acrofacial dysostosis type rodríguez
TRANSCRIPT

American Journal of Medical Genetics 135A:81–85 (2005)
Clinicial ReportAcrofacial Dysostosis Type RodrıguezBoyan Dimitrov,1 Irina Balikova,2 Nely Jekova,3 Lilija Vakrilova,3 Jean-Pierre Fryns,1* and Emil Simeonov2
1Center for Human Genetics, University Hospital Gasthuisberg, University of Leuven, Belgium2Pediatric Clinic, University Hospital ‘‘Alexandrovska’’, Medical University of Sofia, Bulgaria3Neonatology Clinic, University Hospital of Obstetrics and Gynecology ‘‘Maichin Dom’’, Medical University of Sofia, Bulgaria
The acrofacial dysostoses (AFD) are a clinicallyand causally heterogeneous group of conditionscharacterized by mandibulofacial dysostosis anda variety of limb anomalies. Several abnormalitiesaffecting different internal organs and the centralnervous system (CNS) have been described. De-pending on the type of limb defects, two majorgroups have been delineated: (1) with predomi-nantly pre-axial anomalies, Nager type AFD, and(2) with predominantly post-axial involvement,Genee–Wiedemann form of AFD, also known asPOADS, respectively. Other forms of ‘‘true AFD’’have been described as Kelly, Reynolds, Arens(also Tel Aviv form), Rodrıguez (or Madridform), Richieri–Costa, and Patterson–Stevenson–Fontaine types. However, whether they are dis-tinct entities or represent variants of the samecondition remains unclear. Rodırguez AFD wasdescribed as a new lethal form of AFD in threeaffected sibs with severe mandibular hypoplasia,severe predominantly pre-axial limb deficien-cies, absent fibulae and ribs, and internal organanomalies, the most remarkable of which arearrhinencephaly and abnormal lung lobulation.We present a newborn girl with Rodrıguez type ofAFD, who died a few days after the birth due torespiratory failure. The phenotype and the causeof this condition are discussed.� 2005 Wiley-Liss, Inc.
KEY WORDS: acrofacial dysostosis; mandibulo-facial dysostosis; pre-axial limbdefects; post-axial limb defects;Rodrıguez syndrome
INTRODUCTION
The acrofacial dysostoses (AFDs) represent a number ofconditions characterized by the combination of mandibulofa-cial dysostosis and limb defects in varying degrees. Theyappear to represent polytopic developmental field defects dueto causal heterogeneity [Opitz et al., 1993]. In 1993, on a purelyclinical basis, Opitz et al. delineated twomajor groups of AFDs:(1)Nager syndromewithpredominantly pre-axial limbdefects;and (2) Genee–Wiedemann syndrome with much more pro-minent post-axial limb defects. These authors also recognized
other rare forms of AFDs and acrofacial field defects as a partof ‘‘related conditions’’ or chromosomal abnormalities [Opitz,1987; Opitz et al., 1993].
Rodrıguez et al. [1990] described three affected sibs withsevere, apparently lethal AFD with severe mandibular hypo-plasia, predominantly pre-axial limb deficiencies, absentfibulae and ribs, and internal organ anomalies, the most re-markable of which are arrhinencephaly and abnormal lunglobulation.However, one of these sibs hadwhatmay be regard-ed ‘‘severeNager syndrome.’’ Subsequently, a fewadditional orpreviously published cases were thought to be examples ofRodrıguez AFD [Hecht et al., 1987; Fryns and Kleczkowska,1991; Petit et al., 1992;Oostra et al., 1998;Wessels et al., 2002].The subsequent editorial correspondence [Fryns, 1992, 1999;Hecht, 1992; Rodrıguez et al., 1992a,b; Oostra et al., 1999] hasreopened the discussion about the nosology and classificationof severe AFDs.
We present a newborn girl with an acrofacial dysostosisconsistent with that described by Rodrıguez et al. [1990].
