anastasia nikolskaya pir (protein information resource), georgetown university medical center
DESCRIPTION
FUNCTIONAL ANALYSIS OF PROTEIN SEQUENCES: ANNOTATION AND FAMILY CLASSIFICATION. Anastasia Nikolskaya PIR (Protein Information Resource), Georgetown University Medical Center. Problem:. Most new protein sequences come from genome sequencing projects Many have unknown functions - PowerPoint PPT PresentationTRANSCRIPT

Anastasia NikolskayaPIR (Protein Information Resource),
Georgetown University Medical Center
FUNCTIONAL ANALYSIS OF PROTEIN SEQUENCES:ANNOTATION AND FAMILY CLASSIFICATION

2
Most new protein sequences come from genome sequencing projects Many have unknown functions Large-scale functional annotation of these sequences based simply on
BLAST best hit has pitfalls; results are far from perfect
Problem: Overview
Highly curated and annotated protein classification system Automatic annotation of sequences based on protein families
Solution for Large-scale Annotation:
Full-length protein family classification based on evolution Highly annotated, optimized for annotation propagation Functional predictions for uncharacterized proteins Used to facilitate and standardize annotations in UniProt
PIRSF Protein Classification System
Functional Analysis of Protein Sequences: Homology-based (sequence analysis, structure analysis) Non-homology (genome context, phylogenetic distribution)

3
Proteomics and Bioinformatics
BioinformaticsComputational analysis and integration of these data
Making predictions (function etc), reconstructing pathways
Data: Gene expression profiling Genome-wide analysis of gene expression
Data: Protein-protein interaction Data: Structural genomics 3D structures of all protein
families Data: Genome projects (Sequencing) ….

4
What’s In It For Me?
When an experiment yields a sequence (or a set of sequences), we need to find out as much as we can about this protein and its possible function from available data
Especially important for poorly characterized or uncharacterized (“hypothetical”) proteins
More challenging for large sets of sequences generated by large-scale proteomics experiments
The quality of this assessment is often critical for interpreting experimental results and making hypothesis for future experiments
Sequence function

5
Genomic DNA Sequence
5' UTRPromoter Exon1 Intron Exon2 Intron Exon3 3' UTR
A
G
GT
A
G
Gene Recognition
Exon2Exon1 Exon3
C
A
C
A
C
A
A
T
T
A
T
A
Protein Sequence
A
T
G
AA
T
A
A
A
Structure Determination
Protein Structure
Function Analysis
Gene NetworkMetabolic Pathway
Protein FamilyMolecular Evolution
Family Classification
GT
Gene Gene
DNASequence
Gene
Protein
Sequence
Function
Work with Protein, not DNA Sequence

6
The Changing Face of Protein Science
20th century
Few well-studied proteins
Mostly globular with enzymatic activity
Biased protein set
21st centuryMany “hypothetical”
proteins (Most new proteins come from genome sequencing projects, many have unknown functions)
Various, often with no enzymatic activity
Natural protein set
Credit: Dr. M. Galperin, NCBI

7
Knowing the Complete Genome Sequence
All encoded proteins can be predicted and identified The missing functions can be identified and analyzed Peculiarities and novelties in each organism can be
studied Predictions can be made and verified
Advantages:
Challenge: Accurate assignment of known or predicted functions
(functional annotation)

8
Escherichia coli Methanococcus jannaschii
Yeast Human
E. coli M. jannaschii S. cerevisiae H. sapiens Characterized experimentally 2046 97 3307 10189 Characterized by similarity 1083 1025 1055 10901 Unknown, conserved 285 211 1007 2723 Unknown, no similarity 874 411 966 7965 from Koonin and Galperin, 2003, with modifications

9
Experimentally characterized Find up-to-date information, accurate interpretation
Characterized by similarity (“knowns”) = closely related to experimentally characterized Avoid propagation of errors
Function can be predicted (no close sequence similarity, may be distant similarity to characterized proteins) Extract maximum possible information, avoid errors and
overpredictions Most value-added (fill the gaps in metabolic pathways, etc)
“Unknowns” (conserved or unique) Rank by importance
Functional Annotationfor Different Groups of Proteins

