a recipe for geometry optimization of diradicalar singlet states from broken-symmetry calculations
TRANSCRIPT

A Recipe for Geometry Optimization of Diradicalar Singlet Statesfrom Broken-Symmetry CalculationsJean-Paul Malrieu* and Georges Trinquier
Laboratoire de Chimie et Physique Quantiques (CNRS-UMR5626), IRSAMC, Universite Paul-Sabatier (Toulouse III),31062 Toulouse Cedex, France
ABSTRACT: The equilibrium geometries of the singlet andtriplet states of diradicals may be somewhat different, whichmay have an influence on their magnetic properties. Thesingle-determinantal methods, such as Hartree−Fock orKohn−Sham density functional theory, in general rely onbroken-symmetry solutions to approach the singlet-stateenergy and geometry. An approximate spin decontaminationis rather easy for the energy of this state but is rarelyperformed for its geometry optimization. We suggest simpleprocedures to estimate the optimized geometry and energy ofa spin-decontaminated singlet, the accuracies of which aretested on a few organic diradicals. This technique can begeneralized to interactions between higher-spin units or tomultispin systems.
1. INTRODUCTION
In its simple Kohn−Sham version, the density functional theory(DFT) is extremely convenient and popular. Of course, theexchange correlation potential is not known, a very largenumber of variants are practiced, most of them introducingsome parameters, but a reasonable accuracy is attainable fornonexotic closed-shell molecules in their ground state,especially regarding the equilibrium geometries and vibrationalfrequencies. The use of DFT is also very popular for magneticsystems which are of open-shell character, with a few singlyoccupied orbitals.1 The state of highest spin multiplicity andhighest Ms value may be approached by a single determinant,with 2 Ms electrons in 2 Ms singly occupied orbitals withparallel spins, and the single-determinantal approaches are inprinciple applicable to estimate its energy. One may use eitherrestricted or unrestricted formalisms, the former one keeping aclosed-shell character to the doubly occupied orbitals, thesecond one introducing some spin polarization, which meansthat slightly different orbitals are used for electrons of α and βspins. In this case, the determinant is not a pure spin state, i.e.,an eigenfunction of the S2 operator, but the deviation of itsmean value from its expected value is weak. If geometryoptimization of so-described states of highest spin multiplicitydoes not present any conceptual difficulty, a severe problemconcerns the states of lower spin multiplicities, which cannot berepresented as an open-shell single determinant. One mayoptimize an open-shell single determinant of low Ms value, withMs α and β unpaired electrons tending to localize in differentsites. This solution is a broken-symmetry (BS) determinant,2−5
and the corresponding function is a mixture of spineigenfunctions. From the energies of the upper multiplet andBS determinants, it is possible to approximate the value of thelowest S2 eigenvalue, according to one of the more-or-lessapproximate spin-decontamination techniques.6−8 Regardingthe optimal geometry of the low-spin state, it cannot inprinciple be obtained by minimizing the energy of the BSdeterminant, as noticed in various works where principles ofappropriate geometry optimization of the low-spin state areproposed.9 The present work first suggests a simple analyticalprocedure to estimate the optimal geometry and energy of thelow-spin state from the only knowledge of geometries andenergies of the single-determinant solutions of high and lowMs.Next, a more refined treatment will exploit the optimizedgeometries of the triplet and broken-symmetry determinants todefine a collective coordinate along which a spin-decontami-nated energy curve is calculatedhence a better evaluation ofsinglet-state energy minimum. Both treatments are applied on aseries of organic diradicals presenting either a ferromagnetic oran antiferromagnetic coupling between the unpaired electrons.Last, subsequent generalizations to any high-spin units will beassessed.
Received: April 20, 2012Revised: July 5, 2012Published: July 27, 2012
Article
pubs.acs.org/JPCA
© 2012 American Chemical Society 8226 dx.doi.org/10.1021/jp303825x | J. Phys. Chem. A 2012, 116, 8226−8237

2. SITUATION OF THE PROBLEMA. Energy Difference. Let us specify the problem for the
elementary case of two unpaired electrons in two molecularorbitals a and b, which may be localized in different parts of themolecule. If these orbitals are orthogonal, the triplet-state singledeterminant will be written as
Φ = |Π |+ i i abiT
in a restricted formalism, and
Φ′ = |Π ′ |+ i i abiT
in the unrestricted one. The mean values of the S2 operator isequal to 2 for the first determinant, and close to 2 for thesecond one. The i index runs over all doubly occupied orbitals.In the second solution, the difference between the “core”orbitals i and i′ introduces the effect of the exchange fieldcreated by the two unpaired electrons of α spin, in a and b, onthe α-spin electrons of the core. This is a spin-polarizationphenomenon, responsible, for instance, of the appearance of a σspin density in planar conjugated hydrocarbons where unpairedelectrons occupy π orbitals. Starting from a restricted open-shell Fock operator, the operator responsible for spinpolarization of doubly occupied orbitals is (Ka+Kb)/2, whereKa and Kb are the exchange operators associated to the singlyoccupied orbitals a and b.Let us call ET the energy of Φ′T+
= ⟨Φ′ | |Φ′ ⟩+ +E HT T T
The corresponding proper singlet state should be written as ΦS= |Πiii(ab + ba)|/√2, i.e., from a two-determinant wavefunction. One may of course optimize the energy of a singledeterminant ΦBS = |Πii′i′(a′b′)|,10 or more generally, in anunrestricted formalism, ΦBS = |Πii″i‴(a′b′)|. Let us call EBS itsenergy
= ⟨Φ | |Φ ⟩E HBS BS BS
The essentially singly occupied orbitals a′ and b′ have noreason to remain orthogonal, their delocalization introducessome valence-bond (VB) ionic components which areresponsible for the stabilization of the singlet state, accordingto the kinetic exchange (or superexchange) mechanism.11 Thedifference between the essentially closed-shell orbitals, i″ andi‴, introduces the spin polarization of the core, the electrons ofwhich feel the presence of an α-spin electron near a and of a β-spin one near b, through an exchange field (Ka − Kb)/2.
