608 gender specific predilection of microrna biomarkers for colorectal cancer risk stratification
TRANSCRIPT

AG
AA
bst
ract
swere able to form tumors when injected in NOD-SCID IL2Rγ-/- mice. In contrast, the non-TICs (CD44-/CD166-/ALDH1- cells) were unable to form tumors when injected at lowerthan 100,000 cells. The chemical library screen revealed 5 compounds that selectively targetcolon TICs. Surprisingly, an antagonist against the high affinity substance P, neurokininreceptor-1 (NK-1R) displayed the highest effectiveness (>90%) to suppress the growth ofcolon TICs, without affecting non-TICs. Combinational therapy of the NK-1R antagonisttogether with oxaliplatin suppressed tumor growth and prevented relapse in the colon cancermouse models, while oxaliplatin alone was not effective in preventing disease relapse. Highthroughput phospho-kinome analysis revealed that the NK-1R antagonist acted throughsuppression of the NF-κB, AKT and MAPK signaling in colon TICs. Conclusions: Weidentified a CD44+/CD166+/ALDH1+ colon cancer subpopulation with tumor-initiatingproperties and highly resistant to conventional chemotherapy. We also found that a NK-1R antagonist together with oxaliplatin suppresses selectively their growth In Vitro and InVivo. Acknowledgement: We thank the Translational Pathology Core Laboratory, Departmentof Pathology at UCLA for providing human colon tissues.
605
ADAM17 Mediates EGFR Transactivation by Sdf-1α in Colon Cancer Cellsand is Regulated by miR-145Reba Mustafi, Michelle Fletcher, Urszula Dougherty, Joel Pekow, Loren J. Joseph,Alessandro Fichera, Marc Bissonnette
Background - ADAM17, a member of “a desintegrin and metalloprotease” family, controlsepidermal growth factor receptor (EGFR) signals by releasing EGFR ligands from membranebound proforms. As a result, ADAM17 serves as a central convergence point for manyreceptor tyrosine kinases and G-protein coupled receptors (GPCR) that transactivate EGFRby inducing ADAM17-mediated EGFR ligand release. The cytokine stromal cell-derivedfactor-1alpha (sdf-1α) is induced by pro-inflammatory stimuli and up-regulated in severalcancers. Sdf-1α activates leukocytes and epithelial cells via CXCR4, a GPCR. In preliminarystudies we showed that sdf-1α transcripts were significantly up-regulated by a Western diet(Dougherty et al, DDW 2012). Since the effects of Western diet on colonic tumor promotionrequire EGFR, we asked if sdf-1α transactivated EGFR in colon cancer cells. As a secondgoal to gain insights into ADAM17 regulation in colonic tumorigenesis, we examinedADAM17 control by miRNAs. ADAM17 is a bioinformatics predicted target of microRNA-145 (miR-145), miR-148, and miR-152, which are down-regulated in colon cancer.MethodsHCT116 colon cancer cells were pretreated for 2 hrs with vehicle or 5 μM INCB3619, anADAM17-specific inhibitor. Cells were then treated with 10 ng/ml sdf-1α or vehicle for 5min. EGFR transactivation was assessed by Western blotting using antibodies specific forphospho-active ErbB2 and pERK. For miRNA studies, HCT116 cells were transfected witha luciferase reporter regulated by the ADAM17-3'UTR and co-transfected with putativemiRNA regulators or (scrambled) control oligonucleotides. We also mutated ADAM17-3'UTR, abolishing the miR-145 target sequence to examine the effects of miR-145 on mutantADAM17-3'UTR. Luciferase expression was measured 48 hrs after transfection. Results -Sdf-1α induced EGFR signals and this (trans) activation appeared to require ADAM17, asINCB3619 caused 50±6%(n=6, p<0.05) reduction in EGFR signals. In HCT116 cells, trans-fectedmiRNAs predicted to target ADAM17 significantly reduced ADAM-17-3'UTR-luciferaseactivity (fold of scrambled): miR-145, 0.5±0.1-fold control (p<0.05); miR-148, 0.7±0.2-fold control (p<0.05); miR-152: 0.6±0.1-fold control (p<0.05). For miR-145, luciferasesuppression was abrogated by mutation of ADAM17-3'UTR, indicating a direct effect ofmiR-145 on ADAM17. Summary - Sdf-1α transactivates EGFR and we postulate this involvesan ADAM17-dependent mechanism. Furthermore, ADAM17 is negatively regulated by miR-145, miR-148 and miR-152. Since these miRNAs are down-regulated in colon cancer, wespeculate that their loss contributes to up-regulation of ADAM17 and increased EGFRtransactivation by Sdf-1α.