CLINICAL REPORT
A newborn girl died 6 days after birth due to severerespiratory problems. She was born at 36 weeks of gestation,after Caesarean section because of maternal pre-eclampsia,the second pregnancy in a family with a healthy 40-year-oldmother and a 36-year-old father. The first pregnancy endedin spontaneous abortion. Results of biochemical screeningfor Down syndrome in the second trimester were normal. Thefamily history was unremarkable. Birth-weight and -lengthwere 1,300 g and 45 cm, respectively. For severe respiratorydistress she required mechanical respiratory support. Onexamination she had short, down-slanting palpebral fissures,deep-set eyes, hypertelorism, prominent nasal bridge, medi-ally sparse eyebrows, low temporal hair line,microstomia, thinlips, marked micrognathia with protruding pre-maxilla, lowset abnormally modeled ears with absent external auditorymeatus (Fig. 1a,b). There was severe hypotonia with hypo-reflexia. The arms and forearms were very short, withmore severely affected forearms, and the hands appeared tobe attached directly to the upper arms (Fig. 1d). Thumbs wereabsent and there was complete cutaneus syndactyly betweenthe third and fourth fingers and bilateral clinodactyly of fifthfingers. Therewas also a duplication of the terminal phalanx ofthe second left finger, which gave it a thumb-like appearance,and bilateral duplication of the terminal phalanges of thefourth fingers (Fig. 1c,d). She had proximally placed hallucesand the fourth and fifth toes of the right foot were shorter, withpartial syndactyly (Fig. 3a,b). Autopsy did not show any ano-malies of brain, thoracic, or abdominal organs. Skull and limbradiographs demonstrated severe hypoplasia of the mandible,fused temporomandibular joint, hypoplastic scapulae, 11 ribs,hypoplastic shoulder girdle, rudimentary triangular humeri,single, hypoplastic forearm bones, absent first digital rays andcutaneous syndactyly 3–4, duplication of the distal phalanx ofthe second finger and additional ossification centers between
*Correspondence to: Jean-Pierre Fryns, Centre for HumanGenetics, University Hospital Gasthuisberg, Herestraat 49, 3000Leuven, Belgium.E-mail: [email protected]
Received 6 October 2004; Accepted 26 January 2005
DOI 10.1002/ajmg.a.30673
� 2005 Wiley-Liss, Inc.

Fig. 1. a–d: Newborn girl with severe acrofacial dysostosis and typical facial appearance.
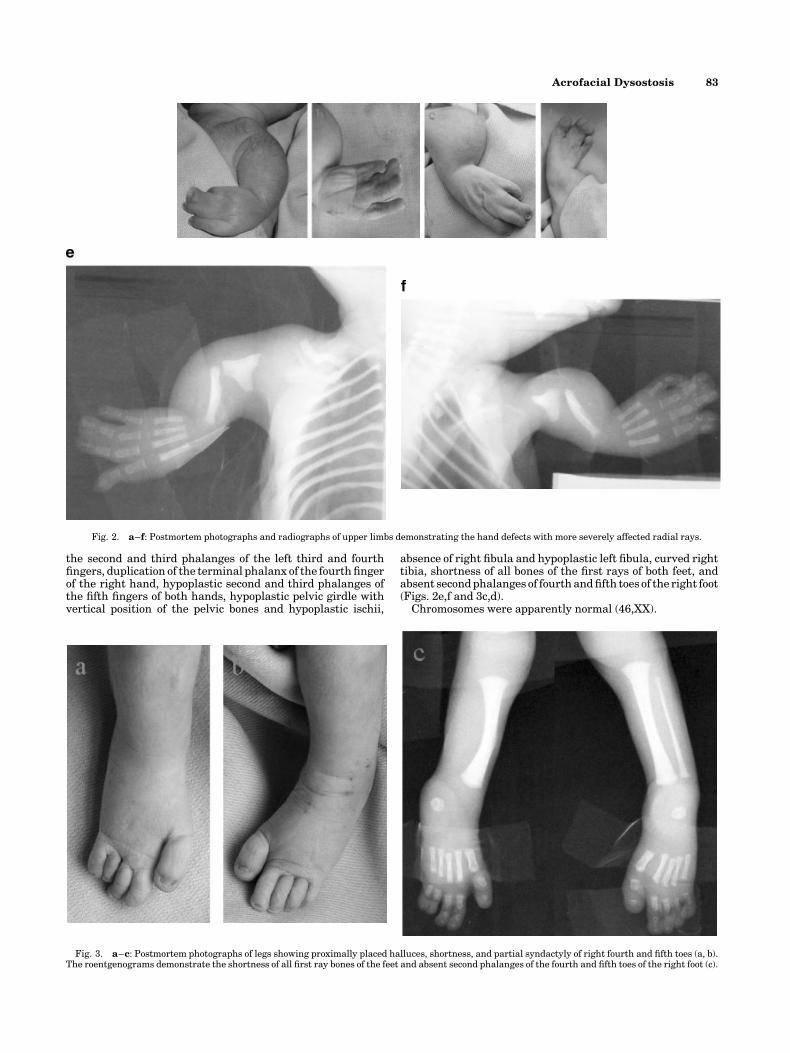
the second and third phalanges of the left third and fourthfingers, duplication of the terminal phalanx of the fourth fingerof the right hand, hypoplastic second and third phalanges ofthe fifth fingers of both hands, hypoplastic pelvic girdle withvertical position of the pelvic bones and hypoplastic ischii,
absence of right fibula and hypoplastic left fibula, curved righttibia, shortness of all bones of the first rays of both feet, andabsent secondphalanges of fourthandfifth toes of the right foot(Figs. 2e,f and 3c,d).
