6 haematology - welcome to drsarma and erythropoietin. clinical assessment a clinical assessment...
TRANSCRIPT

6.1 Background 255
6.2 Red cell disorders 259
6.3 White cell disorders 267
6.4 Platelet disorders 273
6.5 Coagulation disorders 275
6.6 Disseminated intravascular coagulation 279
Self-assessment: questions 280
Self-assessment: answers 284
6.1 Background
This chapter is structured around disorders of:
● red cells● white cells● platelets● coagulation.
Remember, however, that individual diseases comm-only affect more than one of them.
Physiology
The marrow is a large organ, approaching the size of theliver. In adults, most of it is in the flat bones, includingthe sternum, pelvis and vertebrae. White blood cell pre-cursors form 75% of the marrow and most of the restconsists of erythroid precursors. Megakaryocytes (fromwhich platelets are formed) are scattered throughout. Itmay seem surprising that so much of the marrow isdevoted to the white cell series, given that there are 500times as many red cells as white cells in the circulation.However, erythrocytes have a mean life of 120 dayswhereas white cells have a circulating lifespan meas-ured in hours. Even in health, marrow is an extremelyactive tissue which is able to respond to sudden stresseslike haemorrhage and infection. All blood cells arederived from multipotent, uncommitted stem cells.These differentiate into the lines of committed stem cellsfrom which red cells, platelets, monocytes, granulocytesand lymphocytes are formed. The processes of differen-tiation and proliferation are controlled by growthfactors, including interleukins, colony-stimulatingfactors and erythropoietin.
Clinical assessment
A clinical assessment begins as the patient walks intothe room or you go to the bedside:
● look for pallor and signs of bruising/bleeding● is the patient unkempt or thin and is there a smell of
alcohol?● take note of racial origin and gender.
HistorySpecific symptomsAnaemia. The symptoms include:
255
Haematology6
Learning objectives
You should:
● know what to ask about in the haematological history andwhat to look for on examination
● know when to measure and how to interpret a full bloodcount, film, differential white count, platelet count anderythrocyte sedimentation rate (ESR)
● understand the other main haematological investigationsand when to carry them out
—haematinics: iron and total iron-binding capacity, ferritin,vitamin B12, folate and red cell folate
—coagulation tests: international normalised ratio (INR),prothrombin time (PT), activated partial thromboplastin time(APTT), plasma fibrinogen, fibrin degradation products.
● know the indications for, and the information that can begained from bone marrow examination and lymph nodebiopsy.
● know which situations commonly confront a house officerand understand how to manage them
● know enough about the other major haematologicaldiseases to recognise them, make appropriate and timelyreferrals and explain them to your patients.

● tiredness● breathlessness● chest pain● ankle swelling.
Tiredness is thought of as the classical symptom ofanaemia but most tired people are not anaemic andsome patients (particularly elderly ones) have fewsymptoms despite profound anaemia. This is usuallybecause the anaemia has developed slowly and theirlifestyle does not make heavy demands for oxygen.Apart from tiredness, impaired oxygen delivery causesbreathlessness, light-headedness and faints. It worsensangina and can precipitate heart failure, especially if it issuperimposed on coronary artery disease.
Red cell excess (polycythaemia). The symptomatology iscovered on page 266.
Leucopenia. The symptoms of leucopenia include:
● mouth ulceration● infective symptoms.
Mouth ulceration must always be taken seriously in anat-risk patient. Infective symptoms occur with bothneutropenia and lymphopenia.
White cell excess (leukaemia and lymphoma). The symp-tomatology is relatively specific to individual diseases,which are covered on pages 267–273.
Platelet/coagulation disease. Symptoms include:
● easy bruising● bleeding● thromboses.
Thrombocytopenia causes petechial haemorrhages andbruising. When it is severe, it also causes bleeding fromthe gums or into the gut. Impaired coagulation morecommonly causes bleeding into soft tissues than overtexternal bleeding. Haemophilia may cause prolongedbleeding from minor injuries and surgery but often pres-ents with bleeding into joints. Other hereditary clottingdisorders are rare, apart from von Willebrand’s disease,which most often causes skin and mucous membranebleeding because the abnormality results in a defect ofplatelet function. Remember to ask about menorrhagiaand bleeding problems after dental extractions and sur-gery. Thrombophilia (increased tendency to clot) causesrecurrent venous and arterial thromboses.
General historyYou should take a detailed drug history from everypatient. Remember not just the prescription drugs butalso medicines bought over the counter. Take a familyhistory if the patient has haemolytic anaemia, a bleedingtendency or thrombophilia and construct a family tree ifit is positive. Take a dietary and alcohol history. Notethe length of history and take particular note if thesymptoms suggest that more than one aspect of
haematological function is affected (e.g. symptoms ofinfection, bruising and anaemia). Be alert to symptomsof systemic disease, such as weight loss, dyspepsia,change in bowel habit, night sweats and pruritus.
Examination
Your examination (Fig. 64) should include
● a thorough and systematic examination of the skin● careful palpation of all groups of lymph nodes
(including deep palpation of the abdomen)● inspection of the conjunctivae and mouth● palpation of the spleen and liver● a thorough search for signs of infection.
Red cell disordersThe physical signs of anaemia are extremelysubjective. Examine the conjunctivae, which are lessaffected by variations in capillary blood flow than theskin or nail beds. Definite conjunctival pallor is
256
Haematology6
Polycythaemia:
Face: plethoricConjunctivae: suffused
Lymphoma/leukaemia:
LymphadenopathyPruritis (scratch marks)
HepatomegalySplenomegaly
Anaemia:
Conjunctivae: pallorSclerae: jaundice
Mouth: smooth tongue
Thrombocytopenia:
Fundi: haemorrhagesGeneral examination:
signs of bleedingSkin: bruises
petechiae
Neutropenia:
Mouth: ulcersPerineum
Perianal regionGeneral examination
Haemophilia:
Joints: haemarthroses
Signs ofinfection
Fig. 64 Physical signs of haematological disease. Uncommonsigns are omitted.

relatively specific for moderate-to-severe anaemia.Examine the sclerae carefully for jaundice (notdetectable until serum bilirubin is raised to three timesnormal); it is present in haemolytic but not other typesof anaemia. Look at the patient’s face: a plethoriccomplexion with suffusion of the conjunctivae may bea sign of polycythaemia. Examine the abdomen forsplenomegaly, which is a prominent feature of chronicextravascular haemolysis, and is a key finding inpolycythaemia vera.
White cell disordersNeutropenia should be considered in any patient withsevere oral ulceration/candidiasis. Examination forlymphadenopathy requires skill, not just in knowingwhere and how to palpate but in interpreting the find-ings. If there are palpable lymph nodes, note:
● whether they are hard, firm or soft● whether they are tender or non-tender● their approximate size● whether they are mobile and discrete or matted
together.
Axillary and inguinal lymphadenopathy is commonlyfound in normal people, particularly if they have a his-tory of injury or infection. Pathological lymph nodes arelarger, present in the neck and epitrochlear region of theelbow and sometimes coalescent or matted. Lymphnodes that are enlarged because of infection are smallerthan those of lymphoma (usually less than 2 cm) andmore likely to be discrete. The distribution of lymph-adenopathy in Hodgkin’s and non-Hodgkin’s lymph-oma (NHL) is considered on page 271. Assess the size ofthe spleen and liver and search for para-aortic andpelvic nodes. Mild splenomegaly may be caused by aninfection but moderate or massive splenomegaly sug-gests lymphoma, myelofibrosis or chronic leukaemia.Hepatomegaly may be found in myeloproliferativediseases. Tissue infiltration (particularly of the gums)occurs in some leukaemias.
Platelet and coagulation disordersPetechial haemorrhages are a sign of thrombocytope-nia. They are characteristically seen first over thelower legs, where capillary pressure is highest. Laterthey become generalised, and may coalesce as bruises(ecchymoses); particularly over the arms. Inspect thegums for bleeding and the fundi for retinal haemor-rhages, which should be taken very seriously as a signof impending cerebral haemorrhage. Joint swellingcaused by bleeding (haemarthrosis) is an importantfeature of haemophilia and other coagulationdisorders.
Thrombocytosis and other hypercoagulable stateshave no specific physical signs other than those ofarterial or venous thrombosis.
Investigation
There are a large number of investigations in haematol-ogy, some routine and some for more specialised param-eters. Box 10 lists the more common tests and theabbreviations for these tests.
Blood count and film
When you take blood, you can affect the result by:
● excessive cuffing: a prolonged increase in venouspressure artificially increases the haematocrit
● being slow to put blood in the tube or not mixing itthoroughly: coagulation in the tube gives a falselylow platelet count and abnormal coagulation results.
Automated counters measure concentrations (per unitvolume) of red cells, haemoglobin, platelets andleucocytes (lymphocytes, monocytes, neutrophils,eosinophils and basophils). They also measure the per-centage of red cells present as reticulocytes (newlyreleased cells) and calculate indices which describe thesize of red cells and platelets and the degree of haemo-globinisation. Normal values for adults are shown inTable 1 (p. 7). In addition, a dried and stained blood filmcan be inspected to assess the morphology of the cellsand detect abnormal cells. Some of the terms you willcome across on blood film reports are given in Table 60.
257
6Background
Blood count and filmFull blood count; red cell count (RCC), white cell count(WCC)Blood filmDifferential white cell countPlatelet countErythrocyte sedimentation rate (ESR)Mean cell volume (MCV)Red cell width distribution (RDW)
HaematinicsIron and total iron-binding capacityFerritinVitamin B12Folate and red cell folate
CoagulationInternational normalised ratio (INR)Prothrombin time (PT)Activated partial thromboplastin time (APTT)Plasma fibrinogenFibrin degradation products (FDPs)D-dimer
Bone marrowLymph node biopsy
Box 10 Investigations in haematology

Interpreting the haemoglobin and red cell indicesAnaemia is diagnosed when there is a reduction in theconcentration of haemoglobin caused by a reducednumber of red cells or a reduced amount of haemo-globin within the cells. An increase in haemoglobin con-centration resulting from an increased number of redcells per unit volume is termed erythrocytosis.
Whenever you are told that a patient is anaemic, youshould ask for the mean cell volume (MCV), by whichanaemias are classified (explained further on page 259).The cell counter also tells you how similar the red cellsare to one another in size. This is expressed as the redcell distribution width (RDW). A low RDW signifies anormal, homogeneous population of cells. An increasedRDW signifies heterogeneity in cell size as a result of:
● active haemopoiesis, because immature red cells(reticulocytes) are large
● treatment of iron deficiency, resulting in a mixture ofmicrocytic and normocytic cells
● mixed deficiency, giving both small and large‘diseased’ cells.
Reticulocytes can be counted automatically and areexpressed as a percentage of the total red cell count.
White cells and plateletsChanges in cell counts are diagnosed by the automatedcounter. Examination of the film gives an accurate dif-ferential count and detects cells such as leukaemic blastcells. It also detects morphological changes such as thedegree of lobulation of the neutrophil nuclei, whichdepends on their age. In infection, neutrophils are rela-tively immature and may appear as bands rather thanwith the normal multilobulated appearance; a ‘left shift’.Conversely, the neutrophils are hypermature (‘rightshifted’) in megaloblastic anaemia because vitamin B12deficiency impairs haemopoiesis (p. 261). There mayalso be cytoplasmic abnormalities, e.g. vacuolation orheavy granulation of neutrophils during bacterialinfection. Abnormalities of neutrophils, such ashypogranularity, are seen in myelodysplasia.
PlateletsA simple platelet count is the clue to most plateletdiseases.
Erythrocyte sedimentation rate
The erythrocyte sedimentation rate (ESR) is a very crudetest which measures the rate (in mm/h) at which redcells sediment. Abnormal proteins in inflammatory con-ditions and myeloma cause rapid sedimentation. TheESR is, therefore, a non-specific marker of infection,inflammation and neoplasia. It may be non-specificallyraised in anaemia and some non-inflammatory diseasessuch as uraemia. In some hospitals, blood viscosity oracute-phase proteins is measured as more reliablemarkers of inflammation.
Haematinics
The blood count must always be checked before meas-uring haematinics because it is unlikely that a patient hassignificant iron, vitamin B12 or folate deficiency if thecount is normal. An extremely high index of suspicion(e.g. suspected subacute combined degeneration of thecord, p. 236) would be needed to justify these extrameasurements in the face of a normal blood count.Similarly, it is wasteful to measure serum iron or ferritin ifthe diagnosis of iron deficiency is obvious from the clinicalcontext and blood count. With those provisos, measure:
● vitamin B12 and folate in patients with macrocyticanaemia
● iron in patients with microcytosis● all three in a patient with anaemia and a normal MCV
but a high RDW (to exclude a mixed deficiency).258
Haematology6Table 60 Terms used to describe abnormalities of redblood cells
Term Description
Hypochromia Defective haemoglobinisation, as in some anaemias
Anisochromia Variable haemoglobinisation, as in haemolysis
Polychromasia Bluish tinge to the red cells, reflects increased haemopoiesis
Microcytosis Small size (p. 259)
Macrocytosis Large size (p. 259)
Anisocytosis Variable size, as in iron deficiency
Poikilocytosis Abnormal shape; as in myelofibrosis (p. 271)
Erythrocytosis Increased number of red cellsSpherocytosis, Abnormal shape resulting from
elliptocytosis congenital or acquired cell membrane defect
Sickle cells Abnormal shape resulting from abnormal haemoglobin
Target cells Increased surface:volume ratio;as in liver disease
Fragmented or Trauma to red cells (p. 264)damaged cells
InclusionsBasophilic stippling Immature cells resulting from
accelerated haemopoiesisIron granules Seen with special stain in
(siderocytes) sideroblastic anaemia
Howell–Jolly bodies Nuclear remnants seen after splenectomy

