ab initio supermolecule study of charge transfer in the glyoxal–formamide and in the...
TRANSCRIPT

Ab initio Supermolecule Study of Charge Transfer in the Glyoxal-Formamide and in the
HzS-Formamide Systems
PETER OTTO, SANDOR SUHAI, A N D JANOS LADIK Lehrstuhl fur Theoretische Chemie der Friedrich Alexander Unioersitat Erlangen-
Niirnberg, 8520 Erlangen, Germany
Abstract
Ah inirio supermolecule calculations have been performed for the glyoxal-formamide and for the H+formamide charge transfer complexes in their ground states. According to the results obtained with Mulliken's population analysis, glyoxal acts always as an electron acceptor against I'ormumidc which wc have used as model compound for a peptide group. The results indicate also that the amount of transferred charge depends strongly on the intermolecular distance and on the rckitive orientation or the molecular planes. On the other hand we have found that H2S against our expectation acts at most relative geometrical positions as an electron acceptor with respect to Corniamide.
Introduction
Szent-Gyorgyi [ I ] has pointed out that unsaturated ketons and aldehydes, or diketons and dialdehydes, respectively, can act as electron acceptors with regard to proteins, converting these insulators to conductors. Further on, in analogy to the TCNQ-TTF charge transfer system [2] it was suggested that sul- fur-containing compounds or/and sulfur-containing side chains of amido acids may act as electron donors with respect to the polypeptide groups [3]. In the second case the originally unfilled conduction band of the protein molecules would become partially filled, and in this way the system would be converted again from insulator to conductor.
To start a quantum chemical investigation of these interesting ideas as a first step, we have performed ab initio supermolecule calculations for the model charge transfer systems glyoxal-formamide and H$Wormamide, respectively. These investigations have been performed using different relative geometries for the two partner moleculcs and applying different basis sets.
Method and Results
For the glyoxal-formamide system two basis sets have been used, a minimal one (4s/2p (C, N, 0) + 4s (H) contracted to 2s/lp + 1s [4]) and a double zeta type one (7/3 + 4 - 4/2 + 2 [S]). In the case of the HsS-formamide system the same double zeta basis set has been used as for the first model system [5] and for sulfur a 10s/6p - 5s/3p basis [5] has been applied. In one relative geometrical position (see below) the calculation has been repeated with the help
International Journal oTQuanturn Chemistry: Quantum Biology Symposium 4,45 1-457 (1977) fc 1977 by John Wiley & Sons, Inc. 45 I

