a novel gene mutation in a family with x-linked retinoschisis
TRANSCRIPT

+ MODEL
Journal of the Formosan Medical Association (2014) xx, 1e9
Available online at www.sciencedirect.com
ScienceDirect
journal homepage: www.jfma-onl ine.com
ORIGINAL ARTICLE
A novel gene mutation in a family withX-linked retinoschisis
Yu-Hung Lai a,b,c,i, Shun-Ping Huang d,e,i, Shee-Ping Chen f,Pei-Shin Hu g, Shu-Fung Lin e, Min-Muh Sheu e,h,Hwei-Zu Wang a, Rong Kung Tsai e,h,*
a Department of Ophthalmology, Kaohsiung Medical University, Kaohsiung, Taiwanb Department of Ophthalmology, Kaohsiung Medical University Hospital, Kaohsiung Medical University,Kaohsiung, Taiwanc Graduate Institute of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwand Department of Molecular Biology and Human Genetics, Tzu Chi University, Hualien, Taiwane Department of Ophthalmology, Buddhist Tzu Chi General Hospital, Hualien, Taiwanf Stem Cells Center, Buddhist Tzu Chi General Hospital, Hualien, Taiwang Department of Ophthalmology, Changhua Christian Hospital, Changhua, Taiwanh Department of Ophthalmology and Visual Science, Tzu Chi University, Hualien, Taiwan
Received 20 December 2012; received in revised form 1 January 2014; accepted 8 January 2014
KEYWORDSoptic coherencetomography;
electroretinogram;retinoschisin 1;X-linked retinoschisis
Conflicts of interest: The authors h* Corresponding author. Department
Road, Hualien 970, Taiwan.E-mail address: [email protected]
i Y.-H.L. and S.-P.H. contributed eq
Please cite this article in press as: LaMedical Association (2014), http://d
0929-6646/$ - see front matter Copyrhttp://dx.doi.org/10.1016/j.jfma.201
Background/Purpose: To describe the clinical characteristics of a Taiwanese family with X-linked retinoschisis (XLRS) and to investigate the molecular genetics of a novel mutation inthe retinoschisin 1 (RS1) gene.Methods: A total of 15 participants in this XLRS family were analyzed. Complete ophthalmicexaminations and fundus photography were performed on 15 family members. These testsidentified five affected males and two female carriers. Blood samples were collected, andgenomic DNA was extracted. Best-corrected visual acuity, optical coherence tomography(OCT), electroretinogram (ERG), and direct DNA sequence analysis of the RS1 gene were per-formed on 15 family members.Results: Five affected males, with visual acuity ranging from 0.2 to 0.7, had macular schisisand abnormal retinal pigment epithelium pigmentation. The mixed scotopic ERG “b” wavewas more reduced than the “a” wave. OCT revealed typical microcystic schisis cavities withinthe macula area. Direct DNA sequence analysis revealed a single base pair deletion, 97delT, inall the affected individuals. This deletion resulted in a frameshift mutation of the RS1 gene,causing protein truncation. The affected males in this family showed moderately decreasedvisual acuity and dysfunction in both cone cells and phototransduction.
ave no conflicts of interest relevant to this article.of Ophthalmology, Buddhist Tzu Chi General Hospital, Tzu Chi University, 707, Section 3, Chung-Yang
.tw (R.K. Tsai).ually to this work.
i Y-H, et al., A novel gene mutation in a family with X-linked retinoschisis, Journal of the Formosanx.doi.org/10.1016/j.jfma.2014.01.001
ight ª 2014, Elsevier Taiwan LLC & Formosan Medical Association. All rights reserved.4.01.001

2 Y.-H. Lai et al.
+ MODEL
Figure 1 The pedigree of a Taiwafemale carrier; filled square, affect
Please cite this article in press as: LaMedical Association (2014), http://d
Conclusion: We identified a novel RS1 (97delT ) mutation in a Taiwanese family with XLRS. Thisfinding expands the RS1 mutation spectrum and may help to further understand the molecularpathogenesis of XLRS.Copyright ª 2014, Elsevier Taiwan LLC & Formosan Medical Association. All rights reserved.
Introduction
X-linked retinoschisis (XLRS) is a leading cause of juvenilemacular degeneration in males, with prevalence ranging from1:5000 to 1:25,000.1e3 The XLRS patients develop early-onsetbilateral vitreoretinal degeneration and splitting of the retinabetween the nerve fiber and ganglion cell layers. Typically, acartwheel-like pattern is seen in the patient’s fovea. Periph-eral retinoschisis and vitreous hemorrhage commonlyaccompany these XLRS symptoms.3 Visual acuity is highlyvariable and usually reduced to 20/1004,5 in school-age chil-dren but can manifest early in life.6 Visual defects may prog-ress slowlywith age inmost patients,5,7,8 but severe visual losshas also been described in some cases.1,2 Electroretinogram(ERG) examinations in patients with XLRS generally show areduction in b-wave amplitude with relative preservation ofthea-waveamplitude.9Optical coherence tomography (OCT),which provides high-resolution cross-sectional images of themacular region, is helpful for detecting microstructuralchanges in the eyes of XLRS patients.10e13
XLRS is associated with mutations in a single gene,retinoschisin 1 (RS1; NCBI reference sequence NM000330), that has been mapped to Xp22. The RS1 geneconsists of six exons and encodes RS (NCBI referencesequence: NP 000321.1), a 224-amino acidesolublesecretory protein.14,15 The RS1 RNA transcript contains3040 nucleotides.14 The RS1 protein is expressed inphotoreceptor and bipolar cells as a homo-oligomeric
nese family with X-linked juvened male; arrow, proband; slash
i Y-H, et al., A novel gene mutatix.doi.org/10.1016/j.jfma.2014.01
protein complex, and is involved in maintaining retinalintegrity.15,16 Although over 190 unique variants in the RS1gene have been reported and studied in XLRS patients, theseverity of visual impairment of XLRS does not correlatestrongly with mutation type.17e23
In this study, we describe a novel RS1 mutation in aTaiwanese family and report the clinical findings associatedwith this genotype. A sequence analysis revealed a 1-base-pair (bp) deletion in Exon 3 of the RS1 gene that results in aframeshift mutation and protein truncation.
