a bicarbonate bridged diruthenium(i) complex: key evidence for the decarboxylation step in the...
TRANSCRIPT

Inorganica Chimica Acta 372 (2011) 94–99
Contents lists available at ScienceDirect
Inorganica Chimica Acta
journal homepage: www.elsevier .com/locate / ica
A bicarbonate bridged diruthenium(I) complex: Key evidence for thedecarboxylation step in the base-assisted reduction of Ru2Cl4(CO)6
Tapas Ghatak, Arup Sinha, S.M. Wahidur Rahaman, Jitendra K. Bera ⇑Department of Chemistry, Indian Institute of Technology Kanpur, Kanpur 208 016, India
a r t i c l e i n f o a b s t r a c t
Article history:Available online 31 January 2011
Dedicated to Prof. S.S. Krishnamurthy on theoccasion of his 70th birthday.
Keywords:DirutheniumBicarbonateDecarboxylationHydroxycarbonylNaphthyridine
0020-1693/$ - see front matter � 2011 Elsevier B.V. Adoi:10.1016/j.ica.2011.01.067
⇑ Corresponding author.E-mail address: [email protected] (J.K. Bera).
Base-assisted reduction of [Ru(CO)3Cl2]2 in the presence of NP-Me2 (2,7-dimethyl-1,8-naphthyridine) inthf provides an unsupported diruthenium(I) complex [Ru2(CO)4Cl2(NP-Me2)2] (1). Two NP-Me2 and fourcarbonyls bind at equatorial positions and two chlorides occupy sites trans to the Ru–Ru single bond.Reaction of [Ru(CO)3Cl2]2, TlOTf, KOH and NP-Me2 in acetonitrile, in a sealed container, affords a bicar-bonate bridged diruthenium(I) complex [Ru2(CO)2(l-CO)2(l-O2COH)(NP-Me2)2](OTf) (2). The in situ gen-erated CO2 is the source for bicarbonate under basic reaction medium. Isolation of 2 validates thedecarboxylation step in the base-assisted reduction of [RuII(CO)3Cl2]2 ? [RuI
2(CO)4]2+.� 2011 Elsevier B.V. All rights reserved.
1. Introduction complexes of type A, the DFT optimization of a diruthenium–dihy-
The metal–metal singly-bonded diruthenium(I) complex[RuI
2(CO)4(CH3CN)6]2+ has been a useful precursor for advancingthe chemistry of dimetal compounds [1]. Ligand modulation ofthe metal–metal bond [2], C–H bond activation [3], C–C bond for-mation [4] and cyclopropanation reactions [5–7] have been studiedat sites trans to the Ru–Ru bond. Discrete dimers and multinucleararchitectures based on the [RuI
2(CO)4]2+ core are synthesized inlarge numbers [8–17]. Klemperer et al. reported the first synthesisof [RuI
2(CO)4(CH3CN)6][PF6]2 from [Ru(CO)3Cl2]2 in 1993 [18]. Werecently investigated the mechanism and proposed an overall reac-tion given in Eq. (1) [19].
In order to isolate and identify possible intermediates, naph-thyridine based ligands were employed in our investigation. Thebase-assisted reduction involves the initial nucleophilic activationof metal-coordinated carbonyl to hydroxycarbonyl, followed bythermal decarboxylation, and subsequent binuclear reductiveelimination of molecular hydrogen leading to the formation ofthe [RuI
2(CO)4]2+ core (Scheme 1). We offered crucial evidencesin favor of this mechanism which include the isolation and struc-tural characterization of several diruthenium–hydroxycarbonyl
ll rights reserved.
dride species B and the identification of molecular hydrogen as oneof the side products in the reaction [19]. In this present work, weaddressed two follow-up questions: (1) Can paddlewheel‘Ru2(CO)4’ structure be obtained without the deliberate exclusionof chloride? Lavigne’s parallel chemistry afforded mixed chloro/carbonyl ruthenium clusters instead of dimers [20,21]. The objec-tive here is to avoid the use of Tl salt in accessing discrete ‘RuI(-CO)2’ dimer. (2) Although we proposed thermal decarboxylationin the reaction pathway, the evolved CO2 could not be traced inthe basic reaction medium. We made an effort to identify bicar-bonate or related species from the reaction mixture to garner sup-port for the decarboxylation step in the mechanism.