10
Protein Sequence
Function
Automatic assignment based on sequence similarity (best BLAST hit):gene name, protein name, function
Large-scale functional annotation of sequences based simply on BLAST best hit has pitfalls; results are far from perfect
To avoid mistakes, need human intervention (manual annotation)
How are Protein Sequences Annotated?“regular approach”
Quality vs Quantity

11
Experimentally characterized Find up-to-date information, accurate interpretation
Characterized by similarity (“knowns”) = closely related to experimentally characterized Avoid propagation of errors
Function can be predicted (no close sequence similarity, may be distant similarity to characterized proteins) Extract maximum possible information, avoid errors and
overpredictions Most value-added (fill the gaps in metabolic pathways, etc)
“Unknowns” (conserved or unique) Rank by importance
Functional Annotationfor Different Groups of Proteins

12
Misinterpreted experimental results (e.g. suppressors, cofactors) Biologically senseless annotations Arabidopsis: separation anxiety protein-like
Helicobacter: brute force protein
Methanococcus: centromere-binding protein
Plasmodium: frameshift
“Goofy” mistakes of sequence comparison (e.g. abc1/ABC) Multi-domain organization of proteins Low sequence complexity (coiled-coil, transmembrane, non-
globular regions) Enzyme evolution: - Divergence in sequence and function (minor mutation in active site)- Non-orthologous gene displacement: Convergent evolution
Problems in Functional Assignments for “Knowns”

13
Problems in Functional Assignments for “Knowns”: multi-domain organization of proteins
ACT domain
Chorismate mutase domain ACT domain
New sequence
Chorismate mutase
BLAST
In BLAST output, top hits are to chorismate mutases ->The name “chorismate mutase” is automatically assigned to new sequence. ERROR ! (protein gets erroneous name, EC number, assigned to erroneous pathway, etc)

14
Previous low quality annotations lead to
propagation of mistakes
Problems in Functional Assignments for “Knowns”

15
Experimentally characterized Find up-to-date information, accurate interpretation
Characterized by similarity (“knowns”) = closely related to experimentally characterized Avoid propagation of errors
Function can be predicted (no close sequence similarity, may be distant similarity to characterized proteins) Extract maximum possible information, avoid errors and
overpredictions Most value-added (fill the gaps in metabolic pathways, etc)
“Unknowns” (conserved or unique) Rank by importance
Functional Annotationfor Different Groups of Proteins

16
in non-obvious cases: Sophisticated database searches (PSI-BLAST, HMM) Detailed manual analysis of sequence similarities Structure-guided alignments and structure analysis
Often, only general function can be predicted: Enzyme activity can be predicted, the substrate remains unknown
(ATPases, GTPases, oxidoreductases, methyltransferases, acetyltransferases)
Helix-turn-helix motif proteins (predicted transcriptional regulators)
Membrane transporters
Functional Prediction:I. Sequence and Structure Analysis
(homology-based methods)

17
Proteins (domains) with different 3D folds are not homologous (unrelated by origin). Proteins with similar 3D folds are usually (but not always) homologous
Those amino acids that are conserved in divergent proteins within a (super)family are likely to be functionally important (catalytic or binding sites, ect).
Reaction chemistry often remains conserved even when sequence diverges almost beyond recognition
Using Sequence Analysis:Hints

18
Prediction of 3D fold (if distant homologs have known structures!) and of general biochemical function is much easier than prediction of exact biological function
Sequence analysis complements structural comparisons and can greatly benefit from them
Comparative analysis allows us to find subtle sequence similarities in proteins that would not have been noticed otherwise
Using Sequence Analysis:Hints
Credit: Dr. M. Galperin, NCBI

19
Structural Genomics: Structure-Based Functional Predictions
Methanococcus jannaschii MJ0577 (Hypothetical Protein)
Contains bound ATP => ATPase or ATP-Mediated Molecular Switch
Confirmed by biochemical experiments
Protein Structure Initiative: Determine 3D structures of all protein families

20
Crystal Structure is Not a Function!
Credit: Dr. M. Galperin, NCBI

21
Phylogenetic distribution (comparative genomics) Wide - most likely essential Narrow - probably clade-specific Patchy - most intriguing
Domain association – “Rosetta Stone” Genome context (gene neighborhood, operon
organization)
Functional Prediction:II. Computational Analysis Beyond Homology
Clues: specific to niche, pathway type