Materials and methods
Clinical examinations
A 15-member family that included five males affected withXLRS (Fig. 1) was recruited for mutational analysis. Informed,written consent was obtained before blood sample acquisi-tion. This study was approved by the Institutional ReviewBoard at the Tzu Chi General Hospital and was performed inaccordance with the tenets of the Declaration of Helsinki. Allthe study participants underwent ophthalmic examinationsthat included best-corrected visual acuity (BCVA), slit-lampbiomicroscopy, indirect ophthalmoscopy, OCT (Stratus OCT3version 4.0.2; Carl Zeiss Meditec, Dublin, CA, USA), ERG(UTAS-E 4000; LKC Technologies, Gaithersburg, MD, USA), andfundus photography.
ile retinoschisis. Square, male; circle, female; circle with dot,, deceased.
on in a family with X-linked retinoschisis, Journal of the Formosan.001

Table 1 The primer sequences used to amplify XLRS exons 1e6.
Exon Sense Antisense
1 AGATGCTTAATGAGGCCAGG TAGAAGGCTCCTCACAGAAC2 CTCACCATGACTTCTTCCAG CACCATGCCCAGCCAAAATA3 AGGACTGAGTGTGATCACTC CCCGCATTAACATAGGCTTA4 ACAGGTAAAGCCTTCTCCGT AGACGGGGTTTCACTGTGTT5 TAGGAGACAAGGCTCAGACT AAGCGCAGATGATCCACTGT6 AAAAGTTGGGGCTAGCTCCA GCGAAATATAGCCCTGTCCA
XLRS Z X-linked retinoschisis.
Novel gene mutation in X-linked retinoschisis 3
+ MODEL
Polymerase chain reaction
Blood samples were collected from all 15 family members.Genomic DNA was extracted from peripheral blood leuko-cytes using a standard procedure. The sequences of the sixcoding exons of the RS1 gene were amplified by polymerasechain reaction using the primers shown in Table 1. All of thesamples were amplified in 50-mL reaction mixtures thatcontained 50 ng of genomic DNA, 10 mM TriseHCl, 50 mMKCl, 2.5 mM MgCl2, 0.2 mM of each deoxyribonucleotidetriphosphate, 200 mM of each primer, and 0.15 U ofAmpliTaq Gold DNA polymerase (Applied Biosystems, FosterCity, CA, USA; Roche Molecular Systems, Inc., Branchburg,NJ, USA). The thermal cycling profile consisted of an initialdenaturation step at 95 �C for 10 minutes followed by 35
Table 2 A summary of the clinical findings of all 15 family mem
No. Age(y)
Gender BCVA(OD/OS)
Macular/retinal appearance
I-1 70 M 1.0/1.0 NormalII-1 45 M 1.0/1.0 NormalII-2a 46 F 1.0/1.0 NormalII-3 44 M 1.0/1.0 NormalII-4a 45 F 1.0/1.0 NormalII-5 40 M 1.0/1.0 NormalII-6 43 F 1.0/1.0 NormalII-7b 39 M 0.2/0.2 Cartwheel-like appearance an
pigmentation abnormalities inRPE atrophy of the fovea in O
II-8b 37 M 0.2/0.3 Cartwheel-like appearance anpigmentation abnormalities in
III-1 16 M 1.0/1.0 NormalIII-2b 13 M 0.2/0.2 Cartwheel-like appearance an
pigmentation abnormalities inRPE atrophy of the fovea in Oinferior retinoschisis in OU
III-3b 11 M 0.5/0.7 Cartwheel-like appearance anpigmentation abnormalities in
III-4b 9 M 0.3/0.2 Cartwheel-like appearance anpigmentation abnormalities in
III-5 14 M 1.0/1.0 NormalIII-6 10 F 1.0/1.0 Normal
BCVA Z best-corrected visual acuity; ERG Z electroretinography; F ZOU Z oculus unitas; RPE Z retinal pigment epithelium.a Carrier.b Deletion at the 33rd codon in exon 3 of the RS1 gene (97delT ). Th
Nucleotide 1 is A of the ATG initiation codon.
Please cite this article in press as: Lai Y-H, et al., A novel gene mutatiMedical Association (2014), http://dx.doi.org/10.1016/j.jfma.2014.01
cycles of 30-second denaturation at 95 �C, 30-secondannealing at 50 �C, 1-minute extension at 72 �C, and afinal 10-minute extension step at 72 �C.
