anno 7 seminari 2010 di ematologia oncologica - siesonline · sicologico e il reale potenziale...
TRANSCRIPT

NEL PROSSIMO NUMERO
LINFOMI AGGRESSIVI Meccanismi patogenetici • LNH a grandi cellule • LNH mantellare • LNH linfoblastico • LNH T periferici •
Edizioni Medico Scientifiche - Pavia
E D I Z I O N I I N T E R N A Z I O N A L I s r l
Editor in chiefGiorgio Lambertenghi Deliliers
Anno 7Numero 32010 Seminari
di EmatologiaOncologica
Leucemia linfatica cronica
ISSN 2038-2839

Vol. 7 - n. 3 - 2010
Edizioni Internazionali srlDivisione EDIMES
Edizioni Medico-Scientifiche - PaviaVia Riviera, 39 - 27100 Pavia
Tel. +39 0382 526253 r.a. - Fax +39 0382 423120E-mail: [email protected]
Editor in ChiefGiorgio Lambertenghi Deliliers
Fondazione IRCCS Ca’ GrandaOspedale Maggiore Policlinico di Milano
Editorial BoardSergio Amadori
Università degli Studi Tor Vergata, Roma
Mario BoccadoroUniversità degli Studi, Torino
Alberto BosiUniversità degli Studi, Firenze
Federico Caligaris CappioUniversità Vita e Salute, Istituto San Raffaele, Milano
Antonio CuneoUniversità degli Studi, Ferrara
Marco GobbiUniversità degli Studi, Genova
Mario PetriniUniversità degli Studi, Pisa
Giovanni PizzoloUniversità degli Studi, Verona
Giorgina SpecchiaUniversità degli Studi, Bari
Direttore ResponsabilePaolo E. Zoncada
Registrazione Trib. di Milano n. 532del 6 settembre 2007
Eziopatogenesi, diagnosi e clinica 5ANTONIO CUNEO, GIAN MATTEO RIGOLIN,
SARA MARTINELLI, LUCA FORMIGARO,
GIANLUIGI CASTOLDI
Fattori prognostici 21ACHILLE AMBROSETTI, ILARIA NICHELE,
GIOVANNI PIZZOLO
Linfocitosi B monoclonale 39LYDIA SCARFÒ, PAOLO GHIA
Sindrome di Richter 59MARCO FANGAZIO, DAVIDE ROSSI,
GIANLUCA GAIDANO
Terapie innovative 73ROBIN FOÀ, ILARIA DEL GIUDICE,
FRANCESCA R. MAURO
Leucemia linfatica cronica

2
PeriodicitàQuadrimestrale
ScopiSeminari di Ematologia Oncologica è un periodico di aggiorna-mento che nasce come servizio per i medici con l’intenzione direndere più facilmente e rapidamente disponibili in formazioni suargomenti pertinenti l’ematologia oncologica.Lo scopo della rivista è quello di as sistere il lettore fornendo-gli in maniera esaustiva:a) opinioni di esperti qualificati sui più recenti progressi in formachiara, aggiornata e concisa;
b) revisioni critiche di argomenti di grande rilevanza pertinenti gliinteressi culturali degli specialisti interessati;
NORME REDAZIONALI
1) Il testo dell’articolo deve essere editato utilizzando il programmaMicrosoft Word per Windows o Macintosh. Agli AA. è riservata la correzione ed il rinvio (entro e non oltre 5gg. dal ricevimento) delle sole prime bozze del lavoro.
2) L’Autore è tenuto ad ottenere l’autorizzazione di «Copyright»qualora riproduca nel testo tabelle, figure, microfotografie odaltro materiale iconografico già pubblicato altrove. Tale mate-riale illustrativo dovrà essere riprodotto con la dicitura «perconcessione di …» seguito dalla citazione della fonte di pro-venienza.
3) Il manoscritto dovrebbe seguire nelle linee generali la seguentetraccia:
TitoloConciso, ma informativo ed esauriente.Nome, Cognome degli AA., Istituzione di appartenenza senzaabbreviazioni.Nome, Cognome, Foto a colori, Indirizzo, Telefono, Fax, E-mail del1° Autore cui andrà indirizzata la corrispondenza.
IntroduzioneConcisa ed essenziale, comunque tale da rendere in maniera chia-ra ed esaustiva lo scopo dell’articolo.
Parole chiaveSi richiedono 3/5 parole.
Corpo dell’articoloIl contenuto non deve essere inferiore alle 30 cartelle dattiloscritte(2.000 battute cad.) compresa la bibliografia e dovrà rendere lo statodell’arte aggiornato dell’argomento trattato. L’articolo deve esserecorredato di illustrazioni/fotografie, possibilmente a colori, in file adalta risoluzione (salvati in formato .tif, .eps, .jpg). Le citazioni bibliografiche nel testo devono essere essenziali, maaggiornate (non con i nomi degli AA. ma con la numerazione cor-rispondente alle voci della bibliografia), dovranno essere numera-te con il numero arabo (1) secondo l’ordine di comparsa nel testoe comunque in numero non superiore a 100÷120.
BibliografiaPer lo stile nella stesura seguire le seguenti indicazioni o consultareil sito “International Committee of Medical Journal Editors UniformRequirements for Manuscripts Submitted to Biomedical Journals:Sample References”.
Es. 1 - Articolo standard1. Bianchi AG, Rossi EV. Immunologic effect of donor lymphocy-tes in bone marrow transplantation. N Engl J Med. 2004; 232:284-7.
Es. 2 - Articolo con più di 6 autori (dopo il 6° autore et al.)1. Bianchi AG, Rossi EV, Rose ME, Huerbin MB, Melick J, MarionDW, et al. Immunologic effect of donor lymphocytes in bone mar-row transplantation. N Engl J Med. 2004; 232: 284-7.
Es. 3 - Letter1. Bianchi AG, Rossi AV. Immunologic effect of donor lymphocytes[Letter]. N Engl J Med. 2004; 232: 284-7.
Es. 4 - Capitoli di libri1. Bianchi AG, Rossi AV. Immunologic effect of donor lymphocy-tes. In: Caplan RS, Vigna AB, editors. Immunology. Milano:MacGraw-Hill; 2002; p. 93-113.
Es. 5 - Abstract congressi (non più di 6 autori)1. Bianchi AG, Rossi AV. Immunologic effect of donor lymphocytesin bone marrow transplantation [Abstract]. Haematologica.2002; 19: (Suppl. 1): S178.
RingraziamentiRiguarda persone e/o gruppi che, pur non avendo dignità di AA.,meritano comunque di essere citati per il loro apporto alla realizza-zione dell’articolo.
Edizioni Internazionali SrlDivisione EDIMES
EDIZIONI MEDICO SCIENTIFICHE - PAVIA
Via Riviera, 39 • 27100 PaviaTel. 0382526253 r.a. • Fax 0382423120
E-mail: [email protected]
Seminari
di EmatologiaOncologica
Periodico di aggiornamento sulla clinica e terapia
delle emopatie neoplastiche

3
EditorialeEditoriale
GIORGIO LAMBERTENGHI DELILIERSFondazione IRCCS Ca’ Granda, Ospedale Maggiore Policlinico di Milano
Seminari di Ematologia Oncologica dedicaquesto ultimo numero dell’annata 2010 alla leu-cemia linfatica cronica, ritenuta la neoplasia emo-poietica più comunemente osservata nel mondooccidentale, con un’incidenza che tende adincrementarsi per l’aumento dell’aspettativamediana di vita della popolazione. Le conoscen-ze patogenetiche moderne hanno modificato ilconcetto di disordine indolente da accumulo deipiccoli linfociti, e riconosciuto nella storia natu-rale della malattia la cosiddetta linfocitosi Bmonoclonale, condizione preleucemica o in alter-nativa espressione di una stimolazione cronicaoligoclonale legata all’invecchiamento. La leuce-mia linfatica cronica viene oggi ritenuta unamalattia dinamica, legata ad una complessa seriedi eventi che si esprimono con modificazioni delprofilo antigenico, citogenetico e molecolare,facilmente identificabili con le tecnologie in usonei laboratori ematologici. Questa variabilità biologica si correla al decorsoeterogeneo tipico della malattia, con pazienti chenon richiedono per decenni alcun trattamento,
mentre altri hanno una breve spettanza di vita. Daqui l’esigenza di formulare alla diagnosi una pre-visione sul futuro, basandosi sul rilievo di marca-tori biologici, alcuni dei quali sono già entrati nel-l’uso clinico comune, mentre altri più numerosiaspettano conferme da studi in corso. La cono-scenza di questi predittori è oggi un utile com-plemento alla tradizionale stadiazione clinico-ematologica che rimane ancora fondamentale, alfine di riconoscere precocemente i pazienti chesono a rischio di rapida progressione o di trasfor-mazione in sindrome di Richter. I recenti progres-si nella comprensione dei meccanismi che gover-nano la storia naturale della leucemia linfaticacronica si sono tradotti nello sviluppo di nuoviagenti biologici, di più potenti anticorpi monoclo-nali e di strategie immunoterapiche o vaccinali.Gli studi in corso dovranno valutarne il profilo tos-sicologico e il reale potenziale curativo su unamalattia, come la leucemia linfatica cronica, noneradicabile con la terapia convenzionale e fre-quentemente caratterizzata dall’insorgenza di unacondizione di refrattarietà.


5
n INTRODUZIONE
La leucemia linfatica cronica (LLC) è un disordi-ne linfoproliferativo cronico che coinvolge i linfo-citi B CD5-positivi e che rientra tra le neoplasiea cellule B-mature della classificazione WHO (1).È più frequente nei maschi che nelle femmine (1,5-2,0/1), ed ha un’incidenza nei paesi occidentali,riferita a 100.000 abitanti, compresa tra 2-6casi/anno, mentre è rara in Giappone e nei pae-si orientali, ove l’incidenza è <1 caso/100.000 abi-tanti (2) (Figura 1a). L’età media alla diagnosi è attorno ai 70 anni, el’incidenza aumenta da 1 caso/anno/100.000 abi-tanti nella fascia 40-50 anni a 20 casi nella fascia70-80 anni. Oltre il 40% delle LLC è diagnostica-ta ad un’età >75 anni, mentre meno del 10% èdiagnosticata prima dei 50 anni (3) (Figura 1b).
n EZIOLOGIA
L’eziologia della LLC non è nota. Non può esse-re escluso un ruolo per le radiazioni ionizzanti (4),
anche se lo studio della popolazione sopravvis-suta all’incidente nucleare di Chernobyl hamostrato un aumento di incidenza di molte for-me di leucemia, ma non di LLC (5). Alcune atti-vità agricole, con particolare riguardo all’impiegodi pesticidi, si possono associare ad un aumen-to dei casi (6).Vi è evidenza di una possibile associazione coni fattori genetici. La LLC ha un’incidenza bassanelle popolazioni orientali rispetto agli occidenta-li e i gruppi etnici che migrano in altri paesi man-tengono l’incidenza di questa malattia al livello diquella del paese di provenienza. Nei parenti di primo grado di soggetti affetti daLLC il rischio di sviluppo della malattia o di altresindromi linfoproliferative (linfoma di Hodgkin enon-Hodgkin) è superiore rispetto a quello dellapopolazione generale di pari età e sesso (7, 8) esi possono rinvenire espansioni di piccoli cloniB linfocitari con il fenotipo classico della LLC enegatività per CD38, con una frequenza franca-mente maggiore rispetto alla popolazione gene-rale (9). In un’analisi di 24 famiglie con più di due mem-bri affetti si è potuto documentare, accanto alleclassiche anomalie citogenetiche, una elevatafrequenza di delezioni o guadagno di materialegenetico a livello delle bande Xp11.1-p21,Xq21-qter, 2p12-14 e 4q11-21 (10). È possibileche, diversamente dal cancro mammario, ove ungene (BRCA1) ha un importantissimo effetto, ilsubstrato genetico della LLC consista nell’inter-vento di più geni con basso potenziale predispo-nente (11).
Indirizzo per la corrispondenza
Prof. Antonio CuneoIstituto di EmatologiaAzienda Ospedale, Università S. AnnaC.so GIovecca, 203 - 44121 FerraraE-mail: [email protected]
Eziopatogenesi, Eziopatogenesi, diagnosi e clinicadiagnosi e clinicaANTONIO CUNEO, GIAN MATTEO RIGOLIN, SARA MARTINELLI, LUCA FORMIGARO, GIANLUIGI CASTOLDI
Sezione di Ematologia, Dipartimento di Scienze Biomediche e Terapie Avanzate, Università degli Studi di Ferrara
Antonio Cuneo
Parole chiave: leucemia linfatica cronica, patogenesi,lesioni citogenetico-molecolari, diagnosi, evoluzione

6 Seminari di Ematologia Oncologica
n PATOGENESI
La patogenesi della LLC riconosce numerosimomenti (Tabella 1), incentrati sulle particolaritàdella cellula d’origine, sulle sue interazioni con ipo-tetici antigeni e con il microambiente e sullo svi-luppo di una vasta gamma di lesioni genetiche.
Cellule d’origineSi ritiene che la LLC derivi dalla trasformazione diun linfocito B, esprimente l’antigene CD5, corri-spondente alla popolazione B-1, presente nella
cavità peritoneale del topo, che mostra la capaci-tà di produrre anticorpi naturali, mediante una rea-zione T-indipendente (12). L’analogo umano di que-sta sottopopolazione non è stato identificato concertezza. È stata proposta l’origine da un linfocitoB CD5+ della zona mantellare (13), o da un linfo-cito della zona marginale che esprime il CD5, nor-malmente assente in questa sede, in seguito allostato di attivazione del linfocito leucemico. I linfo-citi della LLC esprimono un BCR che presenta ana-logie strutturali con gli anticorpi che reagiscono con-tro autoantigeni ed antigeni polisaccaridici batte-rici, al pari dei linfociti della zona marginale (14). In una parte dei casi (50-80% nelle varie casisti-che) la cellula d’origine presenta >2% di muta-zioni somatiche nella sequenza del gene codifi-cante per la porzione variabile delle catenepesanti delle Ig (IGHV), un processo che fisiolo-gicamente avviene all’interno del centro germina-tivo, grazie all’intervento di enzimi quali l’activa-tion-induced cytidine deaminase (AID), in rispo-sta ad antigeni T-dipendenti. La restante parte del-le LLC presenta una configurazione germline del-la porzione variabile del gene Ig (i.e. <2% di muta-zioni). I linfociti delle LLC “mutate” e “non-muta-te” hanno molte somiglianze: a) presentano un profilo globale di espressio-
ne genica vicino a quello di una cellula B-memoria;
b) esprimono un profilo immunofenotipicoCD23+; CD25+; CD27+, tipico dei linfociti atti-vati che hanno incontrato l’antigene, con bas-sa espressione di molecole normalmentedown-regolate in seguito ad attivazione cellu-lare (CD22; CD79b; IgD).
In alcuni casi le LLC non-mutate presentano unpattern di espressione degli antigeni CD69 eCD71 coerente con una vicinanza temporale allostimolo antigenico induttivo più marcata rispet-to alle LLC “mutate” (15). Si può pertanto rite-nere che il linfocito da cui origina la LLC sia unacellula B-memoria che: 1) ha incontrato l’antigene in una reazione T-
dipendente all’interno del centro germinativonel caso delle LLC “mutate”;
2) è stata stimolata al di fuori del centro germi-nativo da uno stimolo incapace di attivare ilprocesso di ipermutazione somatica del geneIg (autoantigene, antigene polisaccaridici o
a) incidenza nei diversi paesi
Australia
USA
Irlan
daIta
lia
Svizz
era
Canad
a
Fran
cia
Austria
R. C
eca
Danimarca
Spag
na
Regn
o Uni
to
German
ia
Polonia
Svez
ia
Paes
i Bas
si
Bras
ile
Costa R
ica
Giapp
oneCin
a
Ecua
dor
6
5
4
3
2
1
0
Uomini Donne
Tass
o d
i inc
iden
za p
er 1
00.0
00
b) incidenza per età
31
0-4
5-9
10-1
4
15-1
9
20-2
4
25-2
9
30-3
4
35-3
9
40-4
4
45-4
9
50-5
4
55-5
9
60-6
4
65-6
9
70-7
4
75-7
9
80-8
485
+
35
30
25
20
15
10
5
0
25,6
21,9
18,4
14,3
9
6,3
3,71,8
0,70,30000000
Tass
o d
i inc
iden
za p
er 1
00.0
00
Classi di età (anni)
FIGURA 1 - Incidenza della LLC. (a) Tassi standardizzati per età.(b) Tassi età-specifici (2, 3).

7Eziopatogenesi, diagnosi e clinica
superantigene) nelle forme “non-mutate”.
Ruolo della stimolazione antigenica È noto che il clone trasformato nella LLC presen-ta un utilizzo preferenziale di alcune famiglie V, De J (ad esempio VH1-69; VH4-34), che non riflet-te la frequenza di questi riarrangiamenti nellapopolazione B-linfocitaria CD5 normale. Poichéqueste sequenze, assemblate durante la matu-razione B-linfocitaria intramidollare, formano laporzione variabile del gene Ig espressa in super-ficie come B-cell receptor (BCR), si può dedurreche alcuni antigeni sono in grado di ingaggiare icloni esprimenti questi BCR favorendone l’espan-sione e la successiva trasformazione. Questo concetto è stato rafforzato dalla dimo-
strazione di BCR stereotipati, che presentano unastrettissima analogia della porzione del BCR chelega l’antigene nota come complementaritydetermining region (CDR) (16-18). La probabili-tà che due linfociti B normali possano avere unBCR stereotipato è dell’ordine di 10-9-10-12,mentre è stato osservato che fino al 25 % deicasi di LLC può mostrare questo fenomeno (19).Tra gli antigeni in grado di ingaggiare il BCR nel-la LLC si annoverano gli elementi polisaccaridi-ci batterici, il fattore reumatoide, il DNA, la car-diolipina, antigeni espressi sulle cellule apopto-tiche (16). Si è così affermato in questi ultimi anniil concetto di una relazione patogenetica tra sti-molazione antigenica, spesso sostenuta daautoantigeni, e LLC (17). In effetti è stata fornitauna recente elegante dimostrazione di come unaproteina, nota come catena pesante della mio-sina non-muscolare (non muscle myosin heavychain), avente un ruolo nel movimento cellulare,possa essere esposta sulla superficie delle cel-lule apoptotiche e di come la maggior parte del-la LLC non-mutate possa riconoscere tramite isuoi anticorpi questo antigene (20). Esiste inol-tre dimostrazione che il linfocito della LLC puòmantenere la capacità di rispondere all’antigene: a) andando incontro in vivo a switch di classe Ig
(21); b) sviluppando nuove mutazioni del gene IGHV
(22); c) esprimendo l’enzima AID (23), importante nel
processo di ipermutazione somatica; d) modificando il profilo di espressione genica e
attivando il ciclo cellulare (24). È interessante notare che queste caratteristichesono più spiccate nei casi di LLC “non-mutate”CD38+ e ZAP-70+, rispetto alle altre LLC (25, 26).In effetti, nelle LLC “non-mutate”, il BCR-signa-ling è attivo, mentre nelle forme “mutate” è inat-tivo in seguito ad uno stato di anergia funziona-le legato ad una protratta stimolazione antigeni-ca, con conseguente desensibilizzazione delBCR stesso (27). Questa condizione di anergia siassocia ad uno specifico profilo di espressionedi geni coinvolti nel signaling BCR-mediato (28).
Turn-over cellulare Contrariamente a quanto ritenuto nel recente pas-sato, la LLC non può essere oggi considerata una
TABELLA 1 - Momenti patogenetici nella LLC.
Aspetti patogenetici
Cellula d’origine• Linfocito B di probabile origine dalla zona marginale
che esprime il CD5 come marker di attivazione cel-lulare
Caratteristiche della cellula d’origine• Ha incontrato l’antigene in una reazione T-dipenden-
te all’interno del centro germinativo (LLC con IGHV“mutate”) o al di fuori del centro germinativo (LLC conIGHV “non mutate”)
Stimolazione antigenica• Autoantigeni (ad es. catena pesante della miosina
non-muscolare, fattore reumatoide, DNA, cardiolipi-na, antigeni espressi sulle cellule apoptotiche)
• Antigeni polisaccaridici• Attivazione del BCR signaling (LLC “non mutate”)• Stato di anergia a seguito della cronica stimolazione
antigenica (LLC “mutate”)
Divisione cellulare• Ripetuti cicli di replicazione • Accorciamento dei telomeri (> nelle LLC “non muta-
te”) e conseguente instabilità genetica• 0,1-1% di rinnovo del clone cellulare ogni giorno, più
spiccato nella frazione CD38+
Lesioni genetiche e citogenetiche• Delezione miR-15 e miR-16• Delezione DLEU2• Delezione DLEU7• 17p-/TP53 mutato, 11q-/ATM, 14q32/IGH, +12, 6q-,
13q-

8 Seminari di Ematologia Oncologica
patologia da accumulo di linfociti che non vannoincontro ad apoptosi. I linfociti patologici manten-gono la sensibilità ad alcuni stimoli pro-apopto-tici mediati da Fas e dal legame di anticorpi antiIgM che ingaggiano il BCR (29, 30) e proliferanoin vivo ad un ritmo pari allo 0,1-1% dell’intero clo-ne ogni giorno (31). Il ritmo di divisione cellularee di rinnovo è più elevato nella frazione cellulareCD38+ (32).
Interazioni con il microambiente Nel linfonodo esiste un comparto di accumulocostituito da piccoli linfociti ed un comparto, quel-lo dei centri di proliferazione, ricco in paraimmu-noblasti e prolinfociti, ove le cellule mostrano icaratteri dell’attivazione e vanno incontro a divi-sione cellulare. Queste strutture istologiche, checonferiscono un quadro pseudofollicolare al linfo-nodo della LLC, si rinvengono anche nei tessutiinfiammati dei soggetti affetti da patologia autoim-mune (33) e richiamano il concetto del ruolo del-la stimolazione da parte di autoantigeni nella gene-si della LLC. Nei centri di proliferazione i prolinfo-citi e i paraimmunoblasti sono a stretto contattocon linfociti CD4 e cellule follicolari dendritiche. Nel comparto di accumulo i piccoli linfociti inte-ragiscono con le cellule stromali, in un contestodi interazioni cellula-cellula che ne favoriscono lasopravvivenza. In effetti, la stimolazione da par-te del CD40 ha un ruolo nel mantenimento in vitadel clone B-linfocitario, al pari dell’ interazione coni linfociti CD4, in grado di determinare la produ-zione di citochine anti-apoptotiche (IL4, IFN) (34).Nella distribuzione e sopravvivenza delle cellulepatologiche (27) giocano un ruolo importante: a) alcune chemochine e loro recettori, espressi
dal linfocito leucemico (CXCR3 e CXCR5); b) cellule del sangue periferico in grado di diffe-
renziarsi in cellule nutrici (nurselike), che favo-riscono la sopravvivenza e la migrazione dellinfocito all’interno degli spazi midollari attra-verso lo stromal-derived growth factor (35);
c) le cellule dendritiche, attraverso il CD44 e gra-zie all’induzione dell’espressione di una pro-teina BCL2 correlata (Mcl-1) (36).
L’angiogenesi può giocare un ruolo nelle fasi diaccelerazione della malattia o nei sottogruppi piùaggressivi, ove si riscontrano livelli sierici più ele-vati di VEGF (37, 38).
Lesioni citogenetico-molecolari- Geni micro Rna e TCL1 Nella LLC non è ad oggi nota la lesione geneti-ca primaria in grado di innescare il processo ditrasformazione, ma sono disponibili numeroseinformazioni su una serie di lesioni che governa-no il processo di trasformazione (Figura 2). Sono stati localizzati due geni codificanti permicroRNA (i.e miR-15 e miR-16) nella regione13q14, deleta in un 40-50% delle LLC (39).Questi geni mostrano ridotta espressione inseguito a delezione (39) e, in queste condizio-ni, può risultare alterata l’espressione di geni checontrollano la progressione del ciclo cellulare neilinfociti B (40). In effetti, la delezione di miR-15e miR-16 nel topo determina l’insorgenza diun’espansione clonale di linfociti B che presen-ta le caratteristiche biologiche della LLC. La pro-liferazione linfoide è più marcata e aggressiva sela delezione coinvolge, oltre ai suddetti geni amicro Rna, il gene DLEU2 che mappa nella stes-sa regione (40). La concomitante delezione di DLEU7, posiziona-to all’interno della regione di minima delezione,può contribuire alla patogenesi per la perdita/ridu-zione della sua fisiologica funzione di inibizionedi NF-kB (41).Nella LLC, inoltre, vi è una consistente overespres-sione del gene TCL1 che mappa a livello dellabanda 14q32.1, determinato da un meccanismodi demetilazione del promotore (42) e/o da duegeni a micro-Rna, miR-29 and miR-181 (43). Esistela documentazione che il topo transgenico per uncostrutto che contiene l’enhancer del gene Ig edil gene TCL1 sviluppa un’espansione clonale B-CD5+, che con il passare del tempo assume lecaratteristiche della LLC (44). Analogamente, iltopo transgenico che iperesprime miR-29 nei lin-fociti B, può sviluppare la LLC (45).- TelomeriI telomeri sono costituiti da sequenze ripetute diDNA che conferiscono stabilità alla struttura deicromosomi. Con l’invecchiamento della cellulaed in seguito ai cicli replicativi a cui questa vaincontro si assiste ad un accorciamento dei telo-meri, che viene normalmente limitato dall’attivi-tà delle telomerasi. Nella LLC i telomeri dei linfociti patologici sonopiù corti rispetto ai linfociti B normali di sogget-

9Eziopatogenesi, diagnosi e clinica
ti di pari età e sesso. Inoltre l’accorciamento deitelomeri è più spiccato nelle LLC “non-mutate”,nelle quali si rinviene anche una maggiore atti-vità telomerasica (46, 47), e si associa ad unaprognosi sfavorevole e ad un’aumentata proba-bilità di sviluppare la sindrome di Richter (48).Queste osservazioni indicano come la storiareplicativa delle LLC sia differente a seconda del-lo stato mutazionale dei geni Ig e come nei casipiù aggressivi vi sia stato uno stimolo prolifera-tivo nelle fasi di emergenza del clone neoplasti-
co in grado di indurre numerosi cicli di attivazio-ne e replicazione con accorciamento, disfunzio-ne e fusione dei telomeri e conseguente insor-genza di esteso danno del genoma (49).- Profilo citogenetico-molecolareL’introduzione della FISH ha permesso l’individua-zione di aberrazioni cromosomiche in circa l’80%dei casi e ogni paziente viene oggi incluso in unospecifico gruppo in base a una classificazionecitogenetica gerarchica che attribuisce importan-za decrescente alle seguenti lesioni: 17p- >11q->+12 >13q-. I risultanti gruppi citogenetici han-no una frequenza diversa a seconda dello stadiodi malattia, come riportato in Tabella 2. Recentemente, l’introduzione di una stimolazioneefficace delle mitosi mediante oligonucleotidi e IL2ha mostrato che approssimativamente il 30% del-le LLC senza difetti cromosomici mediante anali-si FISH in interfase può presentare una lesione cro-mosomica nel cariotipo. Inoltre, è stato dimostra-to con questa tecnica che cariotipi complessi pote-vano essere documentati in una significativa fra-zione dei casi in associazione con fattori progno-stici e quadro clinico sfavorevole (50). Un quadroriassuntivo del significato delle principali lesionicitogenetiche è presentato in Tabella 3.Nuove sottili aberrazioni sono state identificatemediante sensibilissime tecniche di scansione del-l’intero genoma, grazie alla quale sono state docu-mentate lesioni genetiche in virtualmente tutti i casidi LLC (51).
13q-La delezione 13q14 è la più frequente anomaliacitogenetica nella LLC e si presenta in più del50% dei casi. Questa delezione è stata descrit-
Ripetute divisionicellulariAccorciamentodei telomeri
13q-; +12, 6q-, 14q32, 11q-, 17p-
Traslocazioni cromosomiche
Autoantigeni
Stimolazione antigenica di linfociti B CD5+ con particolari BCR
Delezione DLEU7/DLEU2 Delezione miR-15a/16-1
↓ miR-29 and miR-181 ↑ TCL1 expression
Instabilità genetica
↓ inibizione NF-kB ↑ ciclo cellulare↑ fosforilazione di Rb
FIGURA 2 - Momenti patogenetici e lesioni citogenetico-mole-colari nella LLC.
TABELLA 2 - Frequenza (% di casi) di aberrazioni cromosomiche alla FISH e stato mutazionale IGHV in >4000 casi di LLC arruolatinei protocolli del GCLLSG (*).
Non indicazioni Indicazioni Resistential trattamento al trattamento
13q- isolata 48 36 22+ 12 12 14 1211q- 7 21 2517p- 3 5 31IGHV “non mutato” 34 64 78
(*) Dati pubblicati da Zenz et al., Best Practice Clin Haematol 2007, aggiornati da S. Stilgenbauer, comunicazione personale (ASH meeting, S. Francisco, 2008).

10 Seminari di Ematologia Oncologica
ta come eterozigote in approssimativamente il 75-80% dei casi e omozigote nel restante 20-25%.I pazienti con delezione omozigote del cromo-soma 13q14 presentano una maggiore cineticadi crescita linfocitaria rispetto ai pazienti con dele-zioni eterozigoti. Studi molecolari hanno dimostrato che la regio-ne comunemente deleta comprende 790 kb fra imarcatori D13S1150 e D13S25, tuttavia nessungene in questa regione, incluso il succitato LEU2,mostra mutazioni inattivanti nel restante allele.Nel 2002 Calin et al. (39) hanno identificato unapiccola regione deleta di 29 kb su 13q14, fra gliesoni 2 e 5 del gene LEU2, contenente due genicodificanti per micro-RNA, miR-15A e miR-16-1, l’espressione dei quali è significativamente
deregolata in una frazione di LLC. La delezionedi questi geni per miRNA è stata recentementeconfermata da altri ricercatori che hanno utiliz-zato CGH array ad alta risoluzione in 58 casi diLLC 21 (51).
+12La trisomia del cromosoma 12 è la più frequen-te acquisizione di materiale cromosomico nellaLLC, ove è rinvenuta in un 15% circa dei pazien-ti. Sono stati riportati alcuni casi di LLC con tri-somia parziale del cromosoma 12 con segmen-to duplicato compreso tra le bande cromosomi-che 12q13 e 12q21.2. Questo dato suggerisceche questa regione potrebbe contenere geniimportanti nella patogenesi della LLC ed è inte-
TABELLA 3 - Significato clinicobiologico dei difetti cromosomici ricorrenti nella LLC.
Anomalie Gene coinvolto Citomorfologia Immunofenotipo/ Caratteristiche cliniche e biologicheStato dei geni Ig
17p- TP53 CLL/PL CD38+++/- Prognosi severa, in particolare negli stadiZAP-70+++/- intermedio-avanzati e se associata a IGHV non
Ig non mutati +++/- mutato (sopravvivenza mediana <5 anni). Resistente agli analoghi delle purine. Risposte alla chemioimmunoterapia di breve durata.Responsiva ad alemtuzumab.
11q- ATM CLL tipica CD38+++/- Sviluppo di adenopatie marcate. ZAP-70++/-- Prognosi sfavorevole, migliorata con
Ig non mutati +++/- l’introduzione della terapia con fludarabina,ciclofosfamide e rituximab.
+12 12q13-15 CLL atipica CD38++/- Prognosi intermedia (sopravvivenza medianaZAP-70+++/-- 10-15 anni).
Ig non mutati ++/--
6q- 6q21 CLL atipica CD38+++/-- Elevata conta di globuli bianchi. ZAP70++/-- Prognosi intermedia (sopravvivenza mediana
Ig non mutati ++/-- 10-15 anni).
14q32 IgH + partners vari CLL tipica CD38++/--- Prognosi intermedia, richiede un trattamentoZAP70++/--- precoce.
Ig non mutati ++/---
13q- miR-15a CLL tipica CD38- ZAP-70+/--- Prognosi buona se presente come anomaliamiR-16 Ig non mutati +/--- isolata (sopravvivenza mediana >15 anni).DLEU2
Traslocazioni sconosciuto NA CD38+++/- Prognosi sfavorevole.e cariotipo Ig non mutati +++/-complesso
+++/-: 60-80% positivo; ++/-- 30-59% positivo; +/--- <30% positivo. NA: non applicabile.

11Eziopatogenesi, diagnosi e clinica
ressante notare che il gene CLLU1, upregolatonella LLC con decorso clinico aggressivo, è loca-lizzato sulla banda 12q22 (52). Il gene MDM2, chemappa in 12q14.3-q15 potrebbe essere uprego-lato nelle LLC con trisomia 12. MDM2 è un geneil cui prodotto è un importante regolatore del geneoncosoppressore TP53 e la sua overespressio-ne comporta una inattivazione funzionale del pro-dotto di TP53.
11q-Questa anomalia può essere individuata nel 7-25% dei casi a seconda dello stadio della malat-tia (Tabella 1). Il segmento comunemente deletoinclude il gene dell’atassia teleangectasia (ATM)che è coinvolto nel processo di trasduzione delsegnale attivato in risposta a rotture del DNA. Ilrimanente allele ATM è mutato in circa il 30% del-le LLC con 11q- e i pazienti con difetti omozigo-ti di ATM presentano una malattia più aggressi-va rispetto ai pazienti con solo 11q- (53).L’instabilità genetica si associa alla delezione 11q,come dimostrato dallo sviluppo di alterazioni cro-mosomiche aggiuntive mediante analisi del cario-tipo (54).I pazienti con 11q- mostrano in genere unamalattia contrassegnata da adenopatie estesee un intervallo libero da trattamento e unasopravvivenza più brevi rispetto ad altre LLC (55).In diversi trials clinici la presenza di 11q- si asso-ciava a percentuali di risposta completa più bas-sa e a una breve sopravvivenza libera da pro-gressione (56-58). Tuttavia, l’aggiunta di rituxi-mab alla tradizionale chemioterapia con fluda-rabina e ciclofosfamide nei pazienti giovani hamigliorato la percentuale di risposta completa ela sopravvivenza libera da progressione (59, 60)e vi è evidenza non ancora consolidata che il tra-pianto di midollo osseo allogenico con condizio-namento a ridotta intensità potrebbe superarel’impatto prognostico sfavorevole dell’11q- (61).
17p-/mutazioni di TP53L’anomalia 17p- è frequentemente accompagna-ta da aberrazioni cromosomiche aggiuntive ecariotipo complesso e si associa, virtualmente intutti i casi, a perdita di un allele dell’oncosoppres-sore TP53. In più del 70% delle LLC con dele-zione 17p, è presente mutazione del rimanente
allele TP53 e mutazioni inattivanti del gene TP53,rilevabili mediante metodiche molecolari, sonopresenti in un 2-5% di pazienti che non mostra-no delezione 17p- e si associano, analogamen-te alla delezione, a prognosi severa (62). È sta-ta documentata l’espansione del clone 17p-dele-to o TP53-mutato in seguito alla chemioterapia(62). È stato sviluppato un semplice test citofluo-rimetrico per indagare disfunzioni del pathway dip53 (64).Il decorso clinico nei pazienti con 17p-/TP53mutato è sfavorevole (5, 63), soprattutto neipazienti che presentano stadio intermedio-avan-zato e stato IGHV “non mutato” (65) in quanto lepercentuali di risposta alla chemioimmunoterapia,la sopravvivenza libera da progressione e lasopravvivenza di questi pazienti sono inferioririspetto alle altre classi citogenetiche. L’anticorpo monoclonale alemtuzumab può supe-rare la farmacoresistenza in una parte significa-tiva dei casi ed il trapianto allogenico non mie-loablativo può avere un ruolo in questa forma diLLC (66).
Altri difetti cromosomiciLa delezione 6q- si presenta con un’incidenza del3-7% e si associa a un numero più elevato di glo-buli bianchi all’esordio, morfologia atipica, CD38+,geni IGHV non mutati nel 60% dei casi, più bre-ve intervallo libero da trattamento e ridottasopravvivenza rispetto alle LLC con aberrazionicitogenetiche favorevoli (13q-, normale) (67). Laporzione deleta si trova attorno alla regione 6q21ed è interessante il fatto che la perdita allelica in6q sia stata individuata mediante tipizzazione alle-lica ad alta risoluzione in più del 15% dei casi (68).Traslocazioni del cromosoma 14q32 che coin-volgono il gene IGH si presentano con un’in-cidenza del 4-9%. Partner cromosomici ricor-renti includono 18q21/BCL2 e 19q13/BCL3;altri partners identificati occasionalmente sono2p12/BCL11A, 2p13, 4p16, 4p31, 5q31, 6p21/CCND3, 7q21/CDK6, 8q11, 9q34 e 17p11. Laclassica t(11;14)(q13;q32), indistinguibile dal-la traslocazione associata al linfoma mantel-lare, è stata documentata nella LLC da diver-si gruppi. Questi casi rappresentano una forma atipica diLLC, che condivide alcune caratteristiche con il

12 Seminari di Ematologia Oncologica
linfoma mantellare leucemizzato (69). La t(14;19)è associata ad una forma aggressiva di LLC ati-pica; è spesso associata ad anomalie cromoso-miche aggiuntive, specialmente trisomia 12 e conIGHV non mutati. I casi con t(14;19) e cariotipocomplesso includente anomalie quali 7q- e/o 6q-,17p, riarrangiamenti di 1q, frequentemente pos-sono rappresentare casi di linfomi non Hodgkinleucemizzati (70). È stata fornita evidenza che ildecorso clinico della LLC con traslocazioni14q32 potrebbe essere peggiore rispetto allaLLC con cariotipo favorevole (71, 72).
Evoluzione clonaleUna frazione di LLC acquisisce anomalie cromo-somiche durante la storia naturale della malattia.In uno studio prospettico (73), 11 pazienti su 64(17%) seguiti per una mediana di 42 mesi hannomostrato evoluzione clonale con una del(17)(p13)in 4 casi, del(6)(q21) in 3 casi, del(11)(q23) in 2 casi,+(8)(q24) in 1 caso ed evoluzione da 13q- etero-zigote a omozigote in tre casi. La comparsa tar-
diva di 11q- nella LLC è stata associata con l’evo-luzione della malattia (74).
n DIAGNOSI
La LLC viene oggi diagnosticata nella maggiorparte dei casi in occasione di esami del sangueroutinari che dimostrano la presenza di >5 x 109
linfociti nel sangue periferico. Una minoranza deicasi mostra già alla diagnosi un quadro clinicoconclamato con adenopatie e/o splenomegalia,
TABELLA 4 - Condizioni associate a linfocitosi reattiva.
Infezioni acute Infezioni croniche
Pertosse TubercolosiMononucleosi BrucellosiEpatite SifilideCitomegalovirus RickettsiaToxoplasmosi
FIGURA 3

13Eziopatogenesi, diagnosi e clinica
TABELLA 5 - Caratteristiche di laboratorio distintive dei disordini linfoproliferativi cronici.
Patologia Morfologia Immunofenotipo FISH & Altri aspetti†
CLLTipica Piccoli linfociti con <10% linfociti di grandi *CD5+, CD19+, Del(17p13.1), del(11q22.3), trisomia 12q13;
dimensioni o prolinfociti. CD23+, CD22+debole, del(6q21), traslocazioni 14q32/IgH,Atipica Piccoli linfociti con una quota di grandi CD79b+debole; del(13q14) o cariotipo normale.
linfociti e prolinfociti fino al 55%. sIg+debole, FMC7-.
MBL Piccoli linfociti. Come per CLL. Linfocitosi assoluta B-clonale <5X109/LCLL-like Cariotipo normale, 13q-, +12, raramente 11q-
or 17p-. Infiltrazione linfocitaria midollare <30%, obiettività clinica nei limiti di norma.
MCL Cellule linfoidi di medie dimensioni con nucleo CD5+, CD20+intenso, t(11;14)(q13;q32).leucemizzato irregolare, tipicamente non monomorfe. sIg+intenso, CD23-, ciclina D1+.
B-PLL Prolinfociti >55%: cellule di medie dimensioni CD5+/-, FMC7+, sIg+intenso. Talora t(11;14)(q13;q32), del(17p13.1)con nucleo regolare, cromatina condensata, Importante linfocitosi periferica. un nucleolo centrale prominente. Decorso aggressivo.
HCL Cellule di medie dimensioni con nucleo ovale CD5-, CD11c+intenso, Non anomalie citogenetiche costanti.eccentrico, cromatina piuttosto lassa e CD25+, CD103+, Conta leucocitaria periferica in generecitoplasma con fini proiezioni uniformemente HML-1+,B-ly7+. ridotta con monocitopenia assoluta.distribuite. Citochimica: TRAP+. Annessina V+ (biopsia ossea).
HCL-V Caratteristiche ibride tra B-PLL Come nella HCL eccetto Non anomalie citogenetiche costanti.e HCL classica: cellule analoghe per CD25-. Linfocitosi periferica; le cellule linfocitariealle cellule capellute tipiche ma con hanno proiezioni citoplasmatiche; può mancarenucleoli prominenti e TRAP-. la monocitopenia.
SLVL Piccoli linfociti con nucleo regolare, CD5-/+, CD11c+ (50% dei casi), del (7q), + 3q, t(1q), 6q-, +12, cromatina condensata e corte proiezioni CD25+ (25% dei casi); t(8q), 14q32 translocations,citoplasmatiche a disposizione polare. i casi CD25+ sono in genere 17p- mediante FISH.
negativi per CD11c/103. Frequente la splenomegalia.
FL Piccoli linfociti con scarso citoplasma, CD5-, CD10+, CD20+. t(14;18)(q32;q21).leucemizzato cromatina condensata e nucleo clivato Il midollo osseo mostra infiltrati linfoidi
(centrociti). Possono essere presenti paratrabecolari.anche cellule di grandi dimensioni con 1-3 nucleoli (centroblasti).
T-PLL Identica a B-PLL nel 75% dei casi; Nella maggior parte dei casi CD3+, Inv(14)(q11;q32), t(14;14)(q11;q32)nel 25% i prolinfociti T hanno scarso CD7+, CD4+, CD8-; Più frequenti rispetto a B-PLLcitoplasma, nucleo irregolare e nucleoli in un terzo dei casi CD4+ e CD8+; le linfoadenomegalie e leindistinti (variante a piccole cellule). raramente CD4- e CD8+. localizzazioni cutanee.Frequenti le protrusioni citoplasmatiche (blebs).
Espansione Cellule di medie dimensioni con T-LGL (85%): CD3+, CD4-, CD8+, Non anomalie citogeneticheLGL citoplasma abbondante contenente CD16+, CD56-, CD57+, costanti.
granuli azzurrofili. TCRalfa-beta+. Forme tendenzialmente indolenti.NK-LGL (15%): CD3-, CD4-, CD8+, Citopenie.CD16+, CD56+, CD57-.
Linfocitosi Linfociti spesso binucleati, con CD5-, CD19+, sIg policlonali. Isocromosoma 3q, frequente nelleB-policlonale citoplasma relativamente abbondante. donne, fumatrici e di età media.persistente
*Assegnando 1 punto ciascuno a CD5+, CD23+, CD22 o CD79b debole+, sIg debole +, l’85-90% delle CLL presenta uno score di 4 o 5, il 10-15% presenta score 3, <1% delle CLL ha score <3. †Nessuna anomalia citogenetica è diagnostica per CLL, tuttavia la presenza della traslocazione t(11;14) o l’espressione della ciclina D1 sono in genere diagnostici per MCL. Abbreviazioni: CLL, leucemialinfatica cronica; MBL, monoclonal B-cell lymphocytosis; MCL, linfoma a cellule mantellari; FL, linfoma follicolare; HCL, hairy cell leukemia; HCL-V, hairy cell leukemia, forma variante; PLL, leucemia pro-linfocitica; sIg, espressione delle immunoglobuline di superficie; SLVL, linfoma splenico con linfociti villosi; LL, grandi linfociti; PL, prolinfociti.