12 If theoverlap between a′ and b′ remains small and if spin polarizationis weak enough, the BS determinant may be considered as ahalf-and-half mixture of the desired singlet state ΦS and of theMs=0 component of the triplet state
Φ = |Π − |ii ab ba( ) / 2T i0
The corresponding mean value of the S2 operator
⟨ ⟩ = ⟨Φ | |Φ ⟩S S2BS BS
2BS
is then close to 1. Its energy should be halfway between theenergy of the triplet state and that of the singlet state, so thatthe singlet-state energy may be written as
= + − ⇒ − = −E E E E E E E E2( ) 2( )s T BS T s T BS T
A more sophisticated estimate of the singlet−triplet energydifference has been proposed6,7 by Yamaguchi as
− =−
⟨ ⟩ − ⟨ ⟩E E
E ES S2( )
s TBS T
2T
2BS
This formula has the advantage of being applicable whateverthe diradicalar character of the singlet state, but it may present adefect in the present context. If the Ms=0 solution tends tobecome a closed shell, i.e., a pure singlet, the singlet-to-tripletenergy difference is no longer equal to EBS − ET as it should.Due to the spin polarization of the core orbitals, ⟨S2⟩T is herenot equal to 2, and its deviation also depends on the spinpolarizationa real and physically meaningful phenomenonthat is not to be ignored.Alternatively, one may solve the two equations regarding the
mean values of H and S2 on the BS solution, which is a linearcombination of the sought singlet state and of the Ms=0solution for the triplet
γ δΦ = Φ + Φ =MBS S T( 0)s
δ⟨Φ | |Φ ⟩ = ⟨Φ | |Φ ⟩S SBS2
BS2
T2
T
(assuming that the mean value of S2 is the same for the Ms=0and Ms=1 components of the triplet)
γ δ⟨Φ | |Φ ⟩ = ⟨Φ | |Φ ⟩ + ⟨Φ | |Φ ⟩H H HBS BS2
S S2
T T
which leads to
− =⟨ ⟩ −⟨ ⟩ − ⟨ ⟩
E ES E ES S
( )s T
2T BS T
2T
2BS
With this formula, the gap behaves correctly when the Ms=0solution tends to become a closed shell. Note that the simplerelation
− =−
− ⟨ ⟩E E
E ES
2( )2s T
BS T2
BS
also behaves correctly in this limiting situation and may as wellbe used in this case. At this point, it is convenient to introduce ageneral spin-decontamination factor (SDF), which can be takenat 2, or defined either from ref 6 as
λ′ =⟨ ⟩ − ⟨ ⟩S S
22
T2
BS
or more generally as
λ =⟨ ⟩
⟨ ⟩ − ⟨ ⟩S
S S
2T
2T
2BS (1)
The corrected singlet-to-triplet energy difference thereforewrites as
λ− = −E E E E( )s T BS T
B. Geometries. The geometry optimization of the tripletstate provides an energy minimum ET
min and an optimalgeometry XT
min, corresponding to a set of internal coordinates(bond lengths, bond angles and dihedral angles). The energy ofthe BS solution calculated for this geometry,
* =E E X( )BS BS Tmin
leads to the vertical transition energy from the optimal tripletgeometry (Figure 1)
λ* − = * −E E E E( )S Tmin
BS Tmin
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp303825x | J. Phys. Chem. A 2012, 116, 8226−82378227

One may also determine the optimal geometry XBSmin of the BS
determinant and its energy EBSmin. The energy of the triplet for
this geometry is
* =E E X( )T T BSmin
Of course δET=ET*−ETmin and δEBS=EBS*−EBS
min are positive. If⟨S2⟩BS ≠ 0, the geometry XBS
min is not the optimal geometry ofthe spin-decontaminated singlet state, but one may assume thatthe XT
min → XBSmin geometry change goes in the right direction
and that the spin-decontaminated singlet geometry is lyingsomewhere along this collective geometrical change. Let us focuson this collective motion path, quantified by an advancementparameter Q:
= + −X Q X Q X X( ) ( )Tmin
BSmin
Tmin
Assuming an harmonic behavior of the potential energysurfaces, the quantities δET and δEBS are sufficient to determinethe curvatures of the curves ET(Q) and EBS(Q):
δ= +E Q E Q E( )T Tmin 2
T
δ= + −E Q E Q E( ) ( 1)BS BSmin 2
BS
If the spin-decontamination factor λ does not depend on Q, thesinglet-state energy may be written as
λ δ
λ δ
= + −
+ − +
E Q E Q E
E Q E
( ) ( ( 1) )
(1 )( )S BS
min 2BS
Tmin 2
T (2)
Direct differentiation provides the optimized geometry of thesinglet state
λδλδ λ δ
=− −
QE
E E( 1)Smin BS
BS T (3)
If the curvatures are identical (δEBS = δET), the above relationsimplifies to QS
min = λ. For ideal values of ⟨S2⟩ (⟨S2⟩T = 2, ⟨S2⟩BS
= 1), λ = 2, therefore locating QSmin around 2. For Q = QS
min, theoptimized energy of the singlet state is
λλδ
λδ λ δ= + * − −
− −E E E E
EE E
( )( )
( 1)Smin
Tmin
BS Tmin BS
2
BS T
which gives the adiabatic triplet-to-singlet energy gap
λλδ
λδ λ δ− = * − −
− −E E E E
EE E
( )( )
( 1)Smin
Tmin
BS Tmin BS
2
BS T(4)
Again, this expression can take simplified forms. If λ = 2, itbecomes
δδ δ
− = * − −−
E E E EE
E E2( )
4( )2S
minTmin
BS Tmin BS
2
BS T (5)
If both triplet and BS potential curves exhibit a same curvature(δEBS = δET), it further reduces to
δ− = * − −E E E E E2( ) 4Smin
Tmin
BS Tmin
BS (6)
suggesting a singlet-state relaxation from triplet geometry fourtimes larger than that of the BS solution. Still assuming δEBS =δET, but now taking λ ≠ 2, one gets
λ λ δ− = * − −E E E E E( )Smin
Tmin
BS Tmin 2
BS
All these derivations suppose that the decontamination factorλ does not change significantly with the geometry. If thisparameter is different for the Ms=1 and Ms=0 optimizedgeometries one may either (i) try to use a linear dependence ofλ on the geometry change,
λ λ λ= +Q Q( ) 0 1
(λ0 and λ1 being the values of λ at geometries of triplet and BSsolutions, respectively) or, preferably, (ii) recalculate theenergies of the triplet and BS determinants along the Qcoordinate. This numerical method consists in recalculating the
Figure 1. Schematic representation of potential energy along the collective geometrical coordinate Q.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp303825x | J. Phys. Chem. A 2012, 116, 8226−82378228

spin-decontaminated energy for a series of values of Q between0 and 2 (and beyond if necessary), λ being recalculated for eachvalue of Q:
λ= + −E Q E Q Q E Q E Q( ) ( ) ( )( ( ) ( ))s T BS T (7)
This method consists of a constrained exploration of thepotential-energy surface of the decontaminated singlet. Due tothis constraint, it only leads to an upper bound of the overallenergy minimum. This technique is somewhat more expensive,but it offers the possibility to check the validity of thepredictions based on the preceding analytical formulas.
