an introduction to serology for diagnosis of animal … · web viewin the indirect elisa antigen is...
TRANSCRIPT

Serology: Enzyme-linked immunosorbent assay (ELISA)
Serology:Enzyme-linked immunosorbent assay (ELISA)
Authors: Dr. RW Worthington (Retired)
Licensed under a Creative Commons Attribution license.
INDIRECT ELISAIn the indirect ELISA antigen is immobilised by coating it onto an ELISA microtitre plate. Serum suspected of containing antibody is then added and antibody, if present, attaches to the immobilised antigen and a second antibody (Ab2) that has been conjugated to an enzyme is used to identify the attached antibody.
Antigens may be suspensions of bacteria, tissue culture preparations, pelleted virus particles, extracts of infectious tissues, concentrated supernatants from cultures, proteins produced from cloned cells by recombinant DNA technology, highly purified antigens etc. Protein and lipopolysaccharide antigens usually attach readily to polystyrene ELISA plates by non-covalent bonding. They are dissolved or suspended in pH 9.6 bicarbonate buffer or in phosphate buffered saline and simply pipetted into the wells of the plate. Methods are available for covalently binding antigens to plates but are generally unnecessary for tests for infectious diseases. The plates are then washed to remove any unbound antigen. The wells are then filled with a solution containing either bovine serum albumen or dried milk powder or another suitable protein to block unoccupied binding sites on the polystyrene. After this they are washed, dried or filled with buffer and stored until used. Before use plates are washed repeatedly with buffer solution containing 0.05%-0.1% Tween 20. Washing is usually done with a plate washer that repeatedly fills wells with buffer and then sucks it out again. Where only a few tests are done or a laboratory cannot afford a plate washer, plates can be washed in a bucket containing buffer or water with 0.05% Tween 20. Plates are shaken to remover the blocking solution, tapped on a towel or blotting paper to make sure all the liquid is removed and then dipped into the washing solution, swirled gently, shaken to remove the fluid and again tapped dry on a towel or blotting paper. This sequence is repeated as many times as required.
The use of Tween 20 in buffers is essential as it prevents the non-specific attachment of protein to the polystyrene plates but does not interfere with antibody antigen reactions. It is used at a concentration of 0.05 - 0.1% in washing solutions and conjugate and test serum diluent buffers.
Test sera are suitably diluted in PBS/Tween 20 (PBST) and dispensed into wells on the plate and the plate incubated for the required length of time before washing to remove surplus reagents.
1 | P a g e

Serology: Enzyme-linked immunosorbent assay (ELISA)
At this stage the Ab2 or detector antibody is added to identify any antibodies that have been trapped by the antigen on the plate. The Ab2 is an antibody raised in a different species of animal (antiserum donor animal) to the animals for which test is used. According to the requirements of the test the donor animal may be immunised with IgG, IgM or pooled immunoglobulins. For use in a test for cattle sera, anti-bovine sera are often raised in rabbits or goats. Rabbit antiserum raised against bovine immunoglobulins will cross react substantially with immunoglobulins from related ruminant species. However, these cross reactions are of no consequence for many tests as all that is required is that Ab2 identifies the antibody captured on the plate and this antibody can only have come from the serum being tested. Once a suitable serum has been produced, an immunoglobulin preparation is prepared from it by ammonium sulphate precipitation, ion exchange chromatography or another suitable method.
Figure 1: Principle of indirect ELISA
The Ab2 used is covalently bound (conjugated) to a suitable enzyme; the conjugated Ab2/enzyme is usually referred to as the conjugate. The most commonly used enzyme is horseradish peroxidase but other enzymes have also been used (Table 1). There are several methods of conjugating enzyme and antibody and for optimal results the number of enzyme molecules bound to each antibody molecule should be optimised. However, most conjugates are now commercially available and few diagnostic laboratories prepare their own.
After incubation to allow the conjugate immunoglobulins to attach to the antibodies that have already attached to the antigen, the unbound conjugate is removed by washing. It is assumed that the amount of antibody bound to the antigen is proportional to the enzyme activity on the plate. A substrate is added to the plate and the enzyme catalyses a reaction on the substrate that results in the development of a coloured product. Horseradish peroxidase catalyses a reaction in which a commonly used substrate orthophenylene diamine (OPD) is oxidised by hydrogen peroxide to a yellow/brown oxidation product. The amount of coloured product that develops is measured spectrophotometrically at a wavelength of 490 nm on an ELISA plate reader. If no plate reader is available a reasonable estimate of the amount of reaction can be made by visual comparison of test
2 | P a g e

