aldolase b enhancer in vivo 1 in vivo functional characterization of
TRANSCRIPT

Aldolase B enhancer in vivo
1
In vivo functional characterization of the aldolase B gene enhancer
Claudine Gregori(1), Arlette Porteu(1), Claudia Mitchell(1), Axel Kahn(1),and Anne-Lise Pichard(1, 2)
1. Département de Génétique, Développement et Pathologie Moléculaire,Institut Cochin, INSERM, CNRS et Université René Descartes – 75014 –
PARIS – France
Running title : Aldolase B enhancer in vivo
Address reprint requests to: Institut Cochin, 24 rue du Faubourg St-Jacques, 75014 PARIS, FRANCE
Fax : (33) 1. 44. 41. 24. 21 email : pichard@cochin. inserm. fr
Copyright 2002 by The American Society for Biochemistry and Molecular Biology, Inc.
JBC Papers in Press. Published on May 28, 2002 as Manuscript M204047200 by guest on A
pril 6, 2018http://w
ww
.jbc.org/D
ownloaded from

Aldolase B enhancer in vivo
2
Summary
A 400-bp intronic enhancer fragment in conjunction with the proximal
promoter of the aldolase B gene provided correct tissue-specific expression in
transgenic mice together with hormonal regulation in the liver. We investigated
in vivo and in cultured cells the contribution of the intronic regulatory
sequences and their interaction with the promoter elements in controlling
aldolase B gene expression. Transgene activity was completely abolished by
disruption of the two HNF1 binding sites in the enhancer, whereas mutation of
one HNF1 site had no effect in the liver but strongly decreased activity in the
kidney. Our data show that the HNF1 binding site(s) in the enhancer were key
regulators of aldolase B transgene expression both in the liver and kidney.
Deletion of the C/EBP site in the promoter completely abolished the enhancer
function in HepG2 cells. These results suggest that expression of the aldolase
B gene in the liver requires cooperative interactions between C/EBP and
HNF1. Deletion of the HNF4 binding site in the enhancer suppressed
expression in both liver and kidney in half of the transgenic lines, suggesting
that this element might play a role in chromatin opening at the insertion site.
We firmly establish that the endogenous aldolase B gene’s first response to
glucagon or cyclic AMP exposure was a transient increase in the expression
in the liver, followed by a secondary decline in the transcription, as previously
reported. This response was reproduced by all transgenes studied, indicating
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
3
that neither HNF1 nor HNF4 binding sites in the enhancer were involved in
this biphasic cyclic AMP response.
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
4
Introduction
The aldolase B enzyme catalyzes the reversible cleavage of fructose-1-
phosphate into dihydroxyacetone phosphate and glyceraldehyde, and is
involved in two opposite metabolic pathways, glycolysis and gluconeogenesis.
The expression of the aldolase B gene is subject to tissue-specific, hormonal
and metabolic regulation. The adult liver expresses the aldolase B gene
exclusively, while the kidney and enterocytes co-express both aldolase A and
B genes (1). In the liver, transcription of the aldolase B gene is induced by a
carbohydrate-rich diet and inhibited by fasting and glucagon, while, in the
kidney, it is almost unresponsive to dietary and hormonal regulation (2).
However, even in the liver, the expression of the aldolase B gene is never
completely abolished. In addition, it is restimulated after prolonged starvation
(our unpublished data). This behavior may reflect the dual role of aldolase B in
hepatocytes where it is required for the opposite glycolytic and gluconeogenic
pathways.
Finally, hereditary fructose intolerance is a recessive genetic disease caused
by aldolase B deficiency. Repeated ingestion of noxious sugars by
homozygotes leads to hepatic and renal injury (3), with metabolic
disturbances (including low concentrations of blood glucose) that may prove
fatal (4).
The aldolase B gene proximal promoter (-194 to +14) is sufficient to direct
cell-type-specific expression in cultured hepatoma cells, but in mice a distal
intronic enhancer fragment (+1916 to +2324) is required for transgene
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
5
expression (5). We have previously studied both these regions extensively (6-
9), and have identified the regulatory elements and cognate binding factors
(Fig.1, Fig.3). Their respective contributions to promoter activity and enhancer
function were investigated by transient CAT assays in HepG2 hepatoma cells.
Main findings were: the promoter activity was up-regulated by hepatocyte
nuclear factor 1 (HNF1) and CAAT/enhancer binding protein (C/EBP), but
repressed by hepatocyte nuclear factor 3 (HNF3) (7). The enhancer function
was abolished by mutation or deletion of either of the two HNF1 binding sites
and reduced by deletion of the HNF4 binding site (9).
Dietary regulation of a transgene directed by 1600 bp of the 5’ flanking region
of the aldolase B gene and the 1st intron appeared paradoxical: it was down-
regulated by a high carbohydrate diet and stimulated by prolonged fasting (5).
In order to explain these results we hypothesized that the endogenous gene
possessed distinct glucose-dependent and fasting-dependent responsive
elements, only the latter being present in the transgenic construct.
The objective of the current study was to assess, in a chromosomal in vivo
context, the contribution of the different DNA elements of the intronic
enhancer to the tissue-specific expression and metabolic regulation of the
transgenes.
Our results demonstrate a major contribution of the HNF1 enhancer binding
sites to the tissue-specific expression of the aldolase B gene. Deletion of the
two HNF1 binding sites suppressed transgene expression in both liver and
kidney. In contrast, mutation in one HNF1 site did not significantly affect
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
6
transgene expression in the liver but resulted in a 100-fold loss of activity in
the kidney. Findings suggested that the differential role of the HNF1 sites in
the different organs could be related to the abundance of C/EBP in the liver
compared to the kidney. Indeed, we show that a C/EBP binding site previously
identified in the proximal promoter is crucial for maximal expression in
hepatocytes.
Deletion of the HNF4 site seemed to render the transgene activity more
dependent on the insertion site, silencing it completely in about half of the
transgenic lines. Finally, we demonstrate that transgene activation by fasting
could be mimicked by glucagon and dibutyryl cAMP, but we have so far failed
to delineate a cyclic AMP response element in the enhancer.
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
7
Experimental Procedures
Plasmid constructions.
