annals of clinical & laboratory science, vol. 44, no. 4 ... ·...
TRANSCRIPT

Available online at www.annclinlabsci.org
Pathogenesis of Post Primary Tuberculosis:Immunity and Hypersensitivity in the Development of CavitiesRobert L. Hunter1, Jeffrey K. Actor1, Shen-An Hwang1, Vadim Karev2, and Chinnaswamy Jagannath1
1Department of Pathology and Laboratory Medicine, University of Texas-Houston Medical School Houston, Texas,USA, and 2Scientific-Research Institute of Childhood Diseases of the Federal Medical and Biological Agency, SaintPetersburg, Russia
Abstract. M. Tuberculosis (MTB) is an obligate human parasite even though humans are more resistantthan any of the animals used for study. It is a human parasite because only humans develop post primarytuberculosis (TB) in their lungs that mediates transmission of infection to new hosts. The extreme paucityof human lung tissue with post primary TB has forced scientists to study animal models and human tissuesthat do not have the disease. Consequently, the unique features of post primary TB remain largely unknownand misconceptions are widely accepted. This manuscript presents a revised pathogenesis of post primaryTB based on studies of lung tissues of thousands of patients by multiple authors and related literature. Pri-mary TB stimulates systemic immunity that kills organisms and heals granulomas resulting in both protec-tion from disseminated TB and resistance to new infection. Post primary TB, in contrast, requires systemicimmunity that it subverts to produce local susceptibility in the apex of the lung. It begins in the part of lungwith the lowest ventilation, perfusion and movement and then proceeds to paralyze alveolar macrophages,block the exits and suppress inflammation to further isolate the area with post obstructive pneumonia.This provides a safe place for a small number of MTB to drive prolonged accumulation of host lipids andmycobacterial antigens in an otherwise immune person. After many months, the affected lung suddenlyundergoes caseation necrosis with vanishingly few MTB. The necrotic tissue fragments to produce a cavityor hardens to develop fibrocaseous disease. Evidence suggests that this is triggered by a hypersensitivity reac-tion against cord factor and then progresses as the Koch phenomenon against many antigens. MTB grow inperfusion only in dead tissue or on a cavity wall. We anticipate that a more accurate understanding of thepathogenesis of post primary TB will facilitate focusing modern technologies to produce rapid advances inunderstanding and combating TB.
Keywords: Pulmonary tuberculosis, Mycobacterium tuberculosis, Pathogenesis, Pathology, Cavity andLung
Introduction
Tuberculosis (TB) remains an urgent global healthproblem with approximately 9 million new casesand 1.4 million deaths each year [1]. A very largenumber of people are latently infected withMycobacterium tuberculosis (MTB) and at risk ofdeveloping disease in coming decades. The dualpandemic of TB and HIV/AIDS and the emer-gence of increasingly drug-resistant strains severelyaggravate the problem and hamper control efforts.
Immunity to TB is different from that of any otherinfection. The enigma of MTB is that it is an obli-gate human parasite even though humans are moreresistant than any of the animals used for study. It isa human parasite because only humans developpost primary disease in their lungs [2,3]. Post pri-mary TB produces cavities that support prolifera-tion of vast numbers of MTB in an otherwise im-mune host. Transmission is accomplished bycoughing MTB into the environment.
While much progress has been made in under-standing granulomas produced by animals, little orno progress has been made in understanding thepeculiarities of post primary TB that occur only inhumans [4]. Unanswered questions include: What
Annals of Clinical & Laboratory Science, vol. 44, no. 4, 2014
0091-7370/14/0400-365. © 2014 by the Association of Clinical Scientists, Inc.
Address correspondence to Robert L Hunter MD, PhD; Professor;Department of Pathology and Laboratory Medicine, MSB 2.034,University of Texas-Houston Medical School, 6431 Fannin Street,Houston, TX 77030; phone: 713 500 5301; fax: 713 500 0730;e-mail: [email protected]
365

is the nature of immunity that protects 95% ofpeople from disease? Why does recovery from postprimary TB make people more susceptible to newpost primary infections, but not to other types ofTB? [5,6]. Why does it occur only in the lung?What is the role of hypersensitivity?
Recent research has served to deepen the mystery.Unlike HIV that modifies its antigens to evade im-mune responses, MTB keeps its antigens remark-ably constant with even less variation than essentialstructural genes [7]. Why does TB need exactlythese epitopes? Failure to answer these questionshas frustrated leaders in the field who increasinglybelieve that TB can never be eliminated without avaccine that prevents transmission of infection [8].Lack of understanding of the immunology andpathogenesis of late stage TB is widely regarded asthe major impediment to vaccine development.One can neither rationally design nor test vaccinecandidates without knowledge of the type of im-munity needed for protection.
Since human lung tissue with developing cavitaryTB has seldom been available since the introduc-tion of antibiotics in the 1940’s, researchers havebeen forced to rely on animal models. Unfortunately,nearly all animals eventually die of primary infec-tion that most humans clear in weeks. Consequently,research on animals is producing a detailed under-standing of the early phases of TB. However, with-out human tissues for reference, modern sciencehas not been able to develop models or studies toapproach the late stages of infection that accountfor 80% of clinical disease and nearly 100% oftransmission to new hosts.
The pathology of pulmonary TB has a long history.Two distinct types of pathology, ‘productive’ and‘exudative’, were recognized by Laennec in 1821and confirmed by virtually all investigators throughthe mid 20th century [9-14]. In modern terms ‘pro-ductive’ refers to nodular tubercles or granulomasand ‘exudative’ refers to pneumonia. Multiple at-tempts to explain the pathogenesis of these process-es were published in the late 19th and early 20th
centuries. Some investigators proposed the mecha-nism that is widely accepted today, that cavities de-velop by erosion of granulomas into bronchi
followed by discharge of infected necrotic materialinto the airways. Recognizing that this is an apt de-scription of the disease produced by M. bovis,Medlar examined thousands of tubercles lookingfor evidence of a similar pathogenesis in humans.He found none [12]. Once formed, all humanMTB granulomas remained small.
In the pre antibiotic era, multiple investigators,each of whom had studied over 1000 cases of adultTB, reported that MTB spread through bronchi toproduce tuberculous pneumonia that underwentcaseation and then cavitation [9-14]. Rich reported“It has been found by all who have studied earlypulmonary lesions that they represent areas of case-ous pneumonia rather than nodular tubercles.”[10]. There was much speculation as to what seededthe pneumonia [15]. Rich believed that small erod-ed granulomas released MTB into bronchi.However, it was universally recognized that devel-oping post primary TB was a pneumonic, not agranulomatous process.
Interest in TB declined with the introduction of an-tibiotics in the 1950s. It resumed with resurgenceof the disease in the 1980’s with a new generationof investigators armed with new technologies andanimal models. The rabbit infected with M. boviswas the only common laboratory animal in whichchronic fibrosing TB with cavities could be readilyproduced [15]. However, the lesions and life cycleof M. bovis are different from those of MTB. M.bovis must be transmitted to new hosts during thelifetime of a cow, not the 10-30 years of MTB inpeople. M. bovis does not produce post primary TB[3,16]. It remains a particularly aggressive form ofprimary TB with widespread development of case-ating granulomas that erode into surfaces of thepharynx, bronchi, pleura, GI, urinary tracks andmammary glands to discharge organisms [17]. Thedifferences between lesions produced by MTB andM. bovis were forgotten [3]. As a consequence, theconcept that the caseating granuloma is the charac-teristic lesion of both primary and post primary TBbecame the bed rock paradigm that has guided TBresearch throughout the rise of cellular immunolo-gy, molecular microbiology and genetics. This pa-per challenges that paradigm.
Annals of Clinical & Laboratory Science, vol. 44, no. 4, 2014366
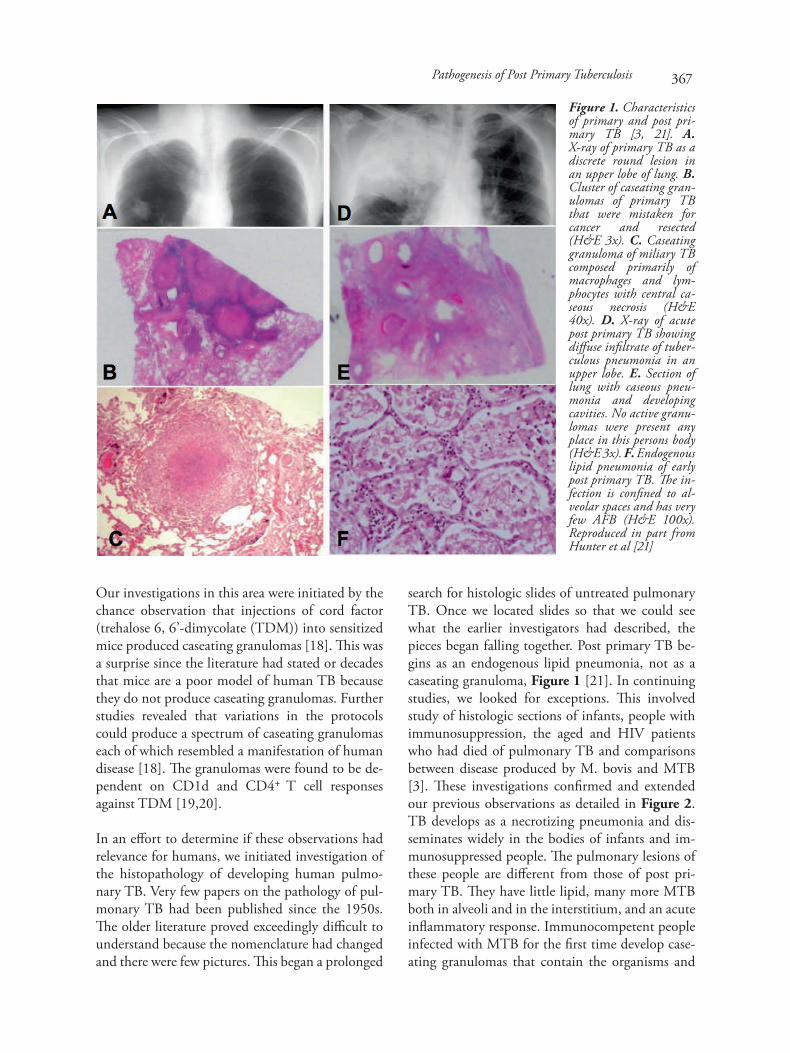
Our investigations in this area were initiated by thechance observation that injections of cord factor(trehalose 6, 6’-dimycolate (TDM)) into sensitizedmice produced caseating granulomas [18]. This wasa surprise since the literature had stated or decadesthat mice are a poor model of human TB becausethey do not produce caseating granulomas. Furtherstudies revealed that variations in the protocolscould produce a spectrum of caseating granulomaseach of which resembled a manifestation of humandisease [18]. The granulomas were found to be de-pendent on CD1d and CD4+ T cell responsesagainst TDM [19,20].
In an effort to determine if these observations hadrelevance for humans, we initiated investigation ofthe histopathology of developing human pulmo-nary TB. Very few papers on the pathology of pul-monary TB had been published since the 1950s.The older literature proved exceedingly difficult tounderstand because the nomenclature had changedand there were few pictures. This began a prolonged
search for histologic slides of untreated pulmonaryTB. Once we located slides so that we could seewhat the earlier investigators had described, thepieces began falling together. Post primary TB be-gins as an endogenous lipid pneumonia, not as acaseating granuloma, Figure 1 [21]. In continuingstudies, we looked for exceptions. This involvedstudy of histologic sections of infants, people withimmunosuppression, the aged and HIV patientswho had died of pulmonary TB and comparisonsbetween disease produced by M. bovis and MTB[3]. These investigations confirmed and extendedour previous observations as detailed in Figure 2.TB develops as a necrotizing pneumonia and dis-seminates widely in the bodies of infants and im-munosuppressed people. The pulmonary lesions ofthese people are different from those of post pri-mary TB. They have little lipid, many more MTBboth in alveoli and in the interstitium, and an acuteinflammatory response. Immunocompetent peopleinfected with MTB for the first time develop case-ating granulomas that contain the organisms and
Pathogenesis of Post Primary Tuberculosis
Figure 1. Characteristicsof primary and post pri-mary TB [3, 21]. A.X-ray of primary TB as adiscrete round lesion inan upper lobe of lung. B.Cluster of caseating gran-ulomas of primary TBthat were mistaken forcancer and resected(H&E 3x). C. Caseatinggranuloma of miliary TBcomposed primarily ofmacrophages and lym-phocytes with central ca-seous necrosis (H&E40x). D. X-ray of acutepost primary TB showingdiffuse infiltrate of tuber-culous pneumonia in anupper lobe. E. Section oflung with caseous pneu-monia and developingcavities. No active granu-lomas were present anyplace in this persons body(H&E 3x). F. Endogenouslipid pneumonia of earlypost primary TB. The in-fection is confined to al-veolar spaces and has veryfew AFB (H&E 100x).Reproduced in part fromHunter et al [21]
367