Serology: Enzyme-linked immunosorbent assay (ELISA)
sera and standards. Provided there is an excess of substrate and the reaction is stopped before substrate is used up or accumulated waste products inhibit the reaction, the colour that develops is proportional to the amount of enzyme, and therefore to the amount of antibody, on the plate. Methods of calculating and expressing the data are discussed in Section 1.3. Some examples of enzymes and substrates used in ELISAs are given in Table 1.
Table 1: Some enzymes and enzyme substrates used in ELISAs.
Enzyme Substrate
Horseradish peroxidase Orthophenylene diamine (OPD) and hydrogen peroxide
Horseradish peroxidase 2,2azino-bis-[3-ethylbenzothiazoline-5-sulphonic acid} (ABTS) and hydrogen peroxide
Horseradish peroxidase Dimethyl aminobenzaldehyde (DMAB) , 3-methyl-2-benzothazolinone hydrochloride (MBTH) and hydrogen peroxide
Horseradish peroxidase Tetramethyl benzidine and hydrogen peroxide
Alkaline phosphatase Nitroblue tetrazolium/5-bromo-chloro-indoxylphosphate
Urease Urea and an agent for the colorimetric detection of ammonium ions
On each plate there must be appropriate controls. A number of negative sera are included on each plate to give the baseline for a negative reaction. Positive controls vary with the test method. There may be a single positive control, low, medium and strong positive controls or a series of dilutions of a positive control depending on how the results are to be calculated. There should also be controls to show that substrate on its own is stable, and that it reacts with conjugate but not with antigen. The antigen control contains antigen (attached to the plate) and substrate. The conjugate control contains antigen, conjugate and substrate.
Variations of the indirect ELISA
There are many minor and major variations to the way ELISA tests are carried out. A few are discussed below:
Use of capture antibody for attaching antigen to plate
In some tests, plates are coated with an antibody to the antigen. After the antibody has attached to the plate, antigen is added and it attaches to the antibody that has been coated onto the plate (). The antibody used for this purpose is called a capture antibody (CAb).
3 | P a g e

Serology: Enzyme-linked immunosorbent assay (ELISA)
Figure 2: Attachment of antigen to an ELISA plate by using a capture antibody.
If the serum or immunoglobulin preparation used as the CAb is polyclonal it will contain a mixture of different types of antibodies with specificities for different epitopes. Therefore, the antigen will be attached in a variety of different stereoscopic presentations and a variety of epitopes will be presented for interaction with test serum antibodies. If a monoclonal antibody is used as CAb it will always attach to a specific antigen epitope and if there is only one of these epitopes on each antigen molecule then antigen will always be attached in the same stereoscopic conformation.
It is important to choose a CAb that will not react with the conjugate. If a bovine serum is used as CAb then the antigen plates are unsuitable for testing bovine sera. The Ab2 (conjugate) used to detect bovine antibodies attaching to the antigen will also attach to the CAb molecules on the plate giving strong false positive reactions in every well. Therefore, in a test for bovine sera the CAb should be from an animal that will not cross-react with theAb2 used in the test. If possible the CAb and the conjugate should be from the same species of animal to ensure that the conjugate will not bind to the CAb.
The use of capture antibodies increases the specificity of the test. Only antigen molecules and not a variety of contaminating molecules found in unpurified antigen preparations, attach to the plate. Using a CAb is equivalent to having an affinity chromatography step in the preparation of the antigen. When a monoclonal CAb is used the specificity is still further improved ensuring that only antigens with a specific epitope are attached.
Alternatives to using second antibodies
Instead of using an antibody as conjugate, other molecules that react specifically with immunoglobulins can be used. One such alternative is the use of Protein G conjugated to horseradish peroxidase. This reagent can be used for the demonstration of antibodies in cattle and sheep sera. Protein G conjugate is available from commercial suppliers. Another protein that attaches specifically to the C fragment of immunoglobulins is Protein A from Staphylococcus aureus. It can also be purchased from commercial suppliers in the form of an enzyme conjugate.
Sandwich ELISA
Sandwich ELISA tests are tests in which the Ab2 or other detection reagent is not conjugated to an enzyme and a third antibody (Ab3), conjugated to an enzyme, is used to detect the Ab2.
4 | P a g e