3. NUMERICAL TESTS
In coordination-chemistry magnetic complexes, when goingfrom triplet to singlet state of a diradical, geometry changes areexpected to be extremely small, and are usually neglected.Actually in these systems the unpaired electrons are welllocalized on the transition metal ions, connected through ratherrigid ligands. As geometry changes are supposed to be larger inorganic diradicals, especially in conjugated hydrocarbons, whereunpaired electrons are spread on the entire π system, it istherefore desirable to study this phenomenon on organicsystems. Because one may suspect that equilibrium-geometrychanges are larger when the magnetic coupling (or singlet-to-triplet transition energy) is large, the selected systems shouldalso present non-negligible singlet−triplet differences. Threeexamples have been selected. The first one is a typicalferromagnetic system, m-xylylene, 1, the ground state of
which is triplet, with a transition energy to the singlet statearound 0.5 eV.13 As an antiferromagnetic coupling, we chose p-benzyne, 2, in which the two unpaired electrons belong to the σsystem, and where the singlet-to-triplet transition energy isaround 0.3 eV.14 Last, we selected an architecture constitutedby two phenalene radicals coupled in an antiferromagneticmanner by an acetylene bridge, 3, a system already studied byNakano et al. and possibly presenting dramatic properties.15 Allthe calculations were performed using the UDFT version of the
Figure 2. Energy curves along the collective geometrical coordinate Q for m-xylylene.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp303825x | J. Phys. Chem. A 2012, 116, 8226−82378229

Gaussian03 quantum chemistry package, with standard B3LYPhybrid functional and 6-311G** basis set.16 Optimizedgeometries were carried out up to energy gradients lowerthan 10−5 au. Optimized geometries correspond to C2v, D2h,and C2h symmetries in the three molecules, respectively. If thesensitivity of magnetic coupling to the percentage of exactexchange incorporated in the exchange−correlation potential isa major problem in UDFT approaches, the methodologicalconclusions of the present work are not affected by this issue.A. m-Xylylene. The vertical energy gap EBS* − ET
min is herecalculated at 0.011 149 au. Let us first consider the case whereboth spin-decontamination factor λ and singlet-state optimizedgeometry QS
min take their ideal values of 2. As δEBS and δET arehere close enough (0.000 745 and 0.000 693 au, respectively),one can take a mean value for both at 0.000 719 au and use eq 6to estimate ES
min − ETmin from EBS* − ET
min. In this way one getsESmin − ET
min = 0.019 422 ua (0.53 eV). Taking the explicit valuesfor δEBS and δET and using eq 5 has little effect on this resultsince it leads to ES
min − ETmin = 0.019 512 ua (0.53 eV). In this
case, the position of QSmin along the coordinate Q can be
evaluated from eq 3, which is now
δδ δ
=−
QE
E E2
2Smin BS
BS T
leading to a value of QSmin = 1.87. Going a step further will use
values of SDF different from 2, either λ′ or λ. Because ⟨S2⟩ hasnot exactly the same values at XT
min (Q=0) and XBSmin (Q=1), one
has to take averaged values of them to evaluate λ′ and λ (for m-xylylene, these changes are tiny, but we shall see later on thatthis is not always the case). In doing so, one gets λ′ = 1.90 andλ = 1.96. Reporting these values into eq 3 leads to values ofQS
min of 1.79 and 1.84, respectively. From eq 4, the adiabaticenergy gap ES
min − ETmin is now estimated at 0.018 653 au (0.51
eV) when λ′ = 1.90, and 0.019 170 au (0.52 eV) when λ = 1.96.Time has now come to check how these predictions compare
to the values obtained from explicit calculations along the Qcollective coordinate. To do so, we consider a simultaneous andregular variation of all internal coordinates from the triplet-stategeometry XT
min (Q=0) to that of the broken-symmetry Ms = 0solution XBS
min (Q=1). For each parameter of the Z-matrix, thedifference between the bounds of the [0, 1] interval along Q isdivided into ten regular elementary increments. Exploring thepotential curve along this collective coordinate up to Q=2 andbeyond therefore requires to increment more than 20 timesfrom the starting triplet geometry. In this way we get explicitcalculations for a set of regularly interpolated and extrapolatedpoints along the collective coordinate. On each of these points,two UDFT calculations are carried out, one for the triplet state(S=1), the other one for the broken-symmetry Ms=0 solution.The corresponding results are reported in the two lower curvesof Figure 2, both exhibiting clear quadratic shape. For each ofthese points, the spin-decontaminated singlet-state energy canbe evaluated straightly from the eq 7, leading to the bold uppercurve in Figure 2. Here, the spin-decontamination factor is nolonger averaged but is obtained using the values of ⟨S2⟩T and⟨S2⟩BS calculated at each point of the curve. Using 2, λ′, or λ asSDF (λ is used in Figure 2) has here little effect on theminimum of the curve, located at Q=1.87, 1.77, or 1.83 in thethree cases respectively, in remarkable agreement with theresults given by the above-discussed model, as summarized inTable 1. The so-extrapolated singlet-state geometrical param-eters corresponding to QS
min are listed in Table 2. On the
overall, they are in good agreement with benchmark valuescalculated at EOM-SF-CCSD level,13b illustrating how thewhole deformation occurring from the triplet UDFT geometryto that of the BSMs=0 solution must be continued by about thesame extent.The adiabatic energy difference ES
min − ETmin is obtained
directly from the minima of the triplet and spin-decontami-nated singlet curves. For SDF taken at 2, λ′, and λ, one getsadiabatic gaps of 0.019 514 au (0.53 eV), 0.018 701 au (0.51eV), and 0.019 202 au (0.052 eV), respectively, in closeagreement with the values given by the above model, andexhibiting small corrections only with respect to simple broken-symmetry calculations, as illustrated in Table 4.
B. p-Benzyne. As a σ diradical with a singlet ground state,p-benzyne is a good test to check whether the above-developedprocedures can be likewise appropriate when the ground stateis singlet. Due to this state of thing, the situation depicted inFigure 1 is now reversed. δEBS and δET still have the samedefinition and are here calculated at 0.000 711 and 0.000 869au, respectively. The starting energy gap is now the vertical gapEBS* − ET
min = 0.005 064 au. From an average value of δEBS andδET, eq 6, which is now written
δ− = * − −E E E E E2( ) 4Tmin
Smin
T BSmin
BS
leads to an adiabatic singlet-to-triplet gap of 0.006 986 au (0.19eV). Using the exact values for δEBS and δET, eq 5 lead to amodified gap of ET
min − ESmin = 0.006 471 au (0.18 eV). Again,
averaging the values of ⟨S2⟩T and ⟨S2⟩BS for Q=0 and Q=1 leads
to mean values for λ′ and λ at 1.90 and 1.91, respectively.Obtained from eq 3, QS
min is now significantly beyond 2, (QSmin =
2.38 and 2.39, respectively), giving a ETmin − ES
min gap from eq 4at 0.006 414 and 0.006 419 au, respectively (0.17 eV).
Table 1. Calculated QSMin along the Collective Geometrical
Coordinate
δET (au) δEBS (au) SDFa modelb explicit curvec
m-xylylene 0.000 693 0.000 745 2.00 1.87 1.871.90 1.79 1.771.96 1.84 1.83
p-benzyne 0.000 869 0.000 711 2.00 2.57 2.431.90 2.38 1.871.91 2.39 1.88
aSpin-decontamination factor, taken at 2, λ′, or λ from top to bottom,respectively, with average ⟨S2⟩ between QT and QBS coordinates.
bSeeeq 3. cSee Figures 3 and 4. Here, λ′ and λ are calculated from ⟨S2⟩values at each point of the curves.
Table 2. Calculated CC Bond Lengths (Å) in m-Xylylene
CCSDa UB3LYP/6-311G**
triplet singlet triplet BS Ms=0 extrapolated singlet
a 1.392 1.389 1.389 1.389 1.389b 1.433 1.402 1.432 1.419 1.406c 1.419 1.400 1.418 1.411 1.403d 1.402 1.452 1.400 1.420 1.440
aFrom EOM-SF-CCSD with UHF/6-31G* reference (ref 13b).