2. Results and discussion
2.1. Synthesis of [Ru2(CO)4Cl2(NP-Me2)2] (1)
We made two important changes in the synthetic protocol out-lined in Eq. (1). The choice of tetrahydrofuran (thf) as the reactionmedium was driven by Lavigne’s isolation of low-valent Ru-chlorocomplexes from the same solvent [22]. Secondly, we employed 2,7-dimethyl-1,8-naphthyridine (NP-Me2) in this work. The paralleldisposition of two nitrogen lone pairs opens up a door for 1,8-naphthyridines to bridge a variety of metal ions [1]. On the con-trary, NP-Me2 forms chelate complexes [23,24] thereby reducingthe possibility of chloride binding at equatorial sites, and thus dis-favoring the formation of multinuclear clusters.
Room temperature treatment of [Ru(CO)3Cl2]2 in thf followedby the addition of ethanolic solution of KOH and NP-Me2 provides

Scheme 1. Reaction course for the formation of the [RuI2(CO)4]2+ core.
Fig. 1. ORTEP diagram (30% probability thermal ellipsoids) of [Ru2(CO)4Cl2(NP-Me2)2] (1) with important atoms labeled. Hydrogen atoms are omitted for the sakeof clarity. Selected bond distances (Å) and angles (�): Ru1–C1 1.844(6), Ru1–N12.169(4), Ru1–Cl1 2.5122(18), Ru1–Ru1 2.8123(11), C1–Ru1–C1 89.2(4), C1–Ru1–N1 165.9(2), N1–Ru1–N1 61.3(2), C1–Ru1–Cl1 94.25(17), N1–Ru1–Cl1 82.99(12),Cl1–Ru1–Ru1 175.62(5), N1–Ru1–Ru1 93.24(11), C1–Ru1–Ru1 88.87(17), C15–N1–
T. Ghatak et al. / Inorganica Chimica Acta 372 (2011) 94–99 95
the diruthenium(I) complex [Ru2(CO)4Cl2(NP-Me2)2] (1) in highyield (80%) (Scheme 2).
The molecular structure of 1, determined by X-ray crystallogra-phy, is provided in Fig. 1 and important metrical parameters arecollected in the corresponding caption. Molecular structure revealsan unbridged diruthenium compound incorporating two NP-Me2,four carbonyls at equatorial sites, and two chlorine atoms at axialsites. Two NP-Me2 ligands are disposed anti to each other as re-vealed in the N1–Ru1–Ru1–N1 dihedral angle 180.0�. Each NP-Me2 chelates the ruthenium atom forming a four member chelatering with N1–Ru1–N1 angle of 61.3(2)�. The molecule has a crystal-lographically imposed C2 symmetry perpendicular to the Ru–Ruvector and a mirror plane passing through the same. The Ru–Rubond length 2.812(1) Å is similar to the other unsupported diruthe-nium(I) compounds [10,18,25,26].
The 1H NMR spectrum of the compound 1 shows two signals forNP protons appearing as doublets at d 8.55 and 7.78 ppm. Themethyl proton signal appears at d 2.36 ppm. The ESI-MS exhibitssignal at m/z 481 which is assigned for [Ru(CO)(NP-Me2)2Cl]+, indi-cating the cleavage of the metal-metal bond under experimentalcondition.
The formation of complex 1 involves the dissociation of the[RuCl2(CO)3]2 in presence of coordinating solvent (thf) to form[Ru(CO)3Cl2(thf)] [27]. It should be noted here that such dissocia-tion was not observed in acetonitrile [19]. Nucleophilic attack ofthe hydroxide to CO leads to the hydroxycarbonyl species [Ru(-CO)2Cl{C(O)OH}(thf)]. The carbonyl carbon center is susceptibleto nucleophilic attack due to feeble back-donation from RuII lead-ing to the instantaneous formation of the Ru–hydroxycarbonyl
Scheme 2. Synthetic scheme for compound 1.