The diagnosis of iron, vitamin B12 and folate deficienciesare considered on pages 260 and 262.
Tests of coagulation and fibrinolysis
These are discussed on page 276.
Bone marrow examination
Bone marrow can be examined either as an aspiratefrom the sternum or iliac crest or as a trephinebiopsy from the iliac crest. Marrow aspirate is useful fordetailed cytology, cell counts and assessment of ironstores. Trephine biopsies are useful for judging cellu-larity, detecting tumour and diagnosing myelofibrosis(p. 271).
Remember that marrow examination is not infalliblein detecting tumour deposits because they may beabsent from the area sampled. Marrow aspiration is apainful procedure which should not be done withoutgood indications. These include:
● to confirm iron deficiency or megaloblastic anaemia,if the diagnosis is in doubt or the patient does notrespond to treatment
● to investigate unexplained anaemia● to investigate suspected leukaemia, myeloma or
other haematological malignancy● to monitor response to the treatment of leukaemia● to diagnose cancer, where there is circumstantial
evidence of marrow involvement● to investigate agranulocytosis or thrombocytopenia● to stain and obtain cultures for suspected
tuberculosis, histoplasmosis and leishmaniasis.
In some conditions—notably myelofibrosis and malig-nant infiltration of the marrow—bone marrow aspira-tion may be unsuccessful. A ‘dry tap’ is pathological andof diagnostic value.
Lymph node biopsy
Lymph node biopsy is the definitive investigation todiagnose lymphoma and identify its histological type. Itis also used to diagnose disseminated cancer and othercauses of lymphadenopathy. The features of lymphnodes that make them ‘suspicious’ are described onpage 257, and other clinical features suggesting lym-phoma are described on page 269. Which node shouldbe biopsied is partly a surgical decision and partlydetermined by their ‘feel’. Lymph node biopsies areusually taken under general anaesthesia. The specimenwill be ruined for some analyses if it is put straight intoformalin. It should be promptly handled by a skilledhistology technician. Remember to request cultureif tuberculosis is suspected.
6.2 Red cell disorders
Erythropoiesis:
● is controlled by erythropoietin, which is secreted bythe kidneys
● is stimulated by hypoxia● requires an adequate supply of folic acid, vitamin B12
and iron● is inhibited by poor nutrition, systemic disease and
local disease within the marrow.
Anaemias
Think of anaemias in terms of:
● decreased red cell production● increased loss.
This classification is expanded in Table 61. Note that thecommonest form, iron-deficiency anaemia, often resultsfrom a combination of both. The MCV is central to thediagnosis of anaemia. Remember that:
● iron deficiency causes microcytosis● vitamin B12 and folate deficiency cause macrocytosis
(other causes are covered on p. 261)● the anaemia of marrow suppression is often
normocytic.
Exceptions to these rules are discussed under individualdiseases. Given the haemoglobin and MCV, white cellcount, platelet and reticulocyte counts and a descriptionof the blood film, you can make a working diagnosis inmost cases.
Iron-deficiency anaemia
Iron-deficiency anaemia is the most common and, to thegeneralist, most important anaemia because it:
● is a significant public health problem259
6Red cell disorders
Learning objectives
You should:
● understand the range of diseases that cause anaemia andhow they do so
● know how to diagnose and treat anaemias
● know the indications for blood transfusion and how toavoid complications
● know what polycythaemia is, what can cause it and how itcauses symptoms and signs.

● may be the presentation of an occult gastrointestinal(GI) carcinoma at a curable stage
● is eminently treatable whatever its cause.
Iron deficiency is caused by an imbalance betweendietary availability and blood loss, so much of the pop-ulation is iron deficient in areas of the developing worldwhere GI parasites are endemic. Active growth, preg-nancy and lactation increase the demand for iron andmay unmask deficiency. The prevalance of depleted ironstores in women of menstrual age may be as high as 20%and that iron deficiency can persist into the post-menopausal years if they have a poor diet and do notreplete their iron stores. Iron deficiency is much lessprevalent in adult men and, therefore, more likely to becaused by an occult carcinoma or other underlyingdisease.
There are sizeable iron stores in the liver, reticulo-endothelial system and marrow, as well as in the ery-throcytes themselves, so negative iron balance mustexist for some time before anaemia develops. For thesame reason, replenishment of iron stores takes longerthan the restoration of haemopoiesis, and iron treatmentshould be continued after the haemoglobin has returnedto normal.
CausesThese are:
● blood loss
● dietary deficiency● malabsorption.
Iron may be malabsorbed as a result of small intestinaldisease or after gastrectomy.
Sources of blood loss, in order of frequency, are:
● menstruation● the GI tract (Ch. 3)● the urinary tract.
In practice, it may be difficult to decide whether menor-rhagia is the sole cause of blood loss. If in doubt, suspectanother disease.
Common causes of GI blood loss are:
● gastritis, peptic ulcer or oesophagitis, particularly inpatients taking aspirin, non-steroidal anti-inflammatory drugs (NSAIDs) or steroids
● diverticular disease● carcinoma of the stomach, caecum or colon● angiodysplasia of the colon, an increasingly
recognised cause in elderly people● oesophageal varices.
A less common cause is Meckel’s diverticulum.Although only a small minority of iron-deficient
patients have GI tumours, it is worthwhile identifyingthem because diagnosis at the stage of a predominantlymucosal lesion may allow curative surgery.
Clinical and haematological signsSymptoms and signs were covered earlier and in Figure64. Early signs on the blood film are variability in sizeand shape of the red cells. In established iron deficiency,they are hypochromic and microcytic and ‘pencil cells’may be seen.
Diagnosis and further investigationIron deficiency is overwhelmingly the most commonUK cause of microcytosis but it also occurs in thalas-saemia trait and, sometimes, in the anaemia of chronicdisease. If there is doubt, iron deficiency is diagnosed bymeasuring serum iron and total iron-binding capacity.These are measured together because binding proteindeficiency may lower the serum iron concentrationwithout signifying true iron deficiency; < 10% satur-ation of iron-binding capacity is diagnostic of irondeficiency. Measurement of serum ferritin is a betterreflection of iron stores (particularly to detect ironoverload, as in haemachromatosis). Absence of stainablemarrow iron stores and a significant improvement inthe blood count with iron replacement are definitiveevidence of iron deficiency.
Stool samples are often tested for occult blood to con-firm or exclude the GI tract as the site of blood loss, butthey may be falsely positive or negative. Dietary assess-ment and tests for malabsorption (p. 127) may be indi-
260
Haematology6Table 61 A classification of the anaemias
Decreased Increased production destruction/
loss
Acute haemorrhage +Haematinic deficiency
Iron deficiency + ±B12 deficiency +Folate deficiency +
Hypoplastic anaemiasIdiopathic +Secondary +
Anaemia of chronic disease +Marrow replacement
Leukaemias +Myeloma +Other tumours +
Congenital haemolysisSpherocytosis and ellipt
ocytosis ± +Sickle cell disease +Thalassaemia +Glucose 6-phosphate +
dehydrogenase deficiency
Acquired haemolysisAutoimmune +Microangiopathic +
Hypersplenism +

cated. The investigations are not complete until a patientwith undiagnosed iron deficiency has had gastroscopy,colonoscopy (or barium studies) to look for a site ofblood loss.
ManagementThere is little to choose between the many formulationsof oral iron. Ferrous sulphate is usually prescribed. Itmay cause nausea, abdominal pain, diarrhoea or consti-pation, in which case the dose should be reduced or adifferent iron salt given. A response should be seenwithin 1 week of starting treatment and a rise in haemo-globin of 10 g/l each week is to be expected. Treatmentis continued for 3 months after the haemoglobin hasreturned to normal to replenish iron stores.
Megaloblastic anaemias
Vitamin B12 (cobalamin) and folic acid are coenzymesfor cellular metabolism, particularly haemopoiesis.Deficiency affects DNA synthesis in all marrow cell linesand the red cell precursors develop ‘megaloblastic’ mor-phological changes. Ineffective erythropoiesis reducesthe production of mature red cells and they are enlarged(macrocytic). Formation of platelets and granulocytes isalso impaired.
Vitamin B12 and folate deficiency are not the onlycauses of macrocytosis; others, in approximate order offrequency, are:
● common causes— alcohol abuse— liver disease— active haemopoiesis (immature red cells are
large): haemolytic anaemias, blood loss
● less common causes— myelodysplasia— aplastic anaemia— antifolate drugs: methotrexate, phenytoin.
However, vitamin B12 and folate deficiency are the onlycommon conditions to produce the typical megaloblas-tic bone marrow appearances. They have identicalhaematological features but differ in their causes andnon-haematological manifestations. This section willfirst present the common features and then the differ-ences between them.
Clinical and haematological featuresPatients typically present with symptoms of anaemiaand malaise. These develop insidiously and the anaemiamay be very severe by the time the diagnosis is made.Elderly people, in particular, may tolerate it surpris-ingly well until they present with heart failure orangina resulting from tissue hypoxia. There may be
● mild jaundice caused by haemolysis of the defectivered cells
● fever● glossitis● splenomegaly (very unusually).
Although moderate neutropenia and thrombocytopeniaare common, they are not usually symptomatic. VitaminB12 deficiency may, uncommonly, present with itsneurological complications (see Ch. 5). There may befeatures of an underlying GI disease in vitamin B12,folate or a mixed deficiency anaemia.
Whatever the cause, the blood film shows:
● pancytopenia● macrocytosis: large oval red cells● hypersegmentation of the neutrophil nuclei (termed
a ‘right shift’)● megaloblasts in peripheral blood in severe cases.
This haematological picture is so characteristic that thediagnosis can generally be made without bone marrowexamination, the definitive way of demonstratingmegaloblastic change.
Vitamin B12 deficiency
Vitamin B12 is present in meat and dairy produce. Itis absorbed as a complex with intrinsic factor (IF)produced by the gastric parietal cells. Pure dietarydeficiency is exceptionally rare because there are largehepatic stores but can occur in alcoholics. Morecommonly, deficiency is caused by
● autoimmune damage to the gastric parietal cells:pernicious anaemia
● destruction of vitamin B12 by bacterial overgrowth indiverticulae, blind loops or fistulae
● disease of the terminal ileum where the B12–IFcomplex is absorbed: usually Crohn’s disease
● pancreatic exocrine deficiency● gastrectomy.
Pernicious anaemiaPernicious anaemia is an organ-specific autoimmunedisease caused by antibodies to gastric parietal cells andintrinsic factor. It
● is commoner in women than men● may develop from adolescence onwards but is most
common in middle to old age.
It is associated with:
● blood group A● failure of gastric acid secretion● an increased risk of gastric acid carcinoma
261
6Red cell disorders