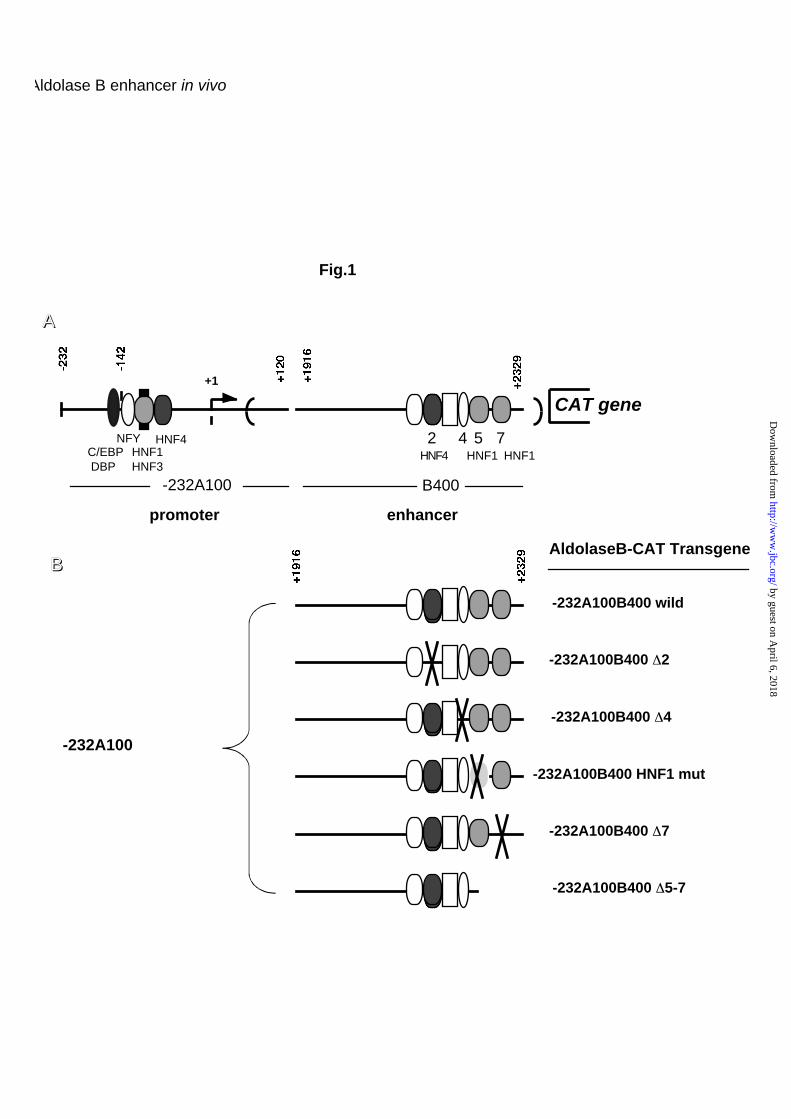
Aldolase B-CAT constructs used have been previously described (9). Fig. 1A
presents a diagram of the aldolase B-CAT gene, indicating the main cis-acting
elements and cognate DNA binding proteins. The enhancer fragment (+1916
to 2329) was maintained in an intronic position, the splice sites are indicated
by the bold brackets. Fig. 1B shows the microinjected constructs indicating the
locations of the block mutations or deletions performed in the aldolase B
enhancer. Fig. 3 schematizes various constructs with deletions or mutations in
the promoter. Here, the enhancer fragment was subcloned in the Cla I site
located downstream of the CAT gene (8).
Generation and analysis of transgenic mice.
The DNA constructs were digested with restriction enzymes Cla I (cutting in 3’
in the vector) and Hind III (cutting in 5’ in the plasmid linker). The fragments of
interest were isolated by electrophoresis, electroeluted and purified by using
elutip-d columns (Schleicher and Schuell), then microinjected into fertilized
mouse eggs according to Gordon and Ruddle, (10). The progeny was
analyzed for the presence of the transgene by Southern blot.
Cell culture and transient transfection.
HepG2 cells were grown in Dubellco’s modified Eagle medium (DMEM) in the
presence of 10% (v/v) fetal calf serum, 1 µM L-triiodothyronine, 1 µM
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
8
dexamethasone, and 10 nM insulin, at 37° in 5% (v/v) CO2. Transfections
were carried out by the calcium phosphate method (11), under experimental
conditions previously described (9). In each experiment 7.5 µg of the CAT
plasmids and 2 µg of the luciferase plasmid were cotransfected. The pRSV
luciferase standardization plasmid was used to monitor variations in
transfection efficacy. The chloramphenicol acetyltransferase (CAT) assay (12)
and luciferase assay (13) were performed as described (8).
Hepatocytes in primary culture
Hepatocytes were isolated from the livers of adult transgenic mice carrying the
-232A100B400 wild-type transgene by perfusion with collagenase (14). Viable
hepatocytes were separated from other cells using isodensity Percoll
centrifugation (15). Hepatocytes (>95% viability) were seeded at a density of
3.5 x106 cells per dish (78 cm2). After cell attachment (4 h), the medium was
replaced by M199 medium with Earle’s salts (GIBCO/BRL) containing 20 mM
glucose, supplemented with 1 µM L-triiodothyronine, 1 µM dexamethasone,
10 nM insulin, at 37° in 5% (v/v) C02 for 3 days. The culture medium was
changed every 24 h. Cells were then cultured in the same media either with or
without 3.3 mg/L glucagon and 3.3 mg/L theophylline.
Dietary and hormonal control
Adult mice heterozygous for the transgene were used for analysis. One group
of mice was fasted 24 h prior to sacrifice; the other group was fasted 24 h
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
9
then refed a high carbohydrate diet ad libitum for 18 h prior to sacrifice.
Organs were stored at - 80°C until use. To determine the effect of cyclic AMP
(cAMP) or glucagon, mice were either starved for 18 h or fed a high
carbohydrate diet ad libitum for 18 h, then injected intraperitoneally every hour
with either dibutyryl cyclic AMP (Bt2cAMP) (30 mg/kg of body weight), or
glucagon (0.5 mg/kg of body weight) with theophylline (30 mg/kg of body
weight), sacrificed after 2 or 4 h of treatment, then tissues were removed and
kept at - 80°C until use.
RNA isolation and Northern blot.
The mRNA extraction from liver, kidney and cultured hepatocytes was
performed using the RNAzol B reagent (Bioprobe systems) according to the
manufacturer’s instructions. Twenty µg of total RNA was electrophoresed
through a 1.5% (w/v) denaturing formaldehyde agarose gel and transferred to
Hybond N+ (Amersham). The cDNA probes were labeled by random priming
using a labeling kit, according to the manufacturer’s instructions. The CAT
cDNA probe was a Cla1-EcorR1 fragment of the PeCAT vector (5). The
murine aldolase B probe was a 379-bp fragment of cDNA, and the
phosphoenolpyruvate kinase (PEPCK) probe was a 1305-bp fragment of
cDNA (gift of Dr. B Antoine (16)).
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
10
Results
In vivo role of the different enhancer elements.
Full and tissue-specific expression of the aldolase B transgenes results from a
cooperation between a proximal promoter and a distal intronic enhancer (5).
The enhancer activity is provided by a 400-bp sequence (+1916 to +2329)
containing binding sites for known liver-enriched nuclear factors HNF1 (+2212
to +2246 and +2275 to +2304), HNF4 (+2146 to +2184) and unknown factor
(+2195 to +2220). We analyzed the contribution of each of the cis-acting
elements to the expression of the aldolase B gene in vivo. For this purpose,
chimeric genes that contained the wild-type aldolase B promoter (-232A100)
linked to either wild-type or mutant aldolase B enhancer in its normal intronic
position, were ligated to the CAT gene (Fig. 1). Figure 2 shows the results of
the expression of the CAT transgenes directed by aldolase B gene regulatory
sequences in the liver and kidney. Expression of the aldolase B-CAT
transgenes was strictly restricted to the liver, kidney and small intestine (not
shown), no ectopic expression was detected in other tissues tested, e.g. brain
and lung (not shown). In agreement with prior data (5), the level of transgene
expression was totally independent of the copy number and strongly
dependent on the integration site. Among the six lines harboring the wild-type
aldolase B-CAT transgene, one was very weakly expressed. The HNF1-mut
transgene carries a 5 bp-block mutation of the 5’ HNF1 binding site (enhancer
element 5). In five lines with this construct, transgene expression was
conserved in the liver (although weak in two of the line) and was very low in
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
11
the kidney, (a decrease on average of 100 fold compared to the wild-type
construct). Among 16 lines harboring a transgene with deletion of enhancer
element 7, i.e. the 3’ HNF1 binding site, only one expressed CAT activity in
the liver and, very weakly, in the kidney (Fig. 2). None of the 9 lines harboring
the transgene with deletion of both HNF1 binding sites (construct ∆ 5 to 8)
expressed CAT activity either in the liver or in the kidney (data not shown).