22
Using Genome Context for Functional Prediction
Embden-Meyerhof and Gluconeogenesis pathway: 6-phosphofructokinase (EC 2.7.1.11)
SEED analysis tool (by FIG)

23
Functional Prediction: Problem Areas
Identification of protein-coding regions Delineation of potential function(s) for distant
paralogs Identification of domains in the absence of
close homologs Analysis of proteins with low sequence
complexity

24
What to do with a new protein sequence Basic:- Domain analysis (SMART = most sensitive; PFAM= most complete, CDD)- BLAST- Curated protein family databases (PIRSF, InterPro, COGs)- Literature (PubMed) from links from individual entries on BLAST output
(look for SwissProt entries first)
If not sufficient:- PSI-BLAST- Refined PubMed search using gene/protein names, synonyms, function and other terms you found- Genome neighborhood (prokaryotes)
• Advanced: - Multiple sequence alignments (manual) - Structure-guided alignments and structure analysis- Phylogenetic tree reconstruction

25
Case Study: Prediction Verified: GGDEF domain
Proteins containing this domain: Caulobacter crescentus PleD controls swarmer cell - stalk cell transition (Hecht and Newton, 1995). In Rhizobium leguminosarum, Acetobacter xylinum, required for cellulose biosynthesis (regulation)
Predicted to be involved in signal transduction because it is found in fusions with other signaling domains (receiver, etc)
In Acetobacter xylinum, cyclic di-GMP is a specific nucleotide regulator of cellulose synthase (signalling molecule). Multidomain protein with GGDEF domain was shown to have diguanylate cyclase activity (Tal et al., 1998)
Detailed sequence analysis tentatively predicts GGDEF to be a diguanylate cyclase domain (Pei and Grishin, 2001)
Complementation experiments prove diguanylate cyclase activity of GGDEF (Ausmees et al., 2001)

26
Most new protein sequences come from genome sequencing projects Many have unknown functions Large-scale functional annotation of these sequences based simply on
BLAST best hit has pitfalls; results are far from perfect Manual annotation of individual proteins is not efficient
Problem:
The Need for Classification
Highly curated and annotated protein classification system Automatic annotation of sequences based on protein families
Solution:
Good quality and large-scale Systematic correction of annotation errors Protein name standardization Functional predictions for uncharacterized proteins
Facilitates:
This all works only if the system is optimized for annotation

27
Levels of Protein ClassificationLevel Example Similarity Evolution
Class / Structural elements No relationships
Fold TIM-Barrel Topology of backbone Possible monophyly
Domain Superfamily
Aldolase Recognizable sequence similarity (motifs); basic biochemistry
Monophyletic origin
Family Class I Aldolase High sequence similarity (alignments); biochemical properties
Evolution by ancient duplications
Orthologous group
2-keto-3-deoxy-6-phosphogluconate aldolase
Orthology for a given set of species; biochemical activity; biological function
Traceable to a single gene in LCA
Lineage-specific expansion
(LSE)
PA3131 and PA3181
Paralogy within a lineage Recent duplication

28
Protein Evolution
With enough similarity, one can trace back to a
common origin
Sequence changes
What about these?
Domain shuffling
Domain: Evolutionary/Functional/Structural Unit

29
PDT?
CM/PDH?
Consequences of Domain Shuffling
PIRSF001500CM (AroQ type) PDT ACT
PIRSF001501
CM (AroQ type)
PIRSF006786
PDH
PIRSF001499
PIRSF005547PDH ACT
PDT ACT PIRSF001424
CM = chorismate mutasePDH = prephenate dehydrogenase PDT = prephenate dehydrataseACT = regulatory domain
PDH?
CM/PDT?
CM?PDHCM (AroQ type)

30
Peptidase M22Acylphosphatase ZnF YrdCZnF- - - -
Whole Protein = Sum of its Parts?
On the basis of domain composition alone, biological function was predicted to be: ● RNA-binding translation factor ● maturation protease
PIRSF006256
Actual function: ● [NiFe]-hydrogenase maturation factor, carbamoyltransferase
Full-length protein functional annotation is best done using annotated full-length protein families