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp303825x | J. Phys. Chem. A 2012, 116, 8226−82378230
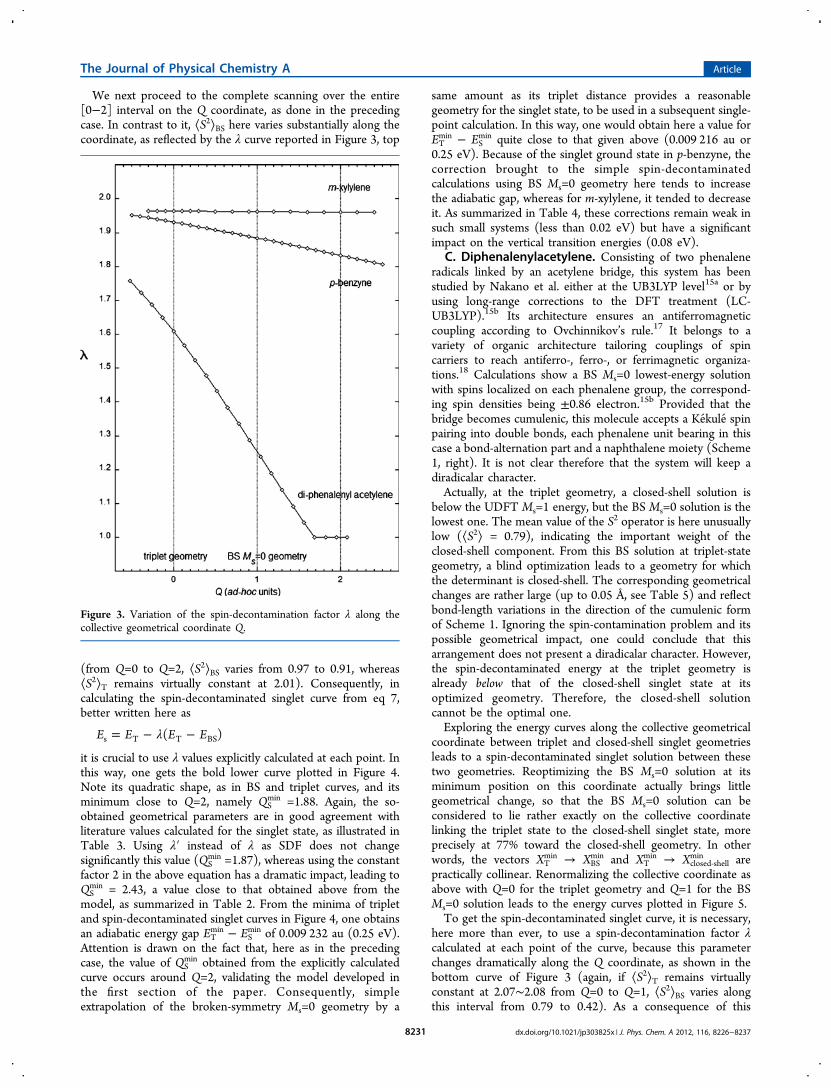
We next proceed to the complete scanning over the entire[0−2] interval on the Q coordinate, as done in the precedingcase. In contrast to it, ⟨S2⟩BS here varies substantially along thecoordinate, as reflected by the λ curve reported in Figure 3, top
(from Q=0 to Q=2, ⟨S2⟩BS varies from 0.97 to 0.91, whereas⟨S2⟩T remains virtually constant at 2.01). Consequently, incalculating the spin-decontaminated singlet curve from eq 7,better written here as
λ= − −E E E E( )s T T BS
it is crucial to use λ values explicitly calculated at each point. Inthis way, one gets the bold lower curve plotted in Figure 4.Note its quadratic shape, as in BS and triplet curves, and itsminimum close to Q=2, namely QS
min =1.88. Again, the so-obtained geometrical parameters are in good agreement withliterature values calculated for the singlet state, as illustrated inTable 3. Using λ′ instead of λ as SDF does not changesignificantly this value (QS
min =1.87), whereas using the constantfactor 2 in the above equation has a dramatic impact, leading toQS
min = 2.43, a value close to that obtained above from themodel, as summarized in Table 2. From the minima of tripletand spin-decontaminated singlet curves in Figure 4, one obtainsan adiabatic energy gap ET
min − ESmin of 0.009 232 au (0.25 eV).
Attention is drawn on the fact that, here as in the precedingcase, the value of QS
min obtained from the explicitly calculatedcurve occurs around Q=2, validating the model developed inthe first section of the paper. Consequently, simpleextrapolation of the broken-symmetry Ms=0 geometry by a
same amount as its triplet distance provides a reasonablegeometry for the singlet state, to be used in a subsequent single-point calculation. In this way, one would obtain here a value forETmin − ES
min quite close to that given above (0.009 216 au or0.25 eV). Because of the singlet ground state in p-benzyne, thecorrection brought to the simple spin-decontaminatedcalculations using BS Ms=0 geometry here tends to increasethe adiabatic gap, whereas for m-xylylene, it tended to decreaseit. As summarized in Table 4, these corrections remain weak insuch small systems (less than 0.02 eV) but have a significantimpact on the vertical transition energies (0.08 eV).