Chromosomes were apparently normal (46,XX).
Fig. 2. a–f: Postmortem photographs and radiographs of upper limbs demonstrating the hand defects with more severely affected radial rays.
Fig. 3. a–c: Postmortem photographs of legs showing proximally placed halluces, shortness, and partial syndactyly of right fourth and fifth toes (a, b).The roentgenograms demonstrate the shortness of all first ray bones of the feet and absent second phalanges of the fourth and fifth toes of the right foot (c).
Acrofacial Dysostosis 83

TABLE
I.AllCurren
tlyPublish
edCasesRep
resentingAcrofacialDysostoses
(AFD)TypeRod
rıguez*
Rod
rıguez
etal.
[1990]
Frynsand
Kleczkow
ska[1991]
Hechtet
al.
[1987]
Petitet
al.
[1992]
Bateset
al.[2002]
Wessels
etal.
[2002]
Present
case
2004
12
34
56
78
910
Gestation
(wee
ks)
?41
40
28–29
31
20
24
21
27
36
Birth
weight(kg)
2.020
2.500
2.850
0.690
1.530
?0.465
Normal
0.718
1,300
Birth
length
(cm)
??
50
?42
??
Normal
?45
OFC
(cm)
??
??
29.2
?23.5
Normal
??
Face Sev
eremicrognathia
þþ
þþ
þþ
þþ
þþ
Malarhypop
lasia
þþ
þþ
þþ
þþ
þþ
Malformed
ears
þþ
þþ
þþ
þþ
þProminen
t,broadnasa
lbridge
þþ
þþ
þþ
þþ
þþ
Dow
n-slantingpalpeb
ralfissures
þþ
þþ
þþ
þþ
þþ
Cleft
palate
þþ
High,arched
�þ
High,arched
þþ
þ�
Atretic
earcanals
þþ
Narrow
canals
Narrow
canals
þ?
þþ
þþ
X-rayfindings
Vertebralanom
alies
��
��
��
þ�
��
Shou
lder
girdle
hypop
lasia
þþ
��
��
??
þþ
Eleven
ribs
þ�
þ�
��
þ�
þþ
Hypolastic
scapulae
þþ
��
��
��
?þ
Short(absent)
humerus
þþ
��
��
þ�
þm
þSingle
(absent)
forearm
þþ
��
��
þ�
hþ
mþ
Radioulnarsynostosis
��
þþ
þ�
��
��
Pre-axialthumbdefects
þ�
þþ
þþ
þOligod
actyly
iOligod
actyly
nþ
Post-axialfinger
defects
þþ
��
�þ
þOligod
actyly
iOligod
actyly
nþ
Cutaneo
usfinger
syndactyly
??
1–2
�1–2
�c
?dþ
3–4
3–4
Pelvic
girdle
hypop
lasia
?þ
��
��
þ?
?þ
Absent/hypop
lastic
isch
ii�
þ�
��
��
?�
þLow
erlimbdefects
a�
��
��
�þ
eþ
jþ
o�
Absent/hypop
lastic
fibulae
�þ
��
��
þþ
jþ
þPre-axialtoedefects
��
?�
��
þOligod
actyly
k�
þPost-axialtoedefects
�þ
?�
��
þOligod
actyly
k�
þToe
syndactyly
??
4–5
��
�?