● an increased incidence of the other organ-specificautoimmune diseases, including vitiligo, diabetesmellitus, hypothyroidism and Addison’s disease.
The diagnosis is made by demonstrating a reducedserum vitamin B12 concentration, and IF antibodies. It isconfirmed by the Schilling test, in which radiolabelledB12 is given on two occasions, first on its own and thenwith oral IF. Urinary excretion of the label is measured.In pernicious anaemia, there is malabsorption which iscorrectable by IF. Other causes of malabsorption are notcorrectable.
Neurological effects of B12 deficiencyFull blood counts are now so widely available that B12deficiency is usually recognised and treated before it hasany significant neurological effects; however, some indi-viduals may develop neurological effects with little orno anaemia. These include:
● optic atrophy● dementia● subacute combined degeneration of the cord (Ch. 5).
TreatmentOne simple rule is that folate and vitamin B12 shouldalways be given together in megaloblastic anaemia, atleast until the haematinic results are known, becausefolate treatment alone can increase haemopoiesis inpatients with pure vitamin B12 deficiency and actuallyprecipitate neurological complications. Patients withvitamin B12 deficiency should be given injections of vita-min B12 weekly for 6 weeks and then 3-monthly indefi-nitely thereafter. The reticulocyte response should bechecked after 7 days. Potassium supplements areneeded in some cases, as well as iron and folate, becausecellular anabolism requires potassium. Whether ornot patients with severe megaloblastic anaemia shouldbe transfused is controversial because even carefultransfusion can precipitate heart failure. If it is done,it should be slow and with diuretic cover and shouldnot aim to restore haemoglobin immediately to normal.
Folate deficiency
Folate is absorbed in the upper small intestine. The bodystores are relatively small so folate deficiency developsearlier in malabsorption syndromes than does iron orvitamin B12 deficiency. For the same reason, folate defi-ciency is more likely to have a purely dietary cause, todevelop during pregnancy or to be precipitated byactive haemopoiesis in haemolytic states. Folate metab-olism is vulnerable to a wide range of drugs and isactually the target of some chemotherapeutic agents,e.g. methotrexate. These are the main causes of folatedeficiency:
● dietary, for example in:— alcoholism— elderly or neglected people
● increased demand— pregnancy— active haemopoiesis, e.g. haemolytic anaemia
● malabsorption— coeliac disease— pancreatic insufficiency— postgastrectomy— Crohn’s disease
● drugs which interfere with folate absorption:phenytoin
● drugs which interfere with folate metabolism:methotrexate, phenytoin, trimethoprim.
Investigation of the cause of folate deficiency willdepend on the context in which it is diagnosed. If thereis a clear dietary or drug cause, no further investigationis needed. Otherwise, investigation for malabsorption isindicated.
DiagnosisA low serum or red cell folate concentration confirmsthe diagnosis. Of the two, red cell folate more accuratelyreflects total body folate stores.
Prevention and treatmentPregnant women are routinely given prophylactic oralfolate. Alcoholic or poorly nourished patients should begiven folate as part of their rehabilitation. Proven folatedeficiency is treated with oral folic acid. As explainedabove, this must never be given alone in macrocyticanaemia unless vitamin B12 deficiency has been exclud-ed. If there is evidence of iron deficiency, oral ironshould also be given.
Aplastic/hypoplastic anaemia
Aplastic anaemia is a rare condition which may becongenital or acquired. It may be primary (no causeidentified) or secondary to:
● drugs, e.g. chloramphenicol, chemotherapy,zidovudine, ganciclovir, phenylbutazone
● irradiation● chemicals● infection, e.g. viral hepatitis● autoimmune disease.
There is usually leucopenia and thrombocytopenia aswell as anaemia, although pure red cell aplasia mayoccur. The aplasia may be transient or chronic, and partial(hypoplastic) or complete (aplastic). The symptoms andsigns depend on the relative degrees of anaemia,thrombocytopenia and neutropenia. The red cells arenormocytic or slightly macrocytic. The marrow trephine
262
Haematology6

biopsy is hypoplastic. The differential diagnosis is withother causes of pancytopenia, particularly marrowreplacement/fibrosis. Severe aplastic anaemia has ahigh mortality. A significant number of patients respondto antilymphocyte globulin, but the best chance of curelies in bone marrow transplantation from an HLA-identical sibling donor. Androgens and corticosteroidshave occasionally been effective. Supportive care of theneutropenic patient (p. 268) and transfusions of plateletand red cells are needed. With time, platelet and red cellantibodies develop and may complicate management.
Leucoerythroblastic anaemia
Leucoerythroblastic anaemia is used to describe a condi-tion in which the blood film shows immature leucocytesand erythroblasts. It has many causes, notably marrowinfiltration with a solid tumour, myeloma, lymphoma,leukaemia or myelofibrosis. It is investigated by marrowaspiration/biopsy.
Anaemia of chronic disease
Anaemia caused by chronic disease is, second to irondeficiency, the most common anaemia you are likely toencounter. Common causes are:
● chronic infection● renal failure● liver disease● malignancy● autoimmune disease.
The typical pattern is a normocytic or microcyticanaemia with reduced serum iron and total iron-bindingcapacity, intact marrow iron stores and reduced eryth-roblast haemosiderin. It is usually caused by suppressederythropoiesis, although other mechanisms such ashaemolysis may contribute. The anaemia is oftenrelatively mild. There is no specific treatment other thanmanagement of the causative disease.
Haemolytic anaemias
Destruction of red cells by a disease process eitherintrinsic or extrinsic to the cell causes:
● shortened red cell survival● increased erythropoiesis● anaemia if erythropoiesis cannot keep pace with red
cell destruction.
There may be morphological changes in the red cells,giving a clue to the cause of haemolysis.
In most haemolytic anaemias, red cells are removedby macrophages of the reticuloendothelial system,chiefly in the spleen. This process is termed extravascu-
lar haemolysis. Breakdown of haemoglobin increasesthe plasma level of unconjugated bilirubin, causingclinically overt jaundice in severe haemolysis.Splenomegaly and pigment gall stones may occur.
When there is rapid breakdown of red cells withinthe circulation (intravascular haemolysis), haemo-globin is liberated. Initially, it is bound to plasma pro-teins called haptoglobins. When the binding capacity ofhaptoglobins is exceeded, free haemoglobin is filtered inthe kidneys and converted to haemosiderin in renaltubular cells. Haemosiderin can be detected in urine.
The haemolytic anaemias are a heterogeneous groupof disorders which can be classified into congenitaland acquired forms. Congenital haemolytic anaemiasinclude:
● haemoglobinopathies— sickle cell disease— thalassaemia
● membrane defects— spherocytosis— elliptocytosis
● red cell enzyme defects— glucose 6-phosphate dehydrogenase deficiency.
Acquired haemolytic anaemias include:
● autoimmune (p. 264)● non-immune
— microangiopathic haemolytic anaemia— prosthetic heart valve— drug or toxin induced.
DiagnosisThe clinical features of haemolytic anaemias are those ofthe anaemia, the sequelae of haemolysis and any under-lying cause. In addition to haematological features,patients with chronic haemolytic anaemia are prone toleg ulceration as well as pigment gall stones.
Laboratory features of haemolysis are:
● evidence of increased erythropoiesis: reticulocytosis,polychromasia, erythroid hyperplasia of bonemarrow
● evidence of increased red cell breakdown: increasedplasma unconjugated bilirubin and urinaryurobilinogen, reduced plasma haptoglobin.
Congenital haemolytic anaemiasThe only congenital haemolytic anaemias covered hereare those that you might expect to encounter as a houseofficer.
Sickle syndromes Sickle syndromes are caused by arecessively inherited amino acid substitution in thehaemoglobin A molecule, which causes it to becomeinsoluble and cross-link under hypoxic conditions. This
263
6Red cell disorders

causes haemolysis, ‘stickiness’ and microvascular occlu-sion.
Africans are most often affected, so heterozygotes arecommon in areas with a high African population. Theymay develop symptoms after surgery or during anysituation that causes hypoxia.
‘Sickle trait’ can be diagnosed by the in vitro ‘sickletest’ and by haemoglobin electrophoresis.
Homozygotes are more severely affected. They havea chronic haemolytic anaemia and may have sicklingcrises precipitated by:
● hypoxia● dehydration● infection● other systemic stresses.
During crises, microvascular occlusion causes infarcts ofbone and soft tissues. Patients experience severe pain,fever and malaise. Splenic infarcts lead to hyposplenismand an increased risk of pneumococcal septicaemia.Salmonella osteomyelitis is common in areas of in-farcted bone. There may be soft tissue complications,including retinopathy, acute renal papillary necrosis,acute pulmonary syndrome, and leg ulcers. Pigmentgall stones are common. Pregnancy is hazardous. Prophy-lactic penicillin is required to prevent pneumococcalsepticaemia.
Sickling crises are treated by
● keeping the patient warm● oxygen● intravenous fluids● opiate analgesia● antibiotics.
The thalassaemias The thalassaemias are a group ofcongenital haemolytic anaemias caused by mutations orgene deletions affecting the α-and β-globin chains (α-and β-thalassaemia, respectively). They vary in severityfrom a chance finding in an asymptomatic individual toa disease that is incompatible with life. Synthesis ofabnormal haemoglobin causes ‘ineffective erythro-poiesis’. There is microcytosis and the cells are irregularin size, shape and degree of haemoglobinisation.
Heterozygotes (thalassaemia trait) usually remainasymptomatic but may become mildly anaemic duringintercurrent infection or pregnancy. Remember thalas-saemia trait in the differential diagnosis of microcyticanaemia. Thalassaemia major is not considered furtherhere.
Acquired haemolytic anaemiaAutoimmune haemolytic anaemia Autoimmunehaemolytic anaemia (AIHA) is most commonly idio-pathic but may be caused by:
● autoimmune disease, e.g. systemic lupuserythematosus (SLE)
● neoplasia, e.g. chronic lymphocytic leukaemia● infection, e.g. infectious mononucleosis, mycoplasma● drugs, e.g. methyldopa.
The anti-red cell antibody involved may cause haemo-lysis at body temperature (‘warm AIHA’: usually IgG)or only at temperatures well below 37˚C (‘cold AIHA’:usually IgM) and the clinical picture varies accordingly.
Cold AIHA (cold haemagglutinin disease).Haemolysis is mediated by complement and usuallyintravascular, classically causing episodes of dark urine(haemoglobinuria) after cold exposure. At the sametime, agglutination of red cells in peripheral capillariesmay cause peripheral cyanosis, Raynaud’s-like symp-toms and even gangrene. Cold AIHA may occur as atransient problem after infectious diseases such as infec-tious mononucleosis or mycoplasma pneumonia. Redcell agglutinates can be seen on a blood film at roomtemperature.
Warm AIHA. Haemolysis is usually extravascular,particularly in the spleen, and leads to jaundice andsplenomegaly. The blood film shows spherocytosis.
Direct antiglobulin test. In either case, the definitivetest for AIHA is the direct antiglobulin test, in whichantibodies are demonstrated on the red cell surface bythe addition of anti-human globulin, which will causeagglutination.
Management of warm AIHA. Warm AIHA can betreated with:
● steroids● splenectomy● immunosuppressive therapy.
Management of cold AIHA. Patients with cold agglu-tinins should be kept warm; they respond less well tosteroids and splenectomy. Blood transfusion may beneeded for haemolytic crises, but cross-matching can bedifficult.
Non-immune acquired haemolytic anaemias The non-immune acquired haemolytic anaemias are caused by:
● fibrin deposited in the microcirculation, as indisseminated intravascular coagulation (DIC)(p. 279)
● heart valves or other mechanical prostheses● toxic causes, including uraemia, lead poisoning and
some drugs● paroxysmal nocturnal haemoglobinuria, a rare
condition in which deficiencies of membraneproteins make the red cells susceptible to the lyticactions of serum complement.
In the case of DIC and mechanical red cell damage, schis-tocytes (fragmented red cells) are seen on the blood film.
264
Haematology6

Blood transfusion
There are two quite distinct indications for blood trans-fusion:
● to restore volume after acute blood loss● to restore the red cell mass in a patient with anaemia
and a normal blood volume (compensated anaemia).
An excellent, ‘official’ guide is available athttp://www.show.scot.nhs.uk/sbts/webpages/01aud.htm.
Acute blood loss
Acute blood loss is caused by trauma, haemorrhage,usually from the GI tract, and surgery. If haemorrhage issevere, there is circulatory collapse. The physical signsare from the blood loss itself and from volume depletion(p. 188).
You should transfuse red cells (with other fluids, ifneeded) as quickly as necessary to restore the circula-tion, monitoring the pulse, blood pressure and urineoutput. Remember that the haemoglobin concentrationis no guide to the severity of acute blood loss because ittakes time for haemodilution to occur.
Transfusion for anaemia
In anaemia, plasma volume is normal despite lack of redcells. This process of compensation takes hours or daysafter acute blood loss. The indications for transfusionare less strong than after acute blood loss because:
● the patient is not shocked● treating the underlying cause of anaemia is a more
permanent (albeit slower) solution than transfusingred cells, which have a short lifespan
● transfusion increases blood volume and may causevolume overload (heart failure).
Transfusion may nevertheless be indicated to:
● relieve symptoms● prepare anaemic patients for surgery
● increase the red cell mass in case of further bleeding.
As a rule of thumb, blood transfusion:
● is rarely indicated if the haemoglobin concentrationis above 100 g/l
● may be indicated between 80 and 100 g/l● is likely to be needed below 80 g/l.
In general, iron, vitamin B12 or folate deficiency shouldbe treated with haematinics. Transfusion for anaemia isparticularly hazardous if the patient is in heart failure,when red cells should be given with an i.v. loop diureticand close observation of the patient’s haemodynamicstate. Blood transfusion is expensive and potentiallyhazardous. It should not be ‘dished out’ carelessly.
Blood groups
The main ABO system consists of two markers inheritedas Mendelian dominant traits from the parents. The redcells of an individual may carry the A antigen (group A),the B antigen (group B), both (group AB) or neither(group O). The antigens are carried on many tissuesother than red cells and individuals are exposed early inlife to those antigens that they do not carry. Thus groupA individuals have agglutinins to the B antigen and viceversa; group O individuals have agglutinins to bothantigens and group AB individuals have agglutinins toneither. The importance of this system to blood transfu-sion is that donated cells may be lysed by host agglu-tinins; lysis of host cells by donor agglutinins is notusually a problem. Table 62 summarises the system andreminds you that group O subjects are ‘universaldonors’ because their cells have neither antigen and willnot be lysed if they are transfused into group A, B or ABrecipients. Group AB subjects are ‘universal recipients’because they have agglutinins against neither antigen.
The rhesus (D) blood group system is also conceptu-ally very simple. About 85% of the Caucasian popula-tion (and a higher proportion of Asians) carry the D (orrhesus) antigen on their red cells. Those who are rhesusnegative may develop antibodies against the D antigen
265
6Red cell disorders
Table 62 The ABO and rhesus blood group systems
Blood Relative Alleles Agglutinins Plasma will Commentgroup frequency (%) present agglutinate
cell types
O 45 Null, null Anti-A, Anti-B A, B, AB Universal donorA 40 A, A or A, null Anti-B B, ABB 10 B, B or B, null Anti-A A, ABAB 5 A, B None None Universal recipientRh+ 85 D+ None NoneRh– 15 D– None or anti-D None or Rh+

