adaptation of a luciferase gene reporter and lac
TRANSCRIPT

APPLIED AND ENVIRONMENTAL MICROBIOLOGY, Mar. 2007, p. 1501–1513 Vol. 73, No. 50099-2240/07/$08.00�0 doi:10.1128/AEM.02454-06Copyright © 2007, American Society for Microbiology. All Rights Reserved.
Adaptation of a Luciferase Gene Reporter and lac ExpressionSystem to Borrelia burgdorferi�†
Jon S. Blevins,1‡ Andrew T. Revel,2‡ Alexandra H. Smith,1Gulnaz N. Bachlani,1 and Michael V. Norgard1*
Department of Microbiology, University of Texas Southwestern Medical Center, Dallas, Texas 75390,1 and Department ofMicrobiology and Molecular Genetics, Harvard Medical School, Boston, Massachusetts 021152
Received 19 October 2006/Accepted 28 December 2006
The development of new genetic systems for studying the complex regulatory events that occur within Borreliaburgdorferi is an important goal of contemporary Lyme disease research. Although recent advancements havebeen made in the genetic manipulation of B. burgdorferi, there still remains a paucity of basic molecularsystems for assessing differential gene expression in this pathogen. Herein, we describe the adaptation of twopowerful genetic tools for use in B. burgdorferi. The first is a Photinus pyralis firefly luciferase gene reporter thatwas codon optimized to enhance translation in B. burgdorferi. Using this modified reporter, we demonstratedan increase in luciferase expression when B. burgdorferi transformed with a shuttle vector encoding the outersurface protein C (OspC) promoter fused to the luciferase reporter was cultivated in the presence of freshrabbit blood. The second is a lac operator/repressor system that was optimized to achieve the tightest degreeof regulation. Using the aforementioned luciferase reporter, we assessed the kinetics and maximal level ofisopropyl-�-D-thiogalactopyranoside (IPTG)-dependent gene expression. This lac-inducible expression systemalso was used to express the gene carried on lp25 required for borrelial persistence in ticks (bptA). Theseadvancements should be generally applicable for assessing further the regulation of other genes potentiallyinvolved in virulence expression by B. burgdorferi.
The zoonotic life cycle of Borrelia burgdorferi, the causativeagent of Lyme disease, is complex, involving both an arthropodtick (Ixodes scapularis) vector and a mammalian host (68). Theability of B. burgdorferi to occupy these two very diverse nichesis governed by a complex regulatory shift that dramaticallyalters the expression of major outer surface proteins (Osps) (3,25, 27, 28, 33, 37, 50, 56, 63, 64, 73). By far the best-charac-terized example of this coordinated response is the reciprocalregulation of OspA and OspC in response to certain environ-mental signals (e.g., pH, cell density, temperature, or blood)(18, 19, 57, 59, 64, 77, 79). OspC is preferentially expressedwhen exposed to conditions akin to those that the bacteriummight encounter in the midgut of a feeding tick or a mamma-lian host. Although there is some discrepancy regarding theprecise role of OspC in the infectious cycle of B. burgdorferi,studies agree that OspC is required for the early events con-tributing to the transition of the bacterium into the mammalianhost (40, 53, 72, 76).
Targeted gene disruption studies have led to the discovery ofthe RpoN/RpoS alternative sigma factor regulatory pathway(16, 34, 42, 83), responsible for modulating key adaptive re-sponses involved in the transition of B. burgdorferi from the tickto the mammal. RpoN, in concert with the response regulatorRrp2 (82), activates the transcription of the alternative sigma
factor gene, rpoS. RpoS, in turn, activates the transcription ofOspC, a member of the group I-regulated lipoproteins (42, 79).RpoS may also function to repress ospA expression, albeitthrough an unknown mechanism (15).
At the present time, the choices for available reporter sys-tems that are suitable for assessing the regulation of selectedgenes in B. burgdorferi are limited. Gene reporter studies, em-ploying primarily green fluorescent protein (gfp)-based systems(15, 16, 20, 31, 32), have been used for elucidating the regu-latory events governing the expression of OspA, OspC, andother members of the group I lipoproteins, particularly inresponse to RpoS activation. To date, only one additional genereporter system, chloramphenicol acetyltransferase (cat), hasbeen utilized in B. burgdorferi (2, 66, 67). Unfortunately, thesestudies utilized a vector that is inherently unstable in B. burg-dorferi, therefore resulting in only transient expression of cat.Another disadvantage of the cat reporter system is that assaysfor measuring cat expression are rather laborious (e.g., en-zyme-linked immunosorbent assay, thin-layer chromatography,radioactive/fluor diffusion, or real-time PCR). One of the morepopular bioluminescent reporters currently being utilized forgene expression studies in both eukaryotes and prokaryotes isbased on Photinus pyralis firefly luciferase (luc) (1, 29, 38, 69).The level of luciferase in a cell lysate is conveniently quanti-tated using a luminometer that measures the light produced asluciferase catalyzes the oxidation of the luciferin reagent; theentire process from harvesting cells to expression analysis re-quires a maximum of 30 min. It is this relative ease and speedby which a luciferase assay can be performed, combined withthe overall high sensitivity of the assay, that make a luc-basedreporter system a preferable alternative to those employing gfpor cat. For these reasons, we sought to determine whether
* Corresponding author. Mailing address: Department of Microbi-ology, University of Texas Southwestern Medical Center, 6000 HarryHines Boulevard, Dallas, TX 75390-9048. Phone: (214) 648-5900. Fax:(214) 648-5905. E-mail: [email protected].
† Supplemental material for this article may be found at http://aem.asm.org/.
‡ J.S.B. and A.T.R. contributed equally to this work.� Published ahead of print on 12 January 2007.
1501
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/a
em o
n 14
Nov
embe
r 20
21 b
y 11
0.14
.202
.42.

luciferase could be used as a transcriptional gene reporter in B.burgdorferi.
An inherent potential difficulty in utilizing all reporter sys-tems in B. burgdorferi lies in the codon usage by this bacterium(60). Because B. burgdorferi has a GC content of only 28.6%,the codon bias for borrelial genes is shifted towards AT-richcodons (36). This bias compromises the translation of reportergenes developed for organisms with a higher GC content (e.g.,Escherichia coli lacZ [GenBank accession NC_000913], 56.3%GC; or P. pyralis luc [GenBank accession U47122.2], 46.8%),thereby potentially making reporter activity an inaccurate rep-resentation of transcriptional activity. Herein, we sought toadapt a P. pyralis firefly luc reporter for use in differential geneexpression analyses in B. burgdorferi, with an emphasis on mod-ifying the codon bias of the luc gene to better match that of B.burgdorferi.
Another genetic tool that has been unavailable to Borreliaresearchers is an inducible expression system. Inducible sys-tems, such as the lac operator/repressor and tet operator/re-pressor, were originally derived from E. coli (9, 10, 41), buthave been utilized in numerous bacterial species with full func-tionality (24, 43–45, 52, 74, 85). The development of inducibleexpression systems will serve to advance the borrelial field, asit will allow researchers to perform a multitude of experiments,including the generation of conditional lethal mutants (44, 45,52, 74) and the ectopic expression of putative virulence/regu-latory factors (24, 43, 52). To this end, Cabello et al. (14)described the first inducible expression system, based on the E.coli Tn10 tet operon, for B. burgdorferi. To further expand thegenetic armamentarium available to Borrelia researchers, wehave adapted the lac operator/repressor system for applicationin B. burgdorferi. The relative simplicity of the lac-induciblesystem makes it ideally suited to application in B. burgdorferi(9, 10). For full function, this system requires only a constitu-tively expressed repressor (LacI), a promoter sequence con-taining a LacI-binding site (operator), and a suitable inducer,such as isopropyl-�-D-thiogalactopyranoside (IPTG). In thepresent study, we demonstrate the first application of theIPTG-inducible lac repressor/operator system to B. burgdorferigenetics.
MATERIALS AND METHODS
Bacterial strains and culture conditions. All strains and plasmids used in thisstudy are described in Table 1. E. coli strains XL1-Blue (Stratagene, La Jolla,CA) and TOP10 (Invitrogen, Carlsbad, CA) were used as cloning hosts. Trans-formation of B. burgdorferi was carried out as described by Yang et al. (84). BptAinducible expression experiments employed the previously characterizedbptA::aadA insertion mutant, BbDTR596 (Table 1) (58). BbDTR630 (Table 1)was utilized for borrelial expression experiments to characterize the luciferasereporter and lac inducible expression system. BbDTR630, which carries anaph[3�]-IIIa aminoglycoside resistance gene on the virulence-associated plasmidlp25 (39, 54, 55, 78), was generated from an isolate of the infectious, nonclonalstrain 297 designated PL133 (Table 1) (58). The integration of the selectablemarker was achieved by first amplifying aph[3�]-IIIa from pMS2 (Table 1) (62)using Takara Ex-Taq polymerase (Takara Bio Inc., Japan) and the primersaph-IIIa 5�-AscI and aph-IIIa 3�-AscI (see Table S1 in the supplemental mate-rial), which also incorporated AscI restriction sites into the 5� and 3� ends of theresistance marker. The AscI-flanked resistance marker then was subcloned intopDTR627 (Table 1) (58), a suicide vector carrying a 3.7-kb region of lp25 with aunique AscI restriction site (engineered 40 bp downstream of the pncA/bbe22gene). The resulting construct, pDTR630 (Table 1), then was transformed intoPL133 to generate BbDTR630. Phenotypic analyses revealed that BbDTR630exhibited in vitro growth characteristics and OspA/OspC regulatory patterns
indistinguishable from wild-type PL133 (79). Furthermore, when the infectivityof BbDTR630 was assessed using the murine (needle-challenge) model of Lymeborreliosis (7, 8), there was no change in 50% infective dose (ID50) valuesrelative to the PL133 parent (data not shown). For luciferase reporter validationstudies, B. burgdorferi clones were cultured in Barbour-Stoenner-Kelley II (BSK-II) medium (6). BSK-H incomplete medium (Sigma Chemical Co., St. Louis,MO) supplemented with 6% rabbit serum (Pel-Freez Biologicals, Rogers, AR)was used for experiments characterizing the inducible expression system. Kana-mycin (Kan) at a final concentration of 160 �g/ml was included in BSK mediumfor the selection of Kanr transformants. To select for streptomycin-resistant(Strepr) transformants, streptomycin was added to cultures at a final concentra-tion of 100 �g/ml. Cell culture density was assessed by enumerating spirochetesusing dark-field microscopy. To ensure accuracy of density determination, trip-licate counts were performed on culture samples.
Modification of pKFSS1 shuttle vector. The existing cp9-based B. burgdorferishuttle vectors, pBSV2 (71) and pKFSS1 (35) (Table 1), contain an unnecessaryzeocin resistance marker that, with its flanking sequence, increases the size of theshuttle vector approximately 1 kb. To remove this extraneous DNA, the specti-nomycin/streptomycin resistance marker (PflgB-aadA) of pKFSS1 first was am-plified by PCR with primers FlgBprom-HindIII-5� and FlgB/aadA-BspHI-3� (seeTable S1 in the supplemental material) and TA-cloned into pGEM-T easy(Promega Corp., Madison, WI). Following confirmation of the TA clones,pKFSS1 was digested with HindIII and BspHI and the shortened PflgB-aadA wassubstituted for the original PflgB-aadA and flanking zeocin resistance marker togenerate pJD1 (Table 1). pKFSS1 and pBSV2 also contain two promoter se-quences that could confound promoter/gene reporter analyses. The first pro-moter sequence is Plac, which was originally derived from the Plac/LacZ/MCSregion of pCR-XL-TOPO (Invitrogen). The second promoter is located in theborrelial plasmid origin of replication. This promoter is upstream of the trun-cated BBC12 open reading frame (ORF) and overlaps with one of the highlyconserved inverted repeats flanking the BBC01-to-BBC03 gene cluster (21, 30,36, 71). Because the precise function of these inverted repeats is not fullyunderstood, mutation of the putative promoter elements was a less-favoredapproach. Therefore, an alternative approach of introducing a transcriptionalterminator upstream of the shuttle vector MCS was employed. A 253-bp frag-ment of the MCS, Plac, was excised from pJD1 (Table 1) by digestion with EagIand KpnI. A Rho-independent intrinsic transcriptional terminator, designedfrom the sequence of cp8.3 (30) and the consensus of Lesnik et al. (46), then wasgenerated by annealing four complementary oligonucleotides; Bb term frag #1,Bb term frag #2, Bb term frag #3, and Bb term frag #4 (see Table S1 in thesupplemental material). The annealed product possessed overhangs capable ofligating to pJD1 digested with EagI and KpnI. The resulting plasmid was desig-nated pJD7 (Table 1 and Fig. 1A).
