a regulator’s perspective on challenges in the development ...c.ymcdn.com/sites/ · pdf...
TRANSCRIPT

A Regulator’s Perspective on Challenges in the Development of Antibody-Drug
Conjugates and Bispecific Antibodies
Marjorie Shapiro, Ph.D.Office of Biotechnology Products/CDER/FDA
CASSS Midwest Discussion GroupJune 8, 2017

2
Disclaimer
The views presented today do not represent official FDA policy, but rather represent my opinion based on my experience as a reviewer of monoclonal antibody and related products at the FDA.
2

3
Outline• Monoclonal Antibody History• Antibody Drug Conjugates (ADC)
– Trends– DSI– mAb and drug/linker perspectives
• Bispecific Antibodies (BsAb)– Platforms– Development considerations and challenges– FIH considerations
• BLA Submission• High Potency Compounds• Additional Slides
– ADCs– BsAbs

4
History of Monoclonal Antibodies
2000 200219941986 1997 19981992
Orthoclone1st mAb
Murine
Oncoscint
Imaging agent
ChimericFragment
ReoPro
RituxanZenapax
Intact chimeric Humanized
Fc-fusion
Enbrel
ADC - CalicheamicinNDA
Mylotarg
Human/phage display
ZevalinHumira
Therapeuticradioimmunoconjugate
Continued…
5
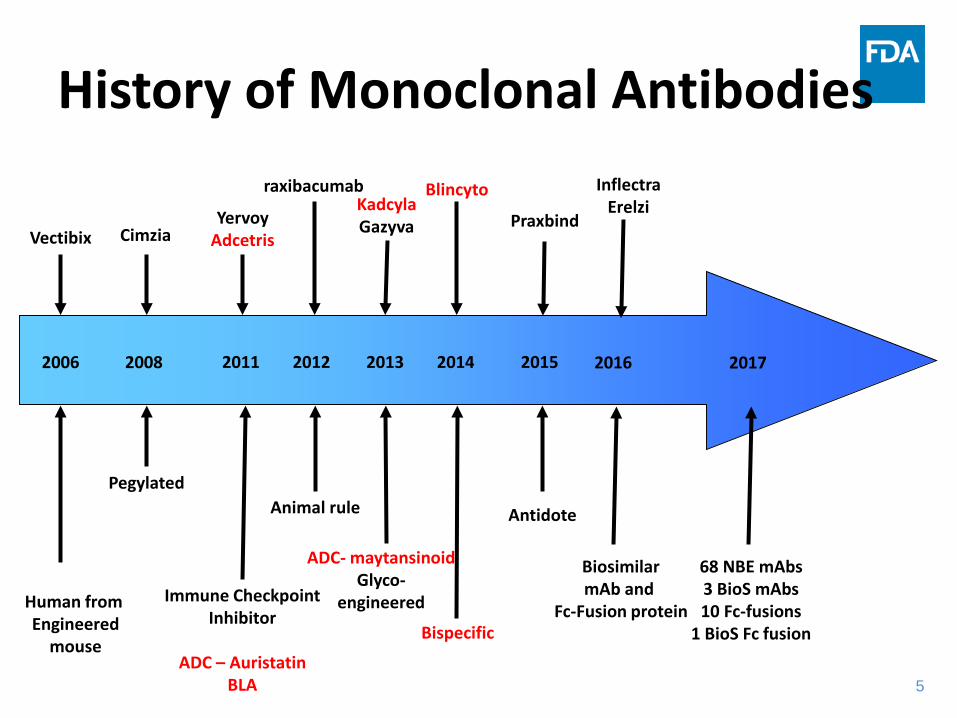
History of Monoclonal Antibodies
Human from Engineered
mouse
2006 20132008 20122011 2014 2015
Vectibix Cimzia
Pegylated
YervoyAdcetris
Immune CheckpointInhibitor
ADC – Auristatin BLA
raxibacumab
Animal rule
KadcylaGazyva
Blincyto
ADC- maytansinoidGlyco-
engineered
Bispecific
68 NBE mAbs3 BioS mAbs10 Fc-fusions
1 BioS Fc fusion
Praxbind
Antidote
2016
BiosimilarmAb and
Fc-Fusion protein
InflectraErelzi
2017
6
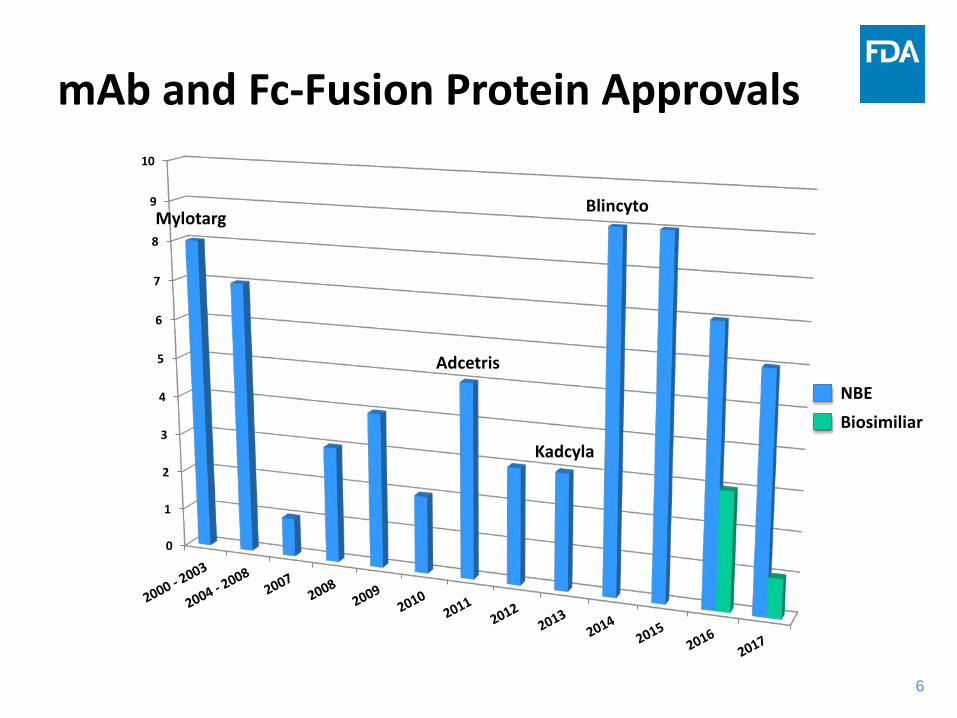
mAb and Fc-Fusion Protein Approvals
0
1
2
3
4
5
6
7
8
9
10
Mylotarg
Adcetris
Kadcyla
Blincyto
NBEBiosimiliar