606
Heterozygous Knockout of β-Catenin in Apc1638n Mice PreventsGastrointestinal Tumor Formation but Predisposes for Mammary LesionsElvira R. Bakker, Elmer Hoekstra, Patrick Franken, Werner Helvensteijn, Wendy vanVeelen, Ernst J. Kuipers, Ron Smits
Apc-driven tumor formation in patients and Apc mutant mouse models is generally attributedto increased levels of β-catenin signaling. We and others have proposed that a specific levelof β-catenin signaling is required to successfully initiate tumor formation, and that eachtissue selects for different dosages of signaling. This is nicely illustrated by the differenttumor phenotypes displayed by different Apc mutant mouse models. ApcMin mice haverelatively high levels of β-catenin signaling and develop intestinal tumors at high multiplicity(>100). Animals carrying the hypomorphic Apc1638Nmutation, associatedwith intermediateβ-catenin signaling, characteristically develop intestinal tumors at lower multiplicity (<10)and in parallel show a high susceptibility for extra-intestinal tumor types such as cutaneouscysts and desmoid tumors. The Apc1572T mouse model, associated with lower levels of β-catenin signaling, is free of intestinal tumors but instead develops mammary tumors withhigh penetrance. This indicates that intestinal tumors are associated with higher levels ofβ-catenin signaling than cysts and desmoids, which in turn are associated with higher β-catenin signaling than mammary tumors. Although the concept of β-catenin signaling dosageand its impact on tumor growth among tissues is gaining acceptance, it has not been formallyproven. In addition, alternative explanations for Apc-driven tumor formation have beenproposed. To obtain direct evidence that different tumor phenotypes are the result of differentβ-catenin levels, we crossed Apc1638N mice with heterozygous β-catenin knockout mice,thereby reducing their β-catenin levels. All Apc1638N control mice (n=19) developed gastro-intestinal tumors (3.1 ± 1.5 per animal). In stark contrast, all Apc1638Nmice with heterozyg-ous knockout of β-catenin (n=21) revealed complete absence of gastrointestinal tumors.Incidence of other Apc1638N-associated lesions was strongly reduced as well, includingdesmoids (8.6 ± 3.0 to 0.2 ± 0.4 in females, 61.4 ± 14.4 to 19.1 ± 8.1 in males) and cysts(5.6 ± 3.8 to 0.4 ± 0.6 in females, 29.8 ± 19.5 to 2.4 ± 1.5 in males). Interestingly, 7 outof 14 female mice with heterozygous β-catenin knockout now developed mammary lesions(1-2 per mouse), which are rarely observed in Apc1638N mice. Taken together, reducing
S-118AGA Abstracts
β-catenin levels in Apc1638N phenocopies tumor predisposition typically observed inApc1572T mice. We hereby provide In Vivo evidence that by simply reducing β-cateninlevels, tumors develop in different tissues. This clearly demonstrates the central role of β-catenin dosage to dictate tissue-specific tumor predisposition in the setting of Apc-drivencancer.