Codon adaptation of Photinus pyralis firefly luciferase from pSP-luc�. Codonoptimization of the luciferase ORF from the pSP-luc� reporter (Table 1) (Pro-mega) was achieved using gene building technology. The DNAbuilder program(Preston Hunter and Stephen Albert Johnston, Center for Innovations in Med-icine, Arizona State University; www.biodesign.asu.edu) converted the aminoacid sequence for the luciferase ORF of pSP-luc� (lucSp
�) to a DNA sequenceusing the codon usage parameters for B. burgdorferi at http://www.kazusa.or.jp/codon (49). Overlapping (20 bp) 50-mer oligonucleotides then were synthe-sized from the optimized DNA sequence (Midland Certified Reagent Co., Mid-land, TX), and the gene was constructed using the assembly PCR technique (70).Briefly, the oligonucleotides were reconstituted to a concentration of 100 �M,pooled, and then diluted to a final concentration of 25 �M. This oligonucleotidepool served as the template in the primary assembly PCR. A second PCRcontaining only the 5�- and 3�-terminal oligonucleotides and an aliquot of thefirst round of PCR then was performed to amplify the full-length gene product.The resulting PCR product was gel purified and ligated into pGEM-T Easy.Transformants were verified by restriction digest and DNA sequence analysis.The confirmed plasmid was designated pJSB80 (Table 1). A list of the oligonu-cleotides used for the PCR assembly reaction is provided in Table S2 in thesupplemental material.
To assess the impact of codon optimization, the pre- and postadapted lucgenes, lucSp
� and lucBb�, respectively, were fused to the promoter of the con-
stitutively expressed flagellar core protein FlaB (PflaB) and ligated into pJD7. Toachieve this, a 251-bp fragment of DNA containing PflaB first was PCR amplifiedfrom B. burgdorferi PL133 using the primers flaBpro5�-XbaI and flaBpro3�-NcoI-2nd (see Table S1 in the supplemental material) and cloned into pGEM-TEasy. Next, the lucSp
� ORF was PCR amplified from pSP-luc� using the primers5�luciferase-2nd and 3� luciferase (see Table S1 in the supplemental material).PflaB and the corresponding luc ORFs then were excised from pGEM-T easy bydigestion with XbaI/NcoI and NcoI/HindIII, respectively, and ligated into pJD7
1502 BLEVINS ET AL. APPL. ENVIRON. MICROBIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/a
em o
n 14
Nov
embe
r 20
21 b
y 11
0.14
.202
.42.
that was linearized by digestion with XbaI and HindIII. The resulting clones wereverified by restriction digestion and DNA sequence analysis. pJD7::PflaB-SPluc� and pJD7::PflaB-Bbluc� are designated herein as pJSB66 and pJSB82(Table 1 and Fig. 1B), respectively. pJSB66 and pJSB82 were transformed intoBbDTR630, to generate BbJSB66 and BbJSB82 (Table 1). To confirm Strepr
transformants, DNA was isolated from Borrelia clones using the Wizard Plus SVmini-prep kit (Promega) and transformed into E. coli. Plasmid DNA was isolatedfrom the resulting E. coli clones and verified by restriction digestion and DNAsequence analysis.
To assess codon optimization, BbJSB66 and BbJSB82 were inoculated intoBSK-H medium containing Strep selection from frozen stocks stored at �70°C.Once these cultures grew to an adequate density (�1 � 107 spirochetes/ml), theywere used to inoculate fresh BSK-H medium, also containing Strep, to a densityof 1 � 103 cells/ml. These cultures were grown for approximately 3 days to a celldensity of between 0.5 � 106 and 5 � 106 spirochetes/ml, at which time four 1-ml
aliquots of each culture were transferred to 1.5-ml microcentrifuge tubes and thespirochetes were collected by centrifugation (10 min at 10,000 � g). The mediumwas aspirated, and pellets were retained for luciferase assays.
Luciferase assays. A commercial luciferase assay system (Promega) was usedin this study. Lysates were prepared by resuspending Borrelia cell pellets in 100�l of cell culture lysis mixture containing 25 mM Tris-phosphate (pH 7.8), 2 mMdithiothreitol, 2 mM 1,2-diaminocyclohexane-N,N,N�,N�-tetraacetic acid, 10%glycerol, 1% Triton X-100, 0.125% lysozyme, and 0.25% bovine serum albumin.Next, the cell suspension was thoroughly mixed by vortexing for 1 min. Tenmicroliters of lysate was aliquoted into a black 96-well assay plate (Corning Inc.,Corning, NY), and immediately prior to beginning measurements, 50 �l ofluciferase assay reagent was added to each well. The reactions were mixed byagitating the plate for 5 s, after which the luciferase activity in each well wasmeasured for 1 s using a Centro LB 960 luminometer (Berthold Technologies,Oak Ridge, TN). Measurements are reported as relative luciferase units (RLU)
TABLE 1. Strains and plasmids used in this study
Strain or plasmid Description Source or reference
StrainsE. coli
XL1-Blue recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac�F� proAB lacIqZM15::Tn10 (Tetr) StratageneTOP10 F� mcrA (mrr-hsdRMS-mcrBC) �80lacZM15 lacX74 recA1 ara139 (ara-leu)7697 galU
galK rpsL (Strr) endA1 nupGInvitrogen
B. burgdorferiPL133 Transformable, nonclonal, infectious derivative of strain 297 58BbDTR630 Infectious, clonal derivative of PL133, lp25::aph�3�-IIIa; Kanr This studyBbDTR596 Infectious, clonal BptA mutant of PL133, bptA::aadA; Strepr 58BbJSB66 BbDTR630 transformed with pJSB66; Strepr Kanr This studyBbJSB82 BbDTR630 transformed with pJSB82; Strepr Kanr This studyBbJSB161 BbDTR630 transformed with pJSB161; Strepr Kanr This studyBbJSB165 BbDTR630 transformed with pJSB165; Strepr Kanr This studyBbJSB175 BbDTR630 transformed with pJSB175; Strepr Kanr This studyBbJSB56 BbDTR630 transformed with pJSB56; Strepr Kanr This studyBbJSB70 BbDTR630 transformed with pJSB70; Strepr Kanr This studyBbJSB104 BbDTR630 transformed with pJSB104; Strepr Kanr This studyBbJSB252 BbDTR630 transformed with pJSB252; Strepr Kanr This studyBbJSB194 BbDTR596 transformed with pJSB194; Strepr Kanr This study
PlasmidspGEM-T easy TA cloning vector; Ampr PromegapMS2 B. burgdorferi/E. coli shuttle vector with aph�3�-IIIa; Kanr 62pDTR627 pGEM-T Easy::lp25 region with AscI; Ampr 58pDTR630 pDTR627::AscI-aph�3�-IIIa-AscI; Ampr Kanr This studypKFSS1 B. burgdorferi/E. coli shuttle vector with PflgB-aadA; Spec/Strepr 35pJD1 pKFSS1 with minimal PflgB-aadA region; Spec/Strepr This studypJD7 pJD1 with Plac excised and terminator inserted; Spec/Strepr This studypSP-luc� Luciferase gene reporter construct; Ampr PromegapJSB80 pGEM-T Easy::Bbluc� (codon-optimized SPluc�); Ampr This studypJSB66 pJD7::PflaB-SPluc�; Spec/Strepr This studypJSB82 pJD7::PflaB-Bbluc�; Spec/Strepr This studypJD44 pJD7-based shuttle vector with aph�3�-IIIa; Kanr 58pJD48 pJD44::promoterless lucBb
�; Kanr This studypJSB161 pJD7::divergently oriented promoterless lucBb
�; Spec/Strepr This studypJSB165 pJD7::divergently oriented PospC-Bbluc�; Spec/Strepr This studypJSB175 pJD7::divergently oriented PflaB-Bbluc�; Spec/Strepr This studypQE-30 Expression vector with lac-inducible T5 promoter; Ampr QIAGENpJSB91 pGEM-T Easy::PpQE30 promoter region; Ampr This studypJSB92 pGEM-T Easy::PpQE30-Bbluc�; Ampr This studypJSB70 pJD7::PpQE30-Bbluc�; Spec/Strepr This studypET-11a Expression vector with T7 tag and lacIq; Ampr NovagenpJSB104 pJD7::PpQE30-Bbluc� and PflaB-BblacI (tandem); Spec/Strepr This studypJSB252 pJD7::PpQE30-Bbluc� and PflaB-BblacI (divergent); Spec/Strepr This studypJSB56 pJD7::promoterless lucBb
�; Spec/Strepr This studypDTR644 pJD44::bbe17pro:bptA (BglII-HindIII); Kanr 58pJSB184 pGEM-T easy::PpQE30-bptA; Ampr This studypJSB186 pJD7::PpQE30-bptA and PflaB-BblacI (tandem); Spec/Strepr This studypJSB194 pJD44::PpQE30-bptA and PflaB-BblacI (tandem); Kanr This study
VOL. 73, 2007 CONTROLLABLE GENE REPORTER FOR B. BURGDORFERI 1503
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/a
em o
n 14
Nov
embe
r 20
21 b
y 11
0.14
.202
.42.

with average background luminescence subtracted from the readings. Unlessotherwise noted, results are presented as the RLU/1 � 105 spirochetes.
Generation of luciferase reporter constructs for validation experiments. Tovalidate the luciferase reporter, two promoters were fused to the lucBb
� ORF.Due to restriction site incompatibility, the promoter-reporter fusions could notbe assembled directly in pJD7. Therefore, a shuttle vector carrying the promot-erless lucBb
� ORF (pJD48; Table 1) was generated first by ligating the HindIII/BglII fragments of pJSB80 (pGEM-T Easy::Bbluc�) and the pJD44 shuttlevector (Table 1 and Fig. 1A). A 140-bp DNA fragment containing the ospCpromoter (PospC) was amplified using PL133 DNA as a template and the primerset 5�OspC up 1723-BglII and 3�ospC prom-NdeI #2 (see Table S1 in thesupplemental material). The PCR fragment was TA cloned into pGEM-T Easy,verified by DNA sequencing, excised with NdeI and BglII, and ligated into pJD48digested with same restriction enzymes. Because BbDTR630 is already Kanr, itwas necessary to replace the aph[3�]-IIIa cassette in the pJD44-based shuttlevectors with PflgB-aadA from pJD7. The marker replacement was achieved firstby digesting pJD7, pJD48, and pJD48-PospC with SpeI and BglII to excise afragment containing the relevant marker. The pJD7-derived PflgB-aadA frag-ment then was ligated into pJD48 and pJD48-PospC to construct pJSB161 andpJSB165 (Table 1 and Fig. 1B), respectively. The flaB promoter for the positivecontrol PflaB-Bbluc� was isolated from pJSB82 (pJD7::PflaB-Bbluc�) digestedwith BamHI and NdeI. The fragment encoding the promoterless lucBb
� wasexcised from pJD48 by digesting with NdeI and SpeI, and, as before, PflgB-aadAwas derived from the BglII/SpeI fragment of pJD7. These fragments then wereligated to create pJSB175 (Table 1 and Fig. 1B). Note that in these plasmids, thelucBb
� reporter and resistance marker are divergently oriented to prevent tran-scriptional readthrough from upstream genes. The B. burgdorferi clones recov-ered after electroporation of BbDTR630 with pJSB161, pJSB165, or pJSB175were designated BbJSB161, BbJSB165, and BbJSB175 (Table 1), respectively. Toverify Strepr clones, DNA was isolated from Borrelia transformants and electro-porated into E. coli. Plasmid DNA was isolated from the resulting E. coli clonesand characterized by restriction digestion and DNA sequence analysis.
