advances in biochemistry - rcin.org.plrcin.org.pl/content/32231/wa488_24011_p939_t42-z3-pb.pdf ·...
TRANSCRIPT

POLSKIE I I TOWARZYSTWO BIOCHEMICZNE
Advances in BiochemistryTOM 42, NR 3,1996
Gratulacje jubileuszowe 223Polaryzacja komórki i cytoki-n e z a ....................................................227Kompleksy chromatynowe . . . . 238Kinazy MAP i A P -1 ....................... 244Cytoplazmatyczna męskosteryl-ność u ro ś lin ..................................253Ekspresja mtDNA drożdży . . . . 259 Roślinny łańcuch oddechowy . . 268Sterowanie poliketydów 276Ary losu Ifataza A — efekty niedoboru ....................................................284Izoenzymy fosfolipazy C 290Sygnał sfingomielinowy 299Sprawozdania..................................310
http://rcin.org.pl

K w arta ln ik „ Postępy B io ch em ii "
w ydaw any z pom ocą finansow ą K om itetu Badań N aukow ych
http://rcin.org.pl

W YDAW CA
EditorPO LSK IE TOWARZYSTWO BIOCHEM ICZNE Polish Biochemical Society ul. Pasteura 302-093 Warszawa Poland Poland
Drukarnia Naukowo-Techniczna Mińska 6503-828 Warszawa
REDAKCJAEditorial Board REDAKTOR NACZELNY Editor in chief ZOFIA ZIELIŃSKA tel. 31-24-03 REDAKTORZY EditorsGRAŻYNA PALAMARCZYK tel. 658-47-02ANDRZEJ JERZM ANOW SKI tel. 659-70-72 w. 3234 ANNA SZAKIEL tel. 23-20-46JOLANTA GRZYBOWSKA tel. 13-05-15BARBARA ZARZYCKA tel. 659-85-71 w. 332
R E C E N Z E N C I ZESZY TU Referees of this issue GRAŻYNA DOBROWOLSKA (Warszawa)LILLA HRYNIEWIECKA (Poznań)RENATA JASIŃSKA (Warszawa)ANDRZEJ JERZMANOWSKI (Warszawa)TADEUSZ KŁOPOTOWSKI (Warszawa)STEFAN MALEPSZY (Warszawa)GRAŻYNA PALAMARCZYK (Warszawa)KRZYSZTOF STAROŃ (Warszawa)PIOTR STĘPIEŃ (Warszawa)KRYSTYNA STRZYKAŁA (Poznań)
ADRES REDAKCJIAddressREDAKCJA KW ARTALNIKA„POSTĘPY B IO CH EM II”PO LSK IE TOWARZYSTWOBIOCHEM ICZNEul. Pasteura 302-093 Warszawatel. (2) 659-85-71 w. 332fax: (22) 22-53-42
SPIS TREŚCI CONTENTS
Listy gratulacyjne z okazji 90-lecia Członków Honorowych Towarzystwa:profesor Stelli Niemierko i profesora Tadeusza Korzybs- kiegoCongratulation on the 90th birthday of Professors Stella Niemierko and Tadeusz Korzybski.................................................................223
Polaryzacja komórki, białka polarne i cytokinezaCell polarity, polar proteins and cytokinesis JANINA KACZANOWSKA, DOROTA WŁOGA, ANDRZEJ KACZANOWSKI ........................................................................................ 227
Rola kompleksów chromatynowych w regulacji ekspresji genówChromatin complexes in regulation of gene expression during developmentTOMASZ T. CALIKOWSKI.................................................................238
Kinazy MAP i ich rola w regulacji poziomu, składu podje- dnostkowego oraz stopnia fosforylacji czynnika trans- krypcyjnego AP-1Mitogen-activated protein kinases (MAPKs) — their role in regulation of protein level, subunit composition and degree of phosphorylation of the transcription factor AP-1 IRENEUSZ W. BIEDERM ANN........................................................244
Molekularne podstawy cytoplazmatycznej męskosteryl- ności u roślin wyższychMolecular basis of cytoplasmic male sterility in higher plants HANNA JAŃSKA, MAGDALENA WOŁOSZYŃSKA.....................253
Rola genomu jądrowego w regulacji funkcjonowania m itochondriów Saccharomyces cerevisiaeNuclear control of mitochondrial functions i Saccharomyces cerevisiaeMAGDALENA BO G UTA....................................................................259
Roślinny łańcuch oddechowyPlant respiratory chain ANNA M. RYCHTER ................. 268
Sterowana biosynteza antybiotyków poliketydowychEngineered biosynthesis of polyketide antibiotics KATARZYNA KUCZEK, MAGDALENA KOTOWSKA, MARIAN MORDARSKI ..................................................................................... 276
Niedobór aktywności arylosuifatazy A jako podłoże leu- kodystrofii metachromatycznejDeficiency of arylsulfatase A activity as a basis of metachromatic leukodystrophyAGNIESZKA ŁUGOWSKA, ANNA TYŁKI-SZYMAŃSKA . . . . 284
http://rcin.org.pl

Izoenzymy fosfolipazy C specyficznie hydrolizującej fos- fatydyloinozytole i regulacja ich aktywnościPhosphatidylinisitol-specific phospholipase C isozymes and regulation of their activityTADEUSZ PAWELCZYK............................................... 290
Sfingomielina i jej m etabolity jako pośredniki w przenoszeniu sygnału w komórceSphingomyelin and its metabolites in cellular signal transduction JOLANTA M. DZIK, ELŻBIETA W AŁAJTYS-RODE.....................299
Sprawozdania i K om unikaty........................................................310
ErrataW zeszycie nr 2 b.r. Postępów Biochemii (str. 219) zostały zniekształcone nazwiska:Kol. Jacka Kuźnickiego z Warszawy,Kol. Jerzego Meduskiego z Kaliforni.Za niedopatrzenie błędów w korekcie Redakcja uprzejmie Kolegów przeprasza.
222 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

Warszawa dn. 09.05.1996.
Wielmożna Pani Profesor Stella Niemierko
Wielce Szanowna Pani Profesor
Z okazji pięknego jubileuszu dziewięćdziesięciolecia urodzin mam zaszczyt złożyć Pani Profesor, Członkowi Honorowemu Polskiego Towarzystwa Biochemicznego i Znakomitemu Biochemikowi, najserdeczniejsze życzenia pomyślności i zdrowia od Zarządu Głównego Polskiego Towarzystwa Biochemicznego i całej społeczności biochemików polskich.
Przywilej reprezentowania Towarzystwa pozwala mi także wyrazić naszą wdzięczność za wielki wkład Pani Profesor w odbudowę po zniszczeniach wojennych, tak ważnej placówki badawczej, jaką jest Instytut imienia M. Nenckiego; za stworzenie w nim dynamicznie rozwijającej się szkoły biochemii układu nerwowego, a przez to zainicjowanie w Polsce badań neurochemicznych.
Łączę wyrazy uznania oraz najgłębszego poważania
Prezes Polskiego Towarzystwa Biochemicznego Profesor dr Liliana Konarska
POSTĘPY BIOCHEM II 42 (3 ) , 1996 223http://rcin.org.pl

http://rcin.org.pl

Warszawa, 4 czerwca, 1996
W ielmożny Pan Profesor Tadeusz Korzybski
Z okazji pięknego jubileuszu dziewięćdziesięciolecia urodzin mam zaszczyt złożyć Panu Profesorowi, Znakomitemu Biochemikowi, Członkowi Założycielowi i Członkowi Honorowemu Polskiego Towarzystwa Biochemicznego, najserdeczniejsze życzenia zdrowia i pomyślności. Życzenia te wraz z gratulacjami składam w imieniu Zarządu Głównego Polskiego Towarzystwa Biochemicznego i całej społeczności biochemików polskich.
Przywilej reprezentowania Towarzystwa pozwala mi także na wyrażenie naszego uznania za znaczący wkład Pana Profesora w rozwój biochemii w naszym kraju, a zwłaszcza wiedzy dotyczącej antybiotyków.
Pamiętamy też o udziale Pana Profesora w organizacji i działalności Polskiego Towarzystwa Biochemicznego, a w szczególności jego Komisji Słownictwa Biochemicznego. Na naszą wdzięczność zasługuje także fakt, że jako uczeń Profesora Jakuba Karola Parnasa umiał Pan utrwalić pamięć o tym wielkim uczonym wśród biochemików kilku pokoleń — poprzez swoje publikacje, odczyty i chętnie udostępniane wspomnienia oraz zgromadzone pamiątki.
Z wyrazami najgłębszego szacunku i poważania
Prezes Polskiego Towarzystwa Biochemicznego Profesor dr Liliana Konarska
POSTĘPY BIOCHEM II 42 (3 ) , 1996 225http://rcin.org.pl

■
.
http://rcin.org.pl

ARTYKUŁY
Artykuł ten dedykujemy Pani Profesor Dr hab. Zofii Zielińskiejw dowód naszego szacunku i przyjaźni
Polaryzacja komórki, białka polarne i cytokineza
Cell polarity, polar proteins and cytokinesis
J A N IN A K A C Z A N O W S K A 1, D O R O T A W Ł O G A 2, A N D R Z E J K A C Z A N O W S K I3
Spis treści:
I. Polarność komórki stanowi instrukcję strukturalną w rozwoju
II. Zewnętrzne sygnały mogą wywołać reorganizację cyto- szkieletu
III. Mechanizmy powstania polarności komórki i wyznaczania położenia bruzdy podziałowej w trakcie pączkowania drożdży Saccharomyces cerevisiaeIII-l. Przebieg pączkowania jest związany z reorganiza
cją cytoszkieletu w trakcie cyklu komórkowego III-2. Septyny drożdży mogą stanowić instrukcję struk
turalną dla wyznaczania miejsca pączkowania III-3. Zewnętrzny bodziec może wywołać reorganiza
cję cytoszkieletu i wyznaczyć miejsce gromadzenia białek biegunowych
IV. Funkcje białek biegunowych w komórkach różnych typów
V. Biegunowość jaja robaka Caenorhabditis elegans i wyznaczanie położenia bruzd podziałowych w embriogenezie
VI. Powstawanie osi w jaju Drosophila melanogaster jako przejaw wzajemnego oddziaływania na siebie oocy- tu i komórek folikularnych
VII. Wnioski i pytania
Wykaz stosowanych skrótów: EGF — (ang. epidermal growth factor) epidermalny czynnik wzrostu; PDGF — (ang. platelet derived growth factor) płytkowy czynnik wzrostu; LPA— (ang. lysophosphatidylic acid) kwas lizofosfatydylowy; Ras— produkt protoonkogenu genu ras pośredniczący w szlaku przekazu bodźca do jądra; Rho — (ang. Ras homologues) grupa białek o podobnej strukturze i funkcji do białka Ras; Rac — (ang. Reorganization o f actin cytoskeleton) białka zbliżone do Ras bezpośrednio związane z reorganizacją cytoszkieletu aktynowego; CDC42 — produkt genu cdc42 Saccharomyces cerevisiae o strukturze i funkcji podobnej do białka Ras; GEF — (ang. guanine nucleotide exchange factor) czynnik regulujący GTPazową funkcję białka Ras; GAP— (ang. GTPase activating protein) aktywator funkcji GTPa- zowej białek Ras; CLbl, Clb 2 — (ang. cyclin binding proteins)
1,3 Prof. dr hab., 2 mgr Zakład Cytofizjologii, Uniwersytet Warszawski, Warszawa 00-927, ul. Krakowskie Przedmieście 26/28
POSTĘPY BIOCHEMII 42 (3 ) , 1996
Contents:
I. Cell polarity is a developmental instructionII. External signals serve as the stimuli for the cytoskeletal
reorganisationIII. Mechanisms of appearance of cell polarity and deter
mination of position of the fission line during budding ofSaccharomyces cerevisiaeIII-l. Morphogenesis of the bud is coupled to reorgani
zation of the cytoskeleton during cell cycle III-2. Yeast septines may function as structural deter-
minans for selection of the sites of budding III-3. External signal induces reorganization of the
cytoskeleton and determines the site of nucleation of the polar proteins
IV. Functions of polar proteins in various cellular systemsV. Polarity of the Caenorhabditis elegans egg and deter
mination of the pattern of fission lines in embryogenesisVI. Development of antero-posterior and ventro-dorsal axes
in the Drosophila melanogaster egg as an interplay of signals between the oocyte and the follicular layer
VII. Conclusions and questions
aktywatory cyklino-zależnej kinazy białkowej Saccharomyces cerevisiae: Myo 2 — produkt genu myo 2 będący homologiem miozyny w Saccharomyces cerevisiae; septyny— (ang. septins) kompleks białek szkieletowych z domeną wiążącą nukleotyd wyznaczających położenie bruzdy podziałowej; bud — zespół genów kontrolujących powstawanie miejsca pączkowania u Saccharomyces cerevisiae; cdc24, cdc43, bem 1, bem 3, rsr 1, spa2, gtpb — geny Saccharomyces cerevisiae kontrolujące tworzenie kompleksu białek biegunowych; GDI — (ang. guanine nucleotide dissociation inhibitor) białko negatywnej kontroli funkcji Ras; ERM— (ang. ezrin, radixin, moesin family) białka cytoszkieletu występujące albo w cytoszkielecie miejsc przylegania komórki do podłoża, albo w* bruździe cytokinetycznej; cdcl5, peanut — geny kodujące białka charakterystyczne dla bruzdy podziałowej; MAP kinaza — (ang. Mitogen activated protein kinase) kinaza serynowo-treoninowa uczestnicząca w szlaku kaskady kinaz przenoszących mitogenny sygnał zewnętrzny z cytoplazmy do jądra; c-jun, c-fos — geny
227http://rcin.org.pl

kodujące powstanie czynnika transkrypcyjnego (AP-1) w wyniku działania mitogenów na komórki; par — geny kodujące białka kontrolujące położenie bruzd podziałowych w trakcie bruzdkowania robaka Caenorhabditis elegans; bicoid, staufen, nanos, oskar, gurken — geny matczyne Drosophila melanogaster kodujące determinanty osi jaja; MTOC — (ang. microtubules organizing center) kompleks białek zdolnych do nukleowania polimeryzacji mikrotubuli, zwykle jest to zespół białek otaczających centriolę tworzącą razem strukturę zwaną centrosomem; rTGF — (ang. tranfor- mation growth factor receptor) białko receptora błonowego przyłączającego czynnik transformujący; torpedo — gen kodujący białko receptorowe eksprymowane w niektórych komórkach folikularnych otaczających jajo Drosophila melanogaster; tor so — gen matczyny kodujący białko o charakterze receptorowej kinazy tyrozynowej eksprymowany w jaju Drosophila melanogaster.
I. Polarność komórki stanowi w trakcie rozwoju instrukcję strukturalną
Celem tego artykułu jest przedstawienie m echanizmów pow staw ania osi ciała kom órek somatycznych i zapłodnionych jaj w odpowiedzi na sygnały zewnętrzne. K om órka, poprzez transdukcję tych sygnałów, reorganizuje w określony sposób swój cytoszkielet oraz urucham ia nowe transkrypcje. Pow stanie nieprzypadkowego rozmieszczenia struk tu r w kom órce w wyniku takiej reorganizacji wzdłuż osi przednio- tylnej i grzbietowo-brzusznej stanowi jej instrukcję strukturalną. Ta instrukcja wyznacza położenie bruzdy podziałowej w trakcie cytokinezy, oraz w przypadku zapłodnionej kom órki jajowej — także organizację przestrzenną zarodka. Blastomery powstałe z przedniej części kom órki jajowej stanowić będą przód ciała zarodka. Blastom ery powstałe z tylnej części kom órki wytworzą tył zarodka. Oznacza to, że układ struktur w zapłodnionej kom órce jajowej determ inuje przyszły plan przestrzennego rozwoju zarodka. M echanizm pow staw ania polarności jaja, a także pewnych kom órek spolaryzowanych (np. neuronów) jest p raw dopodobnie uniwersalny i wiąże się z uporządkow aniem przestrzennym rozmieszczenia kom pleksu tzw. białek biegunowych.
II. Zewnętrzne sygnały mogą wywoływać reorganizację cytoszkieletu
K om órka eukariotyczna może być kom órką apolar- ną. O znacza to, że organizacja struk tu r cytoszkieletal- nych nie wykazuje żadnego uporządkow ania o charakterze osiowym. Oś tak a może pojawić się w niektórych kom órkach pod wpływem czynników zewnętrznych np. chem oatraktantów . W obecności takiego czynnika kom órka reorganizuje swój cytoszkielet i powstaje przód kom órki z nibynóżkam i przesuwającymi ko m órkę w kierunku źródła chem oatrak tan ta [1]. Przykładem m ogą być fibroblasty myszy, k tóre pod wpływem czynników wzrostowych, takich jak epiderm alny czynnik w zrostu (EGF), płytkowy czynnik wzrostu (PD G F), czy też kw asu lizofosfatydylowy (LPA) wy
tw arzają p łatow ate nibynóżki (lammellipodia) i rusztow anie cytoszkieletu w postaci aktynowych kabli (ang. actin cables).
C entralną rolę w przewodzeniu bodźca wyzwalającego przem iany w cytoszkielecie grają różne białka0 charakterze G T P az zbliżonych do białka Ras określanych jako Rho (ang. Ras Homolgues = Rho), odm iana G T P az typu Rac związanych bezpośrednio z reorganizacją cytoszkieletu aktynowego (ang. R eorganization o f Actin Cytoskeleton = Rac) (Rye. 1) i być może kluczowe białko CDC42 (w niektórych publikacjach używ ana jest pisownia Cdc42p) należące do grupy G T P az typu Ras [2, 3]. Te same bodźce m ogą również prowadzić do aktywacji czynników trans- krypcyjnych, a następnie do syntezy nowych białek.
N iektóre poznane do tąd drogi transdukcji sygnałów1 aktywacji poszczególnych szlaków m etabolicznych związane są z różnymi białkam i Ras (Rye. 1). Schemat ten jest jeszcze niekom pletny ponieważ obecnie po szukuje się białek pośredniczących w przenoszeniu sygnału od odpowiedniego receptora kom órkow ego do białek typu Ras, Rac i CDC42. Białka pośredniczące powinny zawierać dom eny SH2 i SH3 oddziaływujące z pobudzonym receptorem oraz dom eny aktywujące białka typu Ras. W procesie aktywacji b iałka Ras występuje wym iana G D P na G TP; zatem pewne białka pośredniczące m uszą spełniać funkcję wymieniacza tych nukleotydów (ang. GEF = Guanine nucleotide Exchange Factor), a inne regulować aktywację hydrolitycznej funkcji G T Pazy (ang. GAP = GTPase Activating Protein). Aktywacja białek Ras z kolei aktywuje pośredniczący system kinaz białkowych aby uruchom ić systemy efektorów (z jednej strony m echanizm y reorganizacji cytoszkieletu, a z drugiej aktywację czynników transkrypcyjnych).
W wyniku zadziałania bodźca powstaje kom pleks białek: uczestniczących w transdukcji sygnału (Rac, Rho, Ras, CD C42 G E F etc.), ośrodka inicjującego reorganizację cytoszkieletu (ang. nucleating center), oraz białek szkieletowych reorganizow anych pod wpływem bodźca (aktyna, b iałka regulujące polim eryzację aktyny, miozyny, m ikrotubule i towarzyszące im białka m otoryczne etc). Taki złożony kom pleks białek może stanowić zespól białek biegunowych decydujących0 polarności komórki. W spółczesne badania skupiają się na poznaniu składu, funkcjonow aniu i trwałości kom pleksu białek biegunowych, a szczególnie na p o znaniu ich roli jako instrukcji strukturalnej w trakcie różnicow ania i embriogenezy.
Po okresie interfazy zarów no kom órka po larna jak1 apo larna m ogą wchodzić w stadium mitozy. W rzeciono kariokinetyczne wyznacza położenie bruzdy p o działowej, k tó ra pow staje prostopadle do osi wrzeciona [4]. Jak powstaje oś wrzeciona? Jaki wpływ m a polarność kom órki na determ inację położenia bruzdy podziałowej? Jakie znaczenie w rozwoju m a wyznaczenie przestrzennego ułożenia bruzd podziałowych? P y tan ia te na razie pozostają bez jednoznacznych od-
228 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

hydroliza fosfatydyloinozitolu
kaskada kinaz
? czynnikitranskrypcyjne
'ekspresja i • ' \ genówk in a z a # \ 1aktywująca \ fc -J U N , ^regulacja
zm iany m itozyw cytoszkielecie aktyny i ERM
Ryc. 1 Uproszczony i hipotetyczny schemat zależności stymulacji białek G, Ras, Rho, Rac i CDC42 z aktywacją reorganizacji cytoszkieletu (grube strzałki) i pobudzaniem aktywności niektórych kinaz białkowych (kompilacja schematów wielu autorów głównie [3,16,44, i 45]). FAK — kinaza specyficzna dla miejsc przylegania komórek do podłoża (ang. foci adhesion kinase), ERM — (ang. ezrin, radixin, moesin) białka cytoszkieletu związane z wytwarzaniem miejsc przylegania do podłoża i występujące w bruździe cytokinetycznej, G s — białko G stymulowane hormonalnie (wzrost stężenia cAMP) i Gj — białko G stymulowane kwasem lizofosfatydylowym (LPA) (obniżenie poziomu cAMP) powoduje aktywację fosfolipazy a następnie aktywację kinazy białkowej C (PKC), aktywację kinazy fos- fatydylonoizytolu (PI3K) i Rac (RAC), oraz aktywację Rho (być może z udziałem niereceptorowych kinaz tyrozynowych (src i JAK); Przy działaniu LPA i czynników wzrostowych (IGF i EGF) obserwuje się aktywację szlaku Ras. Stymulacja szlaku Ras jest sprzężona z aktywacją CDC42 być może przez wspólny czynnik aktywujący GAP.
powiedzi. Jednakże postęp badań nad m echanizm am i pączkow ania drożdży, pow staw ania polarności ja ja rob ak a Caenorhabditis elegans i wyznaczania położenia bruzd w trakcie embriogenezy, oraz badan ia nad m echanizm em pow staw ania osi ciała w ja ju Drosophila melanogaster rokują nadzieję na rozwikłanie tych zależności.
III. Mechanizmy powstania polarności komórki i wyznaczania położenia bruzdy podziałowej w trakcie pączkowania drożdży Saccharomyces cererisiae
III-l. Przebieg pączkowania jest związany z reorganizacją cytoszkieletu w trakcie cyklu komórkowego
Pączkow anie drożdży S. cerevisiae jest asym etrycznym podziałem kom órki. W późnym stadium G1 kom órka rodzicielska osiąga m aksym alne rozm iary po czym przestaje rosnąć i przygotow uje się do replikacji DNA. W okresie wchodzenia w replikację (stadium G l/sD N A ) kom órka rodzicielska zaczyna tworzyć pączek (Ryc. 2a). Pow staje pierw otna polar- ność wzdłuż osi „kom órka rodzicielska-pączek” (Ryc. 3d). Pow staw anie pączka jest zależne od ak tywacji kinazy białkowej zależnej od cykliny (Cln/Cdc28) inicjującej replikację D N A poprzez przyłączenie dwóch białek (C lbl i Clb2 = Cyclin Binding proteins) (Ryc. 2a i b). A więc tworzenie pączka jest bezpośrednio związane z cyklinam i kontrolującym i przebieg cyklu kom órkowego. Pom iędzy kom órką
rodzicielską a pączkiem powstaje charakterystyczny pierścień złożony z białek biegunowych. Ten pierścień wyznacza położenie przyszłej bruzdy cytokinetycznej (Ryc. 2b). Przyrost pączka odbyw a się wyłącznie w obrębie pierścienia. Tu tworzy się palczasty wyrostek z szybko rosnącym czubkiem. W czubku także są białka biegunowe (wzrost apikalny) (Ryc. 2c). Do czubka przemieszcza się również wiele składników cytoszkieletu np. aktyna, m ikrotubule wraz z kinezyną, kaldesm on, kalm odulina i inne białka związane z k o n trolą stanu polimeryzacji aktyny. D o czubka są transportow ane pęcherzyki stanowiące zapas błony kom órkowej; pęcherzyki te drogą egzocytozy na czubku pączka pow odują szybki jego wzrost (Ryc. 3d). W transporcie tym uczestniczy hom olog miozyny ludzkiej kodowanej przez gen M yo 2 drożdża.aktywacja kinazy
cdc 28 +C lb1 /2G 1 - s DNA
aktywacja przez fosfatazę
IClb1/2 cdc 28
degradacja ' cyklin
przejście wzrostu apikalnego w izometryczny
rozpraszaniebiałekbiegunowych
Ryc. 2 Zmiany w rozmieszczeniu białek biegunowych w trakcie pączkowania Saccharomyces cerevisiae. Uproszczony schemat wg [5]. Szczegółowy opis w tekście.
POSTĘPY BIOCHEM II 42 (3 ) , 1996 229http://rcin.org.pl

W następnym stadium cyklu kom órkow ego (stadium przejścia sDNA /G 2) zmienia się typ wzrostu pączka, z biegunowego wydłużającego się palca, na izometryczny, czyli wydłużający się palec staje się równom iernie rosnącą kulą z rów nom iernie sekreto- wanymi pęcherzykam i na całej powierzchni pączka. M om ent tego przejścia z wzrostu apikalnego na izometryczny jest pod kon tro lą genetyczną (ang. api- cal/isometric switch) [5]. Aktywacja czynników Clb 1/2 przez fosfatazę jest w arunkiem niezbędnym do zmiany typu wzrostu pączka. Towarzyszy tem u rozproszenie wszystkich składników białek biegunowych w obrębie całego pączka, a więc przejściowy zanik osiągniętej uprzednio polarności (Ryc. 2d).
Inaktyw acja kom pletu białek Clb 1/2 Cln/Cdc28 (przez proteolizę cykliny) w trakcie fazy M (Ryc. 2e) wiąże się z pow tórnym zgrupowaniem aktyny i innych składników kom pleksu białek apikalnych w regionie bruzdy cytokinetycznej. W tym miejscu tw orzą się teraz dwa równoległe pierścienie szyjki, jeden dla pączka, drugi dla kom órki macierzystej. W trakcie cytokinezy pierścienie te zostaną rozdzielone do k o m órek potom nych wyznaczając miejsce skupienia kom pleksu białek biegunowych (Ryc. 2e i f).
Oznacza to, że w trakcie pączkow ania pierwszym etapem jest wyznaczenie pewnego specjalnego miejsca w którym tworzy się pączek, i gdzie w przyszłości odbędzie się cytokineza, a więc wyznaczenie granicy tylnej pączka. Z kolei w obrębie tego miejsca, zwanego miejscem szyjki (ang. neck filam ent site), w postaci pierścienia podczepionego do błony kom órkowej grupują się zarów no białka biegunowe o charakterze enzymów jak i spolimeryzowane pewne specyficzne dla bruzdy podziałowej białka cytoszkieletowe np. sep- tyny (ang. septins), radyksyna (ang. radixin), obok niespecyficznych białek szkieletowych (np. aktyny). W środku tego pierścienia w yodrębnia się kom pleks białek apikalnych (ang. bud site proteins) nie związanych z septynam i, ale z niespecyficznymi białkam i szkieletowymi, k tóre zgrupują się na rosnącym czubku pączka. A więc kolejno tw orzą się dwa bieguny pączka, z charakterystycznym i kom pletam i białek biegunowych, przejściowo rozpraszanym i w trakcie wczesnej m itozy i przemieszczanymi i kondensow anym i ponow nie z septynam i w trakcie cytokinezy [5-7].
III-2. Septyny drożdży mogą wyznaczyć miejsce tworzenia kolejnego pączka
Białka filam entu szyjki, wraz z wielocukrem o charakterze chityny łatw o zabarw ić barw nikiem zwanym kalkofluorem . Pozw ala to na wybarwienie pierścienia na powierzchni drożdża interfazalnego, jak o blizny, po odbytej uprzednio cytokinezie po oddzieleniu pączka (Ryc. 21). Jeżeli kom órka pączkow ała wielokrotnie, m ożna sprawdzić, czy istnieje określona prawidłow ość przestrzenna w rozmieszczeniu tych pierścieni. A więc m ożna sprawdzić, czy kom plet białek szyjki jest infor
m acją struk tu ralną dla wyznaczenia miejsc pączkow ania w kolejnych cyklach kom órkowych. Badania p ro wadzone w pracow niach D r P r i n g l e ’ a i H e r s - k o w i t z a pozwoliły wyróżnić dwa podstawow e typy wzoru przestrzennego pączkow ania u S. cerevisiae; a) typ osiowy, (Ryc. 3a) i b) typ biegunowy (Ryc. 3b) [1 ,6].
Odżywione drożdże, k tóre często pączkują charakteryzuje osiowy typ pączkowania. W trakcie kolejnych pączkow ań poprzedni pierścień szyjki stanowi instrukcję przestrzenną dla wyznaczenia miejsca tw orzenia następnego pączka w tej samej okolicy. W obec tego po kilku pączkow aniach kom órka m acierzysta wykazuje uprzywilejowaną strefę na powierzchni drożdża w p o staci jakby naszyjnika, bądź lokalnego skupienia kolejnych pierścieni (Ryc. 3a).
Jeżeli jednak kom órka długo nie pączkuje (zostaje zgłodzona), albo w międzyczasie przeszła przez zapłodnienie, wtedy zmienia się reguła wyznaczania miejsca następnego pączkow ania na dwubiegunowy typ pączkow ania (Ryc. 3b). Pączek tworzy się na biegunie przeciwnym w stosunku do miejsca poprzedniego pączkowania. O kazjonalnie także pow stają kom órki
a .ty p osiow y b .ty p dw ubiegunowy
Ryc. 3 Determinacja położenia miejsc tworzenia pączków S. cere- visiae: a. typ osiowy, b. typ dwubiegunowy i metameryczny, i d. i e. porównanie morfogenezy pączka i wyrostka koniuga- cyjnego shmoo. Wg [1, i 45] uproszczone. Objaśnienia w tekście.
230 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

o metamerycznej symetrii wyznaczania miejsc pączkow ania (Ryc. 3b, najniższy rząd). W obec tego przypuszcza się, że b iałka pierścienia szyjki są źródłem sygnału wyznaczającego miejsce pączkow ania (zapewne septyny są związane z białkam i biegunowymi), ale inform acja ta z czasem zanika (np. w trakcie głodzenia po zaprzestaniu pączkow ania b iałka biegunowe zostają rozproszone), a pow tórne ich zgrupow anie odbywa się wraz z nowo polimeryzowanym i septynam i w najdalszym miejscu w stosunku do „starego” pierścienia (po procesie seksualnym).
W yizolowano szereg m utantów procesu pączkow ania grupy bud, („pączka”), czyli genów związanych z wyznaczaniem miejsca pączkow ania u S. cerevisiae. B iałka BU D nie m ają zasadniczego wpływu na przebieg samego pączkow ania. D otyczą wyłącznie instrukcji o miejscu tw orzenia pączka [1, 6, 7]. M utan ty loci bud 1-5 tw orzą pączki w dowolnym miejscu ciała i bez względu na rozmieszczenie poprzednich pieścieni szyjki na komórce. Białka BUD1 i BUD3 (a zapewne i inne białka BUD) grom adzą się w pierścieniu szyjki w trak cie każdego pączkow ania przyłączając b iałka receptorow e (RS1 i SPA2) [3, 6, 7], oraz białko kotwiczące w postaci septyny AFR1 (neck filam ent protein) [8]. Ten jeszcze słabo poznany kom pleks oddziaływ ujących na siebie białek może być źródłem sygnału w którym uczestniczy p roduk t BUD1. Sklonowanie genu budl pozwoliło bowiem wywnioskować, że jest to gen kodujący G T Pazę typu Ras. P rodukty genów bud 2 i 5 są białkam i typu G E F (czynnik wymiany nukleo- tydu G D P na G T P) i G A P (aktyw ator GTPazy). (GAP). A więc b iałka biegunowe drożdży stanow ią istotnie instrukcję topograficzną w wyznaczaniu miejsca pączkow ania i są regulow ane przez mechanizm sygnalizacji z pośrednictw em pewnych typów białek typu Ras [9].
III-3. Zewnętrzny bodziec może wywołać reorganizację cytoszkiełetu i wyznaczyć miejsce gromadzenia białek biegunowych
Zgłodzone kom órki dwóch typów płciowych wydzielają do środow iska ferom ony (gamony) oddziałujące na receptory kom órki o przeciwnym typie płciowym. W odpowiedzi na gam ony, dwie kom órki różnych typów płciowych przypadkow o znajdujące się blisko siebie zaczynają tworzyć rosnące ku sobie palczaste wyrostki (ang. shmoo). W ten sposób d o chodzi do połączenia obu kom órek. Szczegółowe badan ia morfologiczne i m olekularne wykazały, że w rosnących w yrostkach tworzy się kom pleks białek biegunowych, a procesy morfogenezy pączka i w yrostka są bardzo podobne (porównaj Ryc. 3d i 3e). O znacza to, że lokalizacja i tworzenie kom pleksu białek biegunowych w yrostków stanowi rzeczywiście odpow iedź na sygnał (w tym przypadku gam onu sekretowanego przez kom órkę odmiennej płci).
B adania genetyczne i m olekularne pozwoliły na
wykrycie drugiego zespołu m utantów (innych niż m utanty bud) związanych z tworzeniem kom pleksu białek biegunowych [3, 6, 10-13]. M utacje genów cdc24, cdc42, cdc43, bem 1, bem 3 i kilku innych charakteryzuje u tra ta zdolności do wytwarzania pierścienia inicjującego powstawanie pączka (lub shmoo). M utacje tych genów pow odują, że kom órka m acierzysta w trakcie mitozy powiększa równom iernie swoją powierzchnię, a nie tworzy zlokalizowanego miejsca wzrostu pączka. Obecnie wykryto, że w tych m utantach zachodzi apolarna sekrecja pęcherzyków niosących zapas błon, dezorganizacja cytoszkiełetu, nie tw orzą się aktynow e kable, a b iałka norm alnie zlokalizowane wyłącznie w rosnącym czubku (w stadium G l/sD N A ), bądź też w szyjce (w stadiach M i G l) są rozproszone na całej powierzchni kom órki.
Po raz pierwszy model łączący procesy sygnalizacyjne w trakcie pączkow ania drożdża z kon tro lą m orfogenezy przedstawili C h a n t i H e r s k o w i t z [6] (Ryc. 4). W szystkie zebrane dane z ostatnich czterech lat pozw alają przypuszczać, że białko CDC42 jest uniwersalną determinantą bieguna przedniego (apikał- nego) komórki monopolarnej [12-17], CDC42 jest m ałym białkiem wiążącym G T P, o charakterze G T P azy sprzężonej z kon tro lą przem ian w cytoszkielecie[11] pod wpływem czynników w zrostu [2, 3, 16, 17]. Białko to m a charakterystyczny koniec karboksylow y (typu CAAX), które ułatw ia modyfikację tego zakończenia pod wpływem transferazy geranylgeranylowej z formy białka cytosolowej na formę zakotw iczoną w błonie plazmatycznej. G T P aza CDC42 w drożdżach grom adzi się w rosnącym czubku pączka w błonie plazmatycznej; a zarów no podczepienie do błony plazmatycznej, jak i aktywność G T Pazow a są w arunkam i koniecznymi dla wytworzenia pączka. Inny p ro dukt tej grupy genów CDC43 stanow i jednostkę beta transferazy geranyl geranylowej. Należy przypuszczać, że działanie tego enzymu aktywuje CDC42 um ożliwiając m u trwałe zakotwiczenie w błonie plazmatycz- nej [10]. Z kolei CDC24 działa na CDC42 aktywująco poprzez wymianę G D P na G T P w CDC42 (ang. guanine nucleotide exchange factor — GEF). Porów nanie sekwencji am inokwasów w ludzkim hom ologu CD C24 (zwanym DBL i oddziaływującym na ludzki hom olog białka CDC42) z sekwencjami CDC24 drożdży wykazuje w yraźną hom ologię jednej dom eny [14]. Pośrednio, na podstaw ie badań genetycznych sugeruje się, że w białku drożdżowym CD C24 istnieje jeszcze inna dom ena wrażliwa na G T Pazę typu BUD1. To tłumaczy funkcjonalny związek pom iędzy białkam i B U D i białkam i polarnym i CDC42, 43, 24, BEM1 i BEM3. Geny bem 1 i bem 3 kodują b iałka regulujące aktywność CDC42 [7, 14]. P onad to białko BEM1, które jest niezbędne dla utrzym ania apikalnego wzrostu pączka, zawiera dwie dom eny SH3 (a więc m a cechy białka pośredniczącego w transdukcji sygnału). W skład kom pletu białek biegunowych czubka wchodzą również białka receptorowe RSR1 i SPA-2 nie-
POSTĘPY BIOCHEMII 42 (3 ) , 1996 231http://rcin.org.pl

R SR 1 / B U D 1 = G TPB + SP A 2 •/ BU D 1 -► DETERMINACJA MIEJSCA PĄCZKOWANIA
R SR 1 / SPA 2 / BU D 1 CDC 42 GTP aza
CDC 43 ger anyl /ger any 1 tran sferaza BEM 1 = GAP
k askad a k in az (?)
CDC 24 aktywator w ym iany
GDP na GTP
polim eryzacja aktyny i lokalny wzrost
(?)
aktyw acjatranskrypcji(?)
POCZĄTEK TWORZENIAKOMPLETUBIEGUNOWEGO
WZROST PĄCZKA NA DŁUGOŚĆ
R O ZPA D LUB TR A N SL O K A C JASK ŁADNIK ÓW KOMPLETU ► ’’ 9 RI CBIEG UNO W EG O S W liL t i t r i
POW STANIE NIEAK TYW N EG OKOM PLETU „SZYJK I” CY TO K IN EZANA M IEJSC U CYTO K INEZY
Ryc. 4 Schemat zmian w komplecie białek biegunowych w trakcie pączkowania S. cerevisiae. Opis w tekście. Wg [3, 44].
zbędne dla u trzym ania w zrostu polarnego. Z przytoczonych tutaj danych wynika schemat przem ian w białkach biegunowych niezbędnych dla realizacji kolejnych stadiów pączkow ania [3] (Ryc. 4). Schemat ten jest uproszczony i nie uwzględnia kontroli przez działanie inhibitorów aktywacji Ras lub Rho typu G D I (ang. guanine nucleotide dissociation inhibitors) wygaszających m echanizm sygnalizacji [16]. P raw dopodobnie w trakcie morfogenezy drożdży istnieje pew na hierarchiczna kolejność aktywacji G T P az BUD1, C D C42 i R ho 1-4 uczestniczących w selekcji miejsca pączkow ania, a więc w tworzeniu pierścienia szyjki [9, 13, 19, 20].
R ola C D C42 wraz z kom pleksem białek nie ogranicza się do kontro li reorganizacji cytoszkieletu i wyznaczenia miejsca pączkow ania we wczesnej fazie cyklu. Równocześnie ze stym ulacją cyklu kom órkow ego i pow staw aniem pączka, odbyw a się aktywacja kinazy threoninowo-serynowej należącej do grupy M A P kinaz (uczestniczących w ścieżce aktywacji transkrypcji wczesnych genów w cyklu kom órkowym ) [21]. W badaniach in vitro wykazano, że CDC42 jest źródłem aktywacji pewnych charakterystycznych dla koniugacji kinaz białkow ych [15]. W ynik ten oznacza, że CDC42 jako przekaźnik sygnałów feromonowych spełnia jednocześnie potrójną rolę; wyznacznika miejsca inicjacji reorganizacji cytoszkieletu, utrzymania polar- ności wzrostu pączka i aktywatora ekspresji genów [22].
IV. Funkcje białek biegunowych w komórkach różnych typów
W przypadku fibroblastów myszy aktywacja G T P a-
zy typu Rho prow adzi do wytworzenia kabli ak tynowych cytoszkieletu i do wytworzenia miejsc przylegania do podłoża (adhesionfoci), a aktyw acja CDC42 do wytworzenia wiodącej cienkiej nibynóżki zwanej filopodium. M ikroinjekcja białek Rho, Rac i CDC42 do kom órek spoczynkowych stym ulow ała wyjście k o m órek ze stadium G1 i rozpoczęcie sDNA. W trakcie cytokinezy tych kom órek, białko — Rho jest przem ieszczane z cytosolu do bruzdy podziałowej, a tow arzyszą tem u przemieszczanie się aktyny i białek cyto- szkieletowych z grupy ERM (ezryny, radiksyny i moe- zyny). Białka grupy ER M występują, albo w cyto- szkielecie przylg (regionów przylegania kom órek do podłoża) w okresie interfazy, albo są w bruzdzie w trakcie cytokinezy [3].
Istnieją również pośrednie przesłanki, aby sądzić, że niektóre białka kom pleksu białek biegunowych o charakterze septyn są przemieszczane do bruzdy p o działowej w trakcie cytokinezy i m ogą pełnić funkcję zarów no przekaźnika w transdukcji sygnału jak i ośrodka polimeryzacji innych białek cytoszkieletu.
W badaniach nad podziałem drożdża Schizosac- charomyces pombe w obrębie sekwencji nukleotydów genu cdcl5 o charakterze septyny, znaleziono fragm ent hom ologiczny wobec dom eny kodującej SH3, a także dom eny charakterystycznej dla białek na których m oże odbywać się polim eryzacja innych białek. Białko CDC15 jest silnie ufosforylowaną fosfoproteiną w tra kcie całego cyklu, za wyjątkiem m itozy i tylko wtedy staje się ośrodkiem kondensacji F-aktyny i pośrednikiem w podczepieniu cytoszkieletu do błony kom órkowej. Białko C D C 15 może być uniwersalnym pośrednikiem w wyznaczaniu miejsca bruzdy cytokinetycznej
232 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

ponieważ sekwencje hom ologiczne względem sekwencji cdcl5 znaleziono w genomie S. cerevisiae, przywry i myszy [23]. Podobnie w trakcie tworzenia blastoder- my w zarodku Drosophila melanogaster wykryto, że p roduk t prawidłow y genu veanut związany z cyto- kinezą wykazuje znaczne podobieństw o do septyn drożdży [24].
Należy sobie również zdawać sprawę, że podane tutaj wiadom ości dotyczące podobieństw w składzie i organizacji bruzdy podziałowej w kom órkach różnych typów, nie dotyczą kontroli sprzężenia położenia bruzdy podziałowej z wyznaczaniem osi wrzeciona mitotycznego. W iadom ości na ten tem at są skąpe. W iadom o, że w niektórych przypadkach sprzężenie osi wrzeciona m itotycznego i prostopadłego położenia bruzdy może być regulow ane przez interakcję jednego z biegunów wrzeciona z białkam i biegunowym i (Ryc. 3c) [25].
W fibroblastach myszy wykryto specjalizację funkcji poszczególnych G T P az w przekazyw aniu bodźców urucham iających nowe transkrypcje [16]: wyłącznie Ras aktywuje kaskadę kinaz pobudzanych czynnikam i wzrostu (Ryc. 1). N atom iast białka Rac i C D C42 (ale nie Rho) stym ulowały aktywację specyficznej kinazy serynowo-treoninowej fosforylującej czynnik trans- krypcyjny c-Jun. Fosforylacja ta wzmacnia transak- tywację czynnika transkrypcyjnego AP-1 (tzn. kooperujących c-Jun i c-Fos) niezbędnej dla wejścia kom órek w podziały [17]. Rho i CDC42 aktywowały również kinazę serynow o-treoninow ą z m ózgu (neuronalną)
A
[26]. P odobną specjalizację funkcji G TPaz, Rac, Rho i CDC42 wykazano w przebiegu odm iennych typów reorganizacji aktyny [22].
Badania m olekularne dotyczące produktów genów cdc42, rho, rac oraz odkrycie funkcjonalnych hom o- logów tych produktów w genomie ludzkim [27], a także u robaka Caenorhabditis elegans [28] i innych m odelowych kom órkach ssaków pozw alają przypuszczać, że m echanizm uzyskania polarności przez kom órkę jest uniwersalny i bardzo konserwatywny ewolucyjnie. W zależności od bodźca i rodzaju kom órki, białka biegunowe uczestniczą w urucham ianiu no wych transkrypcji, oraz różnych typów reorganizacji cytoszkieletu. Reorganizacja cytoszkieletu może p ro wadzić do wyznaczania k ierunku i stymulacji ruchu kom órki, w innych przypadkach do wytworzenia miejsc przyczepu do podłoża i praw dopodobnie do (słabo jeszcze poznanej) koordynacji pow staw ania osi wrzeciona i położenia bruzdy. Białka polarne (septyny) związane są również z wyznaczaniem miejsca i reorganizacją cytoszkieletu w trakcie tworzenia bruzdy podziałowej w stadium cytokinezy. Kolejne pytanie dotyczy roli białek biegunowych w rozwoju.
V. Biegunowość jaja robaka Caenorhabditis elegans i wyznaczanie położenia bruzd podziałowych w embriogenezie
W trakcie oogenezy M etazoa, albo po zapłodnieniu, bądź też po kilku podziałach bruzdkow ania odbywa się determ inacja przestrzenna rozwoju zarodka. P ro ces ten jest dobrze poznany w przypadku oogenezy robaka C. elegans. 70 m inut po zapłodnieniu oocytu robaka, filamenty aktynowe grom adzą się na przednim końcu jaja, natom iast pęcherzyki P (ang. P-granules) grom adzą się w biegunie tylnym jaja. W trakcie następnych 30 min jąd ro ja ja (pronucleus żeński) zlewa się z jądrem plem nika (pronukleusem męskim). Jądro zygoty przesuwa się do tylnego bieguna ja ja i zaczyna tworzyć wrzeciono m itotyczne wzdłuż osi przednio- tylnej zygoty. Przesunięcie do tyłu wrzeciona m itotycznego powoduje, że bruzda cytokinetyczna rozdziela asymetrycznie większy przedni blastom er (nazywany blastom erem AB) od tylnego mniejszego blastom eru
Ryc. 5 Schemat dwóch podziałów bruzdkowania jaja robaka Caenorhabditis elegans. A i P — oś przednio-tylna jaja w trakcie pierwszego podziału, a. drugi podział bruzdkowania w normalnym zarodku; z blastomeru AB tworzą się dwie komórki ABa i ABp; w blastomerze P I zachodzi rotacja wrzeciona mitotycznego i potomne komórki EMS i P2 uzyskują prawidłową konfigurację (strzałka), b. rozmieszczenie aktywnego produktu PAR1 przez interakcję z białkiem biegunowym (homolog białka CDC42); c. obrót wrzeciona mitotycznego w mutancie par 3 uniemożliwiający otrzymanie prawidłowej konfiguracji blastomerów; występuje symetryczny charakter miejsca przyczepu wrzecion (strzałka), d. fenotypy mutantów par 1,2, 4, 5 o złej konfiguracji czterech blastomerów związanej z brakiem rotacji wrzeciona mitotycznego w blastomerze P I i brakiem miejsca podczepienia wrzeciona do bruzdy AB/PI. Inne objaśnienia w tekście. Rysunek kombinowany wg [28-30].
POSTĘPY BIOCHEMII 42 (3 ) , 1996 233http://rcin.org.pl

P I (Ryc. 5). M igracja jąd ra do tyłu i orientacja położenia bruzd w trakcie bruzdkow ania podlega kontroli genetycznej kilkunastu produktów genów typu par (ang. partition).
Sklonow anie genu par-1 wykazało, że p rodukt tego genu stanowi kinazę serynow o-treoninow ą rozmieszczoną rów nom iernie w obrębie ja ja [29]. W trakcie pierwszego podziału ja ja białko PAR1 przemieszcza się do tylnego blastom eru, a na tylnym biegunie tej kom órki pojawia się skupienie białka homologicznego do CDC42. W drugim podziale bruzdkow ania, wyłącznie w blastom erze P I wrzeciono m itotyczne podlega rotacji o 90°, a położenie bruzdy w tym blastom erze staje się prostopadłe w stosunku do osi jaja. Rotacja wrzeciona m itotycznego kończy się podczepieniem do błony kom órkow ej jednego z biegunów wrzeciona m itotycznego kom órki P I w miejscu znajdującym się na bruzdzie podziałowej (ang. cortical site) rozdzielającej blastom ery AB i P I [30, 31] (Ryc. 5a). W tym okresie, kinaza PAR1 jest aktyw ow ana przez G TPazę CDC42 i powstaje gradient tylno-przedni aktywacji tej kinazy (Ryc. 5b).
M utan ty genu par 1 nie są zdolne do segregowania PAR-1 na końcu ciała, a także zanikają inne cechy biegunowości jaja. Inhibicja funkcji każdego z dwóch enzymów, zarów no kinazy PAR1 jak i G TPazy CD C42 pow oduje w zarodku depolimeryzację zarów no m ikrotubuli jak i rusztow ania aktynowego. W obec tego takie zarodki nie są zdolne do przeprow adzenia rotacji i podczepienia wrzeciona m itotycznego do miejsca bruzdy podziałowej pom iędzy blastom eram i A B /PI. Pow staje wtedy zarodek złożony z czterech kom órek o nieprawidłowej segregacji granul P i zarodek ginie (Ryc. 5d) [29]. Działając inhibitoram i polimeryzacji aktyny na prawidłow e i norm alne genetycznie ja ja uzyskano fenokopie takiego nieprawidłowego b raku rotacji wrzeciona w blastom erze P I [32].
Inne m utanty typu par pow odują również niepraw idłowe wyznaczanie położenia bruzd podziałowych w trakcie cytokinezy i wszystkie prow adzą do złego wzajemnego położenia blastom erów i śmierci zarod ków (Ryc. 5d). Nieco inny charakter m a m utacja par 3. O tóż fenotypy m utan ta par 3 m ają dwa ośrodki podczepiania wrzecion m itotycznych do bruzdy A B /PI po obu stronach przegrody (Ryc. 5c; strzałka), podczas gdy w zarodkach dzikich taki ośrodek na przegrodzie jest tylko w blastom erze tylnym (porównaj schem at Ryc. 5a i 5c). Zatem w fenotypach par 3 rotacji podlegają oba wrzeciona mitotyczne zarów no w blastomerze AB jak i P I. P row adzi to do nieprawidłowego ułożenia względem siebie blastom erów i b raku k o n tak tu kom órki P2 z ABp (Ryc. 5c) i oczywiście zarodek ginie.
Prawdopodobnie białko biegunowe CDC42 aktywuje kinazę PAR1 wyznaczając gradient tylno-przedni aktywacji jaja, a inne białka PAR uczestniczą w wyznaczaniu biegunowości zarodka niezbędnej dla prawidłowego rozwoju robaka [19, 29, 31, 32]. Jednak wyniki
te, aczkolwiek im ponujące, dotyczą tylko embriogene- zy jaj o rozwoju determ inacyjnym .
VI. Powstawanie osi w jaju Drosophila mela- nogaster jako przejaw wzajemnego oddziaływania na siebie oocytu i komórek folikularnych
W jaju Drosophila melanogaster białka biegunowe w ykryto w bruzdach podziałowych w blastoderm al- nych kom órkach, a więc w bardzo późnym stadium bruzdkow ania [24]. D eterm inacja osi ciała odbyw a się wcześniej, w trakcie oogenezy, i jest pod kon tro lą som atycznych kom órek folikularnych otaczających jajo. B adania wykazały, że pow stanie osi ciała jest złożonym procesem kolejnych wzajemnych oddziaływań pom iędzy jajem a otaczającą w arstwą kom órek folikularnych. Te oddziaływania m ają charakter pewnej sekwencji wysyłania sygnałów i odbierania ich, a następnie reakcji na sygnał w postaci reorganizacji cytoszkieletu i pow staw ania nowych transkryptów zarów no w jaju, jak i w poszczególnych kom órkach folikularnych (metaforycznie mówiąc istnieje rozm ow a pom iędzy oocytem, a kom órkam i folikularnymi). A więc instrukcja struk tu ralna w ja ju muszki ow ocowej tw orzy się przez pierw otne zróżnicowane podłoża cytoplazm atycznego ja ja na etapie poprzedzającym pierwszy cykl m itotyczny przez kolejne interakcje ja ja z poszczególnymi grupam i przylegających kom órek folikularnych.
D la wyznaczenia osi przednio-tylnej i grzbietowo- brzusznej w oocycie D. melanogaster m a zasadnicze znaczenie określone rozmieszczenie poszczególnych rodzajów m RNA i białek wielu produktów genów m atczynych. W czesne stadia tworzenia polarności oocytu kontroluje kilka genów matczynych, k tóre podlegają translacji w trakcie oogenezy. m RNA bicoid stanow i determ inantę przedniego końca osi przednio- tylnej, a m RN A staufen, nanos, oskar i parę innych determ inują tylny koniec osi. m RN A gurken i białko G U R K E N determ inują grzbietową część osi grzbieto- wo-brzusznej jaja, a następnie zarodka. Ustalenie tych osi w postaci gradientów skupienia tych m R N A i b iałek wyznacza porządek przestrzenny losów poszczególnych części zarodka (szczegółowe omówienie S t. J o h n s t o n & N i i s s l e i n - Y o l h a r d [33]*). (W obrębie tych części zarodka odbyw a się pierw otna i w tórna segmentacja, ale te późne etapy nie stanow ią przedm iotu tego artykułu). Badania nad oogenezą wykazały jak ważną rolę w tym procesie odgrywa pierwotne funkcjonowanie cytoszkieletu, migracja jąd ra i różne formy kon tak tu jaja z kom órkam i ościennymi. A oto główne stadia tego złożonego procesu [34, 35]:
* W utrzymywaniu tego porządku biorą udział m.in. wyspecjalizowane regulatory transkrypcyjne działające na poziomie chromatyny, o których mówi artykuł na stronie 238 niniejszego numeru „Postępów Biochemii”.
234 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

selekcja p ro -o o cy tu komórkiodżywcze
MTOC
postępujący rozwój 16 komo—k kanały doprowadzającew jajniku Drosophila mRNA i białka
do oocytu
Etapy rozwoju jaja O b j a ś n i e n i a : m p NA grzbietowej* determinanty GURKEN
» - mRNA bicoid
— mRNA determinanta bieguna tylnego jaja
Ryc. 6 Oogeneza Drosophila melanogaster. Według [34] uproszczony, a. schemat selekcji oocytu i komórek odżywczych, b. schemat powstawania kanałów doprowadzających m RNA do oocytu, c. schemat powstawania w owarioli coraz bardziej zaawansowanych zespołów komórek składających się z komórek odżywczych i oocytu otoczonego warstwą komórek folikularnych, d. kolejne stadia rozwoju jaja w zespole komórek i stopniowe pojawianie się, strefowego występowania determinant w jaju. W późnym stadium rozwoju po stronie grzbietowej osłonki jajowej pojawiają się wyrostki (jeden przedstawiony na schemacie). Inne objaśnienia w tekście.
skierowanym i ku centrosom ow i (ang. microtubule organization center, M TO C ) oocytu (Ryc. 7a).
Szczegółowe badania wykazały, że w stadiach 1-6 rozwoju, wiązki m ikrotubuli są dekorow ane dyneiną i wobec tego cytoplazm a jest transportow ana aktywnie z kom órek odżywczych ku centrosom ow i oocytu. W tym okresie jąd ro wraz z centrosom em znajduje się w środku oocytu i tam napływ ają przede wszystkim m RNA oskar, a także m RN A i białka STA U FE N i G U R K E N , a nieco później m RN A bicoid. M imo, że nie m a jeszcze wyraźnej strefowości w jajo, białka STA U FEN i G U R K E N są w tylnej okolicy oocytu [34, 35] (Ryc. 6d).
W yraźnie strefowe rozmieszczenie poszczególnych typów m RN A i białek pojaw ia się w 7 i 8 stadium oogenezy i jest związane ze zm ianą kierunku transportu cytoplazm y względem centrosom u. Centrosom wraz z jądrem przesuwa się ku tyłowi oocytu i wobec tego najpóźniej transportow any m RN A bicoid zostaje energicznie przesunięty do bieguna przedniego jaja, a pozostałe p rodukty do końca tylnego jaja. Szczegółowe badania nad m utantam i tego procesu, i badania nad rolą m ikrotubuli w polaryzow aniu się ja ja [34, 35] zostały potw ierdzone wykryciem udziału kinezyny
a.
Jajnik Drosophila składa się z 16 owarioli; w których pow stają zespoły kom órek otaczających jajo. W raz z rozwojem poszczególne zespoły kom órek tow arzyszące oogenezie są przesuw ane ku tyłowi ovarioli, a w przedniej części pow tarza się proces tworzenia nowego zespołu (Ryc. 6a). Oogeneza Drosophila rozpoczyna się asym etrycznym podziałem kom órki wyjściowej na proksym alnym końcu owarioli na potom ną kom órkę wyjściową i na kom órkę zw aną cytoblastem . C ytoblast przechodzi przez kolejne cztery podziały k tórym towarzyszą niepełne cytokinezy (Ryc. 6a; M l- M4). W rezultacie pow staje cysta 16 siostrzanych kom órek połączonych między sobą m ostam i cytoplaz- m atycznym i zwanymi kanałam i. Dwie kom órki w o b rębie 16-tu są połączone z innymi największą liczbą kanałów a jedna z tych dw óch uprzywilejowanych kom órek staje się oocytem. M echanizm tej determ inacji nie jest znany. T ak więc tylko jedna kom órka stanie się oocytem, a pozostałe 15 kom órek stanow ią poliploidalne kom órki odżywcze (ang. nurse cells), k tóre przekazują poprzez kanały m RN A i cytoplazm ę do oocytu (Ryc. 6b). T aka cysta jest otoczna w arstwą diploidalnych som atycznych kom órek folikularnych i stanow i kom orę jajow ą (ang. egg chamber) przesuw aną do części tylnej owarioli (Ryc. 6c, Ryc. 7a). W ciągu 79 godzin, kolejne 13 stadiów rozwoju prow adzi do pow stania dojrzałej kom órki jajowej (Ryc. 6d). W raz z przesuwaniem się ku tyłowi oocytu wszystkie kanały początkow o gwiaździście połączone między sobą stają się wydłużonymi kanałam i z wiązkam i m ikrotubuli
Wędrówka jądra (gruba strzałka) ku grzbietowej stronie jaja.
Schemat powiększenia fragmentu grzbietowego jaja.
Białko GURKEN sekretowane(—>) odziaływuje ra receptory TORPEDO! >—) powodując determinację grzbietowych komórek folikularnych.
Ryc. 7 Schemat podwójnego pojawiania się kolejno determinacji komórek folikularnych tylnych i grzbietowych przez sek- retowanie białka G U R K EN w trakcie migracji jądra w jaju Drosophila melanogaster.a. determinacja tylnych komórek folikularnych (pogrubione obrysy komórek) w stadium 6-8 rozwoju, b. determinacja grzbietowych komórek folikularnych (pogrubione obrysy komórek) w stadium 8-10 rozwoju, c. schemat oddziaływania jaja w stadium 8-10 na przyległe komórki folikularne (system sygnałów G U RK EN i receptorów TORPEDO). Według danych [40]. Inne objaśnienia w tekście.
Sygnał białka GURKEN determinuje tylne komórki folikularne.
Migracja na m ikrotubulach(strzałki) materiału mRNA do jaja (wstępna segregacja).
b. Zmiana kierunku transportu na mikrotubulach (strzałki), ostateczna segregacja
mRNA biegunów jaja.
sygnał białka GURKEN
determinuje grzbietowe komórki folikularne
POSTĘPY BIOCHEMII 42 (3 ) , 1996 235http://rcin.org.pl

w tym transporcie ku peryferiom ja ja [36].W tym okresie białko GURKEN znajdujące się na
tylnym końcu oocytu jest wydzielane na zewnątrz jaja jako białko sekrecyjne i zaczyna oddziaływać jako bodziec na ościenne komórki folikularne. Odebranie tego sygnału przez tylne komórki folikularne powoduje ich określoną determinację jako tylne komórki folikularne (Ryc. 7a; pogrubione kom órki folikularne). Jak się okazało białko G U R K E N jest analogiem czynnika T G F alfa (ang. transformation growth factor a) i zawiera pow tórzenia sekwencji charakterystycznych dla EG F. G U R K E N uw alniane z tyłu oocytu oddziaływuje aktywująco na białko receptora błonowego (o charakterze T G Fr) w przyległych kom órkach folikularnych [37, 38]. To białko receptora kom órek folikularnych jest kodow ane przez gen torpedo i przyłączenie sygnału G U R K E N pow oduje uruchom ienie syntezy czynników transkrypcyjnych w kom órkach folikularnych i wyznacza ich nieodw racalną determ inację (Ryc. 7c).
Najbardziej jednak niezwykłym odkryciem było stwierdzenie, że genotyp i determinacja losu poszczególnych komórek folikularnych oddziaływują zwrotnie na polarność i losy jaja. O tóż m utanty genu torpedo, dotyczące przecież białek pojawiających się wyłącznie w kom órkach folikularnych, a nie w jaju, nie tworzyły prawidłowej osi przedniotylnej oocytu, a defektywny zarodek obum ierał. T ransplantacja dzikiego (norm alnego) ja ja do kom ory jajowej m utan ta torpedo p row adziła do śmierci zarodka, podczas gdy zm utow ane jajo torpedo im plantow ane do prawidłowej kom ory jajowej z norm alnie zdeterm inow anym i kom órkam i folikular- nymi rozwijało się praw idłow o pom im o mutacji. P o dobne badania wykazały, że m ała grupa kom órek folikularnych przednich kontroluje praw idłow ą lokalizację m RN A bicoid w ja ju Drosophila. Dalsze badania potwierdziły, że determ inacja osi przedniotylnej ja ja wymaga aby zdeterm inow ane kom órki folikularne przednie i tylne wydzieliły specyficzne białka sekrecyjne oddziaływujące zw rotnie na odpowiednie receptory w przyległych częściach jaja. Procesy zw rotnego oddziaływania zdeterm inowanych kom órek folikularnych na poszczególne części oocytu m ają istotnie charakter odbioru sygnału kontrolującego praw idłową polarność jaja, bo np. p roduk t genu torso wcześnie pojawiający się na powierzchni oocytu jest receptorow ą kinazą tyrozynow ą, a jej wczesna i prawidłow a aktywacja warunkuje prawidłowy rozwój zarodka [39].
W następnych stadiach rozwoju (8 do 10), jąd ro oocytu przemieszcza się wraz z przyległą cytoplazm ą zawierająca białko G U R K E N z położenia tylnego ku stronie grzbietowej oocytu (Ryc. 7a; gruba strzałka). M echanizm tego przemieszczania jąd ra nie jest znany. W stadium 10 przemieszczone jąd ro aktywuje pow tórnie sekrecję białka G U R K E N . Przyległa cyto- plazm a bogata w G U R K E N rozpoczyna sygnalizację oddziaływując tym razem na przyległe kom órki folikularne grzbietowe (Ryc. 7b; pogrubione kom órki folikularne). K om órki folikularne odbierają ten sygnał i sta
ją się kom órkam i zdeterm inowanym i (grzbietowymi) (Ryc. 7c). Łatw o uchwytnym przejawem tej grzbietowej determ inacji kom órek folikularnych jest ich zdolność do w ytw arzania charakterystycznego ukształtow ania chorionu (czyli osłonki jaja) w postaci w yrostków (Ryc. 6d; ostatnie stadium rozwoju). Tak zdeterm inow ane grzbietowe kom órki folikularne oddziaływują zw rotnie na przyległą część ja ja rozpoczynając serie kolejnych przem ian w wyniku czego powstaje determ inacja osi grzbietowo-brzusznej jaja. Tak więc obie osie jaja Drosophila: przednio-tylna i grzbietowo- brzuszna powstają w wyniku dwukrotnego powtórzenia sekrecji sygnału GURKEN z oocytu, odbieranego przez komórki folikularne, które po zdeterminowaniu ich różnicowania oddziaływują zwrotnie na ten oocyt nadając mu określoną determinację okolic przedniej, tylnej i grzbietowej [40].
VII. Wnioski i pytania
W szystkie przytoczone tu przykłady wskazują na rolę procesów odbierania odpowiednich bodźców na determ inację polarności ciała kom órek. W wielu przypadkach udało się już powiązać rolę sygnałów z zew nątrz z nieprzypadkow ą reorganizacją cytoszkieletu, a naw et zbadać mechanizm transdukcji sygnału za pośrednictw em białek typu Ras, Rac, Rho i CDC42. Istnieją poważne przesłanki aby sądzić, że odkryte m echanizm y są uniwersalnymi m echanizm am i tw orzenia instrukcji przestrzennej w różnicow aniu i em briogenezie. Jest znaczny postęp w badaniach dotyczących organizacji i funkcjonow ania bruzdy cytokinety- cznej i septyn [41]. Niezwykle istotne są również badania nad wpływem sygnałów na urucham ianie i przełączanie jednego szlaku morfogenezy (np. p o działowej), na inny typ morfogenezy (np. pod wpływem gam onu) [42, 43].
Jak do tąd brak jednak wiadom ości o udziale białek polarnych we wczesnej embriogenezie Drosophila*, i kręgowców. N adal nieznany jest mechanizm regulujący instrukcję przestrzenną dotyczącą migracji aktyny w cyklu kom órkow ym i wyznaczania osi wrzeciona m itotycznego. N ieznane są m echanizm y wczesnych etapów tw orzenia się pierwotnej organizacji cytoszkieletu jaj i migracji struk tu r wewnątrz kom órki np. przesuw ania się jąd ra ja ja Drosophila niezbędnego dla prawidłowej determ inacji osi ciała. Słabo poznany jest m echanizm regulacji kolejności (tzw. zegara) przem ian cytoplazm atycznych w embriogenezie i różnicowaniu. W tej chwili badane są m echanizm y zw rotnych oddziaływań kom órek różnych typów na przebieg ich różnicow ania i uzyskania polarności. Odkrycie właśnie tych m echanizm ów jest obecnym wyzwaniem; nic dziwnego, że właśnie tymi procesam i zajm ują się
* Właśnie ukazała się publikacja na ten temat: M u r p h y AM, M o n t e l l D J (1996) J Celi Biol 133: 617-630
236 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

obecnie ubiegłoroczny noblista D r W i e s c h a u s [44] i niemniej słynny D r N u r s e [45,46] — odkryw ca uniwersalnej roli cyklin w sterow aniu cyklem kom órkowym .
Koszty artykułu są finansowane z grantu KBN Nr 6P04C 07110.
A rtyku ł otrzymano 16 stycznia 1996 r. Zaakceptowano do druku 17 maja 1996 r.
Piśmiennictwo
1. C h e n e v e r t J (1994) M ol Biol Celi 5: 1169-11752. H a l l A (1990) Science 249: 635-6403. T a k a i Y, S a s a k i T, T a n a k a K, N a k a s h i n i H (1995)
Trends Biol Sei 20: 227-2314. R a p p a p o r t R (1961) J Exp Zool 148:81-895. L e w D, R e e d SI (1995) Curr Op Genetics Dev 5: 17-236. C h a n t J, H e r s k o w i t z I (1991) Cell 65: 1203-12127. C h a n t J, C o r r a d o K, P r i n g l e JR, H e r s k o w i t z
I (1991) Cell 65: 1213-12248. K o n o p k a JB, D e M a t t e i C, D a v i s C (1995) M ol Cell
Biol 15: 723-7309. C h a n t J, S t o w e r s L (1995) Cell 81: 1-4
10. A d a m s AE, J o h n s o n DI , L o n g n e c k e r RW, S l o a t BF, P r i n g l e J R (1990) J Cell Biol 111: 131-142.
11. J o h n s o n D, P r i n g l e J (1990) J Cell Biol 111: 143-15212. Z i m a n M, P r e u s s D, M u l h o l l a n d J, O ’ B r i e n J M,
B o t s t e i n D, J o h n s o n D I (1993) M ol Biol Cell 4: 1307- 1316
13. M i l l e r PJ , J o h n s o n D I (1994) M ol Cell Biol 14: 1075- 1083
14. Z h e n g Y, C e r i o n e R, B e n d e r A (1994) J Biol Chem 269: 2369-2372
15. S i m o n M - N , D e V i r g i l i o C, S o u z a B, P r i n g l e JR, A b o A, R e e d S I (1995) Nature (Lond) 376: 702-705
16. R i e d l e y A J (1994) J Cell Sei, Suppl 18: 127-13117. O l s o n MF , A s h w o r t h A, H a l l A (1995) Science 269:
1270-1272
18. R o n D , Z a n i n i M , L e w i s M, W i c k n e r R B , H u n t LT, G r a z i a G, T r o n i c k SR, A a r o n s o n S A, E v a A (1991) New Biol 3: 372-379
19. G l o t z e r M, H y m a n A (1995) Curr Biol 5: 1102-110520. C h e n e v e r t J, V a l t z N, H e r s k o w i t z I (1994) Genetics
136: 1287-129721. M a z z o n i C , Z a r c o v P , R a m b o u r g A , M a n n C (1993)
J Cell Biol 123: 1821-183222. Z i g m o n d S H (1996) Curr Op Cell Biol 8: 66-7823. F r a n k h a u s e r C, R e y m o n d A, C e r u t t i L, U t z i g S,
H o f m a n n K, S i m a n i s V (1995) Cell 82: 435-44424. N e u f e l d TP , R u b i n G M (1994) Cell IT. 371-37925. M u h u a I, K a r p o v a TS, C o o p e r J A (1994) Cell 78:
669-67926. M a n s e r E, L e u n g T, S a l i h u d d i n H, Z h a o Z, L i m
L (1994) Nature (Lond) 367: 40-4327. S h i n j o K, K o l a n d J G, H a r t MJ , N a r a s h i m a h a n
V, J o h n s o n D I, E v a n s T, C e r i o n e R A (1990) Proc Natl Acad Sei USA 87: 9853-9857
28. K a y n e PS, S t e r n b e r g P W (1995) Curr Op Genetics Dev 5: 38-43
29. G u o S, K e m p h u e s K J (1995) Cell 81: 611-62030. S t r o m e S (1993) Cell 72: 3-631. P r i e s s J R (1994) Curr Op Genetics Dev 4: 563-56832. A l l e n VW, K r o p f D L (1992) Development 115: 873-88333. St. J o h n s t o n D, N u s s l e i n - V o l h a r d C (1992) Cell 68:
201-21934. T h e u r k a u f W E (1994) Dev Biol 165: 352-36035. S u l l i v a n W, T h e u r k a u f W E (1995) Curr Op Cell Biol 7:
18-2236. C l a r k I, G i n i g e r E, R u c h o l a - B a k e r H, J a n LY,
J a n N Y (1994) Curr Biol 4: 289-30037. N e u m a n n - S i l b e r b e r g FS, S c h u p b a c h T (1993)
Cell 75: 165-17438. C l i f f o r d R C , S c h u p b a c h T (1989) Genetics 123: 771-78739. D u f f y JB, P e r r i m o n N (1994) Dev Biol 166: 380-39540. G o n z a l e s - R e y e s A, E l l i o t t H, St . J o h n s t o n
D (1995) Nature (Lond) 375: 654-65841. C h a n t J , (1996) Cell 84: 187-19042. H e r s k o w i t z I (1995) Cell 80: 187-19743. K r ö n SJ, G o w N A R (1995) Curr Op Cell Biol 7: 845-85544. S c h e i t e r E D, W i e s c h a u s E (1993) Annu Rev Cell Biol 9:
67-9945. S n e l l V, N u r s e P (1994) E M BO J 13: 2066-207446. C h a n g F. W o o l l a r d A, N u r s e P (1996) J Cell Sei 109:
131-142
Uprzejmie zawiadamiamy PT Koleżanki i PT Kolegów o przeniesieniu siedziby Zarządu Głównego
Polskiego Towarzystwa Biochemicznego na teren Instytutu Biologii Doświadczalnej im. M . Nenckiego
pokój 632 i 633
Obecny adres:
Polskie Towarzystwo Biochemiczne ul. Pasteura 3, 02-093 Warszawa
Tel. bezpośredni 658 2099 Tel. przez centralę 65985 71 w . 352
Fax 22 5342
Dyżury biura Zarządu odbywać się będą jak dotychczas w e w torki w godz 12-18
POSTĘPY BIOCHEMII 42 (3 ) , 1996 237http://rcin.org.pl