Ru1 94.4(3), C11–N1–Ru1 146.7(4), O1–C1–Ru1 176.5(5), N1–Ru1–Ru1–N1118.6(2), N1–Ru1–Ru1–C1 13.9(2), C1–Ru1–Ru1–C1 90.8(4), N1–Ru1–Ru1–N1 180.
complex. The nature of the bonding changes in going from the p-acceptor CO to r-donor j-C–CO2H making the trans chloride sub-stitutionally labile [28–33]. This prompted us to propose a coord-inatively unsaturated species C (Scheme 3) [21]. Under thereaction condition and in presence of NP-Me2, the hydroxycarbon-yl intermediate undergoes dimerisation to form bis-hydroxycar-bonyl species [Ru2(CO)4(NP-Me2)2(l2-g2-CO2H)2]2+. Subsequentloss of two CO2 by thermal decarboxylation results in a transientbis-hydrido bridged dimer [Ru2(l-H)2(CO)4(NP-Me2)2]2+. Rapidreductive elimination of the H2 and concomitant coordination of
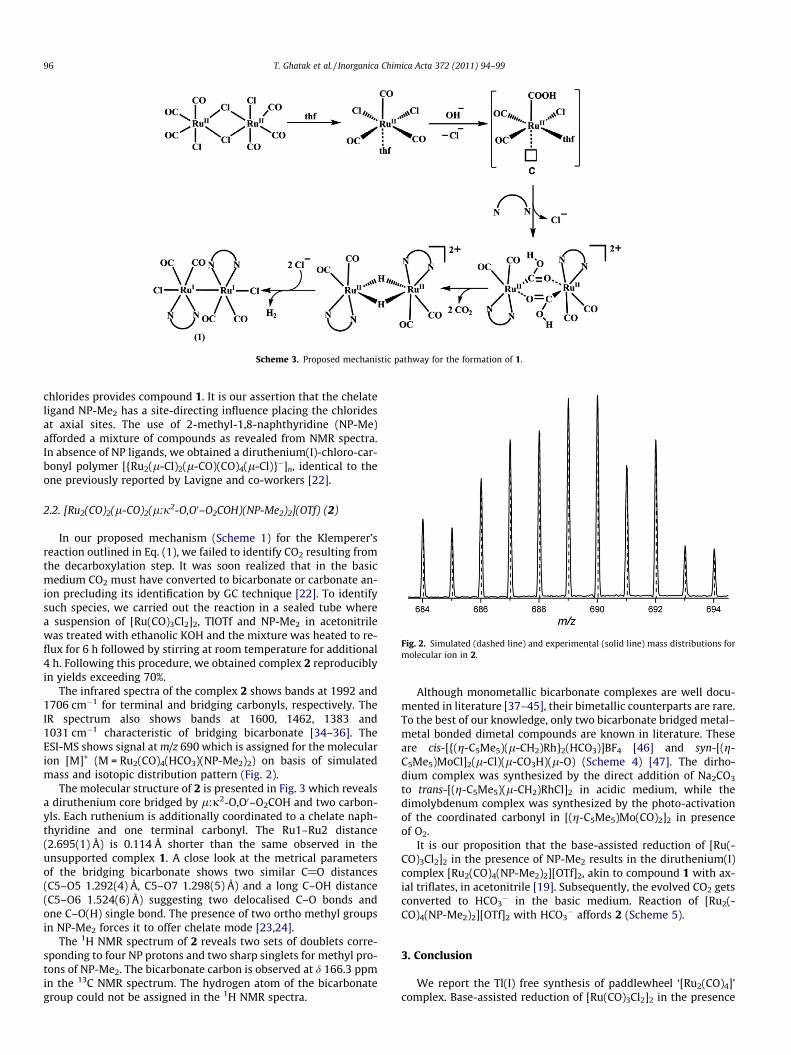
Scheme 3. Proposed mechanistic pathway for the formation of 1.
Fig. 2. Simulated (dashed line) and experimental (solid line) mass distributions formolecular ion in 2.
96 T. Ghatak et al. / Inorganica Chimica Acta 372 (2011) 94–99
chlorides provides compound 1. It is our assertion that the chelateligand NP-Me2 has a site-directing influence placing the chloridesat axial sites. The use of 2-methyl-1,8-naphthyridine (NP-Me)afforded a mixture of compounds as revealed from NMR spectra.In absence of NP ligands, we obtained a diruthenium(I)-chloro-car-bonyl polymer [{Ru2(l-Cl)2(l-CO)(CO)4(l-Cl)}�]n, identical to theone previously reported by Lavigne and co-workers [22].