To validate the luc reporter, the influence of blood supplementation (77) onthe expression of luciferase was assessed in cultures of BbJSB161, BbJSB165,and BbJSB175. BbJSB161, BbJSB165, and BbJSB175 were inoculated fromfrozen stocks stored at �70°C into BSK-II medium supplemented with Strep.When these cultures grew to the appropriate density (approximately 1 � 107
spirochetes/ml), they were used to inoculate 40 ml of fresh BSK-II medium, alsocontaining Strep, to a dilution of 1 � 103 spirochetes/ml. After cultures reacheda density between 0.3 � 106 to 3 � 106 cells/ml, they were divided. One culturewas treated with 6% fresh heparinized rabbit blood (10 ml BD sodium heparinVacutainer; Becton Dickinson Co., Franklin Lakes, NJ) with the buffy coatextracted, while the second culture was treated with 6% heparin-supplementedBSK-II (�14 USP/ml). Cultures were grown for 2 days with intermittent mixingto resuspend the red blood cells (RBCs). At 2 days posttreatment, the cell densityfor each was determined and four 1-ml aliquots were collected. Immediatelyprior to processing, 6% whole blood was added to the aliquots of the culturegrown in the absence of blood; hemoglobin released from RBCs during lysatepreparation quenches the luminescence signal, thereby skewing the readings (22,65). RBCs and bacterial cells then were collected by centrifugation (10 min at10,000 x g), lysed, and luciferase assays were performed as described above. Theremaining portion of the culture was processed for quantitative real-time PCR(qRT-PCR) analysis (see below).
qRT-PCR analysis. Cultures of BbJSB161, BbJSB165, and BbJSB175 cellswere grown and treated as described above. Immediately prior to centrifugationof cultures to collect cells, 6% whole blood was added to the cultures grown inthe absence of blood to standardize purification. To prevent RNase-dependentmRNA degradation, a 1/10 volume of chilled 5% water-saturated phenol (pH�7.0) in ethanol was added to each culture. Cell pellets were stored at �80°Cprior to processing. Total RNA was extracted from cell pellets using Trizol(Invitrogen), as per the manufacturer’s instructions, purified from the Trizol-extracted aqueous phase using RNeasy mini columns (QIAGEN Inc., Valencia,CA). Purified RNA then was treated with DNase, and RNA integrity was de-termined on the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto,CA); RNA integrity numbers were 8 for all samples. Relative levels of ospC,lucBb
�, and flaB transcripts were determined using the Applied Biosystems 7500reverse transcription-PCR (RT-PCR) system and SYBR Green one-step qRT-PCR (Applied Biosystems, Foster City, CA). The primer sets (see Table S1 in thesupplemental material) ospCF391 and ospCR479, lucF607 and lucR688, andflaBF9 and flaBR82 were used for ospC, lucBb
�, and flaB, respectively. Twenty-five nanograms of RNA was added per 25-�l reaction mixture with six replicatesrepresented per sample. Each reaction contained 12.5 �l SYBR Green PCRmaster mix (Applied Biosystems), 0.5 �l of each primer (2.5 mM), and 0.125 �l
FIG. 1. Diagram illustrating the two B. burgdorferi shuttle plas-mids used in this study (A) and the relevant regions and restrictionsites of derivative constructs generated and transformed into B.burgdorferi (B). pJSB66 and pJSB82 were used to assess the impactof codon optimization on luciferase expression. pJSB161, pJSB165,and pJSB175 were created for the lucBb
� reporter (indicated asBbluc�on the figure) validation studies. pJSB56, pJSB70, pJSB104,and pJSB252 were generated for the development and evaluation ofa lac repressor/operator system optimized for use in B. burgdorferi.pJSB194 was used to express the BptA protein under the control ofthe lac-inducible expression system. SPluc�, lucSp
�.
1504 BLEVINS ET AL. APPL. ENVIRON. MICROBIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/a
em o
n 14
Nov
embe
r 20
21 b
y 11
0.14
.202
.42.

Multiscribe reverse transcriptase (50 U/�l; Applied Biosystems). The run pro-tocol consisted of a reverse transcription step (30 min at 48°C), a denaturationstep (10 min at 95°C), and a 40-cycle quantitation program (15 s at 95°C, 60 s at60°C). The results of two independent sets of cultures are reported in theResults. Because the amplification efficiencies of the reference (flaB) and theunknowns (ospC and lucBb
�) were approximately equal, the Ct method ofrelative transcript quantitation could be applied to these samples (Applied Bio-systems 7500 system—SDS software version 1.3.1.22). Control reactions, inwhich reverse transcriptase was excluded, were negative for amplification.
Generation of test constructs for the borrelial lac inducible expression system.The lac inducible promoter from pQE-30 (QIAGEN Inc.), PpQE30, was gener-ated by annealing four complementary oligonucletides; pQE30 prom I, pQE30prom II, pQE30 prom III, and pQE30 prom IV (see Table S1 in the supplemen-tal material). The annealed fragment contained 5� and 3� overhangs that werecomplementary to the ends generated when pJD7 was digested with XbaI andPstI. The PstI site at the 3� end of the promoter then was exchanged for an NdeIsite to allow fusion of the promoter to the lucBb
� reporter gene. This modifica-tion was achieved by PCR amplification of the promoter region in the shuttlevector with a 5� oligonucleotide anchored upstream of PpQE30 in pJD7 and a 3�primer that mutated the PstI site, cp9 ORF seq and pQE30 3� NdeI (see TableS1 in the supplemental material), respectively. This NdeI-modified PpQE30fragment was cloned into pGEM-T Easy to generate pJSB91 (Table 1). ThelucBb
� fragment was excised from pJSB80 (pGEM-T easy::Bbluc�) by digestionwith NdeI and ligated with pJSB91 also digested with NdeI. The resultingtransformants were screened for proper fusion of the promoter and reporterfragments. Once a correct clone (pJSB92; Table 1) was identified, the PpQE30-Bbluc� fusion was excised with AscI/HindIII and ligated into pJD7 that waslinearized with AscI and HindIII. The resulting vector was designated pJSB70(Table 1 and Fig. 1B).
The second requisite component required for the lac inducible expressionsystem, a highly-expressed constitutive LacI repressor, was generated by PCRamplifying a 243-bp DNA fragment encoding the flaB promoter from PL133using the primers 5�PflaB-HindIII and 3�PflaB-Nco/ATG (see Table S1 in thesupplemental material). To further enhance the expression of LacI, the lacI ORFfrom pET-11a (Novagen, San Diego, CA) (Table 1) was codon adapted using thegene-building approach noted earlier. A list of the oligonucleotides synthesizedfor lacI codon optimization is provided in Table S2 in the supplemental material.To fuse PflaB and the gene-built LacI ORF (lacIBb), the two fragments weredigested with HindIII/NcoI and BspHI/BglII and ligated into pJSB70 digestedwith HindIII and BglII. The resulting plasmid is referred to as pJSB104 (Table1 and Fig. 1B). To generate pJSB252 (Table 1 and Fig. 1B), in which theconstitutive LacI is divergently oriented upstream from the inducible lucBb
�, thePflaB-BblacI was PCR amplified using pJSB104 as a template and the primer set5� PflaB-XbaI and 3� BbLacI-BamHI (see Table S1 in the supplemental mate-rial). The PCR fragment was cloned into pGEM-T Easy and verified by DNAsequence analysis. The PflaB-BblacI insert was excised using XbaI and BamHIand then ligated into pJSB70 linearized with the same restriction enzymes. Thepromoterless lucBb
� construct, pJSB56 (Table 1 and Fig. 1B), was generated byligating the XbaI/HindIII-digested lucBb
� fragment from pJSB80 and pJD7 alsodigested with XbaI and HindIII. The B. burgdorferi clones recovered after elec-troporation of BbDTR630 with pJSB56, pJSB70, pJSB104, or pJSB252 are des-ignated BbJSB56, BbJSB70, BbJSB104, and BbJSB252, respectively (Table 1).Strepr transformants were verified by electroporation of E. coli with DNA iso-lated from the Borrelia transformants. Plasmid DNA was purified from theresulting E. coli clones and confirmed by restriction digestion and DNA sequenceanalysis.
For test inductions, BbJSB56, BbJSB70, BbJSB104, and BbJSB252 were in-oculated into BSK-H medium containing Strep from frozen stocks stored at�70°C. Once these cultures grew to a cell density of approximately 1 � 107
spirochetes/ml, they were used to inoculate fresh BSK-H medium, also supple-mented with Strep selection, to a dilution of 1 � 103 spirochetes/ml. Thesecultures were grown to a cell density between 0.5 � 106 and 1 � 106 cells/ml, atwhich time the cultures were induced with various concentrations of IPTG. Atthe appropriate time intervals, the cell density was determined and quadruplicatesamples were processed for luciferase assays as described above (for BbJSB66and BbJSB82).
The bptA inducible expression construct, pJSB194 (Table 1 and Fig. 1B), wasgenerated by excising the BptA ORF from pDTR644 (Table 1) (58) usingNdeI/PstI and ligating it into pJSB91(pGEM-T Easy::Ppqe30 promoter) digestedwith NdeI and NsiI. The resulting construct, containing PpQE30 fused to thebptA ORF, was designated pJSB184 (Table 1). The PpQE30-bptA fusion wasremoved from pJSB184 using AscI and HindIII and combined with pJSB104digested with the same enzymes to generate pJSB186 (Table 1). Because the
BptA test induction was to be carried out in BbDTR596 (Table 1) (58), which isalready Strepr, the induction cassette had to be moved into pJD44. To achieve this,the region containing the PpQE30-bptA fusion and PflaB-BblacI was removed frompJSB186 using XbaI and BglII and inserted into XbaI/BglII-digested pJD44. Theresulting construct, pJSB194 (Table 1 and Fig. 1B), was electroporated intoBbDTR596 to generate BbJSB194 (Table 1). DNA was isolated from Kanr clonesand transformed into E. coli. Plasmid DNA was isolated from E. coli transformantsand verified by restriction digestion and DNA sequence analysis. Inductions withBbJSB194 were carried out as detailed above (for the PpQE30-Bbluc� studies), andculture aliquots were collected at 6 h after the addition of 1 mM IPTG for assess-ment by immunoblotting.
Immunoblot analyses. BbJSB56, BbJSB70, BbJSB104, and BbJSB194 wereinoculated into BSK-H medium containing Strep from frozen stocks stored at�70°C. Once these cultures grew to a density of approximately 1 � 107 spiro-chetes/ml, they were used to inoculate fresh BSK-H medium, also supplementedwith Strep selection, to a dilution of 1 � 103 spirochetes/ml. These cultures weregrown to a cell density of 1 � 106 cells/ml at which time the cultures were divided;one culture remained untreated, while 1 mM IPTG was added to the second. At0, 6, and 24 h postinduction, the cell density was determined and quadruplicatesamples of BbJSB56, BbJSB70, and BbJSB104 were processed for luciferaseassays to confirm proper induction of lucBb
�. Samples also were collected andprocessed for sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting, as previously described (81). Immunoblots weredeveloped using the SuperSignal West Pico chemiluminescent substrate accord-ing to the manufacturer’s instructions (Pierce, Rockford, IL). Unless otherwisenoted, a volume of whole-cell lysate equivalent to 1 � 107 spirochetes was loadedper gel lane. Luc was detected using a commercially available goat polyclonalantibody directed against recombinant luciferase (Promega). The monoclonalanti-LacI antibody from clone 9A5 was supplied by Upstate USA (Charlottes-ville, VA). The chicken immunoglobulin Y anti-FlaB antibody was a generousgift from Kayla Hagman (University of Texas Southwestern Medical Center,Dallas). The generation of the rat anti-BptA antisera was previously reported(58). The molecular mass standard, Bio-Rad Laboratories (Hercules, CA) AllBlue Precision Plus marker, was detected using horseradish peroxidase (HRP)-conjugated StrepTactin (Bio-Rad).