607
Ubiquitin-Conjugating Enzyme 9-Methionine Adenosyltransferase 2A-Bcl2Axis Regulates Apoptosis in Liver and Colon Cancer CellsMaria Lauda Tomasi, Anna Skay, Ivan Tomasi, Pasquale Giordano, Shelly C. Lu
BACKGROUND & AIMS: Ubiquitin-conjugating enzyme 9 (Ubc9) is required for sumoyl-ation and inhibits apoptosis by a Bcl-2-dependent pathway in breast cancer cells but themolecular mechanism is unknown. Both Ubc9 and Bcl2 are overexpressed in colon cancerand protect against apoptosis. S-adenosylmethionine (SAMe) is the methyl donor synthesizedby methionine adenosyltransferase (MAT). MATII, encoded by MAT2A, is overexpressed inliver and colon cancers. SAMe treatment decreases MAT2A expression and is pro-apoptoticin cancer cells. Knockdown of MAT2A is also pro-apoptotic in liver and colon cancer cells.Here we examined whether Ubc9, MAT2A and Bcl2 interact to affect apoptosis.METHODS:Our studies used human colon and liver cancer cell lines RKO and Huh-7, respectively,and liver and colon cancer specimens. Real-time PCR and Western blotting measured geneand protein expression, respectively. Bcl2 promoter activity and protein binding to the Bcl2promoter region were measured using reporter assay and chromatin immunoprecipitation(ChIP) analysis, respectively. MATII sumoylation was examined using anti-SUMO-1 antibodyfor immunoprecipitation followed by Western blotting for MATII. Expression of MAT2A,Ubc9 and SUMO-1 was reduced using specific siRNAs. RESULTS: MAT2A, Bcl2 and Ubc9are all overexpressed in liver and colon cancer specimens. MATII is present in both cytosoland nuclei of liver and colon cancer cells. SAMe and its metabolite methylthioadenosine(MTA) treatment lowered Bcl-2 expression at mRNA and protein levels by 55-80% andinduced apoptosis in Huh-7 and RKO cells. MAT2A protein sequence contains possiblesumoylation sites and this was confirmed using anti-SUMO-1 antibody followed by Westernblotting for MATII. Knockdown of Ubc9 or SUMO-1 reduced sumoylated MATII and totalMAT2A protein (but not mRNA) levels by 25%. Knockdown of MAT2A reduced Bcl-2mRNA level and Bcl-2 promoter activity by 50-65%, while MAT2A overexpression doubledBcl2 promoter activity in both cell types. Chip assay showed that MATII could physicallyinteract with Bcl-2 promoter and this was abolished when Ubc9 is knocked down.CONCLU-SIONS: MATII is sumoylated and this increases protein stability. MATII binds to the Bcl-2 promoter region and this requires Ubc9. MATII acts as a positive regulator of Bcl2 at thetranscriptional level. The ability of MATII to act as a trans-activating factor has neverbeen reported before. Taken together, Ubc9 enhances MATII expression likely throughsumoylation, which in turn activates Bcl2 expression to protect against apoptosis. This is anovel axis that can be targeted for cancer therapy. This work was supported by NIH grantsR01DK51719, R01AT1576 and R01AT004896 (SC Lu), and F32AA020150 (ML Tomasi).Huh-7 and RKO cells were provided by the Cell Culture Core of the USC Research Centerfor Liver Diseases (P30DK48522).
608
Gender Specific Predilection of MicroRNA Biomarkers for Colorectal CancerRisk StratificationDhananjay Kunte, Ramesh K. Wali, Mart DeLaCruz, Wentao Qi, Amir C. Patel, YolandaE. Stypula, Tina P. Gibson, Hemant K. Roy
Background: Despite having profoundly different epidemiology (later age of onset, proximaldistribution) and biology (more MSI-high disease), human colorectal cancer (CRC) screeningrecommendations are identical for men and women. However, we have noted that advancedadenomas appear to have a marked gender preference (JAMA 2009, Can Prev Res 2010).Emerging evidence indicates that microRNAs (miRNAs) are powerful biomarkers of CRC.Our group has focused on use of biomarkers to detect field carcinogenesis and hence achieveCRC risk stratification. We therefore wanted to assess if miRNA biomarkers have significantdifferences between females and males using the well-validated azoxymethane (AOM)-treatedrat model. Methods: Male and female Fisher 344 rats were given two weekly i.p. injectionsof AOM (15 mg/Kg) and were euthanized after 40 wks. At a 16 wk time point, colonicbiopsies were obtained through colonoscopy. Total RNA from the colonic mucosa andbiopsies was isolated using Ribopure kit (Ambion) and reverse transcribed using TaqmanmiRNA RT kit using rodent Megaplex RT Primers (Applied Biosystems). Real time PCR wasperformed with Taqman Low Density Arrays (TLDA) using ABI 7900HT system. Expressionof miR-34a was assessed using Taqman miRNA assay. Data analysis was done using SDSRQ Manager and Realtime Statminer software. Results: The comparative analysis of themiRNA profiles of the uninvolved colonic mucosa from the male and female AOM ratsshowed significant differences. In male rats, 65miRNAs were significantly modulated wherein62 were upregulated and 3 were downregulated. In comparison, in female AOM rats showeddysregulation of a total of 38 miRNAs, out of which 32 were significantly upregulated and6 were downregulated. We also noted that the male and female AOM rats only six miRNAsin common, viz., miR-34a, miR142-5p, miR450a-5p, miR-802, miR-20b-3p and miR-547.At a premalignant time point, we analyzed the expression of miR-34a (induced in bothgenders) and correlated to the presence or absence of tumors in the male and female AOMrats. We noted that miR-34a induction was significantly more in the females with tumorsas compared to those without tumors. In male AOM rats, we did not find any correlationwith the miR-34a induction and the size of the tumor. The higher miRNA modulation inthe male AOM rats corresponded with higher (89%) tumor incidence and tumor burdencompared to 50% in females. Conclusions: We demonstrate that miRNA are powerfulmarkers of field carcinogenesis and thus may be useful for personalization of CRC screeningstrategies. Importantly, we demonstrate for the first time that miRNA dysregulation wasprofoundly different between female andmale. This underscores the necessity to test biomark-ers in a gender-specific fashion not simply for microRNAs but all biomarkers.
