advanced organic chemistry یاهشور یلآ داوم زتنس...
TRANSCRIPT

Advanced Organic
Chemistry
روشهایسنتز مواد آلی
به نام خدا
Dr Morteza MehrdadUniversity of Guilan, Department of
Chemistry,
Rasht, [email protected]
5

Functional Group Interconversion by
Substitution,
Including Protection and Deprotection
2
3

3
3.1. Conversion of Alcohols to Alkylating Agents (Sulfonate Esters, Halides)
3.2. Introduction of Functional Groups by Nucleophilic Substitution at Saturated Carbon
3.2.1. General Solvent Effects
3.2.2. Nitrile Nucleophile
3.2.3. Oxygen Nucleophiles
3.2.4. Nitrogen Nucleophiles
3.2.5. Sulfur Nucleophiles
3.3. Cleavage of Carbon-Oxygen Bonds in Ethers and Esters
3.4. Interconversion of Carboxylic Acid Derivatives
3.4.1. Acylation of Alcohols
3.4.3. Preparation of Amides
3.5. Installation and Removal of Protective Groups
3.5.1. Hydroxy-Protecting Groups
3.5.2. Amino-Protecting Groups
3.5.3. Carbonyl-Protecting Groups
3.5.4. Carboxylic Acid-Protecting Groups
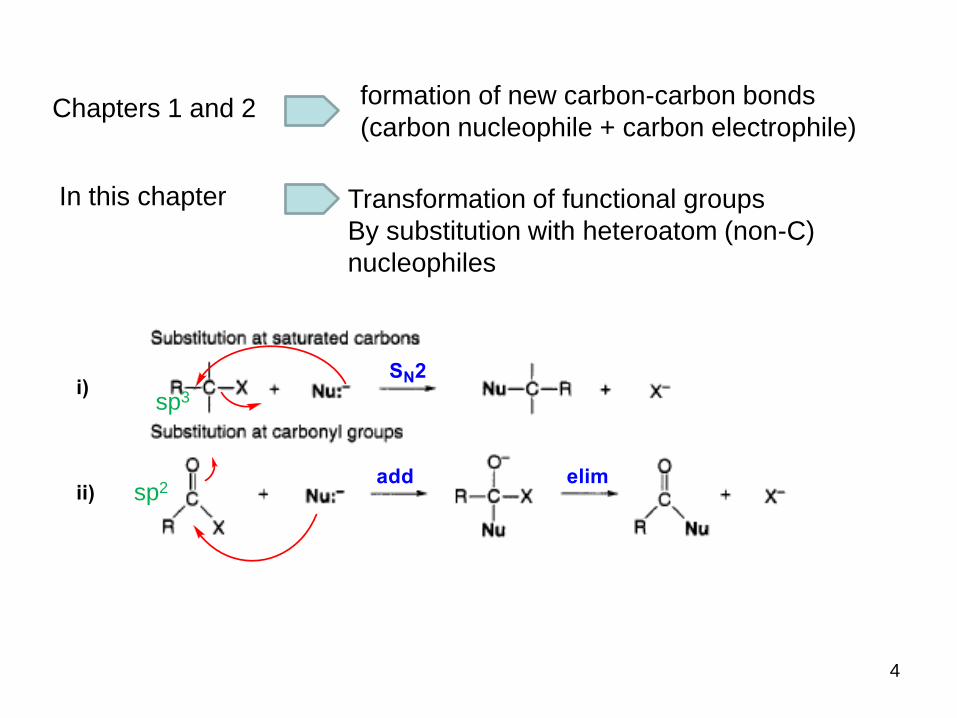
4
sp3
sp2
Transformation of functional groups
By substitution with heteroatom (non-C)
nucleophiles
Chapters 1 and 2formation of new carbon-carbon bonds
(carbon nucleophile + carbon electrophile)
In this chapter
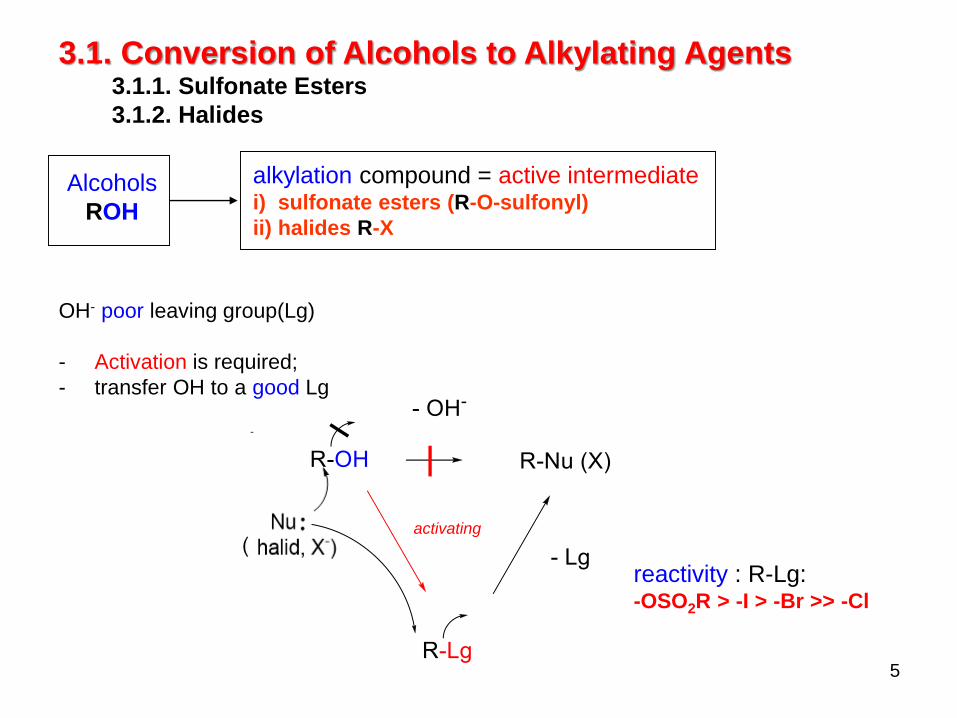
5
OH- poor leaving group(Lg)
- Activation is required;
- transfer OH to a good Lg
-
3.1. Conversion of Alcohols to Alkylating Agents3.1.1. Sulfonate Esters
3.1.2. Halides
activating
reactivity : R-Lg:-OSO2R > -I > -Br >> -Cl
Alcohols
ROH
alkylation compound = active intermediatei) sulfonate esters (R-O-sulfonyl)
ii) halides R-X
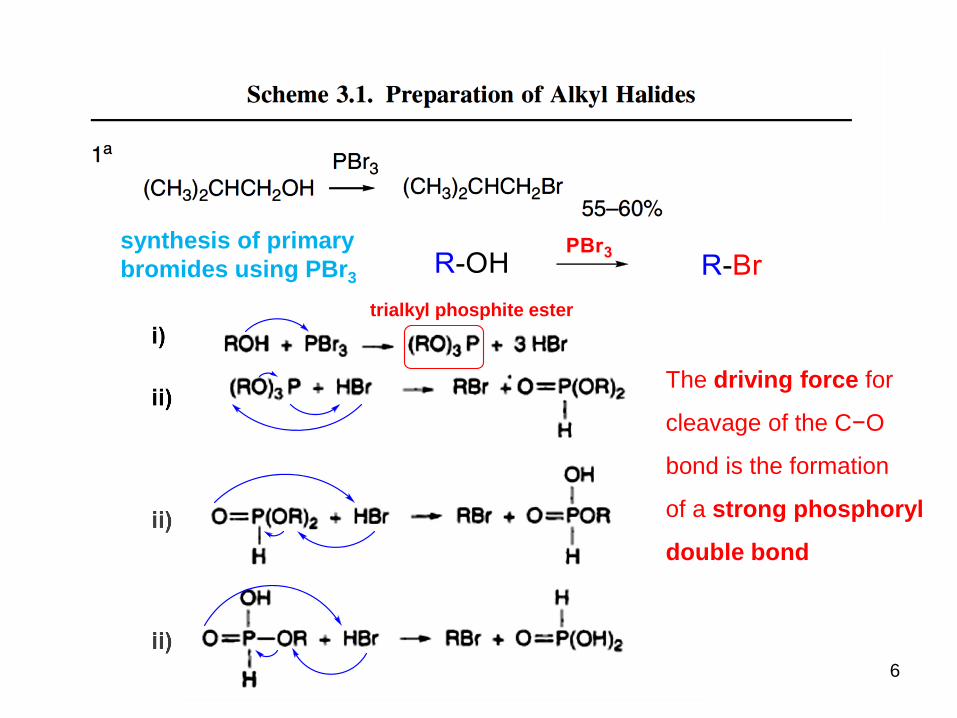
6
trialkyl phosphite ester
The driving force for
cleavage of the C−O
bond is the formation
of a strong phosphoryl
double bond
synthesis of primary
bromides using PBr3
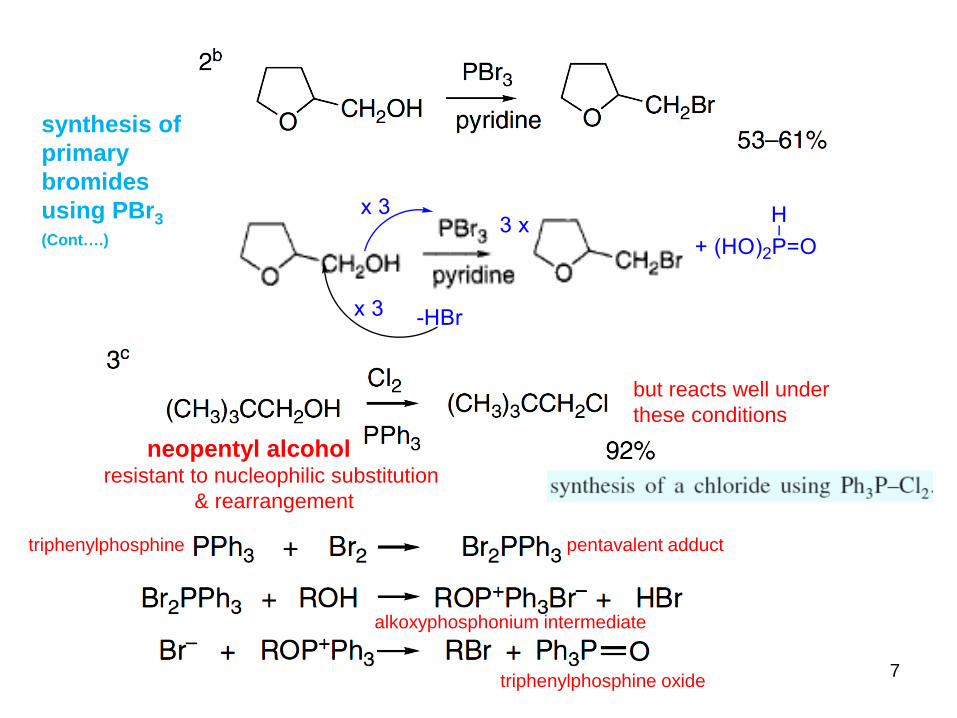
7
triphenylphosphine pentavalent adduct
alkoxyphosphonium intermediate
triphenylphosphine oxide
synthesis of
primary
bromides
using PBr3
(Cont….)