systemic immunity sufficient to control the infec-tion in weeks1. MTB, however, remain dormant intheir bodies. The location and properties of dor-mant MTB are controversial [22]. Much of the lit-erature assumes that they reside in granulomas.However, multiple studies found that granulomasare sterile after 5 years and organisms are morecommonly isolated from normal appearing lungand fat cells [10,11,23-25]. In any case, post pri-mary TB begins by reactivation of dormant MTBor new infection from the environment. It typically
begins after all granulomas have healed. It startswith infection of alveolar cells in persons whomaintain effective immunity to protect the entirerest of their bodies. The lesions develop as an en-dogenous lipid pneumonia [21,26]. Most such le-sions regress spontaneously so that progression toclinical disease is a rare event. If they do not regress,the lesions undergo necrosis to produce a caseouspneumonia that may fragment and be expelled toleave a cavity or remain as a mass that induces gran-ulomas and fibrocaseous disease.
Annals of Clinical & Laboratory Science, vol. 44, no. 4, 2014
1The terms granuloma and pneumonia require definition since the term granuloma is frequently used incorrectly to describe any lesion producedby TB.
Granuloma: A granuloma is a localized nodular inflammation found in tissues. In TB, a granuloma is a nodular delimited aggregation ofmononuclear inflammatory cells within tissue. It is a collection of modified macrophages resembling epithelial cells that are usually surroundedfirst by a rim of lymphocytes and later by fibroblasts.
Pneumonia is an infection of the alveoli (air sacs) of the lungs. It is typically diffuse within the air spaces of a lobule and does not penetrate intothe lung tissue. The alveoli may fill with fluid and/or inflammatory cells in addition to infectious organisms.
Figure 2. Stages of TB in humans. Disseminated: TB develops as a disseminated infection in the lungs and many otherorgans in people without competent immune systems such as infants and adults with AIDS (H&E 40x).Primary: PrimaryTB develops in response to the first infection of immunocompetent individuals. Many animals die of primary TB inmonths, but most humans heal it within weeks. It produces cell-mediated immunity that effectively mediates life long pro-tection from disease in all parts of the body except the vulnerable parts of the lung (H&E 40x). Post primary TB beginsas an endogenous lipid pneumonia in the apices of the lung. Approximately 95% of such lesions resolve spontaneously. Theother 5% go on to produce caseous pneumonia that can either soften and fragment to produce a cavity or harden to producecaseating granulomas and fibrocaseous disease. Greater detail of the pathology of post primary TB is presented in the refer-ences [3, 21]. Reproduced in part from Hunter et al [3, 21].
368
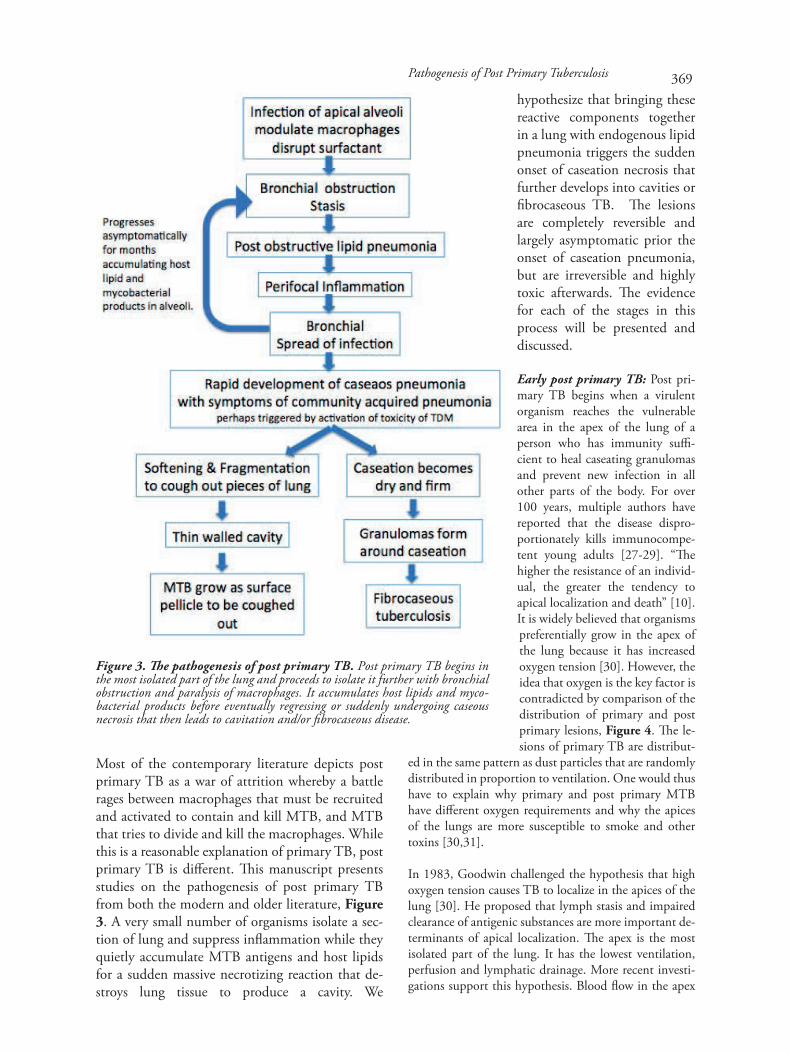
Most of the contemporary literature depicts postprimary TB as a war of attrition whereby a battlerages between macrophages that must be recruitedand activated to contain and kill MTB, and MTBthat tries to divide and kill the macrophages. Whilethis is a reasonable explanation of primary TB, postprimary TB is different. This manuscript presentsstudies on the pathogenesis of post primary TBfrom both the modern and older literature, Figure3. A very small number of organisms isolate a sec-tion of lung and suppress inflammation while theyquietly accumulate MTB antigens and host lipidsfor a sudden massive necrotizing reaction that de-stroys lung tissue to produce a cavity. We
hypothesize that bringing thesereactive components togetherin a lung with endogenous lipidpneumonia triggers the suddenonset of caseation necrosis thatfurther develops into cavities orfibrocaseous TB. The lesionsare completely reversible andlargely asymptomatic prior theonset of caseation pneumonia,but are irreversible and highlytoxic afterwards. The evidencefor each of the stages in thisprocess will be presented anddiscussed.
Early post primary TB: Post pri-mary TB begins when a virulentorganism reaches the vulnerablearea in the apex of the lung of aperson who has immunity suffi-cient to heal caseating granulomasand prevent new infection in allother parts of the body. For over100 years, multiple authors havereported that the disease dispro-portionately kills immunocompe-tent young adults [27-29]. “Thehigher the resistance of an individ-ual, the greater the tendency toapical localization and death” [10].It is widely believed that organismspreferentially grow in the apex ofthe lung because it has increasedoxygen tension [30]. However, theidea that oxygen is the key factor iscontradicted by comparison of thedistribution of primary and postprimary lesions, Figure 4. The le-sions of primary TB are distribut-
ed in the same pattern as dust particles that are randomlydistributed in proportion to ventilation. One would thushave to explain why primary and post primary MTBhave different oxygen requirements and why the apicesof the lungs are more susceptible to smoke and othertoxins [30,31].
In 1983, Goodwin challenged the hypothesis that highoxygen tension causes TB to localize in the apices of thelung [30]. He proposed that lymph stasis and impairedclearance of antigenic substances are more important de-terminants of apical localization. The apex is the mostisolated part of the lung. It has the lowest ventilation,perfusion and lymphatic drainage. More recent investi-gations support this hypothesis. Blood flow in the apex
Pathogenesis of Post Primary Tuberculosis
Figure 3. The pathogenesis of post primary TB. Post primary TB begins inthe most isolated part of the lung and proceeds to isolate it further with bronchialobstruction and paralysis of macrophages. It accumulates host lipids and myco-bacterial products before eventually regressing or suddenly undergoing caseousnecrosis that then leads to cavitation and/or fibrocaseous disease.
369

of the lung is very low and frequently zero because thepressure within alveoli is lower than the pulmonary ar-tery pressure in upright people. The pressure in the pul-monary artery of ~10-12 mm Hg is insufficient to raiseblood from the heart to the apex of the lung of people inthe upright position. Consequently the pressure withinthe alveoli of ~2 mm Hg is sufficient to completely
occlude perfusion.The oxygen tensionin the apices is high-er even though ven-tilation is lower, be-cause there isinsufficient bloodflow to carry it away.
Several pieces of evi-dence support thehypothesis that re-duced blood flow isa factor in pathogen-esis of early post pri-mary TB. First, bedrest was the main-stay of therapy forTB for many yearsbefore introduction
to antibiotics [27,32]. Bed rest increases blood flow inthe apex of the lung because the right heart does nothave to overcome the effect of gravity of a recumbentperson. Second, there are reports that this effect could befurther increased by raising the foot of the bed to pro-duce still greater blood flow in the apex [33]. In addi-tion, pulmonary TB frequently improves when the pa-tients reside in high altitudes where the rarefied airrequires more vigorous respiratory efforts. Finally, TBwas very rare in people with high pulmonary artery pres-sure due to mitral valve stenosis and devastating in peo-ple with decreased pulmonary artery pressure due topulmonic valve stenosis [30].
The technologies for measuring the effects of respirationand heart beat motion of individual portions of the lunghave become highly sophisticated because of the needs ofradiation oncology. When treating lung cancer with ra-diation, it is important to deliver the maximal dose tothe tumor while minimizing radiation of normal tissues.This becomes a difficult feat for a tumor in a movinglung. Four-dimensional CAT scans (4 DCT) visualizethe lung in three dimensions plus time to show move-ment [34]. The movement of the lung is greatest at thebase where the diaphragm moves, moderate in the lowerchest where the ribcage moves but nonexistent in theapices where the ribs are fixed. The ventilation at the api-ces is very low in both upright and recumbent positionsbecause the lungs don’t expand and contract with respi-ration. Ventilation is driven only by pressure changes inthe lung [35]. These data suggests that post primary TBbegins in the apices because they do not move, have lowventilation and perfusion and, as discussed later, can befurther isolated by obstruction of bronchi.
Annals of Clinical & Laboratory Science, vol. 44, no. 4, 2014
Figure 5. MTB localization in alveolar macrophagesin early post primary TB. AFB are found in alveolarmacrophages and nowhere else in the bodies of people withearly post primary TB. Similar localization of AFB isfound in mice with slowly progressing pulmonary TB orreactivated TB. This is a section of mouse with reactivatedTB induced by an i.p. injection of 100 ug of TDM as anoil-in-water emulsion (AFB Stain, 1000x).
Figure 4. Distribution of lesions in primary and post primary TB. The localization oflesions of primary and post primary TB of many people were plotted on an X-ray. Each dotmarks the location of an individual’s lesion. The distribution of primary TB is consistent withchance distribution of air-borne infection while most post primary lesions were apical.(Reproduced with permission from Medlar 1948 [87]).
370

Alveolar Macrophages: A characteristic of early post pri-mary TB is that MTB are located exclusively in alveolarmacrophages [3,11,21]. They do not enter the intersti-tium to produce granulomas or infect any other part ofthe body. Alveolar macrophages have dual functions ofcollecting and eliminating innocuous dust particles andrecognizing and reacting appropriately to pathogens. Itappears as if MTB are treated as innocuous particles inimmune individuals whereas they are treated as patho-gens that are carried into tissue where they induce granu-lomas in immunologically naïve subjects [36]. Whilemuch is unknown about this phenomenon, several piec-es of evidence may be relevant. Both innate and adaptiveimmune responses have been implicated in this response[37]. Killed mycobacterial or purified mycolic acidscause macrophages to become foamy and to accumulatelipids [38]. Oxygenated mycolic acids cause differentia-tion of monocyte-derived macrophages into foamy mac-rophages [39,40]. MTB persisted in these cells in a non-replicative state. Electron microscopy demonstratedbacilli in the lipid droplets [41].
The mouse model seems particularly relevant for thisphase of infection [21,42]. In primary TB, alveolar mac-rophages containing MTB migrate into tissue to formsmall granulomas. In contrast, infected alveolar macro-phages in immune mice remain in alveoli, Figure 5.Acid fast bacilli (AFB) are found only within foamy al-veolar macrophages and no place else in the entire body.MTB may be detected by culture in other organs, butthey are too few in number to be detected by AFB stain-ing. This preferential localization of AFB in alveolarmacrophages happens only in animals with sufficientsystemic immunity to prevent infection in all other partsof the body. Lesions of foamy alveolar macrophages candevelop very rapidly in sensitized individuals. Levine
reported that TBpneumonia can de-velop within one dayafter surgery for TB[13]. Similar largenumbers of foamymacrophages in alveo-li have been observedin BCG immunizedmice at 24 hours afteriv challenge with viru-lent MTB [43].Finally, we observedthat injections of cordfactor into mice in-fected with MTB pro-duces a reduction ofnodular granulomasand an increased ininfected alveolar mac-rophages, Figure 6.
Collectively, these data suggest that TB infects the leastactive part of the lung and then manipulates the macro-phages in order to prevent formation of granulomas andto inhibit their normal functions.
Surfactant: Pulmonary surfactant is a critical regulatorof inflammatory and immune functions within alveoliand very likely has a role in early post primary TB [44].Modulation of surfactant function has major effects onboth macrophage function and fluid balance in alveoli.Mice with genetic modifications affecting surfactant areunable to remove MTB from alveoli to interstitial granu-lomas. This results in rapidly progressive tuberculouspneumonia [45]. Peripheral cell wall lipids of MTB aredirectly inhibitory to surfactant function [46]. Finally,surfactant has been used with chemotherapy in patientswith pulmonary TB. Patients received surfactant inhala-tions for 8 weeks during chemotherapy while the controlpatients received only chemotherapy. The surfactant in-creased sputum production, diminished cough, and has-tened both elimination of MTB from sputum and clo-sure of cavities. Cavernous closure was achieved in72.9% of the surfactant treated group and in 41.4% ofthe controls at 4 months [47].
Bronchial Obstruction: There is much evidence thatbronchial obstruction is an essential component of earlypost primary TB [33]. In 1901, Hektoen reported find-ing obstruction of bronchi in 100% of lesions of pulmo-nary TB [48]. Medlar proposed that post primary TBshould be called bronchogenic TB because it characteris-tically spreads through the bronchi to produce obstruc-tion and TB pneumonia [12]. This is also supported bythe modern radiologic literature. TB frequently presentsradiologically as a wedge shaped lesions in an upper lobe.The point of the wedge is an obstructed bronchus
Pathogenesis of Post Primary Tuberculosis
Figure 6. TDM stimulates foamy alveolar macrophages in murine TB. C57Bl/6 micewere infected i.v. with 106 MTB and injected i.p. one day later with 100ug TDM in an o/wemulsion or the emulsion alone and examined on day 36.A. In the control animal, infection isconfined to small non caseating granulomas in the lung (H&E, 100x).B. In the TDM injectedanimal, granulomas are smaller and much of the lung shows tuberculous pneumonia withfoamy macrophages in alveoli (H&E, 100x). Most AFB are seen in foamy alveolar macro-phages (Insert AFB, 100x).
371
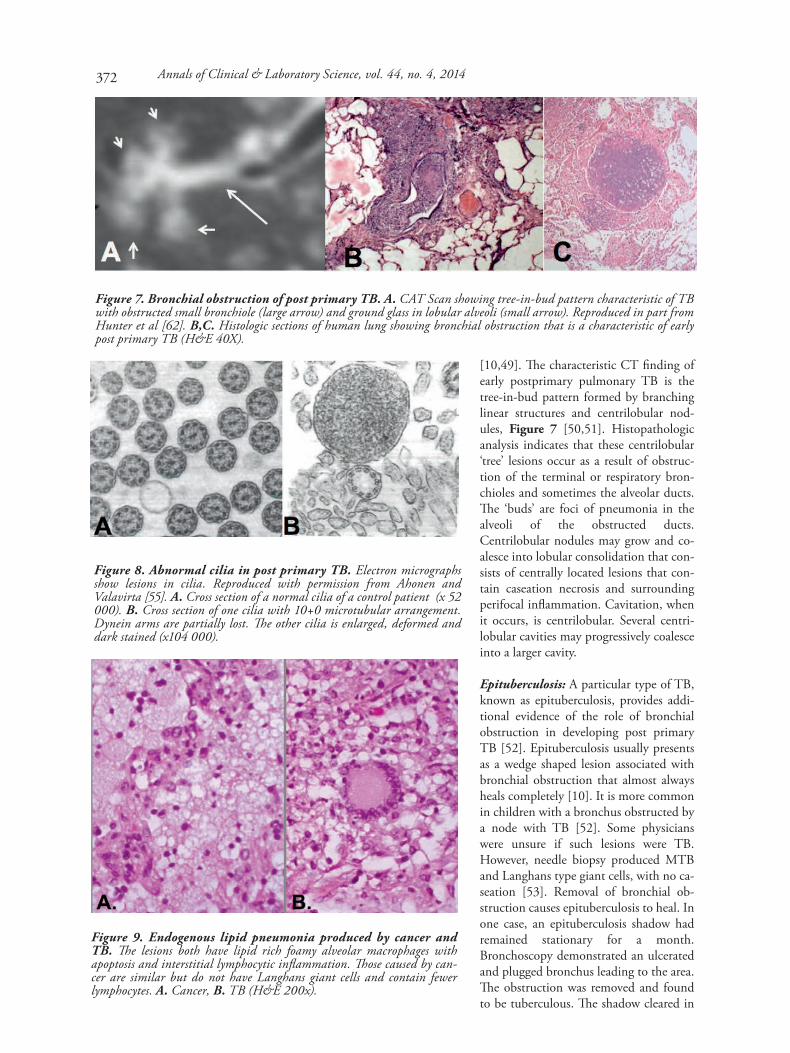
[10,49]. The characteristic CT finding ofearly postprimary pulmonary TB is thetree-in-bud pattern formed by branchinglinear structures and centrilobular nod-ules, Figure 7 [50,51]. Histopathologicanalysis indicates that these centrilobular‘tree’ lesions occur as a result of obstruc-tion of the terminal or respiratory bron-chioles and sometimes the alveolar ducts.The ‘buds’ are foci of pneumonia in thealveoli of the obstructed ducts.Centrilobular nodules may grow and co-alesce into lobular consolidation that con-sists of centrally located lesions that con-tain caseation necrosis and surroundingperifocal inflammation. Cavitation, whenit occurs, is centrilobular. Several centri-lobular cavities may progressively coalesceinto a larger cavity.
Epituberculosis: A particular type of TB,known as epituberculosis, provides addi-tional evidence of the role of bronchialobstruction in developing post primaryTB [52]. Epituberculosis usually presentsas a wedge shaped lesion associated withbronchial obstruction that almost alwaysheals completely [10]. It is more commonin children with a bronchus obstructed bya node with TB [52]. Some physicianswere unsure if such lesions were TB.However, needle biopsy produced MTBand Langhans type giant cells, with no ca-seation [53]. Removal of bronchial ob-struction causes epituberculosis to heal. Inone case, an epituberculosis shadow hadremained stationary for a month.Bronchoscopy demonstrated an ulceratedand plugged bronchus leading to the area.The obstruction was removed and foundto be tuberculous. The shadow cleared in
Figure 8. Abnormal cilia in post primary TB. Electron micrographsshow lesions in cilia. Reproduced with permission from Ahonen andValavirta [55]. A. Cross section of a normal cilia of a control patient (x 52000). B. Cross section of one cilia with 10+0 microtubular arrangement.Dynein arms are partially lost. The other cilia is enlarged, deformed anddark stained (x104 000).
Figure 9. Endogenous lipid pneumonia produced by cancer andTB. The lesions both have lipid rich foamy alveolar macrophages withapoptosis and interstitial lymphocytic inflammation. Those caused by can-cer are similar but do not have Langhans giant cells and contain fewerlymphocytes. A. Cancer, B. TB (H&E 200x).
Annals of Clinical & Laboratory Science, vol. 44, no. 4, 2014
Figure 7. Bronchial obstruction of post primary TB. A. CAT Scan showing tree-in-bud pattern characteristic of TBwith obstructed small bronchiole (large arrow) and ground glass in lobular alveoli (small arrow). Reproduced in part fromHunter et al [62]. B,C. Histologic sections of human lung showing bronchial obstruction that is a characteristic of earlypost primary TB (H&E 40X).
372

