activation of the hprt gene on the inactive x chromosome in...
TRANSCRIPT

Activation of the hprt gene on the inactive X chromosome in transformed
diploid female Chinese hamster cells
STEPHEN G. GRANT#'f and RONALD G. WORTON
Genetics Department and Research Institute, Hospital for Sick Children, 555 University Avenue, Toronto, Ontario M5C 1X8, Canadaand Departments of Medical Genetics and Medical Biophysics, University of Toronto, Toronto, Ontario, Canada
•Author for correspondencef Present address: Department of Molecular and Cellular Biology, Roswell Park Memorial Institute, 666 Elm Street, Buffalo, NY 14263, USA
Summary
Treatment with 5-azacytidine, a potent inhibitor ofDNA methylation, was used to induce activation ofthe selectable hprt gene on the inactive X chromo-some in a diploid female Chinese hamster cell line.The transformed, stably diploid cell line F3B wasselected in media containing the lethal purine ana-logue 6-thioguanine, to generate a phenotypicallyHPRT" mutant, F3BT1, of presumed genotypehprf~ Ihprtt+\ where (+) represents the presum-ably wild-type allele on the inactive X chromosome.Treatment of F3BT1 with 5-azacytidine resulted inphenotypic reversion to HPRT+ at a frequencygreater than 10~3. Similar treatment of6-thioguanine-resistant control lines derived frommale cells, or from CHO (which has no inactive Xchromosome), had no effect on the frequency of
phenotypic reversion, indicating that activation ofthe hprr+) allele, rather than reversion of the hprt~is responsible. This conclusion is substantiated bydocumentation of the low mutagenic capacity of5-azacytidine in this system. Proof that the hpr6+)
allele can be activated by 5-azacytidine treatmentwas obtained in somatic cell hybrids in which hprtgene products from the active and inactive Xchromosomes could be distinguished by isoelectricfocusing. Our results demonstrate that X-linkedgene activation associated with generalized DNAdemethylation occurs with high frequency in trans-formed diploid Chinese hamster cells.
Key words: 5-azacytidine, transformation, cell hybridization,DNA methylation.
Introduction
The somatic cells of mammalian females carry two copiesof the X chromosome, one of which becomes inactivatedearly during development, presumably as a means ofcompensating for the excess dosage of X-linked genes inXX females as opposed to XY males (Lyon, 1961). Thisgenetic inactivation has been shown by DNA transfectionexperiments to be a property of the X chromosome DNAitself (Liskay & Evans, 1980; Chapman et al. 1982). Theonly known modification of DNA in mammalian cells ismethylation of cytosine residues (Vanyushin et al. 1970),usually those present in a symmetrical CG configuration(Doskocil & Sorm, 1962). The pattern of DNA modifi-cation by methylation is retained through replication(Bird, 1978), and the extent of such modification hasbeen shown to correlate inversely with gene expression inthe majority of systems studied (Waalwijk & Flavell,1978; Sutter & Doerfler, 1980; Groudine et al. 1981).The pyrimidine nucleoside analogue 5-azacytidine(SazaCR), which acts as a strong inhibitor of eukaryoticDNA methylases upon incorporation into DNA (Creusot
Journal of Cell Science 92, 723-732 (1989)Printed in Great Britain © The Company of Biologists Limited 1989
et al. 1982; Taylor & Jones, 1982), and hence of DNAmethylation (Jones & Taylor, 1981), has been shown toinduce activation of repressed genes in many systems(Constantinides et al. 1977; Groudine et al. 1981).
Four genes located on the human inactive X chromo-some, HPRT (hypoxanthine-guanine phosphoribosyl-transferase), G6PD (glucose-6-phosphate dehydrogen-ase), PGA' (phosphoglycerate kinase), and GALA (a-galactosidase) have been shown to be derepressed by5azaCR treatment of human-rodent cell hybrids thathave lost most other human chromosomes (Mohandas etal. 1981; Lester et al. 1982; Hors-Cayla et al. 1983). Incontrast, the HPRT gene on the inactive X chromosomein human diploid female fibroblasts has not been affectedby drug selection (Migeon, 1972) or drug treatment(Comings, 1966), including treatment with mutagens(Migeon et al. 1978) or 5azaCR (Wolf & Migeon, 1982).In this report, 5azaCR-induced reactivation of the selec-table hprt gene on the inactive X chromosome is demon-strated in a transformed diploid female Chinese hamstercell line.
723

Table 1. Properties of cell lines used in this study
Cell line Origin Karyotype* Genotype HPRT phenotype
F3BTFHE3MtxR I"
F3BTld
VEOT3CHWLEO5T1DRO1
Female lungFemale earFemale ovary (CHO)
Female lungMale lung (V79)Male ear (CHW)Female ovary (CHO)Female ovary (CHO)
X./X,X./X,
x.cx^x,x.e
x.c
hprt+/hprt<+)
hprt+/hprt(+)
hprt+
hprt~/hprt(+)
hprt~hprt~hprt~hprtBh
HPRT+HPRT+
HPRT+
HPRT"HPRT"HPRT"HPRT"HPRT-
*X, active X chromosome, X, inactive X chromosome.bA/>rt'+' gene on inactive X, presumably wild type allele.cSecond X may be marker Z10 with deletion of Xp including hprt locus (12,59).d Spontaneous 6tgr mutant, all others mutagen induced.eY chromosome lost (51).'Actually t(X,7), hprt localized to der7 (15,35)."Actually t(X,Z3) and t(X,7), hprt localized to t(X,Z3)(5,6).hhprtB produces an electrophoretically distinct HPRT with 5 % activity (6).
Materials and methods
Cell lines and culture conditionsMost experiments were performed with the diploid Chinesehamster cell line F3B, which has a transformed phenotype andarose spontaneously from a primary culture of female Chinesehamster lung fibroblasts. It was generated in this laboratory bythree successive cloning steps, selecting each time for cells witha normal karyotype (Campbell & Worton, 1979). Derivatives ofseveral different transformed Chinese hamster cell lines wereutilized as controls (Table 1). These include CHO (ovarianfibroblast), TFHE3 (female ear fibroblast), V79 (male lungfibroblast) and CHW (male ear fibroblast). The TFHE3 linewas cloned from a primary culture of female ear fibroblasts bypicking a colony with typical transformed appearance. The F3Band TFHE3 lines have normal Chinese hamster karyotypes,whereas the chromosomes of the CHO, V79 and CHW lineshave undergone structural rearrangement as described (Lin etal. 1971; Deaven&Petersen, 1973; Worton et al. 1977; Worton,1978; Thacker, 1981).
Three sublines of CHO were used. One subline, Mtx R " \used as a control for the biochemical assays of HPRT activity,had a wild-type level of HPRT. A second subline. DR01,derived from DR31 (Chasin, 1972), carries a 6-thioguanine-resistant (6tgr) mutation resulting in an electrophoreticallyaltered HPRT protein with 5% activity (Chasin & Urlaub,1976). The other CHO subline, as well as thioguanine-resistantmutants of the F3B, V79 and CHW lines show negligibleHPRT activity. The CHO line, despite its female origin, hasonly a single intact X chromosome (Campbell & Worton, 1977)and only one copy of the X-linked hprt gene (Chu & Mailing,1968). These cell lines are described in detail in Table 1.