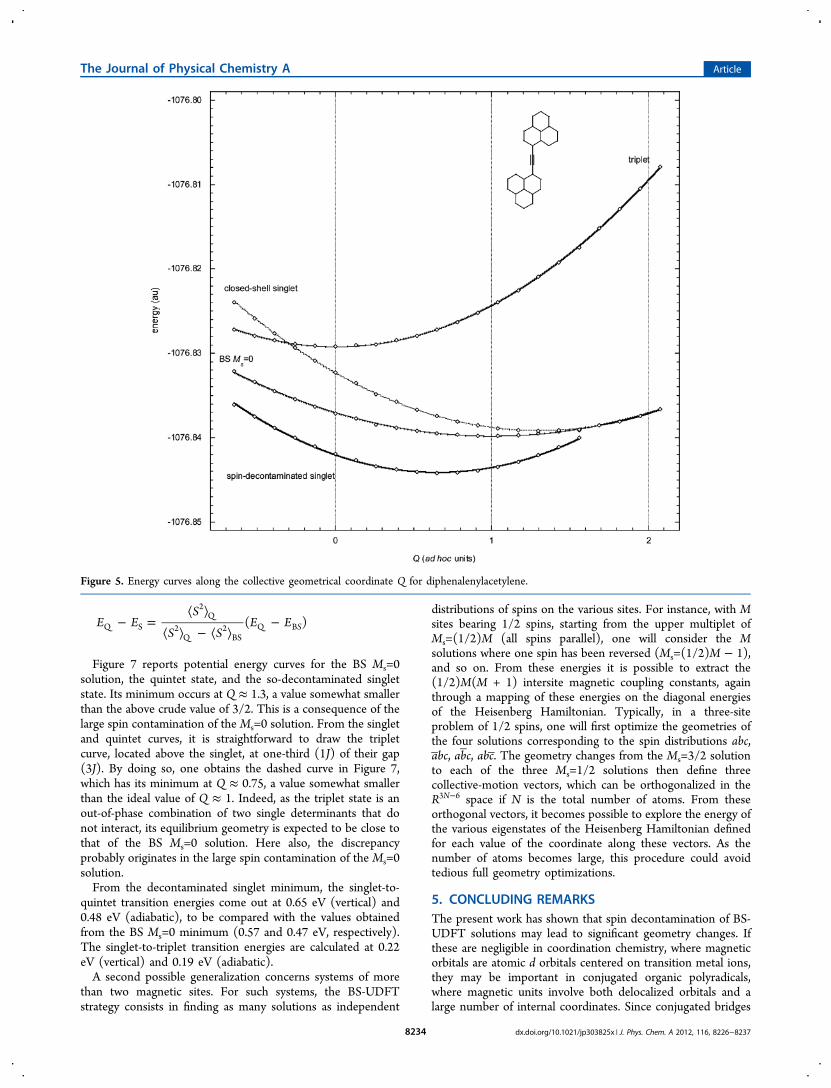
C. Diphenalenylacetylene. Consisting of two phenaleneradicals linked by an acetylene bridge, this system has beenstudied by Nakano et al. either at the UB3LYP level15a or byusing long-range corrections to the DFT treatment (LC-UB3LYP).15b Its architecture ensures an antiferromagneticcoupling according to Ovchinnikov’s rule.17 It belongs to avariety of organic architecture tailoring couplings of spincarriers to reach antiferro-, ferro-, or ferrimagnetic organiza-tions.18 Calculations show a BS Ms=0 lowest-energy solutionwith spins localized on each phenalene group, the correspond-ing spin densities being ±0.86 electron.15b Provided that thebridge becomes cumulenic, this molecule accepts a Kekule spinpairing into double bonds, each phenalene unit bearing in thiscase a bond-alternation part and a naphthalene moiety (Scheme1, right). It is not clear therefore that the system will keep adiradicalar character.Actually, at the triplet geometry, a closed-shell solution is
below the UDFT Ms=1 energy, but the BS Ms=0 solution is thelowest one. The mean value of the S2 operator is here unusuallylow (⟨S2⟩ = 0.79), indicating the important weight of theclosed-shell component. From this BS solution at triplet-stategeometry, a blind optimization leads to a geometry for whichthe determinant is closed-shell. The corresponding geometricalchanges are rather large (up to 0.05 Å, see Table 5) and reflectbond-length variations in the direction of the cumulenic formof Scheme 1. Ignoring the spin-contamination problem and itspossible geometrical impact, one could conclude that thisarrangement does not present a diradicalar character. However,the spin-decontaminated energy at the triplet geometry isalready below that of the closed-shell singlet state at itsoptimized geometry. Therefore, the closed-shell solutioncannot be the optimal one.Exploring the energy curves along the collective geometrical
coordinate between triplet and closed-shell singlet geometriesleads to a spin-decontaminated singlet solution between thesetwo geometries. Reoptimizing the BS Ms=0 solution at itsminimum position on this coordinate actually brings littlegeometrical change, so that the BS Ms=0 solution can beconsidered to lie rather exactly on the collective coordinatelinking the triplet state to the closed-shell singlet state, moreprecisely at 77% toward the closed-shell geometry. In otherwords, the vectors XT
min → XBSmin and XT
min → Xclosed‑shellmin are
practically collinear. Renormalizing the collective coordinate asabove with Q=0 for the triplet geometry and Q=1 for the BSMs=0 solution leads to the energy curves plotted in Figure 5.To get the spin-decontaminated singlet curve, it is necessary,
here more than ever, to use a spin-decontamination factor λcalculated at each point of the curve, because this parameterchanges dramatically along the Q coordinate, as shown in thebottom curve of Figure 3 (again, if ⟨S2⟩T remains virtuallyconstant at 2.07∼2.08 from Q=0 to Q=1, ⟨S2⟩BS varies alongthis interval from 0.79 to 0.42). As a consequence of this
Figure 3. Variation of the spin-decontamination factor λ along thecollective geometrical coordinate Q.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp303825x | J. Phys. Chem. A 2012, 116, 8226−82378231

variation, the above-developed analytic formulas cannot be usedto predict the spin-decontaminated singlet geometry QS
min, thesearch of which must be numerical and proceed frominterpolation of the spin-decontaminated curve between Q=0to Q=1. From Figure 5, this leads to a value of QS
min=0.65. As aconsequence of the vanishing of ⟨S2⟩BS around Q≈1.6, both BSMs=0 and closed-shell singlet curves merge beyond this point.There, λ = 1 so that the spin-decontaminated singlet-state curve
exactly coincides with the closed-shell singlet one. Because thevariations of ⟨S2⟩BS and λ are smooth and regular, the junctionbetween these two curves, not drawn in Figure 5, shouldbehave in the same way.Listed in Table 5, the resulting interpolated bond length for
the singlet state differ by about 0.01 Å from those of the BS Ms= 0 solution. Notice in this table the overall agreement betweenthe present UDFT results and those calculated at LR-UDFTlevel.15b At this QS
min geometry, the diradicalar character,reflected by ⟨S2⟩BS = 0.54, corresponds to a summed spindensity of ±0.60 electron on each phenalene fragment, whereasthis would reduce to ±0.50 electron for the BS Ms=0 structureat Q=1, as illustrated in Figure 6. The interpolated curve ofFigure 5, differing incidentally from the extrapolations aroundQ=2 in the prior cases, now makes it possible to bring ourcorrections to the singlet−triplet energy gaps. The results aresummarized in Table 4, lower part (for the obvious reasonsdiscussed above, the line corresponding to SDF = 2 is heremeaningless and must be disregarded). With respect to thebroken-symmetry approach, our corrected values show anincrease of the adiabatic gap by about 0.05 eV, and a decrease ofthe vertical gap by about 0.08 eV, the latter effect being hereopposite to that observed in the benzyne diradical.
4. POSSIBLE GENERALIZATIONS
Seeking whether this strategy might be useful for othermagnetic architectures beyond interactions between two 1/2spins, two possible extensions come to mind. The first oneconcerns the interaction between sites bearing spins larger than
Figure 4. Energy curves along the collective geometrical coordinate Q for p-benzyne.