but only if exposed to it by blood transfusion or, in preg-nancy, by placental leakage.
Cross-matchingAfter determining the patient’s ABO and rhesos bloodgroups, donor red cells of an appropriate group are thencross-matched, both at room temperature and at 37˚Cagainst patient’s serum, to detect cold and warm anti-bodies. Positive results in these tests mandate furtherinvestigation, and a significant delay before compatibleblood can be made available. There is an increasingmove towards storing the patient’s own blood inpreparation for elective surgery, eliminating the risks ofcross-infection and incompatibility (autologous transfu-sion).
Complications of transfusion
The major risks of HIV and hepatitis B and C infectioncan largely be eliminated by careful screening of donorblood. Other potential complications are:
● minor febrile reactions● haemolytic reactions● severe allergic reactions● transfusion-related lung injury● problems caused by massive transfusions● chronic iron overload.
Minor febrile reactionsMinor febrile reactions are not uncommon, particularlyin patients who have developed platelet and/or whitecell antibodies from repeated transfusions. They may beprevented by leucodepleting the red cells before trans-fusion, or using a filter to trap white cells and plateletsduring transfusion.
Acute, severe haemolytic reactionsSevere acute haemolytic reactions are fortunately rare,invariably caused by ABO incompatibility (e.g. a groupO recipient receiving group A blood) and almost alwayscaused by elementary errors like the incorrect labellingof blood samples or incomplete pretransfusion checks.They are likely to lead to undefendable litigation. Thepatient may develop pyrexia, chest or abdominal painand pass black urine, rapidly progressing to shock,acute renal failure and DIC. If a haemolytic reaction issuspected, you should:
● stop the transfusion immediately● give intravenous hydrocortisone and
chlorphenamine● check the patient’s identity and details of the donor
blood● return the blood to the transfusion laboratory with a
fresh sample of the patient’s blood
● call for senior help and resuscitate as needed.
Severe allergic reactionsSevere allergic reactions occur occasionally. Patientswho are deficient in IgA (1 in 600) and produce anti-IgAantibodies are particularly at risk.
Transfusion-related acute lung injuryAcute lung injury can be caused by aggregation ofneutrophils in lung capillaries precipitated by anti-white cell antibodies in donor plasma; it may result inacute respiratory distress.
Massive transfusionMassive transfusions may lead to particular problems.Hypocalcaemia can result from the chelating action ofcitrate, particularly if there is liver disease, whichimpairs citrate metabolism. Dilution of platelets andclotting factors by stored blood leading to an increasedrisk of bleeding is another potential complication.
Iron overloadRepeated blood transfusions can eventually lead tocardiac, liver and endocrine damage.
Polycythaemia
The term polycythaemia (increased number of red cells)is frequently used to mean erythrocytosis (increasedhaemoglobin concentration). That is not strictly correctbut has become accepted in medical parlance.Polycythaemia and erythrocytosis are therefore bothdefined as an increased red cell mass. You will recogniseit on the blood count as an increased haemoglobin con-centration, red cell count and haematocrit. It may beabsolute, or relative to a reduced plasma volume.
Absolute polycythaemia may be:
● primary: termed polycythaemia vera● secondary to increased erythropoietin.
Causes of secondary polycythaemia include:
● compensatory increased erythropoietin secretion as aresult of:— tissue hypoxia— lung disease— cyanotic heart disease— altitude— hypoventilation (Pickwickian syndrome)
● non-compensatory increased erythropoietin secretionfrom:— renal tumour or cysts— other tumours.
Only lung disease is a common cause of clinical poly-cythaemia.
266
Haematology6

Polycythaemia increases the oxygen-carrying cap-acity of blood but it also increases viscosity and impairsblood flow. Many of its clinical effects are the result ofstasis and impaired tissue oxygenation.
Polycythaemia vera
Polycythaemia vera is a neoplastic stem cell prolifera-tion which increases the formation primarily of red cellsbut also of leucocytes and platelets. Clinical manifesta-tions result from:
● hyperviscosity● tissue hypoxia● vascular occlusive events resulting from stasis and
thrombocytosis.
Paradoxically, patients may also bleed easily becausetheir platelets are dysfunctional.
Clinical presentationPatients may present with:
● systemic symptoms such as pruritus (particularlyafter a warm bath) or malaise
● an arterial or deep venous thrombosis● neurological symptoms, including headache, tinnitus
and visual disturbance● cardiovascular symptoms, including angina and
intermittent claudication● dyspepsia or non-specific abdominal pain signifying
GI mucosal ulceration● episodes of epistaxis or GI haemorrhage● gout.
On examination, there is plethora, cyanosis, injection ofthe conjunctivae and, in most cases, splenomegaly.There may also be hepatomegaly. The blood countshows a haematocrit in the range 50–70%, with leucocy-tosis and an increased platelet count. The red cell mass,measured isotopically, is increased. The marrow isactive but normoblastic. Serum uric acid and vitaminB12 are usually raised.
ManagementTreatment is with:
● venesection to reduce hyperviscosity● phosphorus-32 or an alkylating agent to reduce stem
cell proliferation.
The aim is to reduce both the haematocrit and theplatelet count. Life expectancy is reduced despite treat-ment, and the disease may transform into acuteleukaemia or myelofibrosis.
Relative polycythaemia
Relative polycythaemia is far more common than poly-cythaemia vera. It is a disorder often seen in middle-
aged hypertensive men who drink and smoke too much.The primary abnormality is a reduced plasma volume.The aetiology is unknown.
Faced with an apparently polycythaemic patient:
● measure the haematocrit more than once to be surethe apparent erythrocytosis is not a cuffing artefact
● consider the possibility of secondary polycythaemia,of which the common cause is chronic lung disease;measure the blood gases
● check that the patient is truly polycythaemic (i.e.exclude relative polycythaemia) by measuring the redcell mass.
If the patient has a leucocytosis, thrombocytosis orsplenomegaly, the diagnosis is likely to be poly-cythaemia vera.
6.3 White cell disorders
PhysiologyGranulocytesGranulocytes are the most abundant white cells inperipheral blood.
Neutrophils. Most granulocytes are neutrophils, a keyelement of defence against most bacteria and somefungi. Most neutrophils in the normal person are pres-ent in the blood loosely adherent to the walls of vessels(marginating pool). When the appropriate stimuluscomes along (such as a bacterial infection) they arereleased from the marginating pool and attracted tosites of inflammation by chemotactic factors includingcomplement. Once a neutrophil comes into contact witha microbe or foreign body it attaches itself, ingests it intoa vacuole called a phagosome and kills it.
267
6White cell disorders
Learning objectives
You should:
● understand the causes of neutropenia
● know what infections to be concerned about in theneutropenic patient and what to do if such a patient gets afever
● know the diseases of white cell proliferation and how theyare diagnosed and treated.
A simple way of approaching white cell disorders is to think interms of white cell numbers. They may be:
● reduced, increasing susceptibility to infection
● increased, signifying systemic disease or marrowproliferation.

Eosinophils. These are the next most abundant form.They attack parasites which are too large to be phagocy-tosed, and also have a role in mucosal immunity.
Basophils. These are the least abundant form. Theyrelease histamine and other inflammatory mediatorsduring immediate hypersensitivity reactions.
MonocytesMonocytes are large non-granulated white cells that arereleased from the bone marrow, circulate for severaldays and then enter tissues to become the tissue macro-phages, including pulmonary alveolar macrophages,osteoclasts and the Kupffer cells of the liver. They engulfand kill bacteria in tissues and, as antigen-presentingcells, play a key role in immunity.
LymphocytesLymphocytes are derived from the same stem cells ofthe bone marrow as the other cell lines. During fetaldevelopment, they migrate out to populate the thymus,liver, spleen, lymph nodes and other lymphoid tissues.In adult life, some lymphocytes are formed in the mar-row but most are formed in lymphoid tissues elsewhere.There are two distinct cell lines, morphologically identi-cal but distinguishable by cell-surface markers. The Bcells — plasma cells and memory cells — are responsiblefor humoral immunity. T cells are responsible for cell-mediated immunity and for activating B cells. The sub-types of T cells are not discussed further here but anunderstanding of them is crucial to an understanding ofAIDS (p. 381).
Leucopenia
Leucopenia is a reduction in the number of white bloodcells.Lymphopenia is caused by:
● HIV infection● autoimmune disease● lymphoma● irradiation
Neutropenia is caused by:
● decreased production— drugs, e.g. cytotoxic therapy, carbimazole,— phenylbutazone— vitamin B12 or folate deficiency— irradiation— marrow aplasia, fibrosis, malignant invasion,
acute myeloid leukaemia (AML)— marrow dysplasia (p. 270)— infection
● increased consumption— hypersplenism— antineutrophil antibodies.
Neutropenia is relatively rare except in haematologicalmalignancies and as a result of their treatment, thesubject of the next section.
Management of neutropenia
The normal neutrophil count is around 2 × 109 cells/l.There is an increasing risk of infection when the countfalls below 1 × 109 cells/l and the risk of invasive infec-tions rises substantially when the count falls below 0.5 ×109 cells/l. A longer duration of neutropenia (e.g. morethan 7 days) also puts the patient at substantially greaterrisk of life-threatening infection. Infections include:
● streptococci● Gram-negatives, including Pseudomonas aeruginosa● Staphylococcus epidermidis (usually related to
indwelling central venous lines, and not immediatelylife-threatening)
● mucosal and invasive candidiasis● invasive pulmonary aspergillosis● herpes simplex virus (especially mouth ulcers).
A typical clinical scenario is that you are on call forhaematology patients and summoned to the wardbecause a patient with no circulating neutrophils hasdeveloped a new fever 7–14 days after chemotherapyfor leukaemia. You need to:
● assess whether there are any localising symptomssuch as cough, skin rash, nasal symptoms,abdominal pain, etc.
● examine the mouth, chest, skin and rectal area andany other sites that are symptomatic
● assess whether the patient is in shock or going intorespiratory failure
● order a portable chest radiograph if the illness hasany respiratory features
● check the notes and recent results for any positivemicrobiology
● take at least one blood culture and collect any othermicrobiological specimens that are indicated by thepatient’s symptoms
● start broad-spectrum i.v. antibiotics according to thepolicy of the unit; typically these will includecoverage for Ps. aeruginosa and streptococcalinfections as a minimum (e.g. ceftazidime)
● if there are any unusual features, or the patient isvery ill, you should contact a senior member of staffresponsible for the patient.
Expert microbiological advice is needed if the patientremains profoundly neutropenic and has persistentfever.
Ceasing therapy. Antibiotics can usually be stoppedwhen the neutrophil count has risen above 0.5 × 109
cells/l.268
Haematology6