Sequencing
The sequencing reactions were performed using the BigDyeTerminator v3.1 Cycle Sequencing Kit (Applied Biosystems).Each sequencing reaction was amplified in a 10-mL reactionmixture that contained 10e30 ng of amplified DNA. Thethermal cycling profile consisted of an initial denaturationstep at 95 �C for 10 seconds followed by 25 10-seconddenaturation cycles at 95 �C, 5-second annealing at 50 �C,and 1-minute extension at 60 �C. The primers used for thedirect sequencing reactions were identical to those used for
bers.
Macularschisis
Peripheralschisis
ERG findings
No No d
No No d
No No NormalNo No d
No No NormalNo No d
No No d
d RPEOU;U
Yes No Dysfunction of conecells, rod cells, andphototransduction
d RPEOU
Yes No Dysfunction of conecells and phototransduction
No No d
d RPEOU;D;
Yes Yes Dysfunction of conecells, rod cells, andphototransduction
d RPEOU
Yes No d
d RPEOU
Yes No d
No No d
No No d
female; M Z male; OD Z oculus dexter; OS Z oculus sinister;
e numbering follows the NCBI Reference Sequence NM 000330.3.
on in a family with X-linked retinoschisis, Journal of the Formosan.001
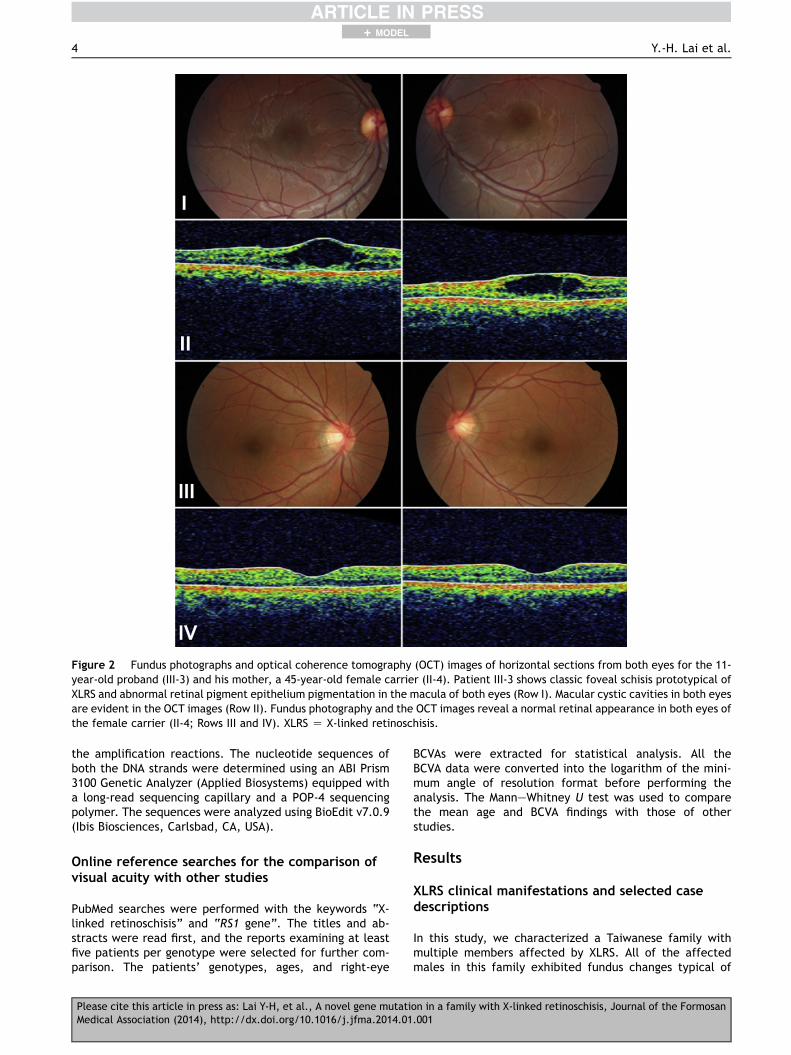
Figure 2 Fundus photographs and optical coherence tomography (OCT) images of horizontal sections from both eyes for the 11-year-old proband (III-3) and his mother, a 45-year-old female carrier (II-4). Patient III-3 shows classic foveal schisis prototypical ofXLRS and abnormal retinal pigment epithelium pigmentation in the macula of both eyes (Row I). Macular cystic cavities in both eyesare evident in the OCT images (Row II). Fundus photography and the OCT images reveal a normal retinal appearance in both eyes ofthe female carrier (II-4; Rows III and IV). XLRS Z X-linked retinoschisis.
4 Y.-H. Lai et al.
+ MODEL
the amplification reactions. The nucleotide sequences ofboth the DNA strands were determined using an ABI Prism3100 Genetic Analyzer (Applied Biosystems) equipped witha long-read sequencing capillary and a POP-4 sequencingpolymer. The sequences were analyzed using BioEdit v7.0.9(Ibis Biosciences, Carlsbad, CA, USA).
Online reference searches for the comparison ofvisual acuity with other studies
PubMed searches were performed with the keywords “X-linked retinoschisis” and “RS1 gene”. The titles and ab-stracts were read first, and the reports examining at leastfive patients per genotype were selected for further com-parison. The patients’ genotypes, ages, and right-eye
Please cite this article in press as: Lai Y-H, et al., A novel gene mutatiMedical Association (2014), http://dx.doi.org/10.1016/j.jfma.2014.01
BCVAs were extracted for statistical analysis. All theBCVA data were converted into the logarithm of the mini-mum angle of resolution format before performing theanalysis. The ManneWhitney U test was used to comparethe mean age and BCVA findings with those of otherstudies.