2.2. [Ru2(CO)2(l-CO)2(l:j2-O,O0–O2COH)(NP-Me2)2](OTf) (2)
In our proposed mechanism (Scheme 1) for the Klemperer’sreaction outlined in Eq. (1), we failed to identify CO2 resulting fromthe decarboxylation step. It was soon realized that in the basicmedium CO2 must have converted to bicarbonate or carbonate an-ion precluding its identification by GC technique [22]. To identifysuch species, we carried out the reaction in a sealed tube wherea suspension of [Ru(CO)3Cl2]2, TlOTf and NP-Me2 in acetonitrilewas treated with ethanolic KOH and the mixture was heated to re-flux for 6 h followed by stirring at room temperature for additional4 h. Following this procedure, we obtained complex 2 reproduciblyin yields exceeding 70%.
The infrared spectra of the complex 2 shows bands at 1992 and1706 cm�1 for terminal and bridging carbonyls, respectively. TheIR spectrum also shows bands at 1600, 1462, 1383 and1031 cm�1 characteristic of bridging bicarbonate [34–36]. TheESI-MS shows signal at m/z 690 which is assigned for the molecularion [M]+ (M = Ru2(CO)4(HCO3)(NP-Me2)2) on basis of simulatedmass and isotopic distribution pattern (Fig. 2).
The molecular structure of 2 is presented in Fig. 3 which revealsa diruthenium core bridged by l:j2-O,O0–O2COH and two carbon-yls. Each ruthenium is additionally coordinated to a chelate naph-thyridine and one terminal carbonyl. The Ru1–Ru2 distance(2.695(1) Å) is 0.114 Å shorter than the same observed in theunsupported complex 1. A close look at the metrical parametersof the bridging bicarbonate shows two similar C@O distances(C5–O5 1.292(4) Å, C5–O7 1.298(5) Å) and a long C–OH distance(C5–O6 1.524(6) Å) suggesting two delocalised C–O bonds andone C–O(H) single bond. The presence of two ortho methyl groupsin NP-Me2 forces it to offer chelate mode [23,24].
The 1H NMR spectrum of 2 reveals two sets of doublets corre-sponding to four NP protons and two sharp singlets for methyl pro-tons of NP-Me2. The bicarbonate carbon is observed at d 166.3 ppmin the 13C NMR spectrum. The hydrogen atom of the bicarbonategroup could not be assigned in the 1H NMR spectra.
Although monometallic bicarbonate complexes are well docu-mented in literature [37–45], their bimetallic counterparts are rare.To the best of our knowledge, only two bicarbonate bridged metal–metal bonded dimetal compounds are known in literature. Theseare cis-[{(g-C5Me5)(l-CH2)Rh}2(HCO3)]BF4 [46] and syn-[(g-C5Me5)MoCl]2(l-Cl)(l-CO3H)(l-O) (Scheme 4) [47]. The dirho-dium complex was synthesized by the direct addition of Na2CO3
to trans-[(g-C5Me5)(l-CH2)RhCl]2 in acidic medium, while thedimolybdenum complex was synthesized by the photo-activationof the coordinated carbonyl in [(g-C5Me5)Mo(CO)2]2 in presenceof O2.
It is our proposition that the base-assisted reduction of [Ru(-CO)3Cl2]2 in the presence of NP-Me2 results in the diruthenium(I)complex [Ru2(CO)4(NP-Me2)2][OTf]2, akin to compound 1 with ax-ial triflates, in acetonitrile [19]. Subsequently, the evolved CO2 getsconverted to HCO3
� in the basic medium. Reaction of [Ru2(-CO)4(NP-Me2)2][OTf]2 with HCO3
� affords 2 (Scheme 5).
3. Conclusion
We report the Tl(I) free synthesis of paddlewheel ‘[Ru2(CO)4]’complex. Base-assisted reduction of [Ru(CO)3Cl2]2 in the presence

Fig. 3. ORTEP diagram (30% probability thermal ellipsoids) of [Ru2(CO)2(l-CO)2(l:j2-O,O0–O2COH)(NP-Me2)2](OTf) (2) with important atoms labeled. Hydrogen atoms,except the hydroxyl hydrogen, are omitted for the sake of clarity. Selected bond distances (Å) and angles (�): Ru1–Ru2 2.6952(6), Ru1–C1 1.872(4), Ru1–C3 2.006(4), Ru1–C42.017(4), Ru1–O5 2.089(3), Ru1–N2 2.244(3), Ru1–N1 2.279(3), Ru2–C2 1.882(4), Ru2–C4 1.998(4), Ru2–C3 2.009(4), Ru2–O7 2.092(3), Ru2–N3 2.243(4), Ru2–N4 2.281(3),C5–O5 1.292(4), C5–O7 1.298(5), C5–O6 1.524(6), C1–Ru1–C3 88.94(18), Ru1–C3–Ru2 84.32(17), Ru2–C4–Ru1 84.35(18), O5–C5–O7 124.7(4), O5–C5–O6 115.2(3), C1–Ru1–C4 90.61(18), C3–Ru1–C4 95.01(17), C1–Ru1–O5 177.97(16), C3–Ru1–O5 90.18(14), C4–Ru1–O5 91.28(14), Ru1–O5–O7–Ru2 0.27(14), C2–Ru2–Ru1–C1 1.53(19), Ru1–C3–Ru2–C4 6.96(17).
