an atypical chondroid syringoma with malignant
TRANSCRIPT

Henry Ford Health System Henry Ford Health System
Henry Ford Health System Scholarly Commons Henry Ford Health System Scholarly Commons
Dermatology Articles Dermatology
1-20-2021
An atypical chondroid syringoma with malignant degeneration: An atypical chondroid syringoma with malignant degeneration:
Utility of comparative genomic hybridization in confirming the Utility of comparative genomic hybridization in confirming the
diagnosis diagnosis
Shereen Zia
Brandon Shaw
Stephanie Chapman
Ben J. Friedman
Follow this and additional works at: https://scholarlycommons.henryford.com/dermatology_articles

C A S E R E P O R T
An atypical chondroid syringoma with malignant degeneration:Utility of comparative genomic hybridization in confirming thediagnosis
Shereen Zia MD1 | Brandon Shaw PhD1 | Stephanie Chapman MD, MS2 |
Ben J. Friedman MD1,2
1Department of Pathology and Laboratory
Medicine, Henry Ford Hospital, Detroit,
Michigan
2Department of Dermatology, Henry Ford
Hospital, Detroit, Michigan
Correspondence
Ben J. Friedman, MD, Department of
Dermatology, Henry Ford Hospital, 3031 West
Grand Boulevard, Detroit, MI 48202.
Email: [email protected]
Abstract
Chondroid syringoma (CS) represents the cutaneous counterpart of mixed tumor
(pleomorphic adenoma) of salivary glands. Definitive diagnosis is made on histopa-
thology and is based on the presence of characteristic epithelial and stromal compo-
nents. We report a case of an atypical CS arising on the extremity of an elderly male
patient. Histomorphologic features of necrosis and cellular atypia raised suspicion for
malignant degeneration, an exceptionally rare circumstance in this context. To further
support the diagnosis of malignancy, array comparative genomic hybridization was
performed from both low and higher grade areas of the tumor. Both regions demon-
strated multiple copy number gains and losses, with additional loss of q7p (TP53), loss
of 19p, and loss of heterozygosity on16q demonstrated in the more atypical foci. To
our knowledge, this is the first case description of malignant degeneration of a CS
with correlative microarray analysis. The findings in this case may prove useful in
confirming the diagnosis in future ambiguous cases.
K E YWORD S
chondroid syringoma, comparative genomic hybridization, mixed tumor
1 | INTRODUCTION
Chondroid syringoma (CS; also known as cutaneous mixed tumor) is a
benign cutaneous neoplasm of sweat gland origin consisting of both
epithelial and mesenchymal components.1 It bears morphologic and
genetic resemblance to pleomorphic adenoma, the most common
tumor of the salivary glands.2 Features that contribute to a diagnosis
of CS include monomorphous epithelial cells arranged in cords and
tubules with a peripheral myoepithelial layer, and occurring within a
myxoid or chondroid stroma.3 Other variably present features include
foci of squamous differentiation and keratocyst formation, fat meta-
plasia, and bone formation. Recently it was discovered that some CSs
may harbor fusions in PLAG1, similar to those seen in pleomorphic
adenoma.2,4 Atypical and malignant forms of CS have been described,
though due to the uncommon nature of these tumors, reliable diag-
nostic criteria on histomorphology are a matter of debate.5,6
Moreover, the molecular events leading to malignant CS are largely
unknown. Interestingly, there have been cases of CS with a decep-
tively bland cytological appearance (analogous to so-called benign
metastasizing pleomorphic adenoma) that have later proven to behave
in a malignant fashion.7,8 In this report, we describe a case of malig-
nant CS arising within an atypical CS in which array comparative
genomic hybridization (aCGH) was found to be especially useful in
supporting a diagnosis of malignancy. To our knowledge, this is the
first report describing cytogenetic alterations in atypical and
malignant CS.
2 | CASE REPORT
An 84-year-old Caucasian male with the past medical history of diabe-
tes and hypertension presented with a slow-growing mass on his right
Received: 12 November 2020 Revised: 11 January 2021 Accepted: 12 January 2021
DOI: 10.1111/cup.13965
J Cutan Pathol. 2021;1–6. wileyonlinelibrary.com/journal/cup © 2021 John Wiley & Sons A/S. Published by John Wiley & Sons Ltd. 1

upper extremity near the axilla that had been present for seven years.
On physical examination, a 9 × 7 cm subcutaneous nodule was seen
at the anteromedial aspect of the right arm. Excision of the mass was
recommended given its large size, though the clinical impression was
initially that of a lipoma.