14 Seminari di Ematologia Oncologica
segni di insufficienza midollare secondaria a dif-fusione della malattia, sintomi sistemici e, rara-mente, localizzazioni extranodali. Dopo averescluso la presenza di infezioni in grado di deter-minare linfocitosi reattiva (Tabella 4), si procedecon gli esami di laboratorio necessari per la dia-gnostica differenziale dei disordini linfoprolifera-tivi cronici (Tabella 5), o con la rara linfocitosi Bpersistente policlonale (75) condizione più fre-quente nelle giovani donne fumatrici che presen-ta tendenza alla distribuzione familiare. Fondamentali sono l’analisi morfologica dello stri-
scio di sangue periferico che mostra piccoli lin-fociti a cromatina addensata (Figura 3) e le ombredi Gumprecht, che rappresentano linfociti rottidurante la preparazione dello striscio a causa diuna loro intrinseca fragilità determinata dalla ridu-zione della proteina del citoscheletro vimentina(76). L’analisi immunofenotipica consente di porre unadiagnosi di certezza in presenza di una espan-sione di elementi CD19+, CD5+, CD23+, conCD22 e/o CD79b debolmente positivo e debo-le espressione delle immunoglobuline di super-ficie (sIg) associata a restrizione delle catene leg-gere (rapporto K/l >3 o <3) e negatività perFMC7. È utile l’applicazione dello score immu-nofenotipico che, attribuendo 1 punto a CD5+,CD23+, CD22/CD79b+ debole, sIg+ debole eFMC7-, identifica in presenza di uno score ≥3oltre il 95% dei casi (77, 78), permettendone ladistinzione rispetto alle altre sindromi linfoproli-ferative (Tabella 5).La diagnostica viene completata con le indaginiindicate in Tabella 6, necessarie per una corret-ta stadiazione e per una corretta programmazio-ne della terapia.
n CLINICA
Manifestazioni principali Circa il 70% dei pazienti viene diagnosticato inseguito ad esami ematici routinari che dimostra-no linfocitosi asintomatica, con obiettività nega-tiva o con adenopatie diffuse a poche sedi (79).Può essere presente già alla diagnosi ipogamma-globulinemia. Negli stadi intermedi compaiono, nelle principa-li sedi superficiali, adenopatie non dolenti, di con-sistenza parenchimatosa, non dura, associate omeno a splenomegalia. Gli stadi avanzati contemplano, per definizione,la presenza di anemia e/o piastrinopenia secon-darie a infiltrazione midollare. Le stadiazioni di Raie di Binet (80, 81) sono riassunte in Tabella 7, oveè anche riportata la sopravvivenza media nei diver-si stadi di malattia. È importante escludere la natu-ra autoimmune dell’anemia e della piastrinopeniaprima di assegnare un paziente allo stadio III-IVdi Rai o C di Binet (82) in quanto è noto che la
TABELLA 6 - Valutazione diagnostica nella LLC.
Test diagnostici
Test per stabilire la diagnosiEmocromo• > 5X109/L linfociti B circolantiConta differenziale al microscopio ottico• Prevalenza di piccoli linfociti; linfociti clivati, grandi lin-
fociti e/o prolinfociti <55%• Ombre di Gumprecht
Immunofenotipo dei linfocitiEspansione clonale di elementi CD5+, CD19+, CD23+con restrizione per le catene leggere delle Ig (scoreimmunofenotipico ≥3)
Valutazione alla diagnosi e/o prima del trattamentoAnamnesi ed esame obiettivo, performance statusConta completa e differenziale delle cellule ematiche Aspirato midollare e biopsia• nel caso l’obiettivo della terapia sia la remissione com-
pleta• per la diagnosi differenziale di eventuali citopenieEsami di laboratorio sierici, inclusa LDH e b2-microglo-bulina, immunoglobuline sieriche, test diretto antiglobu-lineRadiografia del torace ed ecografia dell’addomeIndagine FISH/molecolare• da effettuarsi qualora sia importante ottenere infor-
mazioni prognostiche • ricerca di 17p-/mutazioni TP53; 11q-, traslocazioni
14q32/IgH, trisomia 12, del(6q), del(13q), cariotipo dalinfociti del sangue periferico
• 17p- merita approccio terapeutico diverso, che puòincludere, in casi selezionati, il trapianto allogenico dimidollo, possibilmente nell’ambito di un trial clinico
Stato mutazionale IGHV e CD38• utile perché fornisce informazione prognostica• ZAP-70 non ancora standardizzato

15Eziopatogenesi, diagnosi e clinica
prognosi degli stadi avanzati è migliore se la cito-penia è di origine autoimmune piuttosto che dainfiltrazione midollare (83). Le moderne terapie citostatiche e di supporto, uni-tamente al miglioramento delle condizioni gene-rali di salute della popolazione adulta, hanno deter-minato un significativo allungamento dell’aspet-tativa di vita negli stadi avanzati rispetto ai datistorici (84-86).I sintomi, quando presenti, possono essere rife-riti alla presenza di adenopatie massive o di impo-nente epatosplenomegalia e alla presenza di insuf-ficienza midollare con segni legati all’anemia o allapiastrinopenia. Può manifestarsi astenia nonassociata ad anemia significativa. Sintomi siste-mici, quali febbre >38°C senza cause apparenti,dimagramento >10% di peso corporeo, sudora-zione profusa, prurito, dolori muscolari sono pre-senti alla diagnosi in una minoranza dei casi, men-tre possono comparire più frequentemente nellefasi avanzate di malattia e/o di resistenza al trat-tamento. La malattia può esordire con complican-ze infettive, espressione di deficit del sistemaimmunitario legato alla malattia ed alle terapie.L’anemia emolitica autoimmune può comparire inqualunque fase della malattia; in generale la disre-golazione del sistema immunitario, incentrata suimeccanismi presentati in Tabella 8, può manife-starsi con le patologie a genesi autoimmune elen-cate in Tabella 9 (87).
TABELLA 7 - Classificazione di Rai e Binet.
Gruppo Caratteristiche Sopravvivenza mediana Sopravvivenza medianadi rischio nel Rai report originale in accordo con lo stadio
(n = 125) Rai al Mayo Clinic CLL Database* (n = 2397)
Basso 0 Rai Solo linfocitosi 150 mesi 130 mesiA Binet <3 LN aree coinvolte, non citopenia
Intermedio I Rai + Linfadenopatia 101 106B Binet ≥3 LN aree coinvolte, non citopeniaII Rai + OrganomegaliaB Binet ≥3 LN aree coinvolte, non citopenia
Alto III + Anemia** 19 58IV Rai + trombocitopenia** 19 69C Binet Anemia e/o trombocitopenia
*Tutti i pazienti con LLC sono stati visti al Mayo Clinic Division of Hematology dal 1995 (Shanafelt T, ASH educational book, 2009). **Hb <11 g/dL a causadell’infiltrazione midollare, conta piastrinica <100 x 109/L a causa dell’infiltrazione midollare.
TABELLA 8 - Difetti immunitari nella LLC.
Primari
• Difetti delle cellule B - Ipogammaglobulinemia- Scarsa risposta alla vaccinazione
• Difetti delle cellule T - Quantitativi:
• Aumento di numero- Qualitativi
• Diminuito rapporto CD4/8• Polarizzazione Th 2 • Anomala risposta CD30• Difetto acquisito reversibile del CD40L• Anomalie nell’espressione genica (citoscheletro, gra-
nuli)• Cellule NK
- Mancanza di granuli- Ridotta attività di killing
• Neutrofili - Ridotta funzione fagocitica e battericida- Migrazione e chemiotassi anomale
• Monociti/macrofagi Ridotta citotossicità• Complemento
- Riduzione dei livelli e difetto di attivazione e legame
Secondari
• Insufficienza midollare a causa della malattia avan-zata
• Tossicità della terapia

purine non si è associato ad un incrementosignificativo del rischio di seconde neoplasierispetto a quanto atteso sulla base dei dati storicidisponibili (91). Il rischio di sviluppare mielodisplasiao leucemia acuta mieloide secondaria è basso, mapuò aumentare nei casi trattati con alchilanti e conanaloghi della purine (92). - Trasformazione istologica. La sindrome diRichter, definita dalla comparsa di un linfomaaggressivo, con le caratteristiche del linfomadiffuso a grandi cellule associato a sintomisistemici, versamenti nelle sierose e cachessia, sipuò verificare nel 5-10% dei casi (93). La trasformazione in leucemia a prolinfociti puòessere osservata in una minoranza dei casi.
n CONCLUSIONE
Rispetto alla visione storica che definiva questamalattia come disordine indolente da accumulodi piccoli linfociti, le conoscenze patogenetichehanno consentito di riconoscere nella LLC unamalattia dinamica (14), che trae la sua origine dauna ricca serie di eventi biologici e genetici pri-mari e secondari (94). L’interazione con gli anti-geni, lo stato di attivazione cellulare che ne seguee le complesse interazioni con il microambienteplasmano una malattia dal decorso eterogeneo,talora preceduta da una condizione predisponen-te, la linfocitosi B-monoclonale recentemente defi-nita nei suoi contorni nosografici (95). Grazie aiprogressi nella comprensione dei meccanismi chegovernano la sua storia naturale, l’approcciomoderno alla gestione del paziente si avvale diuna caratterizzazione clinica che prevede lo stu-dio di una serie di marcatori prognostici, molto uti-li per l’adeguata programmazione di terapiesempre più efficaci.
n BIBLIOGRAFIA
1. Muller-Hermelink HK, Montserrat E, Catovsky D,Campo E, Harris NL, Stein H. Chronic lymphocytic leu-kemia/small lymphocytoc lymphoma. In: Swerdlow Eet al, eds. WHO classification of tumours of haematopoi-etic and lymphoid tissue. IARC, Lyon 2008; 180-2.
2. Redaelli A, Laskin BL, Stephens JM, Botteman MF,Pashos CL. The clinical and epidemiological burden
16 Seminari di Ematologia Oncologica
Quadro evolutivo- Infezioni. I pazienti sono soggetti ad infezionirecidivanti negli stadi avanzati anche in relazio-ne a moderni trattamenti che includono analo-ghi delle purine ed anticorpi monoclonali. I qua-dri più frequenti sono sostenuti da Streptococcuspneumoniae, Staphylococcus ed Haemophilusinfluenzae. L’Herpes zoster è frequente e, con l’in-troduzione di nuovi trattamenti, sono da tenere inconsiderarazione le infezioni opportunistiche daLegionella pneumoniae, Pneumocystis jirovecii,Listeria monocytogenes. La polmonite da Citomegalovirus è un problemaemergente, al pari delle infezioni fungine daCandida e Aspergillo (88).- Seconde neoplasie. L’incidenza di secondeneoplasie nella LLC è pari a 1.2-2.2 volte rispettoall’incidenza attesa in una popolazione di pari età(89, 90) e le forme più frequenti sonorappresentate da tumori cutanei, della prostata,della mammella, dei melanomi, del sistemagastrointestinale e del polmone. I principali fattori di rischio sono rappresentatidall’età avanzata, dal sesso maschile e da livellielevati di LDH, beta2-microglobulina e creatinina,mentre non sembra giocare un ruolo il tipo di terapiaeseguita (90). L’utilizzo diffuso degli analoghi delle
TABELLA 9 - Complicanze autoimmuni nella LLC.
Ematologiche
• Anemia emolitica • Coombs positiva• Trombocitopenia immune• Aplasia pura della serie rossa• Neutropenia autoimmune
Non ematologiche
• Angioedema• Pemfigoide bolloso/pemfigo paraneoplastico• Sindrome di Churg-Strauss • Tiroidite autoimmune• Sindrome nefrosica (glomerulonefrite)• Polineuropatia• Sindrome di Sjögren • Lupus eritematoso sistemico• Sindrome di Raynaud • Artrite reumatoide• Colite ulcerosa• Vasculiti

17Eziopatogenesi, diagnosi e clinica
of chronic lymphocytic leukaemia. Eur J Cancer Care.2004; 13: 279-87.
3. Brenner H, Gondos A, Pulte D. Trends in long-term sur-vival of patients with chronic lymphocytic leukemia fromthe 1980s to the early 21st century. Blood. 2008; 111:4916-214.
4. Richardson DB, Wing S, Schroeder J, Schmitz-Feuerhake I, Hoffmann W. Ionizing radiation and chron-ic lymphocytic leukemia.Environ Health Perspect.2005; 113: 1-5.
5. Lin SL, Awan FT, Byrd JC. Chronic lymphocyticleukemia. In Hoffman R, et al. eds: Hematology. Basicprinciple and practice. Churchill Livingstone:Philadelphia PA, USA; 2009; 1327-47.
6. Caporaso N, Marti GE, Goldin L. Perspectives on famil-ial chronic lymphocytic leukemia: genes and the envi-ronment. Semin Hematol. 2004; 41: 201-6.
7. Capalbo S, Trerotoli P, Ciancio A, Battista C, Serio G,Liso V. Increased risk of lymphoproliferative disordersin relatives of patients with B-cell chronic lymphocyt-ic leukemia: relevance of the degree of familial linkage.Eur J Haematol. 2000; 65: 114-7.
8. Goldin LR, Pfeiffer RM, Li X, Hemminki K. Familial riskof lymphoproliferative tumors in families of patientswith chronic lymphocytic leukemia: results from theSwedish Family-Cancer Database. Blood. 2004; 104:1850-4.
9. Rawstron AC, Green MJ, Kuzmicki A, Kennedy B,Fenton JA, Evans PA, et al. Monoclonal B lymphocyteswith the characteristics of “indolent” chronic lympho-cytic leukemia are present in 3.5% of adults with nor-mal blood counts. Blood. 2002; 100: 635-9.
10. Martin AJ, Summersgill BM, Fisher C, Shipley JM, DeanAF. Chromosomal imbalances in familial chronic lym-phocytic leukaemia: a comparative genomic hybridi-sation analysis. Leukemia. 2002; 16: 1229-32.
11. Goldin LR, Slager SL. Familial CLL: genes and envi-ronment. Hematology Am Soc Hematol Educ Program.2007: 339-45.
12. Darzentas N, Hadzidimitriou A, Murray F, Hatzi K,Josefsson P, Laoutaris N, et al. A different ontogene-sis for chronic lymphocytic leukemia cases carryingstereotyped antigen receptors: molecular and compu-tational evidence. Leucemia. 2010; 24: 125-32.
13. Dighiero G. CLL biology and prognosis. HematologyAm Soc Hematol Educ Program. 2005; 278-84.
14. Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocyt-ic leukemia. N Engl J Med. 2005; 352: 804-15.
15. Damle RN, Ghiotto F, Valetto A, Albesiano E, Fais S,Yan XJ, et al. B-cell chronic lymphocytic leukemia cellsexpress a surface membrane phenotype of activated,antigen-experienced B lymphocytes. Blood. 2002; 99:4087-93.
16. Caligaris-Cappio F, Ghia P. Novel insights in chroniclymphocytic leukemia: are we getting closer to under-standing the pathogenesis of the disease? J Clin Oncol.2008; 26: 4497-503.
17. Ghiotto F, Fais F, Valetto A, Albesiano E, Hashimoto S,Dono M, et al. Remarkably similar antigen receptorsamong a subset of patients with chronic lymphocyticleukemia. J Clin Invest. 2004; 113: 1008-16.
18. Tobin G, Thunberg U, Karlsson K, Murray F, Laurell A,Willander K, et al. Subsets with restricted immunoglob-ulin gene rearrangement features indicate a role for anti-gen selection in the development of chronic lympho-cytic leukemia. Blood. 2004; 104:2879-85.
19. Stamatopoulos K, Belessi C, Moreno C, BoudjograhM, Guida G, Smilevska T, et al. Over 20% of patientswith chronic lymphocytic leukemia carry stereotypedreceptors: Pathogenetic implications and clinical cor-relations. Blood. 2007; 109: 259-70.
20. Chu CC, Catera R, Zhang L, Didier S, Agagnina BM,Damle RN, et al. Many chronic lymphocytic leukemiaantibodies recognize apoptotic cells with exposed non-muscle myosin heavy chain IIA: implications forpatient outcome and cell of origin. Blood. 2010; 115:3907-15.
21. Malisan F, Fluckiger AC, Ho S, Guret C, BanchereauJ, Martinez-Valdez H. B-chronic lymphocytic leukemiascan undergo isotype switching in vivo and can beinduced to differentiate and switch in vitro. Blood. 1996;87: 717-24.
22. Gurrieri C, McGuire P, Zan H, Yan XJ, Cerutti A,Albesiano E, et al. Chronic lymphocytic leukemia B cellscan undergo somatic hypermutation and intraclonalimmunoglobulin V(H)DJ(H) gene diversification. J ExpMed. 2002; 196: 629-39.
23. Oppezzo P, Vuillier F, Vasconcelos Y, Dumas G, MagnacC, Payelle-Brogard B, et al. Chronic lymphocyticleukemia B cells expressing AID display dissociationbetween class switch recombination and somatichypermutation. Blood. 2003; 101: 4029-32.
24. Guarini A, Chiaretti S, Tavolaro S, Maggio R, PeragineN, Citarella F, et al. BCR ligation induced by IgM stim-ulation results in gene expression and functionalchanges only in IgV H unmutated chronic lymphocyt-ic leukemia (CLL) cells. Blood. 2008; 112: 782-92.
25. Lanham S, Hamblin T, Oscier D, Ibbotson R, StevensonF, Packham G. Differential signaling via surface IgM isassociated with VH gene mutational status and CD38expression in chronic lymphocytic leukemia. Blood.2003; 101: 1087-93.
26. Chen L, Widhopf G, Huynh L, Rassenti L, Rai KR, WeissA, et al. Expression of ZAP-70 is associated withincreased B-cell receptor signaling in chronic lympho-cytic leukemia. Blood. 2002; 100: 4609-14.
27. Stevenson FK, Caligaris-Cappio F. Chronic lymphocyt-ic leukemia: revelations from the B-cell receptor. Blood.2004; 103: 4389-95.
28. Muzio M, Apollonio B, Scielzo C, Frenquelli M,Vandoni I, Boussiotis V, et al. Constitutive activation ofdistinct BCR-signaling pathways in a subset of CLLpatients: a molecular signature of anergy. Blood. 2008;112: 188-95.

18 Seminari di Ematologia Oncologica
29. Chu P, Deforce D, Pedersen IM, Kim Y, Kitada S, ReedJC, et al. Latent sensitivity to Fas-mediated apopto-sis after CD40 ligation may explain activity of CD154gene therapy in chronic lymphocytic leukemia. ProcNatl Acad Sci USA. 2002; 99: 3854-9.
30. Zupo S, Cutrona G, Mangiola M, Ferrarini M. Role ofsurface IgM and IgD on survival of the cells from B-cell chronic lymphocytic leukemia. Blood. 2002; 99:2277-8.
31. Messmer BT, Messmer D, Allen SL, Kolitz JE, KudalkarP, Cesar D, et al. In vivo measurements document thedynamic cellular kinetics of chronic lymphocyticleukemia B cells. J Clin Invest. 2005; 115: 755-64.
32. Calissano C, Damle RN, Hayes G, Murphy EJ,Hellerstein MK, Moreno C, et al. In vivo intraclonal andinterclonal kinetic heterogeneity in B-cell chronic lym-phocytic leukemia. Blood. 2009; 114: 4832-42.
33. Humby F, Bombardieri M, Manzo A, Kelly S, BladesMC, Kirkham B, et al. Ectopic lymphoid structuressupport ongoing production of class-switched autoan-tibodies in rheumatoid synovium. PLoS Med. 2009;6: e1.
34. Ghia P, Caligaris-Cappio F. The indispensable role ofmicroenvironment in the natural history of low-gradeB-cell neoplasms. Adv Cancer Res. 2000; 79: 157-73.
35. Burger JA, Burger M, Kipps TJ. Chronic lymphocyt-ic leukemia B cells express functional CXCR4chemokine receptors that mediate spontaneousmigration beneath bone marrow stromal cells. Blood.1999; 94: 3658-67.
36. Pedersen IM, Kitada S, Leoni LM, Zapata JM, KarrasJG, Tsukada N, et al. Protection of CLL B cells by afollicular dendritic cell line is dependent on inductionof Mcl-1. Blood. 2002; 100: 1795-801.
37. Molica S, Vacca A, Ribatti D, Cuneo A, Cavazzini F,Levato D, et al. Prognostic value of enhanced bonemarrow angiogenesis in early B-cell chronic lympho-cytic leukemia. Blood. 2002; 100: 3344-51.
38. Maffei R, Martinelli S, Santachiara R, Rossi D,Guarnotta C, Sozzi E, et al. Angiopoietin-2 plasmadosage predicts time to first treatment and overall sur-vival in chronic lymphocytic leukemia. Blood. 2010; 116:584-92.
39. Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S,Noch E, et al. Frequent deletions and down-regulationof micro- RNA genes miR15 and miR16 at 13q14 inchronic lymphocytic leukemia. Proc Natl Acad Sci USA.2002; 99: 15524-9.
40. Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, etal. The DLEU2/miR-15a/16-1 cluster controls B cell pro-liferation and its deletion leads to chronic lymphocyt-ic leukemia. Cancer Cell. 2010; 17: 28-40.
41. Palamarchuk A, Efanov A, Nazaryan N, Santanam U,Alder H, Rassenti L, et al. 13q14 deletions in CLLinvolve cooperating tumor suppressors. Blood. 2010;115: 3916-22.
42. Yuille MR, Condie A, Stone EM, Wilsher J, Bradshaw
PS, Brooks L, et al. TCL1 is activated by chromoso-mal rearrangement or by hypomethylation. GenesChromosomes Cancer. 2001; 30: 336-41.
43. Efanov A, Zanesi N, Nazaryan N, Santanam U,Palamarchuk A, Croce CM, et al. CD5+CD23+leukemic cell populations in TCL1 transgenic miceshow significantly increased proliferation and Akt phos-phorylation. Leukemia. 2010; 24: 970-5.
44. Bichi R, Shinton SA, Martin ES, Koval A, Calin GA,Cesari R, et al. Human chronic lymphocytic leukemiamodeled in mouse by targeted TCL1 expression. ProcNatl Acad Sci USA. 2002; 99: 6955-60.
45. Santanam U, Zanesi N, Efanov A, Costinean S,Palamarchuk A, Hagan JP, et al. Chronic lymphocyt-ic leukemia modeled in mouse by targeted miR-29expression. Proc Natl Acad Sci USA. 2010; 107:12210-5.
46. Damle RN, Batliwalla FM, Ghiotto F, Valetto A,Albesiano E, Sison C, et al. Telomere length and telom-erase activity delineate distinctive replicative featuresof the B-CLL subgroups defined by immunoglobulinV gene mutations. Blood. 2004; 103: 375-82.
47. Grabowski P, Hultdin M, Karlsson K, Tobin G, AleskogA, Thunberg U, et al. Telomere length as a prognosticparameter in chronic lymphocytic leukemia with spe-cial reference to VH gene mutation status. Blood. 2005;105: 4807-12.
48. Rossi D, Lobetti Bodoni C, Genuardi E, Monitillo L,Drandi D, Cerri M, et al. Telomere length is an inde-pendent predictor of survival, treatment requirementand Richter’s syndrome transformation in chronic lym-phocytic leukemia. Leukemia. 2009; 23: 1062-72.
49. Lin TT, Letsolo BT, Jones RE, Rowson J, Pratt G,Hewamana S, et al. Telomere dysfunction andfusion during the progression of chronic lymphocyt-ic leukaemia: evidence for a telomere crisis. Blood.2010; 116: 1899-907
50. Haferlach C, Dicker F, Schnittger S, Kern W, HaferlachT. Comprehensive genetic characterization of CLL: astudy on 506 cases analysed with chromosome band-ing analysis, interphase FISH, IgV(H) status andimmunophenotyping. Leukemia. 2007; 21: 2442-51.
51. Grubor V, Krasnitz A, Troge JE, Meth JL, Lakshmi B,Kendall JT, et al. Novel genomic alterations and clon-al evolution in chronic lymphocytic leukemia revealedby representational oligonucleotide microarray analy-sis (ROMA). Blood. 2009; 113: 1294-303.
52. Buhl AM, Jurlander J, Jørgensen FS, Ottesen AM,Cowland JB, Gjerdrum LM, et al. Identification of agene on chromosome 12q22 uniquely overexpressedin chronic lymphocytic leukemia. Blood. 2006; 107:2904-11.
53. Austen B, Skowronska A, Baker C, Powell JE,Gardiner A, Oscier D, et al. Mutation status of the resid-ual ATM allele is an important determinant of the cel-lular response to chemotherapy and survival inpatients with chronic lymphocytic leukemia containing

19Eziopatogenesi, diagnosi e clinica
an 11q deletion. J Clin Oncol 2007; 25: 5448-57.54. Fegan C, Robinson H, Thompson P, Whittaker JA,
White D. Karyotypic evolution in CLL: identification ofa new sub-group of patients with deletions of 11q andadvanced or progressive disease. Leukemia. 1995; 9:2003-8.
55. Döhner H, Stilgenbauer S, James MR, Benner A,Weilguni T, Bentz M, et al. 11q deletions identify a newsubset of B-cell chronic lymphocytic leukemia charac-terized by extensive nodal involvement and inferior pro-gnosis. Blood. 1997; 89: 2516-22.
56. Catovsky D, Richards S, Matutes E, Oscier D, Dyer MJ,Bezares RF, et al. Assessment of fludarabine pluscyclophosphamide for patients with chronic lympho-cytic leukaemia (the LRF CLL4 Trial): a randomised con-trolled trial. Lancet. 2007; 370: 230-9.
57. Grever MR, Lucas DM, Dewald GW, Neuberg DS, ReedJC, Kitada S, et al. Comprehensive assessment ofgenetic and molecular features predicting outcome inpatients with chronic lymphocytic leukemia: results fromthe US Intergroup Phase III Trial E2997. J Clin Oncol.2007; 25: 799-804.
58. Eichhorst BF, Busch R, Hopfinger G, Pasold R,Hensel M, Steinbrecher C, et al. Fludarabine pluscyclophosphamide versus fludarabine alone in first-linetherapy of younger patients with chronic lymphocyticleukemia. Blood. 2006; 107: 885-91.
59. Tsimberidou AM, Tam C, Abruzzo LV, O’Brien S, WierdaWG, Lerner S, et al. Chemoimmunotherapy may over-come the adverse prognostic significance of 11q dele-tion in previously untreated patients with chronic lym-phocytic leukemia. Cancer. 2009; 115: 373-80.
60. Hallek M, Fischer K, Fingerle-Rowson G, Fink AM,Busch R, Mayer J, et al for the International Group ofInvestigators; German Chronic Lymphocytic LeukaemiaStudy Group. Addition of rituximab to fludarabine andcyclophosphamide in patients with chronic lymphocyt-ic leukaemia: a randomised, open-label, phase 3 tri-al. Lancet. 2010; 376: 1164-74.
61. Sorror ML, Storer BE, Sandmaier BM, Maris M, ShizuruJ, Maziarz R, et al. Five-year follow-up of patients withadvanced chronic lymphocytic leukemia treated withallogeneic hematopoietic cell transplantation after non-myeloablative conditioning. J Clin Oncol. 2008; 26:4912-20.
62. Dicker F, Herholz H, Schnittger S, Nakao A, PattenN, Wu L, et al. The detection of TP53 mutations inchronic lymphocytic leukemia independently predictsrapid disease progression and is highly correlated witha complex aberrant karyotype. Leukemia. 2009; 23:117-24.
63. Zenz T, Kröber A, Scherer K, Häbe S, Bühler A, BennerA, et al. Monoallelic TP53 inactivation is associated withpoor prognosis in chronic lymphocytic leukemia:results from a detailed genetic characterization withlong-term follow-up. Blood. 2008; 112: 3322-9.
64. Carter A, Lin K, Sherrington PD, Pettitt AR. Detection
of p53 dysfunction by flow cytometry in chronic lym-phocytic leukaemia. Br J Haematol. 2004; 127: 425-8.
65. Tam CS, Shanafelt TD, Wierda WG, Abruzzo LV, VanDyke DL, O’Brien S, et al. De novo deletion 17p13.1chronic lymphocytic leukemia shows significant clini-cal heterogeneity: the M. D. Anderson and Mayo Clinicexperience. Blood. 2009; 114: 957-6466.
66. Dreger P, Döhner H, Ritgen M, Böttcher S, Busch R,Dietrich S, et al for the German CLL Study Group.Allogeneic stem cell transplantation provides durabledisease control in poor-risk chronic lymphocyticleukemia: long-term clinical and MRD results of theGerman CLL Study Group CLL3X trial. Blood. 2010;116: 2438-47.
67. Cuneo A, Rigolin GM, Bigoni R, De Angeli C, VeroneseA, Cavazzini F, et al. Chronic lymphocytic leukemia with6q- shows distinct hematological features and inter-mediate prognosis. Leukemia. 2004; 18: 476-83.
68. Novak U, Oppliger Leibundgut E, Hager J, MühlematterD, Jotterand M, Besse C, et al. A high-resolution allelo-type of B-cell chronic lymphocytic leukemia (B-CLL).Blood. 2002; 100: 1787-94.
69. Cuneo A, Bigoni R, Negrini M, Bullrich F, Veronese ML,Roberti MG, et al. Cytogenetic and interphase cyto-genetic characterization of atypical chronic lymphocyt-ic leukemia carrying BCL1 translocation. Cancer Res.1997; 57: 1144-50.
70. Martín-Subero JI, Ibbotson R, Klapper W, Michaux L,Callet-Bauchu E, Berger F, et al. A comprehensivegenetic and histopathologic analysis identifies two sub-groups of B-cell malignancies carrying a t(14;19)(q32;q13) or variant BCL3-translocation. Leukemia.2007; 21: 1532-44.
71. Cavazzini F, Hernandez JA, Gozzetti A, Russo RossiA, De Angeli C, Tiseo R, et al. Chromosome 14q32translocations involving the immunoglobulin heavychain locus in chronic lymphocytic leukaemia identifya disease subset with poor prognosis. Br J Haematol.2008; 142: 529-37.
72. Haferlach C, Dicker F, Weiss T, Schnittger S, Beck C,Grote-Metke A, et al. Toward a comprehensive prog-nostic scoring system in chronic lymphocytic leukemiabased on a combination of genetic parameters.Genes Chromosomes Cancer. 2010; 49: 851-9.
73. Stilgenbauer S, Sander S, Bullinger L, Benner A,Leupolt E, Winkler D, et al. Clonal evolution in chron-ic lymphocytic leukemia: acquisition of high-riskgenomic aberrations associated with unmutated VH,resistance to therapy, and short survival. Haematolo -gica. 2007; 92: 1242-5.
74. Cuneo A, Bigoni R, Rigolin GM, Roberti MG, BardiA, Cavazzini F, et al. Late appearance of the 11q22.3-23.1 deletion involving the ATM locus in B-cell chron-ic lymphocytic leukemia and related disorders.Clinico-biological significance. Haematologica. 2002;87: 44-51.
75. Salcedo I, Campos-Caro A, Sampalo A, Reales E,

20 Seminari di Ematologia Oncologica
Brieva JA. Persistent polyclonal B lymphocytosis: anexpansion of cells showing IgVH gene mutations andphenotypic features of normal lymphocytes from theCD27+ marginal zone B-cell compartment. Br JHaematol. 2002; 116: 662-6.
76. Nowakowski GS, Maurer MJ, Habermann TM, AnsellSM, Macon WR, Ristow KM, et al. Percentage ofsmudge cells on routine blood smear predicts survivalin chronic lymphocytic leukemia. J Clin Oncol. 2010;28: 412-7.
77. Matutes E, Owusu-Ankomah K, Morilla R, GarciaMarco J, Houlihan A, Que TH, et al. The immunolog-ical profile of B-cell disorders and proposal of a scor-ing system for the diagnosis of CLL. Leukemia. 1994;8: 1640-5.
78. Moreau EJ, Matutes E, A’Hern RP, Morilla AM, MorillaRM, Owusu-Ankomah KA, et al. Improvement of thechronic lymphocytic leukemia scoring system with themonoclonal antibody SN8 (CD79b). Am J Clin Pathol.1997; 108: 378-82.
79. Johnson JB, Seftel M, Gibson SB. Chronic lympho-cytic leukemia. In: Greer JP, Forster J, Rodgers GM,Paraskevas F, Glader B, Arber DA, Means RT Jr edi-tors. Wintrobe’s clinical hematology 12th ed, LippincottPhiladephia, PA 2009; 2214-55.
80. Rai KR, Sawitsky A, Cronkite EP, Chanana AD, LevyRN, Pasternack BS. Clinical staging of chronic lympho-cytic leukemia. Blood. 1975; 46: 219-34.
81. Binet JL, Auquier A, Dighiero G, Chastang C, PiguetH, Goasguen J, et al. A new prognostic classificationof chronic lymphocytic leukemia derived from a mul-tivariate survival analysis. Cancer. 1981; 48: 198-206.
82. Oscier D, Fegan C, Hillmen P, Illidge T, Johnson S,Maguire P, et al. Guidelines on the diagnosis and man-agement of chronic lymphocytic leukaemia. Br JHaematol. 2004; 125: 294-317.
83. Zent CS, Ding W, Schwager SM, Reinalda MS, HoyerJD, Jelinek DF, et al. The prognostic significance ofcytopenia in chronic lymphocytic leukaemia/small lym-phocytic lymphoma. Br J Haematol. 2008; 141: 615-21.
84. Shanafelt TD. Predicting clinical outcome in CLL: howand why. Hematology Am Soc Hematol Educ Program.2009: 421-9.
85. Abrisqueta P, Pereira A, Rozman C, Aymerich M, Giné
E, Moreno C, et al. Improving survival in patients withchronic lymphocytic leukemia (1980-2008): theHospital Clinic of Barcelona experience. Blood. 2009;114: 2044-50.
86. Kristinsson SY, Dickman PW, Wilson WH, CaporasoN, Björkholm M, Landgren O. Improved survival inchronic lymphocytic leukemia in the past decade: apopulation-based study including 11,179 patients diag-nosed between 1973-2003 in Sweden. Haematologica.2009; 94: 1259-65.
87. Dearden C. Disease-specific complications of chron-ic lymphocytic leukemia. Hematology Am Soc HematolEduc Program. 2008; 450-6.
88. Montserrat E, Campo E. Small lymphocytic lymphoma/chronic lymphocytic leukemia. In: Canellos GP, ListerTA, Young BD editors. The Lymphomas 2nd edition.Saunders Elsevier, Philadelphia, 2006; 406-14.
89. Hisada M, Biggar RJ, Greene MH, Fraumeni JF Jr,Travis LB. Solid tumors after chronic lymphocyticleukemia. Blood. 2001; 98: 1979-81.
90. Tsimberidou AM, Wen S, McLaughlin P, O’Brien S,Wierda WG, Lerner S, et al. Other malignancies inchronic lymphocytic leukemia/small lymphocytic lym-phoma. J Clin Oncol. 2009; 27: 904-10.
91. Jennifer A Woyach JA. Treatment with F+R ProducesExtended OS and PFS in CLL without Increased Riskof Second Malignancy: Long-Term Follow up of CAL-GB Study 9712 (abstract). ASH 2009 Book ofabstracts: Abs No. 539.
92. Cheson BD, Vena DA, Barrett J, Freidlin B. Secondmalignancies as a consequence of nucleoside analogtherapy for chronic lymphoid leukemias. J Clin Oncol.1999; 17: 2454-60.
93. Rossi D, Cerri M, Capello D, Deambrogi C, Rossi FM,Zucchetto A, et al. Biological and clinical risk factorsof chronic lymphocytic leukaemia transformation toRichter syndrome. Br J Haematol. 2008; 142: 202-15.
94. Damle RN, Calissano C, Chiorazzi N. Chronic lympho-cytic leukaemia: a disease of activated monoclonal Bcells. Best Pract Res Clin Haematol. 2010; 23: 33-45.
95. Shanafelt TD, Ghia P, Lanasa MC, Landgren O,Rawstron AC. Monoclonal B-cell lymphocytosis (MBL):biology, natural history and clinical management.Leukemia. 2010; 24: 512-20.

21
n INTRODUZIONE
La leucemia linfatica cronica (LLC) è in genera-le considerata una malattia indolente: in effetti alladiagnosi si giunge spesso casualmente e inassenza di sintomi, alcuni pazienti rimangonoasintomatici per anni e spesso non richiedono alungo alcun trattamento. In realtà il decorso del-la LLC è estremamente variabile: talora il suoandamento appare non evolutivo, con sopravvi-venze fino a oltre 20 anni, in altri casi è franca-mente aggressivo, con spettanza media di vitainferiore ai 3 anni (1).Da ciò è derivata l’esigenza di formulare alla dia-gnosi una previsione sul futuro decorso dellamalattia e di stabilire la strategia terapeutica piùadatta al singolo paziente. Tale esigenza si è par-ticolarmente rafforzata negli ultimi tempi con lo svi-luppo di numerose nuove e più efficaci armi tera-peutiche. Una precisa e individualizzata formula-zione prognostica risulta paricolarmente impor-tante negli stadi iniziali della malattia, che attual-mente rappresentano la grande maggioranza deicasi di LLC alla diagnosi e per i quali in base allelinee guida tutt’ora accettate (peraltro basate su
studi condotti con farmaci alchilanti) non vi è indi-cazione al trattamento immediato. Negli ultimi annii sostanziali progressi ottenuti nella conoscenzadei meccanismi biologici della LLC hanno porta-to all’individuazione di numerosi nuovi marcato-ri prognostici, alcuni dei quali si sono diffusamen-te affermati nella pratica clinica ad affiancare edintegrare i parametri tradizionali.
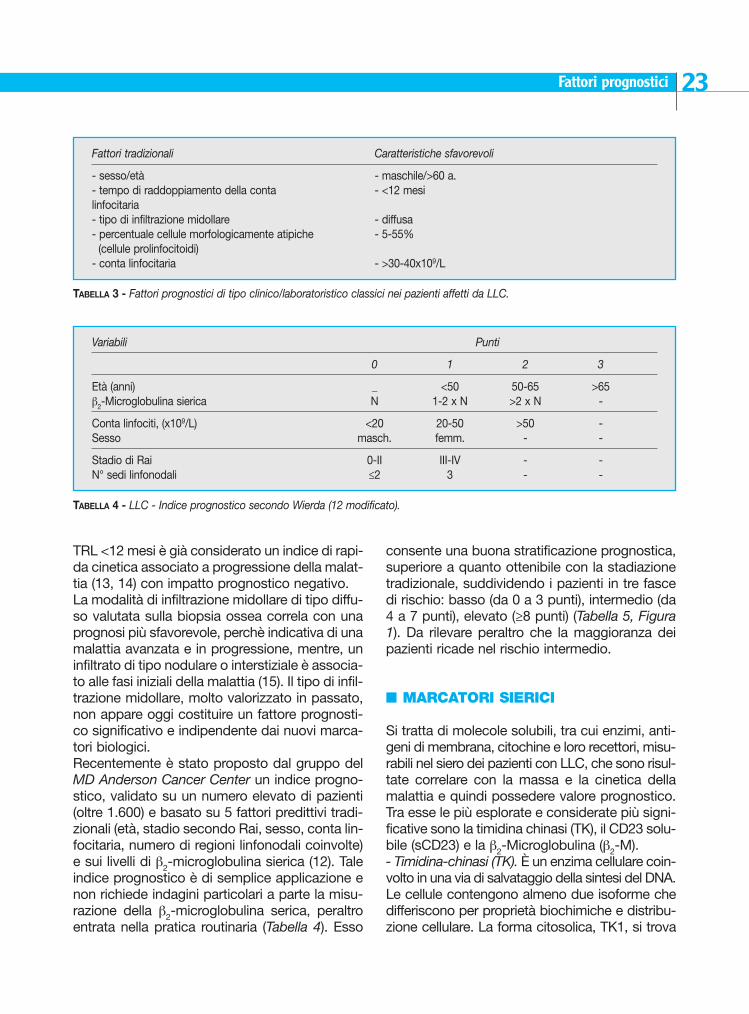
n INDICI PROGNOSTICI TRADIZIONALI (CLINICO-EMATOLOGICI)
Stadiazione I primi indici prognostici applicati su vasta scalanella LLC derivano dagli schemi di stadiazioneproposti oltre 30 anni fa che consentivano di sud-dividere i pazienti in categorie a diversa progno-si sulla base di semplici elementi clinico-emato-logici, è cioè l’obiettività e l’esame emocromoci-tometrico. Tali schemi valutano sostanzialmentela massa tumorale (linfoadenomegalia ed organo-megalia) e la presenza di insufficienza midollareresponsabile di citopenia (anemia e piastrinope-nia) come conseguenza dell’infiltrazione leucemi-ca midollare. Il primo sistema di stadiazione ampiamente accet-tato, proposto da Rai nel 1975 (2), comprende 5stadi, correlati con una diversa sopravvivenza(Tabella 1), e fu modificato successivamente in tregruppi a basso (0), intermedio (I e II) ed alto (III eIV) rischio. Nel 1981 Binet (3) propose un altro siste-ma stadiativo che valorizza l’importanza ai fini pro-gnostici della massa tumorale, intesa come pos-sibile interessamento di 5 diverse aree linfoidi: cer-vicale, ascellare, inguinale, milza e fegato. Sulla
Indirizzo per la corrispondenza
Prof. Achille AmbrosettiDivisione di EmatologiaPoliclinico G.B. RossiP.le L.A. Scuro, 10 - 37134 VeronaE-mail: [email protected]
Fattori prognosticiFattori prognosticiACHILLE AMBROSETTI, ILARIA NICHELE, GIOVANNI PIZZOLODivisione e Cattedra di Ematologia, Azienda Ospedaliera Universitaria di Verona
Achille Ambrosetti
Parole chiave: leucemia linfatica cronica, prognosi,citogenetica, biologia molecolare

22 Seminari di Ematologia Oncologica
base del numero di aree coinvolte e della presen-za o meno di anemia o piastrinopenia vengonodistinti 3 gruppi prognostici (A, B e C) (Tabella 2). È da precisare che la valutazione della massatumorale è fondata in entrambi gli schemi sul sem-plice esame obiettivo e non tiene conto dei datisull’impegno linfonodale profondo ed epatosple-nico ottenibili oggi mediante le tecniche di ima-ging di uso routinario nella pratica clinica.Sebbene la TAC possa evidenziare linfoadenome-galia profonda o splenomegalia non obbiettiva-bile in circa ¼ dei pazienti in stadio 0 di Rai e seb-bene una TAC positiva possa correlare con unmaggior rischio di progressione (4), il suo ruolonella stadiazione della LLC non è ancora accer-tato (5). Per quanto attiene la presenza di anemia e pia-strinopenia andrebbero distinte le forme iporige-nerative conseguenti ad insufficienza midollare daampia infiltrazione linfoide dalle forme da consu-mo su base autoimmune, anch’esse peraltro adimpatto prognostico negativo (6-8). Sebbene tuttora validi e ampiamente utilizzatientrambi i sistemi non risultano tuttavia adegua-ti per una valutazione completa della dinamica del-
la malattia e, soprattutto, non consentono alla pre-sentazione di prevedere il futuro decorso dei sin-goli pazienti nell’ambito dei diversi stadi e, in par-ticolare, negli stadi iniziali (Rai 0 e Binet A) checostituiscono attualmente circa i 2/3 dei pazien-ti alla diagnosi mentre rappresentavano il 37% nel-la casistica di Rai.
Altri fattori prognostici tradizionaliAccanto allo stadio clinico sono stati proposti nelpassato altri fattori prognostici di tipo clinico elaboratoristico, alcuni dei quali con valore indipen-dente dallo stadio, che correlano con una ridot-ta sopravvivenza e una più rapida progressione.Questi includono: sesso, età, conta linfocitaria, tipodi infiltrazione midollare, citomorfologia, tempo diraddoppiamento linfocitario (9-11) (Tabella 3). La conta linfocitaria è un parametro correlato siacon la sopravvivenza che con il tempo alla pro-gressione. Sono stati proposti vari cut-off (30.000-40.000/mmc) (12). Tuttavia, di maggiore e sicuramente rilevanteimpatto prognostico è il tempo di raddoppiamen-to linfocitario (TRL), cioè il tempo necessario affin-chè il numero di linfociti circolanti raddoppi. Un
TABELLA 1 - Stadiazione della LLC secondo Rai (2).
Stadio Rischio Descrizione Sopravvivenza mediana (m)
0 Basso Solo linfocitosi (periferica e midollare) >150
I Intermedio Linfocitosi + adenomegalie 101II Linfocitosi + splenomegalia e/o epatomegalia 71
III Alto Linfocitosi + anemia (Hb <11 gr/dl) 19IV Linfocitosi + piastrinopenia (<100.000/mmc) 19
TABELLA 2 - Stadiazione della LLC secondo Binet (3).
Stadio Descrizione Sopravvivenza mediana (mesi)
A Interessamento di meno di tre aree linfoidi*, assenza di anemia e piastrinopenia >120(Hb >10 gr/dl, PLTS ≥100.000/mmc)
B Interessamento di 3 o più aree linfoidi, assenza di anemia e piastrinopenia 84
C Anemia e/o piastrinopenia (Hb<10gr/dl, PLTS<100.000/mmc) indipendentemente 24dal numero di aree linfoidi coinvolte
*Sono considerate 5 diverse aree linfoidi: a) cervicale; b) ascellare; c) inguinale (in tutti i casi l’interessamento può essere mono- o bilaterale); d) milza; e) fegato.
23Fattori prognostici
TRL <12 mesi è già considerato un indice di rapi-da cinetica associato a progressione della malat-tia (13, 14) con impatto prognostico negativo.La modalità di infiltrazione midollare di tipo diffu-so valutata sulla biopsia ossea correla con unaprognosi più sfavorevole, perchè indicativa di unamalattia avanzata e in progressione, mentre, uninfiltrato di tipo nodulare o interstiziale è associa-to alle fasi iniziali della malattia (15). Il tipo di infil-trazione midollare, molto valorizzato in passato,non appare oggi costituire un fattore prognosti-co significativo e indipendente dai nuovi marca-tori biologici.Recentemente è stato proposto dal gruppo delMD Anderson Cancer Center un indice progno-stico, validato su un numero elevato di pazienti(oltre 1.600) e basato su 5 fattori predittivi tradi-zionali (età, stadio secondo Rai, sesso, conta lin-focitaria, numero di regioni linfonodali coinvolte)e sui livelli di b2-microglobulina sierica (12). Taleindice prognostico è di semplice applicazione enon richiede indagini particolari a parte la misu-razione della b2-microglobulina serica, peraltroentrata nella pratica routinaria (Tabella 4). Esso
consente una buona stratificazione prognostica,superiore a quanto ottenibile con la stadiazionetradizionale, suddividendo i pazienti in tre fascedi rischio: basso (da 0 a 3 punti), intermedio (da4 a 7 punti), elevato (≥8 punti) (Tabella 5, Figura1). Da rilevare peraltro che la maggioranza deipazienti ricade nel rischio intermedio.
n MARCATORI SIERICI
Si tratta di molecole solubili, tra cui enzimi, anti-geni di membrana, citochine e loro recettori, misu-rabili nel siero dei pazienti con LLC, che sono risul-tate correlare con la massa e la cinetica dellamalattia e quindi possedere valore prognostico.Tra esse le più esplorate e considerate più signi-ficative sono la timidina chinasi (TK), il CD23 solu-bile (sCD23) e la b2-Microglobulina (b2-M).- Timidina-chinasi (TK). È un enzima cellulare coin-volto in una via di salvataggio della sintesi del DNA.Le cellule contengono almeno due isoforme chedifferiscono per proprietà biochimiche e distribu-zione cellulare. La forma citosolica, TK1, si trova
TABELLA 3 - Fattori prognostici di tipo clinico/laboratoristico classici nei pazienti affetti da LLC.
Fattori tradizionali Caratteristiche sfavorevoli
- sesso/età - maschile/>60 a.- tempo di raddoppiamento della conta - <12 mesilinfocitaria- tipo di infiltrazione midollare - diffusa- percentuale cellule morfologicamente atipiche - 5-55%
(cellule prolinfocitoidi)- conta linfocitaria - >30-40x109/L
TABELLA 4 - LLC - Indice prognostico secondo Wierda (12 modificato).
Variabili Punti
0 1 2 3
Età (anni) _ <50 50-65 >65b2-Microglobulina sierica N 1-2 x N >2 x N -
Conta linfociti, (x109/L) <20 20-50 >50 -Sesso masch. femm. - -
Stadio di Rai 0-II III-IV - -N° sedi linfonodali ≤2 3 - -

24 Seminari di Ematologia Oncologica
nelle fasi G1 e S del ciclo cellulare, ma è assentenelle cellule quiescenti; è responsabile del 95% del-l’attività sierica della timidin-chinasi (s-TK) riscon-trata ed è probabilmente dipendente dal numerodi cellule neoplastiche in attiva replicazione. s-TKcorrela pertanto con la massa tumorale e con l’at-tività proliferativa delle cellule leucemiche (16-18).Nella LLC i livelli di TK correlano non solo con lostadio clinico, ma anche con altri marcatori pro-gnostici (stato mutazionale, citogenetica, ZAP-70,CD38) (17, 19-21). Recenti studi mostrano chevalori di TK elevati identificano un sottogruppo dipazienti in stadio precoce (Binet A) ad alto rischiodi rapida progressione (19, 20) e si associano aridotta sopravvivenza e aumentato rischio di evo-luzione in sindrome di Richter (20). - sCD23 (CD23 solubile). L’antigene CD23 (recet-tore a bassa affinità per le IgE) è una glicoprotei-
na di membrana, caratteristicamente espressadalle cellule di LLC, che viene clivata in frammen-ti solubili (sCD23) di varie dimensioni, che espli-cano numerose e diverse attività biologiche lamaggior parte delle quali indipendenti dalle IgE(22). I livelli sierici di sCD23 correlano con lo sta-dio clinico, con l’attività della malattia e con lamassa tumorale (23). Valori elevati sono inoltreassociati ad un pattern di infiltrazione diffusa delmidollo osseo alla biopsia osteomidollare, a untempo di raddoppiamento linfocitario ridotto e aelevati livelli sierici di timidin-chinasi (24). In par-ticolare, nei pazienti in stadio A di Binet, il sCD23appare utile per definire una popolazione ad altorischio di progressione, che potrebbe beneficia-re di un approccio terapeutico precoce e aggres-sivo (25). Inoltre, un tempo di raddoppiamento deilivelli di sCD23 inferiore all’anno appare preditti-vo di più rapida progressione e ridotta sopravvi-venza anche tra i pazienti con restanti indici pro-gnostici favorevoli (26).- b2-Microglobulina (b2-M). È una proteina extra-cellulare espressa da tutte le cellule nucleate edè associata alla catena a del complesso maggio-re di istocompatibilità di classe I (MHC I). Vieneliberata nel siero dalle cellule di LLC sia sponta-neamente che in seguito a stimoli (27, 28). I livel-li sierici di b2-M sono risultati correlati allo stadio,
TABELLA 5 - LLC - Sopravvivenza in base allo score di Wierda(12, modificato).
Rischio Punteggio Sopravvivenza Sopravvivenzaa 5 a. a 10 a.
Basso 1-3 97% 80%Intermedio 4-7 80% 52%Alto ≥8 55% 26%
FIGURA 1 - LLC: curve di sopravviven-za in base allo score di Wierda (12,modificato).