Nucleotide sequence accession numbers. The sequences for the codon-opti-mized lucBb
� and lacIBb ORFs have been deposited in GenBank under accessionno. EF043384 and EF043385, respectively.
RESULTS
Adaptation of Photinus pyralis firefly luciferase reporter foruse in B. burgdorferi. To date, there is only one gene reportersystem, gfp, whose utility has been demonstrated for differen-tial gene expression studies in B. burgdorferi (5, 13, 15, 16, 20,31, 32, 47). Because of the limitation in the number of suitablereporter systems, we sought to identify additional transcrip-tional reporters (such as firefly luciferase) that might also beused in Borrelia. However, codon usage analysis of the lucSp
�
gene (derived from pSP-luc�) revealed numerous codons thatare significantly underutilized by B. burgdorferi (see Table S3 inthe supplemental material) (36, 49). Because these “rare”codons could result in inefficient translation, it was possiblethat results from reporter studies obtained using the lucSp
�
ORF might not accurately reflect transcriptional activity. Tocorrect this potential expression bias, the lucSp
� ORF wascodon adapted to optimize luc expression in B. burgdorferi (49,70). Codon optimization resulted in a change in the luc genefrom 46.8% GC to 35.9% GC. To assess the impact of codonoptimization on luciferase expression/activity, the constitutiveflaB promoter (PflaB) was fused to the codon-adapted lucifer-ase ORF (lucBb
�), ligated into the pJD7 shuttle vector(pJSB82; Fig. 1B), and transformed into BbDTR630. An anal-ogous construct containing the lucSp
� ORF also was generated(pJSB66; Fig. 1B) and transformed into BbDTR630. The lu-ciferase activity for the codon-optimized lucBb
� (see Table S3
VOL. 73, 2007 CONTROLLABLE GENE REPORTER FOR B. BURGDORFERI 1505
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/a
em o
n 14
Nov
embe
r 20
21 b
y 11
0.14
.202
.42.

in the supplemental material) ranged from four- to sixfoldgreater than that of the unmodified lucSp
� (Fig. 2).Validation of lucBb
� luciferase gene reporter. Although thecomparison between pJSB66 and pJSB82 (above) indicated thatactive lucBb
� was expressed in B. burgdorferi, it did not address theextent to which lucBb
� could be used to assess differential geneexpression. To validate the lucBb
� reporter, PospC was fused tolucBb
� and introduced into the pJD7 shuttle vector to createpJSB165 (Table 1 and Fig. 1B). A positive control construct(pJSB175; Table 1 and Fig. 1B), in which the constitutive PflaBwas fused to lucBb
�, also was generated. The construct pJSB161(Table 1 and Fig. 1B), which contains the promoterless lucBb
�,served as a negative control. pJSB161, pJSB165, and pJSB175were electroporated into BbDTR630 to generate BbJSB161,BbJSB165, and BbJSB175, respectively. Transformants were ver-ified as described in Materials and Methods.
OspC is preferentially expressed in vitro when B. burgdorferiis cultured under conditions emulating those that the spiro-chete might encounter in the mammalian host or the midgut ofa feeding tick (18, 19, 57, 59, 64, 77, 79). For instance, Yang etal. (79) demonstrated that elevated temperature, decreasedpH, and increased cell density act in concert to enhance ospCexpression. Unfortunately, the impact of these modified cul-ture conditions on the relative level of OspC induction also isdependent on the formulation of the BSK medium (4, 80). An
alternative approach for upregulating OspC expression, there-fore, was employed (77) in which the bacteria were exposed tofresh 6% heparinized whole rabbit blood for 2 days. BbJSB161,BbJSB165, and BbJSB175 were grown, and when the culturesreached the appropriate cell density (0.3 � 106 to 3 � 106
cells/ml), they were divided. One culture was treated with he-parinized blood, whereas the second culture was treated withheparin-supplemented BSK-II medium. The results from arepresentative trial are shown in Fig. 3A. The addition of bloodresulted in an 18.5-fold increase in luciferase activity fromBbJSB165, whereas blood had no effect on the activity ofBbJSB175 (Fig. 3A). The average luciferase activity observedin BbJSB161 was barely above background RLU and was un-affected by blood supplementation (Fig. 3A). The data ob-tained with BbJSB165 indicated that ospC was consistentlyupregulated upon treatment of cultures with blood, as has been
FIG. 2. The impact of codon optimization on the expression offirefly luciferase. The PflaB promoter was fused to either the lucSp
�
(pJSB66) or lucBb� (codon-optimized, pJSB82) ORFs and the result-
ing clones were transformed into B. burgdorferi. After cultures hadgrown to a cell density of approximately 0.5 � 106 to 5 � 106 spiro-chetes/ml, four 1-ml aliquots of each culture were collected for lucif-erase assays. RLU readings were normalized according to cell densityand are presented as the RLU/1 � 105 bacteria � standard deviation.The results from two independent experiments (trials 1 and 2) arepresented.
FIG. 3. Influence of blood supplementation (in BSK medium) onluciferase expression driven by various B. burgdorferi promoters. Cul-tures of BbJSB161 (promoterless lucBb
�), BbJSB165 (PospC-Bbluc�),and BbJSB175 (PflaB-Bbluc�) were treated with either 6% fresh he-parinized rabbit blood (�6% blood) or with 6% heparin-supple-mented BSK medium (�6% blood). Two days posttreatment, sampleswere removed and processed for luciferase assays. (A) Luciferaseactivities (RLU) from quadruplicate samples of each culture werestandardized according to a cell density of 1 � 105 spirochetes, andresults are presented as the mean RLU/1 � 105 bacteria � standarddeviation. (B) qRT-PCR analysis was performed on RNA extractedfrom the culture of BbJSB165 (PospC-Bbluc�) used for luciferaseassays (above). SYBR Green one-step qRT-PCR was used to deter-mine the relative levels of ospC, lucBb
�, and flaB transcripts; flaB wasincluded in the analyses for the purpose of signal standardization. Theresults from six replicate reactions are presented as the mean foldchange (fold [relative to heparin-treated culture]) � standard devia-tion. Two independent experiments were performed, and representa-tive results from one trial are provided in the figure.
1506 BLEVINS ET AL. APPL. ENVIRON. MICROBIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/a
em o
n 14
Nov
embe
r 20
21 b
y 11
0.14
.202
.42.

previously demonstrated (77). Furthermore, this increase wasspecific for the PospC-Bbluc� fusion in BbJSB165, because sup-plementation of cultures of BbJSB161 (promoterless lucBb
�) orBbJSB175 (PflaB-Bbluc�) with whole blood had no measurableeffect on luciferase activity.
Although the results obtained with BbJSB165 suggested thatthe codon-adapted luciferase could be used for differentialgene expression analyses in B. burgdorferi, it was still necessaryto verify that the increase in luciferase activity after cultivation(in the presence of blood) correlated with an increase inPospC-Bbluc� transcription. It also was necessary to deter-mine whether the increase in PospC-Bbluc� expression wasequivalent to the increase in the expression of the native ospCgene present on circular plasmid cp26 (36). Therefore, to fur-ther validate the lucBb
� reporter, total RNA was extractedfrom cultures of BbJSB165 (used for luciferase assays in Fig.3A) and qRT-PCR was performed to assess the relative levelsof ospC and lucBb
� transcripts in the spirochetes culturedunder the two conditions. The addition of blood resulted in a10.8-fold and 13.4-fold upregulation in lucBb
� and ospC ex-pression, respectively (Fig. 3B). Of note, the level of the blood-dependent increase in luciferase activity from PospC-Bbluc�(18.5-fold protein induction; Fig. 3A) compared favorably withthe induction of transcripts for lucBb
� (10.8-fold increase; Fig.3B) or ospC (13.4-fold increase; Fig. 3B), thereby validatingthat the codon-adapted lucBb
� could be used as a reliabletranscriptional reporter.
Development of a lac repressor/operator expression systemin B. burgdorferi. Proper regulation of the E. coli lac inducibleexpression requires three components: (i) the LacI repressorprotein, (ii) a promoter containing LacI operator sequence(s),and (iii) an inducer (9, 10). It has been demonstrated in E. colithat LacI-dependent transcriptional repression is improvedwhen the expression of the repressor is enhanced using a stron-ger promoter (17, 48). To achieve elevated levels of lacI tran-scription in B. burgdorferi, the highly expressed, constitutivePflaB promoter was used to drive expression of the repressorprotein. Because the comparison of the codon utilization of B.burgdorferi and the lacI gene revealed substantial digressionbetween the two usage profiles (see Table S3 in the supple-mental material), the lacI ORF from pET-11a (Novagen) wascodon optimized (lacIBb) to enhance the production of LacI inB. burgdorferi. Codon optimization of lacI resulted in a changefrom 56.2% GC to 40.5% GC. The inducible promoter chosenin this study, designated PpQE30, was derived from the phageT5 promoter (12) of the pQE30 expression construct. To max-imize LacI-dependent repression, this promoter contains twolac operator (lacO) sites (11). The first of these is locatedbetween the �35 and �10 regions, whereas the 5� end of thesecond site overlaps the transcriptional start. The third com-ponent necessary for proper function of the lac inducible sys-tem is an inducer. In the native E. coli lac system, the releaseof repression is achieved by the binding of allolactose, derivedfrom lactose (that enters the cell via the LacY permease), tothe LacI tetramer (9, 10). A synthetic alternative to allolactoseis IPTG, a nonhydrolyzable inducer of the LacI repressor thatis capable of crossing the membrane of numerous prokaryotes(24) and eukaryotes (75), even in the absence of the requisiteLacY permease. This is relevant because there is no LacYhomolog encoded within the B. burgdorferi genome (36). Pre-
liminary studies revealed that the addition of IPTG to culturesof PL133 at concentrations as high as 100 mM, which repre-sents a concentration 10- to 100-fold greater than that typicallyutilized (24, 43, 85) in induction studies, had no deleteriousimpact on spirochete growth (data not shown).
To test the lac inducible system, three constructs were gener-ated in the pJD7 shuttle vector; an overview of these vectors isprovided in Fig. 1B and Table 1. The first construct, pJSB56,contains only the promoterless lucBb
� ORF. The remaining twoshuttle vectors, pJSB70 and pJSB104, both encode PpQE30 fusedto lucBb
�, but pJSB104 also contains the constitutively expressedand codon-adapted repressor PflaB-BblacI. pJSB56, pJSB70, andpJSB104 were electroporated into BbDTR630, thereby generat-ing BbJSB56, BbJSB70, and BbJSB104 (Table 1), respectively.
Test inductions were performed by growing BbJSB104 to adensity between 0.5 � 106 and 1 � 106 spirochetes/ml, at whichtime the cultures were supplemented with various concentra-tions of IPTG. This lower cell density was chosen to minimizethe formation of cell clumps that can occur when borrelialcultures are grown to higher cell densities; such clumping canresult in the underestimation of the bacterial cell counts viadark-field microscopy. This was important because the RLUreadings obtained from the luciferase assays were standardizedaccording to cell count. It should be pointed out, though, thatinductions of BbJSB104 performed at higher initial cell densi-ties (�1 � 107 spirochetes/ml) yielded results similar to thosedescribed for the lower-density cultures (see below).
For induction experiments, samples were collected at 0, 1, 3,6, 9, 12, 15, 24, and 36 h postinduction from BbJSB104 culturessupplemented with three different concentrations of IPTG (0.1mM, 1 mM, and 10 mM). A culture from which IPTG wasexcluded was included in the analysis (for an uninduced con-trol sample). This range of IPTG concentrations was chosen sothat IPTG levels that were 10-fold greater (10 mM) and 10-foldless (0.1 mM) than the concentration typically used for induc-ible expression studies (1 mM) could be assessed (43, 85). Tomeasure the level of induction, samples were collected at therespective times and the luciferase activities were determined.Four independent induction experiments were performed withsimilar results (representative results are provided in Fig. 4A).Treatment of all cultures with IPTG induced luciferase activity,and cultures treated with 1 mM and 10 mM IPTG yieldedmaximal responses (Fig. 4A). The observation that 0.1 mMIPTG induced luciferase activity to only about one-third thelevel of what was observed for cultures treated with either 1mM or 10 mM IPTG supported the dose dependency of IPTGtreatment. In all experiments, maximal levels of luciferase in-duction were observed at approximately 6 to 9 h postinduction(Fig. 4A).