Cells were maintained in monolayer culture in alpha mini-mum essential medium supplemented with 7 % foetal calfserum at 37°C in a 5 % CO2 humidified atmosphere. Mediumused for selections was supplemented with dialysed foetal calfserum.
5azaCR toxicityCytotoxicity of 5azaCR for the F3B cell line was determined intwo ways. First, a survival curve was generated by duplicateplating of 106 cells/dish and stepwise 10-fold dilutions down to102 cells/dish in medium containing 0-5-100/iM-5azaCR.Second, a distinct survival curve was generated for cells given a
24-h exposure to 5azaCR followed by plating in non-selectivemedium.
Mutagenesis and mutant selectionsTwo T75 flasks were seeded with 106 cells in normal mediumand incubated overnight. This medium was then replaced inone flask with fresh normal medium, and in the other with freshmedium containing 200/igml"1 ethyl methanesulphonate(EMS). Twenty hours later, this medium was aspirated, thecells were washed with phosphate-buffered saline (PBS), andgrown in normal medium for 3 days, until the cells reachedconfluency. Cells were then harvested by trypsinization, andseeded at 104, 105 and 106 cells per plate in selective medium.Control plates were also seeded at 102 cells per plate in normalmedium. Medium containing 10j<M-6-thioguanine was used toselect for 6tgr variants, while HAT medium (lOj/gmr1 hypo-xanthine + 1/un-methotrexate + 10/igml"1 thymidine) wasused to select for HATR variants.
5azaCR treatmentFrom 10s to 5 X106 cells were plated into a T7S flask in mediumlacking nucleosides and deoxynucleosides. After overnightincubation this medium was aspirated, the cells were washedwith PBS, and fresh nucleoside-free medium containing005-100;iM-5azaCR was added. Control flasks were mocktreated by changing the medium. After 12-84h the mediumwas again aspirated, the cells were washed with PBS, and thecells plated as below, or fresh medium supplemented withnucleosides and deoxynucleosides was added and the flasks wereincubated for 1-5 days, until the cells were nearing confluency.These cells were then trypsinized, counted, and seeded at Mr,104 and 106 cells per plate in selective medium. Control plateswere also seeded at 10 cells per plate in normal medium. HATmedium was used to select for cells expressing a functional,active, hprt gene. In one experiment medium containing 10 /.IM-6tg was used to select for cells with an HPRT" phenotype.After 14 days, representative colonies were picked into freshselective medium, and all remaining colonies were stained andcounted.
Related compounds were tested for their ability to reactivatethe hprt gene and for their ability to compete with 5azaCR usinga similar protocol. In these experiments cytidine, deoxycyti-dine, 6-azacytidine, 5-methylcytidine, 5-fluorodeoxycytidine,5-bromodeoxyuridine, emetine or hydroxyurea were added to,or substituted for, 5azaCR in the treatment medium.
724 S. G. Grant and R. G. Worton
Hybridization and segregationCell hybrids formed between the F3B and DR01 lines weregenerated by polyethylene glycol-induced cell fusion, followedby selection in HAT medium containing 3mM-ouabain. Suchmedium selected for cells carrying both the dominant hprt+
gene of F3B, and the ouaR gene of DR01 , while killing bothparental lines. These hybrids were then transferred into me-dium containing 6tg to select for cells that had lost the active Xchromosome carrying an active hprt gene donated by the F3Bline.
KaryotypingHybrid and segregant cell lines were harvested and karyotypedby standard techniques (Worton & Duff, 1979). All chromo-somes were analysed, with emphasis on the complement of Xchromosomes, including the translocated fragments specific tothe DR01 line (Worton, 1978).
Biochemical characterizationHPRT activity assays and isoelectric focusing were performedby the methods of Chasin & Urlaub (1976). One semi-confluentflask of cells was harvested and 3X106 cells were divided intosamples and washed with PBS. The cells were then resus-pended in 450^1 of 0-03 M-Tris-HCl buffer, pH7-4, and lysedby alternately freezing and thawing six times. The suspensionwas centrifuged at 4°C in an Eppendorf microfuge for 20 min toremove nuclei and cell membranes. The extract was thenassayed for HPRT activity, applied to a gel for electrophoresisor frozen at — 70°C for later use.
HPRT activity was determined using 50-/il samples of cellextract in 0-02mM-[8-14C]hypoxanthine (NEN, 48Cimol~'),1 mM-5-phosphoribosyl pyrophosphate, 5mM-MgCl2, lOmM-NaN3, 2-4/ igmr1 bovine serum albumin and 0-05 M-Tris-HCl, pH7-4, in a total volume of 100 (A. The tubes wereincubated at 37 °C for 20 min and the reaction was terminated byremoval to ice and the addition of 1 ml cold stop buffer (0-05 M-sodium acetate, pH 50 , 2mM-K2HPO4). The radioactive prod-uct was precipitated by addition of 200 ̂ tl of 0-SM-LaCl,transferred onto Whatman GF/C filters and counted.
Isoelectric focusing was performed with polyacrylamide slabgels lmm thick containing 4-9% acrylamide, 0-13% NJ\fl-methylenebisacrylamide (total acrylamide 5%), 15% sucrose,3 % Ampholines, p H 9 - l l , 1-5% Ampholines, pH6-8 . Thissystem maximized separation of proteins with pi values from 6to 8. Samples of cell extract (20 (x\) were applied and the gel wasrun for 2h at 1600 V, 10 mA, 10 W. HPRT protein wasvisualized autoradiographically by an activity assay similar tothat described above involving phosphoribosylation of [4,5-3H]hypoxanthine.