Scheme 4. Metal-metal bonded dimetal compounds bridged by bicarbonate ligand[46,47].
T. Ghatak et al. / Inorganica Chimica Acta 372 (2011) 94–99 97
of NP-Me2 in thf provides [Ru2(CO)4Cl2(NP-Me2)2] (1). X-ray struc-ture of 1 reveals an unsupported diruthenium(I) complex with twoequitorial NP-Me2 ligands in anti arrangement and two chlorides ataxial sites. Furthermore, a bicarbonate bridged diruthenium(I)complex [Ru2(CO)2(l-CO)2(l-O2COH)(NP-Me2)2](OTf) (2) is iso-lated in the reaction of [Ru(CO)3Cl2]2, TlOTf, KOH and NP-Me2 inacetonitrile. It is proposed that the base-assisted reduced product[Ru2(CO)4(NP-Me2)2][OTf]2 is formed initially; the in situ generatedCO2 reacts with the diruthenium(I) complex in basic medium(KOH) to afford 2 in high and reproducible yield. The chelate bind-ing of NP-Me2 ligand has major influence on the overall structuresof both compounds.
4. Experimental
4.1. General procedures
All reactions with metal complexes were carried out under anatmosphere of purified nitrogen using standard Schlenk-vesseland vacuum-line techniques. Glassware was flame-dried under avacuum prior to use. 1H NMR spectra were obtained on JEOL
Scheme 5. Schematic representa
JNM-LA 500 MHz spectrometers. 1H NMR chemical shifts were ref-erenced to the residual hydrogen signal of the deuterated solvents.Infrared spectra were recorded in the range 4000–400 cm�1 on aBruker Vertex 70 spectrophotometer with KBr pellets and analyseswere performed on a Thermoquest EA1110 CHNS/O analyzer. Thecrystallized compounds were powdered, washed several timeswith dry diethyl ether and dried in vacuum for at least 48 h priorto elemental analyses.
4.2. Materials
Solvents were dried by conventional methods, distilled undernitrogen and deoxygenated prior to use [48]. RuCl3�xH2O was pur-chased from Arora Matthey India, The compounds [Ru(CO)3Cl2]2
[27] and 2,7-dimethyl-1,8-naphthyridine (NP-Me2) [49] were syn-thesized following the literature procedures.
4.3. Synthesis of [Ru2(CO)4Cl2(NP-Me2)2] (1)
A 1 M solution of KOH in ethanol was added to a suspension of[Ru(CO)3Cl2]2 (41 mg, 0.079 mmol) and NP-Me2 (26 mg,0.17 mmol) in 15 mL tetrahydrofuran and the mixture was stirredfor 12 h at room temperature. The resulting brown solution was fil-tered and concentrated. Diethyl ether was added to induce precip-itation. Repeated washing and prolonged drying under vacuumprovided a brown solid. X-ray quality crystals were grown by lay-ering petroleum ether over a dichloromethane solution of the com-pound. Yield: 45 mg (80%). 1H NMR (500 MHz, CD3OD, 292 K): d8.56–8.54 (d, J = 8.6 Hz, 1H, NP), 7.79–7.77 (d, J = 9.0 Hz, 1H, NP),2.36 (s, 3H, Me). 13C NMR (125 MHz, CD3OD, 294 K): 211.5 (CO),194.1 (CO), 165.2 (NCCNP), 138.4 (NCNNP), 126.3 (CHNP), 124.7(CHNP), 118.7 (CCCNP), 21.7 (CH3). IR (KBr) data (cm�1): m(CO):2034 (s), 1999 (s). ESI-MS, m/z 481 corresponds to [Ru(CO)(NP-Me2)2Cl]+. Anal . Calc. for C24H20N4O4Cl2Ru2: C, 41.03; H, 2.87; N,7.98. Found: C, 40.98; H, 2.73; N, 7.81%.
tion for the formation of 2.