25Fattori prognostici
al tempo alla progressione e alla sopravvivenzain numerosi studi retrospettivi (28-30) La b2-M èstata inoltre utilizzata da Wierda tra i parametri checompongono il sistema prognostico da lui pro-posto (12) (v. sopra). Dal momento che i livelli sie-rici di b2-M sono influenzati dalla funzionalità rena-le, è stato recentemente proposto un sistema dicorrezione dei valori in base alla filtrazione glo-merulare (GFR). I valori di b2-M aggiustati in basealla GFR sono risultati indipendenti da ZAP-70come fattore predittivo di progressione (31).- Interleuchine (IL-6, IL-8, IL-10, TNFalfa) e lororecettori solubili (sIL-6R, sTNFRs). Le interleuchi-ne IL-6, IL-8 e IL-10 sono state studiate da soleo in combinazione nella LLC (32-34). Livelli ema-tici elevati sono risultati correlare con stadio avan-zato, valori di LDH e b2-M aumentati e decorsosfavorevole; non è chiaro tuttavia se esse possie-dano un significato prognostico indipendente.Anche i livelli plasmatici di TNFalfa sono risultatiaumentati nella LLC, in associazione con fattoriprognostici clinici e biologici sfavorevoli e conimpatto prognostico indipendente sulla soprav-vivenza (35). - CD20 e CD52 circolanti. Valori aumentati dientrambi questi antigeni in circolo sono risultatiassociati a caratteristiche sfavorevoli della LLC tracui stadio avanzato, b2-M aumentata e ridottasopravvivenza. Per entrambe le molecole solubili,bersagli di immunoterapia con anticorpi monoclo-nali di largo impiego, è stata confermata la possi-bilità che possano interferire con il legame alle cel-lule leucemiche rispettivamente di rituximab e alem-tuzumab, con conseguenti possibili ripercussionisull’efficacia terapeutica e tossicità (36, 37).- CD27 e CD44 solubili. I livelli ematici di ambe-due queste molecole correlano con caratteristi-che di LLC avanzata ed aggressiva. Per il sCD44si è inoltre evidenziato un significato prognosti-co indipendente (38, 39).Numerose altre molecole solubili sono state stu-diate quali possibili marcatori prognostici nella LLC,tra cui: sCD25, sCD8, sICAM-1, sVCAM-1,Trombopoietina (TPO), syndecam-1, Matrix metal-loproteinasi 9 (MMP-9) (40) (vedi anche oltre: mar-catori di angiogenesi e del microambiente). Anchei livelli sierici di LDH (lattato-deidrogenasi) sonoutilizzati come marcatore prognostico. La LDH,considerata indice di turnover cellulare, è comu-
nemente impiegata nella pratica clinica quale indi-ce di massa tumorale e di cinetica proliferativa innumerose neoplasie ematologiche e non. È rara-mente elevato nella LLC e il suo innalzamento puòfar sospettare l’evoluzione in sindrome di Richter.Livelli aumentati di LDH sono comunque corre-lati con ridotta sopravvivenza e con altri marca-tori prognostici sfavorevoli come citogenetica,CD38, ZAP-70 (41-43).
n MARCATORI BIOLOGICI
Nel corso dell’ultima decade, in particolare aseguito della scoperta del valore prognostico del-lo stato mutazionale dei geni IGHV e di alcunealterazioni citogenetiche evidenziate dalla FISHnelle cellule di LLC, si è assistito ad un notevolesviluppo nella ricerca di nuovi marcatori progno-stici di tipo biologico della LLC, in coincidenza coni rapidi progressi nella conoscenza dei meccani-smi patogenetici della malattia. Il numero di marcatori biologici cui è stato via viaattribuito un significato prognostico nella LLC èdecisamente ridondante rispetto a quello ben piùlimitato attualmente utilizzato, comprendentetalune alterazioni citogenetiche (studiate median-te FISH), lo stato mutazionale dei geni IGHV,l’espressione di ZAP-70 e CD38. Da sottolineare tuttavia come questi moderni fat-tori prognostici non siano predittivi per la soprav-vivenza nelle classi di età più avanzate (44).
TABELLA 6 - Principali marcatori solubili nella LLC, correlazionecon l’attività di malattia e significato prognosticosinteticamente riportati in base ai dati delle letteratura.
Molecola Correlazione Valorecon attività prognosticodi malattia indipendentee prognosi
TK (timidina-chinasi) + +sCD23 + +b2-Microglobulina + +sCD44 + +sCD20 + +sCD52 + +sTNF/TNFR + ±LDH + ±

26 Seminari di Ematologia Oncologica
CitogeneticaAnomalie citogenetiche possono essere eviden-ziare in circa l’80% dei casi di LLC. La FISH (fluo-rescence in situ hybridization) rappresenta la tec-nica di riferimento (41, 45) ma anche le tecnichecariotipiche tradizionali con bandeggio conserva-no il loro ruolo (46). Le principali aberrazioni cor-relano con aspetti clinici e/o costituiscono impor-tanti fattori prognostici. Da sottolineare il fatto cheil quadro citogenetico nella LLC non è stabile,potendo evolvere nel decorso della malattia, conpossibile acquisizione di nuove anomalie in cir-ca ¼ dei casi (47).- La delezione 13q14 rappresenta la più frequen-te anomalia strutturale, rilevabile in oltre la metàdei casi di LLC in stadio iniziale e può essere asso-ciata ad altre anomalie. Se isolata, è correlata conun decorso favorevole. Nella delezione del 13qsono coinvolti due geni microRNA (mir-15a e mir-16-1) la cui attività è inversamente correlata conl’espressione del bcl-2, proteina ad azione antia-poptotica; la perdita (o la mutazione) di talimicroRNA comporterebbe pertanto un’aumenta-ta sopravvivenza del clone leucemico (48, 49). - La delezione di 11q22-q23 è presente in circail 10% di casi in stadio iniziale e nel 20-25% deicasi in fase avanzata di malattia. Si associa a sta-dio avanzato, grandi masse linfonodali e anda-mento aggressivo (50). È associata ad instabilità
genetica e circa 1/3 dei pazienti con tale anoma-lia presenta alterazioni cromosomiche aggiuntivenel corso della malattia (51). Vari studi clinici han-no dimostrato minore risposta al trattamento eaccorciata sopravvivenza libera da progressionenei pazienti con 11q- (52-54). Tale significato pro-gnostico sfavorevole appare peraltro almeno inparte superato dall’aggiunta del rituximab alla che-mioterapia (55).- La trisomia del Cr 12 è riscontrabile nel 10-20%dei casi e la sua incidenza non aumenta con la pro-gressione della malattia. Se non accompagnata daaltre anomalie citogenetiche sfavorevoli (11q-, 17p-) non pare influenzare negativamente la soprav-vivenza. I pazienti con trisomia 12 vengono con-siderati a prognosi intermedia (41, 56).- La delezione del Cr17 coinvolge quasi costan-temente la regione 17p13 in cui è localizzato ilgene oncosoppressore TP53. Ecco perché nellamaggior parte dei casi (oltre il 70%) a tale dele-zione si associa una mutazione inattivante di TP53sull’altro allele. La delezione di 17p si riscontra nel3-5% dei pazienti con LLC alla diagnosi o nonancora trattati. La sua frequenza aumenta note-volmente con il progredire della malattia fino al30% circa nei casi refrattari in quanto cloni leu-cemici con 17p- possono essere selezionati daltrattamento, in particolare se basato su alchilan-ti ed analoghi purinici (57). I casi di LLC con 17p-
TABELLA 7 - Caratteristiche delle principali anomalie citogenetiche nella LLC.
Anomalia cromosomica Frequenza Gene coinvolto Prognosi Caratteristiche cliniche
Delezione 13q14 ≥50% miR15a, Buona (se isolata) IndolentemiR16-1?
Trisomia 12 15% Sconosciuto Intermedia Responsiva ad analoghi purinici
Delezione 11q22-23 15% (25% in paz. ATM Sfavorevole Adenopatiarefrattari) massiva
Decorso aggressivoMorfologia atipica
Delezione 17p13/ 5-10% (fino a 30% P53 (spesso Molto sfavorevole Resistente a terapiaMutazione p53 in fase avanzata associata a con alchilanti e
per selezione mutazione p53 analoghi puriniciclonale) nell’altro allele)
Delezione 6q21 3-7% sconosciuto Sfavorevole Linfocitosi elevata, morfologia atipica

27Fattori prognostici
sono scarsamente responsivi o refrattari ai trat-tamenti e hanno sopravvivenza ridotta (attorno ai2 anni dal momento in cui si rende necessario untrattamento). Migliori risultati, ancorchè ancorainsoddisfacenti, si possono ottenere con anti-CD52, alte dosi di metilprednisolone, lenalidomi-de, flavopiridolo e trapianto allogenico di cellulestaminali (58). Tuttavia nell’ambito dei casi con17p- il decorso può essere eterogeneo ed esse-re influenzato anche dallo stadio clinico e dallostato mutazionale (59)Mutazioni di TP53, rilevabili con tecniche mole-colari, sono peraltro riscontrabili anche in pazien-ti che non presentano delezione 17p (circa il 5%dei pazienti non trattati). I casi con mutazione diTP53 presentano caratteristiche cliniche aggres-sive analoghe a quelle dei casi con delezione 17passociata a mutazione di TP53 dell’allele (60). Nederiva l’opportunità di testare per la delezione 17pe mutazione TP53 i pazienti da trattare (61).- La delezione 6q si riscontra nel 3-7% delle LLC.È associata a conta linfocitaria elevata, morfolo-gia atipica, positività del CD38, IGVH non muta-ti e decorso aggressivo (62)- Traslocazioni che coinvolgono il cromosoma14q32 (gene IGH) insieme con diversi altri partnerscromosomici interessano complessivamente il 4-9% dei casi di LLC. Sono ancora limitati i dati cir-ca il significato prognostico di tali anomalie (63).
Stato mutazionale IGHVIn circa la metà dei casi di LLC l’analisi dello sta-to mutazionale dei geni delle regioni variabili del-le catene pesanti delle immunoglobuline (IGHV)evidenzia la presenza di ipermutazioni somatiche,mentre i restanti casi risultano non mutati (64-66).I casi non mutati presentano decorso più aggres-sivo, sopravvivenza inferiore e maggior frequen-za di anomalie citogenetiche sfavorevoli e di altrifattori prognostici negativi (41). Tale diversità diaspetti biologici e clinici ha fatto ipotizzare che ledue categorie di LLC (mutate e non-mutate) rap-presentassero in realtà malattie diverse (67), tut-tavia studi dell’espressione genica hanno dimo-strato in entrambe un profilo sostanzialmente uni-co e caratteristico della malattia, fatta eccezioneper un certo numero di geni tra cui quello dellazeta associated protein 70 (ZAP-70) (68) (v. oltre).Lo stato mutazionale è risultato indipendente dai
fattori prognostici tradizionali, e in particolare dal-lo stadio (69), e predittivo del tempo alla progres-sione e della sopravvivenza anche nei pazienti instadio iniziale. La definizione dello stato mutato o non mutatorichiede tecniche di analisi complesse e costo-se ed è basata su un valore soglia, per lo più defi-nito come il 98% di omologia rispetto al genedella linea germinale. Tale valore soglia è risul-tato all’analisi statistica la discriminante miglio-re in grado di distinguere due gruppi di LLC condecorso assai differente (70). Nei casi con livel-li di omologia ai limiti si consiglia la valutazionecomplessiva dei vari marcatori prognostici. Unaprognosi sfavorevole analoga a quella dei casinon mutati è stata riscontrata anche nei casi diLLC mutati ma esprimenti il gene VH3-21 (71).Lo stato mutazionale viene attualmente consi-derato il marcatore prognostico di riferimento perla LLC.
Marcatori “surrogati” dello stato mutazionaleLa distinzione di due forme di LLC in base allostato mutazionale ne ha sottolineato l’importan-za come parametro prognostico. Tuttavia lacomplessità dell’analisi, i costi e la disponibiltà nongeneralizzata dei laboratori per la sua correttadeterminazione ne hanno finora limitato la diffu-sione e hanno stimolato la ricerca di altri marca-tori biologici di più semplice impiego da usarecome surrogati dello stato mutazionale. Tra di essii più noti e impiegati sono lo studio dell’espres-sione di CD38 e/o ZAP-70 da parte delle celluledella LLC.
CD38È una glicoproteina trans-membrana multifunzio-nale che agisce sia come ecto-enzima sia comerecettore. L’attività enzimatica è esplicitata daldominio extra-cellulare, che possiede attività diadenilato ciclasi (72). Come recettore, mediante il legame a CD31espresso dalle cellule stromali, determina un’ipe-respressione di CD100, una semaforina coinvol-ta nella proliferazione cellulare, per cui induce laproliferazione e aumenta la sopravvivenza dellecellule leucemiche (73). Il CD38 è stato il primomarcatore dimostratosi strettamente correlato con

28 Seminari di Ematologia Oncologica
lo stato mutazionale, tanto da essere stato pro-posto come suo surrogato (66). La presenza dipiù del 30% di linfociti B esprimenti CD38 è sta-ta considerata indicativa di un genotipo non muta-to, e viceversa una positività <30% per il CD38di un genotipo mutato, con una concordanza ini-zialmente riscontrata di oltre il 90%, ma rivelata-si in realtà inferiore negli studi successivi (70, 74).I soggetti con più del 30% di linfociti B esprimen-ti CD38 mostrano una sopravvivenza inferiore euna scarsa risposta ai trattamenti. Peraltro è dasottolineare che la soglia di rappresentatività delCD38 non è accettata in modo univoco e alcuniautori indicano come valore limite il 20% o addi-rittura il 7% dei linfociti B (42, 75). D’altra parte,sembra che la presenza di una popolazione leu-cemica CD38 positiva, ancorché di piccola enti-tà, ma identificabile con certezza, possa deter-minare in modo significativo la prognosi dellamalattia (76). In realtà è stato dimostrato che laconcordanza tra l’espressione di CD38 e lo sta-to mutazionale è solo parziale (variabile tra il 56e il 75%) (40) e questi due parametri biologici risul-tano essere fattori prognostici indipendenti (77).Il CD38 rimane comunque un indiscusso fattoreprognostico nella LLC e numerosi lavori dimostra-no la sua correlazione con un decorso clinico piùaggressivo, scarsa risposta alla terapia e brevesopravvivenza globale e l’associazione con altrimarcatori prognostici sfavorevoli (42, 77). Un limi-te di questo marcatore è che la sua espressionepuò variare nel corso della malattia; non raramen-te (fino ad ¼ circa dei casi) infatti è possibile cheaumenti in seguito a progressione di malattia oterapia (77). Questa variabilità ne limita il valorepredittivo.Attualmente il CD38 più che un surrogato dellostato mutazionale viene generalmente conside-rato un importante marcatore prognostico com-plementare.
ZAP-70 (zeta-associated protein 70)Questo importante marcatore prognostico è sta-to individuato confrontando il profilo genico deicasi di LLC mutati e non: tra i geni diversamen-te espressi nei due gruppi (peraltro con profilogenerale comune e caratteristico) si distinguevain particolare ZAP-70 (68, 78) che codifica per unazeta-associated protein di 70 kDa. Si tratta di unatirosin-chinasi associata al recettore CD3 dei lin-fociti T, coinvolta nella trasduzione del segnale pro-veniente dal TcR (T-cell receptor) quando lega l’an-tigene. Normalmente è espressa dai linfociti T eNK, ma anche in alcune sottopopolazioni B (79)ed in varie neoplasie linfoidi B (80). La maggiorparte dei linfociti B non possiede questa mole-cola, ma utilizza un’altra tirosin-chinasi associa-ta, SyK, per la trasduzione del segnale dal com-plesso BcR (B-cell receptor). Non è nota la ragio-ne per la quale i linfociti di una parte dei casi diLLC esprimono ZAP-70, ma è possibile che talemolecola funga da trasduttore del segnale in lin-fociti B deficitari di SyK (81) e contribuisca apotenziare il segnale dovuto alle IgM di membra-na (82) determinando un vantaggio nella rispostamigratoria e nella sopravvivenza e contribuendoprobabilmente al comportamento clinico aggres-sivo (78, 83). L’espressione di ZAP-70 può essere valutata convari metodi, come Western blotting, PCR quan-titativa, immunoistochimica, tuttavia la tecnica piùlargamente diffusa nella pratica clinica è senz’al-tro la citometria a flusso su sangue periferico (84). Quest’ultima risulta la tecnica più utilizzata nellaroutine, grazie alla sua praticità e velocità, ed èla tecnica impiegata nella maggior parte dei nume-rosi studi che hanno dimostrato l’importanza del-l’espressione di ZAP-70 come marcatore progno-stico nei pazienti con LLC (85-88). La soglia dipositività è stata fissata al 20% dei lifociti B. Unavalida alternativa alla citofluorimetria è rappresen-
TABELLA 8 - LLC: principali marcatori surrogati dello stato mutazionale IGHV.
Marcatore Tecnica Soglia Concordanza Valore con IGHV prognostico
CD38 Citofluorimetria 30% (20%, 7%) 56-75% Indipendente
ZAP-70 Citofluorimetria 20% 70-93% Indipendenteimmunoistochimica +

29Fattori prognostici
tata dall’analisi immunoistochimica su biopsiaosteo-midollare i cui risultati si sono rivelati altret-tanto affidabili (90, 91).La validità di ZAP-70 come fattore prognostico èstata dimostrata da numerosi studi clinici, chehanno messo in evidenza come l’espressione diquesta proteina correli con un andamento clini-co più aggressivo, sopravvivenza più breve enecessità di trattamento precoce (86-88). Inizialmente proposto come surrogato dello sta-to mutazionale IgHV per la sua notevole diversi-tà di espressione nei casi non mutati rispetto aimutati, ZAP-70 si è rivelato un marcatore progno-stico indipendente e fortemente predittivo in ter-mini sia di tempo alla progressione che disopravvivenza totale anche nei pazienti in stadioiniziale (89). In realtà la concordanza tra espres-sione di ZAP-70 e stato mutazionale non è com-pleta, o quasi, come inizialmente segnalato; infat-ti i casi discordanti variano tra il 7 e il 30% aseconda delle casistiche e soprattutto dellemetodiche impiegate (40). Alcuni dei casi ZAP-70 positivi ma IGHV mutatisono risultati esprimere il gene VH3-21 e vicever-sa in alcuni casi non mutati ma ZAP-70 negativisono state rilevate anomalie citogenetiche sfavo-revoli (11q- o 17p-) (92).I casi discordanti presentano comunque in gene-re prognosi intermedia (87).Un limite all’affidabilità di ZAP-70 è rappresenta-
FIGURA 2 - LLC - valutazione dell’espressione di ZAP-70 median-te citofluorimetria su sangue periferico: a) caso ZAP-70 negati-vo (1% di cellule B CD19+/ZAP-70+); b) caso ZAP-70 positivo(86% di cellule B CD19+/ZAP-70+). I linfociti T presenti risulta-no CD19-/ZAP-70+.
FIGURA 3 - LLC - valutazione dell’espressione di ZAP-70 mediante immunoistochimica su biopsia osteomidollare: nel riquadro asinistra area midollare estesamente infiltrata da cellule leucemiche ZAP-70+, a destra risultano positivi solo sparsi linfociti T (Gentileconcessione di Marco Chilosi).

30 Seminari di Ematologia Oncologica
to dalle problematiche inerenti alla standardizza-zione delle metodiche citofluorimetriche, nonancora del tutto risolte e riguardanti in particola-re la scelta del campione e dell’anticorpo e le tec-niche di fissazione e permeabilizzazione. Quantoalla stabilità dell’espressione di questo marcato-re nel corso della malattia, a fronte di alcunesegnalazioni di variazione del dato citofluorime-trico (43), esso viene generalmente ritenutosostanzialmente costante dalla maggioranza deiclinici. Data l’arbitrarietà della soglia, per la mag-gior parte degli studi fissata al 20% dei linfocitiB neoplastici, i casi che presentassero valori vici-ni alla soglia dovrebbero essere attentamentevalutati nel contesto di altri fattori. L’espressionedi ZAP-70 è di fatto attualmente considerata,accanto alla citogenetica (FISH) e allo stato muta-zionale, uno dei marcatori prognostici più rilevan-ti per la LLC.Nello studio condotto su oltre 1.000 pazienti dalConsorzio Internazionale per la LLC (86) volto aconfrontare l’impatto sfavorevole dell’espressio-ne di ZAP-70 sul decorso della LLC emerge chetale fattore non viene influenzato dallo stato muta-zionale IGHV, mentre tra i casi ZAP-70 negativiquelli con IGHV non mutati hanno prognosi peg-giore (tempo al trattamento inferiore). Questo stu-dio se da un lato rafforza il valore prognostico diZAP-70 (non più semplice surrogato), dall’altro evi-denzia come l’impiego combinato di più marca-
tori possa migliorare le capacità prognostiche deisingoli casi. Tuttavia, un altro studio (92) ha identificato lo sta-to mutazionale IGHV, l’uso di V3-21 e le anoma-lie genomiche sfavorevoli, ma non l’espressionedi ZAP-70 come fattori prognostici indipendenti,peraltro evidenziando come quest’ultime fosserofrequenti nei casi discordanti ZAP-70 negativi.Analogamente, valutando la combinazione diFISH, stato mutazionale IGHV ed espressione diCD38 in pazienti in stadio iniziale si è rilevato chela presenza di delezione 17p configura un rischioelevato indipendentemente dallo stato mutazio-nale, mentre la delezione 11q comporta una pro-gnosi intermedia, analoga a quella dei casi IGHVnon mutati (93) (Figura 4). L’impiego combinatodei principali marcatori può verosimilmente con-tribuire a migliorare le capacità predittive nei sin-goli pazienti (43, 87, 88).
Altri marcatori biologici- Lipoprotein-lipasi (LPL). Tra i geni la cui espres-sione ha dimostrato una forte correlazione con lostato mutazionale (68). LPL è un enzima legatoai glicosaminoglicani che compongono l’endote-lio, particolarmente abbondante nel muscolo, neltessuto adiposo e nei macrofagi e gioca un ruo-lo centrale nel metabolismo e trasporto dei lipidi(94). L’espressione del mRNA della LPL è risul-tata aumentata nei casi di LLC con IGVH non
FIGURA 4 - LLC: sopravvivenza in basea stato mutazionale IGHV e citogene-tica sfavorevole (92, modificata).

31Fattori prognostici
mutati e si è evidenziato il suo ruolo come fatto-re prognostico indipendente (94, 95). Si è riscon-trato che l’espressione di LPL è selettiva nei lin-fociti leucemici (94) e che i livelli di espressionesono strettamente correlati allo stato mutaziona-le, con una concordanza variabile tra il 76 e il 90%(40). Da sottolineare che gli elevati livelli di LPLsono associati ad un decorso clinico più aggres-sivo, con un breve intervallo libero da trattamen-to e ridotta sopravvivenza globale (96).- ADAM29 (a disintegrin and metalloproteinase29). Il gene codifica per una proteina coinvoltanelle interazioni cellula-cellula e cellula-matrice,appartenente alla famiglia delle disintegrine emetalloproteinasi trans-membrana. Studi di pro-filo genico hanno evidenziato che i casi di LLCcon IGVH mutati esprimono livelli più elevati diquesto gene (94). È possibile pertanto che l’as-sociazione di LPL e di ADAM29 possa aumen-tarne la specificità come fattori prognostici.Questo marcatore è stato valutato in associazio-ne a LPL: un rapporto aumentato (>1) traespressione (mRNA) di LPL e ADAM29 è risul-tato infatti marcatore prognostico indipendenteper sopravvivenza libera da eventi (94).- AID (Activation-induced cytidine Deaminase). Èun enzima essenziale per il processo di ipermu-tazione somatica delle immunoglobuline. Livelliaumentati di mRNA di AID sono stati riscontratinelle CLL ad alto rischio (non mutate) e nei casipositivi per AID si è osservata una maggior fre-quenza di anomalie citogenetiche sfavorevoli, LDHaumentato e positività di CD38 (97, 98). - CLLU1. Si tratta di un gene (CLL upregulatedgene 1), individuato da un gruppo danese (99) esituato sul cromosoma 12q22 (ma non coinvol-to nella delezione 12), la cui elevata espressioneè risultata caratteristica dei casi di LLC a progno-si sfavorevole. L’aumentata espressione di CLLU1è correlata ad altri marcatori quali stato mutazio-nale, ZAP-70, CD38 e predittiva di tempo al trat-tamento e di sopravvivenza globale inferiori(100). Un aumento dell’espressione del mRNA diCLLU1 è risultato fattore prognostico indipenden-te, ma solo in pazienti di età <70 anni. I dati alriguardo sono tuttavia contrastanti (101).- CD49d. Il CD49d/integrina alfa 4 è una mole-cola di adesione per la matrice extracellulare lacui aumentata espressione sulla superficie delle
cellule di LLC è risultata associata a stadio avan-zato di malattia, positività di CD38 e ZAP-70 e constato IGHV non mutato. Un’elevata espressionedi CD49d (superiore alla soglia del 30%) è risul-tata fattore prognostico indipendente sia per lasopravvivenza che per il tempo al trattamento(102, 103). - Lunghezza dei telomeri ed attività della telome-rasi. La lunghezza dei telomeri, costituiti dasequenze ripetute di DNA atte a stabilizzare lastruttura dei cromosomi, è inversamente propor-zionale alla storia replicativa delle cellule; d’altron-de l’attività telomerasica, volta a contrastare l’ac-corciamento dei telomeri dovuto alle divisioni cel-lulari, è aumentata nelle cellule maggiormente pro-liferanti. Nei linfociti della LLC è stata rilevata unacorrelazione inversa tra lunghezza dei telomeri edattività telomerasica. I casi con telomeri più cor-ti hanno mostrato un andamento più aggressivo,una maggiore frequenza di fattori prognostici sfa-vorevoli (citogenetica, ZAP-70, CD38) (104, 105)ed un rischio più elevato di evoluzione inSindrome di Richter (106).
Marcatori di angiogenesi e del microambienteIn base ai più recenti concetti sulla patogenesi del-la LLC l’aumentata sopravvivenza delle cellule leu-cemiche dipende, oltre che da difetti nei mecca-nismi di apoptosi, anche da fattori estrinseci chederivano dal microambiente e che consistono sia
TABELLA 9 - LLC: marcatori prognostici biologici aggiuntivi.
Marcatore Tecnica Valore prognostico indipendente
LPL: lipoprotein-lipasi/ PCR +/ADAM29
AID: Activation-Induced PCR ±cytidine Deaminase
CLLU1: CLL PCR, FISH ±Upregulated Gene 1
CD49d/ integrina citofluorimetria +alfa 4
Lunghezza telomeri/telomerasi PCR, FISH ±

32 Seminari di Ematologia Oncologica
in interazioni dirette cellula-cellula e cellula-matri-ce, sia mediate da fattori solubili (107). È noto inol-tre come le cellule di LLC, attraverso la produ-zione di fattori proangiogenetici, possano indur-re la formazione di nuovi vasi che favoriscono lasopravvivenza e la disseminazione in circolo del-le cellule stesse (108).Alcune molecole del microambiente sono statemesse in relazione al decorso clinico della malat-tia e vengono oggi comprese nell’ampio panora-ma dei fattori di prognosi. Essi sono APRIL, e ifattori propriamente angiogenetici: angiopoietina-2 (Ang2) e VEGF.- APRIL (A Proliferation Inducing Ligand). È unmembro della superfamiglia del TNF (Tumornecrosis factor), sintetizzato come proteina tran-smembrana omotrimerica di tipo II e rilasciato informa biologicamente attiva nel compartimentoextracellulare prevalentemente in forma solubile.Prodotta da cellule del microambiente ematopo-ietiche e non dalle cellule tumorali stesse, APRILsi lega ai recettori BCMA (B Cell MaturationAntigen), TACI (Transmembrane activator and cal-cium modulator and cyclophilin ligand interactor)e verosimilmente anche HSPGs (heparan sulpha-te proteoglycans), innescando una cascata di tra-sduzione del segnale a valle che converge sullavia canonica di NF-kB (109). Nella LLC APRIL aumenta la sopravvivenza deilinfociti maligni sia attraverso un meccanismoparacrino essendo prodotto dalle cellule del micro-ambiente, sia tramite un circuito autocrino.Recentemente è stata infatti dimostrata un’au-mentata espressione del trascritto di APRIL daparte dei linfociti maligni e la presenza sulla lorosuperficie dei recettori TACI e BCMA (110, 111)Il coinvolgimento di APRIL nella patogenesi del-la LLC ha indotto a studiarne il ruolo prognosti-co. Si è evidenziato che concentrazioni sierichedi APRIL sopra la mediana correlano con ridottasopravvivenza (112, 113).- ANG-2 (Angiopoietina-2) e VEGF. ANG-2 è unaglicoproteina di 75 kDa che si lega al recettore tiro-sin-chinasico Tie-2 posto sulla superficie delle cel-lule endoteliali ed opera in concerto con VEGF nelpromuovere il rimodellamento e la formazione dinuovi vasi. Prodotto e immagazzinato dalle cellule endote-liali, ANG-2 è prodotto anche da cellule neopla-
stiche e compete con il ligando fisiologico di Tie-2, ovvero angiopoietina-1, fattore che promuovela maturazione e l’integrità strutturale delle cellu-le endoteliali in condizioni normali. ANG-2 attivae altera le cellule endoteliali pre-esistenti e VEGFpromuove la migrazione e la proliferazione di nuo-ve cellule endoteliali producendo così nuovi vasi(114). La conoscenza del ruolo angiogenetico diANG-2 e VEGF nelle neoplasie, deriva da studipreliminari relativi a tumori solidi, ma è stato ripor-tato anche recentemente che ANG-2 e VEGF ven-gono prodotti, soprattutto in condizioni di ipos-sia, anche dalle cellule di LLC dove tali fattori pro-muovono la formazione di nuovi vasi in modoparacrino attraverso interazioni reciproche con lecellule endoteliali del microambiente (115). Diverse osservazioni suggeriscono l’ipotesi di unacorrelazione nella LLC tra elevata angiogenesi eaggressività di malattia. In particolare è stato dimo-strato come la densità vascolare midollare,aumentata nei pazienti affetti da LLC rispetto aicontrolli, sia in grado di predire il tempo libero daprogressione negli stadi inziali di malattia (116).Uno studio recente condotto su un’ampia casi-stica di pazienti, ha dimostrato come elevati livel-li di ANG-2 in pazienti affetti da LLC correlino inmodo significativo con lo stadio avanzato dimalattia secondo Binet, livelli aumentati di b
2-microglobulina, IGVH non mutato e citogeneticasfavorevole. Inoltre, ANG-2 rappresenta un fatto-re di rischio indipendente nel predire l’intervallodi tempo al primo trattamento e la sopravviven-za globale. Nello stesso studio è stato dimostra-to come anche livelli di VEGF più elevati siano cor-relati con un tempo al primo trattamento più bre-ve (117). Quest’ultimo dato è in linea con studiprecedenti in cui era emerso come il livello diVEGF fosse in grado di predire la sopravvivenzalibera da malattia in pazienti con malattia in sta-dio A di Binet (118).
n CONCLUSIONI
Per la maggior parte dei numerosi marcatori di tipobiologico proposti negli ultimi anni per la LLC nonsono disponibili dati sufficienti a validarne l’impie-go come fattori prognostici indipendenti. Fannoeccezione la citogenetica, lo stato mutazionale

33Fattori prognostici
IGHV e, sebbene in misura minore, ZAP-70 eCD38, ormai entrati nell’uso clinico comune inmolti centri al fine di orientare le previsioni pro-gnostiche dei pazienti alla diagnosi. Il dato citogenetico può talora indirizzare la scel-ta terapeutica (come nel caso di delezione17p/mutazione p53), ma ancora si discute se ladecisione di avviare un trattamento possa basar-si sul profilo prognostico e non piuttosto sui cri-teri clinici di malattia in progressione (45).
RingraziamentiI dati personali riportati nella review sono statiprodotti nell’ambito di progetti di ricerca finanziatida: Regione Veneto “Ricerca Sanitaria Finalizzata”;“Fondazione G. Berlucchi per la Ricerca sulCancro”; AIRC - Associazione Italiana Ricerca sulCancro e Fondazione CARIVERONA.
n BIBLIOGRAFIA
1. Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocy-tic leukemia. N Engl J Med. 2005; 352: 804-15.
2. Rai KR, Sawitsky A, Cronkite EP, Chanana AD, LevyRN, Pasternack BS. Clinical staging of chronic lym-phocytic leukemia. Blood. 1975; 46: 219-34.
3. Binet JL, Auquier A, Dighiero G, Chastang C, PiguetH, Goasguen J, et al. A new prognostic classificationof chronic lymphocytic leukaemia derived from a mul-tivariate survival analysis. Cancer. 1981; 48: 198-206.
4. Muntañola A, Bosch F, Arguis P, Arellano-Rodrigo E,Ayuso C, Giné E, et al. Abdominal computed tomo-graphy predicts progression in patients with Rai sta-ge 0 chronic lymphocytic leukemia. J Clin Oncol.2007; 25: 1576-80.
5. Blum KA, Young D, Broering S, Lucas MS, FischerB, Lin TS, et al. Computed tomography scans do notimprove the predictive power of 1996 national can-cer institute sponsored working group chronic lym-phocytic leukemia response criteria. J Clin Oncol.2007; 25: 5624-9.
6. Zent CS, Ding W, Schwager SM, Reinalda MS, HoyerJD, Jelinek DF, et al. The prognostic significance ofcytopenia in chronic lymphocytic leukaemia/smalllymphocytic lymphoma. Br J Haematol. 2008; 141:615-21.
7. Visco C, Ruggeri M, Laura Evangelista M, Stasi R,Zanotti R, Giaretta I, et al. Impact of immune throm-bocytopenia on the clinical course of chronic lympho-cytic leukemia. Blood. 2008; 111: 1110-6.
8. Zanotti R, Frattini F, Ghia P, Visco C, Zamò A, PerbelliniO, et al. ZAP-70 expression is associated with increa-
sed risk of autoimmune cytopenias in CLL patients.Am J Hematol. 2010; 85: 494-8.
9. Lee JS, Dixon DO, Kantarjian HM, Keating MJ, TalpazM. Prognosis of chronic lymphocytic leukemia: a mul-tivariate regression analysis of 325 untreated patients.Blood. 1987; 69: 929-36.
10. Vallespí T, Montserrat E, Sanz MA. Chronic lympho-cytic leukaemia: prognostic value of lymphocyte mor-phological subtypes. A multivariate survival analysisin 146 patients. Br J Haematol. 1991; 77: 478-85.
11. Rozman C, Montserrat E, Feliu E, Granena A, MarinP, Nomdedeu B, et al. Prognosis of chronic lympho-cytic leukaemia: a multivariate surival analysis of 150cases. Blood. 1982; 59: 1001-5.
12. Wierda WG, O’Brien S, Wang X, Faderi S, FerrajoliA, Do KA, et al. Prognostic normogram and index foroverall survival in previously untreated patients withchronic lymphocytic leukaemia. Blood. 2007; 109:4679-85.
13. Montserrat E, Sanchez-Bisono J, Vinolas N, RozmanC. Lymphocite doubling time in chronic lymphocyticleukaemia: analysis of its prognostic significance. BrJ Haematol. 1986; 62: 567-75.
14. Molica S, Alberti A. Prognostic value of the lympho-cyte doubling time in chronic lymphocytic leukemia.Cancer. 1987; 60: 2712-6.
15. Rozman C, Montserrat E, Rodriguez-Fernandez JM,Ayas R, Vallespi T, Parody R, et al. Bone marrow histo-logic pattern - the best single prognosis parameterin chronic lymphocytic leukaemia: a multivariate ana-lysis of 329 cases. Blood. 1984; 64: 642-8.
16. Hallek M, Langenmayer I, Nerl C, Knauf W,Dietzfelbinger H, Adorf D, Ostwald M, Busch R,Kuhn-Hallek I, Thiel E, Emmerich B. Elevated serumthymidine kinase levels identify a subgroup at highrisk of disease progression in early, nonsmolderingchronic lymphocytic leukemia. Blood. 1999; 93:1732-7.
17. Di Raimondo F, Giustolisi R, Lerner S, Cacciola E,O’Brien S, Kantarjian H, Keating MJ. Retrospectivestudy of the prognostic role of serum thymidine kina-se level in CLL patients with active disease treatedwith fludarabine. Ann Oncol. 2001; 12: 621-5.
18. Montillo M, Hamblin T, Hallek M, Montserrat E, MorraE. Chronic Lymphocytic Leukaemia: novel progno-stic dactors and their relevance for risk-adapted the-rapeutic strategies. Haematologica 2005; 90: 391-9.
19. Magnac C, Porcher R, Davi F, Nataf J, Payelle-BrogardB, Tang RP, et al. Predictive value of serum thymidi-ne kinase level for Ig-V mutational status in B-CLL.Leukaemia. 2003; 17: 133-7.
20. Matthews C, Catherwood MA, Morris TC, Kettle PJ,Drake MB, Gilmore, et al. Serum TK levels in CLLidentify Binet stage A patients within biologically defi-ned prognostic subgroups most likely to undergodisease progression. European Journal ofHaematology. 2006; 77, 309-17.

34 Seminari di Ematologia Oncologica
21. Konoplev SN, Fritsche HA, O’Brien S, Wierda WG,Keating MJ, Gornet TG, et al. High serum thymidinekinase 1 level predicts poorer survival in patients withchronic lymphocytic leukemia. Am J Clin Pathol. 2010;134: 472-7.
22. Fujiwara H, Kikutani H, Suematsu S, Naka T, YoshidaK, Tanaka T, et al. The absence of IgE antibody-mediated augmentation of immune responses inCD23-deficient mice. Proceedings of the NationalAcademy of Science - USA. 1994; 91, 6835-9.
23. Reinisch W, Willheim M, Hilgarth M, Gasché C, MaderR, Szepfalusi S, et al. Soluble CD23 reliably reflectsdisease activity in B-cell chronic lymphocytic leuke-mia. J Clin Oncol. 1994; 12: 2146-52.
24. Knauf WU, Langenmayer I, Ehlers B, Mohr B, AdorfD, Nerl CH, et al. Serum levels of soluble CD23, butnot soluble CD25, predict disease progression in ear-ly stage B-cell chronic lymphocytic leukemia. LeukLymphoma. 1997; 27: 523-32.
25. Sarfati M, Chevret S, Chastang C, Biron G,Stryckmans P, Delespesse G, et al Prognostic impor-tance of serum soluble CD23 level in chronic lympho-cytic leukemia. Blood. 1996; 88: 4259-64.
26. Meuleman N, Stamatopoulos B, Dejeneffe M, ElHousni H, Lagneaux L, Bron D. Doubling time of solu-ble CD23: a powerful prognostic factor for newly dia-gnosed and untreated stage A chronic lymphocyticleukemia patients. Leukemia. 2008; 22: 1882-90.
27. Demaria S, Schwab R, Gottesman SR, Bushkin Y.Soluble beta 2-microglobulin-free class i heavychains are released from the surface of activated andleukaemia cells by a metalloprotease. Journal ofBiological Chemistry. 1994; 269: 6689-94.
28. Itala M, Pelliniemi TT, Remes K. GM-CSF raises serumlevels of beta 2-microglobulin and thymidine kinasein patients with chronic lymphocytic leukaemia. BritishJournal of Haematology. 1996; 94: 129-32.
29. Hallek M, Wanders L, Ostwald M, Busch R,Senekowitsch R, Stern S, et al. Serum beta(2)-micro-globulin and serum thymidine kinase are independentpredictors of progression-free survival in chronic lym-phocytic leukaemia and immunocytoma. Leukaemiaand Lymphoma. 1996; 22, 439-47.
30. Molica S, Levato D, Cascavilla N, Levato L, MustoP. Clinico-prognostic implications of simultaneousincreased serum levels of soluble CD23 and beta2-microglobulin in B-cell chronic lymphocytic leukae-mia. European Journal of Haematology. 1999; 62:117-22.
31. Delgado J, Pratt G, Phillips N, Briones J, Fegan C,Nomdedeu J, et al. Beta-2-microglobulin is a betterpredictor of treatment-free survival in patients withchronic lymphocytic leukaemia if adjusted accordingto glomerular filtration rate. British Journal ofHaematology. 2009; 145: 801-5.
32. Molica S, Vitelli G, Levato D, Levato L, Dattilo A,Gandolfo GM. Clinico-biological implications of
increased serum levels of interleukin-8 in B-cell chro-nic lymphocytic leukemia. Haematologica. 1999; 84:208-11.
33. Fayad L, Keating MJ, Reuben JM, O’Brien S, Lee BN,Lerner S, Kurzrock R. Interleukin-6 and interleukin-10 levels in chronic lymphocytic leukemia: correla-tion with phenotypic characteristics and outcome.Blood. 2001; 97: 256-63.
34. Wierda WG, Johnson MM, Do KA, Manshouri T, DeyA, O’Brien S, Giles FJ, Kantarjian H, Thomas D, FaderlS, Lerner S, Keating M, Albitar M. Plasma interleu-kin 8 level predicts for survival in chronic lymphocy-tic leukaemia. Br J Haematol. 2003; 120: 452-6.
35. Ferrajoli A, Keating MJ, Manshouri T, Giles FJ, DeyA, Estrov Z, et al. The clinical significance of tumornecrosis factor-alpha plasma level in patients havingchronic lymphocytic leukemia. Blood. 2002; 100:1215-9.
36. Manshouri T, Do KA, Wang X, Giles FJ, O’Brien SM,Saffer H, et al. Circulating CD20 is detectable in theplasma of patients with chronic lymphocytic leuke-mia and is of prognostic significance.Blood. 2003;101: 2507-13.
37. Albitar M, Johnson MM, Giles FJ, Jilani I, O’Brien S,Cortes J, et al. Free circulating solubile CD52 as atumor marker in chronic lymphocytic leucemia andits implication in therapy with anti-CD52 antibodies.Cancer. 2004; 101: 999-1008.
38. Molica S, Vitelli G, Levato D, Crispino G, Dell’Olio M,Dattilo A, et al. CD27 in B-cell chronic lymphocyticleukemia. Cellular expression, serum release and cor-relation with other soluble molecules belonging to ner-ve growth factor receptors (NGFr) superfamily.Haematologica. 1998; 83: 398-402.
39. Eisterer W, Bechter O, Söderberg O, Nilsson K, TerolM, Greil R, et al. Elevated levels of soluble CD44 areassociated with advanced disease and in vitro proli-feration of neoplastic lymphocytes in B-cell chroniclymphocytic leukaemia. Leuk Res. 2004; 28: 1043-51.
40. Van Bockstaele F, Verhasselt B, Philippè J. Prognosticmarkers in chronic lymphocytic leucemia: a compre-hensive review. Blood Rev. 2009; 23: 25-47.
41. Dohner H, Stilgenbauer S, Benner A, Leupolt E,Krober A, Bullinger L, et al. Genomic aberrations andsurvival in chronic lymphocytic leukemia. N Engl JMed. 2000; 343: 1910-6.
42. Durig J, Naschar M, Schmucker U, Renzing-KohlerK, Holter T, Huttmann A, et al. CD38 expression isan important prognostic marker in chronic lympho-cytic leukaemia. Leukemia. 2002; 16: 30-5.
43. Schroers R, Griesinger F, Trümper L, Haase D, KulleB, Klein-Hitpass L, et al. Combined analysis of ZAP-70 and CD38 expression as a predictor of diseaseprogression in B-cell chronic lymphocytic leukemia.Leukemia. 2005; 19: 750-8.
44. Shanafelt TD, Rabe KG, Kay NE, Zent CS, JelinekDF, Reinalda MS. Age at diagnosis and the utility of

35Fattori prognostici
prognostic testing in patients with chronic lympho-cytic leukemia. Cancer. 2010 Jun 24. [Epub aheadof print].
45. Hallek M, Cheson BD, Catovsky D, Caligaris-CappioF, Dighiero G, Döhner H, et al. International Workshopon Chronic Lymphocytic Leukemia. Guidelines for thediagnosis and treatment of chronic lymphocytic leu-kemia: a report from the International Workshop onChronic lynmphocytic leukemia updating the NationalCancer Institute Working Group 1996 guidelines.Blood. 2008; 111: 5446-56.
46. Haferlach C, Dicker F, Schnittger S, Kern W, HaferlachT. Comprehensive genetic characterization of CLL: astudy on 506 cases analysed with chromosome ban-ding analysis, interphase FISH, IgV(H) status andimmunophenotyping. Leukemia. 2007; 21: 2442-51.
47. Shanafelt TD, Witzig TE, Fink SR, Jenkins RB,Paternoster SF, Smoley SA, et al. Prospective eva-luation of clonal evolution during long-term follow-up of patients with untreated early-stage chroniclymphocytic leukemia. J Clin Oncol. 2006; 24: 4634-41.
48. Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S,Noch E. Frequent deletions and down-regulation ofmicro-RNA genes miR15 and miR16 at 13q14 in chro-nic lymphocytic leukemia. Proc Natl Acad Sci USA.2002; 99: 15524-9.
49. Grubor V, Krasnitz A, Troge JE, Meth JL, Lakshmi B,Kendall JT, et al. Novel genomic alterations and clo-nal evolution in chronic lymphocytic leukemia revea-led by representational oligonucleotide microarrayanalysis (ROMA). Blood. 2009; 113: 1294-303.
50. Döhner H, Stilgenbauer S, James MR, Benner A,Weilguni T, Bentz M, et al. 11q deletions identify anew subset of B-cell chronic lymphocytic leukemiacharacterized by extensive nodal involvement andinferior prognosis. Blood. 1997; 89: 2516-22.
51. Stilgenbauer S, Sander S, Bullinger L, Benner A,Leupolt E, Winkler D, et al. Clonal evolution in chro-nic lymphocytic leukemia: acquisition of high-riskgenomic aberrations associated with unmutated VH,resistance to therapy, and short survival.Haematologica. 2007; 92: 1242-5.
52. Catovsky D, Richards S, Matutes E, Oscier D, DyerMJ, Bezares RF, et al. Haematological OncologyClinical Studies Group; NCRI Chronic LymphocyticLeukaemia Working Group. Assessment of fluda-rabine plus cyclophosphamide for patients withchronic lymphocytic leukaemia (the LRF CLL4 Trial):a randomised controlled trial. Lancet. 2007; 370:230-9.
53. Grever MR, Lucas DM, Dewald GW, Neuberg DS,Reed JC, Kitada S, et al. Comprehensive assessmentof genetic and molecular features predicting outco-me in patients with chronic lymphocytic leukemia:results from the US Intergroup Phase III Trial E2997.J Clin Oncol. 2007; 25: 799-804.
54. Eichhorst BF, Busch R, Hopfinger G, Pasold R, HenselM, Steinbrecher C, et al. Fludarabine plus cyclopho-sphamide versus fludarabine alone in first-line the-rapy of younger patients with chronic lymphocytic leu-kemia. German CLL Study Group. Blood. 2006; 107:885-91.
55. Tsimberidou AM, Tam C, Abruzzo LV, O’Brien S,Wierda WG, Lerner S, et al. Chemoimmunotherapymay overcome the adverse prognostic significanceof 11q deletion in previously untreated patients withchronic lymphocytic leukemia. Cancer. 2009; 115,373-80.
56. Glassman AB, Hayes KJ. The value of fluorescencein situ hybridization in the diagnosis and prognosisof chronic lymphocytic leukemia. Cancer GenetCytogenet. 2005; 158: 88-91.
57. Zenz T, Kröber A, Scherer K, Häbe S, Bühler A, BennerA, et al. Monoallelic TP53 inactivation is associatedwith poor prognosis in chronic lymphocytic leukemia:results from a detailed genetic characterization withlong-term follow-up. Blood. 2008; 112: 3322-9.
58. Schetelig J, van Biezen A, Brand R, Caballero D,Martino R, Itala M, et al. Allogeneic hematopoieticstem-cell transplantation for chronic lymphocytic leu-kemia with 17p deletion: a retrospective EuropeanGroup for Blood and Marrow Transplantation analy-sis. J Clin Oncol. 2008; 26: 5094-100.
59. Tam CS, Shanafelt TD, Wierda WG, Abruzzo LV, VanDyke DL, O’Brien S, et al. De novo deletion 17p13.1chronic lymphocytic leukemia shows significant cli-nical heterogeneity: the M. D. Anderson and MayoClinic experience. Blood. 2009; 114: 957-64.
60. Dicker F, Herholz H, Schnittger S, Nakao A, PattenN, Wu L, et al. Kern W, Haferlach T, Haferlach C. Thedetection of TP53 mutations in chronic lymphocyticleukemia independently predicts rapid disease pro-gression and is highly correlated with a complex aber-rant karyotype. Leukemia. 2009; 23: 117-24.
61. Zenz T, Döhner H, Stilgenbauer S. Genetics and risk-stratified approach to therapy in chronic lymphocy-tic leukemia. Best Pract Res Clin Haematol. 2007; 20:439-53.
62. Cuneo A, Rigolin GM, Bigoni R, De Angeli C, VeroneseA, Cavazzini F, et al. Chronic lymphocytic leukemiawith 6q- shows distinct hematological features andintermediate prognosis. Leukemia. 2004; 18: 476-83.
63. Haferlach C, Dicker F, Weiss T, Schnittger S, BeckC, Grote-Metke A, Oruzio D, Kern W, Haferlach T.Toward a comprehensive prognostic scoring systemin chronic lymphocytic eukemia based on a combi-nation of genetic parameters. Genes ChromosomesCancer. 2010; 49: 851-9.
64. Fais F, Ghiotto F, Hashimoto S, Sellars B, Valetto A,Allen SL et al. Chronic lymphocytic leukaemia B cellsexpress restricted sets of mutated and unmutatedantigen receptors. J Clinical Invest. 1998; 102:1515-25.