Results
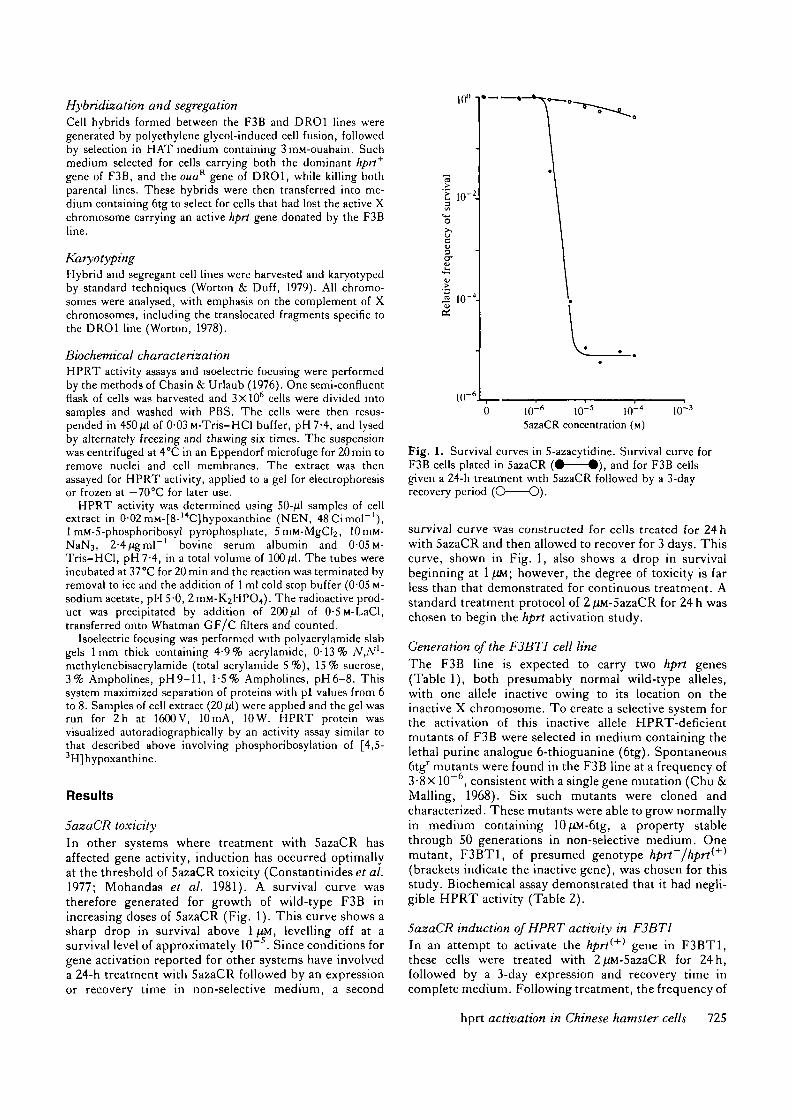
5azaCR toxicityIn other systems where treatment with 5azaCR hasaffected gene activity, induction has occurred optimallyat the threshold of 5azaCR toxicity (Constantinides et al.1977; Mohandas et al. 1981). A survival curve wastherefore generated for growth of wild-type F3B inincreasing doses of 5azaCR (Fig. 1). This curve shows asharp drop in survival above 1/iM, levelling off at asurvival level of approximately 10~5. Since conditions forgene activation reported for other systems have involveda 24-h treatment with 5azaCR followed by an expressionor recovery time in non-selective medium, a second
10" T -
t 1(T2-
3
a"
>
I 1(r
10"1(T6 10"' 10"4
5azaCR concentration (M)i<r3
Fig. 1. Survival curves in 5-azacytidine. Survival curve forF3B cells plated in 5azaCR ( • — — • ) , and for F3B cellsgiven a 24-h treatment with 5azaCR followed by a 3-dayrecovery period (O O).
survival curve was constructed for cells treated for 24 hwith 5azaCR and then allowed to recover for 3 days. Thiscurve, shown in Fig. 1, also shows a drop in survivalbeginning at 1 \OA\ however, the degree of toxicity is farless than that demonstrated for continuous treatment. Astandard treatment protocol of 2 (UM-SazaCR for 24 h waschosen to begin the hprt activation study.
Generation of the F3BT1 cell lineThe F3B line is expected to carry two hprt genes(Table 1), both presumably normal wild-type alleles,with one allele inactive owing to its location on theinactive X chromosome. To create a selective system forthe activation of this inactive allele HPRT-deficientmutants of F3B were selected in medium containing thelethal purine analogue 6-thioguanine (6tg). Spontaneous6tgr mutants were found in the F3B line at a frequency of3-8x10" , consistent with a single gene mutation (Chu &Mailing, 1968). Six such mutants were cloned andcharacterized. These mutants were able to grow normallyin medium containing 10/UM-6tg, a property stablethrough 50 generations in non-selective medium. Onemutant, F3BT1, of presumed genotype hprt~/hprt^(brackets indicate the inactive gene), was chosen for thisstudy. Biochemical assay demonstrated that it had negli-gible HPRT activity (Table 2).
SazaCR induction of HPRT activity in F3BT1In an attempt to activate the hprt^ gene in F3BT1,these cells were treated with 2/iM-5azaCR for 24 h,followed by a 3-day expression and recovery time incomplete medium. Following treatment, the frequency of
hprt activation in Chinese hamster cells 725

Table 2. Biochemical characterization of Chinese hamster cell lines
Cell type
Wild-type
Spontaneous 6tgr mutant
Mutagen-induced 6tgr mutant
SazaCR-induced HATR
reactivant
EMS-induced HATR revertant
Spontaneous 6tgr variantof F3BTla21
Cell line
CHL (primary)F3BMtxRI" (CHO)
F3BT1
VE0T3CHWLE05T1DR01
F3BTlalF3BTla2F3BTla3F3BTla4F3BTlaSF3BTla21F3BTla22F3BTla23F3BTla24F3BTla25
F3BTlrlF3BTlr3
F3BTla21-Tl
HPRT activity*(nmolh~'mg~')±S.D.h
222±4199 ±6214 ±8
0-3±0-l
0-6±010-8 ±0-10-1 ±0-226 ±2
74 ±5183 ±775 ±678 ±8
186 ±4156 ±662 ±4
158 ±7116 ± 4135 ±4
188 ±575 ±6
0-1 ±0-2
'Results given are averages of determinations made on a minimum of two independent extract preparations, each assayed inbStandard deviation.
Table 3. Induction of the HAT* phenotype
Spontaneous
Cells plated Number ofCell line (XlO7) colonies
Frequency*of HAT*
5azaCR-treated
Cells plated Number of(XlO7) colonies
duplicate.
Frequency'of HAT*
F3BT1 6-2 1-6 x 003c
VE0T3CHWLE05T1DR01
0-81-00-90-4
0000
<l-3xlO~7
<l-0xl0~7
< M x l ( T 7
<2-3xlCT7
0-70-80-80-5
0
0000
2-8xl(T3
(0-86-4-4)d
<l-4xl(T7
<1-3X1O~7
<l-2xlO"7
<2-lxl(T7
"Results given are weighted averages of a minimum of two independent experiments. Frequencies preceded by < represent experiments inwhich no HATR colonies were detected. The frequencies presented are therefore upper limits calculated by assuming one colony among the cellsplated.
bThis frequency is based on a single colony detected after staining plates with Methylene Blue. The phenotype of this colony was thereforenot tested, and the frequency given represents an upper limit.
c30xl07 cells were plated in these experiments. The number given represents cells on plates eventually scored for HATR colonies.d Values given are the range of frequencies obtained over 16 independent experiments, X10" .
cells capable of growth in HAT medium increased fromless than 10~7 to over 10~3 (Table 3). To establish thatthe HATR phenotype of these colonies was the result ofhprt gene activation 10 HATR colonies were isolated andtested for HPRT activity using a radiolabelling assay.The results (Table 2) clearly demonstrate that HPRTactivity was present in these cell lines. The wild-type F3Band CHO cell lines had normal HPRT activities in therange of 200nmol phosphoribosylated product h~ mg~protein (Farrell & Worton, 1977), while F3BT1 itself hadnegligible activity (Table 2). The 10 HATR derivatives ofF3BT1 had HPRT activities ranging from 74 to 186 units(37-93% wild-type activity).