Rola kompleksów chromatynowych w regulacji ekspresji genów podczas rozwoju
Chromatin complexes in regulation of gene expression during development
T O M A S Z T. C A L IK O W S K I*
Spis treści:
I. WprowadzenieII. Drosophila melanogaster — organizm modelowy do
badań nad rozwojemIII. Grupa genów Polycomb (Pc-G) — represory ekspresji
genówIII-I. Modele działania Pc-G
IV. Grupa genów trithorax (trx-G) i mechanizmy ich działania
V. Podsumowanie
Wykaz stosowanych skrótów: ANT-C — kompleks genów homeotycznych Antennapedia; BX-C — kompleks genów homeotycznych Bithorax; Ubx — gen homeotyczny Ultra- bithorax; hb — gen segmentacji hunchback; Pc-G — grupa genów Polycomb z Drosophila; trx-G — grupa genów trit- horax z Drosophila.
I. Wstęp
Rozwój organizm u w ielokom órkow ego możliwy jest dzięki istnieniu systemu regulacyjnego, kierującego ekspresją genów w poszczególnych polach morfo- genetycznych zarodka. Ekspresja genów u Eukariota zależy od dostępności m atrycy D N A dla polimerazy RN A i czynników transkrypcyjnych. D N A w kom órce eukariotycznej upakow any jest w złożone kom pleksy nukleoproteinow e — chrom atynę. Podstaw ow ą jed nostką chrom atyny są nukleosom y zbudow ane z b iałek histonow ych i owiniętego wokół nich D N A [32]. S truk tu ra chrom atyny wpływa w istotny sposób na dostępność D N A dla czynników regulacyjnych. N iektóre geny (tak zwane geny milczące, ang. silent genes), pozostają nieaktywne m imo obecności w kom órce białek potrzebnych do ich transkrypcji. G eny te pozostają w stanie zablokow anym również po przejściu kom órki przez mitozę. Tego rodzaju efekty przypisuje się często tw orzeniu specyficznych struk tu r chrom atynowych. Blokowanie ekspresji genów może wynikać nie tylko z występow ania kom pleksów nukleosom ów w określonej pozycji (zwykle po prom otorach genów
* Mgr, Pracownia Biologii Molekularnej Roślin, Instytut Biochemii i Biofizyki PAN, ul. Pawińskiego 5a, 02-106 Warszawa
Contents:
I. IntroductionII. Drosophila melanogaster — a model for studying develop
mentIII. The Polycomb gene group (Pc-G) — the repressors of
gene expressionIII-l. The models of Pc-G’s action
IV. The trithorax gene group (trx-G) and its mode of actionV. Conclusions
[33, 34]), ale również z tworzenia kom pleksów chrom atynow ych wyższego rzędu. Chodzi tu w szczególności o kondensację chrom atyny prowadzącą do utw orzenia nieaktywnej transkrypcyjnie heterochrom atyny.
Podczas rozwoju, w sterow aniu aktywnością za pom ocą struk tu r chrom atynow ych biorą udział dwa złożone systemy regulatorow e — represyjnie działająca grupa genów Polycomb {Pc-G) i aktyw atorow a grupa genów trithorax (trx-G ). Geny Pc-G b iorą udział w tw orzeniu struk tu r silnie skondensowanej nieaktyw nej transkrypcyjnie heterochrom atyny w rejonach D N A zawierającego geny, k tóre m ają ulec trw ałem u wyłączeniu. Geny trx-G przeciwdziałają tej represji rozluźniając chrom atynę i czyniąc ją bardziej dostępną dla czynników aktyw atorow ych [3-5]. P roduk ty obu grup genów zaangażow ane są również w wiele p ozornie nie związanych ze sobą zjawisk takich jak wygaszanie ekspresji genów (silencing) u drożdży [52], kon tro la genów rozwojowych u Drosophila i myszy a także powstawanie niektórych białaczek u człowieka [36, 39, 40].
II. Drosophila melanogaster — organizm modelowy do badań nad rozwojem
Wiele czynników zidentyfikowanych u Drosophila bierze udział w kształtow aniu podstawowego planu ciała już w bardzo wczesnych etapach rozwoju [2]. Geny związane z rozwojem u Drosophila podzielić m ożna na trzy klasy: geny mateczne, geny segmentacji i geny homeotyczne. Różnią się one organizacją m olekularną, sposobem działania i funkcją. I tak geny m ateczne (około 30) działają jako pierwsze podczas oogenezy określając główne osie jaja. G eny te od
238 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

pow iadają za wykształcenie gradientów wzdłuż osi przód-tył i góra-dół zarodka, a tym sam ym za wytworzenie wstępnych wzorów rozwoju. Działanie ich rozpoczyna się jeszcze przed zapłodnieniem. P rzedstawicielami genów matecznych są bicoid, nanos i Toll [1, 2, 6]. W kolejnym etapie rozwoju kaskadow a ekspresja genów segmentacji (około 25) dzieli zygotę na typowy u m uch szereg pow tarzających się segmentów. G eny segmentacji ulegają ekspresji po zap łodnieniu jaja, zaś ich m utacje wpływają na liczbę lub orientację segmentów zarodka. Przedstawicielam i tej grupy są geny hunchback, even skipped i hedgehog. O statn im typem genów rozwojowych są geny hom eo- tyczne działające wzdłuż osi przód-tył. D ostarczają one segmentom, za pom ocą specyficznej kom binacji produktów , inform ację niezbędną do wytworzenia struk tu r i przydatków charakterystycznych dla segmentów. G eny hom eotyczne kon tro lu ją tożsam ość i położenie segmentów, lecz nie wpływają na ich liczbę, orientację lub wielkość. M utacje w obrębie tych genów prow adzą do przekształcenia jednej części ciała w inną. I tak na przykład po tró jna m utacja w genach hom eo- tycznych abx, bx i pbx prow adzi do przekształcenia segm entu T3A (zawierającego przedzm ianki czyli zredukow ane skrzydła) w tkankę typow ą dla segm entu T2 (niosącego skrzydła). W rezultacie powstaje m ucha z dwiem a param i skrzydeł zam iast jak zwykle z jedną [1, 2, 7]. G eny hom eotyczne zgrupow ane są w dwa bardzo duże kom pleksy genowe liczące 300— 350 kb (tysięcy p ar zasad) każdy: kom pleks genów Anten- napedia (ANT-C), ulegający ekspresji w przedniej (tułowiowej) części zarodka oraz kom pleks Bithorax (BX-C) związany z rozwojem segmentów odw łokowych. W rozwój Drosophila zaangażow anych jest 8 genów hom eotycznych, z czego 5 występuje w kom pleksie AN T-C (między innym i geny Antennapedia i Deformed), a 3 w kompleksie BX-C (geny Ultrabit- horax, Abdominal-A i Abdominal-B). Podobnie zorganizow ane kom pleksy genów hom eotycznych występują również u kręgowców, w tym u myszy i człowieka [45]. Ekspresja genów hom eotycznych jest konieczna nie tylko do przejścia kom órek w stan zróżnicowany, ale również — w przeciwieństwie do wczesnych genów m atecznych i segmentacji — do podtrzym ania zróżnicowanego stanu kom órki. D latego też geny hom eotyczne są głównymi regulatoram i rozwoju. W szystkie czynniki działające na wczesnych etapach rozwoju (geny mateczne, segmentacji i hom eotyczne, a także ich produkty) m ają ściśle ograniczony czas działania. N a przykład p roduk t genu segmentacji hunchback (hb) działa jak o represor hom eotycznego genu Ultrabithorax (U bx) przez około cztery godziny em briogenezy ograniczając ekspresję Ubx w przedniej części zarodka (granicą ekspresji Ubx jest parasegm ent 5) [10]. M im o krótkiego okresu działania hb ograniczona ekspresja genu Ubx jest zachow ana podczas dalszych etapów rozwoju. Decyzje rozwojowe będące konsekwencją aktywności pierw otnych regulatorów
m uszą być zatem podtrzym ane przez osobny m echanizm zwany „pamięcią kom órkow ą”. T aka działająca na dużą odległość i przez długi czas regulacja oparta jest u Drosophila na kontroli kondensacji chrom atyny przez duże kom pleksy białkowe, aktywujące lub re- prym ujące długie regiony chrom osom owe. W ażną cechą „pamięci kom órkow ej” jest zdolność do rozpoznaw ania aktywnego lub nieaktywnego stanu ekspresji genów i do jego utrzym ania przez wiele podziałów kom órkowych. W przeciwieństwie do wczesnych regulatorów rozwoju, składniki takiej „pamięci” występują powszechnie we wszystkich kom órkach.
III. Grupa genów Polycomb (Pc-G) — represory ekspresji genów
Geny Pc-G zostały zidentyfikowane jak o represory genów homeotycznych. M utacje w poszczególnych genach tej grupy prow adzą do transform acji przypominających transform acje wywołane m utacjam i w samych genach hom eotycznych [1] jak na przykład pojawienie się nóg pierwszej pary w miejscu nóg drugiej i trzeciej pary. D o tej pory poznano kilkanaście (11-15) genów tej grupy, z których najlepiej zbadane zostały geny Polycomb (Pc) [1, 11], extra sex combs (esc) [12], Additional sex combs (Asx), Posterior sex combs (Psc), Sex comb on midleg (Scm ) [14], Poly comblike (Pci), Super sex combs (Sxc) [15], polyhomeotic (ph) [16], Sex combs extra (See) [17], Enhancer o f zeste (E(z)) [18 ,19] (Tab. 1). Liczbę genów tej grupy szacuje się na około 30-40 [8]. G eny grupy Pc-G są niezbędne do represji genów kontrolujących tożsam ość segmentów (genów homeotycznych) A N T-C i BX-C w ciągu rozwoju. Zaobserwowano, że w zarodkach niosących m utację w genach Pc-G wczesny wzór ekspresji genów hom eotycznych jest identyczny jak w zarodkach „dzikich”, nie mających m utacji w genach Pc-G. W ten sposób wykazano, że Pc-G nie są zaangażow ane w wytworzenie wzorca ekspresji genów hom eotycznych, ale raczej w jego podtrzym anie [20]. Z tego pow odu geny Pc-G uważa się za kluczowe elementy systemu pamięci kom órkowej. Z akłada się, że p rodukty Pc-G mogłyby rozpoznaw ać nieaktywny stan ko n kretnego genu hom eotycznego, będący rezultatem działania wczesnych genów rozwojowych, a następnie utrzym ywałyby ten stan w stabilny i dziedziczny sposób przez wiele podziałów kom órki podczas rozwoju. Elementem kierującym kom pleks białek Pc-G do właściwej sekwencji D N A może być represor wiążący się tylko przejściowo do celu (na przykład wczesny czynnik transkrypcyjny związany z DNA). P raw dopodobnym kandydatem do takiej funkcji może być p roduk t genu hunchback (hb) [42], Stwierdzono, że geny Pc-G kontro lu ją nie tylko geny homeotyczne, ale również znaczną liczbę innych genów (będących często regulatoram i procesów rozwojowych) prow adząc do ich inaktywacji [8, 21]. U stalono na przykład, że jeden z genów Pc-G, E(z), jest niezbędny do utrzym ania
POSTĘPY BIOCHEMII 42 (3 ) , 1996 239http://rcin.org.pl

stabilnej represji dwóch genów segmentacji, które są przejściowo blokow ane przez cząsteczki b iałka hb — knirps i griant. W ykazano, że fragm ent regionu regulacyjnego knirps (1.8 kb) zawierający liczne miejsca w iązania dla hb wykazuje prawidłow y wzór ekspresji genu reporterow ego (podłączonego w opisywanym doświadczeniu do tego regionu) tylko w obecności E(z), co świadczy o wspólnej sekwencji docelowej dla wczesnego represora hb oraz dla kom pleksu Pc-G [41]. D o tej pory scharakteryzow ano na poziom ie m olekularnym trzy geny grupy Pc-G. Polycomb (PC) jest m ałym genem (4 kb) kodującym białko jądrow e o d ługości 390 am inokw asów (44 kD a) [22]. Zarów no sam transkrypt jak i białko rozmieszczone są równom iernie we wszystkich tkankach, chociaż najwięcej białka Pc stwierdza się w takich silnie proliferujących tkankach jak ośrodkow y układ nerwowy, dyski imaginalne i kom órki hodow ane in vitro [23, 27]. Białko Pc zawiera konserw ow any ewolucyjnie m otyw — chro- m odom enę, złożony z 48 am inokwasów i położony blisko N -końca cząsteczki. M otyw ten występuje między innym i w białku H P1 (białko związane z hetero- chrom atyną Drosophila melanogaster) [25] jak również w białkach M31 i M 32 myszy oraz w mysim białku CHD-1 zdolnym do w iązania DNA. M im o tych związków z chrom atyną nie w ykryto ani w iązania białka Pc do D N A in vitro, ani też nie wykazano istnienia w tym białku znanych m otywów wiążących się z DNA. Świadczy to o działaniu Pc poprzez dodatkow e czynniki. Białka Pc, polyhomeotic (ph) i Posterior sex combs (Psc) a także białko Polycomb-like (Pel) wiążą się w około 80-100 różnych, lecz niemal wspólnych dla tych czterech białek, miejscach na chrom osom ach
politenicznych, przy czym białko Pcl wiąże się do chrom osom ów samodzielnie jeszcze w innych miejscach. W yniki takie uzyskano za pom ocą przeciwciał skierow anych przeciwko tym białkom [23, 24]. Białko polyhomeotic (ph) zostało dobrze poznane pod względem m olekularnym ; ph jest dużym białkiem (168 kD a) [24], zawierającym m otyw pojedynczego palca cynkowego, co sugeruje zdolność w iązania do DNA. Również białko Posterior sex combs (170 kD a) zawiera region bogaty w cysteiny zdolny teoretycznie do tw orzenia palców cynkowych i do wiązania D N A [26].
Białko Psc występuje w 75% miejsc chrom osom owych, w których wiążą się białka Pc i ph [29]. W spólna im m unoprecypitacja białek tej grupy wskazuje, że Pc-G działają w dużych m ultim erycznych kom pleksach zawierających co najmniej 10 składników [4]. Bezpośredni dow ód na związek białek represyjnych ze struk tu rą chrom atyny pochodzi z badań nad Enhancer ofzeste (E(z)), jedynym przedstawicielu grupy Pc-G, dla którego opisano term owrażliwą m utację [29]. Inak- tywacja białka E(z) poprzez podniesienie tem peratury hodow li larw Drosophila do wartości niepermisywnej (zmiana z 23°C do 29°C) pow odow ała ogólną dekon- densację (rozluźnienie struktury) chrom osom ów politenicznych. Brak p roduk tu E(z) pow odow ał dysrupcję kom pleksu Pc-G i odłączenie białek Psc, ph i Pc od genów docelowych. W yniki te świadczą o wpływie jak i m ultim eryczne białka Pc-G wywierają na wyższy p o ziom organizacji chrom atyny — usunięcie jednego tylko białka tej grupy prow adzi do widocznych cytologicznie zm ian organizacji chrom atyny.
P odobnie interesujące są wyniki badań nad ssaczym hom ologiem genów Psc i Su(z)2 — mysim genem bmi-1
Tabela 1.Sklonowane geny z grupy Polycomb i ich produkty [3].
Gen Masa cząsteczkowaZnane motywy
białkowe Homologi
Polycomb (Pc) 44 kDa chromodomena Su(var) 2-5 (Drosophila) M33 (mysz)
polyhomeotic (ph) 169 kD a palec cynkowy rae 28 (mysz)
Posterior sex combs (psc)
170 kD a palec RIN G BMI-1 (mysz/ człowiek) mell8 (mysz)
Polycomb-like (Pcl) 95 kDa nieznane —
Enhancer o f zeste E(z)
87 kD a domena trx, palec cynkowy
trx, Su(var) 2-9, ssacze
Suppressor o f zeste 2 (S«[z]2)
144 kD a palec RING BMI-1 (mysz/ człowiek
extra sex combs (esc)
48 kDa powtórzenia WD(oddziaływaniabiałko-białko)
TUP-1 (drożdże) i ssacze
Chromodomena Pc jest niezbędna do działania białka [27], natom iast domeny typu palców cynkowych w białkach ph i E(z) i palców R IN G (Psc, Su(z)2) nie są nieodzowne do funkcjonowania tych białek [26, 28].
240 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

(znany jest również ludzki hom olog tego genu [37]). G en ten jest zaangażow any w pow staw anie białaczek kom órek B i T [35, 36], zaś jego b rak u myszy bm il prow adzi do przekształcenia przednich części szkieletu osiowego w struk tury tylnych kręgów [37]. N adeksp- resja bmi-1 u zwierząt tram genicznych ujaw nia przeciwny fenotyp: tym razem dochodzi do przesunięcia ekspresji licznych genów hom eotycznych (w tym genu H oxc-5 ) ku tyłowi ciała i szeregu deformacji rozwojowych. Deform acje te, to między innym i przem ieszczenie w yrostka ościstego (processus spinosus) z d ru giego kręgu piersiowego na trzeci (zmiana tożsam ości kręgu z T3 na T2), wydłużenie i pogrubienie żeber LI i zlanie się łuków neuronalnych na kręgach szyjnych C l i C2 [38]. Represja genów H ox przez gen bmi-1 przypom ina zatem funkcję genów Psc i Su(z)2 w sto sunku do genów hom eotycznych Drosophila. N a to m iast p roduk t drożdżow ego hom ologu genu extra sex combs (esc), Tup-1, choć nie jest zdolny do bezpośredniego w iązania się z DNA, jest w stanie tworzyć duże multim eryczne kom pleksy zdolne do stabilizowania nukleosom ów w długich szeregach, co pow oduje represję transkrypcyjną [43]. Jest więc możliwe, że podobne zjawisko zachodzi również podczas regulacji genów hom eotycznych.
III-l. M odele działania genów grupy Pc-G
on ić c h r o m a ty n y
k o m p le k s P c -G
e l e m e n t o d p o w ie d z i . n a P c-G ___________
/ \ a k ty w a to r
e n h a n c e r
O inhibicja
Ryc. 1 Dwa modele działania Pc-G na chromatynę [21]
Z aproponow ano dw a możliwe m echanizm y inak- tywacji ekspresji genów przez białka Pc-G. Pierwszy polega na organizacji nici chrom atynow ej przez b iałka Pc-G w struktury wyższego rzędu (Ryc. la) [21]. D N A zawierałby miejsca reagujące na białka Pc-G (elementy odpowiedzi — ang. Pc-G response elements, P R E s), od którego rozpoczynałoby się pakow anie chrom atyny w struk tury wyższego rzędu. W ew nątrz takiego kom pleksu mogłyby zostać ukryte enhancery, niedostępne w ten sposób dla aktyw ujących czynników transkryp- cyjnych. D rugi m echanizm to tworzenie przez kom pleksy Pc-G przedziałów w ewnątrzjądrow ych (Ryc. Ib), w których elementy odpowiedzi (PRE) służą jako punk ty zaczepienia chrom atyny do białek Pc-G. Im więcej D N A zawiera taki kom pleks, tym mocniejsza jest represja D N A [30, 31]. W związku z tym m ogła to być jedna z przyczyn skupienia genów hom eotycznych w dużych blokach (kompleksach) podczas ewolucji. S tabilna represja tych genów jest najłatwiejsza do osiągnięcia wtedy, gdy ich organizacja chrom osom owa pozw ala na zbudow anie dużych kom pleksów represyjnych [21].
IV. Grupa genów trithorax (trx-G) — aktywatorów ekspresji genów
D rugą grupę genów biorących udział w regulacji genów hom eotycznych jest grupa trithorax (trx-G ) [44]. P roduk ty genowe przedstawicieli tej grupy są zdolne do aktywacji genów rozwojowych poprzez
przeciwdziałanie represji wywołanej przez kom pleksy Pc-G, nukleosom y i inne czynniki działające w trans na poziomie chrom atyny. M utacje w genach trx-G ob niżają ekspresję genów hom eotycznych podczas późniejszych etapów rozwoju. N iektóre z tych genów są supresoram i m utacji w genach Pc-G, lecz w przeciwieństwie do tych ostatnich nie tw orzą współdziałających kompleksów, zaś ich oczyszczone p rodukty m ogą (przynajmniej w niektórych przypadkach) wiązać się do D N A in vitro [46]. Białka tej grupy są heterogenne pod względem sekwencji, struk tury oraz miejsca i zapewne m echanizm u działania. Spośród poznanych do tej pory przeszło 18 przedstawicieli grupy [44,47] dokładniej poznano trzy geny i ich produkty. Są to: trithorax (trx) [40, 48], brahma [49, 50] i trithorax-like (tri), którego p roduk t zwany jest też czynnikiem GAGA [51] (Tab. 2).
brahma jest niezbędnym genem Drosophila — m utan ty hom ozygotyczne brahma giną pod koniec em briogenezy [53]. M utacje w brahma są supresoram i transform acji spowodowanych przez m utacje w genach Poly comb. Białko brahma wykazuje uderzające podobieństw o do drożdżowego ak tyw atora trans- krypcyjnego Snf2 /Sm 2, znanego również jako GAM1 i TYE3 [54]. U drożdży Snf2/Swi2 jest czynnikiem koniecznym do transkrypcji wielu regulow anych genów. Snf2/Swi2 nie wiąże się bezpośrednio do DNA, lecz łączy się z białkam i Sw il, Swi3, Snf5, Snfó i pięciom a innymi, tworząc ogrom ny kom pleks białkowy o masie 2 M D a zaangażow any w aktywację
POSTĘPY BIOCHEMII 42 (3 ) , 1996 241http://rcin.org.pl

Tabela 2.Konserwowane domeny w białkach grupy trithorax i ich homologi [4],
Białko Motyw białkowy Homologi
trithorax (trx) domena SET palce PH D
Htrx-1, (H RX, M LL, ALL-1) (człowiek/mysz)
brahma (brm) bromodomena, DNA-załeżna ATPaza, helikaza
Swi2/Snf2 (drożdże) brgl (mysz/człowiek) Hbrm (człowiek)
E(var)3-93D motyw tramtrack —
trithorax-like (tri)/czynnik GAGA
motyw tramtrack, palec cynkowy
---
transkrypcyjną [50]. Podobieństw o białek brahma i Swi2 jest największe w dwu regionach; pierwszym z nich jest dom ena A TPazy zależnej od D N A (500 aminokwasów), zaś drugim m otyw tak zwanej brom o- dom eny (77 aminokwasów), występujący często w eukariotycznych białkach regulatorow ych [4,'50]. P rzypuszcza się, że elementy te m ogą brać udział w rozbijaniu struk tu r nukleosom owych i wspom agać czynniki i aktyw atory transkrypcyjne w wiązaniu do odpowiednich elementów D N A (na przykład wiązanie aktyw atorów transkrypcyjnych GAL4-VP16 i GAL4-AH) [55, 56]. Ludzki hom olog genu brahma, BRG1, bierze również udział w kontroli cyklu kom órkowego poprzez tworzenie kom pleksów z białkiem retinoblastoma; odgrywa także rolę w integracji prowi- rusów [57],
Czynnik GAG A jest aktyw atorem transkrypcji wiążącym się do miejsc w D N A bogatych w powtórzenia (GA)n [46]. Czynnik ten kodow any jest przez gen z grupy trx-G, trithorax-like (tri), i m a 519 am inokwasów [51]. M utacje w genie tri prow adzą do transform acji hom eotycznych przypom inających m utacje w genach hom eotycznych Abd-B i Ubx (na przykład powiększenie przedzm ianek i przekształcenie segmentów odwłoka), czyli prow adzą do obniżenia ekspresji tych genów hom eotycznych. Stwierdzono, że in vitro czynnik GAGA wiąże się do D N A w pobliżu prom otorow ego bloku TATA, a następnie wykorzystując energię w postaci ATP, usuwa nukleosom y umożliwiając w ten sposób wiązanie się polimerazy II RNA i czynnika transkrypcyjnego T FIID [58, 59]. Ponadto czynnik GAG A przeciwdziała represji trans- krypcyjnej będącej rezultatem działania histonu H I [59].
G en trithorax (trx) koduje co najmniej dwie izofor- my białkowe — tr x l o masie 368 kD a i t r x l l o masie 404 kD a, z k tórych obie przechodzą złożony proces obróbki proteolitycznej [44, 48]. B iałka trx zawierają region bogaty w cysteiny, położony w centralnej części białka i zdolny do tw orzenia palców cynkowych [21, 44]. M im o to nie udow odniono bezpośredniego w iązania trx do DNA. W trx występuje również drugi motyw białkowy, położony blisko jego C-końca i wspólny dla wielu białek Drosophila, w tym dla Enhancer o f zeste, przedstawiciela grupy Pc-G. trx wiąże się do chrom o
somów politenicznych w 16 rożnych miejscach. M iejsca te w dużym stopniu pokryw ają się z miejscami wiązania białka Pc-G (na przykład E(z) [48]). W skazuje to na podobieństw a w m echanizm ie działania białek obu tych grup. U Drosophila zarodki i dorosłe owady z m utacją trx wykazują transform acje hom eotyczne podobne do obserwowanych w m utantach Sex combs reduced (Ser), Ubx, Antennapedia (Antp), Abdominal-A i Abdominal-B. Świadczy to, że trx jest konieczny do norm alnej ekspresji tych genów, należących do kom pleksów AN T-C i BX-C [60]. Niezwykle ciekawe jest działanie ludzkiego hom ologu trx, A LL1 (M LL , H R X ). G en M LL-1 (z ang. mixed-lineage leukaemia) położony jest na chrom osom ie 11, zaś jego rearanżacje (głównie typu translokacji) prow adzą do onkogenezy i pow staw ania białaczek u dzieci [61]. G en H R X (M LL ) jest niezwykle długi (około 100 kb), zawiera przynajm niej 21 eksonów, zaś jego produkt, białko0 długości 3910 am inokwasów , m a trzy regiony hom o- logii z trithorax Drosophila. Ocenia się, że 75% ostrych białaczek niemowlęcych związanych jest z rearanżacją genu H R X (lokus llq 2 3 ) [61, 62]. Heterozygotyczne m utacje w genie M L L u myszy prow adzą natom iast do zaham ow ania wzrostu, transform acji hom eotycznych szkieletu osiowego i deformacji m ostka [63]. Brak M L L (-/-) jest em brionalnie letalny. D ysrupcja jednej kopii genu (M L L + /-) pow odow ała przesunięcie g ranic ekspresji genów hom eotycznych H oxa-7 i Hoxa-9 ku tyłowi ciała, ale aktywność tych genów hom eotycznych była zniesiona w zarodkach M LL(-/-). Tak więc hom olog genu trithorax jest konieczny do określenia tożsam ości segmentów ciała i do kontroli ekspresji genów Hox.
M echanizm y działania przedstawicieli grupy trx-G są zróżnicowane. Postuluje się [21], że białka trx-G tw orzą kaskadę regulacyjną — czynniki brahma1 Tri/GAG A mogłyby rozluźniać strukturę chrom atyny z nakładem energii, a trx byłby czynnikiem „ko twiczącym” na poziom ie DNA, koniecznym do przeciwdziałania represyjnym białkom Pc-G. W ażnym m echanizm em działania trx-G może być ponad to wypętlanie DNA, dzięki czemu odległe enhancery lokują się blisko miejsc prom otorow ych [44] i wzmacniają transkrypcję.
242 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

V. Podsumowanie
Om ówione prace dowodzą, że regulacja ekspresji genów rozwojowych obejmuje nie tylko oddziaływanie białek z DNA, ale odbyw? się w jądrze również na innych poziom ach. W m iarę poznaw ania składników kom pleksów regulacyjnych będzie m ożna wyjaśnić w jak i sposób stan transkrypcyjny genów utrzym yw any jest stabilnie przez wiele podziałów kom órkowych. Jest niem al pewne, że w procesach tych struk tu ra chrom atyny odgryw a isto tną i tru d n ą do przecenienia rolę.
Podziękowanie.Chciałbym podziękować panu profesorowi dr hab. An
drzejowi Jerzmanowskiemu za wnikliwe i pomocne uwagi podczas przygotowywania tej pracy.
A rtyku ł otrzymano 5 marca 1996 r.Zaakceptowano do druku 6 maja 1996 r.
Piśmiennictwo
1. L e w i s E B (1978) Nature (Lond) 276: 565-5702. L a w r e n c e P A (1992) W: The making of the fly Blackwell
Scientific Publications, Oxford3. P i r o t t a V (1995) Curr Opin Gen Dev 5: 466-4724. O r l a n d o V, P a r o R (1995) Curr Opin Gen Dev 5: 174-1785. T a m k u n J W (1995) Curr Opin Gen Dev 5: 473-4776. A s h e n b u r n e r A (1989) W: Drosophila. A Laboratory
M anual Cold Spring Harbour, New York, Cold Spring H arbour Laboratory Press
7. S c o t t MP , C a r r o l l S B (1987) Cell 51: 689-6988. E l g i n S C R (1995) W: Chromatin Structure and Gene Regula
tion, IRL Press, Oxford University Press, Oxford9. U r i e l i - S h o v a l S, G r u e n b a u m Y, S e d a t J, R a z i n
A (1982) FEBS Lett 146: 14810. W h i t e R A H , L e h m a n R (1986) Cell 47: 311-32111. D u n c a n I, L e w i s E B (1982) W: Subtelny S, Green FB,
Developmental Order. Its Orgin and Regulation Alan R. Liss, New York
12. S t r u h 1 G (1981) Nature (Lond) 293: 36-4113. D u r a J - M, B r o c k H W, S a n t a m a r i a P (1985) M ol Gen
Genet 198: 213-22014. J u r g e n s G (1985) Nature (Lond) 316: 153-15515. I n g h a m PW (1984) Cell 37: 815-82316. D u n c a n I (1982) Genetics 102: 49-7017. B r e e n TR, D u n c a n I M (1982) Dev Biol 118: 442-45618. J o n e s RS, G e l b a r d W M (1990) Genetics 126: 185-19919. W u C - T , J o n e s R S , L a s k o P F (1989) M ol Gen Genet 218:
559-56420. S i m o n J, C h i a n g A, B e n d e r W (1992) Development 114:
493-50521. P a r o R (1993) Curr Opin Cell Biol 5: 999-100522. P a r o R, H o g n e s s D S (1991) Proc Natl Acad Sci USA 88:
263-26723. P a r o R, Z i n k B (1992) Mech Dev 40: 37-4624. D e C a m i l l i s M, C h e n g NS, P i e r r e D, B r o c k
H (1992) Genes Dev 6: 223-23225. E i s s e n b e r g J C, J a m e s TC, F o s t e r - H a r n e t t D M,
H a r n e t t T, N g a n V, E l g i n S (1990) Proc Natl Acad Sci USA 87: 9923-9927
26. B r u n k BP, M a r t i n EC, A d l e r P N (1991) Nature (Lond) 353: 351-353
27. M e s s m e r S, F r a n c k e A, P a r o R (1992) Genes Dev 6: 1241-1254
28. F r e e m o n t PS, H a n s o n I M, T r o w s d a l e J (1991) Cell 64: 483-484
29. R a s t e l l i L, C h a n CS, P i r o t t a V (1993) EMBO J 12: 1513-1572
30. S i m o n J, C h e n g A, B e n d e r W, S h i m a m u r a MJ , O ’C o n n o r M (1993) Dev Biol 158: 138-144
31. I r v i n e K D , H e l f l a n d SL, H o g n e s s D (1991) Development 111: 407-424
32. K o r n b e r g R D (1974) Science 184: 868-87133. A i m e r A, R u d o l p h H, H i n n e n A, H o r z W (1986)
EMBO J 5: 2689-269634. B r e s n i c k EH, J o h n S, B e r a r d DS, L e F e b r e P,
H a g e r G L (1990) Proc Natl Acad Sci USA 87: 3977-398135. v a n L o h u i z e n M, V e r b e e k S, S c h e i j e n B, W i e n t -
j e n s E, v a n d e r G u l d e n H, B e r n s A (1991) Cell 65: 737-752
36. H a u p t Y, A l e x a n d e r WS, B a r r i G, K l i n k e n SP, A d a m s J M (1991) Cell 65: 753-763
37. v a n d e r L u g t N M , D o m e n J, L i n d e r s K, v a n R o o n M, R o b a n u s - M a a n d a g E, t e R i e l e H, v a n d e r V a l k M, D e s c h a m p s J, S o f r o n i e w M, v a n L o h u i z e n M (1994) Genes Dev 8: 757-769
38. A l k e m a MJ , v a n d e r L u g t N M , B o b e l d i j k RC, B e r n s A, v a n L o h u i z e n M (1995) Nature (Lond) 374: 724-727
39. F o r d AM, R i d g e SA, C a b e r o ME , M a h m o n d M, S t e e l C M, C h a n Lc, G r e a v e s M (1993) Nature (Lond) 363: 358-360
40. T k a c h u k DC, K o h l e r S, C l e a r y M L (1992) Cell 71: 691-700
41. P e l e g r i F, L e h m a n n R (1994) Genetics 136: 1341-135342. B i e n z M (1992) Curr Opin Cell Biol 4: 955-96143. C o o p e r J P, R o t h SY, S i m p s o n R T (1994) Genes Dev 8:
1400-141044. K e n n i s o n J A (1993) Trends Genet 9: 75-7945. L e w i n B (1994) W: Genes V str 1166-1181 Oxford University
Press, Oxford46. B i g g i n MD , T i j a n R (1988) Cell 53: 699-71147. L i n d s l e y DL, Z i m m G G (1992) W: The Genome of
Drosophila melanogaster, Academic Press48. K u z i n B, T i l l i b S, S e d k o v Y, M i z r o c h i L, M a z o
A (1994) Genes Dev 8: 2478-249049. B r i z e l a Bl, E l f r i n g L, B a l l a r d I, T a m k u n J W,
K e n n i s o n J A (1994) Genetics 137: 803-81350. P e t e r s o n CL, D i n g w a l l A, S c o t t M P (1994) Proc
Acad Sci USA 91: 1950-195451. F a r k a s G, G a u s z J, G a l l o n i M, R e u t e r G, G y u r -
k o v i c s M, K a r c h F (1994) Nature (Lond) 371: 806-80852. W i n s t o n F, C a r l s o n M (1992) Trends Genet 8: 38753. K e n n i s o n J A, T a m k u n J W (1988) Proc Natl Acad Sci
USA 85: 8136-814054. T a m k u n J W, D e n n i n g R, S c o t t MP , K i s s i n g e r M,
P a t t a t u c c i AM, K a u f m a n TC, K e n n i s o n J A (1992) Cell 68: 561-572
55. K w o n H, I m b a l z a n o AN, K h a v a n PA, K i n g s t o n RE, G r e e n M R (1994) Nature (Lond) 370: 477-481
56. I m b a l z a n o AN, K w o n H, G r e e n MR , K i n g s o n R E (1994) Nature (Lond) 370: 481-485
57. D u n a i e f JL, S t r o b e r BE, G u h a S, K h a v a r i PA, A l i n K, L u b a n J, B e g e m a n n M, C r a b t r e e GR, G o f f S P (1994) Cell 79: 119-130
58. T s u k i y a m a T, B e c k e r PB , W u C (1994) Nature (Lond) 367: 525-532
59. C r o s t o n GE, K e r r i g a n LA, L i r a L M, M a r s h a k DR, K a d o n a g a J (1991) Science 251: 643-649
60. B r e e n TR, H a r t e P J (1993) Development 117: 119-13461. G u Y, N a k a m u r a T, A l d e r H, P r a s a d R, C a n a a n i
O, C i m i n o G, C r o c e CM, C a n a a n i E (1992) Cell 71: 701-708
62. Z e l e z n i k - L e NJ , H a r d e n AM, R o w l e y J D (1994) Proc Natl Acad Sci USA 91: 10610-10614
63. Y u B D , H e s s JL, H o r n i n g SE, B r o w n G A J , K o r s - m e y e r S J (1995) Nature (Lond) 378: 505-508
POSTĘPY BIOCHEMII 42 (3 ) , 1996 243http://rcin.org.pl

Kinazy MAP i ich rola w regulacji poziomu, składu podjednostkowego oraz stopnia fosforylacji czynnika transkrypcyjnego AP-1
Mitogen-activated protein kinases (MAPKs) — their role in regulation of protein level, subunit composition and degree of phosphorylation of the transcription factor AP-1
IR E N E U S Z W. B IE D E R M A N N *
Spis treści:
I. WstępII. Ogólny schemat kaskady MAPKIII. Kaskady MAPK występujące u kręgowcówIV. Różnicowy wpływ czynników zewnątrzkomórkowych
na aktywność poszczególnych kaskad MAPKV. Mechanizmy aktywujące kaskady MAPKVI. Czynniki transkrypcyjne jako substraty kinaz MAPVII. Regulacja aktywności czynnika transkrypcyjnego AP-1
jako efekt współdziałania różnych kaskad MAPKVIII. Uwagi końcowe
Contents:
I. IntroductionII. General scheme of the MAPK cascadeIII. MAPK cascades of vertebrate cellsIV. Differential effect of extracellular stimuli on activity of
particular MAPK cascadesV. MAPK cascades — mechanisms of activationVI. Transcription factors as the substrates of MAPKsVII. Transcription factor AP-1 regulation as an effect of
cooperation of different MAPKsVIII. Concluding remarks
Wykaz stosowanych skrótów: MAPK, kinaza MAP — kina- za białkowa aktywowana przez czynniki mitogenne, MKP— fosfataza kinazy MAP; MAPKK — kinaza kinazy MAP (kinaza aktywująca kinazę MAP); MAP3K — kinaza kinazy aktywującej kinazę MAP; MAP4K — kinaza kinazy kinazy aktywującej kinazę MAP; MAPKAPK — kinaza białkowa aktywowana przez kinazę MAP; ERK — kinaza regulowana przez sygnały zewnątrzkomórkowe (ang. Extracellular-signal Regulated Kinase), dawniej określana jako kinaza MAP; MEK — kinaza aktywująca ERK; Raf— kinaza aktywująca MEK; PKC — kinaza białkowa C; AP-1 czynnik transkryp- cyjny AP-1 (ang. Activator Protein-1), dimer białek Fos i Jun; JNK — kinaza białka c-Jun (ang. c-Jun N-terminal Kinase); SAPK —- kinaza białkowa aktywowana przez stres, synonim dla JNK; JNKK, SEK — kinaza aktywująca JNK; MEKK— kinaza aktywująca JNKK, dawniej uważana za kinazę aktywującą MEK; FRK — kinaza białka c-Fos (ang. c-Fos Regulating Kinase); p38/RK — kinaza reaktywująca MAP- KAP kinazę-2, (ang. Reactivating Kinase); RKK — kinaza aktywująca p38/RK; GTP — trójfosforan guanozyny; Ras, Racl, Cdc42, RhoA — niskocząsteczkowe GTP-azy (nisko- cząsteczkowe białka wiążące GTP, niskocząsteczkowe białka G); PAK — kinaza aktywowana przez białka Racl i Cdc42 (ang. p21-Cdc42/Rac-activated kinase); SRF — czynnik transkrypcyjny (ang. Serum Responce Factor) uczestniczący w regulacji genu c-fos; TCF — czynnik transkrypcyjny (ang. Ternary Complex Factor) uczestniczący w regulacji genu c-fos; ATF2 — czynnik transkrypcyjny uczestniczący w regulacji genu c-jun.
* Mgr, Pracownia Hodowli Komórek i Tkanek, Instytut Biologii Doświadczalnej im. M. Nenckiego PAN, 02-093 Warszawa, Pasteura 3
I. Wstęp
Funkcjonalnym wyróżnikiem aktywności czynnika transkrypcyjnego AP-1 jest zdolność do specyficznego w iązania się z sekwencją TGA(C/G)TCA, przy czym — o ile sekwencja ta występuje w regionie prom otoro- wym jakiegoś genu — związanie się z nią czynnika transkrypcyjnego AP-1 może prow adzić do wzm ożonej ekspresji p roduk tu tego genu. Należy zauważyć, że term in „czynnik transkrypcyjny AP-1” jest używany jako zbiorowe określenie hom o- i heterodim erów białek rodziny Jun (c-Jun, Jun B, Jun D), oraz hetero dim erów białek Jun z białkam i rodziny Fos (c-Fos, Fos B, Fra-1 i Fra-2). Isto tną cechą genów kodujących białka rodzin Jun i Fos, pozw alającą zaliczyć te geny do tzw. grupy genów odpowiedzi wczesnej (ang. Im- mediate Early Genes, IEG), jest to, że aktyw acja transkrypcji tych genów nie wym aga uprzedniej b iosyntezy białka [1, 2].
Aktywacja czynnika transkrypcyjnego AP-1, czy to poprzez de novo syntezę jego składników, czy też poprzez fosforylację uprzednio istniejących, następuje wskutek zadziałania na kom órkę wielu różnych czynników (hormony, neuroprzekaźniki, czynniki wzrostowe, cytokiny, czynniki rakotw órcze, szok osmotycz- ny, inne) i pośredniczy w różnorodnej, adekwatnej do działającego czynnika, odpowiedzi kom órki (różnicowanie, proliferacja, transform acja now otw orow a, apo- ptoza, inne). Chociaż wobec dużej liczby możliwych
244 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

w ariantów czynnika AP-1 nie pow inno dziwić, że czynnik ten uczestniczy w tak różnorodnych reakcjach kom órek, to jednak wyjaśnienia wymaga mechanizm, dzięki którem u działające czynniki zewnętrzne determ inują poziom, skład podjednostkow y i stopień ufos- forylow ania czynnika AP-1.
D o odpowiedzi na postaw ione wyżej ważne i ciekawe pytanie zbliżyło nas niewątpliwie odkrycie w osta tnich latach nowych kinaz białkowych [3-6], należących do grupy tzw. kinaz aktyw ow anych przez czynniki m itogenne (kinazy M AP, M APK). K inazy M A P są kluczowym ogniwem łańcucha kolejno aktyw ow anych kinaz białkowych, tworzącego kaskadę fosforylacji, zw aną kaskadą M A PK . T a ostatn ia uw ażana jest za wspólny dla kręgowców, drożdży i innych organizmów eukariotycznych podstaw ow y mechanizm sprzęgający zachodzącą na poziomie błony kom órkow ej pierw otną reakcję kom órki na działanie czynników zewnętrznych z właściwą, często w ym agającą zm iany poziom u ekspresji odpow iednich genów, odpowiedzią kom órki na dany bodziec [3, 4],
D la om awianych tutaj zagadnień szczególne znaczenie m a fakt, że dwie z trzech now oodkrytych kinaz M AP, a mianowicie JN K (ang. c-Jun N-terminal Kinase, [5] i F R K (ang. Fos Regulatin Kinase, [6] bezpośrednio fosforylują składniki czynnika AP-1. W arto również nadmienić, że dotychczas znana kinaza M A P, określana ostatnio jak o ER K (ang. Extracellular-signal Regulated Kinase), najpraw dopodobniej uczestniczy w aktywacji czynnika AP-1 [4] oraz, że udział kolejnej kinazy M A P w tym procesie jest postulow any [7].
Zasygnalizowane powyżej fakty jasno sugerują, że pytanie o mechanizm m odulow ania aktywności czynnika transkrypcyjnego AP-1 poprzez działające na kom órkę czynniki zewnętrzne m ożna rozbić na dwie części, mianowicie: (i) jak czynniki zewnętrzne wpływają na aktywność poszczególnych kinaz M A P, oraz (ii) jak różne kinazy M A P m odulują poziom, skład podjednostkow y i stopień fosforylacji czynnika transkrypcyjnego AP-1. Celem niniejszego artykułu jest, poprzedzona skrótow ą charakterystyką om aw ianych kinaz M A P i odpow iadających im kaskad M A PK , próba odpowiedzi na tak sform ułowane pytania.
II. Ogólny schemat kaskady MAPK
Składnikam i występującymi we wszystkich znanych obecnie w ariantach kaskady M A PK , zarów no u kręgowców jak i u drożdży, są: (i) kinaza M A P (M A PK , ang. M itogen-Activated Protein Kinase), (ii) kinaza aktyw ująca kinazę M AP, określana jako M A P K K (ang. M A P Kinase Kinase), oraz (iii) kinaza kinazy aktywującej kinazę M AP, określana jako M A P K K K lub M A P3K (ang. M A P Kinase Kinase Kinase). Ta ostatn ia kinaza, podobnie jak dwie poprzednie, może być aktyw ow ana przez fosforylację, z tym, że funkcję odpowiedzialnej za ten proces kinazy, określanej cza
POSTĘPY BIOCHEMII 42 (3 ) , 1996
sami jako M A P4K, m ogą w poszczególnych realizacjach kaskady M A PK pełnić różne, aktywow ane przez receptory błonowe, kinazy białkowe, w szczególności kinaza białkow a C, kinazy tyrozynowe, czy też bliżej nieokreślone kinazy aktywow ane przy współudziale białek wiążących G T P [3, 4, 8]. Należy tutaj podkreślić, że m echanizm y aktywacji M A P3K nie są do końca wyjaśnione. W szczególności wydaje się, że przynajmniej niektóre M AP3K mogą być aktywowane w procesie nie wykorzystującym fosforylacji (patrz rozdział V).
O ile zarów no M A P3K jest zwykle kinazą specyficzną dla odpowiadającej jej M A PK K , jak i M A P K K jest kinazą specyficzną dla określonej kinazy M AP, o tyle ta ostatn ia może fosforylować różne substraty, również inne kinazy, określane jako „kinazy białkowe aktywowane przez M A P K ” (M A PK A PK ) i uważane za kolejne ogniwo kaskady M A PK . Isto tną grupą substratów kinaz M A P stanow ią czynniki transkrypcyjne [3-6, 8], fosforylowane in vivo po przemieszczeniu się kinazy M A P do jąd ra kom órkow ego [9].
Om ów ione powyżej relacje między kinazam i i ich substratam i zilustrow ano na rycinie 1. Jak widać,
Ryc. 1 Aktywacja kaskady M APK jako podstawowy mechanizm umożliwiający modulację aktywności czynników transkryp- cyjnych w odpowiedzi na pobudzenie receptorów błonowych.Poza przedstawioną tutaj aktywacją MAP3K poprzez fosforylację obecnie znane są inne, prawdopodobnie niezależne od fosforylacji, mechanizmy aktywacji tej kinazy. Ufos- forylowana kinaza MAP może fosforylować substraty cyto- plazmatyczne, lub — po przemieszczeniu się do jądra komórkowego — substraty jądrowe, głównie czynniki transkrypcyjne. Kinazy poziomu MAP4K i M APKAPK nie występują (lub też nie zostały dotychczas zidentyfikowane) w części obecnie znanych kaskad MAPK. Stosowane skróty uwzględniono w wykazie pod spisem treści.
245http://rcin.org.pl

MAPKK
gen MKPinhibitorytranslacji
Ryc. 2 Regulacja aktywności kinazy MAP (MAPK) przez kinazę aktywującą kinazę MAP (MAPKK) i fosfatazę kinazy MAP (MKP).Zaktywowana (ufosforylowana przez MAPKK) kinaza MAP (MAPK) po przemieszczeniu się do jądra kom órkowego fosforyluje czynniki transkrypcyjne (Cz. tr.) związane ze specyficznymi sekwencjami w rejonie promotorowym genu fosfatazy kinazy MAP (MKP). Prowadzi to do syntezy de novo M KP. Nowopowstała fosfataza poprzez defos- forylacje inaktywuje MAPK. Inhibitory translacji przerywając widoczną tutaj pętlę sprzężenia zwrotnego prowadzą do nagromadzenia mRNA będącego produktem transkrypcji genu M K P i innych genów odpowiedzi wczesnej (ang. Immediate Early Genes, 1EG). Opracowano wg [4, 11, 57]. Nie można wykluczyć udziału innych fosfataz białkowych w regulacji M APK [10].
przedstaw iony tam schem at pokazuje jak docierający do błony kom órkow ej sygnał aktywując kaskadę M A PK , urucham ia zależne od niej procesy. N a schemacie tym nie pokazano natom iast, poznanego w znacznie mniejszym stopniu, m echanizm u wyłączającego kaskadę M A PK . W ydaje się, że pewną rolę m ogą odgrywać tutaj fosfatazy białkowe takie jak fosfataza PP-2A i fosfataza tyrozynow a [10]. Istnieją też dane wskazujące, że istotnym składnikiem tego m echanizm u jest specyficzna dla kinazy M A P fosfataza (M K P, ang. M A P Kinase Phosphatase) [11], kodowana przez jeden z genów odpowiedzi wczesnej [4]. Tak więc (Ryc. 2) kinaza M A P, aktyw ując poprzez fosforylację czynników transkrypcyjnych, transkrypcję genów odpowiedzi wczesnej, urucham ia jednocześnie pętlę ujem nego sprzężenia zw rotnego prow adzącą do jej dezaktywacji poprzez defosforylację w wyniku aktywności zsyntetyzowanej de novo fosfatazy kinazy M AP. W chwili obecnej nie są dostatecznie poznane m echanizmy prow adzące do wyłączenia kaskady M A PK na poziomie M A PK K czy M AP3K.
O pisany tutaj ogólny schem at kaskady M A PK jest, z niewielkimi m odyfikacjami, wspólny dla wszystkich obecnie znanych kinaz M AP. K inazy te wykazują także znaczne podobieństw a strukturalne i funkcjonalne. W szczególności należy odnotow ać, że (i) wszystkie kinazy M A P są aktyw ow ane poprzez jednoczesną fosforylację reszty treoniny i tyrozyny oraz (ii) wszyst
kie kinazy M A P fosforylują w substratach reszty seryny lub treoniny występujące w bezpośrednim sąsiedztwie reszty proliny (ang. proline-directed kina- ses) [3].
III. Kaskady MAPK występujące u kręgowców
Przew ażająca większość badań nad kaskadam i M A P K prow adzona jest na dwóch doświadczalnych układach modelowych: drożdżach i hodow anych in vitro kom órkach zwierzęcych (zwłaszcza ssaczych). Poniew aż znaczna część wyników, dotyczących regulacji aktywności czynnika transkrypcyjnego AP-1, pochodzi również z badań wykorzystujących ten ostatni model, ograniczę się w tym opracow aniu do bardziej szczegółowego omówienia czterech głównych klas kinaz M A P występujących u kręgowców. O trzech z nich w spom niano we wstępie do tego artykułu (ERK, JN K , FRK ), czw arta określana bywa najczęściej jako p38/RK.
Pierwsza z wymienionych kinaz, ERK, występuje w dwóch izoformach: E R K I (44 kDa) i ER K 2 (42 kD a) [12]. W arto odnotow ać, że kinazę tę pierwotnie o trzym ano ze stym ulow anych insuliną adypocytów i na podstawie oznaczanej in vitro specyficzności substra to wej nazw ano kinazą białka M AP-2 (ang. Microtubule- Associated Protein-2 kinase) [13]. Później nazwę tej kinazy zm ieniono na M A PK (ang. M itogen-Activated Protein Kinase), i to określenie, chociaż zalecane jest obecnie dla całej rodziny kinaz M AP, bywa używane jak o synonim ER K [8].
P od różnymi nazwam i występuje również JN K . M ianowicie, występujące w kom órkach człowieka izoformy tej kinazy określa się jako JNK 1 (46 kD a) i JN K 2 (55 kD a) [5, 14-16], a izozymy występujące u szczura oznacza się jako SA PK a (55 kD a, ang. Stress-Activated Protein Kinase), SAPK(3 (55 kD a) oraz SAPKy (45 kD a) [17].
K olejna kinaza, odkry ta we wrześniu 1994 r. F R K (88 kD a) jest jeszcze stosunkow o m ało poznana i — jako jedyna z om awianych tutaj kinaz M A P — nie została dotychczas sklonow ana [6].
O statn ia z om aw ianych tutaj kinaz, p38/RK , została zidentyfikow ana jako występujący u ssaków hom olog drożdżowej kinazy M AP, określanej jako H O G 1 (High-Osmolarity Glycerol response 1) [18, 19]. N iezależnie opisano ten enzym jako kinazę zdolną do reaktywacji zdefosforylowanej in vitro M A PK A P ki- nazy-2 (ang. Reactivating Kinase, R K ) [20].
Wyżej wspom niano, że aktyw acja kinaz M A P zachodzi poprzez jednoczesną fosforylację reszty treoniny i tyrozyny. Reszty te, wraz z rozdzielającym je pojedynczym am inokwasem , tw orzą tak zwane miejsce podwójnej fosforylacji. O dpow iadającą tem u miejscu sekwencją jest T hr-G lu-T yr (TEY) w ER K [12], T hr-P ro-T yr (TPY) w JN K [14-17] oraz Thr-G ly-Tyr (TGY) w p38/R K [18]. Te różne w arianty miejsca
246 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

podwójnej fosforylacji rozpoznaw ane są przez specyficzne kinazy aktywujące kinazy M A P (M APKK ). Są nimi, odpowiednio, M E K [21], JN K K [22] (zwana też SEK [23, 24]) i R K K [20],
Znane są dwa izozymy M E K [21], określane jako M EK 1 (44 kD a) i M EK 2 (45 kDa) lub też jako M K K 1 i M K K 2. Scharakteryzow ano też dwie dalsze M A P K K -M K K 3 i M K K 4 [25]. Pierwsza z tych kinaz aktywuje specyficznie p38/R K i przypuszczalnie jest izozymem RK K . D ruga kinaza (M KK4) aktywuje zarów no JN K jak i p38/RK , ale pom im o tego uw ażana jest za izozym JN K K .
Jak się przypuszcza kinazy aktywujące kinazy M A P (M A PK K ) są również aktyw ow ane poprzez podw ójną fosforylację, zachodzącą w tym przypadku w obrębie sekwencji złożonej z dwóch reszt seryny i/lub treoniny rozdzielonych trzem a innymi am inokw asam i [26]. K inazą odpow iedzialną za aktywację M E K (poprzez fosforylację w obrębie sekwencji Ser-M et-Ala-Asn-Ser, SM ANS) jest białko Raf-1 [27, 28], analogiczną funkcję w kaskadzie JN K pełni, przypisywane początkow o do kaskady ERK, białko M E K K [24, 29]. P rzypuszcza się, że któryś z czterech znanych izozymów M E K K pełni funkcję M A P3K w kaskadzie p38/R K [3, 8]. Obecnie nie są znane białka poziom u M A P K K i M A P3K kaskady FRK .
Dwie z om awianych kaskad, ER K i p38/RK , są rozszerzone o kinazę białkow ą aktyw ow aną przez M A PK , odpow iednio M A PK A PK -1 [30] i M A P- K A PK -2 [20, 30]. Pierwsza z tych kinaz, opisana pierw otnie jak o kinaza fosforylująca rybosom alne białko S6, określana jest często jako RSK [31].
O pisana w kilku ostatnich akapitach organizacja występujących u kręgowców wersji kaskady M A PK została podsum ow ana w tabeli 1. Należy zaznaczyć, że w najbliższym czasie lista znanych kinaz M A P i odpow iadających im kaskad M A PK zostanie praw dopodobnie rozszerzona o nowych przedstawicieli.
Powyższy zarys obejmuje niezbędne m inim um informacji o kaskadach M A PK . Więcej szczegółów zainteresow any Czytelnik znajdzie w poświęconych
Tabela 1.Obecnie znane kaskady M APK występujące u kręgowców
kaskadom M A PK pracach przeglądowych [3, 4, 8]. Pom inięte tutaj zupełnie kaskady M A PK drożdży om ówiono w [32].
IV. Różnicowy wpływ czynników zewnątrz- komórkowych na aktywność poszczególnych kaskad MAPK
K ilkukrotny, czasami naw et kilkudziesięciokrotny, wzrost aktywności kinaz M A P m ożna zaobserwować już w kilkanaście sekund po zadziałaniu na kom órki wielu różnych czynników. N iektóre z tych czynników, przykładow o szok osm otyczny [7, 18], prow adzą zwykle do aktywacji wszystkich badanych kinaz M AP; inne — jak naskórkow y czynnik wzrostowy (EGF) — wykazują efekt zależny od rodzaju stosowanych w badaniach kom órek [5]. Znane są też czynniki aktywujące stosunkow o selektywnie wybrane kinazy M AP. Jednakże, ponieważ JN K K (M KK4) może fosforylować zarówno JN K , jak i p38/RK, nie powinno dziwić, że czynniki wywołujące wzrost aktywności JN K pobudzają również w pewnym stopniu p38/RK [25].
Z om awianych kinaz jedynie E R K reaguje znacznym wzrostem aktywności na potraktow anie kom órek znanym aktyw atorem kinazy białkowej C (PKC) — estram i forbolu [17,18]. Z kolei ponad 30-krotny wzrost aktywności JN K obserwuje się w kom órkach poddanych działaniu krótkofalow ego prom ieniow ania ultrafioletowego. W tych samych w arunkach praktycznie nie obserwuje się zm iany aktywności ERK, n a to m iast widoczny jest k ilkukrotny wzrost aktywności p38/R K [5, 7]. D o podobnych wyników prowadzi potraktow anie kom órek kwasem okadajow ym lub anizom ycyną [5, 33]. Pierwsza z tych substancji jest znanym inhibitorem fosfataz białkowych PP1 i PP2A, druga równie znanym inhibitorem translacji. Jest ciekawe, że już przy stężeniach anizomycyny nie wystarczających do pełnego zaham ow ania translacji, obserwuje się wyraźny wzrost aktywności JN K [33].
W ażną grupę substancji aktywujących kinazy M A P stanowią, działające za pośrednictw em swoistych rece
Kaskada ERK Kaskada JN K Kaskada p38/RK Kaskada FRK
MAP3K Raf [27, 28]Raf-1 (70-75 kDa)
MEKK [24, 29]4 izoformy
? ?
MAPKK MEK [21]MEK1 (44 kDa, MKK1) MEK2 (45 kDa, MKK2)
JNKK (SEK) [22-25]JN K KSEK1M KK4
RKK [20, 25] RKK MKK3 M KK4 (?)
?
MAPK ERK [8, 12, 13]ERK I (44 kDa) ERK2 (42 kDa)
JN K (SAPK) [5, 14-17]JNK1 (46 kDa)JN K2 (55 kDa)SAPKa (55 kDa) SAPK(3 (55 kDa) SAPKy (45 kDa)
p38/RK [18-20]p38 (RK, Mpk2)
FRK [6]FRK (88 kDa)
MAPKAPK MAPKAPK-1 (RSK) [30, 31] ---- MAPKAPK-2 [20, 30] ------
POSTĘPY BIOCHEM II 42 (3 ) , 1996 247http://rcin.org.pl