neopentyl alcoholresistant to nucleophilic substitution
& rearrangement
but reacts well under
these conditions
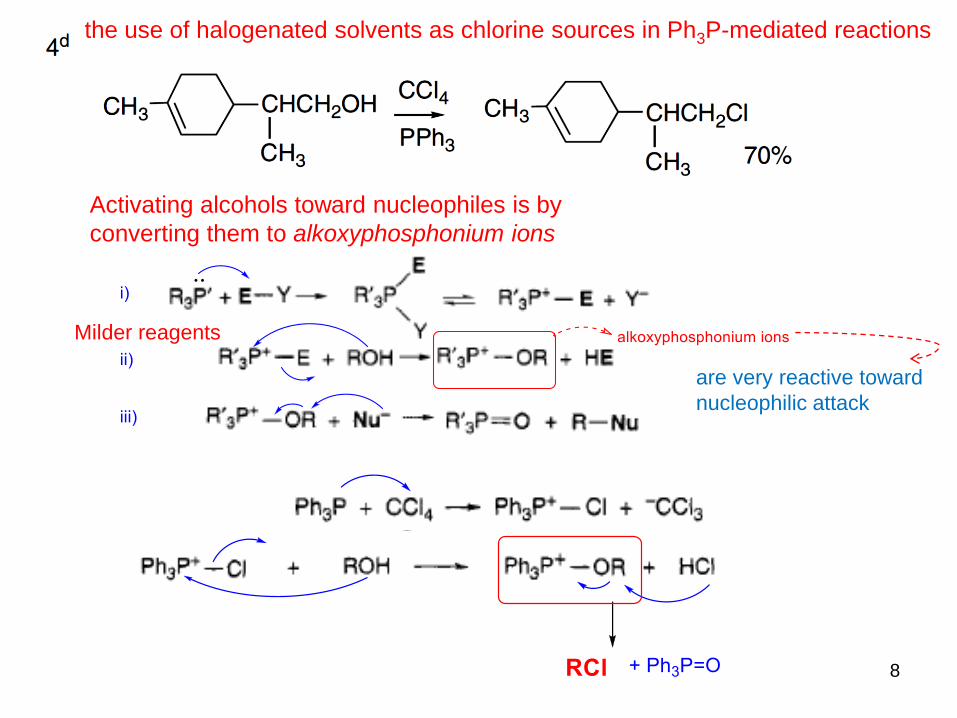
8
Milder reagents
Activating alcohols toward nucleophiles is by
converting them to alkoxyphosphonium ions
are very reactive toward
nucleophilic attack
the use of halogenated solvents as chlorine sources in Ph3P-mediated reactions
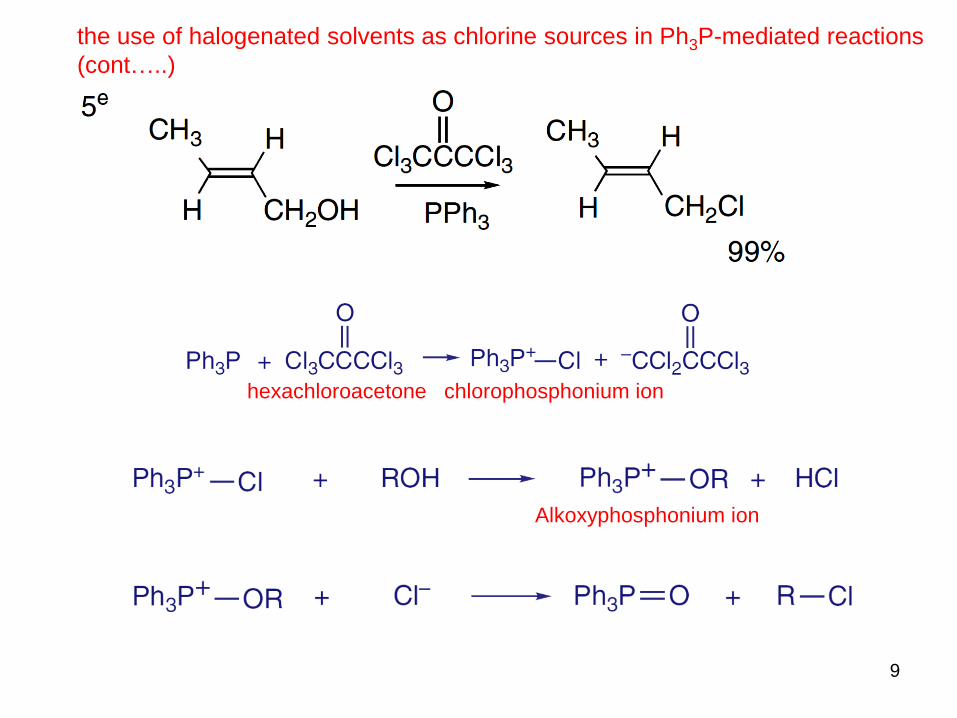
9
hexachloroacetone chlorophosphonium ion
Alkoxyphosphonium ion
the use of halogenated solvents as chlorine sources in Ph3P-mediated reactions
(cont…..)
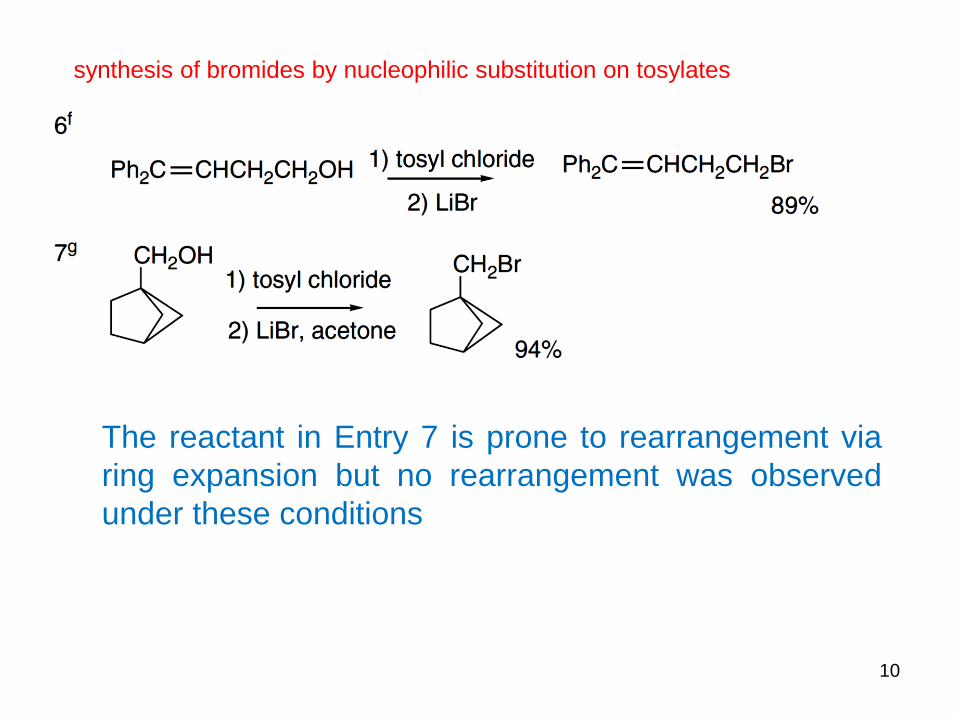
10
The reactant in Entry 7 is prone to rearrangement via
ring expansion but no rearrangement was observed
under these conditions
synthesis of bromides by nucleophilic substitution on tosylates
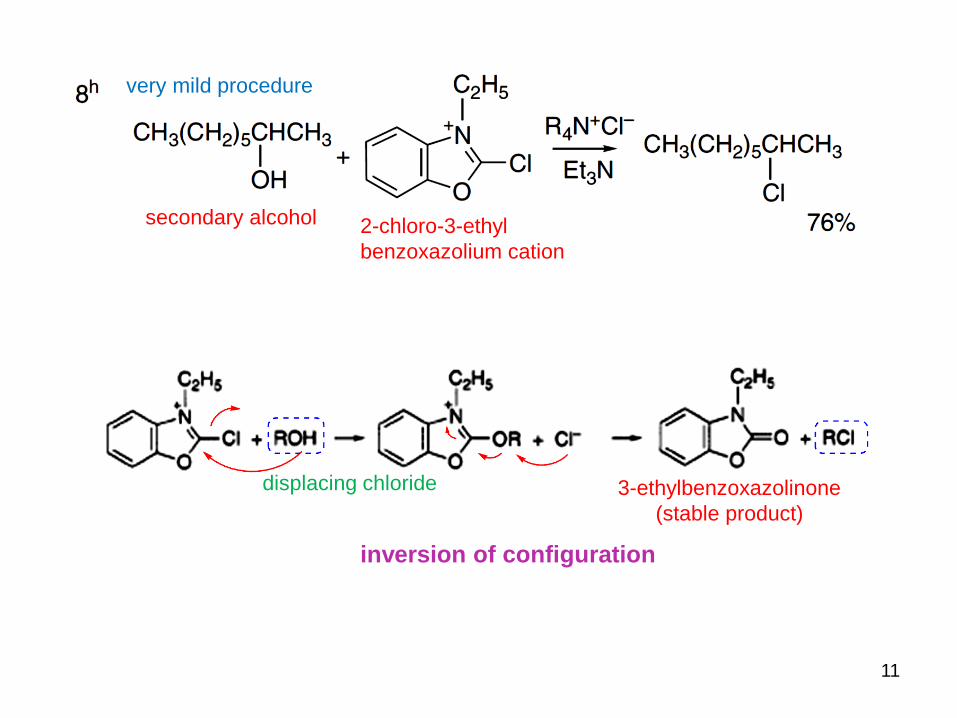
11
2-chloro-3-ethyl
benzoxazolium cation
very mild procedure