Serology: Enzyme-linked immunosorbent assay (ELISA)
For example in a test of bovine serum, a rabbit anti-bovine antibody (not conjugated to an enzyme) might be used to detect bovine antibody bound to the antigen on the plate and then a goat anti-rabbit antibody conjugate might be used to detect the bound rabbit antibodies. It is theoretically possible to add several layers of different antibodies on top of each other in sandwich form.
Theoretically sandwich tests are more sensitive because the number of molecules attaching to the complex is amplified with each new reagent added. For example the bovine antibody that attaches to the antigen has several epitopes on its surface and can bind to a number of molecules of rabbit anti-bovine antibody. Each of these can in turn bind several molecules of the goat anti-bovine conjugate. Therefore at each addition of another detection reagent the number of molecules that binds to the complex is multiplied ().
Figure 3: Amplification of bound enzyme molecules in a two-step sandwich ELISA.
In practice analytical sensitivity is seldom a limiting factor in the use of ELISAs for the diagnosis of infectious diseases and sandwich ELISAs are more likely to be used for the other reasons. For example where the Ab2 is a MAb it is convenient and economical to use it in a non-conjugated form. The bound MAb can then be detected with an anti-mouse conjugate. This same anti-mouse conjugate can be used in all tests in which Ab2 is a mouse monoclonal antibody. Anti-mouse conjugates are readily available from commercial sources.
Another variation of the sandwich technique makes use of the high affinity and specificity of the binding of biotin by the protein avidin. Instead of conjugating theAb2 (or detection reagent) to an enzyme it is conjugated to biotin. Avidin conjugated to an enzyme is then used to detect the antibody biotin complexes (Figure 4). A biotin/anti-mouse antibody in conjunction with an avidin enzyme conjugate can be used to detect all MAb reagents originating from mouse cells.
Figure 4: Use of the biotin/avidin system for the detection of antibody in an indirect ELIS.
5 | P a g e