Leucocytosis
Causes of leucocytosis include:
● neutrophilia— infection, usually bacterial— inflammation— connective tissue disease— myeloproliferative disease— non-haematological malignancy— corticosteroid therapy— diabetic ketoacidosis
● eosinophilia— parasitic infection— allergy— drug reaction— connective tissue disease, e.g. microscopic
polyarteritis (Churg–Strauss syndrome)— cancer
● lymphocytosis— viral infection— connective tissue disease— lymphoproliferative disease
● basophilia: rare and usually caused bymyeloproliferation
● monocytosis— infection, particularly during convalescence— connective tissue disease— myeloproliferative disease.
In many cases, the cause is obvious. In others, detailedhaematological investigation is needed. Remember thecommon causes and remember that diabetic ketoacido-sis and steroid therapy cause leucocytosis in the absenceof infection.
The leukaemias, myeloproliferativedisorders and lymphomas
These neoplastic diseases may seem hard to understandand remember but a few simple principles provide aframework on which the diseases hang:● all of them are neoplastic clonal proliferations but
they differ in their degree of malignancy● the more chronic diseases tend to become
increasingly malignant with time● a distinction can be drawn between
lymphoproliferative and myeloproliferative diseases● the myeloproliferative group, particularly the more
chronic types (polycythaemia vera, essentialthrombocythaemia and chronic granulocyticleukaemia (CGL)) usually involve proliferation ofmore than one cell line
● other marrow tissues that are not derived from stemcells proliferate reactively to myeloproliferation andlymphoproliferation, as in myelofibrosis andlymphoma.
Figure 65 shows the relationship between the diseases.In general, the more malignant the disease, the moreintense the treatment required and the higher the chanceof cure. Low-grade diseases tend to be chronic andincurable.
The acute leukaemiasClassification and risk factorsThere are two broad categories of acute leukaemia,acute lymphoblastic leukaemia (ALL) and acutemyeloid leukaemia (AML), the latter including severalsubtypes, shown in Figure 65. The incidence of ALLpeaks in childhood. It is more common in males. AML isa disease of both children and adults, with a rapidly ris-ing incidence in old age and no gender difference.Radiation exposure, genetic factors, viral infections andtoxins, including chemotherapeutic drugs, have beenimplicated as risk factors for leukaemia, although thecause in an individual case is usually unknown.
Clinical and haematological featuresAcute leukaemia may arise de novo or in patients withchronic myeloproliferative disease or myelodysplasia.The clinical manifestations result from marrow dysfunc-tion and tissue infiltration. They include:
● symptoms and signs of anaemia● bruising, bleeding and purpura● increased susceptibility to infection.
269
6White cell disorders
STEM CELL
LYMPHOPROLIFERATION
MYELOPROLIFERATION
Chronic lymphocytic leukaemia
Lymphoma – Hodgkin's Low-grade non-Hodgkin's
MyelomaHigh-grade non-Hodgkin's lymphomaAcute lymphoblastic leukaemia
Acute myeloid leukaemia including: monocytic myelomonocytic Types promyelocytic
MyelodysplasiaChronic granulocytic leukaemiaPolycythaemia veraPrimary thrombocythaemia
Chronic myelofibrosis
Fig. 65 Schematic representation of the lymphoproliferativeand myeloproliferative diseases. The darkest shadingindicates the highest degree of malignancy.

Tissue infiltration may cause:
● bone and joint pain● gum infiltration● skin rashes.
There is occasionally hepatosplenomegaly and, in ALL,lymphadenopathy. The diagnosis is made on blood filmand bone marrow examination. There is usuallyanaemia and thrombocytopenia. There may be leuco-penia or a leucocytosis with blasts (primitive cells) inperipheral blood. The marrow is hypercellular and infil-trated with blasts. AML and ALL are distinguished andsubclassified on morphological, cytochemical, immuno-logical and cytogenetic characteristics. Rod-shapedcytoplasmic bodies, named Auer rods, are a virtuallypathognomonic feature of AML.
ManagementThe treatment of ALL has been one of the success storiesof oncology because two-thirds of children (feweradults) are cured. Most forms of acute leukaemia areamenable to chemotherapy, but equally important is theintensive supportive treatment with antibiotics andblood products, and sympathetic management of thepatient and his/her family. The aim is to eliminate theabnormal clone from blood, bone marrow and other sitesand allow repopulation with normal haemopoietic cells.This is termed remission. A discussion of the specificdrugs and regimens is beyond the scope of this chapterbut treatment may include the following phases.
1. Remission induction: elimination of neoplastictissue, allowing recovery of normal marrow
2. Consolidation of remission3. Prevention of recurrence in extramedullary sites (as
in ALL) and maintenance of remission4. Treatment of relapses.
There are some important differences between thetreatment of ALL and AML. More intensive myelosup-pression is used in AML; consequently the risks of infec-tion and bleeding are greater. In ALL, there is a high riskthat systemic chemotherapy will not eradicate malig-nant cells from certain extra medullary ‘sanctuary’ sites,including the brain, spinal cord and testes; therefore,additional local treatment (e.g. intrathecal chemo-therapy) is given. Relatively low-dose maintenancetherapy is often continued for 2 to 3 years in ALL,whereas induction in AML is followed by severalfurther courses of very intensive chemotherapy over amuch shorter period of time in an attempt to preventrelapse.
If a suitable donor is available (preferably an HLA-identical sibling), allogeneic bone marrow or periph-eral blood stem cell transplantation may be performedin both AML and ALL, in an attempt to reduce the
chance of relapse. This offers a ‘graft versusleukaemia’ effect, in addition to the benefits of thechemotherapy given, but may at the same time causegraft-versus-host disease, which has a significant mor-bidity and mortality. Alternatively, autologous trans-plantation, using the patient’s own bone marrow orperipheral blood stem cells, collected during remis-sion, may be employed. This avoids the risk of graft-versus-host disease but does not have the benefit of agraft-versus-leukaemia affect, and there is a chancethat the transplanted marrow may still containleukaemic cells.
Over 90% of children and 80% of adults with ALLachieve remission; the 5-year survival for children isover 60%, but unfortunately the majority of adultsrelapse and die of leukaemia. The remission rate withintensive chemotherapy in AML is over 70%, but the5-year survival is only 20%. Many patients are elderlyand not treated intensively, which results in a 5-year sur-vival for all patients with AML of approximately 5%.Increasing age has an adverse prognostic effect on alltypes of leukaemia and a high white count at presenta-tion predicts a bad outcome. Specific cytogenetic defectsin the leukaemic cells are closely related to either a good(e.g. t(8; 21) in AML) or bad (e.g. t(9; 22) in ALL)prognosis.
There is increasing interest in directing specific treat-ments towards molecular genetic defects newly identi-fied in haematological malignancies. A big success storyin this regard has been the treatment of acute promyelo-cytic leukaemia, a subtype of AML that carries a highrisk of early death from bleeding in DIC. The geneticabnormality (t(15; 17)) involves a retinoic acid receptorgene, and it has been found that complete remission canbe achieved by administration of all-trans-retinoic acid.Intensive chemotherapy still has to be used, but the riskof early death and incidence of relapse have beenmarkedly reduced; as a result, the cure rate is nowapproximately 70%.
Myelodysplastic syndrome
Myelodysplastic syndrome describes a group of disor-ders characterised by cytopenias of one or more celllines with a cellular marrow and morphological abnor-malities in both marrow and peripheral blood. Cyto-genetic abnormalities can often be demonstrated. Thecytopenias are presumed the result of ineffectivehaemopoiesis. Myelodysplastic syndrome progresses, ata variable rate, to AML. Management is supportive untilleukaemia develops, when cytotoxic therapy may beindicated although the outlook is poor at this stage.Many patients are elderly and unsuitable for intrusivetreatment. Allogeneic bone marrow transplantation canbe considered in younger patients.
270
Haematology6

Myeloproliferative diseasesChronic granulocytic leukaemiaChronic granulocytic leukaemia (CGL, also known aschronic myeloid leukaemia (CML)), is an uncommondisease which may occur at any age but peaks in the50–60 age group. It is characterised by uncontrolled pro-liferation of myeloid progenitor cells, generally mostnoticeable in granulopoietic cells but also affecting redcells and platelets. The characteristic feature, present in95% of patients, is the Philadelphia chromosome,resulting from a reciprocal translocation between thelong arms of chromosomes 9 and 22 (t(9; 22)).
These are the clinical features and some modes ofpresentation of CGL:
● patients typically present with symptoms ofanaemia, weight loss fatigue, anorexia, drenchingsweats or abdominal discomfort
● palpable splenomegaly in 50%, which may bemassive, and there may be symptomatic splenicinfarction
● there may be symptoms and signs of haemorrhage● the diagnosis may be made by chance on a blood
count.
There is marked leucocytosis including, especially, neu-trophils and myelocytes and, in many cases, basophilsand eosinophils. The platelet count is usually normalor raised at presentation. The bone marrow showsgranulopoietic hyperplasia.
CGL can be distinguished from other causes ofleucocytosis by:
● symptoms and signs● blood film● Philadelphia chromosome● reduced leucocyte alkaline phosphatase score on a
blood film.
CGL progresses over about 3 years to a more malignantphenotype, culminating in an accelerated phase. Thismay be marked by rapid transformation to acuteleukaemia (AML or ALL), or more gradual deteriorationinvolving such features as anaemia, massivesplenomegaly, myelofibrosis, marked basophilia orthrombocytosis. Response to treatment is generally poorand the outlook is bleak.
During the chronic phase, the disease is easily con-trolled by simple oral chemotherapy, usually hydroxy-urea. Nevertheless, the Philadelphia chromosome is not eradicated. Alpha interferon can produce goodhaematological remissions, and occasional cytogeneticremissions. At present, allogeneic bone marrowtransplantation during the chronic phase (if a suitabledonor is available) is the only reliable means ofachieving this key objective. However, new treatment
directed against the gene product of the Philadelphiatranslocation has been developed and may become animportant part of management in the future.
Polycythaemia vera and thrombocythaemiaAlthough polycythaemia vera and thrombocythaemiaare myeloproliferative disorders, they are described inthe red cell (p. 267) and platelet (p. 274) sections of thischapter because their clinical features are determined bythe predominant cell type in peripheral blood ratherthan their progenitor cell.
MyelofibrosisMyelofibrosis describes a disease in which proliferationof fibrous tissue in the marrow is the main feature. Itmay be the final result of other myeloproliferative dis-eases or may present as a primary disorder in middle-aged or elderly people. There is anaemia — which may be leucoerythroblastic (p. 263) — and massivesplenomegaly. It can be impossible to aspirate marrow.Trephine biopsy shows extensive fibrosis. The treatmentis supportive, sometimes with splenectomy to improvered cell, white cell and platelet survival. It may progressto acute leukaemia.
Lymphoproliferative diseasesHodgkin’s diseaseThere are incidence peaks of Hodgkin’s disease in earlyadulthood and old age. The two main clinical character-istics of lymphomas are lymphadenopathy and systemicsymptoms.
A cardinal feature of Hodgkin’s disease (which dis-tinguishes it from NHL) is that, when more than onegroup of nodes is involved, they are always contiguous,suggesting lymphatic spread of malignant cells. The cer-vical nodes are most often involved. With disseminateddisease, there may be hepatosplenomegaly and extra-lymphatic involvement. Systemic symptoms includepruritus, fever, night sweats and weight loss. Anaemia iscommon. There may be neutrophilia and eosinophilia.Impaired cell-mediated immunity predisposes to infec-tion, most typically herpes zoster. Alcohol-induced painat the site of the disease is a quite specific symptom.
The diagnostic feature on lymph node biopsy is thepresence of multinucleated Reed–Sternberg cells.
There are several histological subtypes of Hodgkin’sdisease, which vary, among other things, in the relativeproportion of lymphocytes and reactive elements inthe malignant tissue. Staging, however, is the mostimportant determinant of treatment and prognosis.This is based on the Ann Arbor system’ which, in itssimplest form, is:
stage I: involvement of one group of lymph nodes onlystage II: involvement of more than one group of lymph
271
6White cell disorders

