aneks i charakterystyka produktu...
TRANSCRIPT

1
ANEKS I
CHARAKTERYSTYKA PRODUKTU LECZNICZEGO

2
1. NAZWA PRODUKTU LECZNICZEGO SUTENT 12,5 mg kapsułki twarde 2. SKŁAD JAKOŚCIOWY I ILOŚCIOWY SUBSTANCJI CZYNNYCH Każda kapsułka zawiera jabłczan sunitynibu w ilości odpowiadającej12,5 mg sunitynibu. Substancje pomocnicze: 80,0 mannitolu Pełny wykaz substancji pomocniczych, patrz punkt 6.1. 3. POSTAĆ FARMACEUTYCZNA Kapsułki twarde. Kapsułki żelatynowe z pomarańczowym wieczkiem i pomarańczowym korpusem, z wykonanymi białym atramentem napisami „Pfizer” na wieczku i „STN 12.5 mg” na korpusie, zawierające granulki o barwie od żółtej do pomarańczowej. 4. SZCZEGÓŁOWE DANE KLINICZNE 4.1 Wskazania do stosowania SUTENT jest przeznaczony do leczenia nieoperacyjnych i (lub) przerzutowych nowotworów podścieliskowych przewodu pokarmowego (ang. Gastrointestinal Stromal Tumor-GIST) po niepowodzeniu leczenia metanosulfonianem imatynibu ze względu na oporność lub nietolerancję. SUTENT jest przeznaczony do leczenia zaawansowanego raka nerki i (lub) raka nerki z przerzutami (ang. Metatastaic Renal Cell Cancer - MRCC) po niepowodzeniu leczenia interferonem alfa lub interleukiną-2. Skuteczność leczenia ustalano na podstawie czasu wystąpienia progresji guza oraz wydłużenia czasu całkowitego przeżycia u pacjentów z nowotworami podścieliskowymi przewodu pokarmowego oraz na podstawie wskaźników obiektywnych odpowiedzi u pacjentów z rakiem nerki z przerzutami (patrz punkt 5.1). 4.2 Dawkowanie i sposób podawania Leczenie powinien rozpoczynać lekarz doświadczony w terapii raka nerki lub nowotworów podścieliskowych przewodu pokarmowego. Zalecana dawka preparatu SUTENT wynosi 50 mg doustnie raz na dobę przez 4 kolejne tygodnie, po czym następuje dwutygodniowa przerwa (schemat 4/2), co stanowi pełny cykl 6 tygodni. Można stopniowo dokonywać zmian dawkowania za każdym razem o 12,5 mg, zależnie od indywidualnie ocenianego bezpieczeństwa i tolerancji. Dawka dobowa nie powinna być większa niż 87,5 mg i nie powinna być mniejsza niż 37,5 mg. Należy unikać równoczesnego podawania silnych induktorów CYP3A4, takich jak ryfampicyna (patrz punkty 4.4 i 4.5). Jeżeli nie jest to możliwe, może być konieczne stopniowe zwiększanie dawki preparatu SUTENT, za każdym razem o 12,5 mg (do 87,5 mg na dobę), z równoczesnym starannym monitorowaniem tolerancji. Należy unikać równoczesnego podawania preparatu SUTENT z silnymi inhibitorami CYP3A4, takimi jak ketokonazol (patrz punkty 4.4 i 4.5). Jeżeli nie jest to możliwe, może zaistnieć konieczność zmniejszenia dawek preparatu SUTENT do minimum 37,5 mg na dobę, z równoczesnym starannym monitorowaniem tolerancji. Jeżeli konieczne jest jednoczesne stosowanie innych preparatów, należy wybrać lek wykazujący minimalne działanie indukujące lub hamujące CYP3A4 lub bez takiego działania.

3
Stosowanie u dzieci: Nie przeprowadzono badań dotyczących bezpieczeństwa i skuteczności stosowania preparatu SUTENT u dzieci. Leku nie należy stosować u dzieci dopóki nie będzie dostępnych więcej danych. Stosowanie u pacjentów w podeszłym wieku: Około 25% uczestników badań klinicznych preparatu SUTENT było w wieku 65 lat lub więcej. Nie obserwowano istotnych różnic pod względem bezpieczeństwa lub skuteczności leczenia pomiędzy młodszymi i starszymi pacjentami. Niewydolność wątroby: Nie przeprowadzono badań klinicznych dotyczących stosowania leku u pacjentów z niewydolnością wątroby (patrz punkt 5.2). Niewydolność nerek: Nie przeprowadzono badań klinicznych dotyczących stosowania leku u pacjentów z niewydolnością nerek (patrz punkt 5.2). Preparat SUTENT może być przyjmowany z posiłkiem lub bez posiłku. W przypadku pominięcia jednej z dawek nie należy stosować dodatkowej dawki. Pacjent powinien przyjąć zwykłą przepisaną dawkę następnego dnia. 4.3 Przeciwwskazania Nadwrażliwość na jabłczan sunitynibu lub na którąkolwiek substancję pomocniczą. 4.4 Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania Równoczesne podawanie silnych induktorów izoenzymu CYP3A4, takich jak ryfampicyna, może powodować zmniejszenie stężenia sunitynibu w osoczu. Dlatego należy unikać jednoczesnego stosowania preparatu z lekami z tej grupy. Jeżeli nie jest to możliwe, może być konieczne zwiększenie dawek preparatu SUTENT (patrz punkty 4.2 i 4.5). Równoczesne podawanie silnych inhibitorów izoenzymu CYP3A4, takich jak ketokonazol, może powodować zwiększenie stężenia sunitynibu w osoczu. Jeżeli konieczne jest jednoczesne stosowanie innego preparatu, należy wybrać lek wykazujący minimalne działanie hamujące CYP3A4 lub bez takiego działania. Jeżeli nie jest to możliwe, może być konieczne zmniejszenie dawek preparatu SUTENT (patrz punkty 4.2 i 4.5) Zdarzenia niepożądane ze strony skóry i tkanki podskórnej Częstym zdarzeniem niepożądanym związanym z leczeniem, występującym u około 30% pacjentów, są przebarwienia skóry, być może związane z barwą substancji czynnej (żółtą). Pacjentów należy poinformować, że w trakcie stosowania preparatu SUTENT może też dojść do odbarwienia włosów lub skóry. Do innych możliwych objawów dermatologicznych należą suchość, zgrubienie lub pękanie skóry, pęcherze lub czasem wysypka na powierzchni dłoni i na podeszwach stóp. U około 14% pacjentów stwierdzano ból/podrażnienie w obrębie jamy ustnej. Około 28% pacjentów zgłaszało występowanie zaburzeń smaku. Powyższe zdarzenia nie występowały łącznie, były na ogół odwracalne i zasadniczo nie wiązały się z koniecznością przerwania leczenia. Zdarzenia niepożądane ze strony przewodu pokarmowego Najczęstszymi obserwowanymi zdarzeniami niepożądanymi ze strony przewodu pokarmowego związanymi z leczeniem były nudności, biegunka, zapalenie błony śluzowej jamy ustnej, objawy dyspeptyczne i wymioty. W razie wystąpienia tego typu zaburzeń zastosowane leczenie wspomagające może obejmować stosowanie leków przeciwwymiotnych lub przeciwbiegunkowych.

4
Krwotoki Krwotoki z guza związane z leczeniem występowały u około 2% pacjentów z nowotworami podścieliskowymi przewodu pokarmowego. Zdarzenia te mogą występować nagle, a w przypadku guzów płuc mogą mieć postać ciężkiego, zagrażającego życiu krwotoku płucnego lub krwioplucia. Śmiertelny krwotok płucny wystąpił u 2 pacjentów otrzymujących preparat SUTENT w ramach badania klinicznego nad jego zastosowaniem w leczeniu niedrobnokomórkowego raka płuc (ang. NSCLC) z przerzutami. U obu pacjentów w badaniu histologicznym stwierdzono, raka płaskonabłonkowego. Preparat SUTENT nie jest zarejestrowany do leczenia niedrobnokomórkowego raka płuc. Rutynowa ocena omawianego zdarzenia niepożądanego powinna obejmować wykonanie morfologii krwi i badania przedmiotowego. Krwotok z nosa był najczęściej występującym krwotocznym zdarzeniem niepożądanym związanym z leczeniem, obserwowanym u około połowy pacjentów z guzami litymi, u których wystąpiły powikłania krwotoczne. Żadne z tych zdarzeń nie miało charakteru ciężkiego. Przewód pokarmowy U pacjentów z nowotworami w jamie brzusznej, u których zastosowano preparat SUTENT rzadko występowały ciężkie, czasem śmiertelne powikłania ze strony przewodu pokarmowego, w tym perforacje. Nadciśnienie tętnicze U około 16% pacjentów z guzami litymi stwierdzano związane z leczeniem nadciśnienie tętnicze. U około 2,7% tych pacjentów zmniejszono dawkę lub czasowo odroczono podanie preparatu SUTENT. U żadnego z pacjentów nie przerwano leczenia. Ciężkie nadciśnienie tętnicze (ciśnienie skurczowe >200 mmHg lub ciśnienie rozkurczowe >110 mmHg) wystąpiło u 4,7% pacjentów. Należy badać pacjentów w celu wykrycia nadciśnienia tętniczego i odpowiednio ich kontrolować. U pacjentów z ciężkim nadciśnieniem tętniczym, którego nie udaje się kontrolować farmakologicznie, zaleca się czasowe przerwanie stosowania preparatu. Można je ponownie podjąć po uzyskaniu skutecznej kontroli nadciśnienia. Zaburzenia hematologiczne Odpowiednio u 13,1% i 0,9% pacjentów stwierdzano zmniejszenie bezwzględnej liczby granulocytów obojętnochłonnych 3 i 4 stopnia ciężkości. Odpowiednio u 4% i 0,5% pacjentów obserwowano również zmniejszenie liczby płytek krwi 3 i 4 stopnia ciężkości. Powyższe zdarzenia nie występowały łącznie, były na ogół odwracalne i zasadniczo nie wiązały się z koniecznością przerwania leczenia. U pacjentów, u których stosuje się preparat SUTENT należy wykonać morfologię krwi na początku każdego cyklu leczenia. Zaburzenia ze strony układu krążenia Zmniejszenie frakcji wyrzutowej lewej komory (LVEF) o ≥ 20% i poniżej dolnej granicy normy wystąpił u około 2% pacjentów z nowotworami podścieliskowymi przewodu pokarmowego i 4% pacjentów z rakiem nerki z przerzutami, którym podawano preparat SUTENT oraz 2% pacjentów otrzymujących placebo. Nie wydaje się, aby spadek LVEF był postępujący. Wartości tego parametru często ulegały poprawie w trakcie leczenia. U 0,7% pacjentów z guzami litymi leczonych preparatem SUTENT i 1% pacjentów otrzymujących placebo stwierdzono następujące, związane z leczeniem zdarzenia niepożądane: niewydolność serca, zastoinowa niewydolność serca lub lewokomorowa niewydolność serca. U wszystkich tych pacjentów występowały nowotwory podścieliskowe przewodu pokarmowego. Potencjalny związek pomiędzy zahamowaniem receptora kinazy tyrozynowej (RTK) a czynnością serca nie został wyjaśniony. Z badań klinicznych nad preparatem SUTENT wykluczono pacjentów, u których w ciągu 12 miesięcy przed podaniem leku miały miejsce incydenty związane z układem krążenia, takie jak zawał mięśnia serca (również ciężka/niestabilna dławica piersiowa), przeszczep pomostujący tętnicy wieńcowej/tętnicy obwodowej, objawowa zastoinowa niewydolność serca (ang. CHF), udar mózgu lub przejściowy napad niedokrwienny, bądź też zator tętnicy płucnej. Nie wiadomo, czy pacjenci z tymi współistniejącymi stanami chorobowymi są narażeni na podwyższone ryzyko rozwinięcia się u nich związanej z lekiem niewydolności lewej komory. Należy rozważyć stosunek ryzyka do potencjalnych korzyści ze stosowania preparatu. Pacjentów z tej grupy należy poddawać starannej obserwacji w celu wykrycia ewentualnych klinicznych objawów podmiotowych i przedmiotowych zastoinowej niewydolności serca

5
podczas stosowania preparatu SUTENT. Należy również rozważyć wykonywanie oznaczeń LVEF na początku terapii i okresowo w trakcie leczenia. U pacjentów, u których nie występują kardiologiczne czynniki ryzyka należy rozważyć wykonanie wyjściowej oceny frakcji wyrzutowej. W przypadku wystąpienia objawów klinicznych zastoinowej niewydolności serca zaleca się przerwanie stosowania preparatu SUTENT. U pacjentów bez klinicznych objawów zastoinowej niewydolności serca, ale z frakcją wyrzutową <50% i >20% poniżej wartości wyjściowej należy przerwać stosowanie preparatu lub obniżyć dawkę. Wydłużenie odcinka QT Wydłużenie odcinka QT oceniano w badaniu 24 pacjentów w wieku od 20 do 87 lat z zaawansowanymi nowotworami. Wykazano, że preparat SUTENT, w stężeniach w przybliżeniu dwukrotnie większych niż terapeutyczne, wydłuża odcinek QTcF (z korektą Friederica). U żadnego z pacjentów nie stwierdzono wydłużenia odcinka QT/QTc stopnia ciężkości większego niż 2 (CTCAEvv 3.0), nie odnotowano także zaburzeń rytmu serca. Znaczenie kliniczne zaobserwowanych działań jest niejasne i uzależnione od indywidualnych czynników ryzyka i wrażliwości pacjentów. Należy zachować ostrożność podczas stosowania preparatu SUTENT u pacjentów z wydłużonym odcinkiem QT w wywiadzie, pacjentów przyjmujących leki przeciwarytmiczne oraz u pacjentów z istotnymi chorobami serca w wywiadzie, bradykardią lub zaburzeniami elektrolitowymi. Należy zachować ostrożność podczas jednoczesnego podawania silnych inhibitorów CYP3A4, które mogą powodować zwiększenie stężenia sunitynibu w osoczu, a w razie potrzeby należy zmniejszyć dawkę preparatu SUTENT (patrz punkt 4.5). Żylne zaburzenia zakrzepowo-zatorowe W dwóch badaniach w raku nerki z przerzutami stwierdzono wystąpienie żylnych powikłań zakrzepowo-zatorowych u czterech pacjentów (2%); u dwóch z nich doszło do zatoru tętnicy płucnej (4 stopnia ciężkości), a u dwóch pozostałych – do zakrzepicy żył głębokich (3 stopnia ciężkości). W jednym z tych przypadków przerwano stosowanie leku. W badaniu rejestracyjnym nad nowotworami podścieliskowymi przewodu pokarmowego u siedmiu pacjentów (3%) otrzymujących preparat SUTENT wystąpiły żylne powikłania zakrzepowo-zatorowe natomiast nie wystąpiły one u żadnego z pacjentów z grupy otrzymującej placebo. U pięciu z siedmiu pacjentów powikłania te były 3 stopnia ciężkości, a u dwóch – 1 lub 2 stopnia ciężkości. Czterech pacjentów z nowotworami podścieliskowymi przewodu pokarmowego przerwało leczenie po wystąpieniu pierwszych objawów zakrzepicy żył głębokich. Zator tętnicy płucnej U około 1,1% pacjentów z guzami litymi, którzy otrzymywali preparat SUTENT wystąpił związany z leczeniem zator tętnicy płucnej. Żadne z tych zdarzeń nie doprowadziło do przerwania leczenia, jednak w kilku przypadkach zmniejszono dawkę lub tymczasowo odroczono leczenie. Po podjęciu leczenia nie doszło do ponownego zatoru tętnicy płucnej u tych pacjentów. Niedoczynność tarczycy W dwóch badaniach w raku nerki z przerzutami niedoczynność tarczycy była zgłaszana jako zdarzenie niepożądane u siedmiu pacjentów (4%). Ponadto u czterech pacjentów (2%) stwierdzono podwyższenie stężenia TSH. Ogółem u 7% populacji objętej tymi badaniami stwierdzono kliniczne lub laboratoryjne objawy niedoczynności tarczycy, które wystąpiły w trakcie leczenia. U ośmiu pacjentów (4%) z nowotworami podścieliskowymi przewodu pokarmowego otrzymujących preparat SUTENT i u jednego pacjenta (1%) otrzymującego placebo stwierdzono w czasie leczenia niedoczynność tarczycy wymagającą pilnej terapii. U pacjentów z objawami sugerującymi niedoczynność tarczycy konieczne jest wykonywanie kontrolnych badań laboratoryjnych diagnozujących czynność tego gruczołu i zastosowanie leczenia według obowiązujących standardów. Czynność trzustki U pacjentów z różnymi guzami litymi otrzymujących preparat SUTENT, obserwowano zwiększenie aktywności lipazy i amylazy w surowicy. Wzrost aktywności lipazy był przemijający i na ogół nie towarzyszyły mu objawy podmiotowe i przedmiotowe zapalenia trzustki. Zapalenie trzustki

6
obserwowano u 0,4% pacjentów z guzami litymi. W razie wystąpienia objawów zapalenia trzustki należy zapewnić pacjentom właściwą opiekę medyczną. Napady drgawkowe W badaniach klinicznych z zastosowaniem preparatu SUTENT obserwowano występowanie drgawek u pacjentów z cechami radiologicznymi przerzutów nowotworu do mózgu. Ponadto stwierdzano rzadkie (<1%) przypadki drgawek i objawy radiologiczne zespołu odwracalnej tylnej leukoencefalopatii (RPLS). W żadnym z omawianych przypadków zaburzenia te nie doprowadziły do zgonu. U pacjentów z drgawkami i objawami podmiotowymi/przedmiotowymi sugerującymi obecność RPLS, takimi jak nadciśnienie, ból głowy, zwiększona czujność, zmiany psychiczne i utrata wzroku, w tym ślepota korowa, należy kontrolować występujące zaburzenia poprzez zastosowanie właściwego leczenia, w tym farmakologicznej kontroli nadciśnienia. Zaleca się tymczasowe przerwanie stosowania preparatu SUTENT. Po ustąpieniu objawów lekarz prowadzący może podjąć decyzję o kontynuacji leczenia. 4.5 Interakcje z innymi lekami i inne rodzaje interakcji Leki, które mogą zwiększać stężenie sunitynibu w osoczu: Równoczesne podawanie sunitynibu z silnym inhibitorem izoenzymu CYP3A4, ketokonazolem, powodowało po jednorazowym podaniu jabłczanu sunitynibu u zdrowych ochotników wzrost wartości Cmax i AUC0-∞ kompleksu [sunitynib + podstawowy metabolit] odpowiednio o 49% i 51%. Podawanie preparatu SUTENT jednocześnie z silnymi inhibitorami izoenzymów z rodziny CYP3A4 (np. rytonawir, itrakonazol, erytromycyna, klarytromycyna, sok grejpfrutowy) może wiązać się ze zwiększeniem stężenia sunitynibu. Z tego względu należy unikać podawania go jednocześnie z inhibitorami enzymatycznymi lub należy rozważyć wybranie do leczenia skojarzonego alternatywnego preparatu, o minimalnym działaniu hamującym enzym CYP3A4 lub bez takiego działania. Jeżeli nie jest to możliwe, może zaistnieć konieczność zmniejszenia dawek preparatu SUTENT do minimum 37,5 mg na dobę, na podstawie dokładnie monitorowanej tolerancji (patrz punkt 4.2). Leki, które mogą zmniejszyć stężenie sunitynibu w osoczu: Równoczesne podawanie jabłczanu sunitynibu z silnym induktorem izoenzymu CYP3A4, ryfampicyną, powodowało po jednorazowym podaniu jabłczanu sunitynibu u zdrowych ochotników redukcję wartości Cmax i AUC0-∞ kompleksu [sunitynib + podstawowy metabolit] odpowiednio o 23% i 46%]. Podawanie preparatu SUTENT z silnymi induktorami izoenzymów z grupy CYP3A4 (np. deksametazon, fenytoina, karbamazepina, ryfampicyna, fenobarbital lub ziele dziurawca Hypericum perforatum) może prowadzić do zmniejszenia stężenia sunitynibu. Z tego względu należy unikać jednoczesnego stosowania preparatu z induktorami enzymatycznymi, bądź też należy rozważyć wybranie do leczenia skojarzonego alternatywnego preparatu, o minimalnym działaniu indukującym enzym CYP3A4 lub bez takiego działania. Jeżeli nie jest to możliwe, może zaistnieć konieczność stopniowego zwiększania dawek preparatu SUTENT, za każdym razem o 12,5 mg (do 87,5 mg na dobę) na podstawie dokładnie monitorowanej tolerancji (patrz punkt 4.2). Aby utrzymać docelowe stężenie sunitynibu należy rozważyć wybranie do leczenia skojarzonego preparatów indukujących enzym w mniejszym stopniu. Jeżeli nie jest to możliwe, może być konieczne dostosowanie dawki preparatu SUTENT (patrz punkt 4.2). U pacjentów leczonych preparatem SUTENT rzadko obserwowano krwotoki (patrz punkt 4.4). Pacjenci otrzymujący równocześnie preparaty przeciwzakrzepowe (np. warfaryna, acenokumarol) mogą być poddawani okresowym kontrolom obejmującym wykonanie morfologii krwi (z oznaczeniem liczby płytek krwi), badanie czynników krzepnięcia (czas protrombinowy (PT)/wskaźnik INR) i badaniu przedmiotowemu. 4.6 Ciąża i laktacja Ciąża Nie przeprowadzono badań dotyczących stosowania preparatu SUTENT u kobiet w ciąży. W badaniach na zwierzętach stwierdzono toksyczny wpływ leku na zdolność do rozrodu, objawiający się m.in.

7
wadami wrodzonymi płodów (patrz punkt 5.3). Preparatu nie należy stosować w czasie ciąży lub u kobiet, które nie stosują skutecznej metody antykoncepcyjnej z wyjątkiem przypadku, gdy potencjalne korzyści z leczenia przeważą nad potencjalnym ryzykiem dla płodu. Jeżeli preparat zostanie zastosowany w czasie ciąży lub jeżeli pacjentka zajdzie w ciążę w trakcie jego stosowania, należy ją poinformować o potencjalnym zagrożeniu dla płodu. Kobietom w wieku rozrodczym należy doradzić stosowanie skutecznej metody antykoncepcyjnej podczas przyjmowania preparatu SUTENT. Dane niekliniczne wskazują, że leczenie preparatem SUTENT może wywierać niekorzystny wpływ na płodność kobiet i mężczyzn (patrz punkt 5.3). Laktacja Sunitynib i (lub) jego metabolity przenikają do mleka szczurów. Nie wiadomo, czy sunitynib lub jego główny czynny metabolit przenikają do mleka kobiecego. Ze względu na to, że leki są często wydzielane do mleka kobiecego i ze względu na możliwe występowanie ciężkich działań niepożądanych u niemowląt karmionych piersią, kobiety nie powinny karmić piersią podczas stosowania preparatu SUTENT. 4.7 Wpływ na zdolność prowadzenia pojazdów mechanicznych i obsługiwania urządzeń
mechanicznych w ruchu Nie przeprowadzono badań nad wpływem preparatu na zdolność prowadzenia pojazdów mechanicznych i obsługiwania urządzeń mechanicznych w ruchu. Pacjenci powinni być poinformowani o możliwości wystąpienia zawrotów głowy w trakcie leczenia preparatem SUTENT. 4.8 Działania niepożądane Najczęstszymi ciężkimi zdarzeniami niepożądanymi związanymi z leczeniem preparatem SUTENT u pacjentów z guzami litymi były: zator tętnicy płucnej (1%), trombocytopenia (1%), krwotok z guza (0,9%), gorączka neutropeniczna (0,4%) i nadciśnienie tętnicze (0,4%). Do najczęstszych zdarzeń niepożądanych związanych z leczeniem (wystąpiły u co najmniej 20% pacjentów) o dowolnym stopniu nasilenia należały: uczucie zmęczenia; zaburzenia ze strony przewodu pokarmowego, takie jak biegunka, nudności, zapalenie jamy ustnej, objawy dyspeptyczne i wymioty; przebarwienia skórne; zaburzenia smaku i anoreksja. Uczucie zmęczenia, nadciśnienie tętnicze i neutropenia były najczęstszymi, związanymi z leczeniem zdarzeniami niepożądanymi o maksymalnym stopniu ciężkości 3 u pacjentów z guzami litymi, a podwyższona aktywność lipazy była najczęstszym związanym z leczeniem zdarzeniem niepożądanym o maksymalnym stopniu ciężkości 4. Poniżej wymieniono działania niepożądane, które zostały zgłoszone przez >5% pacjentów z guzami litymi, według klas układowo-narządowych, częstości występowania i stopnia ciężkości. W ramach każdej grupy wyodrębnionej wg częstości występowania działania niepożądane przedstawiono w kolejności od najcięższego do najlżejszego. Według częstości występowania zdarzenia definiowano jako: bardzo częste (> 1/10), częste (od >1/100 do < 1/10), nieczęste (od >1/1000 do <1//100), rzadkie (od >1/10 000 do 1/1000), bardzo rzadkie (< 1/10 000).