SazaCR treatment of control cell linesThe induction of HPRT activity in the F3BT1 cell linesby 5azaCR treatment may have occurred by either of twopossible mechanisms. The expected target for inductionis the presumably wild-type hprt gene located on theinactive X chromosome. A second possibility, however, isreversion of the defective hprt~ allele on the active X. Totest for a possible effect of 5azaCR treatment on reversionat the hprt locus four hprt~ control lines were treated withthe drug under identical conditions to those used forF3BT1. These lines are described in Table 1, andinclude the male-derived lines VEOT3 and CHWL, andthe CHO-derived lines EOST1 and DRO1. Reversion
726 5. G. Grant and R. G. Worton
Table 4. Mutagenic capacity of 5azaCR
Selection
HATR
6 ^
Cellline
VEOT3
EO5T1
F3B
TFHE
Cellsplated(xlO7)
2-8
3-6
1-4
1-0
Spontaneous
Numberof
colonies
3
12
32
9
Mutationfrequency*
1-1X10"8
(1-1-1-813-3X1O"8
(3-9-5-0)
2-3X10"6
(0-8-4-4)90X10" '
Cellsplated(XlO7)
2-1
3-5
1-5
0-8
5azaCR-treated
Numberof
colonies
3
16
45
9
Mutationfrequency*
1-5X10"8
(1-6-2-9)4-6X10"6
(4-7-7-1)
2-9X10"6
(2-0-4-7)1-1x10"*
Cellsplated(XlO7)
1-5
0-9
0-4
0-4
EMS-treated
Numberof
colonies
38
374
363
330
Mutationfrequency*
2-6X10"6
(2-0-3-5)4-IX 10"'(3-7-5-5)
8-8x10"'(6-4-11)
8-lx 10"'(7-5-11) (0-8-1-5) (7-0-9-4)
* Results given are weighted averages of a minimum of two independent experiments. Values in brackets represent the range of frequenciesfrom individual experiments given to the same order of magnitude as the weighted average in each case. These frequency ranges include onlythose experiments in which resistant colonies were detected. The pooled frequency given may not fall within this range when data were includedfrom experiments in which no such colonies were found.
may be studied unambiguously in the male lines, as theylack an inactive X chromosome. The CHO lines arefemale in origin, but cytogenetic studies have indicatedthat the inactive X chromosome has been lost during theevolution of this line in culture (Deaven & Petersen,1973; Worton et al. 1977). The HPRT" phenotype of thecontrol lines was confirmed by biochemical assay(Table 2). While VEOT3, CHWL and EO5T1 all hadnegligible HPRT activities (less than 1 unit), DRO1,although thioguanine resistant, carries the hprtB allele,which produces an electrophoretically distinct HPRTprotein with 5-10% wild-type activity (Chasin &Urlaub, 1976).
5azaCR treatment of these four cell lines had no effecton the frequency of HATR colonies (Table 3). This resultis in accord with previous investigations showing that5azaCR has negligible mutagenic capacity in eukaryotes(Marquardt & Marquardt, 1977; Landolph & Jones,1982), and is consistent with the hypothesis that inacti-vation of the hprt^ gene is, in fact, responsible for5azaCR-induced HAT resistance in F3BT1.
Mutagenic potential of SazaCRSince no HATR colonies were detected from any of thecontrol lines, in either the presence or absence of 5azaCR,VEOT3 and EO5T1 were retested in experiments scaledup 100-fold. These lines were also treated with themutagen ethylmethane sulphonate (EMS). The results,summarized in Table 4, show no increase above thespontaneous mutation frequency in cell populationstreated with 5a2aCR. In contrast, EMS treatment causedan approximate 100- to 1000-fold increase, as expected.These results show that 5azaCR has no detectable effecton the conversion of a defective hprt~ allele to hprt+.Finally, to ensure that the F3B line itself it not unusuallysusceptible to a mutagenic effect of 5azaCR, the wild-type lines F3B and TFHE3 were tested for induction ofthe forward mutation hprt+ to hprt~. The frequency ofthis mutation should be unaffected by the presence of theinactive X chromosome, since mutation at the inactiveallele would not affect the phenotype of the cell. The
results of these experiments, also given in Table 4,demonstrate that 5azaCR has no mutagenic effect at thehprt locus in any of the cell lines tested. As seen inTable 3, F3BT1 has a very low frequency of spontaneousreversion, consistent with those of similar HPRT mu-tants in male cell lines and CHO. Treatment of F3BT1with EMS raised the frequency of HATR colonies to7-9X 10~ , similar to those of mutagenized populations ofVEOT3 or EO5T1 (Table 4).
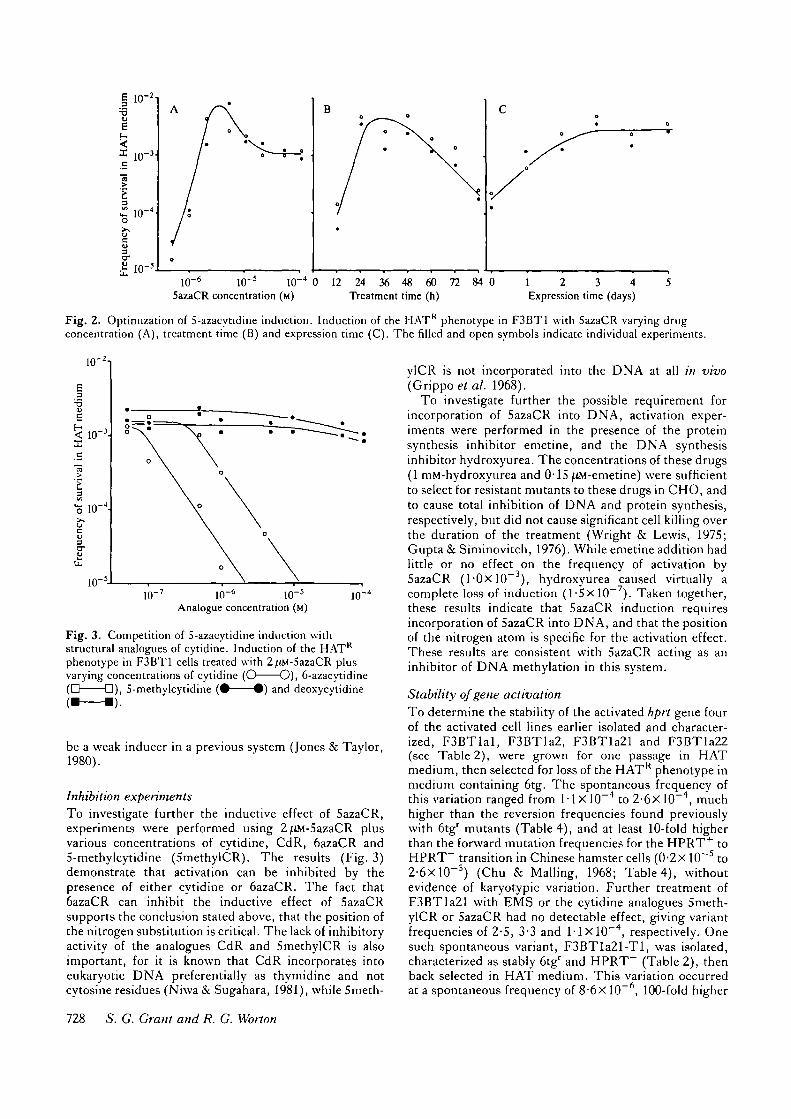
Optimization of 5azaCR inductionTo ensure that the standard induction conditions inferredfrom previous systems, and the toxicity of 5azaCR inF3B, were optimal for observing activation of hprt in theF3BT1 cell line, further experiments were performedvarying 5azaCR concentration, treatment time and ex-pression time. The curves generated by these exper-iments are given in Fig. 2. The standard activationconditions of 2^tM-5azaCR, 24 h treatment time and 3-day expression and recovery time are close to the opti-mum for all three variables.