Gross examination revealed a 7.5 × 7 × 4 cm multilobulated
mass with a thin pseudocapsule. The cut surfaces ranged from pink-
tan and indurated to red, soft, and hemorrhagic (Figure 1). Micro-
scopically, sections revealed a bulky proliferation of epithelial cells
arranged in large clusters and solid cords, embedded in a myxoid
and fibrous stroma (Figures 2 and 3). A surrounding rim of fibrosis
was observed, with the tumor cells seen expanding the dermis and
bulging into the subcutis. A large proportion of the epithelial cells
had associated cytoplasmic vacuolation and clear-cell morphology
(Figure 4). Focal gland formation and areas of chondroid matrix
were appreciated.
In the more central and deeper portions of the tumor, areas of
cystic degeneration and necrosis, irregular gland formation, cellular
atypia, and mitotic activity were identified (Figure 5). An extended
panel of immunohistochemical stains was performed. The cells of
interest were positive for Sox10, CK7, INI-1, S100, AE1/3 and GATA
(patchy), and were negative for hepatocyte antigen, PAX8, CK20,
CD31, HMB45, Melan A and CD34. Taken together, these findings
were consistent with a CS with atypical features worrisome for
F IGURE 1 Gross findings: Pseudoencapsulated multilobulated subcutaneous nodule with considerable hemorrhagic necrosis
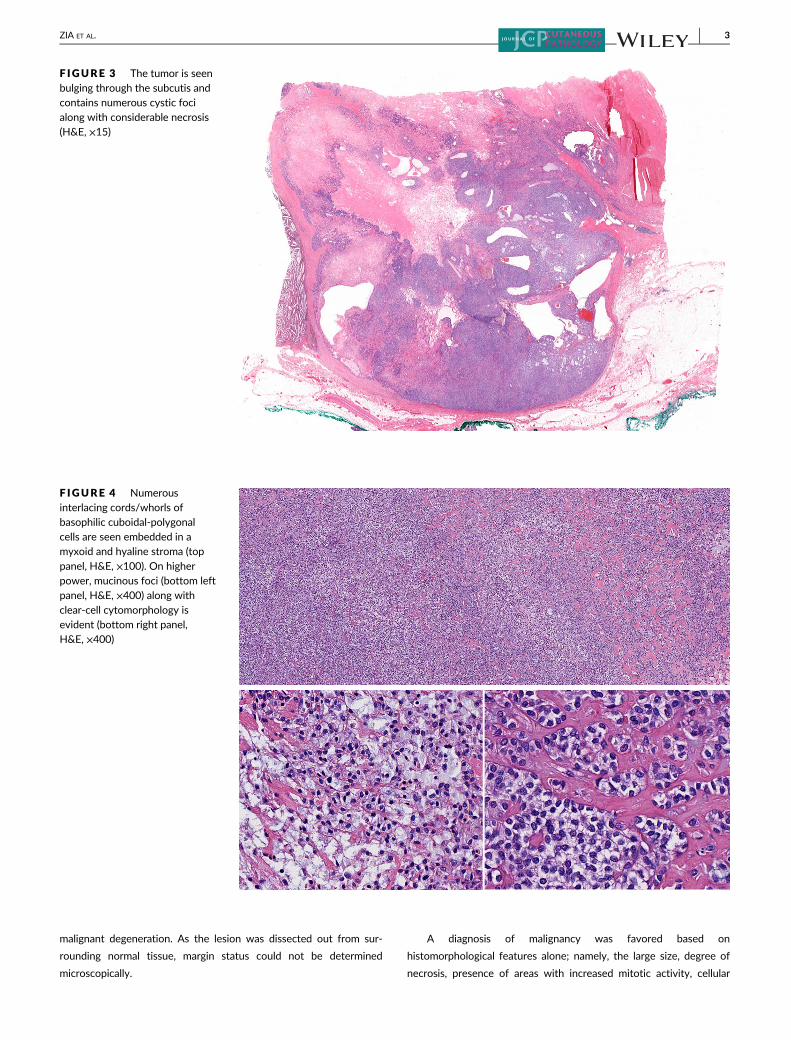
F IGURE 2 Variably sized bluenodules are seen expanding thedermis and are enveloped infibrosis (H&E, ×15)
2 ZIA ET AL.
malignant degeneration. As the lesion was dissected out from sur-
rounding normal tissue, margin status could not be determined
microscopically.