ptorów błonow ych horm ony [13], neuroprzekaźniki [34], czynniki wzrostowe i cytokiny.
Aktywację JN K i p38/R K obserw owano w kom órkach traktow anych, zaliczaną do cytokin, interleuki- ną-1 [14, 17]. Bardziej selektywną aktywację p38/RK wywołuje, działająca za pośrednictw em specyficznego receptora, endotoksyna bakteryjna (lipopolisacharyd, LPS) [18].
Stosunkow o niewiele w iadom o o czynnikach wpływających na aktyw ność FRK . Aktywność tego enzymu nie zmienia się w kom órkach poddanych działaniu prom ieniow ania ultrafioletowego, czy też p o trak tow anych estram i forbolu, natom iast kilkukrotnie wzrasta w kom órkach traktow anych E G F [6]. Ten ostatni czynnik w większości badanych kom órek wywołuje także wzrost aktywności zarów no ERK, jak też — chociaż zwykle w mniejszym stopniu — JN K [5, 33]. Z odw rotną sytuacją, czyli silnym wzrostem aktywności JN K a słabym ERK, m am y do czynienia w kom órkach traktow anych, zaliczanym do cytoklin, czynnikiem m artwicy now otw orów (TN F, ang. Tumor Necrosis Factor) [15, 17].
Chociaż obecnie uważa się, że kinazy M A P pośredniczą w odpowiedzi kom órki na większość czynników zew nątrzkom órkow ych, to jednak powyższe om ówienie właściwie wyczerpuje listę czynników, których wpływ na aktywność różnych kinaz M A P porów nywano. Przyczyną tego stanu rzeczy jest to, że w ciągu niespełna dwóch lat, k tóre upłynęły od odkrycia nowych kinaz M AP, główny wysiłek badaczy został skoncentrow any na próbach ustalenia mechanizm u wiążącego aktywację różnych receptorów błonowych z poszczególnymi kaskadam i M AP. Przejdziemy teraz do omówienia wyników tych badań.
V. Mechanizmy aktywujące kaskady MAPK
Do niedaw na jedynym stosunkow o dobrze poznanym był m echanizm prowadzący do aktywacji kaskady ERK. P od koniec czerwca 1995 r. ukazały się dwie prace [35, 36] wyznaczające główne zarysy m echanizm u aktywującego kaskadę JN K . N adal niewiele w iadom o o m echanizm ach aktywujących kaskady p38/R K i FRK .
Wyżej w spom niano, że do aktywacji kaskady ER K dochodzi w kom órkach traktow anych estram i forbolu, co wskazuje na udział w tym procesie kinazy białkowej C. Faktycznie wykazano, że kinaza białkow a C, zarów no in vitro jak in vivo, jest zdolna do aktywacji białka Raf-1 poprzez bezpośrednią fosforylację [37].
Jest rzeczą ciekawą, że opisano drugi, niezależny od kinazy białkowej C, m echanizm prow adzący do ak tywacji białka Raf-1, a tym samym do „włączenia” kaskady ERK. M echanizm em ten pośredniczy w ak tywacji kaskady E R K przez czynniki wzrostowe działające poprzez receptory wykazujące aktywność k inazy tyrozynowej.
Głównym ogniwem om awianego m echanizm u są
białka rodziny Ras, k tóre wraz z białkam i rodzin Rab, Arf i R ho tw orzą nadrodzinę białek Ras [38, 39]. W szystkie one zaliczane są do tak zwanych niskocząs- teczkowych białek wiążących G T P, określanych też m ianem niskocząsteczkow ych białek G. Jak w iadom o białka G występują w formie nieaktywnej — związanej z G D P , i w formie aktywnej — związanej z G T P. Aktywację białek G, zachodzącą poprzez wymianę związanego z nimi G D P , na G T P, katalizują specyficzne białka określane jako „białka wymieniające nu- kleotydy” (ang. nucleotide exchange protein). Inak- tywacja białek G jest wynikiem hydrolizy G T P, zachodzącej dzięki wykazywanej przez białka G aktyw ności GTP-azy. Proces ten zachodzi przy współudziale specyficznych białek określanych jako G A P (ang. GTP-ase Activator Protein).
Zgodnie z obecnie panującym i poglądam i [40-42], aktyw acja kaskady E R K przez receptory czynników wzrostowych obejmuje następującą sekwencję zdarzeń: (i) związanie liganda z receptorem prow adzi do aktywacji kinazy tyrozynowej receptora, (ii) wskutek autofosforylacji receptor uzyskuje zdolność do w iązania „adaptorow ego” białka Grb2, (iii) to ostatnie białko łączy się z cytozolowym białkiem Sos, k o twicząc je przy cytoplazmatycznej powierzchni błony kom órkowej, (iv) tutaj Sos (będący białkiem wym ieniającym nukleotydy guanidynowe) spotyka białko Ras, stale zakotw iczone przy cytoplazmatycznej pow ierzchni błony, (v) białko Ras, zaktyw owane przez białko Sos, wiąże się z białkiem Raf-1 (vi) wskutek bezpośredniego oddziaływ ania z białkiem Ras dochodzi w bliżej nie znany sposób do aktywacji białka Raf-1 [43,44], co jest rów now ażne z „włączeniem” kaskady ERK.
W ażną rolę w badaniach prowadzących do rozwikłania wyżej omówionego m echanizm u, a także w b adaniach nad m echanizm am i aktywacji innych kaskad M A PK [6, 35, 36], odegrało wykorzystanie dwóch różnych typów m utacji białek G.
Pierwszy typ obejmuje m utacje które pozbaw iając b iałka G aktywności G T Pazy prow adzą do ich perm anentnej aktywności. Ekspresja takiego zm utow anego białka G w badanych kom órkach, możliwa w wyniku transfekcji tych kom órek odpow iednio skonstruowanym plazmidem, jest w ykorzystyw ana do aktywacji procesów zależnych od danego białka G. Aktywacja tak a nie wymaga oczywiście stymulacji kom órek czynnikam i zewnątrzkom órkow ym i. W arto nadmienić, że takie perm anentnie aktywne m utanty białek G są znanymi onkogenam i.
Drugi typ m utacji, określany jak o „dom inujący m utan t negatyw ny”, powoduje zwiększone pow inowactwo danego białka G do G D P. Tak zm utow ane białko nie może zostać zaktyw ow ane przez wymianę G D P na G T P, ale wiąże się z białkiem wymieniającym nukleotydy guanidynowe. D latego też ekspresja dom inującego m utan ta negatywnego białka G prow adzi do wysycenia kom órkow ej puli odpowiedniego białka wymieniającego nukleotydy guanidynow e i tym sa
248 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

mym ham uje aktywację procesów zależnych od obecnego w kom órce niezm utow anego białka G.
W łaśnie stosując oba wymienione typy m utantów białka Ras w ykazano udział tego białka w aktywacji FR K . Stw ierdzono mianowicie, że w kom órkach F9 (linia mysich kom órek now otw orow ych pochodzenia em brionalnego) kinaza ta jest aktyw ow ana przez ekspresję perm anentnie aktywnego m utan ta białka Ras, natom iast w kom órkach A 431 (linia ludzkich kom órek now otw orow ych pochodzenia naskórkowego) ak tywacja F R K przez E G F jest całkowicie znoszona przez ekspresję dom inującego negatywnego m utan ta białka Ras [6].
P odobne doświadczenia wykazały, że w stym ulow anych E G F kom órkach H eL a (linia ludzkich kom órek now otw orow ych pochodzenia nabłonkow ego) ak tywacja JN K jest całkowicie znoszona przez ekspresję dom inującego negatywnego m utan ta b iałka Ras [35]. W tych samych kom órkach ekspresja tak zm utow anego białka Ras nie m iała za to większego wpływu na aktywację JN K w odpowiedzi na czynnik m artwicy now otw orów (TNF). W skazywało to, że białko Ras nie pośredniczy w odpowiedzi kom órek H eLa na TN F. Ten sam zespół badaczy [35] wykazał dalej, że (i) wywołana przez E G F aktyw acja JN K jest częściowo
znoszona przez ekspresję dom inującego negatywnego m utan ta białka R acl należącego do rodziny białek Rho, (ii) ekspresja perm anentnie aktywnego m utanta białka R acl, a także innego przedstawiciela białek Rho, białka Cdc42, prow adzi do aktywacji JN K , (iii) aktywacja JN K przez ekspresję badanych białek Rho jest addytyw na z aktywacją wywołaną przez ekspresję perm anentnie aktywnego m utan ta białka Ras, oraz (iv) ekspresja perm anentnie aktywnych białek R acl i Cdc42 nie aktywuje ERK. Niezależnie [36] podobne wyniki uzyskano w badaniach wykorzystujących kom órki COS-7 (transform ow ane wirusem SV40 ko m órki nerki małpy), przy czym dodatkow o stwierdzono, że (v) ekspresja dom inujących negatywnych m utantów białek R acl i Cdc42 częściowo znosi ak tywację JN K w kom órkach stym ulow anych TN F. P onad to obydwa zespoły stwierdziły, że (vi) białko RhoA (także należące do rodziny białek Rho) nie jest zaangażow ane w aktywację kaskady JN K [35, 36].
Om ów ione w poprzednim akapicie wyniki wskazują więc na udział białek rodziny Rho (R acl i Cdc 42, ale nie RhoA) w aktywacji kaskady JN K . Jednocześnie obydw a zespoły sugerują, że w odróżnieniu od ak tywacji kaskady ERK, gdzie dochodzi do bezpośredniej interakcji białka Ras z białkiem Raf, aktywacja
R E C E P T O R Y B Ł O N O W E
Białko G
MAP4K
MAP3K
MAPKK
MAPK
MEK
ERK FRK
PAK(?)
v
MEKK
JNKK
i
JNK
RhoA
Ryc. 3 Rola niskocząsteczkowych białek G w aktywacji znanych kaskad MAPK występujących u kręgowców. Obecnie uważa się, że niskocząsteczkowe białka G pośredniczą w aktywacji kaskad M APK w odpowiedzi na stymulację większości receptorów błonowych, aktywując odpowiednie M AP3K bezpośrednio lub za pośrednictwem MAP4K. Ze względu na stosowaną metodykę badań (patrz rozdział V) powiązania między różnymi receptorami a poszczególnymi białkami G nie są dokładnie poznane i zostały tutaj przedstawione schematycznie jako przerywane pogrubione linie. Ciągłymi i przerywanymi liniami normalnej grubości przedstawiono (odp. główne i alternatywne) drogi prowadzące do aktywacji poszczególnych kinaz MAP. Hipotetycznej kinazie MAP aktywowanej przez szlak zależny od białka RhoA przypisuje się pewną rolę w regulacji ekspresji genu c-fos (Ryc. 4). Stosowane skróty uwzględniono w wykazie pod spisem treści.
POSTĘPY BIOCHEMII 42 (3 ) , 1996 249http://rcin.org.pl

kaskady JN K nie jest wynikiem bezpośrednim oddziaływania białek Rho z białkiem M E K K . Przypuszcza się raczej, że pośredniczy tutaj białko PA K [45, 46], będące hom ologiem drożdżowego białka STE20, pełniącego funkcję M A P4K w jednej z drożdżowych kaskad M A P K [32].
O m aw iane tutaj zależności między białkam i G a kaskadam i M A PK zilustrow ano na rycinie 3. Funkcje przedstawionej tam hipotetycznej kaskady M A PK aktywowanej przez białko Rho A [7], będą dyskutow ane w rozdziale VII.
VI. Czynniki traskrypcyjne jako substraty kinaz MAP
Istnieją dane wskazujące, że ER K fosforyluje białka c-Fos i c-Jun, jednakże, fosforylacja ta prow adzi do obniżenia aktywności czynnika transkrypcyjnego [47, 48]. P odobny efekt m a fosforylacja białka c-Fos przez aktyw ow aną przez E R K kinazę, określaną jako RSK [47], Z kolei fosforylowanej przez RSK kinazie syn- tetazy glikogenu (GSK-3) przypisuje się analogiczną funkcję wobec białka c-Jun [49]. Należy jednak po d kreślić, że niewiele w iadom o o znaczeniu fizjologicznym om aw ianych w tym akapicie procesów.
W ykazano też, że E R K in vitro fosforyluje, należące do rodziny Fos, b iałka Fra-1 i Fra-2, przy czym fosforylacja ta zachodzi w miejscach istotnych dla aktywacji tych czynników in vivo [50]. Czynnikiem transkrypcyjnym aktyw ow anym przez ER K jest, niewątpliwie, E lk l/T C F [51, 52]. Uważa się, że czynnik ten może być także aktyw ow any przez JN K [52].
Ta ostatn ia kinaza została odkryta jako enzym fosforylujący dwie reszty seryny (Ser63 i Ser73) w białku c-Jun [5, 14-17]. W ykazano, że fosforylacja tych reszt prow adzi do zwiększenia aktywności białka c-Jun jako czynnika transkrypcyjnego. Innym czynnikiem transkrypcyjnym fosforylowanym przez JN K jest ATF-2 [53]. Również w tym przypadku fosforylacja czynnika transkrypcyjnego prow adzi do podniesienia jego aktywności. W arto w tym miejscu zauważyć, że w ykazano iż białka c-Jun i ATF-2 m ogą tworzyć heterodim er [54]. Jego rolę w regulacji genu c-jun om awiam y w następnym rozdziale.
Jedynym obecnie znanym substratem F R K jest białko c-Fos. Fosforylacja tego białka zachodzi na reszcie treoniny (Thr232), znajdującej się w pozycji homologicznej do jednej z reszt seryny (Ser73) fosforylowanej w białku c-Jun przez JN K [6].
Brak jest obecnie danych o czynnikach transkryp- cyjnych fosforylowanych przez p38/RK.
VII. Regulacja aktywności czynnika transkrypcyjnego AP-1 jako efekt współdziałania różnych kaskad MAPK
Przedstaw ione w poprzednim rozdziale dane jasno sugerują, że trzy z om awianych kinaz M AP, m ianow i
cie ERK, JN K i FR K , m ogą poprzez bezpośrednią lub pośrednią fosforylację białek c-Fos i c-Jun m odulować aktyw ność czynnika transkrypcyjnego AP-1.
Jednakże wykazano też, że dwie z tych kinaz (ERK1 JN K ) wpływają z całą pewnością na poziom ekspresji białek c-Fos i c-Jun. Wyżej w spom niano, że substraty JN K , c-Jun i ATF-2, tw orzą dimer. U w aża się, że dim er taki związany jest w regionie prom otorow ym genu c-jun i odpow iada za wzm ożoną transkrypcję tego genu w odpowiedzi na fosforylację białek c-Jun i ATF-2 [54].
Jednym z ważniejszych elementów regulatorow ych genu c-fos jest tak zwane miejsce SRE (ang. Serum Response Element) rozpoznaw ane przez specyficzny czynnik transkrypcyjny określany jako SRF (ang. Serum Responce Factor) [55]. Czynnik ten może wprawdzie sam aktywow ać transkrypcję, jednak ak tywność jego zostaje znacznie zwiększona po utw orzeniu kom pleksu z czynnikiem określanym jako T C F (ang. Ternary Complex Factor) [56]. Jak w spom niano wyżej czynnik E lk l/T C F jest aktywow any przez ER K i JN K [51, 52].
Oczywiście kom pleks czynników S R F/T C F może być regulow any również przez m odulacje aktywności czynnika SRF. O statn io wykazano, że aktywacja czynnika SRF w kom órkach stym ulow anych w ystępującym w osoczu czynnikiem m itogennym — kwasem lizofosfatydylowym — jest zależna od białek G z rodziny Rho (R acl, Cdc42, a także Rho A). Udział w tym procesie kolejnej, nowej kinazy M A P uważa się za wysoce praw dopodobny [7].
Om ów iony tutaj wpływ kinaz M A P na regulację ekspresji genów c-fos i c-jun został przedstaw iony schematycznie na rycinie 4. Schem at ten uwzględnia również fosforylację białek c-Fos i c-Jun przez F R K i JN K . Chociaż pom inięto tam pozostałych przedstawicieli rodzin Fos i Jun (których regulacja przez kinazy M A P nie jest dostatecznie poznana), ze schem atu tego jasno wynika, że zarów no poziom, jak i skład podjednostkow y (w tym przypadku proporcja hom o- dim eru c-Jun i heterodim eru c-Jun/c-Fos) a także stopień ufosforylowania czynnika transkrypcyjnego AP-1 są złożoną funkcją aktywności różnych kinaz M AP.
W arto zwrócić uwagę na jeszcze jeden ciekawy problem . Jest oczywiste, że fosforylacja now opow stałego białka c-Jun jest możliwa jedynie w obecności aktywnej JN K . Tymczasem z porów nania ryciny 4. i 2. jasno wynika, że jednocześnie z białkiem c-Jun syntetyzow ana jest fosfataza (M K P) inaktyw ująca kinazy M A P [11, 57]. Względnie wysoka aktywność JN K w obecności M K P może być utrzym ana jedynie wtedy gdy wciąż aktyw na jest kinaza aktyw ująca JN K (JNKK). Zdaje się to wskazywać, że dodatkow ym czynnikiem regulującym stopień ufosforylowania n o wopowstałych czynników transkrypcyjnych może być czas przez jak i utrzym yw ana jest aktywność odpow iednich kaskad M A PK .
250 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

Ryc. 4 Współdziałanie różnych kinaz MAP w regulacji aktywności czynnika transkrypcyjnego AP-1 (schemat uproszczony).Przedstawiając wpływ różnych kinaz MAP na regulację genów c-fos i c-jun pominięto większość miejsc regulatorowych występujących w prom otorach tych genów. Uwzględniono jedynie miejsce regulatorowe SRE genu c-fos (złożone z sekwencji rozpoznawanych przez czynniki transkrypcyjne SRF i TCF) oraz nietypowe miejsce regulatorowe AP-1 genu c-jun (rozpoznawane przez dimer ATF2/c-Jun). Pytajnikiem (?) oznaczono hipotetyczną kinazę MAP aktywowaną przez szlak zależny od białka RhoA (Ryc. 3) i uczestniczącą prawdopodobnie w regulacji ekspresji genu c-fos [7]. W prawej części ryciny schematycznie zaznaczono, że — przy braku współdziałania z innymi czynnikami transkrypcyjnymi, czy też przy niedostatecznym ufosforylowaniu czynnika AP-1 — samo przyłączenie czynnika AP-1 do odpowiedniej sekwencji regulatorowej nie musi automatycznie prowadzić do transkrypcji regulowanego przez nią genu. Poza pokazanym tutaj heterodimerem c-Jun/c-Fos, znane są inne warianty czynnika transkrypcyjnego AP-1. Stosowane skróty uwzględniono w wykazie pod spisem treści.
VIII. Uwagi końcowe
O gólny schem at kaskady M A P K znany był już od początku lat dziewięćdziesiątych, nie było jednak jasne jak ten pojedynczy szlak może pośredniczyć w przekazywaniu sygnałów między całą paletą różnorodnych receptorów obecnych na powierzchni każdej kom órki a równie liczną i zróżnicow aną pulą obecnych w jej jądrze czynników transkrypcyjnych. Przypuszczano wprawdzie, że kaskada M A P K to raczej jakiś ogólny schemat, a n iejeden konkretny szlak,jednakże dopiero odkrycie, a właściwie sklonowanie, JN K zmieniło to przypuszczenie w pewność.
Chociaż postępu badań k tóry dokonał się w ciągu ostatnich dwóch lat, nie sposób nie doceniać, w arto uzmysłowić sobie, że jest to dopiero początek drogi do zadowalającego poznania wzajemnych relacji między różnym i receptoram i błonowym i i różnymi kaskadam i M A P K oraz różnym i kaskadam i M A PK i różnymi czynnikam i transkrypcyjnym i.
Zauw ażm y chociażby, że sam o wyjaśnienie regulacji
aktywności czynnika transkrypcyjnego AP-1 wymaga szczegółowej znajom ości mechanizm ów regulujących ekspresję trzech genów jun i czterech genów fos. Ekspresja tych genów prow adzi do pow stania osiemnastu możliwych podstawowych w ariantów czynnika AP-1 i znacznie większej liczby w ariantów różniących się stopniem fosforylacji.
PodziękowaniaAutor niniejszej pracy składa serdeczne podziękowania
Panu doc. dr hab. L e s z k o w i K a c z m a r k o w i za wszechstronną opiekę oraz Pani E w i e K u b l i k z a pomoc w wykonaniu rycin.
A rtykuł otrzymano 24 listopada 1995 r. Zaakceptowano do druku 20 marca 1996 r.
Piśmiennictwo
1. A n g e l P, K a r i n M (1991) Biochim Biophys Acta 1072: 129-157
2. K a r i n M (1995) J Biol Chem 270: 16483-16486
POSTĘPY BIOCHEMII 42 (3 ) , 1996 251http://rcin.org.pl

3. D a v i s R J (1994) Trends Biochem Sei 19: 470-4734. C a n o E, M a h a d e v a n L C (1995) Trends Biochem Sei 20:
117— 1225. H i b i M, L i n A, S m e a l T, M i n d e n A, K a r i n M (1993)
Genes Dev 7: 2135-21486. D e n g T, K a r i n M (1994) Nature (lond) 371: 171-1757. H i l l CS, W y n n e J, T r e i s m a n R (1995) Cell 81: 1159-
11708. S e r g e r R, K r e b s E G (1995) FASEB J 9: 726-7359. C h e n RH, S a r n e c k i C , B l e n i s J (1992) M ol Cell Biol 12:
915-92710. A l e s s i DR, G o m e z N M, M o o r h e a d G, L e w i s T,
K e y s e SM, C o h e n P (1995) Current Biology 5: 283-29511. L iu Y, G o r o s p e M, Y a n g C, H o l o b r o o k N J (1995)
J Biol Chem 270: 8377-838012. B o u l t o n TG, Y a n c o p o u l o s G D , G r e g o r y JS,
S l a u g h t e r C, M o o m a w C, H s u J, C o b b M H (1990) Science 246: 64-67
13. R a y LB , S t u r g i l l T W (1987) Proc N atl Acad Sei USA 84: 1502-1506
14. D e r i j a r d B, H i b i M, W u I H, B a r r e t t T, Su B, D e n g T, K a r i n M, D a v i s R J (1994) Cell 76: 1025-1037
15. S l u s s H K , B a r r e t t T, D e r i j a r d B, D a v i s R J (1994) Mol Cell Biol 14: 8376-8384
16. K a l l u n k i T, S u B, T si ge l n y I, S l u s s H K , D e r i j a r d B, M o o r e G, D a v i s RJ , K a r i n M (1994) Gen Dev 8: 2996-3007
17. K y r i a k i s J M, B a n e r j e e P, N i k o l a k a k i E, D a i T, R u b i e EA, A h m a d MF , A v r u c h J, W o o d g e t t J R (1994) Nature (lond) 369: 156-160
18. H a n J, L e e JD , B i b b s L, U l e v i t c h R J (1994) Science 265: 808-811
19. H a n J, R i c h t e r B, Li Z, K r a v c h e n k o Y, U l e w i t c h R J (1995) Biochim Biophys Act 1265: 224-227
20. R o u s e J, C o h e n P, T r i n g o n S, M o r a n g e M, A 1o - n s o - L l a m a z a r e s A, Z a m a n i l l o D, H u n t T, N e b - r e d a A R (1994) Cell 78: 1024-1037
21. C r e w s C M, A l e s s a n d r i n i A, E r i k s o n R L (1992) Science 258: 478-480
22. L in A, M i n d e n A, M a a r t i n e t t o H, C l a r e t FX, L a n g e - C a r t e r C, M e r c u r i o F, J o h n s o n GL, K a r i n M (1995) Science 268: 286-290
23. S a n c h e z I, H u g h e s RT, M a y e r BJ, Y e e K, W o o d g e t t J R, A v r u c h J, K y r i a k i s J M, Z o n L I (1994) Nature (Lond) 372: 794-798
24. Y a u M, D a i T, D e a k JC, K y r i a k i s J M, Z o u LI , W o o d g e t t JR, T e m p l e t o n D J (1994) Nature (Lond) 372: 798-800
25. D e r i j a r d B, R a i n g e a u d J, B a r r e t t T, W u I H, H a n J, U l e v i t c h RJ , D a v i s R J (1995) Science 267: 682-685
26. Z h e n g CF , G u a n K L (1994) EMBO J 13: 1123-113127. K y r i a k i s J M, A p p H, Z h a n g XF, B a n e r j e e P,
B r a u t i g a n DL, R a p p UR, A v r u c h J (1992) Nature (lond) 358: 417-421
28. A l e s s i DR, S a i t o Y, C a m p b e l l D G , S i t h a n a n d a m G, R a p p U, A s h w o r t h A, M a r s c h a l CJ , C o w l e y S (1994) EMBO J 13: 1610-1619
29. M i n d e n A, L i n A, M c M a h o n M, L a n g e - C a r t e r C, D e r i j a r d B, D a v i s RJ , J o h n s o n GL, K a r i n M (1994) Science 266: 1719-1723
30. S t o k o e D, C a m p b e l l D G , N a k i e l n y S, H i d a k a H, L e e v e r s SJ, M a r s h a l l C, C o h e n P (1992) EMBO J 11: 3985-3994
31. A l c o r t a DA, C r e w s C M, S w e e t LJ , B a n k s t o n L, SW, E r i k s o n R L (1989) M ol Cell Biol 9: 3850-3859
32. H e r s k o w i t z I (1995) Cell 80: 187-19733. C a n o E, H a z z a l i n CA, M a h a d e v a n L C (1994) Mol
Cell Biol 14: 7352-736234. M i t c h e l l F M, R u s s e l l M, J o h n s o n G L (1995) Bio
chem J 309: 381-38435. M i n d e n A , L i n A , C l a r e t F X , A b o A , K a r i n M (1995)
Cell 81: 1147-115736. C o so O A , C h i a r i e l l o M, Y u JC, T e r a m o t o H,
C r e s p o P, X u N, M i k i T, G u t k i n d J S (1995) Cell 81: 1137-1146
37. K o l c h W, H e i d e c k e r G, K o c h s G, H u m m e l R, Y a h i d i H, M i s c h a k H, F i n k e n z e l l e r G, M a r m e D,
R a p p U R (1993) Nature (Lond) 364: 249-25238. H a l l A (1993) Curr Opin Cell Biol 5: 265-26839. B o k o c h G M, D e r C J (1993) FASEB J 7: 750-75940. E g a n S E , W e i n b e r g R L (1993) Nature (lond) 365:781-78341. B u d a y L, D o w n w a r d J (1993) Cell 73: 611-62042. A r o n h e i m A, E n g l e b e r g D, Li N, A l - A l a w i N,
S c h l e s s i n g e r J, K a r i n M (1994) Cell 78: 949-96143. L e e v e r s SJ, P a t e r s o n H F , M a r s h a l l C J (1994)
Nature (lond) 369: 411-41444. S t o k o e D, M a c D o n a l d SG, C a d w a l l a d e r K, S y
m o n s M, H a n c o c k J F (1994) Science 264: 1463-146745. M a n s e r E, L e u n g T, S a l i h u d d i n H, Z h a o ZS, L i m
M (1994) Nature (lond) 367: 40-4646. M a r t i n GA, B o l l a g G, M c C o r m i c k F, A b o A (1995)
EMBO J 14: 1970-197847. C h e n RH, A b a t e C, B l e n i s J (1993) Proc Natl Acad Sei
USA 90: 10952-1095648. M i n d e n A, L i n A, S m e a l T, D e r i j a r d B, C o b b M,
D a v i s R, K a r i n M (1994) M ol Cell Biol 14: 6683-668849. S u t h e r l a n d C, C o h e n P (1994) FEBS Lett 338: 37-4250. G r u d a MC , K o v a r y K, M e t z R, B r a v o R (1994)
Oncogene 9: 2537-254751. G i l l e H, K ö r t e n j a n n M, T h o m a e O, M o o m a w C,
S l a u g h t e r C , C o b b M H , S h a w P E (1995) EMBO J 14: 951-962
52. W h i t m a r s h A J, S h o r e P, S h a r r o c k s AD, D a v i s R J (1995) Science 269: 403-407
53. G u p t a S, C a m p b e l l D, D e r i j a r d B, Davis RJ (1995) Science 267: 389-393
54. v a n D a m H, D u y n d a m M, R o t t i e r R, B o s c h A, d e V r i e s - S m i t s L, H e r r l i c h P, Z a n t e m a A, A n g e l P, v a n d e r E b A J (1993) EMBO J 12: 479-487
55. N o r m a n C, R u n s w i c k M, P o l l o c k R, T r e i s m a n R (1988) Cell 55: 989-1003
56. T r e i s m a n R (1994) Curr Opin Genet Dev 4: 96-10157. B r o n d e l l o J M, M c K e n z i e FR, S u n H, T o n k s N K ,
P o u y s s e e g u r J (1995) Oncogene 10: 1895-1904
Wydawca prosi o kontakt tych,
którzy chcieliby wykorzystać łamy „Postępów Biochemii” do reklamowania swych produktów i usług związanych z biochemią, biologią molekularną i biologią komórki.
252 POSTĘPY BIOCHEMII 42 (3 ), 1996http://rcin.org.pl

Molekularne podstawy cytoplazmatycznej męskosterylno- ści u roślin wyższych
Molecular basis of cytoplasmic male sterility in higher plants
H A N N A J A Ń S K A 1 M A G D A L E N A W O Ł O S Z Y Ń S K A 2
Spis treści:
I. WstępII. Organizacja genomu mitochondrialnegoIII. Charakterystyka zmian towarzyszących CMS
III-l. Zmiany w organizacji genomu mitochondrialnego III-2. Zmiany w ekspresji
IV. Geny przywracające płodnośćV. Funkcja i lokalizacja URF13VI. Zastosowanie w hodowliVII. Podsumowanie
Wykaz stosowanych skrótów: CMS — cytoplazmatyczna męskosterylność; kp — tysiąc par zasad; mtDNA — mito- chondrialny DNA; urf, orf — otwarta ramka odczytu; cox I, cox I I — geny kodujące odpowiednio podjednostkę I, II mitochondrialnego kompleksu oksydazy cytochromowej; rrn26 — gen kodujący 26S rybosomalny RNA; trnR (tRNAArg) — gen kodujący RNA transportujący argininę; kDa — kilodalton; R f— gen przywracający płodność; atpó— gen kodujący podjednostkę 6 mitochondrialnej syntazy ATP; URF 13 — białko kodowane przez gen urf 13 występujący w linii CMS-T kukurydzy; HmT — Helmintho- sporium may dis szczep T; Pm — Phyllosticta may dis.
I. Wstęp
Inform acja genetyczna kom órek roślinnych jest podzielona między jądro , m itochondria i chloroplasty, a prawidłow e funkcjonow anie rośliny wym aga współdziałania produktów genów pochodzących z tych organelli. Zaburzenie interakcji jąd ra i m itochondriów jest przyczyną cytoplazmatycznej m ęskosterylności (CMS) u roślin wyższych. Najogólniej cechę tę m ożna określić jako niezdolność rośliny do wytw arzania funkcjonalnego pyłku, a więc do zapylania [1-3]. Rozwój pyłku może być zaburzony na różnych e ta pach. W większości przypadków m orfologia roślin sterylnych jest norm alna, a jedynie m ikrosporogeneza jest niepraw idłow a [1, 4]. Często związane jest to z niewłaściwym funkcjonow aniem tapetum [1, 5]— tkanki pełniącej odżywczą funkcję w rozwoju pyłku [6]. Rzadziej rośliny sterylne nie w ytwarzają pyłku z pow odu zmienionej morfologii kwiatu. W kw iatach
1 Dr, 2 mgr, Instytut Biochemii, UW, ul. Tamka 2,50-137 Wrocław
Contents:
I. IntroductionII. Organization of mitochondrial genomeIII. Characterization of changes associated with CMS
III-l. Changes in mitochondrial genome organization III-2. Changes in expression
IV. Fertility restorer genesV. Function and localization URF13VI. Application in breedingVII. Concluding remarks
sterylnego tytoniu (Nicotiana) pręciki nie pow stają lub są przekształcane w struktury podobne do płatków albo słupka [7]. W rzadkich przypadkach wytworzony pyłek nie jest zdolny do zapylania [3]. Cytoplaz- m atyczną m ęskosterylność zaobserw ow ano u ponad 140 roślin kwiatowych. Sterylne osobniki pojaw iają się niekiedy spontanicznie w populacji roślin, ale większość otrzym ano w rezultacie krzyżow ań wewnątrz- gatunkowych, m iędzygatunkow ych oraz międzyro- dzajowych. Krzyżowanie m iędzygatunkow e prow adzące do alloplazmii, czyli umieszczenia jąd ra kom órkowego jednego gatunku w środow isku cytoplazmy innego gatunku, jest źródłem 3/4 znanych fenotypów CMS.
Nazw a cytoplazm atyczna m ęskosterylność naw iązuje do pozajądrowego, m atecznego dziedziczenia w odróżnieniu od m ęskosterylności warunkow anej przez genom jądrow y nazywanej jądrow ą lub M end- lowską m ęskosterylnością (MST) [3], Uważa się, że m utacje w genomie m itochondrialnym , a nie ch loroplastowym, są przyczyną cytoplazmatycznej m ęskosterylności [1]. Fenotypow e zm iany będące rezultatem tych m utacji są jednak obserwowane tylko w obecności odpowiedniego genom u jądrow ego. Jeśli genom jądrow y zawiera specyficzne restorery (geny przyw racające płodność) to roślina m imo m utacji w genomie m itochondrialnym jest płodna. O becność restorerów nie jest w arunkiem koniecznym do tego, aby rośliny z niezm utowanym genomem m itochondrialnym stały się płodne. O kazało się, że różne typy CM S (pow odowane przez inny rodzaj m utacji w genomie m itochondrialnym) wym agają różnych genów przywracających płodność [8]. W niektórych przypadkach niezbędna
POSTĘPY BIOCHEM II 42 (3 ) , 1996 253http://rcin.org.pl

jest obecność nawet więcej niż jednego genu do odzyskania płodności [8, 9]. Zazwyczaj wpływ re- storerów nie jest trwały — rośliny ponow nie stają się sterylne, gdy usuniem y te geny poprzez odpowiednie krzyżowanie. Sugeruje się, że geny przywracające płodność uczestniczą w posttranskrypcyjnej i/lub po- sttranslacyjnej regulacji ekspresji genów związanych z cechą CMS.
M ęskosterylne rośliny są cennym m ateriałem genetycznym nie tylko do badania oddziaływań między m itochondrium a jądrem , ale również odgrywają isto tną rolę w produkcji nasion mieszańcowych.
II. Organizacja genomu mitochondrialnego
G enom y m itochondrialne roślin należą do najw iększych wśród dotychczas poznanych. Ich wielkość waha się od 200 kpz [10] do 2500 kpz [11]. D la porów nania genom m itochondrialny ssaków ma wielkość około 15 kpz, a drożdży 75 kpz [12]. Innym i słowy najmniejszy genom m itochondrialny roślin wyższych jest ponad 10 razy większy od genom u m itochondrialnego człowieka, a największy jest wielkości połowy chrom osom u E. coli. Zróżnicowanie wielkości wykryto nawet u roślin blisko spokrewnionych. N a przykład m tD N A arbuza (330 kpz) jest ponad siedm iokrotnie mniejszy od m tD N A m elona (2500 kpz) [11]. Jak dotychczas nie znaleziono racjonalnego wytłum aczenia tak dużych różnic w rozm iarach m itochondrialnego DNA.
M im o badań trwających już ponad pół wieku, organizacja roślinnego genom u m itochondrialnego jest ciągle kontrow ersyjna. N adal nie w iadom o, czy m itochondrialne D N A występuje w formie kolistej, czy liniowej [13,14]. K ontrow ersje dotyczą również ilości niezależnie replikujących się cząsteczek D N A (chromosomów) [15-18]. W edług klasycznej teorii, m itochondrialny genom roślin wyższych składa się z wielu kolistych cząsteczek o różnej wielkości. Największa z nich, zw ana chrom osom em głównym (ang. master chromosome), zawiera całą informację genetyczną genom u i jest jedyną cząsteczką m tD N A zdolną do
A
replikacji [19,20], Pozostałe cząsteczki są produktam i rekom binacji hom ologicznych zachodzących między dużymi sekwencjami pow tórzonym i chrom osom u głównego (ang. large repealed sequence) [21, 22]. Duże jednostki pow tórzone są to sekwencje nukleotydow e długości 1-14 kpz występujące przeważnie w dwóch trzech kopiach. W zależności od wzajemnej orientacji sekwencji pow tórzonych pow stają formy izomeryczne (Ryc. 1A) lub cząsteczki koliste o innej wielkości (Ryc. IB). Zdaniem badaczy tego zjawiska rekom binacja hom ologiczna dużych sekwencji pow tórzonych jest procesem odwracalnym , a uczestniczące w niej cząsteczki m tD N A są w stanie równow agi dynamicznej. Liczba, układ i orientacja dużych sekwencji pow tórzonych określa złożoność m itochondrialnego genom u [22-24],
Roślinne genomy m itochondrialne zawierają rów nież małe sekwencje pow tórzone (ang. small repeated sequence) o długości 10-1000 pz. [25]. Rekom binacje zachodzą między nimi rzadko i są źródłem nowych struktur. Tak powstałe cząsteczki D N A występują w niskim stężeniu, ale m ogą być w trakcie ewolucji amplifikowane, co doprow adza do zm iany struktury genom u [26, 27]. N ieodw racalność rekom binacji m ałych sekwencji powtórzonych jest przypuszczalnie spow odow ana ograniczonym praw dopodobieństw em ich ponow nego połączenia się ze względu na wielkość tych sekwencji. Uważa się, że często obserwowane u roślin duplikacje i delecje fragm entów m itochondrialnego D N A są konsekwencją rekom binacji między małymi sekwencjami pow tórzonym i [25-27].
W przeciwieństwie do ogromnej zmienności strukturalnej, genomy m itochondrialne roślin wyższych w ykazują małe zróżnicowanie sekwencji nukleotydo- wych [28]. N aw et u blisko spokrewnionych gatunków roślin obserwuje się różną kolejność genów w m tD N A , pom im o że sekwencje nukleotydowe tych genów są konserwatywne. Geny m ogą być prostym i ram kam i odczytu bądź zawierać in trony [29]. R ezultatem częstych rekom binacji są geny chim eralne składające się z fragm entów D N A pierwotnie nie sąsiadujących ze
Ryc. 1 Rekombinacja homologiczna dużych sekwencji potworzonych zlokalizowanych w kolistej cząsteczce DNA mitochondrialnego.A. Produktem rekombinacji dużych sekwencji potworzonych o przeciwnej orientacji (ang. inverted repeat) jest izomeryczna forma, w której nastąpiła inwersja frag-
mentu b-c.c J H H L b B. Produktam i rekombinacji du-f \ żych sekwencji powtórzonych o teji j samej orientacji (ang. direct repeat)\ J są dwie koliste cząsteczki DNA
zwane subgenomowymi (ang. sub-genomic circles).
254 POSTĘPY BIOCHEMII 42 (3 ), 1996http://rcin.org.pl

sobą, geny te m ogą również zawierać sekwencje o nieznanym pochodzeniu [24]. W D N A genom u mito- chondrialnego roślin zidentyfikowano regiony hom ologiczne z D N A chloroplastow ym i jądrow ym [30,31]. Sądzi się, że obecność tych fragm entów jest wynikiem przenoszenia informacji genetycznej pom iędzy organellami w czasie ewolucji.
M itochondria niektórych roślin m ogą zawierać, specyficzne dla gatunku, liniowe i koliste plazm idy oraz dwuniciowe cząsteczki RNA [2, 32, 33], W niektórych przypadkach próbow ano skorelow ać obecność tych cząsteczek z cechą cytoplazmatycznej męs- kosterylności. M im o że zaobserw ow ano więcej p lazm idów w liniach roślin m ęskosterylnych niż w liniach roślin płodnych, ich związek z CM S nie jest potw ierdzony.
III. Charakterystyka zmian towarzyszących CMS
M itochondrialny genom roślin jest zbyt duży, aby m ożna było lokalizować w nim zm iany specyficzne dla linii m ęskosterylnych poprzez sekwencjonowanie. D latego najczęściej identyfikuje się zm utow ane regiony, porów nując profile restrykcyjne (podrozdział III- 1), transkrypcyjne czy polipeptydowe (podrozdział III-2) roślin sterylnych z płodnymi. W przypadku niektórych gatunków roślin porów nano takie profile właściwe dla okazów płodnych i sterylnych som atycznych hybrydów [34, 35]. Nie każda zaobserw ow ana różnica w profilach musi wskazywać na gen, trans- k rypt lub białko odpowiedzialne za pojawienie się męskosterylności. Jest to spow odow ane zróżnicow aniem organizacji m itochondrialnego genom u roślin wyższych, naw et w obrębie tego samego gatunku. Poszukuje się zatem roślin jak najbliżej spokrew nionych, aby zminimalizować ilość różnic nie zw iązanych ze sterylnością. D latego cennym m ateriałem są rew ertanty pojawiające się czasami spontanicznie wśród roślin sterylnych. W badaniach nad CM S wykorzystuje się również rośliny będące rezultatem krzyżówek linii CM S z linią, której genom jądrow y różni się tylko obecnością genów przywracających płodność. Rośliny te odzyskały płodność, czyli nie m ogą zawierać transkryptów bądź białek pow odujących wystąpienie cytoplazmatycznej męskosterylności lub zawierają ich mniej.
III-l. Zmiany w organizacji genomu mitochondrialnego
Fragm enty genom u m itochondrialnego charak terystyczne dla CM S m ożna identyfikować, porów nując wzory restrykcyjne m tD N A linii sterylnych i płodnych. Izolacja czystego m tD N A jest często trudna i p racochłonna, dlatego najczęściej do wykrycia różnic struktury genom u m itochondrialnego linii sterylnych i p łodnych stosuje się hybrydyzację sond m itochondrial-
nych z całkow itym D N A kom órkow ym strawionym enzymami restrykcyjnym i [36-39]. Jako sondy często stosowane są heterologiczne geny m itochondrialne. M etoda ta jest również używ ana do odróżniania typów CM S w obrębie jednego gatunku [40, 41]. Najpełniejszych jednak danych o zm ianach w o rganizacji genom u m itochondrialnego dostarcza porów nanie m ap fizycznych m tD N A (kolejności fragmentów restrykcyjnych w genomie) linii płodnych i sterylnych [16, 34, 42-44].
Zidentyfikowane regiony genom u m itochondrialnego specyficzne dla linii męskosterylnych bardzo często zawierają unikalne geny chimeralne, w których pow staw aniu b iorą udział w ielokrotne rekombinacje. Geny chim eralne odkryto w m tD N A kukurydzy typu T-CM S (u r fl3 ) [1, 9], fasoli zwykłej (pvs) [4, 45], słonecznika (orfH 522 ) [46], hybrydach petunii (p c f) [1, 47]. D w a typy CM S rzepaku zawierają dwa różne geny chim eralne — o r fl 38 jest charakterystyczny dla typu O gura [35], a po l-u rf dla typu polim a [44].
W innych przypadkach okazało się, że regiony skorelow ane z cechą CM S zawierają zm utow ane geny m itochondrialne. Rearanżacje sekwencji nukleotydo- wych m ogą występować w regionie kodującym genu (np. cox I sorgo [48]) lub w 5' i 3' sekwencjach flankujących (np. 5' flankujący region genu cox I I bu raka cukrowego [49], 3' flankująca sekwencja atpó u ryżu [50]).
Poniżej dokładnie opisano region charakterystyczny dla CM S typu T kukurydzy — najlepiej poznanego systemu cytoplazmatycznej m ęskosterylności na po ziomie m olekularnym (Ryc. 2). Region ten zawiera dwie ram ki odczytu: urfl3 i orf221 (poprzednia nazwa orf25) [1, 9]. G en urfl3 jest chim eralny i składa się z fragm entów 3' sekwencji flankującej i kodującej genu rrn26 oraz fragm entu o nieznanym pochodzeniu. Jest unikalny — występuje tylko u m ęskosterylności typu T kukurydzy. W kierunku 3' w stosunku do chimeral- nego genu znajduje się gen orf221, norm alny m itochondrialny gen zawierający fragm ent sekwencji chloroplastowego genu trnR (tR N A Arg) [51]. Obie ram ki odczytu są regulowane przez tę sam ą sekwencję hom ologiczną do regionu regulatorowego atpó. U w aża się, że przynajmniej 7 zdarzeń rekom binacyjnych brało udział w tworzeniu regionu skorelow anego z męsko- sterylnością u kukurydzy. W pobliżu tego regionu znajduje się rekom binacyjnie aktyw na sekwencja p o wtórzona. Jej bliskie położenie może być przypadkow e bądź odzwierciedlać jej udział w pow staw aniu locus CMS. Regiony m tD N A powiązane z cechą cytoplazmatycznej m ęskosterylności u petunii i fasoli zwykłej również są zlokalizowane w pobliżu długich jednostek rekom binacyjnych [16, 18].
III-2. Zmiany w ekspresji
Przedstawione w poprzednim podrozdziale wyniki badań dotyczyły różnic w organizacji m itochondrial-
POSTĘPY BIOCHEMII 42 (3 ) , 1996 255http://rcin.org.pl

TURF 2113
Ryc. 2 Struktura regionu mtDNA skorelowanego z CMS typu T kukurydzy (TURF 2H3). Duże prostokąty reprezentują otwarte ramki odczytu. Pierwsze 1145 par zasad tego regionu jest homologiczne do 5' sekwencji flankującej atpó. Gen chimeralny urfl3 złożony jest z fragmentów 3' sekwencji flankującej i kodującej rrn26. Orf221 jest normalnym genem mitochondrialnym zawierającym sekwencję homologiczną z odpowiednią sekwencją chloroplastowego genu trnR pochodzącego z tytoniu. Wg [1] zmodyfikowano.I ... 1 sekwencja atpó H H sekwencja rrn26
sekwencja o nieznanym pochodzeniu sekwencja orf221 sekwencja chloroplastowego trnR tytoniu
tytoń trnR (tRNA^r§)
104 pz
o rf 221
nego D N A roślin sterylnych i płodnych. Badania porównawcze prow adzi się również nad ekspresją genom u m itochondrialnego. T ranskrypty pojedynczego genu m itochondrialnego roślin często różnią się pod względem wielkości [32, 52]. Jest to konsekwencją wielu miejsc inicjacji i term inacji w procesie transkrypcji oraz zjawisk posttranskrypcyjnych. T ranskrypty genów chim eralnych czy zm utow anych genów m ito- chondrialnych skorelow anych z cytoplazm atyczną m ęskosterylnością w ykazują również tak ą heterogen- ność. G eny chim eralne są często kotranskrybow ane z jednym [46, 53-56] lub wielu norm alnym i genami m itochondrialnym i [1, 57]. Opisane różnice wzorów transkrypcyjnych roślin sterylnych i płodnych dotyczą pojaw ienia się u roślin sterylnych specyficznych trans- kryptów [9, 35, 36, 38, 39, 53, 58, 59]. Dalsze badania niekiedy ujawniają, że niektóre zaobserwow ane specyficzne transkrypty nie m ają związku z cytoplazm atyczną m ęskosterylnością [58, 60]. Bliższych informacji o transkryptach odpowiedzialnych za sterylność dostarczają porów nania m ap transkrypcyjnych roślin sterylnych z roślinam i o takim samym genomie m itochondrialnym , ale płodnych dzięki obecności restore- rów w genomie jądrow ym (rozdział IV).
O statn io opublikow ano kilka prac sugerujących, że zaburzenia procesu redagow ania (ang. editing) [50, 61, 62] lub składania RNA (ang. splicing) [53] m ogą być przyczyną CMS. Jeden z takich przypadków przedstawimy w rozdziale IV. Zm iany na poziomie transkrypcji zostały opisane u wielu gatunków roślin, ale tylko w kilku przypadkach zidentyfikow ano białka unikalne dla roślin sterylnych. Dwie m etody badawcze umożliwiają taką identyfikację. Jedna z nich opiera się na porów naniu białek powstających w obecności ra
dioaktyw nych am inokwasów w m itochondriach izolowanych ze sterylnych i płodnych roślin. D ruga m etoda wykorzystuje przeciwciała skierowane przeciw syntetycznem u peptydowi skonstruow anem u na po d stawie sekwencji genu skorelow anego z m ęskosterylnością. Stosując te m etody, najwcześniej wykryto białko U R F13 o masie 13 kD a, występujące tylko w CM S typu T kukurydzy [1, 63]. Podobne badania umożliwiły rozpoznanie produktów genów chim eralnych u petunii (gen p c f białko o masie 25 kD a) [1, 64], u rzodkiewki (orf 138, białko o masie 20 kD a) [65], u pszenicy (orf256, białko o masie 7 kD a) [66] i u słonecznika (orfC, białko o masie 15 kDa) [67, 68]. Cechą w spólną tych białek jest ich lokalizacja w błonie m itochondrialnej. Jednak sposób ich połączenia z b łoną jest najpraw dopodobniej różny [1].
IV. Geny przywracające płodność
Jak już wspom inano, fenotypowe ujawnienie się m itochondrialnej m utacji odpowiedzialnej za CM S jest uzależnione od jądrow ej informacji genetycznej. Z badań wynika, że większość genów jądrow ych przyw racających płodność działa posttranskrypcyjnie i/lub posttranslacyjnie [1-3]. Pow odują one zm iany lokalizacji końców transkryptów regionów skorelow anych z cechą CM S [69, 70] i/łub inne stosunki ilościowe między transkryptam i [57]. W konsekwencji obserwuje się obniżenie ilości [64,65,71] lub brak [66] białek charakterystycznych dla cytoplazm m ęsko- sterylnych. Stwierdzono, że redukcja poziom u tych białek może zachodzić tylko w pewnych częściach rośliny, na przykład w tkance rozrodczej fasoli zwykłej [71], w kw iatach i liściach rzodkiewki [65]. Ciągle
256 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

jednak bardzo m ało w iadom o o m echanizm ach opisanych zmian. N iedawno, próbując wyjaśnić działanie genu Rf-1 w procesie przyw racania płodności u ryżu zaproponow ano hipotezę [50], zgodnie z k tó rą gen ten koduje enzym uczestniczący w dojrzewaniu p rekursora m tR N A jednej z kopii genu atpó. K opia ta jest obecna wyłącznie w m itochondriach roślin sterylnych. B adania wykazały, że tylko m tR N A powstały w wyniku działania tego enzymu może być efektywnie redagowany, tak aby następnie m ógł stanowić m atrycę do syntezy funkcjonalnego białka. P od nieobecność re- storera nieprzekształcony transkrypt nie jest praw idłowo redagowany, a syntetyzowane białko jest zmienione i zaburza dojrzewanie pyłku przez obniżenie aktywności ATPazowej.
W śród restorerów wyjątkowym działaniem charakteryzuje się jeden z jądrow ych genów przywracających płodność męskosterylnej linii fasoli zwykłej [72, 73]. Pow oduje on usunięcie regionu m tD N A odpow iedzialnego za sterylność poprzez eliminację cząsteczki zawierającej ten region [16]. W związku z tym, nawet po jego usunięciu, kolejne pokolenia roślin są płodne. Z ostatnich badań wynika, że zm iana struk tury genom u m itochondrialnego kierow ana tym genem zachodzi w czasie rozwoju kw iatu, jest zależna od ekspresji regionu CM S oraz wymaga dw óch alleli dom inujących do trwałego przywrócenia płodności [74].
Obecnie trw ają badania nad lokalizacją genów restorerów [8 ,75], ale żaden z nich nie został jak dotąd sklonow any i zsekwencjonowany.
V. Lokalizacja i funkcja URF13
Białko URF13 jest odpowiedzialne nie tylko za CM S typu T u kukurydzy, ale również za jej w rażliwość na grzybowe toksyny H m T i Pm oraz ich funkcjonalny analog — środek owadobójczy metom yl [76]. Działanie toksyn i m etom ylu pow oduje pęcznienie m itochondriów , rozprzęgnięcie fosforylacji oksydacyjnej, zaham ow anie oddychania, zwiększenie przepuszczalności błony, co w konsekwencji doprow adza do masowego „przecieku” jonów . O bjaw ów tych nie obserwowano w m itochondriach roślin płodnych.
Brak efektywnego sposobu transform acji kukurydzy spow odował zastosow anie innych organizm ów do badan ia roli URF13. T ransform acja E. coli potw ierdziła jego błonow ą lokalizację i odpowiedzialność za wrażliwość na toksyny i m etom yl [77]. Drożdże również stawały się wrażliwe na toksyny i metomyl, ale tylko wtedy gdy p roduk t translacji zakotw iczał się w błonie wewnętrznej m itochondriów [78, 79]. D latego były one transform ow ane genem chim eralnym złożonym z genu u rfl3 i m itochondrialnej sekwencji kierującej. W obecności m etom ylu transform anty drożdżowe wykazywały brak aktywności oddechowej. B adania transgenicznego ty toniu pokazały, że p roduk t genu urfl 3 nie musi być zw iązany z b łoną m itochon- drialną, aby te rośliny wykazywały wrażliwość na
toksyny [3]. Gdy białko U R F13 zlokalizowane jest w m itochondriach, staje się toksyczne dla tytoniu.
Prow adzone są również intensywne badania struktury kom pleksu URF13 z toksynam i (lub metomylem) [80]. Związane z b łoną białko jest receptorem toksyn i w wyniku oddziaływania pom iędzy nimi tw orzą się hydrofilowe pory w wewnętrznej błonie m itochondrialnej.
M im o ogrom nego postępu w zrozum ieniu związku białka URF13 z wrażliwością m itochondriów CM S-T kukurydzy na toksyny i metomyl, sposób, w jak i to białko zaburza rozwój pręcików, jest ciągle nieznany. W edług hipotezy F l a v e l l a [81] pręciki zawierają organospecyficzne białko oddziałujące z URF13 tak jak grzybowe toksyny i metomyl.
VI. Zastosowanie w hodowli
M ęskosterylne rośliny są od daw na wykorzystywane w hodowli odm ian mieszańcowych [82, 83], O trzym ane z nich rośliny przewyższają swoich rodziców wielkością plonów, odpornością na choroby i zmiany środowiskowe. Użycie linii m ęskosterylnych jako żeńskiego partnera w krzyżówce wyklucza niebezpieczeństwo sam ozapylenia, dzięki tem u nie m a potrzeby ręcznego usuw ania pręcików. Bardzo często rośliny pokolenia F I są również męskosterylne. Nie m a to znaczenia, jeśli produktem ważnym dla hodowcy są tkanki wegetatywne (liście tytoniu, bulwy ziemniaka). Jeśli natom iast rośliny są upraw iane dla nasion, jak w przypadku słonecznika czy kukurydzy, płodność mieszańca jest niezbędna. W takiej sytuacji jako p artnera męskiego stosuje się linię zawierającą w swym genomie restorery. Linie CM S po skrzyżowaniu z taką linią dają płodne potom stwo.
N ajstarszym sposobem otrzym ywania roślin męskosterylnych w hodowli jest krzyżowanie konw encjonalne. Stosuje się również som atyczną hybrydyzację, której celem jest przeniesienie cechy CM S z jed nego gatunku do drugiego [3]. W czasie fuzji p ro to plastów może nastąpić rekom binacja rodzicielskich genomów m itochondrialnych — powstaje wtedy nowy typ genomu. D o takiej rekom binacji nie może dojść w czasie krzyżowania konwencjonalnego, ponieważ roślinie potom nej przekazywany jest wyłącznie genom m itochondrialny linii matecznej. O statn io pokazano, że sterylność roślin m ożna wywołać, stosując m etody inżynierii genetycznej [2, 83]. Rośliny transform uje się genem zaburzającym rozwój pręcików, którego ekspresja regulow ana jest przez p rom oto r aktywny jedynie w tej części rośliny. Rybonukleaza, pow stająca wyłącznie w tapetum dzięki specyficznemu dla tej tkanki prom otorow i, pow odow ała męskosterylność transgenicznego tytoniu i rzepaku [84]. Tego samego p rom oto ra użyto do ekspresji inhibitora rybonukleazy w innych roślinach transgenicznych działających w krzyżowaniu tak, jak linie przywracające płodność [85]. Stw orzono w ten sposób sztuczny system z genem
POSTĘPY BIOCHEM II 42 (3 ) , 1996 257http://rcin.org.pl

powodującym sterylność i genem działającym jak restorer.
VII. Uwagi końcowe
M oże pow stać pytanie, dlaczego defekt m itochon- driów, obecnych przecież we wszystkich tkankach rośliny, w sposób specyficzny upośledza pyłek. F ak t ten tłumaczy się wzrostem ilości m itochondriów w czasie rozw oju męskich kom órek rozrodczych [86]. Liczba m itochondriów w zrasta wtedy dw udziestokrotnie w tkance sporogenicznej, a w tapetum — aż czterdziestokrotnie. M utacja w genomie m itochondrialnym m oże więc mieć o wiele bardziej drastyczne konsekwencje dla rozwoju pyłku niż dla innych procesów nie w ym agających tak dużej aktywności m itochondriów [1]. M ożliwe jest również, że zwiększenie liczby m itochondriów doprow adza do wzrostu ilości białka m itochon- drialnego, k tóre jest toksyczne dla kom órek roślinnych. Sugeruje się również, że białka występujące tylko w pręcikach odpow iadają za organospecyficzne ujaw nianie się m utacji m itochondrialnej [81].
M im o niewątpliwego postępu w identyfikacji m ito- chondrialnych regionów, specyficznych transkryptów i białek związanych z CM S, m olekularny mechanizm wywoływania sterylności przez te b iałka jest ciągle hipotetyczny. W yjaśnienie jest trudne ze względu na b rak bezpośredniej m etody transform acji m itochondriów roślinnych. C oraz powszechniej uważa się, że m echanizm ten nie jest uniwersalny, a raczej specyficzny dla każdego typu CMS.
A rtykuł otrzymano 5 grudnia 1995 r.Zaakceptowano do druku 20 marca 1996 r.
Piśmiennictwo
1. H a n s o n M R (1991) Annu Rev Genet 25: 461-4862. N a i r C K K (1993) J Biosci 18: 407-4223. V e d e l F, P 1 a M, V i t a r t V, G u t i e r r e s S, C h e t r i t P,
D e P a e p e R (1994) Plant Physiol Biochem 32: 601-6184. J o h n s C, L u M, L y z n i k A, M a c k e n z i e S (1992) Plant
Cell 4: 435-4495. W a r m k e HE, L e e S L J (1977) J Hered 68: 213-226. T a n a k a I (1993) J Plant Res 106: 55-637. K o f e r W, G l i m e l i u s K, B o n e t t H T (1991) The Plant
Cell 3: 759-7698. W i s e RP , S c h n a b l e P S (1994) Theor Appl Genet 88:
785-7959. D e w e y RE, L e v i n g s s C S I I I , T i m o t h y D H (1986)
Cell 44: 439-44910. P a l m e r J D, H e r b o n L A (1987) Curr Genet 11: 565-57011. W a r d BL, A n d e r s o n RS, B e n d i c h A J (1981) Cell 25:
792-80312. B o n e n L (1991) Curr Opinion in Genet and Develop 1: 515-52213. B e n d i c h A J, S m i t h S (1990) Curr Genet 17: 421-42514. B e n d i c h A J (1993) Curr Genet 24: 279-29015. Y a m a t o Y, O g u r a Y, K a n e g a e T, Y a m a d a Y,
O h y a m a K (1992) Theor Appl Genet 83: 279-28816. J a n s k a H, M a c k e n z i e SA (1993) Genetics 135: 869-87917. A n d r e CP , W a l b o t V (1995) Mol Gen Genet 247: 255-26318. F o l k e r t s O, H a n s o n M r (1991) Genetics 129: 885-89519. P a l m e r J D, S h i e l d s C R (1984) Nature (Lond) 307:
437-440
20. L o n s d a l e D M (1984) Plant Mol Biol 3: 201-20621. S t e r n DB, P a l m e r J D (1984) Nucleic Acids Res 12:
6141-615722. L o n s d a l e D M, B r e a r s T, H o d g e TP , M e l v i l l e SE,
R o t t m a n n W H (1988) Phil Trans R Soc Lond B 319:149-16323. C o n k l i n P, H a n s o n M (1993) Curr Genet 23: 477-48224. F a u r o n C, C a s p e r M, G a o Y, M o o r e B (1995) Trends
Genet 11: 228-23525. A n d r e C, L e v y A, W a l b o t V (1992) Trends Genet 8:
128-13226. S m a l l I D, I s a a c P G, L e a v e r C J (1987) EMBO J 6:
865-86927. S m a l l I D, S u f f o l k R, L e a v e r C J (1989) Cell 58: 69-7628. P a l m e r J D, H e r b o n L A (1988) J Mol Evol 28: 87-9729. W i s s i n g e r B, B r e n n i c k e A, S c h u s t e r W (1992)
Trends Genet 8: 322-32830. P a l m e r J D (1990) Trends Genet 6: 115-12031. W a t a n a b e N, N a k a z o n o M, K a n n o A, T s u t s u m i
N, A t s u s h i H (1994) Curr Genet 26: 512-51832. L e v i n g s I I I CS, B r o w n G G (1989) Cell 56: 171-17933. F i n n e g a n P M, B r o w n G G (1986) Proc Natl Acad Sci
USA 83: 5175-517934. F o l k e r t s O, H a n s o n M (1991) Genetics 129: 885-89535. B o n h o m m e S, B u d a r F, F e r a u l t M, P e l l e t i e r
G (1991) Curr Genet 19: 121-12736. D u d a r e v a NA, P o p o v s k y AV, K a s j a n o v a UV,
Y e p r e v SG, M g l i n e t s AV, S a l g a n i k R I (1991) Theor Appl Genet 83: 217-224
37. H o l f o r d P, C r o f t J H, N e w b u r y H J (1991) Theor Appl Genet 82: 737-744
38. R o u w e n d a l G J A , C r e e m e r s - M o l e n a a r J, K r e n s F A (1992) Theor Appl Genet 83: 330-336
39. S a n e A P, N a t h P, S a n e P Y (1994) J Biosci 19: 43-4540. S a u m i t o n - L a p r a d e P, R o u w e n d a l G J A , C u g u -
e n J, K r e n s FA, M i c h a e l i s G (1993) Theor Appl Genet 85: 529-535
41. R a j e s h w a r i R, S i v a r a m a k r i s h n a n S, S m i t h RL, S u b r a h m a n y a n i N C (1994) Theor Appl Genet 88:441-448
42. S i c u l e l l a L , P a l m e r J D (1988) Nucleic Acid Research 16: 3787-3799
43. F a u r o n C, H a v l i k M (1989) Curr Genet 15: 149-15444. L ’H o m m e Y , B r o w n G G (1993) Nucleic Acid Research 21:
1903-190945. C h a s e CD, O r t e g a V M (1992) Curr Genet 22: 147-15346. K o h l e r RH, H o r n R, L o s s l A, Z e t s c h e K (1991) Plant
Cell 3: 369-37647. Y o u n g EG, H a n s o n M R (1987) Cell 50: 41-4948. B a i l e y - S e r r e s , H a n s o n D K , F o x TD, L e a v e r C J
(1986) Cell 47: 567-57649. S e n d a M, D a g l e s s E, H o d g e R, P a u l W, P e t e r s o n
PA, S a e d l e r H (1991) Curr Genet 19: 175-18150. I w a b u c h i M, K y o z u k a J, S h i m a m o t o K (1993)
EMBO J 12: 1437-144651. P r i o l i L, H u a n g J, L e v i n g s C S I I I (1993) Plant Mol
Biol 23: 287-29552. G r a y M, H a n i c - J o y c e PJ , C o v e l l o P S (1992) Annu
Rev Plant Physiol Mol Biol 43: 145-17553. C h a s e C D (1994) Curr Genet 25: 245-25154. S i n g h M, B r o w n G (1993) Curr Genet 24: 316-32255. A k a g i H, S a k a m o t o M, S h i n j y o C, S h i m a d a H,
F u j i m u r a T (1994) Curr Genet 25: 52-5856. S o n g J, H e d g c o t h C (1994) Genome 37: 203-20957. P r u i t t K D , H a n s o n M R (1991) Mol Gen Genet 227:
348-35558. M a k a r o f f C , A p e l I J, P a l m e r J D (1991) Curr Genet 19:
183-19059. S e n d a M, H a r a d a T, M i k a m i T, S u g i u r a M, K i n o -
s h i t a T (1991) 19: 175-18160. M a k a r o f f C, A p e l I, P a l m e r J D (1990) Plant M ol Biol
15: 735-74661. H e r n o u l d M, S u h a r s o n o S, L i t v a k S, A r a y a A,
M o u r m a s A (1993) Proc Natl Acad Sci 90: 2370-237462. S t a h l R, S u n S, L ’ H o m m e Y, K e t e l a T, B r o w n G G
(1994) Nucl Acids Res 22: 2109-211363. W i s e RP, F l i s s AE, P r i n g DR, G e n g e n b a c h B G
(1987) Plant M ol Biol 9: 121-12664. N i v i s o n H T , H a n s o n M R (1989) Plant Cell 1:1121-1130
258 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

65. K r i s h n a s a m y S, M a k a r o f f C A (1994) Plant M ol Biol 26: 935-946
66. S o n g J , H e g o t h C (1994) Plant M ol Biol 26: 535-53967. H o r n R, R a i n e r H K , Z e t s c h e K (1991) Plant Mol Biol
17: 29-3668. L a v e r H K , R e y n o l d s SJ, M o n e g e r F, L e a v e r C J
(1991) Plant J 1: 185-19369. K e n n e l l J C , P r i n g D R (1989) Mol Gen Genet 216: 16-2470. S i n g h M, B r o w n G G (1991) Plant Cell 3: 1349-136271. A b a d A , M e h r t e n s B, M a c k e n z i e S (1995) Plant Cell 7:
271-28572. M a c k e n z i e S, P r i n g D, B a s s e t t M, C h a s e C (1988)
Proc Natl Acad Sei USA 85: 2714-271773. M a c k e n z i e S, C h a s e C (1990) Plant Cell 2: 905-91274. H e S, L y z n i k A, M a c k e n z i e S (1995) Genetics 139:
955-96275. H e S, Y u Z H , V a l l e j o s CE, M a c k e n z i e (1995) Theor
Appl Genet 90: 7-8
76. L e v i n g s C S I I I , S i e d o w J N (1992) Plant Mol Biol 19: 135-147
77. D e w e y RE, S i e d o w J N, T i m o t h y D H , L e v i n g s CS (1988) Science 239: 293-295
78. G l a b N, W i s e RP, P r i n g DR, J a c q C, S l o n i m s k i P (1990) Mol Gen Genet 223: 24-32
79. H u a n g J, L e e SH, L i u C, M e d i c i R, H a c k E, M y e r s A M (1990) EMBO J 9: 339-347
80. R h o a d s D M, K a s p i Cl , L e v i n g s C S I I I , S i e d o w J N (1994) Proc Natl Acad Sci USA 91: 8253-8257
81. F l a v e l l R (1975) Physiol Plant Pathol 6: 107-11682. D u v i c k D N (1959) Econ Bot 13: 167-19583. R o g e r s HJ , L o n s d a l e D M (1992) Plant Growth Regula
tion 11: 21-2684. M a r i a n i C, B e u e k e l e e r MD , T r u e t t n e r J, L e -
e m a n s J, G o l d b e r g RB (1990) Nature (Lond) 347: 737-74185. M a r i a n i C, B l o c k MD , G o l d b e r g RB, G r e e f W D ,
L e e m a n s J (1992) Nature (Lond) 357: 384-38786. W a r m k e HE, L e e S L (1978) Science 200: 561-63
Rola genomu jądrowego w regulacji funkcjonowania mitochondriów Saccharomyces cerevisiae
Nuclear control of mitochondrial functions in Saccharomyces cerevisiae
M A G D A L E N A B O G U T A *
Spis treści: Contents:
I. Wstęp I. IntroductionII. Mitochondrialny genom Saccharomyces cerevisiae II. Mitochondrial genome in yeastIII. Mutacje mitochondrialne III. Mitochondrial mutationsIY. Dziedziczenie mitochondriów i mechanizm utrzymywa IV. Mitochondrial inheritance and maitenance of mtDNA
nia mtDNA V. Replication and transcription of mtDNAV. Replikacja i transkrypcja mtDNA VI. RNA stability and splicingVI. Stabilność i obróbka transkryptów mitochondrialnych VII. Mitochondrial translationVII. Translacja mitochondrialna VIII. Modification, assembly and degradation of mitochondVIII. Modyfikacja, degradacja i włączanie białek mitochon- rial protein
drialnych w kompleksy IX. Mitochondrial control of nuclear functionsIX. Mitochondria regulują funkcję jądra komórkowego X. ConclusionsX. Podsumowanie
Wykaz stosowanych skrótów: mt — mitochondrialny; bp — para zasad.
I. Wstęp
M itochondria są organellam i obecnymi w prawie wszystkich kom órkach eukariotycznych (wyjątek stanow ią erytrocyty i niektóre ameby). Podstaw ow ą funkcją m itochondriów jest wytwarzanie energii w k a skadzie reakcji utleniania zachodzących w m itochon-
* Dr Instytut Biochemii i Biofizyki PAN, Pawióskiego 5A, 02-106 Warszawa
drialnym łańcuchu oddechowym. W arunkiem kom petencji oddechowej kom órki eukariotycznej jest ekspresja genom u m itochondrialnego. System organizacji i ekspresji m itochondrialnego D N A jest unikalny z punk tu widzenia biologii m olekularnej, choć w obrębie różnych eukariontów m tD N A wykazuje wysokie zróżnicowanie wielkości i ilości genów. Ilość informacji genetycznej zawartej w m tD N A jest znikom o m ała w porów naniu z inform acją pochodzącą z chro- m osom alnego D N A i dotyczącą funkcji m itochon- drialnych. W ysoki stopień złożoności organizacji m tD N A i sprzężenia ekspresji genom u m itochondrialnego i jądrow ego sugerują, że naw et drobne defekty
POSTĘPY BIOCHEMII 42 (3 ) , 1996 259http://rcin.org.pl