98 T. Ghatak et al. / Inorganica Chimica Acta 372 (2011) 94–99
4.4. Synthesis of [Ru2(CO)2(l-CO)2(l:j2-O5,O7–O2COH)(NP-Me2)2](OTf) (2)
In a closed reaction vessel, a 1 M solution of KOH in ethanol wasadded slowly to a suspension of [Ru(CO)3Cl2]2 (45 mg, 0.088 mmol)in acetonitrile. TlOTf (128 mg, 0.361 mmol) and NP-Me2 (29 mg,0.18 mmol) were added to the yellow solution. All reagents wereadded in a span of 5 min; the mixture was heated at reflux for6 h. The solution was then allowed to cool to room temperatureand the stirring was continued for another 4 h. The white precipi-tate was removed by filtration and the filtrate was collected in aSchlenk flask. Bright yellow solution was concentrated and diethyl-ether was added to it to induce precipitation. Repeated washingfollowed by prolonged drying in vacuum provided a yellow solid.Crystals suitable for X-ray study were grown by layering petro-leum ether over a dichloromethane solution of the compound.Yield: 52 mg (70%) 1H NMR (500 MHz, CD3CN, 292 K): d 8.49–8.47 (d, J = 8.8 Hz, 1H), 8.37–8.35 (d, J = 8.8 Hz, 1H), 7.75–7.74 (d,J = 7.7 Hz, 1H), 7.55–7.53 (d, J = 8.1 Hz, 1H), 2.66 (s, 3H, Me), 2.57(s, 3H, Me). 13C NMR (125 MHz, CD3CN, 294 K): 202.7 (CO), 194.5(CO), 166.3 (CO3H), 157.9 (NCCNP), 138.6 (NCNNP), 126.3 (CHNP),122.4 (CHNP), 119.9 (CCCNP), 22.1 (CH3). IR (KBr) data (cm�1):m(CO): 1991 (s), 1706 (s), m(OCO): 1600 (s), 1462 (m, br), 1382(m), 1031 (s), m(OTf): 1261 (s). ESI-MS, m/z 690 corresponds to[M]+ (M = Ru2(CO)4(HCO3)(NP-Me2)2). Anal . Calc. forC26H21N4O7F3SRu2: C, 37.05; H, 2.51; N, 6.65. Found: C, 36.8; H,2.43; N, 6.50%.
4.5. X-ray data collections and refinement
Single crystal X-ray structural studies were performed on a CCDBruker SMART APEX diffractometer equipped with an OxfordInstruments low-temperature attachment. Data were collected at100(2) K using graphite-monochromated Mo Ka radiation(ka = 0.71073 Å). The frames were indexed, integrated and scaledusing SMART and SAINT software package [50], and the data werecorrected for absorption using the SADABS program [51]. Thestructures were solved and refined using SHELX suite of programs
Table 1Crystallographic data and pertinent refinement parameters for compounds 1 and 2.
1 2
Empirical formula C24H20Cl2N4O4Ru2 C26H21F3N4O10Ru2SFormula weight 701.48 840.67Crystal system monoclinic monoclinicSpace group C2/m P21/ca (Å) 16.624(2) 8.891(2)b (Å) 11.411(2) 13.220(3)c (Å) 8.1954(13) 27.113(7)a (�) 90.00 90.00b (�) 105.959(3) 98.984(4)c (�) 90.00 90.00V (Å3) 1494.7(4) 3147.5(13)Z 2 4qcalc (g cm�3) 1.559 1.774l (mm�1) 1.222 0.906F(0 0 0) 692 1664Reflections collected 6553 27840Independent reflections 1936 7849Observed [I > 2r(I)] 1685 5974Number of variables 89 420Goodness-of-fit (GOF) 1.162 1.103Rint 0.0349 0.0603Final R indices [I > 2r(I)]a R1 = 0.0546 R1 = 0.0481
wR2 = 0.1733 wR2 = 0.1143R indices (all data)a R1 = 0.0640 R1 = 0.0703
wR2 = 0.1976 wR2 = 0.1330
a R1 = R||Fo| � |Fc||/R|Fo| with Fo2 > 2r (Fo
2). wR2 = [Rw(|Fo2| � |Fc
2|)2/R|Fo2|2]1/2.