Serology: Enzyme-linked immunosorbent assay (ELISA)
Dot ELISAs
Antigen can be absorbed onto nitrocellulose e.g. spotted onto a nitrocellulose dipstick. Unbound binding sites on the nitrocellulose are then blocked with milk powder or another suitable blocking agent. The dipstick can then be dipped into serum for an appropriate time, removed and washed in 0.05% Tween 20/PBS, dipped into conjugate, washed, dipped into substrate containing diamino-benzidine and hydrogen peroxide, and finally into stopping solution. A positive reaction is seen as a brown spot where the antigen was spotted onto the dipstick. Single or small numbers of tests can be done rapidly, but the method is less suitable for testing large numbers of sera.
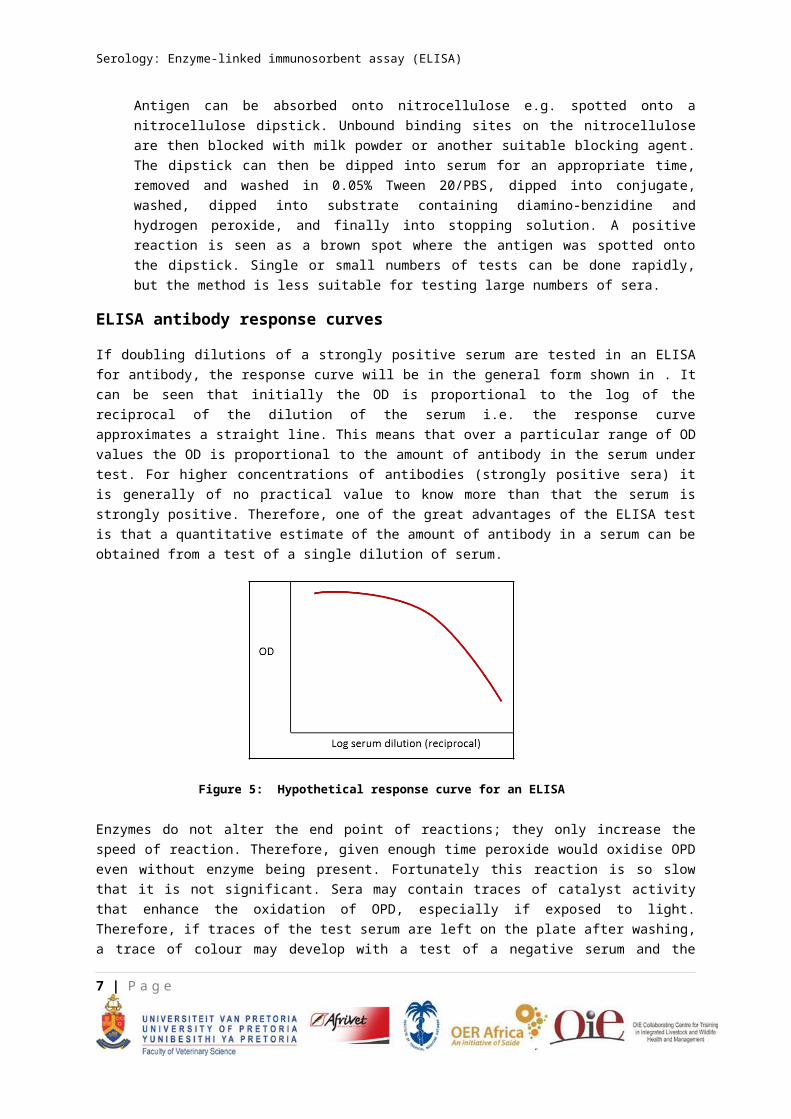
ELISA antibody response curves
If doubling dilutions of a strongly positive serum are tested in an ELISA for antibody, the response curve will be in the general form shown in . It can be seen that initially the OD is proportional to the log of the reciprocal of the dilution of the serum i.e. the response curve approximates a straight line. This means that over a particular range of OD values the OD is proportional to the amount of antibody in the serum under test. For higher concentrations of antibodies (strongly positive sera) it is generally of no practical value to know more than that the serum is strongly positive. Therefore, one of the great advantages of the ELISA test is that a quantitative estimate of the amount of antibody in a serum can be obtained from a test of a single dilution of serum.
Figure 5: Hypothetical response curve for an ELISA
Enzymes do not alter the end point of reactions; they only increase the speed of reaction. Therefore, given enough time peroxide would oxidise OPD even without enzyme being present. Fortunately this reaction is so slow that it is not significant. Sera may contain traces of catalyst activity that enhance the oxidation of OPD, especially if exposed to light. Therefore, if traces of the test serum are left on the plate after washing, a trace of colour may develop with a test of a negative serum and the amount of colour that develops varies from one serum to another. For this reason a number of negative sera are included on each plate and the mean of their ODs is used as the negative control value and is subtracted from the OD reading for each serum. It would be more theoretically correct to test each serum in a well containing antigen and in a well that does not contain antigen thus providing its own negative control. However, for practical reasons and because it makes little difference to the results, this is seldom done except when the antigen may be suspected of containing substances that could
6 | P a g e
Serology: Enzyme-linked immunosorbent assay (ELISA)
bind to antibodies in the serum under test. This is particularly the case in some viral antigens produced on tissue culture. In these cases a positive and a negative antigen are used. The positive antigen is antigen prepared from tissue culture and the negative antigen is antigen prepared in the same way from tissue culture cells that have not been inoculated with virus. The OD of the test using “negative antigen” is subtracted from the OD of the test with normal antigen.
Assuming that 20 wells are used for various controls on a plate it is still possible to do 76 tests on a single plate compared to the 11 tests that can be done with the CFT. This also makes it cost efficient to do all test in duplicate on each plate. Despite this some workers still prefer to test doubling dilutions of each serum in the traditional manner used in other serological tests.
The analytical sensitivity of ELISA tests is very high and all sera must be suitably diluted before testing. The most suitable dilution at which to test sera must be determined for each test. In the Brucella abortus ELISA test sera are diluted 1/200 for testing.
Calculation of results
It is not possible under normal laboratory testing conditions to get identical results in terms of OD when testing a serum repeatedly on different plates or days. For this reason the results are expressed as being relative to the result for a control serum that has been standardised against an international or national control serum, and is tested on the same plate as the serum being tested. Results can be calculated and expressed in a number of ways:
A single dilution of strongly positive serum may be used as control. In this case the control serum is diluted to a dilution that gives a response on the linear portion of the dose response curve for that serum. A dilution is often chosen where the response is slightly below the plateau phase of the curve. However, any dilution that gives a result falling on the linear part of the response curve can be used, provided the same dilution is always used. In this case a result for a serum is expressed as a percentage of the reaction for the control serum. The result then becomes:
Mean OD for aliquots of serum tested−Mean OD of negative control seraMean OD of control serum−Mean OD of negative control sera
×100
The threshold for a suspicious or positive result is given as a percentage of the positive control serum.
The antibody content of the control serum may be expressed in units. A number of dilutions of serum, which when tested give results that fall on the linear portion of the response curve, are tested on each test plate. A best-fit response function of OD against the log of the units of antibody is calculated (assuming it is a straight line fitting the function y = ax + b, where a = the slope of the line and b = the intercept on the y axis). The number of units of antibody in each test sample is then calculated from the mathematical function. Alternatively a graph of the response (OD) against units of antibody for the control serum can be plotted and the results for each serum read from the graph. However, in practice graphing of results is clumsy and impractical for testing large numbers of sera and generally the data is fed directly from the ELISA plate reader into a computer. The computer is programmed to calculate the regression function for the control
7 | P a g e