Results
XLRS clinical manifestations and selected casedescriptions
In this study, we characterized a Taiwanese family withmultiple members affected by XLRS. All of the affectedmales in this family exhibited fundus changes typical of
on in a family with X-linked retinoschisis, Journal of the Formosan.001

Figure 3 Optical coherence tomography (OCT) images of both eyes from Patient III-4, taken at different times. Bilateralmicrocystic schisis cavities in the fovea were observed at the first visit at the age of 4 (top). As the disease progressed and visualacuity decreased, the OCT images showed microcyst coalescence and subsequent flattening of the foveal schisis cavity of the lefteye at the second (middle) and third visit (bottom) at the age of 8.
Novel gene mutation in X-linked retinoschisis 5
+ MODEL
XLRS, including bilateral macular cystic cavities, abnormalretinal pigment epithelium pigmentation, and subnormalERG b-wave amplitudes compared with their a-waveamplitudes. The family’s clinical data are summarized inTable 2. All five affected patients developed macula schisisbut only one patient (III-2) showed peripheral retinoschisis.The ERG examinations in three of the affected patients (II-7, II-8, and III-2) showed decreased b/a ratios by scotopicwhite flash (0.6, 1.1, and 0.6, respectively; normal value:2.3 � 0.34), decreased scotopic dim flash amplitudes inPatients II-7 and III-2 but not in Patient II-8 (0, 0, and271.2 mV, respectively; normal value: 220.5 � 83.9 mV), andin 30-Hz flicker stimulations with decreased amplitudes(54.2, 44.7, and 33.2 mV, respectively; normal value:80.6 � 26.1 mV) and with prolonged implicit times (33.4,33.3, and 33.0 ms, respectively; normal value:28.4 � 1.7 ms).
Case description of Patient III-3
The proband was an 11-year-old boy who was found tohave poor visual acuity during a vision screening at theage of 4. A family history of poor vision or maculardegeneration only in male relatives was noted (uncles,cousin, and a younger brother; Fig. 1). Initially, he wasdiagnosed as having cone dystrophy, macular degenera-tion, or retinitis pigmentosa. He was not diagnosed withX-linked juvenile retinoschisis until he was 6 years and5 months of age. At the time of the diagnosis, his right eyeBCVA and refraction were 0.3 corrected with þ0.25D cyl�0.5D A 90 and his left eye BCVA and refraction were 0.4corrected with þ0.25D and cyl �0.75D A 65.
Please cite this article in press as: Lai Y-H, et al., A novel gene mutatiMedical Association (2014), http://dx.doi.org/10.1016/j.jfma.2014.01
Characteristic folds with a cartwheel-like appearanceradiating outward in a stellate pattern from the foveawere observed in both eyes (Fig. 2, Row I). The OCT ex-amination revealed microcystic schisis cavities within themacula area (Fig. 2, Row II). These findings prompted theother family members to undergo comprehensiveophthalmic examinations. His most recent BCVA of theright eye and left eye was 0.5 and 0.7, respectively, atthe age of 11. His mother (II-4) was a carrier, and hervisual acuity in each eye was 1.0. The fundus photographyand OCT images of both her eyes showed normal macula(Fig. 2, Rows III and IV). Her ERG results were also normal(Fig. 4, middle column).
Patient III-4
Patient III-4 was the 9-year-old younger brother of PatientIII-3. His fovea also showed the cartwheel-like appearancein both eyes. The initial OCT revealed foveal schisis in botheyes (macular thickness: 447 mm in the right eye and400 mm in the left eye; Fig. 3, upper row) at the age of 4. Atthis time, the BCVA and refraction in his right eye was 0.3and þ1.00 þ 0.75 � 109�, respectively; the BCVA andrefraction in his left eye was 0.3 and þ0.25 þ 1.25 � 59�,respectively. Over time, the foveal cyst in the left eyediminished and exhibited some pigmentary changes (at theage of 7, Fig. 3, middle row; at the age of 8, Fig. 3, lowerrow). His visual acuity at the age of 7 was 0.2 (0.5 correctedwith þ0.25D and cyl þ1.0D A 120) in the right eye and 0.2(0.4 corrected with �0.25D and cyl þ1.50D A 56) in the lefteye; at the age of 8, the visual acuity was 0.3 in the righteye (0.5 corrected with þ0.75D and þ1.00D A 120) and 0.1
on in a family with X-linked retinoschisis, Journal of the Formosan.001

Figure 4 The representative electroretinogram (ERG) results of the family members. Left column (XLRS, II-8): a low scotopic andphotopic b/a ratio with decreased 30-Hz flicker amplitudes that implied both phototransduction and cone system dysfunction. Middlecolumn:TheERGof thecarrier’smother (II-4) demonstratednormalfindings. Rightcolumn:normalcontrol. XLRSZX-linkedretinoschisis.
6 Y.-H. Lai et al.
+ MODEL
in the left eye (0.1 corrected with �0.75D and cyl þ1.75D A74). Interestingly, the visual acuity in his left eye worsenedas the schisis cavity flattened based on the findings of OCTexaminations.