Table 3. Calculated CC Bond Lengths (Å) in p-Benzyne
ref 14c UB3LYP/6-311G**
triplet singlet triplet BS Ms=0 extrapolated singlet
aa 1.370 1.358 1.377 1.370 1.363b 1.398 1.419 1.407 1.421 1.435
ab 1.383 1.374b 1.406 1.424
ac 1.387 1.383b 1.403 1.411
aSF-DFT (50,50)/6-31G*. bEOM-SF-CCSD/6-31G*. cCASSCF-(8,8)/6-31G*.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp303825x | J. Phys. Chem. A 2012, 116, 8226−82378232

1/2, the magnetic interaction of which can be studied from theBS-UDFT approach. Let us consider two sites A and B, bearingm and n unpaired electrons, respectively, with maximumground-state spin multiplicity at each center. The correspond-ing magnetic-coupling amplitude can be evaluated from theenergies of two single determinants with m + n open shells thatbelong to the set of vectors on which the HeisenbergHamiltonian HH = JABS A·S B is defined (i.e., the products ofground-state single determinants on each site). These energieswill be identified with its diagonal terms. Two such single
determinants may be considered, namely the solution with Ms
= 1/2(m + n), which gives the energy Em+n of the uppermultiplet of the AB dimer, and the solution of Ms = 1/2(m −n), where m α-electrons are centered on site A and n β-electrons are centered on site B. This solution, of energy Em−n,is not a spin eigenstate, but the corresponding energy differenceenables one to fix the value of the magnetic coupling, throughthe relation Em+n − Em−n = 1/2 JAB mn. Knowing the value ofJAB leads to the full low-energy spectrum of the AB complex.Again, optimizations of the two solutions lead to differentgeometries. From them one may define a collective geometrychange and explore the energies of the various eigenstates ofthe Heisenberg Hamiltonian along this coordinate. Applyingthis to an antiferromagnetic interaction between two S=1 spins,and neglecting spin contamination, one may write EQ − ES =3JAB = 3/2(E4 − E0). Again, if the curvatures of the twosolutions along the Q coordinate are similar, one expects asinglet-state equilibrium geometry around QS
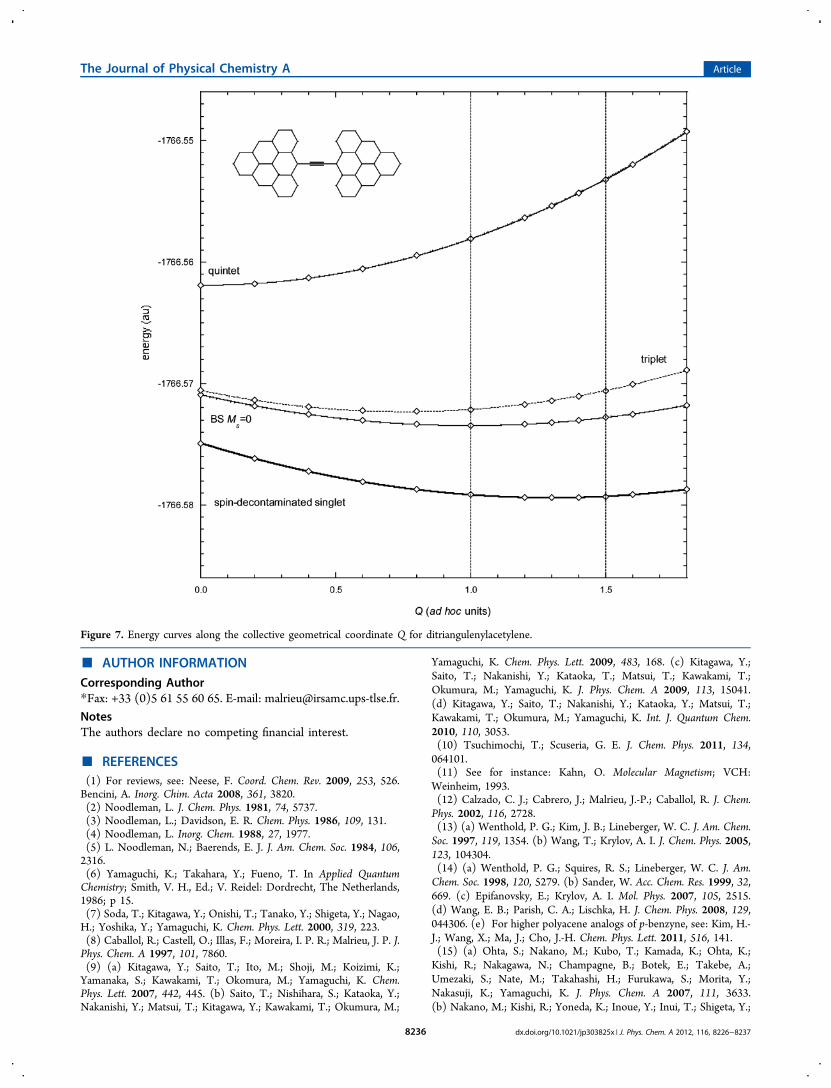
min = 3/2,suggesting a singlet-state relaxation from quintet geometry at9/4 of that of the BS solution.To illustrate this procedure, we can study the larger analog of
3, ditriangulenyl acetylene, in which two triangulene units ofspin S = 1 are linked by an acetylene bridge joining middle-edgecarbons.19 The coupling between the two units is hereantiferromagnetic, as occurring with the phenalenyl groups in3. Similarly, quinonization of the bonds would result in alowering (here from 1 to 1/2) of the spins carried by each unit(Scheme 2). Actually, from the Ms=2 solution to the Ms=0 one,the bond-length changes along the bridge have the right signs,but their amplitudes remain moderate (−0.04, +0.02, −0.04 Åalong the bridge; altogether, only ten or so CC bond lengthsvary by more than 0.01 Å). The mean value of S2 for the Ms=2solution is slightly larger than 6, due to the spin polarization ofthe closed shells (⟨S2⟩ = 6.19). For the Ms=0 solution, it issignificantly smaller than 2, due to the ionic components of thewave function, and the tendency to reduce the number ofunpaired electrons (⟨S2⟩ = 1.67). It diminishes when goingfrom the equilibrium geometry of the quintet (⟨S2⟩ = 1.90) tothe optimal geometry of the Ms=0 solution (⟨S2⟩ = 1.67) andbeyond. If these deviations are taken into account through a λfactor as proceeded above, the energy difference can be writtenas
λ− = = −E E J E E3 ( )Q S AB Q BS
Table 4. Calculated Singlet/Triplet Transition Energies (eV)
broken-symmetry geometry spin-decontaminated geometryb
SDFa vertical adiabatic vertical adiabatic
m-xylylene (3B2 →1A1) 2 0.61 0.55 0.53
λ′ 0.60 0.54 0.52λ 0.59 0.54 0.52
p-benzyne (1Ag →3B1u) 2 0.28 0.25 0.36 0.28
λ′ 0.26 0.24 0.33 0.25λ 0.26 0.24 0.34 0.25
diphenalenylacetylene (1Ag →3Bu) 2 0.86 0.60 0.67 0.61
λ′ 0.52 0.36 0.45 0.39λ 0.54 0.37 0.46 0.41
aSpin decontamination factor (see the text for the definition of λ′ and λ). bDetermined for SDF = λ.