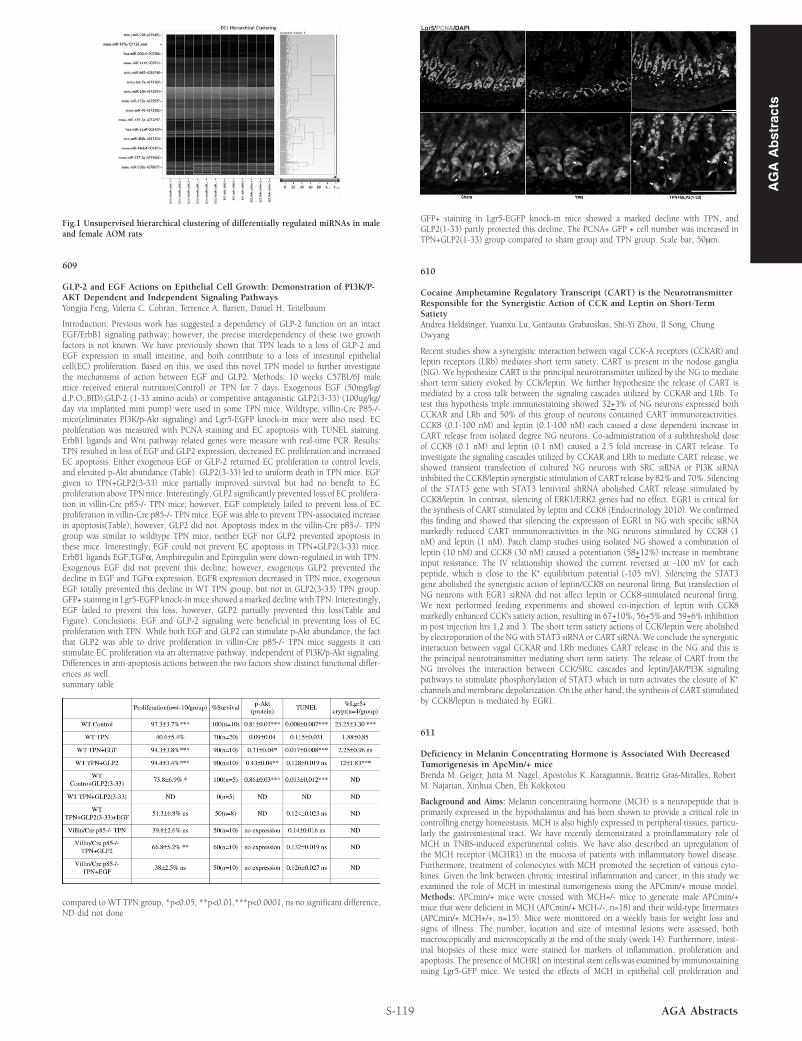
Fig.1 Unsupervised hierarchical clustering of differentially regulated miRNAs in maleand female AOM rats
609
GLP-2 and EGF Actions on Epithelial Cell Growth: Demonstration of PI3K/P-AKT Dependent and Independent Signaling PathwaysYongjia Feng, Valeria C. Cohran, Terrence A. Barrett, Daniel H. Teitelbaum
Introduction: Previous work has suggested a dependency of GLP-2 function on an intactEGF/ErbB1 signaling pathway; however, the precise interdependency of these two growthfactors is not known. We have previously shown that TPN leads to a loss of GLP-2 andEGF expression in small intestine, and both contribute to a loss of intestinal epithelialcell(EC) proliferation. Based on this, we used this novel TPN model to further investigatethe mechanisms of action between EGF and GLP2. Methods: 10 weeks C57BL/6J malemice received enteral nutrition(Control) or TPN for 7 days. Exogenous EGF (50mg/kg/d,P.O.,BID),GLP-2 (1-33 amino acids) or competitive antagonistic GLP2(3-33) (100ug/kg/day via implanted mini pump) were used in some TPN mice. Wildtype, villin-Cre P85-/-mice(eliminates PI3K/p-Akt signaling) and Lgr5-EGFP knock-in mice were also used. ECproliferation was measured with PCNA staining and EC apoptosis with TUNEL staining.ErbB1 ligands and Wnt pathway related genes were measure with real-time PCR. Results:TPN resulted in loss of EGF and GLP2 expression, decreased EC proliferation and increasedEC apoptosis. Either exogenous EGF or GLP-2 returned EC proliferation to control levels,and elevated p-Akt abundance (Table). GLP2(3-33) led to uniform death in TPN mice. EGFgiven to TPN+GLP2(3-33) mice partially improved survival but had no benefit to ECproliferation above TPNmice. Interestingly, GLP2 significantly prevented loss of