Patient II-8
Patient II-8, a 37-year-old man, was the maternal uncle ofPatient III-3. Although his fovea also showed the charac-teristic cartwheel-like appearance in both eyes, the OCTexamination results were normal. However, the ERG ex-aminations revealed low scotopic and photopic b/a ratioand decreased 30-Hz flicker amplitudes that implied conesystem and phototransduction dysfunction (Fig. 4, leftcolumn). The patient’s BCVA was 0.2 in the right eye and0.3 in the left eye. His refraction was þ1.50D and cyl�1.75D A 73 in the right eye and þ0.50D and cyl �1.00D A180 in the left eye.
Mutational analysis
The RS1 coding regions of each family member weresequenced for detecting mutations. The direct DNAsequence analysis showed that all five males with XLRS (II-7,II-8, III-2, III-3, and III-4) had a novel 1-bp deletion mutation
Please cite this article in press as: Lai Y-H, et al., A novel gene mutatiMedical Association (2014), http://dx.doi.org/10.1016/j.jfma.2014.01
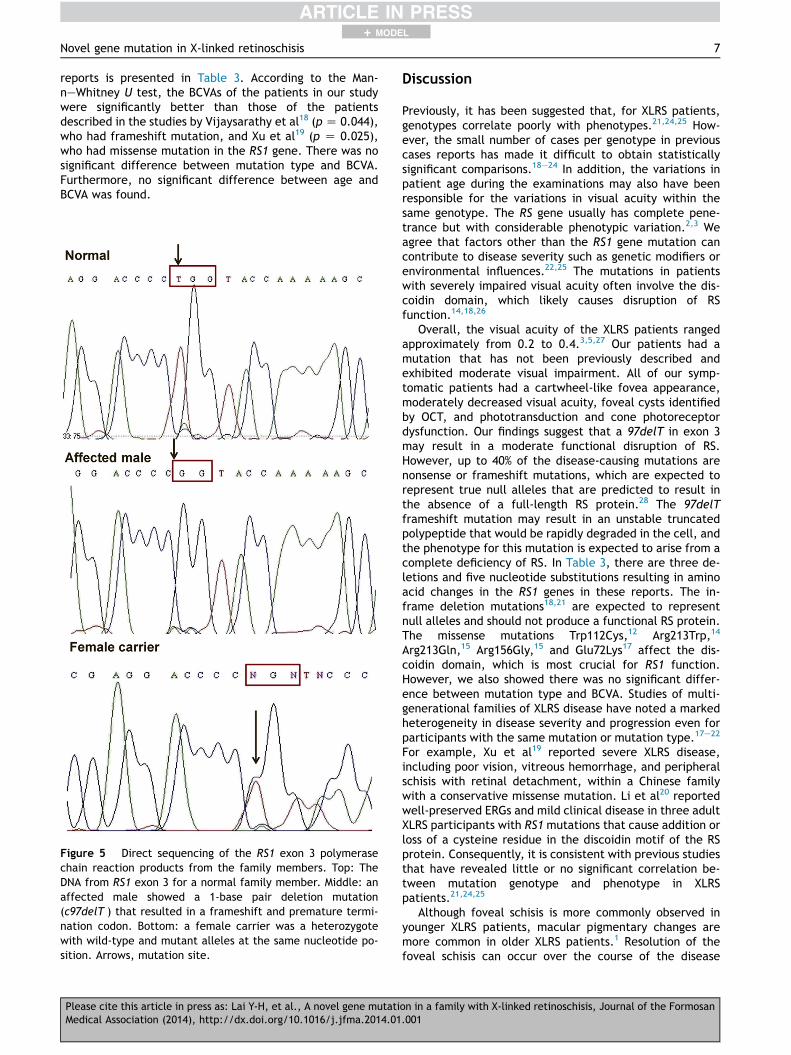
(termed 97delT ) in Exon 3 of the RS1 gene; the numberingsystem followed the NCBI reference sequence NM 000330.3,and nucleotide 1 was the A of the ATG initiation codon. Thisdeletion resulted in a frameshift mutation (Fig. 5) that isexpected to produce a truncated protein consisting of theinitial 32 amino acids of the wild-type RS1 protein followedby 92 aberrant amino acids and a premature terminationcodon (Fig. 6). Thus, the truncated mutant RS1 proteinlacked the final 100 amino acids of the wild-type RS1 proteinmaking it unstable and rapidly degrading in the cell, whichresulted in the absence of a functional RS protein. The twofemale carriers (II-2 and II-4)were heterozygous, having bothwild-type and mutant alleles at the same nucleotide posi-tion. All of the other family members who did not presentwith ophthalmic symptoms had normal genotypes. To thebest of our knowledge, this mutation has not been reportedin the literature; further, it is not listed in the RetinoschisisConsortium database that is maintained by Leiden University(http://www.dmd.nl/rs/index.html).
Correlation between mutation type and BCVA inXLRS patients in the literature
A summary of the comparisons between mutation type,age, and BCVA in XLRS patients from various published
on in a family with X-linked retinoschisis, Journal of the Formosan.001
Novel gene mutation in X-linked retinoschisis 7
+ MODEL
reports is presented in Table 3. According to the Man-neWhitney U test, the BCVAs of the patients in our studywere significantly better than those of the patientsdescribed in the studies by Vijaysarathy et al18 (p Z 0.044),who had frameshift mutation, and Xu et al19 (p Z 0.025),who had missense mutation in the RS1 gene. There was nosignificant difference between mutation type and BCVA.Furthermore, no significant difference between age andBCVA was found.