displacing chloride 3-ethylbenzoxazolinone
(stable product)
secondary alcohol
inversion of configuration
12
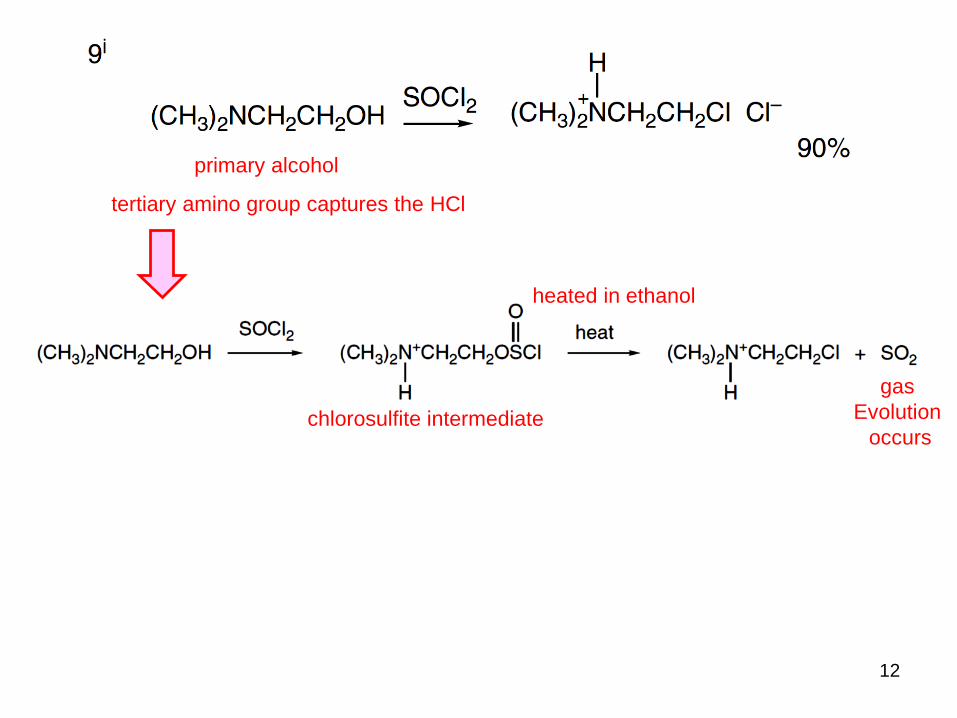
primary alcohol
tertiary amino group captures the HCl
chlorosulfite intermediate
heated in ethanol
gas
Evolution
occurs
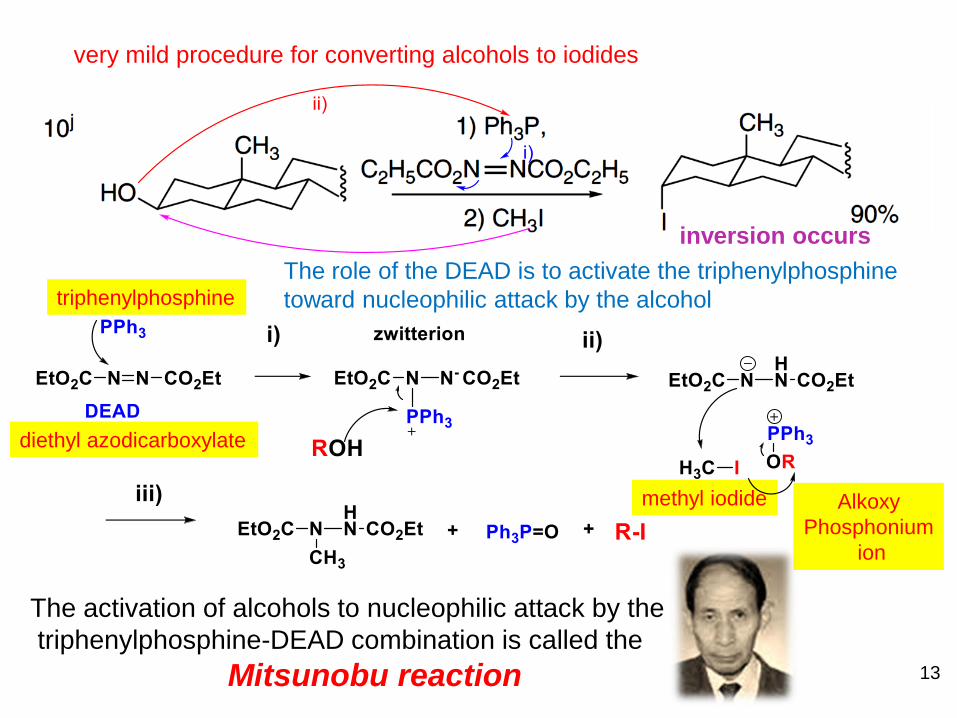
Alkoxy
Phosphonium
ion
methyl iodide
13
very mild procedure for converting alcohols to iodides
triphenylphosphine
diethyl azodicarboxylate
The role of the DEAD is to activate the triphenylphosphine
toward nucleophilic attack by the alcohol
The activation of alcohols to nucleophilic attack by the
triphenylphosphine-DEAD combination is called the
Mitsunobu reaction
inversion occurs
14
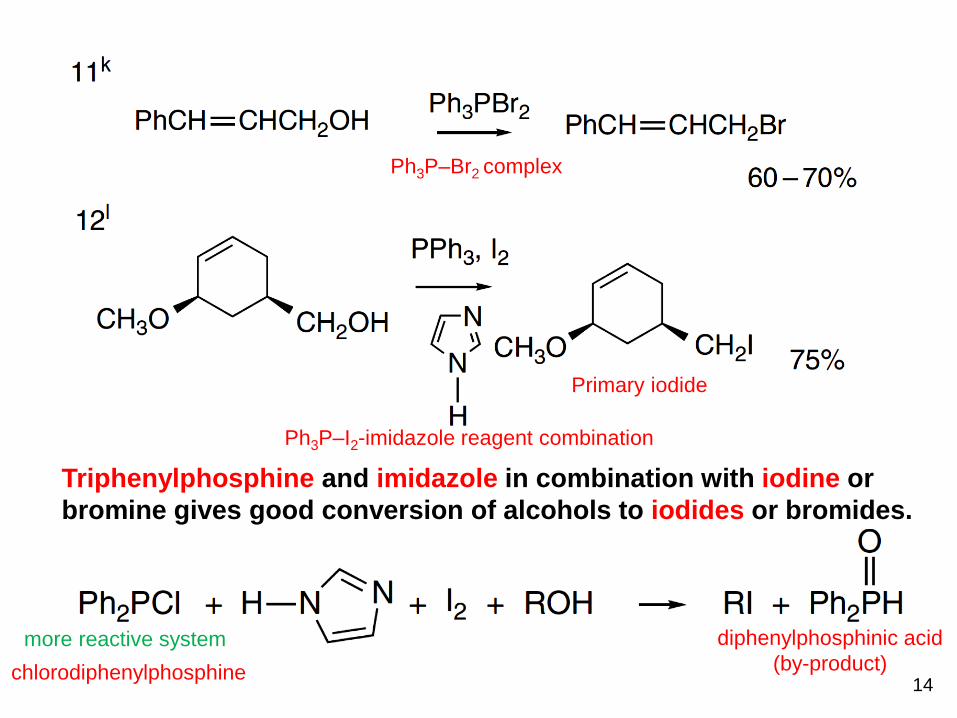
Ph3P–Br2 complex
Ph3P–I2-imidazole reagent combination
Primary iodide
Triphenylphosphine and imidazole in combination with iodine or
bromine gives good conversion of alcohols to iodides or bromides.