Tabela 1.Testowanie mutacji zmieniających funkcję mitochondriów S. cerevisiae.
Typ mutacji Wzrost na glukozie
Wzrost na glicerolu
typ dziki + +jądrowa, w genie kodującymbiałko mitochondrialne + -mitochondrialna + -supresor mutacjimitochondrialnej + —
genetyczne m ogą pow odow ać u organizm ów wyższych krytyczne efekty patologiczne. Szybko rośnie dokum entacja m olekularna chorób człowieka związanych z m tD N A . Zaburzenia funkcji m itochondrialnych u roślin wyższych, k tóre m ogą wynikać z defektów w m tD N A lub nieprawidłowej kom unikacji m itochon- drium -jądro kom órkow e m ogą prowadzić do zjawiska męskiej sterylności, kluczowego dla produkcji rolnej. D latego poznanie m olekularnych mechanizm ów regulacji funkcjonow ania m itochondriów m a też ważne znaczenie praktyczne.
S. cerevisiae jest modelowym organizm em służącym do badań genetyki i funkcji m itochondrialnych gdyż, w odróżnieniu od większości innych organizmów, jest fakultatyw nym anaerobem . W praktyce oznacza to, że jeśli oddychanie nie jest możliwe, organizm jest żywotny, gdyż może czerpać energię z procesu fermentacji. Testowanie funkcji m itochondrialnych może się odbywać poprzez proste m anipulow anie pożywkam i zawierającymi glukozę jako ferm entowalne i glicerol lub etanol jako nieferm entowalne źródło węgla (Tab. 1).
II. Mitochondrialny genom Saccharomyces cerevisiae
D N A m itochondrialny (m tDNA) S. cerevisiae jest kolistą cząsteczką o wielkości 74-85 kb, w zależności od szczepu i stanowi 5-15% całkowitego D N A kom órkowego [1]. K ołow a m apa m tD N A jest przedstaw iona na rycinie 1. Należy jednak pam iętać, że cząsteczki koliste stanow ią tylko część populacji m tD N A S. cerevisiae, k tó ra w w arunkach in vivo zawiera także długie liniowe konkatam ery [2].
Poznano dotychczas sekwencję 92% m tD N A S. cerevisiae. Analogicznie jak w m tD N A innych organizmów, w skład m tD N A drożdży wchodzą dwa geny kodujące rRNA , pełen zestaw genów tR N A oraz geny kodujące niektóre podjednostki enzymów oddechowych (Tab. 2). D w a geny kodujące białka (COB i COXI, patrz Ryc. 1) oraz gen L S U kodujący 21S rR N A posiadają introny. D odatkow e geny, VARI i R P M I, które nie zawsze m ają swoje odpowiedniki w m itochondrialnych genom ach innych organizm ów, kodują białko V a rlp związane z m ałą podjednostką rybosom ów m itochondrialnych oraz RNA wchodzące w skład
m itochondrialnej RNAzy P odpowiedzialnej za o b róbkę tRNA. Sekwencjonowanie ujawniło ponad to dwie ram ki odczytu, z k tórych jedna, EN S2, koduje podjednostkę endonukleazy zaangażowanej w rekom binację m tD N A . Sekwencje intronow e, często w po łączeniu z poprzedzającym i eksonami, kodują endonukleazy odpowiedzialne za propagację in tronów oraz m aturazy wym agane do obróbki m itochondrialnych transkryptów . m tD N A drożdży nie zawiera genów k o dujących podjednostki N A D H dehydrogenazy, obecnych w większości m tD N A innych organizm ów [3].
C harakterystyczną cechą m tD N A drożdży jest niezwykle niska zaw artość par G C (18%), k tóre tw orzą skupiska. Poszczególne geny są rozdzielone długimi sekwencjami AT o nieznanej funkcji. Wysoce hom ologiczne sekwencje bogate w G C o długości 50-100 bp odgrywają praw dopodobnie rolę w procesach rearan- żacji m tD N A [4]. M itochondrialny genom drożdży wykazuje stosunkow o wysoką zm ienność ze względu na możliwość tworzenia delecji o dużym zasięgu [5] oraz duplikacji i transpozycji intronów .
U drożdży, jak też u grzybów nitkowatych, obserwuje się również frapujące zjawisko migracji m tD N A do jąd ra kom órkow ego, k tóre praw dopodobnie od grywa rolę w ewolucji. W genomie jądrow ym drożdży zidentyfikowano sekwencje pochodzące z m tD N A [6]. W ędrówkę D N A z m itochondriów do jąd ra badano eksperym entalnie i zidentyfikow ano jądrow y gen YM E I odpowiedzialny za to zjawisko [7]. Transm isja m ateriału genetycznego z m tD N A do jąd ra kom órkowego wiąże się z procesem starzenia u Neurospora crassa.
Klasyczne m etody transform acji są zawodne przy
Tabela 2.Produkty genów mitochondrialnych Saccharomyces cerevisiae.
gen/intron kodowany produkt
C O XI podjednostką I oksydazy cytochromu cintron ail maturaza ailintron ai2 maturaza ai2intron ai3 endonukleaza IScelllintron ai4 endonukleaza IScellintron ai5a endonukleaza IScelVintron ai5b
COX2 podjednostką II oksydazy cytochromu cCOX3 podjednostką III oksydazy cytochromu cATP6 podjednostką VI ATPazyATP8 podjednostką VIII ATPazyATP9 podjednostką IX ATPazyCOB cytochrom b
intron bi2 m aturaza bi2intron bi3 m aturaza bi3intron bi4 m aturaza bi4
VARI białko rybosomalneENS2 endonukleazaRF1, RF2 ?LSU rybosomalny RNA 21S
intron co endonukleaza IScelSSU rybosomalny RNA 15SR P M I składnik RNA mt RNAzy P25 genów tRNA komplet mt tRNA
260 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

w prow adzaniu D N A do m itochondriów . Sukces osiągnięto bom bardując kom órki cząstkam i m etali p o krytym i DNA. U dało się w ten sposób w prow adzić do m itochondriów fragm enty m tD N A , które były stabilne i ulegały replikacji [8, 9].
III. Mutacje mitochondrialne
W spólną cechą m utantów m itochondrialnych S. cerevisiae jest b rak zdolności do w zrostu na pożywce zawierającej nieferm entowalne źródło węgla. W yróżnia się trzy podstaw ow e typy m utacji związanych z defektam i oddechowymi: mit ~, rho ~ i rho0. M utacje mit~ są to m utacje punktow e lub małe delecje w genach kodujących białka wchodzące w skład enzymów oddechowych. Zasadniczo efektem m utacji mit~ jest b rak syntezy lub aktywności pojedynczego białka. M utacje mit~ w in tronach m itochondrialnych m ogą mieć bardziej skom plikow ane fenotypy [10].
M utacje rho ~ zwane petite, są ogrom nym i delecjami obejm ującym i co najmniej 50% całej cząsteczki m tD N A . Zachow any odcinek ulega kom pensacyjnej amplifikacji, prowadzącej w efekcie do odtw orzenia cząsteczki m tD N A o wielkości odpow iadającej m tD N A szczepu dzikiego. F ragm enty m tD N A zachow ane u m utanów rho~ są wiernie replikowane, choć nie m uszą zawierać miejsc inicjacji replikacji [11]. m tD N A może być także całkowicie wydeletowany i takie szczepy nazywam y rho0.
Podstaw ow ym problem em , nie do końca rozstrzygniętym przez genetyków, jest segregacja wielokopij- nych genom ów m itochondrialnych, k tó ra nie stosuje się do klasycznych praw M endla. Jeśli z dwóch kom órek o genetycznie różnych m tD N A tworzy się zygota lub jeśli w pojedynczej cząsteczce m tD N A powstaje m utacja to m am y do czynienia z kom órką heteroplaz- m atyczną. Bez względu na to w jak i sposób tw orzy się heterogenna populacja m tD N A , obserwuje się szybką segregację m itochondrialnych genom ów w czasie ko lejnych podziałów i utworzenie populacji hom ogennej[12]. Nie dokładnie w iadom o w jak i sposób określony w ariant genom u m t osiąga przewagę selektywną nad całą populacją. Szczególna sytuacja dotyczy hetero- plazm atycznego diploida powstałego w wyniku krzyżówki pom iędzy szczepem rho+ i szczepem rho~ . Jeśli sekwencja am plifikowana w m utancie rho~ zawiera jeden z głównych startów replikacji (Ryc. 1), to może zajść sytuacja, że genom rho~ będzie się replikował szybciej niż rho+. M am y wtedy do czynienia ze zjawiskiem supresyjności petitów.
Stwierdzono, że m tD N A tworzy w kom órce agregaty zwane chondrillam i, k tóre m ożna uznać za niezależnie segregujące „jednostki” m tD N A . Skłonność m tD N A do agregacji zw iązana jest z wydajną rekom binacją. Ilość chondrilli zależy od p roduk tu genu M G T l, endonukleazy przecinającej złącza rekom bina- cyjnych struk tu r DNA. Inaktyw acja genu M GT1 p o woduje zmniejszenie ilości chondrilli, a co za tym idzie
zwiększenie wydajności rekom binacji i zmniejszenie ilości podziałów wym aganych do osiągnięcia hom ogennej populacji m tD N A [13].
IV. Dziedziczenie mitochondriów i mechanizm utrzymywania mtDNA
Transm isja m itochondriów do kom órki potom nej zależy od szeregu genów jądrow ych, których funkcje nie są do końca znane. W kom órce drożdży obserwowano ułożenie m itochondriów wzdłuż włókien ak tyny [14]. Znane są m utanty w genie kodującym aktynę A C T l, w których m orfologia i dziedziczenie m itochondriów są zaburzone [15]. Przypuszcza się, że białko analogiczne do m iozyny pośredniczy w ATP-zależnym wiązaniu m itochondriów do włókien aktyny, a konsekwencją tych oddziaływań jest sterowanie ruchem m itochondriów wzdłuż torów wyznaczonych przez cytoszkielet, czyli kon tro la pozycji m itochondriów w kom órce oraz ich transm isja do kom órki potom nej w czasie podziału. Niezależnie od cytoszkieletu aktynowego sugeruje się istnienie w komórce drożdży struk tury m ikrofilam entów, k tó ra jest zaangażow ana w proces dystrybucji organelli. O pisano m utan ta m dml, k tóry w tem peraturze restryktyw nej wykazuje brak transm isji jąd ra i m itochondriów do kom órki potom nej [16]. G en M D M 1 koduje białko niezbędne dla kom órki, które wykazuje podobieństw o sekwencji do białek ludzkich, będących składnikam i cytoplazmatycznej siateczki mikrofilam entów. Innym dowodem na powiązanie m itochondriów z cytoszkie- letem jest zm ieniona m orfologia i degradacja m tD N A w m utantach w genie M G M 1, k tóry koduje dynaminę, białko wiążące m ikrotubule [17]. Niezależnie stwierdzono, że zm iany w cytoszkielecie wywołane m utacją w genie VPR1/M DP2 kodującym werprolinę wpływają pośrednio na funkcje m itochondrialne zmieniając transport białek do m itochondriów [18].
Drugim , poza cytoszkieletem, elementem zasadniczym dla dystrybucji m itochondriów jest właściwa struk tu ra błon. C harakterystyka m utan ta mdm2 w skazuje na niezbędny udział nienasyconych kwasów tłuszczowych w dziedziczeniu m itochondriów . W tem peraturze restryktywnej nie następuje w m utancie mdm2 transm isja m itochondriów do pączka kom órki po tom nej, podczas gdy inne organelle są przenoszone praw idłowo. Białko M dm 2p jest desaturazą A 9 kwasów tłuszczowych [19].
Aby zapewnić kom órce kom petencję oddechową, transm isji m itochondriów w czasie podziału musi towarzyszyć przenoszenie m tDN A. D okładny m echanizm segregacji m tD N A i m olekularny m echanizm kontroli tego procesu nie są do końca poznane. W iadom o, że stabilność genom u m itochondrialnego rho+ wymaga aktywnej translacji m itochondrialnej, czego nie wymaga dziedziczenie genom u rho~ [20]. N ie w iadom o jednak czym różni się mechanizm zapewniający stabilność genom u rho+, od analogicznego
POSTĘPY BIOCHEMII 42 (3 ) , 1996 http://rcin.org.pl

m echanizm u dla genom u rho~. Istnieje możliwość, że uboczny p ro d u k t translacji m itochondrialnej uczestniczy w replikacji lub transm isji m tD N A rho + , a jest zbędny dla utrzym ania m tD N A rho~ [3]. Podobnie nie wiadom o, na jakiej zasadzie do stabilności m tD N A w ym agana jest obecność p roduk tu genu P IM 1, prote- azy odpowiedzialnej za degradację nieprawidłowo fałdowanych białek im portow anych do m itochond- riów [21]. W ykryto też ostatnio interesujące pow iązanie między syntezą am inokwasów a stabilnością m tD N A . Stwierdzono, że inaktyw acja genu ILV5 kodującego reduktoizom erazę kwasu hydroksyocto- wego, k tó ra katalizuje jeden z etapów syntezy am inokwasów o łańcuchu rozgałęzionym, powoduje niestabilność m tD N A [22].
Istnieje ścisła zależność między żyw otnością kom órki, m itochondriam i i funkcjonalnym m tD N A . Jakko lwiek m itochondria są absolutnie niezbędne dla życia kom órki, u tra ta funkcjonalnego m tD N A jest to lerow ana przez S. cerevisiae [23]. Istnieją jednak m utanty, które nie tolerują u traty lub degradacji m tDN A. Tolerancja ta jest w arunkow ana poprzez oksydacyjną fosforylację; szczepy z m utacją opl w genie kodującym przenośnik A D P /A T P są letalne w połączeniu z genomem m itochondrialnym rho~ [224]. W arunkiem u tra ty funkcjonalnego m tD N A jest zatem aktyw na fosforylacja oksydacyjna. L eta lnośćm utantów rho~ pow odują także m utacje w genie YME1 odpowiedzialnym za migrację m tD N A do jąd ra kom órkow ego [7].
V. Replikacja i transkrypcja mtDNA
W zależności od szczepu drożdży, m tD N A zawiera 7-8 startów replikacji (ori), k tóre zidentyfikowano na podstaw ie podobieństw a sekwencji do analogicznych startów w D N A innych organizm ów [25]. Delecja przynajm niej trzech sekwencji startow ych (ori 1, 2 i 7) pozostaje bez efektu, podczas gdy inne starty (ori 3) są konieczne do przebiegu replikacji [3]. Z identyfikow ana szereg lat tem u m itochondrialna polim eraza D N A nigdy nie została oczyszczona [26]. Praw dopodobnie enzym ten jest kodow any przez jądrow y gen M IP1[27], k tóry wykazuje podobieństw o sekwencji poli- m erazy m tD N A do polim eraz E. coli, bakteriofagów i eukariontów . O napraw ie uszkodzeń w m tD N A niewiele wiadom o; praw dopodobnie białko kodow ane przez gen jądrow y M SH 1, hom olog genu M u tS z E. coli, uczestniczy w napraw ie postreplikacyjnej typu „m ism atch” [28]. Zidentyfikowano dwa białka kodowane w jądrze związane z rekom binacją m tDNA: endonukleazę kodow aną przez gen M G Tl (patrz Rozdz. III) oraz A TP-zależną D N A helikazę kodowaną przez gen PIF1 [29]. W iadom o, że gen PIF1 kontroluje równocześnie długość telom erów w D N A jądrow ym . PIF1 stanow i ciekawy przykład genu, k tóry poprzez w ykorzystanie dwóch alternatyw nych startów translacji koduje dw a białka różniące się sekwencją N -końca — białko dłuższe kierow ane do
m itochondriów i białko krótsze transportow ane do jąd ra kom órkow ego [30].
T ranskrypcja m tD N A zachodzi z udziałem m itochondrialnej polim erazy RNA kodowanej przez gen jądrow y R P 041 . Białko to przypom ina raczej polim erazy RN A bakteriofagów T3, T7 i SP6 niż RNA polim erazy eukariotyczne [31]. M itochondrialna polim eraza RNA współdziała z czynnikiem transkrypcyj- nym kodow anym przez gen M TF1 i przypom inającym bakteryjny czynnik 8 [31]. M iejsca inicjacji transkrypcji w m tD N A drożdży oznaczono na rycinie 1. M ito chondrialne transkrypty są wielogenowe, a poszczególne geny w m tD N A nie posiadają własnych p rom otorów, co umożliwia wielostopniowy system regulacji. Pierw otne transkrypty są cięte, a m otywem rozpoznaw anym przez endonukleazę jest charakterystyczna sekwencja dodekam eru [33], m R N A nie m ają struk tury czapeczki na 5' końcu ani posttranskrypcyjnie syntetyzowanego poliA na 3' końcu [3].
T ranskrypty m t tR N A są dodatkow o przycinane na obu końcach. W procesie obróbki końca 5' m t tR N A bierze udział m itochondrialny analog RN azy P. E nzym ten składa się z części białkowej kodow anej przez jądrow y gen R P M 2 [34] oraz RNA kodow anego przez m itochondrialny gen RPM 1 [35], Poszczególne zasady w m t tR N A są chemicznie m odyfikowane z udziałem enzymów kodow anych przez jądrow e geny CC A l , MOD5, TRM 1 i TRM 2. Poprzez wykorzystanie a lternatywnych startów transkrypcji i translacji geny te kodują izoenzymy, k tóre są sortow ane do różnych kom partm entów kom órkow ych i odpow iadają za an alogiczne modyfikacje tR N A m itochondrialnych i tR N A cytozolowych [36]. Rycina 2 przedstaw ia schematycznie ekspresję genu MOD5 oraz lokalizację wewnątrzkom órkową powstających izoenzymów [37].
VI. Stabilność i obróbka transkryptów mitochondrialnych
3' końce transkryptów m itochondrialnych pow stają poprzez cięcie specyficznej dodekam erowej sekwencji (patrz wyżej). Sekwencja ta pełni praw dopodobnie dodatkow ą rolę wiążąc kom pleks białek, k tóre odpow iadają za stabilność transkryptu [38]. Stw ierdzono, że w skład kom pleksu stabilizującego wchodzą białka kodow ane przez geny jądrow e SUV3 i DSS1 [39-41]. Sekwencje stabilizujące 5' koniec m t tran skryptów nie zostały dotychczas scharakteryzowane.
Zidentyfikow ano szereg genów jądrow ych odpow iedzialnych, w sposób bardziej lub mniej specyficzny, za stabilność transkryptów m itochondrialnych. O pisano szczegółowo gen CBP1, k tórego ekspresja warunkuje specyficznie stabilność m RN A cytochrom u b [42]. D w a inne geny jądrow e, AEP1 i AEP2, kodują białka odpowiedzialne za obróbkę i trwałość m RNA podjed- nostki 9 ATPazy [43]. Z identyfikow ano także m u tan ty jądrow e, takie jak SUV3 czy N A M I, które oprócz destabilizacji transkryptów miały wpływ na wycinanie
262 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

Ryc. 1 Genom mitochondrialny S. cerevi- siae [3]. Geny kodujące rRNA i znane białka oznaczano na czarno. W przypadku genów zawierających introny, na czarno oznaczono kodujące sekwencje eksonowe. Introny pozostawiono białe, a w przypadku intronów, które występują tylko w niektórych szczepach drożdży, zaznaczono miejsca ich insercji. Introny grupy II oznaczono gwiazdką. N a biało oznaczono także geny kodujące hipotetyczne białka RF1, RF2 i ENS2. Geny tRNA oznaczono według odpowiednich aminokwasów w kodzie jednoliterowym. Cyfry podano dla tRNA izoakceptoro- wych. Miejsca występowania sekwencji startu replikacji oznaczono białymi trójkącikami, zaś miejsca startu transkrypcji — czarnymi trójkącikami.
in tronów m itochondrialnych. G en SUV3 koduje heli- kazę RNA, a p roduk t genu N A M I nie wykazuje istotnego podobieństw a do innych białek [40, 44, 45].
Trzy geny m itochondrialne S. cerevisiae, C O X I, COB i L S U zawierają introny (Ryc. 1). Ilość in tronów jest zależna od szczepu; gen C O X I zawiera m aksym alnie 7 intronów , gen COB — m aksym alnie 5 intronów , zaś gen L S U posiada 1 in tron lub występuje w formie bezintronowej. Skonstruow ano szczep laboratoryjny, k tóry nie posiadał w ogóle in tronów m itochondrialnych. B rak in tronów nie wywołuje zauważalnych zm ian fenotypowych i nie zaburza funkcji m itochondrialnych [46].
In trony m itochondrialne dzieli się na dwie grupy (I i II) w zależności od przewidywanej struk tury drugo- rzędowej. W ykazano, że w w arunkach in vitro in trony m itochondrialne są zdolne do sam owycinania, a m echanizm tej reakcji różni się nieznacznie dla intronów obu grup [47]. W iększość in tronów m itochondrialnych grupy I zawiera o tw artą ram kę odczytu (ORF) kodującą bądź białko uczestniczące w wycinaniu (ma- turazę), bądź w rozprzestrzenianiu (endonukleazę)
kodującego je intronu. N ależą one do rodziny białek zawierających charakterystyczne motywy sekwencji aminokwasowej LA G L I-D A D G . In trony grupy II kodują m aturazy, k tóre w ykazują znaczące podobieństwo sekwencji do odwrotnych transkryptaz retro- wirusów [3].
Sekwencje intronow e kodujące m aturazy są zgodne w fazie i ciągłe z sekwencjami poprzedzających ek- sonów, co umożliwia kaskadow y proces wycinania kolejnych intronów przedstaw iony schematycznie na rycinie 3. Wycięcie pierwszego in tronu genu COB, niezależne od m aturazy, powoduje połączenie się dwóch pierwszych eksonów, BI i B2. Translacja obu tych eksonów wraz z sekwencją kodującą stanowiącą 5' koniec drugiego in tronu prow adzi do pow stania m aturazy. K atalityczna ilość m aturazy wystarcza do wycięcia drugiego intronu, co prowadzi do połączenia BI, B2 i B3. Teraz sekwencja kodująca in tronu trzeciego leży bezpośrednio za sekwencją trzech połączonych eksonów i tak może ulegać translacji następna m atura- za, k tó ra bierze udział w wycinaniu trzeciego intronu. Ilość m aturazy w kom órce jest tak niska, że jej
MOD5-M2
MITOCHONDRIA CYTOPLAZMA JĄDRO KOMÓRKOWE
+ +
MOD5-M1 u------------------- PU * . + +
Ryc. 2 Lokalizacja subkomórkowa powstających izoenzymów MOD5 [37]. Dwa kodony inicjacyjne AUG są zgodne w fazie i odległe o 12 aminokwasów. Sekwencje odpowiedzialne za transport do mitochondriów przedstawiono jako czarny blok, sekwencje kierujące do jądra komórkowego zakreskowano.
POSTĘPY BIOCHEMII 4 2 (3 ) , 1996 263http://rcin.org.pl

pre m R N A
I T ir ' «
Ryc. 3 Wstępne etapy dojrzewania mRNA cytochromu b (COB) S. cerevisiae [25]. Eksony B1-B6 zaczerniono, obszary 5' i 3' nie ulegające translacji zakreskowano, zaś sekwencje kodujące w intro- nach pozostawiono białe.
indentyfikacja jest możliwa jedynie w m utantach, w których występują specyficzne defekty w wycinaniu intronów [48].
Część intronów m itochondrialnych grupy I koduje endonukleazy, k tóre um ożliwiają m obilność intronu, co wykazano m etodam i klasycznej genetyki [11]. M echanizm procesów związanych z m obilnością intronów grupy I przedstaw iono schematycznie na rycinie 4 [49]. Delecje i addycje intronów m itochondrialnych znajdują potwierdzenie doświadczalne. Zarów no powielanie in tronów jak ich delecje nie wpływają na funkcję oddechow ą m itochondriów . Stw ierdzono istnienie spontanicznych m itochondrialnych rewertan- tów, w których następuje delecja jednego lub większej ilości intronów . Delecja in tronu następuje dokładnie na granicy intron-ekson. Addycję in tronu grupy I w hom ologiczne miejsce genu bezintronow ego nazwano “in tron hom ing”. Endonukleaza pow stała w wyniku translacji sekwencji intronowej rozpoznaje specyficzne miejsce w D N A hom ologicznego genu nie
posiadającego in tronu i nacina obie nici. Dwuniciow a przerwa jest napraw iana na bazie istniejącej matrycy, k tó rą stanowi gen wraz z intronem . Także i sekwencje sąsiadujące z intronem ulegają powieleniu. W budow any in tron może ulegać ekspresji p rodukując endonu- kleazę.
O statn io opisano także mechanizm addycji dla in tronów grupy II na przykładzie in tronu ai2 genu C O X I. Proces ten nazw ano “retrohom ing”, gdyż przeniesienie in tronu do allelu bezintronowego zachodzi poprzez odw rotną transkrypcję m RN A allelu zawierającego intron. Białko kodow ane przez in tron ai2 spełnia równocześnie 3 funkcje: m aturazy, endonukleazy i odwrotnej transkryptazy, przy czym za każdą z funkcji odpow iada określona dom ena białka [50].
O pisano ciekawą m utację w sekwencji endonukleazy in tronu czwartego genu C O X I, k tó ra kom pensuje defekt m aturazy genu COB [51]. Zatem pojedyncza substytucja am inokw asu wystarczy aby endonukleaza była zdolna do funkcjonow ania jako m aturaza.
DELECJA INTRONU
Gen X
Ryc. 4 Uproszczone schematy procesów związanych z mobilnością intronów mitochondrialnych [49]. Gen Y Gen Y
264 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

Transpozycja in tronu nie została potw ierdzona do świadczalnie. O pisano jednak u drożdży efektywny m echanizm rozprzestrzeniania się sekwencji in trono- wych obejm ujący wstawienie sekwencji intronow ych w niehom ologiczne miejsca poprzez odw rotną transkrypcję i integracją powstałego cD N A do genom u m itochondrialnego [52].
W procesie wycinania in tronów z m t transkryptów bierze także udział szereg białek kodow anych w jądrze i transportow anych do m itochondriów . Większość tych białek zidentyfikow ano m etodam i genetycznymi. N iektóre z nich uczestniczą także w m itochondrialnej syntezie białek. W ykazano na przykład, że m itochon- drialna syntetaza tR N A kodow ana przez gen N A M 2 jest bezpośrednio zaangażow ana w wycinanie określonych in tronów genów COB i C O X I [53, 54]. P roduk ty innych genów jądrow ych regulują syntezę m aturaz, przez co m ają pośredni wpływ na wycinanie in tronów m itochondrialnych [55].
Bezpośredni związek między translacją a procesem wycinania in tronów jest teoretycznie możliwy właśnie jedynie w m itochondrialnym systemie ekspresji genów. M itochondria stanow ią bowiem unikalny system, gdzie oba procesy m ogą zachodzić „fizycznie” w tym samym miejscu podczas gdy oddzielenie jąd ra kom órkowego kom órki eukariotycznej poprzez błonę ją d row ą pow oduje ich rozdzielenie.
VII. Translacja mitochondrialna
N aszą obecną wiedzę o m itochondrialnym systemie translacji drożdży zawdzięczamy głównie eksperym entom genetycznym. B adania biochemiczne są ograniczone, gdyż do tej pory nie udało się odtw orzyć m itochondrialnego systemu translacji in vitro. Szacuje się, że w skład m itochondrialnego ap ara tu translacji wchodzi około 150 różnych białek i cząsteczek RNA [56]. Co najmniej 77 białek wchodzi w skład rybo- som u m itochondrialnego, czyli m itorybosom u [57]. Z a syntezę, obróbkę, modyfikację i aminoacylację m t tR N A odpow iada co najmniej 50 genów. O statn ią grupę białek stanow ią generalne czynniki transla- cyjne oraz specyficzne dla poszczególnych m itochondrialnych transkryptów aktyw atory translacji. Należy podkreślić, że m itochondrialny system translacji wym aga skoordynow anej ekspresji genom u jądrow ego i m tD N A . K om pletny zestaw tRNA, 15S i 21S rRNA, a także V arlp , jedno z białek m itorybosom ów oraz podjednostka enzym u odpowiedzialnego za obróbkę m t tR N A są kodow ane w m t DNA. Wszystkie, poza V arlp , białka m itorybosom ów oraz pozostałe składniki ap ara tu translacji są kodow ane w jądrze kom órkowym, syntetyzowane w cytozolu, a następnie transportow ane do m itochondriów .
M itorybosom y, k tóre dysocjują na podjednostki 54S i 37S, przypom inają pod pewnymi względami rybosom y prokariontów , a mniej są podobne do cytozolowych rybosom ów eukariotycznych [58]. Po
pierwsze szereg zidentyfikowanych białek m itorybosomów wykazuje podobieństw o sekwencji do białek rybosom ów bakteryjnych. Zestaw antybiotyków , k tó re ham ują translację m itochondrialną przypom ina te, k tóre działają na rybosom y bakteryjne. Także m itochondrialne czynniki translacyjne przypom inają b ardziej czynniki translacyjne p rokariontów niż euka- riontów [3]. M itorybosom y zawierają jednak większą liczbę białek, które też często są inne, a czasem podobne, ale większe od analogicznych białek bakteryjnych [59]. Spośród 23 białek m itorybosom alnych, k tórych sekwencje są znane, prawie wszystkie są niezbędne dla funkcjonow ania translacji, ale tylko 10 wykazuje hom ologię do znanych białek rybosom al- nych E. coli [60]. U nikalną cechą m itorybosom ów jest zdolność do oddziaływania ze specyficznymi aktyw atoram i translacji oraz z błoną m itochondrialną.
T ranslacja co najmniej pięciu spośród ośmiu podstawowych m t m RN A wymaga jednego lub większej ilości specyficznych aktyw atorów [25]. Sugeruje się, że b iałka te funkcjonują poprzez oddziaływanie z mito- rybosom em , 5' sekwencją wiodącą m RN A i wewnętrzną błoną m itochondrialną [61]. Rola aktyw atorów polega na rozpoznaniu miejsca startu translacji, co może być utrudnione, gdyż m itochondrialne m RNA nie m ają czapeczki, są długie, a ich drugorzędow a struk tu ra jest m ało zróżnicow ana ze względu na znaczącą przewagę reszt AT. Sugeruje się także, że aktyw atory translacji praw dopodobnie ułatw iają wbudow anie powstającego p roduktu białkowego w kom pleksy oddechowe, które funkcjonują w błonie m itochondrialnej [62, 63]. N atom iast niespecyficznym represorem translacji m itochondrialnej, białkiem, k tó re wiąże się do 5' końca wszystkich m itochondrialnych mRNA, jest dehydrogenaza izocytrynianu. Jest to jeden z podstaw owych enzymów cyklu Krebsa, który, jak się niedaw no okazało posiada zdolność wiązania się z RNA i pełni alternatyw ną funkcję w regulacji translacji m itochondrialnej. Ten dualizm funkcji dehydrogenazy izocytrynianu pozw ala na sprzężenie translacji m itochondrialnej z generalnym metabolizm em kom órkow ym [64]. Z identyfikowano też białka o nieznanej funkcji, które są praw dopodobnie związane z rybosom am i m itochondrialnym i [65].
Poza pewnymi aspektam i struktury m itorybosomów, specyficzność m itochondrialnego systemu translacji polega na stosow aniu kodu genetycznego odmiennego od kodu uniwersalnego [66, 67], U S. cerevisiae kodony AUA i C U N oznaczają odpow iednio m etioninę i treoninę, zaś kodon U G A jest tłum aczony na tryptofan [68]. O graniczona liczba 25 typów m t tR N A uniemożliwia translację według reguły chwiejności sformułowanej przez C r i c k a, a brak izoakceptorow ych tRN A wymusza uproszczoną regułę odczytyw ania [69]. W odróżnieniu od roślin, u drożdży nie m a konieczności transportu cytozolowych tR N A do m itochondriów . Stw ierdzono jedynie im port cytoplazm atycznego tR N A lys, który jednak nie fun
POSTĘPY BIOCHEMII 42 (3 ) , 1996 265http://rcin.org.pl

kcjonuje w aparacie translacyjnym, a jego funkcja w m itochondriach jest nieznana [70].
M itochondrialne rR N A nie zawierają metylowa- nych zasad, a metylacja rybozy jest ściśle ograniczona do jednej guanozyny w pozycji w 21S rRNA, podczas gdy 16S rR N A pozostaje niezmodyfikowany. Za m odyfikację odpow iada gen PET56, k tórego ekspresja warunkuje powstanie m itorybosom ów [71].
U nikalną cechą m itorybosom ów S. cerevisiae jest b rak cząsteczki 5S rRNA.
VIII. Modyfikacja, degradacja i włączanie białek mitochondrialnych w kompleksy
W szystkie produk ty m itochondrialnej translacji, za wyjątkiem V a rlp i białek kodow anych w intronach, są hydrofobowym i białkam i błonowym i wchodzącymi w skład kom pleksów oddechowych zlokalizowanych w wewnętrznej błonie m itochondrialnej. Pom im o, że powiązanie m itorybosom ów i aktyw atorów translacji z b łoną m itochondrialną pow inno ułatwiać włączanie powstających polipeptydów w błonę, zidentyfikow ano białka, k tóre ułatw iają ten proces. G en ABC1 jest niezbędny do utw orzenia kom pleksu b e l [72]. Geny SC O l i O XA1 są niezbędne do pow stania oksydazy cytochrom u c, a b rak ich aktywności prow adzi do destabilizacji określonych podjednostek tego enzymu [73, 74].
K odow ane jądrow o i transportow ane do m itochondriów białka są w większości przypadków pozbawiane N-końcowej sekwencji kierującej. O bróbka ta zachodzi podczas lub zaraz po transporcie z udziałem wysoce specyficznych proteaz zlokalizow anych w m atrix lub wewnętrznej błonie m itochondrialnej. M etalo- p roteaza M P P, złożona z dwóch podjednostek ko d o wanych przez geny M A S1 i M A S2, odcina sekwencje kierujące do m atrix i część sekwencji kierującej do przestrzeni między błonowej [75]. O dpow iednik prote- azy M P P u Neurospora i u roślin wchodzi w skład kom pleksu oddechowego b e l [76], co wskazuje na wspólną regulację biogenezy m itochondriów i ich aktywności energetycznej. P ro teaza IM P odpow iada za odcinanie sekwencji kierującej do przestrzeni mię- dzybłonowej. Enzym ten składa się z katalitycznych podjednostek IM P1 i IM P 2 o odmiennej specyficzności [77].
O prócz specyficznych enzymów, m itochondria po siadają proteazy, k tóre funkcjonują w ATP-zależnym procesie degradacji białek o nieodpowiedniej konformacji lub takich, k tórych synteza nie była kom pletna[78]. W m itochondriach występują hom ologi proteaz La i Cip E. coli, kodow ane odpow iednio przez geny drożdżowe P I M l i HSP78. Białko P im l współdziała z m itochondrialnym białkiem szoku cieplnego w rozpoznaniu i degradacji niewłaściwie pofałdow anych białek, a jednocześnie w arunkuje stabilność m tD N A[79]. Brak białka Hsp78 nie zaburza istotnej funkcji m itochondriów i jego ro la nie jest poznana [80].
Rośnie liczba białek m itochondrialnych A TPazo- podobnych z rodziny nazwanej A A A, których funkcja nie jest, wyjaśniona. D o tej klasy należą p rodukty genów jądrow ych BCS1, AGF3, RCA1 i YME1. Sekwencje wszystkich tych białek posiadają dwie dom eny po około 200 aminokwasów, k tóre zaw ierają charakterystyczne dla A TPaz m otywy odpowiedzialne za wiązanie nukleotydów. Pom im o podobieństw a sekwencji nie m ożna przypisać wymienionym białkom analogicznych funkcji. B cslp i R ca lp m ają udział w form ow aniu m itochondrialnych kom pleksów oddechowych [81, 82]. W spom niane uprzednio w obecnym artykule białko Y m el wpływa na migrację m tD N A do jąd ra kom órkow ego [7]. Agf3 praw dopodobnie pełni równocześnie funkcje białka chroniącego (chaperon) i proteazy, przez co jednocześnie pom agałoby białkom m itochondrialnym przybrać właściwą konfrom ację i degradow ałoby „nienorm alne” polipep- tydy [3].
IX. Mitochondria regulują funkcję jądra komórkowego
K om órka drożdżow a jest w stanie zmieniać ekspresję różnych genów jądrow ych w zależności od stanu fizjologicznego m itochondriów , a także stanu m tD N A . Zjawisko to jest znane jako regulacja typu retro, co podkreśla możliwość kom unikacji organelli w przeciwnym kierunku w stosunku do udokum entow anego obszernie ruchu informacji jąd ro kom órkow e-m ito- chondria. Przykładem regulacji retro jest wpływ niefunkcjonalnych m itochondriów w m utantach rho° na podwyższenie poziom u transkrypcji genu CIT2, k tóry koduje peroksysom alną formę syntazy cytrynianu. Efekt ten jest sterow any przez sekwencje UAS w 5' niekodującej sekwencji genu C IT2 za pośrednictwem białek regulatorow ych kodow anych przez geny RTG1 i RTG 2 [83]. P rodukty genów RTG w arunkują wzrost drożdży na pożywce z kwasem olejowym i są niezbędne do syntezy białek peroksysom alnych. Sugeruje się, że białka Rtg funkcjonują w systemie kom unikacji między m itochondriam i, peroksysom am i i jądrem k o m órkow ym [84].
X. Podsumowanie
W ciągu ostatniego dziesięciolecia wyizolowano kilkaset genów jądrow ych odpowiedzialnych za funkcje m itochondrialne Saccharomyces cerevisiae. P o przez identyfikację genów związanych z im portem białek do m itochondriów , dziedziczenie m itochondriów i ich oddziaływaniem z cytoszkieletem stwierdzono isto tną rolę m itochondriów w procesach wzrostu i podziału kom órki. M etody klonow ania pozwoliły na izolację genów, k tóre w w ypadku nadekspresji są supresoram i znanych m utacji m itochondrialnych. S trategia ta umożliwiła określenie hipotetycznego w spółdziałania produktów różnych genów odpow ie
266 POSTĘPY BIOCHEMII 42 (3 ), 1996http://rcin.org.pl

dzialnych za funkcje m itochondrialne. Wreszcie poprzez realizację projektu sekwencjonowania genom u Saccharomyces cerevisiae zidentyfikow ano szereg genów, których inaktyw acja pow odow ała defekty w funkcjonow aniu m itochondriów . Analiza sekwencji p o zwoliła na stworzenie rodzin białek hom ologicznych gdyż białkom danej rodziny przypisuje się analogiczne funkcje. Przykładem jest rodzina białek m itochon- drialnych AAA, które regulują oddziaływ ania białko- białko w arunkując pow stawanie kom pleksów i czas życia białek. Stw ierdzono też, że m itochondria m ogą wpływać na ekspresję genów jądrow ych. N iewątpliwie charakterystyka oddziaływań jąd ro kom órko we-mito- chondria pozw ala na głębsze zrozum ienie zjawiska kom unikacji między organellam i kom órkowym i.
PodziękowaniaPraca została zrealizowana w ramach projektu grantowego KBN nr 6P04A06208.
A rtyku ł otrzymano 21 lutego 1996 r. Zaakceptowano do druku 15 maja 1996 r.
Piśmiennictwo
1. B o l o t i n - F u k u h a r a M, G r i v e l l L A (1992) Antonie van Leeuwenhoek; J Gen Microbiol 62: 131-153
2. M a l e s z k a R, S k e l l y PJ , C l a r k - W a l k e r G D (1991) EMBO J 10: 3923-3929
3. G r i v e l l L A (1995) Crit Rev Bioch Mol Biol 30: 121-1644. G r o s m a n LI , C r y e r DR, G o l d r i n g ES, M a r m u r
J (1971) J M ol Biol 62: 565-5. B e r n a r d i G (1982) Trends Biochem Sei 7: 404-4086. F a r r e l l y F, B u t ó w R A (1983) Nature (Lond) 301:296-3017. T h o r s n e s s PE, W h i t e K H , F o x T D (1993) Mol Celi
Biol 13: 5418-54268. J o n s t o n SA, A n z i a n o P Q, S h a r k K, S a n f o r d JC,
B u t ó w R A (1988) Science 240: 1538-15419. F o x T D , S a n f o r d JC, M c M u l l i n T W (1988) Proc Natl
Acad Sei USA 85: 728810. K r u s z e w s k a A, B o g u t a M (1988) Post Biol Kom 15:
327-34611. B 1 a n c H, D u j o n B (1980) Proc Natl Acad Sei USA 77: 394212. B i r k y C W (1994) J Hered 85: 355-36513. L o c k s o n D, Z w e i f e l SG, F r e e m a n - C o o k LL, L o -
r i m e r HE, B r e w e r BJ, F a n g m a n W L (1995) Celi 81: 947-955
14. D r u b i n D G , J o n e s H D , W e r t m a n K F (1993) Mol Biol Celi 4: 1277-1294
15. L a z z a r i n o DA, B o l d o g h I, S m i t h MG , R o s a n d J, P o n L (1994) M ol Biol Celi 5: 807-818
16. M c C o n n e l l SJ, Y a f f e M P (1993) Science 260: 687-68917. J o n e s BA, F a n g m a n W L (1992) Genes Dev 6: 380-38918. Z o l a d e k T, V a d u v a G, H u n t e r LA, B o g u t a M, G o
BD, M a r t i n NC , H o p p e r A K (1995) Mol Celi Biol 15: 6884-6894
19. S t e w a r t LC, Y a f f e M P (1991) J Celi Biol 115: 1249-125720. M y e r s AM, P a p e LK, T z a g o l o f f A (?985) EMBO J 4:
2087-209221. W a g n e r LH, A r l t H, v a n D y c k L, L a n g e r T,
N e u p e r t W (1994) EMBO J 13: 5135-514522. Z e l e n a y a - T r o i t s k a y a O, P e r l m a n PS, B u t ó w
R A (1995) EMBO J 14: 3268-327623. S ł o n i m s k i P P , P e r r o d i n G , C r o f t J H (1968)Biochem
Biophys Res Com 30: 232-23924. K o l a r o v J , K o l a r o v a N , N e l s o n N (1990) J Biol Chem
265: 12711-1271625. P o n L, S c h a t z G (1991) The Molecullar Biology of Sac
charomyces cerevisiae 11, Cold Spring H arbor Laboratory Press,
str 333-40626. W i n t e r s b e r g e r U, B l u t s c h H (1976) Eur J Biochem 13:
11-1927. G e n g a A, B i a n c h L, F o u r y F (1986) J Biol Chem 261:
932828. R e e n a n RA, K o l o d n e r R D (1992) Genetics 132: 975-98529. F o u r y F, V a n D y c k E (1985) EMBO J 4: 3225-353030. S c h u l z VP, Z a k i a n VA (1994) Cell 76: 145-15531. S c h i n k e l AH, T a b a k F H (1989) Trends Genet 5: 149-15432. J a n g SH, J a e h n i n g J A (1991) J Biol Chem 266: 22671-
2267733. H o f m a n n TJ , M i n JJ , Z a s s e n h a u s H P (1993) Yeast 9:
1319-133034. D a n g YL, M a r t i n N C (1993) J Biol Chem 268: 19791-
1979635. M i l l e r DL, M a r t i n N C (1983) Cell 34: 911-91736. M a r t i n NC, H o p p e r A K (1994) Biochimie 76: 1161-116737. B o g u t a M, H u n t e r LA, S h e n WC, G i l m a n EC,
M a r t i n NC, H o p p e r A K (1994) Mol Cell Biol 14: 2298-2306
38. M in J J , Z a s s e n h a u s H P (1993) M ol Cell Biol 13: 4167-4173
39. D m o c h o w s k a A, G o l i k P, S t ę p i e ń P P (1995) Curr Genet 28: 108-112
40. S t ę p i e ń P P , K o k o t L, L e s k i T, B a r t n i k E (1995) Curr Genet 27: 234-238
41. S t ę p i e ń P P , M a r g o s s i a n SP, L a n d s m a n D, B u t ó w R A (1992) Proc Natl Acad Sei USA 89: 6813-6817
42. M i t t e l m e i e r T M, D i e c k m a n C L (1993) Mol Cell Biol 13: 4203-4213
43. Z i a j a K, M i c h a e l i s G, L i s o w s k y T (1993) J Mol Biol 229: 909-916
44. G r o u d i n s k y O, B o s q u e t I, W a l l i s MG , S l o n i m s - k y P P , D u j a r d i n G (1993) M ol Gen Genet 240: 419-427
45. G o l i k P, S z c z e p a n e k T, B a r t n i k E, S t ę p i e ń P, Ł a z o w s k a J (1995) Curr Genet 28: 217-224
46. S e r a p h i n B, B o u l e t A, S i m o n M, F a y e G (1987) Proc N atl Acad Sei USA 84: 6810-6814
47. L a m b o w i t z AM, B e l f o r t M (1993) Annu Rev Biochem 62: 587-622
48. Ł a z o w s k a J, J a c q C, S ł o n i m s k i P P (1980) Cell 22: 333-342
49. D u j o n B (1989) Gene 82: 91-11450. C u r c i o MJ , B e l f o r t M (1996) Cell 84:9-1251. D u j a r d i n G, J a c q C, S ł o n i m s k i P P (1982) Nature
(Lond) 298: 628-63252. M u e l l e r M, A l l m a i e r M, E s k e s R, S c h w e y e n R J
(1993) Nature (Lond) 366: 174-17653. L a b o u e s s e M (1990) M ol Gen Genet 224: 209-22154. Z a g ó r s k i W, C a s t a i n g B, H e r b e r t CJ , L a b o u e s s e
M, M a r t i n R, S ł o n i m s k i P P (1991) J Biol Chem 266: 2357-2541
55. P e l H, R e p M, G r i v e l l L A (1992) Nucleic Acid Res 20: 6339-6346
56. P e l H, G r i v e l l L A (1994) M ol Biol Rep 19: 183-19457. B o g u t a M, M i e s z c z a k M, Z a g ó r s k i W (7988) Curr
Genet 13: 129-13558. K i t a k a w a M, I s o n o K (1991) Biochimie 73: 813-82559. B o g u t a M, D m o c h o w s k a A, B o r s u k P, W r ó b e l K,
G a r g o u r i A, Ł a z o w s k a J, S ł o n i m s k i PP , S z c z e ś - n i a k - K r u s z e w s k a A (1992) Moll Cell Biol 12: 402-412
60. F e a r o n K, Mason TL (1992) J Biol Chem 257: 5162-517061. C o n s t a n z o MC , F o x T D (1990) Annu Rev Genet 24:
91-11362. B r o w n N G , C o n s t a n z o MC , F o x T D (1994) Mol Cell
Biol 14: 1045-105363. M u l e r o JJ , F o x T D (1993) Mol Biol Cell 4: 1327-135564. E l z i n g a SD, B e d n a r z A L, v a n O o s t e r u m K, D e k -
k e r PJ , G r i v e l l L A (1993) Nucleic Acids Res 21: 5328-533165. K o n o p i ń s k a A, S z c z ę ś n i a k B, B o g u t a M (1995)
Yeast 11: 1513-151866. F o x T D (1987) Annu Rev Genet 21: 67-9167. B o g u t a M, P u t r a m e n t A (1987) Kosmos 34: 67-8768. M a r t i n NC, P h a m H D , U n d e r b r i n k - L y o n K,
M i l l e r DL, D o n e l s o n J E (1990) Nature (Lond) 285: 579-581
69. M a r t i n R P, S i b l e t AP, G e h r k e CW, K u o K, E d -
POSTĘPY BIOCHEMII 42 (3 ) , 1996 267http://rcin.org.pl

m o n s CG, M c C l o s k e y JA, D i r h e i m e r G (1990) Biochemistry 29: 956-959
70. M a r t i n RP, S c h n e l l e r J M, S t a h l JC, D i r h e i m e r G (1979) Biochemistry 18: 4600-4605
71. S i r u m - C o n n e l l y K, M a s o n T L (1993) Science 262: 1886-1889
72. B o s q u e t I , D u j a r d i n G , S ł o n i m s k i P P (1991)EMBO J 10: 2023-2031
73. B o n n e f f o y N , C h a l v e t F , H a m e l P , S ł o n i m s k i PP , D uj a r d i n G (1994) J M ol Biol 239: 201-212
74. K r u m m e c k G, R o d e l G (1990) Curr Genet 18: 13-1575. Y a f f e MP , S c h a t z G (1984) Proc Natl Acad Sei USA 81:
4819-482376. E r i c k s o n AC, G l a s e r E (1992) Biochem Biophys Acta
1140: 208-214
77. N u n n a r i J, F o x TD, W a l t e r P (1993) Science 262: 1997-2004
78. G o l d b e r g A L (1992) Eur J Biochem 203: 9-2379. S u z u k i CK, S u d a K, W a n g N, S c h a t z G (1994)
Science 264: 273-27680. L e o n a r d t SA, F e a r o n K, D a e s e P N, M a s o n T L
(1993) M ol Cell Biol 13: 6304-631381. N o r b e r g a F G, N o r b e r g a MP , T z a g o l o f f A (1992)
EMBO 11: 3821-382982. T z a g o l o f f A, D i e c k a m n C L (1990) Microbiol Rev 54:
211-22583. L i a o X, B u t ó w R A (1993) Cell 72: 61-7184. C h e ł s t o w s k a A, B u t ó w R A (1995) J Biol Chem 270:
18141-18146
Roślinny łańcuch oddechowy
Plant respiratory chain
A N N A M. R Y C H T E R *
Spis treści:
I. WstępII. Dehydrogenazy NAD(P)H roślinnego łańcucha oddecho
wegoII-I. Zewnętrzne dehydrogenazy NAD(P)HII-l.l. Charakterystyka zewnętrznych dehydrogenaz
NAD(P)H11.1.2. Rola fizjologiczna zewnętrznych dehydrogenaz
NAD(P)HII-2. Wewnętrzne dehydrogenazy NADHII-2.1. Kompleks I, dehydrogenaza NADH (hamowana
przez rotenon)11.2.2. Dehydrogenaza NADH niewrażliwa na rotenon
III. Oksydaza alternatywnaIII-l. Charakterystyka oksydazy alternatywnejIII-2. Regulacja przepływu elektronów przez drogę cyto-
chromową i alternatywnąIII-3. Oznaczanie aktywności drogi alternatywnejIII-4. Rola fizjologiczna drogi alternatywnej
IV. Uwagi końcowe
I. Wstęp
W ydajność energetyczna oddychania wpływa na wiele procesów związanych ze wzrostem i rozwojem roślin. K ońcow y etap oddychania zachodzi w m ito- chondriach, gdzie zlokalizow any jest łańcuch oddechowy i cykl K rebsa. Rośliny oddychają nie tylko w ciemności, ale również na świetle, podczas fotosyntezy. W kom órkach fotosyntetyzujących źródłem sub-
* Prof. dr hab., Zakład Bioenergetyki Roślin, Instytut Biologii Eksperymentalnej Roślin, Uniwersytet Warszawski, 02-106 Warszawa, ul. Pawińskiego 5A.
268
Contents:
I. IntroductionII. Plant respiratory chain NAD(P)H dehydrogenases
II-l. External NAD(P)H dehydrogenasesII-l.l. Characteristics of external NAD(P)H dehydroge
nasesII-1.2. Physiological role of external NAD(P)H dehy
drogenases II-2. Internal NADH dehydrogenases II-2.1. Internal NADH dehydrogenase (inhibited by rote
non), Complex III-2.2. Internal NADH dehydrogenase, rotenon insen
sitiveIII. Alternative oxidase
III-l. Characteristics of alternative oxidaseIII-2. Regulation of electron flux between cytochrome
and alternative pathwaysIII-3. Determination of the activity of alternative path
wayIII-4. Physiological role of alternative pathway
IV. Concluding remarks
stratów oddechowych dla m itochondriów może być nie tylko glikoliza (pirogronian, jabłezan), ale również pośrednio fotosynteza (jabłezan) lub fotooddychanie (glicyna). Dopływ tych substratów może podlegać zm ianom , nie tylko w w arunkach światło-ciemność, ale również na świetle w zależności od intensywności fotosyntezy lub fotooddychania. W kom órkach niefo- tosyntetyzujących ilość substratów również może się zmieniać w zależności od m etabolizm u i od wpływu w arunków środowiskowych, np. przy zm ianach tem peratury.
M itochondria uważa się za centrum przetw arzania energii w kom órce. G łów ną ich funkcją jest utlenianie
POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

substratów związane z transportem elektronów sprzężonych z syntezą ATP. Łańcuch oddechowy składa się z wielobiałkowych kom pleksów biorących udział w przenoszeniu elektronów z różnych substratów na tlen cząsteczkowy. Cztery główne kom pleksy to: kom pleks I — oksydoreduktaza N A D H : ubichinon, kom pleks II — oksydoreduktaza bursztynian: ubichinon, kom pleks III — oksydoreduktaza u b ich ino l: cyto- chrom c i kom pleks IV — oksydaza cytochrom ow a (Ryc. 1). W błonie m itochondrialnej znajduje się rozpuszczony w niej m obilny ubichinon, k tóry łączy ze sobą kom pleksy enzymatyczne dehydrogenaz łańcucha oddechowego z oksydazam i regulując przepływ i dystrybucję elektronów na łańcuch cytochrom ow y (kompleks III, cytochrom y b-c2 oraz kom pleks IV, oksydaza cytochrom owa) lub oksydazę alternatyw ną. Zlokalizow any po zewnętrznej stronie błony wewnętrznej, cytochrom c działa jako m obilny przenośnik elektronów łączący kom pleks III z kom pleksem IV. P odobnie jak w m itochondriach zwierzęcych kom pleksy I, III i IV są ułożone w poprzek błony i uczestniczą w w ytw arzaniu gradientu protonów . G radien t ten wykorzystywany jest przez syntazę A TP (F0 Fj — ATPazę) do syntezy ATP.
W m itochondriach roślinnych, dzięki obecności dodatkow ych kom pleksów enzymatycznych, możliwy jest transport elektronów na tlen z pom inięciem tych fragm entów łańcucha oddechowego, k tóre są związane z translokacją protonów (Ryc. 1). Ze względu na możliwość przenoszenia elektronów bez syntezy ATP, m itochondria roślinne m ogą charakteryzow ać się niższym stopniem sprzężenia (stosunek P/O). Roślinny łańcuch oddechowy jest więc bardziej złożony niż zwierzęcy łańcuch oddechowy, a centralnym jego członem jest ubichinon. M itochondria roślinne różnią
się od m itochondriów zwierzęcych:— rozbudow anym systemem utleniania NAD(P)H; dodatkow ą (oprócz kom pleksu I), wewnątrz mito- chondrialną dehydrogenazą N A D H , niewrażliwą na ro tenon oraz możliwością utleniania cytosolowego, zewnątrz m itochondrialnego N A D (P)H przez związaną z łańcuchem oddechowym zew nętrzną dehydrogenazę NAD(P)H;— obecnością tzw. oksydazy alternatywnej, niewrażliwej na cyjanek, dzięki której możliwy jest transport elektronów na tlen z pominięciem łańcucha cytochro- mowego.— obecnością w m itochondriach enzymu jabłczano- wego (dehydrogenazy jabłczanowej dekarboksylują- cej, specyficznej dla NAD).— zdolnością do utleniania glicyny przez m itochondria izolowane z liści roślin, u k tórych występuje fotoodychanie.
Wszystkie te cechy odzwierciedlają bardziej „plastyczną” funkcję m itochondriów w kom órce roślinnej. W ynika ona z konieczności szybkiego przystosow yw ania się do zmiennych w arunków m etabolicznych oraz w spółdziałania z innymi organellam i przetw arzającymi energię (chloroplasty) lub z peroksyzom am i biorącymi udział w fotooddychaniu. Z tego też względu funkcja m itochondriów w kom órce roślinnej nie może być rozw ażana w oderw aniu od innych organelli oraz od tkanki (np. liści łub korzeni) i w arunków zewnętrznych (np. światło, ciemność) w jakich znajduje się cała roślina. Wiele cech wspólnych z m itochondriam i roślin wyższych wykazują m itochondria am eb [1]. W osta tnich latach ukazały się artykuły przeglądowe opisujące zarów no budowę roślinnego łańcucha oddechowego jak i funkcję m itochondriów [2-6]. B adania z zastosowaniem technik biologii m olekularnej okazały się
KCN
H+ NAD(P)H
f Przestrzeń międzybłonowa
MatriksRyc. 1 Schemat roślinnego łańcucha oddechowego, zmodyfikowano wg [6 i 10], B.w. — wewnętrzna błona mitochondrialna, w której
umieszczony jest łańcuch oddechowy; I, II, III i IV — oznaczono kolejno kompleksy łańcucha oddechowego, strzałkami oznaczono miejsca i kierunek translokacji protonów; podwójnymi strzałkami oznaczono fragmenty łańcucha, z których przeniesienie elektronów nie jest związane z translokacją protonów; grubą kreską oznaczono miejsce hamowania przez rotenon, K CN i SHAM; Q — ubichinon; deh.z. i deh.w. — odpowiednio zewnętrzne i wewnętrzne dehydrogenazy NAD(P)H, niewrażliwe na rotenon; Alt. — oksydaza alternatywna; b, c1# c, a 3, a — cytochromy tworzące tzw. łańcuch cytochromowy.
POSTĘPY BIOCHEM II 42 (3 ) , 1996 269http://rcin.org.pl

niezmiernie przydatne do określenia struktury i miejsca pow iązania niektórych kom ponentów łańcucha oddechowego (oksydaza alternatyw na) oraz m echanizmów regulacji przepływu elektronów. A rtykuł ten jest podsum ow aniem ostatnich badań dotyczących struktury i funkcji dodatkow ych, charakterystycznych dla roślin, fragm entów łańcucha oddechowego.
II. Dehydrogenazy NAD(P)H roślinnego łańcucha oddechowego
Roślinny łańcuch oddechowy m a dwa systemy dehydrogenaz związane z utlenianiem NAD(P)H: zewnętrzne dehydrogenazy N AD (P)H , utleniające NAD (P)H cytosolowe i dehydrogenazy wewnętrzne, utleniające N A D H wytworzone w m atriks. M itochondrialnym dehydrogenazom N A D (P)H w ostatnich latach po święcono szereg artykułów przeglądowych [7-10].
II-l. Zewnętrzne dehydrogenazy N A D (P )H
I I - l . l . Charakterystyka zewnętrznych dehydrogenaz N A D (P )H
Izolow ane m itochondria roślinne, w odróżnieniu od m itochondriów zwierzęcych, aktywnie utleniają do d ane, egzogene NAD (P)H . W ynika to z obecności w roślinnym łańcuchu oddechowym dehydrogenazy utleniającej N A D (P)H cytosolowe i przekazującej elektrony na ubichinon [11,12]. W związku z tym utlenianiu zewnątrz m itochondrialnego N A D H tow arzyszą dwie fosforylacje [9]. K ontrow ersyjne jest zagadnienie czy jest to jedna dehydrogenaza specyficzna zarów no dla N A D H jak i N A D P H czy też są to dwie dehydrogenazy. N a obecność jednej dehydrogenazy wskazywały prace M o l i e r a i w s p . [10]. O statnie badania przy zastosow aniu przeciwciał wykazały jednak obecność co najmniej dw óch dehydrogenaz, różniących się optim um pH i specyficznością substratow ą w stosunku do N A D H i N A D P H [13].
U tlenianie N A D (P)H przez dehydrogenazę zewnętrzną jest zależne od pH. O ptim um utleniania N A D H zachodzi przy pH między Ó.8-7.2, natom iast utlenianie N A D P H zachodzi przy pH nieco niższym [14-17], a więc jest to pH bardzo zbliżone do pH cytosolowego (7.1-7.6) [18]. U tlenianie egzogennego N A D (P)H przez m itochondria roślinne jest stym ulowane przez katio ny, szczególnie zaś przez jony w apnia [14]. Funkcja jak ą pełnią jony C a2+ w regulacji aktywności zewnętrznej dehydrogenazy N A D H nie jest wyjaśniona. Obecnie sądzi się, że jony C a2+ nie stym ulują aktyw ności dehydrogenazy natom iast ułatw iają przekazywanie elektronów na ubichinon znajdujący się w lipidowej frakcji błony, co wskazuje, że miejscem działania C a2 + jest raczej ubichinon [19]. Stym ulacja utleniania N A D H przez jony C a2+ obserw ow ana jest przy pH obojętnym , spada natom iast przy pH poniżej 7 [20]. Usunięcie jonów C a2+ (np. przez dodanie czynników
chelatujących) powoduje zaham ow anie utleniania N A D H przez izolowane m itochondria [20]. G dy dehydrogenaza N A D H uw olniona jest z błony m ito chondrialnej przez sonifikację lub traktow anie detergentem, zależność aktywności od jonów C a2+ spada, aczkolwiek enzym ten w dalszym ciągu redukuje analogi ubichinonu [21, 22]. G odnym odnotow ania jest fakt, że m aksim um stymulacji przypada na stężenia C a2+ bliskie stężeniom występującym w cytosolu u większości roślin, k tóre to stężenie podlega regulacji i zm ienia się podczas w zrostu i rozw oju oraz pod wpływem zmiennych w arunków środowiskowych. Stymulację przez jony C a2+ obniżają też regulatory wzrostu, poliam iny [23].
II-1.2. Rola fizjologiczna zewnętrznych dehydrogenaz N A D (P )H
Zew nętrzne dehydrogenazy N A D (P)H m ogą regulować stężenie N A D (P)H w cytosolu. Regulacja n ad m iaru siły redukcyjnej, przy pow iązaniu dehydrogenazy zewnętrznej N A D (P)H z drogą alternatyw ną, m ogłaby zachodzić bez produkcji ATP. Jednakże podczas utleniania cytosolowego N A D H lub N A D P H elektrony przekazyw ane na ubichinon przepływają na drogę cytochrom ow ą z jednoczesną syntezą dwóch cząsteczek A TP [24-27]. N ie wyjaśniony jest brak dostępu zewnętrznej dehydrogenazy N A D H do drogi alternatywnej u roślin nieterm ogennych [12,23, 27]. Tylko u Araceae, u których poziom oddychania cyjanood- pornego podczas kwitnienia jest bardzo wysoki, utlenianie zewnątrz m itochondrialnego N A D H może zachodzić prawie wyłącznie przy udziale drogi alternatywnej [12, 26].
N A D H utleniane jest bardzo intensywnie przez m itochondria większości roślin, K m dla N A D H waha się pom iędzy 10-100 pM [8], natom iast stężenie N A D H w cytosolu w liściach szpinaku na świetle wynosi zaledwie 0.6 pM [28]. P o nad to szereg substratów (glicyna, jabłczan) utlenianych jest preferencyjnie na świetle przez m itochondria roślinne w porów naniu z N A D H [29, 30]. T ak więc wydaje się, że in vivo utlenianie N A D H cytosolowego jest na dość niskim poziom ie i zachodzi głównie przy udziale drogi cyto- chromowej. Aczkolwiek nie jest wykluczone, że przy dużym wzroście stężenia N A D (P)H w cytosolu udział w jego utlenianiu zewnętrznej dehydrogenazy N A D (P)H może się zwiększać.
II-2. Wewnętrzne dehydrogenazy N A D H
Źródłem N A D H wewnątrz m itochondriów jest głównie cykl K rebsa, dehydrogenaza jabłczanow a dekar- boksylująca, oksydaza glicyny oraz system przenoszący N A D H /N A D z cytosolu. N A D H wytwarzane wew nątrz m itochondriów może być utleniane przez jedną z dwóch dehydrogenaz N A D H łańcucha oddechowego (Ryc. 1) lub transportow ane na zewnątrz m itochon-
270 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