A diagnosis of malignancy was favored based on
histomorphological features alone; namely, the large size, degree of
necrosis, presence of areas with increased mitotic activity, cellular
F IGURE 3 The tumor is seenbulging through the subcutis andcontains numerous cystic focialong with considerable necrosis(H&E, ×15)
F IGURE 4 Numerousinterlacing cords/whorls ofbasophilic cuboidal-polygonalcells are seen embedded in amyxoid and hyaline stroma (top
panel, H&E, ×100). On higherpower, mucinous foci (bottom leftpanel, H&E, ×400) along withclear-cell cytomorphology isevident (bottom right panel,H&E, ×400)
ZIA ET AL. 3
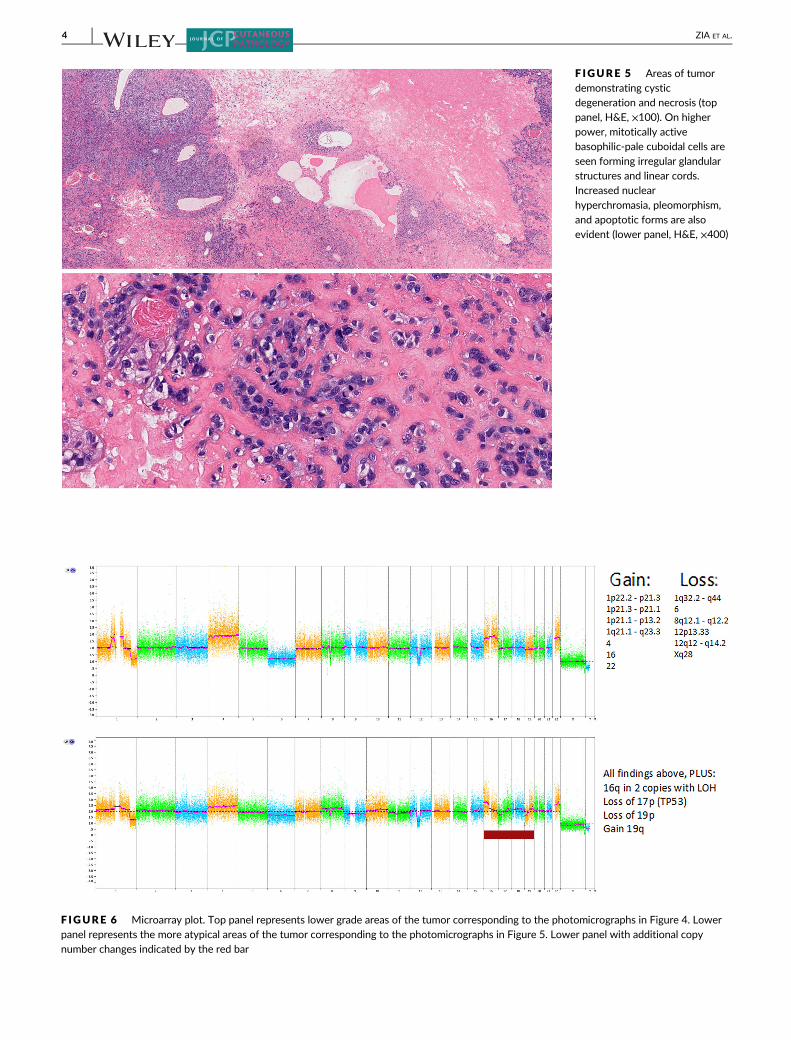
F IGURE 5 Areas of tumordemonstrating cysticdegeneration and necrosis (toppanel, H&E, ×100). On higherpower, mitotically activebasophilic-pale cuboidal cells areseen forming irregular glandularstructures and linear cords.Increased nuclear
hyperchromasia, pleomorphism,and apoptotic forms are alsoevident (lower panel, H&E, ×400)
F IGURE 6 Microarray plot. Top panel represents lower grade areas of the tumor corresponding to the photomicrographs in Figure 4. Lowerpanel represents the more atypical areas of the tumor corresponding to the photomicrographs in Figure 5. Lower panel with additional copynumber changes indicated by the red bar
4 ZIA ET AL.

atypia, and the identification of highly irregular glandular structures.