36 Seminari di Ematologia Oncologica
65. Hamblin TJ, Davis Z, Gardiner A, Oscier DG,Stevenson FK. Unmutated Ig V(H) genes are asso-ciated with a more aggressive form of chronic lym-phocytic leukemia. Blood. 1999; 94: 1848-54.
66. Damle RN, Wasil T, Fais F, et al. Ig V gene statusand CD38 expression as novel prognostic indica-tors in chronic lymphocytic leukemia. Blood. 1999;94: 1840-7.
67. Hamblin T. Chronic lymphocytic leukaemia: one disea-se or two? Ann Hematol. 2002; 81: 299-303.
68. Rosenwald A, Alizadeh AA, Widhopf G, Simon R,Davis RE, Yu X, et al. Relation of gene expression phe-notype to immunoglobulin mutation genotype in B cellchronic lymphocytic leukemia. J Exp Med. 2001; 194:1639-47.
69. Vasconcelos Y, Davi F, Levy V, Oppezzo P, MagnacC, Michel A et al. Binet’s staging system and VHgenes are independent but complementary progno-stic indicators in chronic lymphocytic leukemia. J ClinOncol. 2003; 21: 3928-32.
70. Oscier DG, Gardiner AC, Mould SJ, et al. Multivariateanalysis of prognostic factors in CLL: clinical stage,IGVH gene mutational status, and loss or mutationof the p53 gene are independent prognostic factors.Blood. 2002; 100: 1177-84.
71. Tobin G, Thunberg U, Johnson A, Thörn I, SöderbergO, Hultdin M, et al. Somatically mutated Ig V(H)3-21genes characterize a new subset of chronic lympho-cytic leukemia. Blood. 2002; 99: 2262-4.
72. Deaglio S, Vaisitti T, Aydin S, Ferrero E, Malavasi F.In-tandem insight from basic science combined withclinical research: CD38 as both marker and key com-ponent of the pathogenetic network underlyingchronic lymphocytic leukemia. Blood. 2006; 108:1135-44.
73. Morabito F, Damle RN, Deaglio S, Keating M,Ferrarini M, Chiorazzi N. The CD38 ectoenzyme fami-ly: advances in basic science and clinical practice.Mol Med. 2006; 12: 342-4.
74. Kröber A, Seiler T, Benner A, Bullinger L, Brückle E,Lichter P, et al. V(H) mutation status, CD38 expres-sion level, genomic aberrations, and survival in chro-nic lymphocytic leukemia. Blood. 2002; 100: 1410-6.
75. Ibrahim S, Keating M, Do KA, O’Brien S, Huh YO,Jilani I, et al. CD38 expression as an important pro-gnostic factor in B-cell chronic lymphocytic leukemia.Blood. 2001; 98: 181-6.
76. Ghia P, Guida G, Stella S, Gottardi D, Geuna M, etal. The pattern of CD38 expression defines a distinctsubset of chronic lymphocytic leukemia (CLL)patients at risk of disease progression. Blood. 2003;101: 1262-9.
77. Hamblin TJ, Orchard JA, Ibbotson RE, Davis Z,Thomas PW, Stevenson FK, et al. CD38 expressionand immunoglobulin variable region mutations areindependent prognostic variables in chronic lympho-cytic leukemia, but CD38 expression may vary
during the course of the disease. Blood. 2002; 99:1023-9.
78. Wiestner A, Rosenwald A, Barry TS, Wright G, DavisRE, et al. ZAP-70 expression identifies a chronic lym-phocytic leukemia subtype with unmutated immuno-globulin genes, inferior clinical outcome, and distinctgene expression profile. Blood. 2003; 101: 4944-51.
79. Nolz JC, Tschumper RC, Pittner BT, Darce JR, KayNE, Jelinek DF. ZAP-70 is expressed by a subset ofnormal human B-lymphocytes displaying an activa-ted phenotype. Leukemia. 2005; 19: 1018-24.
80. Admirand JH, Rassidakis GZ, Abruzzo LV, ValbuenaJR, Jones D, Medeiros LJ. Immunohistochemicaldetection of ZAP-70 in 341 cases of non-Hodgkin andHodgkin lymphoma. Mod Pathol. 2004; 17: 954-61.
81. Chen L, Widhopf G, Huynh L, Rassenti L, Rai KR,Weiss A, et al. Expression of ZAP-70 is associatedwith increased B-cell receptor signaling in chroniclymphocytic leukemia. Blood. 2002; 100: 4609-14.
82. Chen L, Apgar J, Huynh L, Dicker F, Giago-McGahanT, Rassenti L, et al. ZAP-70 directly enhances IgMsignaling in chronic lymphocytic leukemia. Blood.2005; 105: 2036-41.
83. Richardson SJ, Matthews C, Catherwood MA,Alexander HD, Carey BS, Farrugia J, et al. ZAP-70expression is associated with enhanced ability torespond to migratory and survival signals in B-cellchronic lymphocytic leukemia (B-CLL). Blood. 2006;107: 3584-92.
84. Crespo M, Villamor N, Giné E, Muntañola A, ColomerD, Marafioti T, et al. ZAP-70 expression in normalpro/pre B cells, mature B cells, and in B-cell acutelymphoblastic leukemia. Clin Cancer Res. 2006; 12:726-3.
85. Orchard JA, Ibbotson RE, Davis Z, Wiestner A,Rosenwald A, Thomas PW, et al. ZAP-70 expressionand prognosis in chronic lymphocytic leukaemia.Lancet. 2004; 363: 105-11.
86. Rassenti LZ, Huynh L, Toy TL, Chen L, Keating MJ,Gribben JG, et al. ZAP-70 compared with immuno-globulin heavy-chain gene mutation status as a pre-dictor of disease progression in chronic lymphocy-tic leukemia. N Engl J Med. 2004; 351: 893-901.
87. Del Principe MI, Del Poeta G, Buccisano F, MaurilloL, Venditti A, Zucchetto A, et al. Clinical significanceof ZAP-70 protein expression in B-cell chronic lym-phocytic leukemia. Blood. 2006; 108: 853-61.
88. Del Giudice I, Morilla A, Osuji N, Matutes E, MorillaR, Burford A, et al. Zeta-chain associated protein 70and CD38 combined predict the time to first treat-ment in patients with chronic lymphocytic leukemia.Cancer. 2005; 104: 2124-32.
89. Rassenti LZ , Jain S, Keating MJ, Wierda WG, GreverMR, Byrd JC, et al. Relative value of ZAP-70, CD38,and immunoglobulin mutation status in predictingaggressive disease in chronic lymphocytic leukemia.Blood. 2008; 112: 1923-30.

37Fattori prognostici
90. Zanotti R, Ambrosetti A, Lestani M Ghia P, PattaroC, Remo C, et al. ZAP-70 expression, as detectedby immunohistochemistry on bone marrow biopsiesfrom early-phase CLL patients, is a strong adverseprognostic factor. Leukemia. 2007; 21: 102-9.
91. Admirand JH, Knoblock RJ, Coombes KR, Tam C,Schlette EJ, Wierda WG, Immunohistochemicaldetection of ZAP70 in chronic lymphocytic leukemiapredicts immunoglobulin heavy chain gene mutationstatus and time to progression. Mod Pathol. 2010;23: 1518-1523.
92. Kröber A, Bloehdorn J, Hafner S, Bühler A, Seiler T,Kienle D, et al. Additional genetic high-risk featuressuch as 11q deletion, 17p deletion, and V3-21 usa-ge characterize discordance of ZAP-70 and VH muta-tion status in chronic lymphocytic leukemia. J ClinOncol. 2006; 24: 969-75.
93. Kröber A, Seiler T, Benner A, Bullinger L, Brückle E,Lichter P, et al. V(H) mutation status, CD38 expres-sion level, genomic aberrations, and survival in chro-nic lymphocytic leukemia. Blood. 2002; 100: 1410-6.
94. Oppezzo P, Vasconcelos Y, Settegrana C, Jeannel D,Vuillier F, et al. French Cooperative Group on CLL.The LPL/ADAM29 expression ratio is a novel progno-sis indicator in chronic lymphocytic leukemia. Blood.2005; 106: 650-7.
95. Heintel D, Kienle D, Shehata M, Kröber A, KroemerE, Schwarzinger I, et al. CLL Study Group. Highexpression of lipoprotein lipase in poor risk B-cellchronic lymphocytic leukemia. Leukemia. 2005; 19:1216-23
96. Nuckel H, Huttmann A, Klein-Hitpass L, Schroers R,Fuhrer A, Sellmann L, et al. Lipoprotein lipase expres-sion is a novel prognostic factor in B-cell chronic lym-phocytic leukemia. Leuk Lymphoma. 2006; 47:1053-61.
97. Albesiano E, Messmer BT, Damle RN, Allen SL, RaiKR, Chiorazzi N. Activation-induced cytidine dea-minase in chronic lymphocytic leukemia B cells:expression as multiple forms in a dynamic, varia-bly sized fraction of the clone. Blood. 2003; 102:3333-9.
98. Heintel D, Kroemer E, Kienle D, Schwarzinger I, GleissA, Schwarzmeier J, et al. German CLL Study Group.High expression of activation-induced cytidine dea-minase (AID) mRNA is associated with unmutatedIGVH gene status and unfavourable cytogenetic aber-rations in patients with chronic lymphocytic leukae-mia. Leukemia. 2004; 18: 756-62.
99. Buhl AM, Jurlander J, Jørgensen FS, Ottesen AM,Cowland JB, Gjerdrum LM, et al. Identification of agene on chromosome 12q22 uniquely overexpres-sed in chronic lymphocytic leukemia. Blood. 2006;107: 2904-11.
100. Josefsson P, Geisler CH, Leffers H, Petersen JH,Andersen MK, Jurlander J, et al. CLLU1 expressionanalysis adds prognostic information to risk predic-
tion in chronic lymphocytic leukemia. Blood. 2007;109: 4973-9.
101. Chen L, Li J, Zheng W, Zhang Y, Wu Y, Li L, et al.The prognostic evaluation of CLLU1 expression levelsin 50 Chinese patients with chronic lymphocytic leu-kemia. Leuk Lymphoma. 2007; 48: 1785-92.
102. Gattei V, Bulian P, Del Principe MI, Zucchetto A,Maurillo L, Buccisano F, et al. Relevance of CD49dprotein expression as overall survival and progressi-ve disease prognosticator in chronic lymphocytic leu-kemia. Blood. 2008; 111: 865-73.
103. Shanafelt TD, Geyer SM, Bone ND, Tschumper RC,Witzig TE, Nowakowski GS, et al. CD49d expressionis an independent predictor of overall survival inpatients with chronic lymphocytic leukaemia: a pro-gnostic parameter with therapeutic potential. Br JHaematol. 2008; 140: 537-46.
104. Damle RN, Batliwalla FM, Ghiotto F, Valetto A,Albesiano E, Sison C, et al. Telomere length and telo-merase activity delineate distinctive replicative featu-res of the B-CLL subgroups defined by immunoglo-bulin V gene mutations. Blood. 2004; 103: 375-82.
105. Terrin L, Trentin L, Degan M, Corradini I, BertorelleR, Carli P, et al. Telomerase expression in B-cell chro-nic lymphocytic leukemia predicts survival and deli-neates subgroups of patients with the same igVHmutation status and different outcome. Leukemia.2007; 21: 965-72.
106. Rossi D, Lobetti Bodoni C, Genuardi E, Monitillo L,Drandi D, Cerri M, et al. Telomere length is an inde-pendent predictor of survival, treatment requirementand Richter’s syndrome transformation in chronic lym-phocytic leukemia. Leukemia. 2009; 23: 1062-72.
107. Burger JA, Ghia P, Rosenwald A, Caligaris-Capio F.The microenvironment in mature B-cell malignancies:a target for new treatment strategie. Blood. 2009; 14:3367-75.
108. Chen H, Treeweeke AT, West DC, et al. In vitro andin vivo production of vascular endothelial growth fac-tor by chronic lymphocytic leukemia cells. Blood.2000; 96: 3181-7.
109. Haiat S, Billard C, Quiney C, Ajchenbaum-CymbalistaF, Kolb JP. Role of BAFF and APRIL in human B-cellchronic lymphocytic leukemia. Immunology. 2006;118: 281-92.
110. Nishio M, Endo T, Tsukada N, Ohata J, Kitada S, ReedJC, et al. Nurselike cells express BAFF and APRILwhich can promote survival of chronic lymphocyticleukemia cells via paracrine pathway distinct from thatof SDF-1a. Blood. 2005; 3: 1012-20.
111. Endo T, Nishio M, Enzler T, Cottam HB, Fukuda T,James DF, et al. BAFF and APRIL support chroniclymphocytic leukemia B-cell survival through activa-tion of the canonical NF-kappaB pathway. Blood.2007; 109: 703-10.
112. Planelles L, Castillo-Gutiérrez S, Medema JP, Morales-Luque A, Merle-Béral H, Hahne M. APRIL but not

38 Seminari di Ematologia Oncologica
BLyS serum levels are increased in chronic lympho-cytic leukemia: prognostic relevance of APRIL for sur-vival. Haematologica. 2007; 92: 1284-5.
113. Bojarska-Junak A, Hus I, Chocholska S, Wasik-Szczepanek E, Sieklucka M, Dmoszyn’ska A, et al.BAFF and APRIL expression in B-cell chronic lym-phocytic leukemia: correlation with biological and cli-nical features. Leuk Res. 2009; 33: 1319-27.
114. Maisonpierre PC, Suri C, Jones PF, Bartunkova S,Wiegand SJ, Radziejewski C, et al. Angiopoietin-2,a natural antagonistic for Tie2 that disrupts in vivoangiogenesis. Science. 1997; 277 (5322): 55-60.
115. Maffei R, Martinelli S, Castelli I, Santachiara R,Zucchini P, Fontana M, et al. Increased angiogene-sis induced by chronic lymphocytic leukemia B cells
is mediated by leukemia-derived Ang2 and VEGF.Leuk Res. 2010; 34: 312-21.
116. Molica S, Vacca A, Ribatti D, Cuneo A, Cavazzini F,Levato D, et al. Prognostic value of enhanced bonemarrow angiogenesis in early B-cell chronic lympho-cytic leukemia. Blood. 2002; 100: 3344-50.
117. Maffei R, Martinelli S, Santachiara R, Rossi D,Guarnotta C, Sozzi E, et al. Angiopoietin-2 plasmadosage predicts time to first treatment and overall sur-vival in chronic lymphocytic leukemia. Blood. 2010;116: 584-90.
118. Molica S. Angiogenesis in B-cell chronic lymphocy-tic leukemia: methods of study, clinical significanceand prognostic implications. Leukemia andLymphoma. 2001; 42: 603-7.

39
n INTRODUZIONE
Nonostante la presenza di popolazioni monoclo-nali di linfociti B nel sangue periferico di soggettiadulti altrimenti sani sia un fenomeno descritto dapiù di 25 anni (1), l’entità diagnostica della linfo-citosi B monoclonale (MBL) è stata solo recente-mente oggetto di inquadramento e definizione (2).Con l’affinamento delle metodiche di laboratorioed in particolare della citofluorimetria multipara-metrica, il riscontro di espansioni B cellulari, inassenza di segni e sintomi di patologia linfopro-liferativa, è divenuto relativamente frequente, cre-ando la necessità di definire meglio la natura ditale condizione. Bisogna infatti considerare che,in più del 75% dei casi, la popolazione B cellu-lare clonale è fenotipicamente indistinguibile dal-le cellule neoplastiche della leucemia linfatica cro-nica (LLC) (2, 3).La LLC è la forma di leucemia più frequente negliadulti del mondo occidentale; secondo datiSEER aggiornati al quinquennio 2003-2007 l’in-
cidenza, ponderata per età, di tale patologia siaggira intorno a 4.2 per 100.000 persone peranno, caratteristicamente più frequente negli indi-vidui di sesso maschile ed in aumento con l’avan-zare dell’età. L’età media alla diagnosi è infatti paria 72 anni, con il 69.2% dei casi diagnosticato inindividui di età superiore a 65 anni, mentre solol’1.7% delle diagnosi viene effettuato in personedi età compresa tra i 20 e i 44 anni (4).Nel corso degli ultimi 50 anni si è assistito adimportanti modificazioni dei criteri diagnostici del-la LLC e grazie al miglioramento delle metodi-che di laboratorio e alla diffusione della citofluo-rimetria, le caratteristiche cliniche all’esordio el’incidenza stessa della malattia sono radicalmen-te cambiate (5). Negli anni ’70, infatti, la diagno-si di LLC in pazienti senza linfoadenopatie, epa-tosplenomegalia o citopenie, che quindi esibiva-no quale unica caratteristica patologica una lin-focitosi a livello del sangue periferico (stadio 0di Rai), si basava su un numero di linfociti circo-lanti di almeno 15.000 per μl (6); le linee guidadel National Cancer Institute (NCI), elaborate cir-ca 20 anni dopo, abbassavano la soglia a 5.000(7, 8). Nelle nuove linee guida dell’InternationalWorkshop on Chronic Lymphocytic Leukemia(IWCLL) pubblicate nel 2008 (9), in considerazio-ne della vasta diffusione di metodiche di citofluo-rimetria in grado di distinguere le diverse sotto-popolazioni linfocitarie ed allo scopo di non cata-logare erroneamente come affetti da LLC ipazienti con linfocitosi T reattiva, il parametro diriferimento per la diagnosi di LLC è divenuto la
Indirizzo per la corrispondenza
Paolo GhiaLaboratorio di Neoplasie Linfoidi BDivisione di Oncologia MolecolareUniversità Vita-Salute San Raffaelec/o 4A3, Via Olgettina, 58 - 20122 MilanoE-mail: [email protected]
Linfocitosi B Linfocitosi B monoclonalemonoclonaleLYDIA SCARFÒ1-3, PAOLO GHIA1,2,41Laboratorio di Neoplasie Linfoidi B, Divisione di Oncologia Molecolare, Università Vita-Salute San Raffaele, Milano;2Unità Linfomi, Dipartimento di Oncologia, Istituto Scientifico San Raffaele, Milano;3Ematologia, Università degli Studi di Ferrara, Ferrara;4Università Vita-Salute San Raffaele, Milano
Paolo Ghia
Parole chiave: linfocitosi B monoclonale, leucemia lin-fatica cronica, invecchiamento del sistema immunitario

40 Seminari di Ematologia Oncologica
presenza, a livello del sangue periferico, di unnumero di linfociti B (CD19+) pari o superiore a5.000 per μl. È rimasto invece inalterato negli anniil requisito del riscontro del tipico immunofeno-tipo della LLC, caratterizzato dall’espressione dimarcatori B-cellulari (alcuni espressi ad intensi-tà ridotta, quali CD20 (10) e le immunoglobuli-ne di superficie), associati alla positività per CD23e CD5 (quest’ultimo tipicamente espresso nei lin-fociti T ed in una piccola sottopopolazione deilinfociti B normali). Grazie a questo assetto feno-tipico caratteristico, la sensibilità della citofluo-rimetria a quattro colori permette attualmente dirilevare una cellula LLC tra 10 000 cellule nor-mali, rivelandosi utile anche per l’analisi dellamalattia minima residua nel contesto di studi cli-nici controllati (11). Per questo motivo, il riscon-tro di espansioni B linfocitarie, anche minime, concaratteristiche immunofenotipiche simili allaLLC, negli individui sani è divenuto sempre piùfrequente ed ha portato alla definizione dell’en-tità diagnostica della MBL, creata nel 2005. Conquesto termine si indica la presenza di linfocitiB monoclonali nel sangue periferico, in assen-za di linfoadenopatie, organomegalia o sintomiB, e con una concentrazione inferiore a 5.000 perμl, il cut-off numerico al di sopra del quale, comesopra enunciato, si deve porre diagnosi di LLC(9). Poichè la MBL LLC-simile è fenotipicamen-te indistinguibile dalla LLC vera e propria, l’as-segnazione all’una od all’altra categoria dipen-de sostanzialmente dal valore numerico dei lin-fociti B.La creazione di una categoria diagnostica riser-vata alla MBL ha posto quindi le basi necessarieper garantire l’uniformità e la possibilità di con-fronto di studi a carattere epidemiologico e bio-logico. La definizione e la caratterizzazione dellalinfocitosi B monoclonale come entità a sé e l’ana-lisi della relazione tra MBL e LLC sono state ogget-to di intensa ricerca negli ultimi anni, che ha pro-dotto una notevole mole di risultati, dando spa-zio ad un acceso dibattito sulla natura della MBLe sul suo potenziale rischio di evoluzione in LLC(12). Un più preciso inquadramento biologico eclinico della MBL riveste infatti un ruolo chiave sot-to almeno tre aspetti: in primo luogo, a seguitodel diffuso miglioramento delle tecniche di rileva-zione, è prevedibile che il riscontro di individui
affetti da MBL nella pratica clinica quotidianadiventi un fenomeno progressivamente più fre-quente, con la gestione del quale il clinico dovràsempre più spesso confrontarsi. Inoltre, come illu-strato in seguito, dato che una quota, seppurminoritaria, di individui affetti da MBL progredi-sce a franca LLC, chiarire i fattori che determi-nano questa evoluzione è importante dal puntodi vista clinico per la necessità di stabilire un fol-low up che sia il più possibile adattato al pazien-te. Infine, come verificatosi in passato per altrecondizioni, una più approfondita comprensionedelle caratteristiche biologiche della potenzialelesione pre-neoplastica può costituire un puntodi partenza privilegiato per capire la biologia del-la malattia.
n DEFINIZIONE E CARATTERISTICHEIMMUNOFENOTIPICHE
Come premesso, la definizione di MBL prevedeil riscontro di un’espansione B cellulare con carat-teristiche immunofenotipiche peculiari e la restri-zione del rapporto kappa/lambda, indice della pre-senza di una popolazione monoclonale (2, 13).Anche se il sottotipo più frequente di MBL è quel-lo fenotipicamente identico alla LLC, bisogna ricor-dare che esistono anche altre due sottocatego-rie (Tabella 1 e Figura 1).- MBL con fenotipo di LLC atipica: positivo perla presenza di CD5 e CD19, con variabile espres-sione di CD23, ma con CD20 e immunoglobuli-ne di superficie espressi ad elevata intensità.Poiché il linfoma mantellare, un sottotipo di lin-foma non Hodgkin a decorso tendenzialmenteaggressivo, condivide le stesse caratteristicheimmunofenotipiche di questa variante, nei casi diMBL LLC-atipica CD23 negativa è importantericercare, nella popolazione B-cellulare aberran-te, la presenza della traslocazione (11; 14), pro-pria del linfoma mantellare, allo scopo di evitareil potenziale mancato riconoscimento di questapatologia;- MBL con fenotipo non-LLC: negativo per CD5,ma anche per altri marcatori tipici di altre malat-tie linfoproliferative quali CD10 (espresso nei lin-fomi follicolari); presenta un immunofenotipo simi-le a quello dei linfomi della zona marginale.

41Linfocitosi B monoclonale
FIGURA 1 - Sottoclassificazione sulla base dell’immunofenotipo della MBL (Gentile concessione di Claudia Fazi, Istituto ScientificoSan Raffaele, Milano).
TABELLA 1 - Criteri diagnostici e sottoclassificazione della MBL [adattato da (13)].
CRITERI DIAGNOSTICI
1. Riscontro documentato di una popolazione B-cellulare clonale1 tramite uno o più dei seguenti parametri:A. Restrizione delle catene leggere:
- rapporto kappa/lambda >3:1 or <0.3:1 oppure- più del 25% dei linfociti B privi dell’espressione o con espressione a bassa intensità delle immunoglobuline di super-
ficie B.Riarrangiamenti IGHV monoclonali delle catene pesanti
2. Presenza di un immunofenotipo malattia-associato2
3. Valore assoluto dei linfociti B <5x109 cellule/L4. Nessuna altra caratteristica suggestiva di disordine autoimmune o linfoproliferativo
A. Esame obiettivo nei limiti di norma (assenza di linfoadenopatie o organomegalie)B.assenza di sintomi B (e.g. astenia, calo ponderale, sudorazioni notturne, febbricola serotina) attribuibili a linfoma non
Hodgkin (LNH)C.Assenza di malattie autoimmuni o infettive in atto
SOTTOCLASSIFICAZIONE:
A. Fenotipo LLC-simile- coespressione di CD5 e CD19, CD20 a bassa intensità (dim) e CD23- restrizione della catena leggera con bassa intensità (dim) di espressione delle immunoglobuline di superficie (cloni MBL
di minima entità possono essere oligoclonali e perciò non esibire restrizione di catene leggere)B.Fenotipo LLC atipica
- coespressione di CD5 e CD19; CD20 espresso ad alta intensità (bright); possibile negatività per CD23- restrizione della catena leggera con intensità di espressione delle immunoglobuline di superficie da moderata a intensa- consigliata ricerca della t(11;14) per escludere il linfoma mantellare
C.Fenotipo Non-LLC - negatività per CD5- espressione di CD20 normale o elevata- restrizione della catena leggera con intensità di espressione delle immunoglobuline di superficie da moderata a intensa
1Se possibile, determinazioni ripetute dovrebbero dimostrare la stabilità della popolazione B-cellulare monoclonale per un periodo di almeno 3 mesi.2In assenza di un immunofenotipo malattia-associato, la restrizione del rapporto kappa/lambda può derivare da un processo reattivo.

42 Seminari di Ematologia Oncologica
n STUDI DI POPOLAZIONE E STUDI CLINICI: QUALI DIFFERENZE?
Quando furono introdotti nel 2005, i criteri di dia-gnosi della MBL prevedevano la determinazio-ne citofluorimetrica di un cluster con un nume-ro minimo di 50 eventi costituiti da linfociti Bmonoclonali, ma non specificavano un metodocitofluorimetrico standard. La frequenza con cuitale parametro può essere riscontrato quindi èmolto variabile e dipende da vari fattor tecniciquali l’approccio citofluorimetrico di elezione, ilnumero di eventi acquisiti e la combinazione dianticorpi impiegata.Un altro importante parametro da tenere in con-siderazione è la tipologia di individui che vengo-no studiati ed in particolare se tale condizione vie-ne riscontrata in:• studi di popolazione: comprendono studi
svolti su soggetti sani o comunque con distur-bi di tipo non ematologico né neoplastico; ipartecipanti a questo tipo di studi mostrano unesame emocromocitometrico nei limiti di nor-ma. Una categoria particolare di studi di popo-lazione, intesi appunto come indagini rivolte adindividui sani con parametri ematici non alte-rati, è rappresentata dalle indagini effettuate neimembri di in famiglie con elevata incidenza diLLC (cosiddette LLC familiari, analizzateappunto in studi familiari);
• studi clinici: effettuati su pazienti con rilievodi linfocitosi all’esame emocromo, che nonsoddisfano i criteri diagnostici per la LLC o peraltri disordini linfoproliferativi, infettivi o autoim-muni.
Nella definizione di MBL risultano con ogni pro-babilità incluse, quindi, entità cliniche e biologi-che differenti, seppur indistinguibili dal punto divista immunofenotipico, come dimostrato dal-l’analisi cumulativa dei dati registrati a livello mon-diale (14). Nella revisione critica dei principali studi pubbli-cati è perciò importante differenziare il tipo di MBLstudiato (a bassa conta vs associata a linfocito-si) ed il contesto di partenza (popolazione gene-rale vs famiglie ad alto rischio per LLC vs ambi-to clinico), allo scopo di inquadrare correttamen-te i molteplici, e talvolta contrastanti, risultati attual-mente disponibili.
Studi di popolazioneI primi studi di popolazione, intesi a determinarel’esistenza e l’incidenza di popolazioni di linfoci-ti B monoclonali, furono pianificati nei primi anni’90 negli Stati Uniti, nell’ambito di studi ambien-tali volti a chiarire il ruolo della residenza in areead elevato inquinamento come fattore predispo-nente per lo sviluppo di un’ampia varietà di distur-bi (15, 16). Tra il 1991 ed il 1994, 1926 parteci-panti, residenti in aree inquinate dal punto di vistaambientale ed in zone più salubri di confronto,furono sottoposti ad una serie di tests, compren-dente l’analisi delle sottopopolazioni linfocitarie (lin-fociti B, T, NK e subsets di specifico interesse),con metodiche di citofluorimetria a 2 colori. Il pan-nello immunofenotipico iniziale non prevedeva laricerca di popolazioni clonali tramite determina-zione del rapporto kappa/lambda, ma gli indivi-dui che presentavano un incremento dei linfocitiB venivano sottoposti a test aggiuntivi per la ricer-ca di un eventuale popolazione clonale. Un clo-ne B fu riscontrato in 11 soggetti, di età compre-sa tra 47 e 72 anni; 2 di questi vennero in segui-to esclusi dall’analisi in quanto risultati affetti daLLC. La prevalenza globale di MBL nella popo-lazione campione di età superiore a 45 anni si rive-lò di conseguenza pari allo 0.6% (9/1499). Sei per-sone (delle 9 iniziali) vennero seguite nel tempo,e in tutti fu confermata la persistenza del clone.Nel corso degli studi successivi, furono riscontra-ti altri 2 casi grazie alla determinazione del rap-porto kappa/lambda nello screening iniziale. Adun duplice follow up a distanza di 3 e 9 anni, 3(37%) rimasero stabili, 3 (37%) risultarono dece-duti per cause non correlate, ad un partecipantecon MBL fu diagnosticata una macroglobuline-mia di Waldenström, stabile nel tempo, ed infineun altro soggetto sviluppò una LLC, causa deldecesso al momento del secondo follow up(2003). Successive analisi di screening su dona-tori di sangue confermarono nel complesso unabassa frequenza della MBL (17, 18), e fu solo qual-che anno più tardi, quindi, che, grazie a metodi-che citofluorimetriche più avanzate, venne corret-tamente inquadrata la reale entità del fenomeno.In particolare, in Inghilterra Rawstron et al. (19)scelsero di analizzare una popolazione di indivi-dui con esame emocromocitometrico normale,ricorsi alle cure mediche presso una struttura

43Linfocitosi B monoclonale
ospedaliera per motivazioni non connesse conpotenziali patologie ematologiche e con anamne-si negativa per neoplasie. Vennero utilizzati duepannelli citofluorimetrici a quattro colori compren-denti le combinazioni di anticorpi controCD20/CD79b/CD19/CD5 e κ/λ/ CD19/CD5, conl’acquisizione di un numero più elevato di even-ti totali (almeno 200.000). Furono valutati 910 cam-pioni di sangue periferico (425 da uomini e 485da donne, di età superiore ai 40 anni), bilanciatiin maniera da riprodurre, seppur su scala ridot-ta, la distribuzione per sesso ed età della popo-lazione britannica. In tale gruppo campione la pre-valenza della MBL con caratteristiche simili allaLLC risultò pari al 3.5%, delineandosi un nettoincremento in proporzione all’età (2.1% negli indi-vidui di età compresa tra 40 e 60 anni, a confron-to con 5.0% nei soggetti di età superiore a 60 anni)ed al sesso (con una chiara predominanzamaschile) (Figura 2). In 9/910 individui (1.0%) fuindividuata, grazie allo sbilanciamento del rappor-to kappa/lambda, una popolazione monoclona-le CD5-negativa. Da segnalare che il numero asso-luto di linfociti B LLC-simili riscontrati risultava mol-to basso, con un valore assoluto mediano di 13
per μl (da un minimo di 3 ad un massimo di 1.458per μl), ed un valore percentuale mediano, rispet-to al totale dei linfociti B, dell’11% (compreso tra3 e 95%).Avvalendosi di un approccio citofluorimetrico ana-logo, Ghia et al. (20) ricercarono la presenza dipopolazioni B cellulari monoclonali in 500 indivi-dui di età superiore a 65 anni (269 donne e 211uomini), sottoposti a prelievo ematico per esamidi routine (essenzialmente emocromo ed asset-to lipidico), in assenza di linfocitosi o altre altera-zioni dei parametri ematici e con anamnesi nega-tiva per patologie oncologiche. L’età media del-la popolazione risultava pari a 73.7 anni (range 65-98 anni) e lo studio fu effettuato in un lasso tem-porale di circa 20 mesi allo scopo di escludereeventuali distorsioni stagionali, potenzialmente ingrado di inficiare i risultati dell’analisi. La frequen-za cumulativa di MBL LLC-simili in questa popo-lazione si presentava pari a 5.5% con un valoremolto simile al 5% rilevato nello studio inglesenegli individui di età superiore a 60 anni, indican-do l’universalità del fenomeno MBL, risultato indi-pendente da fattori geografici. Anche in questapopolazione veniva riscontrato un chiaro incre-
18-40 40-49 50-59 60-69 70-79 80-89 ≥90
80
60
40
20
0
Età (anni)
Inghilterra
Italia
Spagna
Freq
uenz
a M
BL (%
)
FIGURA 2 - Prevalenza della MBL LLC-simile nelle diverse fasce d’età in relazione al numero di fluorescenze utilizzate e di eventiacquisiti. Inghilterra (19, 24): 4 fluorescenze, 200.000 eventi acquisiti; Italia (21): 5 fluorescenze, 500.000 eventi acquisiti; Spagna(23): 8 fluorescenze, 5.000.000 eventi acquisiti.

44 Seminari di Ematologia Oncologica
mento di prevalenza con l’avanzare dell’età, par-ticolarmente evidente negli individui di età supe-riore a 75 anni. In valore percentuale, il clone LLC-simile costituiva tendenzialmente una proporzio-ne molto bassa dei linfociti B (media 1.8%, ran-ge 0.7-4%), con un valore medio di linfociti BCD19+ pari a 165 per μl, compreso tra un mini-mo di 85 ed un massimo di 264 per μl. Più recentemente Ghia et al. (21) hanno analiz-zato per la prima volta la prevalenza della MBLin una popolazione di individui arruolati in un pro-getto non inteso a chiarire uno specifico proble-ma di salute, ma a valutare il ruolo della compo-nente genetica nel potenziale sviluppo di comu-ni malattie (ad esempio, ipertensione arteriosa,osteoporosi, disturbi tiroidei). In tale contesto1.725 individui di età superiore a 18 anni, resi-denti in una valle isolata dell’Italia del Nord, sono
stati sottoposti a caratterizzazione immunofeno-tipica utilizzando un pannello a cinque colori edacquisendo un totale di 500.000 eventi, incremen-tando così la sensibilità della metodica. Il nume-ro totale di MBL rilevate corrispondeva a 128(7.4%) e, come previsto, le MBL LLC-simili rap-presentavano la maggioranza dei casi (89/128)con una prevalenza pari al 5.2% dell’intera coor-te e 6.7% tra gli individui >40 anni di età. Le MBLLLC atipica e le MBL non-LLC risultavanorispettivamente 19/128 e 20/128. È interessan-te notare che l’età media dei soggetti con MBLera pari a 66.9 anni, che la frequenza di tale con-dizione si confermava più elevata negli individuidi sesso maschile e appariva aumentare in manie-ra significativa con l’avanzare dell’età, comedimostrato dal fatto che, nel gruppo di parteci-panti di età superiore a 90 anni, un clone MBL
TABELLA 2 - Parametri clinici e biologici analizzati nei principali studi di popolazione [adattato da (26)].
Parametri Shim Yk Rawstron AC Ghia P Rawstron AC Dagklis A Nieto WGet al.,2007 et al., 2002 et al., 2004 et al., 2008 (27) and Fazi C et al.,
(16) (19) (20) (27) et al., 2009 (21) 2009 (23)
Età media (range) 53.0 anni 57 73.7 anni 74.0§ anni 55.2 anni 62.0 anni (42-70)@ (40-89) (65-98) (62-80) (18-102) (40-97)
Partecipanti 1926 910 500 1520 1725 608Eventi acquisiti N.V. 200.000 200.000 200.000 500.000 5.000.000N° di fluorescenze 2 4 4 4 5 8Prevalenza MBL
LLC-simile• intera coorte 11/1926 32/910 22/500 78/1520 89/1725 73/608
(0.57%) (3.5%) (5.5%) (5.1%) (5.2%) (12.0%)• >60 anni 22/442 22/500# 78/1520 69/770 N.V.
(5.0%) (5.5%) (5.1%) (9.0%) (>20%*)Parametri biologici:Omologia geni IGHV N.V. N.V.• Mutati - 3/3 - 17/20 36/51 2/7
(100%) (85.0%) (70.5%) (28.5%)• Non mutati - 0/3 - 3/20 15/51 5/7
(15.0%) (29.5%) (71.4%)Utilizzo geni IGHV N.V. IGHV3-21 N.V. IGHV3-07, IGHV4-59, N.V.
IGHV3-74 IGHV 3-23, IGHV4-61IGHV 4-34
FISH N.V. N.V. N.V. N.V.• Del13q - - - 15/38 (39.5%) - 10/37 (27.0%)• +12 - - - 4/22 (18.2%) - 2/37 (5.4%)• Del11q - - - 0/21 - 0/37Del17p - - - 0/10 - 0/37
N.V. non valutabile; *calcolato sulla base dei dati in (23);§età mediana; #età >65 anni; @età mediana, 10°-90° percentile.

45Linfocitosi B monoclonale
LLC-simile era rilevabile in circa il 45.0% dei casi(22) (Figura 2). Le popolazioni clonali appartenen-ti ai sottogruppi non-LLC e LLC-atipica erano pre-senti in percentuale significativa anche tra gli indi-vidui più giovani e la loro frequenza risultava menoevidentemente influenzata dall’invecchiamento.Anche in questo contesto, tra i partecipanti conclone MBL LLC-simile il numero di cellule B perμl era compreso nei limiti di norma (valore medio170, range 10-1920 per μl) e le cellule B clonalicostituivano una percentuale variabile, ma ten-denzialmente bassa, dei linfociti B totali, con unamedia del 6.9%; solo 13 casi infatti, mostrava-no una percentuale di cellule clonali superiore al10%. Potenziando ulteriormente la metodica citofluo-rimetrica utilizzata per la rilevazione, ed in parti-colare avvalendosi di una combinazione di ottocolori ed acquisendo fino a 5.000.000 di eventitotali, il gruppo di Salamanca (23) ha recentemen-te evidenziato la presenza di un clone MBL nel12% della popolazione totale, rappresentata da608 individui sani, di età superiore a 40 anni, dimo-strando addirittura l’esistenza di una popolazio-ne monoclonale aberrante in circa il 75% degliindividui di età superiore a 90 anni (Figura 2). Inquesto studio, in conseguenza dell’aumentatasensibilità della tecnica utilizzata, i cloni B costi-tuivano solamente lo 0.38% dei linfociti B totali,variando dallo 0.14 al 4.2% (Tabella 2). La mag-gior parte dei casi individuati (62%), era quindi aldi sotto dei limiti di rilevazione delle metodicheprecedentemente utilizzate e, proprio a causa del-la minima entità delle cellule aberranti, è stato pos-sibile confermare, con metodiche addizionali, laclonalità delle popolazioni individuate solo in 18/73casi, tutti con una percentuale di cellule aberran-ti superiore allo 0.01%.In conclusione, gli studi di popolazione hannochiaramente dimostrato la natura universale del-la MBL, riscontrabile in molteplici condizioni geo-grafiche e demografiche; l’utilizzo di metodichecitofluorimetriche progressivamente più sensibi-li ha inoltre permesso di rilevare un’elevata pre-valenza di questa condizione, strettamente dipen-dente dal numero di fluorescenze utilizzate e deglieventi acquisiti. A questo proposito, tuttavia, èinteressante sottolineare che la prevalenza dellaMBL non aumenta in maniera direttamente pro-
porzionale all’incremento della sensibilità nellametodica citofluorimetrica utilizzata; infatti, nellostudio spagnolo, pur essendo la sensibilità del-l’approccio immunofenotipico circa 10 voltesuperiore, la frequenza della MBL è risultata soloraddoppiata in confronto agli studi precedenti.Questo dato sembrerebbe suggerire che, anchedisponendo in linea teorica di una metodica disensibilità illimitata, non sarebbe possibile indivi-duare un clone MBL in tutti gli individui analizza-ti (22). È d’altra parte innegabile un netto aumen-to di prevalenza di tale condizione con l’invecchia-mento, tanto da poter ipotizzare, in maniera deltutto plausibile, che ogni individuo sia potenzial-mente destinato a sviluppare una MBL, indipen-dentemente da fattori di predisposizione individua-le, se dotato di un’aspettativa di vita sufficiente-mente lunga (22). A supporto di questa idea, gra-zie all’utilizzo di un modello statistico predittivo,è stato recentemente suggerito che, processan-do un più elevato volume di sangue periferico allaricerca di cellule LLC-simili, il 100% degli indivi-dui di età superiore a 70 anni risulterebbe porta-tore di un popolazione B-cellulare aberrante (25).
Studi familiariUn discorso a parte merita il rilievo della condi-zione di MBL nell’ambito di studi familiari. La fami-liarità per LLC rappresenta uno dei più importan-ti fattori di rischio per lo sviluppo della malattia ei familiari di primo grado dei pazienti affetti da LLChanno un rischio relativo di sviluppare una francaleucemia da 2 a 7 volte più elevato rispetto allapopolazione generale (28-31). Alcuni studi hannopertanto valutato la frequenza della MBL in fami-glie ad alto rischio, in cui due o più membri dellastessa famiglia risultano affetti da LLC. Il numerodi casi di MBL in individui appartenenti a tali fami-glie, con emocromo nei limiti è risultato pari a 8/59(13.5%) (32) e 6/33 (18%) (33) in due studi, unoinglese e l’altro americano rispettivamente. In par-ticolare, il rischio complessivo di rilevare un clo-ne LLC-simile in questi individui è 4-6 volte supe-riore a quello della popolazione generale e variain maniera significativa nelle diverse classi d’età,con gli individui più giovani (di età compresa tra16 e 40 anni) portatori di un rischio fino a 17 vol-te superiore rispetto ai coetanei non appartenen-ti a famiglie ad alto rischio (24, 32). Inoltre, tra i sog-

46 Seminari di Ematologia Oncologica
getti appartenenti a famiglie ad alto rischio per LLC,la possibilità di sviluppare una MBL entro l’età di90 anni risulterebbe pari al 61%, a sostegno di unaelevata prevalenza di questa condizione in taleambito, il cui ruolo e le cui potenzialità evolutiverestano però ancora da chiarire (34).
Studi clinici e rischio di progressioneA seguito dell’introduzione dei nuovi criteri dia-gnostici per la LLC, basati su un numero di lin-fociti B superiore a 5.000 per μl, circa il 40% deipazienti classificati in precedenza come LLC sta-dio 0 di Rai, sono improvvisamente rientrati nel-la categoria diagnostica della MBL (35-37).Questo cambiamento ha sottolineato ulteriormen-te la difficoltà di differenziare in modo ottimale lacondizione di LLC con linfocitosi isolata (stadio0 di Rai) dalla MBL ed un semplice valore nume-rico non sembra poter soddisfare questa neces-sità clinica, divenendo quindi oggetto di contro-versia nella comunità scientifica. Allo stesso tem-po, l’analisi di queste coorti di MBL (definite comeMBL con linfocitosi oppure MBL cliniche) ha per-messo di studiare, seppur in maniera retrospet-tiva, l’appropriatezza del valore soglia discriminan-te nella diagnosi, il rischio di progressione alme-no in questa categoria clinicamente rilevabile el’eventuale esistenza di fattori predittivi della evo-luzione leucemica.- Valore soglia per differenziare MBL da LLC.L’attribuzione di un’etichetta diagnostica qualequella di leucemia (con tutto il carico psicologi-co per il paziente) dovrebbe ragionevolmentebasarsi non tanto sulla definizione arbitraria diun valore, ma sul rischio concreto di morbiditàe mortalità derivanti dalla condizione in esamee sull’eventuale necessità di trattamento. In talsenso, i nuovi criteri diagnostici che considera-no la conta linfocitaria B e non quella assolutacome elemento discriminante, vanno nella dire-zione giusta di una maggior affidabilità preditti-va in quanto, come dimostrato anche da recen-ti studi retrospettivi (36, 38), il valore dei linfoci-ti totali, pur correlando con la sopravvivenza libe-ra da trattamento e globale, è in realtà un fatto-re predittivo meno forte rispetto alla concentra-zione di cellule B. Tale conclusione risulta anco-ra più condivisibile considerando che la contalinfocitaria in sé e per sé non rappresenta altro
che la somma delle concentrazioni di diverse sot-topopolazioni, essenzialmente rappresentate dalinfociti B, T ed NK.Al contrario, non vi è alcun vantaggio in terminipredittivi nel valutare più precisamente la concen-trazione delle cellule B clonali, e non semplice-mente dei linfociti B totali (36) in quanto è statodimostrato, che, negli individui con MBL e linfo-citosi (e quindi con una conta linfocitaria tenden-zialmente >5.000 μl), la quota di cellule B policlo-nali rappresenta una frazione molto ridotta deltotale dei linfociti B. Pertanto il valore dei linfoci-ti B individuato tramite valutazione citofluorime-trica dell’espressione di CD19 (marcatore carat-teristico di tale popolazione) riflette in manieraattendibile la popolazione clonale di cellule B inquesta categoria di soggetti e può essere utiliz-zato in maniera affidabile per monitorare il cloneB cellulare aberrante.Vari studi retrospettivi clinici hanno confermato cheil valore di linfociti B alla presentazione costitui-sce un fattore predittivo indipendente dello svi-luppo di una franca linfocitosi con un incremen-to continuo e proporzionale del rischio per ogniaumento di 1.000 cellule B per μl (27, 36, 38-41).Ciò nonostante, questi studi hanno acceso unintenso dibattito sulla soglia più idonea a sepa-rare la categoria MBL dalla LLC stadio 0.Rawstron et al. (27) hanno evidenziato che indi-vidui con un valore di linfociti B <1.900 per μl dopoun follow up mediano di quasi 7 anni tendono amantenere invariata la conta linfocitaria, mentre isoggetti con una conta linfocitaria B >4.000 perμl mostrano, in maniera significativamente più fre-quente, un incremento della linfocitosi. Analogamente, lo studio retrospettivo di Rossi etal. (40) depone a favore del mantenimento dellasoglia di 5.000 per μl. Al contrario, studi retrospet-tivi della Mayo Clinic (36, 38) hanno mostrato cheun valore soglia di 11.000 linfociti B per μl è ingrado di discriminare in maniera più precisa la pro-gnosi, ed in particolare non solo di predire lasopravvivenza libera da trattamento, ma anchequella globale, a differenza del cut-off di 5.000 perμl, capace solamente di predire la necessità diterapia. L’insieme di questi dati conferma la esi-genza di studi prospettici coinvolgenti un ampionumero di partecipanti per meglio definire il limi-te adeguato.