driów przez przenośnik jabłczan/szczaw iooctan obecny w błonie m itochondrialnej [31, 32].
Jeśli różne enzymy wytwarzające N A D H w m atriks miałyby taki sam dostęp do dehydrogenaz łańcucha oddechowego, trudno byłoby zrozum ieć w pewnych w arunkach tzw. preferencyjne utlenianie poszczególnych substratów np. glicyny, jab łczanu [33, 34]. P rzypuszcza się, że poszczególne enzymy redukujące N A D m ogą znajdow ać się w sąsiedztwie odpow iednich dehydrogenaz łańcucha oddechowego i tworzyć kom pleksy zwane m etabolonam i [35].
Stosunek N A D H /N A D w cytosolu w czasie fo tosyntezy, jest około 70-krotnie niższy niż wewnątrz m itochondriów podczas utleniania substratow ego w stanie 3 [36], W skazuje to na ścisłą regulację transportu N A D H z m atriks m itochondriów do cytosolu zabezpieczającą w m atriks właściwe stężenie N A D H niezbędne do wytw arzania A T P w łańcuchu oddechowym.
N A D H w m atriks może być utleniane przez dwie dehydrogenazy — dehydrogenazę N A D H kom pleksu I, ham ow aną przez ro tenon i zw iązaną z syntezą ATP, lub dehydrogenazę nie ham ow aną przez ro tenon i nie uczestniczącą w przemieszczaniu protonów w poprzek błony (brak syntezy ATP). Obie wewnętrzne dehydrogenazy N A D H badano w tzw. pęcherzykach błon m itochondrialnych, w których błona w ewnętrzna była odw rócona na zewnątrz. O trzym uje się je poprzez sonifikację m itochondriów przy wysokim stężeniu soli, a następnie poprzez wirowanie różnicowe [15, 37-39].
II-2.1. Dehydrogenaza N A D H (hamowana przez rotenon), kompleks I
D ehydrogenaza N A D H zw iązana z kom pleksem I jest bardzo podobna do dehydrogenazy kom pleksu I w m itochondriach zwierzęcych [10]. Kom pleks I niedaw no wyizolowano i oczyszczono z m itochondriów b uraka czerwonego [40]. Jest to duży kom pleks białkowy o ciężarze 680 kD a [10] lub 400 kD a [40], składający się z co najmniej 6 polipeptydów i zaw ierający flawinę [41-43]. K m dla N A D H wynosi 8 pM [5], wskazuje to na znacznie wyższe powinow actw o do N A D H niż dehydrogenazy niewrażliwej na rotenon.
II-2.2. Dehydrogenaza N A D H niewrażliwa na rotenon
Obecność wewnętrznej dehydrogenazy N A D H niewrażliwej na ro tenon i ro la jak ą pełni w przenoszeniu elektronów była dyskutow ana przez wiele lat. Z asadniczą trudnością w badaniach jest b rak specyficznego inh ib itora tej dehydrogenazy albowiem zew nętrzna dehydrogenaza N A D H również nie jest ham ow ana przez ro tenon [5, 10]. W ew nętrzna dehydrogenaza N A D H niewrażliwa na ro tenon została wyizolowana z buraka czerwonego, jest ona praw dopodobnie dime-
rem składającym się z dwóch identycznych jednostek o masie 43 kD a [10].
W odróżnieniu od dehydrogenazy kom pleksu I, dehydrogenaza niewrażliwa na ro tenon wykazuje niskie powinowactwo do N A D H [8, 9], a więc może działać tylko, gdy stężenie N A D H w m atriks jest wysokie (Km = 80 pM), podczas gdy K m dehydrogenazy N A D H kom pleksu I jest 10-krotnie niższe, a więc m oże działać przy bardzo niskim stężeniu N A D H w m atriks [44]. W ysokie stężenie N A D H może występować w m atriks, gdy zwiększa się aktywność enzymów cyklu K rebsa lub utlenianie jabłczanu przez obecną w m itochondriach roślinnych dehydrogenazę jabłczanow ą dekarboksylującą oraz przy dużej aktyw ności przenośnika jabłczan/szczaw iooctan. L a n c e [45] sugerował powiązanie działania dehydrogenazy jabłczanowej dekarboksylującej (wytwarzającej wew nątrz m itochondriów pirogronian i N A D H ) z dehydrogenazą niewrażliwą na ro tenon i oksydazą alternatywną. W ten sposób, w m atriks, przy wysokim poziomie N A D H jego utlenianie m ogłoby się odbywać bez syntezy ATP. Obecnie wydaje się jednak, że przyczyną obserwowanej niewrażliwości oddychania na cyjanek przy wysokim stężeniu N A D H w m atriks jest stym ulacja oksydazy alternatywnej przez p irogronian wytwarzany przez dehydrogenazę jab łczanową dekarboksylującą.
Rola fizjologiczna dehydrogenazy N A D H niewrażliwej na ro tenon nie jest do tej pory wyjaśniona, być może działa ona tylko in situ [10, 39].
III. Oksydaza alternatywna
Jedną z najdawniej poznanych, charakterystycznych cech roślinnego łańcucha oddechowego, jest obecność końcowej oksydazy alternatywnej, k tó ra katalizuje przeniesienie elektronów z ubichinonu na tlen, z pom inięciem łańcucha cytochrom owego (Ryc. 1). O ksydaza ta jest nazyw ana również oksydazą niewrażliwą na cyjanek, drogą niewrażliwą na cyjanek i odpowiedzialna jest za tzw. oddychanie cyjanoodporne. Tego typu oddychanie obserwowane było u roślin już od początku stulecia. Najlepiej scharakteryzow ana jest rola fizjologiczna oddychania cyjanoodpornego u roślin z rodziny Araceae, u których oddychaniu podczas procesu kw itnienia towarzyszy wydzielanie dużych ilości ciepła [46-48]. Artykuł poświęcony charakterystyce biochemicznej i roli fizjologicznej oksydazy alternatywnej opublikowany był w „Postępach Biochemii” [49].
III-l. Charakterystyka oksydazy alternatywnej
Pom im o wielu prób, do tej pory m etodam i biochemicznymi, nie udało się wyizolować oksydazy alternatywnej. Częściowo oczyszczony enzym uzyskano z kolb Arum maculatum, Sauromatum guttatum oraz Symplocarpus foetidus [50-56]. Przyczyną braku sukcesów jest po pierwsze trudność uwolnienia kom
POSTĘPY BIOCHEM II 42 (3 ) , 1996 271http://rcin.org.pl

pleksu oksydazy alternatywnej z błony m itochondrialnej, a ponad to b rak odpowiedniego donora elektronów dla oksydazy alternatyw nej in vitro (zastępującego kom pleks I lub II in vivo). Z niewiadom ych przyczyn durochinol, k tóry jest dobrym donorem elektronów dla oksydazy alternatyw nej wyizolowanej z Araceae, m a bardzo słabą aktywność dla oksydazy alternatyw nej izolowanej z roślin nieterm ogennych [5, 56]. Oczywiste ograniczenie badań powoduje więc konieczność w ykorzystania roślin z rodziny Araceae, k tóre kw itną tylko raz w roku. P onad to uw olniona z błon oksydaza alternatyw na jest bardzo niestabilna, w ciągu12-24 godz. wykazuje w temp. 0°C znaczny spadek aktywności [4, 53]. Jedną z przyczyn tej niestabilności jest praw dopodobnie wysoka aktyw ność proteazy sulfhydrylowej w tkance Araceae [56]. W szystkie te trudności sprawiły, że do tej pory nie udało się uzyskać, m etodam i biochemicznymi, hom ogennego p reparatu oksydazy alternatywnej.
O ksydaza alternatyw na wyizolowana z Arum macu- latum lub Sauromatum guttatum czy też z Symplocarpus foetidus jest kom pleksem m ałych białek [53-56]. W yizolowanie z Sauromatum guttatum i nieznaczne oczyszczenie oksydazy alternatywnej pozwoliło E 11 h o - n o w i i M c l n t o s h o w i otrzym ać przeciwciała m ono i poliklonalne dla oksydazy alternatyw nej, które okazały się specyficzne dla trzech polipeptydów 37, 36 i 35 k D a [54, 55, 57]. Zapoczątkow ało to badania oksydazy alternatywnej z zastosow aniem technik b iologii m olekularnej [6, 58]. Przeciwciała te wykazywały krzyżową aktywność ze wszystkimi roślinam i posiadającym i aktywność oksydazy alternatywnej, mogły więc służyć jak o sondy m olekularne do jej wykrywania [59-63]. Przeciwciała te użyto do wyizolowania i zsek- wencjonowania pojedynczego klonu cD N A dla oksydazy alternatywnej z Sauromatum guttatum, Arabido- psis thaliana oraz ty toniu [64-66]. W ykazano, że aktywność oksydazy alternatywnej i związane z nią oddychanie niewrażliwe na cyjanek jest wynikiem działania jednego genu jądrow ego [64], Z ap roponow ano strukturę białka oksydazy alternatywnej jako dim eru, w którego skład, w zależności od rośliny, m ogą wchodzić białka o ciężarze od 32-39 k D a [5, 6, 58]. Cząsteczka oksydazy alternatywnej um ieszczona jest w poprzek wewnętrznej błony m itochondrialnej, końce C łańcucha polipeptydow ego znajdują się po stronie m atriks, a S heliks po stronie przestrzeni między- błonowej [6, 67]. D im er połączony jest m ostkiem disiarczkowym, w formie utlenionej jest on mniej aktyw ny niż w formie zredukowanej. Stan redukcji/utlenienia m ostka disiarczkowego reguluje więc Vmaks. oksydazy alternatyw nej [6, 68].
Już od daw na obserwow ano, że w wyizolowanych m itochondriach aktywność oksydazy alternatywnej jest różna w zależności od substratu, najwyższe aktyw ności obserw ow ano z jabłczanem i bursztynianem , a najniższe z N A D H [24]. Różnice te m ogą zanikać po dodaniu a-keto kwasów organicznych [69]. W m ito-
chondriach soi niewielka aktywność oksydazy alternatywnej obserw ow ana z bursztynianem może być znacznie stym ulow ana przez pirogronian [70]. Kwas pirogronow y jest efektorem obniżającym K m oksydazy alternatyw nej w stosunku do ubichinolu [6, 71]. Aktywność oksydazy alternatywnej wzmaga również wytwarzanie p irogronianu wewnątrz m itochondriów podczas utleniania bursztynianu lub jabłczanu [70]. W ydaje się więc, że w izolowanych m itochondriach różnice aktywności oksydazy alternatywnej przy różnych substratach m ożna tłumaczyć odm ienną ich zdolnością do w ytw arzania endogennego pirogronianu. D opływ pirogronianu z glikolizy aktywuje drogę alternatyw ną, podobnie jak wytwarzanie pirogronianu wewnątrz m itochondriów np. przez enzym jabłczano- wy [69]. Tłum aczy to, jak się zdaje, zaobserw ow ane wcześniej powiązanie aktywności enzymu jabłczano- wego w m itochondriach z działaniem oksydazy alternatywnej [72].
W transgenicznym tytoniu z zwiększoną ekspresją białka oksydazy alternatywnej, m itochondria wyizolowane z liści, wykazywały stosunkow o niską aktywność drogi alternatywnej, aktywność ta zwiększała się do piero po dodaniu czynnika redukującego i p irogronianu [73]. W m itochondriach tych duża część białka oksydazy alternatywnej była w postaci związanego kowalencyjnie utlenionego (mniej aktywnego) dimeru, dodanie zaś czynnika redukującego (DTT), jak rów nież izocytrynianu i jabłczanu pow odow ało redukcję białka oksydazy alternatywnej do związanego nieko- walencyjnie (bardziej aktywnego) dimeru. Przypuszcza się, że czynnikiem regulującym stan utlenienia/redukcji dim eru oksydazy alternatywnej m ogłaby być nie tioredoksyna [6, 68], ale przede wszystkim N A D P H pow stający wewnątrz m itochondriów [73]. W procesie izolow ania m itochondriów dim er może ulegać utlenieniu, dlatego wydaje się, że oksydaza alternatyw na może być dużo bardziej aktyw na in vivo niż w wyizolowanych m itochondriach. W tkance ponad to stan utlenienia/redukcji białka oksydazy alternatywnej może podlegać regulacji poprzez zm ienną ilość siły redukcyjnej wytwarzanej w ewnątrz m itochondriów [68, 73].
Końcow ym produktem redukcji tlenu przez ok sydazę alternatyw ną jest w oda [6, 49, 74]. O ksydaza alternatyw na m a mniejsze powinowactwo do tlenu niż oksydaza cytochrom ow a (Km dla 0 2 oksydazy alternatywnej wynosi 10-20 pM , podczas gdy oksydazy cytochrom owej 0.1-0.6 pM [75]. Jednakże w zakresie stężeń tlenu (100-200 pM ) używanych do pom iaru oddychania, aktywność oksydazy alternatywnej jest praktycznie niezależna od stężenia tlenu [5].
O d daw na sugeruje się obecność w centrum aktyw nym oksydazy alternatywnej żelaza związanego niehe- m owo [6, 49, 76]. Jednak żadna z dotychczasowych analiz wyizolowanego białka oksydazy alternatywnej nie potw ierdziła w sposób jednoznaczny obecności żelaza [5]. O statn io zaproponow ano hipotetyczną strukturę centrum aktywnego oksydazy alternatywnej,
272 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

analogiczną do m etalo-białka m ono oksygenazy m etanu [6, 77].
III-2. Regulacja przepływu elektronów przez drogę cytochromową i alternatywną
D o oznaczania udziału w oddychaniu drogi a lternatywnej i cytochrom owej używano m etody B a h r a i B o n n e r a polegającej na m iareczkow aniu aktyw ności obu dróg odpow iednio inhibitoram i drogi alternatywnej (kwas salicylohydroksam owy) lub cytochromowej (cyjanek potasu) [49, 78-80]. M etoda ta znalazła zastosow anie nie tylko w odniesieniu do izolow anych m itochondriów , ale również do tkanek i całych roślin [80, 81].
Przy użyciu inhibitorów drogi alternatyw nej i cytochromowej wykazano, że droga alternatyw na działa przy wysyconej drodze cytochrom owej [78, 79]. Obie drogi rozgałęziają się na poziomie ubichinonu, m ają jednak w stosunku do niego odm ienne własności kinetyczne. Aktywność drogi cytochrom owej zależna jest od stopnia redukcji ubichinonu w sposób liniowy, natom iast aktyw ność drogi alternatyw nej nie w ykazuje tej zależności i ujawnia się dopiero, gdy pula ubichinonu jest zredukow ana powyżej 35-40% [82, 83]. Tylko u roślin term ogennych zależność aktyw ności drogi alternatywnej od stopnia redukcji ubichinonu jest zależnością liniową [83].
W yniki pom iarów woltam etrycznych poziom u redukcji ubichinonu wykazały, że jego redukcja może być dw ustopniow a [84]. Z aproponow ano m odel regulacji przepływu elektronów przez pulę ubichinonu na drogę alternatyw ną, tlen reagow ałby z oksydazą alternatyw ną, gdy osiągnie ona stan cztero elektronowej redukcji [84]. Potw ierdzałoby to brak translokacji p ro tonów przez oksydazę alternatyw ną [11], w przeciwieństwie do oksydazy cytochrom owej działającej jak o pom pa pro tonow a [85],
N a stan redukcji ubichinonu wpływa aktyw ność dehydrogenaz łańcucha oddechowego. W większości doświadczeń w izolowanych m itochondriach wysyce- nie obu dróg oznaczano dodając jako substrat bur- sztynian [86, 87]. W tych w arunkach droga cytochrom ow a praw dopodobnie nigdy nie osiąga stanu wysycenia, bo np. dodanie N A D H jednocześnie z bur- sztynianem sprawia, że oddychanie poprzez drogę cytochrom ow ą, w tych samych m itochondriach, jest znacznie wyższe [88]. P onad to oznaczenia aktywności obu dróg przy użyciu inhibitorów (tzw. m iareczkow ania inhibitoram i) wykazały, że zaham ow anie drogi alternatywnej może prow adzić do wzrostu oddychania drogą cytochrom ow ą [87, 88]. W skazuje to na brak wysycenia drogi cytochrom owej przy działającej d ro dze alternatywnej oraz na możliwość jednoczesnego przepływu elektronów przez obie drogi i w spółzawodnictwo o elektrony [86, 89]. Biorąc pod uwagę czynniki wpływające na aktywność oksydazy alternatyw nej, wydaje się, że regulacja przepływu elektronów na
drogę alternatyw ną jest o wiele bardziej skom plikow ana niż to początkow o przypuszczano. Pytanie o m echanizm regulacji stale czeka na odpowiedź.
III-3 Oznaczanie aktywności drogi alternatywnej
O pisane powyżej badania wykazały, że oznaczana przy użyciu inhibitorów aktyw ność drogi alternatyw nej, zarów no w całych roślinach jak izolowanych m itochondriach, może być niższa niż występująca in vivo [87-89]. W izolowanych m itochondriach zarówno poziom pirogronianu jak i czynnika redukującego m ostki disiarczkowe dim eru oksydazy alternatywnej może być różny od poziom u tych czynników w m itochondriach w tkance. P onadto , pobieranie tlenu ham owane przez inhibitor drogi alternatywnej nie może być m iarą jej aktywności in vivo, albowiem zaham ow anie drogi alternatywnej może pow odow ać przepływ elektronów na niewysyconą drogę cytochrom ową. Przy użyciu inhibitorów m ożna więc jedynie określać typ oddychania; np. cyjanoodporne lub wrażliwe na kwasy hydroksam ow e (SHAM), podkreślając jednocześnie, że wartość tego oddychania ważna jest wyłącznie dla danych w arunków eksperym entalnych (oddychanie w obecności inhibitora), może natom iast być różna w w arunkach in vivo, (przy braku inhibitora) [87], W yrazem krytyki oznaczania aktywności oksydazy alternatywnej przy użyciu inhibitorów jest artykuł w dziale „viewpoints” jednego z ostatnich num erów Plant Physiology pod znam iennym tytułem „O ksydaza niewrażliwa na cyjanek: ham ow ać czy nie ham ować, oto jest pytanie”. K onkluzja i odpowiedź na postaw ione w tytule pytanie brzmi — nie ham ować, nie oznaczać aktywności oksydazy alternatywnej przy pom ocy inhibitorów [90]. W obec tego jak oznaczać aktywność drogi alternatywnej in vivo?
O statn io rozwinęła się technika oznaczania aktyw ności drogi alternatywnej poprzez oznaczenie stopnia dyskrym inacji (tzn. nierównom iernie niższego) pobieran ia z pow ietrza izotopu 180 2 w stosunku do 160 2 [91-93]. D yskrym inacja pobierania 180 2 przez drogę alternatyw ną jest większa niż przez drogę cytochrom ową. Stopień dyskrym inacji pobierania przez drogę cytochrom ow ą wynosi 17,1-20%, natom iast przez d ro gę alternatyw ną 23,5-25,5%, wartości te w przypadku różnych roślin są nieco odm ienne [91-93]. Przy zastosow aniu tej techniki wykazano, że w izolowanych m itochondriach soi w w arunkach stanu 3 (w obecności ADP), po dodaniu pirogronianu, przy niskim poziomie redukcji ubichinonu, obie drogi współzawodniczą o zredukow aną pulę ubichinonu [93]. Zastosow anie do tych samych w arunków tradycyjnej m etody pom iaru oddychania elektrodą C larka i użycie inhibitorów nie wykazało udziału drogi alternatywnej [93].
W ydaje się, że technika dyskrym inacji pobierania izotopu tlenu jest obecnie jedyną w iarygodną i nieinwazyjną (bez użycia inhibitorów ) m etodą oznaczania in vivo udziału drogi alternatywnej w oddychaniu. Nie
POSTĘPY BIOCHEMII 42 (3 ) , 1996 273http://rcin.org.pl

ulega jednak wątpliwości, że jest nieco bardziej czasochłonna i wym agająca specyficznej aparatu ry (chrom atograf gazowy, spektrom etr masowy).
III-4. Rola fizjologiczna drogi alternatywnej
Rola fizjologiczna drogi alternatywnej jako procesu generującego ciepło wydaje się być bezsporna jedynie u Araceae [47], U innych roślin poziom wytw arzania ciepła przez drogę alternatyw ną jest zbyt mały, aby m iał jakiekolw iek znaczenie fizjologiczne. H ipoteza „przelewu” (overflow) wysunięta przez L a m b e r s a opisywała stany fizjologiczne w jakich może funkcjonow ać droga alternatyw na, przy wy syconej drodze cytochrom owej [94-96]. W pewnych w arunkach (np. wzrost zawartości cukrów w kom órce i wynikający z tego intensywniejszy ich przepływ przez szlak glikoli- tyczny) może wystąpić brak równow agi pomiędzy oksydacyjnym m etabolizm em węgla a możliwością przepływu elektronów przez łańcuch cytochromowy. D roga cytochrom ow a może być wysycona przy spadku A D P, ograniczeniu aktywności jednego ze składników łańcucha oddechowego [66, 73, 79], lub przy deficycie P i [97, 98]. W tedy, przy zwiększonym m etabolizmie oksydacyjnym, następuje uruchom ienie drogi alternatywnej. W ydaje się jednak, że hipoteza ta musi ulec pewnej modyfikacji i uwzględnić udział drogi alternatywnej przy nie całkowicie wysyconej drodze cytochrom owej, a także uwzględnić regulację aktyw ności drogi alternatyw nej tn vivo przez p irogronian i potencjał redukcyjny wewnątrz m itochondriów . Jak wykazały ostatnie badania, w izolowanych m itochondriach tytoniu, w arunkiem uruchom ienia drogi alternatywnej jest spadek poziom u ADP, wzrost siły redukcyjnej wewnątrz m itochondriów , (niezbędnej do redukcji w iązania disiarczkowego) oraz obecność piro- gronianu (potrzebna do stymulacji zredukowanej formy oksydazy alternatywnej) [73].
Inną interesującą hipotezą opisującą rolę fizjologiczną oksydazy alternatyw nej jest jej udział w regulacji poziom u ponadtlenków i nadtlenków w odoru w tk an ce [87, 101]. N adtlenki w odoru m ogą odgrywać rolę w indukcji ekspresji genu oksydazy alternatywnej [87]. W w arunkach stresu spowodowanego chłodem [99- 101], inwazją pasożytów , zranieniem [96], czy też ograniczoną dostępnością fosforanu [97,98] następuje uruchom ienie oddychania niewrażliwego na cyjanek. W tedy, przy ograniczonej aktywności drogi cytochromowej i zwiększonej redukcji ubichinonu, na terenie m itochondriów może dochodzić do w ytw arzania po- nadtlenku lub nadtlenku w odoru [101]. Sugeruje się, że udział w oddychaniu drogi alternatywnej może ograniczać ich w ytwarzanie [87, 101]. Z aproponow ana ostatnio hipoteza regulacji aktywności i indukcji oksydazy alternatyw nej uwzględnia zarów no stym ulację aktywności oksydazy alternatywnej przez kwasy organiczne zapobiegającą zwiększonej redukcji ubichinonu jak i udział nadtlenków w indukcji ekspresji genu
274
odpowiedzialnego za syntezę białka oksydazy alternatywnej [87]. Ta niezmiernie interesująca hipoteza jest obecnie testowana.
IV. Uwagi końcowe
W ciągu ostatnich pięciu lat techniki biologii m olekularnej niewątpliwie bardzo posunęły naprzód b ad ania nad struk tu rą i ro lą fizjologiczną oksydazy a lternatywnej. D ostępność przeciwciał oksydazy alternatywnej oraz klonów cD N A pozw ala śledzić ekspresję oddychania niewrażliwego na cyjanek na poziom ie RN A i białka z jednoczesnym i pom iaram i gazomet- rycznymi (pobieranie tlenu). O trzym anie roślin trans- genicznych ze zwiększoną lub zmniejszoną ekspresją oksydazy alternatyw nej umożliwi określenie jej roli w procesach wzrostu i rozw oju roślin.
Oznaczenie udziału w oddychaniu niefosforylującej drogi alternatywnej oraz m echanizm ów związanych z regulacją tego udziału m a ogrom ne znaczenie dla określenia wydajności energetycznej oddychania [102]. U pewnych roślin (Lolium perenne) obserwuje się negatyw ną korelację między wysokością p lonow ania a intensywnością oddychania. M oże to być m.in. wynikiem różnej intensywności oksydacyjnej fosforylacji [95]. W ydaje się więc, że określenie udziału w oddychaniu drogi alternatyw nej jest o wiele bardziej istotne niż to się do tej pory wydawało, a do tychczasowe wyliczenia wydajności energetycznej oddychania roślin in vivo m ogą być na znacznie zaniżonym poziomie. D o tych zadań niezbędny jest rozwój n o wych m etod pozwalających na precyzyjne określenie udziału obu dróg, drogi alternatyw nej i cytochrom owej w oddychaniu roślin.
Napisanie tego artykułu było możliwe dzięki sfinansowaniu przez Grant KBN 6P2043705 uczestnictwa autorki w Konferencji „Plant Mitochondria from Gene to Function”.
A rtyku ł otrzymano 6 lutego 1996 r.Zaakceptowano do druku 20 marca 1996 r.
Piśmiennictwo
1. Hr y n i e wi e c k a L (1993) Post Biol Komórki 20: 181-1992. Douc e R, Br o u q u i s s e R, J o u r n e t E- P (1987) W:
Davies D.D (red) The Biochemistry of Plants: A Comprehensive Treatise, t 11 Academic Press, San Diego, Ca, str. 177-221
3. D o u c e R (1985) W: Mitochondria in Higher Plants: Structure, Function, and Biogenesis. Academic Press, Orlando, Florida
4. Douc e R, N e u b u r g e r M (1989) Annu Rev Plant Physiol Plant M ol Biol 40: 371-414
5. Mo o r e AL , S i e d o wJ N (1991)BiochimBiophysActa 1059: 121-140
6. S i edow JN, Umb a c h AL (1995) Plant Cell 7: 821-8317. P a l me r JM, Mö l l e r IM (1982) TYends Biochem Sci 7:
258-2618. P a l me r JM, Wa r d JA (1985) Encycl Plant Physiol 18:
173-2019. Möl l e r IM, Lin W (1986) Annu Rev Plant Physiol 37:
309-33410. Mö l l e r IM, R a s mu s s o n AG, F r e d l u n d KM (1993)
J Bioenerg Biomembranes 25: 377-384
POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

11. Mo o r e AL, Bo n n e r WD, Ri ch PR (1978) J Exp Bot 29: 1-1 2
12. C o t t i n g h a m IR, Mo o r e AL (1983) Biochim Biophys Acta 724: 191-200
13. E l t h o n TE (1995) Referat na Konferencji „Plant Mitochondria” From Gene to Function” Duke University, Durham, N.C. USA
14. Mö l l e r IM, P a l me r JM (1981) Biochim Biophys Acta 638: 22-233
15. Mö l l e r IM, P a l me r JM (1981) Physiol Plant 53: 413-42016. Möl l e r I M, Pa l me r J M, J o h n s t o n SP (1983)Biochim
Biophys Acta 725: 289-29717. E d ma n K, Er i c s o n I , Mö l l e r IM (1985) Biochem J 232:
471-47718. K u r k d j i a n A, Gu e r n J (1989) Annu Rev Plant Physiol
Plant Mol Biol 40: 271-30319. So o l e KL, Dr y l B , Wi s k i c h J T (1990) Physiol Plant 78:
205-21020. Möl l e r IM, J o h n s t o n SP, P a l me r JM (1981) Biochem
J 194: 487-49521. Cook ND, Ca mma c k R (1984)E urJ Biochem 141:573-57722. C o t t i n g h a m IR, Mo o r e AL (1984) Biochem J 224:
171-17923. Rugo l o M, Z a n n o n i D (1992) Plant Physiol 99:1037-104324. Lance C, C h a u v e a u M, Di z e n g r e me l P (1985)
Encycl Plant Physiol 18: 202-24725. G a r d e s t r ö m P, E d wa r d s GE (1983) Plant Physiol 11\
24-2926. H uq S, P a l me r JM (1978) Plant Sei Lett 11: 351-35827. H e mr i k a - Wa g n e r AM, Gu d e H, Ma r i s s e n N,
Van der Pi as LHW, Ve r l e u r J.D. (1986) Plant Cell Physiol 27: 499-503
28. He i n eke D, Ri ens B, Gr o s s e H, Ho f e r i c h t e r P, Pe t e r U (1991) Plant Physiol 95: 1131-1137
29. D ry IB, D ay DA, Wi s k i ch JT (1983) FEBS Lett 158: 154-158
30. Be r g ma n A, Er i cs on I (1983) Physiol Plant 59: 421-42731. E b b i g h a u s e n H, Chen J, He i d t HW (1985) Biochim
Biophys Acta 810: 184-18932. Ol i ve r DJ, Wa l k e r GH (1984) Plant Physiol 16: 409-41333. D ay DA, N e u b u r g e r M, Do u c e R (1985) Austr J Plant
Physiol 12: 119-13034. D ry IB, Wi s k i ch JT (1985) Austr J Plant Physiol 12:
329-33935. Wi s k i ch JT, Bryce JH, Day DA, Dr y IB (1990) Plant
Physiol 93: 611-61636. K r ö me r S, He i d t HW (1991) Biochim Biophys Acta 1057:
42-5037. Möl l e r IM, Li den AC, Er i c s o n I, G a r d e s t r ö m
P (1987) Methods in Enzymol 148: 442-45338. Pe t i t PX, G a r d e s t r ö m P, Ra s mu s s o n AG, M ö l
ler IM (1991) Plant Sei 78: 177-18339. Ra s mu s s o n AG, Mö l l e r IM (1991) Physiol Plant 83:
357-36540. Sool e K L, D r y IB, Wi s k i ch JT (1992) Plant Physiol 98:
588-59441. C o t t i n g h a m IR, Cl ee t e r MW J, Ragan Cl, Mo o r e
A L (1986) Biochem J 236: 201-20742. C o t t i n g h a m IR, Mo o r e AL (1988) Biochem J 254:
303-30543. Chen S, Gu i l l o r y PJ (1981) J Biol Chem 256: 8318-832344. Möl l e r IM, P a l me r JM, J o h n a s t o n SP (1983) Bio
chim Biophys Acta 725: 289-29745. Lance C, Ru s t i n P (1984) Physiol Veg 22: 625-64146. Meeus e BJD (1975) Annu Rev Plant Physiol 26: 117-12647. Or d e n t l i c h A, Li nze r RA, Ra s k i n I (1991) Plant
Physiol 97: 1545-155048. Rh o a d s DM, Mc I n t o s h L (1992) Plant Cell 4:1131-113949. Ry c h t e r AM (1982) Post Biochem 28: 89-11150. H uq S, P a l me r JM (1978) FEBS Lett 95: 217-22051. Ri ch PR (1978) FEBS Lett 96: 252-25652. Bo n n e r WD, Ri ch PR (1983) 72: S-1953. Bo n n e r WD, Cl a r k e S D, Ri ch PR (1986) Plant Physiol
80: 838-84254. E l t h o n TE, Mc I n t o s h L (1986) Plant Physiol 82: 1-655. E l t h o n TE, Mc I n t o s h L (1987) Proc Natl Acad Sei USA
84: 8399-8403
56. Be r t h o l d DA, Si edow J N (1993) Plant Physiol 101: 113-119
57. E l t h o n TE, Ni cke l s RL, Mc I n t o s h L (1989) Plant Physiol 89: 1311-1317
58. Mc I n t o s h L (1994) Plant Physiol 105: 781-78659. Hi se r C, Mc I n t o s h L (1990) Plant Physiol 93: 312-31860. L a mb o wi t z AM, S a b o u r i n JR, Be r t r a n d H, N i c
kel s R, Mc I n t o s h L (1988) M ol Cell Biol 9: 1362-136461. Ob e n l a n d D, Di e t h e l m R, Dh i b l e s R, S t e war t
C (1990) Plant Cell Physiol 31: 897-90162. Ke a r n s A, Wh e l a n J, Young S, E l t h o n TE, Day
DA (1992) Plant Physiol 99: 712-71763. Saka j o S, Mi n a g a wa N, K o m i y a m a T, Yos h i mo-
to A (1991) Biochim Biophys Acta 1090: 102-10864. Rh o a d s DM, Mc I n t o s h L (1991) Proc Natl Acad Sci
USA 88 : 2122-212665. K u ma r AM, Soi l D (1992) Proc Natl Acas Sci USA 89:
10842-1084666. Va n l e r b e r g h e GC, Mc I n t o s h L (1994) Plant Physiol
105: 867-87467. Si edow J N, Whe l an J , Kea r ns A, Wi ski ch JT, Day
D A (1992) W: Lambers H, van der Plas LHW (red) Molecular, Biochemical and Physiological Aspects of Plant Respiration. Academic Press, The Hague, The Netherlands, str 19-27
68. Umb a c h AL, Si edow JN (1993) Plant Physiol 103: 845-854
69. Wa g n e r AM, Kr a a k MS, van Emme r i k WAM, van der Pl as LHW (1989) Physiol Plant 27: 837-845
70. Mi l l a r AH, Wi s k i ch JT, Wh e l a n J , Day DA (1993) FEBS Lett 329: 259-262
71. Day DA, Mi l l a r AH, Wi s k i ch J T, Whe l a n J (1994) Plant Physiol 106: 1421-142
72. Rus t i n P, Mo r e a u F, Lance C (1980) Plant Physiol 66: 457-462
73. Va n l e r b e r g h e GC, Day DA, Wi s ki ch J T , Va n l e r be r ghe AE, Mc I n t o s h L (1995) Plant Physiol 109: 353-361
74. K ay CJ, Pa l me r JM (1985) Biochem J 228: 309-31875. R i b a s - C a r b o M, Ber r y JA, Az c o n - Bi e t o J, S i e
dow JN (1994) Biochim Biophys Acta 1188: 205-21276. S c h o n b a u m GR, Bo n n e r WD, St o r ey BT, Bahr
JT (1971) Plant Physiol 47: 124-12877. Si edow JN, Umb a c h A L, Mo o r e AL (1995) FEBS Lett
362: 10-1478. Bahr JT, Bo n n e r WD (1973) J Biol Chem 248: 3441-344579. B a h r J T, B o n n e r W D (1973) J Biol Chem 248: 3446-345080. Mo Her IM, Berczi A, van der Pl a s LHW, L a m
ber s H (1988) Physiol Plant 72: 642-64981. Th e o l o g i e s A, La t i e s GG (1978) Plant Physiol 62:
232-23782. D ry I B, Moor e AL, Day DA, Wi s ki ch JT (1989) Arc/j
Biochem Biophys 273: 148-15783. Day DA, D ry IB, Sool e KL, Wi s h i ch JT, Mo o r e
A L (1991) Plant Physiol 95: 948-95384. Si edow JN, Mo o r e AL (1993) Biochim Biophys Acta 1142:
165-17485. Babcoc k GT, Wi k s t r o m M (1992) Nature (Lond) 356:
301-30986. Wa g n e r AM, Wa g n e r MJ (1995) Plant Physiol 108:
277-28387. Wa g n e r AM, Kr a b K (1995) Physiol Plant 95:318-32588. Ho e f n a g e l MHN, Mi l l a r AH, Wi s k i ch JT, Day
DA (1995) Arch Biochem Biophys 318: 394-40089. At k i n OK, Vi l l a r R, La mb e r s H (1995) Plant Physiol
108: 1179-118390. Day DA, Kr a b K, La mb e r s H, Mo o r e A L, Si edow
JN, Wa g n e r AM, Wi s k i ch JT (1996) Plant Physiol 110: 1-2
91. Weger HG, Guy RD, Tu r p i n DH (1990) Plant Physiol 93: 356-360
92. Ro b i n s o n SA, Ya ki r D, R i b a s - C a r b o M, Gi l es L, Os mo n d CB, Si edow JN, Ber r y JA (1992) Plant Physiol 100: 1087-1091
93. R i b a s - C a r b o M, Ber r y JA, Ya k i r D, Gi l es L, R o b i n s o n SA, Le n n o n AM, Si edow JN (1995) Plant Physiol 109: 829-837
94. La mb e r s H (1982) Physiol Plant 55: 478-485
POSTĘPY BIOCHEMII 42 (3 ) , 1996 275http://rcin.org.pl

95. L a m b e r s H (1980) Plant Cell and Environment 3: 293-30296. L a m b e r s H (1985) W: Douce R, Day DA (red.) Encyklopedia
of Plant Physiology New Series, t. 18 Springer-Yerlag Berlin Eleidelberg, str 418-473
97. R y c h t e r AM, M i k u l s k a M (1990) Physiol Plant 79: 663-667
98. R y c h t e r A M, C h a u v e a u M, B o m s e l J - L, L a n c e C (1992) Physiol Plant 84: 80-86
99. R y c h t e r AM, C i e ś l a E, K a c p e r s k a A (1988) Physiol Plant 73: 299-304
100. Y a n l e r b e r g h e GC, M c I n t o s h L (1992) Plant Physiol 100: 115-119
101. P u r v i s AC, S h e w f e l t R L (1993) Physiol Plant 8 8 : 712-718
102. M o o r e AL, L e a c h G, W h i t e h o u s e DG, v a n d e r B e r g e n C, W a g n e r A, K r a b H (1994) Biochim Biophys Acta 1187: 145-151
Sterowana biosynteza antybiotyków poliketydowych
Engineered biosynthesis of polyketide antibiotics
K A T A R Z Y N A K U C Z E K 1, M A G D A L E N A K O T O W S K A 2, M A R IA N M O R D A R S K I 3
Spis treści:
I. WstępII. Syntazy poliketydoweIII. Geny syntaz poliketydowychIY. Próby sterowania biosyntezą
IV-1. Badanie mutantów zablokowanych na poszczególnych etapach biosyntezy
IV-2. Heterologiczna ekspresja genów IV-3. Badanie funkcji natywnego kompleksu syntazy
podlegającego ekspresji w komórkach Escherichia coli
IY-4. Badanie funkcji wyselekcjonowanego zespołu enzymów PKS w układzie pozakomórkowym
IV-5. Analiza sekwencji genówV. Podsumowanie
Contents:
I. IntroductionII. Polyketide synthasesIII. Genes of polyketide synthasesIV. Attempts of programming of biosynthesis
IV-1. Investigation of mutants blocked on different stages of biosynthesis
IV-2. Heterologous gene expression IV-3. Investigation of synthase complex expressed in
Escherichia coli IV-4. Investigation of enzyme complex in cell free
systems IV-5. Gene sequence analysis
V. Concluding remarks
Wykaz stosowanych skrótów: PKS — syntaza poliketydowa; minPKS — minimalny zestaw genów' syntazy; DEBS — syntaza 6- dezoksyerytronolidu B; SU — jednostka syntazy; AT— acylotransferaza; ACP — białkowy czynnik przenoszący grupy acylowe; KS — syntaza P-ketoacylowa; KR — ketore- duktaza; DH — dehydrogenaza; ER — enoiloreduktaza; TE— tioesteraza; CLF — czynnik determinujący długość łańcucha; ARO — aromataza; CYC — cyklaza; MT— 0-metylotransferaza.
I. Wstęp
N azw a „połiketydy” oznacza klasę związków wytw arzanych w wyniku szeregu reakcji kondensacji reszt acylowych krótkich kwasów karboksylow ych oraz reakcji ich redukcji w sposób podobny do syntezy kwasów tłuszczowych. Jest to heterogenna grupa
3Dr, 2mgr inż.,3prof., Instytut Immunologii i Terapii Doświadczalnej PAN ul. Czerska 12, 53-114 Wrocław.
związków, do której zalicza się roślinne flawonoidy, toksyny grzybowe oraz liczne związki pochodzenia bakteryjnego [1-3] (Ryc. 1). Wiele z nich wykazuje właściwości przeciwbakterjne, przeciwgrzybicze, prze- ciwnowotworowe, a naw et toksyczne wobec pasożytów przew odu pokarm ow ego kręgowców [3-7]. N iektóre połiketydy pochodzenia grzybowego i bakteryjnego odgrywają rolę regulatorów wzrostu i różnicow ania kom órki [8]. Funkcja biologiczna większości z nich nie jest jednak znana. Bogatym źródłem poli- ketydów są prom ieniowce — G ram -dodatnie bakterie glebowe. C harakteryzują się złożonym cyklem rozwojowym, podczas którego wytwarzają wiele m etabolitów w tórnych, m.in. związków poliketydowych. N a jbardziej znanym i z tej grupy są antybiotyki m akro- lidowe oraz tetracykliny. N ależą do nich ponadto: antracykliny, polieny, polietery oraz pochodne chino- nowe [9]. B adania dotyczące m echanizm u biosyntezy poliketydów zm ierzają do utw orzenia kontrolow ane-
276 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

o o OH O
STILBEN
CO,H
AKTYNORODYNA
<, OH OH N(CH3)2 ,OH
Oh IIOH O OH O
CONH,
C O O C H ,
HOOC. ^CH2OH
Ryc. 1 Struktury związków poliketydo- wych: stilben — poliketydowy prekursor fitoaleksyn (związków pochodzenia roślinnego); aflatoksyna BI — toksyna Aspergillus flavus oryzae; tetracenomycyna C — związek o aktywności przeciwnowo- tworowej wytwarzany przez Strep- tomyces glaucescens; aktynorody- na, erytromycyna A, monenzyna A, oksytetracyklina i tylozyna — antybiotyki wytwarzane przez różne szczepy Streptomyces.
go układu dostarczającego związków o zaprogram owanej strukturze.
II. Syntazy poliketydowe
W szystkie syntazy poliketydowe (PKS) są pod względem budow y i m echanizm u działania podobne do syntazy kwasów tłuszczowych. Są to wielofunkcyjne zespoły enzymatyczne. K atalizują pow tarzające się dekarboksylacyjne kondensacje (Claisena) między acylotioestram i koenzym u A: acetylowym, propionyl- owym, m alonylowym lub m etylomaionylowym. R zadziej między estram i innych krótkich kwasów k arb o k sylowych a acylotioestram i połączonym i z cysteiną centrum aktywnego enzymu kondensującego [9] (Ryc 2). P roduk ty kondensacji uzyskują zróżnicowanie strukturalne w wyniku przejścia przez cały, lub tylko część cyklu redukcyjnego, k tóry obejm uje reakcje: ketoredukcji, dehydratacji i redukcji enoilu przy grupie P-ketonowej rosnącego łańcucha poliketydowego. Ł ańcuch węglowy o długości charakterystycznej dla
MONENZYNA A
specyficznego p roduk tu zostaje uwolniony z syntazy wskutek hydrolizy reszty acylowej (tiolizy). Długość łańcucha węglowego i w ybór reszt acylowych budujących łańcuch dodatkow o wpływają na olbrzymie zróżnicowanie strukturalne naturalnie występujących poli- ketydów. Szkielet poliketydowy najczęściej nie jest ostatecznym produktem i ulega dalszym przem ianom , tzw. modyfikacjom post-poliketydowym , co również zwiększa różnorodność powstających związków. D o tych reakcji należą: cyklizacja, tworzenie pierścieni arom atycznych, laktonowych, glikozylacja, metylacja, utlenienie.
Związki poliketydowe m ożna zaliczyć do dwóch typów strukturalnych — poliketydy arom atyczne oraz o strukturze złożonej. Odzwierciedleniem tego po działu są dw a różne typy organizacji kom pleksów enzymatycznych syntazy.
Typ I stanowi jeden lub kilka dużych polipeptydów. K ażdy z nich to m ultifunkcjonalny enzym analogiczny do eukariotycznej syntazy kwasów tłuszczowych i syntazy kw asu 6-metylosalicylowego Pénicillium patulum
POSTĘPY BIOCHEMII 42 (3 ) , 1996 277http://rcin.org.pl

Ryc. 2 Schemat syntezy poliketydów (wg [9], reprodukcja za zgodą Annu Rev Inc). Asymetryczne atomy węgla oznaczono kółkiem. Zmiana koloru z białego na czarny oznacza inwersję konfiguracji. ACP — białkowy czynnik przenoszący grupy acylowe, KS — syntaza P-ketoacy- lowa.
[10]. W ielofunkcyjny polipeptyd składa się z pow tórzonych regionów, zwanych jednostkam i syntazy (syn- thase units - SU), katalizujących kolejne cykle redukcyjne. K ażda jednostka skupia centra katalityczne poszczególnych reakcji doprow adzających do kondensacji i całkowitej lub częściowej redukcji powstającego łańcucha. W szystkie SU zawierają syntazę P-ketoacy- lową (KS), acylotransferazę (AT) i białkowy czynnik przenoszący grupy acylowe (ACP), różnią się liczbą dom en ketoreduktazy (KR), dehydrogenazy (DH) i enoiloreduktazy (ER). D om eną kończącą syntezę szkieletu poliketydowego jest tioesteraza (TE). Każde centrum aktyw ne jest wykorzystywane tylko raz po d czas syntezy jednej cząsteczki produktu .
D o typu I zaliczana jest m.in. syntaza aglikonowej części m akrolidu — erytrom ycyny Saccharopolyspora erythraea oraz inne m akrolidow e syntazy prom ieniow ców i aw erm ektyny Streptomyces avermitilis, tylozyny Streptomyces fradiae, spiram ycyny Streptomyces am- bofaciens oraz karbom ycyny Streptomyces thermotole- rans [11-13].
Syntaza typu II stanowi kom pleks kilku zasoc- jow anych enzymów, z których każdy jest wielokrotnie aktyw ny w ciągu syntezy jednego związku [9]. Te enzymy to KS, AT, ACP, czynnik determ inujący długość łańcucha (chain length determining factor - CLF), KR, cyklaza (CYC) i arom ataza (ARO). D o typu II należy syntaza kwasów tłuszczowych Escheri- chia coli oraz wszystkie znane syntazy arom atycznych poliketydów u prom ieniowców rodzaju Streptomyces.
Ze względów funkcjonalnych został wyróżniony trzeci typ syntazy, występujący wyłącznie u roślin wyższych. C harakteryzuje się on brakiem ACP. Jego funkcje przejm uje koenzym A zestryfikowany kwasami karboksylow ym i [14, 15].
Genetyczne podstaw y program ow ania struktury poliketydu były do niedaw na całkowicie nieznane. Prow adzone obecnie w kilku ośrodkach badan ia zmie-
rzają do ich wyjaśnienia i w ykorzystania w program ow aniu syntez nowych związków in vivo.
III. Geny syntaz poliketydowych
W yodrębniono i sklonow ano geny kilkunastu syntaz poliketydowych promieniowców. G eny każdego z dotychczas scharakteryzow anych zespołów syntaz są zgrupow ane w jednym regionie genomu. Towarzyszą im geny oporności (jeśli poliketyd jest antybiotykiem ) oraz geny enzymów wprow adzających postpolikety- dowe modyfikacje w szkielecie związku.
W przypadku wszystkich zbadanych syntaz typu I geny są długimi jednostkam i transkrypcyjnym i (ok. 10 tysięcy par zasad). K ażda z nich koduje wszystkie enzymy uczestniczące kolejno w cyklach w ydłużania i redukcji łańcucha węglowego. Pełną sekwencję nuk- leotydow ą poznano jedynie w przypadku genów syntazy dezoksyerytronolidu B (6-deoxyerythronolide B synthase - D EBS) czyli aglikonowej części eryt- rom ycym y A. Sekwencja ta obejmuje około 56 tys. par zasad tworzących trzy jednostki transkrypcyjne (otwarte ram ki odczytu): ery A l, eryA II i ery A III (Ryc. 3). K ażda ram ka odczytu zawiera po dwa moduły, z k tó rych każdy koduje ciąg dom en enzymatycznych składających się na jednostkę syntazy. Liczba m odułów w zespole genów syntazy odpow iada liczbie cykli kondensacji i redukcji. T ak więc w zespole genów DEBS w ystępują trzy jednostki transkrypcyjne skupiające po dwa moduły. O dpow iada to łącznie sześciu reakcjom kondensacji i towarzyszących im redukcji podczas syntezy erytromycyny. P onadto , ciąg dom en enzymatycznych kodow anych przez dany m oduł odpow iada dokładnie kolejności reakcji, k tóre zachodzą w określonym cyklu. Tak więc struk tu ra i długość łańcucha poliketydu jest zaprogram ow ana przez liniową sekwencję genów [16].
Analiza D N A izolowanego z różnych szczepów
278 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

MODUŁ 2 MODUŁ 4 MODUŁ 6
M O D U Ł 1 M O D U Ł 3 M ODUŁ 5
(1)
(2)
(3)
(4)
Ryc. 3 Organizacja genetyczna syntazy 6 -dezoksyerytronolidu B (DEBS) i jej modyfikacje (wg [28], zmodyfikowano, reprodukcja za zgodą Am Assoc Adv Sei).(1) -— naturalny produkt DEBS, 6-dezoksyerytronolid B;(2), (3), (4) produkty DEBS zmodyfikowanej przez, odpowiednio, delecję domeny KR z modułu 5, inaktywację domeny ER z modułu 4, przeniesienie domeny TE z modułu 6 na koniec modułu 2.AT — acylotransferaza, ACP — białkowy czynnik przenoszący grupy acylowe, KS — syntaza ß-ketoacylowa, KR — ketoreduktaza, DH — dehydrogenaza, ER — enoiloreduktaza, TE — tioesteraza.
Streptomyces, kodującego enzymy biosyntezy innych m akrolidów takich jak: tylozyna, spiram ycyna, awer- m ektyna, karbom ycyna czy kandycydyna sugeruje podobną organizację m olekularną. Zespoły genów biosyntezy tych m akrolidów obejm ują znacznie większy region genom u w porów naniu z zespołem erytromycyny: aw erm ektyny — 95 tys. par zasad, tylozyny — 80 tys. pz i kandycydyny — 105 tys. pz.
W kompleksie syntazy typu II poszczególne enzymy są kodow ane przez osobne geny. Ułożenie tych genów
jest bardzo podobne we wszystkich syntazach (Ryc. 4). Geny różnych syntaz kodujące KS, AT, C L F i ACP m ają niemal identyczną wielkość i wzajemne położenie [17-24]. W przypadku tego typu organizacji ułożenie genów nie pozw ala wnioskować o kolejności reakcji, a co za tym idzie — o strukturze produktu .
IV. Próby sterowania biosyntezą
Doświadczenia prowadzące do otrzym ania polike-
Ryc. 4 Geny syntaz poliketydowych typu II odpowiedzialnych za syntezę aktynorodyny (act) przez Streptomyces coelicolor, grana- tycyny (gra) — S. violaceoruber, tetraceno- mycyny (tem) — S. glaucescens, frenolicy- ny (fren) — S. roseofulvus, gryzeuzyny (gris) — S. griseus oraz barwnika zarodników (whiE) — S. coelicolor (wg [48], zmodyfikowano, reprodukcja za zgodą Macmillan Magaz Ltd.).KR — ketoreduktaza, KS — syntaza P-ketoacylowa, AT — acylotransferaza, CLF — czynnik determinujący długość łańcucha, ACP — białkowy czynnik przenoszący grupy acylowe, ARO — aromata- za, CYC — cyklaza, M T — o-metylotran- sferaza.
lOOOpz
act < KR KS AT> CLF |aCP̂ >
gra <C KR KS AT> CLF > ACP)>
tem
fren
gris
w hiE
KS AT> CLF |aCP̂ >
KS AT> CLF ACP̂ >
KS AT̂> CLF |aCP̂ >
KS AT> CLF |aCP̂ >
ARO CYC
CYC>ARO/
CYC MT
KR
KR^> ARO~>
>
CYC>
> ARO
SYNTAZA "MINIMALNA"
POSTĘPY BIOCHEM II 42 (3 ) , 1996 279http://rcin.org.pl

tydów nie występujących w naturze są bardzo ściśle powiązane z badaniam i nad mechanizm em biosyntezy. W tych badaniach w ykorzystano wiele m etod eksperym entalnych, k tóre umożliwiły poznanie struktury związków przejściowych, przypisanie funkcji p ro d u k tom poszczególnych genów i określenie specyficzności enzymów. M etody te opisano poniżej.
IV-1. Badanie mutantów zablokowanych na poszczególnych etapach biosyntezy
„K olinearność” genów syntaz typu I i ich p ro d u k tów narzuca sposób potencjalnej zm iany struktury wytwarzanego związku przez modyfikacje genetyczne powodujące unieczynnienie fragm entów określonego m odułu. Stosując takie podejście eksperym entalne D o n a d i o i w s p . [25] dokonali zm ian w obrębie zespołu syntazy dezoksyerytronolidu B, m ających wpływ na redukcję łańcucha węglowego (Ryc. 3). Pow odując delecję genomowego D N A w obrębie całej dom eny ketoreduktazy w podjednostce piątej otrzym ano, w efekcie końcowym, spodziew aną pochodną nie zredukow aną przy węglu piątym , co jednocześnie potwierdziło, że ta ketoreduktaza jest aktyw na przy piątej kolejno reakcji kondensacji. W podobny sposób poprzez inaktywację dom eny enoiloreduktazy z m odułu czwartego uzyskano pochodną zawierającą wiązanie podw ójne w pozycji C-6 — C-7 zam iast norm alnie występującej grupy metylenowej [26]. O bydwa zmodyfikowane związki to prekursory antybiotyków0 szerokim spektrum działania.
Zespół enzymów DEBS wydaje się tolerować, przynajmniej do pewnego stopnia, zm iany w program ie syntezy i prow adzi „obróbkę” zmienionego poliketydu przez dwa lub trzy dalsze cykle. Szkielet poliketydowy jednak rzadko reprezentuje p roduk t końcowy, a zmieniony w wyniku m anipulacji genetycznych poliketyd może nie stanow ić substratu dla enzymów m odyfikujących. Stw arza to potencjalne ograniczenia takich m anipulacji. B adania dotyczące przem ian postpolikety- dowych dowiodły jednak, że enzymy modyfikujące wykazują dość dużą „elastyczność” w stosunku do zm ienionych substratów . N a przykład, podczas syntezy erytrom ycyny z dezoksyerytronolidu B najpierw następuje hydroksylacja w pozycji C-6 prow adzona przez p roduk t genu eryF, a następnie glikozylacja szkieletu w pozycji C-3 i C-5, hydroksylacja przy C-121 O-metylacja jednej z reszt cukrowych. Szczep ze zinaktyw ow anym genem eryF prow adzi wszystkie pozostałe modyfikacje substratu nie mającego grupy hydroksylowej przy węglu szóstym i produkuje 6- dezoksyerytrom ycynę A — antybiotyk znacznie b ardziej stabilny w w arunkach kwaśnych niż erytrom ycyna A [27].
O statn io C o r t e s i w s p . [28] opisali inną modyfikację DEBS polegającą na przeniesieniu aktywnej dom eny katalitycznej w inne miejsce w obrębie kom pleksu syntazy (Ryc. 3). G en kodujący tioesterazę
działającą jak o cyklaza odpowiedzialna za zakończenie syntezy i laktonizację szkieletu został umieszczony na końcu drugiego m odułu DEBS. Spow odow ało to wcześniejsze zakończenie syntezy — po dwóch cyklach kondensacji i redukcji — i utworzenie oczekiwanego związku triketydowego (złożonego z trzech jednostek dwuwęglowych).
Nowe pochodne awerm ektyny otrzym ano w wyniku m utacji szczepu Streptomyces avermitilis [29-31]. W iele z nich m a zwiększone właściwości owadobójcze i toksyczne względem nicieni. M ogą też służyć jako substraty do modyfikacji chemicznych.
Badanie m utantów zablokow anych na poszczególnych etapach biosyntezy było w mniejszym stopniu wykorzystywane do badan ia syntaz poliketydów aro matycznych, ponieważ m utacje w obrębie genów o d powiedzialnych za syntezę szkieletu poliketydowego najczęściej przerywają syntezę na bardzo wczesnym etapie [32]. M etoda ta okazała się przydatna do określenia funkcji enzymów m odyfikujących PK S typu II [33].
IV-2- Heterologiczna ekspresja genów
Heterologiczna ekspresja genów obejmuje kilka różnych typów doświadczeń:a) kom plem entacja m utacji gospodarza za pom ocą plazm idu zawierającego odpow iedni fragm ent genów syntazy z innego organizm u [34],b) wym iana genów na chrom osom ie gospodarza w wyniku podw ójnego crossing-over [35],c) wprow adzenie do kom órek gospodarza — prom ieniowca nie mającego endogennych genów PK S plazm idu zawierającego całą „kasetę ekspresyjną” złożoną genów jednej lub kilku syntaz [36-43],d) wprow adzenie heterologicznych genów syntaz do kom órek gospodarza m ającego własny zestaw genów PK S [44-47].
Dośw iadczenia nad program ow aniem poliketydów typu I wykazały, że specyficzność substratow a p o szczególnych m odułów DEBS jest zw iązana nie tylko z ich liniowym ułożeniem determ inującym kolejność reakcji [42] (Ryc. 3). Syntazy rozpoznają bowiem podstaw ow e cechy struk turalne produktów pośrednich. N a przykład podaw ane z zew nątrz znakow ane związki pośrednie — diketydy w postaci tioestrów były przyjm ow ane jako substraty przez drugi m oduł syntazy wprow adzonej do Streptomyces coelicolor dając prawidłow y p roduk t końcowy. W łączanie diketydów do erytronolidu potw ierdza funkcjonow anie bez zarzu tu syntaz poddanych „obróbce” w heterologicznym gospodarzu. B adania te stw arzają system bezpośredniego testow ania strukturalnych i stereochemicznych podstaw specyficzności przy zastosow aniu analogów substratów .
W prow adzenie genu carE ze Streptomyces ther- motolerans (odpowiedzialnego za przyłączenie reszty kw asu izowalerianowego do karbomycyny) do Strep-
280 POSTĘPY BIOCHEMII 42 (3 ), 1996http://rcin.org.pl

tomyces ambofaciens spow odow ało acylację spiram y- cyny — naturalnego p roduk tu S. ambofaciens [44]. W wyniku w prow adzenia do kom órek Streptomyces galilaeus (wytwarzającego aklacynom ycynę) genu ze Streptomyces peucetius (producenta doksorubicyny), kodującego hydroksylazę aklaw inonu (produktu p o średniego w syntezie obydw u związków) otrzym ano hydroksylow any analog aklacynom ycyny. Związek ten wykazywał wysoką, specyficzną cytotoksyczność in vitro względem linii kom órek białaczkowych i czerniaka [45].
Łącząc kilka pełnych szlaków biosyntezy w kom órkach jednego szczepu uzyskano oprócz naturalnych, zupełnie nowe produkty. Klasycznym już dzisiaj przykładem jest dihydrogranatyrodyna — hybrydow y p ro duk t syntaz: aktynorodyny i granatycyny [46]. O sta tnio dokonano próby połączenia trzech zespołów genów syntaz: tetracenom ycyny, elloramycyny i urdam y- cyny [47].
Analiza struk tury poliketydów produkow anych przez szczepy zawierające różne kom binacje genów pochodzących z jednej lub wielu syntaz umożliwiła określenie funkcji i specyficzności poszczególnych podjednostek budujących syntazy poliketydowe. N a tej podstawie w yodrębniono w zespole genów PK S typu II tzw. „syntazę m inim alną” (m inPKS) obejm ującą KS, AT, C L F i A CP (Ryc. 4). Jest to zestaw trzech genów niezbędnych do utw orzenia na jp rostszego, niezredukow anego szkieletu poliketydowego (Ryc. 5), np. 16-węglowego (SEK4) w przypadku m inPK S aktynorodyny (act) lub 20-węglowego (SEK15) w przypadku m inPK S tetracenom ycyny (tem). Uzyskanie analogu SEK4 zredukow anego w p o zycji C-9 (SEK34) wym agało w prow adzenia d o d atkowo genów K R i ARO. Analogiczne podjednostki różnych syntaz różnią się specyficznością. N a przykład act K R prow adzi redukcję łańcucha zarów no 16- jak i 20-węglowego (SEK4 i SEK15), natom iast act ARO nie jest w stanie utworzyć pierścienia arom atycznego
w SEK15. Stw ierdzono także zjawisko spontanicznej cyklizacji łańcuchów niezredukowanych.
W yzwaniem dla badaczy stało się znalezienie takich kom binacji syntaz m inim alnych i pozostałych podjednostek, które pozw olą na uzyskanie produktów o oczekiwanej strukturze. Om ów ione wyżej badania doprowadziły do sform ułowania wniosków, które m ożna traktow ać jako zasady projektow ania nowych poliketydów arom atycznych przez ingerencję we wczesne etapy ich biosyntezy [48]. Przedstaw ione wskazówki dotyczą długości łańcucha, ketoredukcji, cyklizacji i arom atyzacji poszczególnych pierścieni.
IV-3. Badanie funkcji natywnego kompleksusyntazy podlegającego ekspresji w komórkach Escherichia coli
Jako przykład może posłużyć eksperym ent, w k tó rym jeden z genów syntazy 6-dezoksyerytronolidu B (eryA III) został w prow adzony, w odpowiednim systemie w ektorów ekspresyjnych, do kom órek Escherichia coli. U zyskano w stosunkow o dużych ilościach oraz oczyszczono do stanu bliskiego hom ogen- nem u natywne białko DEBS3. W ystępuje ono w p o staci dimeru, którego podjednostki m ają masę 330 kD A i są wielofunkcyjnymi białkam i o dziewięciu centrach katalitycznych. Białko to nie podlega po- translacyjnej modyfikacji przez enzymy gospodarza. Uzyskanie oczyszczonego białka DEBS stw arza m ożliwość poznania specyficzności substratowej jego domen [48].
IV-4. Badanie funkcji wyselekcjonowanego zespołu enzymów PKS w układzie pozakomór- kowym
Eksperym enty te przeprow adzono m.in. w odniesieniu do zespołu syntazy tetracenom ycyny u Streptomyces glaucescens [5, 50, 51]. W kom órkach tego
SEK4 (act minPKS)
Ryc. 5 Struktury związków poliketydowych otrzymanych w wyniku kombinacji podjednostek syntaz poliketydowych typu II. SEK34
OH
(act minPKS, act KR,
SEK 15 (tem m inPKS)
act A R O )
POSTĘPY BIOCHEMII 42 (3 ) , 1996 281http://rcin.org.pl

szczepu spow odow ano zablokow anie ekspresji własnych genów syntazy tetracenom ycyny. Ekspresji pod legał natom iast wprow adzony z zewnątrz niepełny zestaw genów tej samej syntazy, stanowiący odpow iednio dobraną kom binację. Z kom órek tych uzyskano ekstrakty białkowe — funkcjonalne zestawy enzymów. Poszczególne enzymy m ożna wybiórczo usuwać (metodą immunoprecypitacji), do układu m ożna też dodawać różne substraty szlaku biosyntezy poliketydu. Stosując opisaną m etodę potw ierdzono aktywność „m inim alnego” zestawu białek PK S oraz to, że m ożna skonstruow ać zestaw optym alny pod względem ak tywności przez włączenie białka cyklazy.
M etoda systemu pozakom órkow ego może być stosow ana do badan ia innych enzymów PK S typu II. Umożliwia uzyskanie do dalszych badań oczyszczonych białek oraz rekonstrukcję aktywnej syntazy z wybranych składowych kompleksu.
IY-5. Analiza sekwencji genów
Sekwencje odpowiednich genów różnych syntaz są bardzo zbliżone (ok. 70% identycznych zasad). Również sekwencje białek wyprow adzone na podstawie sekwencji nukleotydow ych są zachowawcze, szczególnie w rejonie przypuszczalnego centrum aktywnego [18,21,52, 53]. Um ożliwia to przypisanie określonych funkcji biologicznych otw artym ram kom odczytu n o wo oznaczanych sekwencji nukleotydow ych [20, 43, 54-58]. Oznaczanie funkcji genów na zasadzie porów nania sekwencji sprawdziło się w przypadku kilkunastu nowo poznanych syntaz. Zespoły kodujących je genów zostały odkryte na podstaw ie podobieństw a kodow anych przez nie białek z podstawowym i enzymami szlaku biosyntezy poliketydów [6, 11, 17-20, 23, 24, 33],
Porów nanie genów różnych syntaz pozw ala wyciągnąć wnioski dotyczące wspólnego schem atu ich o rganizacji. D aje to teoretyczną podstaw ę wszelkich ingerencji w syntezę poliketydów, tj. zarów no tw orzenia związków będących produktem m ozaiki genów, jak i m anipulacji w obrębie poszczególnych genów.
V. Podsumowanie
M edycyna nieustannie poszukuje nowych leków o silniejszym działaniu terapeutycznym , mniejszych skutkach ubocznych, większej trwałości. Związki pochodzenia naturalnego nie zawsze m ają pożądane własności farm akologiczne, a ponad to ich pozyskiw anie i oczyszczanie jest bardzo kosztowne. Trudności te rozwiązuje częściowo synteza chemiczna lub modyfikacja produktów naturalnych.
W raz z rozwojem m etod inżynierii genetycznej wzrasta znaczenie m odyfikowanych genetycznie m ikroorganizm ów jak o producentów nowych leków. D ro b n o ustroje o odpow iednio dobranym zestawie genów m ogą w yprodukow ać substancje, których otrzym anie
w inny sposób jest bardzo trudne lub niemożliwe [59].Pierwsze eksperym enty prow adzące do otrzym ania
„hybrydow ych” związków poliketydowych zostały przeprow adzone przez K o p w o o d a i w s p . w 1985 r. [46]. C oraz dokładniejsze wyjaśnianie m echanizm ów biosyntezy związków poliketydow ych po zwala na przejście od eksperym entalnego łączenia genów różnych syntaz do racjonalnego program ow ania żądanych produktów poprzez ingerencję w struk turę i ułożenie genów odpowiedzialnych za konkretne etapy syntezy [48, 60, 61].
A rtykuł otrzymano 24 listopada 1995 r.Zaakceptowano do druku 25 kwietnia 1996 r.
Piśmiennictwo
1. T r o p f S, L a n z SA, R e n s i n g J, (1994) J Mol Evol 38: 610-618
2. B r o w n D W, S a l v o J J (1994) Appl Environ Microbiol 60: 979-983
3. H o p w o o d DA, S h e r m a n D (1990) Annu Rev Genet 24: 37-66
4. M c N e i l DJ , O c c i JL, G e w a i n K M, M c N e i l T (1994) Ann N Y Acad Sei 721: 123-132
5. S h e n B, H u t c h i n s o n C R (1993) Science 262: 1535-15406. Y e J, D i c k n s ML , P l a t e r R, Li Y, L a w r e n c e J,
S t r o h 1 W R (1994) J Bacteriol 176: 6270-62807. I k e d a H, O m u r a S (1995) J Antibiot 48: 549-5628 . S o l i v e r i J, V i j g e n b o o m E, G r a n o z z i C, P l a s k i t t
KA, C h a t e r K F (1993) J Gen Microbiol 139: 2569-25789. K a t z L, D o n a d i o S (1993) Annu Rev Microbiol 47: 875-912
10. B e c k J, R i p k a S, S i e g n e r A, S c h w e i z e r E (1990) Eur J Biochem 192: 487-489
11. M e r s o n - D a v i e s LA, C u n d l i f f e E (1994) Mol M icrobiol 13: 349-355
12. R i c h a r d s o n MA, K u h s t o s s S, H u b e r M L B , F o r d L, G o d f r e y O, T u r n e r JR, R a o R N (1990) J Bacteriol 172: 3790-3798
13. A r i s a w a A, T s u n e k a w a H, O k a m u r a K, O k a m o - t o R (1995) Biosci Biotech Biochem 59: 582-588
14. L a n z T, T r o p f S, M a r n e r FJ , S c h r ö d e r J, S c h r ö d e r G (1991) J Biol Chem 266: 9971-9976
15. S c h r ö d e r J, S c h r ö d e r G, (1990) Z Naturforsch 45: 1-816. D o n a d i o S, S t a v e r MJ , M c A l p i n e JB, S w a n s o n
SJ, K a t z L (1992) Gene 115: 97-10317. S h e r m a n D H , M a l p a r t i d a F, B i b b MJ , K i e s e r
H M, B i b b M J H o p w o o d D A (1989) EM BO J 8 : 2717-2725
18. B i b b MJ , B i r o S, M o t a m e d i H, C o l l i n s J F , H u t c h i n s o n C R (1989) EM BO Journal 8 : 2727-2736
19. F e r n a n d e z - M o r e n o MA, M a r t i n e z E, B o t o L, H o p w o o d DA, M a l p a r t i d a F (1992) J Biol Chem 267: 19278-19290
20. B i b b MJ , S h e r m a n D H , O m u r a S, H o p w o o d D A (1994) Gene 142: 31-39
21. K im ES, B i b b MJ , B u t l e r MJ , H o p w o o d DA, S h e r m a n D H (1994) Gene 141: 141-142
22. T i n - W e i n Y, B i b b MJ , R e v i l l WP , H o p w o o d D A(1994) J Bacteriol 176: 2627-2634
23. P i e c q M, D e h o t t a y P, B i o t A, D u s a r t J (1994) DNA Sequence - J DNA Sequencing and mapping 4: 219-229
24. H a n L, Y a n g K, R a m a l i n g a m E, M o s h e r RH, V i n i n g L C (1994) Microbiology 140: 3379-3389
25. D o n a d i o S, S t a v e r MJ , M c A l p i n e JB, S w a n s o n SJ, K a t z L (1991) Science 252: 675-679
26. D o n a d i o S, M c A l p i n e JB, S h e l d o n PJ , J a c k s o n M, K a t z L (1993) Proc Natl Acad Sei USA 90: 7119-7123
27. W e b e r J M, L e u n g J O, S w a n s o n SJ, I d l e r KB, M c A l p i n e J B (1991) Science 252: 114-117
282 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