8
Działania niepożądane zgłaszane w badaniach u pacjentów z nowotworami podścieliskowymi przewodu pokarmowego. Klasyfikacja układów i narządów
Częstość występowania
Działania niepożądane
Wszystkich stopni n (%)
Stopnia 3 n (%)
Stopnia 4 n (%)
Bardzo często Niedokrwistość 33 (12,8%) 13 (5,1%) 1 (0,4%) Często Neutropenia 24 (9,3%) 15 (5,8%) 1 (0,4%)
Zaburzenia krwi i układu chłonnego Często Trombocytopenia 23 (8,9%) 6 (2,3%) 1 (0,4%) Zaburzenia endokrynolo- giczne
Często Niedoczynność tarczycy
15 (5,8%) 0 (0,0%) 1 (0,4%)
Zaburzenia metabolizmu i odżywiania
Często Zanik łaknienia 44 (7,1%) 1 (0,4%) 0 (0,0%)
Bardzo często Zaburzenia smaku 48 (18,7%) 0 (0,0%) 0 (0,0%) Zaburzenia układu nerwowego
Bardzo często Bóle głowy 27 (10,5%) 2 (0,8%) 0 (0,0%)
Zaburzenia naczyń
Bardzo często Nadciśnienie tętnicze
43 (16,7%) 18 (7,0%) 0 (0,0%)
Zaburzenia oddechowe, klatki piersiowej i śródpiersia
Często Krwotok z nosa 17 (6,6%) 0 (0,0%) 0 (0,0%)
Zaburzenia nerek i dróg moczowych
Często Zmiana barwy moczu
13 (5,1%) 0 (0,0%) 0 (0,0%)
Bardzo często Biegunka 90 (35,0%) 13 (5,1%) 0 (0,0%) Bardzo często Nudności 69 (26,8%) 2 (0,8%) 0 (0,0%) Bardzo często Zapalenie błony
śluzowej jamy ustnej
49 (19,1%) 2 (0,8%) 0 (0,0%)
Bardzo często Wymioty 46 (17,9%) 1 (0,4%) 0 (0,0%) Bardzo często Objawy
dyspeptyczne 32 (12,5%) 2 (0,8%) 0 (0,0%)
Często Pieczenie języka 17 (6,6%) 0 (0,0%) 0 (0,0%) Często Zaparcia 13 (5,1%) 1 (0,4%) 0 (0,0%) Bardzo często Bóle brzucha* 30 (11,7%) 5 (1,9%) 1 (0,4%) Często Ból w jamie ustnej 16 (6,2%) 0 (0,0%) 0 (0,0%) Często Wzdęcie z
oddawaniem gazów 15 (5,8%) 0 (0,0%) 0 (0,0%)
Często Suchość w jamie ustnej
15 (5,8%) 0 (0,0%) 0 (0,0%)
Zaburzenia żołądkowo-jelitowe
Często Choroba refluksowa przełyku
15 (5,8%) 0 (0,0%) 0 (0,0%)
Bardzo często Przebarwienia skórne
65 (25,3%) 0 (0,0%) 0 (0,0%)
Bardzo często Rumień i zaburzenia czucia dłoni i podeszew stóp
55 (21,4%) 14 (5,4%) 0 (0,0%)
Zaburzenia skóry i tkanki podskórnej
Bardzo często Wysypka*** 39 (15,2%) 2 (0,8%) 0 (0,0%)

9
Klasyfikacja układów i narządów
Częstość występowania
Działania niepożądane
Wszystkich stopni n (%)
Stopnia 3 n (%)
Stopnia 4 n (%)
Często Zmiany koloru włosów
22 (8,6%) 0 (0,0%) 0 (0,0%)
Często Suchość skóry 15 (5,8%) 0 (0,0%) 0 (0,0%) Często Ból w kończynach 21 (8,2%) 1 (0,4%) 0 (0,0%) Często Bóle stawowe 15 (5,8%) 2 (0,8%) 0 (0,0%)
Zaburzenia mięśniowo-szkieletowe, tkanki łącznej i kości
Często Bóle mięśniowe 13 (5,1%) 0 (0,0%) 0 (0,0%)
Bardzo często Uczucie zmęczenia/ osłabienie
135 (52,5%) 25 (9,7%) 0 (0,0%) Zaburzenia ogólne i stany w miejscu podania Bardzo często Zapalenie błon
śluzowych 30 (11,7%) 0 (0,0%) 0 (0,0%)
Często Obrzęk** 21 (8,2%) 1 (0,4%) 0 (0,0%) Często Zmniejszenie
stężenia hemoglobiny
16 (6,2%) 2 (0,8%) 0 (0,0%)
Badania dodatkowe
Często Zwiększenie aktywności fosfokinazy kreatynowej we krwi
14 (5,4%) 0 (0,0%) 0 (0,0%)
Często Zmniejszenie frakcji wyrzutowej
13 (5,1%) 1 (0,4%) 0 (0,0%)
Często Zwiększenie aktywności lipazy
13 (5,1%) 5 (1,9%) 4 (1,6%)
Często Zmniejszenie liczby płytek krwi
13 (5,1%) 2 (0,8%) 1 (0,4%)
Dowolne zdarzenie niepożądane
222 (86,4%) 88 (34,2%) 24 (9,3%)
*Następujące zdarzenia zostały połączone w jedną kategorię: bóle brzucha, bóle w górnej części brzucha i bóle w dolnej części brzucha. **Następujące zdarzenia zostały połączone w jedną kategorię: obrzęki i obrzęki obwodowe. ***Następujące zdarzenia zostały połączone w jedną kategorię: wysypka, wysypka rumieniowa, wysypka plamista i wysypka złuszczająca się Działania niepożądane zgłaszane w badaniach u pacjentów z rakiem nerki z przerzutami Klasyfikacja układów i narządów
Częstość występowania
Działania niepożądane
Wszystkich stopni n (%)
Stopnia 3 n (%)
Stopnia 4 n (%)
Bardzo często Neutropenia 17 (10,1%) 8 (4,7%) 1 (0,6%) Często Niedokrwistość 16 (9,5%) 6 (3,6%) 0 (0,0%) Często Trombocytopenia 15 (8,9%) 5 (3,0%) 2 (1,2%)
Zaburzenia krwi i układu chłonnego
Często Leukopenia 14 (8,3%) 7 (4,1%) 0 (0,0%)
Zaburzenia oka Często Zwiększone łzawienie
9 (5,3%) 0 (0,0%) 0 (0,0%)
Zaburzenia metabolizmu i odżywiania
Bardzo często Zanik łaknienia 47 (27,8%) 1 (0,6%) 0 (0,0%)
Często Odwodnienie 12 (7,1%) 4 (2,4%) 0 (0,0%) Często Zmniejszenie 11 (6,5%) 0 (0,0%) 0 (0,0%)

10
Klasyfikacja układów i narządów
Częstość występowania
Działania niepożądane
Wszystkich stopni n (%)
Stopnia 3 n (%)
Stopnia 4 n (%)
łaknienia Bardzo często Zaburzenia smaku 71 (42%) 0 (0,0%) 0 (0,0%) Bardzo często Bóle głowy 25 (14,8%) 1 (0,6%) 0 (0,0%) Często Zawroty głowy 13 (7,7%) 2 (1,2%) 0 (0,0%)
Zaburzenia układu nerwowego
Często Parestezje 9 (5,3%) 0 (0,0%) 0 (0,0%) Zaburzenia naczyń
Bardzo często Nadciśnienie tętnicze
28 (16,6%) 7 (4,1%) 0 (0,0%)
Często Krwotok z nosa 16 (9,5%) 0 (0,0%) 0 (0,0%) Zaburzenia oddechowe, klatki piersiowej i śródpiersia
Często Duszność 9 (5,3%) 0 (0,0%) 0 (0,0%)
Bardzo często Biegunka 83 (49,1%) 5 (3,0%) 0 (0,0%) Bardzo często Nudności 84 (49,7%) 2 (1,2%) 0 (0,0%) Bardzo często Zapalenie błony
śluzowej jamy ustnej
70 (41,4%) 6 (3,6%) 0 (0,0%)
Bardzo często Objawy dyspeptyczne
69 (40,8%) 1 (0,6%) 0 (0,0%)
Bardzo często Wymioty 52 (30,8%) 2 (1,2%) 0 (0,0%) Bardzo często Zaparcia 34 (20,1%) 0 (0,0%) 0 (0,0%)
Zaburzenia żołądkowo-jelitowe
Bardzo często Pieczenie języka 25 (14,8%) 0 (0,0%) 0 (0,0%) Bardzo często Bóle brzucha* 17 (10,1%) 2 (1,2%) 0 (0,0%)
Często Wzdęcie z oddawaniem gazów
16 (9,5%) 0 (0,0%) 0 (0,0%)
Często Uczucie rozpierania w jamie brzusznej
9 (5,3%) 0 (0,0%) 0 (0,0%)
Często Suchość w jamie ustnej
9 (5,3%) 0 (0,0%) 0 (0,0%)
Bardzo często Przebarwienia skórne
54 (32,0%) 0 (0,0%) 0 (0,0%)
Bardzo często Wysypka** 46 (27,2%) 0 (0,0%) 0 (0,0%) Bardzo często Zmiana barwy
włosów 24 (14,2%) 0 (0,0%) 0 (0,0%)
Bardzo często Rumień i zaburzenia czucia dłoni i podeszew stóp
21 (12,4%) 6 (3,6%) 0 (0,0%)
Często Łysienie 13 (7,7%) 0 (0,0%) 0 (0,0%) Często Złuszczające
zapalenie skóry 10 (5,9%) 2 (1,2%) 0 (0,0%)
Często Obrzęki okołooczodołowe
9 (5,3%) 0 (0,0%) 0 (0,0%)
Bardzo często Suchość skóry 22 (13,0%) 0 (0,0%) 0 (0,0%)
Zaburzenia skóry i tkanki podskórnej
Bardzo często Rumień 20 (11,8%) 0 (0,0%) 0 (0,0%) Bardzo często Ból w kończynach 21 (12,4) 1 (0,6%) 0 (0,0%) Zaburzenia
mięśniowo-szkieletowe, tkanki łącznej i kości
Często Bóle mięśni 15 (8,9%) 1 (0,6%) 0 (0,0%)

11
Klasyfikacja układów i narządów
Częstość występowania
Działania niepożądane
Wszystkich stopni n (%)
Stopnia 3 n (%)
Stopnia 4 n (%)
Bardzo często Uczucie zmęczenia/osłabie- nie
108 (63,9%) 19 (11,2%) 0 (0,0%) Zaburzenia ogólne i stany w miejscu podania Bardzo często Zapalenie błon
śluzowych 30 (17,8%) 1 (0,6%) 0 (0,0%)
Urazy, zatrucia, procedury medyczne i chirurgiczne
Bardzo często Pęcherze 7 (11,1%) 2 (3,2%) 0 (0,0%)
Bardzo często Zwiększenie aktywności lipazy
17 (10,1%) 12 (7,1%) 3 (1,8%)
Często Zaburzenia frakcji wyrzutowej
16 (9,5%) 1 (0,6%) 0 (0,0%)
Badania dodatkowe
Często Zwiększenie aktywności amylazy we krwi
9 (5,3%) 6 (3,6%) 0 (0,0%)
Często Zmniejszenie masy ciała
11 (6,5%) 0 (0,0%) 0 (0,0%)
Często Zmniejszenie liczby leukocytów
10 (5,9%) 3 (1,8%) 0 (0,0%)
Często Zmniejszenie liczby płytek krwi
9 (5,3%) 3 (1,8%) 2 (1,2%)
Dowolne zdarzenie niepożądane
166 (98,2%) 77 (45,6%) 14 (8,3%)
*Następujące zdarzenia zostały połączone w jedną kategorię: bóle brzucha, bóle w górnej części brzucha i bóle w dolnej części brzucha. **Następujące zdarzenia zostały połączone w jedną kategorię: wysypka, wysypka rumieniowa, wysypka grudkowa, wysypka uogólniona, wysypka grudkowa i wysypka ze świądem. 4.9 Przedawkowanie Nie ma doświadczeń z ostrym przedawkowaniem preparatu SUTENT. Nie istnieje swoista odtrutka, a leczenie przedawkowania powinno polegać na zastosowaniu standardowych środków wspomagających. O ile istnieją wskazania, można uzyskać eliminację niewchłoniętego preparatu poprzez wywołanie wymiotów lub płukanie żołądka. 5. WŁAŚCIWOŚCI FARMAKOLOGICZNE Grupa farmakoterapeutyczna: Leki przeciwnowotworowe - Inhibitor kinazy białkowo – tyrozynowej. Kod ATC: LO1XE04 5.1 Właściwości farmakodynamiczne Jabłczan sunitynibu hamuje liczne receptory kinazy tyrozynowej (RTK), które biorą udział we wzroście nowotworów, w patologicznej angiogenezie i w rozsiewie choroby nowotworowej z przerzutami. Sunitynib został zidentyfikowany jako inhibitor receptorów płytkowego czynnika wzrostu (PDGFRα i PDGFRβ), receptorów czynników wzrostu śródbłonka naczyniowego (VEGFR1, VEGFR2 i VEGFR3), receptorów czynnika komórek pnia (KIT), kinazy tyrozynowej podobnej do Fms-3 (FLT3), receptorów czynnika stymulującego powstawanie kolonii (CSF-1R) i receptorów glejopochodnego czynnika

12
neurotroficznego (RET). W testach biochemicznych i komórkowych podstawowy metabolit sunitynibu wykazuje działanie podobne do sunitynibu. BADANIA KLINICZNE Badano bezpieczeństwo kliniczne i skuteczność kliniczną preparatu SUTENT podczas leczenia pacjentów ze złośliwymi nowotworami podścieliskowymi przewodu pokarmowego (GIST) opornymi na imatynib (tj. pacjentów, u których doszło do progresji choroby w trakcie leczenia imatynibem lub po nim) lub nietolerujących imatynibu (tj. pacjentów, u których wystąpiły istotne objawy toksyczności w trakcie leczenia imatynibem, które uniemożliwiły dalsze leczenie) oraz leczenia pacjentów z rakiem nerki z przerzutami po niepowodzeniu leczenia z zastosowaniem cytokin. Skuteczność leczenia ustalano na podstawie czasu wystąpienia progresji guza oraz wydłużenia czasu całkowitego przeżycia u pacjentów z nowotworami podścieliskowymi przewodu pokarmowego oraz na podstawie wskaźników obiektywnych odpowiedzi u pacjentów z rakiem nerki z przerzutami. Nowotwory podścieliskowe przewodu pokarmowego Wstępne otwarte, zakładające zwiększanie dawkowania badanie zostało przeprowadzone u pacjentów z nowotworami podścieliskowymi przewodu pokarmowego po niepowodzeniu stosowania imatynibu (mediana maksymalnej dawki dobowej: 800 mg) ze względu na oporność lub nietolerancję leczenia. Do badania zakwalifikowano dziewięćdziesięciu siedmiu pacjentów, otrzymujących dawki według różnych schematów; 55 pacjentów otrzymywało preparat w dawce 50 mg według zalecanego schematu leczenia: 4 tygodnie przyjmowania leku/2 tygodnie przerwy („schemat 4/2”). W niniejszym badaniu mediana czasu do progresji choroby (Time To Progression, TTP) wynosiła 34,0 tygodnie (95% CI = 22,0–46,0 tygodnia). Przeprowadzono randomizowane, podwójnie ślepe, kontrolowane placebo badanie fazy III preparatu SUTENT u pacjentów z nowotworami podścieliskowymi przewodu pokarmowego, którzy nie tolerowali leczenia imatynibem lub, u których doszło do progresji choroby w trakcie leczenia imatynibem (mediana maksymalnej dawki dobowej 800 mg). W badaniu tym 312 pacjentów w sposób randomizowany (w stosunku 2:1) przydzielono do grup otrzymujących odpowiednio preparat SUTENT w dawce 50 mg lub placebo doustnie, raz na dobę, według schematu 4/2, do momentu wystąpienia progresji choroby lub wycofania z badania z innego powodu (207 pacjentów otrzymywało SUTENT, a 105 pacjentów otrzymywało placebo). Podstawowym punktem końcowym oceny skuteczności w badaniu był TTP definiowany jako czas od randomizacji do pierwszego potwierdzenia obiektywnej progresji guza. Mediana TTP podczas stosowania preparatu SUTENT wynosiła 28,9 tygodnia (95% CI = 21,3–34,1 tygodnia) i była istotnie statystycznie dłuższa niż TTP wynoszący 5,1 tygodnia (95% CI = 4,4–10,1 tygodnie) podczas stosowania placebo. Różnica przeżywalności całkowitej była statystycznie większa na korzyść preparatu SUTENT [współczynnik ryzyka: 0,491 95% (C.I. 0,290- 0,831)]; ryzyko zgonu było 2 razy większe u pacjentów w grupie otrzymującej placebo w porównaniu do grupy leczonej preparatem SUTENT. Odsetki zgonów wynosiły 14% u osób otrzymujących SUTENT w porównaniu do 25% u osób otrzymujących placebo. W momencie przeprowadzania analizy nie osiągnięto jeszcze mediany przeżywalności ogólnej w żadnej z grup terapeutycznych. Rak nerki z przerzutami oporny na leczenie cytokinami (MRCC) Badanie II fazy preparatu SUTENT przeprowadzono u pacjentów opornych na wcześniejsze leczenie cytokinami, tzn. interleukiną-2 lub interferonem-α. Sześćdziesięciu trzech pacjentów otrzymało dawkę początkową 50 mg preparatu SUTENT doustnie, raz na dobę przez 4 kolejne tygodnie, po czym następował dwutygodniowy okres przerwy, który kończył pełny cykl 6 tygodni (schemat 4/2). Podstawowym punktem końcowym oceny skuteczności był wskaźnik obiektywnych odpowiedzi na leczenie (objective response rate, ORR) ustalany na podstawie kryteriów oceny odpowiedzi u pacjentów z guzami litymi (Response Evaluation Criteria in Solid Tumours, RECIST). W omawianym badaniu wskaźnik obiektywnych odpowiedzi wynosił 36,5% (95% C.I. 24,7% – 49,6%), a mediana czasu do progresji choroby (TTP) wynosiła 37,7 tygodnia (95% C.I. 24,0 – 46,4 tygodnia).

13
Przeprowadzono potwierdzające, otwarte, obejmujące jedną grupę pacjentów, wieloośrodkowe badanie oceniające skuteczność i bezpieczeństwo stosowania preparatu SUTENT u pacjentów z rakiem nerki z przerzutami, którzy byli oporni na uprzednio stosowane leczenie cytokinami. Stu sześciu pacjentów otrzymało co najmniej jedną dawkę 50 mg preparatu SUTENT według schematu 4/2. Pierwszorzędowym punktem końcowym oceny skuteczności tego badania był wskaźnik obiektywnych odpowiedzi (ORR). Drugorzędowe punkty końcowe obejmowały TTP, okres utrzymywania się odpowiedzi (duration of response, DR) i przeżycie całkowite (overall survival, OS). W niniejszym badaniu wskaźnik ORR wynosił 38% (95% C.I. 26,8% – 47,5 %). Mediany DR i OS nie zostały jeszcze osiągnięte. Ten produkt leczniczy został dopuszczony do obrotu zgodnie z procedurą „warunkowego dopuszczenia”. Oznacza to, że oczekiwane są dalsze dowody świadczące o korzyści z jego stosowania, w szczególności dane dotyczące wpływu preparatu SUTENT na medianę czasu do wystąpienia progresji u pacjentów z rakiem nerki z przerzutami. Badanie oceniające ten parametr jest w toku. Europejska Agencja ds. Produktów Leczniczych będzie dokonywać przeglądu nowych informacji o produkcie raz do roku i ChPL zostanie uzupełniona tak jak to będzie konieczne. 5.2 Właściwości farmakokinetyczne Właściwości farmakokinetyczne sunitynibu i jabłczanu sunitynibu oceniano w badaniach obejmujących 135 zdrowych ochotników i 266 pacjentów z guzami litymi. Wchłanianie Po podaniu doustnym sunitynibu maksymalne stężenie (Cmax) stwierdza się na ogół po upływie 6 – 12 godzin (Tmax). Pokarm nie wpływa na dostępność biologiczną sunitynibu. Dystrybucja W badaniach in vitro stopień wiązania się sunitynibu i jego podstawowego czynnego metabolitu z ludzkimi białkami osocza wynosił odpowiednio 95% i 90% niezależnie od stężenia. Pozorna objętość dystrybucji (V/F) w przypadku sunitynibu była znaczna – 2230 l, co wskazuje na penetrację leku do tkanek. Metabolizm Obliczone in vitro wartości stałej Ki dla wszystkich badanych izoform enzymu CYP (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 I CYP4A9/11) wskazują na to, że sunitynib i jego podstawowy czynny metabolit prawdopodobnie nie hamują w stopniu istotnym klinicznie metabolizmu preparatów metabolizowanych przez te enzymy. Badania in vitro również wskazują na to, że SUTENT nie indukuje i nie hamuje głównych enzymów CYP, w tym CYP3A4. Biotransformacja Sunitynib jest metabolizowany przede wszystkim przez izoenzym CYP3A4 cytochromu P450, który katalizuje reakcję powstawania głównego czynnego metabolitu podlegającego dalszemu metabolizmowi katalizowanemu przez CYP3A4. Równoczesne podawanie preparatu SUTENT z silnym induktorem CYP3A4 – ryfampicyną powodowało zmniejszenie wartości Cmax i AUC0-∞ sunitynibu o odpowiednio 56% i 78% po podaniu pojedynczej dawki preparatu SUTENT zdrowym ochotnikom. Inne induktory grupy enzymów CYP3A4 (np. deksametazon, fenytoina, karbamazepina, fenobarbital lub ziele dziurawca Hypericum perforatum) podawane jednocześnie z preparatem SUTENT mogą powodować zmniejszenie stężenia sunitynibu. Eliminacja Preparat jest wydalany przede wszystkim z kałem (61%), natomiast przez nerki ulega wydaleniu 16% podanej dawki w postaci niezmienionej i metabolitów. Sunitynib i jego podstawowy czynny metabolit były głównymi związkami pochodnymi leku wykrywanymi w osoczu, moczu i kale, odpowiadając odpowiednio za 91,5%, 86,4% i 73,8% radioaktywności w zebranych próbkach. Pozostałe metabolity o

14
mniejszym znaczeniu zostały zidentyfikowane w moczu i w kale, jednak na ogół nie były wykrywane w osoczu. Całkowity klirens leku po podaniu doustnym (CL/F) wynosił 34–62 l/h. Zaburzenia czynności narządów Niewydolność wątroby: Nie przeprowadzono badań klinicznych nad stosowaniem leku u pacjentów z niewydolnością wątroby. Z badań wykluczano pacjentów z aktywnością AlAT lub AspAT przekraczającą o ponad 2,5 x górną granicę normy lub, gdy podwyższenie to wiązało się z chorobą podstawową – z aktywnością tych enzymów przekraczającą o ponad 5,0 x górną granicę normy. Niewydolność nerek: Nie przeprowadzono badań klinicznych u pacjentów z niewydolnością nerek. Z badań wykluczano pacjentów ze stężeniem kreatyniny w surowicy przekraczającym o ponad 2,0 x górną granicę normy. Populacyjne analizy farmakokinetyki wykazały, klirens pozorny sunitynibu (CL/F) nie jest uzależniony od klirensu kreatyniny w ocenianym zakresie stężeń (42–347 ml/min). Farmakokinetyka w osoczu Po podaniu doustnym zdrowym ochotnikom okres półtrwania sunitynibu i jego podstawowego czynnego metabolitu deetylowego wynosił odpowiednio około 40–60 godzin i 80–110 godzin. W zakresie dawkowania od 25 do 100 mg pole pod krzywą zależności stężenia leku w osoczu od czasu (AUC) i Cmax zwiększa się proporcjonalnie do dawki. Podczas kilkakrotnego podania w ciągu doby sunitynib ulega kumulacji, przy czym jego stężenie zwiększa się 3–4-krotnie, a stężenie jego podstawowego czynnego metabolitu zwiększa się 7–10-krotnie. Stężenie sunitynibu i jego podstawowego czynnego metabolitu w stanie równowagi zostaje osiągnięte w ciągu od 10 do 14 dni. Do 14 dnia łączne stężenie osoczowe sunitynibu i jego podstawowego czynnego metabolitu wynosi 62,9–101 ng/ml, co jest docelowym stężeniem przewidywanym na podstawie danych z badań przedklinicznych jako stężenie hamujące fosforylację receptorów in vitro, prowadzącym do zatrzymania/zmniejszenia wzrostu guzów in vivo. Podstawowy czynny metabolit odpowiada za 23–37% całkowitej ekspozycji na lek. Nie obserwuje się istotnych zmian farmakokinetyki sunitynibu lub jego podstawowego czynnego metabolitu podczas kilkakrotnego podawania leku w ciągu doby lub w trakcie powtarzanych cyklów badanych schematów dawkowania. Parametry farmakokinetyczne były podobne we wszystkich badanych populacjach pacjentów z guzami litymi i u zdrowych ochotników. Populacyjne analizy farmakokinetyki z uwzględnieniem danych demograficznych wskazują na to, że nie są koniecznie korekty dawkowania w zależności od masy ciała lub punktacji wg skali ECOG. Dostępne dane wskazują, że u kobiet klirens pozorny (CL/F) sunitynibu może być o około 30% niższy niż u mężczyzn: różnica ta nie wymaga jednak dostosowywania dawki. 5.3 Przedkliniczne dane o bezpieczeństwie W dziewięciomiesięcznych badaniach nad toksycznością dawek wielokrotnych u szczurów i małp stwierdzono, że podstawowymi narządami, na które wpływa lek, są: przewód pokarmowy (nudności i biegunki u małp), nadnercza (przekrwienie kory i (lub) krwotoki u szczurów i małp, z martwicą i następującym po niej włóknieniem u szczurów), układ limfatyczny i krwiotwórczy (zmniejszenie liczby komórek szpiku kostnego i zanik tkanki limfoidalnej grasicy, śledziony i węzłów chłonnych), zewnątrzwydzielnicza część trzustki (degranulacja komórek pęcherzykowych z martwicą pojedynczych komórek), ślinianki (przerost gronek), stawy (zgrubienie płytki wzrostu), macica (zanik) i jajniki (zmniejszony wzrost pęcherzyków). Wszystkie wyniki uzyskano przy istotnych klinicznie poziomach stężenia osoczowego sunitynibu. Dodatkowe działania preparatu obserwowane w innych badaniach obejmowały: wydłużenie odstępu QTc, zmniejszenie LVEF, przerost przysadki i zanik kanalików jądrowych, rozrost komórek mezangium w nerkach, krwotok z przewodu pokarmowego i do jamy ustnej i przerost komórek płata przedniego przysadki. Uważa się, że zmiany w obrębie macicy (zanik błony śluzowej) i płytki wzrostowej kości (zgrubienie nasad kostnych lub dysplazja chrząstki) są związane z działaniem farmakologicznym sunitynibu. Większość z tych zmian była odwracalna po upływie od 2 do 6 tygodni od zakończenia leczenia. Genotoksyczność

15
Potencjalne działanie genotoksyczne sunitynibu oceniano zarówno in vitro, jak i in vivo. Sunitynib nie wykazywał właściwości mutagennych w badaniach na bakteriach z zastosowaniem aktywacji metabolicznej przez wątrobę szczura. Sunitynib nie indukował strukturalnych aberracji chromosomalnych w ludzkich limfocytach krwi obwodowej in vitro. Obserwowano poliploidię (liczbowe aberracje chromosomalne) w ludzkich limfocytach krwi obwodowej in vitro, zarówno w przypadku zastosowania aktywacji metabolicznej, jak i bez niej. Sunitynib nie wykazywał działania klastogennego w szczurzym szpiku kostnym in vivo u szczurów. Nie oceniano jego podstawowego czynnego metabolitu pod kątem potencjalnej toksyczności genetycznej. Rakotwórczość Nie przeprowadzono badań nad potencjałem rakotwórczym jabłczanu sunitynibu. Toksyczny wpływ na zdolność do rozrodu i rozwój W badaniach toksycznego wpływu na zdolność do rozrodu szczurów nie stwierdzono wpływu preparatu na płodność samic lub samców. Jednak w badaniach toksyczności dawek wielokrotnych przeprowadzonych na szczurach i małpach obserwowano oddziaływanie leku na płodność samic w postaci atrezji pęcherzyków, zwyrodnienia ciałek żółtych, zmian błony śluzowej macicy oraz zmniejszenia masy macicy i jajników przy klinicznie istotnych poziomach ekspozycji układowej. U szczurów obserwowano wpływ leku na płodność samców w postaci zaniku kanalików jąder, zmniejszenia liczby plemników w najądrzach i zmniejszenia ilości koloidu w obrębie gruczołu krokowego i pęcherzyków nasiennych przy poziomach ekspozycji osoczowej 18 razy większych niż stwierdzane w warunkach klinicznych. U szczurów śmiertelność zarodków i płodów przejawiała się istotnym zmniejszeniem liczby żywych płodów, zwiększoną liczbą resorpcji, wzrostem utrat ciąży po zagnieżdżeniu zarodka i całkowitą utratą miotów u 8 z 28 samic ciężarnych przy poziomach stężenia leku w osoczu 5,5 razy większych niż poziomy obserwowane w warunkach klinicznych u ludzi. U królików redukcja masy macicy samic ciężarnych i liczby żywych płodów była związana ze zwiększeniem liczby resorpcji, wzrostem liczby utrat ciąży po zagnieżdżeniu zarodka i całkowitą utratą miotów u 4 z 6 samic ciężarnych przy poziomach stężenia leku w osoczu 3 razy większych niż poziomy obserwowane w warunkach klinicznych u ludzi.
Stosowanie sunitynibu u szczurów w okresie organogenezy prowadziło do zmian rozwojowych, polegających na zwiększonej częstości występowania wad rozwojowych szkieletu płodu, charakteryzujących się przede wszystkim opóźnieniem kostnienia kręgów piersiowych/lędźwiowych. i obserwowanych przy poziomach stężenia leku w osoczu 6 razy większych niż poziomy obserwowane w warunkach klinicznych u ludzi. U królików wpływ leku na rozwój polegał na zwiększeniu częstości występowania rozszczepu wargi przy poziomach stężenia leku w osoczu w przybliżeniu odpowiadających poziomom obserwowanym w warunkach klinicznych u ludzi oraz rozszczepu wargi i rozszczepu podniebienia przy poziomach stężenia leku w osoczu 2,7 razy większych niż poziomy obserwowane w warunkach klinicznych u ludzi. 6. DANE FARMACEUTYCZNE 6.1 Wykaz substancji pomocniczych Zawartość kapsułki Mannitol Kroskarmeloza sodowa Powidon Stearynian magnezu Powłoka kapsułki Żelatyna Czerwony tlenek żelaza (E172)

16
Dwutlenek tytanu (E171) Tusz do nadruku. Szelak Glikol propylenowy Wodorotlenek sodu Powidon Dwutlenek tytanu (E171) 6.2 Niezgodności farmaceutyczne Nie dotyczy. 6.3 Okres trwałości 2 lata 6.4 Specjalne środki ostrożności przy przechowywaniu Brak szczególnych środków ostrożności dotyczących przechowywania. 6.5 Rodzaj i zawartość opakowania Butelki z polietylenu o dużej gęstości (HDPE) z polipropylenowym wieczkiem zawierające po 30 kapsułek. 6.6 Szczególne środki ostrożności dotyczące usuwania Brak szczególnych wymagań. 7. PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE
DO OBROTU Pfizer Ltd Ramsgate Road Sandwich, Kent CT13 9NJ Wielka Brytania 8. NUMER(-Y) POZWOLENIA NA DOPUSZCZENIE DO OBROTU 9. DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU /
DATA PRZEDŁUŻENIA POZWOLENIA Data wydania pierwszego pozwolenia: Data przedłużenia pozwolenia: 10. DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU
CHARAKTERYSTYKI PRODUKTU LECZNICZEGO

17
1. NAZWA PRODUKTU LECZNICZEGO SUTENT 25 mg kapsułki twarde 2. SKŁAD JAKOŚCIOWY I ILOŚCIOWY SUBSTANCJI CZYNNYCH Każda kapsułka zawiera jabłczan sunitynibu w ilości odpowiadającej 25 mg sunitynibu. Substancje pomocnicze: 39,663 mannitolu Pełny wykaz substancji pomocniczych, patrz punkt 6.1. 3. POSTAĆ FARMACEUTYCZNA Kapsułki twarde. Kapsułki żelatynowe z karmelowym wieczkiem i pomarańczowym korpusem, z wykonanymi białym atramentem napisami „Pfizer” na wieczku i „STN 25 mg” na korpusie, zawierające granulki o barwie od żółtej do pomarańczowej. 4. SZCZEGÓŁOWE DANE KLINICZNE 4.1 Wskazania do stosowania SUTENT jest przeznaczony do leczenia nieoperacyjnych i (lub) przerzutowych nowotworów podścieliskowych przewodu pokarmowego (ang. Gastrointestinal Stromal Tumor-GIST) po niepowodzeniu leczenia metanosulfonianem imatynibu ze względu na oporność lub nietolerancję. SUTENT jest przeznaczony do leczenia zaawansowanego raka nerki i (lub) raka nerki z przerzutami (ang. Metatastaic Renal Cell Cancer - MRCC) po niepowodzeniu leczenia interferonem alfa lub interleukiną-2. Skuteczność leczenia ustalano na podstawie czasu wystąpienia progresji guza oraz wydłużenia czasu całkowitego przeżycia u pacjentów z nowotworami podścieliskowymi przewodu pokarmowego oraz na podstawie wskaźników obiektywnych odpowiedzi u pacjentów z rakiem nerki z przerzutami (patrz punkt 5.1). 4.2 Dawkowanie i sposób podawania Leczenie powinien rozpoczynać lekarz doświadczony w terapii raka nerki lub nowotworów podścieliskowych przewodu pokarmowego. Zalecana dawka preparatu SUTENT wynosi 50 mg doustnie raz na dobę przez 4 kolejne tygodnie, po czym następuje dwutygodniowa przerwa (schemat 4/2), co stanowi pełny cykl 6 tygodni. Można stopniowo dokonywać zmian dawkowania za każdym razem o 12,5 mg, zależnie od indywidualnie ocenianego bezpieczeństwa i tolerancji. Dawka dobowa nie powinna być większa niż 87,5 mg i nie powinna być mniejsza niż 37,5 mg. Należy unikać równoczesnego podawania silnych induktorów CYP3A4, takich jak ryfampicyna (patrz punkty 4.4 i 4.5). Jeżeli nie jest to możliwe, może być konieczne stopniowe zwiększanie dawki preparatu SUTENT, za każdym razem o 12,5 mg (do 87,5 mg na dobę), z równoczesnym starannym monitorowaniem tolerancji. Należy unikać równoczesnego podawania preparatu SUTENT z silnymi inhibitorami CYP3A4, takimi jak ketokonazol (patrz punkty 4.4 i 4.5). Jeżeli nie jest to możliwe, może zaistnieć konieczność zmniejszenia dawek preparatu SUTENT do minimum 37,5 mg na dobę, z równoczesnym starannym monitorowaniem tolerancji. Jeżeli konieczne jest jednoczesne stosowanie innych preparatów, należy wybrać lek wykazujący minimalne działanie indukujące lub hamujące CYP3A4 lub bez takiego działania.