Analogue experimentsTo ensure that HPRT activation was a specific effect of5azaCR, F3BT1 was treated under standard activationconditions with analogues of this drug, such as cytidine,deoxycytidine (CdR), 6-azacytidine (6azaCR),5-fluorodeoxycytidine (FCdR) and 5-bromodeoxyuridineat concentrations ranging from 0-05 to 100;UM, thenselected in HAT medium. No HAT-resistant colonieswere observed with any of these compounds (reversionfrequencies less than 10~7). The 6azaCR control isparticularly important, as this compound differs from5azaCR only in the ring position of the extra nitrogenatom. The results indicate that induction is dependentupon substitution at the 5 position of the pyrimidine ring,the position of methyl addition to cytosine residues inDNA. It is also interesting that this is the second study inwhich FCdR failed to induce activity of the inactive hprtgene (Jones et al. 1982), despite the fact it was found to
hprt activation in Chinese hamster cells 727
I'•BEH
10"
>
u. 10-10" 10- 5 io-4 o
5azaCR concentration (M)12 24 36 48 60
Treatment time (h)72 84 0 1 2 3 4
Expression time (days)
Fig. 2. Optimization of S-azacytidine induction. Induction of the HATR phenotype in F3BT1 with SazaCR varying drugconcentration (A), treatment time (B) and expression time (C). The filled and open symbols indicate individual experiments.
10"
E
'•5
10"
10"
10,-5
10" 10" 10" 10"Analogue concentration (M)
Fig. 3. Competition of 5-azacytidine induction withstructural analogues of cytidine. Induction of the HATR
phenotype in F3BT1 cells treated with 2jiM-5azaCR plusvarying concentrations of cytidine (O O), 6-azacytidine( • • ) , 5-methylcytidine ( • • ) and deoxycytidine
be a weak inducer in a previous system (Jones & Taylor,1980).
Inhibition experimentsTo investigate further the inductive effect of 5azaCR,experiments were performed using 2^iM-5azaCR plusvarious concentrations of cytidine, CdR, 6azaCR and5-methylcytidine (5methylCR). The results (Fig. 3)demonstrate that activation can be inhibited by thepresence of either cytidine or 6azaCR. The fact that6azaCR can inhibit the inductive effect of SazaCRsupports the conclusion stated above, that the position ofthe nitrogen substitution is critical. The lack of inhibitoryactivity of the analogues CdR and 5methylCR is alsoimportant, for it is known that CdR incorporates intoeukaryotic DNA preferentially as thymidine and notcytosine residues (Niwa & Sugahara, 1981), while 5meth-
ylCR is not incorporated into the DNA at all in vivo(Grippoef a/. 1968).
To investigate further the possible requirement forincorporation of 5azaCR into DNA, activation exper-iments were performed in the presence of the proteinsynthesis inhibitor emetine, and the DNA synthesisinhibitor hydroxyurea. The concentrations of these drugs(1 mM-hydroxyurea and 0-15 jtiM-emetine) were sufficientto select for resistant mutants to these drugs in CHO, andto cause total inhibition of DNA and protein synthesis,respectively, but did not cause significant cell killing overthe duration of the treatment (Wright & Lewis, 1975;Gupta & Siminovitch, 1976). While emetine addition hadlittle or no effect on the frequency of activation by5azaCR (l-OxlCT3), hydroxyurea caused virtually acomplete loss of induction (l-5xlO~7). Taken together,these results indicate that 5azaCR induction requiresincorporation of 5azaCR into DNA, and that the positionof the nitrogen atom is specific for the activation effect.These results are consistent with 5azaCR acting as aninhibitor of DNA methylation in this system.
Stability of gene activationTo determine the stability of the activated hprt gene fourof the activated cell lines earlier isolated and character-ized, F3BTlal, F3BTla2, F3BTla21 and F3BTla22(see Table 2), were grown for one passage in HATmedium, then selected for loss of the HATR phenotype inmedium containing 6tg. The spontaneous frequency ofthis variation ranged from 1-1X 10 to2-6XlO , muchhigher than the reversion frequencies found previouslywith 6tgr mutants (Table 4), and at least 10-fold higherthan the forward mutation frequencies for the HPRT + toHPRT" transition in Chinese hamster cells (0-2x 10~s to2-6xlO~5) (Chu & Mailing, 1968; Table 4), withoutevidence of karyotypic variation. Further treatment ofF3BTla21 with EMS or the cytidine analogues 5meth-ylCR or 5azaCR had no detectable effect, giving variantfrequencies of 2-5, 3-3 and 1-1X10~ 4 , respectively. Onesuch spontaneous variant, F3BTla21-Tl, was isolated,characterized as stably 6tgr and HPRT" (Table 2), thenback selected in HAT medium. This variation occurredat a spontaneous frequency of 8-6x 10~ , 100-fold higher
728 5. G. Grant and R. G. Worton

than reversion in the original F3BT1 line (Table 3). Thisfrequency was not significantly affected by treatmentwith EMS (5-OxlCT6) but increased to 1-3X10"4 uponstandard treatment with 5azaCR.
Induction in a cell hybrid with an electrophoreticallydistinct mutant HPRT proteinAs a final test of the hypothesis that 5azaCR treatmentaffects the inactive hprt<- gene rather than the mutatedhprf gene in F3BT1, a hybrid system was developedwith an electrophoretically distinguishable mutantHPRT protein. The wild-type F3B line (representedgenotypically as hprt+/hprt^ ) was hybridized to theCHO line DR01 , an ouabain-resistant derivative ofDR31 that carries the hprtB allele (Chasin, 1972; Chasin& Urlaub, 1976). This hybrid, called FDR (hprt+/hprt^/hprtB) was then plated in medium containing 6tgto select for segregants that had lost the active hprt+ gene(the low activity of the hprt® allele allows it to escape thisselection). Previous experiments in this laboratory havedemonstrated that segregation of the HPRT" phenotypeoccurs exclusively by loss or deletion of the active Xchromosome in intraspecific Chinese hamster hybrid cells(Farrell & Worton, 1977). The loss of the wild-typeHPRT activity and retention of the characteristic 5-10 %activity attributable to the hprtB allele was confirmed byHPRT assay (Table 5) and by isoelectric focusing(Fig. 4). Loss or deletion of the active X chromosomewas confirmed karyotypically and by activity assays of theX-linked genes pgk and g6pd (data not shown). Thiskaryotypically reduced hybrid was then treated with5azaCR and selected in HAT medium. As shown inFig. 4, these induced HATR colonies retained theHPRTB gene product without alteration and displayed anew, wild-type gene activity. Similar results were ob-tained for a total of nine activants from two 6tgr segre-gants of two independent FDR hybrids (Table 5).
One such activant was further segregated by a secondround of selection in 6tg. These HPRT" segregants,although they retained the electrophoretic band and 5 %activity characteristic of the hprtB allele, were not induc-ible to HATR with 5azaCR, consistent with the hybrid nolonger carrying an inactive hprt gene. The initial acti-vation of this gene on the inactive X had allowed for theremoval of this chromosome by selection.
These experiments indicate that in the hybrid system5azaCR acts on the hprt gene located on the inactive Xchromosome, and has no detectable effect on the active ormutant allele. This strongly suggests that 5azaCR has thesame mechanism of action on the inactive X chromosomein the diploid F3BT1.