7
Antibody Conjugates
• Radioisotopes• Drugs• Cytokines• Toxins• Peptides • Enzymes• PEG• Biotin/Streptavidin• Other imaging agents
• Intact mAbs• Fab• F(ab’)2• sFv• VH domain mAbs• Bispecific abs
8
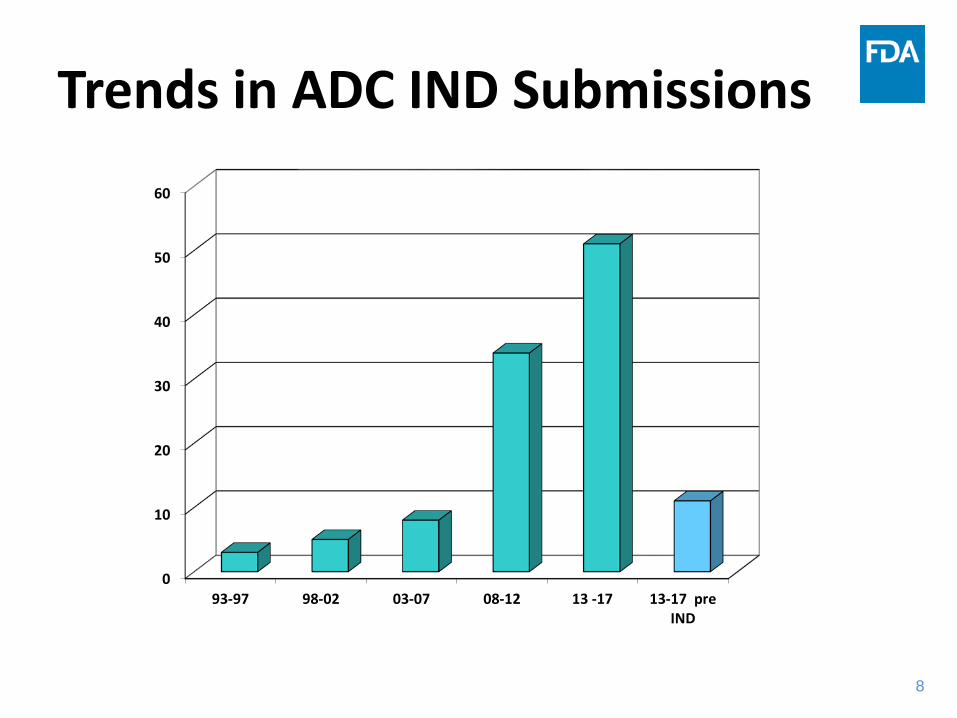
Trends in ADC IND Submissions
0
10
20
30
40
50
60
93-97 98-02 03-07 08-12 13 -17 13-17 preIND

9
Antibody-Drug Conjugate Payloads
Wu and Senter Nature Biotechnology 2005
Sutherland et al. Blood 2012
pyrrolobenzodiazepine dimer
Dokter et al. Molecular Cancer Ther 2013
duocarmycin
Moon et al. J Med Chem 2008
tubulysin
Tumey et al. ACS Med Chem Let 2016

10
Trends in ADC Payloads
0
10
20
30
40
50
60
70
93-97 98-02 03-07 08-12 13 -17
Other
Traditional Chemotherapies
Microtubule Targeting
DNA Targeting