Twelve lines harboring a ∆2 transgene with a deletion of the HNF4 binding
site (enhancer element 2) were generated. In 5 lines, transgene expression
was undetectable; in one it was weak and in the other 6 it was strong. Finally,
expression of a transgene with deletion of enhancer element 4, which binds a
liver-enriched but non-characterized factor (9), was much higher than that of
the wild-type transgene and had a high CAT activity in both liver and kidney in
4 lines and a lower activity in 1 line (Fig. 2).
Hypothetical role of C/EBP in the differential effect of HNF1 site mutations in
the liver and kidney.
The differential effect of the 5’ HNF1 binding site deletion from the enhancer
on transcriptional activity in different cells and tissues could reflect tissue-
specific differences in transcription factor content. We focused on transcription
factors previously demonstrated to be active on aldolase B gene expression.
The C/EBP factor was a good candidate since, its concentration is high in the
liver, low in HepG2 cells and almost zero in the kidney (17). In addition,
C/EBP like HNF1 are potent transactivators of the aldolase B promoter (7). If
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
12
our hypothesis is correct, over-expression of C/EBP in HepG2 cells should
restore transcriptional activity of constructs with mutation or deletion of one of
the enhancer HNF1 binding sites. Indeed, when HepG2 cells were transiently
cotransfected with the wild-type aldolase B-CAT plasmid and a C/EBP
expression vector, CAT activity was stimulated only 5 fold. In contrast, under
the same conditions, the HNF1-mut and ∆7 constructs were stimulated about
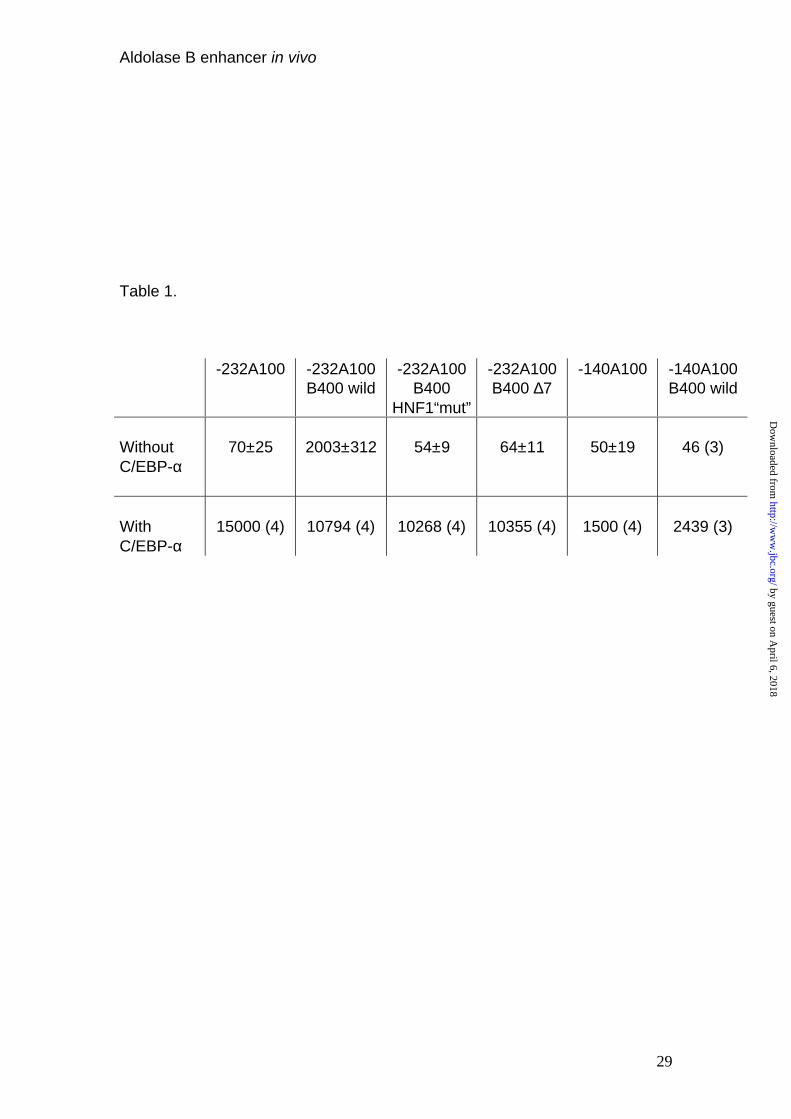
200 fold (Table 1). Next, we wanted to ascertain whether the C/EBP binding
site (-170 to -140) in the aldolase B promoter mediated the observed C/EBP
effect. In fact, a construct devoid of the enhancer sequence and with deletion
of the C/EBP binding site in the promoter (-140A100) responded 10-fold less
to C/EBP than the –232A100 promoter (Table 1). We think that the residual
transactivation was due to promiscuous binding properties of C/EBP, able, at
high concentration, to bind to degenerated, low-affinity sites. Finally, in the
absence of the C/EBP element in the promoter (140A100B400wild) the
enhancer activity was abolished.
These results point to a specific role of C/EBP in the cooperation between the
promoter and the intronic enhancer of the aldolase B gene. In order to
determine whether C/EBP bound to promoter must specifically interact with
other contiguous factors to cooperate with the enhancer, we studied the CAT
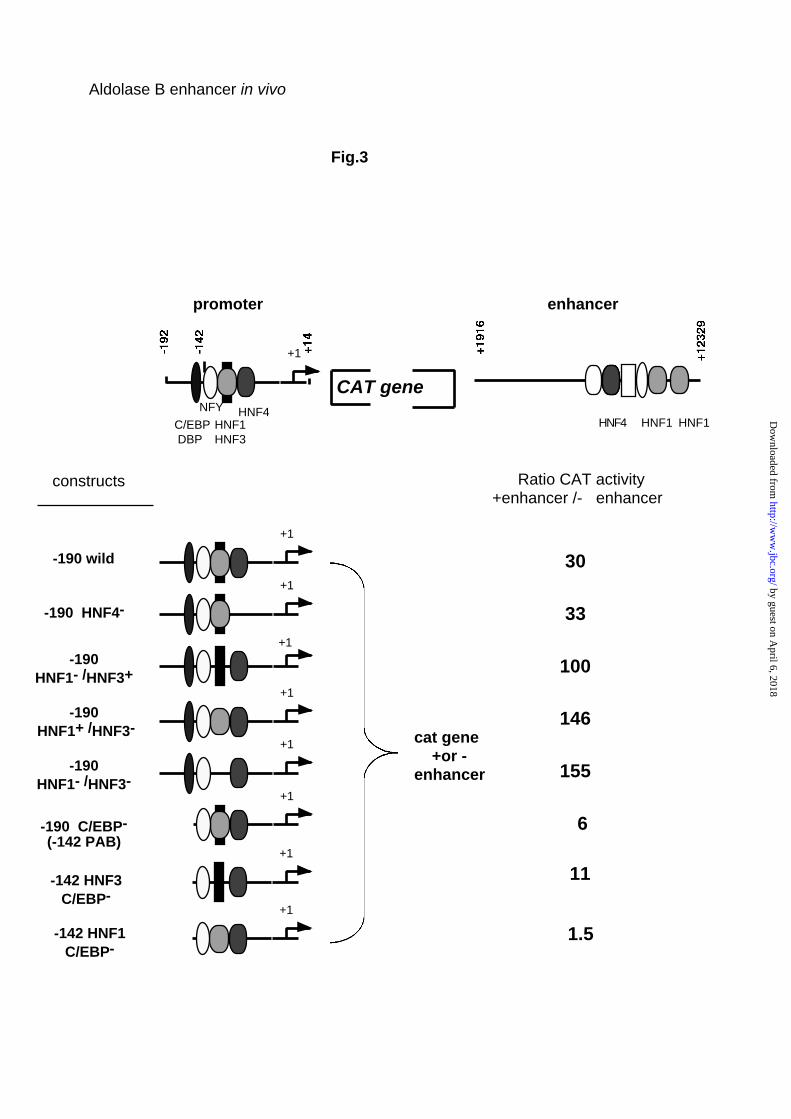
activity of a series of new constructs transfected into HepG2 cells (Fig. 3).