Scheme 1
Table 5. Selected CC Bond Lengths (Å) inDiphenalenylacetylene
LR-UDFTa UDFT
triplet BS Ms=0 triplet BS Ms=0closed-shellsinglet
interpolatedsinglet
a 1.221 1.237 1.215 1.232 1.238 1.226b 1.418 1.382 1.416 1.376 1.365 1.391c 1.409 1.431 1.407 1.431 1.438 1.423d 1.384 1.366 1.381 1.362 1.357 1.369e 1.420 1.436 1.418 1.436 1.441 1.429f 1.439 1.457 1.437 1.457 1.463 1.450
aReference 15b.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp303825x | J. Phys. Chem. A 2012, 116, 8226−82378233
− =⟨ ⟩
⟨ ⟩ − ⟨ ⟩−E E
S
S SE E( )SQ S
2Q
2Q
2BS
Q B
Figure 7 reports potential energy curves for the BS Ms=0solution, the quintet state, and the so-decontaminated singletstate. Its minimum occurs at Q ≈ 1.3, a value somewhat smallerthan the above crude value of 3/2. This is a consequence of thelarge spin contamination of the Ms=0 solution. From the singletand quintet curves, it is straightforward to draw the tripletcurve, located above the singlet, at one-third (1J) of their gap(3J). By doing so, one obtains the dashed curve in Figure 7,which has its minimum at Q ≈ 0.75, a value somewhat smallerthan the ideal value of Q ≈ 1. Indeed, as the triplet state is anout-of-phase combination of two single determinants that donot interact, its equilibrium geometry is expected to be close tothat of the BS Ms=0 solution. Here also, the discrepancyprobably originates in the large spin contamination of the Ms=0solution.From the decontaminated singlet minimum, the singlet-to-
quintet transition energies come out at 0.65 eV (vertical) and0.48 eV (adiabatic), to be compared with the values obtainedfrom the BS Ms=0 minimum (0.57 and 0.47 eV, respectively).The singlet-to-triplet transition energies are calculated at 0.22eV (vertical) and 0.19 eV (adiabatic).A second possible generalization concerns systems of more
than two magnetic sites. For such systems, the BS-UDFTstrategy consists in finding as many solutions as independent
distributions of spins on the various sites. For instance, with Msites bearing 1/2 spins, starting from the upper multiplet ofMs=(1/2)M (all spins parallel), one will consider the Msolutions where one spin has been reversed (Ms=(1/2)M − 1),and so on. From these energies it is possible to extract the(1/2)M(M + 1) intersite magnetic coupling constants, againthrough a mapping of these energies on the diagonal energiesof the Heisenberg Hamiltonian. Typically, in a three-siteproblem of 1/2 spins, one will first optimize the geometries ofthe four solutions corresponding to the spin distributions abc,abc, abc, abc. The geometry changes from the Ms=3/2 solutionto each of the three Ms=1/2 solutions then define threecollective-motion vectors, which can be orthogonalized in theR3N−6 space if N is the total number of atoms. From theseorthogonal vectors, it becomes possible to explore the energy ofthe various eigenstates of the Heisenberg Hamiltonian definedfor each value of the coordinate along these vectors. As thenumber of atoms becomes large, this procedure could avoidtedious full geometry optimizations.
5. CONCLUDING REMARKSThe present work has shown that spin decontamination of BS-UDFT solutions may lead to significant geometry changes. Ifthese are negligible in coordination chemistry, where magneticorbitals are atomic d orbitals centered on transition metal ions,they may be important in conjugated organic polyradicals,where magnetic units involve both delocalized orbitals and alarge number of internal coordinates. Since conjugated bridges
Figure 5. Energy curves along the collective geometrical coordinate Q for diphenalenylacetylene.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp303825x | J. Phys. Chem. A 2012, 116, 8226−82378234

also introduce some nuclear degrees of freedom, the geometrychanges from high- to low-multiplicity eigenstates a prioriconcern a large number of nuclear coordinates. The conceptionof rational (even if approximated) exploration tools of thesegeometry changes and of their impact on the vertical andadiabatic transition energies is certainly welcome. Althoughmore rigorous solutions, involving derivatives of the decon-tamination factor with respect to nuclear coordinates areconceivable and have been proposed,9 the present worksuggests extremely simple recipes to get reasonable estimatesof optimized geometries of spin-decontaminated solutions andof their energies. In most cases, when the decontaminationfactor does not change much between the optimized geo-metries of the Ms=1 and Ms=0 BS solutions, calculation of the
two solutions at these two geometries is sufficient to estimatethe optimal geometry and the energy of the spin-decontami-nated singlet. The accuracy of the prediction is easily checkedby performing a calculation at the analytically predictedgeometry or around it.These statements have been checked and illustrated on three
different diradicalar conjugated hydrocarbons, for which thegeometry changes between the singlet and the triplet states arefar from being negligible. They represent three situations: (i) atriplet ground state; (ii) a singlet ground state of plain open-shell character, not accepting pairing of the electrons; (iii) asystem accepting such a pairing and potentially losing itsdiradicalar character. In the first two cases the geometrychanges are larger than those predicted by simple geometryoptimization of the broken-symmetry solution. They aresignificant and have non-negligible impact on vertical andadiabatic singlet-to-triplet energy differences. Comparisons withwave function theory-based calculations show here a rathergood agreement regarding geometry changes and energeticimpact. In both cases the simple analytic models predicting thegeometry changes and the energy of the spin-decontaminatedsinglet state from unrestricted solutions have proven to be valid,as checked by recalculating the energy of the spin-decontaminated state at its predicted optimal geometry.These simple models are based on a negligible variation ofthe spin-decontamination factor under the concerned geometrychanges. This assumption is manifestly irrelevant in the thirdproblem, where the closed-shell component of the BS solutionis large and increases when the geometry changes from the BSone to the spin-decontaminated one.The above remarks should affect the debate concerning the
possibly diradicalar character of long polyacenes. Calculatedfrom DFT-optimized geometries, singlet-to-triplet energydifferences can be questioned if they rely on poordeterminations of the singlet geometry, either from closed-shell solutions or from spin-contaminated unrestricted BS Ms =0 single-determinantal treatments.20−22 In particular, one maynotice that sophisticated wave function theory-based calcu-lations leading to opposite conclusions rely on different DFT-based geometry optimizations.23−26 Their relevance could beexamined along with the here-proposed procedure. As it is, thepresent strategy may be used directly for systems bridging twohigh-spin units (S > 1/2), such as Ni d8 ions or high-spinhydrocarbons.18,27 For systems involving more than twomagnetic units as in A−B−C architectures, UDFT is frequentlyemployed by mapping different BS-solution energies on thoseof Ising Hamiltonian. The principle of generalization of thepresent strategy to such polyradicalar systems, where severalgeometry changes have to be considered and combined, hasbeen outlined and will be addressed in forthcoming work.A last comment will concern thermodynamic magnetic
properties. We saw that geometry optimization results insignificant variations of vertical and adiabatic differencesbetween the singlet and triplet surfaces. As the curvatures ofthe potential-energy surfaces are different, this adds an extraentropic factor to the spin-degeneracy contribution. Thermo-dynamic magnetic properties such as magnetic susceptibilityshould be affected accordingly, and the use of a simpleHeisenberg Hamiltonian to fit their temperature dependenceshould be questioned.
Figure 6. Summed spin-density in one phenalenyl fragment along thecollective geometrical coordinate Q, calculated for the broken-symmetry Ms = 0 solution. The arrow indicates the spin-decontaminated singlet optimal geometry.