11
Other ADC Trends• Most ADCs use cleavable dipeptide linkers, followed by non-
cleavable linkers, acid labile/disulfide based linkers and some novel linkers.– Linkers with improved stability in systemic circulation include peptide
and non-cleavable thioether linkers
• Currently most ADC conjugation sites are cysteine residues, followed by lysine residues.– Starting to see site specific conjugation through engineered cysteines,
non-natural amino acids, aldehyde tagging and enzymatic conjugation
• There are more CMOs available today who have the capability to manufacture ADCs than existed 5 years ago – More sponsors are also manufacturing their own ADCs (and acting as
CMOs)
12
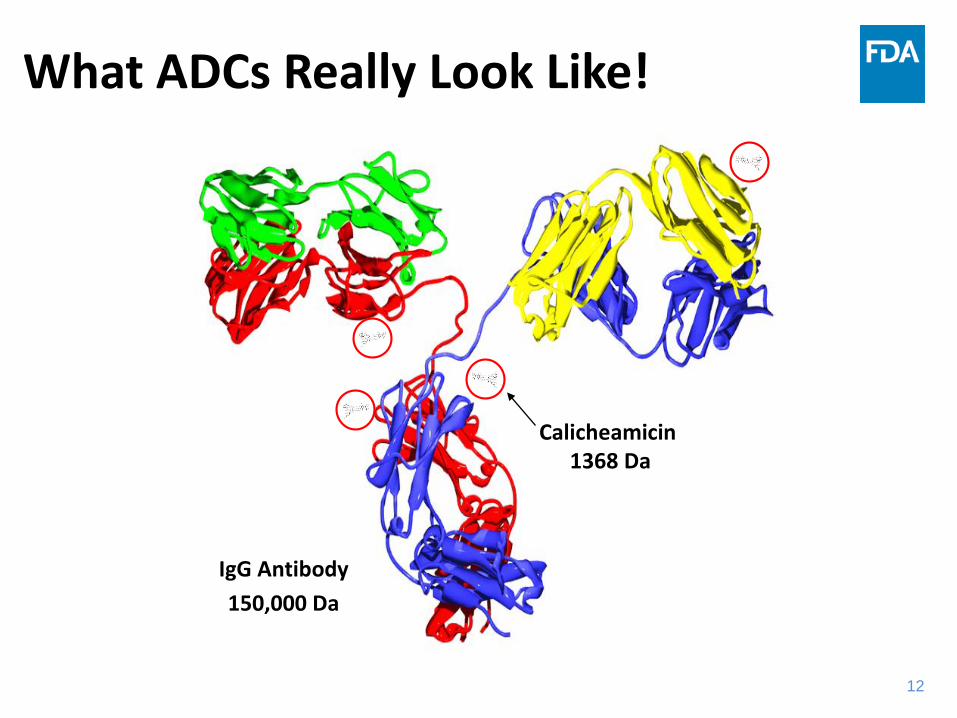
What ADCs Really Look Like!
150,000 DaIgG Antibody
Calicheamicin1368 Da

13
Drug Substance Intermediates• The mAb and drug/linker are drug substance
intermediates– The mAb is not a raw material!
• The expectations for characterization, comparability, release and stability of the mAb intermediate are the same as for the ADC drug substance.
• The expectations for characterization, release and stability of the drug/linker intermediate are the same as for the ADC drug substance.– Discussion of designation for drug/linker starting materials at
the EOP-2 meeting

1414
Antibody- Drug Conjugate: Characterization and Comparability
• Generally more focus on mAb rather than drug-linker
• Primary, secondary and higher order structure• Size and charge variants• Glycosylation• Other PTMs as appropriate• Antigen binding• Other biological activity
•Assessment of impact of conjugation chemistry on: – important biological functions of Ab (binding, effector function,
other)– size and charge (?) variants

1515
Antibody- Drug Conjugate: Identity
• Binding, charge based, peptide map, sequence based.
• The need for multiple assays depends on the properties/characteristics of the mAb/conjugate and if other conjugates are manufactured at the same facility.

1616
Antibody - Drug Conjugate: Purity• Largely the same methods as before conjugation.
• Size and charge variants.
• Conjugation to lysine residues impacts charge based assays.
• Need to control for aggregates and fragments.
• Need to control for free antibody• Need to understand what is stability indicating.• If mAb well characterized and process well controlled, may be
able to eliminate some release tests but this should be a pre-BLA discussion– Pre- and post marketing process changes may require accumulative
data.

1717
Antibody - Drug Conjugate: Potency• MOA = cytotoxicity, but binding to antigen is a necessary
component• Need antigen binding for unconjugated mAb• Need to demonstrate conjugation does not affect antigen
binding• If conjugation process is well controlled, may be able to
eliminate antigen binding assay at time of approval provided cytotoxicity assay is robust. – Need sufficient data, including stability data of ADC, linkage of potency
across lots and discussion with Agency. – Binding assay may be a better stability method.– This will be a BLA review issue, but can discuss at pre-BLA meeting
• Effector function or signal transduction as MOA should be assessed and may need to be included in control strategy.

1818
Characterization of Antibody-Drug Conjugate
• Structural characterization – Molar absorption coefficient– Drug load distribution– Drug/Antibody ratio
• Impurity profile– Free drugs (drug related substances and quenching agents)– Residual solvents and other process related impurities
• Risk based approach to control impurities– Conjugatable vs. non-conjugatable impurities

1919
Drug/Linker Testing and Specifications of Antibody-Drug Conjugate
• Identity – UV absorption
• Assay – Total drug content (UV)
• Purity – Free drug related substances (including quenching agent)– Residual solvents– Heavy metals
• Stability testing – Free drug

2020
Drug/Linker Testing and Specificationsof Antibody-Drug Conjugate
• Phase 1 INDs: – Free drug related impurities in clinical lot should be qualified
relative to data from toxicology studies. – Comparable drug/antibody ratios should be maintained
between the toxicology lot and the clinical lot.
• Phase 3/pivotal clinical studies: Characterization of the impurity profile of drug/linker intermediates, including structure determination of individual impurities at levels >0.1%, is recommended prior to pivotal clinical trials.

2121
Antibody - Drug Conjugate: Purity and Potency
• Drug:Antibody ratio
• Drug loading distribution – homogeneity of the ADC population
• Free Drug
• Free Antibody– biological activity

2222
Antibody - Drug Conjugate: Comparability
• Extent of study depends on life cycle stage.• If changes were made to mAb, cytotoxic drug, or linker
intermediates, should perform comparability on ADC drug substance.– Comparability study should be performed on mAb intermediate as
well as DS.
• Appropriate methods should be used to assess comparability between the toxicology, clinical, and/or commercial batches.
• Small drug perspective – main concern is with drug loading distribution.– DAR may be the same but if DLD is different, this could be a concern
• mAb perspective, if certain tests are dropped for mAb, ADC DS or DP for release, will data be collected for future comparability studies?