These constructs included from 5’ to 3’ the promoter fragment, the CAT gene
and the enhancer. Fig. 3 shows that the enhancer stimulated the activity of the
190 bp wild-type promoter 30 fold.
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
13
Deletion of the HNF4 binding site from the promoter did not change this
stimulation. Deletion or mutation of the PAB element suppressing binding of
either HNF1 or HNF3, or both HNF1 and HNF3 seemed rather to increase the
stimulatory effect of the enhancer. Finally, as expected, all mutants devoid of
the promoter C/EBP binding site were weakly sensitive to the enhancer
action. When the PAB element was replaced by a high affinity HNF1 the
enhancer lost practically all stimulatory activity, most likely because the
C/EBP/HNF1 interaction between promoter and enhancer is replaced by the
same type of interaction inside the promoter.
Dietary regulation of the transgenes.
Northern blot analysis of transgene expression as a function of diet was
performed on at least two different mouse lines for each transgene.
Feeding mice a high carbohydrate diet stimulates aldolase B gene
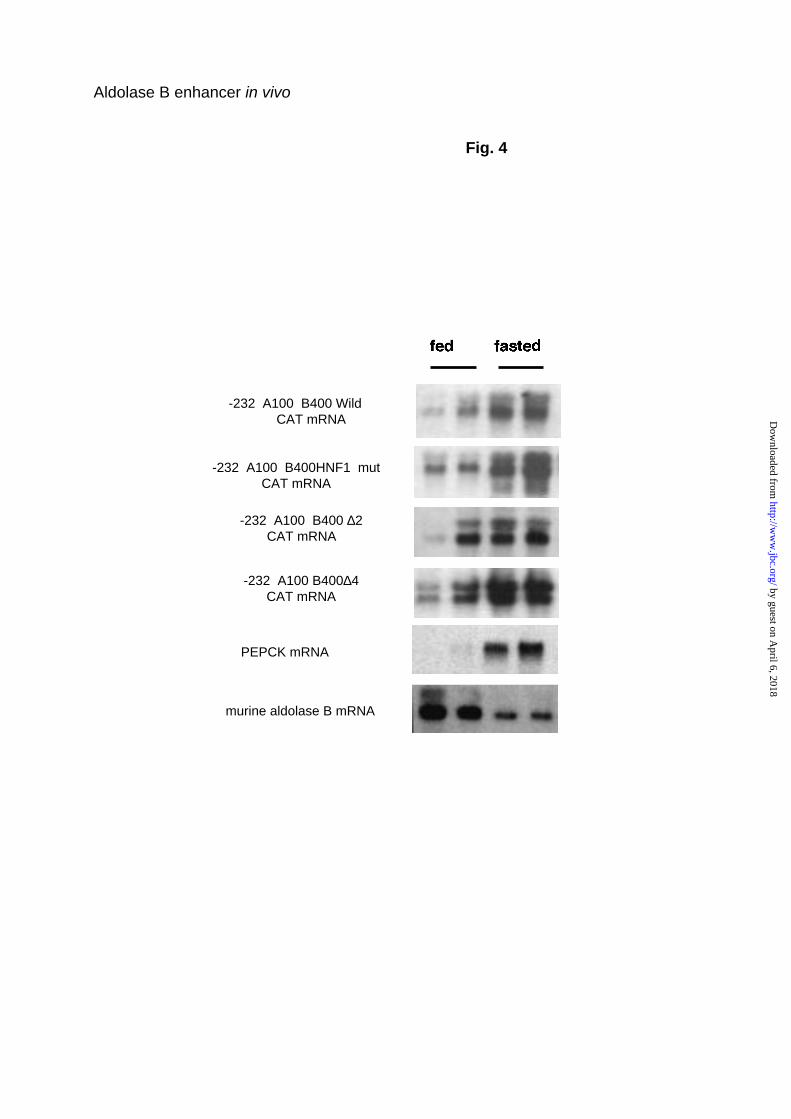
transcription in liver, while starvation decreases its expression. ((2) and Fig.4).
Prior transgenic analysis indicated that a transgene with 1600 bp of the 5’
flanking sequence and the whole 1st intron responds to diet in an opposite
manner (5). Northern blots in Fig.4 demonstrate that exactly the same
expression pattern was reproduced using the “wild-type” construct with only
232 bp of the promoter and the 400 bp intronic enhancer. HNF1 mut, ∆2 and
∆4 transgenes were also induced in mice fasted for 24 h and inhibited in mice
refed a high carbohydrate diet. Therefore, the investigated enhancer
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
14
elements do not seem to be implicated in the long-term dietary regulation of
the aldolase B gene in the liver.
The dietary response of the aldolase B transgenes resembles that of
neoglucogenic genes such as the PEPCK gene. Since such genes are known
to be responsive to glucagon and cAMP, we tested the effects of the hormone
and of cAMP analogues on the expression of the –232A100B400wild-CAT
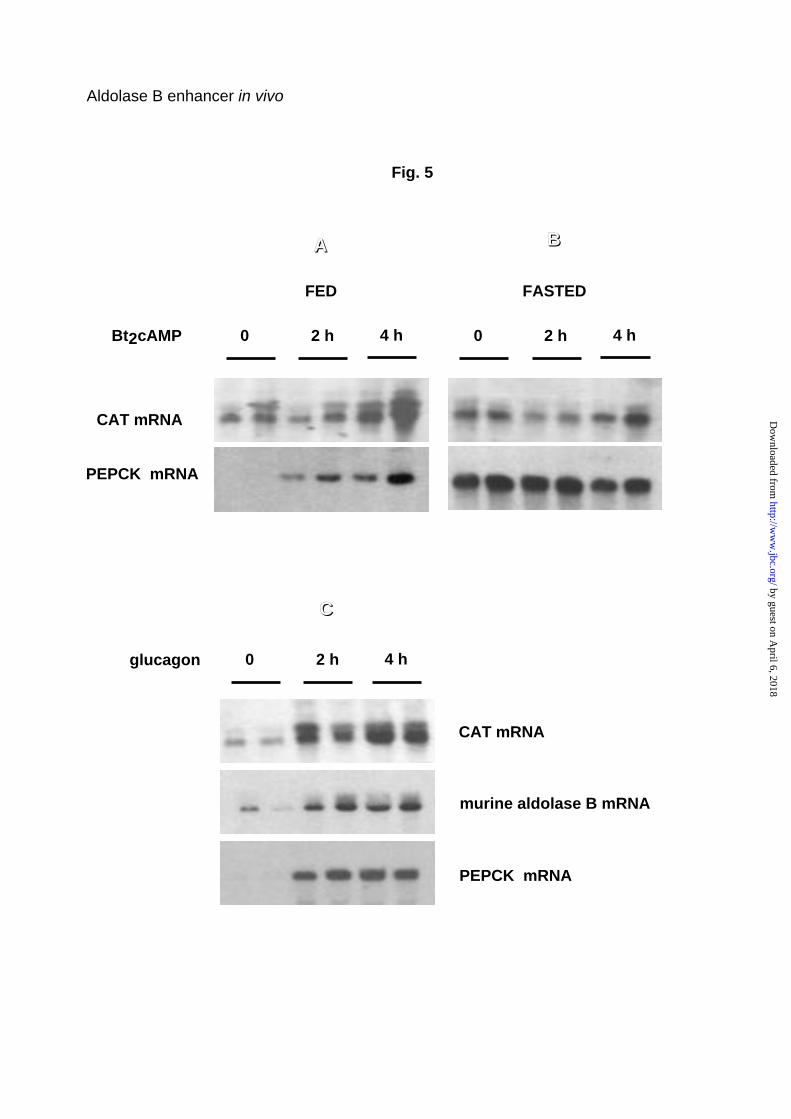
transgene. Figure 5A shows that Bt2 cAMP induced parallel accumulation of
PEPCK and CAT transgenic mRNA 2 and 4 h after injection into
carbohydrate-fed mice. In fasted mice, these messengers were already
abundant before Bt2cAMP treatment, consequently the Bt2cAMP effect was
weak or nil (Fig. 5B).