more reactive system
chlorodiphenylphosphine
diphenylphosphinic acid
(by-product)

15
3.2. Introduction of Functional Groups by Nucleophilic
Substitution at Saturated Carbon
3.2.1. General Solvent Effects
3.2.2. Nitriles
3.2.3. Oxygen Nucleophiles
3.2.4. Nitrogen Nucleophiles
3.2.5. Sulfur Nucleophiles
3.2.6. Phosphorus Nucleophiles
16
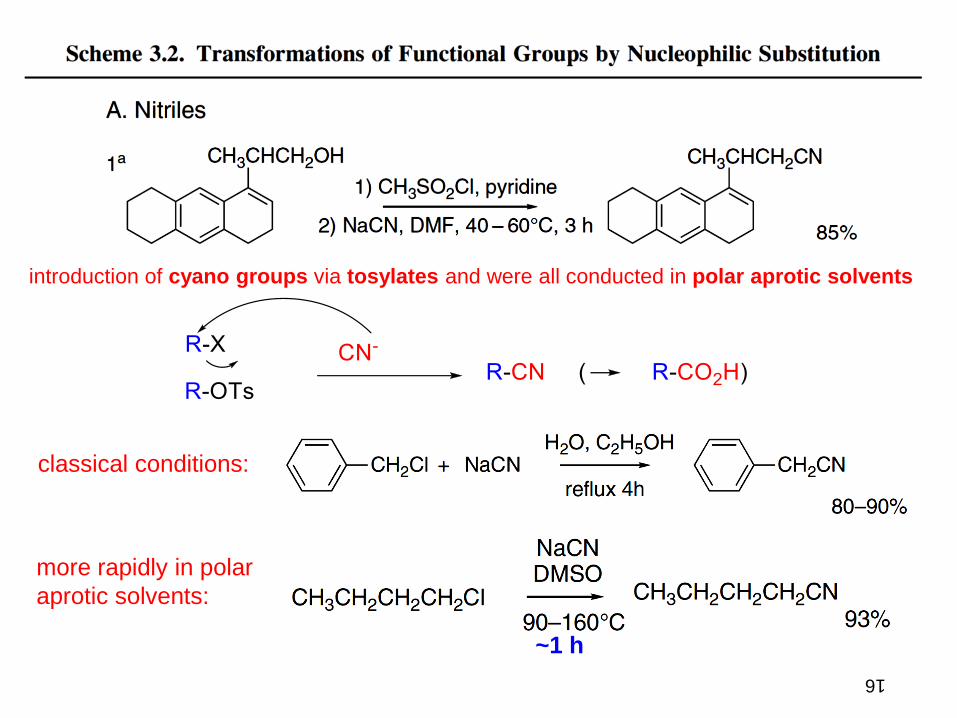
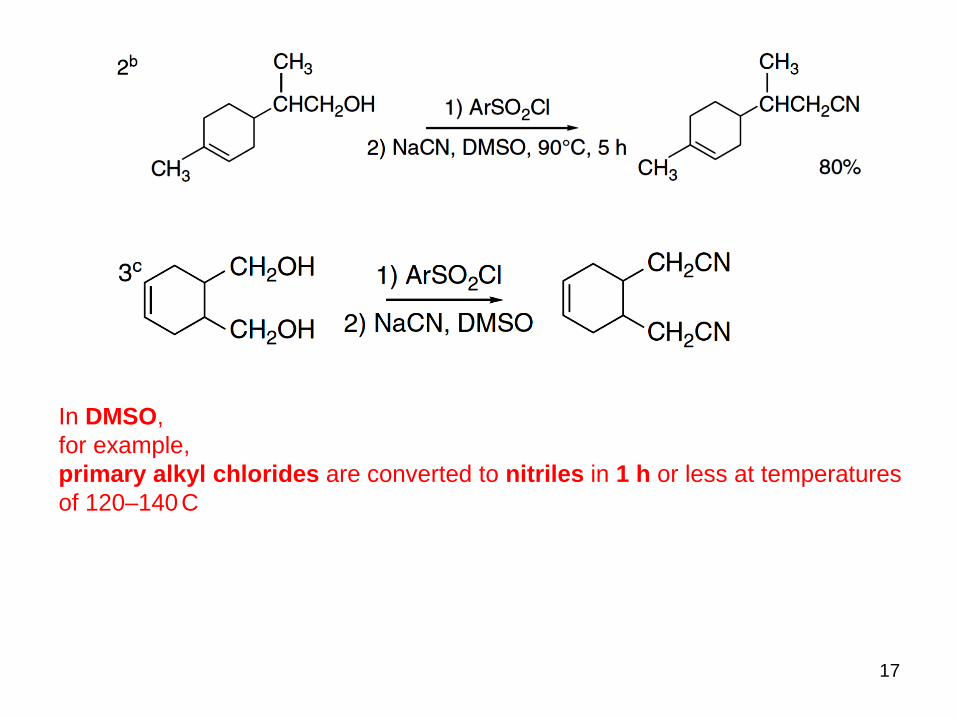
introduction of cyano groups via tosylates and were all conducted in polar aprotic solvents
classical conditions:
more rapidly in polar
aprotic solvents:
~1 h
17
In DMSO,
for example,
primary alkyl chlorides are converted to nitriles in 1 h or less at temperatures
of 120–140C

18
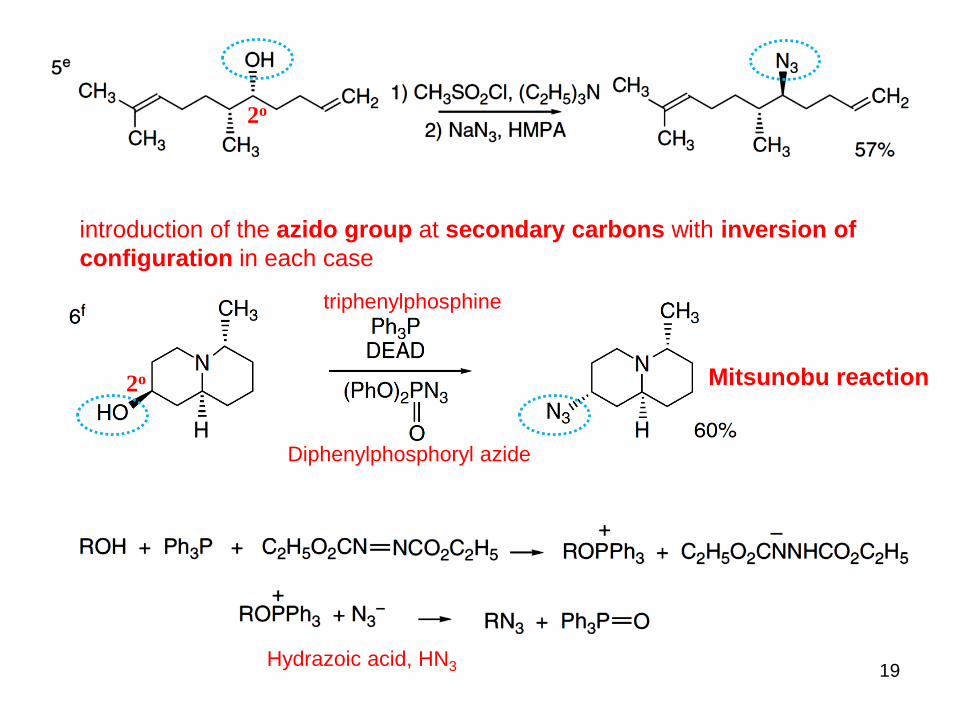
Nitrogen Nucleophiles:
Azides are useful
intermediates
easily reduced to primary amines
undergo cycloaddition reactions
Azido groups are usually introduced into aliphatic compounds by
nucleophilic substitution
under phase transfer conditions
A concentrated aqueous solution of NaN3 was
heated with the alkyl bromide and 5 mol %
methyltrioctylammonium chloride.
19
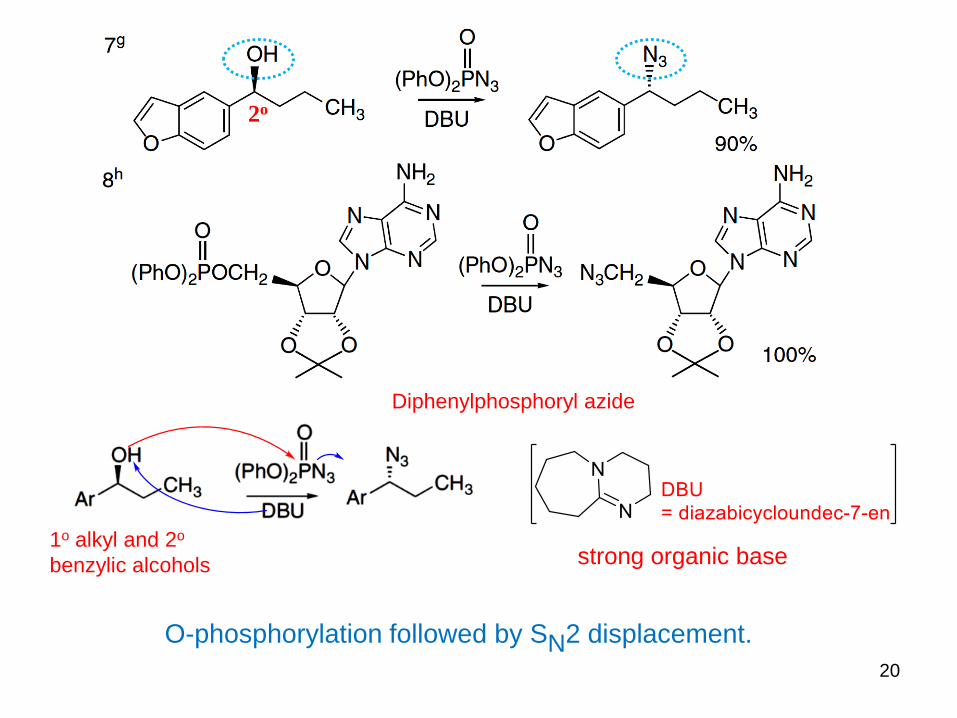
introduction of the azido group at secondary carbons with inversion of
configuration in each case
2o
2o
Diphenylphosphoryl azide
triphenylphosphine
Mitsunobu reaction
Hydrazoic acid, HN3
20
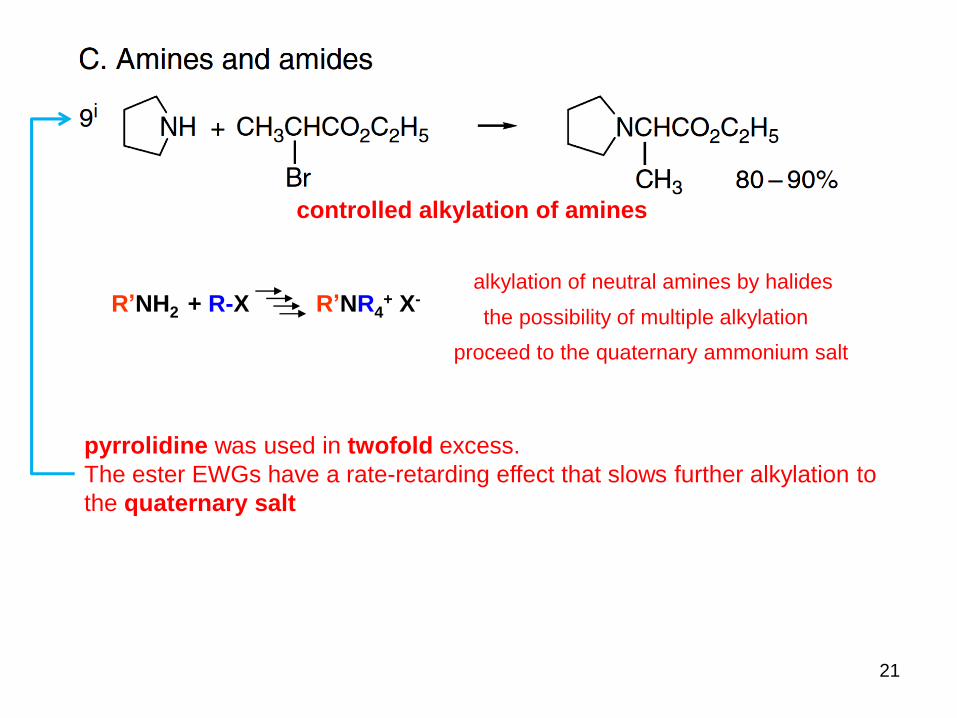
2o
Diphenylphosphoryl azide
1o alkyl and 2o
benzylic alcohols strong organic base
O-phosphorylation followed by SN2 displacement.