six days by x-ray. There are multiple other reports in thepre antibiotic literature where surgical relief of bronchialobstruction produced resolution of pulmonary TB [10].Finally, the role of bronchial obstruction in developingpost primary TB is illustrated by the observation thatextensive unilateral pulmonary TB has been reportedwith segmental atresia of a bronchus [54].
Cilia: Obstruction of small bronchi by developing TBmay be viewed as a failure of ciliary clearance. Most par-ticles that reach alveoli are ingested by alveolar macro-phages that migrate to small bronchi where they contactcilia. The cilia then carry the macrophages and particlesinto the trachea and up to the mouth where they areswallowed. In patients with pulmonary TB, the bron-chial mucosal is generally intact although there may beareas of ulcerated infected tissue extending into the sub-mucosal glands. Ahonen and Valavirta studied cilia ofTB patients by electron microscopy [55]. They demon-strated loss of some ciliary components and major
distortion and swelling of other cilia, Figure 8. Theseunique changes suggest that MTB has a specific effect oncilia.
The effects of tobacco smoke support an important rolefor cilia in the pathogenesis of developing TB [31]. Inepidemiologic studies, cigarette smoking has been con-sistently reported to markedly increase the risk of devel-oping pulmonary TB. It is well established that cigarettesmoke inhibits cilia action. Thus it is likely that paralysisof cilia by the smoke contributes to the pathogenesis ofpost primary TB [56,57].
Post obstructive pneumonia: We reported that post pri-mary TB begins as an endogenous lipid pneumonia[3,21]. We now know that bronchial obstruction fromany cause produces post-obstructive pneumonia charac-terized by gradual accumulation lipid rich foamy macro-phages in alveoli behind the obstruction [58]. The lipidhas been identified as neutral lipids and cholesterol that
Pathogenesis of Post Primary Tuberculosis
Figure 10. Bronchial obstruction by cancer and chemotherapy produced a TB like cavity. A. Anterior view CATscan showing a large mass in the left side of the mediastinum measuring approximately 8.2 x 9 x 9.5 cm (arrow). It encasesthe left upper lobe bronchi, pulmonary artery and pulmonary vessels. Although it does not show well in this reproduction,the pulmonary parenchyma of the left upper lobe had a ground glass appearance characteristic of obstructive pneumonia.(Reproduced with permission in part from Hunter et al [62]). B. Repeat CAT scan 5 weeks later showing a large cavity inthe left upper lobe measuring 12 x 8 cm where the ground glass area had been (arrow). The radiologist’s report stated “Givenits rapid development and the presence of numerous tree-in-bud opacities … the patient needs to be assessed for possibleTB.” C. Posterior photograph of both lungs of the patient at autopsy showing a large thin walled cavity in the left upperlobe (arrow). This cavity, like those caused by TB, extended to the pleural surface, but did not penetrate. D. Section of thewall of the cavity of our cancer case showing a layer of necrotic lung overlying granulation tissue and lipid pneumonia. Thedetached, necrotic fragment of lung (arrow) resembles the fragments of lung that are coughed up by people with developingcavitary TB (H&E Stain 40). E. Giant cell resembling a Langhans Giant cell. (H&E stain 400x). F. Area on necrosisresembling caseation necrosis with cholesterol crystals. (H&E stain 100x).
373

are probably derived from action of macrophages on sur-factant. In 1925, Pagel used fat stains to access theamount of lipid in alveoli during the course of develop-ing post primary TB. He found that stainable lipid in-creased progressively with time [59].
Today, cancer is the most common cause of post obstruc-tive pneumonia [60]. It is an endogenous lipid pneumo-nia with many similarities to early post primary TB. Thedisease produced by cancer has fewer lymphocytes andno Langhans type giant cells, but has a similar pattern oflipid rich foamy macrophages filling alveoli, Figure 9.The lipid is derived from surfactant and is similar to thatfound in tuberculous pneumonia. Post obstructive pneu-monia due to cancer can undergo necrosis resemblingthe caseous necrosis of TB and cavitation. One studyfound cavities in 13% of patients with post obstructivepneumonia caused by squamous cell carcinomas [61]. Inone case, chest x-ray demonstrated ground glass appear-ance of post obstructive pneumonia immediately beforeinitiation of chemotherapy. A similar x-ray one weeklater demonstrated a very large cavity, Figure 10 [62].The patient subsequently died. An autopsy revealed lipidpneumonia with multinucleated giant cells and necrosisresembling caseation in the wall of the cavity with strik-ing similarities to that of recently formed human tuber-culous cavities. Others have reported similar cases [63].These results demonstrate that post obstructive pneumo-nia establishes conditions for the development of caseousnecrosis and cavitation [60].
Accumulation of mycobacterial antigens:Immunohistochemical staining tissues with polyclonalantiserum against whole killed MTB has been developedas a diagnostic test for TB [64]. The amount and distri-bution of MTB antigen staining in tissues varies with thetype of infection. In tissues from people with rapidlyprogressive TB associated with HIV, the staining ofMTB antigen is similar to that of acid fast staining. Thestaining of early post primary TB is different. It typicallydemonstrates far larger amounts mycobacterial antigenthan can be accounted for by organisms observed withacid fast stains. In fact, there is frequently intense stain-ing for mycobacterial products in tissues with no detect-able AFB, Figure 11. The finding of MTB antigens intissues with no AFB is the justification for use of immu-nohistochemistry for diagnosis [64-66]. The staining forMTB antigens in immunocompetent patients is typical-ly diffuse within the cytoplasm of foamy alveolar macro-phages. It is not found extracellularly except in necrosis.These studies demonstrate that mycobacterial antigensaccumulate with the lipid in foci of endogenous lipidpneumonia of post primary TB.
Our earlier studies demonstrated that slowly progressivepulmonary TB in the mouse has many similarities toearly post primary TB in humans [3,21]. This includesobstruction of bronchi an accumulation of foamy mac-rophages containing lipid, few AFB and mycobacterialantigens in portions of the lung distal to the obstruction.In this model, ManLAM, MPT64 and polyclonal
Figure 11. MTB anti-gen in immunocompe-tent and immune com-promised people. A.Immunohistochemicalstain for MTB (IHC) inalveolar macrophages ofan immunocompetentperson with developingpost primary TB (IHCStain 200x). B. Higherpower of same sectionshowing that the antigenis entirely intracellular.(IHC 1000x). These sec-tions were negative byAFB stain demonstratinga paucibacillary infec-tion. C. Many AFB in apatient with HIV anddisseminated TB (AFBstain, 1000x). D. IHCstain for MTB of sectionC reflects AFB in numberand distribution ofMTB. (IHC Stain1000x).
Annals of Clinical & Laboratory Science, vol. 44, no. 4, 2014374
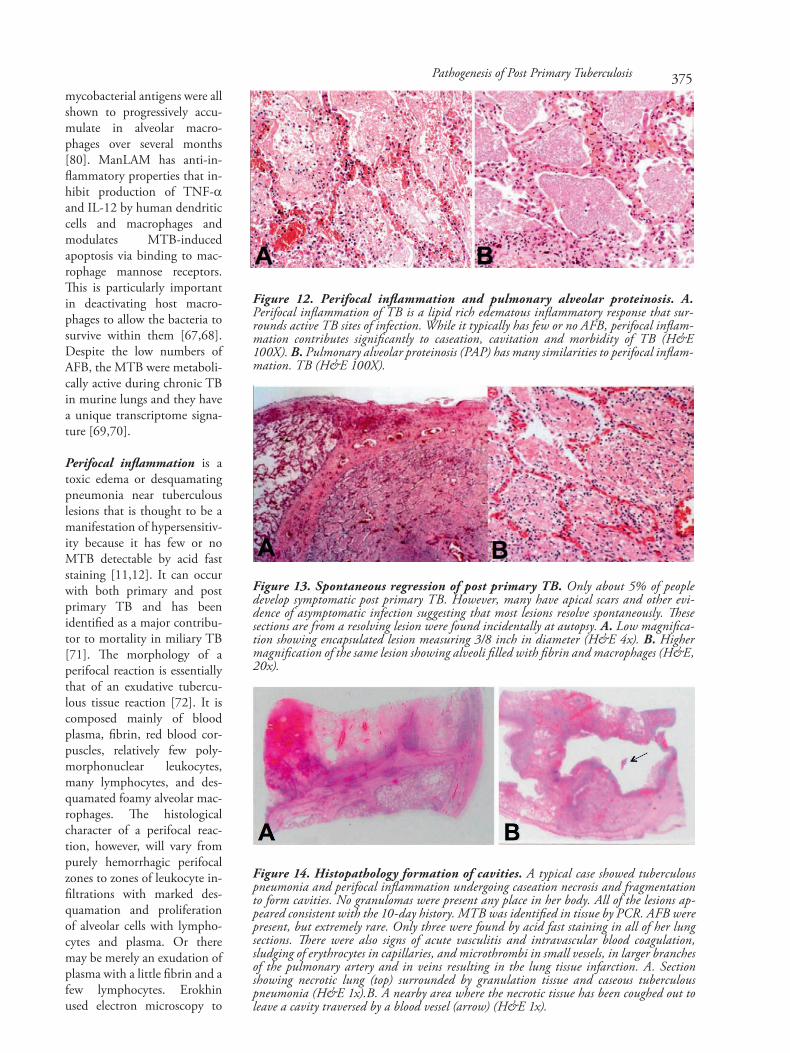
mycobacterial antigens were allshown to progressively accu-mulate in alveolar macro-phages over several months[80]. ManLAM has anti-in-flammatory properties that in-hibit production of TNF-αand IL-12 by human dendriticcells and macrophages andmodulates MTB-inducedapoptosis via binding to mac-rophage mannose receptors.This is particularly importantin deactivating host macro-phages to allow the bacteria tosurvive within them [67,68].Despite the low numbers ofAFB, the MTB were metaboli-cally active during chronic TBin murine lungs and they havea unique transcriptome signa-ture [69,70].
Perifocal inflammation is atoxic edema or desquamatingpneumonia near tuberculouslesions that is thought to be amanifestation of hypersensitiv-ity because it has few or noMTB detectable by acid faststaining [11,12]. It can occurwith both primary and postprimary TB and has beenidentified as a major contribu-tor to mortality in miliary TB[71]. The morphology of aperifocal reaction is essentiallythat of an exudative tubercu-lous tissue reaction [72]. It iscomposed mainly of bloodplasma, fibrin, red blood cor-puscles, relatively few poly-morphonuclear leukocytes,many lymphocytes, and des-quamated foamy alveolar mac-rophages. The histologicalcharacter of a perifocal reac-tion, however, will vary frompurely hemorrhagic perifocalzones to zones of leukocyte in-filtrations with marked des-quamation and proliferationof alveolar cells with lympho-cytes and plasma. Or theremay be merely an exudation ofplasma with a little fibrin and afew lymphocytes. Erokhinused electron microscopy to
Pathogenesis of Post Primary Tuberculosis
Figure 12. Perifocal inflammation and pulmonary alveolar proteinosis. A.Perifocal inflammation of TB is a lipid rich edematous inflammatory response that sur-rounds active TB sites of infection. While it typically has few or no AFB, perifocal inflam-mation contributes significantly to caseation, cavitation and morbidity of TB (H&E100X). B. Pulmonary alveolar proteinosis (PAP) has many similarities to perifocal inflam-mation. TB (H&E 100X).
Figure 13. Spontaneous regression of post primary TB. Only about 5% of peopledevelop symptomatic post primary TB. However, many have apical scars and other evi-dence of asymptomatic infection suggesting that most lesions resolve spontaneously. Thesesections are from a resolving lesion were found incidentally at autopsy. A. Low magnifica-tion showing encapsulated lesion measuring 3/8 inch in diameter (H&E 4x). B. Highermagnification of the same lesion showing alveoli filled with fibrin and macrophages (H&E,20x).
Figure 14. Histopathology formation of cavities. A typical case showed tuberculouspneumonia and perifocal inflammation undergoing caseation necrosis and fragmentationto form cavities. No granulomas were present any place in her body. All of the lesions ap-peared consistent with the 10-day history. MTB was identified in tissue by PCR. AFB werepresent, but extremely rare. Only three were found by acid fast staining in all of her lungsections. There were also signs of acute vasculitis and intravascular blood coagulation,sludging of erythrocytes in capillaries, and microthrombi in small vessels, in larger branchesof the pulmonary artery and in veins resulting in the lung tissue infarction. A. Sectionshowing necrotic lung (top) surrounded by granulation tissue and caseous tuberculouspneumonia (H&E 1x).B. A nearby area where the necrotic tissue has been coughed out toleave a cavity traversed by a blood vessel (arrow) (H&E 1x).
375