47Linfocitosi B monoclonale
- Rischio di progressione a LLC. Il rischio di pro-gressione da MBL clinica a franca LLC o a linfo-ma a piccoli linfociti (SLL) con necessità di trat-tamento è stato valutato in ampie casistiche; i datiderivanti dall’unico studio prospettico effettuatoe avvalorati da 2 dei 3 studi retrospettivi pubbli-cati recentemente suggeriscono un rischio di pro-gressione compreso tra 1.1 e 2% (27, 36, 41). Talevalore ricorda in maniera molto stretta il datoriscontrato in studi di correlazione tra gammopa-tia monoclonale di incerto significato (MGUS) emieloma multiplo (42). La MBL associata a linfo-citosi è infatti una condizione per molti aspetti affi-ne alla MGUS: il rischio di progressione dientrambe si aggira intorno all’1% per anno, la cur-va della sopravvivenza libera da malattia non sem-bra mai raggiungere una fase di plateau, indican-do la necessità di un monitoraggio clinico a tem-po indeterminato e la maggior parte dei decessinegli individui con tali condizioni è imputabile acause non correlate.
Il dato pari all’1-2% l’anno come rischio di pro-gressione in LLC con necessità di trattamento pergli individui con MBL va confrontato con il rischiopari al 5-7% per anno dei pazienti affetti da LLCstadio 0 di Rai. Questa differenza, apparentemen-te limitata, si traduce di fatto in un rischio a 10 annidi necessità di trattamento del 7-14% per gli indi-vidui con MBL, e di ben il 50-70% per i soggetticon LLC Rai 0, legittimando l’attuale adozione diun cut-off di 5.000 linfociti B per μl come sogliaper la diagnosi differenziale, anche se suscettibi-le di migliorie, da verificare in studi prospettici.La questione della relazione tra MBL e LLC puòessere analizzata anche da un punto di vista oppo-sto; in altre parole è giusto chiedersi non solo quan-te MBL progrediscono a LLC, ma anche quanteLLC sono precedute da MBL. La risposta a que-sto quesito è arrivata da uno studio di popolazio-ne americano organizzato nell’ambito di un pro-gramma di prevenzione delle patologie neoplasti-che in cui più di 150.000 individui sani stati segui-
TABELLA 3 - Parametri clinici e biologici analizzati nei principali studi clinici.
Parametri Rawstron AC et al., Shanafelt T et al., Rossi D et al.,2008 (27) 2009 (38) 2009 (40)
Coorte 309 631 277cMBL 309 302 123Conta linfocitaria mediana (x109/l) 6.0 (1.1-16.8) 5.4 [0.3-9.6] 5.1 [1.22-9.9]Linfociti B (x109/l) 3.3 [0.1-4.99] 2.76 [0.02-4.99] 2.8 [0.2-4.9]*Età (anni) 71 [39-99] 69 [34-93] 68 [59-75]*Sesso (M/F) 149/160 175/127 60/63Hb (g/l) 134 [62-203] 138 [127-150] 140 [132-156]*Piastrine (x109/l) 221 [67-487] 226 [184-283] 221 [188-261]∅
Omologia geni IGHV (%) [range] 93.5 [88.2-98.9] n.v. 94.3 [92.0-97.0]∅
Geni IGHV non mutati 2/20 (10%) 25/109 (23%) 21/105 (20%) Geni IGHV utilizzati VH4-34, VH3-23 n.v. VH4-34, VH3-23,
VH3-07 VH1-69, VH3-30 CD38≥30% 58/174 (33.3%) 60/274 (22%) 27/119 (22.7%)FISH 33 pz 126 pz 105 pzDel13q 19 (58%) 56 (44%) 37 (35.2%)+12 7 (21%) 23 (18%) 19 (18.1%)Del11q 2 (6%) 2 (2%) 0Del17p 1 (3%) 4 (3%) 4 (3.8%)Linfocitosi progressiva 51 (27.6%) 74/210 (35.2%) 34/123 (27.6%)Progressione a LLC 28 (15.1%) n.v. 56/123 (45.5%)Necessità di trattamento 13 (7.0%) 7 (2.3%) 19/123 (15.4%)Sopravvivenza libera da trattamento 48 [13.2-121.2] n.v. n.v.mediana (mesi)Follow up mediano (mesi) 80.4 [2.4-141.6] 18 [0-97] 42.7
*Linfociti B LLC simili; ∅25°-75° percentile

48 Seminari di Ematologia Oncologica
ti nel tempo per valutare l’eventuale sviluppo dineoplasie (43). Nel corso di tale studio, effettua-to nel periodo tra il 1992 ed il 2001, 45 individuihanno manifestato negli anni una LLC. L’analisi diun loro campione di sangue prelevato al momen-to dell’arruolamento nello studio ha dimostrato che44/45 pazienti mostravano la presenza di un clo-ne B-cellulare fino a 7 anni prima della diagnosi.È interessante sottolineare che, negli 8 casi congeni immunoglobulinici non mutati, che di normadepongono per un decorso clinico più aggressi-vo e per cui era stata messa in dubbio l’esisten-za di una fase di MBL, almeno 3 mostravano lapresenza di un clone più di 3 anni prima della dia-gnosi di LLC, dimostrando quindi che virtualmen-te tutti i casi di LLC (sia i casi mutati sia quelli nonmutati) sono preceduti da una MBL.- Fattori predittivi di progressione. Dato che l’am-bito della LLC si è recentemente arricchito di unapletora di fattori prognostici, più o meno diffusa-mente validati, ritenuti in grado di predire il rischiodi progressione della malattia, un passaggio logi-co naturale è stato quello di valutare se tali fat-tori (almeno quelli più consolidati) siano applica-bili tout court anche alla MBL. I dati a questo pro-posito sono estremamente controversi, poichémolte casistiche non permettono un’analisi mul-tivariata dei diversi fattori, determinati solo in unapercentuale limitata di pazienti. Sebbene sia sta-to proposto (40) che la percentuale di omologiadei geni IGHV, l’espressione di CD38 e CD49d,la presenza di aberrazioni genetiche rilevabili allaFISH possano costituire tutti fattori in grado disegregare anche la MBL con linfocitosi in duegruppi con decorso clinico differente, l’unico rilie-vo confermato in più di uno studio riguarda la cor-relazione tra l’espressione di CD38 ed un ridottointervallo di tempo prima del trattamento, confer-mando quindi la necessità di una più ampia moledi dati prima di poter trarre conclusioni definitivein tal senso (Tabella 3).
n CARATTERISTICHE BIOLOGICHE DELLA MBL: SOMIGLIANZE E DIFFERENZE CON LA LLC
L’origine e la natura della MBL sono tuttora ogget-to di controversia. In particolare non è ancora chia-
ro se i linfociti B LLC-simili in tale condizione sia-no di natura neoplastica ab initio o possano esse-re considerati la controparte normale della LLC.Diversi studi si sono pertanto proposti di carat-terizzare la MBL dal punto di vista fenotipico,molecolare e citogenetico, allo scopo di indagar-ne il potenziale legame biologico con la LLC.- Espressione genica e molecolare. Come accen-nato in precedenza, in molti casi la MBL condi-vide lo stesso assetto fenotipico della LLC. Questaaffermazione, però, rappresenta in un certo sen-so un circolo vizioso poiché la stessa definizio-ne di MBL LLC-simile si basa sull’espressione diquesti markers (44). Allo scopo di stabilire l’esi-stenza di un potenziale marcatore espresso inmaniera differenziale nelle due condizioni, il pro-filo di espressione di alcuni specifici geni (FMOD,CKAP4, PI3Kc2b, LEF1, PFTK1, Bcl2 and GPM6a)è stato paragonato in una serie di campioni deri-vati da individui con MBL o affetti da LLC e in cel-lule B normali. Questa analisi ha mostrato chia-ramente che le cellule MBL e LLC differiscono dailinfociti B normali e sono accomunate da uno stes-so profilo, ed in particolare, dall’espressione diLEF-1 (45). Tale gene, non rilevabile nella sotto-popolazione B-cellulare CD5+, appare espressosia nella MBL sia nella LLC, suggerendo un ruo-lo potenziale nella transizione da cellule B norma-li a cellule B aberranti con fenotipo LLC-simile. È stato inoltre analizzata l’espressione in citofluo-rimetria di numerosi marcatori cellulari addiziona-li rispetto a quelli utilizzati nelle procedure diagno-stiche. Il gruppo di Leeds, in due studi succes-sivi (46, 47) ha confermato una sostanziale iden-tità del profilo di espressione molecolare tra MBLe LLC, indistinguibili l’una dall’altra, ma ben dif-ferenziabili da altri disordini linfoproliferativi e dal-le cellule B normali. È comunque interessantenotare che, come in parte atteso, una di questeanalisi (47) ha evidenziato una diversa espressio-ne di alcune proteine responsabili del traffickinglinfoide (in particolare di CXCR5 e CCR6, e diCD62L) tra MBL LLC-simile e le forme di LLC adecorso clinico più aggressivo (con delezione del17p e/o delezione dell’11q), permettendo di ipo-tizzare una minor predisposizione alla localizza-zione nei tessuti linfoidi (con il conseguente ridot-to apporto di stimoli da parte del microambien-te) nei casi MBL.

49Linfocitosi B monoclonale
- Geni delle immunoglobuline. Nel campo dellaLLC, l’analisi delle caratteristiche del B-cellreceptor (BCR), una molecola chiave per lasopravvivenza dei linfociti B maturi, di cui le immu-noglobuline costituiscono una componente essen-ziale, è stata foriera di conoscenze illuminanti nonsolo dal punto di vista biologico ma anche sul ver-sante clinico. Il repertorio immunoglobulinico del-la LLC è caratterizzato da una particolare predi-lezione nell’uso di alcuni geni variabili (IGHV) (inparticolare IGHV1-69, IGHV4-34, IGHV3-7,IGHV3-23) (48). Inoltre, i casi con un’omologia disequenza immunoglobulinica pari o superiore al98% rispetto alla forma germ-line (non mutati) esi-biscono un decorso clinico tendenzialmente piùaggressivo, mentre i casi mutati (con un’omolo-gia di sequenza inferiore al 98%) tendono a mani-festare un andamento più indolente, con una pro-lungata sopravvivenza libera da progressione (49,50). Un fenomeno particolarmente interessante ecaratteristico della LLC, è costituito dall’espres-sione dei cosiddetti recettori stereotipati. È statodimostrato (51) che oltre il 30% dei pazienti affet-ti da LLC, geograficamente distanti, condividesequenze simili o identiche a livello della cosid-detta complementarity-determining-region 3(CDR3) che è il principale sito preposto al lega-me con l’antigene. Da un punto di vista biologi-co, tale riscontro suggerisce l’esistenza di un
numero limitato di antigeni in grado di stimolaree quindi selezionare cloni LLC in pazienti diversi(52-54). Numerosi studi hanno quindi analizzatol’espressione delle immunoglobuline degli indivi-dui con MBL, per paragonarle a quelle della LLC,partendo dal presupposto che la dimostrazionedi un analogo repertorio immunoglobulinico e del-la presenza di recettori stereotipati rappresente-rebbe una forte evidenza a favore di un percor-so patogenetico comune. Gli studi effettuati suindividui con MBL LLC-simile e linfocitosi, eviden-ziate in un contesto clinico, mostrano un reper-torio immunoglobulinico analogo a quello dellaLLC conclamata (27), pur con una maggior rap-presentazione di casi mutati. Al contrario, i datinei pazienti con una quota di cellule B aberrantimolto bassa (tipicamente <10 cellule LLC-similiper μl), quale quella che si riscontra nella popo-lazione generale, mostrano uno scenario differen-te. In particolare, negli studi di popolazione (21,55), nei soggetti MBL si assiste ad una sottorap-presentazione dei geni IGHV caratteristici dellaLLC (con ridotta frequenza del gene IGHV4-34 eassenza di IGHV1-69) e ad un più frequente rilie-vo del gene IGHV4-59/61, di più raro riscontro nel-la LLC (Figura 3). È inoltre interessante sottolinea-re che, nelle coorti di soggetti con MBL a bassaconta, la presenza di recettori stereotipati è estre-mamente rara, e solo 2 casi di MBL che condi-
25
20
15
10
5
0
Per
cent
age
(%)
IGHV1-
69
IGHV3-
15
IGHV3-
21
IGHV4-
59/6
1
IGHV4-
39
IGHV4-
34
IGHV4-
31
IGHV4-
30
IGHV3-
73
IGHV3-
7
IGHV3-
66
IGHV3-
64
IGHV3-
53
IGHV3-
49
IGHV3-
48
IGHV3-
30
IGHV3-
23
MBLCLL
FIGURA 3 - Repertorio immunoglobulinico dei soggetti con MBL LLC-simile a bassa conta a confronto con quello dei pazienti conLLC [modificata da (21)].

50 Seminari di Ematologia Oncologica
vidono una struttura del CDR3 omologa a quel-la descritta in pazienti affetti da LLC è riportatain letteratura (21).- Anomalie genetiche. Pur premettendo che laLLC non si associa ad alcuna anomalia citoge-netica specifica, l’analisi tramite ibridazione in situcon sonde fluorescenti (FISH) permette di iden-tificare alterazioni genetiche ricorrenti nell’80% deipazienti (56). Tra queste, la delezione del brac-cio lungo del cromosoma 13 (del13q), riconosciu-ta in oltre il 50% dei casi, è la più frequente, men-tre, in ordine decrescente, si rilevano la delezio-ne del braccio lungo del cromosoma 11 (del11q),presente nel 18% dei casi e correlata ad altera-zioni del gene ATM, la trisomia del cromosoma12 (+12), nel 16%, ed infine la delezione del brac-cio corto del cromosoma 17 (del17p), nel 7%,comportante la perdita di funzione del gene p53.Il riscontro di tali alterazioni ha una documenta-ta valenza prognostica, in quanto necessità ditrattamento, decorso clinico e sopravvivenza glo-bale differiscono nei diversi sottogruppi, con ipazienti con del13q che dimostrano la soprav-vivenza più prolungata ed il decorso clinico piùindolente, mentre il sottogruppo con del17p risul-ta prognosticamente sfavorito e tende ad avereuna sopravvivenza ridotta ed una scarsa rispo-sta ai trattamenti tradizionali (56). Alcuni studi hanno tentato di caratterizzare il pat-tern di anomalie cromosomiche dei casi di MBLLLC-simile, evidenziando forti somiglianze con leforme di LLC a prognosi favorevole. In manieraabbastanza univoca, infatti, sia nelle forme MBLa bassa conta che in quelle in cui il valore asso-luto delle cellule B aberranti è più elevato, l’alte-razione di più frequente riscontro è rappresen-tata dalla delezione del cromosoma 13, in pro-porzione pari a circa il 39% nelle MBL con con-ta leucocitaria nei limiti di norma e a circa il 58%nelle forme associate a linfocitosi (27). Le altreanomalie genetiche, ed in particolare quelle asignificato prognostico sfavorevole come del11qe del17p, sono di riscontro estremamente raro,e sono state individuate soprattutto nelle formecon linfocitosi; anche in tali circostanze, del resto,tendono ad essere evidenziate in una quota mino-ritaria (< del 20%) delle cellule aberranti (57).Infine è stato recentemente dimostrato che 10polimorfismi di singoli nucleotidi (SNPs) valutati
nell’ambito dell’intero genoma conferiscono unincremento del rischio di sviluppare una LLC, dientità modesta ma ben quantificabile (58). Alcunidi questi SNPs corrispondono a geni implicati nel-lo sviluppo e nella funzione linfocitaria, includen-do FARP2 (regolatore dell’attività di MYC), IRF4(coinvolto nella differenziazione plasmacellulare)e SP140 (implicato nella risposta alla stimolazio-ne antigenica). Dato che la condizione di MBL èstata spesso considerata un marcatore surroga-to di predisposizione a sviluppare la LLC l’inci-denza di questi SNPs in 419 casi di MBL (342casi di MBL clinica e 77 MBL derivate da studidi popolazione) è stata confrontata con quellariscontrata in 1.753 controlli sani. Per 6 di essi èstata evidenziata un’associazione statisticamen-te significativa tra genotipo e rischio di MBL (59).Il rischio di sviluppare una MBL sembra aumen-tare in relazione al numero di alleli presenti per iloci in esame e gli individui con più di 6 alleli arischio mostrano un probabilità 3 volte maggio-re di sviluppare una MBL rispetto agli altri indi-vidui (59). È interessante sottolineare che, puressendo il rischio associato ad ognuna dellevarianti alleliche abbastanza basso, la frequen-za di individui portatori di alleli a rischio nellapopolazione europea è piuttosto elevata, fornen-do perciò un contributo significativo allo svilup-po della MBL.- Modelli murini. Il riscontro di popolazioni B-cel-lulari CD5-positive clonali è una caratteristicaabbastanza comune nei topi di diversi ceppi, chesi verifica però dopo un lungo periodo di laten-za, in animali anziani (di età superiore a 15 mesi)senza dar luogo ad un vero e proprio quadro leu-cemico (60). Nei topi New Zealand Black (NZB)e New Zealand White (NZW) tale fenomeno ten-de invece a manifestarsi più precocemente, conlo sviluppo di espansioni linfoproliferative B-cel-lulari monoclonali CD5-positive a livello spleni-co e nel sangue periferico, che evolvono poi inun franco disordine leucemico all’età di 9-12 mesi(61-64). Un nuovo modello murino recentemen-te introdotto è invece caratterizzato dalla dele-zione del locus MDR (minimal deleted region),localizzato a livello del braccio lungo del cromo-soma 13, nella regione 13q14 (65), che si presen-ta deleto in circa il 50% dei pazienti LLC alla dia-gnosi e dei casi di MBL. Mentre i topi MDR-/- svi-

51Linfocitosi B monoclonale
luppano un franco disordine linfoproliferativo LLC-simile in una percentuale vicina al 27%, in circail 5% degli animali si riscontra, un’espansioneCD5+B220low (l’equivalente murino del CD20umano) di proporzioni limitate (pari a circa l’8%delle cellule mononucleate a livello del sangueperiferico), strettamente analoga alla MBL docu-mentata nell’uomo.
n MBL E IMMUNOSENESCENZA
Come evidenziato nell’uomo e corroborato daimodelli murini, la MBL è una condizione associa-ta all’invecchiamento, la cui frequenza aumentanegli individui di età superiore a 60 anni. Il nostrosistema immunitario subisce inevitabilmente unaserie di cambiamenti parafisiologici correlatiall’età (nell’insieme definiti immunosenescenza),che condizionano alcune alterazioni della rispo-sta immunitaria particolarmente frequenti neglianziani, quali la maggior suscettibilità alle infezio-ni e la ridotta risposta alle vaccinazioni (66).Per quanto riguarda i linfociti B (67, 68), il proces-so di senescenza è caratterizzato dalla restrizio-ne del repertorio immunoglobulinico, da una ridot-ta incidenza di mutazioni somatiche a carico del-le immunoglobuline, dall’incremento percentua-le della sottopopolazione linfocitaria B1 (69, 70),e, soprattutto, dall’aumentata incidenza di popo-lazioni oligo- e monoclonali, come testimoniatodal più frequente riscontro di gammopatie mono-clonali di incerto significato in individui di età supe-riore a 60 anni (42). Per quanto riguarda i linfociti T, la comparsa diespansioni oligo- e monoclonali associate ad unarestrizione del repertorio cellulare nel processo disenescenza è stata dimostrata a carico di diver-se sottopopolazioni (71). Le evidenze più stringen-ti riguardano il subset CD8+, ma in più del 50%degli individui di età superiore a 65 anni è stataevidenziata la presenza di popolazioni monoclo-nali anche a carico della sottopopolazione di cel-lule T CD4+CD8+ (cosiddette doppie positive), cherappresenta una piccola quota dei linfociti T tota-li (generalmente pari al 2-3%) (72).Tra le ipotesi più affascinanti proposte per spie-gare questa tendenza all’oligo- monoclonalità chesi manifesta nel corso dell’invecchiamento, una
parte di primo piano spetta al potenziale coinvol-gimento degli agenti infettivi, il cui ruolo è già sta-to dimostrato nelle modificazioni a carico del com-parto T-cellulare. In particolare, le infezioni viralicroniche persistenti, in primis quella da CMV (73),sono state associate alla comparsa di espansio-ni clonali CD8+, che alterano e riducono la varie-tà del repertorio immunitario, conferendo poten-zialmente una maggiore suscettibilità ad altri tipidi infezione. Pur non esistendo ad oggi studi spe-rimentali in grado di comprovare il potenziale nes-so tra insorgenza di MBL e stimoli antigenici cro-nici nella popolazione generale, è stata osserva-ta un’aumentata frequenza di MBL negli individuicon infezione da HCV (74). Tale incremento è con-seguente ad un aumento di tutti e tre i sottotipidi MBL inclusa quella LLC-simile (74). In sintesiquindi la monoclonalità e la comparsa di espan-sioni B-cellulari (nello specifico LLC-simili) potreb-bero non costituire di per sé un evento neopla-stico, ma rappresentare un epifenomeno dellemodificazioni del sistema immunitario correlateall’invecchiamento ed in particolare all’esposizio-ne a stimoli antigenici cronici, sia di natura infet-tiva ma anche potenzialmente self.
n MONITORAGGIO CLINICO
I dati derivanti dal monitoraggio clinico della MBLsono attualmente molto limitati a causa della suarecente definizione. Nella pratica clinica quotidia-na, nella maggior parte dei casi, il riscontro diMBL avviene nell’ambito degli accertamentieseguiti a seguito del rilievo di una linfocitosi odi un’altra anomalia dell’emocromo. Una voltaaccertata la diagnosi, avendo quindi accurata-mente escluso la presenza di un disordine linfo-proliferativo, si ritiene auspicabile proseguire ilmonitoraggio con controlli clinico-laboratoristicia cadenza annuale (13). Poiché la condizione diMBL è almeno 100 volte più frequente nella popo-lazione generale rispetto alla LLC, è infatti evi-dente che solo una minima percentuale di casiè destinata a progredire verso una franca formaleucemica. Nell’impossibilità di determinare alladiagnosi quali individui andranno incontro a pro-gressione, pur rassicurando la singola personasul rischio sostanzialmente basso di sviluppare

52 Seminari di Ematologia Oncologica
una LLC, è importante istruire i soggetti con MBLsui potenziali sintomi e segni d’allarme (linfoade-nopatie, febbre, sudorazioni notturne, perdita dipeso, astenia) e procedere ad un regolare followup. Come accennato, in analogia a quanto avvie-ne per la MGUS, la curva che disegna il rischiodi progressione degli individui con MBL non rag-giunge mai un plateau, comportando perciò lanecessità di un monitoraggio a tempo indeter-minato (Tabella 4).
n ALTRE CONSIDERAZIONI CLINICHEDal punto di vista della pratica clinica, la defini-zione della categoria diagnostica di MBL ha aper-to alcuni rilevanti quesiti, tuttora in attesa di riso-luzione. In primo luogo, non esistendo un codi-ce ICD9 applicabile alla MBL, il medico che effet-
tua la diagnosi si trova a dover scegliere tra la defi-nizione di linfocitosi (ICD-9 288.8) e LLC (204.1);nel primo caso, in alcuni paesi, gli accertamentinecessari non possono essere eseguiti a caricodel sistema sanitario nazionale, mentre nel secon-do (soprattutto nei sistemi sanitari che si basanosulla copertura assicurativa del singolo, come negliStati Uniti) i costi assicurativi sono aumentati e lepratiche per ottenere un’assicurazione sulla vitapossono essere bloccate, pur non potendosi difatto ascrivere tale condizione all’ambito dei disor-dini neoplastici (26).Un altro aspetto pratico meritevole di valutazio-ne è rappresentato dalla potenziale indicazione adeffettuare una valutazione di screening per la pre-senza di una popolazione B cellulare clonale neiderivati ematici e negli organi destinati alla dona-zione. Un’analisi effettuata su campioni derivan-
TABELLA 4 - Raccomandazioni per la valutazione ed il follow-up nella pratica clinica [adattato da (13)].
Raccomandazioni MBL LLC-simile MBL1 LLC-simile MBL LLC atipica orilevata in studi rilevata clinicamente Non-LLC identificatadi popolazione1 in un contesto clinico
Work up Anamnesi2 Sì Sì Sìdiagnostico Esame obiettivo3 Sì Sì Sì
Immunofenotipo dei linfociti Sì Sì SìEmocromo con formula Sì Sì SìTest FISH con sonda per t(11;14) No No Sì4
TC torace/addome/pelvi No No SìBiopsia osteomidollare No No SìTest prognostici per la LLC No No No
Counseling Istruzione dei pazienti sui sintomi Sì Sì Sì& follow-up da monitorare2
Rischio di progressione con Non definito ma basso5 1-2%/anno Non definitonecessità di terapiaAnamnesi2 Controlli di routine Annuale 3-12 mesi6
Esame obiettivo3 Controlli di routine Annuale 3-12 mesi6
Emocromo con formula Annuale 6-12 mesi 6-12 mesi6
TC torace/addome/pelvi No No Secondo il giudizio clinico6
1La MBL LLC-simile identificata nel corso di una valutazione clinica per la linfocitosi viene definita MBL LLC-simile rilevata clinicamente, mentre la MBL LLC-simile identificata nel corso di studi di ricerca in pazienti con conta linfocitaria normale viene definita come MBL LLC-simile rilevata in studi di popolazione.2Attenzione ai sintomi costituzionali (febbre, sudorazioni notturne, calo ponderale, astenia). 3Attenzione alla valutazione linfonodale ed alla determinazione del-l’epatosplenomegalia. 4Nei pazienti con fenotipo LLC atipica CD5+ ma CD23- dovrebbe essere effettuata una valutazione FISH con sonda per la t(11;14),caratteristica del linfoma mantellare. 5Nonostante la limitata disponibilità di dati in merito, la progressione nei soggetti con MBL LLC-simile rilevata in studidi popolazione sembrerebbe essere rara. Dato che diversi studi suggeriscono che il numero di linfociti B correli con il decorso clinico negli individui con MBL,il rischio di progressione in soggetti con MBL LLC-simile rilevata in studi di popolazione è ritenuto inferiore rispetto a quello degli individui con MBL LLC-simile clinicamente rilevabile. 6Per i rari soggetti che soddisfano i criteri della MBL con un fenotipo e studi citogenetici suggestivi di linfoma mantellare o diun altro sottotipo di linfoma aggressivo, si suggerisce un monitoraggio ogni 3-6 mesi con esecuzione di TC almeno ogni 6 mesi. Per gli individui con MBLLLC atipica o Non-LLC, il cui fenotipo ricorda un sottotipo più indolente di linfoma, si raccomanda un monitoraggio ogni 6-12 mesi e l’esecuzione di esamistrumentali di follow up ad una frequenza stabilita secondo il giudizio clinico.

53Linfocitosi B monoclonale
ti da 5.141 donatori di sangue ha rivelato la pre-senza di soli 7 casi di linfocitosi B monoclonale,con una prevalenza quindi sorprendentementebassa, pari allo 0.14% (18); la metodica di scree-ning utilizzata, tuttavia, era dotata di una scarsasensibilità, motivo per il quale al momento nonpossono essere tratte conclusioni definitive ed intale ambito è prematuro raccomandare l’attuazio-ne di metodiche di screening.Ancora controversa è anche la linea di condottada applicare nello screening per MBL dei paren-ti di primo grado di pazienti affetti da LLC, can-didati a trapianto allogenico da donatore familia-re, in considerazione dell’aumentata frequenza delrilievo di MBL in questa condizione (75). Almomento non è stato ancora definito in manieraunivoca se, in tale contesto, sussista l’indicazio-ne ad uno screening per MBL e se eventuali fami-
liari con MBL debbano essere considerati non ido-nei alla donazione di cellule staminali midollari.
n CONCLUSIONI
La condizione di MBL costituisce un’ampiacategoria diagnostica tuttora in via di definizio-ne, caratterizzata verosimilmente dalla presenzadi diverse entità, accomunate dall’espressione diun analogo immunofenotipo. Studi di metanali-si delle diverse coorti di individui MBL con feno-tipo LLC (14), pubblicate a livello mondiale han-no dimostrato che la distribuzione di questa con-dizione può essere fondamentalmente suddivi-sa in due principali sottogruppi, con caratteristi-che biologiche e cliniche differenti:- MBL con quota di cellule B aberranti inferiori a
FIGURA 4 - Ipotetica patogenesi della MBL erelazione con la LLC.

54 Seminari di Ematologia Oncologica
50 per μl: riscontrate prevalentemente negli stu-di di popolazione, in quanto clinicamente non rile-vabili a causa della bassa sensibilità dei test dilaboratorio di routine, caratterizzate da un profi-lo di rischio molecolare e citogenetico in qualchemodo simile alla LLC a prognosi favorevole, macon un repertorio immunoglobulinico del tutto dif-ferente, a volte anche oligoclonale. Per tale con-dizione, qualora riscontrata incidentalmente agliesami di routine, non sussiste alcuna indicazio-ne al monitoraggio, in quanto non indicativa di unaumento significativo del rischio di sviluppare unafranca LLC.- MBL con quota di cellule aberranti superiore a2.000 per μl: di riscontro frequente nel corso degliaccertamenti eseguiti nell’ambito di una linfoci-tosi, dal punto di vista biologico e del repertorioimmunoglobulinico molto simile alla LLC clinica-mente definita, esibisce un rischio di progressio-ne in leucemia franca pari a circa l’1% per anno.Le forme di MBL con quota linfocitaria compre-sa tra 50 e 2.000 per μl rappresentano la mino-ranza dei casi al momento riscontrata nei vari stu-di pubblicati e mostrano un profilo biologico menodefinito, per molti aspetti simile alla LLC clinica-mente manifesta, con l’importante differenza chela conta B cellulare tende a rimanere stabile neltempo e rappresenta il parametro da monitora-re, data l’inattendibilità, in questo sottogruppo inparticolare, delle modificazioni della quota linfo-citaria totale.In conclusione, la condizione di MBL si presta amolteplici chiavi interpretative: da un lato, infatti,le somiglianze biologiche ed immunofenotipiche,il frequente riscontro di monoclonalità e il dimo-strato, seppur basso, rischio di progressione inLLC con necessità di trattamento, depongono afavore della natura pre-leucemica della MBL, inmaniera per certi versi analoga alla transizione traMGUS e mieloma multiplo. Dall’altro, però, trat-tandosi di una condizione la cui prevalenza nel-la popolazione generale è circa 100 volte mag-giore rispetto a quella della LLC, che diviene pro-gressivamente più frequente nel corso dell’invec-chiamento, talvolta con caratteristiche di oligoclo-nalità, è altrettanto ragionevole ipotizzare che talecondizione rappresenti la conseguenza di una sti-molazione antigenica cronica e persistente, di persé quindi non di natura neoplastica, ma più espo-
sta al rischio di eventi genetici addizionali che nepossano condizionare, seppur raramente, l’evo-luzione in franca leucemia, in accordo con ilmodello multi-hit già dimostratosi concretamen-te valido nello spiegare la patogenesi di molte for-me neoplastiche (Figura 4).La natura della relazione biologica tra MBL e LLCrisulta perciò ad oggi solo parzialmente definitaed è campo di attiva indagine, sia in relazione allanecessità di individuare fattori di rischio in gradodi predire il decorso clinico di questa entità el’eventuale evoluzione in franca leucemia, checome affascinante modello di studio per penetra-re a fondo i meccanismi patogenetici alla base delsubstrato biologico della LLC.
n BIBLIOGRAFIA
1. Han T, Ozer H, Gavigan M, Gajera R, Minowada J,Bloom ML, et al. Benign monoclonal B cell lympho-cytosis - a benign variant of CLL: clinical, immunolog-ic, phenotypic, and cytogenetic studies in 20 patients.Blood. 1984; 64: 244-52.
2. Marti GE, Rawstron AC, Ghia P, Hillmen P, HoulstonRS, Kay N, et al. Diagnostic criteria for monoclonal B-cell lymphocytosis. Br J Haematol. 2005; 130: 325-32.
3. Marti G, Abbasi F, Raveche E, Rawstron AC, Ghia P,Aurran T, et al. Overview of monoclonal B-cell lympho-cytosis. Br J Haematol 2007; 139: 701-8.
4. Ghia P, Ferreri AM, Caligaris-Cappio F. Chronic lym-phocytic leukemia. Crit Rev Oncol Hematol. 2007; 64:234-46.
5. Molica S, Levato D. What is changing in the naturalhistory of chronic lymphocytic leukemia? Haematolo -gica. 2001; 86: 8-12.
6. Rai KR, Sawitsky A, Cronkite EP, Chanana AD, LevyRN, Pasternack BS. Clinical staging of chronic lympho-cytic leukemia. Blood. 1975; 46: 219-34.
7. Cheson BD, Bennett JM, Rai KR, Grever MR, Kay NE,Schiffer CA, et al. Guidelines for clinical protocols forchronic lymphocytic leukemia: recommendations of theNational Cancer Institute-sponsored working group. AmJ Hematol. 1988; 29: 152-63.
8. Cheson BD, Bennett JM, Grever M, Kay N, Keating MJ,O’Brien S, et al. National Cancer Institute-sponsoredWorking Group guidelines for chronic lymphocyticleukemia: revised guidelines for diagnosis and treat-ment. Blood. 1996; 87: 4990-7.
9. Hallek M, Cheson BD, Catovsky D, Caligaris-CappioF, Dighiero G, Dohner H, et al. Guidelines for the diag-nosis and treatment of chronic lymphocytic leukemia:a report from the International Workshop on ChronicLymphocytic Leukemia updating the National Cancer

55Linfocitosi B monoclonale
Institute-Working Group 1996 guidelines. Blood. 2008;111: 5446-56.
10. Muller-Hermelink HK ME, Catovsky, D, Campo, E,Harris NL, Stein H. Chronic Lymphocytic Leukemia/small lymphocytic lymphoma. In: Swerdlow SH CE,Harris NL, et al., eds. WHO Classification of Tumoursof Haematopoietic and Lymphoid Tissues, FourthEdition. Lyon: IARC. 2008; 180-2.
11. Rawstron AC, Villamor N, Ritgen M, Bottcher S, GhiaP, Zehnder JL, et al. International standardizedapproach for flow cytometric residual disease moni-toring in chronic lymphocytic leukaemia. Leukemia.2007; 21: 956-64.
12. Caligaris-Cappio F, Ghia P. Novel insights in chroniclymphocytic leukemia: are we getting closer to under-standing the pathogenesis of the disease? J Clin Oncol.2008; 26: 4497-503.
13. Shanafelt TD, Ghia P, Lanasa MC, Landgren O,Rawstron AC. Monoclonal B-cell lymphocytosis (MBL):biology, natural history and clinical management.Leukemia. 2010; 24: 512-20.
14. Rawstron AC, Shanafelt T, Lanasa MC, Landgren O,Hanson C, Orfao A, et al. Different biology and clini-cal outcome according to the absolute numbers ofclonal B-cells in monoclonal B-cell lymphocytosis(MBL). Cytometry B Clin Cytom. 2010; 78 (Suppl. 1):S19-23.
15. Vogt RF, Shim YK, Middleton DC, Buffler PA,Campolucci SS, Lybarger JA, et al. Monoclonal B-celllymphocytosis as a biomarker in environmental healthstudies. Br J Haematol. 2007; 139: 690-700.
16. Shim YK, Vogt RF, Middleton D, Abbasi F, Slade B, LeeKY, et al. Prevalence and natural history of monoclon-al and polyclonal B-cell lymphocytosis in a residentialadult population. Cytometry B Clin Cytom. 2007; 72:344-53.
17. Plapp FV, Rachel, JM, Zucker, ML. Chronic B lympho-cytic leukemia in blood donors. Transfusion. 1999; 39.
18. Rachel JM, Zucker ML, Fox CM, Plapp FV, MenitoveJE, Abbasi F, et al. Monoclonal B-cell lymphocytosisin blood donors. Br J Haematol. 2007; 139: 832-6.
19. Rawstron AC, Green MJ, Kuzmicki A, Kennedy B,Fenton JA, Evans PA, et al. Monoclonal B lymphocyteswith the characteristics of “indolent” chronic lympho-cytic leukemia are present in 3.5% of adults with nor-mal blood counts. Blood. 2002; 100: 635-9.
20. Ghia P, Prato G, Scielzo C, Stella S, Geuna M, GuidaG, et al. Monoclonal CD5+ and CD5- B-lymphocyteexpansions are frequent in the peripheral blood of theelderly. Blood. 2004; 103: 2337-42.
21. Dagklis A, Fazi C, Sala C, Cantarelli V, Scielzo C,Massacane R, et al. The immunoglobulin gene reper-toire of low-count chronic lymphocytic leukemia(CLL)-like monoclonal B lymphocytosis is different fromCLL: diagnostic implications for clinical monitoring.Blood. 2009; 114: 26-32.
22. Scarfo L, Dagklis A, Scielzo C, Fazi C, Ghia P. CLL-
like monoclonal B-cell lymphocytosis: are we all boundto have it? Semin Cancer. Biol 2010; 20: 384-90.
23. Nieto WG, Almeida J, Romero A, Teodosio C, Lopez A,Henriques AF, et al. Increased frequency (12%) of cir-culating chronic lymphocytic leukemia-like B-cell clonesin healthy subjects using a highly sensitive multicolor flowcytometry approach. Blood. 2009; 114: 33-7.
24. de Tute R, Yuille M, Catovsky D, Houlston RS, HillmenP, Rawstron AC. Monoclonal B-cell lymphocytosis(MBL) in CLL families: substantial increase in relativerisk for young adults. Leukemia. 2006; 20: 728-9.
25. Almeida J, Nieto W, Teodosio C, Pedreira C, Nieto A,Romero A, Lopez A, Rodriguez-Caballero A, BarcenaP, Rasillo A, Fernandez-Navarro P, Vega T, Orfao A.Small CLL-like B-cell clones in healthy adult subjects:leukemic precursors versus counterpart of CLL cells?Haematologica. 2010; 95 (Suppl. 2): 317.
26. Rawstron AC. Monoclonal B-cell lymphocytosis.Hematology Am Soc Hematol Educ Program. 2009;430-9.
27. Rawstron AC, Bennett FL, O’Connor SJ, Kwok M,Fenton JA, Plummer M, et al. Monoclonal B-cell lym-phocytosis and chronic lymphocytic leukemia. N EnglJ Med. 2008; 359: 575-83.
28. Yuille MR, Matutes E, Marossy A, Hilditch B, CatovskyD, Houlston RS. Familial chronic lymphocyticleukaemia: a survey and review of published studies.Br J Haematol. 2000; 109: 794-9.
29. Neuland CY, Blattner WA, Mann DL, Fraser MC, TsaiS, Strong DM. Familial chronic lymphocytic leukemia.J Natl Cancer Inst. 1983; 71: 1143-50.
30. Bevan S, Catovsky D, Matutes E, Antunovic P, AugerMJ, Ben-Bassat I, et al. Linkage analysis for major his-tocompatibility complex-related genetic susceptibilityin familial chronic lymphocytic leukemia. Blood. 2000;96: 3982-4.
31. Goldin LR, Bjorkholm M, Kristinsson SY, Turesson I,Landgren O. Elevated risk of chronic lymphocyticleukemia and other indolent non-Hodgkin’s lym-phomas among relatives of patients with chronic lym-phocytic leukemia. Haematologica. 2009; 94: 647-53.
32. Rawstron AC, Yuille MR, Fuller J, Cullen M, KennedyB, Richards SJ, et al. Inherited predisposition to CLLis detectable as subclinical monoclonal B-lymphocyteexpansion. Blood. 2002; 100: 2289-90.
33. Marti GE, Carter P, Abbasi F, Washington GC, Jain N,Zenger VE, et al. B-cell monoclonal lymphocytosis andB-cell abnormalities in the setting of familial B-cellchronic lymphocytic leukemia. Cytometry B ClinCytom. 2003; 52: 1-12.
34. Goldin LR, Lanasa MC, Slager SL, Cerhan JR, VachonCM, Strom SS, et al. Common occurrence of mono-clonal B-cell lymphocytosis among members of high-risk CLL families. Br J Haematol. 2010; 151: 152-8.
35. Shanafelt TD, Kay NE, Call TG, Zent CS, Jelinek DF,LaPlant B, et al. MBL or CLL: which classification bestcategorizes the clinical course of patients with an

56 Seminari di Ematologia Oncologica
absolute lymphocyte count >or= 5 x 10(9) L(-1) but aB-cell lymphocyte count <5 x 10(9) L(-1)? Leuk Res.2008; 32: 1458-61.
36. Shanafelt TD, Kay NE, Jenkins G, Call TG, Zent CS,Jelinek DF, et al. B-cell count and survival: differenti-ating chronic lymphocytic leukemia from monoclonalB-cell lymphocytosis based on clinical outcome.Blood. 2009; 113: 4188-96.
37. Mulligan CS, Thomas ME, Mulligan SP. Lymphocytes,B lymphocytes, and clonal CLL cells: observations onthe impact of the new diagnostic criteria in the 2008Guidelines for Chronic Lymphocytic Leukemia (CLL).Blood. 2009; 113: 6496-7; author reply 6497-8.
38. Shanafelt TD, Kay NE, Rabe KG, Call TG, Zent CS,Maddocks K, et al. Brief report: natural history of indi-viduals with clinically recognized monoclonal B-celllymphocytosis compared with patients with Rai 0chronic lymphocytic leukemia. J Clin Oncol. 2009; 27:3959-63.
39. Fung SS, Hillier KL, Leger CS, Sandhu I, Vickars LM,Galbraith PF, et al. Clinical progression and outcomeof patients with monoclonal B-cell lymphocytosis. LeukLymphoma. 2007; 48: 1087-91.
40. Rossi D, Sozzi E, Puma A, De Paoli L, Rasi S, SpinaV, et al. The prognosis of clinical monoclonal B cell lym-phocytosis differs from prognosis of Rai 0 chronic lym-phocytic leukaemia and is recapitulated by biologicalrisk factors. Br J Haematol. 2009; 146: 64-75.
41. Mulligan CS, Thomas ME, Mulligan SP. Monoclonal B-cell lymphocytosis and chronic lymphocytic leukemia.N Engl J Med. 2008; 359: 2065-6; author reply 2066.
42. Kyle RA, Therneau TM, Rajkumar SV, Larson DR,Plevak MF, Offord JR, et al. Prevalence of monoclon-al gammopathy of undetermined significance. N EnglJ Med. 2006; 354: 1362-9.
43. Landgren O, Albitar M, Ma W, Abbasi F, Hayes RB, GhiaP, et al. B-cell clones as early markers for chronic lym-phocytic leukemia. N Engl J Med. 2009; 360: 659-67.
44. Rawstron AC, Bennett F, Hillmen P. The biological andclinical relationship between CD5+23+ monoclonal B-cell lymphocytosis and chronic lymphocytic leukaemia.Br J Haematol. 2007; 139: 724-9.
45. Gutierrez A Jr., Tschumper RC, Wu X, Shanafelt TD,Eckel-Passow J, Huddleston PM 3rd, et al. LEF-1 isa prosurvival factor in chronic lymphocytic leukemiaand is expressed in the preleukemic state of mon-oclonal B cell lymphocytosis. Blood. 2010; 116: 2375-83.
46. Rawstron AC, de Tute R, Jack AS, Hillmen P. Flow cyto-metric protein expression profiling as a systematicapproach for developing disease-specific assays:identification of a chronic lymphocytic leukaemia-spe-cific assay for use in rituximab-containing regimens.Leukemia. 2006; 20: 2102-10.
47. Rawstron AC, Shingles J, de Tute R, Bennett F, JackAS, Hillmen P. Chronic lymphocytic leukaemia (CLL)and CLL-type monoclonal B-cell lymphocytosis (MBL)
show differential expression of molecules involved inlymphoid tissue homing. Cytometry B Clin Cytom.2010; 78 (Suppl. 1): S42-6.
48. Fais F, Ghiotto F, Hashimoto S, Sellars B, Valetto A,Allen SL, et al. Chronic lymphocytic leukemia B cellsexpress restricted sets of mutated and unmutated anti-gen receptors. J Clin Invest. 1998; 102: 1515-25.
49. Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, AllenSL, et al. Ig V gene mutation status and CD38 expres-sion as novel prognostic indicators in chronic lympho-cytic leukemia. Blood. 1999; 94: 1840-7.
50. Hamblin TJ, Davis Z, Gardiner A, Oscier DG, StevensonFK. Unmutated Ig V(H) genes are associated with amore aggressive form of chronic lymphocytic leukemia.Blood. 1999; 94: 1848-54.
51. Stamatopoulos K, Belessi C, Moreno C, BoudjograhM, Guida G, Smilevska T, et al. Over 20% of patientswith chronic lymphocytic leukemia carry stereotypedreceptors: Pathogenetic implications and clinical cor-relations. Blood. 2007; 109: 259-70.
52. Murray F, Darzentas N, Hadzidimitriou A, Tobin G,Boudjogra M, Scielzo C, et al. Stereotyped patternsof somatic hypermutation in subsets of patients withchronic lymphocytic leukemia: implications for the roleof antigen selection in leukemogenesis. Blood. 2008;111: 1524-33.
53. Messmer BT, Albesiano E, Efremov DG, Ghiotto F, AllenSL, Kolitz J, et al. Multiple distinct sets of stereotypedantigen receptors indicate a role for antigen in promot-ing chronic lymphocytic leukemia. J Exp Med. 2004;200: 519-25.
54. Ghia P, Chiorazzi N, Stamatopoulos K. Microenviron -mental influences in chronic lymphocytic leukaemia:the role of antigen stimulation. J Intern Med. 2008; 264:549-62.
55. Dagklis A, Fazi C, Scarfo L, Apollonio B, Ghia P.Monoclonal B lymphocytosis in the general population.Leuk Lymphoma. 2009; 50: 490-2.
56. Dohner H, Stilgenbauer S, Benner A, Leupolt E, KroberA, Bullinger L, et al. Genomic aberrations and survivalin chronic lymphocytic leukemia. N Engl J Med. 2000;343: 1910-6.
57. Lanasa MC, Allgood SD, Volkheimer AD, GockermanJP, Whitesides JF, Goodman BK, et al. Single-cell analy-sis reveals oligoclonality among ‘low-count’ monoclon-al B-cell lymphocytosis. Leukemia. 2010; 24: 133-40.
58. Crowther-Swanepoel D, Houlston RS. Genetic varia-tion and risk of chronic lymphocytic leukaemia. SeminCancer Biol. 2010; 20: 363-9.
59. Crowther-Swanepoel D, Corre T, Lloyd A, Gaidano G,Olver B, Bennett FL, et al. Inherited genetic suscepti-bility to monoclonal B-cell lymphocytosis. Blood. 2010;DOI 10.1182/blood-2010-07-294975.
60. LeMaoult J, Manavalan JS, Dyall R, Szabo P, Nikolic-Zugic J, Weksler ME. Cellular basis of B cell clonal pop-ulations in old mice. J Immunol 1999; 162: 6384-91.
61. Phillips JA, Mehta K, Fernandez C, Raveche ES. The

57Linfocitosi B monoclonale
NZB mouse as a model for chronic lymphocyticleukemia. Cancer Res. 1992; 52: 437-43.
62. Hamano Y, Hirose S, Ida A, Abe M, Zhang D, KoderaS, et al. Susceptibility alleles for aberrant B-1 cell pro-liferation involved in spontaneously occurring B-cellchronic lymphocytic leukemia in a model of NewZealand white mice. Blood. 1998; 92: 3772-9.
63. Stall AM, Farinas MC, Tarlinton DM, Lalor PA,Herzenberg LA, Strober S, et al. Ly-1 B-cell clones sim-ilar to human chronic lymphocytic leukemias routine-ly develop in older normal mice and young autoimmune(New Zealand Black-related) animals. Proc Natl AcadSci USA. 1988; 85: 7312-6.
64. Seldin MF, Conroy J, Steinberg AD, D’Hoosteleare LA,Raveche ES. Clonal expansion of abnormal B cells inold NZB mice. J Exp Med. 1987; 166: 1585-90.
65. Klein U, Lia M, Crespo M, Siegel R, Shen Q, Mo T, etal. The DLEU2/miR-15a/16-1 cluster controls B cell pro-liferation and its deletion leads to chronic lymphocyt-ic leukemia. Cancer Cell. 17: 2 8-40.
66. Wick G, Grubeck-Loebenstein B. The aging immune sys-tem: primary and secondary alterations of immune reac-tivity in the elderly. Exp Gerontol. 1997; 32: 401-13.
67. Ghia P, Melchers F, Rolink AG. Age-dependentchanges in B lymphocyte development in man andmouse. Exp Gerontol. 2000; 35: 159-65.
68. Klinman NR, Kline GH. The B-cell biology of aging.Immunol Rev. 1997; 160: 103-14.
69. LeMaoult J, Szabo P, Weksler ME. Effect of age onhumoral immunity, selection of the B-cell repertoire
and B-cell development. Immunol Rev. 1997; 160:115-26.
70. Nicoletti C. Antibody protection in aging: influence ofidiotypic repertoire and antibody binding activity toa bacterial antigen. Exp Mol Pathol. 1995; 62: 99-108.
71. Colombatti A, Doliana R, Schiappacassi M, ArgentiniC, Tonutti E, Feruglio C, et al. Age-related persistentclonal expansions of CD28(-) cells: phenotypic andmolecular TCR analysis reveals both CD4(+) andCD4(+)CD8(+) cells with identical CDR3 sequences.Clin Immunol Immunopathol. 1998; 89: 61-70.
72. Ghia P, Prato G, Stella S, Scielzo C, Geuna M,Caligaris-Cappio F. Age-dependent accumulation ofmonoclonal CD4+CD8+ double positive T lympho-cytes in the peripheral blood of the elderly. Br JHaematol. 2007; 139: 780-90.
73. Khan N, Shariff N, Cobbold M, Bruton R, AinsworthJA, Sinclair AJ, et al. Cytomegalovirus seropositivitydrives the CD8 T cell repertoire toward greater clon-ality in healthy elderly individuals. J Immunol. 2002;169: 1984-92.
74. Fazi C, Dagklis A, Cottini F, Scarfo L, Bertilaccio MT,Finazzi R, et al. Monoclonal B cell lymphocytosis inhepatitis C virus infected individuals. Cytometry B ClinCytom. 78 (Suppl. 1): S61-8.
75. Del Giudice I, Mauro FR, De Propris MS, Starza ID,Armiento D, Iori AP, et al. Identification of monoclon-al B-cell lymphocytosis among sibling transplantdonors for chronic lymphocytic leukemia patients.Blood. 2009; 114: 2848-9.