nodes on one side of the diaphragmstage III: involvement of lymph nodes on both sides ofthe diaphragmstage IV: presence of extra lymphatic disease (e.g. bonemarrow, liver).
The disease is also subclassified as:A: no systemic symptoms.B: presence of fever, night sweats or significant weight
loss.Staging therefore involves:
● a careful history and examination● liver function tests● computed tomographic (CT) scanning of chest,
abdomen and pelvis● bone marrow trephine biopsy.
Less favourable prognosis is associated with:
● lymphocyte depleted histology● higher stage● B subtype symptoms● increasing age● very bulky lymphadenopathy (e.g. massive
mediastinal enlargement).
Broadly speaking, treatment is with local radiotherapyfor stages IA and IIA disease, and with chemotherapy,with or without radiotherapy, for higher stages.Depending on the above factors, Hodgkin’s disease hasa 50–90% 5-year survival.
Non-Hodgkin’s lymphomaNHL is a more heterogeneous group of disorders withvarying malignancy and prognoses. It differs fromHodgkin’s disease in four ways:
● higher prevalence● older mean age at diagnosis● non-contiguous, multicentric spread● extranodal involvement is more common.
Several types of NHL are known to be caused byviruses, including Epstein–Barr virus (Burkitt’s lym-phoma), human T cell leukaemia-1 virus and humanimmunodeficiency virus (HIV). Like Hodgkin’s disease,NHL may present with lymphadenopathy and systemicsymptoms, although the symptomology is morediverse.
Remember that lymphadenopathy must always betaken seriously. In younger people, it is more likely to becaused by infection than malignancy. In older people,there is a high likelihood of cancer (usually localisedlymphadenopathy), lymphoma or chronic lymphocyticleukaemia (generalized lymphadenopathy). The diag-nosis of NHL is made by biopsy. It is staged as forHodgkin’s disease.
There are two broad histological categories:
● low-grade NHL: this is indolent, but may becomemore aggressive with time; it is essentially withoutcure but survival may be prolonged (> 5 years)
● high-grade NHL: This form carries a much higherearly mortality but is more responsive to treatment,with an approximately 30% 5-year disease-freesurvival.
Treatment Because NHL is usually more widespreadthan Hodgkin’s disease at presentation, treatment ismore often with chemotherapy, although localiseddisease is treated with radiotherapy. Stem cell or bonemarrow transplantation is used in some patients.
Chronic lymphocytic leukaemiaChronic lymphocytic leukaemia (CLL) is the least malig-nant of the leukaemias and is characteristically a diseaseof elderly people. It is more common in men thanwomen. It may be a chance finding on a blood film ormay present with lymphadenopathy or anaemia.Typically, there is hepatosplenomegaly. The blood filmshows (sometimes massively) increased numbers ofmature lymphocytes. Because the lymphocytes are Bcells in over 95% of patients, there may be:
● a ‘paraprotein’ (see Myeloma, below)● depression of normal immunoglobulins and
increased susceptibility to infection● autoimmune phenomena such as haemolytic
anaemia or thrombocytopenia.
Cytotoxic therapy or corticosteroids may be given forsymptoms or complications, although many patientsneed no treatment. Progressive disease causes:
● worsening lymphocytosis● increasing hepatosplenomegaly and
lymphadenopathy● eventually, bone marrow failure.
There is a 50% five-year survival.
Multiple myeloma
Multiple myeloma is a malignant proliferation ofplasma cells (B cells), which are relatively highly differ-entiated and secrete a monoclonal immunoglobulin.Myeloma is predominantly a disease of old people,although it may arise at any time in adult life. Theplasma cells may secrete:
● IgG, with or without free light chains (50% ofpatients)
● IgA, with or without free light chains (20%)● free light chains only (20%)● Other patterns of paraprotein (10%).
272
Haematology6

The clinical features result from:
● plasma cell proliferation● systemic effects of the paraprotein● impairment of normal haematological function.
There is bone destruction caused by humorally medi-ated activation of osteoclasts without the normalosteoblastic response. This may cause diffuse osteo-penia or, radiologically, punched-out lesions at the siteof plasma cell deposits. Pathological fractures mayresult. Such deposits may present as mass lesions:plasmacytomas. Bone destruction mobilises calcium;consequently hypercalcaemia (p. 355) is a commoncomplication of myeloma.
Apart from its skeletal features, two other character-istics of myeloma are hyperviscosity and renal failure.Hyperviscosity occurs because the paraprotein altersthe surface charge of the red cells and thus leads toaggregation. It may impair cerebral and peripheralblood flow, typically causing lethargy, confusion andimpaired consciousness. It does not usually present aclinical problem in myeloma but is seen more often inWaldenström’s macroglobulinaemia, a rare plasma cellproliferative disease with an IgM paraprotein.
Renal failure in myeloma results from:
● a direct nephrotoxic effect of free light chains● hypercalcaemia● hyperuricaemia and urate nephropathy● infection● amyloid deposition in the kidneys● plasma cell infiltration of the kidneys.
The effects upon normal haematological functioninclude:
● increased susceptibility to bacterial infection(typically septicaemia or pneumonia) as a result ofreduced leucocyte numbers of function together withreduced secretion of normal immunoglobulins
● anaemia or pancytopenia because of marrowreplacement or suppression of haemopoiesis.
Clinical presentationPatients may present with bone pain or fractures (partic-ularly involving the spine and ribs), weight loss,anaemia, infective episodes and renal failure. Other typ-ical symptoms include thirst, polyuria, nocturia (owingto hypercalcaemia and/or renal failure), constipation(from hypercalcaemia), lethargy and confusion.
The diagnosis is based upon finding:
● a high ESR (except in those with only light chainsecretion)
● the paraprotein, by immunoelectrophoresis of bloodand/or urine
● reduced concentrations of normal immunoglobulins(immune paresis)
● radiological evidence of generalised osteopenia orlocal bone destruction
● increased plasma cell numbers in a bone marrowaspirate.
Other laboratory features include:
● anaemia, thrombocytopenia and leucopenia● renal failure● hypercalcaemia.
It should be noted that the term Bence–Jonesproteinuria is of historical interest only. It describes thebehaviour of light chains in boiled urine, now super-seded by immunoelectrophoresis of concentrated urine.
ManagementPatients may become caught in a vicious spiral of hyper-calcaemia, volume depletion and renal failure. The firststep for such patients is i.v. fluid herapy (describedunder acute renal failure, p. 175). Infection should besought and treated, pain controlled, and hypercalcaemiatreated with corticosteroids and/or i.v. bisphosphonateif fluid alone does not control it. Plasma exchange mayoccasionally be needed to control hyperviscosity. Long-term treatment is with chemotherapy such as oralmelphalan, an alkylating agent, or more aggressive reg-imens in younger patients. Bone pain may be controlledwith radiotherapy. Despite treatment, the 50% survivalis about 2 years.
6.4 Platelet disorders
Physiology
The role of platelets is to ‘plug’ defects in damagedvessels, initiate coagulation and promote healing. They:
● adhere to the vessel wall● become activated● degranulate● aggregate.
273
6Platelet disorders
Learning objectives
You should:
● be able to understand the clinical presentations of plateletdisorders
● understand the indications for platelet transfusions
● understand how increased platelet numbers can causethrombophilia.

This can best be thought of as a cascade process, whichbecomes self-perpetuating as they activate one another.
The cascade may be triggered by:
● damage to the vessel wall, which exposes platelets tocollagen and von Willebrand factor (vWF)
● blood coagulation, which leads to thrombinformation
● the activation of other platelets, which causesdischarge of ADP, thromboxane A2 and platelet-derived growth factor
● inflammation, which leads to release of platelet-activating factor from neutrophils and monocytes.
The process of adherence and aggregation is promotedby the prostaglandin thromboxane A2. This is derivedfrom arachidonic acid within platelets and held in checkby prostacyclin, which is synthesised from arachidonicacid in the endothelium.
Adherence leads to platelet degranulation. The con-tents of the granules attract other platelets, causeplatelet aggregation and trigger blood coagulation.Uncontrolled platelet adhesion and thrombosis are pre-vented by secretion of prostacyclin and activation ofprotein C (p. 275) from adjacent healthy endothelium.
Platelet numbers can easily be measured with anautomated counter. Their function can be assessedcrudely by measuring how long a patient bleeds after‘nicking’ the skin (bleeding time) and more accuratelyby platelet aggregation tests in the laboratory.
Conceptually, platelet disorders are straightforward.Patients may have too many or too few platelets orplatelets that do not work properly. Changes in plateletnumber are caused by increased or reduced throm-bopoiesis or platelet consumption. Moderate thrombo-cytopenia (e.g. count < 50 × 109 cells/l) or decreasedplatelet function may result in easy bruising. Severethrombocytopenia (e.g. count < 10 × 109 cells/l) cancause mucosal or intracerebral haemorrhage, in whichcase urgent platelet transfusion is indicated. Transfusedplatelets have such a short life that transfusions mayneed to be repeated daily. Thrombocytosis or increasedplatelet activity raise the coagulability of blood, causingarterial and venous thromboses. Thrombocytosis istreated with antiplatelet drugs or chemotherapy.
Thrombocytopenia
Decreased platelet production is usually caused by one ofthe major haematological diseases or its treatment.Other causes include:
● bone marrow infiltration● myelosuppressive drugs● radiation exposure● megaloblastic anaemia.
Increased platelet loss may be caused by:
● increased activity of the spleen causing‘sequestration’ of platelets
● entrapment of platelets in fibrin deposited in smallvessels (see DIC, p. 279)
● immunity or autoimmunity.
The two immune mechanisms of platelet destructionare:
● antibodies formed against platelet antigens● antigen–antibody complexes (e.g. precipitated by a
drug), which bind to platelets and accelerate theirdestruction.
Idiopathic (immune) thrombocytopenic purpura,marrow suppression and DIC are the most importantconditions to know about.
Idiopathic thrombocytopenic purpura
Idiopathic thrombocytopenic purpura may occuracutely, often in infants or young adults, sometimessoon after a viral infection and with a self-limitedcourse. It may also have a less acute presentation,typically in a young adult or middle-aged woman, and achronic course.
The presentation is with purpura and bleeding,which may be severe. In its subacute form, it may be achance laboratory finding. The disease is frequentlybenign but may lead to serious bleeding, such as GIhaemorrhage, or (rarely) to cerebral haemorrhage.Platelet antibodies can be demonstrated in somepatients and the marrow is active, signifying com-pensatory thrombopoiesis. Steroids or high-doseimmunoglobulin infusions may be used. In the chronicform, steroid therapy may induce a lasting remission.If it does not and thrombocytopenia is severe, splene-ctomy may be effective by removing the site ofplatelet destruction.
Thrombocytosis
Thrombocytosis may result from decreased platelet con-sumption after splenectomy, although it is usually onlytransient. Increased production may be secondary toany marrow stimulus such as bleeding, haemolysis sur-gery or as a reaction to inflammation or malignancy. Theone primary disease is essential thrombocythaemia, anuncommon myeloproliferative disease characterised byincreased numbers of variably active platelets. As hasbeen explained above, patients may either develop arte-rial/venous thromboses or bleed. It is characteristicallya disease of middle or old age. As well as thrombocyto-sis, there may be normocytic anaemia and leucocytosis
274
Haematology6

(caused by myeloproliferation) and either splenomegalyor hyposplenism owing to splenic infarction. Themarrow shows megakaryocyte proliferation withother myeloproliferative features. Treatment is withantiplatelet drugs or chemotherapy. Leukaemic trans-formation or myelofibrosis may be late complications.
Antiplatelet therapy
Particularly in ischaemic heart disease and transientischaemic attacks, antiplatelet therapy is of provenvalue in preventing arterial thromboses. It may also pro-tect against venous thrombo-embolism. Aspirin is themost effective drug. It works by irreversible inhibitionof cyclo-oxygenase, the key enzyme in prostaglandinsynthesis. Since cyclo-oxygenase inhibition preventssynthesis of endothelial prostacyclin as well as plateletthromboxane, aspirin has prothrombotic as well asantithrombotic effects. These effects are dose related andthe balance can be shifted towards antithrombosis bygiving lower doses. The main side-effects are dyspepsia,peptic ulceration and GI haemorrhage. Other plateletinhibitors such as sulfinpyrazone and dipyridamolemay also be used but there is less evidence that they areclinically effective. Platelet glycoprotein receptor antag-onists are a new class of drug currently undergoing con-trolled clinical trial for treatment of cardiovasculardisease.
Whether antiplatelet or anticoagulant therapy ismore appropriate depends on the pathophysiology ofthe disease in question: platelet activation is central to the arterial thrombo-embolism of cerebrovascular (p. 225) and ischaemic heart disease (p. 14), so aspirin isthe treatment of choice to prevent them. Intracardiacthrombosis and venous thrombo-embolism are moredependent on the coagulation pathways and are, there-fore, treated with anticoagulants.
6.5 Coagulation disorders
Physiology
The functions of the coagulation and fibrinolytic systemare to:
● maintain the fluidity of blood● plug damaged vessels● prevent the uncontrolled propagation of blood clot● remove clot as healing proceeds.
Coagulation is a complex process, triggered by damageto tissue or the vessel wall. This exposes collagen orreleases tissue thromboplastin, which activate the coag-ulation cascade, summarised in Figure 66. Platelet acti-vation also triggers coagulation. The final result isconversion of fibrinogen to an insoluble fibrin plug. Thisis catalysed by thrombin formed from its precursor pro-thrombin. Within the cascade, there are the intrinsic andextrinsic systems. These converge on a common path-way that activates the final two steps of thrombin andfibrin formation. Calcium is essential to coagulation.The coagulation factors are synthesised in the liver; anumber of them are dependent on vitamin K.
There are also mechanisms that restrain coagulation,notably the thrombin inhibitor antithrombin III. ProteinC is another. This protein is activated by thrombin for-mation and feeds back to suppress the coagulation cas-cade. Protein S is a cofactor for that inhibitory pathway.
Once formed, clots are lysed by plasmin with therelease of fibrin degradation products (FDPs). The con-version of plasminogen to plasmin is promoted bytissue plasminogen activator (tPA). The dynamic
275
6Coagulation disorders
Learning objectives
You should:
● understand how coagulation defects are acquired
● know how warfarin and heparin work, when to use them,and their potential dangers
● understand the concept of hypercoagulability(thrombophilia) and its causes.
Collagen Calcium, platelet phospholipid & coagulation co-factors
Tissuethromboplastin
Common pathway
ThrombinProthrombin
Fibrinogen Fibrin
Plasmin
Plasminogen
Streptokinase
Tissueplasminogenactivator
Fibrinremoval
Fibrinformation
Extrinsic system
Intrinsic system
Fig. 66 The coagulation and fibrinolytic systems.