Serology: Enzyme-linked immunosorbent assay (ELISA)
serum dilutions and then calculates the number of units of antibody in each test serum (see Section 1.4.4).
In some cases a weak positive serum is included as a control and sera giving reactions equal or greater than the weak positive serum are classed as positive. In this case strong and medium level positives may also be included on the plate as further reference points.
Test protocol for a typical indirect ELISA
The protocol given below is an example of a protocol used for doing ELISA tests for Brucella abortus. Many different test protocols can be found in the literature and method chosen for a particular test will vary with the availability of reagents and equipment, the purpose of the test and the personal preferences of the operators doing the test.
Test reagents
Coating buffer
Na2HPO42H2O 7.74 g
KH2 PO4 0.88 g
distilled water up to l,000 ml
0.05M phosphate-buffered saline/ Tween 20 (PBST20)
Na2HPO42H2O 6.41 g
KH2 PO4 1.91 g
NaCl 9.00 g
Tween 20 0.50 ml
Distilled water up to 1,000 ml
Ortho phenylene diamine (OPD) substrate
Citric acid 9.6 g
Na2HPO42H2O 17.8 g
OPD 0.5 g
Adjust pH to 5.2 while making up with distilled water to 1,000 ml
8 | P a g e

Serology: Enzyme-linked immunosorbent assay (ELISA)
OPD solution can be divided into in 20 ml aliquots and stored at –200C in the dark until used. Coloured solutions of OPD should not be used. Before use 20 l of 30% wv hydrogen peroxide is added to 20ml of OPD solution
Stopping solution
±2.0 M H2SO4, i.e. 6.0 % concentrated H2SO4
9 | P a g e