2323
Antibody - Drug Conjugate: Control Strategy• Need a scientifically sound risk assessment when establishing
control strategy for DSIs– What attributes may be common to small molecule and mAb
intermediates• Visible particles• Sub-visible particles• Bioburden• Endotoxin
• If the control strategy for intermediates is too broad, DS may fail.
• If control strategy for DS is too narrow, may not have acceptable intermediate lots to manufacture DS
• Need to understand CQAs of DSIs, DS and DP.
• Need to link DP EOSL criteria back through DP release, DS EOSL and release and DSI EOSL and release criteria

2424
Antibody - Drug Conjugate: Setting Specifications
• 1 lot of drug/linker + 1 lot of mAb = ≥1 lot of ADC drug substance
• Use as many combinations of distinct drug/ linker/mAb lots as possible during clinical development
• Plan different combinations for conformance lots
• A good topic for a pre-BLA discussion

25
Bispecific Antibodies (BsAbs)

26
Examples of Biologic Multi-Target Therapies
• Combinations– Separate
manufacture of DS and DP
– May be co-developed or developed individually
– May be administered separately
• Same day• Different days
• Cocktails– Individual mAbs
pooled during DS DP manufacture
– May not be feasible to test separately (anti-viral combinations)
– A fixed dose of combination is administered
Bispecifics– Single molecular
entity with two specificities
– Cannot separate arms to test each specificity separately
– Fixed dose is administered
26

27
Rationale for the Development of Bispecifics
• Despite great advances with therapeutic monoclonal antibodies across indications, there remains unmet medical needs
• There can be a strong scientific rationale to engage more than one target
• Single dosage form is more convenient than co-administration
• Advances in biotechnology have improved the ease of manufacturing bispecific molecules
27

28
Trends in BsAb IND Submissions
0
2
4
6
8
10
12
14
INDpreIND
There were some BsAb INDs in the 1990s when I first joined the agency

29
Bispecific mAbs (BsAbs)• First therapeutic BsAbs
were made through fusions of two hybridomas –termed quadromas
• Endogenous IgG4 can form bispecifics in vivo
Lindhofer et al. Journal of Immunology 1995Van der Neut Kolfschoten et al. Science 2007

30
BsAbs Today – Five Basic Formats
• Intact Ig Structure• Appended Ig Structure• Bispecific Fragments• Bispecific Fusion Proteins• Bispecific Conjugates
• Altogether there are currently >60 BsAb formats– Most formats are still in preclinical development– Several are in clinical development