Figure 5 Direct sequencing of the RS1 exon 3 polymerasechain reaction products from the family members. Top: TheDNA from RS1 exon 3 for a normal family member. Middle: anaffected male showed a 1-base pair deletion mutation(c97delT ) that resulted in a frameshift and premature termi-nation codon. Bottom: a female carrier was a heterozygotewith wild-type and mutant alleles at the same nucleotide po-sition. Arrows, mutation site.
Please cite this article in press as: Lai Y-H, et al., A novel gene mutatiMedical Association (2014), http://dx.doi.org/10.1016/j.jfma.2014.01
Discussion
Previously, it has been suggested that, for XLRS patients,genotypes correlate poorly with phenotypes.21,24,25 How-ever, the small number of cases per genotype in previouscases reports has made it difficult to obtain statisticallysignificant comparisons.18e24 In addition, the variations inpatient age during the examinations may also have beenresponsible for the variations in visual acuity within thesame genotype. The RS gene usually has complete pene-trance but with considerable phenotypic variation.2,3 Weagree that factors other than the RS1 gene mutation cancontribute to disease severity such as genetic modifiers orenvironmental influences.22,25 The mutations in patientswith severely impaired visual acuity often involve the dis-coidin domain, which likely causes disruption of RSfunction.14,18,26
Overall, the visual acuity of the XLRS patients rangedapproximately from 0.2 to 0.4.3,5,27 Our patients had amutation that has not been previously described andexhibited moderate visual impairment. All of our symp-tomatic patients had a cartwheel-like fovea appearance,moderately decreased visual acuity, foveal cysts identifiedby OCT, and phototransduction and cone photoreceptordysfunction. Our findings suggest that a 97delT in exon 3may result in a moderate functional disruption of RS.However, up to 40% of the disease-causing mutations arenonsense or frameshift mutations, which are expected torepresent true null alleles that are predicted to result inthe absence of a full-length RS protein.28 The 97delTframeshift mutation may result in an unstable truncatedpolypeptide that would be rapidly degraded in the cell, andthe phenotype for this mutation is expected to arise from acomplete deficiency of RS. In Table 3, there are three de-letions and five nucleotide substitutions resulting in aminoacid changes in the RS1 genes in these reports. The in-frame deletion mutations18,21 are expected to representnull alleles and should not produce a functional RS protein.The missense mutations Trp112Cys,12 Arg213Trp,14
Arg213Gln,15 Arg156Gly,15 and Glu72Lys17 affect the dis-coidin domain, which is most crucial for RS1 function.However, we also showed there was no significant differ-ence between mutation type and BCVA. Studies of multi-generational families of XLRS disease have noted a markedheterogeneity in disease severity and progression even forparticipants with the same mutation or mutation type.17e22
For example, Xu et al19 reported severe XLRS disease,including poor vision, vitreous hemorrhage, and peripheralschisis with retinal detachment, within a Chinese familywith a conservative missense mutation. Li et al20 reportedwell-preserved ERGs and mild clinical disease in three adultXLRS participants with RS1 mutations that cause addition orloss of a cysteine residue in the discoidin motif of the RSprotein. Consequently, it is consistent with previous studiesthat have revealed little or no significant correlation be-tween mutation genotype and phenotype in XLRSpatients.21,24,25
Although foveal schisis is more commonly observed inyounger XLRS patients, macular pigmentary changes aremore common in older XLRS patients.1 Resolution of thefoveal schisis can occur over the course of the disease
on in a family with X-linked retinoschisis, Journal of the Formosan.001

Figure 6 The predicted amino acid sequence for the mutant RS1 gene. A comparison between the wild-type and the mutant RS1predicted amino acid sequences after T deletion at codon 97 (arrow) showed that the mutation resulted in a truncated proteinconsisting of the initial 32 amino acids of the wild-type RS1 protein followed by 92 aberrant amino acids and a prematuretermination codon.
Table 3 Comparisons of mutation type, age, and visual acuity in previously published reports on X-linked retinoschisis.
Study N Age(mean � SD)
logMAR BCVA � SD(Snellen equivalent)
Exon Nucleotide change Predicted aminoacid change
Present 5 21.8 � 14.9* 0.58 � 0.18 (0.3) 3 97delT Truncatedprotein
Simonelli et al 200317 6 22.3 � 7.1 0.59 � 0.62 (0.3) 5 336G > T Trp112CysVijayasarathy et al 201018 5 33.6 � 16.8 0.96 � 0.25 (0.1)** 5 354delC, 354e371
Ins GGTGTGCCTGGCTCTCCATruncatedprotein
Xu et al 201019 7 18.3 � 12.6 1.03 � 0.35 (0.1)*** 6 637C > T Arg213TrpEksandh et al 200021 12 30.8 � 21.3 0.73 � 0.45 (0.2) 1 Exon 1 deletion UnknownLi et al 200720 5 27.0 � 6.4 0.40 � 0.17 (0.4) 5 466G > A Arg156GlyLi et al 200720 5 17.6 � 6.5 0.59 � 0.11 (0.3) 6 638G > A Arg213GlnRiveiro-Alvarez et al 200922 5 27.0 � 20.7 0.68 � 0.33 (0.2) 4 214G > A Glu72Lys
Light perception, hand motion, and finger counting were arbitrarily set to 0.01, 0.02, and 0.03, respectively.All comparisons were performed using the ManneWhitney U test.* No statistically significant differences in age between groups.**p Z 0.044, compared with the present study.***p Z 0.025, compared with the present study.logMAR BCVA Z best-corrected visual acuity in the logarithm of the minimum angle of resolution format. N Z case number;SD Z standard deviation.