Serology: Enzyme-linked immunosorbent assay (ELISA)
Conjugate
Protein G conjugated to horseradish peroxidase, from a commercial supplier.
Antigen
A suspension of Brucella abortus cells as used in the CFT and agglutination tests.
Control sera
The national or laboratory standard, positive control serum and six negative control sera are used.
Titration of antigen when optimal conjugate dilution is known
Suitable dilutions of a commercially available Brucella abortus agglutination antigen are made up in coating buffer e.g. doubling dilutions from 1/50 to 1/1,600.
150 l of the first dilution of antigen is dispensed into vertical rows 1 and 2 on the plate. The second dilution is similarly dispensed into rows 3 and 4 etc. (Figure 6).
Figure 6: Plate set-up. Dispensing of antigen dilutions for antigen titration. a1 = 150 l of the first antigen dilution. a2 = 150 l of the second antigen dilution. a3-a4 = 150 l of the
subsequent antigen dilutions.
The plates are stored at 40C overnight and then washed repeatedly. A positive control serum is diluted to a level at which it would be expected to give a
response near the top of the linear part of the response curve, in a standard test e.g. 1/800. Three doubling dilutions are made from it (1/1,600, 1/3,200, and 1/6,400) in PBST.
100 l of negative control serum, diluted 1/200, is dispensed into each well on the horizontal rows F, G and H.
100 l of the first dilution of positive control serum is dispensed into all wells in row E. (1/800 in the example). 100 l of the second dilution of positive serum (1/1,600) is
10 | P a g e

Serology: Enzyme-linked immunosorbent assay (ELISA)
dispensed to the wells in Row D. 100 l of the third (1/3,200) to the wells in Row C and the fourth (1/6,400) to row B. 100 l of PBST is dispensed into row A (Figure 7).
Figure 7: Plate set-up for antigen titration. Dispensing control sera: n = 100 l of negative control serum, p1 = 100 l of the first dilution of positive serum, p2 = 100 l of the first second dilution of
positive serum, p3 = 100 l of the first third dilution of positive serum, p4 = 100 l of the first fourth dilution of positive serum, b = 100 l of PBST.
The plate is incubated at 40C overnight and then washed repeatedly. 100 l of suitably diluted conjugate is dispensed into all wells. If the conjugate dilution is
not known it must be determined (see below). The plate is incubated at 370C for 2 hours and then washed repeatedly. 100 l of substrate is dispensed into all wells. The plate is covered and left in the dark for 7
minutes. 100 l of stopping solution are dispensed into all wells, in the same order as was used
when dispensing substrate onto the plate. The plate is read on the plate reader at 490 nm.
The ratio of the mean OD of the four positive sample dilutions to the mean of the OD of the negative samples (sometimes called the signal to noise ratio) is calculated for each serum and antigen dilution. The antigen dilution giving the highest mean ratio for the four dilutions of positive serum is taken as the working dilution of antigen.
The titration can be repeated, as necessary, using different dilutions of antigen to more exactly determine the optimal antigen level.
Titration of conjugate when optimal antigen dilution is known.
When the optimal antigen dilution is known, plates coated with antigen at optimal dilution are used to titrate the conjugate. The titration is exactly as for antigen (Section 1.4.2) except that varying dilutions of conjugate are used instead of varying dilutions of antigen. The dilution of
11 | P a g e