demonstrate destruction of alveolar epithelium, particu-larly Type 2 cells [29]. Tissue with perifocal inflamma-tion is seldom seen in surgical resections since it clearsrapidly with antibiotic therapy [10,12]. Reversal of peri-focal inflammation may be observed in X-rays within 72hours of the initiation of therapy [73].
Perifocal inflammation, at first glance, appears to be anepiphenomenon: something that takes place as a resultof TB, not a primary contributor to it. However, severalpieces of information suggest that it may be central to hepathogenesis of TB. First, perifocal inflammation under-goes caseous necrosis to contribute a major portion oflung afflicted with caseous pneumonia. Next, it can beinduced in tuberculous people by injections of tubercu-lin and is associated with clinical exacerbation of disease.This is the Koch phenomenon to be discussed later.Finally, the fact that it resolves quickly with therapy in-dicates that it requires live MTB.
Histologically, perifocal inflammation of TB resemblespulmonary alveolar proteinosis (PAP), Figure 12. This isa condition in which alveoli are filled with lipid richfluid and foamy macrophages and lymphocytes but fewother inflammatory cells. It can be caused by a deficiencyin surfactant protein B or by an autoimmune diseasewith altered GM-CSF signaling [74,75]. PAP can becaused by a number of infections including TB. We sus-pect that some of these were cases of perifocal inflamma-tion of TB rather than cases of alveolar proteinosis com-plicated by TB [76,77]. The disease can frequently betreated simply by lavage that washes out the lipid richmaterial from alveoli.
PAP frequently occurs in association with endogenouslipid pneumonia [78]. Most cases analyzed showed mor-phological features of both PAP and endogenous lipidpneumonia. Type II pneumocytes, macrophages, andneutrophils play a significant part in the pathogenesis ofboth. Historically, endogenous lipid pneumonia hasbeen reported to be caused by bronchial obstruction bycancer, but it also can occur with infection and otherdiseases that are not associated with bronchial obstruc-tion. Additionally, in PAP, the alveoli are usually filledwith protein and lipid material resembling surfactant[78,79].
Regression of post primary TB: The large majority ofearly post primary TB cases regress prior to developmentof symptoms. Only about 5% of people develop disease[42]. Since some people resolve infections repeatedly, itis likely that over 99% of early post primary infectionsresolve spontaneously. Consequently, it is a rare event forthe process to progress to clinical disease. Scars found inthe apices of the lungs of people born in the preantibi-otic era have been attributed to healed TB that regressedin the very early stages, Figure 13.
Onset of clinical disease: Patients with post primary TB whodevelop disease typically do so within one or two years follow-ing infection [42,80]. The onset of clinical pulmonary TB isextraordinarily diverse causing much difficulty with diagnosis[27]. It can present as acute pneumonia, chronic dyspeptic andanemic symptoms or bronchitic with so little symptoms or fe-ver that TB is discovered only accidentally by sputum. Themost instructive cases, however, are those who undergo a rapidonset of caseous pneumonia, Figure 14. Investigators fromLaennec in the early 19th century through Erokhin in the early21st century described this mode of presentation [29,81].Available evidence suggests that the disease process is similar,but less extensive and less well synchronized, in patients withless severe onset.
Caseous pneumonia typically begins with sudden acute onsetof chill, fever, and rapidly advancing symptoms of severe in-toxication [27,29]. Patients often complain of pain in thechest, dyspnea and coughing with expectoration of sometimesrust-colored sputum. Physical examination in the first days ofdisease reveals intensive deadening of percussion sounds, bron-chial breathing with crackling rales of high pitch and sonority.The disease at this early stage is typically indistinguishable fromcommunity acquired bacterial pneumonia [29,82]. The X-raysshow only diffuse density of lung lobes or lobules, which be-comes intensive over a few days. Diagnosis is especially difficultbecause of the lack of AFB in sputum [83]. In a recent study,only 12.8% of caseous pneumonia cases had AFB in their spu-tum in first two weeks of the disease [29]. This number in-creased to 39.2% by the end of the first month, and to 67% bythe end of the second month.
Patients with caseous pneumonia did have two characteristiclaboratory findings [29]. The first was profound hyper coagula-tion with development of disseminated intravascular coagula-tion (DIC). This is a well known complication of tuberculosis[84]. The second was lymphopenia of CD4+ cells. Patientswith caseous pneumonia had significant relative and absolutedecrease in CD4+-lymphocytes versus patients with chronicpulmonary TB and age matched controls. At the same time,the numbers of CD8+ cells in caseous pneumonia patients didnot differ from those found in patients with chronic pulmo-nary TB. The CD4+/CD8+ ratio was 0.7±0.03 for patientswith caseous pneumonia versus 1.4±0.09 for patients withchronic pulmonary TB and 1.4±0.07 for normal controls.
The Koch Phenomenon: The clinical presentation of caseouspneumonia has striking resemblance to the Koch phenomenon[85]. Koch proposed that tuberculin could be a specific curefor TB. In early attempts at therapy, large doses of tuberculinwere injected subcutaneously. The treatment caused severe lo-cal and systemic reactions in patients with relatively mild dis-ease. It induced perifocal reactions that progressed rapidly tocaseation and cavitation. Of the 230 patients with advancedcavitary disease who received this treatment, 30 died [86].Koch, himself, had a severe constitutional reaction followingthe injection of old tuberculin [72].
Annals of Clinical & Laboratory Science, vol. 44, no. 4, 2014376

More recent studies have confirmed and extended theseresults. Perifocal reactions occur only around existing ac-tive tuberculous lesions. The systemic symptoms includefever often up to 40°C or higher with toxemia [85].Injections of tuberculin have the potential to increase theseverity of TB markedly [72]. The morphology of an in-flammatory reaction induced by tuberculin around a tu-berculous focus is essentially that of a perifocal reactionof active TB. It is composed mainly of blood plasma, fi-brin, red blood corpuscles, relatively few polymorpho-nuclear leucocytes, many lymphocytes, desquamated al-veolar epithelial cells and alveolar macrophages.
Growth and cultivability of MTB in tissue: The litera-ture frequently states that massive numbers of MTBoverwhelm host defenses to produce caseation necrosis[15]. This is at odds with the observations that the onsetof caseous pneumonia is a pauci bacillary process withvery few MTB. Many papers report that some early le-sions are paucibacillary while others have many AFB[11,12]. A likely explanation is that most of these inves-tigators are reporting on chronic pulmonary TB that hasmultiple stages simultaneously. In addition, MTB dogrow in massive numbers producing necrosis in immunecompromised people such as those with HIV. This is notthe case in immunocompetent people. Canetti andMedlar who independently examined thousands of casesreport that AFB are found in large numbers only in re-cently necrotic tissue or on cavity walls [11,12,87]. Theyare never found in large numbers in viable tissue of im-munocompetent people. This is our observation as well,Figure 15. An unexplained, but consistent finding, isthat the vast majority of AFB in closed necrotic tissueswill not grow in culture nor will they infect guinea pigs[11,88]. In contrast, nearly 100% of AFB present in
cavities will grow in culture [11,12,89]. It has been as-sumed that MTB that do not grow in culture are dead.Recent investigations demonstrate that many such or-ganisms can be resuscitated and that proliferation ofMTB is tightly controlled by both host and parasite[135]. Organisms that proliferate massively in any partof the body except a cavity endanger their host andthereby themselves. Increasing data indicates that MTBcontrols its proliferation to insure our survival and todevelop conditions for its transmission to new hosts.
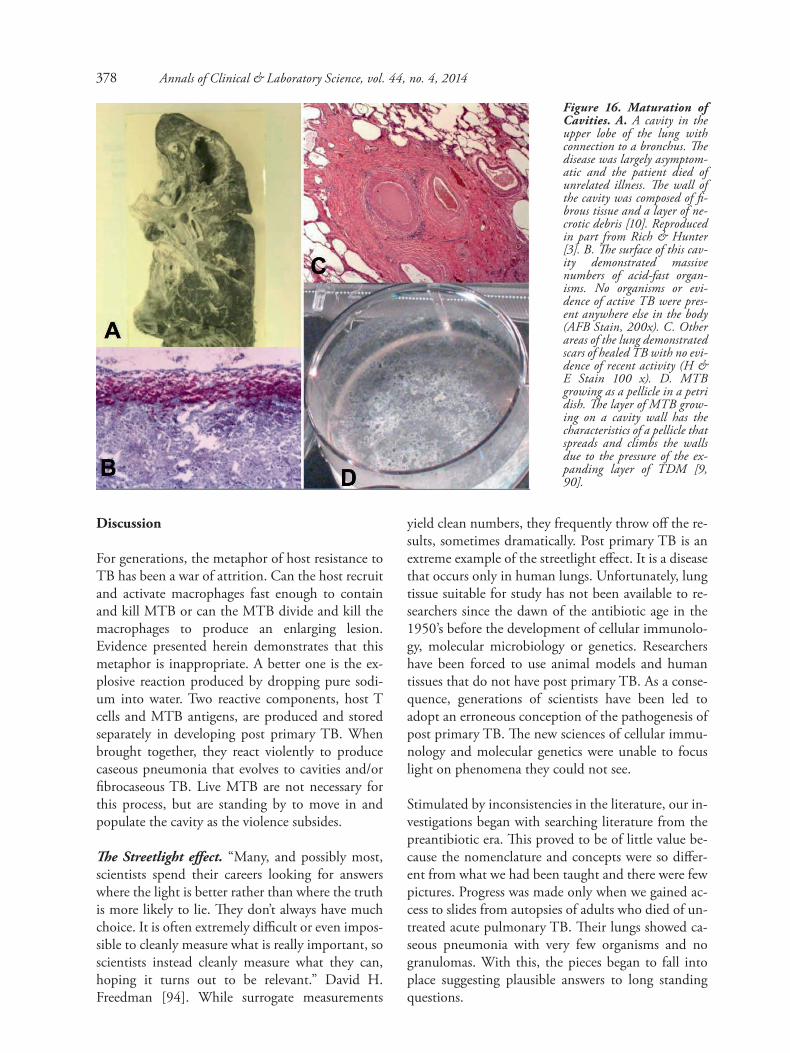
Production of cavities: Once a lesion develops caseouspneumonia, it may further develop in one of two direc-tions. First, it may soften and fragment to the point thatpieces of lung can be coughed out to produce a cavity.The fragmentation and removal of necrotic lung may benearly complete in some patients. In these cases, the re-sultant cavities begin with a shaggy wall of debris. Overtime this resolves and they develop a thin fibrous wallwith little or no inflammation and bacteria grow only onthe inner surface of the cavity. Laennec noticed that suchcavities are always centered on bronchi and that they arelined with a smooth waxy material that pushes its wayinto the bronchi. Histologically, the inner surface is amass of organisms that grow as a pellicle [3,90]. MTBgrown in liquid media without surfactants grows prefer-entially as a surface pellicle which forces its way acrossthe surface and climbs the sides of the vessel, Figure 16.This is due to rigidity of the monolayer of TDM [90].
Fibrocaseous TB: The caseous pneumonic material thatis not coughed out will dehydrate and become surround-ed by granulomatous inflammation [11]. However, un-like granulomas of primary TB in which caseation ne-crosis develops within granulomas, the granulomas inpost primary TB develop around preexisting foci of case-ous pneumonia. If a caseous pneumonic lesion is small,it forms the nidus of a caseating granuloma superficiallysimilar to those of primary TB, Figure 17. However, theunderlying alveolar structure can be observed with con-nective tissue stains. If a caseous pneumonic is large, theformation of granuloma around it results in fibrocaseousTB that is the most common type of chronic disease.
TB is the largest cause of pulmonary fibrosis worldwide[91,92]. It has been reported that the rabbit is the onlyanimal model that will produce fibrocaseous TB.However, such lesions can be produced in mice by injec-tions of mycobacteria in an oil emulsion of TDM [18].In addition, it appears that fibrocaseous TB is a reactionto dehydrated caseous necrosis; a lipid matrix containingmycobacterial antigens. Freund’s complete adjuvantconsisting of mycobacteria in an oil-in-water emulsionreproduces this rather well [93].
Pathogenesis of Post Primary Tuberculosis
Figure 15. AFB in viable and necrotic lung. Section oflung of a person with necrotizing tuberculous pneumonia.In immunocompetent people, very few and frequently noAFB are ever found in viable tissue (Right). Large numbersof AFB are found only in newly necrotic tissue (ArrowLeft). However, AFB in necrotic tissue seldom grow in cul-ture [11, 12]. Their numbers decline as the nuclear debrisis removed (AFB Stain 100x).
377
Discussion
For generations, the metaphor of host resistance toTB has been a war of attrition. Can the host recruitand activate macrophages fast enough to containand kill MTB or can the MTB divide and kill themacrophages to produce an enlarging lesion.Evidence presented herein demonstrates that thismetaphor is inappropriate. A better one is the ex-plosive reaction produced by dropping pure sodi-um into water. Two reactive components, host Tcells and MTB antigens, are produced and storedseparately in developing post primary TB. Whenbrought together, they react violently to producecaseous pneumonia that evolves to cavities and/orfibrocaseous TB. Live MTB are not necessary forthis process, but are standing by to move in andpopulate the cavity as the violence subsides.
The Streetlight effect. “Many, and possibly most,scientists spend their careers looking for answerswhere the light is better rather than where the truthis more likely to lie. They don’t always have muchchoice. It is often extremely difficult or even impos-sible to cleanly measure what is really important, soscientists instead cleanly measure what they can,hoping it turns out to be relevant.” David H.Freedman [94]. While surrogate measurements
yield clean numbers, they frequently throw off the re-sults, sometimes dramatically. Post primary TB is anextreme example of the streetlight effect. It is a diseasethat occurs only in human lungs. Unfortunately, lungtissue suitable for study has not been available to re-searchers since the dawn of the antibiotic age in the1950’s before the development of cellular immunolo-gy, molecular microbiology or genetics. Researchershave been forced to use animal models and humantissues that do not have post primary TB. As a conse-quence, generations of scientists have been led toadopt an erroneous conception of the pathogenesis ofpost primary TB. The new sciences of cellular immu-nology and molecular genetics were unable to focuslight on phenomena they could not see.
Stimulated by inconsistencies in the literature, our in-vestigations began with searching literature from thepreantibiotic era. This proved to be of little value be-cause the nomenclature and concepts were so differ-ent from what we had been taught and there were fewpictures. Progress was made only when we gained ac-cess to slides from autopsies of adults who died of un-treated acute pulmonary TB. Their lungs showed ca-seous pneumonia with very few organisms and nogranulomas. With this, the pieces began to fall intoplace suggesting plausible answers to long standingquestions.
Figure 16. Maturation ofCavities. A. A cavity in theupper lobe of the lung withconnection to a bronchus. Thedisease was largely asymptom-atic and the patient died ofunrelated illness. The wall ofthe cavity was composed of fi-brous tissue and a layer of ne-crotic debris [10]. Reproducedin part from Rich & Hunter[3]. B. The surface of this cav-ity demonstrated massivenumbers of acid-fast organ-isms. No organisms or evi-dence of active TB were pres-ent anywhere else in the body(AFB Stain, 200x). C. Otherareas of the lung demonstratedscars of healed TB with no evi-dence of recent activity (H &E Stain 100 x). D. MTBgrowing as a pellicle in a petridish. The layer of MTB grow-ing on a cavity wall has thecharacteristics of a pellicle thatspreads and climbs the wallsdue to the pressure of the ex-panding layer of TDM [9,90].
Annals of Clinical & Laboratory Science, vol. 44, no. 4, 2014378