8 Y.-H. Lai et al.
+ MODEL
progression. Therefore, individuals with XLRS oftendemonstrate coalescence of the microcysts and a subse-quent flattening of the foveal schisis cavity.3,5,8,9,29
Consistent with these ophthalmoscopic findings, thefollow-up OCT examination revealed an improvement in thefoveal schisis of one patient (III-4). In contrast to a previousreport, our patient’s visual acuity actually worsenedfollowing the foveal schisis regression.30 The XLRS disrup-tions in visual function could not be identified solely by themacular thickness assessments of the OCT examinations.31
The ERG examinations of our XLRS patients showed bothphototransduction and photoreceptor dysfunction. Typi-cally, the ERG readings in XLRS patients show a reduced b-wave amplitude with relative preservation of the a-waveamplitude.8,9 With age and retinal pigment epithelial cell
Please cite this article in press as: Lai Y-H, et al., A novel gene mutatiMedical Association (2014), http://dx.doi.org/10.1016/j.jfma.2014.01
atrophy, both a- and b-wave amplitudes can decrease.8,11
This ERG dysfunction is found throughout the retina and isnot limited to the areas of schisis.8,9,32 Abnormal photore-ceptor (both rods and cones) function was also noted in theERG results of our patients, implying that both rod and conephotoreceptors were involved in XLRS functionalabnormalities.33,34
In conclusion, we have reported a novel gene mutation(97delT ) involving Exon 3 of RS1 in a Taiwanese family withXLRS. This mutation resulted in moderate visual deficits inthe five affected family members. Understanding themechanism of molecular genetics of this family with XLRSexpands the RS1 mutation spectrum and will aid in theinvestigation of molecular pathogenesis of XLRS. However,elucidation of the specific mechanism of this mutation will
on in a family with X-linked retinoschisis, Journal of the Formosan.001

Novel gene mutation in X-linked retinoschisis 9
+ MODEL
require further study of the wild-type and mutant RS1proteins in the retina.
Acknowledgments
None.
References
1. Falcone PM, Brockhurst RJ. X-chromosome-linked juvenileretinoschisis: clinical aspects and genetics. Int OphthalmolClin 1993;33:193e202.
2. George ND, Yates JR, Moore AT. X linked retinoschisis. Br JOphthalmol 1995;79:697e702.
3. George ND, Yates JR, Moore AT. Clinical features in affectedmales with X-linked retinoschisis. Arch Ophthalmol 1996;114:274e80.
4. Forsius H, Krause U, Helve J, Vuopala V, Mustonen E,Vainio-Mattila B, et al. Visual acuity in 183 cases of X-chro-mosomal retinoschisis. Can J Ophthalmol 1973;8:385e93.
5. Roesch MT, Ewing CC, Gibson AE, Weber BH. The naturalhistory of X-linked retinoschisis. Can J Ophthalmol 1998;33:149e58.
6. George ND, Yates JR, Bradshaw K, Moore AT. Infantilepresentation of X linked retinoschisis. Br J Ophthalmol 1995;79:653e7.
7. Apushkin MA, Fishman GA, Rajagopalan AS. Fundus findings andlongitudinal study of visual acuity loss in patients with X-linkedretinoschisis. Retina 2005;25:612e8.
8. Kellner U, Brummer S, Foerster MH, Wessing A. X-linkedcongenital retinoschisis. Graefes Arch Clin Exp Ophthalmol1990;228:432e7.
9. Peachey NS, Fishman GA, Derlacki DJ, Brigell MG.Psychophysical and electroretinographic findings in X-linkedjuvenile retinoschisis. Arch Ophthalmol 1987;105:513e6.
10. Azzolini C, Pierro L, Codenotti M, Brancato R. OCT images andsurgery of juvenile macular retinoschisis. Eur J Ophthalmol1997;7:196e200.
11. Stanga PE, Chong NH, Reck AC, Hardcastle AJ, Holder GE.Optical coherence tomography and electrophysiology inX-linked juvenile retinoschisis associated with a novel muta-tion in the XLRS1 gene. Retina 2001;21:78e80.
12. Yu J, Ni Y, Keane PA, Jiang C, Wang W, Xu G. Foveomacularschisis in juvenile X-linked retinoschisis: an optical coherencetomography study. Am J Ophthalmol 2010;149:973e8. e2.
13. Kjellstrom S, Vijayasarathy C, Ponjavic V, Sieving PA,Andreasson S. Long-term 12 year follow-up of X-linkedcongenital retinoschisis. Ophthalmic Genet 2010;31:114e25.
14. Sauer CG, Gehrig A, Warneke-Wittstock R, Marquardt A,Ewing CC, Gibson A, et al. Positional cloning of the geneassociated with X-linked juvenile retinoschisis. Nat Genet1997;17:164e70.