452 OTTO, SUHAI, A N D LADIK
0
Figure 1 . Standard geometries of formamide, glyoxal, and H2S.
of a still larger basis (9s/5p + 6s - 6s/3p + 3s for C, N, 0, and H [ 6 ] , re- spectively, and 12s/9p - 6s/4p for S [7]). Since we wanted to obtain the charges in three decimals, we have chosen as convergence criteria for all elements of the charge-bond order matrix
(pij(n) - p . 1.J . ( n - ~ ) l 5 10-4 (1) In the case of the self-consistent field (SCF) procedure we were faced with serious convergence difficulties. In the case of the HzS-formamide complex we could overcome these difficulties by taking the diagonal blocks of the starting charge-bond order matrix of the complex from the final SCF results of the constituent single molecules and taking zeros in the off-diagonal blocks. In the case of the glyoxal-formamide system, however, this procedure was not suc- cessful at medium distances. Therefore, in the first eight iteration steps we turned on stepwise the intermolecular interaction in the complex by multiplying the off-diagonal blocks of the Fock matrix by factors 0.3,0.4, - - , 1 .O, respectively. In this way in about thirty iteration steps we were able to obtain convergent results for both systems, fulfilling the above given condition.
For the single molecules standard geometries [8] have been applied which are shown in Figure 1. In the case of the formamide-glyoxal system we have used
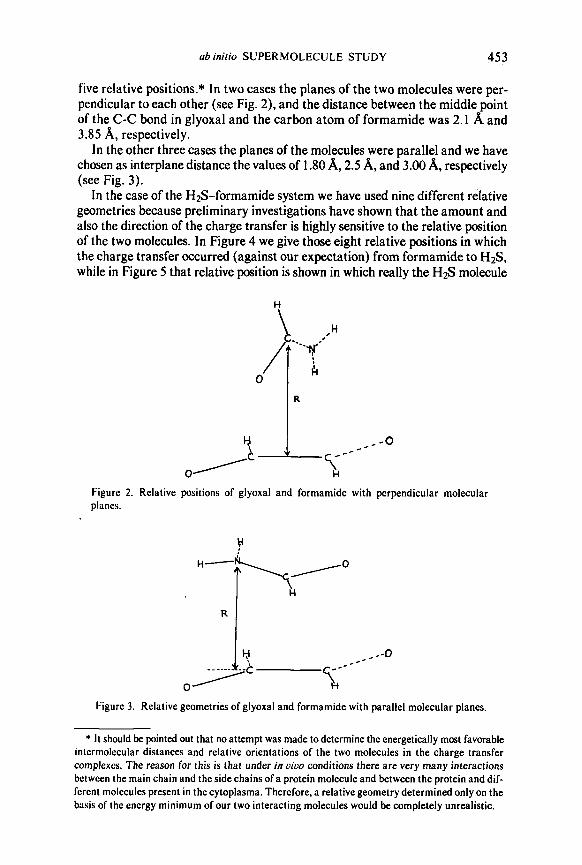
ab inirio SUPERMOLECULE STUDY 453
five relative positions.* In two cases the planes of the two molecules were per- pendicular to each other (see Fig. 2), and the distance between the middle point of the C-C bond in glyoxal and the carbon atom of formamide was 2.1 A and 3.85 A, respectively.
In the other three cases the planes of the molecules were parallel and we have chosen as interplane distance the values of 1 .SO A, 2.5 A, and 3.00 A, respectively (see Fig. 3).
In the case of the HzS-formamide system we have used nine different relative geometries because preliminary investigations have shown that the amount and also the direction of the charge transfer is highly sensitive to the relative position of the two molecules. In Figure 4 we give those eight relative positions in which the charge transfer occurred (against our expectation) from formamide to H2S, while in Figure 5 that relative position is shown in which really the H2S molecule
H
-0 * - -
0 / \--- H 1
Figure 2. Relative positions of glyoxal and formamide with perpendicular molecular planes.
Figure 3. Relative geometries of glyoxal and formamide with parallel molecular planes.
* I 1 should be pointed out that no attempt was made to determine the energetically most favorable intermolecular distances and relative orientations of the two molecules in the charge transfer complexes. The reason for this is that under in uiuo conditions there are very many interactions between the main chain and the side chains of a protein molecule and between the protein and dif- ferent molecules present in the cytoplasma. Therefore, a relative geometry determined only on the basis of the energy minimum of our two interacting molecules would be completely unrealistic.

454 OTTO. SUHAI, A N D LADIK
is the donor. In each case the number on the arrow between the two molecules indicates the amount of charge (in electronic charges) transferred between the two partner molecules in the direction of the arrow. In Figure 5 we have written two numbers on the arrow, one has been calculated with the aid of the same basis set as in the eight previous cases (see above) while the number in parentheses is the result of the calculation with the still larger basis set of Huzinaga [6] and Veillard [7]. Further we give in the same figure the changes of the charges (again in electronic charges) on the different atoms due to charge transfer. In all cases the numbers in parentheses refer to the larger basis set.
To determine the amount of charge transferred from one molecule to the other we have used Mulliken's population analysis [9]. The amount of charge trans- ferred has been computed with the aid of the expression
6
i= I C [(Qi'.".)supermol - (Qi'.".)single ,011 (2)
where (Q;f.a.)supermol and (Q;f.a.)single mol stand for the total charge (in electronic charges) for the ith atom of formamide in the supermolecule and in the single molecule, respectively.
In Table I we show the amount of transferred charge in the formamide glyoxal system by different relative geometries using different basis sets. The detailed charge distributions in the single molecules as well as in the supermolecules for the different cases can be requested from the authors.
Discussion
As we can see from Table I, the amount of the charge transferred from formamide to glyoxal is strongly dependent on the intermolecular distance and on the relative orientation of the planes of the molecules. The second part of this statement can be understood if we compare the value of -0.094 of the perpen- dicular case at R = 2.10 A (which corresponds to a Cglyoxal-Oformamidc distance of 2.00 A) calculated with the minimal basis with the value of -0.14 in the parallel case at R = 2.10 A which can be estimated by the interpolation of the calculated values at other distances. On the other hand the results show that the size of the basis set has a not very significant effect.
In the case of the H$3-formamide system we can find again (see Fig. 4) that the relative orientation and the distance of the two molecules also affect very
TABLE I . Amount of charge transferred from formamide to glyoxal (in electronic charges) by different relative geometries using different basis sets.
Min. Basis Double {Basis
Pcrpendicular R = 2.lOA -0.094 -0.097 molecular R = 3.85 A 0.000 0.000 planes
Para I lel R = 1.808, -0.2 I9 -0.233 molecular R = 2.50 8, -0.030 planes R = 3.00 A -0.007 -0.023