21
controlled alkylation of amines
R’NH2 + R-X R’NR4+ X-
alkylation of neutral amines by halides
the possibility of multiple alkylation
pyrrolidine was used in twofold excess.
The ester EWGs have a rate-retarding effect that slows further alkylation to
the quaternary salt
proceed to the quaternary ammonium salt
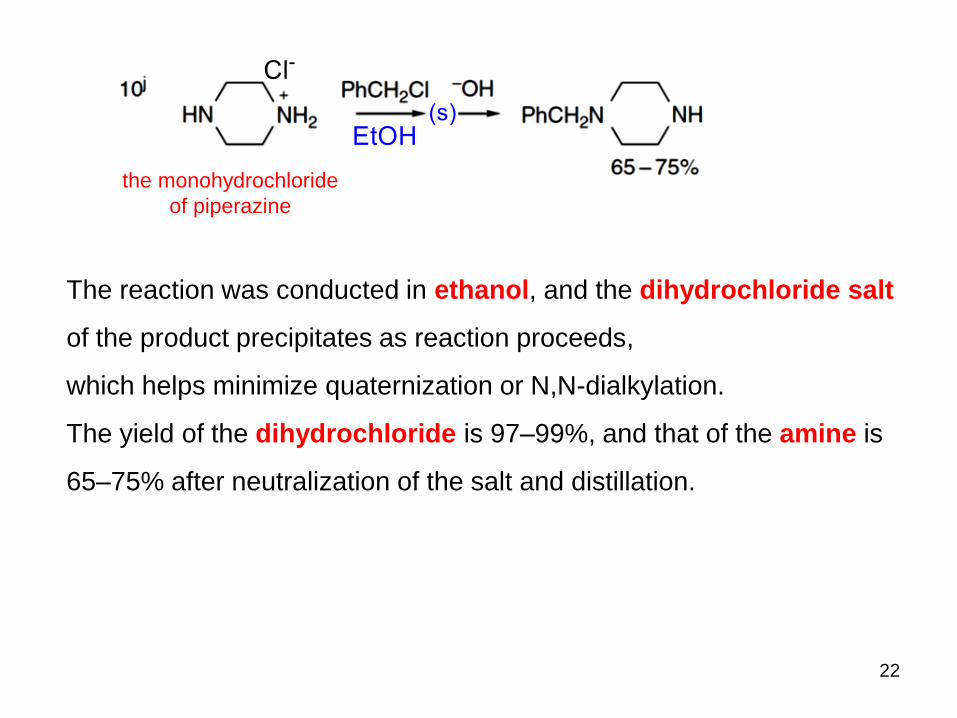
22
The reaction was conducted in ethanol, and the dihydrochloride salt
of the product precipitates as reaction proceeds,
which helps minimize quaternization or N,N-dialkylation.
The yield of the dihydrochloride is 97–99%, and that of the amine is
65–75% after neutralization of the salt and distillation.
the monohydrochloride
of piperazine
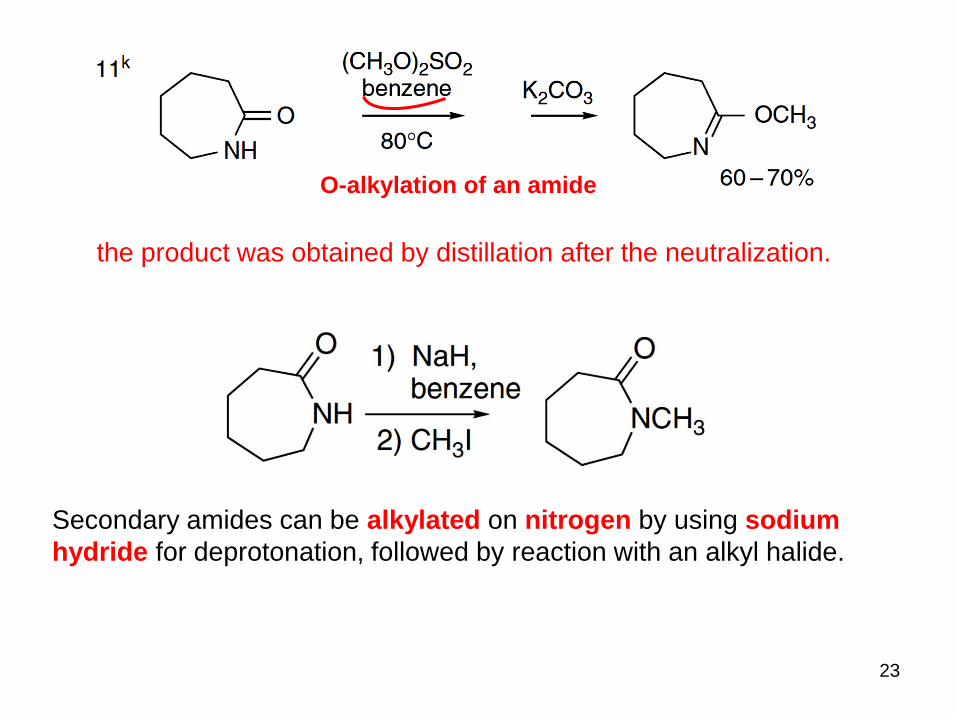
23
O-alkylation of an amide
the product was obtained by distillation after the neutralization.
Secondary amides can be alkylated on nitrogen by using sodium
hydride for deprotonation, followed by reaction with an alkyl halide.
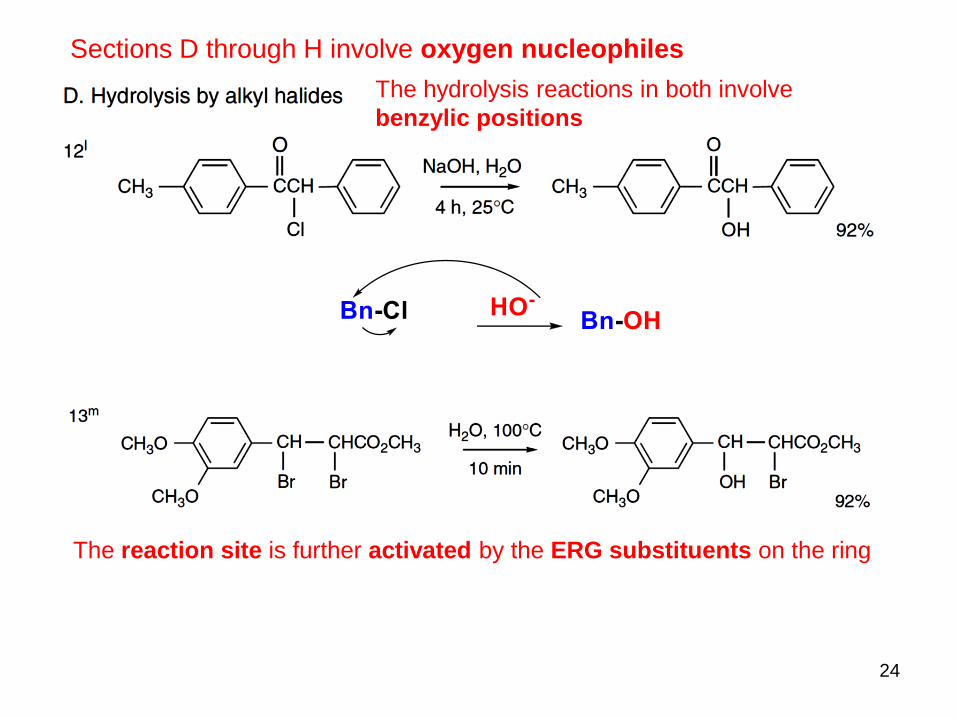
24
Sections D through H involve oxygen nucleophiles
The hydrolysis reactions in both involve
benzylic positions
The reaction site is further activated by the ERG substituents on the ring
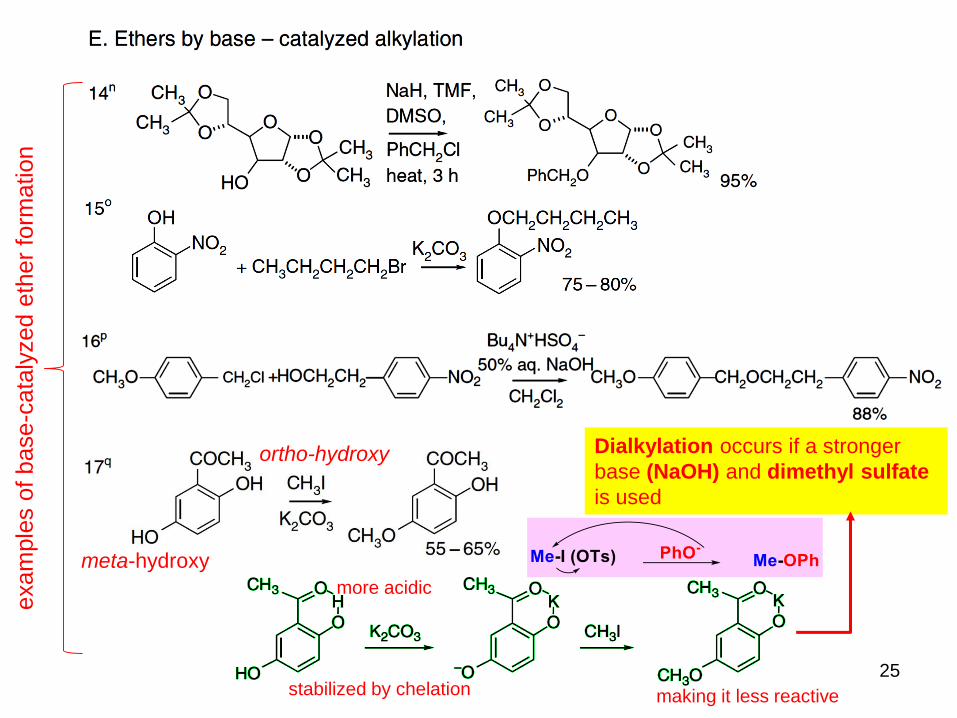
25
exam
ple
s o
f base-c
ata