As expected, the effect of Bt2cAMP in stimulating the transgene was
mimicked by glucagon (Fig. 5C). The response to glucagon was conserved for
the ∆2 and HNF1-mut transgenes, characterized by a deleted HNF4 binding
site and a mutated HNF1 binding site, respectively (not shown). Although the
endogenous aldolase B gene has been described rather as a typical
“glycolytic gene”, up-regulated by carbohydrates and down-regulated by
fasting and glucagon (2), the results observed with the aldolase B-CAT
transgenes prompted us to reevaluate more precisely the response of the
endogenous gene to glucagon. Fig. 5C shows that, in fact, endogenous
aldolase B mRNA accumulated 2 and 4 h after glucagon administration to
carbohydrate-fed animals, in parallel with accumulation of the PEPCK mRNA.
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
15
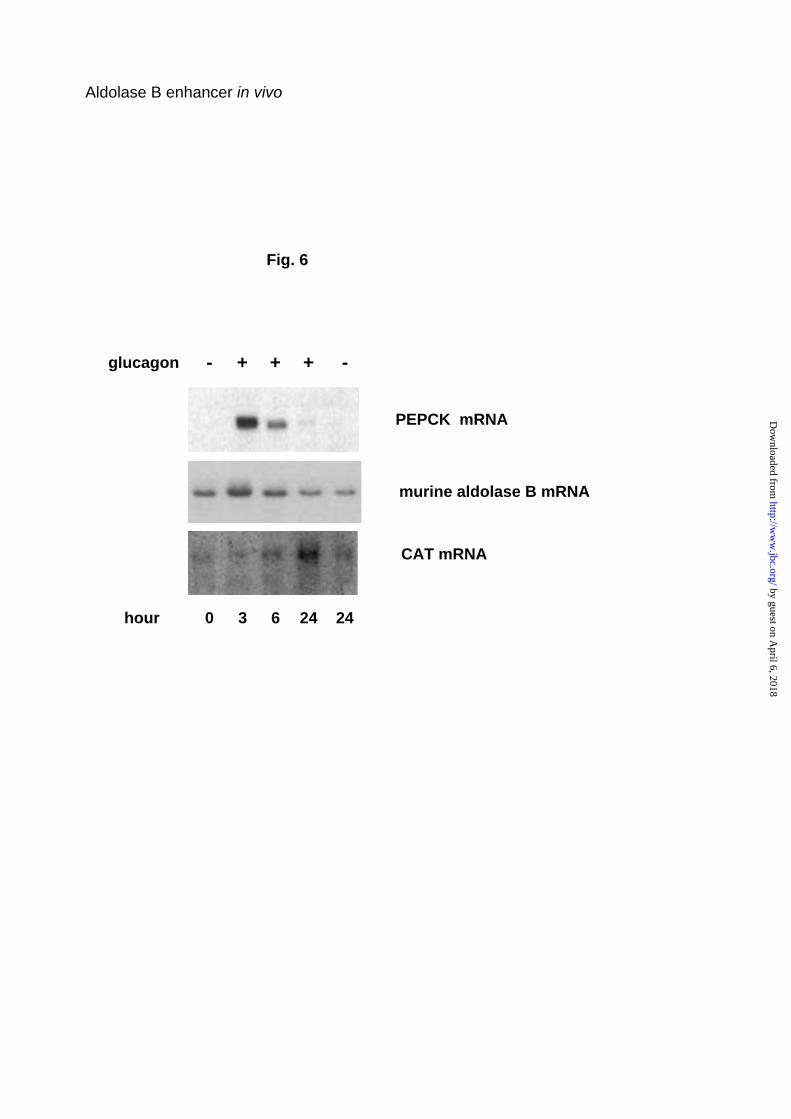
In primary hepatocytes isolated from the liver of mice bearing the
-232A100B400wild-CAT transgene and cultured for 3 days in the presence of
25 mM glucose and 20 nM insulin, addition of glucagon led first to aldolase B
mRNA accumulation, with a peak at 3 h and thereafter a decline. Aldolase B,
and PEPCK mRNA inductions were parallel. In contrast, the response of the
aldolase B-CAT transgene was delayed, with a peak at 24 h (Fig. 6).
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
16
Discussion
Tissue-specific transcription of the aldolase B gene is governed by a short
200-bp promoter stimulated by a 400-bp intronic enhancer (9), (5). In previous
studies using transient transfection assays, both the promoter and the
enhancer were shown to be cell-specific (18), (9). The nuclear factors mainly
involved in liver-specific gene transcription include HNF1 (19), HNF3 (20),
HNF4 (21), HNF6 (22), C/EBP (23) and DBP (24). Both the aldolase B gene
promoter and the enhancer fragments contain HNF1 and HNF4 binding sites;
in addition, a binding site for C/EBP is present in the promoter. Therefore the
aldolase B gene was an attractive model for identifying, in vivo, the promoter
and enhancer elements necessary for tissue-specific, dietary-regulated gene
expression.
In the present study we have shown that mutation of the 5’ HNF1 binding site
in the enhancer, preventing binding of HNF1, reduced expression of the
transgene (-232A100B400HNF1mut) in the kidney but did not alter its
transcription in the liver. A similar role for HNF1 in the differential control of
PEPCK gene transcription in the liver and kidney has been reported (25). In
contrast, the aldolase B gene was normally transcribed in the liver (26) and in
the kidney of HNF1 knock-out mice (Pontoglio & Yaniv, personal
communication). However, these results are not inconsistent with our own
data since the two isoforms HNF1α and HNF1β, binding to the same element,
are coexpressed in the liver and the kidney, HNF1β being increased by a
compensatory mechanism in HNF1α -/- (26).