ab inirio SUPERMOLECULE STUDY 455
Figure 4. Those investigated relative geometrical positions of the HzS-formamide system in which the H2S molecules act as acceptors.
strongly the amount of the charge transferred (against our expectation) from formamide to H2S (values between 0.30 and 0.01). Only in the case of one rel- ative geometry (Fig. 5 ) did we find that the H2S molecule is the donor and the formamide is the acceptor. As we can see from the figure, in this case the amount of the transferred charge is more sensitive to the size of the basis set than in the case of the glyoxal-formamide system. (The change in the numerical values from

456 OTTO, SUHAI, AND LADlK
Figure 4. (continued from previous page.)
0.045 to 0.059 is not very large but one has to take into account that the two basis sets used were a double zeta type one and a still larger one.)
To summarize our results we can draw the conclusion that also according to this ab initio SCF LCAO supermolecule calculation glyoxal can certainly act as an electron acceptor against a peptide group, but it is uncertain that a single sulfur-containing compound will donate electrons to the main protein chain. To learn more about this possibility, one has to perform calculations in which both model molecules are essentially larger. In this respect we could follow the
Figure 5. That investigated relative position of the H2S-formarnide system in which H2S is the donor.

ab initio SUPERMOLECULE STUDY 457
suggestion of Szent-Gyorgyi [ 101 that as possible acceptor compound, N- methylacetamide
0 II
(CH3- N-C-CH,) H
and as possible donor, 1 -hydroxy- 1 -methyl-sulfid-acetone
(HF-S-CH-C-CH,,) I 11 OH 0
should be used. We plan to perform these investigations in the future applying again different relative geometries.
Acknowledgment
The authors would like to express their gratitude to Prof. A. Szent-Gyorgyi for suggesting to them the problem and for many very interesting discussions. Special thanks are due to Dr. K. Laki for many illuminating considerations. They are much indebted to Dr. G. Diercksen for helpful discussions and for putting to their disposal his MUNICH(D3 ab initio program. They would like to express their gratitude to the Deutsche Forschungsgemeinschaft and to the National Foundation for Cancer Research for financial support and to the Computing Center of the Max Planck Institute for Plasmaphysics, Garching, for giving them time on their IBM 360/91 computer.
Bibliography
[I] A. Szent-Gyorgyi, Int. J. Quantum Chem. QBS 3,45 (1976). [2] L. B. Coleman, M. J. Cohen, D. J. Sandman, F. G. Yamagishi, A. F. Garito, and A. J. Heeger,
[3] A. Szent-Gyorgyi, K. Laki, and J. Ladik. unpublished. [4] B. Mely and A. Pullman, Theor. Chim. Acta 13,278 (1969). [ 5 ] B. Roos and P. Siegbahn, Theor. Chim. Acta 17,209 (1970). [6] S. Huzinaga, J. Chem. Phys. 42, 1293 (1964). [7] A. Veillard, Theor. Chim. Acta 12,405 (1968). [8] Tables of Interatomic Distances and Configurations in Molecules and Ions, L. E. Sutton, Ed.,
Spec. Publ. I I . , (Chem. Society, London, 1958). (91 R. S. Mulliken, J. Chem. Phys. 23, 1833 (1955).
Received February 5, 1977
Solid State Comm. 12, I125 (1973).