lyzed e
ther
form
ation
meta-hydroxy
ortho-hydroxy
more acidic
stabilized by chelation making it less reactive
Dialkylation occurs if a stronger
base (NaOH) and dimethyl sulfate
is used
26
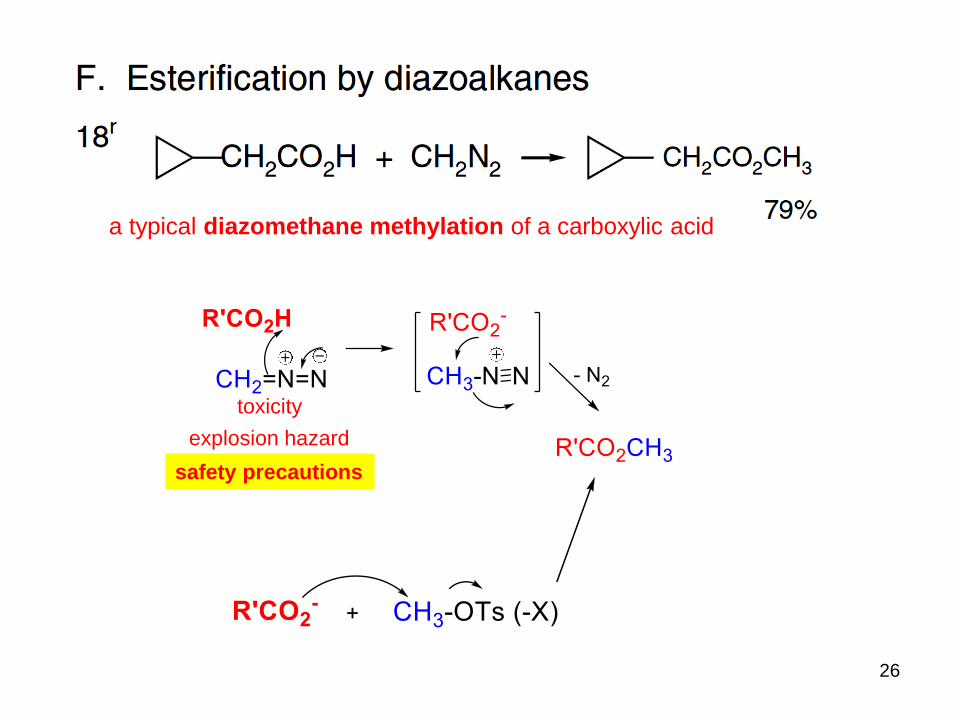
a typical diazomethane methylation of a carboxylic acid
toxicity
explosion hazard
safety precautions
27
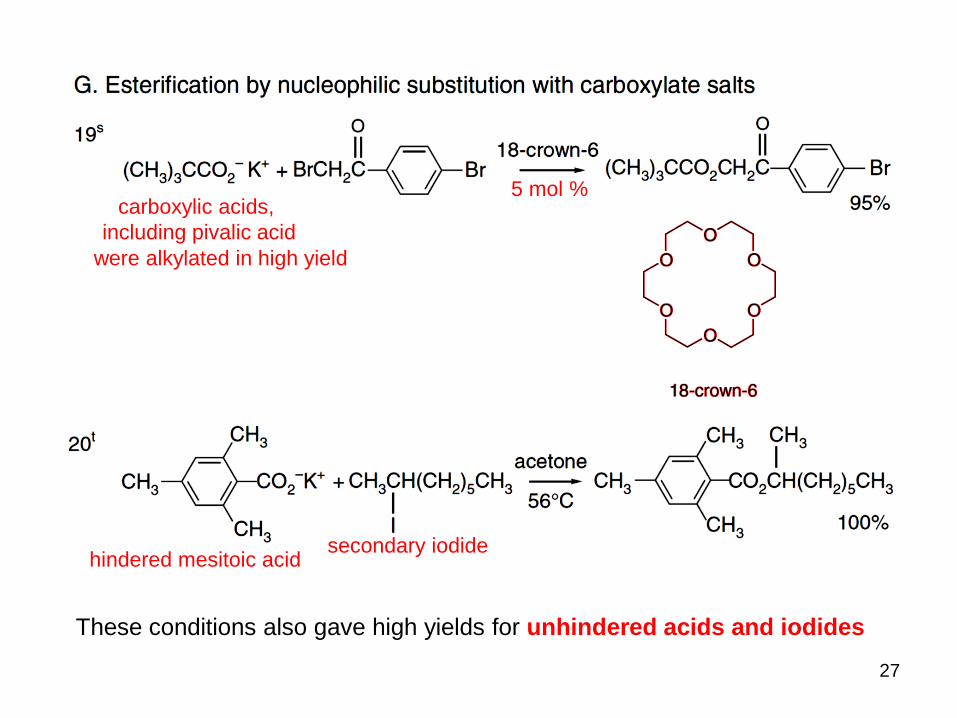
5 mol % carboxylic acids,
including pivalic acid
were alkylated in high yield
These conditions also gave high yields for unhindered acids and iodides
hindered mesitoic acidsecondary iodide
28
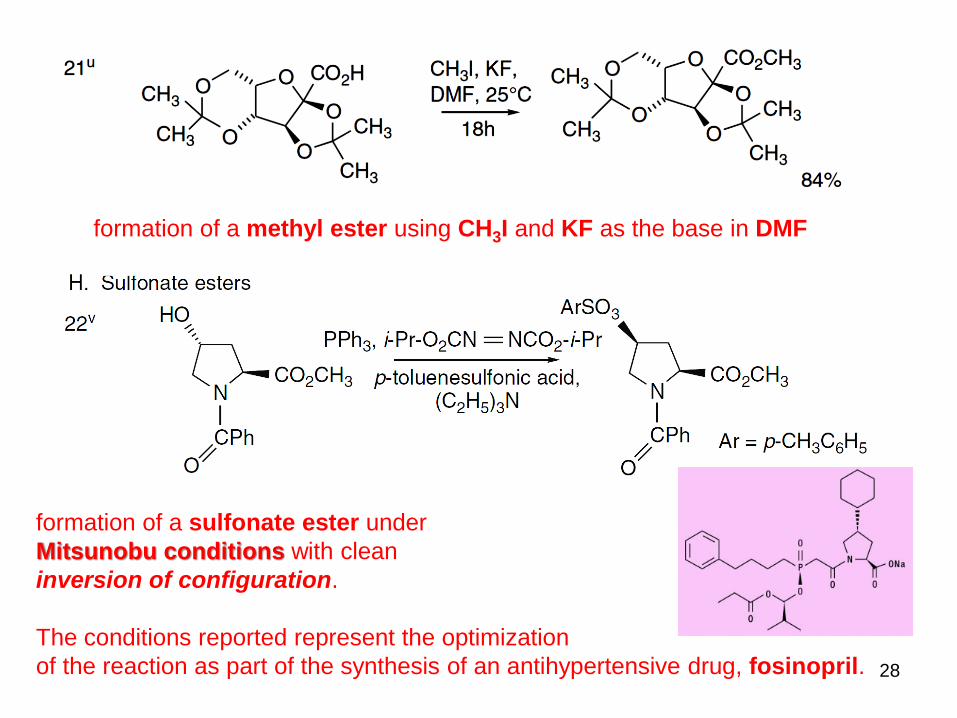
formation of a sulfonate ester under
Mitsunobu conditions with clean
inversion of configuration.
The conditions reported represent the optimization
of the reaction as part of the synthesis of an antihypertensive drug, fosinopril.
formation of a methyl ester using CH3I and KF as the base in DMF
29
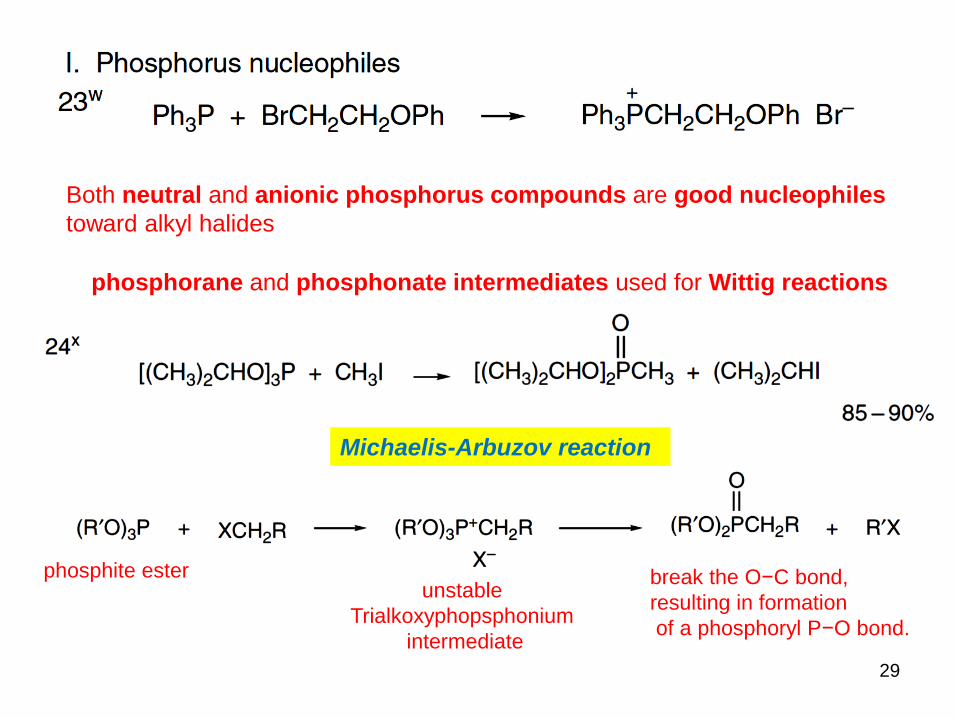
Both neutral and anionic phosphorus compounds are good nucleophiles
toward alkyl halides
phosphorane and phosphonate intermediates used for Wittig reactions
phosphite ester
Michaelis-Arbuzov reaction
unstable
Trialkoxyphopsphonium
intermediate
break the O−C bond,
resulting in formation
of a phosphoryl P−O bond.