However, a small degree of doubt was raised based on the lack of dis-
cernable host tissue invasion and the possibility that the necrosis was
procedure-induced/trauma-related. Therefore, aCGH was performed
from two separate regions within the tumor: (a) more conventional-
appearing low grade area (top plot, Figure 6) more atypical-appearing
higher grade area (lower plot, Figure 6). Multiple copy number gains
and deletions were identified in both sampled areas, consistent with
atypical/unstable tumor clones. Specifically gains were seen in
1p22.2p21.3, 1p21.3p21.1, 1p21.1p13.2, 1q21.1q23.3, 4p16.3q35.2,
16p13.3q22.1, 16q22.1q24.3, 22q11.1q13.33, with losses seen in
1q32.2q44, 6p25.3q27, 8q12.1q12.2, 12p13.33, 12q12q14.2, and
Xq28 (homozygous loss). The deletions seen at 8q12 and 12q12
regions further supported the diagnosis of a myoepithelial neoplasm.9
Additionally, the more atypical portion of the tumor demonstrated
evidence of clonal evolution with additional loss of 17p13.3p11.2
(TP53), loss of 19p13.3p13.11, and loss of heterozygosity (LOH) on
16q11.2q24.3. Based on this additional information, a low-grade
malignant CS was the favored diagnosis. Correlation with
intraoperative findings was recommended to ensure that the tumor
was adequately excised. A positron emission tomography-computed
tomography scan failed to reveal any evidence of metastatic disease.
After discussion of the potential risks/benefits with the patient, the
decision was made to forgo a wider excision and to proceed with
close clinical surveillance alone. The patient has remained free of
detectable recurrence for seven months.
3 | DISCUSSION
CS is considered a rare tumor of sweat glands, first described by
Hirsch and Helwig in 1961.3 The reported incidence of CS is 0.01% to
0.098%.10,11Although there is no distinct clinical feature to aid in defi-
nite diagnosis, it usually presents as a firm, asymptomatic, skin col-
ored, slow-growing, solitary dermal, or subcutaneous nodule. The
typical size ranges from 0.5 to 3 cm.11-13 The most common site of
involvement is the head and neck, and it predominantly affects
middle-aged men.10
Histopathologically, the lesion is characterized by an admixture of
epithelial and mesenchymal elements; hence the term “mixed tumor.”The proposed histopathologic criteria by Hirsch and Helwig included five
features: (a) nests of cuboidal or polygonal cells; (b) intercommunicating
tubuloalveolar structures lined with two or more rows of cuboidal cells;
(c) ductal structures composed of one or two rows of cuboidal cells;
(d) occasional keratinous cysts; and (e) a matrix of varying composition.
The stroma exhibits varying density and can be chondroid, myxoid,
fibrous, hyaline, or osseous. The ductal and tubuloalveolar structures
consist of two types of cells: the darker, cuboidal to polyhedral cells lin-
ing the inner layer of these structures exhibit epithelial lineage, whereas
the light, low-cuboidal cells forming the outer layer of these tubular and
ductal structures manifest myoepithelial differentiation. Varela-Duran
et al proposed the pluripotentiality of the myoepithelial cells, citing their
role in the production of the chondroid areas of the tumor.14 Later on,
Mills determined that CSs are clonal neoplasms consisting of replicating
cells with the potential to differentiate toward epithelium or mesen-
chyme, which explains the tremendous diversity in histopathologic
appearance.15
The malignant variant of CS is extremely infrequent and may arise
from malignant transformation of an otherwise ordinary CS.16 Fea-
tures previously attributed to malignant CS include a greater propen-
sity to occur on the extremities, larger size, the presence of cellular
atypia and increased cellularity, increased mitotic rate, invasion of sur-
rounding tissue, and tumoral necrosis.5,6 Metastasis to regional lymph
nodes, as well as to distant sites such as bone and visceral organs, has
also been described.5,17 The term “cutaneous mixed tumor with
atypical features” was previously proposed for tumors with architec-
tural features of malignancy, though without evidence of metasta-
sis.5 Wide excision of the primary tumor is generally regarded as the
preferred treatment for the primary tumor, with radiation therapy
and chemotherapy potentially having a role in the setting of metasta-
sis (though the optimal efficacious systemic therapy is unknown
given the rarity of the tumor overall and lack of randomized con-
trolled trials).18
Progressive accumulation of genomic changes is a crucial step in
the alteration of normal biological mechanisms and eventual devel-
opment of malignancy within a tumor. aCGH is a molecular cytoge-