18
Stosowanie u dzieci: Nie przeprowadzono badań dotyczących bezpieczeństwa i skuteczności stosowania preparatu SUTENT u dzieci. Leku nie należy stosować u dzieci dopóki nie będzie dostępnych więcej danych. Stosowanie u pacjentów w podeszłym wieku: Około 25% uczestników badań klinicznych preparatu SUTENT było w wieku 65 lat lub więcej. Nie obserwowano istotnych różnic pod względem bezpieczeństwa lub skuteczności leczenia pomiędzy młodszymi i starszymi pacjentami. Niewydolność wątroby: Nie przeprowadzono badań klinicznych dotyczących stosowania leku u pacjentów z niewydolnością wątroby (patrz punkt 5.2). Niewydolność nerek: Nie przeprowadzono badań klinicznych dotyczących stosowania leku u pacjentów z niewydolnością nerek (patrz punkt 5.2). Preparat SUTENT może być przyjmowany z posiłkiem lub bez posiłku. W przypadku pominięcia jednej z dawek nie należy stosować dodatkowej dawki. Pacjent powinien przyjąć zwykłą przepisaną dawkę następnego dnia. 4.3 Przeciwwskazania Nadwrażliwość na jabłczan sunitynibu lub na którąkolwiek substancję pomocniczą. 4.4 Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania Równoczesne podawanie silnych induktorów izoenzymu CYP3A4, takich jak ryfampicyna, może powodować zmniejszenie stężenia sunitynibu w osoczu. Dlatego należy unikać jednoczesnego stosowania preparatu z lekami z tej grupy. Jeżeli nie jest to możliwe, może być konieczne zwiększenie dawek preparatu SUTENT (patrz punkty 4.2 i 4.5). Równoczesne podawanie silnych inhibitorów izoenzymu CYP3A4, takich jak ketokonazol, może powodować zwiększenie stężenia sunitynibu w osoczu. Jeżeli konieczne jest jednoczesne stosowanie innego preparatu, należy wybrać lek wykazujący minimalne działanie hamujące CYP3A4 lub bez takiego działania. Jeżeli nie jest to możliwe, może być konieczne zmniejszenie dawek preparatu SUTENT (patrz punkty 4.2 i 4.5) Zdarzenia niepożądane ze strony skóry i tkanki podskórnej Częstym zdarzeniem niepożądanym związanym z leczeniem, występującym u około 30% pacjentów, są przebarwienia skóry, być może związane z barwą substancji czynnej (żółtą). Pacjentów należy poinformować, że w trakcie stosowania preparatu SUTENT może też dojść do odbarwienia włosów lub skóry. Do innych możliwych objawów dermatologicznych należą suchość, zgrubienie lub pękanie skóry, pęcherze lub czasem wysypka na powierzchni dłoni i na podeszwach stóp. U około 14% pacjentów stwierdzano ból/podrażnienie w obrębie jamy ustnej. Około 28% pacjentów zgłaszało występowanie zaburzeń smaku. Powyższe zdarzenia nie występowały łącznie, były na ogół odwracalne i zasadniczo nie wiązały się z koniecznością przerwania leczenia. Zdarzenia niepożądane ze strony przewodu pokarmowego Najczęstszymi obserwowanymi zdarzeniami niepożądanymi ze strony przewodu pokarmowego związanymi z leczeniem były nudności, biegunka, zapalenie błony śluzowej jamy ustnej, objawy dyspeptyczne i wymioty. W razie wystąpienia tego typu zaburzeń zastosowane leczenie wspomagające może obejmować stosowanie leków przeciwwymiotnych lub przeciwbiegunkowych.

19
Krwotoki Krwotoki z guza związane z leczeniem występowały u około 2% pacjentów z nowotworami podścieliskowymi przewodu pokarmowego. Zdarzenia te mogą występować nagle, a w przypadku guzów płuc mogą mieć postać ciężkiego, zagrażającego życiu krwotoku płucnego lub krwioplucia. Śmiertelny krwotok płucny wystąpił u 2 pacjentów otrzymujących preparat SUTENT w ramach badania klinicznego nad jego zastosowaniem w leczeniu niedrobnokomórkowego raka płuc (ang. NSCLC) z przerzutami. U obu pacjentów w badaniu histologicznym stwierdzono raka płaskonabłonkowego. Preparat SUTENT nie jest zarejestrowany do leczenia niedrobnokomórkowego raka płuc. Rutynowa ocena omawianego zdarzenia niepożądanego powinna obejmować wykonanie morfologii krwi i badania przedmiotowego. Krwotok z nosa był najczęściej występującym krwotocznym zdarzeniem niepożądanym związanym z leczeniem, obserwowanym u około połowy pacjentów z guzami litymi, u których wystąpiły powikłania krwotoczne. Żadne z tych zdarzeń nie miało charakteru ciężkiego. Przewód pokarmowy U pacjentów z nowotworami w jamie brzusznej, u których zastosowano preparat SUTENT rzadko występowały ciężkie, czasem śmiertelne powikłania ze strony przewodu pokarmowego, w tym perforacje. Nadciśnienie tętnicze U około 16% pacjentów z guzami litymi stwierdzano związane z leczeniem nadciśnienie tętnicze. U około 2,7% tych pacjentów zmniejszono dawkę lub czasowo odroczono podanie preparatu SUTENT. U żadnego z pacjentów nie przerwano leczenia. Ciężkie nadciśnienie tętnicze (ciśnienie skurczowe >200 mmHg lub ciśnienie rozkurczowe >110 mmHg) wystąpiło u 4,7% pacjentów. Należy badać pacjentów w celu wykrycia nadciśnienia tętniczego i odpowiednio ich kontrolować. U pacjentów z ciężkim nadciśnieniem tętniczym, którego nie udaje się kontrolować farmakologicznie, zaleca się czasowe przerwanie stosowania preparatu. Można je ponownie podjąć po uzyskaniu skutecznej kontroli nadciśnienia. Zaburzenia hematologiczne Odpowiednio u 13,1% i 0,9% pacjentów stwierdzano zmniejszenie bezwzględnej liczby granulocytów obojętnochłonnych 3 i 4 stopnia ciężkości. Odpowiednio u 4% i 0,5% pacjentów obserwowano również zmniejszenie liczby płytek krwi 3 i 4 stopnia ciężkości. Powyższe zdarzenia nie występowały łącznie, były na ogół odwracalne i zasadniczo nie wiązały się z koniecznością przerwania leczenia. U pacjentów, u których stosuje się preparat SUTENT należy wykonać morfologię krwi na początku każdego cyklu leczenia Zaburzenia ze strony układu krążenia Zmniejszenie frakcji wyrzutowej lewej komory (LVEF) o ≥ 20% i poniżej dolnej granicy normy wystąpił u około 2% pacjentów z nowotworami podścieliskowymi przewodu pokarmowego i 4% pacjentów z rakiem nerki z przerzutami, którym podawano preparat SUTENT oraz 2% pacjentów otrzymujących placebo. Nie wydaje się, aby spadek LVEF był postępujący. Wartości tego parametru często ulegały poprawie w trakcie leczenia. U 0,7% pacjentów z guzami litymi leczonych preparatem SUTENT i 1% pacjentów otrzymujących placebo stwierdzono następujące, związane z leczeniem zdarzenia niepożądane: niewydolność serca, zastoinowa niewydolność serca lub lewokomorowa niewydolność serca. U wszystkich tych pacjentów występowały nowotwory podścieliskowe przewodu pokarmowego. Potencjalny związek pomiędzy zahamowaniem receptora kinazy tyrozynowej (RTK) a czynnością serca nie został wyjaśniony. Z badań klinicznych nad preparatem SUTENT wykluczono pacjentów, u których w ciągu 12 miesięcy przed podaniem leku miały miejsce incydenty związane z układem krążenia, takie jak zawał mięśnia serca (również ciężka/niestabilna dławica piersiowa), przeszczep pomostujący tętnicy wieńcowej/tętnicy obwodowej, objawowa zastoinowa niewydolność serca (ang. CHF), udar mózgu lub przejściowy napad niedokrwienny, bądź też zator tętnicy płucnej. Nie wiadomo, czy pacjenci z tymi współistniejącymi stanami chorobowymi są narażeni na podwyższone ryzyko rozwinięcia się u nich związanej z lekiem niewydolności lewej komory. Należy rozważyć stosunek ryzyka do potencjalnych korzyści ze stosowania preparatu. Pacjentów z tej grupy należy poddawać starannej obserwacji w celu wykrycia ewentualnych klinicznych objawów podmiotowych i przedmiotowych zastoinowej niewydolności serca

20
podczas stosowania preparatu SUTENT. Należy również rozważyć wykonywanie oznaczeń LVEF na początku terapii i okresowo w trakcie leczenia. U pacjentów, u których nie występują kardiologiczne czynniki ryzyka należy rozważyć wykonanie wyjściowej oceny frakcji wyrzutowej. W przypadku wystąpienia objawów klinicznych zastoinowej niewydolności serca zaleca się przerwanie stosowania preparatu SUTENT. U pacjentów bez klinicznych objawów zastoinowej niewydolności serca, ale z frakcją wyrzutową <50% i >20% poniżej wartości wyjściowej należy przerwać stosowanie preparatu lub obniżyć dawkę. Wydłużenie odcinka QT Wydłużenie odcinka QT oceniano w badaniu 24 pacjentów w wieku od 20 do 87 lat z zaawansowanymi nowotworami. Wykazano, że preparat SUTENT, w stężeniach w przybliżeniu dwukrotnie większych niż terapeutyczne, wydłuża odcinek QTcF (z korektą Friederica). U żadnego z pacjentów nie stwierdzono wydłużenia odcinka QT/QTc stopnia ciężkości większego niż 2 (CTCAEvv 3.0), nie odnotowano także zaburzeń rytmu serca. Znaczenie kliniczne zaobserwowanych działań jest niejasne i uzależnione od indywidualnych czynników ryzyka i wrażliwości pacjentów. Należy zachować ostrożność podczas stosowania preparatu SUTENT u pacjentów z wydłużonym odcinkiem QT w wywiadzie, pacjentów przyjmujących leki przeciwarytmiczne oraz u pacjentów z istotnymi chorobami serca w wywiadzie, bradykardią lub zaburzeniami elektrolitowymi. Należy zachować ostrożność podczas jednoczesnego podawania silnych inhibitorów CYP3A4, które mogą powodować zwiększenie stężenia sunitynibu w osoczu, a w razie potrzeby należy zmniejszyć dawkę preparatu SUTENT (patrz punkt 4.5). Żylne zaburzenia zakrzepowo-zatorowe W dwóch badaniach w raku nerki z przerzutami stwierdzono wystąpienie żylnych powikłań zakrzepowo-zatorowych u czterech pacjentów (2%); u dwóch z nich doszło do zatoru tętnicy płucnej (4 stopnia ciężkości), a u dwóch pozostałych – do zakrzepicy żył głębokich (3 stopnia ciężkości). W jednym z tych przypadków przerwano stosowanie leku. W badaniu rejestracyjnym nad nowotworami podścieliskowymi przewodu pokarmowego u siedmiu pacjentów (3%) otrzymujących preparat SUTENT wystąpiły żylne powikłania zakrzepowo-zatorowe natomiast nie wystąpiły one u żadnego z pacjentów z grupy otrzymującej placebo. U pięciu z siedmiu pacjentów powikłania te były 3 stopnia ciężkości, a u dwóch – 1 lub 2 stopnia ciężkości. Czterech pacjentów z nowotworami podścieliskowymi przewodu pokarmowego przerwało leczenie po wystąpieniu pierwszych objawów zakrzepicy żył głębokich. Zator tętnicy płucnej U około 1,1% pacjentów z guzami litymi, którzy otrzymywali preparat SUTENT wystąpił związany z leczeniem zator tętnicy płucnej. Żadne z tych zdarzeń nie doprowadziło do przerwania leczenia, jednak w kilku przypadkach zmniejszono dawkę lub tymczasowo odroczono leczenie. Po podjęciu leczenia nie doszło do ponownego zatoru tętnicy płucnej u tych pacjentów. Niedoczynność tarczycy W dwóch badaniach w raku nerki z przerzutami niedoczynność tarczycy była zgłaszana jako zdarzenie niepożądane u siedmiu pacjentów (4%). Ponadto u czterech pacjentów (2%) stwierdzono podwyższenie stężenia TSH. Ogółem u 7% populacji objętej tymi badaniami stwierdzono kliniczne lub laboratoryjne objawy niedoczynności tarczycy, które wystąpiły w trakcie leczenia. U ośmiu pacjentów (4%) z nowotworami podścieliskowymi przewodu pokarmowego otrzymujących preparat SUTENT i u jednego pacjenta (1%) otrzymującego placebo stwierdzono w czasie leczenia niedoczynność tarczycy wymagającą pilnej terapii. U pacjentów z objawami sugerującymi niedoczynność tarczycy konieczne jest wykonywanie kontrolnych badań laboratoryjnych diagnozujących czynność tego gruczołu i zastosowanie leczenia według obowiązujących standardów. Czynność trzustki U pacjentów z różnymi guzami litymi otrzymujących preparat SUTENT obserwowano zwiększenie aktywności lipazy i amylazy w surowicy. Wzrost aktywności lipazy był przemijający i na ogół nie towarzyszyły mu objawy podmiotowe i przedmiotowe zapalenia trzustki. Zapalenie trzustki

21
obserwowano u 0,4% pacjentów z guzami litymi. W razie wystąpienia objawów zapalenia trzustki należy zapewnić pacjentom właściwą opiekę medyczną. Napady drgawkowe W badaniach klinicznych z zastosowaniem preparatu SUTENT obserwowano występowanie drgawek u pacjentów z cechami radiologicznymi przerzutów nowotworu do mózgu. Ponadto stwierdzano rzadkie (<1%) przypadki drgawek i objawy radiologiczne zespołu odwracalnej tylnej leukoencefalopatii (RPLS). W żadnym z omawianych przypadków zaburzenia te nie doprowadziły do zgonu. U pacjentów z drgawkami i objawami podmiotowymi/przedmiotowymi sugerującymi obecność RPLS, takimi jak nadciśnienie, ból głowy, zwiększona czujność, zmiany psychiczne i utrata wzroku, w tym ślepota korowa, należy kontrolować występujące zaburzenia poprzez zastosowanie właściwego leczenia, w tym farmakologicznej kontroli nadciśnienia. Zaleca się tymczasowe przerwanie stosowania preparatu SUTENT. Po ustąpieniu objawów lekarz prowadzący może podjąć decyzję o kontynuacji leczenia. 4.5 Interakcje z innymi lekami i inne rodzaje interakcji Leki, które mogą zwiększać stężenie sunitynibu w osoczu: Równoczesne podawanie sunitynibu z silnym inhibitorem izoenzymu CYP3A4, ketokonazolem, powodowało po jednorazowym podaniu jabłczanu sunitynibu u zdrowych ochotników wzrost wartości Cmax i AUC0-∞ kompleksu [sunitynib + podstawowy metabolit] odpowiednio o 49% i 51%. Podawanie preparatu SUTENT jednocześnie z silnymi inhibitorami izoenzymów z rodziny CYP3A4 (np. rytonawir, itrakonazol, erytromycyna, klarytromycyna, sok grejpfrutowy) może wiązać się ze zwiększeniem stężenia sunitynibu. Z tego względu należy unikać podawania go jednocześnie z inhibitorami enzymatycznymi lub należy rozważyć wybranie do leczenia skojarzonego alternatywnego preparatu, o minimalnym działaniu hamującym enzym CYP3A4 lub bez takiego działania. Jeżeli nie jest to możliwe, może zaistnieć konieczność zmniejszenia dawek preparatu SUTENT do minimum 37,5 mg na dobę, na podstawie dokładnie monitorowanej tolerancji (patrz punkt 4.2). Leki, które mogą zmniejszyć stężenie sunitynibu w osoczu: Równoczesne podawanie jabłczanu sunitynibu z silnym induktorem izoenzymu CYP3A4, ryfampicyną, powodowało po jednorazowym podaniu jabłczanu sunitynibu u zdrowych ochotników redukcję wartości Cmax i AUC0-∞ kompleksu [sunitynib + podstawowy metabolit] odpowiednio o 23% i 46%]. Podawanie preparatu SUTENT z silnymi induktorami izoenzymów z grupy CYP3A4 (np. deksametazon, fenytoina, karbamazepina, ryfampicyna, fenobarbital lub ziele dziurawca Hypericum perforatum) może prowadzić do zmniejszenia stężenia sunitynibu. Z tego względu należy unikać jednoczesnego stosowania preparatu z induktorami enzymatycznymi, bądź też należy rozważyć wybranie do leczenia skojarzonego alternatywnego preparatu, o minimalnym działaniu indukującym enzym CYP3A4 lub bez takiego działania. Jeżeli nie jest to możliwe, może zaistnieć konieczność stopniowego zwiększania dawek preparatu SUTENT, za każdym razem o 12,5 mg (do 87,5 mg na dobę) na podstawie dokładnie monitorowanej tolerancji (patrz punkt 4.2). Aby utrzymać docelowe stężenie sunitynibu należy rozważyć wybranie do leczenia skojarzonego preparatów indukujących enzym w mniejszym stopniu. Jeżeli nie jest to możliwe, może być konieczne dostosowanie dawki preparatu SUTENT (patrz punkt 4.2). U pacjentów leczonych preparatem SUTENT rzadko obserwowano krwotoki (patrz punkt 4.4). Pacjenci otrzymujący równocześnie preparaty przeciwzakrzepowe (np. warfaryna, acenokumarol) mogą być poddawani okresowym kontrolom obejmującym wykonanie morfologii krwi (z oznaczeniem liczby płytek krwi), badanie czynników krzepnięcia (czas protrombinowy (PT)/wskaźnik INR) i badaniu przedmiotowemu. 4.6 Ciąża i laktacja Ciąża Nie przeprowadzono badań dotyczących stosowania preparatu SUTENT u kobiet w ciąży. W badaniach na zwierzętach stwierdzono toksyczny wpływ leku na zdolność do rozrodu, objawiający się m.in.

22
wadami wrodzonymi płodów (patrz punkt 5.3). Preparatu nie należy stosować w czasie ciąży lub u kobiet, które nie stosują skutecznej metody antykoncepcyjnej z wyjątkiem przypadku, gdy potencjalne korzyści z leczenia przeważą nad potencjalnym ryzykiem dla płodu. Jeżeli preparat zostanie zastosowany w czasie ciąży lub jeżeli pacjentka zajdzie w ciążę w trakcie jego stosowania, należy ją poinformować o potencjalnym zagrożeniu dla płodu. Kobietom w wieku rozrodczym należy doradzić stosowanie skutecznej metody antykoncepcyjnej podczas przyjmowania preparatu SUTENT. Dane niekliniczne wskazują, że leczenie preparatem SUTENT może wywierać niekorzystny wpływ na płodność kobiet i mężczyzn (patrz punkt 5.3).
Laktacja
Sunitynib i (lub) jego metabolity przenikają do mleka szczurów. Nie wiadomo, czy sunitynib lub jego główny czynny metabolit przenikają do mleka kobiecego. Ze względu na to, że leki są często wydzielane do mleka kobiecego i ze względu na możliwe występowanie ciężkich działań niepożądanych u niemowląt karmionych piersią, kobiety nie powinny karmić piersią podczas stosowania preparatu SUTENT. 4.7 Wpływ na zdolność prowadzenia pojazdów mechanicznych i obsługiwania urządzeń
mechanicznych w ruchu Nie przeprowadzono badań nad wpływem preparatu na zdolność prowadzenia pojazdów mechanicznych i obsługiwania urządzeń mechanicznych w ruchu. Pacjenci powinni być poinformowani o możliwości wystąpienia zawrotów głowy w trakcie leczenia preparatem SUTENT. 4.8 Działania niepożądane Najczęstszymi ciężkimi zdarzeniami niepożądanymi związanymi z leczeniem preparatem SUTENT u pacjentów z guzami litymi były: zator tętnicy płucnej (1%), trombocytopenia (1%), krwotok z guza (0,9%), gorączka neutropeniczna (0,4%) i nadciśnienie tętnicze (0,4%). Do najczęstszych zdarzeń niepożądanych związanych z leczeniem (wystąpiły u co najmniej 20% pacjentów) o dowolnym stopniu nasilenia należały: uczucie zmęczenia; zaburzenia ze strony przewodu pokarmowego, takie jak biegunka, nudności, zapalenie jamy ustnej, objawy dyspeptyczne i wymioty; przebarwienia skórne; zaburzenia smaku i anoreksja. Uczucie zmęczenia, nadciśnienie tętnicze i neutropenia były najczęstszymi związanymi z leczeniem zdarzeniami niepożądanymi o maksymalnym stopniu ciężkości 3 u pacjentów z guzami litymi, a podwyższona aktywność lipazy była najczęstszym związanym z leczeniem zdarzeniem niepożądanym o maksymalnym stopniu ciężkości 4. Poniżej wymieniono działania niepożądane, które zostały zgłoszone przez >5% pacjentów z guzami litymi, według klas układowo-narządowych, częstości występowania i stopnia ciężkości. W ramach każdej grupy wyodrębnionej wg częstości występowania działania niepożądane przedstawiono w kolejności od najcięższego do najlżejszego. Według częstości występowania zdarzenia definiowano jako: bardzo częste (> 1/10), częste (od >1/100 do < 1/10), nieczęste (od >1/1000 do <1//100), rzadkie (od >1/10 000 do 1/1000), bardzo rzadkie (< 1/10 000). Działania niepożądane zgłaszane w badaniach u pacjentów z nowotworami podścieliskowymi przewodu pokarmowego. Klasyfikacja układów i narządów
Częstość występowania
Działania niepożądane
Wszystkich stopni n (%)
Stopnia 3 n (%)
Stopnia 4 n (%)
Bardzo często Niedokrwistość 33 (12,8%) 13 (5,1%) 1 (0,4%) Często Neutropenia 24 (9,3%) 15 (5,8%) 1 (0,4%)
Zaburzenia krwi i układu chłonnego Często Trombocytopenia 23 (8,9%) 6 (2,3%) 1 (0,4%)

23
Klasyfikacja układów i narządów
Częstość występowania
Działania niepożądane
Wszystkich stopni n (%)
Stopnia 3 n (%)
Stopnia 4 n (%)
Zaburzenia endokrynolo- giczne
Często Niedoczynność tarczycy
15 (5,8%) 0 (0,0%) 1 (0,4%)
Zaburzenia metabolizmu i odżywiania
Często Zanik łaknienia 44 (7,1%) 1 (0,4%) 0 (0,0%)
Bardzo często Zaburzenia smaku 48 (18,7%) 0 (0,0%) 0 (0,0%) Zaburzenia układu nerwowego
Bardzo często Bóle głowy 27 (10,5%) 2 (0,8%) 0 (0,0%)
Zaburzenia naczyń
Bardzo często Nadciśnienie tętnicze
43 (16,7%) 18 (7,0%) 0 (0,0%)
Zaburzenia oddechowe, klatki piersiowej i śródpiersia
Często Krwotok z nosa 17 (6,6%) 0 (0,0%) 0 (0,0%)
Zaburzenia nerek i dróg moczowych
Często Zmiana barwy moczu
13 (5,1%) 0 (0,0%) 0 (0,0%)
Bardzo często Biegunka 90 (35,0%) 13 (5,1%) 0 (0,0%) Bardzo często Nudności 69 (26,8%) 2 (0,8%) 0 (0,0%) Bardzo często Zapalenie błony
śluzowej jamy ustnej
49 (19,1%) 2 (0,8%) 0 (0,0%)
Bardzo często Wymioty 46 (17,9%) 1 (0,4%) 0 (0,0%) Bardzo często Objawy
dyspeptyczne 32 (12,5%) 2 (0,8%) 0 (0,0%)
Często Pieczenie języka 17 (6,6%) 0 (0,0%) 0 (0,0%) Często Zaparcia 13 (5,1%) 1 (0,4%) 0 (0,0%) Bardzo często Bóle brzucha* 30 (11,7%) 5 (1,9%) 1 (0,4%) Często Ból w jamie ustnej 16 (6,2%) 0 (0,0%) 0 (0,0%) Często Wzdęcie z
oddawaniem gazów 15 (5,8%) 0 (0,0%) 0 (0,0%)
Często Suchość w jamie ustnej
15 (5,8%) 0 (0,0%) 0 (0,0%)
Zaburzenia żołądkowo-jelitowe
Często Choroba refluksowa przełyku
15 (5,8%) 0 (0,0%) 0 (0,0%)
Bardzo często Przebarwienia skórne
65 (25,3%) 0 (0,0%) 0 (0,0%)
Bardzo często Rumień i zaburzenia czucia dłoni i podeszew stóp
55 (21,4%) 14 (5,4%) 0 (0,0%)
Bardzo często Wysypka*** 39 (15,2%) 2 (0,8%) 0 (0,0%) Często Zmiany koloru
włosów 22 (8,6%) 0 (0,0%) 0 (0,0%)
Zaburzenia skóry i tkanki podskórnej
Często Suchość skóry 15 (5,8%) 0 (0,0%) 0 (0,0%) Często Ból w kończynach 21 (8,2%) 1 (0,4%) 0 (0,0%) Zaburzenia
mięśniowo- Często Bóle stawowe 15 (5,8%) 2 (0,8%) 0 (0,0%)