�4–5
4–5
Autopsy
Card
iacmalformation
sþ
þ�
þ�
�þ
��
�CNSmalformation
sb�
��
��
�þ
f�
��
Arh
inen
cephaly
þþ
��
��
��
þ�
Absentlunglobulation
�þ
��
��
þ�
þ�
Ren
alanom
aly-
��
��
�þ
þþ
�Gen
italanom
aly
��
��
��
þþ
��
Karyotype
�46,XY
46,XY
�46,XX
�46,XX
46,XX
46,XX
46,XX
Sex
MM
MM
FF
FF
FF
Earlylethality
þþ
þþ
þPT
PT
PT
PT
þCon
sanguinity
��
��
��
þ�
�Possible
mod
eof
inheritance
AR
AR
AR
Sporadic
AR
AR
ARg
Sporadic
lSporadic
Sporadic
(þ),present;(�
),absent;(?),unknow
nor
itis
not
men
tion
edin
thetext;M,male;F,female;PT,pregnancy
wasterm
inated;AR,autosomalrecessive.
*Thecase
ofOostraet
al.[1998]is
not
included
because
ofinsu
fficien
tdetailed
description
ofthephen
otype.
aOther
limbdefects
not
inthis
table.
bCNSmalformation
sdifferentfrom
arrhinen
cephaly.
c Patien
t2of
Hechthadproxim
alfusion
oftherightfourthandfifthmetacarp
als.
dProxim
alfusion
oftherightfourthandfifthmetacarp
als.
eAbsence
ofalllogbon
esof
lower
limbs.
f Thirdven
tricle
hydrocephaly
dueto
aqued
uctalsten
osis
andagen
esis
ofcorp
uscallosum.
gTheARmod
eof
inheritance
inthis
patien
twasconfirm
edbyFryns[1999].
hSev
eresh
ortnessof
ulnaeandradii.
i Distalphalanxduplication
ofboththumbs,distaltw
ophalanxduplication
ofthirddigits,andon
lythreemetacarp
als.
j Shortandbow
edtibiaeandseverelysh
ortened
fibulae.
kIn
thetextis
written
thattherewereon
lyfourmetatarsals.
l Parents
werefirstcousinsandanARinheritance
could
besu
spected.
mAsingle,dysp
lastic,andangulatedbon
ewasdetectedbetwee
nthehandsandthesh
oulders.
nTherighthandhadfourdigitsandhypop
lastic
fourthray,andthelefthandhadon
lythreefingerswithou
trecognizable
thumb.
oShortandbow
edtibiae.

DISCUSSION
Over the last 14 years, a few additional reports of patients(Table I) with a phenotype of severe, lethal AFD similar to thesiblings presented by Rodrıguez et al. [1990] were published.Fryns and Kleczkowska [1991] described a newborn boy withsevere AFD, pre-axial upper limb malformations, and con-genital heart defect resembling the third patient of Rodrıguez.In 1992, Hecht [1992] commented that a previously publishedautosomal recessive case of AFD [Hecht et al., 1987] present-ed striking similarities to the sibs reported by Rodrıguezet al. In a correspondence to the editor, Rodrıguez et al.[1992a,b] disagreed with this diagnosis because of the absenceof typical additional skeletal and internal abnormalities. Atthat time, Petit et al. [1992] confirmed the existence of thisnew form of AFD. The proposita seemed to be similar to patient2 of Rodrıguez et al. [1990]. A second affected sib was brieflyreported a few years later [Fryns, 1999], supporting the hypo-thesis of autosomal recessive inheritance, or germinal mosai-cism of a dominant gene.
In 1999, a case with ‘‘severe Nager syndrome with orofacialclefting and phocomelia’’ published by Oostra et al. [1998] wasrecognized as an example of Rodrıguez syndrome [Fryns, 1999;Oostra et al., 1999].
Bates et al. [2002] described what was thought to be ‘‘a newlethal form of AFD.’’ However, the clinical findings arecomparable to those of the patients of Rodrıguez et al. [1990],Petit et al. [1992], and Wessels et al. [2002]. Moreover in thatinstance, there was consanguinity and autosomal recessiveinheritance seemed reasonable.
Recently,Wessels et al. [2002] described a pre-natally ascer-tained female fetus with Rodrıguez AFD and stressed theimportance of internal abnormalities (arrhinencephaly andabnormal lung lobulation) in the diagnosis.
The findings in our patient completely overlap those ob-served in Rodrıguez AFD.