Primary TB stimulates systemic immunity. This ac-tivates macrophages to ingest and kill MTB and tocontain them within granulomas that eventuallyheal. The result is protection from disseminated TBand effective resistance to new infection.Granulomas don’t occur in developing post prima-ry TB because, there is sufficient immunity fromprimary TB to prevent growth of MTB in viabletissue. This protects the entire body except for thevulnerable region of the lung that has special mech-anisms to facilitate transmission of MTB to newhosts.
Post primary TB does not result from a lack of sys-temic immunity, but rather from local susceptibili-ty in the apex of the lung. It begins in the part oflung with the lowest ventilation, perfusion andmovement and proceeds to paralyze alveolar micro-phages, blockade the exits and suppress inflamma-tion to further isolate the area in order to provide aplace for prolonged accumulation of host lipids andmycobacterial products. At some point, there is asudden onset of a necrotizing hypersensitivity reac-tion to the accumulated products. This appears tobe a combination of perifocal inflammation due toprotein antigens and caseous necrosis due to cordfactor. Once induced, the caseous pneumonia mayeither fragment to produce cavities or harden toproduce fibrocaseous TB. There has been much
speculation as to what causes the softening andfragmentation of caseous material [15]. Granulomasin post primary TB occur only in response to case-ous pneumonia that fails to fragment [11,12].
This conception of the pathogenesis of TB is basedupon a mixture of facts, correlations and specula-tions. While some of the ideas presented in this pa-per will surely change with advancing knowledge,the facts will remain and must be considered. Thefollowing statements are supported by multiple in-vestigations each of which involves hundreds orthousands of patients and should be considered tobe facts.
1. In post primary TB, granulomas develop in re-sponse to caseation necrosis and are not the cause ofit [10-12].2. Cavities develop from fragmentation of case-ous pneumonia, not from eroding granulomas[10-13].3. Caseous pneumonia with developing cavities istypically paucibacillary (contains very few MTB)[11,12,27,29].4. Developing post primary TB is characterizedby bronchial obstruction that produces post ob-structive pneumonia that can cavitate [12,13,48,51,95,96].5. TB antigens accumulate within cells in areaswith very few AFB [21,64-66,97].
The value of scientific theory is often judged byhow well it explains natural phenomena. The pro-posed pathogenesis of post primary TB suggestsanswers to long standing questions.
• How can MTB be an obligate human para-site when people are more resistant than any ofthe animals studied? MTB is an obligate humanparasite because it can only be transmitted to newhosts by people. Humans are more resistant to TBbecause most develop effective immunity againstprimary TB in weeks whereas most animals diewithin months of progressive disease. This benefitsthe organism because it maintains the health of thehost who can then spread infection frequently fordecades [27,98]. Only humans develop post pri-mary TB that progresses to pulmonary cavitiesfrom which infection can be transmitted to newhosts.
Pathogenesis of Post Primary Tuberculosis
Figure 17. Chronic Fibrocaseous TB. Aging caseouspneumonia that fails to fragment becomes surrounded bygranulomatous tissue and fibrosis. The central necrotic coreretains the structure of alveoli. It became surrounded by anactive granulomatous process with epitheloid cells, giant cellsand lymphocytes, but no foamy macrophages. Fibrosis devel-ops gradually. This is a characteristic lesion of late post pri-mary TB (Trichrome Stain, 40 x magnification).
379

• What is the nature of the immunity that pro-tects most people from post primary TB? Peopledo not develop post primary TB unless they havesufficient immunity to heal primary TB and pre-vent new infection in every part of their bodies ex-cept the vulnerable area in the apex of the lung. Theappropriate question, therefore, is: How do MTBmanipulate an otherwise effective host responses inthis particular part of the lung? MTB selects theleast active part of lung and uses an otherwise effec-tive immune response to isolate it further and mod-ulate macrophages to accumulate host and myco-bacterial products quietly in preparation for anabrupt and massive necrotizing reaction. Since pro-gression to disease is a rare event, resistance can bedue to any of the many factors that could preventdeveloping conditions for the necrotizing reaction.
• How can multiple pulmonary lesions in asingle lung act independently as if the others didnot exist? Patients with chronic TB frequently havemultiple stages of TB lesions in their lungs that de-velop independently of one another [10,11,15,27].Many stages of lesions in post primary TB may co-exist in a single lung because they are controlledlocally, particularly by bronchial obstruction thatproduces stasis and facilitates storage of lipids andantigens. Obstruction is a local process that can be-gin in different parts of the lung at different times.In contrast, the lesions in persons with primary(miliary) or disseminated TB all look similar to oneanother because they are controlled by systemicimmunity.
• Why doesn’t recovery from post primary TBproduce immunity? Recovery from most infectionsresults in immunity. TB is different. “To havepassed through a period of high mortality risk con-fers not protection, but added hazard in later life.”(The White Plague, Dubos [28]). Recent studiesreport that people who have recovered from TB arehighly susceptible to new infections rather than re-activation of previous infections [5,99]. Such peo-ple are only susceptible to new post primary TB,and not to primary or disseminated TB. Immunitytypically remains sufficient to protect the rest oftheir bodies. Susceptibility in such persons is prob-ably due to production of more vulnerable areas oflung due to disruption of bronchial structures andclearance mechanisms by the previous lesions.
Damage to bronchi probably facilitates mainte-nance of bronchial obstruction by TB. Modifiedimmune responses may contribute, but there is lit-tle data.
• What is the role of immunosuppression inpost primary TB? Suppression of T cell functionby steroid drugs, HIV or old age reactivates latentinfection and increases susceptibility to disseminat-ed TB. This is frequently extrapolated to imply thatall reactivation of TB is initiated by immunosup-pression. However, there is little evidence that peo-ple with pulmonary TB are immunosuppressed.They are not more susceptible to other viral or bac-terial infections. They are not even more susceptibleto other intracellular bacterial infections [100]. Inaddition, people with suppression of T cell func-tion are less likely to develop cavities and spread thedisease to others than immunocompetent youngadults [101-103]. In fact, young adults with thestrongest immune responses are the most likely tohave apical localization of infection, to producecavities and to die of acute disease [10,27]. A strongimmune response is necessary to produce a cavity.Once a cavity is produced, a strong immune re-sponse is needed to keep the massive number ofMTB produced in the cavity from spreading infec-tion to the rest of the body and killing its host.Immunosuppression may be necessary to reactivatelatent MTB and to get organisms to the apex of thelung. From then on its role is unclear.
• What are the roles of hypersensitivity andimmunity? It has long been known that much ofthe tissue damage in TB cannot be attributed di-rectly to the organisms, but is a hypersensitivitythat resembles the Koch phenomenon [10,72,86,104]. Possible roles of hypersensitivity in immunityhave been widely discussed [15,37]. The studiespresented herein suggest that hypersensitivity andimmunity are separate and distinct phenomena.Primary TB stimulates immunity, the ability to re-cruit and activate macrophages sufficiently to con-trol and kill MTB and prevent new infection. Thisimmunity may be weakened by immunosuppres-sion such as HIV to allow disseminated infection.However, it typically persists and protects the bodyfrom new infection in every place except for thevulnerable region of the lung where MTB has es-tablished a unique environment for production of
Annals of Clinical & Laboratory Science, vol. 44, no. 4, 2014380

cavities that facilitate transmission to new hosts.Immunity protects the host from systemic infec-tion. Hypersensitivity reactions destroy lung tissueto produce a cavity. The organism needs both inorder to survive. If there is no cavity, MTB cannever escape to a new host. If here is no systemicimmunity, MTB will produce disseminated infec-tion and kill the host thereby killing itself.
MTB is an obligate human parasite because it canonly be transmitted from host to host by humans.Consequently, all of its genes have been selected toenhance survival and transmission in people.Everything that it makes or does has been selectedfor that one purpose. MTB is an ancient humanpathogen that can persist indefinitely in very smallpopulations of as little as 200 people [105]. Thelifecycle of MTB is to infect a person, frequently achild, and then hide for 10-30 years before reacti-vating to produce a cavity in the lung from whichorganisms are coughed up over a period of decadesto infect new individuals.
Highly conserved immunologic epitopes. Manymicrobial antigens are highly variable in order toevade immune responses. MTB is different. Musserreported remarkably little variation in antibody epi-topes of MTB [106]. In 2012, Comas et al reportedthat the T epitopes of MTB are hyper conserved.[7]. More than 95% of the 491 individual epitopesanalyzed had identical amino acid sequences. Theresearchers speculated that MTB needs these epit-opes because they induce particularly toxic reac-tions that are necessary for tissue destruction andthe formation of cavities that mediate transmissionof infection.
Several pieces of evidence support this hypothesis.First, infected humans are much more sensitive totuberculin than the guinea pig or other animals[85]. Tuberculin test sites in humans with TB fre-quently become necrotic. Necrosis does not occurin animals or in people with positive skin-tests in-duced by BCG or tuberculoid leprosy [107]. MTBshares epitopes with some saprophytic mycobacte-ria. These shared mycobacterial epitopes evoke mildskin-test reactivity in TB patients [107]. Cases havebeen reported in which tuberculin skin tests pro-duced pulmonary perifocal inflammation that was
fatal [72]. Tuberculous pneumonia has developedovernight following surgery for TB [13]. Perifocalreactions in the form of a sudden increase in thesize of pulmonary lesions were probably the resultof dissemination of tuberculoprotein. [72]. Cordfactor produced by MTB is far more toxic that thatproduced by other mycobacteria [108]. MTB cordfactor also has hypersensitivity epitopes that are notpresent on BCG [109]. Furthermore, mixing theMTB with oil in Freund’s adjuvant induces a farmore severe and long lasting toxicity in tuberculoushumans than that the MTB materials alone. This isprobably relevant since caseation necrosis consistsof mycobacterial antigens in a lipid matrix.
Given the nature of natural selection, epitopes arehyper conserved because they help the organismsurvive in their only natural host, people. To dothis, they must have two properties. The first is toproduce particularly necrotizing tissue reactions.The second is that they must remain hidden untilneeded. Premature release of MTB antigens wouldnot stimulate a large enough reaction to produce acavity from which MTB could escape to infect newhosts. There is evidence that hiding of antigens isaccomplished via multiple mechanisms. First, theisolation and blockade of sections of the lung pro-vides a protected environment for MTB. Second,antigens detectable by immunohistochemistry arelocated exclusively in live cells in alveoli. Third, themycobacteria produce anti-inflammatory agentslike man-LAM and sulfolipid [80, 110]. Finally, aswill be discussed later, cord factor is non-toxic untilits toxicity is activated by interaction with appro-priate lipids. It seems reasonable to hypothesize,therefore, that the ability to cause sudden onset ofcaseous pneumonia depends both on the toxicity ofthe hypersensitivity reactions and the ability to hidethe antigens until sufficient quantities are availableto produce a cavity able to mediate transmission ofinfection to new hosts.
Cord factor as the trigger for caseous pneumonia:In the 1950’s, Middlebrook identified two proper-ties of virulent MTB that distinguished them fromavirulent mycobacteria [111]. The first was forma-tion of elongated aggregates of organisms, calledserpentine cords. The second was staining withneutral red. Cord formation is due to cord factor or
Pathogenesis of Post Primary Tuberculosis 381