24
Klasyfikacja układów i narządów
Częstość występowania
Działania niepożądane
Wszystkich stopni n (%)
Stopnia 3 n (%)
Stopnia 4 n (%)
szkieletowe, tkanki łącznej i kości
Często Bóle mięśniowe 13 (5,1%) 0 (0,0%) 0 (0,0%)
Bardzo często Uczucie zmęczenia/ osłabienie
135 (52,5%) 25 (9,7%) 0 (0,0%) Zaburzenia ogólne i stany w miejscu podania Bardzo często Zapalenie błon
śluzowych 30 (11,7%) 0 (0,0%) 0 (0,0%)
Często Obrzęk** 21 (8,2%) 1 (0,4%) 0 (0,0%) Często Zmniejszenie
stężenia hemoglobiny
16 (6,2%) 2 (0,8%) 0 (0,0%)
Badania dodatkowe
Często Zwiększenie aktywności fosfokinazy kreatynowej we krwi
14 (5,4%) 0 (0,0%) 0 (0,0%)
Często Zmniejszenie frakcji wyrzutowej
13 (5,1%) 1 (0,4%) 0 (0,0%)
Często Zwiększenie aktywności lipazy
13 (5,1%) 5 (1,9%) 4 (1,6%)
Często Zmniejszenie liczby płytek krwi
13 (5,1%) 2 (0,8%) 1 (0,4%)
Dowolne zdarzenie niepożądane
222 (86,4%) 88 (34,2%) 24 (9,3%)
*Następujące zdarzenia zostały połączone w jedną kategorię: bóle brzucha, bóle w górnej części brzucha i bóle w dolnej części brzucha. **Następujące zdarzenia zostały połączone w jedną kategorię: obrzęki i obrzęki obwodowe. ***Następujące zdarzenia zostały połączone w jedną kategorię: wysypka, wysypka rumieniowa, wysypka plamista i wysypka złuszczająca się Działania niepożądane zgłaszane w badaniach u pacjentów z rakiem nerki z przerzutami Klasyfikacja układów i narządów
Częstość występowania
Działania niepożądane
Wszystkich stopni n (%)
Stopnia 3 n (%)
Stopnia 4 n (%)
Bardzo często Neutropenia 17 (10,1%) 8 (4,7%) 1 (0,6%) Często Niedokrwistość 16 (9,5%) 6 (3,6%) 0 (0,0%) Często Trombocytopenia 15 (8,9%) 5 (3,0%) 2 (1,2%)
Zaburzenia krwi i układu chłonnego
Często Leukopenia 14 (8,3%) 7 (4,1%) 0 (0,0%)
Zaburzenia oka Często Zwiększone łzawienie
9 (5,3%) 0 (0,0%) 0 (0,0%)
Zaburzenia metabolizmu i odżywiania
Bardzo często Zanik łaknienia 47 (27,8%) 1 (0,6%) 0 (0,0%)
Często Odwodnienie 12 (7,1%) 4 (2,4%) 0 (0,0%) Często Zmniejszenie
łaknienia 11 (6,5%) 0 (0,0%) 0 (0,0%)
Bardzo często Zaburzenia smaku 71 (42%) 0 (0,0%) 0 (0,0%) Bardzo często Bóle głowy 25 (14,8%) 1 (0,6%) 0 (0,0%) Często Zawroty głowy 13 (7,7%) 2 (1,2%) 0 (0,0%)
Zaburzenia układu nerwowego
Często Parestezje 9 (5,3%) 0 (0,0%) 0 (0,0%)

25
Klasyfikacja układów i narządów
Częstość występowania
Działania niepożądane
Wszystkich stopni n (%)
Stopnia 3 n (%)
Stopnia 4 n (%)
Zaburzenia naczyń
Bardzo często Nadciśnienie tętnicze
28 (16,6%) 7 (4,1%) 0 (0,0%)
Często Krwotok z nosa 16 (9,5%) 0 (0,0%) 0 (0,0%) Zaburzenia oddechowe, klatki piersiowej i śródpiersia
Często Duszność 9 (5,3%) 0 (0,0%) 0 (0,0%)
Bardzo często Biegunka 83 (49,1%) 5 (3,0%) 0 (0,0%) Bardzo często Nudności 84 (49,7%) 2 (1,2%) 0 (0,0%) Bardzo często Zapalenie błony
śluzowej jamy ustnej
70 (41,4%) 6 (3,6%) 0 (0,0%)
Bardzo często Objawy dyspeptyczne
69 (40,8%) 1 (0,6%) 0 (0,0%)
Bardzo często Wymioty 52 (30,8%) 2 (1,2%) 0 (0,0%) Bardzo często Zaparcia 34 (20,1%) 0 (0,0%) 0 (0,0%)
Zaburzenia żołądkowo-jelitowe
Bardzo często Pieczenie języka 25 (14,8%) 0 (0,0%) 0 (0,0%) Bardzo często Bóle brzucha* 17 (10,1%) 2 (1,2%) 0 (0,0%)
Często Wzdęcie z oddawaniem gazów
16 (9,5%) 0 (0,0%) 0 (0,0%)
Często Uczucie rozpierania w jamie brzusznej
9 (5,3%) 0 (0,0%) 0 (0,0%)
Często Suchość w jamie ustnej
9 (5,3%) 0 (0,0%) 0 (0,0%)
Bardzo często Przebarwienia skórne
54 (32,0%) 0 (0,0%) 0 (0,0%)
Bardzo często Wysypka** 46 (27,2%) 0 (0,0%) 0 (0,0%) Bardzo często Zmiana barwy
włosów 24 (14,2%) 0 (0,0%) 0 (0,0%)
Bardzo często Rumień i zaburzenia czucia dłoni i podeszew stóp
21 (12,4%) 6 (3,6%) 0 (0,0%)
Często Łysienie 13 (7,7%) 0 (0,0%) 0 (0,0%) Często Złuszczające
zapalenie skóry 10 (5,9%) 2 (1,2%) 0 (0,0%)
Często Obrzęki okołooczodołowe
9 (5,3%) 0 (0,0%) 0 (0,0%)
Bardzo często Suchość skóry 22 (13,0%) 0 (0,0%) 0 (0,0%)
Zaburzenia skóry i tkanki podskórnej
Bardzo często Rumień 20 (11,8%) 0 (0,0%) 0 (0,0%) Bardzo często Ból w kończynach 21 (12,4) 1 (0,6%) 0 (0,0%) Zaburzenia
mięśniowo-szkieletowe, tkanki łącznej i kości
Często Bóle mięśni 15 (8,9%) 1 (0,6%) 0 (0,0%)
Bardzo często Uczucie zmęczenia/osłabie- nie
108 (63,9%) 19 (11,2%) 0 (0,0%) Zaburzenia ogólne i stany w miejscu podania Bardzo często Zapalenie błon
śluzowych 30 (17,8%) 1 (0,6%) 0 (0,0%)

26
Klasyfikacja układów i narządów
Częstość występowania
Działania niepożądane
Wszystkich stopni n (%)
Stopnia 3 n (%)
Stopnia 4 n (%)
Urazy, zatrucia, procedury medyczne i chirurgiczne
Bardzo często Pęcherze 7 (11,1%) 2 (3,2%) 0 (0,0%)
Bardzo często Zwiększenie aktywności lipazy
17 (10,1%) 12 (7,1%) 3 (1,8%)
Często Zaburzenia frakcji wyrzutowej
16 (9,5%) 1 (0,6%) 0 (0,0%)
Badania dodatkowe
Często Zwiększenie aktywności amylazy we krwi
9 (5,3%) 6 (3,6%) 0 (0,0%)
Często Zmniejszenie masy ciała
11 (6,5%) 0 (0,0%) 0 (0,0%)
Często Zmniejszenie liczby leukocytów
10 (5,9%) 3 (1,8%) 0 (0,0%)
Często Zmniejszenie liczby płytek krwi
9 (5,3%) 3 (1,8%) 2 (1,2%)
Dowolne zdarzenie niepożądane
166 (98,2%) 77 (45,6%) 14 (8,3%)
*Następujące zdarzenia zostały połączone w jedną kategorię: bóle brzucha, bóle w górnej części brzucha i bóle w dolnej części brzucha. **Następujące zdarzenia zostały połączone w jedną kategorię: wysypka, wysypka rumieniowa, wysypka grudkowa, wysypka uogólniona, wysypka grudkowa i wysypka ze świądem. 4.9 Przedawkowanie Nie ma doświadczeń z ostrym przedawkowaniem preparatu SUTENT. Nie istnieje swoista odtrutka, a leczenie przedawkowania powinno polegać na zastosowaniu standardowych środków wspomagających. O ile istnieją wskazania, można uzyskać eliminację niewchłoniętego preparatu poprzez wywołanie wymiotów lub płukanie żołądka. 5. WŁAŚCIWOŚCI FARMAKOLOGICZNE Grupa farmakoterapeutyczna: Leki przeciwnowotworowe - Inhibitor kinazy białkowo – tyrozynowej. Kod ATC: LO1XE04 5.1 Właściwości farmakodynamiczne Jabłczan sunitynibu hamuje liczne receptory kinazy tyrozynowej (RTK), które biorą udział we wzroście nowotworów, w patologicznej angiogenezie i w rozsiewie choroby nowotworowej z przerzutami. Sunitynib został zidentyfikowany jako inhibitor receptorów płytkowego czynnika wzrostu (PDGFRα i PDGFRβ), receptorów czynników wzrostu śródbłonka naczyniowego (VEGFR1, VEGFR2 i VEGFR3), receptorów czynnika komórek pnia (KIT), kinazy tyrozynowej podobnej do Fms-3 (FLT3), receptorów czynnika stymulującego powstawanie kolonii (CSF-1R) i receptorów glejopochodnego czynnika neurotroficznego (RET). W testach biochemicznych i komórkowych podstawowy metabolit sunitynibu wykazuje działanie podobne do sunitynibu. BADANIA KLINICZNE

27
Badano bezpieczeństwo kliniczne i skuteczność kliniczną preparatu SUTENT podczas leczenia pacjentów ze złośliwymi nowotworami podścieliskowymi przewodu pokarmowego (GIST) opornymi na imatynib (tj. pacjentów, u których doszło do progresji choroby w trakcie leczenia imatynibem lub po nim) lub nietolerujących imatynibu (tj. pacjentów, u których wystąpiły istotne objawy toksyczności w trakcie leczenia imatynibem, które uniemożliwiły dalsze leczenie) oraz leczenia pacjentów z rakiem nerki z przerzutami po niepowodzeniu leczenia z zastosowaniem cytokin. Skuteczność leczenia ustalano na podstawie czasu wystąpienia progresji guza oraz wydłużenia czasu całkowitego przeżycia u pacjentów z nowotworami podścieliskowymi przewodu pokarmowego oraz na podstawie wskaźników obiektywnych odpowiedzi u pacjentów z rakiem nerki z przerzutami. Nowotwory podścieliskowe przewodu pokarmowego Wstępne otwarte, zakładające zwiększanie dawkowania badanie zostało przeprowadzone u pacjentów z nowotworami podścieliskowymi przewodu pokarmowego po niepowodzeniu stosowania imatynibu (mediana maksymalnej dawki dobowej: 800 mg) ze względu na oporność lub nietolerancję leczenia. Do badania zakwalifikowano dziewięćdziesięciu siedmiu pacjentów, otrzymujących dawki według różnych schematów; 55 pacjentów otrzymywało preparat w dawce 50 mg według zalecanego schematu leczenia: 4 tygodnie przyjmowania leku/2 tygodnie przerwy („schemat 4/2”). W niniejszym badaniu mediana czasu do progresji choroby (Time To Progression, TTP) wynosiła 34,0 tygodnie (95% CI = 22,0–46,0 tygodnia). Przeprowadzono randomizowane, podwójnie ślepe, kontrolowane placebo badanie fazy III preparatu SUTENT u pacjentów z nowotworami podścieliskowymi przewodu pokarmowego, którzy nie tolerowali leczenia imatynibem lub, u których doszło do progresji choroby w trakcie leczenia imatynibem (mediana maksymalnej dawki dobowej 800 mg). W badaniu tym 312 pacjentów w sposób randomizowany (w stosunku 2:1) przydzielono do grup otrzymujących odpowiednio preparat SUTENT w dawce 50 mg lub placebo doustnie, raz na dobę, według schematu 4/2, do momentu wystąpienia progresji choroby lub wycofania z badania z innego powodu (207 pacjentów otrzymywało SUTENT, a 105 pacjentów otrzymywało placebo). Podstawowym punktem końcowym oceny skuteczności w badaniu był TTP definiowany jako czas od randomizacji do pierwszego potwierdzenia obiektywnej progresji guza. Mediana TTP podczas stosowania preparatu SUTENT wynosiła 28,9 tygodnia (95% CI = 21,3–34,1 tygodnia) i była istotnie statystycznie dłuższa niż TTP wynoszący 5,1 tygodnia (95% CI = 4,4–10,1 tygodnie) podczas stosowania placebo. Różnica przeżywalności całkowitej była statystycznie większa na korzyść preparatu SUTENT [współczynnik ryzyka: 0,491 95% (C.I. 0,290- 0,831)]; ryzyko zgonu było 2 razy większe u pacjentów w grupie otrzymującej placebo w porównaniu do grupy leczonej preparatem SUTENT. Odsetki zgonów wynosiły 14% u osób otrzymujących SUTENT w porównaniu do 25% u osób otrzymujących placebo. W momencie przeprowadzania analizy nie osiągnięto jeszcze mediany przeżywalności ogólnej w żadnej z grup terapeutycznych. Rak nerki z przerzutami oporny na leczenie cytokinami (MRCC) Badanie II fazy preparatu SUTENT przeprowadzono u pacjentów opornych na wcześniejsze leczenie cytokinami, tzn. interleukiną-2 lub interferonem-α. Sześćdziesięciu trzech pacjentów otrzymało dawkę początkową 50 mg preparatu SUTENT doustnie, raz na dobę przez 4 kolejne tygodnie, po czym następował dwutygodniowy okres przerwy, który kończył pełny cykl 6 tygodni (schemat 4/2). Podstawowym punktem końcowym oceny skuteczności był wskaźnik obiektywnych odpowiedzi na leczenie (objective response rate, ORR) ustalany na podstawie kryteriów oceny odpowiedzi u pacjentów z guzami litymi (Response Evaluation Criteria in Solid Tumours, RECIST). W omawianym badaniu wskaźnik obiektywnych odpowiedzi wynosił 36,5% (95% C.I. 24,7% – 49,6%), a mediana czasu do progresji choroby (TTP) wynosiła 37,7 tygodnia (95% C.I. 24,0 – 46,4 tygodnia). Przeprowadzono potwierdzające, otwarte, obejmujące jedną grupę pacjentów, wieloośrodkowe badanie oceniające skuteczność i bezpieczeństwo stosowania preparatu SUTENT u pacjentów z rakiem nerki z przerzutami, którzy byli oporni na uprzednio stosowane leczenie cytokinami. Stu sześciu pacjentów otrzymało co najmniej jedną dawkę 50 mg preparatu SUTENT według schematu 4/2.

28
Pierwszorzędowym punktem końcowym oceny skuteczności tego badania był wskaźnik obiektywnych odpowiedzi (ORR). Drugorzędowe punkty końcowe obejmowały TTP, okres utrzymywania się odpowiedzi (duration of response, DR) i przeżycie całkowite (overall survival, OS). W niniejszym badaniu wskaźnik ORR wynosił 38% (95% C.I. 26,8% – 47,5 %). Mediany DR i OS nie zostały jeszcze osiągnięte. Ten produkt leczniczy został dopuszczony do obrotu zgodnie z procedurą „warunkowego dopuszczenia”. Oznacza to, że oczekiwane są dalsze dowody świadczące o korzyści z jego stosowania, w szczególności dane dotyczące wpływu preparatu SUTENT na medianę czasu do wystąpienia progresji u pacjentów z rakiem nerki z przerzutami. Badanie oceniające ten parametr jest w toku. Europejska Agencja ds. Produktów Leczniczych będzie dokonywać przeglądu nowych informacji o produkcie raz do roku i ChPL zostanie uzupełniona tak jak to będzie konieczne. 5.2 Właściwości farmakokinetyczne Właściwości farmakokinetyczne sunitynibu i jabłczanu sunitynibu oceniano w badaniach obejmujących 135 zdrowych ochotników i 266 pacjentów z guzami litymi. Wchłanianie Po podaniu doustnym sunitynibu maksymalne stężenie (Cmax) stwierdza się na ogół po upływie 6 – 12 godzin (Tmax). Pokarm nie wpływa na dostępność biologiczną sunitynibu. Dystrybucja W badaniach in vitro stopień wiązania się sunitynibu i jego podstawowego czynnego metabolitu z ludzkimi białkami osocza wynosił odpowiednio 95% i 90% niezależnie od stężenia. Pozorna objętość dystrybucji (V/F) w przypadku sunitynibu była znaczna – 2230 l, co wskazuje na penetrację leku do tkanek. Metabolizm Obliczone in vitro wartości stałej Ki dla wszystkich badanych izoform enzymu CYP (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 I CYP4A9/11) wskazują na to, że sunitynib i jego podstawowy czynny metabolit prawdopodobnie nie hamują w stopniu istotnym klinicznie metabolizmu preparatów metabolizowanych przez te enzymy. Badania in vitro również wskazują na to, że SUTENT nie indukuje i nie hamuje głównych enzymów CYP, w tym CYP3A4. Biotransformacja Sunitynib jest metabolizowany przede wszystkim przez izoenzym CYP3A4 cytochromu P450, który katalizuje reakcję powstawania głównego czynnego metabolitu podlegającego dalszemu metabolizmowi katalizowanemu przez CYP3A4. Równoczesne podawanie preparatu SUTENT z silnym induktorem CYP3A4 – ryfampicyną powodowało zmniejszenie wartości Cmax i AUC0-∞ sunitynibu o odpowiednio 56% i 78% po podaniu pojedynczej dawki preparatu SUTENT zdrowym ochotnikom. Inne induktory grupy enzymów CYP3A4 (np. deksametazon, fenytoina, karbamazepina, fenobarbital lub ziele dziurawca Hypericum perforatum) podawane jednocześnie z preparatem SUTENT mogą powodować zmniejszenie stężenia sunitynibu. Eliminacja Preparat jest wydalany przede wszystkim z kałem (61%), natomiast przez nerki ulega wydaleniu 16% podanej dawki w postaci niezmienionej i metabolitów. Sunitynib i jego podstawowy czynny metabolit były głównymi związkami pochodnymi leku wykrywanymi w osoczu, moczu i kale, odpowiadając odpowiednio za 91,5%, 86,4% i 73,8% radioaktywności w zebranych próbkach. Pozostałe metabolity o mniejszym znaczeniu zostały zidentyfikowane w moczu i w kale, jednak na ogół nie były wykrywane w osoczu. Całkowity klirens leku po podaniu doustnym (CL/F) wynosił 34–62 l/h. Zaburzenia czynności narządów

29
Niewydolność wątroby: Nie przeprowadzono badań klinicznych nad stosowaniem leku u pacjentów z niewydolnością wątroby. Z badań wykluczano pacjentów z aktywnością AlAT lub AspAT przekraczającą o ponad 2,5 x górną granicę normy lub, gdy podwyższenie to wiązało się z chorobą podstawową – z aktywnością tych enzymów przekraczającą o ponad 5,0 x górną granicę normy. Niewydolność nerek: Nie przeprowadzono badań klinicznych u pacjentów z niewydolnością nerek. Z badań wykluczano pacjentów ze stężeniem kreatyniny w surowicy przekraczającym o ponad 2,0 x górną granicę normy. Populacyjne analizy farmakokinetyki wykazały, klirens pozorny sunitynibu (CL/F) nie jest uzależniony od klirensu kreatyniny w ocenianym zakresie stężeń (42–347 ml/min). Farmakokinetyka w osoczu Po podaniu doustnym zdrowym ochotnikom okres półtrwania sunitynibu i jego podstawowego czynnego metabolitu deetylowego wynosił odpowiednio około 40–60 godzin i 80–110 godzin. W zakresie dawkowania od 25 do 100 mg pole pod krzywą zależności stężenia leku w osoczu od czasu (AUC) i Cmax zwiększa się proporcjonalnie do dawki. Podczas kilkakrotnego podania w ciągu doby sunitynib ulega kumulacji, przy czym jego stężenie zwiększa się 3–4-krotnie, a stężenie jego podstawowego czynnego metabolitu zwiększa się 7–10-krotnie. Stężenie sunitynibu i jego podstawowego czynnego metabolitu w stanie równowagi zostaje osiągnięte w ciągu od 10 do 14 dni. Do 14 dnia łączne stężenie osoczowe sunitynibu i jego podstawowego czynnego metabolitu wynosi 62,9–101 ng/ml, co jest docelowym stężeniem przewidywanym na podstawie danych z badań przedklinicznych jako stężenie hamujące fosforylację receptorów in vitro, prowadzącym do zatrzymania/zmniejszenia wzrostu guzów in vivo. Podstawowy czynny metabolit odpowiada za 23–37% całkowitej ekspozycji na lek. Nie obserwuje się istotnych zmian farmakokinetyki sunitynibu lub jego podstawowego czynnego metabolitu podczas kilkakrotnego podawania leku w ciągu doby lub w trakcie powtarzanych cyklów badanych schematów dawkowania. Parametry farmakokinetyczne były podobne we wszystkich badanych populacjach pacjentów z guzami litymi i u zdrowych ochotników. Populacyjne analizy farmakokinetyki z uwzględnieniem danych demograficznych wskazują na to, że nie są koniecznie korekty dawkowania w zależności od masy ciała lub punktacji wg skali ECOG. Dostępne dane wskazują, że u kobiet klirens pozorny (CL/F) sunitynibu może być o około 30% niższy niż u mężczyzn: różnica ta nie wymaga jednak dostosowywania dawki. 5.3 Przedkliniczne dane o bezpieczeństwie W dziewięciomiesięcznych badaniach nad toksycznością dawek wielokrotnych u szczurów i małp stwierdzono, że podstawowymi narządami, na które wpływa lek, są: przewód pokarmowy (nudności i biegunki u małp), nadnercza (przekrwienie kory i (lub) krwotoki u szczurów i małp, z martwicą i następującym po niej włóknieniem u szczurów), układ limfatyczny i krwiotwórczy (zmniejszenie liczby komórek szpiku kostnego i zanik tkanki limfoidalnej grasicy, śledziony i węzłów chłonnych), zewnątrzwydzielnicza część trzustki (degranulacja komórek pęcherzykowych z martwicą pojedynczych komórek), ślinianki (przerost gronek), stawy (zgrubienie płytki wzrostu), macica (zanik) i jajniki (zmniejszony wzrost pęcherzyków). Wszystkie wyniki uzyskano przy istotnych klinicznie poziomach stężenia osoczowego sunitynibu. Dodatkowe działania preparatu obserwowane w innych badaniach obejmowały: wydłużenie odstępu QTc, zmniejszenie LVEF, przerost przysadki i zanik kanalików jądrowych, rozrost komórek mezangium w nerkach, krwotok z przewodu pokarmowego i do jamy ustnej i przerost komórek płata przedniego przysadki. Uważa się, że zmiany w obrębie macicy (zanik błony śluzowej) i płytki wzrostowej kości (zgrubienie nasad kostnych lub dysplazja chrząstki) są związane z działaniem farmakologicznym sunitynibu. Większość z tych zmian była odwracalna po upływie od 2 do 6 tygodni od zakończenia leczenia. Genotoksyczność Potencjalne działanie genotoksyczne sunitynibu oceniano zarówno in vitro, jak i in vivo. Sunitynib nie wykazywał właściwości mutagennych w badaniach na bakteriach z zastosowaniem aktywacji metabolicznej przez wątrobę szczura. Sunitynib nie indukował strukturalnych aberracji chromosomalnych w ludzkich limfocytach krwi obwodowej in vitro. Obserwowano poliploidię (liczbowe aberracje chromosomalne) w ludzkich limfocytach krwi obwodowej in vitro, zarówno w

30
przypadku zastosowania aktywacji metabolicznej, jak i bez niej. Sunitynib nie wykazywał działania klastogennego w szczurzym szpiku kostnym in vivo u szczurów. Nie oceniano jego podstawowego czynnego metabolitu pod kątem potencjalnej toksyczności genetycznej. Rakotwórczość Nie przeprowadzono badań nad potencjałem rakotwórczym jabłczanu sunitynibu. Toksyczny wpływ na zdolność do rozrodu i rozwój W badaniach toksycznego wpływu na zdolność do rozrodu szczurów nie stwierdzono wpływu preparatu na płodność samic lub samców. Jednak w badaniach toksyczności dawek wielokrotnych przeprowadzonych na szczurach i małpach obserwowano oddziaływanie leku na płodność samic w postaci atrezji pęcherzyków, zwyrodnienia ciałek żółtych, zmian błony śluzowej macicy oraz zmniejszenia masy macicy i jajników przy klinicznie istotnych poziomach ekspozycji układowej. U szczurów obserwowano wpływ leku na płodność samców w postaci zaniku kanalików jąder, zmniejszenia liczby plemników w najądrzach i zmniejszenia ilości koloidu w obrębie gruczołu krokowego i pęcherzyków nasiennych przy poziomach ekspozycji osoczowej 18 razy większych niż stwierdzane w warunkach klinicznych. U szczurów śmiertelność zarodków i płodów przejawiała się istotnym zmniejszeniem liczby żywych płodów, zwiększoną liczbą resorpcji, wzrostem utrat ciąży po zagnieżdżeniu zarodka i całkowitą utratą miotów u 8 z 28 samic ciężarnych przy poziomach stężenia leku w osoczu 5,5 razy większych niż poziomy obserwowane w warunkach klinicznych u ludzi. U królików redukcja masy macicy samic ciężarnych i liczby żywych płodów była związana ze zwiększeniem liczby resorpcji, wzrostem liczby utrat ciąży po zagnieżdżeniu zarodka i całkowitą utratą miotów u 4 z 6 samic ciężarnych przy poziomach stężenia leku w osoczu 3 razy większych niż poziomy obserwowane w warunkach klinicznych u ludzi.
Stosowanie sunitynibu u szczurów w okresie organogenezy prowadziło do zmian rozwojowych, polegających na zwiększonej częstości występowania wad rozwojowych szkieletu płodu, charakteryzujących się przede wszystkim opóźnieniem kostnienia kręgów piersiowych/lędźwiowych. i obserwowanych przy poziomach stężenia leku w osoczu 6 razy większych niż poziomy obserwowane w warunkach klinicznych u ludzi. U królików wpływ leku na rozwój polegał na zwiększeniu częstości występowania rozszczepu wargi przy poziomach stężenia leku w osoczu w przybliżeniu odpowiadających poziomom obserwowanym w warunkach klinicznych u ludzi oraz rozszczepu wargi i rozszczepu podniebienia przy poziomach stężenia leku w osoczu 2,7 razy większych niż poziomy obserwowane w warunkach klinicznych u ludzi. 6. DANE FARMACEUTYCZNE 6.1 Wykaz substancji pomocniczych Zawartość kapsułki
Mannitol Kroskarmeloza sodowa Powidon Stearynian magnezu
Pomarańczowa powłoka kapsułki Żelatyna Czerwony tlenek żelaza (E172) Dwutlenek tytanu (E171) Karmelowa powłoka kapsułki. Żelatyna Dwutlenek tytanu (E171)

