:9483–9492.
31. Maresova, L., T. J. Pasieka, E. Homan, E. Gerday, and C. Grose. 2005.Incorporation of three endocytosed varicella-zoster virus glycoproteins, gE,gH, and gB, into the virion envelope. J. Virol. 79:997–1007.
32. Marin, M., D. Guris, S. S. Chaves, S. Schmid, and J. F. Seward. 2007.Prevention of varicella: recommendations of the Advisory Committee onImmunization Practices (ACIP). MMWR Recomm. Rep. 56:1–40.
33. Miller, N., and L. M. Hutt-Fletcher. 1988. A monoclonal antibody to glyco-protein gp85 inhibits fusion but not attachment of Epstein-Barr virus. J. Vi-rol. 62:2366–2372.
34. Moffat, J. F., M. D. Stein, H. Kaneshima, and A. M. Arvin. 1995. Tropism ofvaricella-zoster virus for human CD4� and CD8� T lymphocytes and epi-dermal cells in SCID-hu mice. J. Virol. 69:5236–5242.
35. Montalvo, E. A., and C. Grose. 1986. Neutralization epitope of varicellazoster virus on native viral glycoprotein gp118 (VZV glycoprotein gpIII).Virology 149:230–241.
36. Mori, Y., P. Akkapaiboon, S. Yonemoto, M. Koike, M. Takemoto, T. Sad-aoka, Y. Sasamoto, S. Konishi, Y. Uchiyama, and K. Yamanishi. 2004.Discovery of a second form of tripartite complex containing gH-gL of humanherpesvirus 6 and observations on CD46. J. Virol. 78:4609–4616.
37. Nemeckova, S., V. Ludvikova, L. Maresova, J. Krystofova, P. Hainz, and L.Kutinova. 1996. Induction of varicella-zoster virus-neutralizing antibodies inmice by co-infection with recombinant vaccinia viruses expressing the gH orgL gene. J. Gen. Virol. 77:211–215.
38. Nicola, A. V., M. Ponce de Leon, R. Xu, W. Hou, J. C. Whitbeck, C. Krum-menacher, R. I. Montgomery, P. G. Spear, R. J. Eisenberg, and G. H. Cohen.1998. Monoclonal antibodies to distinct sites on herpes simplex virus (HSV)glycoprotein D block HSV binding to HVEM. J. Virol. 72:3595–3601.
39. Niizuma, T., L. Zerboni, M. H. Sommer, H. Ito, S. Hinchliffe, and A. M.Arvin. 2003. Construction of varicella-zoster virus recombinants from parentOka cosmids and demonstration that ORF65 protein is dispensable forinfection of human skin and T cells in the SCID-hu mouse model. J. Virol.77:6062–6065.
40. Nonoyama, S., F. O. Smith, I. D. Bernstein, and H. D. Ochs. 1993. Strain-dependent leakiness of mice with severe combined immune deficiency. J. Im-munol. 150:3817–3824.
41. Ober, B. T., B. Teufel, K. H. Wiesmuller, G. Jung, E. Pfaff, A. Saalmuller,and H. J. Rziha. 2000. The porcine humoral immune response againstpseudorabies virus specifically targets attachment sites on glycoprotein gC.J. Virol. 74:1752–1760.
42. Pasieka, T. J., L. Maresova, and C. Grose. 2003. A functional YNKI motif inthe short cytoplasmic tail of varicella-zoster virus glycoprotein gH mediates
clathrin-dependent and antibody-independent endocytosis. J. Virol.77:4191–4204.
43. Pasieka, T. J., L. Maresova, K. Shiraki, and C. Grose. 2004. Regulation ofvaricella-zoster virus-induced cell-to-cell fusion by the endocytosis-compe-tent glycoproteins gH and gE. J. Virol. 78:2884–2896.
44. Peng, T., M. Ponce-de-Leon, H. Jiang, G. Dubin, J. M. Lubinski, R. J.Eisenberg, and G. H. Cohen. 1998. The gH-gL complex of herpes simplexvirus (HSV) stimulates neutralizing antibody and protects mice against HSVtype 1 challenge. J. Virol. 72:65–72.