trehalose 6’6 dimycolate (TDM) [112,113].Neutral red staining is largely due to sulfolipid thatis an inhibitor of the toxicity of cord factor[110,114,115]. It is probably more than coinci-dence that two long recognized characteristics ofvirulent MTB, cord formation and neutral redstaining relate to TDM and its sulfolipid inhibitor.
TDM is the leading candidate for triggering sud-den onset of caseous necrosis because its potentability to stimulate release of TNF from macro-phages depends on its physical conformation.While it is found in all mycobacteria, the quantitiesexcreted, chemical structure, toxicity and antigenic-ity of that produced by MTB are unique. TDM isthe only chemically defined substance ever to beshown to produce caseation necrosis [18]. It is themost abundant and by orders of magnitude mostthe toxic lipid of MTB [116]. While MTB containsmany lipids, TDM is unique in being free on thebacterial surface in large quantity. Extraction of lip-ids from the surface of viable virulent MTB yieldsnearly pure TDM [116,117]. Removal of TDMfrom the surface of MTB reduces virulence formice. Adding purified TDM back restores the viru-lence [118]. TDM generated controversy immedi-ately after its discovery because its toxic effects wereonly demonstrable when it was injected with oilthat was considered ‘unphysiologic’ [117,119].More recent studies have suggested that rather thanbeing unphysiologic, interactions of TDM withlipids are central to the pathogenesis of TB [90].
The most striking feature of TDM is that it hasmultiple distinct sets of biologic activities that de-pend upon its physical configuration. TDM is solarge and insoluble that it remains as a thick coat onthe organisms. As such, it prevents phagosome lyso-some fusion, prevents acidification of phagosomes,protects organisms from killing by macrophagesand impedes antigen presentation [120,121]. In sa-line, TDM is nontoxic; the LD50 for mice is great-er than 50,000 µg, the highest dose ever injected.TDM removed from MTB and placed as a singlemolecular monolayer on the surface of lipid drop-lets has a completely different physical structureand set of biologic activities [122,123]. It spontane-ously forms a two dimensional crystalline mono-layer on oil-water interfaces that is the most rigidlipid monolayer known [122-125]. The monolayerspontaneously forms on any interface of water with
a hydrophobic surface. When virulent MTB con-tact lipid droplets, they instantly enter the dropletshedding the TDM that assumes the monolayerconfiguration on the water-lipid interface. Themonolayer of TDM is stable with a half life of 4days as a single molecular monolayer in mice[123,124]. It is highly toxic with an LD50 of 30 µgand produces foreign body granulomas in naïvemice and hypersensitive granulomas in immunizedmice [19,126,127]. The hypersensitivity reactionsare dependent on CD1d and CD4+ T cells [19].Injections of 10 µg of TDM emulsion every otherday induce hemorrhagic pneumonitis, cachexia anddeath of C57BL1/6 mice [117]. Much larger doses,50-100 µg are rarely lethal.
The key property of TDM that makes it a candidatetrigger for caseous pneumonia is its ability tochange configuration to suddenly stimulate releaseof large amounts of TNF. Geisel et al demonstratedthat the ability of TDM to induce release of TNFfrom macrophages increases with particle size[128]. TDM on the surface of 90 micron diameterbeads induced release over 2600 pg/ml TNF whilethe same amount on the same surface area of 1 mi-cron beads induces less than 100 pg/ml. In retro-spect, similar observations had been made earlier.Bloch demonstrated that injection of TDM in oil isfar more toxic than injection of the same materialsas an oil-in -water emulsion [112,116]. Yarkonidemonstrated that oil emulsions of TDM withlarge particle size were far more effective as cancerimmunotherapeutic agents than those with thesame qualities in small droplets [129]. We demon-strated that small beads coated with TDM are in-gested by macrophages and induced non-allergicgranulomas. TDM on a plate (very large bead), incontrast, causes macrophages to adhere, spread andlyse within minutes [130].
The hypothesis is that post primary TB isolates asection of lung to facilitate the buildup of host lip-ids and mycobacterial products to generate condi-tions for a hypersensitive reaction that producescaseation pneumonia and cavitation [107,131].Lipid progressively accumulates with increasingdroplet size in post obstructive pneumonia. Thetrigger for initiating caseous pneumonia may be anincrease in the size of lipid droplets coated withTDM to the point that they activate release of largeamounts of TNF that drives a positive feedback for
Annals of Clinical & Laboratory Science, vol. 44, no. 4, 2014382

the rest of the lesions. In support of this, we haveobserved that injection of a TDM emulsion intomice with slowly progressive pulmonary TB induc-es production of larger lipid droplets, greater in-flammation, necrosis and proliferation oforganisms.
Animal Experimental Models: Most studies usinganimal models of TB have focused on understand-ing the early stages of granuloma formation. A ma-jor gap is a failure to address later stages leading totransmission [132]. This gap cannot be addressedwithout a better understanding of pathogenesis.Our observations suggest that most, if not all, ani-mals develop components of both primary and postprimary TB. For example, the late stages of TB inrabbits, mice and guinea pigs are all similar to hu-man TB in that disease is not driven by increasingnumbers of MTB, but by a host response to myco-bacterial products [104]. Humans, however, arethe only species in which the lesions progress toproduce cavities that mediate transmission of infec-tion to new hosts. Most current literature assumesthat caseating granulomas are the characteristic le-sion of both primary and post primary TB.
A central point of this paper is that granulomas arecharacteristic of primary TB, but are not involvedin developing post primary disease. The conceptthat the TB in guinea pigs is more human like thanthat in mice because it develops large caseatinggranulomas is incorrect. Large caseating granulo-mas are characteristic only of progressive primaryTB in infants. Mice are regularly criticized becausethey fail to produce caseating granulomas when, infact, slowly progressive TB in mice is a rather goodmodel of parts of the early stages of post primaryTB. In addition, many reviewers and funding agen-cies insist that animals be infected by low dose aero-sol infection to reproduce the conditions of humaninfection. This reproduces only the first few weeksof infection and has little or no relevance for postprimary TB that occurs decades later. As exempli-fied by our studies with caseating granulomas inmice, one can develop protocols to reproduce inanimals the conditions and lesions that exist in hu-mans at different stages of infection [18]. A recentpublication used this approach to produce cavitarylesions in rabbits [133]. While there is no completeanimal model of post primary TB, we believe that
many models could yield valuable insights into par-ticular aspects the disease. The challenge is to de-fine models that address relevant aspects of the hu-man disease.
Vaccine candidates: Many investigators believethat a vaccine that acts against pulmonary TB is theonly hope of eventually eradicating MTB [8]. Thecurrent vaccine, BCG, provides a degree of protec-tion against disseminated TB, saving the lives of ap-proximately 50 thousand children every year.However, it has no measurable impact on post pri-mary TB or transmission of infection. The funda-mental problem is that one can’t rationally developan effective vaccine for post primary TB withoutunderstanding protective immunity [134].
Vaccine development projects typically focus on re-ducing the numbers of MTB in tissue and prolong-ing the life of animals. This is appropriate for pri-mary TB, but not for post primary disease becausethe disease has very few MTB during developmen-tal stages and needs none to produce disease. Therehas long been realization that the immune responseis responsible for tissue damage as well as control-ling proliferation of MTB. Targeting latent MTBor those active in early post primary disease couldbe effective. However, current, animal models ofprotection from primary TB are not helpful. Theyonly serve to try to induce animals to produce thetype of response that most humans develop sponta-neously in weeks. The challenge for preventing postprimary TB is to find ways to prevent MTB fromco-opting and using our strongest response for itsadvantage. This may be possible since progressionof developing post primary TB is a rare event. Atleast 95% of early infections regress spontaneously.The people who develop disease do not have weakeror less effective immune responses. MTB uses andsubverts immunity to produce an isolated area oflung in which it can slowly accumulate host lipidsand mycobacterial antigens necessary to produce anecrotizing hypersensitivity reaction. A vaccine thatblocks any one of multiple factors required for thisprocess could produce regression.
The Streetlight Effect states that one can only ef-fectively study things that can be seen [94]. Thecapabilities of modern science have increased phe-nomenally since tissues with post primary TB
Pathogenesis of Post Primary Tuberculosis 383

ceased to be readily obtainable due to introductionof antibiotics and declining autopsy rates. This pa-per presents a synthesis of old and new investiga-tions to produce a revised conception of the patho-genesis of post primary TB. Progression of infectionfrom implantation of MTB in a lung to clinicaldisease is a rare event because most infections re-solve spontaneously. Examination of the processsuggests many places where it might be interruptedthereby preventing clinical disease and transmissionof infection to new hosts. We anticipate that thiswill move the streetlight to facilitate use of moderntechnologies to rapidly address previously refracto-ry questions of the world’s most lethal bacterialpathogen.
Acknowledgements
Supported in part by USPHS grants HL 68537, AI78420,AI49534, HL080313.
References
1. World_Health_Organization. Global tuberculosis report2013. 2013.
2. Russell, D. G., C. E. Barry, 3rd, and J. L. Flynn. Tuberculosis:what we don’t know can, and does, hurt us. Science 2010. 328:852-856.
3. Hunter, R. L. Pathology of post primary tuberculosis of thelung: An illustrated critical review. Tuberculosis (Edinb) Epublication ahead of print July 4, 2011.
4. Russell, D. G. Who puts the tubercle in tuberculosis? Nat RevMicrobiol 2007. 5: 39-47.
5. den Boon, S., S. W. van Lill, M. W. Borgdorff, D. A. Enarson,S. Verver, E. D. Bateman, E. Irusen, C. J. Lombard, N. W.White, C. de Villiers, and N. Beyers. High prevalence of tuber-culosis in previously treated patients, Cape Town, SouthAfrica. Emerg Infect Dis 2007. 13: 1189-1194.
6. van Rie, A., T. C. Victor, M. Richardson, R. Johnson, G. D.van der Spuy, E. J. Murray, N. Beyers, N. C. Gey van Pittius,P. D. van Helden, and R. M. Warren. Reinfection and MixedInfection Cause Changing Mycobacterium tuberculosis Drug-resistance Patterns. Am J Respir Crit Care Med 2005.
7. Comas, I., J. Chakravartti, P. M. Small, J. Galagan, S.Niemann, K. Kremer, J. D. Ernst, and S. Gagneux. Human Tcell epitopes of Mycobacterium tuberculosis are evolutionarilyhyperconserved. Nature genetics 2010. 42: 498-503.
8. Brennan, M. J., and J. Thole. Tuberculosis vaccines: a strategicblueprint for the next decade. Tuberculosis (Edinb) 2012. 92Suppl 1: S6-13.
9. Laennec, R. A treatise on diseases of the chest in which they aredescribed according to their anatomical characters, and theirdiagnosis established on a new principle by means of acoustickinstruments. 1821 T&G Underwood, London (reprinted 1979by The Classics of Medicine Library. Birmingham AL).
10. Rich, A. The Pathogenesis of Tuberculosis, Second Edition.1951 Charles C Thomas, Springfield, Illinois.
11. Canetti, G. The tubercle bacillus in the pulmonary lesion ofman. Histobacteriology and its bearing on the therapy of pul-monary tuberculosis. 1955 Springer Publishing Compani Inc.,New Yoyk.
12. Medlar, E. M. The behavior of pulmonary tuberculous lesions;a pathological study. Am Rev Tuberc 1955. 71: 1-244.
13. Levine, E. R. Chapter 7, Classification of reinfection pulmo-nary tuberculosis. In The Fundamentals of PulmonaryTuberculosis and its Complications for Students, Teachers andPracticing Physicians. E. Hayes, ed. Charles C Thomas,Springfield. 1949. 97-113.
14. Pagel, W., F. Simmonds, N. MacDonald, and E. Nassau.Chapter 3. The Morbid Anatomy and Histology of Tuberculosis,An Introduction in Simple Terms. In In PulmonaryTuberculosis, Bacteriology, Pathology, Management,Epidemiology and Prevention. Oxford University Press,London. 1964. 36-63.
15. Dannenberg, A. M., Jr. Pathogenisis of human pulmonary tu-berculosis. Insights from the rabbit model. 2006 ASM press,Washington, DC.
16. Creighton, C. Bovine Tubercuosis in Man: An account of thePathology of Suspected Cases. 1881 MacMillan and Co.,London.
17. Liebana, E., L. Johnson, J. Gough, P. Durr, K. Jahans, R.Clifton-Hadley, Y. Spencer, R. G. Hewinson, and S. H.Downs. Pathology of naturally occurring bovine tuberculosisin England and Wales. Vet J 2008. 176: 354-360.
18. Hunter, R. L., M. Olsen, C. Jagannath, and J. K. Actor.Trehalose 6,6’-dimycolate and lipid in the pathogenesis of case-ating granulomas of tuberculosis in mice. Am J Pathol 2006.168: 1249-1261.
19. Guidry, T. V., R. L. Hunter, Jr., and J. K. Actor. Mycobacterialglycolipid trehalose 6,6’-dimycolate-induced hypersensitivegranulomas: contribution of CD4+ lymphocytes. Microbiology2007. 153: 3360-3369.
20. Guidry, T. V., M. Olsen, K. S. Kil, R. L. Hunter, Jr., Y. J. Geng,and J. K. Actor. Failure of CD1D-/- mice to elicit hypersensi-tive granulomas to mycobacterial cord factor trehalose 6,6’-di-mycolate. J Interferon Cytokine Res 2004. 24: 362-371.
21. Hunter, R. L., C. Jagannath, and J. K. Actor. Pathology ofpostprimary tuberculosis in humans and mice: contradictionof long-held beliefs. Tuberculosis (Edinb) 2007. 87: 267-278.
22. Ehlers, S. Lazy, dynamic or minimally recrudescent? On theelusive nature and location of the mycobacterium responsiblefor latent tuberculosis. Infection 2009. 37: 87-95.
23. Lin, P. L., C. B. Ford, M. T. Coleman, A. J. Myers, R.Gawande, T. Ioerger, J. Sacchettini, S. M. Fortune, and J. L.Flynn. Sterilization of granulomas is common in active and la-tent tuberculosis despite within-host variability in bacterialkilling. Nat Med 2014. 20: 75-79.
24. Neyrolles, O., R. Hernandez-Pando, F. Pietri-Rouxel, P.Fornes, L. Tailleux, J. A. Barrios Payan, E. Pivert, Y. Bordat, D.Aguilar, M. C. Prevost, C. Petit, and B. Gicquel. Is adiposetissue a place for Mycobacterium tuberculosis persistence? PloSone 2006. 1: e43.
25. Agarwal, P., S. R. Khan, S. C. Verma, M. Beg, K. Singh, K.Mitra, A. N. Gaikwad, M. S. Akhtar, and M. Y. Krishnan.Mycobacterium tuberculosis persistence in various adipose de-pots of infected mice and the effect of anti-tubercular therapy.Microbes Infect 2014. 16: 571-580.
26. Hadda, V., and G. C. Khilnani. Lipoid pneumonia: an over-view. Expert review of respiratory medicine 2010. 4: 799-807.
27. Osler, W. Chapter 26, Tuberculosis. In The principles andpractice of medicine. D. Appleton and Company, New York.1892. 184-255.
28. Dubos, R., and G. Dubos. The White Plague. Tuberculosis,Man, and Society. 1987 Rutgers Univesity Press, Brunswick,NJ.
29. Erokhin, V. V., V. Y. Mishin, V. I. Chukanov, and D. V.Guiller. Caseous pneumonia, Pathologic anatomy, pathogene-sis, diagnosis, clinical course and treatment, A manual for prac-titioners 2008 Meditsina Publishers, Moscow.
30. Goodwin, R. A., and R. M. Des Prez. Apical localization ofpulmonary tuberculosis, chronic pulmonary histoplasmosis,and progressive massive fibrosis of the lung. Chest 1983. 83:801-805.
Annals of Clinical & Laboratory Science, vol. 44, no. 4, 2014384