31
Practical classification of proteins:setting realistic goals
We strive to reconstruct the natural classification of proteins to the fullest possible extent
BUT
Domain shuffling rapidly degrades the continuity in the protein structure (faster than sequence divergence degrades similarity)
THUS
The further we extend the classification, the finer is the domain structure we need to consider
SO
We need to compromise between the depth of analysis and protein integrity
OR … Credit: Dr. Y. Wolf, NCBI

32
Domain Classification
Allows a hierarchy that can trace evolution to the deepest possible level, the last point of traceable homology and common origin
Can usually annotate only general biochemical function
Full-length protein Classification
Cannot build a hierarchy deep along the evolutionary tree because of domain shuffling
Can usually annotate specific biological function (preferred to annotate individual proteins)
Can map domains onto proteins
Can classify proteins even when domains are not defined
Complementary Approaches

33
Levels of Protein ClassificationLevel Example Similarity Evolution
Class / Structural elements No relationships
Fold TIM-Barrel Topology of backbone Possible monophyly
Domain Superfamily
Aldolase Recognizable sequence similarity (motifs); basic biochemistry
Monophyletic origin
Family Class I Aldolase High sequence similarity (alignments); biochemical properties
Evolution by ancient duplications
Orthologous group
2-keto-3-deoxy-6-phosphogluconate aldolase
Orthology for a given set of species; biochemical activity; biological function
Traceable to a single gene in LCA
Lineage-specific expansion
(LSE)
PA3131 and PA3181
Paralogy within a lineage Recent duplication

34
Full-length protein classification
PIRSF
Domain classification
Pfam
SMART
CDD
Mixed
•TIGRFAMS
•COGs
Based on structural fold
•SCOP
Protein Classification Databases
InterPro: integrates various types of classification databases
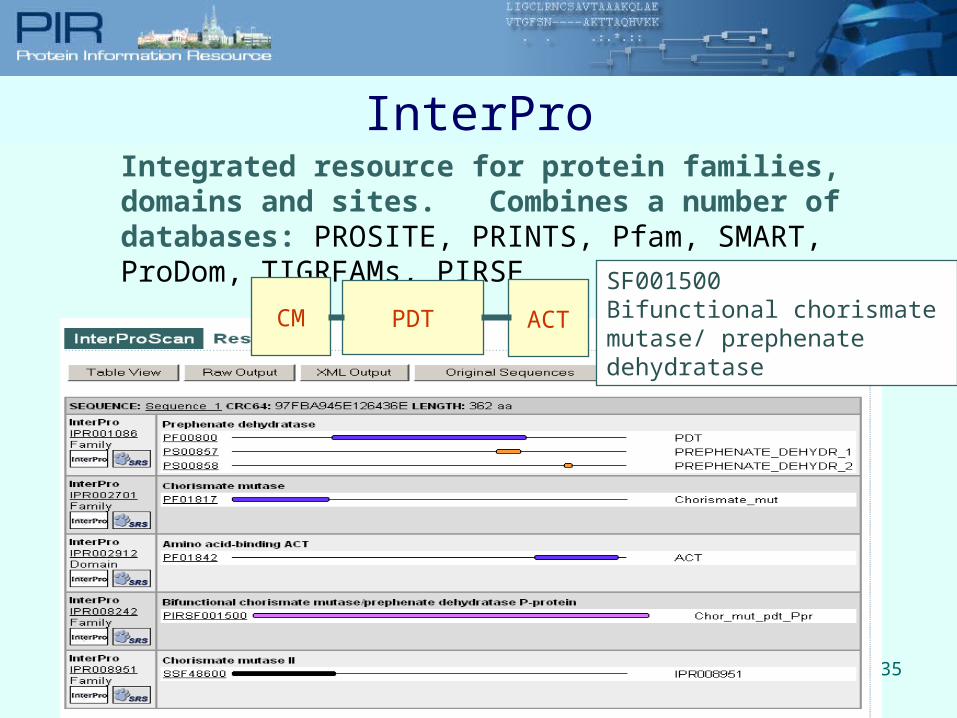
35
Integrated resource for protein families, domains and sites. Combines a number of databases: PROSITE, PRINTS, Pfam, SMART, ProDom, TIGRFAMs, PIRSF
SF001500Bifunctional chorismate mutase/ prephenate dehydratase
InterPro
CM PDT ACT