Serology: Enzyme-linked immunosorbent assay (ELISA)
conjugate used should be that giving the best mean ratio of OD positive serum to OD negative serum for the four dilutions of positive serum.
Antigen/conjugate checkerboard titration
When a new test is being set up and neither antigen nor conjugate dilution is known two checkerboard titrations are done, one using positive control serum and one using negative control serum. Dilutions of antigen are varied in the vertical rows of the plate and the dilutions of conjugate are varied in the horizontal rows of the plate. The positive and negative serum checkerboards are done in alternate rows on the same plate as described below. The dilutions of antigen and conjugate selected for further standardisation are those giving the highest ratio of OD positive serum to OD negative serum.
Make six dilutions of antigen e.g. doubling dilutions 1/50 to 1/1,600. Coat all the wells in vertical rows 1 and 2 with 150 l of the first dilution of antigen as
described in Section 1.4.2. Similarly coat the rows 3 and 4 with the second dilution etc. (Figure 8).
Figure 8: Plate set-up for a conjugate/antigen checkerboard titration. Coating the plate with antigen. a1 = 150 l of first antigen dilution. a2 = 150 l of second dilution of antigen. a3-a6 = 150 l of
subsequent antigen dilutions.
A suitable dilution of positive control serum in PBST, expected to give a response near to the midpoint of the linear region of the response curve in the standard test, is made up e.g. 1/3,200. 100 l is dispensed to wells H-B in vertical rows 1, 3, 5, 7, 9 and 11. 100 l of PBST is dispensed to all wells in horizontal row A.
A suitable dilution of negative control serum as used for testing sera in the standard test (1/200) is made up in PBST and dispensed to wells H-B in vertical rows 2, 4, 6, 8, 10 and 12 (Figure 9).
12 | P a g e

Serology: Enzyme-linked immunosorbent assay (ELISA)
Figure 9: Plate set-up for conjugate/antigen checkerboard titration. Dispensing of control sera. B = 100 l of PBST. P = 100 l of positive control serum. N = 100 l of negative control serum.
The plate is covered and incubated and left overnight at 40C and then washed repeatedly. Six dilutions of conjugate are made up in PBST e.g. 1/1,000, 1/2,000, 1/4,000 1/6,000,
1/8,000, 1/10,000. 100 l of the first dilution of conjugate are dispensed to all wells in horizontal rows H and A. 100 l of the second dilution of conjugate are dispensed to the wells of vertical row G, 100 l of the third dilution to the wells on row F etc. (Figure 10).
Figure 10: Plate set-up for conjugate/antigen checkerboard titration. Dispensing conjugate dilutions. c1 = 100 l of the first dilution of conjugate. c2 = 100 l of the second dilution of
conjugate. c3–c7 = 100 l of subsequent dilutions of conjugate.
The plates are covered and incubated at 370C for 2 hours and then washed repeatedly with PBST.
13 | P a g e

Serology: Enzyme-linked immunosorbent assay (ELISA)
100 l of substrate is added to all wells and the plate is allowed to stand for 7 minutes in the dark.
100 l of stopping solution are added to all wells in the same order as was used when dispensing conjugate onto the plate.
The ODs of all wells are read on an ELISA plate reader.
The dilutions of antigen and conjugate giving the highest ratio of OD positive serum to OD negative serum are used for further standardisation tests. If necessary the titration can be repeated using different dilutions of reagents.
When the titrations have been completed a test, done in the standard format, using the determined reagent dilutions is run on a suitable panel of positive and negative sera. The panel should consist of about 20 positive sera giving responses from low to high ODs, preferably from field cases and not from experimentally infected animals and 10 negative sera. The standard test using the previous batches of reagents is run in parallel on the same panel of sera. If the tests with newly titrated reagents and old reagents do not agree further titrations and adjustments to the test must be made until the overall agreement between the tests reaches the defined acceptable standards. After this the test should be run in parallel with the standard test using the previous batches of reagents over several hundred routine test samples, before it is finally used for routine testing.
Protocol for an indirect ELISA
A description of an indirect ELISA is given below:
The plate is coated with antigen at the determined dilution as described in Section 1.4.1. All samples to be tested and the six negative control sera are diluted 1/200 in PBST (10 l
of serum in 1.99 ml l of PBST). Four doubling dilutions of the positive control serum, that when tested are known to fall in the linear portion of the response curve, are made in PBST. E.g. 1/800, 1/1,600, 1/3,200, 1/6,400.
100 l of 6 negative control sera are dispensed into wells H1, H2, G1, G2, F1, and F2. 100 l of the first dilution of positive control serum (1/800) are dispensed into wells B1 and
B2. 100 l of the second dilution of positive control serum (1/1,600) are dispensed into wells C1
and C2. 100 l of the third dilution of positive control serum (1/3,200) are dispensed into wells D1
and D2. 100 l of the fourth dilution of positive control serum (1/6,400) are dispensed into wells E1
and E2. 100 l of each serum to be tested are dispensed into two adjacent wells in the block of
wells from B3 to H12. 100l of PBST is dispensed into all wells in horizontal row A (Figure 11). The plate is covered and incubated overnight at 40C. The plate is washed repeatedly in PBST.
14 | P a g e