equilibrium between the formation of fibrin and itsremoval maintains haemostasis.
Warfarin prolongs coagulation by preventing thehepatic synthesis of vitamin K-dependent clotting fac-tors. Heparin potentiates antithrombin III. Fibrinolyticdrugs include streptokinase and recombinant tPA, bothof which potentiate plasmin formation and the break-down of fibrin.
Tests of coagulation and fibrinolysis
Two tests are in general use (Fig. 67): the prothrombintime (PT) and activated partial thromboplastin time(APTT). The PT tests the extrinsic system, commonpathway and fibrin formation. It is sensitive to de-ficiency of the vitamin K-dependent clotting factors andis prolonged by:
● malnutrition● warfarin● hepatocellular dysfunction● fat malabsorption● consumption of clotting factors, in DIC.
When the prothrombin time is used to measure theeffect of warfarin, it is performed with standardisedreagents (so that it is reproducible from one laboratoryto another) and expressed as the international nor-malised ratio (INR).
The APTT tests the intrinsic system, common path-way and fibrin formation. It is affected by:
● liver disease● circulating inhibitors of coagulation, including
heparin● DIC.
The APTT is prolonged by deficiency of the vitamin K-dependent clotting factors but less so than the PT. If pro-longation of either the PT or APTT is caused by defi-ciency of a factor, or factors, it can be ‘corrected’ in thelaboratory by mixing normal plasma (replete with thosefactors) with the patient’s plasma. If the prolonged PT or
APTT is caused by an inhibitor, it cannot be ‘corrected’in this way. The APTT is more sensitive than the PT tocirculating inhibitors and is used to monitor heparintherapy.
The only fibrinolytic test that is widely used is meas-urement of FDPs. This detects excess fibrinolysis but isso insensitive that it can only detect DIC. There is also asensitive test for a specific breakdown product of fibrin,D-dimer.
Coagulation defects
Inherited coagulation defects
Only von Willebrand disease is common enough to bedescribed here.
Von Willebrand diseaseVon Willebrand factor (vWF) is a large protein releasedby endothelium that plays an important role in plateletadhesion and aggregation; it is also necessary as acarrier for factor VIII in plasma. Von Willebrand diseaseis by far the commonest inherited disorder of coagula-tion and a variety of types have now been described.These differ in whether vWF is present in reduced quan-tity (type I, type III) or is functionally defective (type II,subtypes A, B, M, N). The commonest (type I) is inherit-ed as an autosomal dominant trait. Typically, there is alifelong history of mild-to-moderate bleeding, usuallyfrom mucosal surfaces, but patients may not be awarethat they have a bleeding disorder until they undergosurgery or have an accident, when bleeding may beexcessive.
Diagnosis and classification of von Willebrand dis-ease is not always easy, because results of laboratoryinvestigations often vary day-to-day. Classically,patients have a prolonged bleeding time and APTT, butboth may be normal. Generally, quantitative abnormal-ities are detected by measuring plasma vWF concentra-tion; functional defects produce abnormal plateletaggregation in response to ristocetin (ristocetin cofactoractivity). Treatment depends upon the situation and thetype of von Willebrand disease but may involve tranex-amic acid (an inhibitor of fibrinolysis), desmopressin (asynthetic analogue of antidiuretic hormone (argininevasopressin; see p. 299), which causes release of vWFfrom endothelial stores), vWF concentrate or, occasion-ally (type IIB), platelet transfusions.
Acquired coagulation defects
The causes of acquired coagulation disorders are listedunder the coagulation tests above. You should remem-ber that any GI disease that affects fat absorption maycause vitamin K deficiency. Hepatocellular dysfunction
276
Haematology6
Intrinsicsystem
Extrinsicsystem
FibrinPlasmin
Commonpathway
Fibrin degradationproducts D-dimer
Activated partialthromboplastin time
Prothrombin time
Fig. 67 Tests of coagulation and fibrinolysis.

impairs coagulation factor synthesis to such an extentthat the PT is a very sensitive test of hepatocellular fail-ure. Clinical implications of this are considered underliver disease (p. 139).
The effects of warfarin and heparin are discussed inthe next section and ‘consumption coagulopathy’ is dis-cussed under DIC (p. 279). Vitamin K is given parenter-ally to treat malabsorption and to reverse the action ofwarfarin. It may also improve clotting factor synthesisin mild liver disease. Otherwise, the treatment forcoagulation factor deficiencies is infusion of fresh frozenplasma.
Anticoagulation and fibrinolytic therapy
Anticoagulation and fibrinolytic therapies are primarilyused in cardiovascular disease. Anticoagulation is con-sidered here and fibrinolysis under acute myocardialinfarction (p. 17).
HeparinUnfractioned heparin consists of natural polysaccha-rides of various molecular weights, which have analmost immediate anticoagulant effect. It markedlypotentiates the action of antithrombin III and inhibitsserine proteases such as activated Factor X (Xa). It has tobe given parenterally, and either by continuous infusionor as several injections per day because it has a shorthalf-life. It prevents the formation of new clot and shiftsthe balance of fibrin formation/lysis in the direction oflysis, but it is not primarily a fibrinolytic drug. It is indi-cated whenever an immediate anticoagulant effect isrequired. It may also be used to prevent thrombosis(usually venous). There are two common schedules:
● prophylaxis: heparin 5000 units subcutaneously 8- or12-hourly
● treatment: heparin 5000 units as an i.v. loading dosefollowed by 1000–2000 units hourly by continuousi.v. infusion.
Sensitivity to heparin varies from patient to patient. Youshould check the APTT every 6–12 hours until it is pro-longed approximately twofold (depending upon theindication) and stable. Warfarin can be introduced whilethe patient is on heparin. Heparin should be continuedat least until the patient is stabilised on warfarin, and fora minimum of 5 days in venous thrombo-embolism.Prolonged heparin treatment (> 5 days) can causethrombocytopenia. More prolonged treatment (as inpregnancy) can cause osteoporosis. Unfractionatedheparin is now being superceded by the newer low-molecular-weight heparin (LMWH) preparations,which may eventually replace it entirely. These com-pounds act mainly by inhibition of factor Xa and have amuch more predictable effect for a standard dose in dif-
ferent patients. This enables treatment to be given byonce or twice daily subcutaneous injections, often with-out need for laboratory monitoring. It also allows out-patient treatment of venous thrombo-embolism inselected patients.
WarfarinWarfarin and other coumarins are used for long-termanticoagulation. They take about 48 hours to becomeeffective, governed by the half-lives of the vitamin K-dependent factors. Warfarin is strongly protein bound.Anything that affects this binding will affect sensitivityto it. Liver function and levels of the vitamin K-dependent factors also affect sensitivity to warfarin tosuch an extent that patients may be ‘autoanticoagu-lated’. For this reason, you must always check the PTbefore starting therapy. Warfarin is given as loadingdoses over 2 days and a maintenance dose adjusted toprolong the PT (reported as the INR, see p. 276) by afactor of 1.5–4 (depending upon the indication). Theeffect of warfarin is unpredictable in alcoholism andliver disease. Important drug interactions are:
● potentiation of warfarin by displacement fromprotein binding, e.g. salicylates, sulphonamides
● potentiation of warfarin by inhibiting warfarinmetabolism, e.g. metronidazole, amiodarone,cimetidine, isoniazid
● potentiation of the effect of warfarin on the liver, e.g.tetracycline
● reduced effect of warfarin by inducing warfarinmetabolism, e.g. phenobarbitol, carbamazepine,rifampicin.
Indications for anticoagulationThere are a number of indications for anticoagulationtherapy;● short-term prophylaxis (usually low-dose heparin)
— prolonged recumbency— immobilisation, e.g. traction— severe heart failure— malignant pelvic disease or impediment to venous
flow— postoperatively, especially after major lower limb
orthopaedic surgery— in pregnancy in women at high risk of venous
thrombo-embolism.● acute (usually heparin)
— venous thrombo-embolism: deep venousthrombosis, pulmonary embolism
— arterial disease: peripheral arterial thrombosis orembolism, cardiac mural thrombo-embolism,unstable angina
● chronic (usually warfarin)— continued treatment of acute indications
277
6Coagulation disorders

— prophylactically in cardiac dysrhythmia (e.g.atrial fibrillation, especially if intermittent orassociated with mitral stenosis), poor cardiacfunction (cardiomyopathy), prosthetic heart valveor vascular prosthesis, after full-thickness anteriormyocardial infarct.
Contraindications to anticoagulationThere are both absolute and relative contraindications toanticoagulation therapy:
● absolute— cerebral haemorrhage— GI, urinary tract or other haemorrhage— active peptic ulceration
● relative— liver disease— alcoholism— likely poor compliance— concomitant drug therapy likely to cause
instability.
Side-effects of anticoagulationEven if the contraindications listed above are observed,over-anticoagulation may occur and directly causehaemorrhage from the mucosae, into the skin or into thetissues (e.g. cerebral haemorrhage). That should be rareprovided the dosage schedule and degree of anticoagu-lation are appropriately chosen and monitored.Warfarin can be reversed within a few hours by givingi.v. vitamin K or immediately by infusion of fresh frozenplasma or clotting factor concentrate. Heparin actionwill wear off after about 4 hours but can be reversedrapidly by i.v. protamine.
Sometimes, a decision has to be taken to give antico-agulation therapy to a patient at risk of haemorrhagebecause the risk of not providing anticoagulation is evenmore unacceptable. Your responsibilities as a prescriberare to:
● vet all patients’ suitability for anticoagulation interms of compliance and understanding
● screen for underlying diseases which may complicatetreatment
● advise patients about the risks and benefits oftreatment and the need to abstain from alcohol anddrugs (including over-the-counter drugs) which maycomplicate anticoagulation
● ensure adequate monitoring of the INR.
Thrombophilic states
Thrombophilic states may be inherited or acquired.
Inherited thrombophilia
The most common cause of inherited thrombophilia is apoint mutation in the gene encoding factor V (factor V
Leiden), which causes resistance to activated protein Cand allows thrombosis to progress uninhibited.Antithrombin III deficiency is an autosomal dominantcondition that is incompatible with life in its homozy-gous form. The prevalence of heterozygosity is about 1per 2000. Protein C and protein S deficiency are simi-larly inherited but are less common. A prothromboticvariant of the prothrombin gene is also well docu-mented.
Acquired thrombophilia
Antiphospholipid syndrome is an uncommon autoim-mune disorder in which an autoantibody triggers coag-ulation. It may be associated with lupus (lupusanticoagulant) and can cause recurrent fetal loss inyoung women. Anticardiolipin antibodies are oftenpresent. Lesser degrees of thrombophilia occur in anyillness that increases fibrinogen levels, in pregnancy andin women taking the oral contraceptive, (which is par-ticularly prone to cause thrombosis in women with thefactor V Leiden mutation).
Clinical presentationInherited thrombophilia presents with thrombosis at anearly age, recurrent thrombosis, thrombosis at unusualsites (e.g. mesenteric vein) and/or a positive familyhistory. The thromboses are usually venous but maybe arterial.
Screening for thrombophiliaThe following are indications to screen a patient forthrombophilia:
● venous or arterial thrombo-embolism in a youngperson (aged < 40 years)
● recurrent venous thrombosis● family history of venous thrombo-embolism● recurrent fetal loss.
InvestigationYou should check a full blood count to exclude poly-cythaemia (p. 266) and thrombocytosis (p. 274): measureplasma fibrinogen and the PT and APTT. Other investi-gations include anticardiolipin antibodies and directmeasurements of activated protein C resistance, proteinC, protein S and antithrombin III. These should be donebefore starting anticoagulants.
ManagementSignificant thrombophilia requires anticoagulation tocover high-risk situations (e.g. pregnancy, surgery) and,sometimes, life long.
278
Haematology6