Of particular interest was the observation that after 9 h ofIPTG induction, luciferase activities in cultures treated with 1or 10 mM IPTG began to decrease (Fig. 4A). The averagedecrease in the luciferase activities between consecutive sam-ples collected at 3-h time intervals ranged from 9 to 19%(13.4% average) (Fig. 4A), with an overall average decrease of62% when comparing the 9- and 24-h postinduction samples(Fig. 4A). Despite this decrease in the RLU from samples ofthe 1 mM and 10 mM cultures, the activity measured at 24 hpostinduction (260 � 14 RLU) was still more than 800-foldhigher than the level observed in the 24-h uninduced sample
VOL. 73, 2007 CONTROLLABLE GENE REPORTER FOR B. BURGDORFERI 1507
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/a
em o
n 14
Nov
embe
r 20
21 b
y 11
0.14
.202
.42.
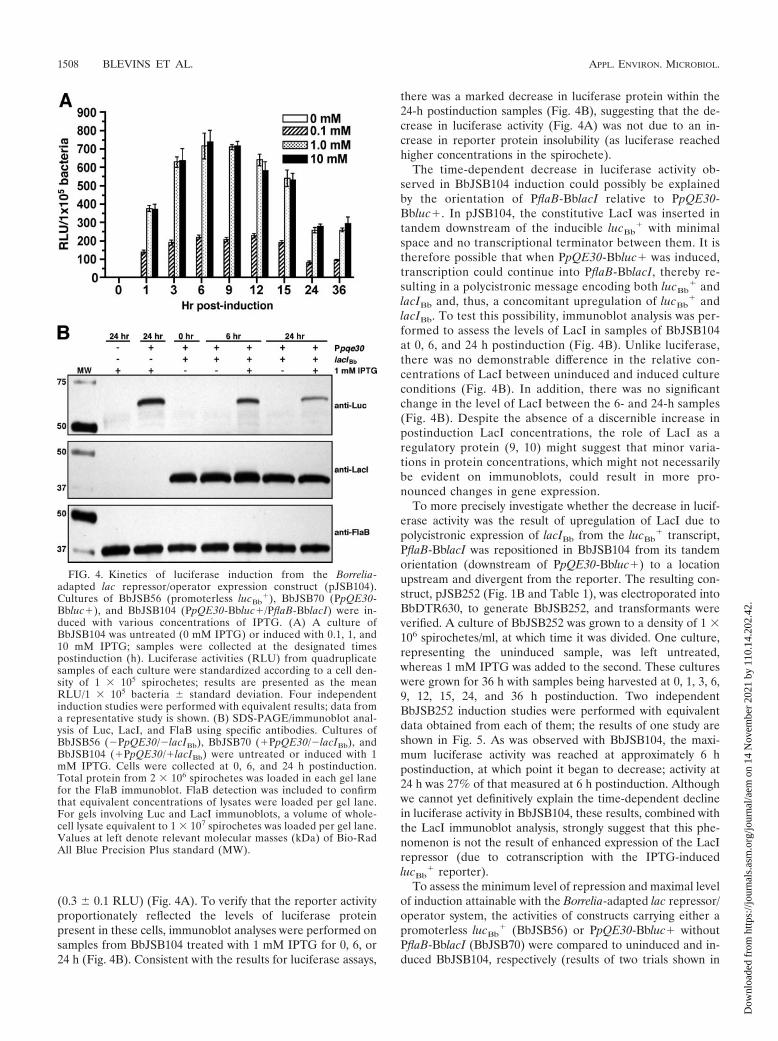
(0.3 � 0.1 RLU) (Fig. 4A). To verify that the reporter activityproportionately reflected the levels of luciferase proteinpresent in these cells, immunoblot analyses were performed onsamples from BbJSB104 treated with 1 mM IPTG for 0, 6, or24 h (Fig. 4B). Consistent with the results for luciferase assays,
there was a marked decrease in luciferase protein within the24-h postinduction samples (Fig. 4B), suggesting that the de-crease in luciferase activity (Fig. 4A) was not due to an in-crease in reporter protein insolubility (as luciferase reachedhigher concentrations in the spirochete).
The time-dependent decrease in luciferase activity ob-served in BbJSB104 induction could possibly be explainedby the orientation of PflaB-BblacI relative to PpQE30-Bbluc�. In pJSB104, the constitutive LacI was inserted intandem downstream of the inducible lucBb
� with minimalspace and no transcriptional terminator between them. It istherefore possible that when PpQE30-Bbluc� was induced,transcription could continue into PflaB-BblacI, thereby re-sulting in a polycistronic message encoding both lucBb
� andlacIBb and, thus, a concomitant upregulation of lucBb
� andlacIBb. To test this possibility, immunoblot analysis was per-formed to assess the levels of LacI in samples of BbJSB104at 0, 6, and 24 h postinduction (Fig. 4B). Unlike luciferase,there was no demonstrable difference in the relative con-centrations of LacI between uninduced and induced cultureconditions (Fig. 4B). In addition, there was no significantchange in the level of LacI between the 6- and 24-h samples(Fig. 4B). Despite the absence of a discernible increase inpostinduction LacI concentrations, the role of LacI as aregulatory protein (9, 10) might suggest that minor varia-tions in protein concentrations, which might not necessarilybe evident on immunoblots, could result in more pro-nounced changes in gene expression.
To more precisely investigate whether the decrease in lucif-erase activity was the result of upregulation of LacI due topolycistronic expression of lacIBb from the lucBb
� transcript,PflaB-BblacI was repositioned in BbJSB104 from its tandemorientation (downstream of PpQE30-Bbluc�) to a locationupstream and divergent from the reporter. The resulting con-struct, pJSB252 (Fig. 1B and Table 1), was electroporated intoBbDTR630, to generate BbJSB252, and transformants wereverified. A culture of BbJSB252 was grown to a density of 1 �106 spirochetes/ml, at which time it was divided. One culture,representing the uninduced sample, was left untreated,whereas 1 mM IPTG was added to the second. These cultureswere grown for 36 h with samples being harvested at 0, 1, 3, 6,9, 12, 15, 24, and 36 h postinduction. Two independentBbJSB252 induction studies were performed with equivalentdata obtained from each of them; the results of one study areshown in Fig. 5. As was observed with BbJSB104, the maxi-mum luciferase activity was reached at approximately 6 hpostinduction, at which point it began to decrease; activity at24 h was 27% of that measured at 6 h postinduction. Althoughwe cannot yet definitively explain the time-dependent declinein luciferase activity in BbJSB104, these results, combined withthe LacI immunoblot analysis, strongly suggest that this phe-nomenon is not the result of enhanced expression of the LacIrepressor (due to cotranscription with the IPTG-inducedlucBb
� reporter).To assess the minimum level of repression and maximal level
of induction attainable with the Borrelia-adapted lac repressor/operator system, the activities of constructs carrying either apromoterless lucBb
� (BbJSB56) or PpQE30-Bbluc� withoutPflaB-BblacI (BbJSB70) were compared to uninduced and in-duced BbJSB104, respectively (results of two trials shown in
FIG. 4. Kinetics of luciferase induction from the Borrelia-adapted lac repressor/operator expression construct (pJSB104).Cultures of BbJSB56 (promoterless lucBb
�), BbJSB70 (PpQE30-Bbluc�), and BbJSB104 (PpQE30-Bbluc�/PflaB-BblacI) were in-duced with various concentrations of IPTG. (A) A culture ofBbJSB104 was untreated (0 mM IPTG) or induced with 0.1, 1, and10 mM IPTG; samples were collected at the designated timespostinduction (h). Luciferase activities (RLU) from quadruplicatesamples of each culture were standardized according to a cell den-sity of 1 � 105 spirochetes; results are presented as the meanRLU/1 � 105 bacteria � standard deviation. Four independentinduction studies were performed with equivalent results; data froma representative study is shown. (B) SDS-PAGE/immunoblot anal-ysis of Luc, LacI, and FlaB using specific antibodies. Cultures ofBbJSB56 (�PpQE30/�lacIBb), BbJSB70 (�PpQE30/�lacIBb), andBbJSB104 (�PpQE30/�lacIBb) were untreated or induced with 1mM IPTG. Cells were collected at 0, 6, and 24 h postinduction.Total protein from 2 � 106 spirochetes was loaded in each gel lanefor the FlaB immunoblot. FlaB detection was included to confirmthat equivalent concentrations of lysates were loaded per gel lane.For gels involving Luc and LacI immunoblots, a volume of whole-cell lysate equivalent to 1 � 107 spirochetes was loaded per gel lane.Values at left denote relevant molecular masses (kDa) of Bio-RadAll Blue Precision Plus standard (MW).
1508 BLEVINS ET AL. APPL. ENVIRON. MICROBIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/a
em o
n 14
Nov
embe
r 20
21 b
y 11
0.14
.202
.42.

Table 2). Cultures were grown to a density between 0.5 � 106
and 6 � 106 spirochetes/ml, at which time, they were dividedand 1 mM IPTG was added to one of them. The remainingportion of culture was left untreated (uninduced sample).After 6 h of induction, samples were harvested and luciferaseassays were performed. In both trials (Table 2), the luciferaseactivity observed in the uninduced BbJSB104 was roughlyequivalent to the RLU values of the clone carrying the pro-moterless lucBb
� (BbJSB56), suggesting that a maximal level ofrepression was obtained. When the average RLU of theBbJSB70 was compared to that of the induced BbJSB104, theactivity of the induced BbJSB104 was 37% of that for BbJSB70(transformed with the construct lacking lacIBb), which was notsurprising considering the high level of LacI repressor ex-pressed in BbJSB104.
Utilization of the lac inducible system to express the B.burgdorferi BptA protein. The bbe16 gene (36), located on thevirulence-associated lp25 plasmid (39, 54, 55, 78), encodes asurface protein required for borrelial persistence in ticks(BptA) (58). Revel et al. (58) demonstrated that bptA insertionmutants of PL133 were incapable of maintaining normal spi-rochetal loads in the ticks, particularly in the weeks immedi-ately following feeding. The molecular mechanism by whichBptA achieves this function has yet to be elucidated. As proofof application of the lac operator/repressor system, the bptA
ORF was fused to the PpQE30 promoter and introduced,along with PflaB-BblacI, into the pJD44 shuttle vector to gen-erate pJSB194 (Fig. 1B and Table 1). This construct was elec-troporated into the bptA::aadA insertional mutant BbDTR596and is referred to herein as BbJSB194. Induction assessmentwas performed by growing a culture of BbJSB194 to the ap-propriate density (0.5 � 106 to 6 � 106 spirochetes/ml), di-vided, and supplemented with either 1 mM IPTG or left un-treated. At 6 h postinduction, the cells were collected andprocessed for SDS-PAGE and immunoblot analysis for FlaBand BptA. A culture of similarly treated BbDTR596 was in-cluded as a negative control. The representative results fromduplicate studies are shown in Fig. 6. As predicted, the addi-tion of 1 mM IPTG for 6 h strongly induced BptA expression.
DISCUSSION
Inducible expression systems, such as lac and tet repressor/operator systems (9, 10, 41), are invaluable tools in the dissec-tion of the molecular events contributing to bacterial generegulation and pathogenesis (24, 43–45, 74). However, no suchsystem had been utilized in B. burgdorferi until Cabello et al.(14) described the successful application of the tet induciblesystem in B. burgdorferi. This system was comprised of theTn10 TetR repressor expressed from PflaB, a hybrid borrelialpromoter containing dual TetR binding sites, and the induceranhydrotetracycline. The lac operator/repressor system (9, 10),whose simplicity is equal to that of the tet inducible system, was
FIG. 5. Kinetics of luciferase expression from the modified lacrepressor/operator expression construct (pJSB252). A culture ofBbJSB252 (analogous to pJSB104, but with PflaB-BblacI reorientedupstream and divergent from lucBb
�) was untreated (0 mM IPTG) orinduced with 1 mM IPTG. Samples were collected at the designatedtimes (h), and luciferase assays were performed. Luciferase activities(RLU) from quadruplicate samples of each culture were standardizedaccording to a cell density of 1 � 105 spirochetes; results are presentedas the mean RLU/1 � 105 bacteria � standard deviation. Two inde-pendent induction studies were performed with equivalent results; onlydata from one experiment are shown.