30
useful method for introducing thiol groups
The solid thiourea is a convenient source of sulfur
thiouronium ion is readilyhydrolyzed by base/
avoids competition from
formation of a dialkyl sulfide
Anions derived from thiols are strong nucleophiles and are easily alkylated by halides.
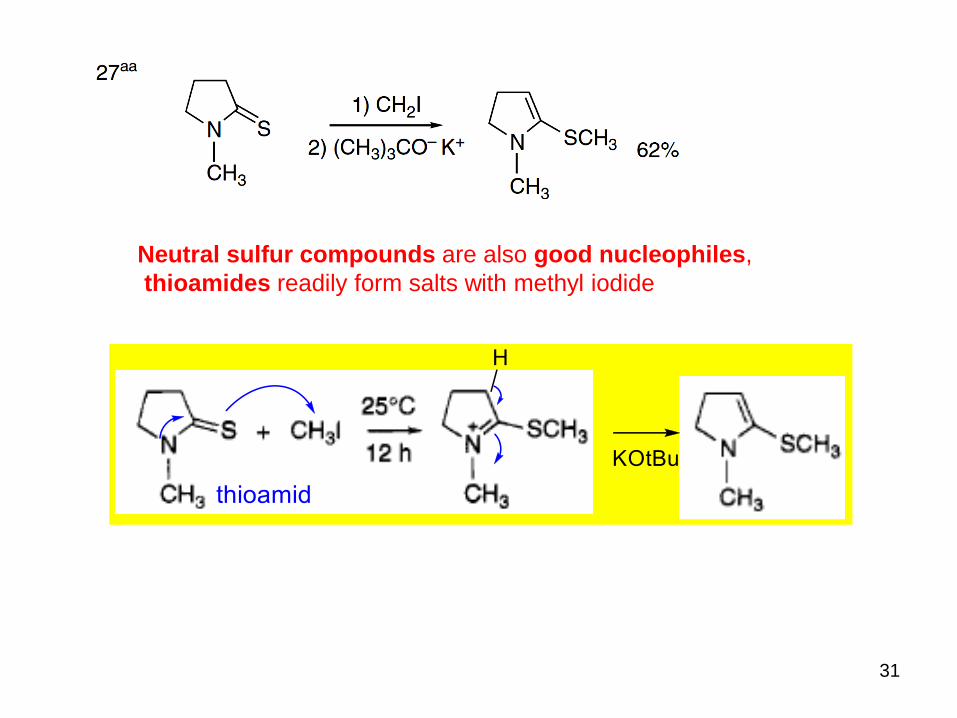
31
Neutral sulfur compounds are also good nucleophiles,
thioamides readily form salts with methyl iodide

32
3.3. Cleavage of Carbon-Oxygen Bonds in Ethers
and Esters
The cleavage of carbon-oxygen bonds in ethers or esters by nucleophilic
substitution is frequently a useful synthetic transformation.
carboxy is only slightly better
alkoxide group is a poor leaving group
As a result,
these reactions usually require assistance from a protic or
Lewis acid

33
use of boron tribromide for ether cleavage
The reactions are conducted at dry ice-acetone temperature and
the exposure to water on workup hydrolyzes residual O−B bonds
adduct formed
attack of bromide ion
The cleavage step
can occur by either an
SN2 or an SN1 process
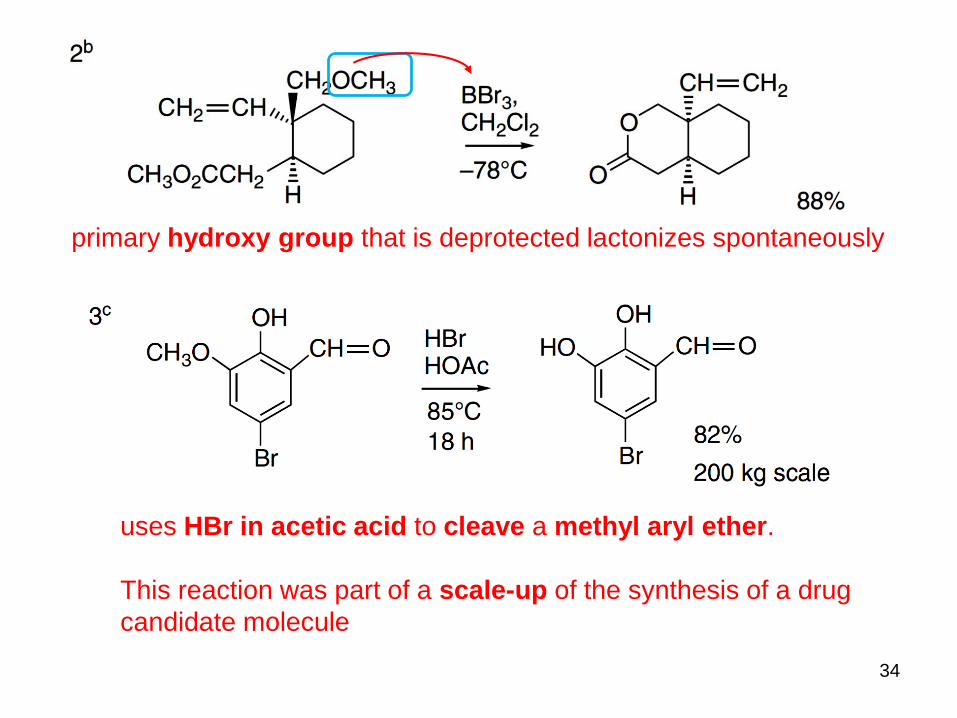
34
primary hydroxy group that is deprotected lactonizes spontaneously
uses HBr in acetic acid to cleave a methyl aryl ether.
This reaction was part of a scale-up of the synthesis of a drug
candidate molecule
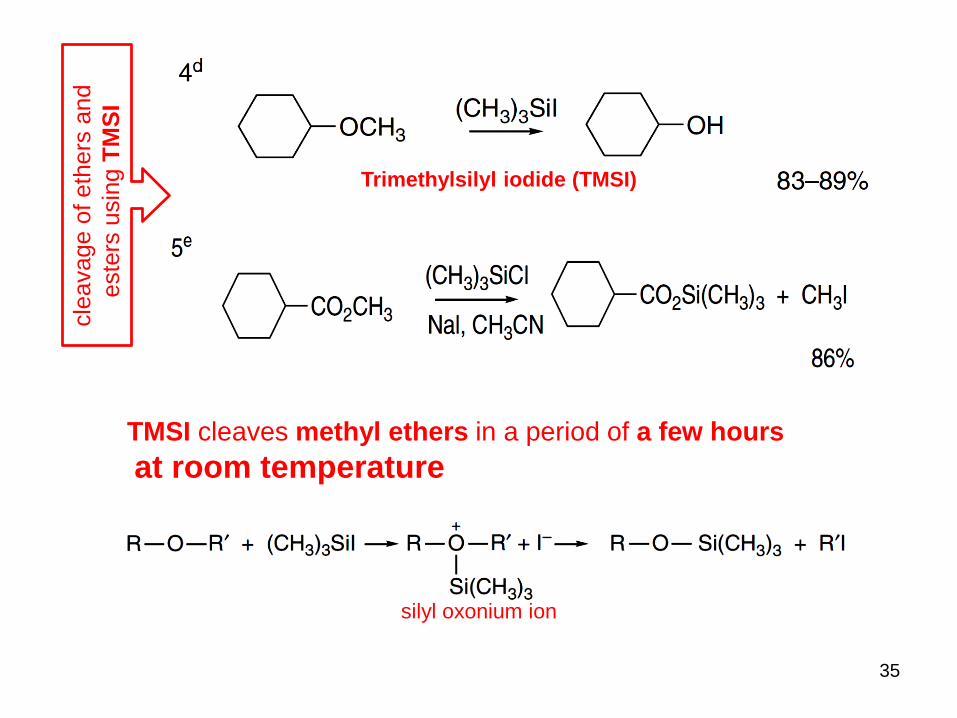
35
cle
avage o
f eth
ers
and
este
rs u
sin
g T
MS
I
Trimethylsilyl iodide (TMSI)