31. Pai, M., A. Mohan, K. Dheda, C. C. Leung, W. W. Yew, D. J.Christopher, and S. K. Sharma. Lethal interaction: the collid-ing epidemics of tobacco and tuberculosis. Expert review ofanti-infective therapy 2007. 5: 385-391.
32. Murray, J. F. Bill Dock and the location of pulmonary tubercu-losis: how bed rest might have helped consumption. Am JRespir Crit Care Med 2003. 168: 1029-1033.
33. Broch, B. L. Chapter 14, The role of the bronchial tree in thepathogenesis of pulmonary tuberculosis. In The Fundamentalsof Pulmonary Tuberculosis and its Complications for Students,Teachers and Practicing Physicians. E. Hayes, ed. Charles CThomas, Springfield. 1949. 245-253.
34. Ehrhardt, J., R. Werner, A. Schmidt-Richberg, and H.Handels. Statistical modeling of 4D respiratory lung motionusing diffeomorphic image registration. IEEE transactions onmedical imaging 2011. 30: 251-265.
35. Du, K., J. E. Bayouth, K. Cao, G. E. Christensen, K. Ding, andJ. M. Reinhardt. Reproducibility of registration-based mea-sures of lung tissue expansion. Medical physics 2012. 39:1595-1608.
36. Lam, C. W., J. T. James, R. McCluskey, and R. L. Hunter.Pulmonary toxicity of single-wall carbon nanotubes in mice 7and 90 days after intratracheal instillation. Toxicol Sci 2004.77: 126-134.
37. O’Garra, A., P. S. Redford, F. W. McNab, C. I. Bloom, R. J.Wilkinson, and M. P. Berry. The immune response in tubercu-losis. Annu Rev Immunol 2013. 31: 475-527.
38. Korf, J., A. Stoltz, J. Verschoor, P. De Baetselier, and J. Grooten.The Mycobacterium tuberculosis cell wall component mycolicacid elicits pathogen-associated host innate immune responses.Eur J Immunol 2005. 35: 890-900.
39. Peyron, P., J. Vaubourgeix, Y. Poquet, F. Levillain, C. Botanch,F. Bardou, M. Daffe, J. F. Emile, B. Marchou, P. J. Cardona, C.de Chastellier, and F. Altare. Foamy macrophages from tuber-culous patients’ granulomas constitute a nutrient-rich reservoirfor M. tuberculosis persistence. PLoS Pathog 2008. 4:e1000204.
40. Korf, J. E., G. Pynaert, K. Tournoy, T. Boonefaes, A. VanOosterhout, D. Ginneberge, A. Haegeman, J. A. Verschoor, P.De Baetselier, and J. Grooten. Macrophage reprogramming bymycolic acid promotes a tolerogenic response in experimentalasthma. Am J Respir Crit Care Med 2006. 174: 152-160.
41. Melo, R. C., and A. M. Dvorak. Lipid body-phagosome inter-action in macrophages during infectious diseases: host defenseor pathogen survival strategy? PLoS Pathog 2012. 8: e1002729.
42. North, R. J., and Y. J. Jung. Immunity to tuberculosis. AnnuRev Immunol 2004. 22: 599-623.
43. Nuermberger, E. L., T. Yoshimatsu, S. Tyagi, W. R. Bishai, andJ. H. Grosset. Paucibacillary tuberculosis in mice after prioraerosol immunization with Mycobacterium bovis BCG. InfectImmun 2004. 72: 1065-1071.
44. Kondo, A., N. Oketani, M. Maruyama, Y. Taguchi, Y.Yamaguchi, H. Miyao, I. Mashima, M. Oono, K. Wada, T.Tsuchiya, H. Takahashi, and S. Abe. [Significance of serumsurfactant protein-D (SP-D) level in patients with pulmonarytuberculosis]. Kekkaku 1998. 73: 585-590.
45. Chroneos, Z. C., K. Midde, Z. Sever-Chroneos, and C.Jagannath. Pulmonary surfactant and tuberculosis.Tuberculosis (Edinb) 2009. 89 Suppl 1: S10-14.
46. Wang, Z., U. Schwab, E. Rhoades, P. R. Chess, D. G. Russell,and R. H. Notter. Peripheral cell wall lipids of Mycobacteriumtuberculosis are inhibitory to surfactant function. Tuberculosis(Edinb) 2008. 88: 178-186.
47. Lovacheva, O. V., V. V. Erokhin, N. V. Chernichenko, G. V.Evgushchenko, L. N. Lepekha, and O. A. Rozenberg. [Resultsof use of surfactant in complex therapy of patients with de-structive pulmonary tuberculosis]. Probl Tuberk Bolezn Legk2006. 12-17.
48. Hektoen, L., and D. Reisman. A Text-Book of Pathology forthe use of students and practitoners of medicine and surgery.1901 W.B. Saunders & Company, London and Philadelphia.
49. Coats, J., and L. K. Sutherland. Manual Of Pathology, Chapter5. Healing Of Phthisis. 1900 Longmans, Green, And Co,London.
50. Im, J. G., H. Itoh, K. S. Lee, and M. C. Han. CT-pathologycorrelation of pulmonary tuberculosis. Crit Rev Diagn Imaging1995. 36: 227-285.
51. Lee, J. Y., K. S. Lee, K. J. Jung, J. Han, O. J. Kwon, J. Kim, andT. S. Kim. Pulmonary tuberculosis: CT and pathologic correla-tion. J Comput Assist Tomogr 2000. 24: 691-698.
52. Kondo, S., T. Miyagawa, and M. Ito. [Reevaluation of patho-genesis of epituberculosis in infants and children with tubercu-losis]. Kekkaku 2007. 82: 569-575.
53. Fish, R., and W. Pagel. The morbid anatomy of epituberculosis.J Path Bact 1938. 47: 593-601.
54. Amita, R., S. Sandhyamani, and M. Unnikrishnan. Extensiveunilateral pulmonary tuberculosis with segmental atresia ofprincipal bronchus. Lung India : official organ of Indian ChestSociety 2014. 31: 94-95.
55. Ahonen, A., and K. Valavirta. Ultrastructure of cilia in pulmo-nary tuberculosis. European journal of respiratory diseases.Supplement 1983. 128 (Pt 2): 460-463.
56. Reed, G. W., H. Choi, S. Y. Lee, M. Lee, Y. Kim, H. Park, J.Lee, X. Zhan, H. Kang, S. Hwang, M. Carroll, Y. Cai, S. N.Cho, C. E. Barry, 3rd, L. E. Via, and H. Kornfeld. Impact ofdiabetes and smoking on mortality in tuberculosis. PloS one2013. 8: e58044.
57. Davies, P. D., W. W. Yew, D. Ganguly, A. L. Davidow, L. B.Reichman, K. Dheda, and G. A. Rook. Smoking and tubercu-losis: the epidemiological association and immunopathogene-sis. Trans R Soc Trop Med Hyg 2006. 100: 291-298.
58. Farver, C., A. E. Fraire, and J. F. Tomashefski. Dail andHammar’s Pulmonary Pathology, Volume 1: NonneoplasticLung Disease 1998 Springer Verlag New York
59. Pagel W, and P. W. Zur Histochemie der Lungentuberkulose,mit besonderer Berucksichtung der Fettsubstanzen undLipoide. (Fat and lipoid content to tuberculous tissue.Histochemical investigation.). . Virchows Arch Pathol Anat1925. 256: 629-640.
60. Johnson, J. L., and J. J. Ellner. Chapter 57. Cavitary PulmonaryDisease. In Clinical Infectious Diseases: A Practical Approach.R. K. Root, ed. Oxford University Press, New Hork. 1999.539-549.
61. Akinosoglou, K. S., K. Karkoulias, and M. Marangos.Infectious complications in patients with lung cancer.European review for medical and pharmacological sciences2013. 17: 8-18.
62. Hunter, R. L. On the pathogenesis of post primary tuberculo-sis: the role of bronchial obstruction in the pathogenesis ofcavities. Tuberculosis (Edinb) 2011. 91 Suppl 1: S6-10.
63. Pattnaik, M. K., P. C. Majhi, A. K. Nayak, and D. Senapati. Arare presentation of a huge mature mediastinal teratoma withright lung cavitation. BMJ case reports 2014. 2014.
64. Humphrey, D. M., and M. H. Weiner. Mycobacterial antigendetection by immunohistochemistry in pulmonary tuberculo-sis. Hum Pathol 1987. 18: 701-708.
65. Ridley, M. J., and D. S. Ridley. Histochemical demonstrationof mycobacterial antigen, specific antibody and complement inthe lesions of tuberculosis. The Histochemical journal 1986.18: 551-556.
66. Pedersen, J. S., I. Clarke, and J. Mills. Improved detection ofmycobacteria species in formalin-fixed tissue sections.Histopathology 2011. 59: 993-1005.
67. Guerardel, Y., E. Maes, V. Briken, F. Chirat, Y. Leroy, C.Locht, G. Strecker, and L. Kremer. Lipomannan and lipoarabi-nomannan from a clinical isolate of Mycobacterium kansasii:
Pathogenesis of Post Primary Tuberculosis 385