FIG. 6. Control of BptA expression using the Borrelia-adapted lacrepressor/operator expression system. Cultures of BbDTR596 (bptA-)and BbJSB194 (bptA-/PpQE30-bptA) were untreated or induced with 1mM IPTG. Six hours postinduction, cells were collected and preparedfor SDS-PAGE/immunoblot analysis; total protein from 1 � 107 spi-rochetes was loaded in each gel lane. Antibody against recombinantBptA was used to assess induction. Equivalent protein loading per gellane was verified by probing for FlaB. Values at left denote relevantmolecular masses (kDa) of Bio-Rad All Blue Precision Plus standard(MW). Two independent induction studies were performed with equiv-alent results; data from one representative experiment are shown.
TABLE 2. Influence of the PpQE30 promoter and lacIBb on expression of codon-optimized luciferase in B. burgdorferi
Plasmid
Expression (RLU/105 spirochetes) with IPTG concna:
Trial 1 Trial 2
0 mM 1.0 mM 0 mM 1.0 mM
Promoterless lucBb� 0.71 � 0.45 0.25 � 0.21 5.46 � 2.95 7.58 � 5.89
PpQE30-Bbluc� 1,405.27 � 55.01 1,325.93 � 88.80 1,167.36 � 37.07 1,044.42 � 47.87PpQE30-Bbluc�/PflaB-BblacI 0.93 � 0.19 504.33 � 31.62 1.30 � 0.40 408.23 � 12.71
a Results (from quadruplicate samples collected at 6 h postinduction) are reported as the mean RLU/105 spirochetes � standard deviation.
VOL. 73, 2007 CONTROLLABLE GENE REPORTER FOR B. BURGDORFERI 1509
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/a
em o
n 14
Nov
embe
r 20
21 b
y 11
0.14
.202
.42.

the focus of our current study. The first component generatedwas the codon-optimized LacI repressor, which was tran-scribed from the PflaB promoter. The increased luciferaseactivity observed with the codon-adapted lucBb
� reporter sug-gested that the expression of LacI could be maximized throughthe codon optimization of the LacI ORF, ultimately enhancingLacI-dependent repression (17, 48). The IPTG-inducible T5promoter (12) derived from pQE30, then was fused to thelucBb
� reporter (PpQE30-Bbluc�), which would allow thequantitiation of induction levels obtained upon treatment ofcultures with IPTG. To circumvent unfavorable codon bias,gene-building technology was employed to generate a lucifer-ase gene that more closely matched the codon usage of B.burgdorferi. Although the results from the comparison of thepre- and postadapted reporters showed that changing thecodon bias was not required for expression in B. burgdorferi,the four- to sixfold increase in luciferase activity with lucBb
�
suggested that codon optimization did maximize expression byrelieving some translational deficiencies.
The maximum level of PpQE30-Bbluc� induction by IPTGoccurred between 6 and 9 h postinduction, with an average400-fold increase in luciferase activity relative to the uninducedcultures. It also was determined that treatment of cultures with1 mM IPTG, a concentration typically used for inducible ex-pression studies (43, 85), resulted in luciferase activity equiv-alent to activities obtained from cultures treated with a 10-foldhigher concentration of IPTG. Because B. burgdoreri lacks anapparent LacY (permease), it remains unknown how extracel-lular and intracellular concentrations of IPTG potentially cor-relate; as such, the utility of IPTG induction remains empiricalat this time. Although the observation that the luciferase ac-tivity in the cultures containing 1 and 10 mM IPTG began todecrease after 9 h of induction was unexpected, it should benoted that the activities measured in the induced 24-h sampleswere still over 800-fold greater than that of the corresponding24-h uninduced culture.
Because there was no transcriptional terminator insertedbetween the 3� end of lucBb
� and the 5� end of lacIBb in thisvector, it initially was suspected that the decrease in luciferaseactivity over time could have been due to an increase in LacIBb
expression as a result of read-through from the lucBb� tran-
script; LacIBb was downstream of the inducible lucBb� in
pJSB104. However, immunoblot analysis revealed no signifi-cant changes in postinduction levels of LacI and the analogouspJSB252 construct, in which the repressor was reoriented up-stream and divergent from lucBb
�, showed the same decreasein luciferase activity between 6 and 9 h postinduction. Toaddress the possibility that this decrease was due to inactiva-tion of the IPTG during the induction, BbJSB104 culturesinitially treated with 1 mM IPTG were supplemented with 10and 100 mM IPTG at 12 or 24 h postinduction. There was noincrease in luciferase activity at 3, 6, or 12 h following thesecondary induction with fresh IPTG (data not shown),thereby suggesting that the decrease in luciferase activity wasnot a function of declining IPTG concentrations. Another pos-sible explanation for the decline in luciferase activity could berelated to the reduced biosynthetic capabilities of B. burgdorferi(23, 36). Upon the treatment of BbJSB104 with IPTG, theoverexpression of LucBb
� might result in the depletion of thelimited intracellular stores of the requisite metabolites at a rate
greater than said stores could be replenished. However, thisscenario seems to be less likely because the depletion of intra-cellular stores would presumably have a negative impact on thegrowth rate and this was not found (uninduced and inducedcultures grew at similar rates) (data not shown). The decreasein luciferase activity could also represent an increase in therelative insolubility of luciferase as the intracellular concentra-tions of the LucBb
� reaches levels that promote formation ofinsoluble/misfolded protein aggregates. The time-dependentloss of luciferase activity also could emanate from a disruptionof protein synthesis or the activation of proteases that degradeluciferase. However, LacI and FlaB levels remained constantwhen luciferase concentrations declined, which argues againsteither a global diminution of protein synthesis or the activationof proteases. Whereas we cannot yet explain the decrease inluciferase expression over time, the collective data suggest thatthe induction of luciferase from the lac inducible system doesnot adversely affect cell viability.
The lac inducible expression system developed in this studywas designed with a particular emphasis on a high level ofLacI-dependent regulation. To prevent inappropriate expres-sion in the absence of IPTG, the expression of the LacI re-pressor was optimized (described above) and the lac-inducibleT5 promoter from pQE30 was employed. This promoter waschosen for two reasons: (i) dual lacO sites provide the highestdegree of repression (11), and (ii) the sequences of the �35and �10 regions of this promoter are also fairly divergent fromthe consensus sequence of a typical bacterial RpoD/�70-depen-dent promoter (12). The use of a suboptimal/weak promoterensures that only a minimal amount of transcription will occurin the event of a momentary release of LacI-dependent repres-sion. In fact, the luciferase activities measured for BbJSB104or BbJSB70 (both contain PpQE30-Bbluc�) induced with 1mM IPTG were approximately 5% and 14%, respectively, ofthe luciferase activity of BbJSB82 (PflaB-Bbluc�) (data notshown). It should also be noted that the higher luciferaseactivity in BbJSB70 (lacks PflaB-BbLacI), by comparison tothat induced in BbJSB104 (contains PflaB-BbLacI), might im-ply that that relief of repression was incomplete. This partialinduction could be due to (i) an inability to obtain saturatinglevels of IPTG within the cell (via passive diffusion), (ii) im-paired active transport from a transporter (unknown) becom-ing saturated with IPTG (as extracellular levels approach 1mM), or (iii) the presence of LacI repressor at concentrationsin excess of those required to occupy all lacO sites.
To validate the newly generated lucBb� reporter for use in B.
burgdorferi differential gene expression studies, two promoterswere fused to lucBb
� and inserted into a borrelial shuttle vec-tor. Employing a blood supplementation technique to activateospC transcription (77), we were able to reproducibly demon-strate a significant increase in transcript levels (for both ospCand lucBb
�) as well as luciferase activity upon the addition ofblood to cultures of the clone expressing the PospC-Bbluc�fusion (BbJSB165). Although there was some discordance be-tween the induction of PospC-Bbluc� and ospC (relative in-crease in mRNA levels for the reporter transcript [10.8-fold]was slightly less than the induction of ospC [13.4-fold]),the disparity could have been due to relative differences in theinherent mRNA stability of the two targets. Because thelucBb
� ORF is 1,649 bp in length, whereas the OspC gene is
1510 BLEVINS ET AL. APPL. ENVIRON. MICROBIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/a
em o
n 14
Nov
embe
r 20
21 b
y 11
0.14
.202
.42.

only 633 bp, it is possible that the increased size of the lucBb�
mRNA could make it more susceptible to degradation. Thissize difference might also affect the processivity of RNA poly-merase; this would result in relatively fewer full-length copiesof the longer lucBb� transcript by comparison with the shorterospC mRNA. Yet another possible explanation for the discrep-ancy in the qRT-PCR would be differences in amplificationefficiency between ospC and lucBb
� during the analysis.Comparison of ospC transcript levels and PospC luciferase
activity revealed approximately equivalent increases in relativeexpression levels, thereby providing convincing validation ofour newly developed lucBb
� reporter. There was a slight vari-ance with respect to the increase in ospC transcript levels andthe increase in luciferase activity, 13.4-fold and 18.5-fold, re-spectively, when the culture of BbJSB165 was supplementedwith blood. This could be due to the stable accumulation of theLucBb
� protein (i.e., a higher rate of ospC transcript turnoverby comparison to the reporter protein), but this represents apotential limitation that is common to most transcriptionalreporter systems. Regardless of the minor inconsistency ob-served between the qRT-PCR and luciferase assay results, therelative complexity and reagent expense of alternative methodsfor measuring transcriptional changes (e.g., Northern blotanalysis or qRT-PCR) makes in vitro gene reporters, such asluciferase, a more favorable alternative for comparative ex-pression analyses.
The lp25-encoded BptA protein of B. burgdorferi was re-cently identified by our lab as being required for the ability ofthe spirochete to persist in the tick vector (58). To validate theborrelial lac repressor/operator expression system, the lucBb
�
in pJSB104 was replaced with the ORF for BptA protein (togenerate pJSB194). BbJSB194 was generated by transformingthis construct into a bptA insertion mutant, and the expressionof BptA was assessed by immunoblot analysis; induction ofcultures with 1 mM IPTG resulted in significant upregulationof BptA. Unfortunately, the molecular mechanism by whichBptA contributes to tick colonization still remains to be eluci-dated, but we now are poised to apply this controlled expres-sion system toward further functional analysis of the role ofBptA in the life cycle of B. burgdorferi.
The successful development of a lac repressor/operator sys-tem marks an advancement in B. burgdorferi molecular biology.The system described in this study is tailored for applications inwhich the tightest degree of regulation is desired, such asmodulating the expression of a catalytically active protein (e.g.,site-specific recombinase), a protein that is toxic when overex-pressed, or an antisense RNA designed to inactivate an essen-tial gene. However, this system could be modified to make itsuitable for maximal expression applications by replacing theweaker pQE30-derived inducible T5 promoter with an E. colitrp/lac hybrid (tac) promoter (26). The tac promoter contains�10 and �35 sequences that are precise matches to the con-sensus sequence for �70-dependent prokaryotic promoters,therefore making it a stronger promoter by comparison to T5.The spac-I promoter (85) is a Bacillus subtilis-derived lac-inducible promoter that has been applied to a wide variety ofbacterial systems and has recently been modified to contain anoptimized synthetic lacO site (24, 61). Another approach,which is analogous to that employed for the previously de-scribed borrelial tet-inducible system (14), involves the integra-
tion of lac operator(s) into a highly-expressed B. burgdorferipromoter. As all of the aforementioned modifications entailthe use of stronger promoters to improve expression levels, thisoften necessitates the use of only one lacO to prevent disrupt-ing the native �10 and �35 sequences. Therefore, to ensurethat the maximal level of LacI-dependent repression is at-tained, it would be necessary to use lacO sites that are opti-mized by sequence and spatial orientation to improve theirrelative binding affinity for the LacI tetramer (61). In additionto enhancing the LacI binding site, it is also possible to im-prove the level of repression by increasing the intracellularconcentration of the repressor protein (17, 48). This could beachieved by fusing the LacIBb ORF to a promoter that isstronger than PflaB, such as PflgB (51), and stably integratingthe fusion into the chromosome (14) or the virulence-associ-ated lp25 (58).