EC prolifera-tion in villin-Cre p85-/- TPN mice; however, EGF completely failed to prevent loss of ECproliferation in villin-Cre p85-/- TPN mice. EGF was able to prevent TPN-associated increasein apoptosis(Table); however, GLP2 did not. Apoptosis index in the villin-Cre p85-/- TPNgroup was similar to wildtype TPN mice, neither EGF nor GLP2 prevented apoptosis inthese mice. Interestingly, EGF could not prevent EC apoptosis in TPN+GLP2(3-33) mice.ErbB1 ligands EGF,TGFα, Amphiregulin and Epiregulin were down-regulated in with TPN.Exogenous EGF did not prevent this decline; however, exogenous GLP2 prevented thedecline in EGF and TGFα expression. EGFR expression decreased in TPN mice, exogenousEGF totally prevented this decline in WT TPN group, but not in GLP2(3-33) TPN group.GFP+ staining in Lgr5-EGFP knock-inmice showed amarked decline with TPN. Interestingly,EGF failed to prevent this loss, however, GLP2 partially prevented this loss(Table andFigure). Conclusions: EGF and GLP-2 signaling were beneficial in preventing loss of ECproliferation with TPN. While both EGF and GLP2 can stimulate p-Akt abundance, the factthat GLP2 was able to drive proliferation in villin-Cre p85-/- TPN mice suggests it canstimulate EC proliferation via an alternative pathway, independent of PI3K/p-Akt signaling.Differences in anti-apoptosis actions between the two factors show distinct functional differ-ences as well.summary table
compared to WT TPN group, *p<0.05, **p<0.01,***p<0.0001, ns no significant difference,ND did not done
S-119 AGA Abstracts
GFP+ staining in Lgr5-EGFP knock-in mice showed a marked decline with TPN, andGLP2(1-33) partly protected this decline. The PCNA+ GFP + cell number was increased inTPN+GLP2(1-33) group compared to sham group and TPN group. Scale bar, 50μm.