59
n INTRODUZIONE
La Sindrome di Richter (SR) rappresenta la tra-sformazione clinico-patologica della leucemia lin-fatica cronica (LLC) in linfoma aggressivo, piùcomunemente in linfoma diffuso a grandi celluleB. Questa condizione venne descritta per la pri-ma volta nel 1928 da Maurice N. Richter, da cuiprese il nome (1). La trasformazione della LLC inSR deve essere distinta dalle altre tipologie di pro-gressione di malattia, che sono definite dalle lineeguida IWCLL-NCI (2). La diagnosi di SR richiedeobbligatoriamente una prova istologica, in assen-za della quale la SR può essere soltanto sospet-tata clinicamente, ma non documentata (3, 4). Latrasformazione della LLC in linfoma diffuso a gran-di cellule B deve essere inoltre distinta dalla tra-sformazione prolinfocitica e da altre neoplasie lin-foidi che mostrano un’incidenza aumentata neipazienti affetti da LLC (4, 5).In questa rassegna, la definizione di SR è appli-cata esclusivamente alla trasformazione da LLCa linfoma diffuso a grandi cellule B (che rappre-
senta più del 90% dei casi di SR). Nonostantequesta restrizione, la definizione di SR rimane ete-rogenea e comprende almeno due differenti con-dizioni biologiche nettamente distinte l’una dal-l’altra: a) la trasformazione clonalmente correlata delle
cellule di LLC a linfoma diffuso a grandi cel-lule B, condizione che si verifica nella maggio-ranza dei casi di SR;
b) lo sviluppo di linfoma diffuso a grandi celluleB non clonalmente correlato alla fase di LLC(3, 6-11).
Nella SR clonalmente correlata, il nesso patoge-netico tra LLC e la fase di linfoma diffuso a gran-di cellule B è palese ed è sostanziato dall’acqui-sizione di nuove lesioni molecolari al momentodella trasformazione clinico-patologica. Viceversa,nel caso dello sviluppo di SR clonalmente non cor-relata, la trasformazione potrebbe essere favori-ta da alterazioni immunologiche del paziente affet-to da LLC, o da altri meccanismi che, al momen-to, non sono noti.Con l’avvento dei nuovi farmaci immunosoppres-sivi utilizzati per la terapia della LLC, e in parti-colar modo con l’introduzione dell’anticorpomonoclonale anti-CD52 (alemtuzumab), è statoosservato sporadicamente lo sviluppo di linfomiclinicamente aggressivi caratterizzati da positivi-tà del clone neoplastico per infezione da Epstein-Barr virus (EBV) e da spiccate analogie con i lin-fomi dei pazienti immunodeficienti (12, 13).Questi casi di linfomi aggressivi associati a tera-pia con alemtuzumab non devono essere confu-si con la SR, ma piuttosto rappresentano un nuo-vo tipo di linfoma associato a immunodeficienzache si sviluppa dopo terapie che riducono il com-
Indirizzo per la corrispondenza
Prof. Gianluca GaidanoDivisione di EmatologiaDipartimento di Medicina Clinica e SperimentaleUniversità degli Studi del Piemonte Orientale “Amedeo Avogadro”Via Solaroli, 17 - 28100 NovaraE-mail: [email protected]
Sindrome di RichterSindrome di RichterMARCO FANGAZIO, DAVIDE ROSSI, GIANLUCA GAIDANODivisione di Ematologia, Dipartimento di Medicina Clinica e Sperimentale, Università degli Studi del Piemonte Orientale “Amedeo Avogadro” e Azienda Ospedaliero-Universitaria Maggiore della Carità, Novara, Italia
Gianluca Gaidano
Parole chiave: sindrome di Richter, leucemia linfaticacronica, linfoma diffuso a grandi cellule B, predittori cli-nici e biologici di trasformazione

60 Seminari di Ematologia Oncologica
partimento T linfocitario in pazienti già immuno-compromessi a causa della malattia di base e/oa causa della pregressa chemioterapia.La propensione a trasformazione in linfomaaggressivo non è esclusiva della LLC. Sebbenecon diversa incidenza, tale propensione si osser-va anche in altri disordini linfoproliferativi indolen-ti della serie B linfocitaria, in particolare il linfomafollicolare e il linfoma della zona marginale.Nonostante questa evidenza, ad oggi non è sta-to possibile chiarire se esista un comune percor-so molecolare alla base della trasformazione in lin-foma diffuso a grandi cellule B a partire da tuttequeste differenti condizioni cliniche, oppure se latrasformazione da un disordine linfoproliferativoB indolente ad uno aggressivo segua strade diver-se a seconda del tipo iniziale di malattia.
n EPIDEMIOLOGIA
La SR è considerata una patologia rara ed unacomplicanza infrequente della LLC. Questa per-cezione, nonostante sia condivisa da molti ema-tologi, non è supportata da evidenze epidemio-logiche derivate da studi riguardanti specificata-mente la SR. Dal momento che la diagnosi di SRrichiede la valutazione istologica, l’eterogenicitàtra la sua incidenza osservata in diverse serie dipazienti affetti da LLC dipende in larga misura dal-la diversa prassi della biopsia linfonodale tra i varicentri (3, 4). In particolare, mentre la rivalutazio-ne bioptica è pratica comune ad ogni progressio-ne di malattia nel linfoma follicolare (14), non sipuò dire lo stesso per quanto riguarda la LLC. Perquesto motivo, la sottostima nella diagnosi di SRpotrebbe influenzare, almeno in parte, la perce-zione della SR come una condizione rara.Studi recenti hanno valutato l’incidenza di SR inuna serie consecutiva di pazienti affetti da LLCche sono stati omogeneamente sottoposti a con-trollo bioptico della lesione indice in caso di: a) linfoadenopatia ≥5 cm; b) rapido incremento della dimensione linfonoda-
le (raddoppiamento del diametro trasversomaggiore in un periodo < 3 mesi);
c) comparsa di lesione extranodale; d) comparsa di sintomi B; e) marcata elevazione di LDH (3).
Questi criteri sono stati utilizzati per generare ilsospetto clinico di SR, e per ottenere la conse-guente conferma istologica (3). La rigorosa appli-cazione di questi criteri per la biopsia in una serieconsecutiva di pazienti affetti da LLC ha rivela-to che l’incidenza cumulativa di SR a 5 e 10 annidalla diagnosi è superiore rispettivamente a 10e 15% (3).In questa stessa serie di pazienti, l’incidenza diSR non si è dimostrata aumentare in modo signi-ficativo con il progressivo fallimento delle varielinee di terapia della LLC (3). Questa osservazio-ne può essere ascritta alla precoce pratica biop-tica adottata, che potrebbe identificare precoce-mente la SR prima di esporre i pazienti a multi-ple linee di terapia, che, essendo dirette controla progressione di LLC, potrebbero risultare inef-ficaci o solo parzialmente efficaci nel caso di tra-sformazione. Nella casistica sovrariportata, iltempo mediano alla trasformazione in SR era di23 mesi dalla diagnosi di LLC (3). Questo dato è confermato da altre serie indipen-denti di pazienti riportate in letteratura (4), e sug-gerisce che, almeno in una frazione di pazienti,la predisposizione a trasformazione da LLC a SRsia intrinseca alle caratteristiche genetiche delclone di LLC già al tempo della diagnosi di LLC(3, 4). A differenza dell’opinione comune che vede la SRcome un evento molto tardivo nella storia clinicadi LLC, una frazione significativa di pazienti svi-luppa la malattia precocemente dopo la diagno-si. In alcuni casi, la diagnosi di SR e LLC sonoconcomitanti. Queste osservazioni suggerisconol’importanza dell’identificazione dei pazienti arischio di trasformazione in SR già al momentodella diagnosi di LLC.
n FATTORI PREDITTIVI È stato dimostrato che l’estensione della malat-tia influenza la prognosi dei pazienti affetti da SR(15). Di conseguenza, il precoce riconoscimentodella SR può essere utile clinicamente e può esse-re favorito dallo stretto monitoraggio dei pazien-ti affetti da LLC che presentino fattori clinici o bio-logici di rischio di trasformazione. Recenti studihanno identificato, in serie retrospettive di pazien-ti, fattori di rischio clinici e biologici che potreb-

61Sindrome di Richter
bero essere utili strumenti per individuare ipazienti a rischio di sviluppo di SR consentendo-ne una diagnosi precoce (3). È importante nota-re che i predittori di trasformazione da LLC a SRsono diversi, almeno in parte, rispetto ai predit-tori di progressione di LLC stabiliti dalle linee gui-da IWCLL-NCI (2, 3). Questa osservazione sug-gerisce che la trasformazione da LLC a SR e laprogressione di LLC senza trasformazione isto-logica siano eventi clinici distinti che richiedono
un approccio diagnostico differente, benchécomplementare, per un’ottimale valutazione glo-bale della categoria di rischio del singolo pazien-te.Numerosi fattori di rischio biologici sono statianalizzati come predittori di trasformazione in SR,tra cui: l’omologia ≥98% del gene codificante perla regione variabile della catena pesante delleimmunoglobuline (IGHV), l’utilizzo di specifici geniIGHV, l’utilizzo di recettore delle cellule B (BCR)stereotipato, il cariotipo FISH, lo stato mutazio-nale di TP53, il genotipo di BCL2, la lunghezzadel telomero e l’espressione di CD38, ZAP70 eCD49d (3, 16-19). Di questi markers biologici,solo alcuni si sono dimostrati essere fattori dirischio indipendenti per lo sviluppo di SR, in par-ticolare: a) espressione di CD38 (Figura 1); b) assenza di del13q14; c) utilizzo di specifici geni IGHV (IGHV4-39); d) utilizzo di BCR stereotipato;e) lunghezza del telomero (3, 16-19) (Figura 2). Oltre a questi però, esistono altri marcatori bio-logici che sono stati identificati come fattori dirischio di trasformazione da LLC a SR: è interes-sante notare come il profilo genetico dell’ospitegiochi un ruolo fondamentale in tal senso (20, 21)(Figura 2). È stato anche dimostrato come ladimensione linfonodale ≥3 centimetri, un parame-tro di malattia solida, sia l’unico fattore di rischioclinico di trasformazione da LLC a SR seleziona-to da due modelli di analisi multivariata (3) (Figura
FIGURA 2 - Fattori di rischio clinici emolecolari che, alla diagnosi di LLC,predicono la trasformazione a SR. Al tempodella diagnosi di LLC, molteplici marcatoripredicono un aumentato rischio ditrasformazione a SR, tra cui: espressione diCD38, dimensioni della linfoadenopatia,lunghezza del telomero, assenza didel13q14, caratteristiche immunogenetichedel clone LLC (BCR stereotipato), genotipiGG/CG dello SNP rs6449182 di CD38, egenotipo TT dello SNP rs2306029 di LRP4.
FIGURA 1 - Predizione di trasformazione a SR in base ad espres-sione di CD38 alla diagnosi di LLC. La curva di Kaplan-Meiermostra il valore di CD38 nel predire la trasformazione a SR nel-la serie di LLC (n=360) della Divisione di Ematologia dell’Universitàdel Piemonte Orientale Amedeo Avogadro (anni 2000-2010).

62 Seminari di Ematologia Oncologica
2). Contrariamente, nella stessa analisi multiva-riata, i parametri di malattia leucemica (conta lin-focitaria, stadio RAI avanzato, splenomegalia, per-centuale e pattern di coinvolgimento del midolloosseo) non sono emersi come fattori di rischio cli-nici indipendenti di trasformazione a SR (3). Comeatteso, i parametri di malattia leucemica sono risul-tati predittivi di breve tempo alla progressione dimalattia nei pazienti affetti da LLC senza trasfor-mazione a SR. La discrepanza nella predizione diprogressione versus trasformazione rafforza ulte-riormente la distinzione tra progressione di LLCe trasformazione in SR (3). Da un punto di vista biologico, il fatto che sial’espressione di CD38 sia la dimensione linfono-dale ≥3 cm siano predittori indipendenti di SR sug-gerisce che l’attività di un circuito molecolare neilinfonodi coinvolti da LLC possa essere importan-te per il rischio dei singoli pazienti e per la pato-genesi di SR. Molte evidenze suggeriscono unastretta associazione tra espressione di CD38 ecoinvolgimento linfonodale nella LLC (22-25).Infatti, l’espressione di CD38: a) è maggiore nelle cellule di LLC del linfonodo
rispetto alle cellule di LLC del sangue perife-rico o del midollo osseo;
b) è richiesta per la migrazione delle cellule di LLCnei tessuti linfoidi;
c) si associa a malattia prevalentemente nodale(22-25).
Queste stesse caratteristiche predicono la trasfor-mazione a SR (3). Il linfonodo può fornire un micro-ambiente ottimale per la proliferazione e la trasfor-mazione blastica delle cellule di LLC che espri-mono CD38 (26-28). Curiosamente, l’attivazionein vitro della via del segnale di CD38 induce la tra-sformazione delle cellule di LLC in plasmablasti,la cui morfologia mima le grandi cellule propriedella trasformazione in SR (26). L’identificazione dell’espressione di CD38 comefattore di rischio per lo sviluppo di SR ha stimo-lato ulteriori ricerche sulla correlazione di questamolecola con la trasformazione di LLC in SR.L’espressione di CD38 è regolata a molteplici livel-li (27). L’estremità 5’ dell’introne 1 del gene codi-ficante per CD38 è nota per essere un importan-te regolatore dell’espressione di CD38 in celluleemopoietiche (29). Nella stessa regione genica èstato mappato un polimorfismo di un singolo
nucleotide (single nucleotide polymorphism, SNP)che comporta la sostituzione C>G in posizionenucleotidica 184 (30). La presenza dell’allele mino-re G, in condizione di eterozigosi o di omozigo-si, correla con lo sviluppo di SR (20). Rispetto agliomozigoti CC, i pazienti affetti da LLC che sonoomozigoti GG hanno un aumento relativo del30.6% del rischio di sviluppare SR, mentre i sog-getti eterozigoti GC mostrano una probabilità inter-media pari al 12.4% (20). Inoltre, a 5 anni, i pazien-ti omozigoti GG mostrano un incremento dellaprobabilità di sviluppare SR, che è approssima-tivamente doppia rispetto a quella dei soggetti ete-rozigoti GC (20). Queste osservazioni dimostra-no che il profilo genetico dell’ospite riveste un ruo-lo rilevante nel predire lo sviluppo di SR nei pazien-ti affetti da LLC.Proprio quest’ultima osservazione ha fatto sì chevenissero intrapresi nuovi studi atti a identifica-re le variabili del background genetico dell’ospi-te coinvolte nella trasformazione da LLC a SR.L’analisi di un elevato numero di SNP ha rileva-to che anche il profilo genetico del gene LRP4(rs2306029) è un importante predittore di trasfor-mazione (21). In particolare, i pazienti omozigo-ti per l’allele minore T del polimorfismo LRP4rs2306029 hanno mostrato una probabilità di tra-sformazione a 5 anni significativamente maggio-re rispetto ai pazienti portatori in omozigosi o ineterozigosi dell’allele comune C (21). È interes-sante notare come il gene LRP4 codifichi per unaproteina coinvolta nella via di trasduzione delsegnale di Wnt/beta-catenina, che è nota peressere attivata nelle cellule di LLC (31). Il coin-volgimento di LRP4 nella patogenesi di altremalattie fa supporre un possibile ruolo dell’inte-razione tra LRP4 e Wnt/beta-catenina nellapatogenesi della SR (32). Il cariotipo FISH eseguito alla diagnosi di LLC hadimostrato di poter predire la trasformazione inSR. In particolare, l’assenza di del13q14 costitui-sce un fattore di rischio indipendente di sviluppo(3). È ipotizzabile che le differenze patogenetichetra LLC con o senza del13q14 siano alla base del-la differente predisposizione alla trasformazionein SR di queste due sottocategorie di LLC. Si ritie-ne infatti che del13q14 giochi un ruolo importan-te nella patogenesi della malattia e che la sua pre-senza attivi vie di trasduzione del segnale cellu-

63Sindrome di Richter
lare che sono differenti da quelle attivate in suaassenza (33, 34). Il concetto che la propensionealla trasformazione in linfoma diffuso a grandi cel-lule B possa essere influenzata dal profilo mole-colare della precedente fase indolente dellamalattia non è esclusivo della LLC. Infatti, anchenel linfoma MALT è stato dimostrato che la tra-sformazione in linfoma diffuso a grandi cellule Bdipende dalle caratteristiche del pre-esistente clo-ne B-cellulare (35). Altre alterazioni FISH, tra lequali del17p13, del11q23, +12 o la traslocazio-ne della banda 14q32, non sono risultate essereassociate a un aumentato rischio di SR (3, 36).Anche la selezione da parte dell’antigene attra-verso il BCR può facilitare la trasformazione di LLCin SR. Il ruolo del BCR nel determinare lo svilup-po di SR può dipendere anche dalle caratteristi-che del gene IGHV utilizzato dal clone tumorale.Infatti, l’utilizzo di uno specifico gene IGHV, chia-mato IGHV4-39, è un fattore di rischio indipen-dente per lo sviluppo di SR (3, 18). In aggiunta,la frequenza di IGHV4-39 nella SR è significati-vamente maggiore rispetto alle LLC che non van-no incontro a trasformazione (18). L’utilizzo diIGHV4-39 è selettivamente predittivo di SR, e nonè stato associato ad altri outcome di LLC (3, 37). Una frazione rilevante di pazienti affetti da LLC (cir-ca il 30%) mostra un elevato grado di somiglian-za strutturale tra i BCR, dovuta all’elevato gradodi omologia tra le regioni VH CDR3 dei geni codi-ficanti per le catene pesanti delle immunoglobu-line (38-41). Questo fenomeno è conosciuto comeBCR stereotipato e suggerisce un importante ruo-lo della stimolazione antigenica nello sviluppo del-la malattia (37). Recentemente, il nostro gruppoha dimostrato come la presenza di BCR stereo-tipato al momento della diagnosi di LLC costitui-sca un fattore di rischio per la trasformazione aSR (18). Comparando le caratteristiche immuno-genetiche di 69 casi di SR con quelle di 714 casidi LLC, la presenza di BCR stereotipato è risul-tato significativamente maggiore nella popolazio-ne di SR rispetto a LLC non trasformate (49.3%rispetto a 21.3%) (18). Applicando un approccioattuariale a questa coorte di pazienti, è stato dimo-strato che l’utilizzo di BCR stereotipato al momen-to della diagnosi di LLC è un predittore indipen-dente di sviluppo di SR (18). Il rischio di trasfor-mazione dei pazienti portatori del BCR stereoti-
pato è indipendente dallo stato mutazionale deigeni IGHV (18).La disponibilità di numerosi prognosticatori indi-pendenti di SR rappresenta la base per costrui-re in futuro uno score di rischio clinico applica-bile al singolo paziente al momento della diagno-si di LLC.
n ISTOPATOLOGIA
Classicamente, la SR è rappresentata dal linfo-ma diffuso a grandi cellule B (3, 4, 9-11, 42-46).La malattia coinvolge più frequentemente i linfo-nodi, ma non sono infrequenti le localizzazioniextra-nodali a livello del tratto gastroenterico, cuta-neo, epatico, tonsillare e nel midollo osseo (3, 4,9-11, 42-46).In alcuni casi, il coinvolgimento della SR non ègeneralizzato a tutti i siti, ma è ristretto ad una sin-gola lesione nodale o extranodale. Pertanto, daun punto di vista pratico-clinico, la biopsia deveessere diretta alla lesione indice, ovvero la lesio-ne che mostra il maggiore diametro tramite ima-ging radiologico. Le caratteristiche PET della lesio-ne, in particolare il valore del SUV, possono aiu-tare per la scelta del sito bioptico, dal momentoche le regioni affette da SR hanno un valore diSUV simile a quello del linfoma diffuso a grandicellule B de novo (47). L’utilizzo della PET nelladiagnosi di SR e nella predizione del coinvolgi-mento degli organi da parte della SR è in studioe non è ancora del tutto standardizzato (47).I casi di SR diagnosticati come linfoma diffuso agrandi cellule B generalmente consistono in stra-ti confluenti di grandi linfociti B neoplastici chesono chiaramente distinguibili dalla proliferazio-ne di piccoli linfociti con scarso citoplasma e cro-matina addensata che sono tipici della fase del-la LLC. I casi di LLC con numerosi centri prolife-rativi o aumentata proporzione di prolinfociti, masenza l’aspetto di linfoma diffuso a grandi cellu-le B, non devono essere diagnosticati come SR.Inoltre, i casi che si presentano come linfoma dif-fuso a grandi cellule B esprimenti il CD5 devonoessere distinti dal linfoma diffuso a grandi cellu-le B de novo CD5 positivo, che rappresenta unadistinta entità patologica (48).Morfologicamente, tra le SR si possono ricono-

64 Seminari di Ematologia Oncologica
scere entrambe le varianti di linfoma diffuso agrandi cellule B: centroblastica e immunoblasti-ca. Basandosi sull’algoritmo di Hans per il profi-lo immunoistochimico dei linfomi diffusi a grandicellule B de novo (49), la maggior parte di SR(approssimativamente l’80%) mostrano un feno-tipo non-germinal center identificato da positivi-tà per IRF4, mentre solo il 20% dei casi mostraun fenotipo germinal center-like definito dal-l’espressione di CD10 e/o BCL-6 (11). La propor-zione di casi di SR con fenotipo germinal center-like rispetto ai casi con fenotipo non-germinal cen-ter differisce marcatamente rispetto a quellariscontrata nei linfomi diffusi a grandi cellule B denovo (11, 49). È da notare, comunque, che l’al-goritmo di Hans è stato sviluppato per i linfomidiffusi a grandi cellule B de novo, e che i markersimmunoistochimici impiegati dall’algoritmo noncoprono la grande eterogeneità di linfomi diffusia grandi cellule B che si presentano come SR (49).Attualmente, mancano studi sul profilo di espres-sione genica della SR, che potrebbero fornire indi-zi importanti per una corretta comprensione del-la biologia e dell’istogenesi della malattia.La trasformazione della LLC in linfoma diffusoa grandi cellule B è frequentemente, sebbenenon sempre, accompagnata dalla perdita del-l’espressione degli antigeni CD5 e CD23, chesono invariabilmente espressi dal clone neopla-stico nella fase di LLC (11). Il CD20 è general-mente espresso dalle cellule linfomatose di SRe rappresenta un bersaglio dell’immunoterapiacon rituximab (11).Come detto prima, il termine SR in questa ras-segna è ristretto alla trasformazione di LLC in lin-foma diffuso a grandi cellule B che è da consi-derarsi la classica forma di SR. Quando usato inmodo più ampio, il termine SR può comprende-re anche altre varianti clinico-patologiche. Il lin-foma di Hodgkin classico può presentarsi in raricasi di LLC, e riflette la morfologia e il profilo diespressione antigenica del linfoma di Hodgkininsorto come malattia primaria (4, 11).
n PATOGENESI MOLECOLARE
I meccanismi molecolari di trasformazione da LLCa SR sono per la maggior parte sconosciuti. Nella
SR classica, rappresentata dal linfoma diffuso agrandi cellule B, studi immunogenotipici hannorivelato che la maggior parte dei casi di SR(approssimativamente l’80%) sono clonalmentecorrelati al pre-esistente clone di LLC (3-11). Siritiene che queste SR siano dovute all’accumulodi lesioni genetiche e/o epigenetiche che guida-no lo sviluppo di una popolazione cellulareaggressiva originante dal clone di LLC. Il restan-te 20% dei casi di SR non è correlato al clone ori-ginale della LLC (3-11). I meccanismi patogene-tici in questo contesto non possono essere diret-tamente correlati all’accumulo di lesioni geneti-che del clone LLC, ma piuttosto potrebbero esse-re correlati ad alterazioni del profilo genetico edalla funzione immunologica dell’ospite, oppure adisfunzioni del microambiente cellulare cheaumentino la probabilità di sviluppo di linfoma dif-fuso a grandi cellule B. Lo sviluppo di linfoma dif-fuso a grandi cellule B non clonalmente correla-to alla LLC può essere parte di un fenomeno piùgeneralizzato che, per ragioni ancora sconosciu-te, aumenta la predisposizione dei pazienti affet-ti da LLC allo sviluppo di una seconda neoplasialinfoide o, più in generale, di un secondo tumo-re (5).Ad oggi, la conoscenza delle lesioni geneticheassociate alla trasformazione da LLC a linfoma dif-fuso a grandi cellule B clonalmente correlato èancora limitata. L’acquisizione di mutazioni diTP53 e/o del17p13 sono eventi molecolari fre-quenti nelle SR (50, 51), così come in altri tipi ditrasformazione da neoplasia B indolente a neo-plasia B aggressiva (52, 53). Curiosamente,rispetto al frequente coinvolgimento nella pato-genesi della SR, le mutazioni di TP53 alla diagno-si di LLC non aumentano il rischio successivo disviluppo di SR (3).L’enzima activation-induced cytidine deaminase(AID) è essenziale per la fisiologica ipermutazio-ne somatica delle cellule B normali, e può con-durre a ipermutazione somatica aberrante (ASHM)di molteplici proto-oncogeni coinvolti nella linfo-magenesi B-cellulare (54, 55). La ASHM dicMYC, RhoH/TTF, PAX5 e PIM1 rappresenta lalesione genetica più frequente ad oggi conosciu-ta nei linfomi diffusi a grandi cellule B de novo (56-58). Nonostante i livelli di AID aumentino al tem-po della trasformazione in SR (9, 59), gli studi di

65Sindrome di Richter
ASHM nella SR hanno prodotto risultati contra-stanti (59, 60). L’apparente bassa incidenza diASHM nella SR distingue la trasformazione daLLC a linfoma diffuso a grandi cellule B rispettoalla trasformazione da linfoma follicolare a linfo-ma diffuso a grandi cellule B. Quest’ultima, infat-ti, è frequentemente accompagnata da un altonumero di eventi ascrivibili ad ASHM che è ope-rativa al momento della trasformazione istologi-ca (60).Gli studi citogenetici finora non hanno rivelato ano-malie ricorrenti selettivamente associate a SR (61-65). Il riscontro di trisomia 12 nella SR non è spe-cifico per la malattia, dal momento che la triso-mia 12 è di frequente riscontro in diverse altre neo-plasie B cellulari, inclusa la LLC (66). A differen-za degli altri tipi di linfoma non-Hodgkin B, la tra-slocazione 14q32 non contribuisce ai meccani-smi molecolari coinvolti nella trasformazione daLLC a SR (36). In accordo con ciò, nella SR nonsi riscontrano traslocazioni di BCL1, BCL2 e BCL6(3). La mancanza del coinvolgimento della traslo-cazione di 14q32 nello sviluppo di SR suggerisceche la patogenesi della SR sia diversa da quelladegli altri linfomi non-Hodgkin aggressivi, inclu-so il linfoma diffuso a grandi cellule B de novo,in cui un meccanismo patogenetico frequente èla deregolazione di proto-oncogeni tramite la giu-stapposizione ai loci delle IG (36, 67, 68). Gli stu-di di citogenetica molecolare hanno dimostratoche la SR si associa ad un alto grado di comples-sità genomica rispetto alla LLC, sebbene nessu-na delle lesioni genetiche ad oggi riscontrate siaspecifica per SR (69).Nella patogenesi della SR è stato postulato ancheun possibile ruolo dell’infezione da EBV. Tuttaviail fatto che la maggior parte dei casi di SR sianegativa per l’infezione da EBV non è a favore diquesta ipostesi (3). La presenza di sequenze diEBV è stata documentata in alcuni, tuttavia nontutti, pazienti affetti da SR precedentemente trat-tati con fludarabina nella fase di LLC (70, 71). Èstato ipotizzato che l’infezione da EBV in questicasi sia conseguenza della deregolazione immu-nunitaria causata dall’analogo nucleosidico.Complessivamente le osservazioni sulla patoge-nesi della SR indicano che questa patologia nonpuò essere facilmente riassunta da lesioni gene-tiche identificate in altri disordini B cellulari. Si
impone quindi la necessità di nuovi studi finaliz-zati a chiarire le basi molecolari della malattia.Auspicabilmente, gli studi genomici attualmentein corso potranno rivelare nuove alterazioni mole-colari e anomalie nell’espressione genica coinvol-te nello sviluppo della SR.
n CARATTERISTICHE CLINICHE
La diagnosi di SR richiede la dimostrazione isto-logica della trasformazione da LLC a linfomaaggressivo. Quando possibile, è preferibile sot-toporre la lesione indice a biopsia. Solamente incasi eccezionali, quando la biopsia non possaessere eseguita a causa del performance statusdel paziente o per difficoltà tecniche all’accessodel sito anatomico della lesione indice, la dimo-strazione citologica ottenuta tramite biopsia adago sottile può essere accettabile per porre dia-gnosi di SR.La prognosi dei pazienti affetti da SR è general-mente considerata altamente sfavorevole. Tuttavia,in una coorte di 148 pazienti affetti da SR diagno-sticata tramite biopsia o agoaspirato è stato dimo-strato che la sopravvivenza della SR non è uni-forme tra i pazienti, con range che va da pochesettimane a 15 anni (15). La prognosi può esse-re predetta dal SR score, che non predice la pos-sibilità di sviluppo di SR nei pazienti affetti da LLC,ma piuttosto predice la prognosi della SR una vol-ta avvenuta la trasformazione (15).Lo SR score predice il rischio di morte del pazien-te affetto da SR in base a 5 diversi fattori sfavo-revoli che predicono una ridotta sopravvivenza: a) Zubrod performance status >1; b) elevati livelli di LDH; c) conta piastrinica ≤100000; d) lesioni di dimensioni ≥5 cm; e) più di 2 precedenti linee di terapia (15). Poichè il rischio relativo associato a ciascuno deifattori di rischio che costituiscono il SR score ècomparabile, il rischio relativo di morte è ottenu-to dalla somma del numero dei fattori di rischioche sono presenti alla diagnosi di SR. I pazientisono perciò assegnati ad uno di 4 gruppi di rischiosulla base del numero dei fattori di rischio pre-sentati: 0 o 1, basso rischio; 2, rischio interme-dio-basso; 3, rischio intermedio-alto; 4 o 5, alto

66 Seminari di Ematologia Oncologica
rischio. Nello SR score originario, la sopravviven-za era 1.12 anni nei pazienti con SR score 0-1;0.90 anni nei pazienti con SR score 2; 0.33 anninei pazienti con SR score 3; e 0.14 anni neipazienti con SR score 4-5 (15). Curiosamente, ifattori di rischio valutati dall’InternationalPrognostic Index (numero di siti extranodali dimalattia, età e stadio di malattia) non sono rile-vanti nel SR score, confermando il fatto che SRe linfoma diffuso a grandi cellule B de novo sonopatologie tra loro differenti non solo biologicamen-te, ma anche clinicamente.
n TRATTAMENTO
Nessuno degli attuali approcci terapeutici puòessere considerato terapia standard o comun-que soddisfacente per la SR. Sebbene la mag-gior parte dei casi di SR sia istologicamente clas-sificata come linfoma diffuso a grandi cellule B,i risultati della terapia sono molto meno promet-tenti rispetto a quelli ottenuti nel linfoma diffu-so a grandi cellule B de novo, e in molti casi iltrattamento è destinato a fallire (72-76). Gli scar-si risultati nella SR rispetto al linfoma diffuso agrandi cellule B de novo sono motivati da treragioni intuitive: a) la SR si associa frequentemente a marcatori
biologici di chemiorefrattarietà, tra cui l’inatti-vazione di TP53;
b) la massa tumorale alla diagnosi è elevata, acausa di frequente ritardo diagnostico;
c) la fragilità dei pazienti affetti da SR costituisceun fattore limitante la scelta terapeutica.
Come per gli altri linfomi trasformati, per esem-pio il linfoma diffuso a grandi cellule B che origi-na da una precedente diagnosi di linfoma follico-lare, i casi di SR sono intrinsecamente più che-mioresistenti, e mostrano un maggior rischio dialterazioni geniche, quali del17p13 e mutazioni diTP53, che rendono questa malattia refrattaria aiprogrammi chemioterapici convenzionali (3, 68).L’esteso coinvolgimento della malattia, la grandemassa tumorale e la rapida cinetica sono carat-teristiche comuni della SR, caratterizzandone piùdel 50% dei casi (3, 15). È stato dimostrato chela massa tumorale è un predittore indipendentedi ridotta sopravvivenza nei pazienti affetti da SR
(15). La fragilità, comune nel contesto dei pazien-ti affetti da SR, deriva dallo scarso performancestatus e dalla scarsa funzionalità midollare e dal-l’immunodeficienza (3, 15), e rappresenta un pre-dittore indipendente di ridotta sopravvivenza neipazienti affetti da SR (15). La fragilità dei pazien-ti preclude, in un’elevata frazione di casi, l’uso diterapia ad alte dosi associata a trapianto di cel-lule staminali, limitando quindi le opzioni terapeu-tiche e il possibile beneficio derivato dall’uso diregimi terapeutici mieloablativi (15). A causa diqueste caratteristiche cliniche e biologiche dellaSR, la sopravvivenza mediana con chemiotera-pia convenzionale con o senza rituximab risultainferiore a 12 mesi (72-76).I regimi chemioterapici utilizzati nella SR sono ete-rogenei e sono stati sviluppati inizialmente per altridisordini linfoidi, in particolare per i linfomi non-Hodgkin aggressivi o per la leucemia linfoblasti-ca acuta. In epoca pre-rituximab, i regimi utiliz-zati per la SR includevano, ma non erano limita-ti a, CHOP-bleo, MACOP-B, ASHAP, VAD ehyper-CVXD (74-77). Rituximab-CHOP, che èconsiderato terapia standard per il linfoma diffu-so a grandi cellule B de novo, è generalmenteadottato nella SR come regime terapeutico discelta, sebbene i risultati siano lontani dall’esse-re soddisfacenti (15, 78). È stata anche utilizza-ta la combinazione di rituximab con regimi fra-zionati, per esempio rituximab-hyper-CVXD (76).La combinazione di rituximab con CHOP o hyper-CVXD ha indotto risposte nel 47% dei pazienti,versus un tasso di risposta pari al 34% utilizzan-do la sola chemioterapia (76). Sebbene le diffe-renze non raggiungano la significatività statisti-ca, i dati suggeriscono che l’immunochemiote-rapia contenente rituximab possa offrire alcunibenefici nella terapia della SR.Recentemente è stato proposto un trial di fase I-II volto a testare, nei pazienti con SR, un regimepoli-chemioterapico denominato OFAR, contenen-te oxaliplatino, fludarabina, citarabina e rituximab(79). Il razionale alla base dello sviluppo del regi-me OFAR prende origine da 3 osservazioni: a) dati pre-clinici hanno dimostrato una citotos-
sicità sinergica tra oxaliplatino e gli analoghinucleosidici fludarabina e citarabina (80, 81);
b) la somministrazione di fludarabina prima dicitarabina aumenta la concentrazione intracel-

67Sindrome di Richter
lulare del metabolita attivo, la citarabina trifo-sfato (81);
c) i regimi basati sull’oxaliplatino contenenticitarabina ad alte dosi hanno mostrato attivi-tà nei linfoma diffuso a grandi cellule B de novorecidivato, così come in altri tipi di linfoma (82,83).
Il 50% dei pazienti affetti da SR e trattati con ilregime OFAR ha ottenuto una risposta comple-ta o parziale (79). OFAR è stato generalmente bentollerato, e le tossicità di grado 3-4 sono state prin-cipalmente ematologiche. A dispetto di questi risultati promettenti, la dura-ta mediana della risposta ottenuta con il regimeOFAR è stata di soli 10 mesi, con un tasso disopravvivenza a 6 mesi del 60% (79). I pazientiche hanno ottenuto una risposta completa o unarisposta parziale hanno mostrato una più lungasopravvivenza rispetto ai pazienti la cui malattianon ha risposto a OFAR (79). Poichè i risultati conl’immunoterapia convenzionale, incluso OFAR,non possono essere considerati soddisfacenti, ènecessario che vengano esplorati nuovi approc-ci terapeutici per la SR. Un’opzione è rappresen-tata dall’utilizzo di terapie di consolidamento o dimantenimento una volta che si sia ottenuta unarisposta completa o una risposta parziale con R-CHOP, OFAR, o altri regimi. Nonostante la scar-sa attività come agenti singoli per la terapia diinduzione della SR (84, 85), l’immunoterapia, conrituximab e alemtuzumab, o la radioimmunotera-pia possono essere opzioni attraenti per la tera-pia di consolidamento e di mantenimento neipazienti affetti da SR nel contesto di approcci mul-tistep, già esplorati nella LLC e nei linfomi non-Hodgkin (86-93). Gli approcci basati su alemtuzumab o radioim-munoterapia possono essere considerati speri-mentali al momento, ed è necessario che sianoinvestigati in trials clinici dedicati.I pazienti affetti da SR più giovani sono candida-ti al trapianto autologo o allogenico di cellule sta-minali emopoietiche (94). Il trapianto allogenico dicellule staminali emopoietiche può essere consi-derato particolarmente promettente (15). Uno stu-dio effettuato dallo MD Anderson Cancer Centerha mostrato che la sopravvivenza cumulativa a3 anni è pari al 75% nei pazienti che avevano otte-nuto una risposta completa o una risposta par-
ziale e che poi erano stati sottoposti a trapiantoallogenico di cellule staminali emopoietiche,rispetto a una sopravvivenza del 27% per i pazien-ti che inizialmente hanno risposto alla terapia, manon sono stati sottoposti al trapianto allogenico(15). Il trapianto allogenico di cellule staminali,come atteso, non è risultato di beneficio neipazienti refrattari ai regimi di induzione (15). Comeper altre malattie aggressive oncoematologiche,quindi, l’ottenimento della risposta in fase di indu-zione è un obiettivo molto importante anche nelcaso di SR.
n STATO ATTUALE E PROSPETTIVE FUTURE
Gli studi degli ultimi anni hanno documentato unnumero crescente di marcatori molecolari efenotipici che, già al tempo della diagnosi di LLC,permettono di attribuire il rischio di sviluppare SRin un qualche momento della storia naturale del-la malattia (3, 18-21). Questi markers sono rap-presentati dall’espressione e dal genotipo diCD38, dal cariotipo FISH, dall’utilizzo di un BCRstereotipato, dalla lunghezza del telomero, dal pro-filo genetico dell’ospite e dall’utilizzo di specificigeni IGHV (3, 18-21). La conoscenza di questi predittori biologici puòessere utile al fine di adottare una stretta sorve-glianza clinica e una politica bioptica più aggres-siva nei pazienti affetti da LLC che sono a rischiodi trasformazione, al fine di riconoscere preco-cemente lo sviluppo di SR. Vari predittori di tra-sformazione fanno parte delle indagini diagno-stiche che sono messe comunque in atto almomento della diagnosi di LLC, tra cui la cito-metria a flusso per CD38 e le indagini moleco-lari sui geni IGHV che sono ormai entrati nella pra-tica clinica. I predittori molecolari finora identifi-cati provengono da coorti retrospettive, e devo-no perciò idealmente essere validati in studi pro-spettici. Le nuove tecnologie disponibili, permet-tendo lo studio dell’intero genoma, possonoessere utili a identificare nuovi e specifici markersdi trasformazione da LLC a SR. La messa a punto dello SR score per la defini-zione della prognosi della SR ha rappresentato unimportante avanzamento nell’identificazione dei

68 Seminari di Ematologia Oncologica
fattori prognostici post-trasformazione nei pazien-ti affetti da SR (15). Attualmente, lo stato dell’arte prevede che ipazienti affetti da SR vengano trattati con tera-pie citoriduttive a base di rituximab in combina-zione con una poli-chemioterapia citotossica conl’intento di ottenere una risposta. Ai pazienti inrisposta dopo il trattamento di induzione, puòessere offerta l’opzione trapiantologica (allo-tra-pianto) se percorribile in relazione all’età delpaziente, al performance status ed alla disponi-bilità di un donatore (15). Non è stato indagato,ad oggi, il ruolo dell’immunoterapia come terapiadi consolidamento per le SR che rispondano allaterapia di induzione, ma che non che sianosuscettibili di trapianto, sul modello di modalitàterapeutiche esplorate nella LLC (es. alemtuzu-mab) e nei linfomi (90Y-ibritumomab, 131I-tositumo-mab). Allo stesso modo, non è stato ancora valu-tato l’impatto dei nuovi farmaci (es. lenalidomide,ofatumomab, flavopiridolo) nell’induzione dellarisposta di SR. Per rispondere a questi quesitisarebbero necessari studi pilota e, qualora pos-sibili, trial clinici multicentrici.Molti sono gli interrogativi che rimangono anco-ra aperti nella SR. Nonostante il termine SR siacorrentemente applicato sia ai linfomi diffusi agrandi cellule B clonalmente correlati sia a quel-li non clonalmente correlati (3, 4), è probabile chele due condizioni siano distinte l’una dall’altra eche abbiano differente prognosi e, potenzialmen-te, richiedano differenti trattamenti. Analogamente,non è noto se le SR ad esordio precoce e le SRad esordio tardivo siano biologicamente e clini-camente differenti. L’ostacolo a una terapia mirata della SR è rap-presentato dalla scarsa conoscenza delle lesio-ni molecolari, genetiche o epigenetiche respon-sabili della trasformazione da LLC a SR. La dia-gnosi precoce di SR nei pazienti affetti da LLC èparticolarmente importante (15). Questa osserva-zione impone di perfezionare l’individuazione difattori di rischio per SR, nonché di definire stru-menti diagnostici non invasivi utili a generare ilsospetto di SR con alta sensibilità e specificità.A questo scopo, gli sforzi devono essere direttia generare un algoritmo diagnostico-prognosti-co che utilizzi i fattori di rischio clinici e biologicioggi noti per la SR.
n BIBLIOGRAFIA
1. Richter MN. Generalized reticular cell sarcoma of lymphnodes associated with lymphatic leukaemia. Am JPathol. 1928; 4: 285-92.
2. Hallek M, Cheson BD, Catovsky D, Caligaris-CappioF, Dighiero G, Döhner H, et al. Guidelines for the diag-nosis and treatment of chronic lymphocytic leukemia:a report from the InternationalWorkshop on ChronicLymphocytic Leukemia updating the National CancerInstitute-Working Group 1996 guidelines. Blood. 2008;111: 5446-56.
3. Rossi D, Cerri M, Capello D, Deambrogi C, Rossi FM,Zucchetto A, et al. Biological and clinical risk factorsof chronic lymphocytic leukaemia transformation toRichter syndrome. Br J Haematol. 2008; 142: 202-15.
4. Tsimberidou A-M, Keating MJ. Richter syndrome.Biology, incidence, and therapeutic strategies. Cancer.2005; 103: 216-28.
5. Maddocks-Christianson K, Slager SL, Zent CS,Reinalda M, Call TG, Habermann TM, et al. Risk fac-tors for development of a second malignancy inpatients with chronic lymphocytic leukemia. Br JHaematol. 2007; 139: 398-404.
6. Cherepakhin V, Baird SM, Meisenholder GW, Kipps TJ.Common clonal origin of chronic lymphocytic leukemiaand high grade lymphoma of Richter’s syndrome.Blood. 1993; 82: 3141-7.
7. Matolcsy A, Inghirami G, Knowles DM. Moleculargenetic demonstration of the diverse evolution ofRichter’s syndrome (chronic lymphocytic leukemia andsubsequent large cell lymphoma). Blood. 1994; 83:1363-72.
8. Nakamura N, Kuze T, Hashimoto Y, Hoshi S, TominagaK, Sasaki Y, et al. Analysis of the immunoglobulin heavychain gene of secondary diffuse large B-cell lymphomathat subsequently developed in four cases with B-cellchronic lymphocytic leukemia or lymphoplasmacytoidlymphoma (Richter syndrome). Pathol Int. 2000; 50:636-43.
9. Timár B, Fülöp Z, Csernus B, Angster C, Bognár A,Szepesi A, et al. Relationship between the mutation-al status of VH genes and pathogenesis of diffuse largeB-cell lymphoma in Richter’s syndrome. Leukemia.2004; 18: 326-30.
10. Smit LA, van Maldegem F, Langerak AW, van derSchoot CE, de Wit MJ, Bea S, et al. Antigen recep-tors and somatic hypermutation in B-cell chronic lym-phocytic leukemia with Richter’s transformation.Haematologica. 2006; 91: 903-11.
11. Mao Z, Quintanilla-Martinez L, Raffeld M, Richter M,Krugmann J, Burek C, et al. IgVH mutational status andclonality analysis of Richter’s transformation. Am J SurgPathol. 2007; 31: 1605-14.
12. O’Brien SM, Kantarjian HM, Thomas DA, Cortes J,Giles FJ, Wierda WG, et al. Alemtuzumab as treatmentfor residual disease after chemotherapy in patients with

69Sindrome di Richter
chronic lymphocytic leukemia. Cancer. 2003; 98:2657-63.
13. Janssens A, Berth M, De Paepe P, Verhasselt B, VanRoy N, Noens L, et al. EBV negative Richter’s syndromefrom a coexistent clone after salvage treatment withalemtuzumab in a CLL patient. Am J Hematol. 2006;81: 706-12.
14. Montoto S, Davies AJ, Matthews J, Calaminici M,Norton AJ, Amess J, et al. Risk and clinical implicationsof transformation of follicular lymphoma to diffuse largeB-cell lymphoma. J Clin Oncol. 2007; 25: 2426-33.
15. Tsimberidou A-M, O’Brien S, Khouri I, Giles FJ,Kantarjian HM, Champlin R, et al. Clinical outcomesand prognostic factors in patients with Richter’s syn-drome treated with chemotherapy or chemoim-munotherapy with or without stem-cell transplantation.J Clin Oncol. 2006; 24: 2343-51.
16. Rossi D, Rasi S, Capello D, Gaidano G. Prognosticassessment of BCL2-938C>A polymorphism in chron-ic lymphocytic leukemia. Blood. 2008; 111: 466-8.
17. Rossi D, Zucchetto A, Rossi FM, Capello D, Cerri M,Deambrogi C, et al. CD49d expression is an independ-ent risk factor of progressive disease in early stagechronic lymphocytic leukemia. Haematologica. 2008;93: 1575-9.
18. Rossi D, Spina V, Cerri M, Rasi S, Deambrogi C, DePaoli L, et al. Stereotyped B-cell receptor is an inde-pendent risk factor of chronic lymphocytc leukemiatransformation to Richter syndrome. Clin Cancer Res.2009; 15: 4415-22.
19. Rossi D, Lobetti Bodoni C, Genuardi E, Monitillo L,Drandi D, Cerri M, et al. Telomere length is an inde-pendent predictor of survival, treatment requirementand Richter’s syndrome transformation in chronic lym-phocytic leukemia. Leukemia. 2009; 23: 1062-72.
20. Aydin S, Rossi D, Bergui L, D'Arena G, Ferrero E,Bonello L, et al. CD38 gene polymorphism and chron-ic lymphocytic leukemia: a role in Richter syndrome?Blood. 2008; 111: 5646-53.
21. Rasi S, Spina V, Bruscaggin A, Vaisitti T, Tripodo C,Forconi F, et al. A variant of the LRP4 gene affectsthe risk of chronic lymphocytic leukemia transforma-tion to Richter syndrome. Br J Haematol. 2011; 152:284-94.
22. Del Poeta G, Maurillo L, Venditti A, Buccisano F,Epiceno AM, Capelli G, et al. Clinical significance ofCD38 expression in chronic lymphocytic leukemia.Blood. 2001; 98: 2633-9.
23. Ibrahim S, Keating M, Do KA, O'Brien S, Huh YO, JilaniI, et al. CD38 expression as an important prognosticfactor in B-cell chronic lymphocytic leukemia. Blood.2001; 98: 181-6.
24. Jaksic O, Paro MM, Kardum Skelin I, Kusec R, PejsaV, Jaksic B. CD38 on B-cell chronic lymphocyticleukemia cells has higher expression in lymph nodesthan in peripheral blood or bone marrow. Blood. 2004;103: 1968-9.
25. Deaglio S, Vaisitti T, Aydin S, Bergui L, D’Arena G,Bonello L, et al. CD38 and ZAP-70 are functionallylinked and mark CLL cells with high migratory poten-tial. Blood. 2007; 110: 4012-21.
26. Deaglio S, Capobianco A, Bergui L, Dürig J, MorabitoF, Dührsen U, et al. CD38 is a signalling molecule inB-cell chronic lymphocytic leukemia cells. Blood. 2003;102: 2146-55.
27. Deaglio S, Vaisitti T, Aydin S, Ferrero E, Malavasi F. In-tandem insights from basic science combined with clin-ical research: CD38 as both marker and key compo-nent of the pathogenetic network underlying chroniclymphocytic leukemia. Blood. 2006; 108: 1135-44.
28. Patten PE, Buggins AG, Richards J, Wotherspoon A,Salisbury J, Mufti GJ, et al. CD38 expression in chron-ic lymphocytic leukemia is regulated by the tumormicroenvironment. Blood. 2008; 111: 5173-81.
29. Kishimoto H, Hoshino S, Ohori M, Kontani K, NishinaH, Suzawa M, et al. Molecular mechanisms of humanCD38 gene expression by retinoic acid. Identificationof retinoic acid response element in the first intron. JBiol Chem. 1998; 273: 15429-34.
30. Ferrero E, Saccucci F, Malavasi F. The human CD38gene: polymorphism, CpG island, and linkage to theCD157 (BST-1) gene. Immunogenetics. 1999; 49: 597-604.
31. Lu D, Zhao Y, Tawatao R et al. Activation of the Wntsignaling pathway in chronic lymphocytic leukemia.Proc Natl Acad Sci USA. 2004; 101: 3118-23.
32. Li Y, Pawlik B, Elcioglu N, Kayserili H, Yigit G, PercinF, et al. LRP4 mutations alter Wnt/beta-catenin signal-ing and cause limb and kidney malformations in Cenani-Lenz syndrome. Am J Hum Genet. 2010; 86: 696-706.
33. Calin GA, Ferracin M, Cimmino A, Di Leva G, ShimizuM, Wojcik SE, et al. A microRNA signature associat-ed with prognosis and progression in chronic lympho-cytic leukemia. N Engl J Med. 2005; 353: 1793-801.
34. Kienle DL, Korz C, Hosch B, Benner A, Mertens D,Habermann A, et al. Evidence for distinct pathomech-anisms in genetic subgroups of chronic lymphocyticleukemia revealed by quantitative expression analysisof cell cycle, activation, and apoptosis-associatedgenes. J Clin Oncol. 2005; 23: 3780-92.
35. Starostik P, Patzner J, Greiner A, Schwarz S, Kalla J,Ott G, et al. Gastric marginal zone Bcell lymphomasof MALT type develop along 2 distinct pathogeneticpathways. Blood. 2002; 99: 3-9.
36. Deambrogi C, Cresta S, Cerri M, Rasi S, De Paoli L,Ramponi A, et al. 14q32 translocations and risk ofRichter’s transformation in chronic lymphocyticleukemia. Br J Haematol. 2008; 144: 131-3.
37. Caligaris-Cappio F, Ghia P. Novel insights in chroniclymphocytic leukemia: are we getting closer to under-standing the pathogenesis of the disease? J Clin Oncol.2008; 26: 4497-503.
38. Stamatopoulos K, Belessi C, Moreno C, BoudjograhM, Guida G, Smilevska T, et al. Over 20% of patients