Discussion
The experiments described in this report were designedto determine whether a gene on the inactive X chromo-some in a diploid female cell might be subject toactivation by the DNA methylation inhibitor 5azaCR.Many of the previous studies on activation of the mam-malian X chromosome have utilized interspecific somatic
Table 5. Biochemical characterization of hybrids,segregants and reactivants
Cell type
Wild-type parent6tgr parent
HybridHybrid mini-culture
6 1 / segregant
5azaCR-induced HATR
reactivant
Hybrid mini-culture
6tgr segregant
SazaCR-induced HATR
reactivant
HybridHybrid mini-culture
6tgr segregant
5azaCR-induced HATR
reactivant
Hybrid mini-culture
6tgr segregant
SazaCR-induced HATR
reactivant
Cell line
F3BDRO1
FDR-1FDR1-1
FDR1-1T3
lT3allT3a2lT3a3lT3a4
FDR1-2
FDR1-2T4
2T4a22T4a4
FDR-2FDR2-1
FDR2-1T1
21Tla221Tla4
FDR2-2
FDR2-2T2
22T2a3
HPRT activity*(nmolh"1 mg~')
±S.D.b
199 ±626±2
165 ±6175 ±6
20 ±2
97 ±8101 ±2112±6177 ±6
178 ±4
28±2
130 ±6147 ±4
208±6214 ±6
13 ±2
164 ±4203 ±6
193 ±4
21 ±1
121 ±4
* Results given are averages of determinations made on a minimumof two independent extract preparations, each assayed in duplicate.
bStandard deviation.
cell hybrid systems, in which the major practical advan-tage was that the activated gene could be unambiguouslyassigned to the inactive X by the species specificity of thegene product (Graves, 1982; Lester et al. 1982; Mohan-das et al. 1981; Hors-Cayla et al. 1983). In contrast,studies of human diploid fibroblasts by Wolf et al. (1982)showed that the inactive HPRT'"1"' gene did not respondto 5azaCR treatment. These contrasting results suggestedthat there might be fundamental differences in thestability of the inactive state and that these differencesmight be related to species of origin, to the transformedstate of the cells or to the chromosomal complement.More recent studies have demonstrated that the inactivehprt gene is activated by 5azaCR in the heteroploidhuman HeLa cell line (Ivarie & Morris, 1986) and inmouse near-diploid undifferentiated EC cells (Paterno etal. 1985). Our own studies were designed to examinewhether such activation is possible in a strictly diploidsomatic cell system.
The subject of this study has been the transformeddiploid Chinese hamster cell line F3B and its thiogua-nine-resistant derivative, F3BT1. The latter, presum-ably, has genotype hprt~'^+\ permitting direct selectionin HAT medium of F3BT1 derivatives that have re-expressed the ( + ) allele. Our results demonstrate thattreatment of F3BT1 with 5azaCR results in a phenotypic
hprt activation in Chinese hamster cells 729

reversion to HATR at high frequency. This inductiveeffect is not demonstrable in control cell lines lacking aninactive X chromosome. These facts, in conjuction withthe demonstrated lack of mutagenic capacity of 5azaCR inthis system, indicate that the most likely role for 5azaCRis in the activation of the hprt gene on the inactive Xchromosome.
The frequency of 5azaCR-induced activation inF3BT1 is comparable to that reported for the hprt geneon the human inactive X chromosome in the interspecificcell hybrids decribed by Mohandas et al. (1984). Spon-taneous activation of X-linked genes in these hybridsoccurred at a frequency of 10~6 (Hellkuhl & Grzeschik,1978; Kahan & DeMars, 1975, 1980), whereas spon-taneous activation of X-linked genes in diploid cells isvery rare, with only a single report in the literature(Migeon et al. 1982). The high frequency of activation inF3BT1 compared to that in human diploid cells mayreflect a fundamental difference between human androdent cells, especially since rodent cells have a smallerproportion of methylated bases in their DNA than dohuman cells (Ehrlich et al. 1982; Gama-Sosae* al. 1983).The frequency of activation may also be related to thestringency of the selection conditions, since in HeLa cellsa greater frequency of phenotypic revertants was demon-strated after selection in azaserine rather than HATmedium (Ivarie & Morris, 1986). Finally, the efficiencyof 5azaCR in activating X-linked genes may be influencedby other cellular properties such as the differentiatedstate of the cells. In this regard we have found thatactivation also occurs in untransformed Chinese hamster
primary cell lines, but that the frequency of the activationevent is dependent on the transformation state of the cells(S. G. Grant & R. G. Worton, 1989).
The results of the analogue and inhibition experimentsare consistent with hprt gene activation being dependenton the incorporation of 5azaCR into DNA. It haspreviously been shown that 5azaCR is a strong inhibitorof eukaryotic maintenance methlyase enzymes (Taylor &Jones, 1982), causing disruption of the inherited meth-lyation pattern (Compere & Palmiter, 1981; Groudine etal. 1981) and a drastic reduction in the proportion ofmethylated bases in the newly synthesized strand (Jones& Taylor, 1981).
As shown in Table 1 the actual level of 5azaCR-induced gene activity varies between different activatedclones. Such variation is characteristic of 5azaCR treat-ment (Mohandas et al. 1984), with no newly activatedgene showing 100% activity. It has been suggested thateven active genes on the inactive X chromosome arepartially repressed (Migeon et al. 1982a,b), and the samemay hold for genes activated by 5azaCR (Mohandas et al.1984; Miller, 1985).
Many studies have demonstrated the inhibitory effectsof 5azaCR on DNA methylation in vivo, and the induc-tive effects of 5azaCR on inactive genes in differentiatedtissues and in repressed endogenous viruses. In general, astrong correlation exists between the degree of hypo-methylation and gene expression. Specific studies on themethylation of the human HPRT gene in the active andinactive states, and after 5azaCR-induced activation,have identified a cluster of methylatable sites in the
HPRTactivity
199
26
165
175
20
97
101
112
177
pi 6-5 pi 7-8
Fig. 4. Isoelectric focusing of HPRT enzyme of hybrids, reduced hybrids, and activants. HPRT from the wild-type line F3Bhas a pi of 6-5; that from the mutant DRO1 has a pi of 7-8. Shown are an original hybrid FDR-1, and a mini-culture ofFDRl-1 begun from a sample of less than 100 cells, both showing a heterozygous HPRT + ' B pattern. The 6tgr segregant FDR1-1T3, which has lost the X chromosome carrying the wild-type HPRT activity, expresses only HPRTB. Four HATR activantsselected after treatment with SaaaCR (lT3al-4) display both the normal and mutant proteins, as expected if the inactive wild-type allele was reactivated, and the mutant allele was unaffected by the treatment.