31
Żółty tlenek żelaza (E172) Czerwony tlenek żelaza (E172) Czarny tlenek żelaza (E172) Tusz do nadruku. Szelak Glikol propylenowy Wodorotlenek sodu Powidon Dwutlenek tytanu (E171) 6.2 Niezgodności farmaceutyczne Nie dotyczy 6.3 Okres trwałości 2 lata 6.4 Specjalne środki ostrożności przy przechowywaniu Brak szczególnych środków ostrożności dotyczących przechowywania. 6.5 Rodzaj i zawartość opakowania Butelki z polietylenu o dużej gęstości (HDPE) z polipropylenowym wieczkiem zawierające po 30 kapsułek. 6.6 Szczególne środki ostrożności dotyczące usuwania Brak szczególnych wymagań. 7. PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE
DO OBROTU Pfizer Ltd Ramsgate Road Sandwich, Kent CT13 9NJ Wielka Brytania

32
8. NUMER(-Y) POZWOLENIA NA DOPUSZCZENIE DO OBROTU 9. DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU /
DATA PRZEDŁUŻENIA POZWOLENIA Data wydania pierwszego pozwolenia: Data przedłużenia pozwolenia: 10. DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU
CHARAKTERYSTYKI PRODUKTU LECZNICZEGO

33
1. NAZWA PRODUKTU LECZNICZEGO SUTENT 50 mg kapsułki twarde 2. SKŁAD JAKOŚCIOWY I ILOŚCIOWY SUBSTANCJI CZYNNYCH Każda kapsułka zawiera jabłczan sunitynibu w ilości odpowiadającej50 mg sunitynibu. Substancje pomocnicze: 79,326 mg mannitolu Pełny wykaz substancji pomocniczych, patrz punkt 6.1. 3. POSTAĆ FARMACEUTYCZNA Kapsułki twarde. Kapsułki żelatynowe z karmelowym wieczkiem i karmelowym korpusem, z wykonanymi białym atramentem napisami „Pfizer” na wieczku i „STN 50 mg” na korpusie, zawierające granulki o barwie od żółtej do pomarańczowej. 4. SZCZEGÓŁOWE DANE KLINICZNE 4.1 Wskazania do stosowania SUTENT jest przeznaczony do leczenia nieoperacyjnych i (lub) przerzutowych nowotworów podścieliskowych przewodu pokarmowego (ang. Gastrointestinal Stromal Tumor-GIST) po niepowodzeniu leczenia metanosulfonianem imatynibu ze względu na oporność lub nietolerancję. SUTENT jest przeznaczony do leczenia zaawansowanego raka nerki i (lub) raka nerki z przerzutami (ang. Metatastaic Renal Cell Cancer - MRCC) po niepowodzeniu leczenia interferonem alfa lub interleukiną-2. Skuteczność leczenia ustalano na podstawie czasu wystąpienia progresji guza oraz wydłużenia czasu całkowitego przeżycia u pacjentów z nowotworami podścieliskowymi przewodu pokarmowego oraz na podstawie wskaźników obiektywnych odpowiedzi u pacjentów z rakiem nerki z przerzutami (patrz punkt 5.1). 4.2 Dawkowanie i sposób podawania Leczenie powinien rozpoczynać lekarz doświadczony w terapii raka nerki lub nowotworów podścieliskowych przewodu pokarmowego. Zalecana dawka preparatu SUTENT wynosi 50 mg doustnie raz na dobę przez 4 kolejne tygodnie, po czym następuje dwutygodniowa przerwa (schemat 4/2), co stanowi pełny cykl 6 tygodni. Można stopniowo dokonywać zmian dawkowania za każdym razem o 12,5 mg, zależnie od indywidualnie ocenianego bezpieczeństwa i tolerancji. Dawka dobowa nie powinna być większa niż 87,5 mg i nie powinna być mniejsza niż 37,5 mg. Należy unikać równoczesnego podawania silnych induktorów CYP3A4, takich jak ryfampicyna (patrz punkty 4.4 i 4.5). Jeżeli nie jest to możliwe, może być konieczne stopniowe zwiększanie dawki preparatu SUTENT, za każdym razem o 12,5 mg (do 87,5 mg na dobę), z równoczesnym starannym monitorowaniem tolerancji. Należy unikać równoczesnego podawania preparatu SUTENT z silnymi inhibitorami CYP3A4, takimi jak ketokonazol (patrz punkty 4.4 i 4.5). Jeżeli nie jest to możliwe, może zaistnieć konieczność zmniejszenia dawek preparatu SUTENT do minimum 37,5 mg na dobę, z równoczesnym starannym monitorowaniem tolerancji. Jeżeli konieczne jest jednoczesne stosowanie innych preparatów, należy wybrać lek wykazujący minimalne działanie indukujące lub hamujące CYP3A4 lub bez takiego działania.

34
Stosowanie u dzieci: Nie przeprowadzono badań dotyczących bezpieczeństwa i skuteczności stosowania preparatu SUTENT u dzieci. Leku nie należy stosować u dzieci dopóki nie będzie dostępnych więcej danych. Stosowanie u pacjentów w podeszłym wieku: Około 25% uczestników badań klinicznych preparatu SUTENT było w wieku 65 lat lub więcej. Nie obserwowano istotnych różnic pod względem bezpieczeństwa lub skuteczności leczenia pomiędzy młodszymi i starszymi pacjentami. Niewydolność wątroby: Nie przeprowadzono badań klinicznych dotyczących stosowania leku u pacjentów z niewydolnością wątroby (patrz punkt 5.2). Niewydolność nerek: Nie przeprowadzono badań klinicznych dotyczących stosowania leku u pacjentów z niewydolnością nerek (patrz punkt 5.2). Preparat SUTENT może być przyjmowany z posiłkiem lub bez posiłku. W przypadku pominięcia jednej z dawek nie należy stosować dodatkowej dawki. Pacjent powinien przyjąć zwykłą przepisaną dawkę następnego dnia. 4.3 Przeciwwskazania Nadwrażliwość na jabłczan sunitynibu lub na którąkolwiek substancję pomocniczą. 4.4 Specjalne ostrzeżenia i środki ostrożności dotyczące stosowania Równoczesne podawanie silnych induktorów izoenzymu CYP3A4, takich jak ryfampicyna, może powodować zmniejszenie stężenia sunitynibu w osoczu. Dlatego należy unikać jednoczesnego stosowania preparatu z lekami z tej grupy. Jeżeli nie jest to możliwe, może być konieczne zwiększenie dawek preparatu SUTENT (patrz punkty 4.2 i 4.5). Równoczesne podawanie silnych inhibitorów izoenzymu CYP3A4, takich jak ketokonazol, może powodować zwiększenie stężenia sunitynibu w osoczu. Jeżeli konieczne jest jednoczesne stosowanie innego preparatu, należy wybrać lek wykazujący minimalne działanie hamujące CYP3A4 lub pozbawiony takiego działania. Jeżeli nie jest to możliwe, może być konieczne zmniejszenie dawek preparatu SUTENT (patrz punkty 4.2 i 4.5) Zdarzenia niepożądane ze strony skóry i tkanki podskórnej Częstym zdarzeniem niepożądanym związanym z leczeniem, występującym u około 30% pacjentów, są przebarwienia skóry, być może związane z barwą substancji czynnej (żółtą). Pacjentów należy poinformować, że w trakcie stosowania preparatu SUTENT może też dojść do odbarwienia włosów lub skóry. Do innych możliwych objawów dermatologicznych należą suchość, zgrubienie lub pękanie skóry, pęcherze lub czasem wysypka na powierzchni dłoni i na podeszwach stóp. U około 14% pacjentów stwierdzano ból/podrażnienie w obrębie jamy ustnej. Około 28% pacjentów zgłaszało występowanie zaburzeń smaku. Powyższe zdarzenia nie występowały łącznie, były na ogół odwracalne i zasadniczo nie wiązały się z koniecznością przerwania leczenia. Zdarzenia niepożądane ze strony przewodu pokarmowego Najczęstszymi obserwowanymi zdarzeniami niepożądanymi ze strony przewodu pokarmowego związanymi z leczeniem były nudności, biegunka, zapalenie błony śluzowej jamy ustnej, objawy dyspeptyczne i wymioty. W razie wystąpienia tego typu zaburzeń zastosowane leczenie wspomagające może obejmować stosowanie leków przeciwwymiotnych lub przeciwbiegunkowych.

35
Krwotoki Krwotoki z guza związane z leczeniem występowały u około 2% pacjentów z nowotworami podścieliskowymi przewodu pokarmowego. Zdarzenia te mogą występować nagle, a w przypadku guzów płuc mogą mieć postać ciężkiego, zagrażającego życiu krwotoku płucnego lub krwioplucia. Śmiertelny krwotok płucny wystąpił u 2 pacjentów otrzymujących preparat SUTENT w ramach badania klinicznego nad jego zastosowaniem w leczeniu niedrobnokomórkowego raka płuc (ang. NSCLC) z przerzutami. U obu pacjentów w badaniu histologicznym stwierdzono raka płaskonabłonkowego. Preparat SUTENT nie jest zarejestrowany do leczenia niedrobnokomórkowego raka płuc. Rutynowa ocena omawianego zdarzenia niepożądanego powinna obejmować wykonanie morfologii krwi i badania przedmiotowego. Krwotok z nosa był najczęściej występującym krwotocznym zdarzeniem niepożądanym związanym z leczeniem, obserwowanym u około połowy pacjentów z guzami litymi, u których wystąpiły powikłania krwotoczne. Żadne z tych zdarzeń nie miało charakteru ciężkiego. Przewód pokarmowy U pacjentów z nowotworami w jamie brzusznej, u których zastosowano preparat SUTENT rzadko występowały ciężkie, czasem śmiertelne powikłania ze strony przewodu pokarmowego, w tym perforacje. Nadciśnienie tętnicze U około 16% pacjentów z guzami litymi stwierdzano związane z leczeniem nadciśnienie tętnicze. U około 2,7% tych pacjentów zmniejszono dawkę lub czasowo odroczono podanie preparatu SUTENT. U żadnego z pacjentów nie przerwano leczenia. Ciężkie nadciśnienie tętnicze (ciśnienie skurczowe >200 mmHg lub ciśnienie rozkurczowe >110 mmHg) wystąpiło u 4,7% pacjentów. Należy badać pacjentów w celu wykrycia nadciśnienia tętniczego i odpowiednio ich kontrolować. U pacjentów z ciężkim nadciśnieniem tętniczym, którego nie udaje się kontrolować farmakologicznie, zaleca się czasowe przerwanie stosowania preparatu. Można je ponownie podjąć po uzyskaniu skutecznej kontroli nadciśnienia. Zaburzenia hematologiczne Odpowiednio u 13,1% i 0,9% pacjentów stwierdzano zmniejszenie bezwzględnej liczby granulocytów obojętnochłonnych 3 i 4 stopnia ciężkości. Odpowiednio u 4% i 0,5% pacjentów obserwowano również zmniejszenie liczby płytek krwi 3 i 4 stopnia ciężkości. Powyższe zdarzenia nie występowały łącznie, były na ogół odwracalne i zasadniczo nie wiązały się z koniecznością przerwania leczenia. U pacjentów, u których stosuje się preparat SUTENT należy wykonać morfologię krwi na początku każdego cyklu leczenia Zaburzenia ze strony układu krążenia Zmniejszenie frakcji wyrzutowej lewej komory (LVEF) o ≥ 20% i poniżej dolnej granicy normy wystąpił u około 2% pacjentów z nowotworami podścieliskowymi przewodu pokarmowego i 4% pacjentów z rakiem nerki z przerzutami, którym podawano preparat SUTENT oraz 2% pacjentów otrzymujących placebo. Nie wydaje się, aby spadek LVEF był postępujący. Wartości tego parametru często ulegały poprawie w trakcie leczenia. U 0,7% pacjentów z guzami litymi leczonych preparatem SUTENT i 1% pacjentów otrzymujących placebo stwierdzono następujące, związane z leczeniem zdarzenia niepożądane: niewydolność serca, zastoinowa niewydolność serca lub lewokomorowa niewydolność serca. U wszystkich tych pacjentów występowały nowotwory podścieliskowe przewodu pokarmowego. Potencjalny związek pomiędzy zahamowaniem receptora kinazy tyrozynowej (RTK) a czynnością serca nie został wyjaśniony. Z badań klinicznych nad preparatem SUTENT wykluczono pacjentów, u których w ciągu 12 miesięcy przed podaniem leku miały miejsce incydenty związane z układem krążenia, takie jak zawał mięśnia serca (również ciężka/niestabilna dławica piersiowa), przeszczep pomostujący tętnicy wieńcowej/tętnicy obwodowej, objawowa zastoinowa niewydolność serca (ang. CHF), udar mózgu lub przejściowy napad niedokrwienny, bądź też zator tętnicy płucnej. Nie wiadomo, czy pacjenci z tymi współistniejącymi stanami chorobowymi są narażeni na podwyższone ryzyko rozwinięcia się u nich związanej z lekiem niewydolności lewej komory. Należy rozważyć stosunek ryzyka do potencjalnych korzyści ze stosowania preparatu. Pacjentów z tej grupy należy poddawać starannej obserwacji w celu wykrycia ewentualnych klinicznych objawów podmiotowych i przedmiotowych zastoinowej niewydolności serca

36
podczas stosowania preparatu SUTENT. Należy również rozważyć wykonywanie oznaczeń LVEF na początku terapii i okresowo w trakcie leczenia. U pacjentów, u których nie występują kardiologiczne czynniki ryzyka należy rozważyć wykonanie wyjściowej oceny frakcji wyrzutowej. W przypadku wystąpienia objawów klinicznych zastoinowej niewydolności serca zaleca się przerwanie stosowania preparatu SUTENT. U pacjentów bez klinicznych objawów zastoinowej niewydolności serca, ale z frakcją wyrzutową <50% i >20% poniżej wartości wyjściowej należy przerwać stosowanie preparatu lub zmniejszyć dawkę. Wydłużenie odcinka QT Wydłużenie odcinka QT oceniano w badaniu 24 pacjentów w wieku od 20 do 87 lat z zaawansowanymi nowotworami. Wykazano, że preparat SUTENT, w stężeniach w przybliżeniu dwukrotnie większych niż terapeutyczne, wydłuża odcinek QTcF (z korektą Friederica). U żadnego z pacjentów nie stwierdzono wydłużenia odcinka QT/QTc stopnia ciężkości większego niż 2 (CTCAEvv 3.0), nie odnotowano także zaburzeń rytmu serca. Znaczenie kliniczne zaobserwowanych działań jest niejasne i uzależnione od indywidualnych czynników ryzyka i wrażliwości pacjentów. Należy zachować ostrożność podczas stosowania preparatu SUTENT u pacjentów z wydłużonym odcinkiem QT w wywiadzie, pacjentów przyjmujących leki przeciwarytmiczne oraz u pacjentów z istotnymi chorobami serca w wywiadzie, bradykardią lub zaburzeniami elektrolitowymi. Należy zachować ostrożność podczas jednoczesnego podawania silnych inhibitorów CYP3A4, które mogą powodować zwiększenie stężenia sunitynibu w osoczu, a w razie potrzeby należy zmniejszyć dawkę preparatu SUTENT (patrz punkt 4.5). Żylne zaburzenia zakrzepowo-zatorowe W dwóch badaniach w raku nerki z przerzutami stwierdzono wystąpienie żylnych powikłań zakrzepowo-zatorowych u czterech pacjentów (2%); u dwóch z nich doszło do zatoru tętnicy płucnej (4 stopnia ciężkości), a u dwóch pozostałych – do zakrzepicy żył głębokich (3 stopnia ciężkości). W jednym z tych przypadków przerwano stosowanie leku. W badaniu rejestracyjnym nad nowotworami podścieliskowymi przewodu pokarmowego u siedmiu pacjentów (3%) otrzymujących preparat SUTENT wystąpiły żylne powikłania zakrzepowo-zatorowe natomiast nie wystąpiły one u żadnego z pacjentów z grupy otrzymującej placebo. U pięciu z siedmiu pacjentów powikłania te były 3 stopnia ciężkości, a u dwóch – 1 lub 2 stopnia ciężkości. Czterech pacjentów z nowotworami podścieliskowymi przewodu pokarmowego przerwało leczenie po wystąpieniu pierwszych objawów zakrzepicy żył głębokich. Zator tętnicy płucnej U około 1,1% pacjentów z guzami litymi, którzy otrzymywali preparat SUTENT wystąpił związany z leczeniem zator tętnicy płucnej. Żadne z tych zdarzeń nie doprowadziło do przerwania leczenia, jednak w kilku przypadkach zmniejszono dawkę lub tymczasowo odroczono leczenie. Po podjęciu leczenia nie doszło do ponownego zatoru tętnicy płucnej u tych pacjentów. Niedoczynność tarczycy W dwóch badaniach w raku nerki z przerzutami niedoczynność tarczycy była zgłaszana jako zdarzenie niepożądane u siedmiu pacjentów (4%). Ponadto u czterech pacjentów (2%) stwierdzono podwyższenie stężenia TSH. Ogółem u 7% populacji objętej tymi badaniami stwierdzono kliniczne lub laboratoryjne objawy niedoczynności tarczycy, które wystąpiły w trakcie leczenia. U ośmiu pacjentów (4%) z nowotworami podścieliskowymi przewodu pokarmowego otrzymujących preparat SUTENT i u jednego pacjenta (1%) otrzymującego placebo stwierdzono w czasie leczenia niedoczynność tarczycy wymagającą pilnej terapii. U pacjentów z objawami sugerującymi niedoczynność tarczycy konieczne jest wykonywanie kontrolnych badań laboratoryjnych diagnozujących czynność tego gruczołu i zastosowanie leczenia według obowiązujących standardów. Czynność trzustki U pacjentów z różnymi guzami litymi otrzymujących preparat SUTENT, obserwowano zwiększenie aktywności lipazy i amylazy w surowicy. Wzrost aktywności lipazy był przemijający i na ogół nie towarzyszyły mu objawy podmiotowe i przedmiotowe zapalenia trzustki. Zapalenie trzustki

37
obserwowano u 0,4% pacjentów z guzami litymi. W razie wystąpienia objawów zapalenia trzustki należy zapewnić pacjentom właściwą opiekę medyczną. Napady drgawkowe W badaniach klinicznych z zastosowaniem preparatu SUTENT obserwowano występowanie drgawek u pacjentów z cechami radiologicznymi przerzutów nowotworu do mózgu. Ponadto stwierdzano rzadkie (<1%) przypadki drgawek i objawy radiologiczne zespołu odwracalnej tylnej leukoencefalopatii (RPLS). W żadnym z omawianych przypadków zaburzenia te nie doprowadziły do zgonu. U pacjentów z drgawkami i objawami podmiotowymi/przedmiotowymi sugerującymi obecność RPLS, takimi jak nadciśnienie, ból głowy, zwiększona czujność, zmiany psychiczne i utrata wzroku, w tym ślepota korowa, należy kontrolować występujące zaburzenia poprzez zastosowanie właściwego leczenia, w tym farmakologicznej kontroli nadciśnienia. Zaleca się tymczasowe przerwanie stosowania preparatu SUTENT. Po ustąpieniu objawów lekarz prowadzący może podjąć decyzję o kontynuacji leczenia. 4.5 Interakcje z innymi lekami i inne rodzaje interakcji Leki, które mogą zwiększać stężenie sunitynibu w osoczu: Równoczesne podawanie sunitynibu z silnym inhibitorem izoenzymu CYP3A4, ketokonazolem, powodowało po jednorazowym podaniu jabłczanu sunitynibu u zdrowych ochotników wzrost wartości Cmax i AUC0-∞ kompleksu [sunitynib + podstawowy metabolit] odpowiednio o 49% i 51%. Podawanie preparatu SUTENT jednocześnie z silnymi inhibitorami izoenzymów z rodziny CYP3A4 (np. rytonawir, itrakonazol, erytromycyna, klarytromycyna, sok grejpfrutowy) może wiązać się ze zwiększeniem stężenia sunitynibu. Z tego względu należy unikać podawania go jednocześnie z inhibitorami enzymatycznymi lub należy rozważyć wybranie do leczenia skojarzonego alternatywnego preparatu, o minimalnym działaniu hamującym enzym CYP3A4 lub bez takiego działania. Jeżeli nie jest to możliwe, może zaistnieć konieczność zmniejszenia dawek preparatu SUTENT do minimum 37,5 mg na dobę, na podstawie dokładnie monitorowanej tolerancji (patrz punkt 4.2). Leki, które mogą zmniejszyć stężenie sunitynibu w osoczu: Równoczesne podawanie jabłczanu sunitynibu z silnym induktorem izoenzymu CYP3A4, ryfampicyną, powodowało po jednorazowym podaniu jabłczanu sunitynibu u zdrowych ochotników redukcję wartości Cmax i AUC0-∞ kompleksu [sunitynib + podstawowy metabolit] odpowiednio o 23% i 46%]. Podawanie preparatu SUTENT z silnymi induktorami izoenzymów z grupy CYP3A4 (np. deksametazon, fenytoina, karbamazepina, ryfampicyna, fenobarbital lub ziele dziurawca Hypericum perforatum) może prowadzić do zmniejszenia stężenia sunitynibu. Z tego względu należy unikać jednoczesnego stosowania preparatu z induktorami enzymatycznymi, bądź też należy rozważyć wybranie do leczenia skojarzonego alternatywnego preparatu, o minimalnym działaniu indukującym enzym CYP3A4 lub bez takiego działania. Jeżeli nie jest to możliwe, może zaistnieć konieczność stopniowego zwiększania dawek preparatu SUTENT, za każdym razem o 12,5 mg (do 87,5 mg na dobę) na podstawie dokładnie monitorowanej tolerancji (patrz punkt 4.2). Aby utrzymać docelowe stężenie sunitynibu należy rozważyć wybranie do leczenia skojarzonego preparatów indukujących enzym w mniejszym stopniu. Jeżeli nie jest to możliwe, może być konieczne dostosowanie dawki preparatu SUTENT (patrz punkt 4.2). U pacjentów leczonych preparatem SUTENT rzadko obserwowano krwotoki (patrz punkt 4.4). Pacjenci otrzymujący równocześnie preparaty przeciwzakrzepowe (np. warfaryna, acenokumarol) mogą być poddawani okresowym kontrolom obejmującym wykonanie morfologii krwi (z oznaczeniem liczby płytek krwi), badanie czynników krzepnięcia (czas protrombinowy (PT)/wskaźnik INR) i badaniu przedmiotowemu. 4.6 Ciąża i laktacja Ciąża Nie przeprowadzono badań dotyczących stosowania preparatu SUTENT u kobiet w ciąży. W badaniach na zwierzętach stwierdzono toksyczny wpływ leku na zdolność do rozrodu, objawiający się m.in.

38
wadami wrodzonymi płodów (patrz punkt 5.3). Preparatu nie należy stosować w czasie ciąży lub u kobiet, które nie stosują skutecznej metody antykoncepcyjnej z wyjątkiem przypadku, gdy potencjalne korzyści z leczenia przeważą nad potencjalnym ryzykiem dla płodu. Jeżeli preparat zostanie zastosowany w czasie ciąży lub, jeżeli pacjentka zajdzie w ciążę w trakcie jego stosowania, należy ją poinformować o potencjalnym zagrożeniu dla płodu. Kobietom w wieku rozrodczym należy doradzić stosowanie skutecznej metody antykoncepcyjnej podczas przyjmowania preparatu SUTENT. Dane niekliniczne wskazują, że leczenie preparatem SUTENT może wywierać niekorzystny wpływ na płodność kobiet i mężczyzn (patrz punkt 5.3).
Laktacja
Sunitynib i (lub) jego metabolity przenikają do mleka szczurów. Nie wiadomo, czy sunitynib lub jego główny czynny metabolit przenikają do mleka kobiecego. Ze względu na to, że leki są często wydzielane do mleka kobiecego i ze względu na możliwe występowanie ciężkich działań niepożądanych u niemowląt karmionych piersią, kobiety nie powinny karmić piersią podczas stosowania preparatu SUTENT. 4.7 Wpływ na zdolność prowadzenia pojazdów mechanicznych i obsługiwania urządzeń
mechanicznych w ruchu Nie przeprowadzono badań nad wpływem preparatu na zdolność prowadzenia pojazdów mechanicznych i obsługiwania urządzeń mechanicznych w ruchu. Pacjenci powinni być poinformowani o możliwości wystąpienia zawrotów głowy w trakcie leczenia preparatem SUTENT. 4.8 Działania niepożądane Najczęstszymi ciężkimi zdarzeniami niepożądanymi związanymi z leczeniem preparatem SUTENT u pacjentów z guzami litymi były: zator tętnicy płucnej (1%), trombocytopenia (1%), krwotok z guza (0,9%), gorączka neutropeniczna (0,4%) i nadciśnienie tętnicze (0,4%). Do najczęstszych zdarzeń niepożądanych związanych z leczeniem (wystąpiły u co najmniej 20% pacjentów) o dowolnym stopniu nasilenia należały: uczucie zmęczenia; zaburzenia ze strony przewodu pokarmowego, takie jak biegunka, nudności, zapalenie jamy ustnej, objawy dyspeptyczne i wymioty; przebarwienia skórne; zaburzenia smaku i anoreksja. Uczucie zmęczenia, nadciśnienie tętnicze i neutropenia były najczęstszymi związanymi z leczeniem zdarzeniami niepożądanymi o maksymalnym stopniu ciężkości 3 u pacjentów z guzami litymi, a podwyższona aktywność lipazy była najczęstszym związanym z leczeniem zdarzeniem niepożądanym o maksymalnym stopniu ciężkości 4. Poniżej wymieniono działania niepożądane, które zostały zgłoszone przez >5% pacjentów z guzami litymi, według klas układowo-narządowych, częstości występowania i stopnia ciężkości. W ramach każdej grupy wyodrębnionej wg częstości występowania działania niepożądane przedstawiono w kolejności od najcięższego do najlżejszego. Według częstości występowania zdarzenia definiowano jako: bardzo częste (> 1/10), częste (od >1/100 do < 1/10), nieczęste (od >1/1000 do <1//100), rzadkie (od >1/10 000 do 1/1000), bardzo rzadkie (< 1/10 000). Działania niepożądane zgłaszane w badaniach u pacjentów z nowotworami podścieliskowymi przewodu pokarmowego. Klasyfikacja układów i narządów
Częstość występowania
Działania niepożądane
Wszystkich stopni n (%)
Stopnia 3 n (%)
Stopnia 4 n (%)
Bardzo często Niedokrwistość 33 (12,8%) 13 (5,1%) 1 (0,4%) Często Neutropenia 24 (9,3%) 15 (5,8%) 1 (0,4%)
Zaburzenia krwi i układu chłonnego Często Trombocytopenia 23 (8,9%) 6 (2,3%) 1 (0,4%)
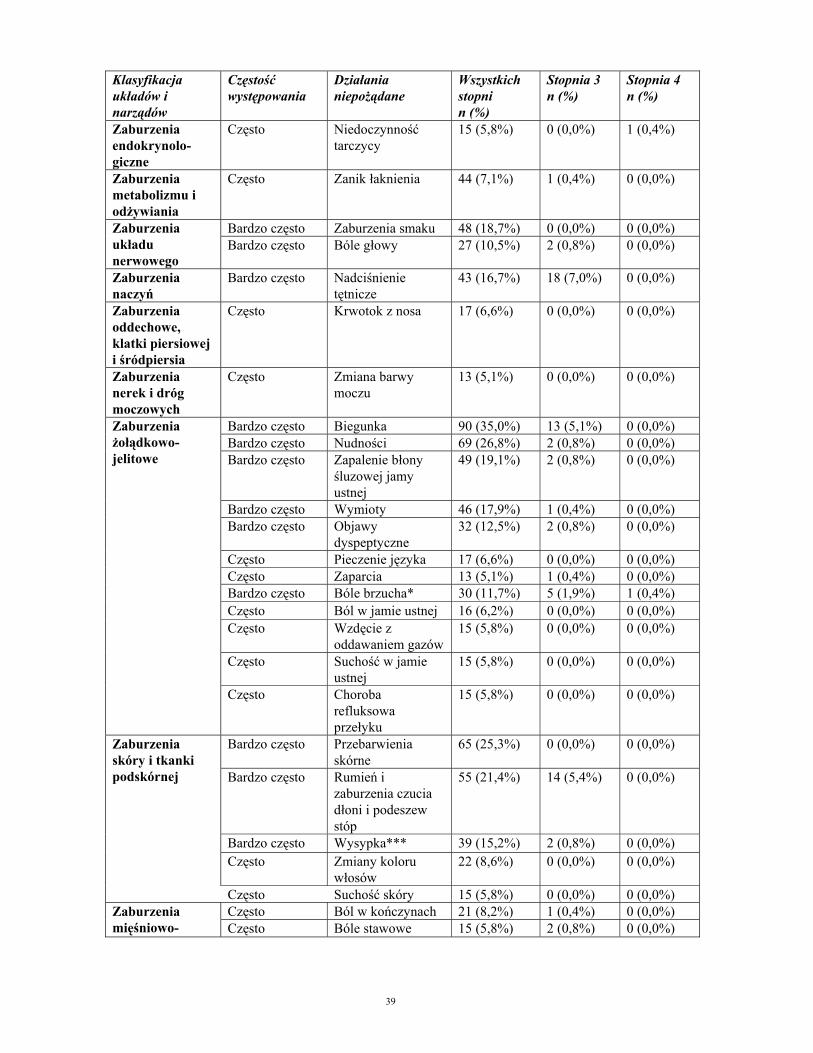
39
Klasyfikacja układów i narządów
Częstość występowania
Działania niepożądane
Wszystkich stopni n (%)
Stopnia 3 n (%)
Stopnia 4 n (%)
Zaburzenia endokrynolo- giczne
Często Niedoczynność tarczycy
15 (5,8%) 0 (0,0%) 1 (0,4%)
Zaburzenia metabolizmu i odżywiania
Często Zanik łaknienia 44 (7,1%) 1 (0,4%) 0 (0,0%)
Bardzo często Zaburzenia smaku 48 (18,7%) 0 (0,0%) 0 (0,0%) Zaburzenia układu nerwowego
Bardzo często Bóle głowy 27 (10,5%) 2 (0,8%) 0 (0,0%)
Zaburzenia naczyń
Bardzo często Nadciśnienie tętnicze
43 (16,7%) 18 (7,0%) 0 (0,0%)
Zaburzenia oddechowe, klatki piersiowej i śródpiersia
Często Krwotok z nosa 17 (6,6%) 0 (0,0%) 0 (0,0%)
Zaburzenia nerek i dróg moczowych
Często Zmiana barwy moczu
13 (5,1%) 0 (0,0%) 0 (0,0%)
Bardzo często Biegunka 90 (35,0%) 13 (5,1%) 0 (0,0%) Bardzo często Nudności 69 (26,8%) 2 (0,8%) 0 (0,0%) Bardzo często Zapalenie błony
śluzowej jamy ustnej
49 (19,1%) 2 (0,8%) 0 (0,0%)
Bardzo często Wymioty 46 (17,9%) 1 (0,4%) 0 (0,0%) Bardzo często Objawy
dyspeptyczne 32 (12,5%) 2 (0,8%) 0 (0,0%)
Często Pieczenie języka 17 (6,6%) 0 (0,0%) 0 (0,0%) Często Zaparcia 13 (5,1%) 1 (0,4%) 0 (0,0%) Bardzo często Bóle brzucha* 30 (11,7%) 5 (1,9%) 1 (0,4%) Często Ból w jamie ustnej 16 (6,2%) 0 (0,0%) 0 (0,0%) Często Wzdęcie z
oddawaniem gazów 15 (5,8%) 0 (0,0%) 0 (0,0%)
Często Suchość w jamie ustnej
15 (5,8%) 0 (0,0%) 0 (0,0%)
Zaburzenia żołądkowo-jelitowe
Często Choroba refluksowa przełyku
15 (5,8%) 0 (0,0%) 0 (0,0%)
Bardzo często Przebarwienia skórne
65 (25,3%) 0 (0,0%) 0 (0,0%)
Bardzo często Rumień i zaburzenia czucia dłoni i podeszew stóp
55 (21,4%) 14 (5,4%) 0 (0,0%)
Bardzo często Wysypka*** 39 (15,2%) 2 (0,8%) 0 (0,0%) Często Zmiany koloru
włosów 22 (8,6%) 0 (0,0%) 0 (0,0%)
Zaburzenia skóry i tkanki podskórnej
Często Suchość skóry 15 (5,8%) 0 (0,0%) 0 (0,0%) Często Ból w kończynach 21 (8,2%) 1 (0,4%) 0 (0,0%) Zaburzenia
mięśniowo- Często Bóle stawowe 15 (5,8%) 2 (0,8%) 0 (0,0%)