36
The Ideal System…
Comprehensive: each sequence is classified either as a member of a family or as an “orphan” sequence
Hierarchical: families are united into superfamilies on the basis of distant homology, and divided into subfamilies on the basis of close homology
Allows for simultaneous use of the full-length protein and domain information (domains mapped onto proteins)
Allows for automatic classification/annotation of new sequences when these sequences are classifiable into the existing families
Expertly curated membership, family name, function, background, etc.
Evidence attribution (experimental vs predicted)

37
PIRSF Classification System
PIRSF: Reflects evolutionary relationships of full-length proteins A network structure from superfamilies to subfamilies
Definitions: Homeomorphic Family: Basic Unit Homologous: Common ancestry, inferred by sequence similarity Homeomorphic: Full-length similarity & common domain architecture Hierarchy: Flexible number of levels with varying degrees of sequence
conservation Network Structure: allows multiple parents
Advantages: Annotate both general biochemical and specific biological functions Accurate propagation of annotation and development of standardized
protein nomenclature and ontology
http://pir.georgetown.edu/

38
PIRSF001499: Bifunctional CM/PDH (T-protein)
PIRSF006786: PDH, feedback inhibition-insensitive
PIRSF005547: PDH, feedback inhibition-sensitive
PF02153: Prephenatedehydrogenase (PDH)
PIRSF017318: CM of AroQ class, eukaryotic type
PIRSF001501: CM of AroQ class, prokaryotic type
PIRSF026640: Periplasmic CM
PIRSF001500: Bifunctional CM/PDT (P-protein)
PIRSF001499: Bifunctional CM/PDH (T-protein)
PF01817: Chorismatemutase (CM)
PIRSF006493: Ku, prokaryotic type
PIRSF500001: IGFBP-1
…
PIRSF500006: IGFBP-6
PIRSF Homeomorphic Subfamily
• 0 or more levels
• Functional specialization
PIRSF018239: IGFBP-related protein, MAC25 type
PIRSF001969: IGFBP
PIRSF003033: Ku70 autoantigen
PIRSF016570: Ku80 autoantigen
PIRSF Homeomorphic Family• Exactly one level
• Full-length sequence similarity and common domain architecture
PIRSF Superfamily
• 0 or more levels
• One or more common domains
PF00219: Insulin-like growth factor binding protein
(IGFBP)
PIRSF800001: Ku70/80 autoantigenPF02735: Ku70/Ku80 beta-barrel domain
Domain Superfamily• One common Pfam
domain
PIRSF001499: Bifunctional CM/PDH (T-protein)
PIRSF006786: PDH, feedback inhibition-insensitive
PIRSF005547: PDH, feedback inhibition-sensitive
PF02153: Prephenatedehydrogenase (PDH)
PIRSF017318: CM of AroQ class, eukaryotic type
PIRSF001501: CM of AroQ class, prokaryotic type
PIRSF026640: Periplasmic CM
PIRSF001500: Bifunctional CM/PDT (P-protein)
PIRSF001499: Bifunctional CM/PDH (T-protein)
PF01817: Chorismatemutase (CM)
PIRSF006493: Ku, prokaryotic type
PIRSF500001: IGFBP-1
…
PIRSF500006: IGFBP-6
PIRSF Homeomorphic Subfamily
• 0 or more levels
• Functional specialization
PIRSF018239: IGFBP-related protein, MAC25 type
PIRSF001969: IGFBP
PIRSF003033: Ku70 autoantigen
PIRSF016570: Ku80 autoantigen
PIRSF Homeomorphic Family• Exactly one level
• Full-length sequence similarity and common domain architecture
PIRSF Superfamily
• 0 or more levels
• One or more common domains
PF00219: Insulin-like growth factor binding protein
(IGFBP)
PIRSF800001: Ku70/80 autoantigenPF02735: Ku70/Ku80 beta-barrel domain
Domain Superfamily• One common Pfam
domain
PIRSF Classification SystemA protein may be assigned to only one homeomorphic family, which may have zero or more child nodes and zero or more parent nodes. Each homeomorphic family may have as many domain superfamily parents as its members have domains.