28. C o r t e s J, W i e s m a n K E H , R o b e r t s GA, B r o w n MJ B , S t a u n t o n J, L e a d l a y P F (1995) Science 268: 1487-1489
29. P a n g CH, M a t s u z a k i K, I k e d a H, T a n a k a H, O m u r a S (1995) J Antibiot 48: 59-66
30. P a n g CH, M a t s u z a k i K, I k e d a H, T a n a k a H, O m u r a S (1995) J Antibiot 48: 92-94
31. I k e d a H, T a k a d a Y, P a n g CH, M a t s u z a k i K, T a n a k a H, O m u r a S (1995) J Antibiot 48: 95-97
32. M e u r e r G, H u t c h i n s o n C R (1995) J Bacteriol 177: 477-481
33. S u m m e r s RG, W e n d t - P i e n k o w s k i E, M o t a m e d i H, H u t c h i n s o n C R (1992) J Bacteriol 174: 1810-1820
34. S h e r m a n D H , K i m ES, B i b b MJ , H o p w o o d D A (1992) J Bacteriol 174: 6184-6190
35. K h o s l a C, E b e r t - K h o s l a S, H o p w o o d D A (1992) M ol Microbiol 6 : 3237-3249
36. M c D a n i e l R, E b e r t - K h o s l a S, H o p w o o d DA, K h o s l a C (1993) J Am Chem Soc 115:11671-11675
37. M c D a n i e l R, E b e r t - K h o s l a S, H o p w o o d DA, K h o s l a C (1993) Scienced 262: 1546-1550
38. F u H, E b e r t - K h o s l a S, H o p w o o d DA, K h o s l a C (1994) J A Chem Soc 116: 4166-4170
39. F u H, M c D a n i e l R, H o p w o o d DA, K h o s l a C (1994) Biochemistry 33: 9321-9326
40. K im ES, C r a m e r K D , S h r e v e AL, S h e r m a n D H(1995) J Bacteriol 177: 1202-1207
41. K a o C M , K a t z L , K h o s l a C (1994) Science 236: 509-51242. C a ñ e DE, G a u n g l i n L, K h o s l a C, K a o C M, K a t z
L (1995) J Antibiot 48: 647-65143. G r i m m A, M a d d u r i K, Al i A, H u t c h i n s o n C R
(1994) 151: 1-1044. E p p JK , H u b e r M L B , T u r n e r JR, G o o d s o n T,
S c h o n e r B E (1989) Gene 85: 293-301
45. H w a n g CK, K i m HS, H o n g YS, K i m YH, H o n g SK, K i m SJ, L e e J J (1995) Antimicro Agents Chemother 39: 1616-1620
46. H o p w o o d DA, M a l p a r t i d a F, K i e s e r H M, I k e d a H, D u n c a n J (1985) Nature (Lond) 314: 642-644
47. D e c k e r H, H a a g S, U d v a r n o k i G, R o h r J (1985) Angew Chem Int Ed Engl 34: 1107-1110
48. M c D a n i e l R, E b e r t - K h o s l a S, H o p w o o d DA, K h o s l a C (1995) Nature (lond) 375: 549-554
49. R o b e r t s GA, S t a u n t o n J, L e a d l a y P F (1993) Eur J Biochem 214: 305-311
50. D e c k e r H, M o t a m e d i H, H u t c h i n s o n C R (1993) J Bacteriol 175: 3876-3886
51. D e c k e r H, H u t c h i n s o n C R (1993) J Bacteriol 175: 3887-3892
52. D o n a d i o S, K a t z L (1992) Gene 111:51-6053. B e v i t t DJ , C o r t e s J, H a y d o c k SF, L e a d l a y P F
(1992) Eur J Biochem 204: 39-4954. A r r o w s m i t h TJ , M a l p a r t i d a F, S h e r m a n DH,
B i r c h A, H o p w o o d DA, R o b i n s o n J A (1992) M ol Gen Genet 234: 254-264
55. A r i s a w a A, K a w a m u r a N, T s u n e k a w a H, O k a - m u r a K, T o n e H, O k a m o t o R (1993) Biosci Biotech Biochem 57: 2020-2025
56. D i c k e n s ML, Ye J, S t r o h l W R (1995) J Bacteriol 177: 536-543
57. F e r n a n d e z - M o r e n o MA, M a r t i n e z E, C a b a l l e r o JL, I c h i n o s e K, H o p w o o d DA, M a l p a r t i d a F (1994) J Biol Chem 40: 24854-24863
58. K u c z e k K, M o r d a r s k i M, G o o d f e l l o w M (1994) FEM S Microbiol Lett 118: 317-326
59. H u t c h i n s o n C R (1994) Bio/Technology 12: 375-38060. M a n n J (1995) Nature (Lond) 375: 533-53461. H o p w o o d D A (1995) FEM S Microbiol Rev 16: 233-234
Do członków Polskiego Towarzystwa Biochemicznego
Zarząd P.T.Bioch. uprzejmie prosi wszystkich członków o wpłatę zaległych i bieżących składek członkowskich. Zachęcamy także do zaprenumerowania „Postępów Biochemii”.
POSTĘPY BIOCHEM II 42 (3 ) , 1996 283http://rcin.org.pl

Niedobór akty wności arylosulfatazy A jako podłoże leuko- dystrofii metachromatycznej
Deficiency of arylsulfatase A activity as a basis of metachromatic leucodystrophy
A G N IE S Z K A Ł U G O W S K A 1, A N N A T Y L K I-S Z Y M A Ń S K A 2
Spis treści:
I. WstępII. Árylosulfataza A i substrat jej działaniaIII. Defekt biochemiczny przyczyną leukodystrofii meta
chromatycznejIV. Aktywator SAP-BV. Pseudodeficyt arylosulfatazy AVI. Defekt posttranslacyjnej modyfikacji sulfatazVII. Molekularne podstawy defektów arylosulfatazy A
Wykaz stosowanych skrótów: LDM — leukodystrofia meta- chromatyczna; ASA — arylosulfataza A; Tf-a ASA — przeciwciała przeciw ASA, połączone z transferyną; SAP-B — ang. sphingolipid activator protein-B; GABA — kwas y-aminomasłowy; ASA-PD — pseudodeficyt arylosulfatazy A; MSD — ang. multiple sulfatase deficiency, brak wielu sulfataz; pz — pary zasad; nt — nukleotyd.
I. Wstęp
Leukodystrofia m etachrom atyczna (LD M ) jest genetycznie uw arunkow anym schorzeniem m etabolicznym, spow odow anym deficytem aktywności arylosulfatazy A (ASA) (EC 3.1.6.1.) prow adzącym do spich- rzania galaktozylosulfatydu (siarczanu cerebrozydu) w lizosom ach kom órki. M imo, że spichrzanie galaktozylosulfatydu dotyczy wszystkich kom órek organizmu, to jednak zm iany patologiczne przebiegają głównie w mielinie ośrodkow ego układu nerwowego oraz mielinie nerwów obwodowych. G alaktozylosulfatyd oraz laktozylosulfatyd (w mniejszym stopniu) spich- rzają się również w nerkach, pęcherzyku żółciowym oraz innych narządach wewnętrznych. Sulfatydy są wydalane w dużych ilościach z moczem i żółcią. W preparatach histologicznych sulfatydy tw orzą struk tury kuliste, barw iące się m etachrom atycznie pod wpływem fioletu krezylu (m etachrom azja) [1].
1 Mgr, Instytut Pomnik Centrum Zdrowia Dziecka, Zakład diagnostyki Laboratoryjnej, Al. Dzieci Polskich 20, 04-736 Warszawa,
2 doc. dr hab n. med., Instytut Pomnik Centrum Zdrowia Dziecka, Zespół Chorób Metabolicznych, Al. Dzieci Polskich 20; 04-736 Warszawa
Contents:
I. IntroductionII. Arylsulfatase A and substrate of its actionIII. Biochemical defect in metachromatic leukodystrophyIV. SAP-B activatorV. Arylsulfatase A pseudodeficiencyVI. Defect of posttranslational modification of sulfatasesVII. Molecular bases of arylsulfatase A defects
U pacjentów z leukodystrofią m etachrom atyczną obserwuje się postępującą demielinizację, k tó ra jest przyczyną objawów neurologicznych, takich jak: zaburzenia chodu, ataksja, niedowład kończyn, zanik nerwów wzrokowych, napady drgawek, demencja.
W yróżnia się trzy typy leukodystrofii m etachrom atycznej na podstawie wieku w ystąpienia pierwszych objawów oraz przebiegu choroby [1]: późnoniem o- wlęcy (pierwsze objawy w wieku 1-2 lat), młodzieńczy (3-16 lat), dorosłych (powyżej 16 lat). Postęp choroby u dorosłych jest powolny i charakteryzuje się obecnością objawów psychotycznych.
II. Arylosulfataza A i substrat jej działania
Arylosulfataza A (ASA) jest spotykana we wszystkich tkankach i płynach ustrojowych. Białko enzymu zostało wyizolowane z różnych źródeł, m.in. ludzkiej wątroby, łożyska i moczu, tkanek jeżowca, szczura, świni [2-4].
ASA jest kw aśną glikoproteiną o niskim pi, co wiąże się z dużą zaw artością kw asu glutam inowego i asp araginowego. Zaw iera również duże ilości proliny. Pow yżej pH 6.5 enzym istnieje jak o m onom er o masie cząsteczkowej ok. 100 kD a. ASA może polimeryzować w zależności od pH środow iska tw orząc dim er przy pH 4.5. Enzym pochodzący z ludzkiego m oczu składa się z dwóch podjednostek o m asach 54 i 63 kD a, n a to m iast pochodzący z ludzkiej wątroby, łożyska i fibro- blastów składa się z dwóch podjednostek o m asach 55 i 64 k D a [5, 6].
W kom órce ASA jest um iejscowiona w lizosomach, gdzie praw dopodobnie tworzy dimery.
284 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

Podobnie, jak w przypadku innych enzymów lizoso- malnych, ASA jest syntetyzow ana na szorstkim retiku- lum endoplazm atycznym jako N-glikozylowany prekursor. N astępnie przenika ona przez retikulum endo- plazm atyczne i ap ara t Golgiego, gdzie do N-związa- nych łańcuchów oligosacharydow ych dołączane są reszty fosforanowe i siarczanowe [7]. Przed tran sp o rtem prekursora do kom partm entu prelizosom alnego (kwaśnego endosom u) w aparacie Golgiego zachodzi synteza m arkera m annozo-6-fosforanowego. Jest on rozpoznaw any przez receptor enzymu w błonie kw aśnego endosom u, lizosomu, jak również w błonie ko mórkowej. S truk turą rozpoznaw aną jest reszta kwasu fosforowego związanego z szóstym atom em węgla reszty m annozowej, k tó ra jest połączona z łańcuchem oligosacharydow ym wiązaniem a l-> 2 . Synteza m arkera zachodzi w dwóch etapach. W pierwszym — N- acetyloglukozoam inotransferaza przenosi N-acetylog- lukozoam ino-l-fosforan z U D P-N -acetyloglukozoa- miny na C 6 mannozy. W drugim etapie następuje odszczepienie N -acetyloglukozoam iny i związanie reszty fosforanowej z C 6 m annozy [8]. Stw ierdzono, że w hodow anych fibroblastach skóry powstaje prekur- sorowy polipeptyd o masie 62 kD a, który za pośrednictwem receptora wiążącego m annozo-6-fosforan przemieszcza się do kwaśnego, prelizosom alnego endosom u [9]. Przed osiągnięciem lizosomu, od C-końca prekursora odcinanych jest ok. 50 aminokwasów .
D i e s n e r i w s p. stosując przeciwciała skierow ane przeciw ASA, połączone z transferyną (Tf-aASA) ud o wodnili, że now osyntetyzow ana ASA przechodzi przez kom partm enty kom órkow e zawierające receptor transferyny do dojrzałego lizosom u [10]. W kom part- m entach zawierających receptor transferyny, tzn. w kwaśnych endosom ach i błonie kom órkow ej, now osyntetyzow ana ASA wiąże się z koniugatam i Tf-aASA, pow raca na powierzchnię kom órki, część enzym u jest uw alniana do środow iska zew nątrzkom órkow ego.
C entrum aktyw ne ASA zawiera histydynę oraz dwie (lub więcej) argininy. Aniony: S 0 42~, P 0 43 - , S 0 32 - , F _ oraz kationy: Ag + , C u 2+ i H g2+ w stężeniach m ilim olarnych są inhibitoram i aktyw ności enzymu. Reakcja katalizow ana przez ASA podlega również inhibicji pod wpływem hydrokortyzonu, 2-hydroksy-
Ryc. 1 Schematyczna budowa galaktozylosulfatydu i miejsce działania arylosulfatazy A.
POSTĘPY BIOCHEMII 42 (3 ) , 1996
dopam iny oraz związków karbonylow ych w obecności C u 2 + .
Arylosulfataza A (EC 3.1.6.1) jest enzymem umiejscowionym we wnętrzu lizosomów. W raz z białkiem SAP-B (ang. sphingolipid activator protein-B) wchodzi w skład 3-sulfohydrolazy cerebrozydo-3-siarczanowej (EC 3.1.6.8.) [1]. ASA katalizuje I etap degradacji sulfolipidów — odszczepia grupę chemiczną zawierającą resztę siarczanową. Term in „sulfolipidy” odnosi się do wszystkich lipidów zawierających w swej cząsteczce atom siarki. Spośród tej grupy związków o rganicznych substratam i ASA są:— galaktozylosulfatyd, czyli siarczan cerebrozydu;— laktozylosulfatyd;— seminolipid, czyli sulfogalaktoglicerolipid;— siarczan psychozyny.Sulfatydy są estram i siarczanowymi galaktocerebrozy- du z grupą siarczanow ą przyłączoną w położeniu C3-hydroksyl cząsteczki galaktozy. Schem atyczną budowę galaktozylosulfatydu przedstawia rycina 1.
G alaktozylosulfatyd jest ważnym składnikiem mie- liny. Związek ten znajduje się na powierzchni mieliny i praw dopodobnie jest zw iązany z białkam i podstaw owymi i białkam i proteolipidów poprzez działanie sił jonowych. P onad to galaktozylosulfatyd uczestniczy w aktyw nym transporcie N a + jako kofaktor dla N a-K zależnej ATP-azy. G alaktozylosulfatyd aktywuje kal- m odulinozależną fosfodiesterazę cyklicznych nukleo- tydów, znajduje się w miejscach wiążących opiaty oraz jest jednym ze składników miejsca rozpoznaw ania GABA.
III. Defekt biochemiczny przyczyną leuko- dystrofii metachromatycznej
Brak aktywności ASA, k tóry uznano za pierw otną przyczynę leukodystrofii m etachrom atycznej, obserwowano u chorych w: moczu, leukocytach krwi obwodowej, hodow anych fibroblastach skóry, surowicy, łzach, ślinie, hodow anych limfocytach, hodow anych kom órkach szpiku kostnego, hodow anych kom órkach płynu owodniowego.
Przeciwciała przeciw ludzkiej ASA reagują krzyżowo z polipeptydam i syntetyzowanym i przez hodow ane
CHaOH
T4 - H s ą ,
A SA
285
H H
c - c - c H r o ,a
OH
http://rcin.org.pl

fibroblasty skóry pacjentów trzech typów L D M [11]. W m ateriale od pacjentów z leukodystrofią m eta- chrom atyczną typu późnoniem owlęcego stwierdzono obecność glikoprotein reagujących krzyżowo z przeciwciałami anty-ASA zarów no w sposób pozytywny (co oznacza, że polipeptyd enzymu podlega syntezie), jak i nie reagujących pozytywnie (co świadczy o tym, że synteza polipeptydu enzymu nie zachodzi). K om órki fibroblastów od pacjentów z LD M typów: m łodzieńczego i dorosłych syntetyzują i wydzielają polipeptydy ASA w tem pie wynoszącym 20-70% tem pa kom órek kontrolnych. Jednak nowosyntetyzowane, zm utow ane cząsteczki enzymu są gwałtownie degradow ane po przedostaniu się do w nętrza lizosom u [12]. Po zastosow aniu inhibitorów proteinaz cysternowych obserwuje się m ateriał reagujący krzyżowo z przeciwciałami anty-ASA oraz częściową aktywność ASA w fibroblastach niektórych pacjentów z LD M typu młodzieńczego lub dorosłych.
Podczas badania aktywności procesów katabolizm u siarczanu cerebrozydu (tzn. galaktozylosulfatydu) in situ w kulturach tkankow ych spostrzeżono, że w ko m órkach pochodzących od pacjentów z L D M typów późnych wykrywa się pewną resztkową aktywność ASA i zależność czasową między zdolnością hodow anych kom órek do degradow ania dostarczanego do pożywki sulfatydu, a wiekiem wystąpienia pierwszych objawów klinicznych u pacjenta [1]. T aka obserwacja tłumaczy zjawisko pseudodeficytu, w którym niska aktywność enzymu, wynosząca 10-30% średnich w artości norm alnych, byłaby wystarczająca do zachow ania norm alnego m etabolizm u kom órki [12-14]. C o n z e l m a n n i S a n d h o f f [15-17] wysunęli hipotezę zakładającą, że powyżej pewnego krytycznego progu aktywności enzymu kom órka może pozbyw ać się nadm iaru substratów , takich np. jak sulfatyd. Stężenie danego substratu w lizosomie określa rów now aga dynam iczna zachow ana między tem pem napływu katabolitu do lizosom u a kinetycznymi param etram i enzymu degradującego ów substrat (Km, Vmax). W edług h i p o t e z y C o n z e l m a n n a i S a n d h o f f a przejście od stanu fizjologicznej norm y do patologicznego spi- chrzania substratu (np. sulfatydu) m ogłoby następować w sytuacji, gdy aktywność resztkowa enzymu byłaby niższa od założonego krytycznego progu ak tywności. H ipoteza ta stanowi próbę wyjaśnienia biochemicznych podstaw w ystępow ania pierwszych objawów klinicznych w różnym wieku u pacjentów z leukodystrofią m etachrom atyczną różnych typów.
IV. Aktywator SAP-B
H ydroliza niektórych sfingolipidów wym aga działania m ałych glikoprotein zwanych SAP (ang. sphin- golipid activator proteins) lub sapozynam i. Białka te działają jak detergenty rozpuszczając hydrofobowe substraty lipidowe lub bezpośrednio aktyw ują niektóre enzymy lizosomalne. ASA może hydrolizować
swoje substraty tylko przy współdziałaniu z SAP-B [18]. Białkowy aktyw ator SAP-B wpływa na rozpuszczalność siarczanu cerebrozydu i czyni go dostępnym dla enzymu [19]. Rzadko spotykana form a leukodyst- rofii m etachrom atycznej jest spow odow ana brakiem aktyw atora SAP-B. D o 1992 r. opisano zaledwie sześciu pacjentów [20]. O braz kliniczny przypom ina typ m łodzieńczy lub późnoniem owlęcy leukodystrofii m etachrom atycznej [21, 22], U pacjentów z brakiem białka SAP-B stwierdza się wydalanie sulfatydów w m oczu i równocześnie aktywność ASA m ierzoną w leukocytach lub fibroblastach w norm ie lub tylko nieznacznie obniżoną.
Cztery białka aktyw atorow e zwane sapozynam i A, B, C i D pow stają ze wspólnego prekursora — prosa- pozyny. P rosapozyna jest glikoproteiną złożoną z 524 aminokwasów . Z budow ana jest ona z czterech hom ologicznych dom en białkowych i obszarów rozdzielających [23, 24]. Prosapozyna jest syntetyzow ana jako glikoproteinow y prekursor o masie 65 kD a. W tej postaci przedostaje się ona do ap ara tu Golgiego, gdzie podlega glikozylacji dając złożony polipeptyd o masie 73 kD a. Niew ielka część puli polipeptydu 73 kD A jest wydzielana do cytozolu. Jednak znaczna część puli polipeptydu 73 kD a przechodzi etap enzymatycznej obróbki w lizosom ach [25]. W wyniku proteolizy z prosapozyny pow stają cztery aktywne białka sapozy- nowe; SAP-B powstaje z drugiej dom eny [26].
V i e l h a b e r i S a n d h o f f stw ierdzili,ż e endocy- toza cząsteczek prosapozyny do w nętrza lizosomów odbyw a się w sposób całkowicie niezależny od receptorów m annozo-6-fosforanu [27].
G en kodujący prosapozynę znajduje się w chrom osomie 10 [28]. K odujący D N A (cDNA) prosapozyny sklonow ano niezależnie w zespołach O ’ B r i e n a i F ü r s t a w 1988 r. [23, 24]. Region kodujący m a długość ok. 1,6 tys. pz. Region nie podlegający tran slacji od końca 3’ m a długość ok. 1,2 tys. pz Sapozyny A, B, C, D w ykazują porów nyw alną homologię, k tó rą szczególnie odzwierciedla zachowanie reszt cysternowych i potencjalnych miejsc N-glikozylacji. G en ko d u jący prosapozynę został sklonowany, chociaż inform acje o budowie eksonu 1 są nadal niekom pletne [29,30]. G en kodujący prosapozynę m a długość ok. 20 tys. pz i praw dopodobnie składa się z 13-tu eksonów. D om ena SAP-B jest kodow ana przez eksony 5-7. D otychczas opisano trzy m utacje w genie prosapozyny uszkadzające białko SAP-B: T217I [31, 32], C241S [33, 19], oraz 777 + 1895C > A [34, 35].
V. Pseudodeficyt arylosulfatazy A
N iską aktyw ność ASA obserwuje się również w około 5-15% ogólnej populacji. W grupie tej znajdują się osoby klinicznie zdrowe [36], jak również „nietypowi” pacjenci z innym i niż L D M schorzeniam i neurologicznymi lub psychiatrycznym i [37]. Zjawisko obniżenia aktywności ASA do 10-30% średnich wartości n o r
286 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

m alnych uznano za polimorfizm w obrębie genu ASA i nazw ano pseudodeficytem. Używając zwykle stosowanych substratów nie m ożna rozróżnić pseudodeficy- tu ASA (ASA-PD) od L D M tylko na podstawie aktywności enzymu. U osób z pseudodeficytem nie obserwuje się w ydalania sulfatydów w moczu. Częste występowanie ASA-PD w ym aga szczególnej procedury w pre- i postnatalnym diagnozow aniu leukodyst- rofii m etachrom atycznej.
VI. Defekt posttranslacyjnej modyfikacji sulfataz
Stwierdzono również inny defekt związany z b ra kiem aktywności nie tylko ASA, lecz również arylosul- fataz B i C oraz innych sulfataz biorących udział w degradacji m ukopolisacharydów . Schorzenie to określono jako brak wielu sulfataz (ang. múltiple sulfatase deficiency, M SD ). B rak wielu sulfataz opisano dotychczas u ok. 50 pacjentów z cechami klinicznymi L D M typu późnoniem owlęcego oraz m ukopolisacha- rydoz.
W edług najnowszych doniesień sulfatazy wym agają dla swojej aktywności pewnej posttranslacyjnej m odyfikacji, k tó ra w przypadku M SD przebiega niepraw idłowo. W przypadkach arylosulfataz A i B opisano nieznany do tąd rodzaj posttranslacyjnej obróbki b iałek tych enzymów, k tó ra polega na zam ianie cysteiny na kwas 2-am ino-3-oksopropionow y. P orów nano sekwencję am inokw asow ą wszystkich znanych sulfataz u Eukaryota. Stwierdzono, że cysteina znajduje się w tym fragmencie sulfataz, k tóry jest zachow any we wszystkich przebadanych enzymach (fragment konserwatywny). N atom iast w nieaktywnych polipeptydach ASA i ASB, pochodzących z fibroblastów od pacjenta z M SD, nie zauw ażono obecności reszty cysternowej. F ak t ten potw ierdza przypuszczenie, że brak cysteiny we fragmencie konserw atywnym jest przyczyną deficytu aktywności sulfataz u chorych z przejawam i M SD [38, 39].
VII. Molekularne podstawy defektów arylo- sulfatazy A
G en kodujący ASA przypisano m etodą hybrydyzacji kom órek som atycznych do chrom osom u 22 [40]. Obecnie w iadom o, że gen ASA znajduje się na długim ram ieniu chrom osom u 22 w regionie prążka q l3 . G en kodujący białko ak tyw atora SAP-B i jego prekursora — prosapozyny przypisano do chrom osom u 10.
W 1989 r. S t e i n i w s p. sklonowali cD N A kodujący ludzką ASA. Jednak pełną i obecnie przyjętą strukturę genu ASA opublikow ał K r e y s i n g i w s p . [41,42]. G en kodujący ASA jest dość mały — długości 3,2 tys. pz i składa się z 8 eksonów. Region 5' nie podlegający translacji zawiera miejsce cap m R N A przy ok. -370nt, a ponad to cztery sekwencje G C -box w p o łożeniu -10, -40, -50, -60, k tóre są typowymi pozycjami
wiązania się z transkrypcyjnym czynnikiem prom oto- rowym Spl. Tylko G C -box w położeniu -10 jest zorientow any w dobrym kierunku. Region 5' nie podlegający translacji nie zawiera sekwencji typu TATA lub CAAT-box, co jest dość rzadkim zjawiskiem wśród genów Eukaryota. Region kodujący podlegający translacji rozpoczyna się kodonem inicjującym ATG, za którym następuje sekwencja 18-amino- kwasowego peptydu sygnałowego i 489-ciu am inokwasów białka enzymu. R am ka odczytu kończy się kodonem term inacyjnym TGA. W odległości ok. lOOnt od kodonu TG A znajduje się sygnał poliadenylacji A AT A AC (1621nt) i ok. 20nt za nim ogon poliA. Region 3' nie podlegający translacji m a długość 140 pz W norm alnych ludzkich fibroblastach pow stają 3 różne rodzaje m RNA dla ASA: 2.1,3.7,4.8 tys. pz [41]. 2.1 tys. pz m RN A stanowi ok. 30-40%, m RNA długości 3.7 oraz 4.8 tys. pz stanow ią ok. 60-70% całości m RNA dla ASA.
W yliczona m asa cząsteczkowa białka kodow anego przez ASA-cDNA wynosi 62 kD a, co pozostaje w zgodzie z wynikami otrzym anym i na drodze biosyntezy [6]. Oprócz polipeptydu o masie cząsteczkowej 62 kD A wykrywa się również formy ASA o masie cząsteczkowej 54-59 kD A [6, 43]. Przypuszcza się, że polipeptyd o masie cząsteczkowej 62 kD A jest p rekursorem ASA, który podlega obróbce proteolitycznej dając formy o mniejszej masie cząsteczkowej [44]. W genie kodującym ASA występują sekwencje dla 3 miejsc możliwej N-glikozylacji cząsteczki białka enzymu, z których faktycznie dwa są wykorzystywane. S trukturę genu ASA przedstaw ia rycina 2.
W 1989 r. G i e s e l m a n n i w s p . [45] sklonowali i zsekwencjonowali gen kodujący ASA w m ateriale pochodzącym od osoby będącej hom ozygotą pseudo- deficytu ASA. W ykazano obecność dwóch tranzycji A -»G . Jedna z nich zam ienia asparaginę w położeniu 1049nt cD N A na serynę, co pow oduje u tratę miejsca N-glikozylacji cząsteczki białka ASA oraz zmniejszenie masy cząsteczkowej enzymu o 2.5 kD a, co jednak nie jest zasadniczą przyczyną pseudodeficytu ASA.
D ruga tranzycja A ->G zmienia sekwencję pierwszego sygnału poliadenylacji za kodonem „stop” z AA- TAAC na AGTAAC. M utacja ta powoduje brak obecności małego 2.1 tys. pz m RNA, który odpow iada za ok. 90% poli(A +) m RN A arylosulfatazy A i obniżenie aktywności enzymu do 10-30% normy. N iska aktywność ASA u osób z pseudodeficytem wynika zatem z częściowo zaham ow anej syntezy białka enzymu, a nie z zaburzenia jego stabilności [46].
Dotychczas w genie kodującym ASA wykryto blisko 60 m utacji związanych z leukodystrofią m etachrom a- tyczną; wśród nich jest: 6 delecji, 5 m utacji typu „splice site” i 47 tranzycji nukleotydowych [46]. W yróżnia się dwa typy m utacji w genie ASA [47-50]. M utacja typu 0 (lub I) powoduje całkowite zaham ow anie syntezy p roduktu genu ASA. M utacje typu 0 w stanie hom o-
POSTĘPY BIOCHEMII 42 (3 ) , 1996 287http://rcin.org.pl

ATG
O
E2P400 E2P436
E2S609 E3P799
E7S2195 E8P238I
E8D2506
E4P1070 E3P898
E3P934 E2D447
E2P445E5P1574
E5P1533
TGAAATAAC
E7P2I33
3.1kb
Ryc. 2 Schemat genu kodującego arylosulfatazę A. Przykładowo pokazano 15 mutacji.Pola czarne wskazują eksony; linie — introny, pola zakreskowane — 5' i 3' regiony nie podlegające translacji. ATG oznacza kodon inicjujący, TGA — kodon terminacyjny translacji. AATAAC wskazuje sygnał poliadenylacji.
zygotycznym prow adzą do późnoniemowlęcej formy LD M . M utacje typu R (lub A) cechują się niską aktywnością resztkow ą enzymu. W stanie hom ozygo- tycznym m utacje typu R prow adzą do postaci do ro słych LD M . Heterozygotyczność m utacji typu 0 i R występuje najczęściej u pacjentów z typem młodzieńczym LDM .
Dwa spośród zm utow anych alleli — 459 + I G > A oraz P426L — stanow ią razem ok. 50% wykrywanych m utacji u pacjentów z LD M [48, 49, 51]. M utacja 459 + IG > A (typ 0) polega na zamianie G -> A w pozycji n t 609 genu, czego skutkiem jest u tra ta donorowego miejsca cięcia dla posttranskrypcyjnej obróbki m RNA — składania (splicing). U pacjentów będących hom o- zygotam i tego allelu prawie nie wykrywa się ASA mRNA. M utacja P426L (typ R) polega na tranzycji C ->T w pozycji n t 2381 genu, co pow oduje zamianę Pro426 w Leu. P ro d u k t zm utow anego genu wykazuje pewną aktywność enzymatyczną, ale jest bardzo niestabilny.
H o n k e zauważył, że m utacje L D M grupują się w regionie 5' genu, szczególnie w eksonach 2 i 3 [52]. O koło połowy wszystkich m utacji umiejscowiono w łaśnie w tych dwóch eksonach [46]. G dy porów nano sekwencje am inokw asow e sulfataz izolowanych od różnych ssaków stwierdzono, że stopień hom ologii maleje od regionu N-końcowego w kierunku regionu C-końcowego. Największy stopień hom ologii w ykazano porów nując sekwencje kodow ane przez ekson 2. Jeśli stopień hom ologii sekwencji am inokwasów odzwierciedla funkcjonalne znaczenie danego regionu cząsteczki enzymu, to grupow anie się m utacji w pew
nych określonych rejonach genu nie jest niczym niezwykłym.
Leukodystrofia m etachrom atyczna jest chorobą dziedziczoną autosom alnie, recesywnie. O gólną częstość występowania L D M m ożna oszacować tylko w przybliżeniu, gdyż jest ona różna w zależności od badanej populacji i wykrywalności klinicznej choroby (rozpoznawalności). W północnej Szwecji częstość występow ania późnoniem owlęcego typu L D M ustalono na 1:40000 [53], we Francji — 1:130000 [54], wśród Żydów habanickich w Izraelu — 1:75 [55], w USA— 1:100000 urodzeń [1], w stanie W ashington— 1:40000 [46]. Częstość występowania allelu pseu- dodeficytu arylosulfatazy A (ASA-PD) w ogólnej po pulacji ustalono na 7.3-15% [1, 57-59], natom iast hom ozygot P D /P D na 0.5-2% [1].
A rtykuł otrzymano 6 listopada 1995 r. Zaakceptowano do druku 30 kwietnia 1996 r.
Piśmiennictwo
1. K o l o d n y EH, F l u h a r t y A L (1995) W: Scriver CR, Beaudet AL, Sly WS, Valle D (red) The Metabolie and Molecular Bases of Inherited Disease, wyd. 7, Me Graw-Hill, New York, str 2693-2739
2. S a s a k i H, Y a m a d a K, A k a s a k a K, K a w a s a k i H, S u z u k i K, S a i t o A, S a t o M, S h i m a d a H (1988) Eur J Biochem 177: 9
3. V a n d e r P a l R H , K l e i n W, V a n G o l d e L M, L o - p e s - C a r d o z o M (1990) Biochim Biophys Acta 1043: 91
4. S e l m i S, M a i r e J, R o u s e t t B (1989) Arch Biochem Biophys 273: 170
5. D r a p e r RK, F i s k u m G M , E d m o n d J (1976) Arch Biochem Biophys 177: 525
288 POSTĘPY BIOCHEMII 42 (3 ), 1996http://rcin.org.pl

6. W a h e e d A, H a s i l i k A, v o n F i g u r a K (1982) Hoppe- Seyler’s Z Physiol Chem 363: 425-430
7. W a h e e d A, v a n E t t e n R L (1985) Biochim Biophys Acta 847: 53
8. V e r b e r t A, C a c a n B, C e c c h e l i n i R (1987) Biochemie 69: 91-99
9. K e l l y BM, Y u CZ, C h a n g P L (1989) Eur J Cell Biol 48: 71
10. D i e s n e r F, S o m m e r l a d e HJ , B r a u l k e T (1993) Biochem Biophys Res Commun 197: 1-7
11. K a p p l e r J, L e i n e k u g e l P, C o n z e l m a n n E, K l e i - j e r WJ , K o h l s c h ü t t e r A, T ö n n e s e n T, R o c h e l M, F r e y c o n F, P r o p p i n g P (1991) Hum Genet 8 6 : 463-470
12. B a c h G, D a g a n A, H e r z B, G a t t S (1987) Clin Genet 31: 211-217
13. B a c h G, N e u f e l d E F (1983) Biochem Biophys Res Commun 112: 198-205
14. F l u h a r t y AL, M e e KW, K i h a r a H (1983) Biochem Biophys Res Commun 112: 191-197
15. C o n z e l m a n n E, S a n d h o f f K (1983/84) Dev Neurosci 6 : 58-71
16. C o n z e l m a n n E, S a n d h o f f K (1991) Dev Neurosci 13: 197-204
17. L e i n e k u g e l P, M i c h e l S, C o n z e l m a n n E, S a n d - h o f f K (1992) Hum Genet 88:513-523
18. F i s c h e r G, J a t z k e w i t z H (1977) Biochim Biophys Acta 481: 561-572
19. S c h l o t e W, H a r z e r K, C h r i s t o m a n o u H, P a t o n BC, K u s t e r m a n n - K u h n B, S c h m i d t B, S e e g e r J, B e n d t U, S c h u s t e r J, L a n g e n b e c k U (1991) Eur J Ped 150: 584-591
20. W e n g e r DA, W i l l i a m s C, C o n s i d i n e E, R a f i M A(1992) Int Pediatr 7: 34-39
21. S h a p i r o LJ , A l e c k KA, K a b a c k MM , I t a b a s h i H, D e s n i c k RJ , B r a n d N, S t e v e n s e n RL, F l u h a r t y A L, K i h a r a H (1979) Pediatr Res 13: 1179
22. W e n g e r DA, D e G a l a G, W i l l i a m s C, T a y l o r HA, S t e v e n s o n RE, P r u i t t JR, M i l l e r J, G a r e n P D, B a i e n t i n e J D (1989) Am J Med Genet 33: 255-265
23. O ’ B r i e n J S, K r e t z KA, D e w j i N N , W e n g e r DA, E s c h F, F l u h a r t y A L (1988) Science 234: 1098-1101
24. F ü r s t W, M a c h l e i d t W , S a n d h o f f K (1988) Biol Chem Hoppe-Seyler 369: 317-328
25. F ü r s t W, S a n d h o f f K (1992) Biochim Biophys Acta 1126:1-16
26. F u j i b a y a s h i S , W e n g e r D A (1986)BiochimBophysActa 875: 554-562
27. V i e l h a b e r G , S a n d h o f f K (1993) 9th ESGLD Workshop, Delphi
28. D e w j i N N , W e n g e r DA, O ’ B r i e n J S (1987) P N AS USA 84: 8652-8656
29. H o l t s c h m i d t H, S a n d h o f f K , F ü r s t W, K w o n HY, S c h n a b e l D, S u z u k i K (1991) FEBS Lett 280: 267-270
30. R o r m a n EG, S c h e i n k e r Y, G r a b o w s k i G A (1992) Genomics 13: 312-318
31. K r e t z K, G i n n s E, C a r s o n G, M o r i m o t o S, K i s h i m o t o Y, F l u h a r t y A L, O ’ B r i e n J S (1989) Am Soc Hum Genet, abstrakt
32. R a f i MA, Z h a n g XL, D e G a l e G, W e n g e r D A (1990) Biochem Biophys Res Commun 166: 1017-1023
33. H o l t s c h m i d t H, S a n d h o f f K , K w o n HY, N a k a n o T, S u z u k i K (1990) J Biol Chem 266: 7556-7560
34. Z h a n g X L , R a f i M A , D e G a l a G , W e n g e r D A (1990) P N A S USA 87: 1426-1430
35. Z h a n g XL, R a f i MA, D e G a l a G, W e n g e r D A (1991) Hum Genet 87: 211-215
36. D u b o i s G, H a r z e r K, B a u m a n N (1977) Am J Hum Genet 29: 191-194
37. H e r s k a M, M o s c o v i c h D G , K a l i a n M, G o t t l i e b D, B a c h G (1987) Am J Med Genet 26: 629-635
38. R o m m e r s k i r c h (1992) PN A S USA 89: 2561-256539. S c h m i d t B, S e l m e r T, I n g e n d o c h A, v o n F i g u r a
K (1995) Cell, in press40. D e L u c a C, B r o w n J A, S h o w s T B (1979) P N A S USA
76: 195741. S t e i n C, G i e s e l m a n n V, K r e y s i n g J, S c h m i d t B,
P o h l m a n n R, W a h e e d A, M e y e r EH, O ’ B r i e n JS,
v o n F i g u r a K (1989) J Biol Chem 264: 1252-125942. K r e y s i n g J, v o n F i g u r a K, G i e s e l m a n n V (1990)
Eur J Biochem 191: 627-63143. H o h e n s c h u t z C, F r i e d l W, S c h l ö r K H, W a h e e d
A, C o n z e l m a n n E, S a n d h o f f K , P r o p p i n g P (1988) Am J Med Genet 31: 169-175
44. G i e s e l m a n n V, v o n F i g u r a K (1990) J Inher Metab Dis 13: 560-571
45. G i e s e l m a n n V, P o l t e n A, K r e y s i n g J, v o n F i g u r a K (1989) P N A S USA 86 : 9436-9440
46. G i e s e l m a n n V, Z l o t o g o r a J, H a r r i s A, W e n g e r DA, M o r r i s C P (1994) Hum Mutation 4: 233-242
47. G i e s e l m a n n V, P o l t e n A, K r e y s i n g J, K a p p l e r J, F l u h a r t y A, B o h n e W, v o n F i g u r a K (1991) Brain Dysfunction 4: 235-243
48. G i e s e l m a n n V, P o l t e n A, K r e y s i n g J, K a p p l e r J, F l u h a r t y A, v o n F i g u r a K (1991) Dev Neurosci 13: 222-227
49. P o l t e n A, F l u h a r t y A L, F l u h a r t y CB, K a p p l e r J, v o n F i g u r a K, G i e s e l m a n n V (1991) N Engl J Med 324: 18-22
50. K a p p l e r J , v o n F i g u r a K, G i e s e l m a n n V (1992)Ann Neurol 31: 256-261
51. B a r t h ML, F e n s o m A, H a r r i s A (1993) Hum Genet 91: 73-77
52. H o n k e K, K o b a y a s h i T, F u j i T, G a s a S, X u M, T a k a m a r u Y, K o n d o R, T s u j i S, M a k i t a A (1993) Hum Genet 92: 451-456
53. G u s t a v s o n K, H a g b e r g B (1971) Acta Pediatr Scand 60: 585-590
54. G u i b a u d P , G a r c i a I, G u y o n n e t C L (1973) Lyon Med 229: 1215
55. Z l o t o g o r a J, B a c h G, B ö s e n b e r g C, B a r a k Y, v o n F i g u r a K, G i e s e l m a n n V (1995) Hum M utat 5: 137-43
56. F a r r e l l D F (1981) Hum Genet 59: 129-13457. H o h e n s c h u t z C, E i c h P, F r i e d l W, W a h e e d A,
C o n z e l m a n n E, P r o p p i n g P (1989) Hum Genet 82: 45-48
58. N e l s o n PY, C a r e y WF , M o r r i s C P (1991) Hum Genet 87: 87-88
59. C h a b a s A, C a s t e l l v i S, B a y e s M, B a l c e l l s S, G r i n b e r g D, Y i l a g e l i n L, M a r f a n y G, L i s s e n s W, G o n z a l e s - D u a r t e R (1993) Clin Genet 44: 320-323
Please contact the Editors if you wish to advertise in „Postępy Biochemii" the products and services related to biochemistry, molecular biology and cell biology.
POSTĘPY BIOCHEMII 42 (3 ) , 1996 289http://rcin.org.pl

Izoenzymy fosfolipazy C specyficznie hydrolizującej fosfatydyloinozytole i regulacja ich aktywności
Phosphatidylinositol-specific phospholipase C isozymes and regulation of their activity
T A D E U S Z P A W E L C Z Y K *
Spis treści:
I. WstępII. Izoenzymy fosfolipazy CIII. Właściwości katalityczne fosfolipazy CIV. Regulacja aktywności izoenzymów fosfolipazy C
IV-1. Regulacja aktywności izoenzymów fosfolipazyC typu p
IV-2. Regulacja aktywności izoenzymów fosfolipazy C typu y
IV-3. Regulacja aktywności izoenzymów fosfolipazy C typu 8
IV-4. Regulacja aktywności fosfolipazy C przez kinazę białkową C i kinazę białkową A
V. Wewnątrzkomórkowe rozmieszczenie izoenzymów fosfolipazy C
VI. Rola fosfolipazy C w patofizjologii
Wykaz stosowanych skrótów: PLC—fosfolipaza C;DAG—diacylogliceroł; Ins—inozytol; IP 3—inozytolotrisfo- sforan; PI—fosfatydyloinozytol; PIP—fosfatydyloinozytolo 4-fosforan; PIP2—fosfatydyloinozytolo 4,5-bisfosforan; PDGF—płytkowopochodny czynnik wzrostu; EGF—czynnik wzrostu naskórka; CSF—czynnik stymulujący komórki macierzyste (colony-stimulating factor); NGF—czynnik wzrostu nerwu; PMA-octan mirystoilo forbolu; PKC—kina- za białkowa C; SM—sfingomielina.
I. Wstęp
Białka receptorow e zlokalizowane w błonie kom órkowej umożliwiają kom órce odbieranie sygnałów z otaczającego ją środowiska. Związanie cząstki sygnałowej z receptorem prow adzi do pow stania i uwolnienia do w nętrza kom órki odpowiedniej substancji przekaźnikowej, k tó ra poprzez stymulację lub zahamowanie określonych przem ian wewnątrz kom órki zmienia jej m etabolizm lub funkcje. Klasycznym przykładem takiego przekaźnika jest cykliczny A M P p o wstający w reakcji katalizow anej przez cyklazę adeny- lanową. Związanie horm onu z receptorem prowadzi do aktywacji białka G i zw iązania G T P z podjednost- ką a b iałka G s lub G ;. Pow stały kom pleks G a-G T P wpływa na aktyw ność cyklazy adenylanowej.
O dm iennym m echanizm em przekazyw ania informacji w kom órce jest kaskada przem ian zapocząt-
* Dr hab., Katedra i Zakład Biochemii Klinicznej, Akademia Medyczna, ul. Dębinki 7, 80-211 Gdańsk
Contents:
I. IntroductionII. Isoenzymes of phospholipase CIII. Catalytic properties of phospholipase CIV. Regulation of phospholipase C isoforms
IV-1. Regulation of phospholipase C p isoformsIV-2. Regulation of phospholipase C y isoformsIV-3. Regulation of phospholipase C 8 isoformsIV-4. Regulation of phospholipase C activity by protein
kinase C and protein kinase AV. Subcellular distribution of phospholipase C isoenzymesVI. Role of phospholipase C in pathophysiology
kow ana hydrolizą fosfatydyloinozytoli w wewnętrznej warstwie błony kom órkowej (Ryc. 1). Związanie substancji sygnałowej z receptorem pow oduje aktywację fosfolipazy C specyficznie hydrolizującej fosfolipidy inozytolowe [1 ,2]. W rezultacie dochodzi do hydrolizy fosfatydyloinozytolo-4,5-bisfosforanu (P IP 2) i pow stania diacyloglicerolu (DAG) oraz inozytolotrisfosfora- nu (IP 3). I P 3 jako związek rozpuszczalny w środow isku w odnym dyfunduje z błony kom órkow ej do cyto- plazmy, a następnie po związaniu ze swoistymi receptoram i w błonach retikulum endoplazm atycznego indukuje uwolnienie w apnia z w ew nątrzkom órkow ych zapasów [2-4]. W zrost stężenia C a2+ stymuluje szereg przem ian kom órkow ych zależnych od tego jonu. M iędzy innym i dochodzi do aktywacji kinaz białkowych zależnych od C a2+ i kalm oduliny. Fosfolipaza C oraz kinaza białkow a C również aktyw ow ana jest przez C a2 + . D iacylogliceroł jak o związek nierozpuszczalny w środowisku wodnym pozostaje w błonie kom órkowej i przemieszczając się w niej może oddziaływać z kinazą białkow ą C, której jest fizjologicznym ak tyw atorem [5]. D otychczas poznano 11 izoenzymów kinazy białkowej C [6]. Substratam i kinazy białkowej C in vivo są białka związane z przekazywaniem informacji w kom órce, białka szlaków m etabolicznych oraz regulatorow e białka związane z ekspresją genów [7 ,8]. Pow stały w wyniku działania fosfolipazy C IP 3 ulega przem ianom w wyniku których następuje odtw orzenie P IP 2 [9, 10].
290 POSTĘPY BIOCHEMII 42 (3 ), 1996http://rcin.org.pl
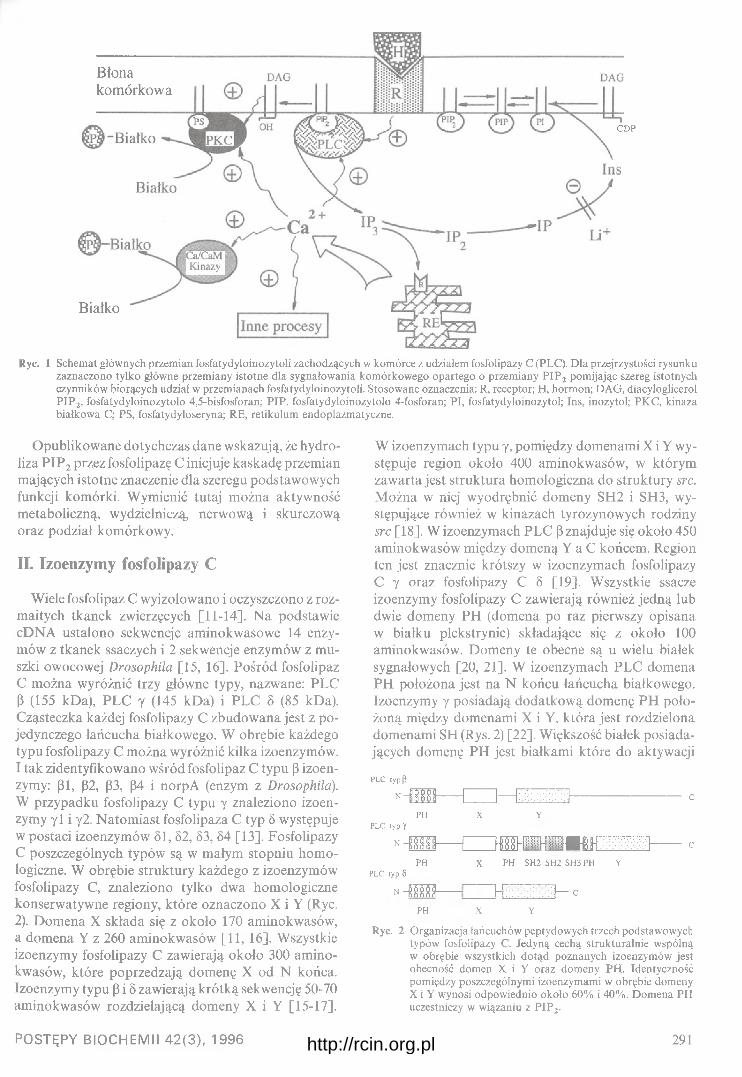
B łonak om órkow a
CDP
B iałko
Ryc. 1 Schemat głównych przemian fosfatydyloinozytoli zachodzących w komórce z udziałem fosfolipazy C (PLC). Dla przejrzystości rysunku zaznaczono tylko główne przemiany istotne dla sygnałowania komórkowego opartego o przemiany P IP 2 pomijając szereg istotnych czynników biorących udział w przemianach fosfatydyloinozytoli. Stosowane oznaczenia: R, receptor; H, hormon; DAG, diacyłoglicerol P IP 2, fosfatydyloinozytolo 4,5-bisfosforan; PIP, fosfatydyloinozytolo 4-fosforan; PI, fosfatydyloinozytol; Ins, inozytol; PKC, kinaza białkowa C; PS, fosfatydyloseryna; RE, retikułum endoplazmatyczne.
O publikow ane dotychczas dane wskazują, że hydroliza P IP 2 przez fosfolipazę C inicjuje kaskadę przem ian mających istotne znaczenie dla szeregu podstawowych funkcji kom órki. W ymienić tutaj m ożna aktywność m etaboliczną, wydzielniczą, nerwową i skurczową oraz podział kom órkow y.
II. Izoenzymy fosfolipazy C
Wiele fosfolipaz C wyizolowano i oczyszczono z rozm aitych tkanek zwierzęcych [11-14]. N a podstawie cD N A ustalono sekwencje am inokwasow e 14 enzymów z tkanek ssaczych i 2 sekwencje enzymów z m uszki owocowej Drosophila [15, 16]. Pośród fosfolipaz C m ożna wyróżnić trzy główne typy, nazwane: PLC (3 (155 kDa), PL C y (145 kDa) i PLC 5 (85 kDa). Cząsteczka każdej fosfolipazy C zbudow ana jest z p o jedynczego łańcucha białkowego. W obrębie każdego typu fosfolipazy C m ożna wyróżnić kilka izoenzymów. I tak zidentyfikow ano wśród fosfolipaz C typu (3 izoenzymy: (31, (32, (33, (34 i norpA (enzym z Drosophila). W przypadku fosfolipazy C typu y znaleziono izoenzymy y l i y2. N atom iast fosfolipaza C typ 6 występuje w postaci izoenzymów 61, 82, 83, 84 [13]. Fosfolipazy C poszczególnych typów są w m ałym stopniu hom ologiczne. W obrębie struktury każdego z izoenzymów fosfolipazy C, znaleziono tylko dw a homologiczne konserwatyw ne regiony, k tóre oznaczono X i Y (Ryc. 2). D om ena X składa się z około 170 am inokwasów , a dom ena Y z 260 am inokw asów [11, 16]. W szystkie izoenzymy fosfolipazy C zawierają około 300 am inokwasów, k tóre poprzedzają dom enę X od N końca. Izoenzymy typu (3 i 8 zawierają k ró tką sekwencję 50-70 am inokw asów rozdzielającą dom eny X i Y [15-17].
W izoenzymach typu y, pomiędzy dom enam i X i Y występuje region około 400 am inokwasów, w którym zaw arta jest struk tu ra hom ologiczna do struktury src. M ożna w niej wyodrębnić dom eny SH2 i SH3, występujące również w kinazach tyrozynowych rodziny src [18]. W izoenzymach PLC (3 znajduje się około 450 am inokwasów między dom eną Y a C końcem. Region ten jest znacznie krótszy w izoenzym ach fosfolipazy C y oraz fosfolipazy C 8 [19]. W szystkie ssacze izoenzymy fosfolipazy C zawierają również jedną lub dwie dom eny PH (dom ena po raz pierwszy opisana w białku plekstrynie) składające się z około 100 am inokwasów. Dom eny te obecne są u wielu białek sygnałowych [20, 21]. W izoenzymach PLC dom ena PH położona jest na N końcu łańcucha białkowego. Izoenzymy y posiadają dodatkow ą dom enę P H położoną między dom enam i X i Y, k tó ra jest rozdzielona dom enam i SH (Rys. 2) [22]. W iększość białek posiadających dom enę PH jest białkam i które do aktywacji
PLC typ |3
N - f f i S — IZZ]— MMM cPH X Y
PLC typ Y
"-iiffl— 1 [BMWWWHlMilł cPH X PH SH2 SH2 SH3 PH Y
PLC typ 8
N -E ffl \Hw w ąw fl- cPH X Y
Ryc. 2 Organizacja łańcuchów peptydowych trzech podstawowych typów fosfolipazy C. Jedyną cechą strukturalnie wspólną w obrębie wszystkich dotąd poznanych izoenzymów jest obecność domen X i Y oraz domeny PH. Identyczność pomiędzy poszczególnymi izoenzymami w obrębie domeny X i Y wynosi odpowiednio około 60% i 40%. Domena PH uczestniczy w wiązaniu z P IP 2.
POSTĘPY BIOCHEMII 42 (3 ) , 1996 291http://rcin.org.pl

wym agają asocjacji z b łoną kom órkow ą. W ykazano, że w obrębie dom eny P H znajdują się miejsca o dużym powinowactwie do P IP 2 [24,25]. W skazywać to może na P IP 2 jako miejsce wiązania pewnej klasy białek sygnałowych.
W piśmiennictwie m ożna również znaleźć inform acje na tem at fosfolipazy C o ciężarze cząsteczkowym 57-70 k D a [23, 26-29]. Enzym ten oznaczono symbolem PL C a. N a podstawie struktury cD N A opublikow ano sekwencję am inokw asow ą tego enzymu [27], brakuje jednak do tąd informacji o ekspresji aktywnego enzymu. S truk tu ra sklonowanej fosfolipazy C a nie jest podobna do struk tur innych znanych fosfolipaz C [27], jest natom iast niemal identyczna ze struk tu rą uprzednio wyizolowanego z m ikrokosm ków w ątroby bliżej nie scharakteryzow anego białka o m asie 58 kD a, k tóre nie wykazuje aktywności fosfolipazy C [30]. M ożna przypuszczać, że izolowane małe białka o aktywności fosfolipazy C są w rzeczywistości fragm entam i proteolizy znanych fosfolipaz C. W ykazano, że we frakcji błonowej przeciwciała przeciw fosfolipa- zie C 5 reagują z białkam i o m asach 45 i 43 k D a [31]. Stw ierdzono również, że fosfolipaza C 8 ulega łatwo proteolizie w miejscu między dom enam i X i Y. P o wstałe fragm enty o m asach 30 i 40 kD a posiadają zdolność oddziaływ ania ze sobą z wytworzeniem stru ktury o masie 70 kD a posiadającej aktywność fosfolipazy C [32].
III. Właściwości katalityczne fosfolipazy C
Aktywność fosfolipazy C zależy od C a2 + zarów no w w arunkach in vitro ja k i in vivo [33-37]. W zależności od substratu i typu fosfolipazy C, wpływ C a2+ na aktywność enzymu obserwuje się zarów no w zakresie stężeń m ikrom ołow ych jak i milimolowych. D otychczas nie m a danych na tem at bezpośredniego wiązania C a2 + przez fosfolipazę C. M utacje lub delecje w prow adzone w obrębie dom eny PL C 5 (144-236) zawierającej m otyw E F (struktura uczestnicząca w wiązaniu w apnia) prow adzą do znacznego obniżenia aktywności enzymu, lecz nie znoszą zależności aktywności od C a2+, co zdaje się wskazywać na istnienie większej liczby miejsc wiążących w apń [38]. Inni autorzy postulują, że miejscem wiązania w apnia w fosfolipazie C jest dom ena Y, k tó ra w części jest hom ologiczna z dom enam i wiążącymi C a2 + w fosfolipazie A2 i kina- zie białkowej C [39].
Substratem fosfolipaz C wszystkich typów są trzy fosfolipidy zawierające inozytol, tj.: fosfatydyloinozy- tol (PI), fosfatydyloinozytolo 4-fosforan (PIP) i fos- fatydyloinozytolo 4,5-bisfosforan (P IP 2), przy czym P IP 2 i P IP są substratam i preferowanymi. Pow inow actwo poszczególnych fosfolipaz C do P IP 2 przedstawia się następująco: PL C p > P L C 5 > P L C y [41]. Inne fosfatydyloinozytolofosforany występujące w kom órce w niewielkich ilościach, jak fosfatydyloinozytolo 3-fos- foran, fosfatydyloinozytolo 3,4-bisfosforan i fosfatydy-
loinozytolo 3,4,5-trisfosforan, nie są hydrolizowane przez żaden z izoenzymów fosfolipazy C [40, 41]. Jak w spom niano powyżej, produktam i reakcji katalizow anej przez fosfolipazę C jest diacyloglicerol oraz IP 3. W ykazano, że IP 3 powstaje w postaci cyklicznej oraz jako p roduk t niecykliczny [43]. Najwięcej cyklicznego inozytolotrisfosforanu powstaje w reakcji katalizow anej przez PL C P, a najmniej w reakcji katalizowanej przez PL C y [43]. Analiza stereochem iczna pow staw ania IP 3 wykazała, że w reakcji katalizowanej przez fosfolipazę C najpierw powstaje cykliczny produkt, k tóry jest uwalniany bądź przem ieniany do związku niecyklicznego [44]. W ykazano, że kom órki z mniejszą zaw artością cyklicznego IP 3 rosną w hodowli kom órkowej do gęstości niższej niż kom órki z wyższą zaw artością tego związku, co sugeruje, że stężenie cyklicznego IP 3 może regulować proliferację kom órek [1].
IV. Regulacja aktywności izoenzymów fosfolipazy C
IV-1. Regulacja aktywności izoenzymów fosfolipazy C typu p
Toksyna krztuśca podobnie jak toksyna cholery może katalizować ADP-rybozylację białek G co powoduje zmianę ich właściwości. Wpływ toksyny krztuśca na przebieg niektórych przem ian kom órkow ych wskazuje pośrednio na udział białek G w tych przem ianach. D latego, obserwowane w niektórych kom órkach zaham ow anie przez toksynę krztuśca horm onalnej stymulacji fosfolipazy C, wskazuje na udział białek G w przekazyw aniu informacji od receptora do fosfolipazy C [45, 46]. W ykazano również, że w kom órkach niewrażliwych na tę toksynę, nie podlegające m etabolizm owi analogi G T P, oraz fluorek glinowy zwiększają efektywność hydrolizy endogennego P IP 2 [46-50]. F ak t ten wskazuje na udział innego rodzaju białek G w regulacji aktywności fosfolipazy C. N aturę białek G niewrażliwych na toksynę krztuśca zbadano niedaw no, i zakwalifikowano je jako podrodzinę G q. O trzym ano i określono sekwencję szeregu cD NA odpow iadających nieznanym uprzednio podjednost- kom a podrodziny G q [51-53]. Obecnie znanych jest 15 różnych podjednostek Goc, które na podstawie sekwencji am inokwasowej zostały pogrupow ane w cztery podrodziny; G s, G i/G 0, G q i G 12. W białkach p o d rodziny G q w ystępują podjednostki Gocq, Goc11? Goc14, G a j5 i G a 16 [15]. W ykazano, że G a q i G a j j aktyw ują fosfolipazę C (31, natom iast nie m ają wpływu na aktywność fosfolipazy C y l i fosfolipazy C 51 [54, 55]. Białko G a 31 połączone z receptorem purynergicznym P 2y aktywuje fosfolipazę C p i w erytrocytach indyka [56, 57]. Expresja G a q, G a n , G a 14 i G a 16 w kom órkach linii Cos-7 i następnie użycie błon tych kom órek w badaniach nad aktyw acją fosfolipazy C p i i p2 wykazało, że wszystkie cztery białka G stym ulują aktywność obu izoenzymów. N ajbardziej efektywnymi
292 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

Ryc. 3 Przypuszczalny mechanizm aktywacji fosfolipazy C (3 przez białko ocq. Znak zapytania wskazuje, że nie jest znany mechanizm przemieszczenia fosfolipazy C |3 z cytoplazmy do miejsca związania z białkiem aq. N a rysunku przedstawiona jest PLC pi jako izoenzym najbardziej podatny na stymulację przez aq. Jak to zaznaczono w tekście, inne izoenzymy PLC p również ulegają aktywacji przez a q lecz w znacznie mniejszym stopniu.
w aktywacji fosfolipazy C (31 okazały się białka G a q, G a 11? natom iast fosfolipaza C (32 jest najbardziej stym ulow ana przez G a 16 [58]. W yniki te wskazują na specyficzność oddziaływ ania białek G z poszczególnymi izoenzymami fosfolipazy C (3. D ośw iadczenia z trzem a subtypam i receptora a adrenergicznego tj. a 1A, a 1B i a lc wykazały, że każdy z tych receptorów oddziaływa ze swoistym izoenzymem fosfolipazy C (3 poprzez odpowiednie białko G [59]. Spośród receptorów, k tóre po aktywacji stym ulują fosfolipazę C poprzez białka G a q i G a 1Ł wymienić m ożna receptory trom boksanu A 2, bradykininy, angiotensyny, histam iny, wazopresyny i acetylocholiny (Ryc. 3) [61-64].
H orm onalna aktyw acja fosfolipazy C w kom órkach jajn ika chom ika, m ięśniówki gładkiej naczyń, kom órkach hodow lanych HL-60 granulocytów i kom órkach m ezangialnych nerki ham ow ana jest przez toksynę krztuśca [45, 65-67]. Dośw iadczenia przeprow adzone z użyciem ludzkich granulocytów linii HL-60 w ykazały, że za aktywację fosfolipazy C typu p w kom órkach tych odpow iedzialna jest dim eryczna podjednostka (3y białka G (Ryc. 4) [68]. Dalsze badan ia wykazały, że podjednostka py aktywuje fosfolipazę C p2 [69, 70]. Receptoram i których aktyw acja prow adzi do stym ulacji PL C P za pom ocą dimerycznego białka Py są receptory m uskarynow e podtyp m2 i m4 oraz receptor dla interleukiny 8 [71]. W oddziaływ aniu PL C P z białkam i G biorą udział różne dom eny białkowe tego enzymu. Podjednostka a b iałka G q oddziaływuje z regionem C końcow ym PL C P leżącym za dom eną Y, natom iast dim er Py posiada powinow actw o do N ko ń cowego fragm entu PL C p zawierającego dom enę P H [72,73]. O becność osobnych miejsc w obrębie których dochodzi do oddziaływ ania PL C p z białkam i G w skazuje, że izoenzymy PL C p m ogą być aktyw ow ane
POSTĘPY BIOCHEM II 42 (3 ) , 1996
addytywnie przez podjednostki białek G. Podatność poszczególnych izoenzymów PL C P na aktywację przez białka G jest różna. W rażliwość izoenzymów PLC P na stym ulujące działanie G qa maleje w kolejności p l > p3 > P4 > p2 [74, 75]. Z kolei dimeryczne białko Py najbardziej aktywuje PL C P3, słabiej PL C P2 i najsłabiej PL C p i [76, 77]. Izoenzym PL C p4 nie jest aktywow any przez białko py [75]. Przytoczone powyżej dane wskazują na różnorodność m echanizm ów biorących udział w kontro li aktywności PLC typu P, co przy zróżnicowanym poziom ie ekspresji poszczególnych izoenzymów w różnych typach kom órek zabezpiecza precyzyjną regulację aktywności tego enzymu.
IV-2. Regulacja aktywności izoenzymów fosfolipazy C typu y
Polipeptydow e czynniki wzrostu, takie jak płyt- kow opochodny czynnik wzrostu (PD G F), czynnik wzrostu naskórka (EGF), czynnik stym ulujący kom órki macierzyste (CSF-1), czynnik w zrostu nerwów (N G F) oraz insulina wywierają swój wpływ na kom órkę docelową poprzez przyłączenie do receptorów obecnych w błonie kom órkowej. W szystkie te receptory oprócz podobnej budowy posiadają w swojej cytoplazmatycznej dom enie aktyw ność kinazy tyrozynowej.
Aktywacja receptora np. P D G F lub E G F powoduje autofosforylację jego dom eny cytoplazmatycznej, czem u towarzyszy przemieszczenie fosfolipazy C z cytoplazm y do przestrzeni błonowej [78, 79]. Dalsze badania wykazały, że dochodzi do zw iązania fosfolipazy C typu y z ufosforylowaną dom eną receptora. W śród pięciu autofosforylowanych miejsc receptora E G F miejscem wiążącym fosfolipazę C y l jest Tyr-992 [80, 81]. Po aktywacji receptora N G F , P D G F lub E G F obserwuje się również fosforylację fosfolipazy
PLC (31
Ryc. 4 Mechanizmu aktywacji fosfolipazy C (3 przez dimeryczną podjednostkę py białka G. Znak zapytania wskazuje, że nie jest znany mechanizm przemieszczenia PLC p z cytoplazmy do miejsca związania z podjednostką py białka G. Na rysunku przedstawiona jest PLC P3 jako izoenzym najbardziej podatny na stymulację przez (3y. Izoenzymy pi i P2 znacznie słabiej są aktywowane przez Py. Izoenzym P4 nie jest aktywowany przez Py.
293http://rcin.org.pl