40
Klasyfikacja układów i narządów
Częstość występowania
Działania niepożądane
Wszystkich stopni n (%)
Stopnia 3 n (%)
Stopnia 4 n (%)
szkieletowe, tkanki łącznej i kości
Często Bóle mięśniowe 13 (5,1%) 0 (0,0%) 0 (0,0%)
Bardzo często Uczucie zmęczenia/ osłabienie
135 (52,5%) 25 (9,7%) 0 (0,0%) Zaburzenia ogólne i stany w miejscu podania Bardzo często Zapalenie błon
śluzowych 30 (11,7%) 0 (0,0%) 0 (0,0%)
Często Obrzęk** 21 (8,2%) 1 (0,4%) 0 (0,0%) Często Zmniejszenie
stężenia hemoglobiny
16 (6,2%) 2 (0,8%) 0 (0,0%)
Badania dodatkowe
Często Zwiększenie aktywności fosfokinazy kreatynowej we krwi
14 (5,4%) 0 (0,0%) 0 (0,0%)
Często Zmniejszenie frakcji wyrzutowej
13 (5,1%) 1 (0,4%) 0 (0,0%)
Często Zwiększenie aktywności lipazy
13 (5,1%) 5 (1,9%) 4 (1,6%)
Często Zmniejszenie liczby płytek krwi
13 (5,1%) 2 (0,8%) 1 (0,4%)
Dowolne zdarzenie niepożądane
222 (86,4%) 88 (34,2%) 24 (9,3%)
*Następujące zdarzenia zostały połączone w jedną kategorię: bóle brzucha, bóle w górnej części brzucha i bóle w dolnej części brzucha. **Następujące zdarzenia zostały połączone w jedną kategorię: obrzęki i obrzęki obwodowe. ***Następujące zdarzenia zostały połączone w jedną kategorię: wysypka, wysypka rumieniowa, wysypka plamista i wysypka złuszczająca się Działania niepożądane zgłaszane w badaniach u pacjentów z rakiem nerki z przerzutami Klasyfikacja układów i narządów
Częstość występowania
Działania niepożądane
Wszystkich stopni n (%)
Stopnia 3 n (%)
Stopnia 4 n (%)
Bardzo często Neutropenia 17 (10,1%) 8 (4,7%) 1 (0,6%) Często Niedokrwistość 16 (9,5%) 6 (3,6%) 0 (0,0%) Często Trombocytopenia 15 (8,9%) 5 (3,0%) 2 (1,2%)
Zaburzenia krwi i układu chłonnego
Często Leukopenia 14 (8,3%) 7 (4,1%) 0 (0,0%)
Zaburzenia oka Często Zwiększone łzawienie
9 (5,3%) 0 (0,0%) 0 (0,0%)
Zaburzenia metabolizmu i odżywiania
Bardzo często Zanik łaknienia 47 (27,8%) 1 (0,6%) 0 (0,0%)
Często Odwodnienie 12 (7,1%) 4 (2,4%) 0 (0,0%) Często Zmniejszenie
łaknienia 11 (6,5%) 0 (0,0%) 0 (0,0%)
Bardzo często Zaburzenia smaku 71 (42%) 0 (0,0%) 0 (0,0%) Bardzo często Bóle głowy 25 (14,8%) 1 (0,6%) 0 (0,0%) Często Zawroty głowy 13 (7,7%) 2 (1,2%) 0 (0,0%)
Zaburzenia układu nerwowego
Często Parestezje 9 (5,3%) 0 (0,0%) 0 (0,0%)

41
Klasyfikacja układów i narządów
Częstość występowania
Działania niepożądane
Wszystkich stopni n (%)
Stopnia 3 n (%)
Stopnia 4 n (%)
Zaburzenia naczyń
Bardzo często Nadciśnienie tętnicze
28 (16,6%) 7 (4,1%) 0 (0,0%)
Często Krwotok z nosa 16 (9,5%) 0 (0,0%) 0 (0,0%) Zaburzenia oddechowe, klatki piersiowej i śródpiersia
Często Duszność 9 (5,3%) 0 (0,0%) 0 (0,0%)
Bardzo często Biegunka 83 (49,1%) 5 (3,0%) 0 (0,0%) Bardzo często Nudności 84 (49,7%) 2 (1,2%) 0 (0,0%) Bardzo często Zapalenie błony
śluzowej jamy ustnej
70 (41,4%) 6 (3,6%) 0 (0,0%)
Bardzo często Objawy dyspeptyczne
69 (40,8%) 1 (0,6%) 0 (0,0%)
Bardzo często Wymioty 52 (30,8%) 2 (1,2%) 0 (0,0%) Bardzo często Zaparcia 34 (20,1%) 0 (0,0%) 0 (0,0%)
Zaburzenia żołądkowo-jelitowe
Bardzo często Pieczenie języka 25 (14,8%) 0 (0,0%) 0 (0,0%) Bardzo często Bóle brzucha* 17 (10,1%) 2 (1,2%) 0 (0,0%)
Często Wzdęcie z oddawaniem gazów
16 (9,5%) 0 (0,0%) 0 (0,0%)
Często Uczucie rozpierania w jamie brzusznej
9 (5,3%) 0 (0,0%) 0 (0,0%)
Często Suchość w jamie ustnej
9 (5,3%) 0 (0,0%) 0 (0,0%)
Bardzo często Przebarwienia skórne
54 (32,0%) 0 (0,0%) 0 (0,0%)
Bardzo często Wysypka** 46 (27,2%) 0 (0,0%) 0 (0,0%) Bardzo często Zmiana barwy
włosów 24 (14,2%) 0 (0,0%) 0 (0,0%)
Bardzo często Rumień i zaburzenia czucia dłoni i podeszew stóp
21 (12,4%) 6 (3,6%) 0 (0,0%)
Często Łysienie 13 (7,7%) 0 (0,0%) 0 (0,0%) Często Złuszczające
zapalenie skóry 10 (5,9%) 2 (1,2%) 0 (0,0%)
Często Obrzęki okołooczodołowe
9 (5,3%) 0 (0,0%) 0 (0,0%)
Bardzo często Suchość skóry 22 (13,0%) 0 (0,0%) 0 (0,0%)
Zaburzenia skóry i tkanki podskórnej
Bardzo często Rumień 20 (11,8%) 0 (0,0%) 0 (0,0%) Bardzo często Ból w kończynach 21 (12,4) 1 (0,6%) 0 (0,0%) Zaburzenia
mięśniowo-szkieletowe, tkanki łącznej i kości
Często Bóle mięśni 15 (8,9%) 1 (0,6%) 0 (0,0%)
Bardzo często Uczucie zmęczenia/osłabie- nie
108 (63,9%) 19 (11,2%) 0 (0,0%) Zaburzenia ogólne i stany w miejscu podania Bardzo często Zapalenie błon
śluzowych 30 (17,8%) 1 (0,6%) 0 (0,0%)
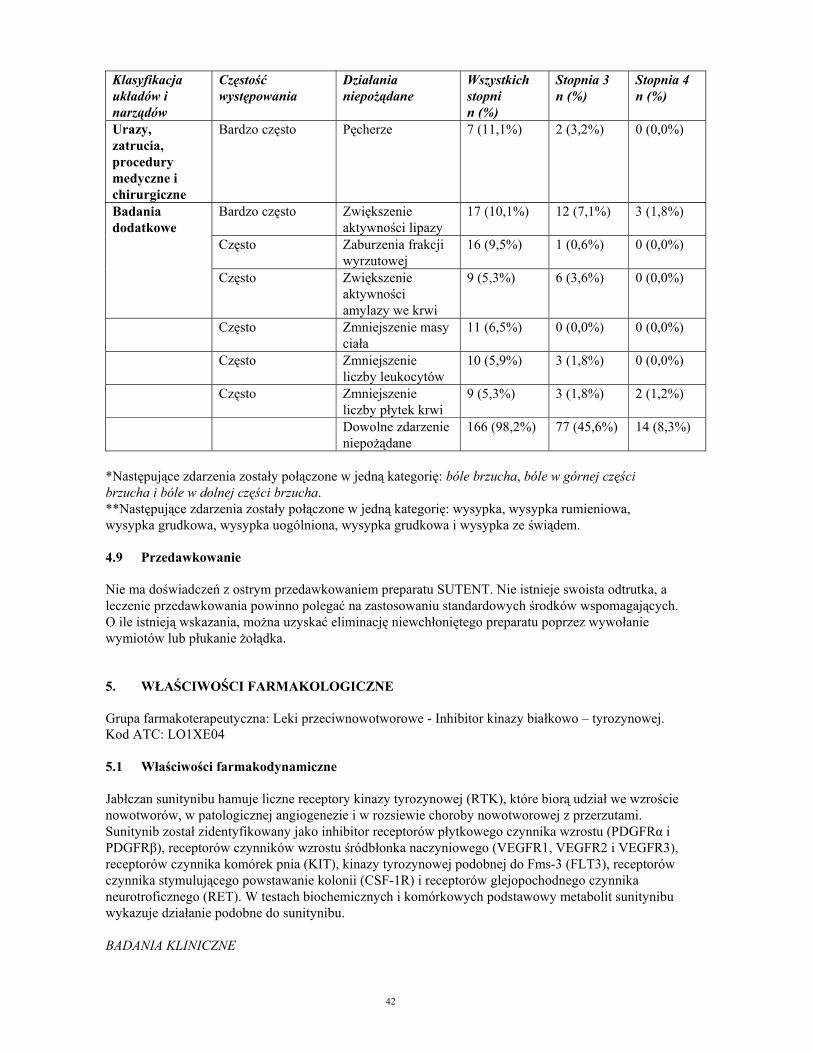
42
Klasyfikacja układów i narządów
Częstość występowania
Działania niepożądane
Wszystkich stopni n (%)
Stopnia 3 n (%)
Stopnia 4 n (%)
Urazy, zatrucia, procedury medyczne i chirurgiczne
Bardzo często Pęcherze 7 (11,1%) 2 (3,2%) 0 (0,0%)
Bardzo często Zwiększenie aktywności lipazy
17 (10,1%) 12 (7,1%) 3 (1,8%)
Często Zaburzenia frakcji wyrzutowej
16 (9,5%) 1 (0,6%) 0 (0,0%)
Badania dodatkowe
Często Zwiększenie aktywności amylazy we krwi
9 (5,3%) 6 (3,6%) 0 (0,0%)
Często Zmniejszenie masy ciała
11 (6,5%) 0 (0,0%) 0 (0,0%)
Często Zmniejszenie liczby leukocytów
10 (5,9%) 3 (1,8%) 0 (0,0%)
Często Zmniejszenie liczby płytek krwi
9 (5,3%) 3 (1,8%) 2 (1,2%)
Dowolne zdarzenie niepożądane
166 (98,2%) 77 (45,6%) 14 (8,3%)
*Następujące zdarzenia zostały połączone w jedną kategorię: bóle brzucha, bóle w górnej części brzucha i bóle w dolnej części brzucha. **Następujące zdarzenia zostały połączone w jedną kategorię: wysypka, wysypka rumieniowa, wysypka grudkowa, wysypka uogólniona, wysypka grudkowa i wysypka ze świądem. 4.9 Przedawkowanie Nie ma doświadczeń z ostrym przedawkowaniem preparatu SUTENT. Nie istnieje swoista odtrutka, a leczenie przedawkowania powinno polegać na zastosowaniu standardowych środków wspomagających. O ile istnieją wskazania, można uzyskać eliminację niewchłoniętego preparatu poprzez wywołanie wymiotów lub płukanie żołądka. 5. WŁAŚCIWOŚCI FARMAKOLOGICZNE Grupa farmakoterapeutyczna: Leki przeciwnowotworowe - Inhibitor kinazy białkowo – tyrozynowej. Kod ATC: LO1XE04 5.1 Właściwości farmakodynamiczne Jabłczan sunitynibu hamuje liczne receptory kinazy tyrozynowej (RTK), które biorą udział we wzroście nowotworów, w patologicznej angiogenezie i w rozsiewie choroby nowotworowej z przerzutami. Sunitynib został zidentyfikowany jako inhibitor receptorów płytkowego czynnika wzrostu (PDGFRα i PDGFRβ), receptorów czynników wzrostu śródbłonka naczyniowego (VEGFR1, VEGFR2 i VEGFR3), receptorów czynnika komórek pnia (KIT), kinazy tyrozynowej podobnej do Fms-3 (FLT3), receptorów czynnika stymulującego powstawanie kolonii (CSF-1R) i receptorów glejopochodnego czynnika neurotroficznego (RET). W testach biochemicznych i komórkowych podstawowy metabolit sunitynibu wykazuje działanie podobne do sunitynibu. BADANIA KLINICZNE

43
Badano bezpieczeństwo kliniczne i skuteczność kliniczną preparatu SUTENT podczas leczenia pacjentów ze złośliwymi nowotworami podścieliskowymi przewodu pokarmowego (GIST) opornymi na imatynib (tj. pacjentów, u których doszło do progresji choroby w trakcie leczenia imatynibem lub po nim) lub nietolerujących imatynibu (tj. pacjentów, u których wystąpiły istotne objawy toksyczności w trakcie leczenia imatynibem, które uniemożliwiły dalsze leczenie) oraz leczenia pacjentów z rakiem nerki z przerzutami po niepowodzeniu leczenia z zastosowaniem cytokin. Skuteczność leczenia ustalano na podstawie czasu wystąpienia progresji guza oraz wydłużenia czasu całkowitego przeżycia u pacjentów z nowotworami podścieliskowymi przewodu pokarmowego oraz na podstawie wskaźników obiektywnych odpowiedzi u pacjentów z rakiem nerki z przerzutami. Nowotwory podścieliskowe przewodu pokarmowego Wstępne otwarte, zakładające zwiększanie dawkowania badanie zostało przeprowadzone u pacjentów z nowotworami podścieliskowymi przewodu pokarmowego po niepowodzeniu stosowania imatynibu (mediana maksymalnej dawki dobowej: 800 mg) ze względu na oporność lub nietolerancję leczenia. Do badania zakwalifikowano dziewięćdziesięciu siedmiu pacjentów, otrzymujących dawki według różnych schematów; 55 pacjentów otrzymywało preparat w dawce 50 mg według zalecanego schematu leczenia: 4 tygodnie przyjmowania leku/2 tygodnie przerwy („schemat 4/2”). W niniejszym badaniu mediana czasu do progresji choroby (Time To Progression, TTP) wynosiła 34,0 tygodnie (95% CI = 22,0–46,0 tygodnia). Przeprowadzono randomizowane, podwójnie ślepe, kontrolowane placebo badanie fazy III preparatu SUTENT u pacjentów z nowotworami podścieliskowymi przewodu pokarmowego, którzy nie tolerowali leczenia imatynibem lub, u których doszło do progresji choroby w trakcie leczenia imatynibem (mediana maksymalnej dawki dobowej 800 mg). W badaniu tym 312 pacjentów w sposób randomizowany (w stosunku 2:1) przydzielono do grup otrzymujących odpowiednio preparat SUTENT w dawce 50 mg lub placebo doustnie, raz na dobę, według schematu 4/2, do momentu wystąpienia progresji choroby lub wycofania z badania z innego powodu (207 pacjentów otrzymywało SUTENT, a 105 pacjentów otrzymywało placebo). Podstawowym punktem końcowym oceny skuteczności w badaniu był TTP definiowany jako czas od randomizacji do pierwszego potwierdzenia obiektywnej progresji guza. Mediana TTP podczas stosowania preparatu SUTENT wynosiła 28,9 tygodnia (95% CI = 21,3–34,1 tygodnia) i była istotnie statystycznie dłuższa niż TTP wynoszący 5,1 tygodnia (95% CI = 4,4–10,1 tygodnie) podczas stosowania placebo. Różnica przeżywalności całkowitej była statystycznie większa na korzyść preparatu SUTENT [współczynnik ryzyka: 0,491 95% (C.I. 0,290- 0,831)]; ryzyko zgonu było 2 razy większe u pacjentów w grupie otrzymującej placebo w porównaniu do grupy leczonej preparatem SUTENT. Odsetki zgonów wynosiły 14% u osób otrzymujących SUTENT w porównaniu do 25% u osób otrzymujących placebo. W momencie przeprowadzania analizy nie osiągnięto jeszcze mediany przeżywalności ogólnej w żadnej z grup terapeutycznych. Rak nerki z przerzutami oporny na leczenie cytokinami (MRCC) Badanie II fazy preparatu SUTENT przeprowadzono u pacjentów opornych na wcześniejsze leczenie cytokinami, tzn. interleukiną-2 lub interferonem-α. Sześćdziesięciu trzech pacjentów otrzymało dawkę początkową 50 mg preparatu SUTENT doustnie, raz na dobę przez 4 kolejne tygodnie, po czym następował dwutygodniowy okres przerwy, który kończył pełny cykl 6 tygodni (schemat 4/2). Podstawowym punktem końcowym oceny skuteczności był wskaźnik obiektywnych odpowiedzi na leczenie (objective response rate, ORR) ustalany na podstawie kryteriów oceny odpowiedzi u pacjentów z guzami litymi (Response Evaluation Criteria in Solid Tumours, RECIST). W omawianym badaniu wskaźnik obiektywnych odpowiedzi wynosił 36,5% (95% C.I. 24,7% – 49,6%), a mediana czasu do progresji choroby (TTP) wynosiła 37,7 tygodnia (95% C.I. 24,0 – 46,4 tygodnia). Przeprowadzono potwierdzające, otwarte, obejmujące jedną grupę pacjentów, wieloośrodkowe badanie oceniające skuteczność i bezpieczeństwo stosowania preparatu SUTENT u pacjentów z rakiem nerki z przerzutami, którzy byli oporni na uprzednio stosowane leczenie cytokinami. Stu sześciu pacjentów otrzymało co najmniej jedną dawkę 50 mg preparatu SUTENT według schematu 4/2.

44
Pierwszorzędowym punktem końcowym oceny skuteczności tego badania był wskaźnik obiektywnych odpowiedzi (ORR). Drugorzędowe punkty końcowe obejmowały TTP, okres utrzymywania się odpowiedzi (duration of response, DR) i przeżycie całkowite (overall survival, OS). W niniejszym badaniu wskaźnik ORR wynosił 38% (95% C.I. 26,8% – 47,5 %). Mediany DR i OS nie zostały jeszcze osiągnięte. Ten produkt leczniczy został dopuszczony do obrotu zgodnie z procedurą „warunkowego dopuszczenia”. Oznacza to, że oczekiwane są dalsze dowody świadczące o korzyści z jego stosowania, w szczególności dane dotyczące wpływu preparatu SUTENT na medianę czasu do wystąpienia progresji u pacjentów z rakiem nerki z przerzutami. Badanie oceniające ten parametr jest w toku. Europejska Agencja ds. Produktów Leczniczych będzie dokonywać przeglądu nowych informacji o produkcie raz do roku i ChPL zostanie uzupełniona tak jak to będzie konieczne. 5.2 Właściwości farmakokinetyczne Właściwości farmakokinetyczne sunitynibu i jabłczanu sunitynibu oceniano w badaniach obejmujących 135 zdrowych ochotników i 266 pacjentów z guzami litymi. Wchłanianie Po podaniu doustnym sunitynibu maksymalne stężenie (Cmax) stwierdza się na ogół po upływie 6 – 12 godzin (Tmax). Pokarm nie wpływa na dostępność biologiczną sunitynibu. Dystrybucja W badaniach in vitro stopień wiązania się sunitynibu i jego podstawowego czynnego metabolitu z ludzkimi białkami osocza wynosił odpowiednio 95% i 90% niezależnie od stężenia. Pozorna objętość dystrybucji (V/F) w przypadku sunitynibu była znaczna – 2230 l, co wskazuje na penetrację leku do tkanek. Metabolizm Obliczone in vitro wartości stałej Ki dla wszystkich badanych izoform enzymu CYP (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 I CYP4A9/11) wskazują na to, że sunitynib i jego podstawowy czynny metabolit prawdopodobnie nie hamują w stopniu istotnym klinicznie metabolizmu preparatów metabolizowanych przez te enzymy. Badania in vitro również wskazują na to, że SUTENT nie indukuje i nie hamuje głównych enzymów CYP, w tym CYP3A4. Biotransformacja Sunitynib jest metabolizowany przede wszystkim przez izoenzym CYP3A4 cytochromu P450, który katalizuje reakcję powstawania głównego czynnego metabolitu podlegającego dalszemu metabolizmowi katalizowanemu przez CYP3A4. Równoczesne podawanie preparatu SUTENT z silnym induktorem CYP3A4 – ryfampicyną powodowało zmniejszenie wartości Cmax i AUC0-∞ sunitynibu o odpowiednio 56% i 78% po podaniu pojedynczej dawki preparatu SUTENT zdrowym ochotnikom. Inne induktory grupy enzymów CYP3A4 (np. deksametazon, fenytoina, karbamazepina, fenobarbital lub ziele dziurawca Hypericum perforatum) podawane jednocześnie z preparatem SUTENT mogą powodować zmniejszenie stężenia sunitynibu. Eliminacja Preparat jest wydalany przede wszystkim z kałem (61%), natomiast przez nerki ulega wydaleniu 16% podanej dawki w postaci niezmienionej i metabolitów. Sunitynib i jego podstawowy czynny metabolit były głównymi związkami pochodnymi leku wykrywanymi w osoczu, moczu i kale, odpowiadając odpowiednio za 91,5%, 86,4% i 73,8% radioaktywności w zebranych próbkach. Pozostałe metabolity o mniejszym znaczeniu zostały zidentyfikowane w moczu i w kale, jednak na ogół nie były wykrywane w osoczu. Całkowity klirens leku po podaniu doustnym (CL/F) wynosił 34–62 l/h. Zaburzenia czynności narządów

45
Niewydolność wątroby: Nie przeprowadzono badań klinicznych nad stosowaniem leku u pacjentów z niewydolnością wątroby. Z badań wykluczano pacjentów z aktywnością AlAT lub AspAT przekraczającą o ponad 2,5 x górną granicę normy lub, gdy podwyższenie to wiązało się z chorobą podstawową – z aktywnością tych enzymów przekraczającą o ponad 5,0 x górną granicę normy. Niewydolność nerek: Nie przeprowadzono badań klinicznych u pacjentów z niewydolnością nerek. Z badań wykluczano pacjentów ze stężeniem kreatyniny w surowicy przekraczającym o ponad 2,0 x górną granicę normy. Populacyjne analizy farmakokinetyki wykazały, klirens pozorny sunitynibu (CL/F) nie jest uzależniony od klirensu kreatyniny w ocenianym zakresie stężeń (42–347 ml/min). Farmakokinetyka w osoczu Po podaniu doustnym zdrowym ochotnikom okres półtrwania sunitynibu i jego podstawowego czynnego metabolitu deetylowego wynosił odpowiednio około 40–60 godzin i 80–110 godzin. W zakresie dawkowania od 25 do 100 mg pole pod krzywą zależności stężenia leku w osoczu od czasu (AUC) i Cmax zwiększa się proporcjonalnie do dawki. Podczas kilkakrotnego podania w ciągu doby sunitynib ulega kumulacji, przy czym jego stężenie zwiększa się 3–4-krotnie, a stężenie jego podstawowego czynnego metabolitu zwiększa się 7–10-krotnie. Stężenie sunitynibu i jego podstawowego czynnego metabolitu w stanie równowagi zostaje osiągnięte w ciągu od 10 do 14 dni. Do 14 dnia łączne stężenie osoczowe sunitynibu i jego podstawowego czynnego metabolitu wynosi 62,9–101 ng/ml, co jest docelowym stężeniem przewidywanym na podstawie danych z badań przedklinicznych jako stężenie hamujące fosforylację receptorów in vitro, prowadzącym do zatrzymania/zmniejszenia wzrostu guzów in vivo. Podstawowy czynny metabolit odpowiada za 23–37% całkowitej ekspozycji na lek. Nie obserwuje się istotnych zmian farmakokinetyki sunitynibu lub jego podstawowego czynnego metabolitu podczas kilkakrotnego podawania leku w ciągu doby lub w trakcie powtarzanych cyklów badanych schematów dawkowania. Parametry farmakokinetyczne były podobne we wszystkich badanych populacjach pacjentów z guzami litymi i u zdrowych ochotników. Populacyjne analizy farmakokinetyki z uwzględnieniem danych demograficznych wskazują na to, że nie są koniecznie korekty dawkowania w zależności od masy ciała lub punktacji wg skali ECOG. Dostępne dane wskazują, że u kobiet klirens pozorny (CL/F) sunitynibu może być o około 30% niższy niż u mężczyzn: różnica ta nie wymaga jednak dostosowywania dawki. 5.3 Przedkliniczne dane o bezpieczeństwie W dziewięciomiesięcznych badaniach nad toksycznością dawek wielokrotnych u szczurów i małp stwierdzono, że podstawowymi narządami, na które wpływa lek, są: przewód pokarmowy (nudności i biegunki u małp), nadnercza (przekrwienie kory i (lub) krwotoki u szczurów i małp, z martwicą i następującym po niej włóknieniem u szczurów), układ limfatyczny i krwiotwórczy (zmniejszenie liczby komórek szpiku kostnego i zanik tkanki limfoidalnej grasicy, śledziony i węzłów chłonnych), zewnątrzwydzielnicza część trzustki (degranulacja komórek pęcherzykowych z martwicą pojedynczych komórek), ślinianki (przerost gronek), stawy (zgrubienie płytki wzrostu), macica (zanik) i jajniki (zmniejszony wzrost pęcherzyków). Wszystkie wyniki uzyskano przy istotnych klinicznie poziomach stężenia osoczowego sunitynibu. Dodatkowe działania preparatu obserwowane w innych badaniach obejmowały: wydłużenie odstępu QTc, zmniejszenie LVEF, przerost przysadki i zanik kanalików jądrowych, rozrost komórek mezangium w nerkach, krwotok z przewodu pokarmowego i do jamy ustnej i przerost komórek płata przedniego przysadki. Uważa się, że zmiany w obrębie macicy (zanik błony śluzowej) i płytki wzrostowej kości (zgrubienie nasad kostnych lub dysplazja chrząstki) są związane z działaniem farmakologicznym sunitynibu. Większość z tych zmian była odwracalna po upływie od 2 do 6 tygodni od zakończenia leczenia. Genotoksyczność Potencjalne działanie genotoksyczne sunitynibu oceniano zarówno in vitro, jak i in vivo. Sunitynib nie wykazywał właściwości mutagennych w badaniach na bakteriach z zastosowaniem aktywacji metabolicznej przez wątrobę szczura. Sunitynib nie indukował strukturalnych aberracji chromosomalnych w ludzkich limfocytach krwi obwodowej in vitro. Obserwowano poliploidię (liczbowe aberracje chromosomalne) w ludzkich limfocytach krwi obwodowej in vitro, zarówno w