Serology: Enzyme-linked immunosorbent assay (ELISA)
100 l of PBST are dispensed into wells A1 – A6 and 100 l of conjugate is dispensed into all remaining wells.
The plate is incubated at 370C for 2 hours and then washed repeatedly in PBST. 100 l of substrate is added to all wells and the plate is left in the dark for 7 minutes. 100 l of stopping solution are added to all wells in the same order as was used when
dispensing conjugate onto the plate. The OD of all wells is read on the ELISA plate reader.
Figure 11: Plate set-up for the standard indirect ELISA test. Dispensing of control sera. n1-n6 = 100 l of six negative control serum. P1 = 100 l of the first dilution of positive control serum. P2
= 100 l of the second dilution of positive control serum. P3 = 100 l of the third dilution of positive control serum. P4 = 100 l of the fourth dilution of positive control serum. B = 100 l of
PBST. S = 100l of serum samples (two wells for each sample).
The data is calculated as follows:
The mean OD of the negative controls (F1, F2, G1, G2, H1 and H2) is calculated and subtracted from OD readings of all other wells in horizontal rows B-H. These corrected values are used in further calculations.
A number of units of antibody are assigned to the positive control serum. The units can be arbitrarily assigned to the control serum. However, in the case of the Brucella abortus test it is convenient to have the units equal to the IUs of agglutinating or complement fixing antibody in the control serum. If in the example used above the control serum contained 800 international complement fixation test units (ICFTU) of antibody/ml, then the four dilutions of sera tested contain 1.0, 0.5, 0.25 and 0.125 ICFTU/ml. The hypothetical theoretically perfect data given in Table 2 and plotted in , might have been found in a test.
15 | P a g e

Serology: Enzyme-linked immunosorbent assay (ELISA)
Table 2: Hypothetical data for perfect fit case when testing a control serum at four different dilutions.
Control serum Units/ml Serum dilution Units/ml in serum tested Log10 units/ml in serum tested Response (OD)
800 1/800 1.000 0 1.20
800 1/1,600 0.500 -0.3010 0.90
800 1/3,200 0.250 -0.6020 0.60
800 1/6,400 0.125 -0.9030 0.30
Figure 12: Regression plot of data in
The equation describing a straight-line regression is y = ax + b with a = the slope of the line and b = the intercept on the y-axis. If y = OD and x = log antibody units/ml of antibody then for the data in Table 2 gives the plot shown in Figure 11. The intercept on the y-axis is 1.2 and the slope of the line is 0.3/0.3010 = 0.9967. Therefore the function becomes y = 0.9967x + 1.2. For any OD (y value) the equation can be solved for x and multiplying the antilog of x by the dilution of the serum tested will give the units of antibody in the test serum e.g. for an OD of 0.90 in the above data:
0.90 = 0.9967x + 1.2
x = (0.90 – 1.2)/0.9967
x = -.3010
16 | P a g e

Serology: Enzyme-linked immunosorbent assay (ELISA)
and 1,600 (antilog -.3010) = 800 units of antibody which is the correct answer for this control serum.
Therefore, the units of antibody for any serum tested will be: antilog ((OD – b)/a) multiplied by the dilution of the serum tested.
This may seem unnecessarily complicated but once the computer is programmed the results are generated automatically and no calculation is involved.
17 | P a g e