15. Grayson C, Reid SN, Ellis JA, Rutherford A, Sowden JC,Yates JR, et al. Retinoschisin, the X-linked retinoschisis pro-tein, is a secreted photoreceptor protein, and is expressed andreleased by Weri-Rb1 cells. Hum Mol Genet 2000;9:1873e9.
16. Molday LL, Hicks D, Sauer CG, Weber BH, Molday RS. Expressionof X-linked retinoschisis protein RS1 in photoreceptor and bi-polar cells. Invest Ophthalmol Vis Sci 2001;42:816e25.
17. Simonelli F, Cennamo G, Ziviello C, Testa F, de Crecchio G,Nesti A, et al. Clinical features of X linked juvenile
Please cite this article in press as: Lai Y-H, et al., A novel gene mutatiMedical Association (2014), http://dx.doi.org/10.1016/j.jfma.2014.01
retinoschisis associated with new mutations in the XLRS1 genein Italian families. Br J Ophthalmol 2003;87:1130e4.
18. Vijayasarathy C, Sui R, Zeng Y, Yang G, Xu F, Caruso RC, et al.Molecular mechanisms leading to null-protein product fromretinoschisin (RS1) signal-sequence mutants in X-linked reti-noschisis (XLRS) disease. Hum Mutat 2010;31:1251e60.
19. Xu J, Gu H, Ma K, Liu X, Snellingen T, Sun E, et al. R213Wmutation in the retinoschisis 1 gene causes X-linked juvenileretinoschisis in a large Chinese family. Mol Vis 2010;16:1593e600.
20. Li X, Ma X, Tao Y. Clinical features of X linked juvenile reti-noschisis in Chinese families associated with novel mutations inthe RS1 gene. Mol Vis 2007;13:804e12.
21. Eksandh LC, Ponjavic V, Ayyagari R, Bingham EL, Hiriyanna KT,Andreasson S, et al. Phenotypic expression of juvenile X-linkedretinoschisis in Swedish families with different mutations inthe XLRS1 gene. Arch Ophthalmol 2000;118:1098e104.
22. Riveiro-Alvarez R, Trujillo-Tiebas MJ, Gimenez-Pardo A,Garcia-Hoyos M, Lopez-Martinez MA, Aguirre-Lamban J, et al.Correlation of genetic and clinical findings in Spanish patientswith X-linked juvenile retinoschisis. Invest Ophthalmol Vis Sci2009;50:4342e50.
23. Inoue Y, Yamamoto S, Okada M, Tsujikawa M, Inoue T,Okada AA, et al. X-linked retinoschisis with point mutations inthe XLRS1 gene. Arch Ophthalmol 2000;118:93e6.
24. Sergeev YV, Caruso RC, Meltzer MR, Smaoui N, MacDonald IM,Sieving PA. Molecular modeling of retinoschisin with functionalanalysis of pathogenic mutations from human X-linked reti-noschisis. Hum Mol Genet 2010;19:1302e13.
25. Shinoda K, Ishida S, Oguchi Y, Mashima Y. Clinicalcharacteristics of 14 Japanese patients with X-linked juvenileretinoschisis associated with XLRS1 mutation. OphthalmicGenet 2000;21:171e80.
26. Functional implications of the spectrum of mutations found in234 cases with X-linked juvenile retinoschisis. The Reti-noschisis Consortium. Hum Mol Genet 1998;7:1185e92.
27. Pimenides D, George ND, Yates JR, Bradshaw K, Roberts SA,Moore AT, et al. X-linked retinoschisis: clinical phenotype andRS1 genotype in 86 UK patients. J Med Genet 2005;42:e35.
28. Molday RS, Kellner U, Weber BH. X-linked juvenileretinoschisis: clinical diagnosis, genetic analysis, andmolecular mechanisms. Prog Retin Eye Res 2012;31:195e212.
29. Tantri A, Vrabec TR, Cu-Unjieng A, Frost A, Annesley Jr WH,Donoso LA. X-linked retinoschisis: a clinical and moleculargenetic review. Surv Ophthalmol 2004;49:214e30.
30. Jo YJ, Kim KN, Kim JY. Spontaneous resolution of foveal cystsassociated with X-linked retinoschisis as observed by opticalcoherence tomography. Can J Ophthalmol 2010;45:414e5.
31. Apushkin MA, Fishman GA, Janowicz MJ. Correlation of opticalcoherence tomography findings with visual acuity and macularlesions in patients with X-linked retinoschisis. Ophthalmology2005;112:495e501.
32. Weber BH, Schrewe H, Molday LL, Gehrig A, White KL,Seeliger MW, et al. Inactivation of the murine X-linked juvenileretinoschisis gene, Rs1h, suggests a role of retinoschisin inretinal cell layer organization and synaptic structure. Proc NatlAcad Sci USA 2002;99:6222e7.
33. Alexander KR, Barnes CS, Fishman GA. High-frequencyattenuation of the cone ERG and ON-response deficits in X-linked retinoschisis. Invest Ophthalmol Vis Sci 2001;42:2094e101.
34. Alexander KR, Fishman GA, Grover S. Temporal frequencydeficits in the electroretinogram of the cone system in X-linkedretinoschisis. Vision Res 2000;40:2861e8.
on in a family with X-linked retinoschisis, Journal of the Formosan.001