730 5. C. Grant and R. G. Worton

G + C rich 5' regulatory region whose level of methyl-ation strongly correlates with expression (Wolf et al.1984; Yen et al. 1984, 1986). Methylation of sites withinthe body of the gene is more variable, with certain sitesactually hypermethylated on the inactive X, althoughmethylation of such sites is not altered upon 5azaCR-induced activation of the gene. This 5' CG cluster hasalso been identified as a site of nuclease hypersensitivitywhen present on the active X chromosome or afterreactivation (Wolf & Migeon, 1985). The mouse Hprtgene shows a similar pattern of a 5' hypomethylatedregion and a 3' methylated region specific to the gene onthe active X chromosome (Lock et al. 1986). Recently,however, it has been shown that methylation of theregulatory region of the Hprt gene actually occurs afterthe gene inactivation event that is associated with Xinactivation, both in mouse embryos and in a cell culturemodel (Lock et al. 1987). This result is consistent withthe findings that there are no changes in DNA methyl-ation associated with inactivation of X-linked genes inmarsupials (Kaslow & Migeon, 1987; Graves, 1987) or inthe extraembryonic lineages of eutherian mammals(Kratzer et al. 1983). DNA methylation is now thoughtto be a secondary mechanism involved in stabilizing themaintenance of X inactivation through DNA replication(Grant & Chapman, 1988). Thus the species-specificsusceptibility to 5azaCR-induced hprt reactivation wehave demonstrated in transformed diploid female Chi-nese hamster cells may have implications beyond thosemechanisms of gene inactivation that are known atpresent.
This work was supported by a research grant (MA 7224) fromthe Medical Research Council of Canada.
References
BIRD, A. P. (1978). Use of restriction enzymes to 8tudy eukaryoticDNA methylation. II. The summary of methylated sites supportssemi-conservative copying of the methylation pattern. .7. molec.Biol. 118, 49-60.
CAMPBELL, C. E. & WORTON, R. G. (1977). Chromosome replicationpatterns in an established cell line (CHO). Cvtogenet. Cell Genet.19, 303-319.
CAMPBELL, C. E. & WORTON, R. G. (1979). Evidence obtained byinduced mutation frequency analysis for functional hemizygosity atthe emt locus in CHO cells. Somat. Cell Genet. 5, 51-65.
CHAPMAN, V. M., KRATZER, P. G., SIRACUSA, L. A., QUARANTILLO,B. A., EVANS, R. & LISKAY, R. M. (1982). Evidence for DNAmodification in the maintenance of X-chromosome inactivation ofadult mouse tissues. Proc. natn. Acad. Sci. U.SA. 79, 5357-5361.
CHASIN, L. A. (1972). Non-linkage of induced mutations in Chinesehamster cells. Nature, new Biol. 240, 50-52.
CHASIN, L. A. & URLAUB, G. (1976). Mutant alleles forhypoxanthine phosphonbosyltransferase: codominant expression,complementation, and segregation in hybrid Chinese hamster cells.Somat. Cell Genet. 2, 453-467.
CHU, E. H. Y. & MALLING, H. V. (1968). Mammalian cell genetics.II. Chemical induction of specific locus mutations in Chinesehamster cells in vitro. Proc. natn. Acad. Sci. U.SA. 61,1306-1312.
COMINGS, D. E. (1966). The inactive X chromosome. Lancet li,1137-1138.
COMPERE, S. J. & PALMITER, R. D. (1981). DNA methylationcontrols the inducibility of the mouse metallothionein-1 gene inlymphoid cells. Cell 25, 233-240.
CONSTANTINIDES, P. G., JONES, P. A. & GEVERS, W. (1977).Functional striated muscle cells from non-myoblast precursorsfollowing 5-azacytidine treatment. Nature, Loud. 267, 364—366.
CREUSOT, F., ACS, G. & CHRISTMAN, J. K. (1982). Inhibition ofDNA methyltransferase and induction of Friend erythroleukemiacell differentiation by 5-azacytidine and 5-aza-2'-deoxycytidine.J. biol. Chem. 257, 2041-2048.
DEAVEN, L. L. & PETERSEN, D. F. (1973). The chromosomes ofCHO, an aneuploid Chinese hamster cell line: G-band, C-band,and autoradiographic analyses. Chromosoma 41, 129-144.
DOSKOCIL, J. & SORM, F. (1962). Distribution of 5-methylcytosine inpyrimidine sequences of deoxyribonucleic acids. Biochim. biophvs.Ada 55, 953-955.
EHRLICH, M., GAMA-SOSA, M. A., HUANG, L.-H., MIDGETT, R.,
Kuo, M. K. C , MCCUNE, R. A. & GEHRKE, C. (1982). Amountand distribution of 5-methylcytosine in human DNA fromdifferent types of tissues of cells. Mud. Acids Res. 10, 2709-2721.
FARRELL, S. A. & WORTON, R. G. (1977). Chromosome loss isresponsible for segregation at the HPRT locus in Chinese hamstercell hybrids. Somat. Cell Genet. 3, 539-551.
GAMA-SOSA, M. A., MIDGETT, R. M., SLAGEL, V. A., GITHENS, S.,
Kuo, K. C , GEHRKE, C. W. & EHRLICH, M. (1983). Tissue-specific differences in DNA methylation in various mammals.Biochim. biophvs. Acta 740, 212-219.
GRANT, S. G. & CHAPMAN, V. M. (1988). Mechanisms of X-chromosome regulation. A. Rev. Genet. 22, 199-233.
GRAVES, J. A. M. (1982). 5-Azacytidine-induced re-expression ofalleles on the inactive X chromosome in a hybrid mouse cell line.Expl Cell Res. 141, 99-105.
GRAVES, J. A. M. (1987). The evolution of mammalian sexchromosomes and dosage compensation: clues from marsupials andmonotremes. Trends Genet. 3, 252-256.
GRIPPO, P., IACCARINO, M., PARISI, E. & SCARANO, E. (1968).
Methylation of DNA in developing sea urchin embryos. J. molec.Biol. 36, 195-208.
GROUDINE, M., EISENMAN, R. & WEINTRAUB, H. (1981). Chromatin
structure of endogenous retroviral genes and activation by aninhibitor of DNA methylation. Nature, Land. 292, 311-317.
GUPTA, R. S. & SIMINOVTTCH, L. (1976). The isolation andpreliminary characterization of somatic cell mutants resistant to theprotein synthesis inhibitor - emetine. Cell 9, 213-219.
HELLKUHL, B. & GRZESCHIK K.-H. (1978). Partial reactivation of ahuman inactive X chromosome in human-mouse somatic cellhybrids. Cvtogenet. Cell Genet. 22, 527-530.
HORS-CAYLA, M. C , HEUERTZ, S. & FREZAL, J. (1983).
Coreactivation of four inactive X-genes in a hamster X humanhybrid and persistence of late replication of reactivated Xchromosome. Somat. Cell Genet. 9, 645-657.
IVARIE, R. & MORRIS, J. A. (1986). Activation of a nonexpressedhypoxanthine phosphoribosyltransferase allele in mutant H23HeLa cells by agents that inhibit DNA methylation. Molec. cell.Biol. 6, 97-104.
JONES, P. A. & TAYLOR, S. M. (1980). Cellular differentiation,cytidine analogs and DNA methylation. Cell 20, 85-93.
JONES, P. A. & TAYLOR, S. M. (1981). Hemimethylated duplexDNAs prepared from 5-azacytidine-treated cells. Nucl. Acids Res.9, 2933-2947.
JONES, P. A., TAYLOR, S. M., MOHANDAS, T. & SHAPIRO, L. J.
(1982). Cell cycle-specific reactivation of an inactive X-chromosome locus by 5-azadeoxycytidine. Proc. natn. Acad. Sci.U.SA. 79, 1215-1219.
KAHAN, B. & DEMARS, R. (1975). Localized derepression on thehuman inactive X chromosome in mouse-human cell hybrids. Proc.natn. Acad. Sci. U.SA. 72, 1510-1514.