31
BsAb: Ig Structure – monovalent for each ag
• Quadroma – catumaxomab approved in EU for malignant ascites in EpCam+ tumors
New platforms solved problems of incorrect H and L chain pairing• Knobs-into-hole (KIH) – unfavorable pairing for CH homodimers, favors heterodimers• Other structure based rational design platforms (CH3 region) favoring heterodimer formation
– symmetric to asymmetric steric complementarity, Charge to charge swap, charge to steric complementarity, electrostatic interaction
• Cross mAb – KIH with CL/ CH1 domain swap to ensure correct H/L chain pairing• Dual Affinity (DAF) – in vitro mutagenesis and selection to bind two different
antigens/epitopesSpeiss et al. Molecular Immunology 2015Spiess et al. Molecular Immunology 2015
32
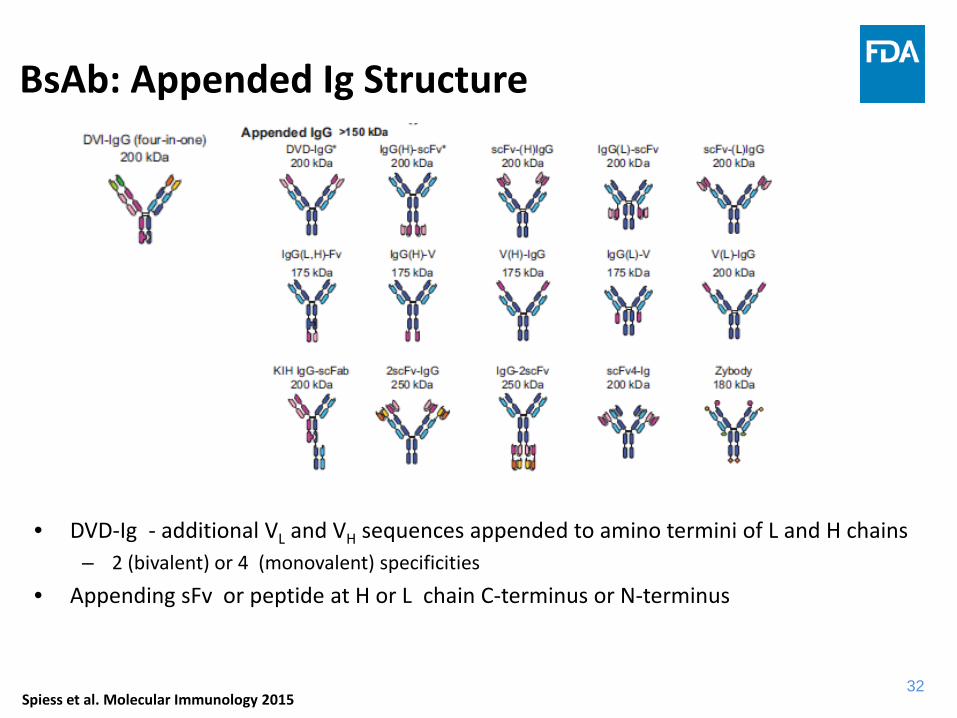
BsAb: Appended Ig Structure
• DVD-Ig - additional VL and VH sequences appended to amino termini of L and H chains– 2 (bivalent) or 4 (monovalent) specificities
• Appending sFv or peptide at H or L chain C-terminus or N-terminus
Speiss et al. Molecular Immunology 2015Spiess et al. Molecular Immunology 2015
33
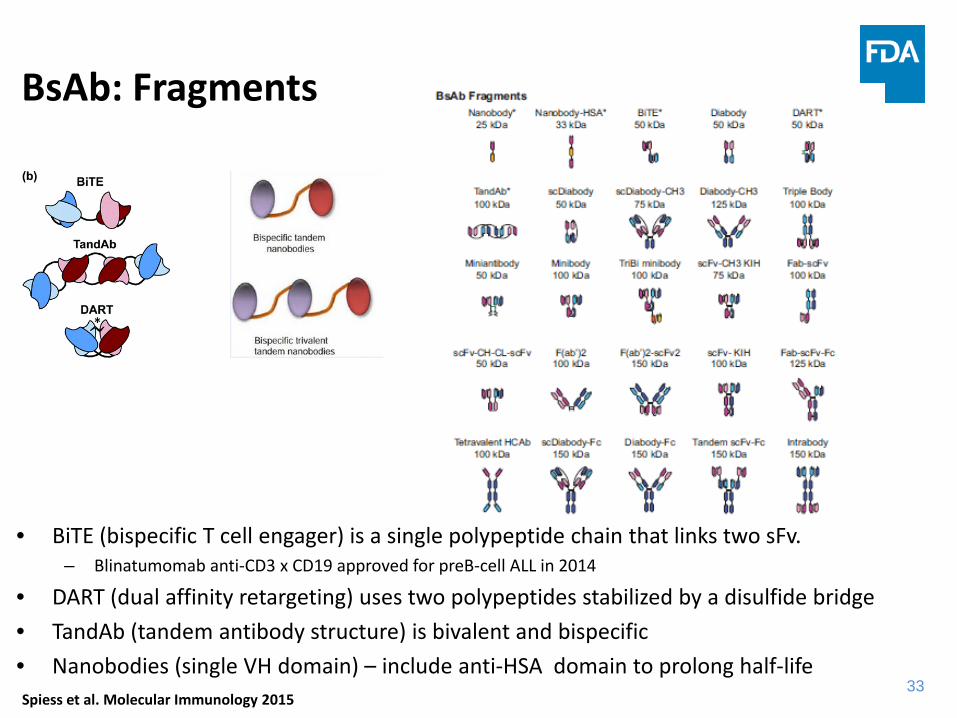
BsAb: Fragments
• BiTE (bispecific T cell engager) is a single polypeptide chain that links two sFv.– Blinatumomab anti-CD3 x CD19 approved for preB-cell ALL in 2014
• DART (dual affinity retargeting) uses two polypeptides stabilized by a disulfide bridge• TandAb (tandem antibody structure) is bivalent and bispecific• Nanobodies (single VH domain) – include anti-HSA domain to prolong half-life
Speiss et al. Molecular Immunology 2015Spiess et al. Molecular Immunology 2015

34
Some Development Challenges for BsAbs - May be Platform Specific
• Manufacturability• Antigen specificity, affinity,
density– compatible on/off rates?
• Potency assay– Fixed ratio of specificities– Simultaneous binding to both
antigens– Do both antigens need to be
bound by the same molecule?• If designed to engage effector
cells– anti-CD3 homodimers?
• Stability– Low solubility and tendency to
aggregate– design of construct may be
empirical (order of VH and VL)• Half-life (if no Fc)
– Extend by fusion to Fc, albumin or PEG conjugation, V region engineering (lower pI)
• Small constructs– Balance short half life with more
rapid distribution and tissue penetration
• Continuous dosing– Sterility assurance
34

35
Trade offs for different BsAb platforms
• Fragment-based– Short half life– Continuous infusion
(sterility assurance)
– More rapid tissue distribution and better penetration
– Less complex structure (no glycan)
– Easier to manufacture and characterize
• Intact structure– Longer half life– Effector function
– Less tumor penetration– More complex structure– More characterization
needed– Control strategy more
complex
35

36
Key measurements – May Depend on Presence or Absence of Fc
• Short half life for small constructs– More rapid distribution and tissue penetration
• Binding to effector and target cells– Does binding to T cell or effector cell cause release of cytokines or
activation in absence of binding to target cell• Binding to soluble antigens
– Anti-cytokine mAbs can increase half life of cytokine– Need appropriate affinity and off-rate to stay bound until cleared– Smaller formats with shorter half life would be cleared more quickly
• Target mediated disposition– Is soluble antigen present?
• Immunogenicity – Novel structures result in potentially novel epitopes– Pre-existing abs may exist to novel epitopes and interfere with the
identification of treatment emergent ADA
36

37
• There is a spectrum of considerations that may impact the optimal study design and clinical program – Proposed indication – Proposed targets– Relationship between the targets – Scientific rationale and available data to support rationale
Clinical Program Considerations for BsAbs
80%
10%
10%
OncologyRheumatology/PulmonaryOther (4 divisions)
Comments, advice, concerns you may get about a particular BsAb will depend on the indication