ACKNOWLEDGMENTS
We thank Rafal Tokarz, Christian Eggers, and Melissa Caimano forassistance regarding the blood supplementation technique. We alsothank David Rasko, Jason Huntley, and Jason Mock for technicaladvice and assistance during manuscript preparation.
This work was supported by grant AI-59062 from the National In-stitute of Allergy and Infectious Diseases, National Institutes ofHealth. J.S.B. was supported by National Institutes of Health traininggrant T32-AI07520 and Ruth L. Kirschstein National Research ServiceAward F32-AI058487 from the National Institutes of Health.
REFERENCES
1. Alam, J., and J. L. Cook. 1990. Reporter genes: application to the study ofmammalian gene transcription. Anal. Biochem. 188:245–254.
2. Alverson, J., S. F. Bundle, C. D. Sohaskey, M. C. Lybecker, and D. S.Samuels. 2003. Transcriptional regulation of the ospAB and ospC promotersfrom Borrelia burgdorferi. Mol. Microbiol. 48:1665–1677.
3. Anguita, J., S. Samanta, B. Revilla, K. Suk, S. Das, S. W. Barthold, and E.Fikrig. 2000. Borrelia burgdorferi gene expression in vivo and spirochetepathogenicity. Infect. Immun. 68:1222–1230.
4. Babb, K., N. El-Hage, J. C. Miller, J. A. Carroll, and B. Stevenson. 2001.Distinct regulatory pathways control expression of Borrelia burgdorferi infec-tion-associated OspC and Erp surface proteins. Infect. Immun. 69:4146–4153.
5. Babb, K., J. D. McAlister, J. C. Miller, and B. Stevenson. 2004. Molecularcharacterization of Borrelia burgdorferi erp promoter/operator elements. J.Bacteriol. 186:2745–2756.
6. Barbour, A. G. 1984. Isolation and cultivation of Lyme disease spirochetes.Yale J. Biol. Med. 57:521–525.
7. Barthold, S. W., K. D. Moody, G. A. Terwilliger, R. O. Jacoby, and A. C.Steere. 1988. An animal model for Lyme arthritis. Ann. N. Y. Acad. Sci.539:264–273.
8. Barthold, S. W., D. H. Persing, A. L. Armstrong, and R. A. Peeples. 1991.Kinetics of Borrelia burgdorferi dissemination and evolution of disease afterintradermal inoculation of mice. Am. J. Pathol. 139:263–273.
9. Beckwith, J. R. 1978. Lac: the genetic system, p. 11–30. In J. H. Miller andW. S. Reznikoff (ed.), The operon. Cold Spring Harbor Laboratory, ColdSpring Harbor, NY.
10. Beckwith, J. R. 1987. The lactose operon, p. 1444–1452. In F. C. Neidhardt, J. L.Ingraham, K. B. Low, B. Magasanik, M. Schaechter, and H. E. Umbarger (ed.),Escherichia coli and Salmonella typhimurium: cellular and molecular biology, vol.2. American Society for Microbiology, Washington, DC.
11. Besse, M., B. von Wilcken-Bergmann, and B. Muller-Hill. 1986. Synthetic lacoperator mediates repression through lac repressor when introduced up-stream and downstream from lac promoter. EMBO J. 5:1377–1381.
12. Bujard, H., R. Gentz, M. Lanzer, D. Stueber, M. Mueller, I. Ibrahimi, M. T.Haeuptle, and B. Dobberstein. 1987. A T5 promoter-based transcription-translation system for the analysis of proteins in vitro and in vivo. MethodsEnzymol. 155:416–433.
13. Bykowski, T., K. Babb, K. von Lackum, S. P. Riley, S. J. Norris, and B.Stevenson. 2006. Transcriptional regulation of the Borrelia burgdorferi anti-genically variable VlsE surface protein. J. Bacteriol. 188:4879–4889.
14. Cabello, F. C., L. Dubytska, A. V. Bryksin, J. V. Bugrysheva, and H. P.Godfrey. 2006. Genetic studies of the Borrelia burgdorferi bmp gene family, p.235–249. In N. ARW (ed.), The molecular biology of spirochetes. IOS Press,Amsterdam, The Netherlands.
VOL. 73, 2007 CONTROLLABLE GENE REPORTER FOR B. BURGDORFERI 1511
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/a
em o
n 14
Nov
embe
r 20
21 b
y 11
0.14
.202
.42.

15. Caimano, M. J., C. H. Eggers, C. A. Gonzalez, and J. D. Radolf. 2005.Alternate sigma factor RpoS is required for the in vivo-specific repression ofBorrelia burgdorferi plasmid lp54-borne ospA and lp6.6 genes. J. Bacteriol.187:7845–7852.
16. Caimano, M. J., C. H. Eggers, K. R. Hazlett, and J. D. Radolf. 2004. RpoSis not central to the general stress response in Borrelia burgdorferi but doescontrol expression of one or more essential virulence determinants. Infect.Immun. 72:6433–6445.
17. Calos, M. P. 1978. DNA sequence for a low-level promoter of the lacrepressor gene and an ‘up’ promoter mutation. Nature 274:762–765.
18. Carroll, J. A., R. M. Cordova, and C. F. Garon. 2000. Identification of 11pH-regulated genes in Borrelia burgdorferi localizing to linear plasmids. In-fect. Immun. 68:6677–6684.
19. Carroll, J. A., C. F. Garon, and T. G. Schwan. 1999. Effects of environmentalpH on membrane proteins in Borrelia burgdorferi. Infect. Immun. 67:3181–3187.
20. Carroll, J. A., P. E. Stewart, P. Rosa, A. F. Elias, and C. F. Garon. 2003. Anenhanced GFP reporter system to monitor gene expression in Borrelia burg-dorferi. Microbiology 149:1819–1828.
21. Casjens, S., N. Palmer, R. van Vugt, W. M. Huang, B. Stevenson, P. Rosa, R.Lathigra, G. Sutton, J. Peterson, R. J. Dodson, D. Haft, E. Hickey, M.Gwinn, O. White, and C. M. Fraser. 2000. A bacterial genome in flux: thetwelve linear and nine circular extrachromosomal DNAs in an infectiousisolate of the Lyme disease spirochete Borrelia burgdorferi. Mol. Microbiol.35:490–516.
22. Colin, M., S. Moritz, H. Schneider, J. Capeau, C. Coutelle, and M. C.Brahimi-Horn. 2000. Haemoglobin interferes with the ex vivo luciferaseluminescence assay: consequence for detection of luciferase reporter geneexpression in vivo. Gene Ther. 7:1333–1336.
23. Cordwell, S. J. 1999. Microbial genomes and “missing” enzymes: redefiningbiochemical pathways. Arch. Microbiol. 172:269–279.
24. Dancz, C. E., A. Haraga, D. A. Portnoy, and D. E. Higgins. 2002. Induciblecontrol of virulence gene expression in Listeria monocytogenes: temporalrequirement of listeriolysin O during intracellular infection. J. Bacteriol.184:5935–5945.
25. Das, S., S. W. Barthold, S. S. Giles, R. R. Montgomery, S. R. Telford III, andE. Fikrig. 1997. Temporal pattern of Borrelia burgdorferi p21 expression inticks and the mammalian host. J. Clin. Investig. 99:987–995.
26. de Boer, H. A., L. J. Comstock, and M. Vasser. 1983. The tac promoter: afunctional hybrid derived from the trp and lac promoters. Proc. Natl. Acad.Sci. USA 80:21–25.
27. de Silva, A. M., and E. Fikrig. 1997. Arthropod- and host-specific geneexpression by Borrelia burgdorferi. J. Clin. Investig. 99:377–379.
28. de Silva, A. M., S. R. Telford, 3rd, L. R. Brunet, S. W. Barthold, and E.Fikrig. 1996. Borrelia burgdorferi OspA is an arthropod-specific transmission-blocking Lyme disease vaccine. J. Exp. Med. 183:271–275.
29. de Wet, J. R., K. V. Wood, D. R. Helinski, and M. DeLuca. 1985. Cloning offirefly luciferase cDNA and the expression of active luciferase in Escherichiacoli. Proc. Natl. Acad. Sci. USA 82:7870–7873.
30. Dunn, J. J., S. R. Buchstein, L.-L. Butler, S. Fisenne, D. S. Polin, B. N. Lade,and B. J. Luft. 1994. Complete nucleotide sequence of a circular plasmidfrom the Lyme disease spirochete, Borrelia burgdorferi. J. Bacteriol. 176:2706–2717.
31. Eggers, C. H., M. J. Caimano, and J. D. Radolf. 2004. Analysis of promoterelements involved in the transcriptional initiation of RpoS-dependent Bor-relia burgdorferi genes. J. Bacteriol. 186:7390–7402.
32. Eggers, C. H., M. J. Caimano, and J. D. Radolf. 2006. Sigma factor selectivityin Borrelia burgdorferi: RpoS recognition of the ospE/ospF/elp promoters isdependent on the sequence of the �10 region. Mol. Microbiol. 59:1859–1875.
33. Fikrig, E., S. W. Barthold, W. Sun, W. Feng, S. R. Telford III, and R. A.Flavell. 1997. Borrelia burgdorferi P35 and P37 proteins, expressed in vivo,elicit protective immunity. Immunity 6:531–539.
34. Fisher, M. A., D. Grimm, A. K. Henion, A. F. Elias, P. E. Stewart, P. A. Rosa,and F. C. Gherardini. 2005. Borrelia burgdorferi �54 is required for mam-malian infection and vector transmission but not for tick colonization. Proc.Natl. Acad. Sci. USA 102:5162–5167.
35. Frank, K. L., S. F. Bundle, M. E. Kresge, C. H. Eggers, and D. S. Samuels.2003. aadA confers streptomycin resistance in Borrelia burgdorferi. J. Bacte-riol. 185:6723–6727.
36. Fraser, C. M., S. Casjens, W. M. Huang, G. G. Sutton, R. Clayton, R. Lathigra,O. White, K. A. Ketchum, R. Dodson, E. K. Hickey, M. Gwinn, B. Dougherty,J. F. Tomb, R. D. Fleischmann, D. Richardson, J. Peterson, A. R. Kerlavage, J.Quackenbush, S. Salzberg, M. Hanson, R. van Vugt, N. Palmer, M. D. Adams,J. Gocayne, J. Weidman, T. Utterback, L. Watthey, L. McDonald, P. Artiach, C.Bowman, S. Garland, C. Fuji, M. D. Cotton, K. Horst, K. Roberts, B. Hatch,H. O. Smith, and J. C. Venter. 1997. Genomic sequence of a Lyme diseasespirochaete, Borrelia burgdorferi. Nature 390:580–586.
37. Gilmore, R. D., Jr., M. L. Mbow, and B. Stevenson. 2001. Analysis of Borreliaburgdorferi gene expression during life cycle phases of the tick vector Ixodesscapularis. Microbes Infect. 3:799–808.
38. Gould, S. J., and S. Subramani. 1988. Firefly luciferase as a tool in molecularand cell biology. Anal. Biochem. 175:5–13.
39. Grimm, D., C. H. Eggers, M. J. Caimano, K. Tilly, P. E. Stewart, A. F. Elias,J. D. Radolf, and P. A. Rosa. 2004. Experimental assessment of the roles oflinear plasmids lp25 and lp28-1 of Borrelia burgdorferi throughout the infec-tious cycle. Infect. Immun. 72:5938–5946.
40. Grimm, D., K. Tilly, R. Byram, P. E. Stewart, J. G. Krum, D. M. Bueschel,T. G. Schwan, P. F. Policastro, A. F. Elias, and P. A. Rosa. 2004. Outer-surface protein C of the Lyme disease spirochete: a protein induced in ticksfor infection of mammals. Proc. Natl. Acad. Sci. USA 101:3142–3147.