[52] while additional crystallographic calculations were performedby the programs PLATON [53]. Figures were drawn using ORTEP32[54]. The hydrogen atom of bicarbonate (H6) and rest of the hydro-gen atoms were included into geometrically calculated positions inthe final stages of the refinement and were refined according to‘riding model’. Pertinent details of data collections and final refine-ment parameters for 1 and 2 are collected in Table 1. Allnon-hydrogen atoms were refined with anisotropic thermalparameters. The ‘SQUEEZE’ option in PLATON was used to removea disordered solvent molecule from the overall intensity data ofcompounds 1 and 2. CCDC – 793937 (1) and 793938 (2) containthe supplementary crystallographic data for this paper. These datacan be obtained free of charge from the Cambridge Crystallo-graphic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Appendix A. Supplementary material
Supplementary data associated with this article can be found, inthe online version, at doi:10.1016/j.ica.2011.01.067.
References
[1] J.K. Bera, N. Sadhukhan, M. Majumdar, Eur. J. Inorg. Chem. (2009) 4023.[2] S.K. Patra, N. Sadhukhan, J.K. Bera, Inorg. Chem. 45 (2006) 4007.[3] S.K. Patra, J.K. Bera, Organometallics 25 (2006) 6054.[4] S.K. Patra, J.K. Bera, Organometallics 26 (2007) 2598.[5] G. Maas, Chem. Soc. Rev. 33 (2004) 183.[6] G. Maas, T. Werle, M. Alt, D. Mayer, Tetrahedron 49 (1993) 881.[7] Y. Sevryugina, B. Weaver, J. Hansen, J. Thompson, H.M.L. Davies, M.A.
Petrukhina, Organometallics 27 (2008) 1750.[8] B. Therrien, G. Süss-Fink, Coord. Chem. Rev. 253 (2009) 2639.[9] S.K. Patra, M. Majumdar, J.K. Bera, J. Organomet. Chem. 691 (2006) 4779.
[10] S. Chardon-Noblat, G.H. Cripps, A. Deronzier, J.S. Field, S. Gouws, R.J. Haines, F.Southway, Organometallics 20 (2001) 1668.
[11] G.H. Cripps, A. Pellissier, S. Chardon-Noblat, A. Deronzier, R.J. Haines, J.Organomet. Chem. 689 (2004) 484.
[12] K.-B. Shiu, C.-H. Li, T.-J. Chan, S.-M. Peng, M.-C. Cheng, S.-L. Wang, F.-L. Liao,M.Y. Chiang, Organometallics 14 (1995) 524.
[13] S.J. Sherlock, M. Cowle, E. Singleton, M.M. de V. Steyn, Organometallics 7(1988) 1663;R.W. Hilts, M. Cowie, Inorg. Chem. 29 (1990) 3349.
[14] J.S. Field, R.J. Haines, C.J. Parry, J. Chem. Soc., Dalton Trans. 1 (1997) 2843.[15] K.-B. Shiu, L.-T. Yang, S.-W. Jean, C.-H. Li, R.-R. Wu, J.-C. Wang, S.-L. Liou, M.Y.
Chiang, Inorg. Chem. 35 (1996) 7845.[16] K.-B. Shiu, W.-M. Lee, C.-L. Wang, S.-L. Wang, F.-L. Liao, J.-C. Wang, L.-S. Liou,
S.-M. Peng, G.-H. Lee, M.Y. Chiang, Organometallics 15 (1996) 2979.[17] K.-B. Shiu, S.-L. Wang, F.-L. Liao, M.Y. Chiang, S.-M. Peng, G.-H. Lee, J.-C. Wang,
L.-S. Liou, Organometallics 17 (1998) 1790.[18] W.G. Klemperer, B. Zhong, Inorg. Chem. 32 (1993) 5821.[19] M. Majumdar, A. Sinha, T. Ghatak, S.K. Patra, N. Sadhukhan, S.M.W. Rahaman,
J.K. Bera, Chem. Eur. J. 16 (2010) 2574.[20] G. Lavigne, Eur. J. Inorg. Chem. (1999) 917.[21] M. Faure, L. Maurette, B. Donnadieu, G. Lavigne, Angew. Chem., Int. Ed. 38
(1999) 518.[22] L. Maurette, B. Donnadieu, G. Lavigne, Angew. Chem., Int. Ed. 38 (1999) 3707.[23] J. Kuzelka, J.R. Farrell, S.J. Lippard, Inorg. Chem. 42 (2003) 8652.[24] M. Andrews, R.H. Laye, S.J.A. Pope, Transition Met. Chem. 34 (2009) 493.[25] P. Homanen, M. Haukka, M. Ahlgrén, T.A. Pakkanen, P.N.W. Baxter, R.E.