C y [82-84]. W kom órkach posiadających wszystkie trzy typy fosfolipazy C stym ulacja receptora N G F powoduje fosforylację tylko typu y a nie P lub 5 [85, 86]. Stym ulacja receptora insulinowego lub CSF-1 nie wpływa na aktywność fosfolipazy C i nie obserwuje się fosforylacji żadnego izoenzymu fosfolipazy C [87, 88], pom im o dużego podobieństw a strukturalnego tych dwóch receptorów do receptorów dla takich czynników w zrostu jak: E G F, N G F , P D G F lub EG F.
W obrębie cząsteczki fosfolipazy C y zidentyfikow ano cztery miejsca fosforylacji. W wyniku stymulacji receptora E G F w fosfolipazie C y l ulegają fosforylacji reszty tyrozynowe w pozycji 771, 783, 1254 oraz 472 [82, 89]. N atom iast w cząsteczce fosfolipazy C y2 znaleziono tylko dwa miejsca fosforylacji Tyr-753 i Tyr-759 [19]. Fosforylacja fosfolipazy C y nie wpływa na aktywność enzymu w oznaczeniach in vitro z egzogennym P IP 2 jako substratem . Przy użyciu techniki m utacji punktowej wykazano, że fosforylacja Tyr-783 i Tyr-1254 niezbędna jest do aktywacji fosfolipazy C y l in vivo [55, 70]. W ykazano, że tylko enzym ufos- forylowany posiada zdolność do hydrolizow ania substratu związanego z białkiem typu profaliny [90, 91]. Przypuszcza się, że w w arunkach in vivo znaczna część substratu dla fosfolipazy C czyli P IP 2 występuje w formie związanej z profaliną i innym i białkam i cytoszkieletu [109].
Przedstaw ione powyżej dane wskazują, że fosforylacja izoenzymów fosfolipazy C typu y jest koniecznym w arunkiem ich aktywacji. W kom órkach niektórych typów w przenoszeniu sygnału od receptora E G F bierze udział białko G. W hepatocytach indukow ane przez E G F wytwarzanie IP 3 oraz diacyloglicerolu ham ow ane jest przez toksynę krztuśca, nie jest zaham ow ana natom iast fosforylacja fosfolipazy C y [92- 94]. P onad to obserwuje się indukow ane przez E G F wiązanie białka G ia z fosfolipazą C y. M ikrow strzyk- nięcie do pojedynczej kom órki hepatocyta przeciwciał przeciwko G ia ham uje indukow any przez E G F wzrost C a2+ [94]. D ane te wskazują, że fosforylacja fos-
Biona
PH
Ryc. 5 Mechanizm stymulacji fosfolipazy C y przez receptor zawierający w swojej strukturze kinazę tyrozynową. W niektórych typach komórek w procesie tym uczestniczy również białko Gia. Y, tyrozyna; Y-P, fosfotyrozyna.
folipazy C y w odpowiedzi na stymulację receptora E G F jest w arunkiem koniecznym, ale nie w ystarczającym do aktywacji enzymu. N a rycinie 5 przedstawiono przypuszczalny m echanizm aktywacji fosfolipazy C y z udziałem receptorow ej kinazy tyrozynowej.
Fosforylację reszt tyrozynow ych prow adzącą do aktywacji izoenzymów fosfolipazy C typu y obserwuje się również w wyniku działania kinaz tyrozynowych nie związanych z receptorem , k tóre są aktyw ow ane w wyniku pobudzenia receptorów w limfocytach, w zasadochłonnych kom órkach białaczkowych, mo- nocytach i kom órkach N K (Ryc. 6) [95-99]. Fos- folipaza C y 1 jest fosforylowana w limfocytach T w od-
ANTYGEN
PH
Ryc. 6 Aktywacja fosfolipazy C y przez receptor antygenowy z udziałem cytozolowej kinazy tyrozynowej. Znak zapytania wskazuje, że nie jest znany mechanizm oddziaływania receptora z cytoplazmatyczną kinazą tyrozynową oraz późniejsze przemieszczenie ufosforylowanej PLC y do błony komórkowej. Y, tyrozyna; Y-P, fosfotyrozyna.
powiedzi na aktywację kom pleksu receptorów (TCR) [100,101]. Z a fosforylację reszt tyrozynow ych w PL C y odpow iadają kinazy należące do rodziny src, p56lck i p59fyn w limfocytach T oraz p56lck i p53lyn w lim focytach B [102]. W limfocytach B głównym substratem kinaz tyrozynow ych jest PL C y2 [103]. Zw iązanie antygenu z receptorem IgE w białaczkowych kom órkach zasadochłonnych RBL indukuje fosforylację reszt tyrozynow ych w PL C y l [91]. N a tu ra i m echanizm oddziaływ ania niereceptorow ych kinaz ty ro zynowych, fosfolipazy C i receptorów leukocytów, jak na razie, nie są znane.
IV-3. Regulacja aktywności izoenzymów fosfolipazy C typu 8
A ktywność fosfolipazy C typu 5 stym ulow ana jest przez C a2+, poliam iny oraz białka zasadowe [34,105]. Przypuszcza się, że rolę fizjologicznego regulatora
unaza
294 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

fosfolipazy C 8 może odgrywać sperm ina, której poziom w kom órce zmienia się wraz z rozwojem kom órki [106, 107]. Jak dotąd , nie znany jest żaden receptorow y mechanizm aktywacji tej fosfolipazy C. Żaden izoenzym fosfolipazy C 8 nie jest substratem kinaz tyrozynowych. Białka G regulujące fosfołipazę C (3 nie m ają wpływu na aktyw ność fosfolipazy C 8. Być może regulacja aktywności PL C 8 jest zw iązana z innymi białkam i należącymi do rodziny białek Ras. O statnio wykazano, że PL C 81 jest aktyw ow ana przez białko p l22-R hoG A P będące aktyw atorem G T Pazo- wym dla białka Rho A [108]. W skazywać to może na udział PL C 81 w regulacji organizacji białek cytoszkieletu. W ykazano, że w erytrocytach około 70% P IP 2 uczestniczy w w iązaniu białek cytoszkieletu tzw. frakcji 4.1 z błoną kom órkow ą [108]. A ktywacja PLC 81 i hydroliza P IP 2 może zatem pociągać zmianę architektury i oddziaływ ania białek cytoszkieletu. W ykazano, że izoenzym 81 wiąże się z błonam i fos- folipidowymi zawierającymi P IP 2 lub sfingomielinę (SM) [110, 111]. N a podstawie stężeń fosfolipidów potrzebnych do związania fosfolipazy C 81 w 50% oraz zawartości P IP 2 i SM w kom órce m ożna wnioskować, że znacząca część tego enzymu może występować in vivo w formie związanej z b łoną kom órkow ą [111]. Potw ierdzeniem tej hipotezy może być doniesienie o wybiórczym w iązaniu się fosfolipazy C 81 z miejscami syntezy P IP 2 w obrębie błon miocytów psa [112]. B adania z użyciem fosfolipazy C 81 oczyszczonej z w ątroby szczura oraz fosfolipazy C 81 z ludzkich fibroblastów indukowanej w E. coli pokazały, że aktywność tego izoenzym u silnie zależy od sfingomieli- ny oraz jej m etabolitów [113, 114]. W ykazano, że sfingomielina jest silnym inhibitorem PL C 81, podczas gdy sfingozyna (metabolit sfingomieliny oraz silny inhibitor kinazy białkowej C) stymuluje ten enzym [113].
D ane powyższe wskazywać m ogą na powiązanie regulacji aktywności PL C 81 z m etabolizm em sfingomieliny. D o zm ian zawartości sfingomieliny w komórce dochodzi zarów no w w arunkach fizjologicznych jak i w niektórych stanach patologicznych [115- 120]. Zainteresow anie sfingolipidami utrzym uje się od m om entu odkrycia, że produkty rozpadu tej klasy lipidów są aktywne biologicznie. Sfingolipidy i lizosfin- golipidy wpływają na ważne funkcje kom órki oraz przejawiają właściwości przeciwnowotw orowe w wielu kom órkach zwierzęcych [21]. D ane na tem at m etabolizmu sfingomieliny w kom órce, zdają się wskazywać na udział tych przem ian w przekazyw aniu informacji wewnątrz kom órki [122, 123]. Zależność regulacji fosfolipazy C 81 od m etabolizm u sfingomieliny może być ogniwem wiążącym szlak przenoszenia informacji, w którym kluczow ą rolę odgrywa fosfolipaza C typu 8 ze szlakiem przekazyw ania informacji opartym o przem iany sfingomieliny.
IV-4. Regulacja aktywności fosfolipazy C przez kinazę białkową C i kinazę białkową A
W kom órkach niektórych typów aktyw acja kinazy białkowej C oraz kinazy A osłabia stym ulow aną receptorow o aktyw ność fosfolipazy C. W skazywać to może na kontrolę aktywności fosfolipazy C na zasadzie sprzężenia zwrotnego. W ykazano, że w kom órkach glejaka C6Bul, kom órkach 3T3 oraz ludzkich białacz- kowych limfocytach T fosfolipaza C typu y może być fosforylowana na reszcie serynowej (Ser-1248) w sposób zależny od cA M P przez kinazę białkow ą A [124, 127]. Fosfolipaza C typu P i 8 nie ulega takiej fosforylacji. Fosfolipaza C y może być również negatywnie regulow ana przez kinazę białkow ą C. W kom órkach mięśni gładkich linii Ju rk a t potraktow anych estrem forbolu (octan m irystoilo forbolu — PM A) obserwuje się fosforylację reszty serynowej 1248 fosfolipazy C y l [124]. Fosforylacja taka zmienia oddziaływanie fosfolipazy C y z niereceptorow ym i kinazami tyrozynowymi lub fosfatazą tyrozynow ą [128]. Inną drogą, na której kinaza białkow a białkowa C reguluje aktywność fosfolipazy C y jest fosforylacja receptora, odpowiedzialnego za stymulację PL C y. Fosforylacja Thr-654 receptora E G F przez PK C obniża zdolność receptorowej kinazy tyrozynowej do fosforylacji fosfolipazy C y l, co zapobiega jej ak tywacji [129].
Również fosfolipaza C typu p fosforylowana jest przez kinazę białkow ą C. Potrak tow anie kom órek PC 12, C6Bul lub N IH 3T3 estrem forbolu powoduje fosforylację Ser-887 fosfolipazy C p i, natom iast nie dochodzi do fosforylacji fosfolipazy C typu y i 8 [130]. Również w w arunkach in vitro oczyszczona fosfolipaza C p l fosforylowana jest przez P K C w pozycji Ser-887 [130]. Dotychczas nie jest znana ro la i znaczenie tej fosforylacji.
V. Wewnątrzkomórkowe rozmieszczenie izoenzymów fosfolipazy C
Funkcjonow anie cyklu przem ian fosfoinozytoli w błonie kom órkowej od szeregu lat jest tem atem intensywnych badań [131-133]. Obecność aktywności fosfolipazy C stwierdzono również w jądrach kom órek fibroblastów linii N IH 3T3, linii Swiss 3T3 oraz kom órek w ątroby [134-136]. B adania im m unocyto- chemiczne wykazały, że poszczególne izoenzymy fosfolipazy C występują w różnych przestrzeniach kom órki oraz, że stym ulacja odpow iednich receptorów pow oduje przemieszczenie poszczególnych typów fosfolipazy C w obrębie kom órki. W kom órkach fibroblastów linii Swiss 3T3 fosfolipaza C typu y obecna jest w cytoplazmie, a fosfolipaza C typu P w jądrze kom órkow ym [136]. Stym ulacja kom órek 3T3 in- sulinopodobnym czynnikiem w zrostu (IGF) indukuje wytwarzanie IP 3 prawie wyłącznie w jądrze kom órkowym. W kom órkach tych nie stw ierdzono obecności fosfolipazy C 8 w cytoplazmie i frakcji jądrow ej [136]. Im m unocytochem iczna analiza kom órek now otw orowych rdzenia nadnerczy linii PC 12 wykazała, że fos-
POSTĘPY BIOCHEMII 42 (3 ) , 1996 295http://rcin.org.pl

folipaza C y występuje w cytoplazmie oraz jądrze kom órkow ym , natom iast fosfolipaza C 8 obecna jest tylko w cytoplazmie. Z kolei fosfolipaza C P znajduje się wyłącznie w jądrze tych kom órek [138]. Badania z użyciem izolowanych jąder kom órek w ątroby w skazują, że jedynym typem fosfolipazy C występującym w jądrze tych kom órek jest fosfolipaza C typu p i, jakkolw iek frakcja jąd row a stanowi tylko 5% kom órkowej zaw artości tego izoenzymu [31]. W szczurzych fibroblastach płodowych fosfolipaza C y obecna jest w formie wolnej w cytoplazmie oraz w formie związanej z filam entam i aktyny. Stym ulacja tych kom órek przez P D G F pow oduje zwiększone nagrom adzenie PL C y w miejscach k on tak tu błony kom órkowej z filam entam i aktyny [139].
D ane te wskazują, że rozmieszczenie poszczególnych izoenzymów fosfolipazy C zależy od typu k o m órki. P o n ad to m ożna przypuszczać, że w różnych organellach kom órki cykl inozytolowy jest k o n tro lo wany przez różne izoenzymy fosfolipazy C.
VI. Rola fosfolipazy C w patofizjologii
Z uwagi na to, że fosfolipaza C odgryw a znaczącą rolę w aktywacji szeregu funkcji w kom órce m ożna przypuszczać, że w wielu stanach patologicznych d o chodzi do zaburzeń w funkcjonow aniu tego enzymu. Stwierdzono, że w nadciśnieniu sam oistnym aktyw ność fosfolipazy C w kom órkach kanalika proksym al- nego nerki szczura jest podwyższona [140]. D ośw iadczenia z użyciem skraw ków kory nerki szczura wykazały, że odpowiedź nerki na dopam inę, m ierzona wzrostem aktyw ności fosfolipazy C jest niższa u szczurów z nadciśnieniem sam oistnym [140] . U szczurów z nadciśnieniem wywołanym przez podaw anie octanu deoksykortikosteronu jednocześnie z hypertonicznym roztw orem soli aktyw ność fosfolipazy C jest podwyższona w kom órkach rdzenia nerki, natom iast obniżona w korze nerki oraz w kom órkach ścian tętniczych [141]. Pom iary ilości P I3 i diacyloglicerolu w kom órkach aorty u szczurów z nadciśnieniem sam oistnym wykazały, że ilość IP 3 i D A G u tych zwierząt jest znam iennie wyższa niż u szczurów no rmalnych. Aktywność fosfolipazy C 81 w kom órkach aorty zwierząt z nadciśnieniem sam oistnym jest wyższa, a poziom P IP 2 obniżony [142]. P onad to enzym ten jest bardziej wrażliwy na stym ulujące działanie niskich stężeń C a2+ [142]. Z a podwyższoną w rażliwość fosfolipazy C 81 na stym ulujące działanie wapnia odpow iedzialna jest jak wydaje się, m utacja punktow a w cząsteczce enzymu [142,143]. Aktywność fosfolipazy C y stym ulow ana jest przez szereg czynników w zrostu co wskazywać może na jej udział w przekazyw aniu kom órce sygnału do podziału. Mi- krowstrzyknięcie fosfolipazy C y lub |3 do fibroblastów indukuje syntezę D N A [144], k tó ra jest zaham ow ana po m ikrow strzyknięciu przeciwciał przeciw tym enzymom [145]. W ykazano, że szereg kom órek now o
tworowych posiada podw yższoną aktywność fosfolipazy C. K om órki now otw oru piersi [146] oraz ludzkiego czerniaka [147] zaw ierają podwyższony poziom fosfolipazy C y. D ane te sugerują, że podział kom órek rakow ych może być stym ulow any przez podwyższony poziom fosfolipazy C. Szereg chorób neurodegenera- cyjnych u ludzi związanych jest z akum ulacją PL C 8 w regionach m ózgu zmienionych chorobow o [148]. U pacjentów cierpiących na chorobę Alzheimera stwierdzono, że enzym ten jest nadm iernie nagrom adzony w zwojach neurofibryli i neuronach otaczających plam ki starcze. Analiza techniką „W estern b io t” wykazała, że fosfolipaza C 8 skoncentrow ana jest we frakcji sparow anych filam entów hełikalnych mózgu [149. 150]. Stwierdzono, że aktyw ność specyficzna tego enzymu jest znacznie obniżona w mózgu pacjentów z chorobą Alzheimera [151].
A rtyku ł otrzymano 28 marca 1996 r.Zaakceptowano do druku 16 maja 1996 r.
Notatka uzupełniająca
Już po wysłaniu tego artykułu do druku, w Naturę* ukazał się artykuł poświęcony strukturze fosfolipazy C 81. Dane otrzymane w wyniku badań krystalograficznych wykazały, że enzym ten posiada w swojej N końcowej części cztery domeny EF, które są umieszczone parami w dwóch pętlach. Domena EF1 i EF2 uczestniczy w wiązaniu wapnia, podczas gdy domena EF3 i EF4 tworząca pętlę drugą nie posiadała związanego wapnia. Autorzy pracy na podstawie analizy struktury kryształu PLC 81 proponują mechanizm oddziaływania enzymu z błoną plazmatyczną, w którym to procesie brałyby udział domeny PH oraz domena C2. W zaproponowanym modelu domena PH zlokalizowana na N końcu cząsteczki miałaby odpowiadać za wiązanie enzymu do błony lipidowej, natomiast domena C2 (626-756 aa) odpowiadałaby za właściwą orientację enzymu w stosunku do substratu. W podsumowaniu autorzy stwierdzają, że architektura domen białkowych PLC 81 wskazuje na typ cząsteczki zdolnej do odwracalnej asocjacji z błoną plazmatyczną, co jest cechą charakterystyczną szeregu białek sygnałowych takich jak kinaza białkowa C, cytoplazmatycz- na fosfolipaza A2 czy fosfatydyloinizytolo 3-kinaza.
Piśmiennictwo
1. M a j e r u s P W (1992) Annu Rev Biochem 61: 225-2502. R a n a RS, H o k i n L E (1990) Physiol Rev 70: 115-1643. I r v i n e R F (1990) FEBS Lett 263: 5-94. I r v i n e R F (1991) Bioassays 13: 419-4275. N i s h i z u k a Y (1988) Nature (London) 334: 661-6656 . N i s h i z u k a Y (1995) FASEB J 9: 489-4967. K i k k a k a w a U, N i s h i z u k a Y (1986) Ann Rev Cell Biol 2:
149-1788. M e e k D W, S t r e e t A J (1992) Biochem J 287: 1-159. D o w n e s CP , M a c p h e e C H (1990) Eur J Biochem 193:
1-1810. M a t o J M (1990) W: Phospholipid metabolism in cellular
signaling. C. R. S. Press, Boca Raton, Ann Arbor, Boston, str 9-17
*E s s e n L-O, P e r i s i c O, C h e u n g R, K a t a n M, W i l l i a m s R L (1996) Nature ( London) 380, 595-602
296 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

11. R h e e SG, K i m H, S u h P - G , C h o i W C (1991) Biochem Soc Trans 19: 337-341
12. R h e e SG , S u h P G , R y S, L e e S Y (1989) Science 244: 546-550
13. C o c k c r o f t S , T h o m a s G M H (1992) BiochemJ 288:1-1414. D e n n i s EA, R h e e S, B i l l a M M , H a n n u n YA (1991)
FASEB J 5: 2068-207715. K r i z R , L i n L - L , S u l t z m a n L , E l l i s C , H e l d i n C - H ,
D a w s o n T, K n o p f J (1990) W: Proto-Oncogenes in Cell Development (Ciba Foundation Symposium 150) str 112-127, Wiley, Chichester, England
16. R h e e S G , C h o K D (1992) Adv Second Messenger Phosphop- rotein Res 26: 35-60
17. R h e e S G (1991) Trends Biochem Sci 16: 297-30118. K o c h AC, A n d e r s o n DA, M o r a n MF , E l l i s C,
P a w s o n T (1991) Science 252: 668-67419. R h e e SG, C h o i K D (1992) J Biol Chem 227: 12393-1239620. M u s a c c h i o A, G i b s o n T, R i c e P, T h o m p s o n J,
S a r a s t e M (1993) Trends Biochem Sci 18: 343-34821. P a r k e r PJ , H e m m i n g s BA, G i e r s c h i k P (1994)
Trends Biochem Sci 19: 54-5522. L e e SB, R h e e S G (1995) Current Opinion in Cell Biol 7:
183-18923. T a k e n a w a T, N a g a i Y (1981) J Biol Chem 256:6769-677524. H a r l a n JE, H a j d u k P J , Y o o n HS, F e s i k S W (1994)
Nature (Lond) 371: 168-17025. C i f u e n t e s MC , H o n k a n e n L, R e b e c c h i M J (1993)
J Biol Chem 268: 11586-1159326. N a k a n i s h i O, H o m m a Y, K a w a s a k i H, E m o r i Y,
S u z u k i K, T a k e n a w a T (1988) Biochem J 256: 453-45927. B e n n e t t CF , C r o o k e S T (1987) J Biol Chem 262:
13789-1379728. T o m k i n s TA, M o s c a r e l l o M A (1991) J Biol Chem 266:
4228-423629. G r i e n d l i n g K K , T a u b m a n MB, A k e r s M, M e n d -
l o w i t z M, A l e x a n d e r R W (1991) J Biol Chem 266: 15498-15504
30. M a r t i n JL, P u m f o r d NR, L a R o s a AC, M a r t i n BM, G o n z a g a H M S , B e a v e n MA, P o h l L R (1991) Biochem Biophys Res Commun 178: 679-685
31. D i v e c h a N , R h e e S G , L e t c h e r A J, I r v i n e R F (1993) Biochem J 289: 617-620
32. E l l i s MV, C a r n e A, K a t a n M (1993) Eur J Biochem 213: 339-347
33. B i d e n TJ , P e t e r - R i e s c h B, S c h l e g e l W, W o l l h e - i m C B (1987) J Biol Chem 262: 3567-3571
34. F u k u i T , L u t z RJ, L o w e n s t e i n J M (1988) J Biol Chem 263: 17730-17737
35. S a s a k a w a N, N a k a k i R, Y a m a m o t o S, K a t o R (1987) FEBS Lett 223: 413-416
36. W a t s o n SP, K a i J, S a s a g u r i T (1990) Br J Pharmacol 99: 212-216
37. W h i t a k e r M, A i t c h i s o n M (1985) FEBS Lett 182: 119-124
38. N a k u s h i m a S, B a n n o Y, W a t a n a b e T , N a k a m u r a Y, M i z u t a n i T, S a k a i H, S u g i m o t o Y, N o z a w a Y (1995) Biochem Biophys Res Comm 211: 365-369
39. C l a r k J D, L i n L- L , K r i z RW, R a m e s h a CS, S u l t z m a n LA, L i n AY, M i l o n a N, K n o p f J W (1991) Cell 65: 1043-1051
40. L i p s DL , M a j e r u s P W, G o r g a FR, Y o u n g AT, B e n j a m i n D L (1989) J Biol Chem 264: 8759-8763
41. S e r u n i a n LA, H a b e r MT , F u k u i T, K i m J W, R h e e SG, L o w e n s t e i n J M, C a n t l e y L C (1989) J Biol Chem 264: 17809-17815
42. R y u S H, S u h P G , C h o K S , L e e K Y , R h e e S G (1987) Proc N atl Acad Sci USA 84: 6649-6653
43. K im JW , R y u SH , R h e e S G (1989) Biochem Biophys Res Commun 163: 177-182
44. B r u z i k KS, M o r o c h o AM, J h o n D- Y, R h e e SG, T s a i M - D (1992) Biochemistry 31: 5183-5193
45. A s h k e n a z i A, P e r a l t a E G, W i n s l o w J W, R a m a - c h a n d r a n J, C a p o n D J (1989) Cell 56: 478-493
46. C h i b a T , R a f f o u l K , Y a m a d a T (1987) J Biol Chem 252: 8467-8469
47. B a l d a s s a r e JJ , K n i p p MA, H e n d e r s o n PA, F i s
h e r G J (1988) Biochem Biophys Res Commun 154: 351-35748. C o c k c r o f t S, G o m p e r t e s B D (1985) Nature (London)
314: 534-53649. D e c k m y n H, T u S- M, M a j e r u s P W (1986) J Biol Chem
261: 16553-1655850. L i t o s c h I, W a l l i s C, F a i n J N (1985) J Biol Chem 260:
5464-547151. S i m o n M I , S t r a t h m a n M P , G a u t a m N (1991)Science
252: 802-80852. S m r e k a AV, H e l p e r J R, B r o w n KO, S t e r n w e i s
P C (1991) Science 250: 804-80753. T a y l o r SJ, S m i t h J A, E x t o n J H (1990) J Biol Chem 265:
17150-1715654. P a r k D, I h o n D- Y, K r i z R, K n o p f J, R h e e S G (1992)
J Biol Chem 267: 16048-1605555. T a y l o r SJ, C h a e HZ, R h e e SG, E x t o n J H (1991)Va-
ture (London) 350: 516-51856. M o r r i s A J, W a l d o GL, D o w n e s CP , H a r d e n T K
(1990) J Biol Chem 265: 13508-1351457. W a l d o GL, B o y e r JL, M o r r i s AJ, H a r d e n T K
(1991) J Biol Chem 266: 14217-1422558. L e e C H , P a r k D , W u D , R h e e S G , S i m o n M I (1992)
J Biol Chem 267: 16044-1604759. W u D, K a t z A, L e e C - H , S i m o n M I (1992) J Biol Chem
267: 25798-2580260. W o n g SK, P a r k e r E M, R o s s E M (1990) J Biol Chem
265: 6219-622461. B e r n s t e i n G, B l a n k JL, S m r e k a A V, H i g a s h i j i -
m a T, S t e r n w e i s PC, E x t o n J H, R o s s E M (1992) J Biol Chem 267: 8081-8088
62. G u t o w s k i S, S m r e k a A V, N o w a k L, W u D, S i m o n M, S t e r n w e i s P C (1991) J Biol Chem 266: 20519-20524
63. S h e n k e r A, G o l d s m i t h P, U n s o n CG, S p i e g e l A M (1991) J Biol Chem 266: 9303-9313
64. W a n g e RL, S m r e k a A V, S t e r n w e i s PC, E x t o n J H(1991) J Biol Chem 266: 11409-11412
65. A s h k e n a z i A, W i n s l o w J W, P e r a l t a EG, P e t e r s o n GL, S c h i m e r l i k MI , C a p o n DJ , R a m a c h a n d - r a n J (1987) Science 238: 672-675
66 . B i r n b a u m e r L, A b r a m o w i t z J, B r o w n A M (1990) Biochim Biophys Acta 1031: 163-224
67. L a B e l l e EF, M u r r a y B M (1990) FEBS Lett 268: 91-9468 . C a m p s M, H o u C, S i d i r o p o u l o s D, S t o c k JB,
J a k o b s K H , G i e r s c h i k P (1992) Eur J Biochem 206: 821-831
69. C a m p s M, C a r o z z i A, S c h n a b e l P, S c h e e r A, P a r k e r PJ , G i e r s c h i k P (1992) Nature (London) 360: 684-686
70. K im H K , K im JW , Z i l b e r s t e i n A, M a r g o l i s BL, K i m CK, S c h l e s s i n g e r J, R h e e S G (1991) Cell 65: 435-441
71. R h e e S G (1994) Signal-activated phospholipases(LiscovithM, Austin RG wyd) Landes Company
72. L e e SB, S h i n SH, H e l p e r JR, G i l m a n AG, R h e e S G (1993) J Biol Chem 268: 25952-25957
73. T o u h a r a K, I n g e l s e J, P i t c h e r J A, S h a w G, L e f - k o w i t z R J (1994) J Biol Chem 269: 10217-10220
74. J h o n D- Y, L e e H - H , P a r k D, L e e C - W, L e e K - H , Y o o O J, R h e e S G (1993) J Biol Chem 268: 6654-6661
75. L e e C - W , L e e K - H , L e e S B , P a r k D , R h e e S G (1994)J Biol Chem 269: 25335-25338
76. P a r k D , J h o n D - Y , L e e C - W , L e e K - H , R h e e S G(1993) J Biol Chem 268: 4573-4576
77. S m r e k a AV, S t e r n w e i s P C (1993) J Biol Chem 268: 9667-9674
78. K im U - H , K i m H- S , R h e e S G (1990) FEBS Lett 270: 33-36
79. T o d d e r u d G, W a h l M, R h e e SG, C a r p e n t e r G (1990) Science 249: 296-298
80. R o t i n D, M a r g o l i s BL, M o h a m m a d i M, D a l y G, D a u m G, Li N, B u r g e s s W, F i s c h e r EH, U l l r i c h A, S c h l e s s i n g e r J (1992) EMBO J 11: 559-567
81. V e g a QC, C o c h e t C, F i l h o l O, C h a n g CP, R h e e SG, G i l l G N (1992) Mol Cell Biol 12: 128-135
82. K im JW , S im S S , K i m U - H , N i s h i b e S , W a h l MI , C a r p e n t e r G, R h e e S G (1990) J Biol Chem 265: 3940- 3943
POSTĘPY BIOCHEM II 42 (3 ) , 1996 297http://rcin.org.pl

83. M e i s e n h e l d e r J, S u h P - G , R h e e SG, H u n t e r T (1989) Cell 57: 1103-1122
84. N i s h i b e S, W a h l MI , R h e e SG, C a r p e n t e r G (1989) J Biol Chem 264: 10335-10338
85. H o m m a Y, S a k a m o t o H, T s u n o d a M, A o k i M, T a k e n a w a T, O o y a m a T (1993) J Biochem 290: 649-653
86 . K im U - H , F i n k DJ r , K i m HS, P a r k DJ , C o n t r e r a s ML , G u r o f f G, R h e e S G (1991) J Biol Chem 266: 1356-1362
87. D o w n i n g JR, M a r g o l i s BL, Z i l b e r s t e i n A, A s h - m u n RA, S h e e r UA, S c h l e s s i n g e r J (1989) EMBO J 8 : 3345-3350
88 . N i s h i b e S , W a h l MI , W e d e r g a e t n e r P B , K i m JJ, R h e e SG, C a r p e n t e r G (1990) Proc N atl Acad Sci USA 87: 424-428
89. W a h l M I , N i s h i b e S , K i m J W , R h e e S G , C a r p e n t e r G (1990) J Biol Chem 265: 3944-3948
90. G o l d s c h m i d t - C l e r m o n t PJ , K i m J W, M a c h e s - k y L M, R h e e SG, P o l l a r d T D (1991) Science 251: 1231-1233
91. G o l d s c h m i d t - C l e r m o n t PJ , M a c h e s k y LM, B a l d a s s a r e JJ , P o l l a r d T D (1990) Science 247: 1575- 1578
92. Y a n g - L , B a f f y G, R h e e SG, M a n n i g DR, H a n s e n CA, W i l l i a m s o n J R (1991) J Biol Chem 266: 22451-22458
93. L i a n g MN , G a r r i s o n J C (1991) J Biol Chem 266: 13342-13349
94. Y a n g - L , C a m o r a t t o AB, B a f f y G, R a j S, M a n n i n g DR, W i l l i a m s o n J R (1993) J Biol Chem 268: 3739-3746
95. A z z o n i L, K a m o u n M, S a l c e d o T W, K a n a k a r a j P, P e r u s s i a B (1992) J Exp Med 176: 1745-1750
96. B i a n c h i n i L, T o d d e r u d G, G r i n s t e i n S (1993) J Biol Chem 268: 3357-3363
97. K u m a d a T, F u j i m i y a H, M i y a t a H, B a n n o Y, N o z a w a Y (1992) Arerugi 41: 1710-1716
98. L i a o F, S h i n HS, R h e e S G (1992) Proc N atl Acad Sci USA 89: 3659-3663
99. T i n g A T , K a r n i t z L M , S c h o o n R A , A b r a h a m RT, L e i b s o n P J (1992) J Exp Med 176: 1751-1755
100. S e c r i s t J P , K a r n i t z L, A b r a h a m T T (1991) J Biol Chem 266: 12135-12139
101. W e e S, S c h i e v e n GL, K i r i h a r a J M, T s u TT, L e d b e t t e r JA, A r u f f o A (1993) J Exp Med 177: 219-223
102. C o o k e MP , A b r a h a m K M , F o r b u s h KA, P e r l m u t t e r R M (1991) Cell 65: 281-291
103. H e m p e l WM, S c h a t z m a n RC, D e F r a n c o A L(1992) J Immunol 148: 3021-3027
104. P a r k DJ , M i n H K , R h e e S G (1991) J Biol Chem 266: 24237-24240
105. H a b e r T M, F u k u i T, L e b o w i t z MS, L o w e n s t e i n J M (1991) Arch Biochem Biophys 288: 243-249
106. M o r g a n D M L (1990) Biochem Soc Trans 18: 1080-1084107. P e g g A E (1988) Cancer Res 48: 759-774108. H o m m a Y, E m o r i Y (1995) EMBO J 14: 286-291109. G a s c a r d P, P a w e l c z y k T, L o w e n s t e i n J M, C o
h e n C M (1993) Eur J Biochem 211: 671-681110. R e b e c c h i M, P e t e r s o n A, M c L a u g h l i n S (1992)
Biochemistry 31: 12742-12747111. P a w e l c z y k T, L o w e n s t e i n J M (1993) Biochem J 291:
693-696112. W o l f R A (1992) Am J Physiol 263: C1021-C1028113. P a w e l c z y k T, L o w e n s t e i n J M (1992) Arch Biochem
Biophys 297: 328-333114. P a w e l c z y k T, L o w e n s t e i n J M (1993) Biochem Phar
macol 45: 493-497115. B a r e n h o l z Y, T h o m p s o n T E (1980) Biochim Biophys
Acta 604: 129-158116. B a r e n h o l z Y, G a t t S (1982) Phospholipids (Hawthorne
JN, Ansell GB wyd.) Elsevier Biomedical Press, Amsterdam117. L e v i n E, J o h n s o n R M, A l b e r t S (1958) Arch Biochem
Biophys 73: 247-254118. H o s t e t l e r KY, Z e n n e r BD, M o r r i s H P (1979)
Cancer Res 39: 2978-2983119. E n g e l m a n B, S t r e i c h S, S c h o n t h i e r U M, R i c h
t e r WO , D u h m J (1992) Biochim Biophys Acta 1165: 32-37120. F u Y F , D o n g YZ, Li H, W a n g W (1992) Chin Med
J Engl 105: 803-808121. H a n n u n YA, B e l l R M (1989) Science 243: 500-507122. D r e s s i e r KA, M a t h i a s S, K o l e s n i c k R N (1992)
Science 255: 1715-1718123. M a t i a s S, Y o u n e s A, K a n C - C , O r l o w I, J o s e p h
C, K o l e s n i c k R N (1993) Science 259: 519-522124. B r u n s C, M a r m e D (1987) FEBS Lett 212: 40-44125. B u r n a t o w s k a - H l e d i n MA, S p i e l m a n W S (1989)
J Clin Invest 83: 84-89126. K a n a i d e H, M a t s u m o t o T, N a k a m u r a M (1986):
Biochem Biophys Res Commun 140: 195-203127. S h a y m n JA, M o r r i s e y JJ, M o r r i s o n A R (1987)
J Biol Chem 262: 17083-17087128. P a r k D J, M i n H K , R h e e S G (1992) J Biol Chem 261:
1496-1501129. D e c k e r SJ, E l l i s C, P a w s o n T, Y e l u T (1990) J Biol
Chem 265: 7009-7015130. G o M, Y o k o y a m a M, A k i t a H, F u k u z a k i H (1988)
Biochem Biophys Res Commun 153: 51-58131. K w i a t k o w s k a J (1989) Post Biochem 35: 253-263132. B a r a ń s k a J, C z a r n y M (1991) Post Biochem 37:129-132133. P o d d a n a H, B a r a ń s k a J (1991) Post Biochem 37:
129-132134. D i v e c h a N, B a n f i c H, I r v i n e R F (1991) EMBO J 10:
3202-3214135. S m i t h CD, W e l l s W W (1983) J Biol Chem 258:9368-9373136. M a r t e l l i AM, G i l m o u r RS, B e r t a g n o l o V, N e r i
L M, M a n z o l i L, C o c o L (1992) Nature (London) 358: 242-245
137. P a y r a s t r e B, N i e v e r s M, B r e t o n M, V e r k l e i j A, V a n b e r g e n e n H e n e g o u w e n P M P (1992) J Biol Chem 261: 5078-5084
138. M a z z o m i M, B e r t a g n o l o Y, N e r i L M, C a r i n i C, M a r c h i s i o M, M i l a n i D, M a n z o l i FA, C a p i t a n i S (1992) Biochem Biophys Res Comm 187: 114-120
139. M c B r i d e K, R h e e SG, J a k e n S (1991) Prot Natl Acad Sci USA 88 : 7111-7115
140. C h e n CJ, V y a s SJ, E i c h b e r g J, L o k h a n d w a l a M F (1992) Hypertension 19: 102-108
141. U e h a r a Y, N u m a b e A, T a k a d a S, H i r a k a w a N, M a t s u o k a H, I k e d a Y, Y a g i S, S a g i m o t o T (1991) Jpn Circ J 55: 509-515
142. K a t o H, F u k a m i K, S h i b a s a k i F, H o m m a Y, T a k e n a w a T (1992) J Biol Chem 261: 6483-6487
143. K a t s u y a T, H i g a k i J, M i k i T, K o h a r a K, Y a g i s a - w a H, T a n e s e H, M i k a n i H, S e r i k a w a T, N o j i m a H, O g i h a r a T (1992) Biochem Biophys Res Commun 187: 1359-1366
144. S m i t h MR , R y u S - H, S u h P - G , R h e e SG, K u n g H - F (1989) Proc Natl Acad Sci USA 86 : 3659-3663
145. S m i t h MR , L i u Y- I , K i m H, R h e e SG, K u n g H - F(1990) Science 247: 1074-1077
146. A r t e a g a CL, J o h n s o n MD , T o d d e r u d G, C o f - f r e y RJ , C a r p e n t e r G, P a g e D L (1991) Proc Natl Acad Sci USA 88 : 10435-10439
147. P e r e l l a F W, J a n k i e w i c z R, D a n d r o w E A (1991) Biochim Biophys Acta 1076: 209-214
148. S h i m o h a m a S, P e r r y G, R i c h e y P, T a k e n a w a T, W h i t e h o u s e PJ , M i y o s h i K, S u e n a g a T, M a t s u m o t o S, N i s h i m u r a M, K i m u r a J (1993) Neurosci Lett 162: 183-186
149. S h i m o h a m a S, H o m m a Y, S u e n a g a T, F u j i m o t o S, T a n i g u c h i T, A r a k i W, Y a m a o k a Y, T a k e n a w a T, K i m u r a J (1991) Am J Pathol 139: 737-742
150. S h i m o h a m a S, P e r r y G, R i c h e y P, P a p r o t n i k D, T a k e n a w a T, F u k a m i K, W h i t e h o u s e PJ , K i m u r a J (1995) Brain Res 669: 217-224
151. S h i m o h a m a S , F u j i m o t o S , M a t s u s h i m a H , T a k e n a w a T, T a n i g u c h i T, P e r r y G, W h i t e h o u s e PJ , K i m u r a J (1995) J Neurochem 64: 2629-2634
Prenumerata „Postępów Biochemii" w 1996 r.Prenumerata dla instytucji — 60 złIndywidualna — 28 zł50% zniżki dla członków PTBioch.
298 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

Szanownej Pani Profesor Zofii Zielińskiejpracę tę dedykują Autorki
Sfingomielina i jej metabolity jako pośredniki w przenoszeniu sygnału w komórce
Sphingomyelin and its metabolites in cellular signal transduction
JOLANTA M. DZIK1, ELŻBIETA WAŁAJTYS-RODE2
Spis treści:
I. WstępII. Wpływ metabolitów sfingomieliny na różnicowanie i pro
liferację komórekII-l. Wpływ pochodnych sfingozyny na poziom kwasu
fosfatydowegoIII. Rola metabolitów sfingomieliny w apoptozieIV. Wpływ sfingozyno-l-P na ruchliwość komórek nowo
tworowychV. Regulacja homeostazy wapniowej w komórce przez
metabolity sfingomielinyVI. Rola metabolitów sfingomieliny w przenoszeniu syg
nałów cytokin i czynników wzrostu w komórkach VI-1. Receptory dla TNFVI-2. Receptor Fas/APO-1VI-3. Różne drogi przenoszenia sygnału pochodzącego
od nSM-azy i aSM-azy VI-4. Receptor PDGF
VII. Uwagi końcowe
Contents:
I. IntroductionII. Regulation of proliferation and differentiation by sphin
gomyelin metabolitesII-l. Influence of sphingosine derivatives on the level of
phosphatidic acidIII. Sphingomyelin metabolites in apoptosisIV. Sphingosine-l-P involvement in cancer cell motilityV. Regulation of cellular calcium homeostasis by sphin
gomyelin metabolitesVI. Sphingomyelin metabolites and cytokine and growth
factor signal transductionVI-1. TNF receptors VI-2. Fas/APO-1VI-3. Diversity of nSM-ase and aSM-ase induced signal
pathways VI-4. PDGF receptor
VII. Concluding remarks
Wykaz stosowanych skrótów: DAG — diacyloglicerol; PC— fosfatydylocholina; PC-PLC — fosfolipaza C swoista dla fosfatydylocholiny; PLA2 — fosfolipaza A2; PLD — fosfolipaza D; SM — sfingomielina; SP — sfingozyna; SPP— sfmgozyno-1-fosforan; SPC — sfmgozylofosfocholina; aSM-aza i nSM-aza — sfingomielinazy o optimach działania w pH odpowiednio kwaśnym i obojętnym; PA — kwas fosfatydowy; IP3 — inozytolotrisfosforan; PMA = TPA,12-o-tetradekanoilo-forbolo-13-octan — ester forbolu, EGF— nabłonkowy czynnik wzrostu; FGF — fibroblastowy czynnik wzrostu; PDGF — płytkowy czynnik wzrostu; PDGF-R — receptor dla PDGF; TNF — czynnik martwicy nowotworów; TNF-R — receptor dla TNF; N Fk B, AP-1— czynniki transkrypcyjne; I-kB — inhibitor NFkB; PKC— kinaza białkowa C; MAPK — kinazy białkowe aktywowane mitogenami; MEK — kinazy MAPK; SAPK, JM K — kinazy białkowe aktywowane stresem; Raf— kinaza białkowa Raf; CAPK — kinaza białkowa aktywowana ceramidem; PDK — „ukierunkowane proliną” seryno- wo/treoninowe kinazy białkowe; CAPP — fosfataza białkowa aktywowana ceramidem; PP-2A — fosfataza białkowa 2A; TRADD — białko homologiczne do domeny śmierci receptora TNF-R55; Fas/APO-1 — receptor z rodziny receptorów TNF.
1 Dr, Instytut Biologii Doświadczalnej im. M. Nenckiego PAN, ul. Pasteura 3, 02-093 Warszawa,
2 prof. dr hab., Zakład Bioinżynierii Instytutu Biotechnologii i Antybiotyków, ul. Starościńska 5,02-516 Warszawa
I. Wstęp
W latach osiemdziesiątych ubiegłego stulecia T h u - d i c h u m wyizolował i scharakteryzow ał składnik wyciągu eterowego mózgu, który nazwał sfmgomieli- ną. Początkow o łączono obecność tego związku z funkcjonowaniem mózgu, ale po stwierdzeniu powszechnego występowania we frakcji lipidów błonowych i w mielinie zaczęto uważać, iż pełni on funkcje strukturalne. W następstwie odkrycia przez O k a z a - k i e g o i w s p. [1], że w itam ina D 3 stymuluje aktyw ność sfingomielinazy działającej w zakresie obojętnego pH (nSM-aza), pow odując nagrom adzanie ceram idu w kom órkach HL-60, rozpoczęto intensywne badania nad m etabolizm em sfingomieliny jako potencjalną drogą przenoszenia sygnału w komórce. W krótce potem stwierdzono, że do czynników aktywujących nSM -azę należą również działające plejotropow o w o rganizmie cytokiny T N F -a, IN F-y i IL-1 [2]. W ażnym następnym etapem badań było wykazanie przez H a n n u n a i jego grupę [3], że krótkołańcuchow e, przechodzące przez błony kom órkow e ceramidy, wywołują wiele biologicznych efektów identycznych do pow odowanych aktyw acją nSM -azy, co sprawiło, że
POSTĘPY BIOCHEMII 42 (3 ) , 1996 299http://rcin.org.pl

ceram idy zostały uznane za w tórne przekaźniki sygnału. Najlepiej dotychczas scharakteryzow anym układem jest aktyw acja nSM -azy przez T N F -a , dokonana przez zespół K o l e s n i c k a [praca przeglądowa: 4], O statnie badania wiążące działanie T N F -a z aktyw acją błonowych kinaz białkowych i fosfatazy białkowej należącej do typu fosfataz P-2A niewątpliwie stawiają sfingomielinę i jej m etabolity w centrum zainteresow ania badaczy nowych dróg przekazywnia sygnałów w kom órce [praca przeglądow a 5].
W „Postępach Biochemii” ukazały się dotychczas dwa artykuły przeglądowe dotyczące roli sfingomieli- ny [6, 7] do k tórych kierujemy czytelników, traktując naszą pracę jako uzupełnienie wiadom ości o tej gwałtownie rozwijającej się dziedzinie badań.
II. Wpływ metabolitów sfingomieliny na różnicowanie i proliferację komórek
Analogicznie do różnych fosfolipaz, k tóre wytwarzają w ew nątrzkom órkow e w tórne przekaźniki będące m etabolitam i glicerofosfolipidów zaproponow ano, że sfingomielinaza działająca w obojętnym zakresie pH (nSM -aza) [fosfodiesteraza sfingomielinowa (EC 3.1.4.12)] może stanowić pierwszy etap w szlaku syg
nałowym (Ryc. 1) [9, 10], Enzym ten jest zlokalizow any wraz ze swym substratem na zewnętrznym listku błony plazmatycznej [11].
Stwierdzono, że hydroliza sfingomieliny m a znaczenie w różnicow aniu ludzkich prom ielocytów białacz- kowych HL-60 indukow anym przez witam inę D 3 [1, 12], czynnik m artwicy now otw orów (TN F-a) i inter- feron-y [2]. P onad to S p i e g e l i w s p . [13] w ykazali, że egzogenna sfingomielinaza stym ulow ała pro liferację mysich fibroblastów 3T3, zaś dodana razem z takim i czynnikam i wzrostowym i jak insulina, n a błonkow y czynnik w zrostu (EG F) lub bom bezyna, potęgow ała ich działanie. Traktow anie kom órek sfin- gom ielinazą pow odow ało znaczący spadek poziom u sfingomieliny, czemu towarzyszył wzrost poziom u ceram idu. Egzogenne, krótkołańcuchow e ceram idy stym ulowały włączanie radioaktyw nej tym idyny i działały synergistycznie z wyżej wspom nianym i czynnikam i wzrostu. Zatem ceram idy m ogą być zaan gażowane także w regulację proliferacji kom órek, a nie tylko jak o pośrednik w ich różnicowaniu. Z badań nad wpływem ceram idów i sfingozyny na proliferacje i wzrost ludzkich keratynocytów D JM -1 wynika, że syntetyczne krótkołańcuchow e ceram idy prom ow ały różnicowanie tych kom órek, podczas gdy sfingozyna
DE NOVO
PALMITOILO-Co A + SERYNA
Ryc. 1 Drogi metabolizmu sfingolipidów. N a podstawie prac przeglądowych [6-8], z uwzględnieniem następujących modyfikacji, odzwierciedlających obecne poglądy: (i) wprowadzenie podwójnego wiązania 4-trans odbywa się już po reakcji acylacji, zatem wolna sfingozyna nie jest pośrednikiem w syntezie sfingolipidów de novo; (ii) biosynteza sfingomieliny polega na przeniesieniu fosfocholiny z fosf- atydylocholiny na ceramid i uwolnieniu diacyloglicerolu; (iii) sfingozyna, która powstaje w wyniku hydrolizy ceramidu, może być przemieniana z powrotem do ceramidu lub degradowana, co obejmuje fosforylację do sfingozyno-l-P oraz lityczny rozpad do fosforanu etanoloaminy i trans-2-heksadekanalu. Zaznaczono wpływ czynników regulatorowych. Dla uproszczenia pominięto niektóre substraty, produkty, a także nazwy enzymów.
300 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

stym ulowała wzrost [14].Sfingozyna, wraz z innymi długołańcuchow ym i za
sadami: stearyloam iną, 3-ketosfinganiną i sfinganiną hamuje specyficznie kinazę białkow ą C (PKC) [15], kluczowy enzym zaangażow any we wzrost i transformację now otw orow ą kom órek. Działanie jej jest więc przeciwstawne działaniu diacyloglicerolu, k tóry jest aktyw atorem P K C [10, 16,17]. Z h a n g i w s p . [18] pokazali, że niskie stężenia sfingozyny < 20 pM stym ulują proliferację mysich fibroblastów spoczynkowych 3T3, a także, że sfingozyna potęguje odpowiedź kom órek na czynniki wzrostowe działające niezależnie od PK C. Działanie sfingozyny może polegać na m odulacji aktywności enzymów potencjalnie zaangażow anych w przenoszenie sygnałów: fosfolipazy D (PLD) [19, 20], fosfohydrolazy kwasu fosfatydowego [21], 80 kD izoenzymu kinazy diacyloglicerolowej [22] i kilku cytosolowych kinaz białkowych [23, 24].
Spośród m etabolitów sfingomieliny obok sfingozyny także N,N-dim etylosfingozyna, SPP i sfingozylo- fosfocholina m ają wpływ na proliferację kom órek drogą niezależną od PK C . N ,N-dim etylosfingozyna jest silniejszym niż sfingozyna inhibitorem kinazy białkowej C, a ponad to aktywuje kinazy tyrozyno we: kinazę src i kinazę receptora E G F [25, 26]. Sfin- gozyno-l-P jest silnym m itogenem dla mysich fibroblastów 3T3 [27]. W odpowiedzi na m itogenne stężenia sfingozyny poziom S PP w zrastał prawie dziesięciokrotnie. M im o że SP i SPP działały synergistycznie z insuliną i E G F , nie obserw owano synergizmu ani addytywności, gdy zastosow ano optym alne dawki SP (20 pM ) i SPP (5 pM), co wskazuje, że oba związki m odulują proliferację kom órek tą sam ą drogą. P o n ad to, kom petycyjny inhibitor kinazy sfingozynowej, DL- treo-dihydrosfingozyna [28], ham ow ał zarów no p o wstawanie SPP jak i syntezę D N A [29].
Sfingozylofosfocholina jest silnym stym ulatorem wzrostu wielu typów kom órek [30]. Jej działanie jest silniejsze niż insuliny czy E G F, lecz równie efektywne jak surowicy płodowej [31]. Z badań wynika, że SPC nie współzawodniczy z estram i forbolu o miejsce w iązania do P K C w fibroblastach 3T3, a działa synergistycznie z TPA stym ulując mitogenezę, co sugeruje, że SPC nie działa poprzez ham ow anie kinazy białkowej C. N ie m ożna wykluczyć jednak pewnej roli tego enzymu w mitogenezie indukowanej przez SPC, gdyż obserw ow ano wyraźny spadek syntezy D N A po zaham owaniu PK C. Zatem SPC może działać zarówno drogą zależną jak i niezależną od kinazy białkowej C.
II-1. Wpływ pochodnych sfingozyny na poziom kwasu fosfatydowego
Poszukując dróg, jakim i sfingozyna może indukować proliferację kom órek zw rócono uwagę na fakt, że jej wpływowi towarzyszyło dw ukrotne podniesienie poziom u kwasu fosfatydowego (PA) — silnego m ito- genu dla mysich fibroblastów 3T3 [20]. W zrost wew
nątrzkom órkow ego poziom u PA może być spow odowany stym ulacją fosfolipazy D, ham ow aniem fosfohydrolazy kwasu fosfatydowego, zwiększeniem acylacji glicerolo-3-P i podwyższeniem fosforylacji diacyloglicerolu katalizow anej przez kinazę diacyloglicerolo- wą. W ykazano, że sfingozyna stym uluje aktywność fosfolipazy D w wielu typach kom órek: N G 108-15, fibroblastach 3T3, fibroblastach N T H 3T3 i w kom órkach śródbłonka naczyń krw ionośnych płuc [19]. In vitro stw ierdzono ham ow anie fosfohydrolazy kwasu fosfatydowego przez sfingozynę [32]. W ludzkich kom órkach białaczkowych Ju rka t T bogatych w 80 kD izoenzym kinazy diacyloglicerolowej sfingozyna pow odow ała akum ulację PA [33].
W wielu typach kom órek kwas fosfatydowy wywołuje efekty podobne działaniu czynników wzrostowych: podnosi w artość pH cytoplazmy, indukuje ekspresję protoonkogenów c-fos i c-myc i pow oduje hydrolizę fosforanów fosfatydyloinozytolu [34], P o nadto działa synergistycznie z czynnikam i wzrostowymi: E G F, F G F lub cielęcą surowicą płodow ą oraz estram i forbolu [20]. U w aża się, że PA jest powiązany z działaniem białka Ras [35], k tóre pośredniczy we wczesnych etapach przenoszenia sygnałów stym ulujących wzrost kom órek [36]. W ykazano również, że P D G F i TPA stym ulują wytwarzanie PA w wyniku aktywacji fosfolipazy D [37]. Jest wysoce p raw dopodobne, że kwas fosfatydowy funkcjonuje jako wew nątrzkom órkow y w tórny przekaźnik w kaskadzie sygnałów m itogennych [38].
Działanie PA m a ważne implikacje dla biologicznej funkcji sfingozyny. W iadom o, że w fibroblastach 3T3 i innych typach kom órek PA ham uje nagrom adzanie się c-A M P i może zapoczątkow yw ać hydrolizę lipidów fosfatydyloinozytolowych, co pow oduje zwiększenie stężenia trisfosfoinozytolu (IP 3) [39]. Podobne skutki wywołują m itogenne (10 pM) stężenia sfinogozyny [20]. Sfinogozyna także indukuje uwalnianie w apnia z m agazynów w ew nątrzkom órkow ych w nienaruszonych fibroblastach naw et pod nieobecność wapnia zew nątrzkom órkow ego [40], co zostanie om ówione w dalszej części artykułu. Zarów no indukow anie uwalniania w apnia jak i proliferacja kom órek są specyficzne dla izom eru D-erytro-sfingozyny.
Sfingozylofosfocholina również stymuluje nagrom adzanie się kwasu fosfatydowego praw dopodobnie przez aktywacje fosfolipazy D [31]. Jednakże nie jest ono tak znaczące jak w przypadku SP i SPP i nie wydaje się żeby PA był zaangażow any w m echanizm działania SPC. W kom órkach traktow anych SPC nie obserwuje się zm iany ilości c-AM P, k tóry pod wpływem SP lub SPP obniża się drastycznie [20], co wskazuje, że ten klasyczny przekaźnik sygnałów nie m a wpływu na mitogenezę i mobilizacje w apnia wywoływaną działaniem SPC. Zaobserw ow ano natom iast, że m itogenne stężenia SPC pow odują uwalnianie kwasu arachidonow ego z fosfolipidów błon mysich fibroblastów 3T3. Kwas arachidonow y w obecności
POSTĘPY BIOCHEMII 42 (3 ) , 1996 301http://rcin.org.pl

w apnia aktywuje występujący w tych kom órkach izoenzym PKC-oc [41, 42]. D odanie sfinogozyny h a m owało zarów no stym ulowane przez SPC uwalnianie kwasu arachidonow ego jak i m itogenne działanie SPC, co potw ierdza rolę kwąsu arachidonow ego w tym procesie.
III. Rola metabolitów sfingomieliny w apop- tozie
Rozwój organizm ów w ielokom órkow ych jest regulowany przez procesy proliferacji i różnicow ania oraz program ow anej śmierci kom órek, zwanej apoptozą [praca przeglądowa: 43]. K om órki ulegają apoptozie w odpowiedzi na różnorodne bodźce fizjologiczne i farmakologiczne. N asza wiedza o sygnałach indukujących proces apoptozy w kom órkach jest ciągle niekom pletna.
Obserwacje, że w kom órkach białaczkowych T N F pow oduje fragm entację D N A prow adzącą do ap o p tozy, zwróciły uwagę na udział w tym procesie jednego z produktów hydrolizy sfingomieliny — ceram idu [44]. Syntetyczny ceram id C2 dodany do kom órek U937 białaczki m onoblastycznej (w jakich T N F pow oduje apoptozę) wywoływał internukleosom ałną fragmentację DNA , co jest uważane za wczesny objaw apoptozy. W kom órkach HL-60, U937, L929/LM i W EHI164/13, działanie sfingomielinazy i ceram idu było znoszone przez fosfolipazę C i syntetyczne diacy- loglicerole, co świadczy, że ceram id działa antagoni- stycznie do DAG. Stwierdzono, że fosfolipaza A2, kwas arachidonow y i fosfolipaza D nie powodowały uszkodzeń D N A [45] . Działanie cytotoksyczne ceram idu jest stereospecyficzne i ogranicza się do D- erytro-ceram idu i odpow iadających m u syntetycznych agonistów; dihydroceram id jest nieaktyw ny [46]. Przebieg reakcji cytotoksycznych indukow anych przez ceram id był analogiczny do wywołanych działaniem T N F w tych samych liniach kom órkow ych i w podobnych w arunkach. Q u i n t a n s i w s p . [47] opisali udział ceram idu w apoptozie kom órek W E- HI231 powodowanej przez przeciwciała, kortykoste- roidy i prom ieniow anie UV. Są też dow ody na rolę ceramidu w procesie apoptozy kom órek T indukowanej zakażeniem wirusem HIV [48]. Również egzogenny ceramid indukow ał apoptozę w tych kom órkach [49].
Pozbawienie kom órek białaczkowych M olt4 surowicy, pow odow ało zatrzym anie przebiegu cyklu ko m órkowego i apoptozę, czemu towarzyszył d ram atyczny i długotrw ały wzrost poziom u w ew nątrzkom órkowego ceram idu. Te same efekty pow odow ał egzogennie dodany ceramid. P raw dopodobnie w zatrzym aniu cyklu kom órkow ego na granicy faz G o /G l przez ceram id pośredniczy defosforylacja p roduk tu genu Rb (uważanego za ważny regulator cyklu kom órkowego), gdyż linie kom órkow e, którym brak Rb lub w których on nie funkcjonuje na skutek działania indukow anych adenowirusem białek wiążących Rb
[50], nie podlegają ham ow aniu przebiegu cyklu k o m órkowego wywoływanemu przez ceramid. Co więcej, estry forbolu lub D A G nie przeciwdziałają zależnemu od ceram idu ham ow aniu cyklu kom órkowego, n a to m iast zapobiegają apoptozie indukowanej ceramidem, co sugeruje występowanie różnych dróg przenoszenia sygnału przez ceram id w tych procesach. Możliwe, że to, czy proces apoptozy w kom órce będzie przebiegał zależy nie tyle od bezwzględnych stężeń ceram idu i diacyloglicerolu, ale od ich stosunku [51].
O statn io S a w a i i w s p . wykazali [52], że w ludzkich kom órkach białaczkowych HL-60 ceram id indukuje aktywacje c-jun/AP-1 we wczesnym etapie apoptozy. A utorzy udowodnili, że poziom m -RNA genu c-jun, poziom białka c-Jun i aktyw ność wiązania AP-1 do fragm entów D N A (wiążących czynnik AP-1 po stymulacji estram i forbolowymi) w zrastają pod wpływem ceram idu. K urkum ina (inhibitor aktywacji czynnika AP-1) znosiła zaham ow anie w zrostu kom órek i fragm entację DNA , które obserwow ano pod wpływem ceram idu. Podobnie działały nukleotydy antysensow ne dla c-jun. W yniki te wskazują, że w ko m órkach H L-60 czynnik transkrypcyjny AP-1 m a kluczowe znaczenie w przebiegu procesu apoptozy i że w ew nątrzkom órkow y przekaźnik sfingolipidowy — ceram id m oduluje przenoszenie sygnału indukującego apoptozę poprzez aktywację tego czynnika. Z k o lei B r a c h i w s p . [53] wykazali powiązanie ak tywacji szlaku sfingomielinowego przez T N F i n a grom adzania ceram idu z pojawieniem się aktywności wiążącej czynnika AP-1 w kom órkach.
D roga za pośrednictw em której ceram id powoduje aktywację czynnika transkrypcyjnego AP-1 nie jest do końca poznana. Spośród wielu kinaz serynow o/treoni- nowych, kinazy M A P są aktywow ane przez ceram id [54]. O statn io postuluje się istnienie grupy kinaz aktyw ow anych stresem (SAPK, JN K kinazy lub k inazy N -końca białka c-Jun) [55]. W łaśnie te kinazy m ogą stym ulow ać aktyw ność transkrypcyjną c-jun poprzez fosforylację N -końcow ych am inokwasów [56]. W ydaje się, że ceram id może pośredniczyć w przenoszeniu sygnału do indukcji apoptozy aktywując czynnik transkrypcyjny AP-1 za pośrednictw em JN K , ponieważ T N F -a i prom ieniow anie UV zwiększają poziom ceram idu i aktyw ują kinazy JN K . W yniki te pozw alają wnioskować, że ceram id pośredniczy w reakcjach stresowych w kom órkach.
Najnow sze badania wykazują udział aktyw ow anych przez ceram id kinaz [57,58] i fosfatazy białkowej [59, 60], k tóre m ogą pośredniczyć w apoptozie stym ulowanej przez ceram id (Ryc. 2). U dział C A PP został potw ierdzony w badaniach W o l f f a i w s p . [61], którzy wykazali, że kwas okadajow y (inhibitor CAPP) ham uje apoptozę indukow aną ceram idem i znosi ham ow anie ekspresji protoonkogenu c-myc.
Należy podkreślić, że pośredniki przenoszenia sygnału do apoptozy m ogą być różne zależnie od typu kom órek. W ludzkich neutrofilach traktow anych T N F
302 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