41. Hillen, W., and C. Berens. 1994. Mechanisms underlying expression of Tn10encoded tetracycline resistance. Annu. Rev. Microbiol. 48:345–369.
42. Hubner, A., X. Yang, D. M. Nolen, T. G. Popova, F. C. Cabello, and M. V.Norgard. 2001. Expression of Borrelia burgdorferi OspC and DbpA is con-trolled by a RpoN-RpoS regulatory pathway. Proc. Natl. Acad. Sci. USA98:12724–12729.
43. Jackson, D. W., K. Suzuki, L. Oakford, J. W. Simecka, M. E. Hart, and T.Romeo. 2002. Biofilm formation and dispersal under the influence of theglobal regulator CsrA of Escherichia coli. J. Bacteriol. 184:290–301.
44. Ji, Y., B. Zhang, S. F. Van Horn, P. Warren, G. Woodnutt, M. K. Burnham,and M. Rosenberg. 2001. Identification of critical staphylococcal genes usingconditional phenotypes generated by antisense RNA. Science 293:2266–2269.
45. Kamionka, A., R. Bertram, and W. Hillen. 2005. Tetracycline-dependent con-ditional gene knockout in Bacillus subtilis. Appl. Environ. Microbiol. 71:728–733.
46. Lesnik, E. A., R. Sampath, H. B. Levene, T. J. Henderson, J. A. McNeil, andD. J. Ecker. 2001. Prediction of rho-independent transcriptional terminatorsin Escherichia coli. Nucleic Acids Res. 29:3583–3594.
47. Miller, J. C., K. von Lackum, M. E. Woodman, and B. Stevenson. 2006.Detection of Borrelia burgdorferi gene expression during mammalian infec-tion using transcriptional fusions that produce green fluorescent protein.Microb. Pathog. 41:43–47.
48. Muller-Hill, B., L. Crapo, and W. Gilbert. 1968. Mutants that make more lacrepressor. Proc. Natl. Acad. Sci. USA 59:1259–1264.
49. Nakamura, Y., T. Gojobori, and T. Ikemura. 2000. Codon usage tabulatedfrom international DNA sequence databases: status for the year 2000. Nu-cleic Acids Res. 28:292.
50. Ohnishi, J., J. Piesman, and A. M. de Silva. 2001. Antigenic and geneticheterogeneity of Borrelia burgdorferi populations transmitted by ticks. Proc.Natl. Acad. Sci. USA 98:670–675.
51. Ojaimi, C., C. Brooks, S. Casjens, P. Rosa, A. Elias, A. Barbour, A. Jasin-skas, J. Benach, L. Katona, J. Radolf, M. Caimano, J. Skare, K. Swingle, D.Akins, and I. Schwartz. 2003. Profiling of temperature-induced changes inBorrelia burgdorferi gene expression by using whole genome arrays. Infect.Immun. 71:1689–1705.
52. Page, A. L., H. Ohayon, P. J. Sansonetti, and C. Parsot. 1999. The secretedIpaB and IpaC invasins and their cytoplasmic chaperone IpgC are requiredfor intercellular dissemination of Shigella flexneri. Cell. Microbiol. 1:183–193.
53. Pal, U., X. Yang, M. Chen, L. K. Bockenstedt, J. F. Anderson, R. A. Flavell,M. V. Norgard, and E. Fikrig. 2004. OspC facilitates Borrelia burgdorferiinvasion of Ixodes scapularis salivary glands. J. Clin. Investig. 113:220–230.
54. Purser, J. E., M. B. Lawrenz, M. J. Caimano, J. K. Howell, J. D. Radolf, andS. J. Norris. 2003. A plasmid-encoded nicotinamidase (PncA) is essential forinfectivity of Borrelia burgdorferi in a mammalian host. Mol. Microbiol. 48:753–764.
55. Purser, J. E., and S. J. Norris. 2000. Correlation between plasmid contentand infectivity in Borrelia burgdorferi. Proc. Natl. Acad. Sci. USA 97:13865–13870.
56. Ramamoorthy, R., and M. T. Philipp. 1998. Differential expression of Bor-relia burgdorferi proteins during growth in vitro. Infect. Immun. 66:5119–5124.
57. Ramamoorthy, R., and D. Scholl-Meeker. 2001. Borrelia burgdorferi proteinswhose expression is similarly affected by culture temperature and pH. Infect.Immun. 69:2739–2742.
58. Revel, A. T., J. S. Blevins, C. Almazan, L. Neil, K. M. Kocan, J. de la Fuente,K. E. Hagman, and M. V. Norgard. 2005. bptA (bbe16) is essential for thepersistence of the Lyme disease spirochete, Borrelia burgdorferi, in its nat-ural tick vector. Proc. Natl. Acad. Sci. USA 102:6972–6977.
59. Revel, A. T., A. M. Talaat, and M. V. Norgard. 2002. DNA microarrayanalysis of differential gene expression in Borrelia burgdorferi, the Lymedisease spirochete. Proc. Natl. Acad. Sci. USA 99:1562–1567.
60. Rosa, P. A., K. Tilly, and P. E. Stewart. 2005. The burgeoning moleculargenetics of the Lyme disease spirochaete. Nat. Rev. Microbiol. 3:129–143.
61. Sadler, J. R., H. Sasmor, and J. L. Betz. 1983. A perfectly symmetric lacoperator binds the lac repressor very tightly. Proc. Natl. Acad. Sci. USA80:6785–6789.
62. Sartakova, M. L., E. Y. Dobrikova, D. A. Terekhova, R. Devis, J. V. Bugry-sheva, O. V. Morozova, H. P. Godfrey, and F. C. Cabello. 2003. Novelantibiotic-resistance markers in pGK12-derived vectors for Borrelia burgdor-feri. Gene 303:131–137.
63. Schwan, T. G., and J. Piesman. 2000. Temporal changes in outer surfaceproteins A and C of the Lyme disease-associated spirochete, Borrelia burg-
1512 BLEVINS ET AL. APPL. ENVIRON. MICROBIOL.
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/a
em o
n 14
Nov
embe
r 20
21 b
y 11
0.14
.202
.42.

dorferi, during the chain of infection in ticks and mice. J. Clin. Microbiol.38:382–388.
64. Schwan, T. G., J. Piesman, W. T. Golde, M. C. Dolan, and P. A. Rosa. 1995.Induction of an outer surface protein on Borrelia burgdorferi during tickfeeding. Proc. Natl. Acad. Sci. USA 92:2909–2913.
65. Smith, A. D., and J. P. Trempe. 2000. Luminometric quantitation of Photinuspyralis firefly luciferase and Escherichia coli beta-galactosidase in blood-contaminated organ lysates. Anal. Biochem. 286:164–172.
66. Sohaskey, C. D., C. Arnold, and A. G. Barbour. 1997. Analysis of promotersin Borrelia burgdorferi by use of a transiently expressed reporter gene. J.Bacteriol. 179:6837–6842.
67. Sohaskey, C. D., W. R. Zuckert, and A. G. Barbour. 1999. The extendedpromoters for two outer membrane lipoprotein genes of Borrelia spp.uniquely include a T-rich region. Mol. Microbiol. 33:41–51.
68. Steere, A. C. 2001. Lyme disease. N. Engl. J. Med. 345:115–125.69. Steidler, L., W. Yu, W. Fiers, and E. Remaut. 1996. The expression of the
Photinus pyralis luciferase gene in Staphylococcus aureus Cowan I allows thedevelopment of a live amplifiable tool for immunodetection. Appl. Environ.Microbiol. 62:2356–2359.
70. Stemmer, W. P., A. Crameri, K. D. Ha, T. M. Brennan, and H. L. Heyneker.1995. Single-step assembly of a gene and entire plasmid from large numbersof oligodeoxyribonucleotides. Gene 164:49–53.
71. Stewart, P. E., R. Thalken, J. L. Bono, and P. Rosa. 2001. Isolation of acircular plasmid region sufficient for autonomous replication and transfor-mation of infectious Borrelia burgdorferi. Mol. Microbiol. 39:714–721.
72. Stewart, P. E., X. Wang, D. M. Bueschel, D. R. Clifton, D. Grimm, K. Tilly,J. A. Carroll, J. J. Weis, and P. A. Rosa. 2006. Delineating the requirementfor the Borrelia burgdorferi virulence factor OspC in the mammalian host.Infect. Immun. 74:3547–3553.
73. Suk, K., S. Das, W. Sun, B. Jwang, S. W. Barthold, R. A. Flavell, and E.Fikrig. 1995. Borrelia burgdorferi genes selectively expressed in the infectedhost. Proc. Natl. Acad. Sci. USA 92:4269–4273.
74. Sun, J., L. Zheng, C. Landwehr, J. Yang, and Y. Ji. 2005. Identification of anovel essential two-component signal transduction system, YhcSR, in Staph-ylococcus aureus. J. Bacteriol. 187:7876–7880.
75. Syroid, D. E., R. I. Tapping, and J. P. Capone. 1992. Regulated expression
of a mammalian nonsense suppressor tRNA gene in vivo and in vitro usingthe lac operator/repressor system. Mol. Cell. Biol. 12:4271–4278.
76. Tilly, K., J. G. Krum, A. Bestor, M. W. Jewett, D. Grimm, D. Bueschel, R.Byram, D. Dorward, M. J. VanRaden, P. Stewart, and P. Rosa. 2006. Borreliaburgdorferi OspC protein required exclusively in a crucial early stage ofmammalian infection. Infect. Immun. 74:3554–3564.
77. Tokarz, R., J. M. Anderton, L. I. Katona, and J. L. Benach. 2004. Combinedeffects of blood and temperature shift on Borrelia burgdorferi gene expressionas determined by whole genome DNA array. Infect. Immun. 72:5419–5432.
78. Xu, Y., C. Kodner, L. Coleman, and R. C. Johnson. 1996. Correlation ofplasmids with infectivity of Borrelia burgdorferi sensu stricto type strain B31.Infect. Immun. 64:3870–3876.
79. Yang, X., M. S. Goldberg, T. G. Popova, G. B. Schoeler, S. K. Wikel, K. E.Hagman, and M. V. Norgard. 2000. Interdependence of environmental fac-tors influencing reciprocal patterns of gene expression in virulent Borreliaburgdorferi. Mol. Microbiol. 37:1470–1479.
80. Yang, X., T. G. Popova, M. S. Goldberg, and M. V. Norgard. 2001. Influenceof cultivation media on genetic regulatory patterns in Borrelia burgdorferi.Infect. Immun. 69:4159–4163.
81. Yang, X., T. G. Popova, K. E. Hagman, S. K. Wikel, G. B. Schoeler, M. J.Caimano, J. D. Radolf, and M. V. Norgard. 1999. Identification, character-ization, and expression of three new members of the Borrelia burgdorferi Mlp(2.9) lipoprotein gene family. Infect. Immun. 67:6008–6018.
82. Yang, X. F., S. M. Alani, and M. V. Norgard. 2003. The response regulatorRrp2 is essential for the expression of major membrane lipoproteins inBorrelia burgdorferi. Proc. Natl. Acad. Sci. USA 100:11001–11006.
83. Yang, X. F., M. C. Lybecker, U. Pal, S. M. Alani, J. Blevins, A. T. Revel, D. S.Samuels, and M. V. Norgard. 2005. Analysis of the ospC regulatory elementcontrolled by the RpoN-RpoS regulatory pathway in Borrelia burgdorferi. J.Bacteriol. 187:4822–4829.
84. Yang, X. F., U. Pal, S. M. Alani, E. Fikrig, and M. V. Norgard. 2004.Essential role for OspA/B in the life cycle of the Lyme disease spirochete. J.Exp. Med. 199:641–648.
85. Yansura, D. G., and D. J. Henner. 1984. Use of the Escherichia coli lacrepressor and operator to control gene expression in Bacillus subtilis. Proc.Natl. Acad. Sci. USA 81:439–443.
VOL. 73, 2007 CONTROLLABLE GENE REPORTER FOR B. BURGDORFERI 1513
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/a
em o
n 14
Nov
embe
r 20
21 b
y 11
0.14
.202
.42.