70 Seminari di Ematologia Oncologica
with chronic lymphocytic leukemia carry stereotypedreceptors: pathogenetic implications and clinical cor-relations. Blood. 2007; 109: 259-70.
39. Bomben R, Dal Bo M, Capello D, Forconi F, Maffei R,Laurenti L, et al. Molecular and clinical features ofchronic lymphocytic leukaemia with stereotyped B cellreceptors: results from an Italian multi center study. BrJ Haematol. 2009; 144: 492-506.
40. Sutton LA, Kostareli E, Hadzidimitriou A, Darzentas N,Tsaftaris A, Anagnostopoulos A, et al. Extensive intr-aclonal diversification in a subgroup of chronic lympho-cytic leukemia patients with stereotyped IGHV4-34receptors: implications for ongoing interactions withantigen. Blood. 2009; 114: 4460-8.
41. Darzentas N, Hadzidimitriou A, Murray F, Hatzi K,Josefsson P, Laoutaris N, et al. A different ontogene-sis for chronic lymphocytic leukemia cases carryingstereotyped antigen receptors: molecular and compu-tational evidence. Leukemia. 2010; 24: 125-32.
42. Armitage JO, Dick FR, Corder MP. Diffuse histiocyticlymphoma complicating chronic lymphocytic leukemia.Cancer. 1978; 41: 422-7.
43. Ott MM, Ott G, Roblick U, Linke B, Kneba M, de LeonF, et al. Localized gastric non-Hodgkin’s lymphoma ofhigh-grade malignancy in patients with preexistingchronic lymphocytic leukemia or immunocytoma.Leukemia. 1995; 9: 609-14.
44. Ratnavel RC, Dunn-Walters DK, Boursier L, Frazer-Andrews E, Orchard G, Russell-Jones R, et al. B-celllymphoma associated with chronic lymphocyticleukaemia: two cases with contrasting aggressive andindolent behaviour. Br J Dermatol. 1999; 140: 708-14.
45. Parrens M, Sawan B, Dubus P, Lacombe F, Marit G,Vergier B, et al. Primary digestive Richter’s syndrome.Mod Pathol. 2001; 14: 452-7.
46. Omoti CE, Omoti AE. Richter syndrome: a review ofclinical, ocular, neurological and other manifestations.Br J Haematol. 2008; 142: 709-16.
47. Bruzzi JF, Macapinlac H, Tsimberidou AM, Truong MT,Keating MJ, Marom EM, et al. Detection of Richter’stransformation of chronic lymphocytic leukemia byPET/CT. J Nucl Med. 2006; 47: 1267-73.
48. Matolcsy A, Chadburn A, Knowles DM. De novo CD5-positive and Richter’s syndrome-associated diffuselarge B cell lymphomas are genotypically distinct. AmJ Pathol. 1995; 147: 207-16.
49. Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD,Delabie J, Ott G, et al. Confirmation of the molecularclassification of diffuse large B-cell lymphoma byimmunohistochemistry using a tissue microarray.Blood. 2004; 103: 275-82.
50. Gaidano G, Ballerini P, Gong JZ, Inghirami G, Neri A,Newcomb EW, et al. p53 mutations in human lymphoidmalignancies: association with Burkitt lymphoma andchronic lymphocytic leukemia. Proc Natl Acad Sci USA.1991; 88: 5413-7.
51. Lee JN, Giles F, Huh YO, Manshouri T, O'Brien S,
Kantarjian HM, et al. Molecular differences betweensmall and large cells in patients with chronic lympho-cytic leukemia. Eur J Hematol. 2003; 71: 235-42.
52. Lo Coco F, Gaidano G, Louie DC, Offit K, Chaganti RS,Dalla-Favera R. p53 mutations are associated with his-tologic transformation of follicular lymphoma. Blood.1993; 82: 2289-95.
53. Lossos IS. Higher-grade transformation of follicular lym-phoma - a continuous enigma. Leukemia. 2005; 19:1331-3.
54. Klein U, Dalla-Favera R. Germinal centres: role in B-cell physiology and malignancy. Nat Rev Immunol.2008; 8: 12-33.
55. Pasqualucci L, Bhagat G, Jankovic M, Compagno M,Smith P, Muramatsu M, et al. AID is required for ger-minal center-derived lymphomagenesis. Nat Genetics.2008; 40: 108-12.
56. Pasqualucci L, Neumeister P, Goossens T, NanjangudG, Chaganti RS, Küppers R, et al. Hypermutation ofmultiple proto-oncogenes in B-cell diffuse large-celllymphomas. Nature. 2001; 412: 341-6.
57. Gaidano G, Pasqualucci L, Capello D, Berra E,Deambrogi C, Rossi D, et al. Aberrant somatic hyper-mutation in multiple subtypes of AIDS-associated non-Hodgkin lymphoma. Blood. 2003; 102: 1833-41.
58. Libra M, Capello D, Gloghini A, Laura P, Berra E, CerriM, et al. Analysis of aberrant somatic hypermutation(SHM) in non-Hodgkin lymphomas of patients with HCVinfection. J Pathol. 2005; 206: 87-91.
59. Reiniger L, Bödör C, Bognár Á, Balogh Z, Csomor J,Szepesi A, et al. Richter’s and prolymphocytic trans-formation of chronic lymphocytic leukemia are asso-ciated with high mRNA expression of activation-induced cytidine deaminase and aberrant somatichypermutation. Leukemia. 2006; 20: 1089-95.
60. Rossi D, Berra E, Cerri M, Deambrogi C, Barbieri C,Franceschetti S, et al. Aberrant somatic hypermuta-tion in transformation of follicular lymphoma and chron-ic lymphocytic leukemia to diffuse large B-cell lym-phoma. Haematologica. 2006; 91: 1405-9.
61. Han T, Sadamori N, Ozer H, Gajera R, Gomez GA,Henderson ES, et al. Cytogenetic studies in 77patients with chronic lymphocytic leukemia: correla-tions with clinical, immunologic, and phenotypic data.J Clin Oncol. 1984; 2: 1121-32.
62. Hébert J, Jonveaux P, d’Agay MF, Berger R.Cytogenetic studies in patients with Richter’s syndrome.Cancer Genet Cytogenet. 1994; 73: 65-8.
63. Brynes RK, McCourty A, Sun NC, Koo CH. Trisomy12 in Richter’s transformation of chronic lymphocyticleukemia. Am J Clin Pathol. 1995; 104: 199-203.
64. Caraway NP, Du Y, Zhang H-Z, Hayes K, Glassman AB,Katz RL. Numeric chromosomal abnormalities insmall lymphocytic and transformed large cell lym-phomas detected by fluorescence in situ hybridizationof fine-needle aspiration biopsies. Cancer Cytopathol.2000; 90: 126-32.

71Sindrome di Richter
65. Santulli B, Kazmierczak B, Napolitano R, Caliendo I,Chiappetta G, Rippe V, et al. A 12q13 translocationinvolving the HMGI-C gene in Richter transformationof a chronic lymphocytic leukemia. Cancer GenetCytogenet. 2000; 119: 70-3.
66. Zenz T, Mertens D, Döhner H, Stilgenbauer S.Molecular diagnostics in chronic lymphocyticleukaemia-pathogenetic and clinical implications. LeukLymphoma. 2008; 49: 864-73.
67. Rossi D, Gaidano G. Molecular heterogeneity of dif-fuse large Bcell lymphoma: implications for diseasemanagement and prognosis. Hematology. 2002; 7: 239-52.
68. Lenz G, Staudt LM. Aggressive lymphomas. N Engl JMed. 2010; 362: 1417-29.
69. Beà S, López-Guillermo A, Ribas M, Puig X, Pinyol M,Carrió A, et al. Genetic imbalances in progressed B-cell chronic lymphocytic leukemia and transformedlarge-cell lymphoma (Richter’s syndrome). Am JPathol. 2002; 161: 957-68.
70. Cohen Y, Da’as N, Libster D, Amir G, Berrebi A, PolliackA. Large-cell transformation of chronic lymphocyticleukemia and follicular lymphoma during or soon aftertreatment with fludarabine-rituximab containing regi-mens: natural history or related therapy complication.Eur J Hematol. 2002; 68: 80-3.
71. Thornton PD, Bellas C, Santon A, Shah G, Pocock C,Wotherspoon AC, et al. Richter’s transformation ofchronic lymphocytic leukemia. The possible role of flu-darabine and the Epstein-Barr virus in its pathogene-sis. Leukemia Res. 2005; 29: 389-95.
72. Harousseau JL, Flandrin G, Tricot G, Brouet JC,Seligmann M, Bernard J. Malignant lymphoma super-vening in chronic lymphocytic leukemia and related dis-orders. Richter’s syndrome: a study of 25 cases.Cancer. 1981; 48: 1302-8.
73. Robertson LE, Pugh W, O’Brien S, Kantarjian H, Hirsch-Ginsberg C, Cork A, et al. Richter’s syndrome: a reporton 39 patients. J Clin Oncol. 1993; 11: 1985-9.
74. Dabaja BS, O’Brien SM, Kantarjian HM, Cortes JE,Thomas DA, Albitar M, et al. Fractionated cyclophos-phamide, vincristine, liposomal daunorubicin (daunox-ome), and dexamethasone (hyperCVXD) regimen inRichter’s syndrome. Leuk Lymphoma. 2001; 42: 329-37.
75. Tsimberidou AM, O’Brien SM, Cortes JE, Faderl S,Andreeff M, Kantarjian HM, et al. Phase II study of flu-darabine, cytarabine (Ara C), cyclophosphamide, cis-platin and GM-CSF (FACPGM) in patients withRichter’s syndrome or refractory lymphoproliferative dis-orders. Leuk Lymphoma. 2002; 43: 767-72.
76. Tsimberidou AM, Kantarjian HM, Cortes J, Thomas DA,Faderl S, Garcia-Manero G, et al. Fractionatedcyclophosphamide, vincristine, liposomal daunorubicin,and dexamethasone plus rituximab and granulocyte-macrophage-colony stimulating factor (GM-CSF) alter-nating with methotrexate and cytarabine plus rituximaband GM-CSF in patients with Richter syndrome or flu-
darabine-refractory chronic lymphocytic leukemia.Cancer. 2003; 97: 1711-20.
77. Seymour JF, Grigg AP, Szer J, Fox RM. Cisplatin, flu-darabine, and cytarabine: a novel, pharmacologicallydesigned salvage therapy for patients with refractory,histologically aggressive or mantle cell non-Hodgkin’slymphoma. Cancer. 2002; 94: 585-93.
78. Coiffier B, Lepage E, Briere J, Herbrecht R, Tilly H,Bouabdallah R, et al. CHOP chemotherapy plus ritux-imab compared with CHOP alone in elderly patientswith diffuse large-B-cell lymphoma. N Engl J Med.2002; 346: 235-42.
79. Tsimberidou AM,Wierda WG, Plunkett W, Kurzrock R,O'Brien S, Wen S, et al. Phase I-II study of oxaliplatin,fludarabine, cytarabine, and rituximab combinationtherapy in patients with Richter’s syndrome or fludara-bine- refractory chronic lymphocytic leukemia. J ClinOncol. 2008; 26: 196-203.
80. Wang YF, Curtis JE, Lipton J, Minkin S, McCulloch EA.Cytosine arabinoside (ara-C) and cis-dichlorodi-ammineplatinum II (cisplatin) alone and in combination:effects on acute myeloblastic leukemia blast cells inculture and in vivo. Leukemia. 1991; 5: 522-7.
81. Yang LY, Li L, Keating MJ, Plunkett W. Arabinosyl-2-fluoroadenine augments cisplatin cytotoxicity andinhibits cisplatin-DNA cross-link repair. Mol Pharmacol.1995; 47: 1072-9.
82. Machover D, Delmas-Marsalet B, Misra SC, Gumus Y,Goldschmidt E, Schilf A, et al. Dexamethasone, high-dose cytarabine, and oxaliplatin (DHAOx) as salvagetreatment for patients with initially refractory orrelapsed non- Hodgkin’s lymphoma. Ann Oncol.2001; 12: 1439-43.
83. Chau I, Webb A, Cunningham D, Hill M, Rao S, AgeliS, et al. An oxaliplatin-based chemotherapy in patientswith relapsed or refractory intermediate and high-gradenon-Hodgkin’s lymphoma. Br J Haematol. 2001; 115:786-92.
84. Faderl S, Thomas DA, O’Brien S, Garcia-Manero G,Kantarjian HM, Giles FJ, et al. Experience with alem-tuzumab plus rituximab in patients with relapsed andrefractory lymphoid malignancies. Blood. 2003; 101:3413-5.
85. Tsimberidou AM, Murray JL, O’Brien S, Wierda WG,Keating MJ. Yttrium-90 ibritumomab tiuxetan radioim-munotherapy in Richter syndrome. Cancer. 2004; 100:2195-200.
86. Hainsworth JD, Litchy S, Barton JH, Houston GA,Hermann RC, Bradof JE, et al. Single-agent rituximabas first-line therapy and maintenance treatment forpatients with chronic lymphocytic leukemia or smalllymphocytic lymphoma: a phase II trial of the MinniePearl Cancer research Network. J Clin Oncol. 2003;21: 1746-51.
87. O’Brien S, Kantarjian HM, Thomas DA, Cortes J, GilesFJ, Wierda WG, et al. Alemtuzumab as treatment forresidual disease after chemotherapy in patients with

72 Seminari di Ematologia Oncologica
chronic lymphocytic leukemia. Cancer. 2003; 98:2657-63.
88. Thieblemont C, Bouafia F, Hornez E, Dumontet C,Tartas S, Antal D, et al. Maintenance therapy with amonthly injection of alemtuzumab prolongs responseduration in patients with refractory B-cell chronic lym-phocytic leukemia/small lymphocytic lymphoma (B-CLL/SLL). Leuk Lymphoma. 2004; 45: 711-4.
89. Wendtner CM, Ritgen M, Schweighofer CD, Fingerle-Rowson G, Campe H, Jäger G, et al. Consolidation withalemtuzumab in patients with chronic lymphocyticleukemia (CLL) in first remission - experience on safe-ty and efficacy within a randomized multicenter phaseIII trial of the German CLL Study group (GCLLSG).Leukemia. 2004; 18: 1093-101.
90. Montillo M, Tedeschi A, Miqueleiz S, Veronese S, CairoliR, Intropido L, et al. Alemtuzumab as consolidation aftera response to fludarabine is effective in purging resid-ual disease in patients with chronic lymphocyticleukemia. J Clin Oncol. 2006; 24: 2337-42.
91. Del Poeta G, Del Principe MI, Buccisano F, Maurillo L,Capelli G, Luciano F, et al. Consolidation and mainte-nance immunotherapy with rituximab improve clinicaloutcome in patients with B-cell chronic lymphocyticleukemia. Cancer. 2008; 112: 119-28.
92. Hainsworth JD, Vazquez ER, Spigel DR, Raefsky E,Bearden JD, Saez RA, et al. Combination therapy withfludarabine and rituximab followed by alemtuzumab inthe first-line treatment of patients with chronic lympho-cytic leukemia or small lymphocytic lymphoma: a phase2 trial of the Minnie Pearl cancer Research Network.Cancer. 2008; 112: 1288-95.
93. Zinzani PL, Tani M, Fanti S, Stefoni V, Musuraca G,Castellucci P, et al. A phase II trial of CHOPchemotherapy followed by yttrium 90 ibritumomabtiuxetan (Zevalin) for previously untreated elderly dif-fuse large B-cell lymphoma patients. Ann Oncol. 2008;19: 769-73.
94. Rodriguez J, Keating MJ, O’Brien S, Champlin RE,Khouri IF. Allogeneic haematopoietic transplantationfor Richter’s syndrome. Br J Haematol. 2000; 110:897-9.
95. Chanan-Khan A, Miller KC, Mursial L, Lawrence D,Padmanabhan S, Takeshita K, et al. Clinical efficacyof lenalidomide in patients with relapsed or refractorychronic lymphocytic leukemia: results of a phase IIstudy. J Clin Oncol. 2006; 24: 5343-9.
96. Byrd JC, Lin TS, Dalton JT, Wu D, Phelps MA, FischerB, et al. Flavopiridol administered using a pharmaco-logically derived schedule is associated with markedclinical efficacy in refractory, genetically high-riskchronic lymphocytic leukemia. Blood. 2007; 109: 399-404.
97. Coiffier B, Lepretre S, Pedersen LM, Gadeberg O,Fredriksen H, van Oers MH, et al. Safety and efficacyof ofatumumab, a fully human monoclonal anti-CD20antibody, in patients with relapsed or refractory B-cellchronic lymphocytic leukemia: a phase 1–2 study.Blood. 2008; 111: 1094-100.
98. Ferrajoli A, Lee BN, Schlette EJ, O’Brien SM, Gao H,Wen S, et al. Lenalidomide induces complete and par-tial remissions in patients with relapsed and refracto-ry chronic lymphocytic leukemia. Blood. 2008; 111:5291-7.

73
n INTRODUZIONE
La leucemia linfatica cronica (LLC) è la forma di leu-cemia più comunemente osservata nei paesi occi-dentali e nella popolazione adulta (1). La sua pre-valenza è in aumento in quanto l’aspettativamediana di vita è generalmente in aumento. Si trat-ta di una forma di leucemia a decorso molto varia-bile. Accanto a casi che presentano una malattiastabile per molti anni, se non per tutta la vita, vene sono altri caratterizzati da malattia rapidamen-te progressiva che richiede precocemente un trat-tamento. La forte diversità del comportamento cli-nico della LLC va ricercata nelle sue caratteristichebiologiche che condizionano in modo importantesia il profilo di crescita cellulare che la risposta tera-peutica. Tra queste caratteristiche, vanno menzio-nate alcune alterazioni citogenetiche evidenziabilicon la metodica FISH, le mutazioni che interessa-no i geni che codificano per le catene pesanti del-le immunoglobuline (IgVH), l’espressione dell’anti-gene CD38 e di ZAP-70 (2-6). Insieme alla delezio-ne 17p, la mutazione del gene TP53 è l’alterazio-ne genetica che si associa alla prognosi più sfa-vorevole essendo correlata ad una resistenza allachemioterapia e ad una ridotta durata di soprav-vivenza (7-9).
Benché negli ultimi 20 anni vi sia stato un notevo-le ampliamento dell’armamentario terapeutico del-la LLC e sia stato ottenuto non solo un incremen-to delle risposte terapeutiche, ma anche una miglio-re qualità delle risposte, la LLC ancor oggi è con-siderata una patologia non eradicabile con la tera-pia convenzionale ed i pazienti presentano nel tem-po ricorrenti recidive di malattia. La storia natura-le della malattia è frequentemente caratterizzata dal-l’insorgenza di una condizione di refrattarietà allaterapia che identifica una situazione clinica progno-sticamente assai sfavorevole (10).Si pone quindi non raramente il problema dellagestione terapeutica di pazienti refrattari o anchedoppi refrattari ovvero, refrattari sia alla fludarabi-na che all’anticorpo monoclonale campath, epazienti refrattari alla fludarabina e con malattia lin-fonodale bulky (diametro delle linfoadenomegalie>5 cm) che li rende non eleggibili ad un trattamen-to con campath. Tam et al. (13) hanno valutato retro-spettivamente la prognosi di pazienti con questecaratteristiche seguiti presso il MD AndersonCancer Center di Houston (MDACC). Solo il 23%dei pazienti otteneva una risposta terapeutica alleterapie successive, le risposte erano parziali condurata mediana non superiore a 3 mesi e la pro-babilità di sopravvivenza mediana era di 9 mesi.L’instaurarsi di una refrattarietà alle terapie conven-zionali rappresenta una condizione di difficilegestione clinico-terapeutica in cui emerge lanecessità di poter disporre di nuovi farmaci noncross-resistenti indispensabili per cercare di prolun-gare la sopravvivenza del paziente con LLC. I nume-rosi studi che hanno attualmente come oggetto lavalutazione di nuovi anticorpi monoclonali e di nuo-ve molecole (Tabella 1) rappresentano la rispostaa questa pressante esigenza. In questa rassegna,
Indirizzo per la corrispondenza
Prof. Robin FoàDipartimento di Biotecnologie Cellulari ed EmatologaUniversità “Sapienza”Via Benevento, 6 - 00161 RomaE-mail: [email protected]
Terapie innovativeTerapie innovativeROBIN FOÀ, ILARIA DEL GIUDICE, FRANCESCA R. MAUROEmatologia, Dipartimento di Biotecnologie Cellulari ed Ematologia, Università “Sapienza” Roma
Robin Foà
Parole chiave: LLC, terapia, immunoterapia, agenti bio-logici, vaccini

74 Seminari di Ematologia Oncologica
verrà offerta una descrizione delle nuove moleco-le di cui viene attualmente valutata la potenzialitàterapeutica in questa forma di leucemia.
n NUOVI ANTICORPI MONOCLONALI
Lo sviluppo di nuovi anticorpi monoclonali è statodiretto alla realizzazione di anticorpi specifici pernuovi antigeni o per regioni diverse dello stesso anti-gene, o è stato anche indirizzato alla formulazionedi anticorpi la cui la struttura è stata modificata inmodo da migliorarne l’attività in termini di antibo-dy-dependent cell-mediated cytotoxicity (ADCC) edi capacità di indurre direttamente il processo diapoptosi. Molti dei nuovi anticorpi monoclonali sonogià stati sottoposti a sperimentazione clinica,mentre alcuni sono attualmente ancora in fase pre-clinica.
OfatumumabOfatumumab è un anticorpo monoclonale diseconda generazione e completamente umanizza-to diretto contro l’antigene CD20. Questo anticor-po riconosce un epitopo dell’antigene CD20 diver-so da quello riconosciuto dall’anticorpo monoclo-nale rituximab. Rispetto a quest’ultimo, si mostra
più fortemente attivo nell’indurre ADCC anche incellule con ridotta densità di espressione dell’an-tigene CD20 come i linfociti della LLC (11). Il pri-mo studio in cui è stata valutata l’attività terapeu-tica dell’anticorpo monoclonale ofatumumab è sta-to lo studio di fase I/II proposto da Coiffier et al.(12). In questo studio sono stati inclusi pazienti pre-cedentemente trattati che sono stati suddivisi in 3coorti. La dose iniziale di ofatumumab è stata nel-le 3 coorti rispettivamente di 100, 300 e 500 mg.Successivamente, nei pazienti delle 3 coorti sonostate somministrate 3 dosi settimanali di ofatumu-mab al dosaggio di 500, 1000 e 2000 mg, rispet-tivamente. È stata documentata una risposta di tipoparziale nel 44% dei pazienti trattati con una soprav-vivenza libera da progressione di malattia di 3 mesicirca.Wierda et al. (13) hanno condotto uno studio mul-ticentrico internazionale in cui è stata valutata l’at-tività terapeutica di ofatumumab in pazienti affettida LLC e refrattari al precedente trattamento. Sonostati trattati 138 pazienti, 59 pazienti refrattari siaalla fludarabina che all’anticorpo monoclonalealemtuzumab (FA-ref) e 79 pazienti refrattari alla tera-pia con fludarabina e non eleggibili ad un trattamen-to con alemtuzumab perchè caratterizzati dalla pre-senza di una malattia linfonodale bulky, ovvero dalinfoadenomegalie di diametro superiore a 5 cm (BF-ref). In questo studio, sono state somministrate 8dosi settimanali di ofatumumab. La prima sommi-nistrazione prevedeva una dose di 300 mg di ofa-tumumab, mentre le successive dosi sono state di2000 mg ciascuna. Terminata questa prima fase ditrattamento, i pazienti hanno ricevuto mensilmen-te 4 somministrazioni di ofatumumab, sempre alladose di 2000 mg. È stata ottenuta una risposta inuna percentuale relativamente elevata di pazienti,nel 55% dei pazienti definiti come FA-ref e nel 47%di quelli definiti come BF-ref. Si tratta di un risulta-to importante se si considera l’insuccesso terapeu-tico che solitamente contraddistingue questi sot-togruppi di pazienti. Le probabilità mediane disopravvivenza e di sopravvivenza libera da progres-sione di malattia sono risultate, rispettivamente, di14 e 6 mesi per pazienti del gruppo FA-ref e di 15mesi e 6 mesi per i pazienti del gruppo BF-ref. Latolleranza all’infusione dell’anticorpo monoclonaleè stata buona. Le reazioni infusionali hanno solita-mente interessato le prime somministrazioni di ofa-
TABELLA 1 - Nuove molecole ed anticorpi monoclonali nellaterapia della LLC.
Agente Meccanismo d’azione
Anticorpi monoclonaliOfatumumab Anti-CD20GA-101 Anti-CD20Veltuzumab Anti-CD20Lumiliximab Anti-CD23
Lenalidomide Immunomodulante
Flavopiridolo Inibitore delle kinasi ciclino-dipendenti
Inibitori di Bcl-2Oblimersen Antisenso di Bcl-2Obatoclax Inibitore di Bcl-2ABT-263 Inibitore di Bcl-2
Inibitori delle tirosin-chinasiDasatinib Inibitore delle Src e
ABL chinasiFostamatinib Inibitore di SYKVatalanib e pazopanib Inibitori del recettore
del VEGF

75Terapie innovative
tumumab e sono state ben controllate dalla tera-pia. L’incidenza di infezioni di grado >III è stata del29% e la tossicità ematologica in termini di granu-locitopenia di grado >III ha interessato il 42% deipazienti. I risultati di questo studio indicano che l’an-ticorpo monoclonale ofatumumab somministratocome singolo agente ha una chiara efficacia tera-peutica in pazienti con LLC refrattari alla terapia eda prognosi altamente sfavorevole.Sulla base di questi risultati, la FDA e l’EMA han-no recentemente approvato l’impiego dell’anticor-po monoclonale ofatumumab nei pazienti affetti daLLC e refrattari a terapie comprendenti sia la flu-darabina che l’anticorpo monoclonale alemtuzu-mab. Sono attualmente in corso 2 studi controlla-ti multicentrici che si propongono di valutare l’effi-cacia di questo anticorpo monoclonale quandosomministrato in combinazione con altri agenti diconosciuta attività nella LLC. Il primo studio è unostudio randomizzato in cui sono inclusi pazienti nonprecedentemente trattati. In questo studio vieneconfrontata l’attività terapeutica di 6 cicli mensili diun trattamento con clorambucile somministratocome singolo agente (C) con quella di 6 cicli men-sili dell’associazione clorambucile e ofatumumab(O-C). Il secondo studio è uno studio randomizza-to che confronta, in pazienti già precedentementetrattati, l’attività di 6 cicli mensili dell’associazionefludarabina, ciclofosfamide (FC) con quella di 6 ciclimensili dell’associazione fludarabina, ciclofosfami-de e ofatumumab (O-FC). Il GIMEMA propone inve-ce uno studio multicentrico in cui sarà valutata l’ef-ficacia terapeutica della associazione bendamusti-na e ofatumumab (Bend-Ofa) in pazienti con LLCin recidiva o refrattari dopo un primo trattamento.
GA-101Attualmente è in corso la valutazione dell’attività dinuovi anticorpi glicoingegnerizzati e tra questi meri-ta particolare interesse l’anticorpo monoclonale anti-CD20 RG7159 (GA-101).La glicoingegnerizzazione è una procedura di bio-tecnologia che si propone di stimolare la capacitàdi un anticorpo monoclonale di indurre una rispo-sta immunitaria contro le cellule tumorali. GA-101è un anticorpo umanizzato di tipo II, in cui il pro-cesso di glicoingegnerizzazione ha significativamen-te incrementato in vitro la capacità di legame alrecettore FC e quindi l’attività ADCC. Ne risulta per-
tanto un’attività ADCC maggiore rispetto all’anticor-po monoclonale rituximab (14-17). Questo tipo dianticorpo è inoltre in grado di promuovere diretta-mente il processo apoptotico (18). Morchhauser etal. (19) hanno valutato l’attività terapeutica di que-sto anticorpo monoclonale in 13 pazienti con LLCrefrattari o in recidiva dopo un numero mediano di3 precedenti trattamenti che, nella maggior partedei casi, avevano compreso l’anticorpo monoclo-nale rituximab. Un terzo dei pazienti presentavacaratteristiche citogenetiche prognosticamentesfavorevoli. La dose somministrata di GA-101 è sta-ta incrementata da 400 mg a 2000 mg. È stata otte-nuta una risposta nel 62% dei casi. L’effetto col-laterale più frequentemente osservato è stato la gra-nulocitopenia e le reazioni infusionali sono state perseverità ed incidenza non dissimili da quelle abi-tualmente osservate dopo somministrazione di ritu-ximab. Il gruppo tedesco per lo studio della LLC(GCLLSG) ha recentemente promosso uno studiomulticentrico randomizzato che si propone di con-frontare in pazienti con LLC, non precedentemen-te trattati e con comorbidità, l’efficacia terapeuticadi 3 diversi approcci terapeutici: clorambucile, clo-rambucile più rituximab e, infine, clorambucile piùGA-101.
VeltuzumabVeltuzumab è un anticorpo monoclonale anti-CD20di recente sviluppo la cui molecola è stata realiz-zata in modo da incrementarne l’affinità di legameal frammento FCGR3a in presenza di polimorfismia bassa affinità di legame. In uno studio attualmen-te in corso (20) questo anticorpo monoclonale dinuova generazione è stato somministrato per viasottocutanea in pazienti con LLC che hannomostrato una riduzione significativa della linfocito-si periferica.
LumiliximabLumiliximab è un anticorpo monoclonale chimeri-co diretto contro l’antigene CD23, una glicoprotei-na transmembrana espressa dalla maggioranza del-le cellule leucemiche di LLC. In un protocollo di dosefinding non è stata documentata una dose limitingtoxicityquando l’anticorpo monoclonale lumiliximabveniva somministrato fino a raggiungere la dose di500 mg/m2 tre volte alla settimana per 4 settima-ne (21). Inoltre, in studi preclinici, l’aggiunta di lumi-

76 Seminari di Ematologia Oncologica
liximab ha incrementato l’attività citolitica sia dellafludarabina che di rituximab. Queste osservazionihanno rappresentato il razionale per il suo impie-go clinico in combinazione con la schedula tera-peutica fludarabina, ciclofosfamide, rituximab(FCR-Lumiliximab).Byrd et al. (22) hanno riportato i risultati di uno stu-dio terapeutico in cui 31 pazienti con LLC prece-dentemente trattati hanno ricevuto un trattamentocon 6 cicli di FCR-Lumiliximab. Questo anticorpoè stato somministrato alla dose di 375 mg/m2 (3pazienti) e di 500 mg/m2 (8 pazienti). La percentua-le di risposte è stata del 65% con il 52% di rispo-ste complete ed il profilo di tossicità è risultato deltutto accettabile. La sopravvivenza libera da pro-gressione di malattia è stata di 29 mesi. Sulla basedi questi dati incoraggianti è stato promosso unostudio randomizzato multicentrico inteso a confron-tare l’attività terapeutica di FCR-lumiliximab conquella di FCR.
n NUOVE MOLECOLE
LenalidomideLa lenalidomide è una molecola con le proprietà fun-zionali di un agente immunomodulante. Questoanalogo, più attivo della talidomide, è oggi impie-gato nel trattamento del mieloma multiplo e dellesindromi mielodisplastiche con delezione del 5q-.Vi è evidenza di una sua attività terapeutica anchein malattie linfoproliferative quali i linfomi e la LLC,benché i suoi meccanismi d’azione, probabilmen-te molteplici, non siano stati ancora ben definiti.In un primo studio di fase II, Chanan Khan et al.(23) hanno impiegato in 45 pazienti con LLC pre-cedentemente trattati, e metà dei quali refrattari allafludarabina, lenalidomide alla dose di 25 mg/die per3 settimane consecutive al mese ottenendo unapercentuale di risposte del 47%, con il 9% di rispo-ste complete. In un secondo studio, Ferrajoli et al.(24) hanno trattato 44 pazienti con lenalidomidesomministrata giornalmente; la dose più frequen-temente tollerata è stata di 10 mg. In questo studio la percentuale di risposte è statadel 38%, con il 7% di risposte complete. In entram-bi gli studi, le tossicità più frequentemente osser-vate sono state la mielotossicità e la fatigue. Menofrequentemente, sono state osservate casi di sin-
drome da lisi tumorale e l’insorgenza di una tumorflare reaction. Quest’ultima è una condizionecaratterizzata da un aumento di volume dolorosodelle linfoghiandole con segni locali di flogosi. Unincremento più graduale della dose di lenalidomi-de può contribuire a ridurre l’incidenza di questeultime complicanze. Ferrajoli et al. (25) hanno riportato in uno studiosuccessivo che i pazienti con alterazioni citoge-netiche a significato prognostico sfavorevole,quali la delezione 11q- e la delezione 17p-, nonmostravano una risposta dissimile rispetto aquella ottenuta da pazienti che non mostravanoqueste caratteristiche genetiche.In uno studio più recente, Ferrajoli et al. (26) han-no trattato con lenalidomide alla dose massimagiornaliera di 10 mg pazienti affetti da LLC menogiovani, di età superiore o uguale a 65 anni otte-nendo una risposta nel 54% dei pazienti trattati.Sempre Ferrajoli et al. (27) hanno riportato i risul-tati preliminari di uno studio in cui alla lenalidomi-de è stato associato l’anticorpo monoclonale ritu-ximab. L’associazione ha dato risultati migliori rispetto aquelli precedentemente ottenuti con lenalidomidesomministrata come singolo agente, con una per-centuale di risposte del 64% ed anche la tossici-tà in termini di tumor flare reaction sembra esse-re stata più contenuta. La dose ottimale di lena-lidomide, la schedula di somministrazione ed il suoruolo come singolo agente in terapia di induzio-ne o di mantenimento o in combinazione con altrichemioterapici e/o anticorpi monoclonali neces-sita ancora di valutazione. Al momento attuale, visono in corso molti studi terapeutici per valutarequesti aspetti ed il ruolo terapeutico della lenali-domide nella LLC. Due studi sponsorizzati da Celgene si propongo-no di valutare il ruolo di lenalidomide somministra-ta come terapia di mantenimento e di confronta-re l’efficacia terapeutica della combinazione clo-rambucile-lenalidomide con quella del solo cloram-bucile. Il gruppo GIMEMA sta attualmente valu-tando in pazienti con LLC precedentemente trat-tati l’attività terapeutica della combinazione fluda-rabina, ciclofosfamide e lenalidomide, ed inpazienti anziani, non precedentemente trattati, l’at-tività terapeutica della associazione clorambucilee lenalidomide.

77Terapie innovative
Flavopiridolo
Il flavopiridolo è un agente che inibisce le chinasiciclino-dipendenti (CDK) arrestando il ciclo cellu-lare e che down-regola l’espressione di proteine adeffetto anti-apoptotico (Mcl-1, X-linked). Inoltre, ilflavopiridolo ha diverse altre attività con effetto pro-apoptotico, quali la riduzione della fosforilazione edattività trascrizionale della RNA polimerasi II e l’at-tivazione della caspasi 3 distalmente al gene TP53.Sulla base di studi di farmacocinetica è stato dimo-strato che per preservare l’attività di flavopiridoloin vivo è necessaria una somministrazione di que-sto agente in un bolo della durata di 30 minuti segui-ta da una successiva infusione della durata di 4 ore.I primi studi clinici (28, 29) hanno mostrato che lasomministrazione di questo agente si associava adelevata tossicità in termini di sindrome da lisi tumo-rale e da rilascio di citochine soprattutto nei pazien-ti con marcata linfocitosi periferica. Lin e collabo-ratori (30) hanno trattato con flavopiridolo 116pazienti con LLC, la maggioranza dei quali resisten-ti alla fludarabina. È stata documentata una rispo-sta nel 53% dei casi con una mediana di soprav-vivenza libera da progressione di malattia di 12 mesi.La risposta terapeutica è stata osservata anche inpazienti con caratteristiche prognostiche partico-larmente sfavorevoli quali la delezione 17p- e la pre-senza di adenomegalie importanti bulky. L’incidenzadi pazienti con mielotossicità e che hanno svilup-pato una sindrome da lisi tumorale è stata impor-tante, ma l’inserimento di steroidi ha reso più tol-lerabile il trattamento riducendo i casi con sindro-me da rilascio delle citochine.
Inibitori di Bcl-2 - Oblimersen. È una proteina antisenso, un oligo-nucleotide che riconosce il mRNA specifico per lasintesi della proteina Bcl-2. Sulla base di un effet-to sinergico con la fludarabina dimostrato in vitro,è stato condotto uno studio in cui 241 pazienti sonostati randomizzati a ricevere 6 cicli della schedulafludarabina e ciclofosfamide (FC) da sola o in com-binazione con oblimersen (31). I pazienti che han-no ricevuto oblimersen in associazione ad FC han-no ottenuto una percentuale significativamentesuperiore di risposte complete e di risposte parzia-li di tipo nodulare, 17%, rispetto al 7% nei pazien-ti trattati con sola FC.- Obatoclax. È una molecola inibente Bcl-2. In uno
studio di fase 1 è stata somministrata come sin-golo agente in 26 pazienti con LLC mostrando atti-vità terapeutica (32). In questa esperienza, gli effet-ti collaterali, in particolare quelli neurologici (sonno-lenza, euforia), fatigue, incremento delle transami-nasi non sono stati tuttavia trascurabili.- ABT-263. Questo agente mimetico di BH3 ha lacapacità di legarsi ed inibire molte proteine ad effet-to anti-apoptotico appartenenti alla famiglia di Bcl-2. In uno studio di fase 1 in cui ABT-263 è statoimpiegato come singolo agente in 55 pazienti condiagnosi di linfoma linfocitico o di LLC, questa mole-cola ha mostrato una chiara attività terapeutica (33). La trombocitopenia, espressione della inibizione diBcl-xL, è stato l’effetto collaterale più importante.Al momento attuale, sono in corso studi terapeu-tici intesi a valutare l’efficacia di schedule che asso-ciano ABT-263 all’anticorpo monoclonale rituximab.
n INIBITORI DELLE TIROSIN CHINASI
Imatinib e DasatinibLa LLC presenta un profilo caratteristico e moltoomogeneo di iperepressione di numerosi geni codi-ficanti per varie protein chinasi (PK), come dimo-strato in un recente studio di profilo di espressio-ne genica mediante microarray (34). In particolare,sono altamente iperespressi 16 geni, che codifica-no per tirosin chinasi (SYK, LYN, BLK, LCK, JAK1,CSK e FGR), serin-treonin chinasi (PIM2, PFTK1,TLK1, MAP4K1, PDPK1, PRKCB1 e STK10) e pro-teine con entrambe le attività (GRK6 e WEE1). Alcunidi essi sono membri di importanti famiglie genichecoinvolte nella trasmissione del segnale intracellu-lare, come le Src chinasi, MAPK e JAK chinasi. Ilprofilo genico delle PK non risulta correlato ai clas-sici fattori prognostici della LLC, suggerendo chel’iperespressione dei geni delle PK caratterizza lamalattia in quanto tale piuttosto che i suoi sottoti-pi. La sensibilità delle cellule di LLC all’imatinib, ini-bitore della tirosin chinasi ABL, è stata testata instudi in vitro (35, 36), che hanno dimostrato una ete-rogeneità nell’effetto apoptotico indotto dall’inibi-tore, correlato con il livello di espressione di ABL.Lin et al. (35) hanno infatti dimostrato che la pro-teina ABL, iperespressa nelle cellule di LLC rispet-to ai linfociti B normali, presenta variabili livelli diespressione che correlano con la massa di malat-

78 Seminari di Ematologia Oncologica
tia, lo stadio clinico e lo stato non-mutato dei genidelle immunoglobuline. Questi dati pre-clinici hanno portato ad ipotizzareche gli inibitori delle PK di seconda generazionepossano avere un ruolo nel trattamento dei pazien-ti con LLC. Il dasatinib è un doppio inibitore cheagisce sia sulle Src chinasi che su ABL, ed è impie-gato nel trattamento della leucemia mieloide cro-nica e delle leucemie linfoblastiche acute Ph+, dimo-strando grande efficacia anche nei casi con muta-zioni del gene ABL, ad eccezione della mutazioneT315I (37).Studi in vitromostrano che il dasatinib è funzional-mente attivo anche su cellule primarie di LLC, ridu-cendone la vitalità (34).Il dasatinib riduce l’espressione di Mcl-1 e Bcl-x(L)ed è capace di indurre apoptosi in cellule di LLCin vitro, specialmente quelle con stato non-muta-to delle immunoglobuline (38); la combinazione didasatinib e fludarabina in vitro su cellule primarieha mostrato un incremento dell’effetto apoptoticoindotto da ciascun farmaco (39).Mentre la fosforilazione delle chinasi Src risulta ini-bita dal dasatinib sia nei casi di LLC che mostra-no apoptosi in vitro che nei casi resistenti, al con-trario l’attività di SYK (spleen tyrosine kinase) e del-la fospolipasi-Cgamma2 correla con la rispostaapoptotica delle cellule al dasatinib. L’inibizione diSYK, forse il bersaglio terapeutico più importan-te, potrebbe quindi predire la risposta cellulare aldasatinib, rappresentando un potenziale marcato-re di sensibilità dei pazienti con LLC a tale tratta-mento (40).
Inibitori di SYKI segnali indotti dalla stimolazione del B-cell recep-tor (BCR) sono determinanti per la sopravvivenza/apoptosi della cellula di LLC. SYK è una tirosin chinasi fondamentale per la tra-duzione del segnale del BCR. Il gene codificanteper SYK è iperespresso nelle cellule di LLC ancherispetto ai linfociti B normali (34, 41), così come risul-ta incrementata l’attività della proteina e di altrimediatori del segnale del BCR nella LLC rispettoai linfociti B normali.Gli inibitori di SYK sono in grado di indurre apop-tosi in vitro in cellule primarie di LLC, effetto pre-sente soprattutto in LLC con geni delle immuno-globuline non-mutati e ZAP-70+. Tali inibitori usa-
ti in combinazione con la fludarabina aumentanol’effetto citotossico in vitro della fludarabina da sola(41).In particolare, l’inibitore di SYK R406 è in grado diabrogare l’effetto protettivo sulla sopravvivenza dicellule di LLC indotto dalla stimolazione con anti-IgM e da cellule nutrici (42), di inibire la capacità dimigrazione nonché la produzione di citochine CCL3e CCL4 da parte di cellule di LLC, interferendo quin-di con i segnali di homing e sopravvivenza che dalmicroambiente vengono trasmessi alle cellulemediante il BCR (43).Un protocollo di fase I/II (44) ha impiegato il pri-mo inibitore di SYK per via orale, il fostamatinib,in pazienti con linfomi non-Hodgkin a cellule B inrecidiva. Nella fase I, la dose-limiting toxicity è rap-presentata da neutropenia, diarrea e trombocito-penia. Il dosaggio di 200 mg x 2/die è stato scel-to per la fase II. Sono stati trattati 68 pazienti in 3coorti: linfoma diffuso a grandi cellule B, linfomafollicolare ed altri linfomi (comprendenti linfomamantellare, linfoma MALT, linfoma marginale, lin-foma linfoplasmocitico e linfoma linfocitico/LLC (11casi)). Le tossicità più frequenti includono diarrea,astenia, citopenie, ipertensione e nausea. Rispostecliniche sono state osservate nel 55% dei linfomilinfocitici/LLC, 22% dei linfomi a grandi cellule, 10%dei linfomi follicolari e 11% dei linfomi mantellari.La sopravvivenza libera da progressione è stata di4,2 mesi.
Inibitori del recettore di VEGFIl vascular endothelial growth factor (VEGF) è unimportante fattore del microambiente che sembraavere un’influenza sulla sopravvivenza delle cellu-le di LLC indipendentemente dai suoi effetti sullaangiogenesi.Il vatalanib ed il pazopanib, inibitori orali del recet-tore del VEGF che ha attività tirosin chinasica, sonostati recentemente testati in vitro su cellule prima-rie di LLC (45). Entrambi riducono la fosforilazionedel recettore del VEGF e inducono l’apoptosi di cel-lule di LLC, mentre i linfociti B normali subisconosoltanto un lieve effetto. Provocano una down rego-lazione delle proteine anti-apoptotiche XIAP e MCL1e mostrano effetti sinergici con altri agenti chemio-terapici convenzionali. L’attività anti-leucemica è sta-ta dimostrata anche in vivo su topi xenografted conlinea cellulare JVM-3.