39
Creation and Curation of PIRSFsUniProtKB proteins
Preliminary Homeomorphic Families
Orphans
Curated Homeomorphic Families
Final Homeomorphic Families
Add/remove members
Name, refs, description
Automatic clustering
Computer-assisted Manual Curation
Automatic Procedure Unassigned proteins
Au
tom
atic
pla
ce
me
nt
Create hierarchies (superfamilies/subfamilies)
Map domains on Families
Merge/split clusters
New proteins
Protein name rule/site rule
Computer-Generated (Uncurated) Clusters
Preliminary Curation (4,700 PIRSFs) Membership Signature
Domains
Full Curation (3,300 PIRSFs) Family Name,
Description, Bibliography
PIRSF Name Rules
Build and test HMMs

40
Taxonomic distribution of PIRSF can be used to infer evolutionary history of the proteins in the PIRSF
PIRSF Family Report: Curated Protein Family Information
Phylogenetic tree and alignment view allows further sequence analysis

43
PIRSF Protein Classification: Platform for Protein Analysis and
Annotation
Improves automatic annotation quality Serves as a protein analysis platform for broad range of
users
Matching a protein sequence to a curated protein family rather than searching against a protein database
Provides value-added information by expert curators, e.g., annotation of uncharacterized hypothetical proteins (functional predictions)

44
Family-Driven Protein AnnotationObjective: Optimize for protein annotation
PIRSF Classification Name Reflects the function when possible Indicates the maximum specificity that still describes the entire group Standardized format Name tags: validated, tentative, predicted, functionally
heterogeneous
HierarchySubfamilies increase specificity (kinase -> sugar kinase -> hexokinase)

45
Define conditions under which family properties propagate to individual proteins
Propagate protein name, function, functional sites, EC, GO terms, pathway
Enable further specificity based on taxonomy or motifs Account for functional variations within one PIRSF, including:- Lack of active site residues necessary for enzymatic activity- Certain activities relevant only to one part of the taxonomic tree- Evolutionarily-related proteins whose biochemical activities are
known to differ
Family-Driven Protein Annotation:Site Rules and Name Rules
Goal: Automatic annotation of sequences based on protein families
to address the quality versus quantity problem

53
Most new protein sequences come from genome sequencing projects Many have unknown functions Large-scale functional annotation of these sequences based simply on
BLAST best hit has pitfalls; results are far from perfect
Problem:Overview
Highly curated and annotated protein classification system Automatic annotation of sequences based on protein families
Solution for Large-scale Annotation:
Functional Analysis of Protein Sequences: Homology-based (sequence analysis, structure analysis) Non-homology (genome context, phylogenetic distribution)
Automatic annotation of sequences based on protein families Systematic correction of annotation errors Name standardization in UniProt Functional predictions for uncharacterized proteins
Facilitates:

54
Impact of Protein Bioinformatics and Genomics
Single protein level Discovery of new enzymes and superfamilies Prediction of active sites and 3D structures
Pathway level Identification of “missing” enzymes Prediction of alternative enzyme forms Identification of potential drug targets
Cellular metabolism level Multisubunit protein systems Membrane energy transducers Cellular signaling systems
55
PIR Team Dr. Cathy Wu, Director Protein Science team
Dr. Darren Natale (lead) Dr. Peter McGarvey Dr. Cecilia Arighi Dr. Anastasia Nikolskaya Dr. Winona Barker Dr. Sona Vasudevan Dr. Zhang-zhi Hu Dr. CR Vinayaka Dr. Raja Mazumder Dr. Lai-Su Yeh
Bioinformatics team Dr. Hongzhan Huang (lead) Yongxing Chen, M.S. Dr. Leslie Arminski Baris Suzek, M.S. Dr. Hsing-Kuo Hua Xin Yuan, M.S. Dr. Robel Kahsay Jian Zhang, M.S.
Students Natalia Petrova
UniProt Collaborators Dr. Rolf Apweiler (EBI) Dr. Amos Bairoch (SIB)
UniProt is supported by the National Institutes of Health, grant # 1 U01 HG02712-01