Benfield, J.A. Connor, J. Organomet. Chem. 552 (1998) 205.[26] .. Haukka, J. Kiviaho, M. Ahlgrén, T.A. Pakkanen, Organometallics 14 (1995)
825.[27] M.I. Bruce, F.G.A. Stone, J. Chem. Soc. A (1967) 1238.[28] D.H. Gibson, B. Sleadd, M.S. Mashuta, J.F. Richardson, Organometallics 16
(1997) 4421.[29] D.L.S. Weaver, Inorg. Chem. 9 (1970) 2250.[30] T.G. Appleton, H.C. Clark, L.E. Manzer, Coord. Chem. Rev. 10 (1973) 335.[31] M.A. Bennett, R.N. Johnson, G.B. Robertson, I.B. Tomkins, P.O. Whimp, J. Am.
Chem. Soc. 98 (1976) 3514.[32] O.P. Anderson, A.B. Packard, Inorg. Chem. 17 (1978) 1333.[33] R. Bardi, A.M. Piazzesi, A. Del Pra, G. Cavinato, L. Toniolo, Inorg. Chim. Acta 102
(1985) 99.[34] D.W. Lee, C.M. Jensen, D.M. Morales, Organometallics 22 (2003) 4744.[35] T. Yoshida, D.L. Thorn, T. Okano, J.A. Ibers, S. Otsuka, J. Am. Chem. Soc. 101
(1979) 4212.[36] A.J. Edwards, S. Elipe, M.A. Esteurelas, F.J. Lahoz, L.A. Oro, C. Valero,
Organometallics 16 (1997) 3828.[37] A. Romero, A. Santos, A. Vegas, Organometallics 7 (1988) 1988.[38] Z.-W. Mao, G. Liehr, R.V. Eldik, J. Am. Chem. Soc. 122 (2000) 4839.[39] C.R. Choudhury, S.K. Dey, S. Mitra, V. Gramlich, Dalton Trans. (2003) 1059.

T. Ghatak et al. / Inorganica Chimica Acta 372 (2011) 94–99 99
[40] M.A. McLoughlin, N.L. Keder, W.T.A. Harrison, R.J. Flesher, H.A. Mayer, W.C.Kaska, Inorg. Chem. 38 (1999) 3223.
[41] R.F.R. Jazzar, P.H. Bhatia, M.F. Mahon, M.K. Whittlesey, Organometallics 22(2003) 670.
[42] S.F. Hossain, K.M. Nichol, J.C.S. Chem. Comm. (1981) 268.[43] L. Broge, I. Søtofte, C.E. Olsen, J. Springborg, Inorg. Chem. 40 (2001) 3124.[44] D.J. Darensbourg, M.W. Holtcamp, B. Khandelwal, J.H. Reibenspie, Inorg. Chem.
34 (1995) 5390.[45] L.L. Graiyiiski, J. Lisowski, J. Am. Chem. In. 109 (1987) 4428.[46] N.J. Meanwell, A.J. Smith, H. Adams, S. Okeya, P.M. Maitlis, Organometallics 2
(1983) 1705.[47] F. Bottomley, J. Chen, Organometallics 11 (1992) 3404.
[48] D.D. Perrin, W.L.F. Armarego, D.R. Perrin, Purification of Laboratory Chemicals,2nd ed., Pergamon Press, 1980.
[49] D.R. Sinha, A.B. Lal, J. Indian Chem. Soc. LVI (1979) 164.[50] SAINT+ Software for CCD difractometers; Bruker AXS: Madison, WI, 2000.[51] G.M. Sheldrick, SADABS Program for Correction of Area Detector Data,
University of Göttingen, Göttingen, Germany, 1999.[52] SHELXTL Package v. 6.10; Bruker AXS: Madison, WI, 2000; G.M. Sheldrick,
SHELXS-86 and SHELXL-97; University of Göttingen: Göttingen, Germany,1997.
[53] L. Spek, PLATON, University of Utrecht, Netherlands, 2001.[54] L.J. Farrugia, J. Appl. Crystallogr. 30 (1997) 565.