netic technique used in detecting chromosomal abnormalities and
locating regions of genomic imbalances. Results from our aCGH
assay further confirmed the lineage of the tumor by identifying early
known genetic events. In the context of myoepithelial neoplasms,
such as CS and pleomorphic adenoma, deletions of the 8q21 region
and the 12q12q14.2 regions are commonly reported findings.9 The
8q21 loss results in the loss of the first exon of the PLAG1 gene and
is most consistent with a PLAG1 gene (nuclear oncoprotein)
rearrangement with an unknown partner gene resulting in PLAG1
overexpression.2,4,19 The loss of 12q12q14.2 results in dysregulation
of the HMGA2 gene, which encodes for a transcriptional gene
regulator.20,21
Furthermore, our results show that aCGH may have a role in the
clinical setting when faced with a CS with atypical features to help
support or refute a diagnosis of malignant degeneration. This may be
particularly useful when only some, but not all the morphologic fea-
tures of malignant CS are identified on prepared sections or when
there is considerable atypia confined focally within the lesion. In this
case, the presence of considerable copy number changes (n = 14)
combined with the morphologic atypia observed and demonstrable
clonal evolution with loss of 17p (TP53) and LOH on 16q (likely
reflecting loss of another tumor suppressor gene) support a diagnosis
of malignant transformation. In particular, loss of TP53 is known to be
associated with disease progression and an adverse prognostic impact
in multiple settings including other cutaneous adnexal neoplasms.22,23
Additional larger studies examining the cytogenetic changes in benign
CS, atypical CS, and bona fide malignant CS are needed before firm
conclusions can be drawn regarding the exact number/threshold or
consistent genetic events that are associated with aggressive behavior
in this context.
ZIA ET AL. 5

4 | MATERIALS AND METHODS
4.1 | Summary of cytogenetic analysis
Following review by a pathologist, 10 slides were cut at 10 μm thickness
from a representative formalin-fixed and paraffin-embedded (FFPE) tissue
block and tumor was macrodissected using a H&E-stained slide as a guide.
DNA was extracted and purified according to the manufacturer's protocols
(Qiagen QIAmp DNA Mini Kit). Extracted DNA was quantified following
the manufacturer's instructions and diluted to a final concentration of
12 ng/μL with a total of 80 ng utilized. Chromosomal microarray analysis
of tumor samples was performed using the OncoScan FFPE Assay, which
utilizes Molecular Inversion Probe (MIP) technology to obtain accurate
genome-wide copy number and LOH profiles. The assay contains 22 000
probes across the genome and targeted cancer regions, allowing for detec-
tion of copy number abnormalities at 50 to 100 kb resolution in�900 can-
cer genes and at 300 kb resolution in other chromosomal regions. Patient
hybridization results are compared to data derived from over 300 FFPE
samples from unaffected tissues. All data were analyzed and reported using
the February 2019 Genome Reference Consortium Human Build 38 patch
release 13 (GRCh38.p13).
4.2 | Reportable range
Deletions larger than 1 megabase, duplications larger than 2 mega-
bases, and copy neutral LOH larger than 5 megabases are generally
reported; however, complex genomic alterations may be reported in
aggregate, and well-documented pathogenic constitutional and/or
acquired abnormalities of any size may also be reported. The genome
coordinates described are best estimates and may not represent pre-
cise breakpoints, especially for abnormalities detected in a low per-
centage of cells. The detection threshold for mosaicism is variable,
depending on the size of the segment. Common population number
variants cited in the Database of Genomic Variants are not reported.
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the
corresponding author upon reasonable request.
ORCID
Ben J. Friedman https://orcid.org/0000-0002-2266-8197
REFERENCES
1. Villalon G, Monteagudo C, Martin JM, Ramon D, Alonso V, Jorda E.
Chondroid syringoma: a clinical and histological review of eight cases.
Actas Dermosifiliogr. 2006;97(9):573-577.
2. Bahrami A, Dalton JD, Krane JF, Fletcher CD. A subset of cutane-
ous and soft tissue mixed tumors are genetically linked to their sal-
ivary gland counterpart. Genes Chromosomes Cancer. 2012;51(2):
140-148.
3. Hirsch P, Helwig EB. Chondroid syringoma. Mixed tumor of skin, sali-
vary gland type. Arch Dermatol. 1961;84:835-847.
4. Panagopoulos I, Gorunova L, Andersen K, et al. NDRG1-PLAG1 and
TRPS1-PLAG1 fusion genes in chondroid syringoma. Cancer Genomics
Proteomics. 2020;17(3):237-248.