TMSI cleaves methyl ethers in a period of a few hours
at room temperature
silyl oxonium ion
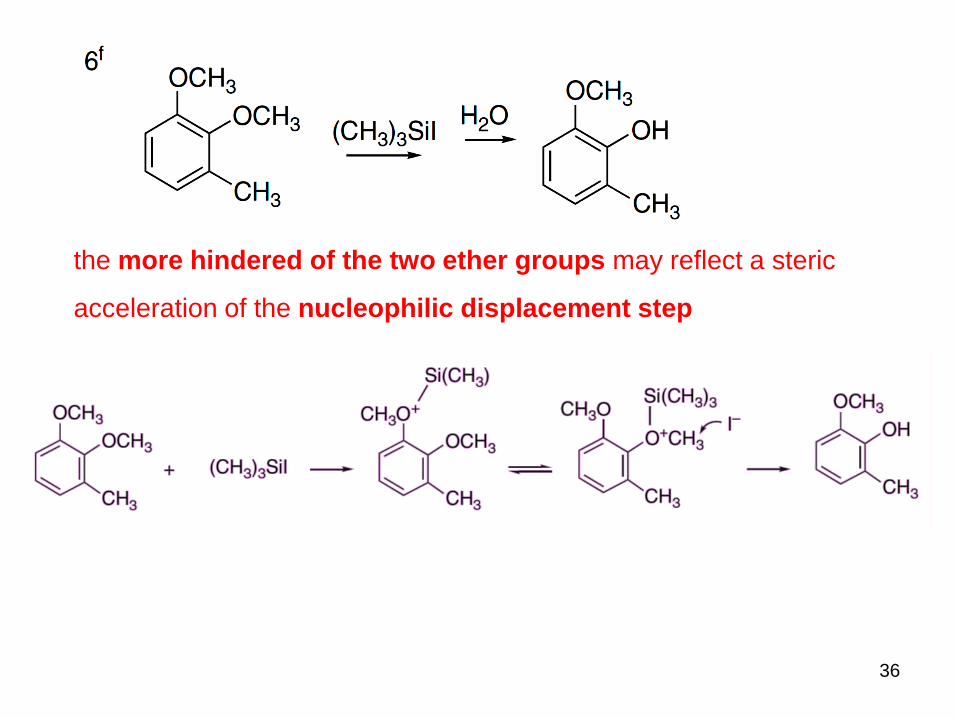
36
the more hindered of the two ether groups may reflect a steric
acceleration of the nucleophilic displacement step
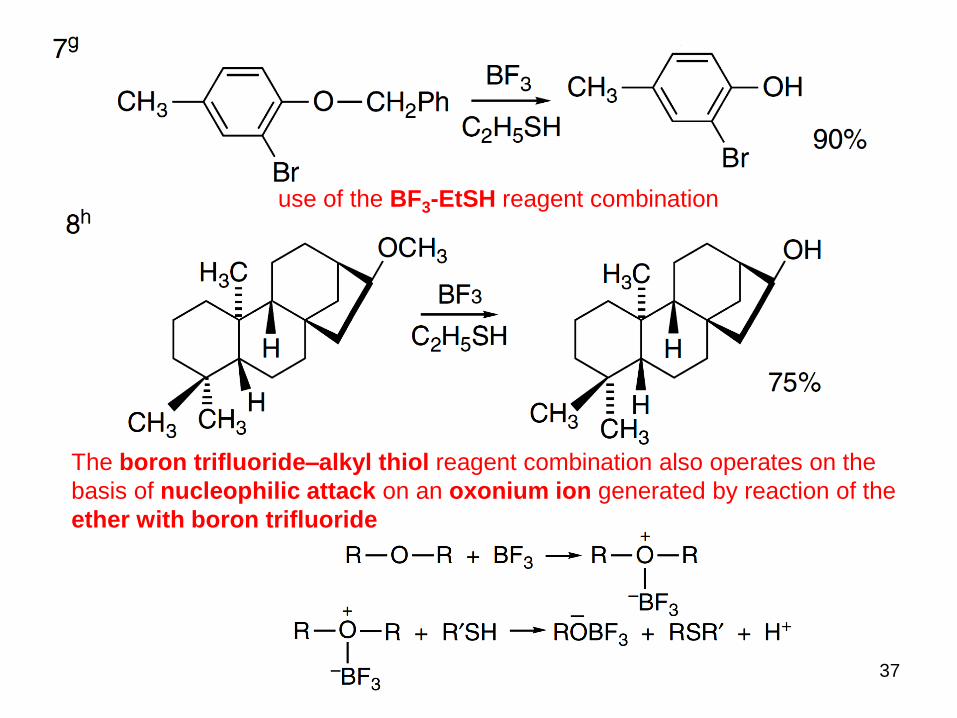
37
use of the BF3-EtSH reagent combination
The boron trifluoride–alkyl thiol reagent combination also operates on the
basis of nucleophilic attack on an oxonium ion generated by reaction of the
ether with boron trifluoride
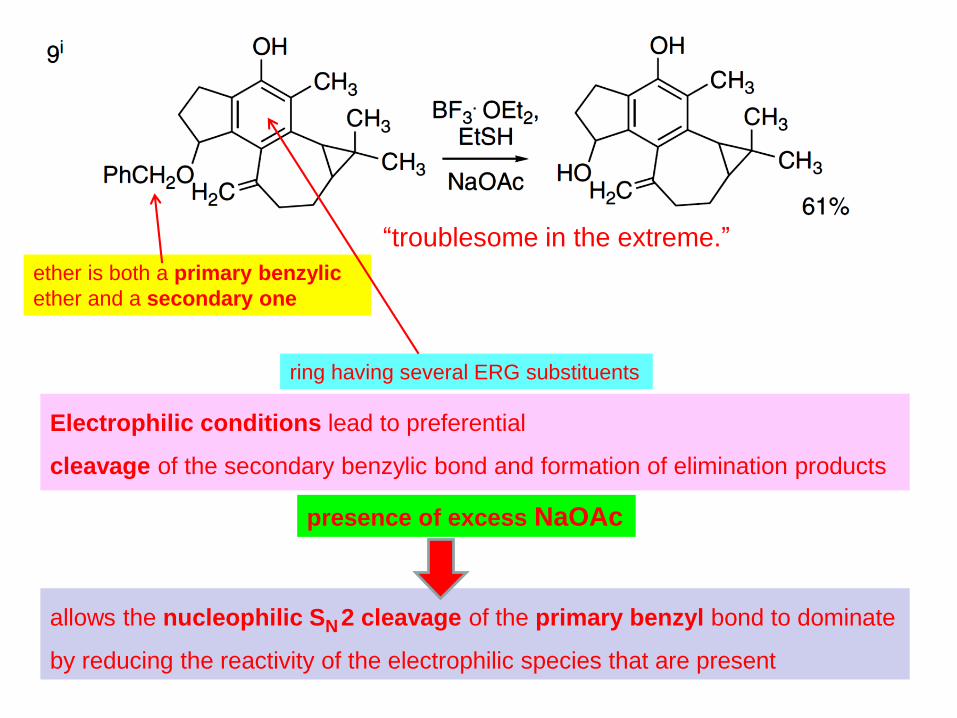
38
“troublesome in the extreme.”
ether is both a primary benzylic
ether and a secondary one
ring having several ERG substituents
Electrophilic conditions lead to preferential
cleavage of the secondary benzylic bond and formation of elimination products
presence of excess NaOAc
allows the nucleophilic SN 2 cleavage of the primary benzyl bond to dominate
by reducing the reactivity of the electrophilic species that are present
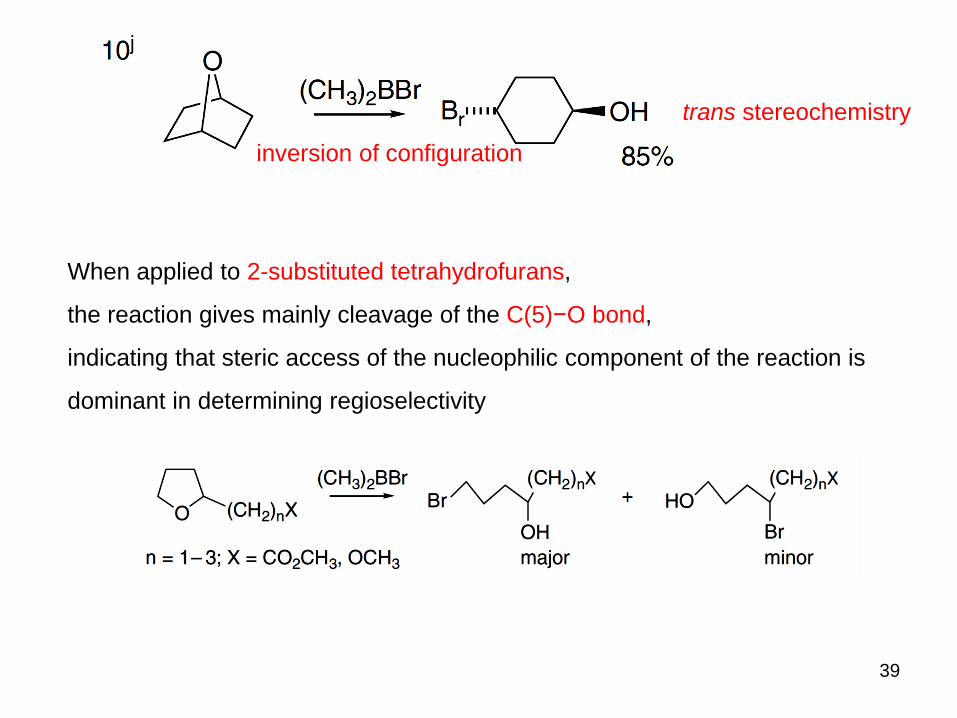
39
inversion of configuration
trans stereochemistry
When applied to 2-substituted tetrahydrofurans,
the reaction gives mainly cleavage of the C(5)−O bond,
indicating that steric access of the nucleophilic component of the reaction is
dominant in determining regioselectivity
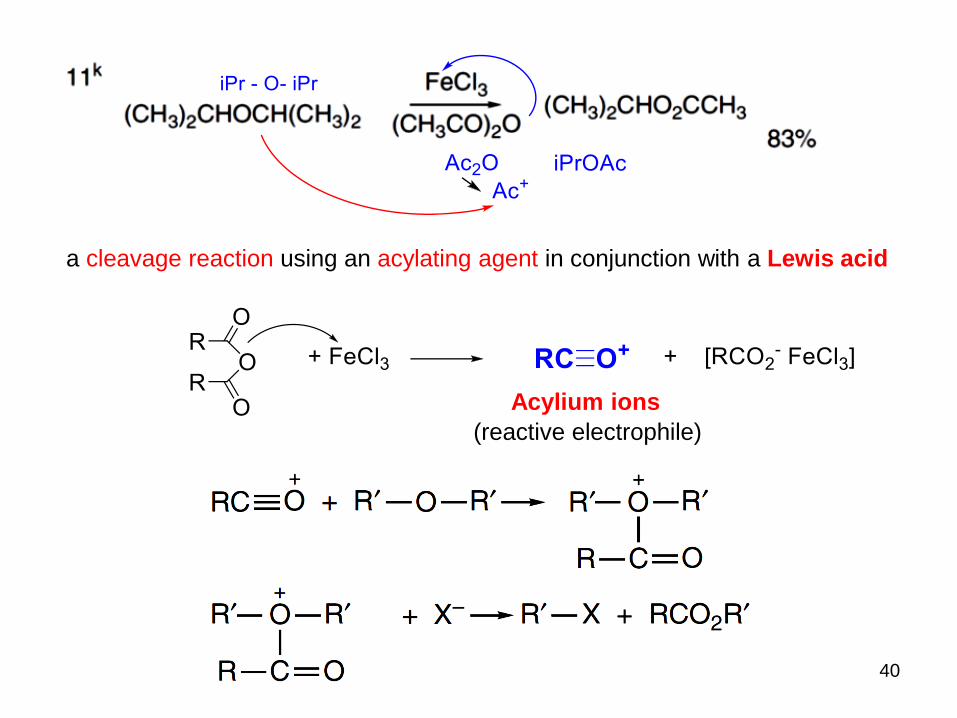
40
a cleavage reaction using an acylating agent in conjunction with a Lewis acid
Acylium ions
(reactive electrophile)