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
17
Deletion of the 2nd HNF1 site in the enhancer (element 7) led to an inactive
transgene in 11 out of the 12 lines tested. In the only line expressing the –232
A100B400∆7 transgene, expression was weak, but easily detectable in the
liver and almost nil in the kidney, which confirms that here again abrogation of
the HNF1 binding site has a more severe effect in the kidney than in the liver.
Since the mutation in the 1st HNF1 site changed 5 bp while deletion of the 2nd
site excised 47 bp from the transgene, we cannot deduce from our results that
the two HNF1 binding sites in the enhancer play different roles. Indeed, it
could be that the 47-bp sequence binds factors other than HNF1 or destroys a
binding site 5’ to the deletion, although we did not detect such a site either by
in vivo footprinting (9), or by gel shift analysis (not shown).
A transgene deleted from the HNF4 site was silent in both livers and kidneys
in half of the mouse lines. It is possible that the HNF4 mutation affects the
recruitment of chromatin-remodeling factors (27), (28). Accordingly, the –
232A100B400∆2 transgene could remain silent when inserted into a closed
chromatin region, and would be active when integrated into a permissive,
open chromatin region. Although both HNF1 (29), (30) and HNF4 binding sites
(31), (32) have been reported to play a role in the response of some genes to
cAMP, the corresponding elements present in the enhancer do not seem to be
involved in the dietary and hormonal regulation of the aldolase B gene.
Indeed, the response of our mutated transgenes to fasting and cAMP was
conserved.
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
18
To explain the differential effect of enhancer HNF1 binding site mutations
according to the cells and tissues, we hypothesized that HNF1 is involved in a
cooperative interaction of the enhancer with another DNA element able to
bind a factor differentially abundant in the liver, hepatoma cells and kidneys.
C/EBPα, present and abundant in adult hepatocytes, at low concentration in
hepatoma cells, and essentially absent from kidney (33) is a good candidate.
Mouse models also suggest a major role for C/EBPα in liver-specific
expression of aldolase B. The liver of albino lethal mice shows reduce
aldolase B and C/EBP levels (34), whereas other transcription factors are
normally expressed. In contrast, aldolase B is normally expressed in the
kidney of these animals. Aldolase B gene expression is also depressed in the
liver of C/EBP α-/- mice (35). A C/EBP binding site (-170 to –140) has been
characterized in the aldolase B promoter (6), and both C/EBP and HNF1 have
been shown to be potent transactivators of the promoter (7). To confirm that
the different levels of C/EBP in different tissues could account for the tissue-
specific cis-effects of the enhancer HNF1 binding sites, we studied by
transient transfection in HepG2 cells the inducibility by C/EBPα of aldolase
B–CAT constructs carrying either the wild-type enhancer or enhancers devoid
of one of the two HNF1 binding sites. The constructs with mutations of the
HNF1 binding site(s) in the enhancer were highly responsive to
overexpression of C/EBP (100-fold induction) compared to the 5-fold induction
of the construct with an intact enhancer. Furthermore, deletion of the C/EBP
site in the promoter impaired the enhancer function, whereas mutations or
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
19
deletions of the other known sites (binding HNF1, HNF3 or HNF4), alone or in
combination, did not affect the enhancer function. These findings suggest that
liver-specific expression of the aldolase B gene requires cooperation between
the C/EBP site in the promoter and at least one HNF1 site in the enhancer. In
contrast, expression in the kidney (lacking C/EBP) requires the presence of
the two enhancer HNF1 binding sites. Cooperation between HNF1 and C/EBP
in the control of gene expression in the liver has already been reported for the
PEPCK gene (25), the phenylalanine hydroxylase gene (36), the
apolipoprotein B gene (37) and the albumin gene (38).
Our data indicate that neither HNF1 nor HNF4 binding sites mediate the
aldolase B gene regulation by fasting and cAMP. However C/EBP was a
possible alternative candidate since its involvement in the response to cAMP
has been established for several genes, in particular the PEPCK gene (39),
(40). Unfortunately, although the wild-type aldolase B-CAT construct was
perfectly responsive to glucagon in hepatocytes isolated from transgenic mice,
the same construct transiently transfected into HepG2 cells was insensitive to
cAMP. This insensitivity could not be reversed by cotransfection with
expression vectors C/EBPα, and (or) the PKA catalytic subunit. Consequently,
further experiments will be needed to determine whether C/EBP and C/EBP-
binding sites are involved in the regulation of the aldolase B gene by fasting,
glucagon and cAMP.
The observed early stimulation of both transgenes and the endogenous
aldolase B gene by glucagon in hepatocytes in primary culture prompted us to
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
20
reevaluate our previous presentation of this gene as being negatively
regulated by glucagon and cAMP (2). In fact, our former results were taken
from mRNA measurements performed 12 to 24 h after glucagon
administration. We now find that the early positive response to glucagon,
maximal at the 3rd h, is followed by a decline that reached undetectable levels
at the 24th h. Accordingly, the transcription rate of the aldolase B gene
measured by run-on-assay showed first a transient increase in transcription
10 min after glucagon injection, followed by transcriptional inhibition (41).
Hormonal regulation of the aldolase B gene reflects its dual role in both
glycolysis and gluconeogenesis. Glucagon and cAMP are involved in
hypoglycemic and stress conditions in which a rapid increase in glucose
production is essential. This requires, in particular, a transient increase in
aldolase B gene transcription, leading to an increase in mRNA and enzyme
abundance. However, this gene must also be stimulated to allow for a proper
adaptation to a shift from a carbohydrate-poor to a carbohydrate-rich diet. As
previously discussed, the regulatory domains responsible for the response to
cAMP (and fasting) are retained in the –232A100B400CAT transgene, while
the region required for the positive response to carbohydrates is absent from
this construct.
In conclusion, we report in this paper the complex, tissue-specific functional
interplay between the promoter and intronic enhancer of the aldolase B gene,
which can be considered to be both a glycolytic and gluconeogenic gene.
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
21
References
1. Weber, A., Marie, J., Cottreau, D., Simon, M. P., Besmond, C., Dreyfus,
J. C., and Kahn, A. (1984) J Biol Chem 259, 1798-802
2. Munnich, A., Besmond, C., Darquy, S., Reach, G., Vaulont, S., Dreyfus,
J. C., and Kahn, A. (1985) J Clin Invest 75, 1045-52
3. Ali, M., Rellos, P., and Cox, T. M. (1998) J Med Genet 35, 353-65
4. Cox, T. M. (1994) Faseb J 8, 62-71
5. Sabourin, J. C., Kern, A. S., Gregori, C., Porteu, A., Cywiner, C.,
Chatelet, F. P., Kahn, A., and Pichard, A. L. (1996) J Biol Chem 271,
3469-73
6. Raymondjean, M., Pichard, A. L., Gregori, C., Ginot, F., and Kahn, A.
(1991) Nucleic Acids Res 19, 6145-53
7. Gregori, C., Kahn, A., and Pichard, A. L. (1993) Nucleic Acids Res 21,
897-903
8. Gregori, C., Kahn, A., and Pichard, A. L. (1994) Nucleic Acids Res 22,
1242-6
9. Gregori, C., Porteu, A., Lopez, S., Kahn, A., and Pichard, A. L. (1998) J
Biol Chem 273, 25237-43
10. Gordon, J. W., and Ruddle, F.H. (1983) Methods Enzymol. 101, 411-
433
11. Graham, F. L., and Eb, A. J. v. d. (1973) Virology 52, 456-67
12. Gorman, C. M., Moffat, L. F., and Howard, B. H. (1982) Mol Cell Biol 2,
1044-51
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
22
13. de Wet, J. R., Wood, K. V., DeLuca, M., Helinski, D. R., and
Subramani, S. (1987) Mol Cell Biol 7, 725-37
14. Mitchell, C., Mignon, A., Guidotti, J. E., Besnard, S., Fabre, M.,
Duverger, N., Parlier, D., Tedgui, A., Kahn, A., and Gilgenkrantz, H.