5. Bates AW, Baithun SI. Atypical mixed tumor of the skin: histologic, immu-
nohistochemical, and ultrastructural features in three cases and a review of
the criteria for malignancy. Am J Dermatopathol. 1998;20(1):35-40.
6. Harrist TJ, Aretz TH, Mihm MC Jr, Evans GW, Rodriquez FL. Cutane-
ous malignant mixed tumor. Arch Dermatol. 1981;117(11):719-724.
7. Ishimura E, Iwamoto H, Kobashi Y, Yamabe H, Ichijima K. Malignant
chondroid syringoma. Report of a case with widespread metastasis
and review of pertinent literature. Cancer. 1983;52(10):1966-1973.
8. Tarsitano A, Foschini MP, Farneti P, Pasquini E, Marchetti C. Metasta-
sizing "benign" pleomorphic salivary adenoma: a dramatic case-report
and literature review. J Craniomaxillofac Surg. 2014;42(8):1562-1565.
9. Stenman G. Fusion oncogenes and tumor type specificity—insights
from salivary gland tumors. Semin Cancer Biol. 2005;15(3):224-235.
10. Yavuzer R, Basterzi Y, Sari A, Bir F, Sezer C. Chondroid syringoma: a diag-
nosis more frequent than expected. Dermatol Surg. 2003;29(2):179-181.
11. Tokyol C, Aktepe F, Yavas BD, Yildiz H, Aycicek A. Chondroid
syringoma: a case report. Acta Cytol. 2010;54(5 suppl):973-976.
12. Sulochana S, Manoharan M, Anitha. Chondroid syringoma—an
unusual presentation. J Clin Diagn Res. 2014;8(7):FD13-FD14.
13. Limaiem F, Bouslama S, Haddad I, et al. Chondroid syringoma: report
of four cases. Skinmed. 2015;13(2):104-106.
14. Varela-Duran J, Diaz-Flores L, Varela-Nunez R. Ultrastructure of chondroid
syringoma: role of the myoepithelial cell in the development of the mixed
tumor of the skin and soft tissues. Cancer. 1979;44(1):148-156.
15. Mills SE. Mixed tumor of the skin: a model of divergent differentia-
tion. J Cutan Pathol. 1984;11(5):382-386.
16. Fernandez-Flores A, Cassarino DS. Malignant cutaneous mixed tumor with
sebaceous differentiation. Rom J Morphol Embryol. 2017;58(3):977-982.
17. Sun TB, Chien HF, Huang SF, Shih TT, Chen MT. Malignant chondroid
syringoma. J Formos Med Assoc. 1996;95(7):575-578.
18. Takahashi H, Ishiko A, Kobayashi M, Tanikawa A, Takasu H, Md MT.
Malignant chondroid syringoma with bone invasion: a case report and
review of the literature. Am J Dermatopathol. 2004;26(5):403-406.
19. Antonescu CR, Zhang L, Shao SY, et al. Frequent PLAG1 gene
rearrangements in skin and soft tissue myoepithelioma with ductal
differentiation. Genes Chromosomes Cancer. 2013;52(7):675-682.
20. Wasserman JK, Dickson BC, Smith A, Swanson D, Purgina BM,
Weinreb I. Metastasizing pleomorphic adenoma: recurrent
PLAG1/HMGA2 rearrangements and identification of a novel
HMGA2-TMTC2 fusion. Am J Surg Pathol. 2019;43(8):1145-1151.
21. El Hallani S, Udager AM, Bell D, et al. Epithelial-myoepithelial carci-
noma: frequent morphologic and molecular evidence of preexisting
pleomorphic adenoma, common HRAS mutations in PLAG1-intact and
HMGA2-intact cases, and occasional TP53, FBXW7, and SMARCB1
alterations in high-grade cases. Am J Surg Pathol. 2018;42(1):18-27.
22. Zahn J, Chan MP, Wang G, et al. Altered Rb, p16, and p53 expression
is specific for porocarcinoma relative to poroma. J Cutan Pathol.
2019;46(9):659-664.
23. Rashid M, van der Horst M, Mentzel T, et al. ALPK1 hotspot mutation
as a driver of human spiradenoma and spiradenocarcinoma. Nat
Commun. 2019;10(1):2213.
How to cite this article: Zia S, Shaw B, Chapman S,
Friedman BJ. An atypical chondroid syringoma with malignant
degeneration: Utility of comparative genomic hybridization in
confirming the diagnosis. J Cutan Pathol. 2021;1–6. https://
doi.org/10.1111/cup.13965
6 ZIA ET AL.