Scheme 2
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp303825x | J. Phys. Chem. A 2012, 116, 8226−82378235
■ AUTHOR INFORMATIONCorresponding Author*Fax: +33 (0)5 61 55 60 65. E-mail: [email protected].
NotesThe authors declare no competing financial interest.
■ REFERENCES(1) For reviews, see: Neese, F. Coord. Chem. Rev. 2009, 253, 526.Bencini, A. Inorg. Chim. Acta 2008, 361, 3820.(2) Noodleman, L. J. Chem. Phys. 1981, 74, 5737.(3) Noodleman, L.; Davidson, E. R. Chem. Phys. 1986, 109, 131.(4) Noodleman, L. Inorg. Chem. 1988, 27, 1977.(5) L. Noodleman, N.; Baerends, E. J. J. Am. Chem. Soc. 1984, 106,2316.(6) Yamaguchi, K.; Takahara, Y.; Fueno, T. In Applied QuantumChemistry; Smith, V. H., Ed.; V. Reidel: Dordrecht, The Netherlands,1986; p 15.(7) Soda, T.; Kitagawa, Y.; Onishi, T.; Tanako, Y.; Shigeta, Y.; Nagao,H.; Yoshika, Y.; Yamaguchi, K. Chem. Phys. Lett. 2000, 319, 223.(8) Caballol, R.; Castell, O.; Illas, F.; Moreira, I. P. R.; Malrieu, J. P. J.Phys. Chem. A 1997, 101, 7860.(9) (a) Kitagawa, Y.; Saito, T.; Ito, M.; Shoji, M.; Koizimi, K.;Yamanaka, S.; Kawakami, T.; Okomura, M.; Yamaguchi, K. Chem.Phys. Lett. 2007, 442, 445. (b) Saito, T.; Nishihara, S.; Kataoka, Y.;Nakanishi, Y.; Matsui, T.; Kitagawa, Y.; Kawakami, T.; Okumura, M.;
Yamaguchi, K. Chem. Phys. Lett. 2009, 483, 168. (c) Kitagawa, Y.;Saito, T.; Nakanishi, Y.; Kataoka, T.; Matsui, T.; Kawakami, T.;Okumura, M.; Yamaguchi, K. J. Phys. Chem. A 2009, 113, 15041.(d) Kitagawa, Y.; Saito, T.; Nakanishi, Y.; Kataoka, Y.; Matsui, T.;Kawakami, T.; Okumura, M.; Yamaguchi, K. Int. J. Quantum Chem.2010, 110, 3053.(10) Tsuchimochi, T.; Scuseria, G. E. J. Chem. Phys. 2011, 134,064101.(11) See for instance: Kahn, O. Molecular Magnetism; VCH:Weinheim, 1993.(12) Calzado, C. J.; Cabrero, J.; Malrieu, J.-P.; Caballol, R. J. Chem.Phys. 2002, 116, 2728.(13) (a) Wenthold, P. G.; Kim, J. B.; Lineberger, W. C. J. Am. Chem.Soc. 1997, 119, 1354. (b) Wang, T.; Krylov, A. I. J. Chem. Phys. 2005,123, 104304.(14) (a) Wenthold, P. G.; Squires, R. S.; Lineberger, W. C. J. Am.Chem. Soc. 1998, 120, 5279. (b) Sander, W. Acc. Chem. Res. 1999, 32,669. (c) Epifanovsky, E.; Krylov, A. I. Mol. Phys. 2007, 105, 2515.(d) Wang, E. B.; Parish, C. A.; Lischka, H. J. Chem. Phys. 2008, 129,044306. (e) For higher polyacene analogs of p-benzyne, see: Kim, H.-J.; Wang, X.; Ma, J.; Cho, J.-H. Chem. Phys. Lett. 2011, 516, 141.(15) (a) Ohta, S.; Nakano, M.; Kubo, T.; Kamada, K.; Ohta, K.;Kishi, R.; Nakagawa, N.; Champagne, B.; Botek, E.; Takebe, A.;Umezaki, S.; Nate, M.; Takahashi, H.; Furukawa, S.; Morita, Y.;Nakasuji, K.; Yamaguchi, K. J. Phys. Chem. A 2007, 111, 3633.(b) Nakano, M.; Kishi, R.; Yoneda, K.; Inoue, Y.; Inui, T.; Shigeta, Y.;
Figure 7. Energy curves along the collective geometrical coordinate Q for ditriangulenylacetylene.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp303825x | J. Phys. Chem. A 2012, 116, 8226−82378236

Kubo, T.; Champagne, B. J. Phys. Chem. A 2011, 115, 8767. (c) Fordifferently-coupled two-phenalene systems, see: Nakano, M.;Nakagawa, N.; Kishi, R.; Ohta, S.; Nate, M.; Takahashi, H.; Kubo,T.; Kamada, K.; Ohta, K.; Champagne, B.; Botek, E.; Morita, Y.;Nakasuji, K.; Yamaguchi, K. J. Phys. Chem. A 2007, 111, 9102. (d) Forstaggered dimeric pairs of phenalenyl radicals, see: Takano, Y.;Taniguchi, T.; Isobe, H.; Kubo, T.; Morita, Y.; Yamamoto, K.;Nakasuji, K.; Takui, T.; Yamaguchi, K. J. Am. Chem. Soc. 2002, 124,11122.(16) Frisch, M. J.; et al. Gaussian 03, revision B.05; Gaussian, Inc.:Wallingford, CT, 2004.(17) Ovchinnikov, A. A. Theor. Chim. Acta 1978, 47, 297. Alsoknown in solid-state physics as the Lieb’s theorem: Lieb, E. H. Phys.Rev. Lett. 1989, 62, 1201.(18) Trinquier, G.; Suaud, N.; Guihery, N.; Malrieu, J.-P. Chem. Phys.Chem. 2011, 12, 3020.(19) For the weak energy-impact nonplanar distortion of this system,see ref 18, p 3029.(20) Bendikov, M.; Duong, H. M.; Starkey, K.; Houk, K. N.; Carter,E. A.; Wudl, F. J. J. Am. Chem. Soc. 2004, 126, 7416.(21) Jiang, D.; Dai, S. J. Phys. Chem. A 2008, 112, 332.(22) Dos Santos, M. C. Phys. Rev. B 2006, 74, 045426.(23) Hachmann, J.; Dorando, J. J.; Aviles, M.; Chan, G. K.-L. J. Chem.Phys. 2007, 127, 134309.(24) Hajgato, B.; Szieberth, D.; Geerlings, P.; De Proft, F.; Deleuze,M. S. J. Chem. Phys. 2009, 131, 224321.(25) Hajgato, B.; Huzak, M.; Deleuze, M. S. J. Phys. Chem. A 2011,115, 9282.(26) Huzak, M.; Deleuze, M. S.; Hajgato, B. J. Chem. Phys. 2011, 135,104704.(27) Trinquier, G.; Suaud, N; Malrieu, J.-P. Chem. Eur. J. 2010, 16,8762.
The Journal of Physical Chemistry A Article
dx.doi.org/10.1021/jp303825x | J. Phys. Chem. A 2012, 116, 8226−82378237