46
przypadku zastosowania aktywacji metabolicznej, jak i bez niej. Sunitynib nie wykazywał działania klastogennego w szczurzym szpiku kostnym in vivo u szczurów. Nie oceniano jego podstawowego czynnego metabolitu pod kątem potencjalnej toksyczności genetycznej. Rakotwórczość Nie przeprowadzono badań nad potencjałem rakotwórczym jabłczanu sunitynibu. Toksyczny wpływ na zdolność do rozrodu i rozwój W badaniach toksycznego wpływu na zdolność do rozrodu szczurów nie stwierdzono wpływu preparatu na płodność samic lub samców. Jednak w badaniach toksyczności dawek wielokrotnych przeprowadzonych na szczurach i małpach obserwowano oddziaływanie leku na płodność samic w postaci atrezji pęcherzyków, zwyrodnienia ciałek żółtych, zmian błony śluzowej macicy oraz zmniejszenia masy macicy i jajników przy klinicznie istotnych poziomach ekspozycji układowej. U szczurów obserwowano wpływ leku na płodność samców w postaci zaniku kanalików jąder, zmniejszenia liczby plemników w najądrzach i zmniejszenia ilości koloidu w obrębie gruczołu krokowego i pęcherzyków nasiennych przy poziomach ekspozycji osoczowej 18 razy większych niż stwierdzane w warunkach klinicznych. U szczurów śmiertelność zarodków i płodów przejawiała się istotnym zmniejszeniem liczby żywych płodów, zwiększoną liczbą resorpcji, wzrostem utrat ciąży po zagnieżdżeniu zarodka i całkowitą utratą miotów u 8 z 28 samic ciężarnych przy poziomach stężenia leku w osoczu 5,5 razy większych niż poziomy obserwowane w warunkach klinicznych u ludzi. U królików redukcja masy macicy samic ciężarnych i liczby żywych płodów była związana ze zwiększeniem liczby resorpcji, wzrostem liczby utrat ciąży po zagnieżdżeniu zarodka i całkowitą utratą miotów u 4 z 6 samic ciężarnych przy poziomach stężenia leku w osoczu 3 razy większych niż poziomy obserwowane w warunkach klinicznych u ludzi.
Stosowanie sunitynibu u szczurów w okresie organogenezy prowadziło do zmian rozwojowych, polegających na zwiększonej częstości występowania wad rozwojowych szkieletu płodu, charakteryzujących się przede wszystkim opóźnieniem kostnienia kręgów piersiowych/lędźwiowych. i obserwowanych przy poziomach stężenia leku w osoczu 6 razy większych niż poziomy obserwowane w warunkach klinicznych u ludzi. U królików wpływ leku na rozwój polegał na zwiększeniu częstości występowania rozszczepu wargi przy poziomach stężenia leku w osoczu w przybliżeniu odpowiadających poziomom obserwowanym w warunkach klinicznych u ludzi oraz rozszczepu wargi i rozszczepu podniebienia przy poziomach stężenia leku w osoczu 2,7 razy większych niż poziomy obserwowane w warunkach klinicznych u ludzi.

47
6. DANE FARMACEUTYCZNE 6.1 Wykaz substancji pomocniczych Zawartość kapsułki Mannitol Kroskarmeloza sodowa Powidon Stearynian magnezu Powłoka kapsułki Żelatyna Dwutlenek tytanu (E171) Żółty tlenek żelaza (E172) Czerwony tlenek żelaza (E172) Czarny tlenek żelaza (E172) Tusz do nadruku. Szelak Glikol propylenowy Wodorotlenek sodu Powidon Dwutlenek tytanu (E171) 6.2 Niezgodności farmaceutyczne Nie dotyczy 6.3 Okres trwałości 2 lata 6.4 Specjalne środki ostrożności przy przechowywaniu Brak szczególnych środków ostrożności dotyczących przechowywania. 6.5 Rodzaj i zawartość opakowania Butelki z polietylenu o dużej gęstości (HDPE) z polipropylenowym wieczkiem zawierające po 30 kapsułek. 6.6 Szczególne środki ostrożności dotyczące usuwania Brak szczególnych wymagań. 7. PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE
DO OBROTU Pfizer Ltd Ramsgate Road Sandwich, Kent CT13 9NJ Wielka Brytania

48
8. NUMER(-Y) POZWOLENIA NA DOPUSZCZENIE DO OBROTU 9. DATA WYDANIA PIERWSZEGO POZWOLENIA NA DOPUSZCZENIE DO OBROTU /
DATA PRZEDŁUŻENIA POZWOLENIA Data wydania pierwszego pozwolenia: Data przedłużenia pozwolenia: 10. DATA ZATWIERDZENIA LUB CZĘŚCIOWEJ ZMIANY TEKSTU
CHARAKTERYSTYKI PRODUKTU LECZNICZEGO

49
ANEKS II
A. WYTWÓRCA(Y) SUBSTANCJI BIOLOGICZNIE CZYNNEJ(YCH) ORAZ WYTWÓRCA (Y) ODPOWIEDZIALNY(I) ZA ZWOLNIENIE SERII
B. WARUNKI POZWOLENIA NA DOPUSZCZENIE DO OBROTU
C. SZCZEGÓLNE OBOWIĄZKI DO SPEŁNIENIA PRZEZ PODMIO
ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU

50
A. WYTWÓRCA(Y) ODPOWIEDZIALNY ZA ZWOLNIENIE SERII Nazwa i adres wytwórcy(ów) odpowiedzialnego(ych) za zwolnienie serii Pfizer Italia S.r.l. Via del Commercio – Zona Industriale - IT- 63046 Marino del Tronto (Ascoli Piceno) Włochy B. WARUNKI POZWOLENIA NA DOPUSZCZENIE DO OBROTU • KATEGORIA DOSTĘPNOŚCI Produkt leczniczy wydawany na podstawie zastrzezonej recepty (patrz Aneks I: Charakterystyka produktu leczniczego, punkt 4.2). • WARUNKI LUB OGRANICZENIA W ODNIESIENIU DO BEZPIECZNEGO I
SKUTECZNEGO UŻYWANIA PRODUKTÓW LECZNICZYCH Nie dotyczy. • INNE WARUNKI Plan Zarządzania Ryzykiem Podmiot odpowiedzialny posiadający pozwolenie na dopuszczenie do obrotu zobowiązuje się do przeprowadzenia badań i wykonania dodatkowych czynności w celu zgłaszania i rejestracji zdarzeń niepożądanych określonych w Planie Nadzoru nad Bezpieczeństwem Stosowania. Uaktualniony Plan Zarządzania Ryzykiem, zgodnie z wytycznymi CHMP dotyczącymi systemów zarządzania ryzykiem dla produktów leczniczych stosowanych u ludzi, powinien być przedłożony razem z dokumentem PSUR w czasie 60 dni od osiągnięcia ważnego (Nadzór nad Bezpieczeństwem Stosowania lub Plan Minimalizacji Ryzyka) etapu lub, gdy dostępne będą wyniki badania klinicznego lub na żądanie właściwych władz. C. SZCZEGÓLNE OBOWIĄZKI DO SPEŁNIENIA PRZEZ PODMIOT ODPOWIEDZIALNY POSIADAJĄCY POZWOLENIE NA DOPUSZCZENIE DO OBROTU Podmiot odpowiedzialny posiadający pozwolenie na dopuszczenie do obrotu powinien przeprowadzić następujący program badań w wyznaczonym czasie. Wyniki tych badań powinny być podstawą do oszacowania profilu korzyść/ryzyko w trakcie rozpatrywania wniosku o rerejestrację. Ocena pod względem klinicznym: Podmiot odpowiedzialny posiadający pozwolenie na dopuszczenie do obrotu zobowiązuje się do przedstawienia do września 2006 r. wyników toczącego się obecnie badania u pacjentów z rakiem nerki z przerzutami wrażliwych na cytokiny.

51
ANEKS III
OZNAKOWANIE OPAKOWAŃ I ULOTKA DLA PACJENTA

52
A. OZNAKOWANIE OPAKOWAŃ

53
INFORMACJE ZAMIESZCZANE NA OPAKOWANIACH ZEWNĘTRZNYCH Tekturowe pudełko zewnętrzne – 12,5 mg kapsułki 1. NAZWA PRODUKTU LECZNICZEGO SUTENT 12,5 kapsułki twarde Sunitynib 2. ZAWARTOŚĆ SUBSTANCJI CZYNNEJ (CZYNNYCH) Każda kapsułka zawiera jabłczan sunitynibu w ilości odpowiadającej12,5 mg sunitynibu 3. WYKAZ SUBSTANCJI POMOCNICZYCH W skład kapsułki wchodzą mannitol i glikol propylenowy 4. POSTAĆ FARMACEUTYCZNA I ZAWARTOŚĆ OPAKOWANIA 30 kapsułek twardych 5. SPOSÓB I DROGA (DROGI) PODANIA Do stosowania doustnego. Należy zapoznać się z treścią ulotki przed zastosowaniem leku. 6. OSTRZEŻENIE DOTYCZĄCE PRZECHOWYWANIA PRODUKTU LECZNICZEGO W
MIEJSCU NIEDOSTĘPNYM I NIEWIDOCZNYM DLA DZIECI Lek przechowywać w miejscu niedostępnym i niewidocznym dla dzieci. 7. INNE OSTRZEŻENIA SPECJALNE, JEŚLI KONIECZNE Lek należy stosować zgodnie z zaleceniami lekarza. 8. TERMIN WAŻNOŚCI EXP 9. WARUNKI PRZECHOWYWANIA Brak szczególnych środków dotyczących przechowywania.

54
10. SPECJALNE ŚRODKI OSTROŻNOŚCI DOTYCZĄCE USUWANIA NIEZUŻYTEGO PRODUKTU LECZNICZEGO LUB POCHODZĄCYCH Z NIEGO ODPADÓW, JEŚLI WŁAŚCIWE
11. NAZWA I ADRES PODMIOTU ODPOWIEDZIALNEGO Pfizer Ltd Ramsgate Road Sandwich Kent CT13 9NJ Wielka Brytania 12. NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU 13. NUMER SERII Nr serii: {numer} 14. KATEGORIA DOSTĘPNOŚCI Lek wydawany na receptę. 15. INSTRUKCJA UŻYCIA 16. INFORMACJA PODANA BRAJLEM SUTENT 12,5 mg

55
MINIMUM INFORMACJI ZAMIESZCZANYCH NA MAŁYCH OPAKOWANIACH BEZPOŚREDNICH Butelka HDPE – 12,5 mg kapsułki 1. NAZWA PRODUKTU LECZNICZEGO i DROGA (DROGI) PODANIA SUTENT 12,5 mg kapsułki twarde Sunitynib Do stosowania doustnego. 2. SPOSÓB PODAWANIA Należy zapoznać się z treścią ulotki przed zastosowaniem leku. 3. TERMIN WAŻNOŚCI EXP 4. NUMER SERII Nr serii: {numer} 5. ZAWARTOŚĆ OPAKOWANIA Z PODANIEM MASY, OBJĘTOŚCI LUB LICZBY
JEDNOSTEK 30 kapsułek

56
INFORMACJE ZAMIESZCZANE NA OPAKOWANIACH ZEWNĘTRZNYCH Tekturowe pudełko zewnętrzne – 25 mg kapsułki 1. NAZWA PRODUKTU LECZNICZEGO SUTENT 25 mg kapsułki twarde Sunitynib 2. ZAWARTOŚĆ SUBSTANCJI CZYNNEJ (CZYNNYCH) Każda kapsułka zawiera jabłczan sunitynibu w ilości odpowiadającej12,5 mg sunitynibu 3. WYKAZ SUBSTANCJI POMOCNICZYCH W skład kapsułki wchodzą mannitol i glikol propylenowy 4. POSTAĆ FARMACEUTYCZNA I ZAWARTOŚĆ OPAKOWANIA 30 kapsułek twardych 5. SPOSÓB I DROGA (DROGI) PODANIA Do stosowania doustnego. Należy zapoznać się z treścią ulotki przed zastosowaniem leku. 6. OSTRZEŻENIE DOTYCZĄCE PRZECHOWYWANIA PRODUKTU LECZNICZEGO W
MIEJSCU NIEDOSTĘPNYM I NIEWIDOCZNYM DLA DZIECI Lek przechowywać w miejscu niedostępnym i niewidocznym dla dzieci. 7. INNE OSTRZEŻENIA SPECJALNE, JEŚLI KONIECZNE Lek należy stosować zgodnie z zaleceniami lekarza. 8. TERMIN WAŻNOŚCI EXP 9. WARUNKI PRZECHOWYWANIA Brak szczególnych środków dotyczących przechowywania 10. SPECJALNE ŚRODKI OSTROŻNOŚCI DOTYCZĄCE USUWANIA NIEZUŻYTEGO
PRODUKTU LECZNICZEGO LUB POCHODZĄCYCH Z NIEGO ODPADÓW, JEŚLI WŁAŚCIWE

57
11. NAZWA I ADRES PODMIOTU ODPOWIEDZIALNEGO Pfizer Ltd Ramsgate Road Sandwich Kent CT13 9NJ Wielka Brytania 12. NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU 13. NUMER SERII Nr serii: {numer} 14. KATEGORIA DOSTĘPNOŚCI Lek wydawany na receptę. 15. INSTRUKCJA UŻYCIA 16. INFORMACJA PODANA BRAJLEM SUTENT 25 mg

58
MINIMUM INFORMACJI ZAMIESZCZANYCH NA MAŁYCH OPAKOWANIACH BEZPOŚREDNICH Butelka HDPE – 25 mg kapsułki 1. NAZWA PRODUKTU LECZNICZEGO i DROGA (DROGI) PODANIA SUTENT 25mg kapsułki twarde Sunitynib Do stosowania doustnego. 2. SPOSÓB PODAWANIA Należy zapoznać się z treścią ulotki przed zastosowaniem leku. 3. TERMIN WAŻNOŚCI EXP 4. NUMER SERII Nr serii: {numer} 5. ZAWARTOŚĆ OPAKOWANIA Z PODANIEM MASY, OBJĘTOŚCI LUB LICZBY
JEDNOSTEK 30 kapsułek

59
INFORMACJE ZAMIESZCZANE NA OPAKOWANIACH ZEWNĘTRZNYCH Tekturowe pudełko zewnętrzne – 50 mg kapsułki 1. NAZWA PRODUKTU LECZNICZEGO SUTENT 50 mg kapsułki twarde Sunitynib 2. ZAWARTOŚĆ SUBSTANCJI CZYNNEJ (CZYNNYCH) Każda kapsułka zawiera jabłczan sunitynibu w ilości odpowiadającej12,5 mg sunitynibu 3. WYKAZ SUBSTANCJI POMOCNICZYCH W skłąd kapsułki wchodzą mannitol i glikol propylenowy 4. POSTAĆ FARMACEUTYCZNA I ZAWARTOŚĆ OPAKOWANIA 30 kapsułek twardych 5. SPOSÓB I DROGA (DROGI) PODANIA Do stosowania doustnego. Należy zapoznać się z treścią ulotki przed zastosowaniem leku. 6. OSTRZEŻENIE DOTYCZĄCE PRZECHOWYWANIA PRODUKTU LECZNICZEGO W
MIEJSCU NIEDOSTĘPNYM I NIEWIDOCZNYM DLA DZIECI Lek przechowywać w miejscu niedostępnym i niewidocznym dla dzieci. 7. INNE OSTRZEŻENIA SPECJALNE, JEŚLI KONIECZNE Lek należy stosować zgodnie z zaleceniami lekarza. 8. TERMIN WAŻNOŚCI EXP 9. WARUNKI PRZECHOWYWANIA Brak szczególnych środków dotyczących przechowywania 10. SPECJALNE ŚRODKI OSTROŻNOŚCI DOTYCZĄCE USUWANIA NIEZUŻYTEGO
PRODUKTU LECZNICZEGO LUB POCHODZĄCYCH Z NIEGO ODPADÓW, JEŚLI WŁAŚCIWE

60
11. NAZWA I ADRES PODMIOTU ODPOWIEDZIALNEGO Pfizer Ltd Ramsgate Road Sandwich Kent CT13 9NJ Wielka Brytania 12. NUMER POZWOLENIA NA DOPUSZCZENIE DO OBROTU 13. NUMER SERII Nr serii: {numer} 14. KATEGORIA DOSTĘPNOŚCI Lek wydawany na receptę. 15. INSTRUKCJA UŻYCIA 16. INFORMACJA PODANA BRAJLEM SUTENT 50 mg

61
MINIMUM INFORMACJI ZAMIESZCZANYCH NA MAŁYCH OPAKOWANIACH BEZPOŚREDNICH Butelka HDPE – 50 mg kapsułki 1. NAZWA PRODUKTU LECZNICZEGO i DROGA (DROGI) PODANIA SUTENT 50 mg kapsułki twarde 2. SPOSÓB PODAWANIA Należy zapoznać się z treścią ulotki przed zastosowaniem leku. 3. TERMIN WAŻNOŚCI EXP 4. NUMER SERII Nr serii: {numer} 5. ZAWARTOŚĆ OPAKOWANIA Z PODANIEM MASY, OBJĘTOŚCI LUB LICZBY
JEDNOSTEK 30 kapsułek

62
B. ULOTKA DLA PACJENTA

63
ULOTKA DLA PACJENTA: INFORMACJE DLA UŻYTKOWNIKA
SUTENT kapsułki twarde 12,5 mg
Sunitynib Należy zapoznać się z treścią ulotki przed zażyciem leku. - Należy zachować tę ulotkę, aby w razie potrzeby móc ją ponownie przeczytać. - Należy zwrócić się do lekarza lub farmaceuty, gdy potrzebna jest rada lub dodatkowa informacja. - Lek ten został przepisany ściśle określonej osobie i nie należy go przekazywać innym, gdyż może
im zaszkodzić, nawet jeśli objawy ich choroby są takie same. - Jeśli nasili się którykolwiek z objawów niepożądanych lub wystąpią jakiekolwiek objawy
niepożądane niewymienione w ulotce, należy powiadomić lekarza. Spis treści ulotki: 1. Co to jest SUTENT i w jakim celu się go stosuje 2. Informacje ważne przed zażyciem leku SUTENT 3. Jak zażywać SUTENT 4. Możliwe działania niepożądane 5. Jak przechowywać lek SUTENT 6. Inne informacje 1. CO TO JEST SUTENT I W JAKIM CELU SIĘ GO STOSUJE SUTENT jest produktem leczniczym stosowanym w leczeniu nowotworów. Hamuje aktywność specjalnej grupy białek, o których wiadomo, że uczestniczą we wzroście i rozsiewie komórek nowotworowych. Może on zostać przepisany wyłącznie przez lekarza doświadczonego w stosowaniu leków przeznaczonych do leczenia raka nerki lub nowotworów podścieliskowych przewodu pokarmowego. SUTENT jest produktem leczniczym stosowanym w leczeniu raka nerki, w przebiegu którego dochodzi do zmian nowotworowych w komórkach kanalików nerkowych. Lek SUTENT stosuje się również w leczeniu złośliwych nowotworów podścieliskowych przewodu pokarmowego (ang. gastrointestinal stromal tumor - GIST). Nowotwór podścieliskowy przewodu pokarmowego może rozwijać się w żołądku lub jelitach. Rozwija się w wyniku niekontrolowanego rozrostu komórek tkanek podporowych tych narządów. Lek SUTENT hamuje wzrost tych komórek. Należy się zwracać do lekarza z wszelkimi pytaniami na temat sposobu działania leku SUTENT lub na temat przyczyn przepisania tego leku. 2. INFORMACJE WAZNE PRZED ZASTOSOWANIEM LEKU SUTENT Należy dokładnie przestrzegać wszystkich zaleceń lekarza, nawet jeżeli różnią się od ogólnych informacji zawartych w niniejszej ulotce. Kiedy nie zażywać leku SUTENT Jeśli u pacjenta stwierdzono uczulenie na sunitynib lub którykolwiek z pozostałych składników leku SUTENT. Kiedy zachować szczególną ostrożność stosując SUTENT
- Jeśli występuje choroba wątroby lub nerek, - Jeśli występuje wysokie ciśnienie tętnicze krwi, - W stwierdzonej lub podejrzewanej ciąży (szczegóły podano poniżej),

64
- W czasie karmienia piersią (szczegóły podano poniżej). W przypadku istnienia którejkolwiek z powyższych okoliczności należy o nich poinformować lekarza przed rozpoczęciem stosowania leku SUTENT.
Stosowanie leku SUTENT z innymi lekami Należy powiedzieć lekarzowi lub farmaceucie o wszystkich przyjmowanych ostatnio lekach, również tych, które wydawane są bez recepty i preparatach ziołowych (wszelkie leki, które mogą zwiększać stężenie leku SUTENT, takie jak ketokonazol, rytonawir, itrakonazol, erytromycyna, klarytromycyna, a także wszelkie leki, które mogą zmniejszać stężenie leku SUTENT, takie jak deksametazon, fenytoina, karbamazepina, ryfampicyna, fenobarbital czy ziele dziurawca Hypericum perforatum). Stosowanie leku SUTENT z jedzeniem i piciem Lek SUTENT można przyjmować podczas posiłków lub między posiłkami, jednak leku SUTENT nie należy przyjmować jednocześnie z sokiem grejfrutowym. Ciąża i karmienie piersią Należy poinformować lekarza o stwierdzonej lub podejrzewanej ciąży. Kobiety ciężarne nie powinny stosować leku SUTENT, o ile nie jest to bezwzględnie konieczne. Lekarz omówi z pacjentką potencjalne ryzyko związane z przyjmowaniem leku w czasie ciąży. Kobietom, które mogą zajść w ciążę, zaleca się stosowanie skutecznej antykoncepcji w trakcie leczenia lekiem SUTENT. Kobiety karmiące piersią powinny o tym poinformować lekarza. Nie należy karmić piersią podczas przyjmowania leku SUTENT. Prowadzenie pojazdów i obsługa maszyn W przypadku wystąpienia zawrotów głowy lub znacznego zmęczenia należy zachować szczególną ostrożność podczas prowadzenia samochodu i obsługi urządzeń mechanicznych w ruchu. 3. JAK ZAŻYWAĆ LEK SUTENT Lekarz przepisze dawkę właściwą dla danego pacjenta. Zaleca się, aby przyjmować lek SUTENT przez 28 dni (4 tygodnie), po czym powinna nastąpić 14-dniowa (dwutygodniowa) przerwa (bez leku), co razem daje sześciotygodniowy cykl leczenia. Lekarz zdecyduje o liczbie cykli leczenia odpowiedniej dla danego pacjenta. Zażycie większej niż zalecana dawki leku SUTENT W przypadku przyjęcia większej liczby kapsułek niż zalecana należy bezzwłocznie porozumieć się z lekarzem. Może być konieczna interwencja medyczna. Pominięcie zażycia dawki leku SUTENT Nie należy stosować dawki podwójnej w celu uzupełnienia pominiętej dawki. 4. MOŻLIWE DZIAŁANIA NIEPOŻĄDANE Jak każdy lek, SUTENT może powodować działania niepożądane, chociaż nie u każdego one występują. Bardzo częste działania niepożądane występujące u więcej niż 10 pacjentów na 100:
• ból/podrażnienie w obrębie jamy ustnej, bolesność ust, zaburzenia smaku, zaburzenia żołądkowe, nudności, wymioty, biegunka, zaparcia, bóle brzucha, utrata apetytu,
• przebarwienia skórne, zmiana koloru włosów, wysypka na dłoniach i podeszwach stóp, pęcherze, suchość skóry
• uczucie zmęczenia, nadciśnienie tętnicze, migrena • zmniejszenie liczby krwinek czerwonych i krwinek białych

65
Inne możliwe działania występujące u od 1 do 10 osób na 100: • zmniejszenie aktywności tarczycy, zmniejszenie przepływu kwi przez serce • krwawienie z nosa, nieprawidłowe zabarwienie moczu, nadmierne łzawienie • bóle stawów, bóle mięśni, gromadzenie płynów w tkankach, w tym także w okolicy oczu • zaburzenia czucia w skórze, skrócony oddech • utrata włosów, utrata masy ciała
Jeśli nasili się którykolwiek z objawów niepożądanych lub wystąpią jakiekolwiek objawy niepożądane nie wymienione w ulotce, należy powiadomić lekarza. 5. JAK PRZECHOWYWAĆ LEK SUTENT
- Lek przechowywać w miejscu niedostępnym i niewidocznym dla dzieci. - Brak specjalnych zaleceń dotyczących przechowywania niniejszego produktu leczniczego. - Nie stosować leku po upływie terminu ważności (EXP) zamieszczonego na opakowaniu
zewnętrznym i opakowaniu bezpośrednim. - Nie stosować leku jeśli opakowanie zostało uszkodzone lub widoczne są ślady wcześniejszych
prób otwierania. Leków nie należy wyrzucać do kanalizacji lub domowych pojemników na odpadki. Należy zapytać farmaceutę co zrobić z lekami, których się już nie potrzebuje. Takie postępowanie pomoże chronić środowisko. 6. INNE INFORMACJE Co zawiera lek SUTENT: - Substancją czynną leku jest sunitynib (w postaci soli jabłczanowej). - Inne składniki leku to: mannitol, sól sodowa kroskarmelozy, powidon i magnezu stearynian.
Otoczka kapsułki składa się z: żelatyny, czerwonego tlenku żelaza (E172) i dwutlenku tytanu (E 171). Tusz do nadruków zawiera szelak, glikol propylenowy, sodu wodorotlenek, powidon i dwutlenek tytanu.
Jak wygląda lek SUTENT i co zawiera opakowanie Lek SUTENT jest dostępny w postaci twardych kapsułek żelatynowych z pomarańczowym wieczkiem i pomarańczowym korpusem, z wykonanymi białym tuszem napisami „Pfizer” na wieczku i „STN 12.5 mg” na korpusie. Lek jest dostępny w butelkach po 30 kapsułek. Podmiot odpowiedzialny Pfizer Limited Ramsgate Road Sandwich, Kent CT13 9NJ Wielka Brytania Wytwórca Pfizer Italia S.r.l. Via del Commercio – Zona Industriale - 63046 Marino del Tronto (Ascoli Piceno) Włochy W celu uzyskania bardziej szczegółowych informacji należy zwrócić się do przedstawiciela podmiotu odpowiedzialnego.