TNFR 55 Fas/APO-1
Ryc. 2 Udział metabolitów sfingomieliny w postulowanym mechaniźmie przenoszenia sygnału receptorów TN F i Fas/APO-1. Receptor TN F (TNF-R55), aktywując w błonie komórkowej sfingomielinazę (nSM-azę), powoduje uwolnienie ceramidu, który z kolei aktywuje specyficzną kinazę białkową (CAPK) inicjującą kaskadę przenoszenia sygnałów od kinazy Raf, przez kinazę M APK (MEK) lub kinazy aktywowane strestem (SAPK), do kinazy MAP (MAPK). M APK stymuluje aktywność fosfolipazy A2 (PLA2) oraz czynników transkrypcji AP-1 i NFkB, stymulujących proliferację i apoptozę. Ceramid indukuje również aktywność specyficznej fosfatazy białkowej (CAPP), uczestniczącej w procesie apoptozy. Białko TRADD, homologiczne do domeny śmierci TNF-R55, pośredniczy w apoptozie oraz aktywacji i translokacji NFkB. Jednocześnie TNF-R55 stymuluje aktywność lizosomalnej aSM-azy przez DAG uwolniony po aktywacji PC-PLC. Ceramid powstający w tym przedziale komórkowym jest odpowiedzialny za przenoszenie sygnału wyłącznie do apoptozy. Fas/APO-1 aktywuje również obie sfingomielinazy, ale potwierdzono tylko przenoszenie sygnału do apoptozy (generowanego analogicznie jak po aktywacji TNF-R55), natom iast nie stwierdzono indukcji ekspresji genów.
[62] sfingozyna indukow ała apoptozę. C eram id i SPP nie działały w tych kom órkach. Dimetylosfingozyna i H-7 (inhibitor PK C ) także indukow ały apoptozę co sugeruje, że zjawisko to m oże być związane z ham ow aniem P K C przez sfinogozynę. W niestym ulow anych ludzkich neutrofilach stężenie ceram idu jest bardzo wysokie (ok. 200 pM ) co poddaje w wątpliwość znaczenie egzogennie dodaw anego ceram idu. W ynika z tego, że w tych kom órkach endogennym m odu lato rem pośredniczącym wywoływaniu apoptozy przez T N F może być sfingozyna.
IY. Wpływ sfingozyno-l-P na ruchliwość komórek nowotworowych
Ze względu na fakt, że S PP podobnie jak czynniki wzrostu, zwiększa poziom w apnia i kwasu fosfatydo- wego w kom órkach, zbadano wpływ SPP na wzrost i różnicowanie dwóch linii kom órkow ych raka piersi: M C F-7 i M DA-M B-231 [63]. K om órki M C F-7 m ają funkcjonujące receptory estrogenowe i in
vitro ich wzrost zależy od estrogenu, zaś kom órki M DA-M B-231 nie m ają takich receptorów i są oporne na działanie antyestrogenów [64]. SPP w stężeniu 1-10 pM ham ow ał wzrost kom órek obu linii. Pod wpływem SPP kom órki M DA-M B-231 zmieniały fenotyp na bardziej prawidłow y i charakteryzow ały się zmniejszoną znacznie inwazyjnością [63]. Sfingozyna nie m iała wpływu na te cechy. W yniki badań nad wpływem S PP na ruchliwość chem otaktyczną i in- wazyjność kilku linii kom órkow ych, mysiego i ludzkiego czerniaka oraz ludzkiego kostniakom ięsaka do w odzą ham ującego wpływu S PP na inwazyjność ko m órek nowotworow ych. Z drugiej strony, SPP nie wpływał na ruchliwość ludzkich kom órek śródbłonka i w łókniakom ięsaka [65]. Zreferowane wyżej wyniki badań nad pozytywnym oddziaływaniem SPP z kom órkam i niektórych linii now otw orow ych sugerują, że ta nietoksyczna ufosforylowana pochodna sfinogozy- ny może zmniejszać inwazyjność now otw orów . Takie działanie pośrednika sfingomielinowego szlaku przesyłania sygnału może otwierać nowe możliwości tera-
POSTĘPY BIOCHEMII 42 (3 ) , 1996 303http://rcin.org.pl

pii przeciwno wotworowej.
V. Regulacja homeostazy wapniowej w komórce przez metabolity sfingomieliny
Regulacja mobilizacji w ew nątrzkom órkow ego C a2+ należy niewątpliwie do istotnych oddziaływań m etabolitów sfingomieliny, k tóre m ogą mieć znaczenie dla procesów fizjologicznych i patologicznych [8]. Liczne badania przeprow adzone na różnych m odelach: izolowanych kom órkach, uprzepuszczalnianych kom órkach i izolowanych pęcherzykach retikulum endoplazm atycznego wskazywały, że sfingozyna może działać na mobilizację jonów C a2+ zarów no przez aktywację fosfolipazy C i hydrolizę fosforanów fos- fatydyloinozytolu [66] jak i bez włączania tego m echanizm u [67, 68].
Najwcześniej ustalono rolę egzogennej sfingozyny w uw alnianiu jonów w apnia z wewnętrzkom órkowej puli zarów no zależnej jak i niezależnej od IP 3 [praca przeglądowa: 69] oraz w ham ow aniu napływu C a2 + przez błonę kom órkow ą [70]. Działanie to może być realizowane poprzez stymulację m etabolizm u fosforanów fosfatydyloinozytolu i nagrom adzanie IP 3 [60] lub przez ufosforylowany produk t sfingozyny, SPP, jak to stw ierdzono w hodow li kom órek mięśni gładkich uprzepuszczalnianych saponiną [40]. Ta ostatn ia droga jest niezależna od hydrolizy fosforanów fosfatydyloinozytolu i uw alniania kw asu arachidonow ego [66, 71], jakkolw iek kilku badaczy uzyskało dane, że egzogenny S PP może pow odow ać produkcję I P 3 [72]. Być może SPP działa bezpośrednio na kanały w apniowe pozostające pod kon tro lą receptorów rianidy- nowych lub IP 3 [71].
Badacze z grupy S a b b a d i n i e g o wykazali, że w stężeniu subm ikrom olarnym sfingozyna ham uje uwalnianie C a2+ z błon retikulum sarkoplazm atycz- nego (SR) mięśni szkieletowych w wyniku oddziaływania na receptor rianidynowy, zmniejszając jego powinow actw a do rianidyny, bez udziału fosforylacji tego receptora. Sfingozyna pow odow ała również wyraźne ham ow anie uw alniania w apnia pod wpływem fizjologicznych czynników stymulujących. W wyższych stężeniach zarów no sfingozyna jak i SPC indukow ały bezpośrednio uwalnianie w apnia z SR [73]. Jest m ożliwe, że sfingozyna stanowi fizjologiczny regulator poziom u w apnia w sercu.
Również SPC jest silnie działającym czynnikiem uwalniającym jony w apnia z w ew nątrzkom órkow ej puli zależnej od IP 3 [40]. W kom órkach Swiss 3T3 D e s a i i w s p . [31] nie stwierdzili akum ulacji inozytolofosforanów po stymulacji przez SPC. Inni autorzy jednak wykazali [72], że w kom órkach HL-60 SPC w dawce poniżej 30 pM aktywuje fosfolipazę C, pow odując następnie mobilizację C a2 + . Sfingozyno- 1-P działa praw dopodobnie poprzez niezidentyfikowany jeszcze receptor sprzężony z białkiem lub białkam i G ham ow anym i toksyną ksztuśca.
W ydaje się, że drogi uw alniania w ew nątrzkom órkowego C a2+ przez sfingozynę, SPP i SPC są różne, zależnie od rodzaju kom órek i stopnia ich zróżnicow ania. O k a j i m a i K o n d o [72] wysuwają hipotezę, że tylko w niezróżnicowanych kom órkach SPC działa poprzez aktywację fosfolipazy C. Należy zaznaczyć, że jakkolw iek SPC może być produkow ana w pewnych w arunkach experym entalnych oraz w patologii np. w śledzionie chorych z syndrom em N iem ann-Picka oraz w kom órkach now otw orow ych [31], nie potw ierdzono nagrom adzania tego związku w w arunkach fizjologicznych.
Należy zaznaczyć, że nie znaleziono wpływu ceram idu ani na uwalnianie w apnia ani na akum ulację IP 3 w kom órkach [66]. N atom iast stw ierdzono, że cera- m id oraz SPP i SPC ham ują wzrost w ew nątrzkom órkowego [C a2 + ] i wywoływanego aktyw acją receptora E G F w ludzkich kom órkach A431, co sugeruje, że odpowiedź na sygnał E G F może być m odulow ana przez sfingolipidowe w tórne przekaźniki sygnału [74].
M echanizm uw alniania C a2+ z puli w ew nątrzkom órkowej przez sfingolipidy nie jest jeszcze wyjaśniony. W yniki O k a j i m a i K o n d o [72] sugerują, że SPC powoduje wzrost cytosolowego C a2+ na drodze m echanizm u włączającego aktywację PL C poprzez receptor sprzężony z wrażliwym na toksynę ksztuśca białkiem G. O statn io K i n d m a n i w s p . [69] opisali nowy rodzaj kanału wapniowego zależnego od sfingolipidów i zlokalizow anego w endoplaz- m atycznym retikulum kom órek bazocytowej białaczki szczura oraz w ludzkich kom órkach śródbłonkow ych [75, 76].
VI. Rola metabolitów sfingomieliny w przenoszeniu sygnałów cytokin i czynników wzrostu w komórkach
Isto tne dla rozwoju badań nad sfingomieliną prace O k a z a k i e g o i w s p . [1, 12] wykazały, że różnicowanie kom órek ludzkiej białaczki prom ielocyto- wej H L-60 pod wpływem 1-a, 25-dihydroksywitam iny D 3 zachodzi ze wzrostem poziom u ceram idu i SPC w wyniku stymulacji nSM -azy. K im i w s p . przebadali następnie wpływ innych czynników indukujących różnicowanie kom órek HL-60, co doprow adziło do odkrycia, że m etabolizm sfingomieliny może być w ażnym m echanizm em pośredniczącym w przekazywaniu sygnału T N F -a i IN F-y w procesie różnicow ania kom órek. Procesowi tem u towarzyszyło ham ow anie ekspresji m R N A dla protoonkogenu c-myc.
O statn ie badan ia dostarczają coraz więcej danych dotyczących wyjaśnienia m echanizm u przenoszenia sygnału cytokin w kom órkach docelowych. Dwie cytokiny IL-1 i T N F -a , wykazujące plejotropow e działanie o podobnym i nakładającym się zakresie oddziaływań biologicznych, pow odują szybkie i przejściowe tworzenie dwu w tórnych lipidowych przekaźników sygnałów: diacyloglicerolu produkow anego
304 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

przez specyficzną dla fosfatydylocholiny fosfolipazę C (PC-PLC) i ceram idu, tw orzonego przez dwie różne sfingomielinazy: nSM -azę, zlokalizow aną w błonie komórkowej i aSM -azę, zlokalizow aną w lizosom ach i endosom ach [p raca przeglądowa: 77].
Do najlepiej poznanych należy niewątpliwie udział sfingomielinaz i ich m etabolitów w przenoszeniu sygnału przez receptory należące do grupy receptorów czynnika martwicy now otw orów (TNF). Schem at tych reakcji jest pokazany na rycinie 2.
Czynnik m artw icy now otw orów indukuje wzrost i różnicowanie wielu rodzajów kom órek oraz jest ważnym parakrynnym i endokrynnym m odulatorem reakcji zapalnych i im munologicznych. Pow oduje również nekrozę pewnych typów now otw orów i jest cytotoksyczny w sposób synerergistyczny z IN F -y dla wielu kom órek now otw orow ych in vitro. Jest też jednym z czynników wywołujących apoptozę [prace przeglądowe: 78, 79]. P lejotropow e działanie T N F w procesach im m unologicznych i zapalnych sprawia, że jest on ważnym czynnikiem w patogenezie wielu chorób. Poznanie m echanizm ów przekazyw ania sygnałów przez tę cytokinę jest więc istotne nie tylko ze względów poznawczych, ale także jako istotny element terapii chorób autoim m unologicznych i raka.
VI-1. Receptory dla TNF
Receptory dla T N F występują w błonach kom órkowych prawie wszystkich jądrzastych kom órek. Scharakteryzow ano i sklonow ano dwa różne typy receptorów dla T N F [prace przeglądowe: 79, 80]. Receptory TN F-R 55 i TN F-R 75 różnią się m asą cząsteczkową i stałą dysocjacji dla T N F, której w arto ści wynoszą odpow iednio 500 pM i 100 pM . Ekspresja genów kodujących te receptory jest w różny sposób regulow ana i niska konstytucyjna ekspresja TN F-R 55 i indukow ana ekspresja TN F-R 75 prow adzą do zróżnicowanych odpowiedzi fizjologicznych. Biologicznie czynną form ą T N F są jego trimery. Aktywacja obu receptorów zachodzi poprzez ich oligomeryzację po przyłączeniu hom otrim eru T N F -a lub T N F -(3 (lim- fotoksyny-a). O bie cytokiny aktyw ują obydw a typy receptorów T N F, po czym następuje szybka in ternalizacja i degradacja tych kom pleksów w endosomach. TN F-R 75 wykazuje podobną aktywność biologiczną jak TN F-R 55, zwłaszcza w kom órkach T i k o m órkach ze zwiększoną ekspresją białka p75, ale w większości kom órek ro la TN F-R 75 polega na wiązaniu T N F i przekazyw aniu tego ligandu na recepto r TN F-R55, charakteryzujący się mniejszym pow inowactwem do ligandu. Stw ierdzono występowanie specyficznych regionów receptorów T N F odpow iedzialnych za wytwarzanie różnych sygnałów. Region błonowy receptora pow oduje aktywację nSM -azy p ro wadzącą do aktywacji „ukierunkow anych p ro liną”, serynow o/treoninow ych kinaz białkowych (P D P — proline directed protein kinases), pow odujących
aktywację kinazy Raf, fosfolipazy A2 i czynnika transkrypcji AP-1 [81, 82]. Z kolei odcinek około 80 am inokw asów końca C receptora, tzw. dom ena sygnału śmierci, jest odpowiedzialny za sygnał prow adzący do cytotoksyczności i apoptozy [83, 84], stymulacji syntezy N O i praw dopodobnie do aktywacji aSM -azy i czynnika transkrypcji N F kB [81].
O statnio opisano białka, nazwane FA D D i TR A D D zawierające dom eny homologiczne wobec dom en śmierci w receptorach odpowiednio Fas/A PO -1 i TNF-R55.
Zwiększenie ekspresji T R A D D indukuje apoptozę i aktywuje N F kB, c o sprawia, że białko to jest uważane za ogniwo sygnału TN F-R 55 dla obu tych procesów [praca przeglądowa: 85].
VI-2. Receptor Fas/APO-1
Błonowy receptor Fas/A PO -1 należy do rodziny receptorów T N F i posiada hom ologiczny region w części zawierającej dom enę śmierci, analogicznie do T N F- R55, k tóry generuje sygnał do apoptozy [83]. Receptor Fas/A PO -1 jest jednym z najistotniejszych czynników regulacji czynności układu im munologicznego, ponieważ umożliwia on ograniczenie w zrostu klonów i n a grom adzania limfocytów po wniknięciu antygenu do narządów limfatycznych. Jest on również odpowiedzialny za procesy eliminacji, przez kom órki w yposażone w ligandy dla tego receptora, zaktywowanych i proliferujących limfocytów obwodowych, a także kom órek, k tóre uległy transform acji nowotworowej lub infekcji wirusowej [86]. Podobnie jak w przypadku TN F-R55, aktyw acja F as/A P O -l-R przez ligandy m oże również prowadzić do aktywacji kom órek [87].
VI-3. Różne drogi przenoszenia sygnału pochodzącego od nSM-azy i aSM-azy
W yróżnienie regionów TN F-R 55 odpowiedzialnych za aktywację nSM -azy i aSM -azy i różna lokalizacja tych enzymów w kom órce sugeruje różne funkcje tych dwu enzymów w przenoszeniu sygnału TN F-R 55, co znalazło potw ierdzenia w badaniach W i e g m a n n a i w s p. [81]. Zgrom adzone obecnie dane pozwoliły na zaproponow anie sekwencji reakcji zilustrow anych na rycinie 2.
Po aktywacji nSM -azy powstający ceram id aktyw uje zw iązaną z błonam i specyficzną kinazę białkową (CAPK), k tó ra fosforyluje receptor nabłonkow ego czynnika w zrostu (EGF-R) w kom órkach A431 [88]. W zrost aktywności tej kinazy obserwowano również w pierwszych m inutach po podziałaniu T N F -a na kom órki HL-60. Szybka aktywacja, zdolność analogów ceram idu do wywoływania efektów ligandu oraz rekonstytucja kaskady reakcji w układzie bezkom ór- kowym pozwoliły uznać ten szlak sfingomielinowy za isto tną drogę przenoszenia sygnału pochodzącego od T N F [4]. C A PK należy do grupy kinaz P D P , które
POSTĘPY BIOCHEMII 42 (3 ) , 1996 305http://rcin.org.pl

fosforylują fragm ent X -Ser/Tre-Pro-X substratu. D o grupy tej należą również kinazy M A P (M APK) i k inazy cdc2. C A PK wymaga do fosforylacji najmniejszego fragm entu zawierającego Leu-Tre-Pro [89, 90].
Równocześnie wykazano, że T N F -a aktywuje k inazę M A P w fibroblastach [91], a w kom órkach HL-60 traktow anych sfingomieliną lub ceram idem dochodzi do fosforylacji i aktywacji kinazy p42 M A P [54]. K inazy M A P są kinazam i serynowo/treoninowym i, k tóre odgryw ają ważną rolę w przenoszeniu sygnału m itogennego pochodzącego od wielu receptorów stym ulow anych przez różne ligandy, włączając w to kinazy tyrozynowe receptorów i receptory sprzężone z białkiem G. Kinazy M AP, do których należą najlepiej poznane 42 k D a E R K I czyli p42 M A PK i 44 kD a ER K 2 czyli p44 M A PK , są aktywow ane na skutek fosforylacji reszt tyrozyny i treoniny przez specyficzne kinazy M A PK : M EK1 i M EK2. Z kolei kinazy M E K są regulow ane przez fosforylację seryny w ich cząsteczce przez kinazy kinazy M E K i kinazę Raf. Zróżnicow anie kinaz kinazy M E K uważa się za główny czynnik umożliwiający zróżnicowanie dróg sygnałowych prow adzących do M A PK [92-94].
W następnych badaniach stwierdzono, że stym ulacja kom órek HL-60 przez T N F pow oduje w przeciągu sekund fosforylację kinazy Raf, kinazy serynow o/treo- ninowej, k tó ra bierze udział w przenoszeniu sygnału m itogennych cytokin, co sugeruje, że R af może pośredniczyć w przenoszeniu sygnału generowanego przez tę cytokinę [82]. P odobny efekt uzyskano działając na kom órki ceram idem lub sfingomielinazą. G rupa K o - l e ś n i c k a wykazała, że C A PK tworzy kom pleks z R af i fosforyluje tą kinazę powodując wzrost jej aktywności wobec M E K kinazy w kom órkach HL-60. W ten sposób droga przenoszenia sygnału sfingomieli- nowego została pow iązana ze szlakiem M A PK [95]. B e l k a i w s p. [82] uzyskali wyniki sugerujące, że nSM -aza, a nie aSM -aza, bierze udział w wywoływanej przez T N F -a fosforylacji i aktywacji kinazy Raf.
K inaza M A P z kolei fosforyluje i aktywuje cyto- solową fosfolipazę A2 [96] w odpowiedzi na ceramid utw orzony przez nSM -azę [81]. Aktywacja PL A 2 wydaje się być odpow iedzialna za nagrom adzanie kwasu arachidonow ego i jego m etabolitów , leuko- trienów i prostaglandyn, pow odujących reakcje zapalne w wyniku działania TN F.
B adania in vitro wpływu ceram idu na fosforylację białek doprow adziły do wykrycia serynow o/treonino- wej fosfatazy białkowej (CAPP) aktywowanej bezpośrednio i w specyficzny sposób przez ceram id [59]. Fosfataza ta należy do grupy fofataz PP2A w skład których wchodzą trzy podjednostki: A — o niezidentyfikowanej funkcji, B — regulatorowej i C — katalitycznej, m ogących występować w formie m ono-di-, lub trim eru. Ceram id aktyw uje wyłącznie formę trim eru działając na podjednostkę B. C A PP jest ham ow ana przez niskie stężenia kwasu okadaj owego, specyficznego inh ib itora tego typu fosfataz [60]. N anom olarne
stężenia kwasu okadaj owego pow odują zaham ow anie wywoływanej przez ceram id apoptozy i ekspresji c- myc w kom órkach HL-60, podobnie jak to obserwowano po trak tow aniu T N F -a , co potw ierdza rolę C A PP w tych procesach [61].
M etabolity sfingomieliny b iorą również udział w przenoszeniu sygnału do aktywacji i translokacji czynnika transkrypcji N F kB. K r o n k e i w s p . [97] wykazali, że egzogenna sfingomielinaza i ceram id pow oduje degradację I-kB, fizjologicznego inh ibitora N F kB i aktywację tego czynnika transkrypcji. Nie jest jasne czy aktywacja kinazy R af wystarcza do aktywacji i translokacji N F kB jak to sugerowały badania F i n c o i B a l d w i n a [98]. O statn io uzyskano dane świadczące o tym, że ceram id nagrom adzony w kom órkach w wyniku aktywacji aSM -azy przez DAG, utw orzony przez zaktyw ow aną PC -PLC , jest odpowiedzialny za aktywację N F kB. Aktywacja aSM - azy jest zależna od tej samej dom eny receptora, k tó ra jest odpow iedzialna za aktywację P C -PL C [81]. A utorzy ci sugerują, że aktyw acja aSM -azy zachodzi w en- dosom ach, w których znajduje się D A G utw orzony po aktywacji P C -PL C i internalizow any razem z T N F - R55.
C oraz więcej danych dotyczy w ystępowania nowej drogi przenoszenia sygnału, k tó ra jest specyficznie w łączana w odpowiedzi na stres np. pod wpływem T N F lub uszkodzenie tkanek w wyniku hipoksji, prom ieniow ania UY czy chemioterapii. Czynniki te aktyw ują zarów no nSM -azę i aSM -azę jak i aktyw owane stresem kinazy białkowe (SAPK, JNK). W e s t - w i e k i w s p . [99, 100] wykazali aktywację SAPK przez T N F, co pow oduje stymulację zależnej od AP-1 transkrypcji genów i indukcję translokacji N F kB do jąd ra i transkrypcji genów zależnych od tego czynnika. Potencjalnym pośrednikiem tych procesów jest ceramid pow stający w wyniku aktywacji sfinogomielinaz. S tw ierdzono natom iast tylko niewielką stymulację aktywności M A PK przez T N F -a i ham ow anie tych kinaz przez ceram id [100].
Stosunkow o mniej danych dotyczy udziału sfin- golipidów w przenoszeniu sygnału Fas/A PO -1. C i - f o n e i w s p . [101] wykazali aktywację nSM -azy, aSM -azy, PL A 2 i PC PLC po stymulacji Fas/A PO -1 i udział ceram idu w inicjacji apoptozy przez ten receptor. U zyskano również dane o aktywacji kinazy M A P i udziale kinazy R af w tym procesie [101]. N atom iast nie udało się stwierdzić aktywacji N F kB przez sygnał Fas/A PO -1 [102]. W ydaje się, że sygnał receptora Fas/A PO -1 do apoptozy przenoszony przez ceram id nie wym aga i nie jest zależny od ekspresji genów [103].
VI-4. Receptor PDGF
B adania przeprow adzone przez K e s t e r a i w s p . [104] wskazują na powiązanie sfingozyny z procesem mitozy niezależnym od kinazy białkowej C. N a m odelu
306 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

in vitro kom órek m ezangialnych oraz w układzie bezkom órkowym wykazano, że płytkowy czynnik wzrostu stymuluje tworzenie m itogennych m etabolitów sfingolipidów poprzez aktywację ceram idazy działającej w zasadow ym zakresie pH , degradującej cera- midy, oraz sfingomielinazy dostarczającej ceram idu, podczas gdy cytokiny IL-1 i T N F -a aktyw ują sfin- gomielinazę pozostając bez wpływu na ceram idazę [104]. A utorom tym udało się uzyskać oddzielenie zależnej od czynnika w zrostu m itogenezy od pow odowanych przez cytokiny różnicow ania i apoptozy k o mórek, zależnie od rodzaju m etabolitu sfingolipidowe- go nagrom adzanego w wyniku specyficznej aktywacji odpowiednich enzymów szlaku sfingomielinowego.
W yniki O l i v e r a i S p i e g e l [105] pokazały, że P D G F powoduje gwałtowny i przejściowy wzrost poziom u sfingozyny i SPP w fibroblastach Swiss 3T3, podczas gdy nie obserw ow ano tych zm ian w obecności EG F. A utorki stwierdziły przejściowy wzrost aktyw ności cytosolowej kinazy sfingozynowej, co w konsekwencji reguluje w ew nątrzkom órkow y poziom SPP i potw ierdza rolę tego czynnika w przenoszeniu sygnału P D G F (Ryc. 4). Również w kom órkach mięśni gładkich naczyń P D G F podnosi poziom sfinogozyny przy równoczesnym obniżeniu w ew nątrzkom órkow ego poziom u ceramidu. P onad to L-cykloseryna, inhibitor syntezy sfingolipidów, a także D L-treo-dihyd- rosfingozyna, inhibitor kinazy sfingozynowej ham ują stym ulow aną przez P D G F syntezę DNA, pozostając bez wpływu na prolifercję kom órek wywoływaną E G F [106]. Z badań S u i w s p . [107] wynika, że SPP indukuje syntezę D N A i podziały kom órkow e przez aktywację czynnika transkrypcyjnego AP-1. Inhib itor kinazy sfingozynowej (DL-treo-dihydrosfingozyna) znosił zdolność wiązania AP-1 do DNA. S PP pow oduje również gwałtowny wzrost stężenia kw asu fosfa- tydowego oraz wzrost stężenia w ew nątrzkom órkow ego w apnia [C a2+]¡, jak to zostało opisane wcześniej. Kwas fosfatydowy jest ważnym aktyw atorem Ras [108] i kaskady kinaz M AP, a także stym uluje zdolność AP-1 do wiązania D N A [107]. P roponow any przez S p i e g e l i w s p. [8] schem at udziału m etabo- liów sfingomieliny w przenoszeniu sygnału P D G F jest pokazany na rycinie 3.
O statn io H u d s o n i w s p . [74] wykazali, że w ludzkich kom órkach raka naskórka A431 sfin- gozyna nie m a wpływu na w ew nątrzkom órkow e stężenie wolnego w apnia [C a2+] ; natom iast znacznie stymuluje napływ w apnia stym ulow any przez EG F. T owarzyszy tem u fosforylacja reszt serynowych dwu niezidentyfikowanych białek, k tóre praw dopodobnie są włączone w przenoszenie sygnału receptora EG F. Inni badacze stwierdzili, że ceram id stym uluje fosforylację receptora E G F w tych samych kom órkach, co sugeruje udział ceram idu w tym procesie [109]. B adania K e s t e r a i w s p . [104] sugerują, że P D G F stym uluje nSM -azę w m ezangialnych kom órkach kłębkowych nerek szczura poprzez fosforylację reszt
PDGF
Ryc. 3 Udział metabolitów sfingomieliny w przenoszeniu sygnału indukowanego aktywacją receptora PD GF. Przyłączenie PD G F do receptora powoduje fosforylację tyrozyny białka receptorowego, co umożliwia przyłączenie białek adapterowych Grb2, fosfolipazy C.f (PLCy) i białka aktywującego GTP-azę (GAP). Wiązanie Grb2 prowadzi do aktywacji układu Ras i rozpoczyna kaskadę kinaz białkowych od kinazy Raf do MAPK, powodując aktywację czynnika transkrypcyjnego AP-1. Jednocześnie PD G F-R aktywuje fosfolipazę D (PLD), kinazę sfingozynową (SPK), która katalizuje powstanie sflngozyno-l-P (SPP). SPP również aktywuje PLD, powodując nagromadzenie kwasu fosfaty- dowego (PA), prowadzące do wzrostu cytosolowego stężenia wapnia, niezależnie od inozytolotrisfosforanu (IP3) gromadzonego w wyniku aktywacji PLCr SPP hamuje także polimeryzację aktyny w komórkach nowotworowych.
tyrozynowych. Obserwacja ta stanowi punkt wyjścia do spekulacji na tem at różnorodnych odpowiedzi kom órek na działanie czynników w zrostu i cytokin.
VII. Uwagi końcowe
Podsum ow ując, m ożna stwierdzić, że sfingozyna, SPP i ceram idy posiadają właściwości predestynujące je do roli w tórnych pośredników przekazyw ania sygnału pom iędzy błoną kom órkow ą, w której są zlokalizowane receptory czynników w zrostu i cytokin, pulą w ew nątrzkom órkow ego wapnia, kaskadam i kinaz i fosfataz białkowych i czynnikam i regulującymi ekspresję genów w procesach prow adzących do podziałów, różnicow ania i apoptozy kom órek. M etabolity sfingomieliny wywołują zróżnicowane odpowiedzi kom órkowe, występują w kom órce w niskich stężeniach, które po stymulacji ulegają gwałtownem u i przejś
POSTĘPY BIOCHEMII 42 (3 ) , 1996 307http://rcin.org.pl

ciowemu podwyższeniu pow odując uwalnianie w apnia z puli wewnątrzkom órkowej przez m echanizm niezależny od IP 3, aktywację P D P kinaz i specyficznej fosfatazy oraz czynników transkrypcji. M ogą one rów nież wzmacniać lub osłabiać kaskadę reakcji poprzez efekt pozytywnej lub negatywnej kontroli zwrotnej.
A rtyku ł otrzymano 25 kwietnia 1996 r. Zaakceptowano do druku 16 maja 1996 r.
Piśmiennictwo
1. O k a z a k i T, B e l l R M, H a n n u n Y A (1989) J Biol Chem 264: 19076-19080
2. K im M Y , L i n a r d i c C, O b e i d L, H a n n u n Y (1991) J Biol Chem 266: 484-489
3. H a n n u n Y A (1994) J Biol Chem 269: 3125-31384. K o l e s n i c k RN, G o l d e D W (1994) Cell 77: 325-3285. B e y a e r t R, F i e r s W (1994) FEBS Lett 340: 9-166 . K w i a t k o w s k a J (1994) Post Biochem 40: 130-1347. R a k o w s k a M, Z b o r o w s k i J (1992) Post Biol Kom 19:
369-3848. S p i e g e l S, M i l s t i e n S (1995) J Membrane Biol 146:
225-2379. M a t h i a s S K o l e s n i c k R N (1993) Adv Lipid Res 25:
65-9010. H a n n u n NY, B e l l R M (1989) Science 243: 500-50711. K o v a l M, P a g a n o R M (1991) Biochim Biophys Acta 1082:
113-12512. O k a z a k i T, B i e l a w s k a A, B e l l R M, H a n n u n YA
(1990) J Biol Chem 265: 15823-1583113. O l i v e r a A, B u c k l e y NE, S p i e g e l S (1992) J Biol Chem
267: 26121-2612714. W a k i t a H, T o g u r a Y, Y a g i H, N i s h i m u r a K,
F u r u k a w a F, T a g i k a w a M (1994) Arch Dermatol Res 286: 350-354
15. H a n n u n YA, L o o m i s CR, M e r r i l A H Jr , B e l l R M (1986) J Biol Chem 261: 12604-12609
16. M e r r i l l A H (1991) J Bioenerg Biomembr 23: 83-10417. H a n n u n YA, B e l l R M (1987) Science 235: 670-67418. Z h a n g H, B u c k l e y NE , G i b s o n K, S p i e g e l S (1990)
J Biol Chem 265: 78-8119. D e s a i N N , Z h a n g H, O l i v e r a A, M a t t i e ME,
S p i e g e l S (1992) J Biol Chem 267: 23122-2312820. Z h a n g H, D e s a i N N , M u r p h e y J M, S p i e g e l
S (1990) J Biol Chem 265: 21309-2131621. J a m a l Z, M a r t i n A, M u n o z A G, B r i n d l e y D N
(1991) J Biol Chem 266: 2988-299622. Y a m a d a K, S a k a n e F, I m a i S- I , T a k e m u r a
H (1993) Biochim Biophys Acta 1169: 217-22423. A r n o l d RS, N e w t o n A C (1991) Biochemistry 30: 7747-
775424. P u s h k a v e r a MY, K h a n WA, A l e s s e n k o A V, S a -
h y o u n N, H a n n u n YA (1992) J Biol Chem 267: 15246- 15251
25. I g a r a s h i Y, H a k a m o r i S, T o y o k u n i T, D e a n B, F u j i t a S, S u i m o t o M, O g a w a T, E l G h e n d y , R a c k e r E (1989) Biochemistry 28: 6796-6800
26. I g a r a s h i Y, K i t a m u r a K, T o y o k u n i T, D e a n B, F e n d e r s o n B, O g a w a T, H a k a m o r i S (1990) J Biol Chem 265: 5385-5389
27. Z h a n g H, D e s a i N N , O l i v e r a A, S e k i T, B r o o k e r G, S p i e g e l S (1991) J Cell Biol 114: 155-167
28. B u e h r e r BM, B e l l R M (1992) J Biol Chem 267: 3154- 3159
29. O l i v e r a A, S p i e g e l S (1993) Nature (Lond) 365: 557-56030. D e s a i N N , S p i e g e l S (1991) Biochem Biophys Res Comm
181: 361-36631. D e s a i N N , C a r l s o n R O , M a t t i e M E , O l i v e r a A (19
Buckley NE, S e k i T, B r o o k e r G, S p i e g e l S (1993) J Cell Biol 121: 1385-1395
32. L a v i e Y, P i t e r m a n O, L i s c o v i t c h M (1990) FEBS Lett 277: 7-10
33. S a k a n e F, Y a m a d a K, K a n o c h H (1989) FEBS Lett 255: 409-413
34. M u r a y a m a T, U i M (1987) J Biol Chem 262: 5522-552935. Y u C l, T s a i MH , S t a c e y D W (1988) Cell 52: 63-1136. B a r b a c i d M (1981) Annu Rev Biochem 56: 119-82137. B e n - A v P, L i s c o v i t c h M (1989) FEBS Lett 259: 64-6638. E x t o n J H (1990) J Biol Chem 165: 1-439. M u r a y a m a T, U i M (1992) J Biol Chem 267: 12463-1246740. G h o s h T K, B i a n J, G i 11 D L (1990) Science (WASH. DC)
248: 1653-165641. M c C a f f r e y P G, R o s n e r M R (1987) Biochem Biophys
Res Comm 146: 140-14642. N i z h i z u k a Y (1992) Science 256: 607-61443. S i k o r a E (1994) Post Biochem 40: 150-16044. H a n n u n YA, O b e i d L M (1995) TIBS 20: 73-7745. J a r v i s WD , K o l e s n i c k RN, F o r n a r i FA, T a y l o r
RS, G e w i r t z DA, G r a n t S (1994) Proc N atl Acad Sei 71: 73-77
46. B i e l a w s k a A, C r a n e H M , L i o t t a D, O b e i d L M, H a n n u n Y A (1993) J Biol Chem 268: 26226-26232
47. Q u i n t a n s J, K i l k u s J, M c S h a n CL, G o t t s c h a l k AR, D a w s o n G (1994) Biochem Biophys Res Commun 202: 710-714
48. V a n Y e l d h o v e n PP , M a t t h e w s TJ , B o l o g n e s i D P, B e 11 R M (1992) Biochem Biophys Res Commun 187: 209-216
49. O b e i d L M i H a n n u n Y A (1995) J Cell Biochem 58: 191-198
50. D b a i b o GS, P u s h k a r e v a MY, J a y a d e v S, S c h w a r z J K, H o r o w i t z J M, O b e i d L M, H a n n u n Y A (1995) Proc Natl Acad Sei USA 92: 1347-1351
51. O b e i d L M, L i n a r d i c C M, K a r o l a k LA, H a n n u n Y A (1993) Science 258: 1769-1771
52. S a w a i H, O k a z a k i T, Y a m a m o t o H, O k a n o H, T a k e d a Y, T a s h i m a M, S a w a d a H, O k u m a M, I s h i k u r a H, U m a h a r a H, D o m a e N (1995)J Biol Chem 270: 27326-27331
53. B r a c h MA, G r u s s H J, S o t t C, H e r r m a n n F (1993) Mol Cell Biol 13: 4284-4290
54. R a i n e s M A, K o l e s n i c k RN, G o l d e D W (1993) J Biol Chem 268: 14572-14575
55. D e r i j a r d B, H i b i M, W u I - H, B a r r e t t T, S u B, D e n g T, K a r i n M, D a v i s R J (1994) Cell 76: 1025-1037
56. M i n d e n A, L i n A, S m e a l T, D e r i j a r d B, C o b b M, D a v i s R, K a r i n M (1994) M ol Cell Biol 14: 6683-6688
57. M a t h i a s S , D r e s s i e r K A , K o l e s n i c k R N (1991)Proc N atl Acad Sei USA 88 : 10009-10013
58. L iu J, M a t h i a s S, Y a n g Z, K o l e s n i c k R N (1994) J Biol Chem 269: 3047-3052
59. D o b r o w s k y RT, H a n n u n Y A (1992) J Biol Chem 261: 5048-5051
60. D o b r o w s k y RT, K a m i b a y a s h i C, M u m b y MC , H a n n u n Y A (1993) J Biol Chem 268: 15523-15530
61. W o l f f R A, D o b r o w s k y RT, B i e l a w s k a A, O b e i d L M, H a n n u n Y A (1994) J Biol Chem 269: 19605-19609
62. O h t a H (1994) FEBS Lett 355: 267-27063. S p i e g e l S, O l i v e r a A, Z h a n g H, T h o m p s o n EW,
S u Y, B e r g e r A (1994) Breast Cane Res Treatm 31: 337-34864. L i p p m a n ME , D i c k c o n R B (1989) Rec Prog Horm Res
45: 383-43965. S a d a h i r a Y, R u a n F, H a k o m o r i S, I g a r a s h i
Y (1992) Proc N atl Acad Sei USA 89: 9686-969066 . C h a o C P , L a u l e d e r k i n S J F , B a l l o u L R (1994)JBiol
Chem 269: 5849-585667. W o n g K, K w a n Y L (1993) Cell Calcium 14: 493-50568 . Y u l e DI , W u D, E s s i n g t o n TE, S h a y m a n JA,
W i l i a m s J A (1993) J Biol Chem 268: 12353-1235869. O l i v e r a A, Z h a n g H, C a r l s o n RO, M a t t i e ME ,
S c h m i d t RR, S p i e g e l S (1994) J Biol Chem 269: 17924- 17930
70. B r e i t t m a y e r J - P , B e r n a r d A, A u s s e i C (1994) J Biol Chem 269: 5054-5058
71. M a t t i e M, B r o o k e r G, S p i e g e l S (1994) J Biol Chem 269: 3181-3188
72. O k a j i m a F, K o n d o Y (1995) J Biol Chem 270: 26332- 26340
73. S a b b a d i n i RA, B e t t o R, T e r e s i A, F a c h e c h i -
308 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

- C a s s a n o G, S a l v i a t i G (1992) J Biol Chern 267: 15475- 15484
74. H u d s o n PL, P e d e r s e n W A, S a l t s m a n WS, L i s - c o v i t c h M, M a c L a u g h l i n DT, D o n a h o e P K, B l u n s z t a j n J K (1994) J Biol Chem 269: 21885-21890
75. K i n d m a n LA, K i m S, M c D o n a l d TV, G a r d n e r ’ P (1994) J Biol Chem 269: 13088-13091
76. K im S, L a k h a n i V, C o s t a DJ , S h a r a r a Al , F i t z J G, H u a n g L W, P e t e r s K G , K i n d m a n L A (1995) J Biol Chem 270: 5266-5269
77. S c h u t z e S, M a c h l e i d t T, K r o n k e M (1994) J Leukoc Biol 56: 533-541
78. V i l c e k J, L e e T H (1991) J Biol Chem 266: 7313-731679 . W a l a j t y s - R o d e E (1995) Kosmos 44: 451-46480. V a n d e n a b e l l e P, D e c l e r q W, B e y a e r t R, F i e r s
W (1995) Trends in Cell Biology 5: 382-39981. W i e g m a n n K, S c h u t z e S, M a c h l e i d t T, W i t t e D,
K r o n k e M (1994) Cell 78: 1005-101582. B e l k a C, W i e g m a n n K, A d a m D, H o l l a n d R,
N e u l o c h M, H e r r m a n F, K r o n k e M, B r a c h M A(1995) EMBO J 14: 1156-1165
83. T a r t a g l i a LA, A y r e s T M, W o n g G H W , G o e d d e l D V (1993) Cell 74: 845-853
84. I t o h N, N a g a t a S (1993) J Biol Chem 268: 10932-1093785. T e w a r i M, D i x i t V M (1996) Curr Opin Genet Dev 6 : 39-4486 . N a g a t a S, G o l d s t e i n P (1995) Science 267: 1449-145687. A l d e r s o n MR , A r m i t a g e RJ , M a r a s k o w s k y E,
T o u g h T W, R o u x E, S c h o o l e y K, R a m s d e l l F, L y n c h D H (1993) J Exp Med 176: 2231-2235
88. D a v i s R J (1993) J Biol Chem 268: 14553-1455689. K e n n e l l y PJ , K r e b s E G (1991) J Biol Chem 266:
1555-155890. J o s e p h CK, B y u n HS, B i t t m a n R, K o l e s n i c k R N
(1993) J Biol Chem 268: 20002-2000691. V a n L i n t J, A g o s t i n i s P, V a n d e v o o r d e V, H a e -
g e m a n G, F i e r s W, M e r l e v e d e W, V a n d e n h e e d e J R (1992) J Biol Chem 267: 17997-17801
92. E g a n SE, G i d d i n g s BW, B r o o k s MW, B u d a y I, S i z e l a n d AM, W e i n b e r g R A (1993) Nature (Lond) 363: 45-51
93. G a l e NW, K a p l a n S, L o w e n s t e i n EJ , S c h l e s s i n - g e r J, B a r - S a g i D (1993) Nature (Lond) 363: 88-92
94. R o z a k i s - A d c o c k M, F e r n l e y R, W a d e J, P a w - s o n T, B o w t e l l D (1993) Nature (Lond) 363: 83-87
95. Y a o B, Z h a n g Y, D e l i k a t S, M a t h i a s S, B a s u S, K o l e s n i c k R (1995) Nature (Lond) 378: 307-310
96. L in L L , W a r t m a n n M, L i n AY, K n o p f I L, S e t h A, D a v i s R J (1993) Cell 72: 269-278
97. M a h l e i d t T, W i e g m a n n K, H e n k e l T, S c h ü t z e S, B a e u e r l e P, K r ö n k e M (1994) J Biol Chem 269: 13760- 13765
98. F i n k o TS, B a l d w i n AS, J r (1993) J Biol Chem 268: 17676-17679
99. W e s t w i c k J K, W e i t z e l C, M i n d e n A, K a r i n M, B r e n n e r D A (1994) J Biol Chem 269: 26396-26401
100. W e s t w i c k J K, B i e l a w s k a A, D b a i b o G, H a n n u n YA, B r e n n e r D A (1996) J Biol Chem 270: 22689-22692
101. C i f o n e MG , R o n c a o l i P, D e M a r i a R, C a m a r d a G, S a n t o n i A, R u b e r t i G, T e s t i R (1995) EMBO J 14: 5859-5868
102. S c h u l z e - O s t h o f f K , K r a m m e r P, D r o g e W (1994) EMBO J 13: 4587-4596
103. W o n g G H W , G o e d d e l D V (1994) J Immunol 152: 1751-1755
104. C o r o n e o s E, M a r t i n e z M, M c K e n n a S, K e s t e r M (1995) J Biol Chem 270: 23305-23309
105. O l i v e r a A, S p i e g e l S (1993) Nature (Lond) 365: 557-560106. J a k o b s LS, K e s t e r M (1993) Am J Physiol 265: c740-c747107. SuY, R o s e n t h a l D, S m u l s o n M, S p i e g e l S (1994)
J Biol Chem 269: 16512-16517108. T s a i M, Y u C, S t a c e y D W (1990) Science 250: 982-985109. G o l d k o r n T, D r e s s i e r KA, M u i n d i J, R a d i n NS,
M e n d e l s o h n J, M e n a l d i n o D, L i o t t a D, K o l e s n i c k R N (1991) J Biol Chem 266: 16092-16098
Nagrody Polskiego Towarzystwa Biochemicznegow 1996 roku
Z przyjemnością informuję naszych Czytelników o przyznaniu przez Zarząd Główny Towarzystwa:
dorocznej nagrody im. Jakuba Karola Parnasa za najlepszą pracę doświadczalną wykonaną całkowicie w laboratoriach krajowych i opublikowaną w minionym roku;
dorocznej nagrody im. Bolesława Skarżyńskiego za najlepszy artykuł opublikowany w kwartalniku Postępy Biochemii w roku poprzednim;
dorocznej nagrody za najlepszy wykład akademicki wygłoszony w b.r. w ramach Konkursu organizowanego przez Sekcję Dydaktyki P.T. Bioch.;
po raz pierwszy nagrody im. Antoniego Dmochowskiego za szczególne osiągnięcia dydaktyczne w dziedzinie biochemii.
Nagroda im. J. K. Parnasa:
Kol. Kol. Alicja Wawrzynów i Maciej Żylicz z Katedry Biologii Molekularnej Uniwersytetu Gdańskiego za pracę pt. " Divergent effects of ATP on binding of the DnaK nad DnaJ chaperones to each other, or to their various native and denaturated protein substrates” , J. Biol. Chem. 270 (33), 19300— 19306, 1995.
Nagroda im. B. Skarżyńskiego:
Kol. Barbara Grzelakowska-Sztabert z Instytutu Biologii Doświadczalnej im. M. Nenckiego w Warszawie za artykuł pt. „Regulacja cyklu komórkowego — udział białkowych inhibitorów kinaz cyklinozależ- nych", Post. Bioch. 41 (2), 80—93, 1995.
POSTĘPY BIOCHEMII 42 (3 ) , 1996 309http://rcin.org.pl

Nagroda za wykład akademicki:
Kol. Hanna Czeczot z Katedry i Zakładu Biochemii Akademii Medycznej w Warszawie za wykład pt. „Biochemia nasienia".Kol. Zygmunt Pojda z Wojskowego Instytutu Higieny i Epidemiologii w Warszawie, za wykład pt. „Cytokiny w regulacji odpowiedzi układu krw iotw órczego".
Laureaci konkursu na najlepszy wykład akademicki w roku 1996: dr Hanna Czeczot, prof. dr hab. Zygmunt Pojda.
Nagroda im. A. Dmochowskiego:Kol. Leokadia Kłyszejko-Stefanowicz z Uniwersytetu Łódzkiego za podręcznik pt. „Cytobiochem ia", opublikowany w 1995 r. przez PWN.
Kol. Grzegorz Bartosz z Uniwersytetu Łódzkiego za podręcznik pt. „D w a oblicza tlenu" opublikowany w 1995 r. przez PWN (wyróżnienie).
Wszystkim Laureatom składamy serdeczne gratulacje i najlepsze życzenia.
Redakcja
SPRAWOZDANIA I KOMUNIKATY
Sprawozdanie z VII Sympozjum Fluorowego
W dniach 26-27 kwietnia 1996 r. odbyło się w Szczecinie siódme sympozjum z cyklu Metabolizm Fluoru zorganizowane przez Katedrę i Zakład Biochemii i Chemii Pomorskiej Akademii Medycznej w Szczecinie oraz szczeciński oddział Polskiego Towarzystwa Biochemicznego. W obradach uczestniczyły 64 osoby. Tematem wiodącym sympozjum była analityka związków fluoru. Po raz pierwszy oprócz części teoretycznej zorganizowano warsztaty będące częścią praktyczną sympozjum.
310 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

Spotkanie rozpoczęło się referatem Prof. dr hab. Zygmunta Machoya pt. „25 lat kompleksowych badań metabolizmu fluoru w Szczecinie", podsumowującym stan badań prowadzonych w Katedrze Biochemii i Chemii w Szczecinie oraz omawiającym wcześniej organizowane sympozja fluorowe.
Kolejne referaty dotyczyły analityki związków fluoru. W pierwszym Prof. dr hab. Wojciech Czarnowski z Katedry Toksykologii AM w Gdańsku, przedstawił różne sposoby przygotowania próbek materiału biologicznego do analizy fluoru, zagadnienie niezwykle ważne dla tych, którzy zajmują się tą problematyką. W drugim referacie, przedstawionym przez dr Tadeusza Ogońskiego z Katedry Biochemii i Chemii PAM w Szczecinie zostały omówione metody stosowane w analityce fluoru ze szczególnym uwzględnieniem dwóch z nich — potencjometrycznej i chromatografii gazowej — stosowanych rutynowo w PAM w Szczecinie. Kolejny referat nt. oznaczania fluoru potencjalnie przyswajalnego z żywności wygłosiła dr hab. Anna Wędzisz z Zakładu Bromatologii IBŚB AM w Łodzi. Część referatową zakończyło wystąpienie mgr Andrzeja Sudera, przedstawiciela firmy Jenoptik Polska z Poznania, na temat zastosowania wyrobów firmy Eppendorf w mikroanalityce. Wyroby te (pipety, wyroby polietylenowe) ze względu na bardzo wysoką jakość oraz dużą niezawodność bardzo dobrze nadają się do prac wymagających wysokiej dokładności dając pełną gwarancję uzyskania precyzyjnych i powtarzalnych wyników.
W drugiej części sympozjum przedstawiono streszczenia z 17 prac doświadczalnych. Wszystkie prezentowane prace zostały uprzednio poddane recenzji oraz ogłoszone drukiem w wydawnictwie pt. „Analityka związków fluoru" (ISBN 83-86342-19-6) wydanym przez organizatorów pod redakcją Tadeusza Ogońskiego, Doroty Samujło oraz Zygmunta Machoya. Tematyka prezentowanych prac obejmowała zagadnienia z zakresu biochemii oraz ekologii związków fluoru ze szczególnym uwypukleniem zagadnień metodycznych. Prace zostały wykonane w następujących ośrodkach akademickich Polski — Gdańsk, Kraków, Lublin, Łódź, Poznań, Sosnowiec, Szczecin, Tychy.
Sympozjum zgromadziło również wiele osób zainteresowanych aktualnym stanem wiedzy w dziedzinie badań nad metabolizmem fluoru w Polsce lub uczestnictwem w warsztatach praktycznych w dniu 27 kwietnia 1996 r. Ku zadowoleniu organizatorów w zajęciach praktycznych wzięło udział 20 osób. Uczestnikom warsztatów przedstawiono zestawy aparatury oraz omówiono sposoby przygotowania próbek do oznaczania fluoru w materiale biologicznym. Pracownicy Katedry omawiali zasady postępowania analitycznego oraz prezentowali praktyczne możliwości wykorzystania chromatografii gazowej oraz metod potencjometrycznych (elektrod fluoroselektywnych) do oznaczania fluoru w różnorodnym materiale biologicznym. W podsumowaniu sympozjum Pracownia Analityki Fluoru Katedry Biochemii i Chemii PAM w Szczecinie, przyjęła na siebie zobowiązanie okresowego organizowania sympozjów fluorowych oraz utworzenia w Szczecinie ośrodka koordynującego całokształt prac z zakresu metodyki badań związków fluoru na terenie Polski.
Tadeusz Ogoński Kierownik Katedry
SEKCJA KWASÓW NUKLEINOWYCH POLSKIEGO TOWARZYSTWA BIOCHEMICZNEGO
W grudniu 1995 r. Zarząd Główny Polskiego Towarzystwa Biochemicznego, na mój wniosek, powołał do życia Sekcję Kwasów Nukleinowych. Celem tej inicjatywy jest integracja środowiska badaczy podejmujących tematykę kwasów nukleinowych, ich funkcji, struktury i właściwości. Powinna to być również płaszczyzna rzeczowych dyskusji nie tylko teoretycznych ale również szczegółowych problemów warsztatowych. Formą działalności sekcji będą spotkania i konferencje naukowe organizowane w różnych ośrodkach naukowych. Będą one dotyczyły różnych aspektów biologii molekularnej kwasów nukleinowych. Będzie to również platforma prezentacji i promocji młodych biochemików oraz laureatów konkursów organizowanych przez Polskie Towarzystwo Biochemiczne.
W celu przyspieszenia obiegu informacji otwarta została w sieci Internet strona WWW (Word Wide Web), na której umieszczane będą wszelkie wiadomości i komunikaty dotyczące aktualnych zamierzeń i działalności sekcji. Jest możliwość nadsyłania i umieszczania na tej stronie własnych wypowiedzi, uwag i komentarzy na temat aktualnych zadań badawczych, udoskonaleń technicznych, ciekawych wyników. Jest możliwość zamieszczania wszelkich sugestii, notatek, informacji o konferencjach, nowych książkach, metodach, wizytach gości zagranicznych, własnych pracach opublikowanych, seminariach lokalnych oraz wszelkich innych sprawach, które mogą być interesujące dla biochemików kwasów nukleinowych i nie tylko. Można podzielić się swoimi przemyśleniami dotyczącymi ogólnych problemów nauki. Materiały te mogą stanowić podstawę do publikacji na WWW biuletynu informacyjnego naszej sekcji. Strona WWW i jej adres widoczne są na ryc. 1.
POSTĘPY BIOCHEMII 42 (3 ) , 1996 311http://rcin.org.pl

F0e Edit View Go Bookmarks Options Directory Window Help ___■
.<fa OBack I Forward'
Ili li Ifll i i IMHome
AEdit R̂old Open
f tFind Stop
i j j j l.
¡0\ Location: j http: //rose. man. poznan. pl/~rnszyman/skn_ptbio. html......
___ ■■■■ i u
Polskie Towarzystwo BiochemiczneSekcja Kwasów Nukleinowych
Przewodniczący: Prof, dr hab. Jan Barci szewski
Instytut Chemii Bioorganicznej PAN Noskowskiego 12/14 61-704 Poznan
tel: (061) 52-85-03 fax: (061) 52-05-32
e-m a il: jba rcisz(cbibch.poznan.pl
mk
mmiS i
«J
Informuję jednocześnie, że pierwsze spotkanie naukowe już się odbyło. Konferencja miała na celu podkreślenie ważności i uczczenie 30 rocznicy odkrycia kodu genetycznego. Oprócz referatów dotyczących problemów rozpoznawania kwasów nukleinowych i białek, które wygłosili: Dr hab. G. Węgrzyn, Prof. dr hab. R. Adamiak, Dr hab. J. Zakrzewska-Czerwińska, Prof. dr hab. T. Twardowski, Dr hab. Z. Szweykowska- Kulińska, Doc. dr hab. K. Szyfteri Dr A. Przykorska, odbyła się sesja otwarta, w której każdy z uczestników mógł wciągu 5 minut przedstawić własne wyniki badawcze. W sesji tej wystąpili: Doc. dr hab. W. Baer-Dubowska, Prof. dr hab. R. Oliński, Prof. dr hab. A. Małkiewicz, Dr M. Figlerowicz, Mgr M. Oczkowski, Mgr K. Lesniewicz i Mgr W. Kaczmarek.Zapraszam do czytania stron WWW.
Jan Bar c i szewski
312 POSTĘPY BIOCHEMII 42 (3 ) , 1996
Uprzejmie informujemy, że nasze pismo Acta Biochimica Polonica jest indeksowane przez Biochemistry & Biophysics Citation Index (ISI, U.S.A.), BIOSIS (U.S.A.), Chemical Abstract (Columbus, U.S.A.), Current Awareness in Biological Sciences (England), Excerpta Medica (Elsevier, Holland), Medline (U.S.A.).
http://rcin.org.pl

SEKCJA MIKROSKOPII ELEKTRONOWEJi
ODDZIAŁ POZNAŃSKI TOWARZYSTWA PATOLOGÓW
organizują Sympozjum Mikroskopii Elektronowej. Odbędzie się ono 6 grudnia 1996 r. w Katedrze i Zakładzie Patomorfologii Klinicznej A.M. w Poznaniu, ul. Przybyszewskiego 49. Planujemy sympozjum jednodniowe w godzinach od 9.00 do 17.00.
Tematem przewodnim będą; ultrastruktura płuca w fizjologii i patologii oraz diagnostyka ultra- strukturalna. Przewidujemy także tematy wolne. Termin nadsyłania zgłoszeń uczestnictwa czynnego i biernego upływa z dniem 30 października.
Streszczenia należy nadesłać także do dnia 30 października. Streszczenia będą drukowane jako wydawnictwo Patologii Polskiej w języku angielskim. Akceptujemy także streszczenia polskie (czasopismo posiada własnego tłumacza).
Koszt uczestnictwa wyniesie 15 zł. Noclegi w cenie ok. 80 zł możemy zabezpieczyć w Hotelu „Olimpia” w pobliżu miejsca obrad.
Serdecznie zapraszamy do wzięcia udziału.
Prof, dr hab. W. Salw a-Żurawska Prof, dr hab. W. Biczysko
Prace prosimy nadsyłać na adres:Pracownia Mikroskopii Elektronowej Katedra i Zakład Patomorfologii Klinicznej Akademia Medyczna w Poznaniu ul. Przybyszewskiego 49 60-355 Poznań
tel. 0-61/673-481lub 0-61/67-68-41 wewn. 481, 482, 483 fax. 0-61/673-481
Nr konta bankowego Oddziału Poznańskiego Polskiego Towarzystwa Patologów:PKO BP I Oddz. w Poznaniu nr 63513-9827-132
W . Mejbaum-KatzenellenbogerTs Seminars on M olecular Biology 4. Nuclear Organization and Intracellular Transport (ARF and Protein 14-3-3)Wroclaw/Szklarska Poręba, June 23—25Info: Prof. Jan SzopaUniwersytet Wrocławski, Inst. Biochemiiul. Przybyszewskiego 63/7751-148 Wrocław, PolandFax (48) +71 252-930
POSTĘPY BIOCHEMII 42 (3 ) , 1996 313http://rcin.org.pl

Wskazówki dla autorów
Wydawany przez Polskie Towarzystwo Biochemiczne kwartalnik „Postępy Biochemii" publikuje prace przeglądowe omawiające bieżące osiągnięcia, koncepcje i kierunki badawcze w dziedzinie biochemii i nauk pokrewnych; publikuje też noty z historii biochemii, zasady polskiego słownictwa biochemicznego, recenzje nadesłanych książek oraz sprawozdania ze zjazdów, konferencji i szkół, w których biorą udział członkowie Towarzystwa.
Prace przeznaczone do publikacji w „Postępach Biochemii" mogą mieć charakter artykułów monograficznych (do 20 stron tekstu licząc piśmiennictwo i tabele), minireviews (do 10 stron tekstu), oraz krótkich not o najnowszych osiągnięciach i poglądach (do 5 stron tekstu).
Autorzy artykułu odpowiadają za prawidłowość i ścisłość podawanych informacji oraz poprawność cytowania piśmiennictwa. Ujęcie prac winno być syntetyczne, a przedstawione zagadnienie zilustrowane za pomocą tabel, rycin, (wykresy, schematy, reakcje), wzorów i fotografii.
Wskazany jest podział artykułów monograficznych na rozdziały i podrozdziały, których rzeczowe tytuły tworzą spis treści. Zgodnie z przyjętą konwencją rozdziały noszą cyfry rzymskie, podrozdziały odpowiednio rzymskie i arabskie np. 1-1, I-2. Poprawność logiczna i stylistyczna tekstu warunkuje jego jednoznaczność i czytelność. Autorzy przeto winni unikać składni obcojęzycznej, gwary laboratoryjnej, a także ograniczać stosowanie doraźnie tworzonych skrótów, nawet jeżeli bywają używane w pracach specjalistycznych. Każda z nadesłanych do Redakcji prac podlega ocenie specjalistów i opracowaniu redakcyjnemu. Redakcja zastrzega sobie możliwość skrócenia tekstu i wprowadzenie zmian nie wpływających na treść pracy, deklaruje też gotowość konsultowania tekstu z Autorami.
Przekazanie artykułu do redakcji jest równoznaczne z oświadczeniem, że nadesłana praca nie była i nie będzie publikowana w innym czasopiśmie, jeżeli zostanie ogłoszona w „Postępach Biochemii". W przypadku, gdy Autor(zy) zamierza(ją) włączyć do swego artykułu ilustracje publikowane przez autorów prac cytowanych, należy uzyskać i przekazać nam odpowiednią zgodę na przedruk.
Redakcja prosi A utorów o przestrzeganie następujących wskazówek szczegółowych:
TEKST: Maszynopis powinien być napisany jednostronnie czcionką wielkości standartowej, z podwójną interlinią, z lewym marginesem ok. 4 cm.W tekście nie należy stosować żadnych podkreśleń, ani rozstrzelonego druku. Ewentualne sugestie co do charakteru czcionki drukarskiej mogą Autorzy zaznaczyć ołówkiem na marginesach maszynopisu. W przypadku stosowania w tekście liter alfabetu greckiego trzeba na marginesie wpisać ołówkiem ich fonetyczne brzmienie.
Strona informacyjna maszynopisu jest nienumerowana, zawiera imiona i nazwisko (a) autora (ów), nazwy, adresy wraz z numerem telefonu zakładów (w języku polskim i angielskim), w których pracują autorzy, adres do korespondencji, nr telefonu
i ewentualnie fax, adresy prywatne autorów, tytuł artykułu w języku polskim i angielskim oraz — w prawym dolnym rogu — liczbę tabel, rycin, wzorów i fotografii oraz skrót tytułu pracy (do 25 znaków).
Strona 1 (ty tu łow a) zawiera imiona i nazwiska autorów, tytuł pracy w języku polskim i angielskim, rzeczowy spis treści też w obu językach, tytuł naukowy każdego z autorów i ich miejsce pracy z adresem pocztowym oraz wykaz stosowanych skrótów. Kolejno numerowane dalsze strony obejmują tekst pracy, piśmiennictwo, tabele, podpisy i objaśnienia rycin, wzorów i fotografii.
PIŚMIENNICTW O: Wykaz piśmiennictwa obejmuje prace w kolejności ich cytowania w tekście, zaznacza się je liczbami porządkowymi ujętymi w nawiasy kwadratowe, np. [3 ,7 ,9— 26]. Odnośniki bibliograficzne winny mieć nową uproszczoną formę. Sposób cytowania czasopism (1), monografii (2), rozdziałów z książek jednotomowych (3), rozdziałów z tomów serii opracowanej przez tych samych redaktorów (4), rozdziałów z tomów serii opracowanych przez różnych redaktorów (5) wskazują poniżej podane przykłady:
1. Hildenbrandt GR, Aronson NN (1980) Biochim Biophys Acta 631: 499-502
2. Bostock CJ, Sumner AT (1978) The Eukaryotic Chromosome, Elsevier, North-Holand, Amsterdam
3. Norberth T, Piscator M (1979) W: Friberg L, Nordberg GF, Von VB (red) Handbook on the Toxicology of Metals. Elsevier, North-Holland, Amsterdam, str. 541-553
4. Delej J, Kesters K (1975) W: Florkin M, Stotz EH (red) Comprehensive Biochemistry t 29B. Elsevier, North-Holland Amsterdam, str. 1 -77
5. Franks NP, Lieb WR (1981) W: Knight CG (red) Research Monographs in Cell and Tissue Physiology, t 7. Elsevier, North-Holland, Amsterdam, str. 243-272
ILUSTRACJE: Ryciny winny być gotowe do reprodukcji. Fotografie czarno-białe (kontrastowe), powinny być wykonane na papierze matowym. Po porozumieniu z Redakcją można proponować reprodukcję fotografii barwnych. Pozostałe ryciny należy wykonać tuszem na białym papierze lub kalce technicznej. Wskazane jest, aby ryciny były dwukrotnie większe od przyszłej reprodukcji, tj. w skali 2 :1 . Cyfry i litery służące do opisu rysunku powinny mieć wysokość nie mniejszą niż 5 mm. Na rysunkach nie należy umieszczać opisów słownych, lecz posługiwać się skrótami. Osie wykresów winny być opatrzone napisem łatwo zrozumiałym. Decyzję o stopniu zmniejszenia ryciny podejmuje wydawca. Ilustracji nie należy włączać w tekst maszynopisu, lecz odpowiednio ponumerować: tabele i ryciny noszą cyfry arabskie, wzory zaś rzymskie. Na marginesie tekstu należy zaznaczyć ołówkiem preferowane miejsce umieszczenia tabeli, ryciny czy wzoru. Tabele odpowiednio porubrykowane, winny być opatrzone jednoznacznym tytułem i ewentualnie także niezbędnymi objaśnieniami. Słowne objaśnienia znaków graficznych można umieścić w podpisie pod ryciną, rysunkowe zaś jedynie na planszy ryciny. Tytuły i objaśnienia rycin sporządza się w postaci oddzielnego wykazu. Ilustracje należy podpisać nazwiskiem pierwszego z autorów i pierwszym słowem tytułu pracy oraz oznaczyć „góra-dół" (ołówkiem, na odwrocie). Ze względu na wewnętrzną spoistość artykułu wskazane jest konstruowanie oryginalnych rycin i zbiorczych tabel na podstawie danych z piśmiennictwa.
Maszynopis i załączniki (w dwu egzemplarzach), właściwie zabezpieczone przed uszkodzeniem w czasie transportu, należy przesłać na adres:
Redakcja kwartalnika „Postępy Biochemii"Polskie Towarzystwo Biochemiczne ul. Pasteura 3,02-093 Warszawa
314 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

U W A G A
Zawiadamiamy o zmianie numeru konta prenumeraty Postępów Biochemii. Nowy numer konta: PBK XIII O/Warszawa 370044-1225-2720-3-69
Aby zaprenumerować „P ostępy Biochemii” w 1996 r. należy wpłacić odpowiednią kwotę na konto bankowe w ydawcy (Polskiego Towarzystwa Biochemicznego) za pomocą przekazu zamieszczonego na odwrocie. Zamówione egzemplarze będziemy wysyłać pocztą na adres podany nam na przekazie. Ponieważ odcinek przekazu docierający do nas jest jednocześnie zamówieniem, prosimy o bardzo wyraźne napi
sanie imienia, nazwiska (lub nazwy instytucji) i dokładnego adresu wraz z kodem pocztowym (DRUKOWANYMI LITERAMI) na wszystkich trzech odcinkach przekazu.
Prenumerata krajowa dla instytucji:60.00 złPrenumerata krajowa indywidualna:28.00 zł (50% zniżki dla członków Polskiego Towarzystwa Biochemicznego).
Prenum erując„PostępyB iochem ii”wspieraszsw ojeczasopismo!
POSTĘPY BIOCHEMII 4 2 (3 ) , 1996 315http://rcin.org.pl

----------------------------------Pokwitowanie dla wpłacającego
z ł , ............................................ .
wpłacający
imię, nazwisko, dokładny adres z kodem pocztowym
na rachunek
Polskie Towarzystwo Biochemiczne 02-093 Warszawa, ul. Pasteura 3 P.B.K. XIIl/O W-wa. Al. Jerozolimskie 37 00 44-1225-2720-3-69
Odcinek dla posiadacza rachunku
zł....... ..... ........słownie....................................
w płacający
im ię, nazwisko, dokładny adres z kodem pocztowym
na rachunek
Polskie Towarzystwo Biochemiczne02-093 Warszawa, ul. Pasteura 3 P.B.K. X Ill/O W-wa. Al. Jerozolimskie 37 00 44-1225-2720-3-69
Odcinek dla poczty lub banku
z ł ...........................................słownie............................... .
w p łacający ...................................
im ię, nazwisko, dokładny adres z kodem pocztowym
na rachunek
Polskie Towarzystwo Biochemiczne02-093 Warszawa, ul. Pasteura 3 P.B.K. X Ill/O W-wa. Al. Jerozolimskie 37 00 44-1225-2720-3-69
stem pel stem pel stem pelPobrano opłatę
Zł.................
Pobrano opłatę
Z ł............................
Pobrano opłatę
Zł.................podpis przyjmującego podpis przyjmującego podpis przyjmującego
316 POSTĘPY BIOCHEMII 42 (3 ) , 1996http://rcin.org.pl

http://rcin.org.pl

http://rcin.org.pl