610
Cocaine Amphetamine Regulatory Transcript (CART) is the NeurotransmitterResponsible for the Synergistic Action of CCK and Leptin on Short-TermSatietyAndrea Heldsinger, Yuanxu Lu, Gintautas Grabauskas, Shi-Yi Zhou, Il Song, ChungOwyang
Recent studies show a synergistic interaction between vagal CCK-A receptors (CCKAR) andleptin receptors (LRb) mediates short term satiety. CART is present in the nodose ganglia(NG). We hypothesize CART is the principal neurotransmitter utilized by the NG to mediateshort term satiety evoked by CCK/leptin. We further hypothesize the release of CART ismediated by a cross talk between the signaling cascades utilized by CCKAR and LRb. Totest this hypothesis triple immunostaining showed 32+3% of NG neurons expressed bothCCKAR and LRb and 50% of this group of neurons contained CART immunoreactivities.CCK8 (0.1-100 nM) and leptin (0.1-100 nM) each caused a dose dependent increase inCART release from isolated degree NG neurons. Co-administration of a subthreshold doseof CCK8 (0.1 nM) and leptin (0.1 nM) caused a 2.5 fold increase in CART release. Toinvestigate the signaling cascades utilized by CCKAR and LRb to mediate CART release, weshowed transient transfection of cultured NG neurons with SRC siRNA or PI3K siRNAinhibited the CCK8/leptin synergistic stimulation of CART release by 82% and 70%. Silencingof the STAT3 gene with STAT3 lentiviral shRNA abolished CART release stimulated byCCK8/leptin. In contrast, silencing of ERK1/ERK2 genes had no effect. EGR1 is critical forthe synthesis of CART stimulated by leptin and CCK8 (Endocrinology 2010). We confirmedthis finding and showed that silencing the expression of EGR1 in NG with specific siRNAmarkedly reduced CART immunoreactivities in the NG neurons stimulated by CCK8 (1nM) and leptin (1 nM). Patch clamp studies using isolated NG showed a combination ofleptin (10 nM) and CCK8 (30 nM) caused a potentiation (58+12%) increase in membraneinput resistance. The IV relationship showed the current reversed at -100 mV for eachpeptide, which is close to the K+ equilibrium potential (-105 mV). Silencing the STAT3gene abolished the synergistic action of leptin/CCK8 on neuronal firing. But transfection ofNG neurons with EGR1 siRNA did not affect leptin or CCK8-stimulated neuronal firing.We next performed feeding experiments and showed co-injection of leptin with CCK8markedly enhanced CCK's satiety action, resulting in 67+10%, 56+5% and 59+6% inhibitionin post injection hrs 1,2 and 3. The short term satiety actions of CCK/leptin were abolishedby electroporation of the NGwith STAT3 siRNA or CART siRNA.We conclude the synergisticinteraction between vagal CCKAR and LRb mediates CART release in the NG and this isthe principal neurotransmitter mediating short term satiety. The release of CART from theNG involves the interaction between CCK/SRC cascades and leptin/JAK/PI3K signalingpathways to stimulate phosphorylation of STAT3 which in turn activates the closure of K+
channels and membrane depolarization. On the other hand, the synthesis of CART stimulatedby CCK8/leptin is mediated by EGR1.
611
Deficiency in Melanin Concentrating Hormone is Associated With DecreasedTumorigenesis in ApcMin/+ miceBrenda M. Geiger, Jutta M. Nagel, Apostolos K. Karagiannis, Beatriz Gras-Miralles, RobertM. Najarian, Xinhua Chen, Efi Kokkotou
Background and Aims: Melanin concentrating hormone (MCH) is a neuropeptide that isprimarily expressed in the hypothalamus and has been shown to provide a critical role incontrolling energy homeostasis. MCH is also highly expressed in peripheral tissues, particu-larly the gastrointestinal tract. We have recently demonstrated a proinflammatory role ofMCH in TNBS-induced experimental colitis. We have also described an upregulation ofthe MCH receptor (MCHR1) in the mucosa of patients with inflammatory bowel disease.Furthermore, treatment of colonocytes with MCH promoted the secretion of various cyto-kines. Given the link between chronic intestinal inflammation and cancer, in this study weexamined the role of MCH in intestinal tumorigenesis using the APCmin/+ mouse model.Methods: APCmin/+ mice were crossed with MCH+/- mice to generate male APCmin/+mice that were deficient in MCH (APCmin/+ MCH-/-, n=18) and their wild-type littermates(APCmin/+ MCH+/+, n=15). Mice were monitored on a weekly basis for weight loss andsigns of illness. The number, location and size of intestinal lesions were assessed, bothmacroscopically and microscopically at the end of the study (week 14). Furthermore, intest-inal biopsies of these mice were stained for markers of inflammation, proliferation andapoptosis. The presence of MCHR1 on intestinal stem cells was examined by immunostainingusing Lgr5-GFP mice. We tested the effects of MCH in epithelial cell proliferation and
AG
AA
bst
ract
s