novel structural features and apoptosis-inducing properties. JBiol Chem 2003. 278: 36637-36651.
68. Chan, J., X. D. Fan, S. W. Hunter, P. J. Brennan, and B. R.Bloom. Lipoarabinomannan, a possible virulence factor in-volved in persistence of Mycobacterium tuberculosis withinmacrophages. Infect Immun 1991. 59: 1755-1761.
69. Talaat, A. M., S. K. Ward, C. W. Wu, E. Rondon, C. Tavano,J. P. Bannantine, R. Lyons, and S. A. Johnston. Mycobacterialbacilli are metabolically active during chronic tuberculosis inmurine lungs: insights from genome-wide transcriptional pro-filing. J Bacteriol 2007. 189: 4265-4274.
70. Rogerson, B. J., Y. J. Jung, R. LaCourse, L. Ryan, N. Enright,and R. J. North. Expression levels of Mycobacterium tubercu-losis antigen-encoding genes versus production levels of anti-gen-specific T cells during stationary level lung infection inmice. Immunology 2006. 118: 195-201.
71. Bhalla, A., M. Mahapatra, R. Singh, and S. D’Cruz. Acutelung injury in miliary tuberculosis. Indian Journal ofTuberculosis 2002. 49: 125-128.
72. Lincoln, E. M., and W. Grethmann. The potetial dangers oftuberculin tests. J Pediatrics 1939. 15: 682-696.
73. Massaro, D., and S. Katz. Rapid Clearing in HematogenousPulmonary Tuberculosis. Archives of internal medicine 1964.113: 573-577.
74. Trapnell, B. C., B. C. Carey, K. Uchida, and T. Suzuki.Pulmonary alveolar proteinosis, a primary immunodeficiencyof impaired GM-CSF stimulation of macrophages. Currentopinion in immunology 2009. 21: 514-521.
75. Carey, B., and B. C. Trapnell. The molecular basis of pulmo-nary alveolar proteinosis. Clin Immunol 2010. 135: 223-235.
76. Ramirez, J. Pulmonary alveolar proteinosis. Treatment in acase complicated by tuberculosis. Am Rev Respir Dis 1967. 95:491-495.
77. Huang, X. Y., C. Yu, X. M. Xu, W. T. Yang, X. Zhang, P. P.Liu, and L. X. Wang. Pulmonary alveolar proteinosis associat-ed with tuberculosis and aspergilloma formation. Chinesemedical journal 2012. 125: 3191-3192.
78. Betancourt, S. L., S. Martinez-Jimenez, S. E. Rossi, M. T.Truong, J. Carrillo, and J. J. Erasmus. Lipoid pneumonia: spec-trum of clinical and radiologic manifestations. AJR Am JRoentgenol 2010. 194: 103-109.
79. Sulkowska, M., and S. Sulkowski. Endogenous lipid pneumo-nia- and alveolar proteinosis-type changes in the vicinity ofnon-small cell lung cancer: histiopathologic, immunohisto-chemical, and ultrastructural evaluation. Ultrastructural pa-thology 1998. 22: 109-119.
80. Mustafa, T., S. Phyu, R. Nilsen, R. Jonsson, and G. Bjune. Amouse model for slowly progressive primary tuberculosis.Scand J Immunol 1999. 50: 127-136.
81. Chapman, R. C., and S. M. Claydon. Mycobacterium tubercu-losis: a continuing cause of sudden and unexpected death inwest London. Journal of clinical pathology 1992. 45:713-715.
82. Alzeer, A., A. Mashlah, N. Fakim, N. Al-Sugair, M. Al-Hedaithy, S. Al-Majed, and G. Jamjoom. Tuberculosis is thecommonest cause of pneumonia requiring hospitalization dur-ing Hajj (pilgrimage to Makkah). J Infect 1998. 36: 303-306.
83. Gonzalez Saldana, N., M. Macias Parra, M. Hernandez Porras,P. Gutierrez Castrellon, V. Gomez Toscano, and H. JuarezOlguin. Pulmonary Tuberculous: Symptoms, diagnosis andtreatment. 19-year experience in a third level pediatric hospital.BMC infectious diseases 2014. 14: 401.
84. Eldour, A. A. A., M. Elfatih, R. A. A. Salih, and H. G. Ahmed.Blood Coagulation Changes among Sudanese Patients withPulmonary Tuberculosis. International Journal of Science andResearch 2014. 3: 176-719.
85. Birnbaum, M. Prof. Koch’s Method to Cure TuberculosisPopularly Treated. In PROJECT GUTENBERG EBOOKMETHOD TO CURE TUBERCULOSIS. http://www.gutenberg.net EBook #27181. 2008.
86. Moreira, A. L., L. Tsenova, M. H. Aman, L. G. Bekker, S.Freeman, B. Mangaliso, U. Schroder, J. Jagirdar, W. N. Rom,M. G. Tovey, V. H. Freedman, and G. Kaplan. Mycobacterialantigens exacerbate disease manifestations in Mycobacteriumtuberculosis-infected mice. Infect Immun 2002. 70:2100-2107.
87. Medlar, E. M. The pathogenesis of minimal pulmonary tuber-culosis; a study of 1,225 necropsies in cases of sudden and un-expected death. Am Rev Tuberc 1948. 58: 583-611.
88. Wayne, L. G., and C. D. Sohaskey. Nonreplicating persistenceof mycobacterium tuberculosis. Annu Rev Microbiol 2001. 55:139-163.
89. Hurford, J. V., and W. H. Valentine. Viable tubercle bacilli inclosed lesions. Tubercle 1957. 38: 194-198.
90. Hunter, R. L., M. R. Olsen, C. Jagannath, and J. K. Actor.Multiple roles of cord factor in the pathogenesis of primary,secondary, and cavitary tuberculosis, including a revised de-scription of the pathology of secondary disease. Ann Clin LabSci 2006. 36: 371-386.
91. Rook, G. A., K. Dheda, and A. Zumla. Immune responses totuberculosis in developing countries: implications for new vac-cines. Nat Rev Immunol 2005. 5: 661-667.
92. Dheda, K., H. Booth, J. F. Huggett, M. A. Johnson, A. Zumla,and G. A. Rook. Lung remodeling in pulmonary tuberculosis.J Infect Dis 2005. 192: 1201-1209.
93. Chapel, H. M., and P. J. August. Report of nine cases of acci-dental injury to man with Freund’s complete adjuvant. ClinExp Immunol 1976. 24: 538-541.
94. Freedman, D. H. Why Scientific Studies Are So Often Wrong:The Streetlight Effect. Discover 2010. July-August.
95. Hatipoglu, O. N., E. Osma, M. Manisali, E. S. Ucan, P. Balci,A. Akkoclu, O. Akpinar, C. Karlikaya, and C. Yuksel. Highresolution computed tomographic findings in pulmonary tu-berculosis. Thorax 1996. 51: 397-402.
96. Collins, J., D. Blankenbaker, and E. J. Stern. CT patterns ofbronchiolar disease: what is “tree-in-bud”? AJR Am JRoentgenol 1998. 171: 365-370.
97. Karimi, S., M. Shamaei, M. Pourabdollah, M. Sadr, M.Karbasi, A. Kiani, and M. Bahadori. Histopathological find-ings in immunohistological staining of the granulomatous tis-sue reaction associated with tuberculosis. Tuberculosis researchand treatment 2014. 2014: 858396.
98. Corbett, E. L., A. Zezai, Y. B. Cheung, T. Bandason, E. Dauya,S. S. Munyati, A. E. Butterworth, S. Rusikaniko, G. J.Churchyard, S. Mungofa, R. J. Hayes, and P. R. Mason.Provider-initiated symptom screening for tuberculosis inZimbabwe: diagnostic value and the effect of HIV status. BullWorld Health Organ 2010. 88: 13-21.
99. van Rie, A., R. Warren, M. Richardson, T. C. Victor, R. P. Gie,D. A. Enarson, N. Beyers, and P. D. van Helden. Exogenousreinfection as a cause of recurrent tuberculosis after curativetreatment. N Engl J Med 1999. 341: 1174-1179.
100. Huaman, M. A., C. T. Fiske, T. F. Jones, J. Warkentin, B. E.Shepherd, L. A. Ingram, F. Maruri, and T. R. Sterling.Tuberculosis and the risk of infection with other intracellularbacteria: a population-based study. Epidemiology and infec-tion 2014. 1-9.
101. Ledru, E., S. Ledru, and A. Zoubga. Granuloma formation andtuberculosis transmission in HIV-infected patients. ImmunolToday 1999. 20: 336-337.
102. Kenyon, T. A., T. Creek, K. Laserson, M. Makhoa, N.Chimidza, M. Mwasekaga, J. Tappero, S. Lockman, T. Moeti,
Annals of Clinical & Laboratory Science, vol. 44, no. 4, 2014386

and N. Binkin. Risk factors for transmission of Mycobacteriumtuberculosis from HIV-infected tuberculosis patients,Botswana. Int J Tuberc Lung Dis 2002. 6: 843-850.
103. Wang, C. S., H. C. Chen, C. J. Yang, W. Y. Wang, I. W.Chong, J. J. Hwang, and M. S. Huang. The impact of age onthe demographic, clinical, radiographic characteristics andtreatment outcomes of pulmonary tuberculosis patients inTaiwan. Infection 2008. 36: 335-340.
104. Dannenberg, A. M., Jr., and F. M. Collins. Progressive pulmo-nary tuberculosis is not due to increasing numbers of viablebacilli in rabbits, mice and guinea pigs, but is due to a continu-ous host response to mycobacterial products. Tuberculosis(Edinb) 2001. 81: 229-242.
105. McGrath, J. W. Social networks of disease spread in the lowerIllinois valley: a simulation approach. Am J Phys Anthropol1988. 77: 483-496.
106. Musser, J. M., A. Amin, and S. Ramaswamy. Negligible geneticdiversity of mycobacterium tuberculosis host immune systemprotein targets: evidence of limited selective pressure. Genetics2000. 155: 7-16.
107. Rook, G. A., and R. Hernandez-Pando. The pathogenesis oftuberculosis. Annu Rev Microbiol 1996. 50: 259-284.
108. Fujita, Y., Y. Okamoto, Y. Uenishi, M. Sunagawa, T. Uchiyama,and I. Yano. Molecular and supra-molecular structure relateddifferences in toxicity and granulomatogenic activity of myco-bacterial cord factor in mice. Microb Pathog 2007. 43: 10-21.
109. McMullen, A. M., S. A. Hwang, K. O’Shea, M. L. Aliru, andJ. K. Actor. Evidence for a unique species-specific hypersensi-tive epitope in Mycobacterium tuberculosis derived cord factor.Tuberculosis (Edinb) 2013. 93 Suppl: S88-93.
110. Okamoto, Y., Y. Fujita, T. Naka, M. Hirai, I. Tomiyasu, and I.Yano. Mycobacterial sulfolipid shows a virulence by inhibitingcord factor induced granuloma formation and TNF-alpha re-lease. Microb Pathog 2006. 40: 245-253.
111. Middlebrook, G., R. G. Dubos, and C. Pierce. Virulence andmorphological characteristics of mammalian tubercle bacilli. J.Exp. Med. 1947. 86: 175-184.
112. Bloch, H. Studies on the virulence of tubercle bacilli: Isolationand biological properties of a constituent of virulent organisms.J. Exp. Med. 1950. 91: 197-219.
113. Hunter, R. L., N. Venkataprasad, and M. R. Olsen. The role oftrehalose dimycolate (cord factor) on morphology of virulentM. tuberculosis in vitro. Tuberculosis (Edinb) 2006. 86:349-356.
114. Goren, M., O. Brokl, B. Das, and E. Lederer. Sulfolipid I ofMycobacterium tuberculosis, strain H37Rv. Nature of the acylsubstituents. Biochemistry 1971. 10: 72-81.
115. Andreu, N., I. Gibert, M. Luquin, P. E. Kolattukudy, and T.Sirakova. Neutral red staining of cells of a sulfolipid-deficientMycobacterium tuberculosis pks2 mutant proves that sulfolip-ids are not responsible for this cytochemical reaction. J ClinMicrobiol 2004. 42: 1379-1380.
116. Goren, M. Cord Factor Revisited: A tribute to the late HubertBloch. Tubercle 1975. 56: 65-71.
117. Goren, M. B., and P. J. Brennan. Chapter 4. Mycobacteriallipids: chemistry and biologic activities. In Tuberculosis. G. P.Youmans, ed. W.B. Saunders, Philadelphia, PA. 1979. 63-193.
118. Indrigo, J., R. L. Hunter, Jr., and J. K. Actor. Influence of tre-halose 6,6’-dimycolate (TDM) during mycobacterial infectionof bone marrow macrophages. Microbiology 2002. 148:1991-1998.
119. Spitznagel, J. K., and R. J. Dubos. A fraction of tubercle bacillipossessing primary toxicity. J Exp Med 1955. 101: 291-311.
120. Indrigo, J., R. L. Hunter, Jr., and J. K. Actor. Cord factor tre-halose 6,6’-dimycolate (TDM) mediates trafficking events
during mycobacterial infection of murine macrophages.Microbiology 2003. 149: 2049-2059.
121. Kan-Sutton, C., C. Jagannath, and R. L. Hunter, Jr. Trehalose6,6’-dimycolate on the surface of Mycobacterium tuberculosismodulates surface marker expression for antigen presentationand costimulation in murine macrophages. Microbes Infect2009. 11: 40-48.
122. Behling, C. A., B. Bennett, K. Takayama, and R. L. Hunter.Development of a trehalose 6,6’-dimycolate model which ex-plains cord formation by Mycobacterium tuberculosis. InfectImmun 1993. 61: 2296-2303.
123. Retzinger, G. S., S. C. Meredith, R. L. Hunter, K. Takayama,and F. J. Kezdy. Identification of the physiologically active stateof the mycobacterial glycolipid trehalose 6,6’-dimycolate andthe role of fibrinogen in the biologic activities of trehalose6,6’-dimycolate monolayers. J Immunol 1982. 129: 735-744.
124. Retzinger, G. S., S. C. Meredith, K. Takayama, R. L. Hunter,and F. J. Kezdy. The role of surface in the biological activities oftrehalose 6,6’- dimycolate. Surface properties and developmentof a model system. J Biol Chem 1981. 256: 8208-8216.
125. Schabbing,R.W.,A.Garcia, andR.L.Hunter.Characterizationof the trehalose 6,6’-dimycolate surface monolayer by scanningtunneling microscopy. Infect Immun 1994. 62: 754-756.
126. Bekierkunst, A. Acute granulomatous response produced inmice by trehalose-6,6-dimycolate. J Bacteriol 1968. 96:958-961.
127. Bekierkunst, A., and E. Yarkoni. Granulomatous hypersensi-tivity to trehalose 6,6’-dimycolate (cord factor) in mice infect-ed with BCG. Infect Immun 1973. 7: 631-638.
128. Geisel, R. E., K. Sakamoto, D. G. Russell, and E. R. Rhoades.In Vivo Activity of Released Cell Wall Lipids of Mycobacteriumbovis Bacillus Calmette-Guerin Is Due Principally to TrehaloseMycolates. J Immunol 2005. 174: 5007-5015.
129. Yarkoni, E., and H. J. Rapp. Toxicity of emulsified trehalose6,6’ dimycolate (cord factor) in mice depends on size distribu-tion of mineral oil droplets. Infect. Immun. 1978. 20:856-860.
130. Syed, S. S., and R. L. Hunter, Jr. Studies on the toxic effects ofquartz and a mycobacterial glycolipid, trehalose 6,6’-dimyco-late. Ann Clin Lab Sci 1997. 27: 375-383.
131. Ashenafi, S., G. Aderaye, A. Bekele, M. Zewdie, G. Aseffa, A.T. Hoang, B. Carow, M. Habtamu, M. Wijkander, M.Rottenberg, A. Aseffa, J. Andersson, M. Svensson, and S.Brighenti. Progression of clinical tuberculosis is associated witha Th2 immune response signature in combination with elevat-ed levels of SOCS3. Clin Immunol 2014. 151: 84-99.
132. Young, D. Animal models of tuberculosis. Eur J Immunol2009. 39: 2011-2014.
133. Kubler, A., B. Luna, C. Larsson, N. C. Ammerman, B. B.Andrade, M. Orandle, K. W. Bock, Z. Xu, U. Bagci, D. J.Molura, J. Marshall, J. Burns, K. Winglee, B. A. Ahidjo, L. S.Cheung, M. Klunk, S. K. Jain, N. P. Kumar, S. Babu, A. Sher,J. S. Friedland, P. T. Elkington, and W. R. Bishai.Mycobacterium tuberculosis dysregulates MMP/TIMP bal-ance to drive rapid cavitation and unrestrained bacterial prolif-eration. J Pathol 2014 Sept epub ahead of print.
134. Frick, M. The TB Vaccines Pipeline. In HIV.HCV.TB PipelineReport. Treatment Action Group http://www.pipelinereport.org, New York. 2013. 263-283.
135. Kondratieva, T., T. Azhikina, B. Nikonenko, A. Kaprelyants,and A. Apt. Latent tuberculosis infection: What we knowabout its genetic control? Tuberculosis (Edinb) 2014.
Pathogenesis of Post Primary Tuberculosis 387