(2000) Hum Mol Genet 9, 1597-602.
15. Kreamer, B. L., Staecker, J. L., Sawada, N., Sattler, G. L., Hsia, M. T.,
and Pitot, H. C. (1986) In Vitro Cell Dev Biol 22, 201-11.
16. Vallet, V., Bens, M., Antoine, B., Levrat, F., Miquerol, L., Kahn, A., and
Vandewalle, A. (1995) Exp Cell Res 216, 363-70.
17. Birkenmeier, E. H., Gwynn, B., Howard, S., Jerry, J., Gordon, J. I.,
Landschulz, W. H., and McKnight, S. L. (1989) Genes Dev 3, 1146-56
18. Gregori, C., Ginot, F., Decaux, J. F., Weber, A., Berbar, T., Kahn, A.,
and Pichard, A. L. (1991) Biochem Biophys Res Commun 176, 722-9
19. Courtois, G., Baumhueter, S., and Crabtree, G. (1988) Proc. Natl.
Acad. Sci. USA 85, 7937-7941
20. Costa, R. H., Grayson, D. R., and Darnell, J. E., Jr. (1989) Mol Cell Biol
9, 1415-25
21. Sladek, F. M., Zhong, W. M., Lai, E., and Darnell, J. E. (1990) Genes
Dev. 4, 2353-2365
22. Lemaigre, F. P., Durviaux, S.M., Truong, O., Lannoy, V.J., Hsuan, J.J.,
and Rousseau, G.G. (1996) Proc. Natl. Acad. Sci. USA 93, 9460-9464
23. Landshultz, W. H., Johnson, P. F., Adashi, E. Y., Graaves, B.J., and
McKighnt, S. L. (1988) Genes Dev. 2, 786-800
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
23
24. Lichtsteiner, S., and Schibler, U. (1989) Cell 57, 1179-87
25. Patel, Y. M., Yun, J. S., Liu, J., McGrane, M. M., and Hanson, R. W.
(1994) J Biol Chem 269, 5619-28
26. Pontoglio, M., Barra, J., Hadchouel, M., Doyen, A., Kress, C., Bach, J.
P., Babinet, C., and Yaniv, M. (1996) Cell 84, 575-85
27. Vignali, M., Hassan, A. H., Neely, K. E., and Workman, J. L. (2000) Mol
Cell Biol 20, 1899-910.
28. Zhong, H., Voll, R. E., and Ghosh, S. (1998) Mol Cell 1, 661-71.
29. Soubt, M. K., Marksitzer, R., Menoud, P. A., and Nagamine, Y. (1998)
Mol Cell Biol 18, 4698-706
30. Streeper, R. S., Svitek, C. A., Goldman, J. K., and O'Brien, R. M.
(2000) J Biol Chem 275, 12108-18
31. You, M., Fischer, M., Cho, W. K., and Crabb, D. (2002) Arch Biochem
Biophys 398, 79-86.
32. Angrand, P. O., Coffinier, C., and Weiss, M. C. (1994) Cell Growth
Differ 5, 957-66.
33. Xanthopoulos, K. G., Prezioso, V. R., Chen, W. S., Sladek, F. M.,
Cortese, R., and Darnell, J. E., Jr. (1991) Proc Natl Acad Sci U S A 88,
3807-11
34. Ruppert, S., Boshart, M., Bosch, F. X., Schmid, W., Fournier, R. E., and
Schutz, G. (1990) Cell 61, 895-904
35. Flodby, P., Barlow, C., Kylefjord, H., Ahrlund-Richter, L., and
Xanthopoulos, K. G. (1996) J Biol Chem 271, 24753-60
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
24
36. Faust, D. M., Catherin, A. M., Barbaux, S., Belkadi, L., Imaizumi-
Scherrer, T., and Weiss, M. C. (1996) Mol Cell Biol 16, 3125-37
37. Brooks, A. R., and Levy-Wilson, B. (1992) Mol Cell Biol 12(3), 1134-48
38. Wu, K. J., Wilson, D. R., Shih, C., and Darlington, G. J. (1994) J Biol
Chem 269, 1177-82.
39. Crosson, S. M., and Roesler, W. J. (2000) J Biol Chem 275, 5804-9.
40. Roesler, W. J. (2001) Annu Rev Nutr 21, 141-65
41. Vaulont, S., Munnich, A., Marie, J., Reach, G., Pichard, A. L., Simon,
M. P., Besmond, C., Barbry, P., and Kahn, A. (1984) Biochem Biophys
Res Commun 125, 135-41.
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
25
Footnotes
The abbreviations used are: bp, base pair(s); kb, kilobase pair(s); CAT,
chloramphenicol acetyltransferase; HNF, hepatic nuclear factor; C/EBP,
CCAAT/enhancer binding protein;
PEPCK, phosphoenolpyruvate carboxykinase,
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
26
Legends
Table 1. Effect of C/EBP- overexpression on activity of aldolase B CAT
vectors containing an impaired HNF1 binding site in the enhancer. Hep G2
cells were transfected with 7.5 µg of aldolase B CAT vectors, 2.5 µg of
luciferase plasmid and, when indicated, 2.5 µg of expression vector for
C/EBP-α. CAT activity was normalized according to transfection efficiency by
measuring the luciferase activity. When more than four independent
experiments were performed, results are given as means ± SE; otherwise, the
means are given, with the number of experiments in parentheses.
Fig. 1. Diagram of the aldolase B-CAT transgenes. (A) Map of the non-
mutated transgene (wild) with the location of the regulatory elements and
transcription factors known to bind to these sites. (B) Map of the various
transgenes with precise indication of the deleted or mutated elements.
Fig. 2. Level of expression of aldolase B CAT transgenes in the liver and
kidney of transgenic mice. CAT activity was assayed as described in liver
(black) and kidney (grey) homogenates from chow-fed animals. Each value
represents the mean of independent assays on different mice (at least 3). The
CAT activity in the kidney of –232A100B400HNF1mut transgenic mice is
shown on a large scale in the inset.
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
27
Fig. 3. Analysis of the element in the promoter required for enhancer function.
At the top is an illustration of the wild construct with the location of the
regulatory elements and transcription factors known to bind to these sites. 7.5
µg of the indicated constructs mutated in the promoter linked or not to the
enhancer fragment were transfected into HepG2 cells with 2.5 µg of the
reference plasmid RSV luciferase. CAT activity was normalized with respect
to the transfection efficiency by measuring luciferase activity. Results give the
ratio of the CAT activity of the construct with and without the enhancer
fragment.
Fig. 4. Northern blot analysis of aldolase B and CAT transcripts in livers of
transgenic mice under different nutritional conditions. 20 µg of total liver
mRNAs were electrophoresed in formaldehyde-agarose gel, transferred onto
a Hybond N+ membrane and hybridized with the indicated probes. Animals
were fasted 24 h (fasted state) then fed a 75% carbohydrate diet for 18 h (fed
state). The northern blots are representative of two different experiments.