KAHAN, B. & DEMARS, R. (1980). Autonomous gene expression onthe human inactive X chromosome. Somat. Cell Genet. 6,309-323.
KASLOW, D. C. & MIGEON, B. R. (1987). DNA methylationstabilizes X chromosome inactivation in eutherians but notmarsupials: evidence for multistep maintenance of mammalian Xdosage compensation. Proc. natn. Acad. Sci. U.SA. 84, 6210-6214.
KRATZER, P. G., CHAPMAN, V. M., LAMBERT, H., EVANS, R. &
LISKAY, R. M. (1983). Differences in the DNA of the inactive X-chromosomes of fetal and extraembryonic tissues of mice. Cell 33,
hprt activation in Chinese hamster cells 731

37-42.LANDOLPH, J. R. & JONES, P. A. (1982). Mutagenicity of 5-aza-
cytidine and related nucleosides in C3H/10T1/2 clone 8 and V79cells. Cancer Res. 42, 817-823.
LESTER, S. C , KORN, N. J. & DEMARS, R. (1982). Derepression of
genes on the human inactive X chromosome: evidence ofdifferences in locus-specific rates of transfer of active and inactivegenes after DNA-mediated transformation. Somat. Cell Genet. 8,265-284.
LIN, C. C , CHANG, T. D. & NIEWCZAS-LATE, V. (1971). The
establishment and chromosome analysis of new cell line of Chinesehamster from spontaneous transformation in vitro. Can. J. Genet.Cytol. 13, 9-13.
LlSKAY, R. M. & EVANS, R. J. (1980). Inactive X chromosome DNAdoes not function in DNA-mediated cell transformation for thehypoxanthine phosphoribosyltransferase gene. Proc. natn. Acad.Sci. U.SA. 77, 4895-4898.
LOCK, L. F., MELTON, D. W., CASKEY, C. T. & MARTIN, G. R.
(1986). Methylation of the mouse hprt gene differs on the activeand inactive X chromosomes. Molec. cell. Biol. 6, 914-924.
LOCK, L. F., TAKAGI, N. & MARTIN, G. R. (1987). Methylation of
the Hprt gene on the inactive X chromosome occurs afterchromosome inactivation. Cell 48, 39-46.
LYON, M. F. (1961). Gene action in the X-chromosome of the mouse(Mus musculus L.). Nature, Land. 190, 372-373.
MARQUARDT, H. & MARQUARDT, H. (1977). Induction of malignanttransformation and mutagenesis in cell cultures by cancerchemotherapeutic agents. Cancer Mi, 1930-1934.
MIGEON, B. R. (1972). Stability of X chromosomal inactivation inhuman somatic cells. Nature, Land. 239, 87-89.
MIGEON, B. R., SPRENKLE, J. A. & Do, T. T. (1978). Studies ofhuman-mouse cell hybrids with respect to X-chromosomeinactivation. In Genetic Mosaics and Chimeras in Mammals (ed.L. B. Russell), pp. 329-337. New York: Plenum Press.
MIGEON, B. R., WOLF, S. F., MARENI, C. & AXELMAN, J. (1982).
Derepression with decreased expression of the G6PD locus on theinactive X chromosome in normal human cells. Cell 29, 595-600.
MILLER, O. ] . (1985). Dosage compensation in mammals: why doesa gene on the inactive X yield less product than one on the activeX} Hum. Genet. 69, 97-101.
MOHANDAS, T., SPARKES, R. S., BISHOP, D. E., DESNICK, R. J. &
SHAPIRO, L. J. (1984). Frequency of reactivation and variability inexpression of X-linked enzyme loci. Atn.J. Hum. Genet. 36,916-926.
MOHANDAS, T., SPARKES, R. S. & SHAPIRO, L. J. (1981).
Reactivation of an inactive human X chromosome: evidence for Xinactivation by DNA methylation. Science 211, 393-396.
NIWA, O. & SUGAHARA, T. (1981). 5-Azacytidine induction of mouseendogenous type C virus and suppression of DNA methylation.Proc. natn. Acad. Sci. U.SA. 78, 6290-6294.
PATERNO, G. D., ADRA, C. N. & MCBURNEY, M. W. (1985). X
chromosome reactivation in mouse embryonal carcinoma cells.Molec. cell. Biol. 5, 2705-2712.
SUTTER, D. & DOERFLER, W. (1980). Methylation of integratedadenovirus type 12 DNA sequences in transformed cells isinversely correlated with viral gene expression. Proc. natn. Acad.Sci. U.SA. 77, 253-256.
TAYLOR, S. M. & JONES, P. A. (1982). Mechanism of action ofeukaryotic DNA methyltransferase. Use of 5-azacytosine-containing DNA. J. molec. Biol 162, 679-692.
THACKER, J. (1981). The chromosomes of a V79 Chinese hamsterline and a mutant subline lacking HPRT activity. Cvtogenet. CellGenet. 29, 16-25.
VANYUSHIN, B. F., TKACHERA, S. & BELOZERSKY, A. N. (1970).
Rare bases in animal DNA. Nature, Land. 255, 948-949.WAALWUK, C. & FLAVELL, R. A. (1978). DNA methylation at a
CCGG sequence in the large intron of the rabbit /3-globin gene:tissue-specific variations. Nucl. Acids Res. 5, 4631-4641.
WOLF, S. F., JOLLY, D. J., LUNNEN, K. D., FRIEDMANN, T. &
MlGEON, B. R. (1984). Methylation of the hypoxanthinephosphoribosyltransferase locus on the human inactive Xchromosome: implications for X-chromosome inactivation. Proc.natn. Acad. Sci. U.SA. 81, 2806-2810.
WOLF, S. F. & MIGEON, B. R. (1982). Studies of X chromosomeDNA methylation in normal human cells. Nature, Land. 295,667-671.
WOLF, S. F. & MIGEON, B. R. (1985). Clusters of CpG dinucleotidesimplicated by nuclease sensitivity as control elements ofhousekeeping genes. Nature, Land. 314, 467-469.
WoRTON, R. G. (1978). Karyotype heterogeneity in CHO cell lines.Cvtogenet. Cell Genet. 21, 105-110.
WoRTON, R. G. & DOFF, C. (1979). Karyotyping. In Methods inEnzymology vol. 58 (ed. W. B. Jacoby & 1. H. Pastan),pp. 322-344. New York: Academic Press.
WORTON, R. G., Ho, C. C. & DUFF, C. (1977). Chromosomestability in CHO cells. Somat. Cell Genet. 3, 27-45.
WRIGHT, J. A. & LEWIS, \V. H. (1975). Evidence of a common site ofaction for the antitumor agents, hydroxyurea and guanazole. J.cell. Physiol. 83, 437-440.
YEN, P. H., MOHANDAS, T. & SHAPIRO, L. J. (1986). Stability ofDNA methylation of the human hypoxanthine phos-phoribosyltransferase gene. Somat. Cell molec. Genet. 12, 153-161.
YEN, P. H., PATEL, P., CHINAULT, A. C , MOHANDAS, T. &
SHAPIRO, L. J. (1984). Differential methylation of hypoxanthinephosphonbosyltransferase genes on active and inactive human Xchromosomes. Proc. natn. Acad. Sci. U.SA. 81, 1759-1763.
{Received 20 October 1988 - Accepted 6 January 1989)
732 S. G. Grant and R. G. Worton