On the basis of the above cases some hallmark findingsemerge in this form of lethal AFD: (1) typical facial anomalieswith severe mandibulofacial dysostosis, absence of auditorycanals, and cleft palate; (2) pre-axial and post-axial handand foot defects with more prominent pre-axial involvement;(3) and additional skeletal anomalies such as 11 ribs, severedysgenesis, and/or agenesis of long bones of upper limbs,anomalies of pelvic bones (hypoplastic/absent ischii), andhypoplasia or agenesis of fibulae. A variety of abnormalitiesaffecting CNS (arrhinencephaly, callosal agenesis), lungs(hypoplastic/hypolobated lungs), heart, and urogenital systemhave been described and further support the diagnosis of AFDtype Rodrıguez. However, the differential diagnosis in the
group of lethal AFDs is not so unequivocal and in the ob-servations reported by Hecht et al. [1987], Fryns andKleczkowska [1991], and Oostra et al. [1998], the diagnosis ofRodrıguezAFDshouldbe taken inaccount at leastwith respectto appropriate genetic counseling of the families. However,until we will get deeper insight into the underlying causalmechanism in this group of lethal AFDs, the debate between‘‘splitters and lumpers’’ in this ‘‘field’’ will remain open.
REFERENCES
Bates AW, Hall CM, Morgan H, Rosser EM, Scheimberg I. 2002. Lethalacrofacial dysostosis, pre- and post-axial defects of the hands, andbilateral renal agenesis. Clin Dysmorphol 11:63–66.
Fryns JP. 1992. Reply to Dr. Rodrıguez et al. Am J Med Genet 42:852.
Fryns JP. 1999. On the nosology of severe acrofacial dysostosis with limbdeficiency. Am J Med Genet 82:282–283.
Fryns JP, Kleczkowska A. 1991. New lethal acrofacial dysostosis syndrome.Am J Med Genet 39:223–224.
Goldstein DJ, Mirkin LD. 1988. Nager acrofacial dysostosis: Evidence forapparent heterogeneity. Am J Med Genet 30:741–746.
Hecht JT. 1992.New lethal acrofacial dysostosis syndrome. AmJMedGenet42:400.
Hecht JT, Immken LL, Harris LF, Malini S, Scott CI Jr. 1987. The Nagersyndrome. Am J Med Genet 27:965–969.
Oostra RJ, Baljet B, Hennekam RC. 1998. Severe acrofacial dysostosis withorofacial clefting and tetraphocomelia diagnosed in the plaster cast of a100-year-old anatomical specimen. Am J Med Genet 78:195–197.
Oostra RJ, Baljet B, Hennekam RC. 1999. Reply to letter to the Editor ofJean-Pierre Fryns ‘‘on the nosology of severe acrofacial dysostosis withlimb deficiency’’. Am J Med Genet 82:283.
Opitz JM. 1987. Nager ‘‘syndrome’’ versus ‘‘anomaly’’ and its nosology withthe postaxial acrofacial dysostosis syndrome of Genee and Wiedemann.Am J Med Genet 27:959–963.
Opitz JM, Mollica F, Sorge G, Milana G, Cimino G, Caltabiano M. 1993.Acrofacial dysostoses: Review and report of a previously undescribedcondition: The autosomal or X-linked dominant Catania form of acro-facial dysostosis. Am J Med Genet 47:660–678.
Petit P, Moerman P, Fryns JP. 1992. Acrofacial dysostosis syndrome typeRodrıguez: A new lethal MCA syndrome. Am J Med Genet 42:343–345.
Rodrıguez JI, Palacios J, Urioste M. 1990. New acrofacial dysostosissyndrome in 3 sibs. Am J Med Genet 35:484–489.
Rodrıguez JI, Palacios J,UriosteM. 1992a.Response toDr.Hecht. AmJMedGenet 42:401.
Rodrıguez JI, Palacios J,UriosteM. 1992b.Acrofacial dysostosis syndromes.Am J Med Genet 42:851.
Wessels MW, Den Hollander NS, Cohen-Overbeek TE, Lesnik ObersteinMS, Nash RM, Wladimiroff JW, Niermeijer MF, Willems PJ. 2002.Prenatal diagnosis and confirmation of the acrofacial dysostosissyndrome type Rodrıguez. Am J Med Genet 113:97–100.
Acrofacial Dysostosis 85