79Terapie innovative
n VACCINI
La LLC presenta alcune peculiari caratteristiche chela rendono una malattia ottimale a cui applicareapprocci immunoterapici. In primo luogo, la LLCpresenta spesso dopo la diagnosi una fase inizia-le ad andamento indolente che rappresenta unafinestra temporale ideale per far maturare una rispo-sta immune contro le cellule tumorali. In secondoluogo, le cellule tumorali sono facilmente accessi-bili dal sangue venoso periferico (svp). In terzo luo-go, le cellule di LLC esprimono antigeni HLA di clas-se I e II e hanno la potenzialità di funzionare comecellule presentanti l’antigene (APC).Inoltre, numerose evidenze dimostrano come la LLCsia responsiva all’azione del sistema immunitario,come dimostrato dall’effetto di graft-versus-leuke-mia dopo il trapianto di cellule staminali allogeni-che (46-49) o l’infusione di linfociti del donatore (50,51), come pure dall’evenienza di remissioni spon-tanee di malattia (52). Inoltre, la presenza di linfo-citi T autologhi ed allogenici con attività anti-LLCè stata dimostrata da numerosi lavori (49, 53).Tuttavia l’approccio immunoterapico nella LLC pre-senta alcune problematiche. Infatti le cellule di LLCsono di per sé incapaci di indurre una rispostaimmunitaria efficace per la mancanza o i bassi livel-li di molecole di adesione e costimolatorie (es. CD80e CD86) e perché sono direttamente responsabilidi meccanismi immunosoppressivi di varia natura(vedi paragrafo successivo), che determinano lo sta-to di immunodeficienza umorale e cellulare dellamalattia. Inoltre, le chemioterapie e gli anticorpimonoclonali impiegati per trattare la LLC hanno unaulteriore azione immunosoppressiva. L’importanza di sviluppare approcci immunotera-pici nella LLC deriva dal fatto che, sebbene le rispo-ste cliniche e la sopravvivenza dei pazienti sianosignificativamente migliorate con i regimi immuno-chemioterapici attualmente utilizzati, la malattiarimane non curabile. L’unico approccio curativo èil trapianto di cellule staminali allogeniche, che peròè applicabile soltanto ad una minoranza deipazienti.Scopo della immunoterapia della LLC è quello diindurre una risposta immune cellulare ed umora-le contro le cellule leucemiche in grado di conte-nerle/eliminarle e prevenire indefinitamente la reci-diva. La strategia di controllo immunoterapico auto-
logo della LLC troverebbe il suo spazio ideale sianelle fasi iniziali della malattia che nelle fasi di malat-tia minima residua dopo la terapia. I presuppostidella immunoterapia autologa risiedono nellaespressione e presentazione di antigeni (Ag)tumorali in un sistema immune dell’ospite capa-ce di riconoscerli e nella generazione di una effet-tiva risposta immune capace di eliminare le cellu-le tumorali.
Il sistema immunitario Le cellule LLC hanno la potenzialità di funzionarecome APC in quanto esprimono Ag tumorali chepossono essere presentati nel contesto dellemolecole di HLA di classe I e II. Tuttavia, nei pazien-ti affetti da LLC non vi è alcuna immunità autolo-ga efficace contro la malattia (54). Ciò è legato a tre fattori: a) la limitata capacità della cellula di LLC di agirecome APC;
b) difetti indotti dalle cellule di LLC sulla normaleattività effettrice dei linfociti T e delle altre cel-lule accessorie;
c) incremento nel numero dei linfociti T regolatori(Tregs).
I linfociti di LLC, nonostante esprimano le moleco-le del sistema HLA di classe I e II ed alcune mole-cole di adesione (CD54 o ICAM-1, CD27, CD40),mancano completamente o esprimono bassi livel-li di molecole costimolatorie (es. CD80 e CD86)essenziali per l’induzione di una risposta immuni-taria efficace. Inoltre, le cellule di LLC secernono citochine ad atti-vità immunosoppressiva come l’interleuchina (IL)-4, IL-6 e IL-10 ed il TGF-β, con la conseguente sop-pressione della attivazione, espansione e funzioneeffettrice dei linfociti T. Recentemente è stato dimostrato che la down rego-lazione del gene della β2-microglobulina (β2M), lacatena β del complesso del sistema HLA di clas-se I, nelle cellule di LLC in confronto con linfocitiB normali e di LLC in remissione spontanea, sup-porta l’ipotesi che tale alterazione possa essereassociata con la progressione della malattia, in lineacon i meccanismi di tumor escape esercitati daitumori solidi nei confronti dei linfociti T citotossici(52).Sono state descritte numerose anomalie nell’am-bito delle cellule accessorie non-leucemiche dei

80 Seminari di Ematologia Oncologica
pazienti con LLC, che hanno suggerito uno statodi cronica incompleta attivazione in vivo, con con-seguente anergia dei linfociti T (55-58). Sono statiriportati anche difetti funzionali nella popolazioneNK (59,60), come pure difetti funzionali ed aberra-zioni fenotipiche a carico delle cellule dendritiche(DC) (61,62). Più recentemente, è stato dimostrato che le cellu-le di LLC sono direttamente responsabili dell’indu-zione di una azione immunosoppressiva sui linfo-citi T (63-65).In primo luogo, i difetti dei linfociti T sono stati carat-terizzati accuratamente analizzando il profilo diespressione genica di linfociti CD4+ e CD8+ puri-ficati dal svp di pazienti con LLC a confronto conlinfociti T di donatori sani della stessa età. Questaanalisi ha dimostrato che vi sono geni differenzial-mente espressi, principalmente coinvolti nella dif-ferenziazione cellulare e nella formazione del cito-scheletro nei linfociti T CD4+, e nella formazionedel citoscheletro, traffico delle vescicole e vie di cito-tossicità nei linfociti T CD8+ (63). L’actina del cito-scheletro appare essenziale per controllare l’attiva-zione immune e l’attività effettrice dei linfociti T.Inoltre, le cellule di LLC possono attivamente sop-primere la funzione del citoscheletro nei linfociti T,inducendo una difettosa formazione della sinapsiimmunologica di F-actina e compromettendo il rico-noscimento delle cellule tumorali autologhe (64,65). Tale difetto è inducibile in linfociti T sia autologhiche allogenici di individui sani, mediante contattodiretto degli stessi con le cellule di LLC. Co-col-ture di cellule di LLC con linfociti T autologhi conlenalidomide rendono reversibile il difetto dellasinapsi immunologica presente nella malattia, ripa-rando il reclutamento delle proteine del citosche-letro alla sinapsi immunologica dei linfociti T edincrementando la citotossicità dei linfociti T CD8+contro le cellule di LLC autologhe (64,65).Recentemente, è stata descritta una aumentata fre-quenza e funzione soppressiva dei linfociti Tregsnei pazienti con LLC, specialmente nei pazienti nontrattati o con malattia progressiva (66). La lenali-domide e la pomalidomide hanno dimostrato unaattività inibitoria in vitro sulla proliferazione e fun-zione dei linfociti Tregs (67). Questi effetti potrebbero rappresentare una com-ponente cruciale delle proprietà adiuvanti della lena-lidomide e della pomalidomide, potenzialmente
applicabile anche nel contesto dell’approccio vac-cinale alla LLC.
Antigeni tumorali L’antigene tumorale ideale (tumor associated Ag,TAA) per la generazione di un vaccino dovrebbeavere 3 caratteristiche fondamentali: essere espres-so unicamente sulle cellule tumorali e non su tes-suti normali allo scopo di ridurre il rischio di rea-zioni autoimmuni; essere espresso su tutte le cel-lule tumorali; essere essenziale per la sopravviven-za della cellula tumorale per prevenire l’emergen-za di cloni Ag-negativi che potrebbero sfuggire alcontrollo immune. Tali TAA, degradati, processatie presentati come peptidi nel contesto di specifi-che molecole HLA, sono potenzialmente in gradodi generare una risposta citotossica da parte di lin-fociti T.Nella LLC non esiste un singolo TAA che rispon-da a tutti questi requisiti e che possa essere appli-cato a tutti i pazienti per una strategia vaccinale.Tra gli Ag LLC-specifici meglio caratterizzati, van-no menzionati:- l’idiotipo delle immunoglobuline, che è paziente-specifico ma poco immunogenico (68,69), su cuisono stati eseguiti tentativi di manipolazione perincrementarne la immunogenicità (70);
- proteine espresse in maniera aberrante dalle cel-lule di LLC ed identificate mediante tecniche direcombinant expression cloning (SEREX) (71);
- CD23 (72), fibromodulina (73), formin-related pro-tein in leukocytes 1 (FMNL1) (74,75), murine dou-ble minute 2 oncoprotein (MDM2) (76), oncofe-tal antigen immature laminin receptor protein(OFA-iLRP) (77,78), orphan receptor type 1 tyro-sine kinase ROR1 (79,80), receptor for hyaluron-ic acid-mediated motility (RHAMM/CD168) (81), survivina (82,83), hTERT (84).
Tutti i TAA sopramenzionati possono generare pep-tidi in grado di indurre risposte citotossiche media-te da linfociti T.In un recente trial clinico di fase I, 6 pazienti conLLC sono stati vaccinati per 4 volte ad intervallibisettimanali con un peptide derivante dalRHAMM/CD168 (HLA-A2 ristretto) con sommini-strazione di GM-CSF come adiuvante. Tale approc-cio non ha presentato tossicità e ha indotto in 4pazienti una risposta T CD8+ specifica contro l’Agtumorale ed una certa riduzione nella linfocitosi (85).

81Terapie innovative
Vaccini cellulari e protocolli cliniciPoichè non è facile trovare un singolo TAA idealeper una strategia vaccinale applicabile a tutti ipazienti con LLC, sono nati approcci alternativi cheevocano una risposta immune anti-tumorale impie-gando cellule tumorali autologhe o lisati cellulari intoto, con i quali tutti gli antigeni tumorali rappresen-tano multipli bersagli potenzialmente in grado digenerare una risposta immune. Infatti, la maggiorparte dei protocolli clinici di vaccinazione condot-ti nella LLC hanno impiegato vaccini cellulari.In considerazione del fatto che le cellule di LLC sonoAPC deficitarie, sono stati studiati vari approcci perrenderle più immunogeniche. Per ciascun approc-cio vengono menzionati i corrispondenti protocol-li clinici.
• Sottoporre le cellule di LLC ex vivo a stress ossi-dativi allo scopo di indurre il rilascio di heat-shockproteins e radicali liberi che incrementino la immu-nogenicità delle cellule di LLC in vivo. Pazienti infase indolente di malattia sono stati vaccinati pervia intramuscolare con cellule di LLC autologhe trat-tate con agenti ossidanti in un protocollo di fase I.Non sono state osservate tossicità significative.Cinque dei 18 pazienti hanno presentato risposteparziali che erano associate con un’aumentata atti-vità T anti-tumorale, ma la durata di tale rispostaè stata breve (1-4 mesi) (86).• Impiegare APC più potenti, come le DC, che pos-sono essere manipolate ex vivo, cioè caricate conlisati cellulari tumorali, RNA tumorale, corpi apop-totici o direttamente fuse con cellule di LLC (ibridi)
TABELLA 2 - Protocolli clinici vaccinali nella LLC.
Vaccino Trial Fase di N° di Sommini- Risposta Risposta Referenzamalattia pazienti strazioni immune clinica*
Peptide dal TAA Fase I 6 4 ogni 2 sett. Aumento linfociti 4/6 riduz. 85RHAMM/CD168 + GM-CSF T CD8+ Ag-specifici linfocitosi(HLA-A2 ristretto) CD107a+IFNγ+
Granzyme B+
LLC autologhe Fase I/II Indolente 18 12 in 6 sett. 9/18 risposta immune 5/18 PR 86trattate con 12 in 16 giorni T a cellule LLC 6/18 SDagenti ossidanti 4 in 6 sett. autologhe 7/18 PD
DC allogeniche Fase I Indolente 9 5 ogni 1/9 risposta linfociti T 8/9 SD 88caricate con lisati 2-3 sett. specifici per RHAMM 1/9 PDtumorali o corpiapoptotici
DC autologhe Fase I/II Indolente 12 8 4/12 risposta linfociti 5/12 riduz. 89caricate con T specifici per RHAMM linfocitilisati tumorali o fibromodulina. 3/12 SD
Nei rispondenti: 4/12 PDincremento IL-12 e riduzione Tregs
LLC autologhe Fase I Progressiva 11 1 Incremento IL-12 e 9/11 riduz. 92trasdotte con IFN gamma. linfociti eAd-CD154 Aumento linfociti T linfonodi
specifici anti-LLC 2/11 PD
Linfociti T Fase I/II Progressiva 17 1 Incremento linfociti Miglioramento 97 Xcellerate crasi ematica
11/14 riduzione dei linfonodi10/12 riduzione della splenome-galia. No rispostanella linfocitosi
*PR risposta parziale; SD malattia stabile; PD malattia progressiva

82 Seminari di Ematologia Oncologica
allo scopo di presentare l’Ag ai linfociti T e deter-minarne l’attivazione e la proliferazione. Studi pre-clinici hanno paragonato le 4 strategie e dimostra-to come i corpi apoptotici di cellule di LLC sianosuperiori agli altri per caricare le DC autologhe (87).Protocolli di fase I/II hanno esplorato l’impiego diDC per vaccinare pazienti con LLC. In un primo stu-dio, sono state impiegate DC allogeniche caricatecon lisati tumorali o corpi apoptotici (88) di celluledi LLC e somministrate per via sottocutanea a 9pazienti con malattia in stadio iniziale per 5 volte.Non sono state rilevate tossicità significative néfenomeni autoimmuni. Un sottogruppo di pazien-ti ha presentato una riduzione nel numero di cel-lule leucemiche durante la vaccinazione ed in uncaso un incremento nella conta dei linfociti T reat-tivi contro l’Ag leucemico RHAMM. Nel secondostudio, sono state impiegate DC autologhe carica-te con lisati tumorali (89) e somministrate per viaintradermica a 12 pazienti con LLC in stadio inizia-le per 8 volte. In alcuni pazienti è stato rilevato unaumento dei linfociti T che reagivano controRHAMM e fibromodulina come pure effetti cliniciminori, ma non è stata osservata alcuna rispostaclinica significativa (88, 89).È in corso uno studio di fase I/II sulla fattibilità, effi-cacia e immunogenicità di un vaccino con DC cari-cate con corpi apoptotici (Apo-DC) e generate daprecursori monocitari arricchiti da svp. Le Apo-DCsono somministrate per almeno 5 volte a 3 coortidi pazienti con LLC: la prima riceve Apo-DC sol-tanto; la seconda Apo-DC+ basse dosi ripetute diGM-CSF; la terza basse dosi di ciclofosfamideseguite da Apo-DC + GM-CSF (90).• Trasferire nelle cellule di LLC geni con proprietàimmunostimolatoria mediante vettori virali (es.adenovirus), come ad esempio geni codificanti permolecole accessorie di superficie come CD80 eCD154 (CD40L) o per citochine stimolatorie comeIL-2 e IL-12. Ad esempio, cellule di LLC infettatecon un vettore adenovirale codificante per ilCD40L ricombinante (Ad-CD154), sono risultatecapaci di stimolare ed espandere linfociti T citotos-sici specifici per le cellule leucemiche autologhe invitro (91). Il vettore virale ideale deve possedere unaelevata efficienza di trasferimento genico nella mag-gior parte delle cellule leucemiche, con alti livelli diespressione dei transgeni selezionati; non devereplicarsi e causare infezione attiva; non dovrebbe
esprimere Ag virali immunodominanti che possa-no competere con la risposta anti-leucemica.Numerosi studi sono volti ad ottimizzare il trasfe-rimento genico in cellule di LLC mediante vari tipidi virus o mediante elettroporazione delle cellule inpresenza di plasmidi di DNA umano contenenti igeni di interesse.Un protocollo di fase I ha esplorato la tollerabilità,tossicità ed attività di una singola dose di celluledi LLC autologhe trasdotte con Ad-CD154. Tale trat-tamento è stato associato alla comparsa di sinto-mi simil-influenzali, un transitorio incremento deivalori delle transaminasi e transitoria trombocito-penia, senza mai evidenziare una dose-limiting toxi-city o citopenie autoimmuni. Il trattamento ha indot-to elevati livelli plasmatici di IL-12 e IFN-γ, associa-ti a riduzione, seppur transitoria, del numero dellecellule di LLC entro le 48 ore dalla somministrazio-ne. Entro 1-2 settimane si assisteva ad una ridu-zione della dimensione dei linfonodi, della duratadi alcune settimane. Entro un mese dalla sommi-nistrazione si assisteva ad un notevole incremen-to nel numero assoluto dei linfociti T CD4+ e CD8+,nonchè dei linfociti T anti-LLC specifici (92). La rapi-da clearance delle cellule di LLC, non giustificatadai tempi di induzione della risposta immunitariacitotossica, è probabilmente provocata dalla indu-zione dei recettori apoptotici (CD95 e DR5) sulle cel-lule leucemiche e da meccanismi dell’immunitàinnnata indotti dalla metodica transgenica (92, 93).In un altro protocollo clinico di fase I, è stata valu-tata la somministrazione sottocutanea di cellule diLLC autologhe modificate per esprimere CD154 edIL-2. Sette su 9 pazienti hanno presentato unaumento transitorio della reattività T contro celluleautologhe di LLC, ma non è stato osservato alcuneffetto clinico (94).Un altro approccio è rappresentato dal vaccino TRI-COM, basato sul trasferimento genico di una tria-de di molecole di adesione e costimolatorie (CD80,ICAM-1 e LFA-3) in cellule di LLC mediante un vet-tore virale, allo scopo di aumentarne l’espressionesulla superficie cellulare ed incrementare l’attivitàdi APC. Le cellule di LLC modificate sono in gra-do di indurre una efficace attività citotossica in vitroda parte di linfociti T contro cellule di LLC autolo-ghe e non modificate (95).• Linfociti T raccolti mediante aferesi da pazienti conLLC possono essere attivati ed espansi dalle 100

83Terapie innovative
alle 1000 volte ex vivomediante coltura con bigliemagnetiche rivestite di anticorpi contro CD3 e CD28in presenza di IL-2 (Xcellerate T cells), potendo recu-perare la capacità di rispondere ad antigeni. I lin-fociti T Xcellerate autologhi sono stati valutati in unprotocollo di fase I/II in cui sono stati somministra-ti a dosi scalari fino a 100 x109 in una singola dose.Non vi è stata nessuna tossicità, con un incremen-to dei linfociti T nel svp dose-dipendente, una ridu-zione della splenomegalia e delle adenopatie. Nonè stata osservata una riduzione delle cellule di LLCnel svp, sebbene vi sia stato un miglioramento neivalori di emoglobina, piastrine e neutrofili (96, 97).• Una forma alternativa di presentazione dell’Agsi basa sull’impiego di cellule di linfoma fuse conun ibridoma (cellule TRIOMA) e modificate peresprimere una Ig diretta contro recettori di super-ficie di APC (98). Le cellule TRIOMA, che esprimo-no multipli Ag tumorali e rappresentano pertantovaccini polivalenti, costituiscono il bersaglio delleAPC che processeranno e presenteranno gli Agtumorali ai linfociti T. Ad esempio, DC pulsate concellule TRIOMA attivano efficacemente i linfociti Tcontro le cellule di LLC in vitro e gli Ag bersagliodi tali linfociti T sono stati identificati nel BCL-2,MDM2 ed ETV5 (99).In conclusione, i numerosi studi pre-clinici e clini-ci sulla strategia immunoterapica e vaccinale nel-la LLC condotti nell’ultima decade hanno dimostra-to che è possibile indurre una risposta immune anti-leucemica, che tuttavia non si è ancora tradotta inun impatto clinico sostanziale.Ulteriori sforzi volti ad eseguire un monitoraggioaccurato e standardizzato della risposta immunedopo la vaccinazione, a correggere i difetti immu-nitari che accompagnano la malattia e ad impie-gare nuovi adiuvanti o farmaci immunomodulantipotranno migliorare tali affascinanti strategie tera-peutiche.
n CONCLUSIONI
Sono attualmente oggetto di valutazione nell’am-bito di studi sperimentali nuovi agenti biologici e piùpotenti anticorpi monoclonali che hanno mostratoun’attività terapeutica in pazienti con LLC refratta-ri alle terapie convenzionali. Molti di essi hanno datorisultati promettenti ma sono ancora in fase di svi-
luppo molto precoce. Di alcuni rimane da chiarireil profilo tossicologico, frequentemente caratteriz-zato dalla comparsa di effetti collaterali non prece-dentemente noti. Se gli studi iniziali hanno dimo-strato per molte molecole una certa attività, altri stu-di sono necessari per definire la modalità più appro-priata di somministrazione indirizzata non solo amantenerne l’efficacia, ma anche a garantirne la tol-lerabilità. Inoltre, per molte di queste nuove mole-cole, una volta dimostrata l’attività come singoloagente, ne va valutato il potenziale terapeutico inschedule di associazione in cui sono combinate confarmaci di nota efficacia nella LLC.È indubbio che la grande attenzione diretta alla defi-nizione delle caratteristiche biologiche della LLC siè tradotta in modo tangibile anche nello sviluppodi nuovi agenti terapeutici per questa forma di leu-cemia.
n BIBLIOGRAFIA
1. Dighiero G, Hamblin TJ. Chronic lymphocytic leukaemia.Lancet. 2008; 371: 1017-9.
2. Hamblin TJ, Davis Z, Gardiner A, Oscier DG, StevensonFK. Unmutated Ig V(H) genes are associated with a moreaggressive form of chronic lymphocytic leukemia. Blood.1999; 94: 1848-54.
3. Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, AllenSL, et al. Ig V gene mutation status and CD38 expres-sion as novel prognostic indicators in chronic lympho-cytic leukemia. Blood. 1999; 94: 1840-7.
4. Döhner H, Stilgenbauer S, Benner A, Leupolt E, KröberA, Bullinger L, Döhner K, Bentz M, Lichter P. Genomicaberrations and survival in chronic lymphocyticleukemia. N Engl J Med. 2000; 343: 1910-6.
5. Crespo M, Bosch F, Villamor N, Bellosillo B, ColomerD, Rozman M, et al. ZAP-70 expression as a surrogatefor immunoglobulinvariable- region mutations in chron-ic lymphocytic leukemia. N Engl J Med. 2003; 348:1764-75.
6. Rassenti LZ, Huynh L, Toy TL, Chen L, Keating MJ,Gribben JG, et al. ZAP-70 compared with immunoglob-ulin heavy-chain gene mutation status as a predictorof disease progression in chronic lymphocytic leukemia.N Engl J Med. 2004; 351: 893-901.
7. Cordone I, Masi S, Mauro FR, Soddu S, Morsilli O,Valentini T, et al. p53 expression in B-cell chronic lym-phocytic leukemia: a marker of disease progression andpoor prognosis. Blood. 1998; 91: 4342-9.
8. Oscier DG, Gardiner AC, Mould SJ, Glide S, Davis ZA,Ibbotson RE, et al. Multivariate analysis of prognosticfactors in CLL: clinical stage, IGVH gene mutational sta-

84 Seminari di Ematologia Oncologica
tus, and loss or mutation of the p53 gene are independ-ent prognostic factors. Blood. 2002; 100: 1177-84.
9. Zenz T, Kröber A, Scherer K, Häbe S, Bühler A, BennerA, Denzel T, Winkler D, Edelmann J, Schwänen C,Döhner H, Stilgenbauer S. Mono-allelic TP53 inactiva-tion is associated with poor prognosis in CLL: Resultsfrom a detailed genetic characterization with long termfollow-up. Blood. 2008; 112: 3322-9.
10. Tam CS, O’Brien S, Lerner S, Khouri I, Ferrajoli A, FaderlS, et al. The natural history of fludarabine-refractorychronic lymphocytic leukaemia patients who fail alem-tuzumab or have bulky lymphadenopathy. LeukLymphoma. 2007; 48: 1931-9.
11. Teeling JL, French RR, Cragg MS, van den Brakel J,Pluyter M, Huang H, Chan C, Parren PW, Hack CE,Dechant M, Valerius T, van de Winkel JG, Glennie MJ.Characterization of new human CD20 monoclonal anti-bodies with potent cytolytic activity against non-Hodgkinlymphomas. Blood. 2004; 104: 1793-800.
12. Coiffier B, Lepretre S, Pedersen LM, Gadeberg O,Fredriksen H, van Oers MH, et al. Safety and efficacyof ofatumumab, a fully human monoclonal anti-CD20antibody, in patients with relapsed or refractory B-cellchronic lymphocytic leukemia: a phase 1-2 study. Blood.2008; 111: 1094-100.
13. Wierda WG, Kipps TJ, Mayer J, Stilgenbauer S, WilliamsCD, Hellmann A, et al. Ofatumumab as single-agentCD20 immunotherapy in fludarabine-refractory chron-ic lymphocytic leukemia. J Clin Oncol. 2010; 28: 1749-55.
14. Niederfellner GJ, Lammens A, Schwaiger A, GeorgesG, Wiechmann K, Franke A, et al. Crystal structureanalysis reveals that the novel type II anti-CD20 anti-body GA101 interacts with a similar epitope as ritux-imab and ocrelizumab but in a fundamentally differentway. [abstract] Blood. 2009; 114: N°3726.
15. Alduaij W, Potluri S, Ivanov A, Honeychurch J, BeersSA, Chan, et al. New-Generation Anti-CD20 MonoclonalAntibody (GA101) Evokes Homotypic Adhesion andActin-Dependent, Lysosome-Mediated Cell Death in B-Cell Lymphoma. [abstract] Blood. 2009; 114: N°725.
16. Zenz T, Volden M, Mast T, Sarno A, Winkler D, SchnaiterA, et al. In vitro activity of the type II anti-CD20 anti-body GA101 in refractory, genetic high-risk CLL.[abstract] Blood. 2009; 114: N°2379.
17. Patz M, Forcob N, Müller B, Klein C, Umana P, HallekM, et al. Depletion of chronic lymphocytic leukemia cellsfrom whole blood samples mediated by the anti-CD20antibodies Rituximab and GA101. [abstract] Blood.2009; 114: N°2365.
18. Ivanov A, Beers SA, Walshe CA, Honeychurch J, AlduaijW, Cox KL, et al. Monoclonal antibodies directed toCD20 and HLA-DR can elicit homotypic adhesion fol-lowed by lysosome-mediated cell death in human lym-phoma and leukemia cells. J Clin Invest. 2009; 119:2143-59.
19. Morschhauser F, Cartron G, Lamy T, Milpied NJ,
Thieblemont C, Tilly H, et al. Phase I study ofRO5072759 (GA101) in relapsed/refractory chronic lym-phocytic leukemia. [abstract] Blood. 2009; 114: N°364.
20. Allen SL, Rai KR, Elstrom R, Negrea OG, Farber CM,Abbasi R, et al. Subcutaneous injections of low dosesof veltuzumab (humanized anti-CD20 antibody): objec-tive responses in B-cell malignancies. [abstract] J ClinOncol. 2009; 27 (suppl): N°8530.
21. Byrd JC, O’Brien S, Flinn IW, Kipps TJ, Weiss M, RaiK, et al. Phase I study of lumiliximab with detailed phar-macokinetic and pharmacodynamic measurements inpatients with relapsed or refractory chronic lymphocyt-ic leukemia. Clin Cancer Res. 2007; 13: 4448-55.
22. Byrd JC, Kipps TJ, Flinn IW, Castro J, Lin TS, WierdaW, John C, et al. Phase 1/2 study of lumiliximab com-bined with fludarabine, cyclophosphamide,and ritux-imab in patients with relapsed or refractory chronic lym-phocytic leukaemia. Blood. 2010; 115: 489-95.
23. Chanan-Khan A, Miller KC, Musial L, Lawrence D,Padmanabhan S, Takeshita K, et al. Clinical efficacy oflenalidomide in patients with relapsed or refractorychronic lymphocytic leukemia: results of a phase II study.J Clin Oncol. 2006; 24: 5343-9.
24. Ferrajoli A, Lee BN, Schlette EJ, O'Brien SM, Gao H,Wen S, et al. Lenalidomide induces complete and par-tial remissions in patients with relapsed and refractorychronic lymphocytic leukemia. Blood. 2008; 111:5291-7.
25. Ferrajoli A, Keating MJ, Wierda WG, et al. Lenalidomideis active in patients with relapsed/refractory chronic lym-phocytic leukemia (CLL) carrying unfavorable chromo-somal abnormalities. [abstract] Blood. 2007; 110:N°754.
26. Ferrajoli A, O’Brien S, Wierda W, Faderl S, Estrov Z,Yerrow K, et al. Lenalidomide as initial treatment of eld-erly patients with chronic lymphocytic leukemia (CLL).[abstract] Blood. 2008; 112: N°45.
27. Ferrajoli A, Badoux XC, O’Brien S, Wierda WC, FaderlS, Estrov Z, Pasia M, et al. Combination therapy withlenalidomide and rituximab in patients with relapsedchronic lymphocytic leukemia (CLL). [abstract] Blood.2009; 114: N°206.
28. Byrd JC, Lin TS, Dalton JT, Wu D, Phelps MA, FischerB, et al. Flavopiridol administered using a pharmaco-logically derived schedule is associated with marked clin-ical efficacy in refractory, genetically high-risk chronic lym-phocytic leukemia. Blood. 2007; 109: 399-404.
29. Phelps MA, Lin TS, Johnson AJ, Hurh E, Rozewski DM,Farley KL, et al. Clinical response and pharmacokinet-ics from a phase I study of an active dosing scheduleof flavopiridol in relapsed chronic lymphocytic leukemia.Blood. 2009; 113: 2637-45.
30. Lin TS, Ruppert AS, Johnson AJ, Fischer B, HeeremaNA, Andritsos LA, et al. Phase II study of flavopiridolin relapsed chronic lymphocytic leukemia demonstrat-ing high response rates in genetically high-risk disease.J Clin Oncol. 2009; 27: 6012-8.

85Terapie innovative
31. O’Brien S, Moore JO, Boyd TE, Larratt LM, SkotnickiA, Koziner B, et al. Randomized phase III trial of flu-darabine plus cyclophosphamide with or withoutoblimersen sodium (Bcl-2 antisense) in patients withrelapsed or refractory chronic lymphocytic leukemia. JClin Oncol. 2007; 25: 1114-20.
32. O'Brien SM, Claxton DF, Crump M, Faderl S, Kipps T,Keating MJ, et al. Phase I study of obatoclax mesylate(GX15-070), a small molecule pan-Bcl-2 family antag-onist, in patients with advanced chronic lymphocyticleukemia. Blood. 2009; 113: 299-305.
33. Wilson W, O’Connor O, Roberts AW, Czuczman M,Brown J, Xiong H, et al. ABT-263 activity and safety inpatients with relapsed or refractory lymphoid malignan-cies in particular chronic lymphocytic leukemia (CLL)/small lymphocytic lymphoma (SLL) [abstract]. J ClinOncol. 2009; 27 (suppl): N°8574.
34. Tavolaro S, Chiaretti S, Messina M, Peragine N, DelGiudice I, Marinelli M, et al. Gene expression profile ofprotein kinases reveals a distinctive signature in chron-ic lymphocytic leukemia and in vitro experiments sup-port a role of second generation protein kinaseinhibitors. Leuk Res. 2010; 34: 733-41.
35. Lin K, Glenn MA, Harris RJ, Duckworth AD, Dennett S,Cawley JC, et al. c-Abl expression in chronic lympho-cytic leukemia cells: clinical and therapeutic implications.Cancer Res. 2006;66: 7801-9.
36. Aloyz R, Grzywacz K, Xu ZY, Loignon M, Alaoui-JamaliMA, Panasci L. Imatinib sensitizes CLL lymphocytes tochlorambucil. Leukemia. 2004; 18: 409-14.
37. Brave M, Goodman V, Kaminskas E, Farrell A, TimmerW, Pope S, et al. Sprycel for chronic myeloid leukemiaand Philadelphia chromosome-positive acute lym-phoblastic leukaemia resistant to or intolerant of ima-tinib mesylate. Clin Cancer Res. 2008; 14: 352-9.
38. Veldurthy A, Patz M, Hagist S, Pallasch CP, WendtnerCM, Hallek M, et al. The kinase inhibitor dasatinibinduces apoptosis in chronic lymphocytic leukaemia cellsin vitro with preference for a subgroup of patients withunmutated IgVH genes. Blood. 2008; 112: 1443-52.
39. Amrein L, Hernandez TA, Ferrario C, Johnston J, GibsonSB, Panasci L, et al. Dasatinib sensitizes primary chron-ic lymphocytic leukaemia lymphocytes to chlorambu-cil and fludarabine in vitro. Br J Haematol. 2008; 143:698-706.
40. Song Z, Lu P, Furman RR, Leonard JP, Martin P, TyrellL, et al. Activities of SYK and PLCgamma2 predictapoptotic response of CLL cells to SRC tyrosine kinaseinhibitor dasatinib. Clin Cancer Res. 2010; 16: 587-99.
41. Buchner M, Fuchs S, Prinz G, Pfeifer D, Bartholomé K,Burger M, et al. Spleen tyrosine kinase is overexpressedand represents a potential therapeutic target in chron-ic lymphocytic leukemia. Cancer Res. 2009; 69: 5424-32.
42. Quiroga MP, Balakrishnan K, Kurtova AV, Sivina M,Keating MJ, Wierda WG, et al. B-cell antigen receptorsignaling enhances chronic lymphocytic leukemia cell
migration and survival: specific targeting with a novelspleen tyrosine kinase inhibitor, R406. Blood. 2009; 114:1029-37.
43. Buchner M, Baer C, Prinz G, Dierks C, Burger M, ZenzT, et al. Spleen tyrosine kinase inhibition preventschemokine- and integrin-mediated stromal protectiveeffects in chronic lymphocytic leukemia. Blood. 2010;115: 4497-506.
44. Friedberg JW, Sharman J, Sweetenham J, Johnston PB,Vose JM, Lacasce A, et al. Inhibition of Syk with fos-tamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia.Blood. 2010; 115: 2578-85.
45. Paesler J, Gehrke I, Gandhirajan RK, Filipovich A,Hertweck M, Erdfelder F, et al. The vascular endothe-lial growth factor receptor tyrosine kinase inhibitors vata-lanib and pazopanib potently induce apoptosis in chron-ic lymphocytic leukaemia cells in vitro and in vivo. ClinCancer Res. 2010; 16: 3390-8.
46. Dreger P, Döhner H, Ritgen M, Böttcher S, Busch R,Dietrich S, et al. Allogeneic stem cell transplantation pro-vides durable disease control in poor-risk chronic lym-phocytic leukemia: long-term clinical and MRD resultsof the German CLL Study Group CLL3X trial. Blood.2010; 116: 2438-47.
47. Dreger P, Corradini P, Kimby E, Michallet M, Milligan D,Schetelig J, et al. Indications for allogeneic stem celltransplantation in chronic lymphocytic leukemia: theEBMT transplant consensus. Leukemia. 2007; 21: 12-7.
48. Kollgaard T, Petersen SL, Hadrup SR, Masmas TN,Seremet T, Andersen MH, et al. Evidence for involve-ment of clonally expanded CD8+ T cells in anticancerimmune responses in CLL patients following nonmye-loablative conditioning and hematopoietic cell transplan-tation. Leukemia. 2005; 19: 2273-80.
49. Nishida T, Hudecek M, Kostic A, Bleakley M, WarrenEH, Maloney D, et al. Development of tumor-reactiveT cells after nonmyeloablative allogeneic hematopoi-etic stem cell transplant for chronic lymphocyticleukemia. Clin Cancer Res. 2009; 15: 4759-68.
50. Marks DI, Lush R, Cavenagh J, Milligan DW, Schey S,Parker A, et al. The toxicity and efficacy of donor lym-phocyte infusions given after reduced-intensity condi-tioning allogeneic stem cell transplantation. Blood. 2002;100: 3108-14.
51. Marina O, Hainz U, Biernacki MA, Zhang W, Cai A,Duke-Cohan JS, et al. Serologic markers of effectivetumor immunity against chronic lymphocytic leukemiainclude nonmutated B-cell antigens. Cancer Res.2010; 70: 1344-55.
52. Del Giudice I, Chiaretti S, Tavolaro S, De Propris MS,Maggio R, Mancini F, et al. Spontaneous regression ofchronic lymphocytic leukemia: clinical and biologic fea-tures of 9 cases. Blood. 2009; 114: 638-46.
53. Rezvany MR, Jeddi-Tehrani M, Rabbani H, Lewin N,Avila-Cariño J, Osterborg A, et al. Autologous T lym-

86 Seminari di Ematologia Oncologica
phocytes may specifically recognize leukaemic B cellsin patients with chronic lymphocytic leukaemia. Br JHaematol. 2000; 111: 608-17.
54. Krackhardt AM, Harig S, Witzens M, Broderick R, BarrettP, Gribben JG. T-cell responses against chronic lym-phocytic leukemia cells: implications for immunother-apy. Blood. 2002; 100: 167-73.
55. Semenzato G, Pezzutto A, Foà R, Lauria F, RaimondiR. T lymphocytes in B-cell chronic lymphocyticleukemia: characterization by monoclonal antibodiesand correlation with Fc receptors. Clinical Immunologyand Immunopathology 1983; 26: 155-61.
56. Foà R. Pathogenesis of the immunodeficiency in B-cellchronic lymphocytic leukemia. In: B.D. Cheson editor.Chronic Lymphocytic Leukemia. New York: MarcelDekker, Inc. USA; 1993; 147-66.
57. Prieto A, Garcia-Suarez J, Reyes E, Lapena P,Hernandez M, Alvarez-Mon M. Diminished DNA syn-thesis in T cells from B chronic lymphocytic leukemiaafter phytohemagglutinin, anti- CD3, and phorbolmyristate acetate mitogenic signals. ExperimentalHematology. 1993; 21: 1563-69.
58. Rossi E, Matutes E, Morilla R, Owusu-Ankomah K,Heffernan AM, Catovsky D. Zeta chain and CD28 arepoorly expressed on T lymphocytes from chronic lym-phocytic leukemia. Leukemia. 1996; 10: 494-7.
59. Foà R, Lauria F, Lusso P, Giubellino MC, Fierro MT,Ferrando ML, et al. Discrepancy between phenotypicand functional features of natural killer T-lymphocytesin B-cell chronic lymphocytic leukaemia. Br J Haematol.1984; 58: 509-16.
60. Foà R, Fierro MT, Lusso P, Raspadori D, Ferrando ML,Matera L, et al. Reduced natural killer T-cells in B-cellchronic lymphocytic leukaemia identified by three mon-oclonal antibodies: Leu-11, A10, AB8.28. Br J Haematol.1986; 62: 151-4.
61. Orsini E, Guarini A, Chiaretti S, Mauro FR, Foà R. Thecirculating dendritic cell compartment in patients withchronic lymphocytic leukemia is severely defective andunable to stimulate an effective T-cell response. CancerRes. 2003; 63: 4497-506.
62. Orsini E, Pasquale A, Maggio R, Calabrese E, MauroFR, Giammartini E, et al. Phenotypic and functional char-acterization of monocyte-derived dendritic cells in chron-ic lymphocytic leukaemia patients: influence of neoplas-tic CD19 cells in vivo and in vitro. Br J Haematol. 2004;125: 720-28.
63. Gorgun G, Holderried TA, Zahrieh D, Neuberg D,Gribben JG. Chronic lymphocytic leukemia cells inducechanges in gene expression of CD4 and CD8 T-cells.J Clin Invest. 2005; 115: 1797-805.
64. Ramsay AG, Johnson AJ, Lee AM, Gorgün G, Le DieuR, Blum W, et al. Chronic lymphocytic leukemia T cellsshow impaired immunological synapse formation thatcan be reversed with an immunomodulating drug. J ClinInvest. 2008; 118: 2427-37.
65. Gorgun G, Ramsay AG, Holderried TA, Zahrieh D, Le
Dieu R, Liu F, et al. E(mu)-TCL1 mice represent a mod-el for immunotherapeutic reversal of chronic lympho-cytic leukemia-induced T-cell dysfunction. Proc NatlAcad Sci USA 2009; 106: 6250-55.
66. Motta M, Rassenti L, Shelvin BJ, Lerner S, Kipps TJ,Keating MJ, et al. Increased expression of CD152(CTLA-4) by normal T lymphocytes in untreatedpatients with B-cell chronic lymphocytic leukemia.Leukemia. 2005; 19: 1788-93.
67. Galustian C, Meyer B, Labarthe MC, Dredge K,Klaschka D, Henry J, et al. The anti-cancer agentslenalidomide and pomalidomide inhibit the proliferationand function of T regulatory cells. Cancer ImmunolImmunother. 2009; 58: 1033-45.
68. Trojan A, Schultze JL, Witzens M, Vonderheide RH,Ladetto M, Donovan JW, et al. Immunoglobulin frame-work-derived peptides function as cytotoxic T-cell epi-topes commonly expressed in B-cell malignancies. NatMed. 2000; 6: 667-72.
69. Harig S, Witzens M, Krackhardt AM, Trojan A, BarrettP, Broderick R, et al. Induction of cytotoxic T-cellresponses against immunoglobulin V region-derivedpeptides modified at human leukocyte antigen-A2 bind-ing residues. Blood. 2001; 98: 2999-3005.
70. Zirlik KM, Zahrieh D, Neuberg D, Gribben JG. CytotoxicT cells generated against heteroclitic peptides kill pri-mary tumor cells independent of the binding affinity ofthe native tumor antigen peptide. Blood. 2006; 108:3865-70.
71. Krackhardt AM, Witzens M, Harig S, Hodi FS, Zauls AJ,Chessia M, et al. Identification of tumor-associated anti-gens in chronic lymphocytic leukemia by SEREX. Blood.2002; 100: 2123-31.
72. Bund D, Mayr C, Kofler DM, Hallek M, Wendtner CM.CD23 is recognized as tumor-associated antigen (TAA)in B-CLL by CD8+ autologous T lymphocytes. ExpHematol. 2007; 35: 920-30.
73. Mayr C, Bund D, Schlee M, Moosmann A, Kofler DM,Hallek M, et al. Fibromodulin as a novel tumor-associ-ated antigen (TAA) in chronic lymphocytic leukemia(CLL), which allows expansion of specific CD8+ autol-ogous T lymphocytes. Blood. 2005; 105: 1566-73.
74. Favaro PM, de Souza Medina S, Traina F, Bassères DS,Costa FF, Saad ST. Human leukocyte formin: a novelprotein expressed in lymphoid malignancies and asso-ciated with Akt. Biochem Biophys Res Commun. 2003;311: 365-71.
75. Schuster IG, Busch DH, Eppinger E, Kremmer E,Milosevic S, Hennard C, et al. Allorestricted T cells withspecificity for the FMNL1-derived peptide PP2 havepotent antitumor activity against hematologic and oth-er malignancies. Blood. 2007; 110: 2931-9.
76. Mayr C, Bund D, Schlee M, Bamberger M, Kofler DM,Hallek M, et al. MDM2 is recognized as a tumor-asso-ciated antigen in chronic lymphocytic leukemia by CD8+autologous T lymphocytes. Exp Hematol. 2006; 34: 44-53.

87Terapie innovative
77. Siegel S, Wagner A, Kabelitz D, Marget M, Coggin JJr, Barsoum A, et al. Induction of cytotoxic T-cellresponses against the oncofetal antigen-immaturelaminin receptor for the treatment of hematologic malig-nancies. Blood. 2003; 102: 4416-23.
78. Siegel S, Wagner A, Friedrichs B, Wendeler A, WendelL, Kabelitz D, et al. Identification of HLA-A*0201-pre-sented T cell epitopes derived from the oncofetal anti-gen-immature laminin receptor protein in patients withhematological malignancies. J Immunol. 2006; 176:6935-44.
79. Baskar S, Kwong KY, Hofer T, Levy JM, Kennedy MG,Lee E, et al. Unique cell surface expression of recep-tor tyrosine kinase ROR1 in human B-cell chronic lym-phocytic leukemia. Clin Cancer Res. 2008; 14: 396-404.
80. Fukuda T, Chen L, Endo T, Tang L, Lu D, Castro JE, etal. Antisera induced by infusions of autologous Ad-CD154-leukemia B cells identify ROR1 as an oncofe-tal antigen and receptor for Wnt5a. Proc Natl Acad SciUSA. 2008; 105: 3047-52.
81. Giannopoulos K, Li L, Bojarska-Junak A, Rolinski J,Dmoszynska A, Hus I, et al. Expression of RHAMM/CD168 and other tumor-associated antigens in patientswith B-cell chronic lymphocytic leukemia. Int J Oncol.2006; 29: 95-103.
82. Schmidt SM, Schag K, Müller MR, Weck MM, AppelS, Kanz L, et al. Survivin is a shared tumor-associatedantigen expressed in a broad variety of malignanciesand recognized by specific cytotoxic T cells. Blood.2003; 102: 571-6.
83. Reker S, Meier A, Holten-Andersen L, Svane IM, BeckerJC, thor Straten P, et al. Identification of novel survivin-derived CTL epitopes. Cancer Biol Ther. 2004; 3: 173-9.
84. Kokhaei P, Palma M, Hansson L, Osterborg A, MellstedtH, Choudhury A. Telomerase (hTERT 611-626) servesas a tumor antigen in B-cell chronic lymphocyticleukemia and generates spontaneously antileukemic,cytotoxic T cells. Exp Hematol. 2007; 35: 297-304.
85. Giannopoulos K, Dmoszynska A, Kowal M, Rolinski J,Gostick E, Price DA, et al. Peptide vaccination elicitsleukemia-associated antigen-specific cytotoxic CD8+T-cell responses in patients with chronic lymphocyticleukemia. Leukemia. 2010; 24: 798-805.
86. Spaner DE, Hammond C, Mena J, Foden C, DeabreuA. A phase I/II trial of oxidized autologous tumor vac-cines during the “watch and wait” phase of chronic lym-phocytic leukemia. Cancer Immunol Immunother.2005; 54: 635-46.
87. Kokhaei P, Choudhury A, Mahdian R, Lundin J,Moshfegh A, Osterborg A, et al. Apoptotic tumor cellsare superior to tumor cell lysate, and tumor cell RNAin induction of autologous T cell response in B-CLL.Leukemia. 2004; 18: 1810-5.
88. Hus I, Roliński J, Tabarkiewicz J, Wojas K, Bojarska-Junak A, Greiner J, et al. Allogeneic dendritic cells
pulsed with tumor lysates or apoptotic bodies asimmunotherapy for patients with early-stage B-cellchronic lymphocytic leukemia. Leukemia. 2005; 19:1621-7.
89. Hus I, Schmitt M, Tabarkiewicz J, Radej S, Wojas K,Bojarska-Junak A, et al. Vaccination of B-CLL patientswith autologous dendritic cells can change the frequen-cy of leukemia antigen-specific CD8+ T cells as wellas CD4+CD25+FoxP3+ regulatory T cells toward anantileukemia response. Leukemia. 2008; 22: 1007-17.
90. Palma M, Adamson L, Hansson L, Kokhaei P, RezvanyR, Mellstedt H, et al. Development of a dendritic cell-based vaccine for chronic lymphocytic leukemia.Cancer Immunol Immunother. 2008; 57: 1705-10.
91. Kato K, Cantwell MJ, Sharma S, Kipps TJ. Gene trans-fer of CD40-ligand induces autologous immune recog-nition of chronic lymphocytic leukemia B cells. J ClinInvest. 1998; 101: 1133-41.
92. Wierda WG, Cantwell MJ, Woods SJ, Rassenti LZ,Prussak CE, Kipps TJ. CD40-ligand (CD154) gene ther-apy for chronic lymphocytic leukemia. Blood. 2000; 96:2917-24.
93. Wierda WG, Castro JE, Aguillon R, Sampath D,Jalayer A, McMannis J, et al. A phase I study of immunegene therapy for patients with CLL using a membrane-stable, humanized CD154. Leukemia. 2010; 24: 1893-900.
94. Biagi E, Rousseau R, Yvon E, Schwartz M, Dotti G,Foster A, et al. Responses to human CD40 ligand/human interleukin-2 autologous cell vaccine in patientswith B-cell chronic lymphocytic leukemia. Clin CancerRes. 2005; 11: 6916-23.
95. Palena C, Foon KA, Panicali D, Yafal AG, ChinsangaramJ, Hodge JW, et al. Potential approach to immunother-apy of chronic lymphocytic leukemia (CLL): enhancedimmunogenicity of CLL cells via infection with vectorsencoding for multiple costimulatory molecules. Blood.2005; 106: 3515-23.
96. Bonyhadi M, Frohlich M, Rasmussen A, Ferrand C,Grosmaire L, Robinet E, et al. In vitro engagement ofCD3 and CD28 corrects T cell defects in chronic lym-phocytic leukemia. J Immunol. 2005; 174: 2366-75.
97. Castro J, Wierda W, Kipps J, Keating M, Bole RN,Anderson B, et al. A Phase I/II Trial of Xcellerated TCellsTM in Patients with Chronic LymphocyticLeukemia. [abstract] Blood. 2004; 104: N°2508.
98. Wahl U, Nössner E, Kronenberger K, Gangnus R, PohlaH, Staege MS, et al. Vaccination against B-cell chron-ic lymphocytic leukemia with trioma cells: preclinicalevaluation. Clin Cancer Res. 2003; 9: 4240-6.
99. Kronenberger K, Nössner E, Frankenberger B, Wahl U,Dreyling M, Hallek M, et al. A polyvalent cellular vac-cine induces T-cell responses against specific self-anti-gens overexpressed in chronic lymphocytic B-cellleukemia. J Immunother. 2008; 31: 723-30.