Fig. 5. Effects of cAMP and glucagon on the level of expression of the
-232A100B400 wild-CAT transgene and the endogenous aldolase B gene in
the liver. Mice were fed a 75% carbohydrate diet for 18 h (panel A and C) or
fasted for 24 h (panel B) prior to intraperitoneal injection of either Bt2cAMP or
glucagon as indicated under Experimental Procedures. Total liver RNA were
prepared at the indicated time after the first injection and northern blots were
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Aldolase B enhancer in vivo
28
analyzed as described in Fig. 4 legend. PEPCK served as a positive control of
the hormonal status.
Fig. 6. Effect of glucagon on aldolase B gene expression in primary cultured
hepatocytes. Hepatocytes isolated from –232A100B400 wild-CAT transgenic
mice were cultured for 3 days without glucagon in medium defined under
Experimental Procedures, then incubated 3 to 24 h in the presence or
absence of glucagon and theophylline. Total RNAs were extracted and
analyzed for the level of transgenic CAT mRNA, endogenous aldolase B and
PEPCK mRNAs. The northern blots are representative of two culture
experiments with two different lines of mice.
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from
Aldolase B enhancer in vivo
29
Table 1.
-232A100 -232A100B400 wild
-232A100B400
HNF1“mut”
-232A100B400 ∆7
-140A100 -140A100B400 wild
WithoutC/EBP-α
70±25 2003±312 54±9 64±11 50±19 46 (3)
WithC/EBP-α
15000 (4) 10794 (4) 10268 (4) 10355 (4) 1500 (4) 2439 (3)
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from
BB
AA
+1
HNF4C/EBP DBP
HNF1HNF3
NFY
promoter enhancer
-232A100
CAT gene
B400
2 4 5 7HNF4 HNF1 HNF1
AldolaseB-CAT Transgene
-232A100B400 wild
-232A100B400 2
-232A100B400 4
-232A100B400 7
-232A100B400 5-7
-232A100B400 HNF1 mut
-232A100
Fig.1
Aldolase B enhancer in vivo
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from
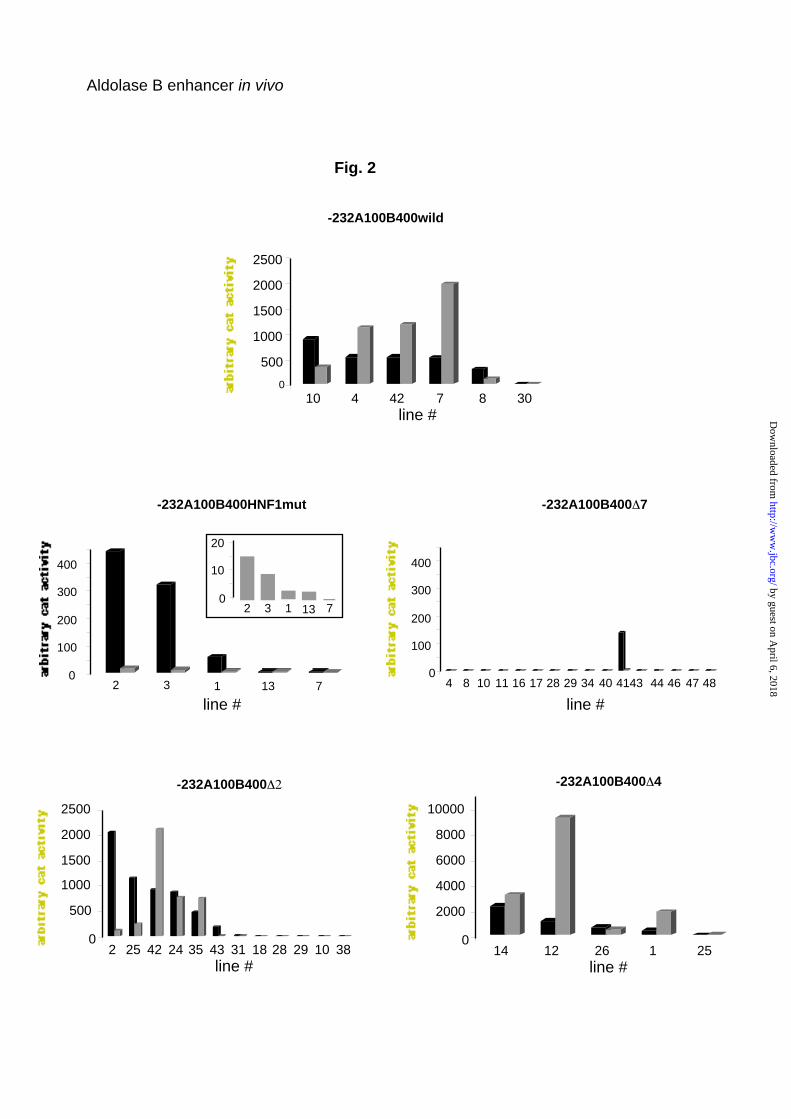
Fig. 2
-232A100B400wild
0
500
1000
1500
2000
2500
10 4 42 7 8 30line #
-232A100B400 7
4 8 10 11 16 17 28 29 34 40 41 44 46 47 480
100
200
300
400
43
line #
line #
0
2000
4000
6000
8000
10000
14 12 26 1 25
-232A100B400 4-232A100B400
0
500
1000
1500
2000
2500
2 25 42 24 35 43 31 18 28 29 10 38line #
Aldolase B enhancer in vivo
-232A100B400HNF1mut
line #2 3 1 13 7
0
100
200
300
400
2 3 1 13 70
10
20
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from
Ratio CAT activity+enhancer /- enhancer
30
33
100
146
155
6
+1
HNF4C/EBP DBP
HNF1HNF3
NFY
CAT gene
HNF4 HNF1 HNF1
promoter enhancer
+1
+1
+1
+1
+1
+1
-190 wild
-190 HNF4-
-190HNF1- /HNF3+
-190 HNF1+ /HNF3-
-190HNF1- /HNF3-
-190 C/EBP-(-142 PAB)
constructs
cat gene +or -enhancer
+1
+1
-142 HNF3C/EBP-
-142 HNF1C/EBP-
11
1.5
Fig.3
Aldolase B enhancer in vivo
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from
-232 A100 B400HNF1 mutCAT mRNA
-232 A100 B400 WildCAT mRNA
-232 A100 B400 ∆2CAT mRNA
-232 A100 B400∆4CAT mRNA
PEPCK mRNA
murine aldolase B mRNA
Fig. 4
Aldolase B enhancer in vivo
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from
glucagon
CAT mRNA
murine aldolase B mRNA
PEPCK mRNA
CC
2 h0 4 h
Bt2cAMP
FED FASTED
CAT mRNA
PEPCK mRNA
AA BB
2 h0 4 h 2 h0 4 h
Fig. 5
Aldolase B enhancer in vivo
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from
CAT mRNA
murine aldolase B mRNA
PEPCK mRNA
hour 0 3 6 24 24
glucagon - + + + -
Fig. 6
Aldolase B enhancer in vivo
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Claudine Gregori, Arlette Porteu, Claudia Mitchell, Axel Kahn and Anne-Lise PichardIn vivo functional characterization of the aldolase B gene enhancer
published online May 28, 2002J. Biol. Chem.
10.1074/jbc.M204047200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on April 6, 2018
http://ww
w.jbc.org/
Dow
nloaded from