66
Belgique / België /Belgien Pfizer S.A. / N.V. Tél/Tel: +32 (0)2 554 62 11
Luxembourg/Luxemburg Pfizer S.A. Tél/Tel: +32 (0)2 554 62 11
Česká republika Pfizer s.r.o. Tel.: +420-283-004-151
Magyarország Pfizer Kft. Tel.: +36-1-488-37-00
Danmark Pfizer ApS Tlf: +45 44 20 11 00
Malta V.J. Salomone Pharma Ltd. Tel. +356 212201
Deutschland Pfizer Pharma GmbH Tel: +49 (0)721 61 01 90 00
Nederland Pfizer BV Tel: +31 (0)10 406 42 00
Eesti Pfizer Luxembourg SARL, Eesti filiaal Tel.: +372 6 405 328
Norge Pfizer AS Tlf: +47 67 52 61 00
Ελλάδα Pfizer Hellas A.E. Τηλ: +30 210 7517981-3
Österreich Pfizer Corporation Austria Ges.m.b.H. Tel: +43 (0)1 521 15-0
España Pfizer S.A. Tél: +34 91 490 99 00
Polska Pfizer Polska Sp. z o.o. Tel.:+ 48 22 335 61 00
France Pfizer Tél: +33 (0)1 58 07 34 40
Portugal Laboratórios Pfizer, Lda. Tel: +351 21 423 5500
Ireland Pfizer Healthcare Ireland Tel: +353 1800 633 363
Slovenija Pfizer Luxembourg SARL Pfizer, podružnica za svetovanje s področja farmacevtske dejavnosti, Ljubljana Tel.: + 386 1 52 11 400
Vistor hf. PharmaNor hf. Sími: +354 535 7000
Slovenská republika Pfizer Luxembourg SARL, organizačná zložka Tel.:+ 421 2 5941 8500
Italia Pfizer Italia S.r.l. Tel: +39 06 33 18 21
Suomi/Finland Pfizer Oy Puh./Tel: +358 (0)9 43 00 40
Kύπρος Geo. Pavlides & Araouzos Ltd. Tηλ.:+ 357 22 818087
Sverige Pfizer AB Tel: +46 (0)8 550 52000
Latvija Pfizer Luxembourg SARL filiale Latvija Tel.: + 371 70 35 775
United Kingdom Pfizer Limited, Tel: +44 (0)1737 331111

67
Lietuva Pfizer Luxembourg SARL filialas Lietuvoje Tel. + 370 52 51 4000
Data zatwierdzenia ulotki: Ten lek otrzymał „warunkowe dopuszczenie do obrotu”. Oznacza to, że oczekuje się dostarczenia więcej danych o leku, w szczególności dotyczących leczenia raka nerki. Wykazano, że stosowanie leku SUTENT powoduje zmniejszenie guza. Jednak oczekiwane są informacje na temat czasu utrzymywania się takiego działania. Europejska Agencja ds. Produktów Leczniczych dokona przeglądu nowych informacji o leku raz do roku i treść ulotki zostanie uzupełniona tak jak to będzie konieczne. Szczegółowa informacja o tym leku jest dostępna na stronie internetowej Europejskiej Agencji ds. Produktów Leczniczych (EMEA): http://emea.eu.int. Można tam również znaleźć linki do stron internetowych o rzadkich chorobach i sposobach leczenia.

68
ULOTKA DLA PACJENTA: INFORMACJE DLA UŻYTKOWNIKA
SUTENT kapsułki twarde 25 mg
Sunitynib Należy zapoznać się z treścią ulotki przed zażyciem leku. - Należy zachować tę ulotkę, aby w razie potrzeby móc ją ponownie przeczytać. - Należy zwrócić się do lekarza lub farmaceuty, gdy potrzebna jest rada lub dodatkowa informacja. - Lek ten został przepisany ściśle określonej osobie i nie należy go przekazywać innym, gdyż może
im zaszkodzić, nawet jeśli objawy ich choroby są takie same. - Jeśli nasili się którykolwiek z objawów niepożądanych lub wystąpią jakiekolwiek objawy
niepożądane niewymienione w ulotce, należy powiadomić lekarza. Spis treści ulotki: 1. Co to jest SUTENT i w jakim celu się go stosuje 2. Informacje ważne przed zażyciem leku SUTENT 3. Jak zażywać SUTENT 4. Możliwe działania niepożądane 5. Jak przechowywać lek SUTENT 6. Inne informacje 1. CO TO JEST SUTENT I W JAKIM CELU SIĘ GO STOSUJE SUTENT jest produktem leczniczym stosowanym w leczeniu nowotworów. Hamuje aktywność specjalnej grupy białek, o których wiadomo, że uczestniczą we wzroście i rozsiewie komórek nowotworowych. Może on zostać przepisany wyłącznie przez lekarza doświadczonego w stosowaniu leków przeznaczonych do leczenia raka nerki lub nowotworów podścieliskowych przewodu pokarmowego. SUTENT jest produktem leczniczym stosowanym w leczeniu raka nerki, w przebiegu którego dochodzi do zmian nowotworowych w komórkach kanalików nerkowych. Lek SUTENT stosuje się również w leczeniu złośliwych nowotworów podścieliskowych przewodu pokarmowego (ang. gastrointestinal stromal tumor - GIST). Nowotwór podścieliskowy przewodu pokarmowego może rozwijać się w żołądku lub jelitach. Rozwija się w wyniku niekontrolowanego rozrostu komórek tkanek podporowych tych narządów. Lek SUTENT hamuje wzrost tych komórek. Należy się zwracać do lekarza z wszelkimi pytaniami na temat sposobu działania leku SUTENT lub na temat przyczyn przepisania tego leku. 2. INFORMACJE WAZNE PRZED ZASTOSOWANIEM LEKU SUTENT Należy dokładnie przestrzegać wszystkich zaleceń lekarza, nawet jeżeli różnią się od ogólnych informacji zawartych w niniejszej ulotce. Kiedy nie zażywać leku SUTENT Jeśli u pacjenta stwierdzono uczulenie na sunitynib lub którykolwiek z pozostałych składników leku SUTENT. Kiedy zachować szczególną ostrożność stosując SUTENT
- Jeśli występuje choroba wątroby lub nerek, - Jeśli występuje wysokie ciśnienie tętnicze krwi, - W stwierdzonej lub podejrzewanej ciąży (szczegóły podano poniżej),

69
- W czasie karmienia piersią (szczegóły podano poniżej). W przypadku istnienia którejkolwiek z powyższych okoliczności należy o nich poinformować lekarza przed rozpoczęciem stosowania leku SUTENT.
Stosowanie leku SUTENT z innymi lekami Należy powiedzieć lekarzowi lub farmaceucie o wszystkich przyjmowanych ostatnio lekach, również tych, które wydawane są bez recepty i preparatach ziołowych (wszelkie leki, które mogą zwiększać stężenie leku SUTENT, takie jak ketokonazol, rytonawir, itrakonazol, erytromycyna, klarytromycyna, a także wszelkie leki, które mogą zmniejszać stężenie leku SUTENT, takie jak deksametazon, fenytoina, karbamazepina, ryfampicyna, fenobarbital czy ziele dziurawca Hypericum perforatum). Stosowanie leku SUTENT z jedzeniem i piciem Lek SUTENT można przyjmować podczas posiłków lub między posiłkami, jednak leku SUTENT nie należy przyjmować jednocześnie z sokiem grejfrutowym. Ciąża i karmienie piersią Należy poinformować lekarza o stwierdzonej lub podejrzewanej ciąży. Kobiety ciężarne nie powinny stosować leku SUTENT, o ile nie jest to bezwzględnie konieczne. Lekarz omówi z pacjentką potencjalne ryzyko związane z przyjmowaniem leku w czasie ciąży. Kobietom, które mogą zajść w ciążę, zaleca się stosowanie skutecznej antykoncepcji w trakcie leczenia lekiem SUTENT. Kobiety karmiące piersią powinny o tym poinformować lekarza. Nie należy karmić piersią podczas przyjmowania leku SUTENT. Prowadzenie pojazdów i obsługa maszyn W przypadku wystąpienia zawrotów głowy lub znacznego zmęczenia należy zachować szczególną ostrożność podczas prowadzenia samochodu i obsługi urządzeń mechanicznych w ruchu. 3. JAK ZAŻYWAĆ LEK SUTENT Lekarz przepisze dawkę właściwą dla danego pacjenta. Zaleca się, aby przyjmować lek SUTENT przez 28 dni (4 tygodnie), po czym powinna nastąpić 14-dniowa (dwutygodniowa) przerwa (bez leku), co razem daje sześciotygodniowy cykl leczenia. Lekarz zdecyduje o liczbie cykli leczenia odpowiedniej dla danego pacjenta. Zażycie większej niż zalecana dawki leku SUTENT W przypadku przyjęcia większej liczby kapsułek niż zalecana należy bezzwłocznie porozumieć się z lekarzem. Może być konieczna interwencja medyczna. Pominięcie zażycia dawki leku SUTENT Nie należy stosować dawki podwójnej w celu uzupełnienia pominiętej dawki. 4. MOŻLIWE DZIAŁANIA NIEPOŻĄDANE Jak każdy lek, SUTENT może powodować działania niepożądane, chociaż nie u każdego one występują. Bardzo częste działania niepożądane występujące u więcej niż 10 pacjentów na 100:
• ból/podrażnienie w obrębie jamy ustnej, bolesność ust, zaburzenia smaku, zaburzenia żołądkowe, nudności, wymioty, biegunka, zaparcia, bóle brzucha, utrata apetytu,
• przebarwienia skórne, zmiana koloru włosów, wysypka na dłoniach i podeszwach stóp, pęcherze, suchość skóry
• uczucie zmęczenia, nadciśnienie tętnicze, migrena • zmniejszenie liczby krwinek czerwonych i krwinek białych

70
Inne możliwe działania występujące u od 1 do 10 osób na 100: • zmniejszenie aktywności tarczycy, zmniejszenie przepływu kwi przez serce • krwawienie z nosa, nieprawidłowe zabarwienie moczu, nadmierne łzawienie • bóle stawów, bóle mięśni, gromadzenie płynów w tkankach, w tym także w okolicy oczu • zaburzenia czucia w skórze, skrócony oddech • utrata włosów, utrata masy ciała
Jeśli nasili się którykolwiek z objawów niepożądanych lub wystąpią jakiekolwiek objawy niepożądane nie wymienione w ulotce, należy powiadomić lekarza. 5. JAK PRZECHOWYWAĆ LEK SUTENT
- Lek przechowywać w miejscu niedostępnym i niewidocznym dla dzieci. - Brak specjalnych zaleceń dotyczących przechowywania niniejszego produktu leczniczego. - Nie stosować leku po upływie terminu ważności (EXP) zamieszczonego na opakowaniu
zewnętrznym i opakowaniu bezpośrednim. - Nie stosować leku jeśli opakowanie zostało uszkodzone lub widoczne są ślady wcześniejszych
prób otwierania. Leków nie należy wyrzucać do kanalizacji lub domowych pojemników na odpadki. Należy zapytać farmaceutę co zrobić z lekami, których się już nie potrzebuje. Takie postępowanie pomoże chronić środowisko. 6. INNE INFORMACJE Co zawiera lek SUTENT: - Substancją czynną leku jest sunitynib (w postaci soli jabłczanowej). - Inne składniki leku to: mannitol, sól sodowa kroskarmelozy, powidon i magnezu stearynian.
Otoczka kapsułki składa się z: żelatyny, żółtego tlenku żelaza (E172), czerwonego tlenku żelaza (E172), czarnego tlenku żelaza (E172) i dwutlenku tytanu (E 171). Tusz do nadruków zawiera szelak, glikol propylenowy, sodu wodorotlenek, powidon i dwutlenek tytanu.
Jak wygląda lek SUTENT i co zawiera opakowanie Lek SUTENT jest dostępny w postaci twardych kapsułek żelatynowych z karmelowym wieczkiem i pomarańczowym korpusem, z wykonanymi białym tuszem napisami „Pfizer” na wieczku i „STN 25 mg” na korpusie. Lek jest dostępny w butelkach po 30 kapsułek. Podmiot odpowiedzialny Pfizer Limited Ramsgate Road Sandwich, Kent CT13 9NJ Wielka Brytania Wytwórca Pfizer Italia S.r.l. Via del Commercio – Zona Industriale - 63046 Marino del Tronto (Ascoli Piceno) Włochy W celu uzyskania bardziej szczegółowych informacji należy zwrócić się do przedstawiciela podmiotu odpowiedzialnego.

71
Belgique / België /Belgien Pfizer S.A. / N.V. Tél/Tel: +32 (0)2 554 62 11
Luxembourg/Luxemburg Pfizer S.A. Tél/Tel: +32 (0)2 554 62 11
Česká republika Pfizer s.r.o. Tel.: +420-283-004-151
Magyarország Pfizer Kft. Tel.: +36-1-488-37-00
Danmark Pfizer ApS Tlf: +45 44 20 11 00
Malta V.J. Salomone Pharma Ltd. Tel. +356 212201
Deutschland Pfizer Pharma GmbH Tel: +49 (0)721 61 01 90 001
Nederland Pfizer BV Tel: +31 (0)10 406 42 00
Eesti Pfizer Luxembourg SARL, Eesti filiaal Tel.: +372 6 405 328
Norge Pfizer AS Tlf: +47 67 52 61 00
Ελλάδα Pfizer Hellas A.E. Τηλ: +30 210 7517981-3
Österreich Pfizer Corporation Austria Ges.m.b.H. Tel: +43 (0)1 521 15-0
España Pfizer S.A. Tél: +34 91 490 99 00
Polska Pfizer Polska Sp. z o.o. Tel.:+ 48 22 335 61 00
France Pfizer Tél: +33 (0)1 58 07 34 40
Portugal Laboratórios Pfizer, Lda. Tel: +351 21 423 5500
Ireland Pfizer Healthcare Ireland Tel: +353 1800 633 363
Slovenija Pfizer Luxembourg SARL Pfizer, podružnica za svetovanje s področja farmacevtske dejavnosti, Ljubljana Tel.: + 386 1 52 11 400
Ísland Vistor hf. Sími: +354 535 7000
Slovenská republika Pfizer Luxembourg SARL, organizačná zložka Tel.:+ 421 2 5941 8500
Italia Pfizer Italia S.r.l. Tel: +39 06 33 18 21
Suomi/Finland Pfizer Oy Puh./Tel: +358 (0)9 43 00 40
Kύπρος Geo. Pavlides & Araouzos Ltd. Tηλ.:+ 357 22 818087
Sverige Pfizer AB Tel: +46 (0)8 550 52000
Latvija Pfizer Luxembourg SARL filiale Latvijà Tel.: + 371 70 35 775
United Kingdom Pfizer Limited, Tel: +44 (0)1737 331111

72
Lietuva Pfizer Luxemburg SARL filialas Lietuvoje Tel. + 370 52 51 4000
Data zatwierdzenia ulotki: Ten lek otrzymał „warunkowe dopuszczenie do obrotu”. Oznacza to, że oczekuje się dostarczenia więcej danych o leku, w szczególności dotyczących leczenia raka nerki. Wykazano, że stosowanie leku SUTENT powoduje zmniejszenie guza. Jednak oczekiwane są informacje na temat czasu utrzymywania się takiego działania. Europejska Agencja ds. Produktów Leczniczych dokona przeglądu nowych informacji o leku raz do roku i treść ulotki zostanie uzupełniona tak jak to będzie konieczne. Szczegółowa informacja o tym leku jest dostępna na stronie internetowej Europejskiej Agencji ds. Produktów Leczniczych (EMEA): http://emea.eu.int. Można tam również znaleźć linki do stron internetowych o rzadkich chorobach i sposobach leczenia.

73
ULOTKA DLA PACJENTA: INFORMACJE DLA UŻYTKOWNIKA
SUTENT kapsułki twarde 50 mg
Sunitynib Należy zapoznać się z treścią ulotki przed zażyciem leku. - Należy zachować tę ulotkę, aby w razie potrzeby móc ją ponownie przeczytać. - Należy zwrócić się do lekarza lub farmaceuty, gdy potrzebna jest rada lub dodatkowa informacja. - Lek ten został przepisany ściśle określonej osobie i nie należy go przekazywać innym, gdyż może
im zaszkodzić, nawet jeśli objawy ich choroby są takie same. - Jeśli nasili się którykolwiek z objawów niepożądanych lub wystąpią jakiekolwiek objawy
niepożądane niewymienione w ulotce, należy powiadomić lekarza. Spis treści ulotki: 1. Co to jest SUTENT i w jakim celu się go stosuje 2. Informacje ważne przed zażyciem leku SUTENT 3. Jak zażywać SUTENT 4. Możliwe działania niepożądane 5. Jak przechowywać lek SUTENT 6. Inne informacje 1. CO TO JEST SUTENT I W JAKIM CELU SIĘ GO STOSUJE SUTENT jest produktem leczniczym stosowanym w leczeniu nowotworów. Hamuje aktywność specjalnej grupy białek, o których wiadomo, że uczestniczą we wzroście i rozsiewie komórek nowotworowych. Może on zostać przepisany wyłącznie przez lekarza doświadczonego w stosowaniu leków przeznaczonych do leczenia raka nerki lub nowotworów podścieliskowych przewodu pokarmowego. SUTENT jest produktem leczniczym stosowanym w leczeniu raka nerki, w przebiegu którego dochodzi do zmian nowotworowych w komórkach kanalików nerkowych. Lek SUTENT stosuje się również w leczeniu złośliwych nowotworów podścieliskowych przewodu pokarmowego (ang. gastrointestinal stromal tumor - GIST). Nowotwór podścieliskowy przewodu pokarmowego może rozwijać się w żołądku lub jelitach. Rozwija się w wyniku niekontrolowanego rozrostu komórek tkanek podporowych tych narządów. Lek SUTENT hamuje wzrost tych komórek. Należy się zwracać do lekarza z wszelkimi pytaniami na temat sposobu działania leku SUTENT lub na temat przyczyn przepisania tego leku. 2. INFORMACJE WAZNE PRZED ZASTOSOWANIEM LEKU SUTENT Należy dokładnie przestrzegać wszystkich zaleceń lekarza, nawet jeżeli różnią się od ogólnych informacji zawartych w niniejszej ulotce. Kiedy nie zażywać leku SUTENT Jeśli u pacjenta stwierdzono uczulenie na sunitynib lub którykolwiek z pozostałych składników leku SUTENT. Kiedy zachować szczególną ostrożność stosując SUTENT
- Jeśli występuje choroba wątroby lub nerek, - Jeśli występuje wysokie ciśnienie tętnicze krwi, - W stwierdzonej lub podejrzewanej ciąży (szczegóły podano poniżej),

74
- W czasie karmienia piersią (szczegóły podano poniżej). W przypadku istnienia którejkolwiek z powyższych okoliczności należy o nich poinformować lekarza przed rozpoczęciem stosowania leku SUTENT.
Stosowanie leku SUTENT z innymi lekami Należy powiedzieć lekarzowi lub farmaceucie o wszystkich przyjmowanych ostatnio lekach, również tych, które wydawane są bez recepty i preparatach ziołowych (wszelkie leki, które mogą zwiększać stężenie leku SUTENT, takie jak ketokonazol, rytonawir, itrakonazol, erytromycyna, klarytromycyna, a także wszelkie leki, które mogą zmniejszać stężenie leku SUTENT, takie jak deksametazon, fenytoina, karbamazepina, ryfampicyna, fenobarbital czy ziele dziurawca Hypericum perforatum). Stosowanie leku SUTENT z jedzeniem i piciem Lek SUTENT można przyjmować podczas posiłków lub między posiłkami, jednak leku SUTENT nie należy przyjmować jednocześnie z sokiem grejfrutowym. Ciąża i karmienie piersią Należy poinformować lekarza o stwierdzonej lub podejrzewanej ciąży. Kobiety ciężarne nie powinny stosować leku SUTENT, o ile nie jest to bezwzględnie konieczne. Lekarz omówi z pacjentką potencjalne ryzyko związane z przyjmowaniem leku w czasie ciąży. Kobietom, które mogą zajść w ciążę, zaleca się stosowanie skutecznej antykoncepcji w trakcie leczenia lekiem SUTENT. Kobiety karmiące piersią powinny o tym poinformować lekarza. Nie należy karmić piersią podczas przyjmowania leku SUTENT. Prowadzenie pojazdów i obsługa maszyn W przypadku wystąpienia zawrotów głowy lub znacznego zmęczenia należy zachować szczególną ostrożność podczas prowadzenia samochodu i obsługi urządzeń mechanicznych w ruchu. 3. JAK ZAŻYWAĆ LEK SUTENT Lekarz przepisze dawkę właściwą dla danego pacjenta. Zaleca się, aby przyjmować lek SUTENT przez 28 dni (4 tygodnie), po czym powinna nastąpić 14-dniowa (dwutygodniowa) przerwa (bez leku), co razem daje sześciotygodniowy cykl leczenia. Lekarz zdecyduje o liczbie cykli leczenia odpowiedniej dla danego pacjenta. Zażycie większej niż zalecana dawki leku SUTENT W przypadku przyjęcia większej liczby kapsułek niż zalecana należy bezzwłocznie porozumieć się z lekarzem. Może być konieczna interwencja medyczna. Pominięcie zażycia dawki leku SUTENT Nie należy stosować dawki podwójnej w celu uzupełnienia pominiętej dawki. 4. MOŻLIWE DZIAŁANIA NIEPOŻĄDANE Jak każdy lek, SUTENT może powodować działania niepożądane, chociaż nie u każdego one występują. Bardzo częste działania niepożądane występujące u więcej niż 10 pacjentów na 100:
• ból/podrażnienie w obrębie jamy ustnej, bolesność ust, zaburzenia smaku, zaburzenia żołądkowe, nudności, wymioty, biegunka, zaparcia, bóle brzucha, utrata apetytu,
• przebarwienia skórne, zmiana koloru włosów, wysypka na dłoniach i podeszwach stóp, pęcherze, suchość skóry
• uczucie zmęczenia, nadciśnienie tętnicze, migrena • zmniejszenie liczby krwinek czerwonych i krwinek białych

75
Inne możliwe działania występujące u od 1 do 10 osób na 100: • zmniejszenie aktywności tarczycy, zmniejszenie przepływu kwi przez serce • krwawienie z nosa, nieprawidłowe zabarwienie moczu, nadmierne łzawienie • bóle stawów, bóle mięśni, gromadzenie płynów w tkankach, w tym także w okolicy oczu • zaburzenia czucia w skórze, skrócony oddech • utrata włosów, utrata masy ciała
Jeśli nasili się którykolwiek z objawów niepożądanych lub wystąpią jakiekolwiek objawy niepożądane nie wymienione w ulotce, należy powiadomić lekarza. 5. JAK PRZECHOWYWAĆ LEK SUTENT
- Lek przechowywać w miejscu niedostępnym i niewidocznym dla dzieci. - Brak specjalnych zaleceń dotyczących przechowywania niniejszego produktu leczniczego. - Nie stosować leku po upływie terminu ważności (EXP) zamieszczonego na opakowaniu
zewnętrznym i etykiecie. - Nie stosować leku jeśli opakowanie zostało uszkodzone lub widoczne są ślady wcześniejszych
prób otwierania. Leków nie należy wyrzucać do kanalizacji lub domowych pojemników na odpadki. Należy zapytać farmaceutę co zrobić z lekami, których się już nie potrzebuje. Takie postępowanie pomoże chronić środowisko. 6. INNE INFORMACJE Co zawiera lek SUTENT: - Substancją czynną leku jest sunitynib (w postaci soli jabłczanowej). - Inne składniki leku to: mannitol, sól sodowa kroskarmelozy, powidon i magnezu stearynian.
Otoczka kapsułki składa się z: żelatyny, żółtego tlenku żelaza (E172), czerwonego tlenku żelaza (E172), czarnego tlenku żelaza (E172) i dwutlenku tytanu (E 171). Tusz do nadruków zawiera szelak, glikol propylenowy, sodu wodorotlenek, powidon i dwutlenek tytanu.
Jak wygląda lek SUTENT i co zawiera opakowanie Lek SUTENT jest dostępny w postaci twardych kapsułek żelatynowych z karmelowym wieczkiem i karmelowym korpusem, z wykonanymi białym tuszem napisami „Pfizer” na wieczku i „STN 50 mg” na korpusie. Lek jest dostępny w butelkach po 30 kapsułek. Podmiot odpowiedzialny Pfizer Limited Ramsgate Road Sandwich, Kent CT13 9NJ Wielka Brytania Wytwórca Pfizer Italia S.r.l. Via del Commercio – Zona Industriale - 63046 Marino del Tronto (Ascoli Piceno) Włochy W celu uzyskania bardziej szczegółowych informacji należy zwrócić się do przedstawiciela podmiotu odpowiedzialnego.

76
Belgique / België /Belgien Pfizer S.A. / N.V. Tél/Tel: +32 (0)2 554 62 11
Luxembourg/Luxemburg Pfizer S.A. Tél/Tel: +32 (0)2 554 62 11
Česká republika Pfizer s.r.o. Tel.: +420-283-004-151
Magyarország Pfizer Kft. Tel.: +36-1-488-37-00
Danmark Pfizer ApS Tlf: +45 44 20 11 00
Malta V.J. Salomone Pharma Ltd. Tel. +356 212201
Deutschland Pfizer Pharma GmbH Tel: +49 (0)721 61 01 90 001
Nederland Pfizer BV Tel: +31 (0)10 406 42 00
Eesti Pfizer Luxembourg SARL, Eesti filiaal Tel.: +372 6 405 328
Norge Pfizer AS Tlf: +47 67 52 61 00
Ελλάδα Pfizer Hellas A.E. Τηλ: +30 210 7517981-3
Österreich Pfizer Corporation Austria Ges.m.b.H. Tel: +43 (0)1 521 15-0
España Pfizer S.A. Tél: +34 91 490 99 00
Polska Pfizer Polska Sp. z o.o. Tel.:+ 48 22 335 61 00
France Pfizer Tél: +33 (0)1 58 07 34 40
Portugal Laboratórios Pfizer, Lda. Tel: +351 21 423 5500
Ireland Pfizer Healthcare Ireland Tel: +353 1800 633 363
Slovenija Pfizer Luxembourg SARL Pfizer, podružnica za svetovanje s področja farmacevtske dejavnosti, Ljubljana Tel.: + 386 1 52 11 400
Ísland Vistor hf. Sími: +354 535 7000
Slovenská republika Pfizer Luxembourg SARL, organizačná zložka Tel.:+ 421 2 5941 8500
Italia Pfizer Italia S.r.l. Tel: +39 06 33 18 21
Suomi/Finland Pfizer Oy Puh./Tel: +358 (0)9 43 00 40
Kύπρος Geo. Pavlides & Araouzos Ltd. Tηλ.:+ 357 22 818087
Sverige Pfizer AB Tel: +46 (0)8 550 52000
Latvija Pfizer Luxembourg SARL filiale Latvijà Tel.: + 371 70 35 775
United Kingdom Pfizer Limited, Tel: +44 (0)1737 331111

77
Lietuva Pfizer Luxemburg SARL filialas Lietuvoje Tel. + 370 52 51 4000
Data zatwierdzenia ulotki: Ten lek otrzymał „warunkowe dopuszczenie do obrotu”. Oznacza to, że oczekuje się dostarczenia więcej danych o leku, w szczególności dotyczących leczenia raka nerki. Wykazano, że stosowanie leku SUTENT powoduje zmniejszenie guza. Jednak oczekiwane są informacje na temat czasu utrzymywania się takiego działania. Europejska Agencja ds. Produktów Leczniczych dokona przeglądu nowych informacji o leku raz do roku i treść ulotki zostanie uzupełniona tak jak to będzie konieczne. Szczegółowa informacja o tym leku jest dostępna na stronie internetowej Europejskiej Agencji ds. Produktów Leczniczych (EMEA): http://emea.eu.int. Można tam również znaleźć linki do stron internetowych o rzadkich chorobach i sposobach leczenia.