38
Compatibility and administration issues with dilute, potent therapeutics (FIH studies)
• Minimal anticipated biological effect level (MABEL) approach used for FIH dosing– mAbs are generally well tolerated and toxicity is related to exaggerated
pharmacology– Sometimes SAEs are seen in FIH studies (TGN1412)– MABEL especially important when there is no relevant animal model
• Typically T cell-engaging BsAbs utilize low doses and FIH dose cohorts start very low– Blinatumomab approved dosing
• ≥45 kg: 9 µg/day days 1-7, 28 µg/day days 8-28 first cycle• <45 kg: 5 µg/m2/day (not to exceed 9 µg/day) days 1-7, 15 µg/m2/day (not to
exceed 28 µg/day) days 8-28 first cycle
• Bioanalytical method may not be sensitive enough to confirm predicted exposure

39
Compatibility and administration issues with dilute, potent therapeutics
• For BsAbs with a potential for cytokine release, the administration instructions need to be precise and clear– Consider that protein may bind to plastic and filters in infusion sets
– “Prime” versus “prime to waste” instructions result in incorrect levels of administered BsAb, which could lead the investigator to think that the first dose was well tolerated. In reality, patients may receive no BsAb or a lower dose than intended.
– Methods to assess protein content in compatibility studies need to be sufficiently sensitive to detect low amounts of product to confirm exposure – especially if PK method can’t
– At some point, patients in a dosing cohort may experience CR as a SAE, but the actual dose will be unknown

40
Additional Considerations to Initiate a BsAb IND• Mostly the same as for a conventional mAb
– Does the CMC package support that the product is safe to administer to patients?
• Need a good understanding of the structure, proposed MOA and manufacturing process– Structure/function relationship– Relevant/appropriate potency assay– Purity/impurities
• Monospecific abs and half antibodies– Stability
• Fragments • Aggregates• Other product specific issues
– Appropriate phase specific control strategy

41
Additional Considerations to Initiate a BsAb IND
– Identity testing• Can methods distinguish heterodimers from homodimers?
– If Fc-containing construct is engineered for reduced/no binding to FcγR, provide data to support this
– Short half-life products with prolonged administration
• Toxicity and effectiveness of anti-microbial agents• Impact on stability of BsAb.• Control of anti-microbial agent for lot release of vehicle
solution

42
What Makes a Good ADC or BsAb BLA Data Package?• Same as for a conventional mAb
– Well written and organized, proofread– Justified established conditions based on development and
validation studies– Understanding of structure/function relationships that allows
appropriate ranking of quality attributes– Thorough stability studies, forced degradation studies to further
understand potential CQAs and to inform control strategy • Do you need a release specification for a certain PTM?
– Appropriate control strategy– Good analytical methods with appropriate validation– Appropriate primary reference standard representative of
materials used in pivotal study and a working reference standard
– A minimum of 6 months DS and DP real time stability data from proposed commercial process
This is not intended to be a complete list of what is needed for a good BLA package

43
What Makes a Good ADC BLA Data Package?
• Separate 3.2.S folders for DSIs and DS
• DMF acceptable for drug/linker, especially if the drug/linker has already been used in an approved product and is sourced from the same place.
• Need to understand relationship of DP CQAs to DS and DSI CQAs and how each step of the manufacturing processes for each impact the final DP CQA.
• DP release and EOSL criteria need to relate back to DS and DSI release criteria.

44
CMC Development Considerations for Breakthrough Therapy Products
• Can process development keep pace with clinical development?
• Will the commercial process be ready for commercialization?– consistently and reliably deliver the clinical performance and other
characteristics stated on the label
• Will it be sufficient to meet market demand?
• Sponsors should request CMC-specific meetings as soon as possible upon receiving BT designation.
• Be prepared to discuss what data will be available at the time of BLA submission• Rolling submission where Module 3 is not the last module submitted
is preferable

4545
Highly Potent Substances in Multi-Product Facilities
• Understand your responsibility as sponsor; understand the responsibility of the CMO.
• Contract Manufacturers– Limited number of facilities capable of handling highly potent
biological drug substances – The number of CMOs that can manufacture ADCs is growing– If not already licensed as multi-product facility, expect it will become a
multi-product facility during life of your product
• For multi-product facilities handling highly potent substances, a risk assessment is expected to identify cross-contamination risks. Segregations and controls mitigating the identified risks should be implemented. – Responsibility of CMO!
• Will be reviewed on inspection• Recommend CMO submit a DMF that includes this risk assessment as well as
change-over procedures– Good communication with customer (You!)

4646
Highly Potent Substances in Multi-Product Facilities – ADCs and some T cell engaging BsAbs
• ISPE Baseline Guide Volume 7: Risk-Based Manufacture of Pharmaceutical Products: A Guide to Managing Risks Associated with Cross-Contamination 2010
• Clinical development versus post-marketing– CMO must notify sponsor when a new product is brought into the
manufacturing suite– BLA sponsor must submit a BLA supplement when a new product is
introduced into the manufacturing suite (PAS for first introduction, CBE-30 for subsequent introductions).
– Supplement should include data demonstrating that the identity test for the approved product can distinguish it from the newly introduced product
– If CMO has a DMF, an amendment should be submitted with specific details related to introduction of the new product

47
Acknowledgements• Joanna Zhou - OBP• Wen Jin Wu – OBP• Kathleen Clouse - OBP• Xiao-Hong Chen – ONDP
Extra slides – BsAb clinical experience/considerations• Al Deisseroth – DHP/OND• Raj Nair – DPARP/OND