45. Reichelt, M., J. Brady, and A. M. Arvin. 2009. The replication cycle ofvaricella-zoster virus: analysis of the kinetics of viral protein expression,genome synthesis, and virion assembly at the single-cell level. J. Virol. 83:3904–3918.
46. Rodriguez, J. E., T. Moninger, and C. Grose. 1993. Entry and egress ofvaricella virus blocked by same anti-gH monoclonal antibody. Virology 196:840–844.
47. Rusten, T. E., T. Vaccari, K. Lindmo, L. M. Rodahl, I. P. Nezis, C. Sem-Jacobsen, F. Wendler, J. P. Vincent, A. Brech, D. Bilder, and H. Stenmark.2007. ESCRTs and Fab1 regulate distinct steps of autophagy. Curr. Biol.17:1817–1825.
48. Sadaoka, T., K. Yamanishi, and Y. Mori. 2006. Human herpesvirus 7 U47gene products are glycoproteins expressed in virions and associate withglycoprotein H. J. Gen. Virol. 87:501–508.
49. Subramanian, R. P., and R. J. Geraghty. 2007. Herpes simplex virus type 1mediates fusion through a hemifusion intermediate by sequential activity ofglycoproteins D, H, L, and B. Proc. Natl. Acad. Sci. USA 104:2903–2908.
50. Takahashi, M. N., W. Jackson, D. T. Laird, T. D. Culp, C. Grose, J. I. HaynesII, and L. Benetti. 2009. Varicella-zoster virus infection induces autophagy inboth cultured cells and human skin vesicles. J. Virol. 83:5466–5476.
51. Tigalonowa, M., J. R. Bjerke, and R. Matre. 1991. Fc gamma-receptors onLangerhans’ cells and keratinocytes in suspension from normal skin charac-terized using soluble immune complexes and monoclonal antibodies. ActaDermatol. Venereol. 71:99–103.
52. Van de Walle, G. R., H. W. Favoreel, H. J. Nauwynck, P. Van Oostveldt, andM. B. Pensaert. 2001. Involvement of cellular cytoskeleton components inantibody-induced internalization of viral glycoproteins in pseudorabies virus-infected monocytes. Virology 288:129–138.
53. Wang, D., and T. Shenk. 2005. Human cytomegalovirus virion protein com-plex required for epithelial and endothelial cell tropism. Proc. Natl. Acad.Sci. USA 102:18153–18158.
54. Wang, X., W. J. Kenyon, Q. Li, J. Mullberg, and L. M. Hutt-Fletcher. 1998.Epstein-Barr virus uses different complexes of glycoproteins gH and gL toinfect B lymphocytes and epithelial cells. J. Virol. 72:5552–5558.
55. Weigle, K. A., and C. Grose. 1983. Common expression of varicella-zosterviral glycoprotein antigens in vitro and in chickenpox and zoster vesicles.J. Infect. Dis. 148:630–638.
56. Westra, D. F., G. M. Verjans, A. D. Osterhaus, A. van Kooij, G. W. Welling,A. J. Scheffer, T. H. The, and S. Welling-Wester. 2000. Natural infection withherpes simplex virus type 1 (HSV-1) induces humoral and T cell responses tothe HSV-1 glycoprotein H:L complex. J. Gen. Virol. 81:2011–2015.
57. Whitbeck, J. C., M. I. Muggeridge, A. H. Rux, W. Hou, C. Krummenacher,H. Lou, A. van Geelen, R. J. Eisenberg, and G. H. Cohen. 1999. The majorneutralizing antigenic site on herpes simplex virus glycoprotein D overlaps areceptor-binding domain. J. Virol. 73:9879–9890.
58. Zerboni, L., C. C. Ku, C. D. Jones, J. L. Zehnder, and A. M. Arvin. 2005.Varicella-zoster virus infection of human dorsal root ganglia in vivo. Proc.Natl. Acad. Sci. USA 102:6490–6495.
152 VLECK ET AL. J. VIROL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 30
Dec
embe
r 20
21 b
y 41
.157
.41.
148.