48
Additional Slides - ADCs

4949
Characterization and Comparability of mAb Intermediate
The expectations are the same for the mAb intermediate as they are for a final drug substance.
• Primary Structure• Secondary/Tertiary
Structure• Fragments/aggregates• Charge
• Glycosylation
• Other post translational modifications
• Antigen binding
• Biological activity as appropriate

5050
mAb Effector Function and Biological Activity
• Most ADCs utilize IgG1 or IgG4 mAbs
• If mAb is engineered to reduce effector function or reduce IgG4 half antibody formation, characterization should demonstrate this.
• The mAb should be characterized for effector function and other biological activity. • The target, tumor location and rate of internalization may play a role.
• If the mAb has effector function or other biological activity, this activity may contribute to the overall MOA of the ADC

5151
mAb Impurities • Product related impurities
– Charge and size variants– Identify, may need to characterize biological function– Understand impact on ADC– Understand impact of conjugation reaction on variant
• mAb process related impurities– Clearance of Virus and DNA– Calculate based on maximum human dose of DP– Removal of impurities should be assessed during mAb
manufacture unless subsequent steps are needed to provide further reduction.
– Risk assessments may be acceptable initially for some process related impurities.

5252
Current mAb Conjugation Sites
• Cysteine (polar)– 4 interchain disulfide bonds in IgG1/IgG4– 8 conjugation sites
• Lysines (basic) – 25/22 lysines in IgG1/IgG4 constant region– 8 lysines in κ constant region– Additional lysines in VH or VL regions
• Number depends on germline sequence and somatic mutations

5353
Testing and Specifications
• Phase 1 INDs: specification for cytotoxicity assay should not be broader than dose escalation scheme– Or ensure that the single clinical lot to be used in the
dose ranging study is sufficient to complete the study.– Qualify cytotoxicity assay with multiple runs of
reference standard to achieve better results to support more realistic range
– However, DAR also is a reflection of potency
• If an ADC is believed to have multiple MOAs, include additional potency assay(s).– mAb intermediate release and/or DS or DP release.

5454
Starting Materials for Drug/Linker
• Fermentation and semi-synthetic compounds– Microbial strains
• Peptides– Amino acids and their derivatives
• Chemically synthesized compounds– Appropriately characterized and stable molecules– Impurity profile established (carry-over vs. non carry-over)– Multiple chemical and purification steps preferred– Controlled process to remove/reduce impurities
• Discussion of designation for SMs at the EOP-2 meeting

5555
Characterization of Drug/Linker
• The expectations are the same for the drug/linker intermediates as they are for a final drug substance.
• Structural characterization – UV, IR, NMR, MS, elemental analysis, etc.
• Impurity profile– Drug/linker related impurities – Process impurities– Structural determination for the impurities present at
levels higher than 0.1%

5656
Drug/Linker Testing and Specifications
• Appearance• Identity
– IR, UV, NMR, MS, etc.– Optical rotation if applicable– Melting point if applicable
• Assay (HPLC)

5757
Drug/Linker Testing and Specifications(as intermediates)
• Purity– HPLC (drug-related impurities/degradants)– Residual solvents– Heavy metals and/or residue on ignition– Water content– Chiral HPLC if applicable
• Stability testing– Physical and chemical stability– Photo-stability– Freeze thaw studies

5858
Future Trends• Site specific conjugation
– non-natural amino acids – aldehyde tagging– engineered cysteine residues– enzymatic conjugation to engineered tags
• New drug platforms – kinase inhibitors– different drugs per mAb
• New/improved linker technologies• Optimizing payloads • Extracellular release at tumor environment• Extracellular targets• Non-oncology indications

5959
Conclusions • Antibody-drug conjugates are both drug and biologic
molecules!• Regardless of the regulatory pathway, characterization,
comparability, release and stability assays need to be appropriate for the molecule.
• Current ADCs are highly potent substances. Appropriate measures are needed regarding potential cross-contamination in multi-product facilities.
• Good communication between Sponsor and Quality review team (OBP/ONDP/OPF/OPRO) helps overall review process.

60
Additional Slides - BsAbs

61
FDA experience with some BsAbs in oncology indications• Most are designed to engage effector cell (T cell or FcγR
expressing cells) with tumor target• Factors associated with toxicity
– Total body tumor burden is a risk factor for a poor response
– Specific patterns of toxicity are associated with specific targets
– Prior therapy or other features which reduce the numbers of resident effector cells in patients may reduce response
– Higher tumor burden may increase the severity of CRS if T cells are not limiting
61Al Deisseroth - OHOP

62
FDA experience with some BsAbs in oncology indications• Factors limiting efficacy
– Host specific factors: qualitative and quantitative defects in the T cells and accessory cells in older individuals or patients with certain tumor types,
– Disease specific factors: heterogeneity of specific tumor types with respect to expression of tumor specific antigen or resistance phenotypes to T cells,
– Resistance factors specific to the T cell (check points etc), – Problems intrinsic to the bispecifics
• ADA seen more with some tumor targets• Poor penetration of solid tumors
62Al Deisseroth - OHOP

63
Cytokine Release Syndrome: Pre-Clinical Studies
• Monitor in vitro ability to induce cytokine release and proliferation of whole blood samples or PMBCs from multiple donors
• Dose titration of product in both soluble and plate bound form
• Measure IFNγ, TNFα, IL6 and other relevant cytokines• Agonistic mAbs can exhibit a bell shaped dose response
curve– Test levels that levels that lead to low (e.g., 10-20%),
medium and complete receptor saturation • Include immobilized product, both wet and dry coated on
the plate and a positive control antibody (OKT3)63Stebbings et al. (2007), J. Immunology 179: 3325-3331 and Roomer P.S. et al. (2011) Blood 118:6772-6782

64
Cytokine Release SyndromeFor bi-functional antibodies targeting T cells
• Cytokine release and proliferation with human PBMCs – Use product optimized stimulation conditions, 6-12 donors and
relevant positive control• Assess cytokine release and proliferation of PBMCs in the
presence of targeted tumor antigen• Assess/characterize the activity of both CD4+ and CD8+ T
cells, cytokines, activation marker up-regulation, Granzyme B, etc
• Assess off-target T cell activation/ cell killing• Characterize target cell killing and cytotoxicity pathway (e.g.,
caspase activation)• Bi-functional binding/cellular potency assays.
64

65
Potential Safety Concerns When Combining Biologic Products
• Previous clinical trial data in rheumatoid arthritis (RA) demonstrated safety concerns when combining biologic products without added benefit – Etanercept (TNF-inhibitor) and anakinra (IL-1
inhibitor)– Etanercept and abatacept (selective costimulation
modulator) • Molecular structure could potentially lead to increased
immunogenicity (novel epitopes, size)
65
66
Evaluation of Bispecific Targets
• There may not be a way to directly evaluate the contribution of each individual target in a bispecific
• Possible to compare a bispecific to an approved comparator that targets one or more similar pathways
– Recognizing that the monospecific product may have a different risk/benefit than that of the bispecific
66

67
Clinical Considerations and Study Design
• Optimal study design may be impacted by clinical considerations
– Potential considerations:• Does the bispecific need to provide additional efficacy over
approved monoclonal antibodies with similar targets?
• Are there safety issues compared to approved monoclonal antibodies with similar targets?
67
68
Biologic Multi-target Therapies
Bispecifics
Both targets are novel At least one proven target

69
Clinical Studies (1)Novel Targets• Neither target has an FDA-
approved monospecific product for the target and indication
• Theoretical Example– Bispecific for asthma
that has novel targets
Potential Study Design
• Generation of monospecific products for comparative assessment is impractical
69
Study armsBispecific
Comparator
70
Biologic Multi-target Therapies
Bispecifics
Both targets are novel At least one proven target
71
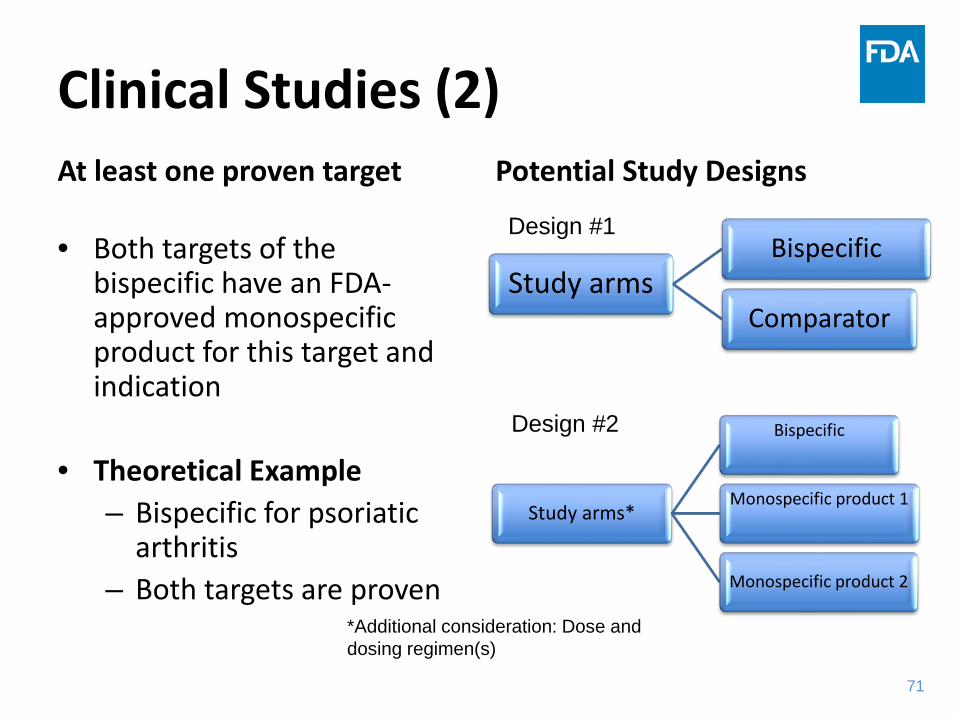
Clinical Studies (2)At least one proven target
• Both targets of the bispecific have an FDA-approved monospecific product for this target and indication
• Theoretical Example– Bispecific for psoriatic
arthritis – Both targets are proven
Potential Study Designs
71
Study armsBispecific
Comparator
Study arms*
Bispecific
Monospecific product 1
Monospecific product 2
Design #1
Design #2
*Additional consideration: Dose and dosing regimen(s)

72
Active Comparator Considerations
• In certain situations, it may be reasonable to compare a bispecific to an approved monospecific product comparator(s)
– Safety concerns example• If co-administration of two biologics is associated with increased risk
of infections without adding benefit
– Efficacy concerns example• If one target is expected to be much more efficacious than the other
72

73
Conclusions
• Bispecific products present novel regulatory considerations – There is a spectrum of issues that may impact the optimal
study design and clinical program
– In certain situations, it may be reasonable to compare a bispecific to a monospecific product(s) with corresponding targets