6.6 Disseminated intravascularcoagulation
Pathophysiology
DIC describes widespread activation of the coagulationpathways secondary to a severe illness. There isintravascular fibrin deposition, which may causemicrovascular occlusion. Clotting factors and plateletsare consumed and may become so depleted that thepatient bleeds. Fibrin in the microvasculature may causemechanical damage to the red cells (microangiopathichaemolysis) leading to haemolytic anaemia, with schis-tocytes (fragmented cells, p. 258) on the blood film.
CausesDIC can occur as a result of a number of underlyingcauses:
● severe sepsis (p. 403)● incompatible blood transfusion (p. 266)● trauma
— particularly crush injury● systemic diseases
— disseminated malignancy— acute pancreatitis— acute promyelocytic leukaemia
● obstetric situations— retained fetal products— antepartum haemorrhage— amniotic fluid embolism.
These factors all have in common the systemic release oftoxins which trigger coagulation.
Clinical effects
The dominant clinical features are usually those of theunderlying illness. Often a fall in the platelet count is thefirst sign that the patient is developing DIC. The accom-
panying features of DIC can be worked out if you thinksystem by system of the effects of small vessel occlusion:
● CNS: impaired consciousness, fits, focal neurologicalsigns
● lungs: acute respiratory distress syndrome● kidneys: acute renal failure● gut: bowel infarction● skin: ischaemic ulceration, digital gangrene.
In addition, there may be haemorrhage into any or all ofthese tissues. Petechial skin haemorrhages, purpura andbleeding from the mucosae may develop.
Investigations
The investigations follow directly from the pathophysi-ology. The diagnostic features of DIC are:
● thrombocytopenia (< 100 × 109 cells/l)● prolonged PT and APTT● raised FDPs and D-dimer● reduced plasma fibrinogen● signs of microangiopathic haemolytic anaemia
(p. 263).
Treatment and natural history
The development of DIC is a bad prognostic sign in adisease that often already has a bad prognosis. The mainaim of treatment is to control the underlying disease;without that, treating the DIC is unlikely to be of value.The treatment of DIC consists of replacing the coagula-tion factors (as fresh frozen plasma) and platelets. Thisnarrative has described severe, fulminating DIC. Low-grade DIC may develop in association with malignantdisease, vasculitis and other illnesses. Again, treatmentis aimed at the underlying disease, and prognosis isdetermined by the disease itself more than the DIC.
Other related diseases
Detailed knowledge of the haemolytic uraemic syn-drome, thrombotic thrombocytopenic purpura andother such rare diseases is not ‘core’. You should simplybe aware that the pathophysiological processes ofconsumption coagulopathy and microangiopathichaemolysis can occur in settings other than classicalDIC.
279
6Disseminated intravascular coagulation
Learning objectives
You should:
● know the causes of DIC
● understand its clinical effects and treatment.

Haematology6
280
Multiple choice questions
1. The following statements are true:a. Hypocalcaemia causes prolongation of the
prothrombin timeb. The prothrombin time is a sensitive test of
hepatocellular dysfunctionc. The activated partial thromboplastin time (APTT)
is prolonged by unfractionated heparin therapyd. The effect of heparin is reversed by vitamin Ke. Deep venous thrombosis can be reliably
diagnosed by measuring fibrin degradationproducts (FDPs)
2. In a patient with severe thrombocytopenia:a. Rectal bleeding is usually the first symptomb. Examination of the optic fundi should be
performed regularlyc. There is a risk of cerebral haemorrhaged. Corticosteroids are given to prevent
haemorrhage, whatever the causee. Platelet transfusions can be expected to cause
prompt normalisation of the platelet count
3. The following may cause a microcytic anaemia:a. Sickle cell diseaseb. The thalassaemiasc. Anaemia of chronic diseased. Anticonvulsant therapye. Haemolysis, whatever the cause
4. In lymphoma:a. If a newly presenting patient has generalised
lymphadenopathy, the diagnosis is more likely tobe non-Hodgkin’s lymphoma than Hodgkin’sdisease
b. Early stages of Hodgkin’s disease can be cured byradiotherapy alone
c. Lymphocyte predominance is a favourablehistological sign in Hodgkin’s disease
d. Pain in lymph nodes after alcohol is very typicalof non-Hodgkin’s lymphoma
e. Bone marrow transplantation has greatlyimproved the prognosis of low-grade non-Hodgkin’s lymphoma
5. The following statements are true:a. A neutrophil count of only 0.8 × 109 cells/l is a
major risk for infectionb. A neutrophil count in a febrile patient of 25 × 109
cells/l reflects mostly the production of newneutrophils from the bone marrow
c. In a patient with less than 0.1 × 109 cells/lneutrophils and a fever, treatment with antibioticsshould await the results of blood culture
d. Neutropenia is common in AIDSe. Neutropenia can be caused by carbimazole
therapy
Case history questions
Case history 1
1. Suggest two important questions which you shouldask
2. Name two haematological investigations which itwould be appropriate to perform
3. Name two possible contraindications toanticoagulation in this case
4. If a decision were made to proceed withanticoagulation, describe two pieces of advice thatyou would give to the patient and his wife
Case history 2
1. Which of the following actions would beappropriate:
Self-assessment: questions
Your consultant is concerned that a 73-year-old manin atrial fibrillation is at risk of stroke and asks you toanticoagulate him. You are aware that the patient isvaguely confused and the nursing staff on the wardhave received a telephone call from a neighbour stat-ing that he has become increasingly reclusive. Hehas often been noted to be unsteady on his feet andhas once been found lying in the road. At visiting timeyou have a chance to meet his wife to discuss the plan to use anticoagulation therapy and to givefurther information.
You are called on a Sunday to see a 63-year-old manwith non-Hodgkin’s lymphoma and a fever of 38.6˚C.He is in reverse barrier nursing because his totalwhite cell count is 0.2 × 109/l and his platelets are15 × 109/l despite daily platelet transfusions. Hereceived intensive chemotherapy (fludarabine) 15 days previously and has been leukopenic for 7 days.You put on your gown, mask and gloves to examinethe patient and find that he has no symptoms exceptthose of the fever, mild headache, sweating and anuncomfortable feeling around his bottom.

6
281
a. Order a chest X-rayb. Take a blood culturec. Do a blood countd. Do a digital rectal examinatione. Start him on oral amoxycillin
2. What actions should you now take?
a. Look at his white cell and platelet counts fromthat morning
b. Arrange for transfer to an intensive care unit(ITU)
c. Examine him again carefullyd. Treat him for a possible fungal infectione. Do blood cultures, including a fungal blood
culture
3. Should you:a. Speak to his consultant at homeb. Arrange a chest X-rayc. Arrange an induced sputumd. Call the physiotherapist ine. Arrange for a special mattress to help his
developing pressure sore4. Give a differential diagnosis and your thoughts on
management
Data interpretation
1. Suggest a cause for the findings described in Table63 for each of six patients.
2. A 22-year-old patient is admitted to hospital 48hours after a paracetamol overdose, complaining ofhaematuria. The prothrombin time is 60 s (control 18 s). What is the likely diagnosis?
3. A 70-year-old man is referred to hospital because heis anaemic (Hb 82 g/dl, MCV 90 fl, platelets 150 ×109 cells/l ESR 130 mm/h. Name a possiblehaematological diagnosis. What biochemical testsshould be performed?
Picture questions
1. This skull radiograph (Fig. 68) was taken from a73-year-old patient who presented in acute renalfailure.a. What abnormality do you see?b. How could the skull radiograph be relevant to
the renal failure? Be as precise as you can.c. Name one rapid biochemical test which might
establish a link.d. What would be your immediate management?
2. A previously fit 28-year-old woman has lost weight.She has night sweats, pruritus and weight loss. Shehas noticed pain in the cervical region after drinkingalcohol. Her GP arranged a chest radiograph whichwas thought to show hilar enlargement. Figure 69shows thoracic computed scans before (a) and after(b) an intravenous contrast agent. T, trachea; A,aortic arch, B, left brachiocephalic vein.a. What abnormality is shown?b. Suggest, in order of likelihood, a differential
diagnosis
Self-assessment: questions
The senior nurse on the ward encourages you to starthim on the ‘usual’ antibiotics (ceftazidime and gen-tamicin). His fever is still elevated 6 hours laterdespite antibiotics and he looks more toxic. The con-sultant suggests that you add metronidazole to hisregimen. Four days later you are also on call and areasked to see him again. He has had fever up to38.3˚C for 4 days despite ceftazidime, gentamicin,metronidazole and 2 days of vancomycin. He nowhas a dry cough, in addition to a painful bottom.
Table 63 Data obtained for six patients
A B C D E F
Hb (g/dl) 42 78 185 91 87 84MCV (fl) 123 69 96 85 80 86WBC (× 109/l) 2.3 13.6 15.3 8.7 63.0 1.7Platelets (× 109/l) 60 200 600 180 140 48
Fig. 68 A skull radiograph of a patient in acute renal failure.
On examination, you find crackles at the left base thatdo not clear on coughing and a black and red peri-anal area about 2 cm across with surrounding ery-thema.
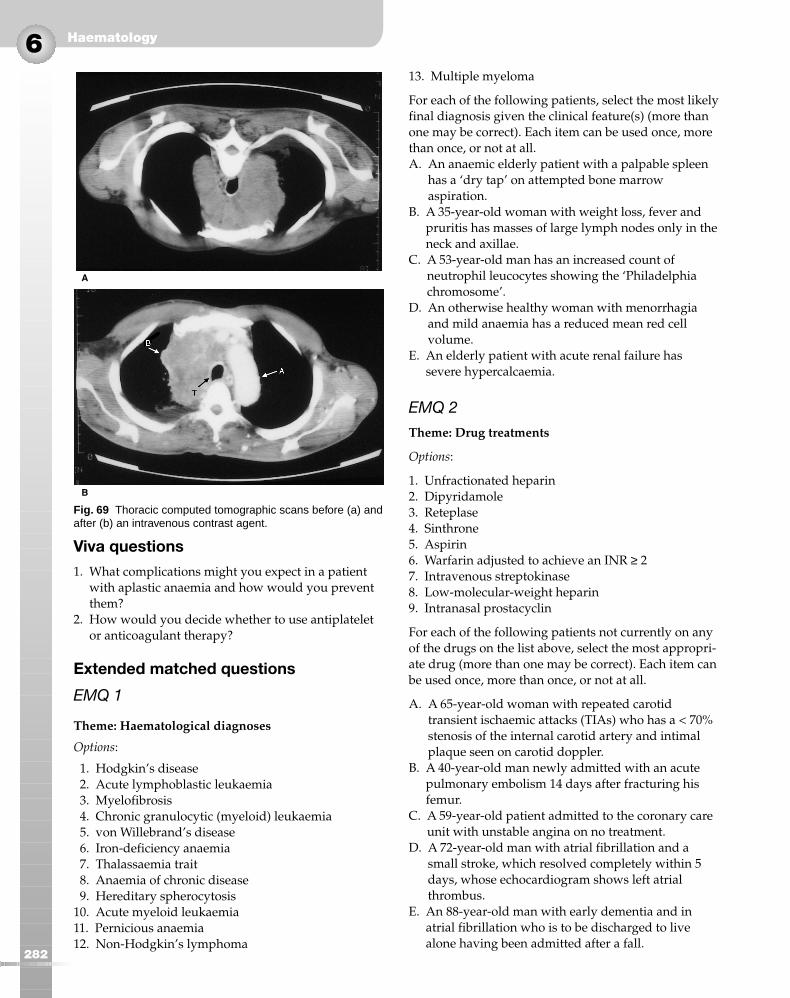
Haematology6
282
Viva questions
1. What complications might you expect in a patientwith aplastic anaemia and how would you preventthem?
2. How would you decide whether to use antiplateletor anticoagulant therapy?
Extended matched questions
EMQ 1
Theme: Haematological diagnoses
Options:
1. Hodgkin’s disease2. Acute lymphoblastic leukaemia3. Myelofibrosis4. Chronic granulocytic (myeloid) leukaemia5. von Willebrand’s disease6. Iron-deficiency anaemia7. Thalassaemia trait8. Anaemia of chronic disease9. Hereditary spherocytosis
10. Acute myeloid leukaemia11. Pernicious anaemia12. Non-Hodgkin’s lymphoma
13. Multiple myeloma
For each of the following patients, select the most likelyfinal diagnosis given the clinical feature(s) (more thanone may be correct). Each item can be used once, morethan once, or not at all.A. An anaemic elderly patient with a palpable spleen
has a ‘dry tap’ on attempted bone marrowaspiration.
B. A 35-year-old woman with weight loss, fever andpruritis has masses of large lymph nodes only in theneck and axillae.
C. A 53-year-old man has an increased count ofneutrophil leucocytes showing the ‘Philadelphiachromosome’.
D. An otherwise healthy woman with menorrhagiaand mild anaemia has a reduced mean red cellvolume.
E. An elderly patient with acute renal failure hassevere hypercalcaemia.
EMQ 2
Theme: Drug treatments
Options:
1. Unfractionated heparin2. Dipyridamole3. Reteplase4. Sinthrone5. Aspirin6. Warfarin adjusted to achieve an INR ≥ 27. Intravenous streptokinase8. Low-molecular-weight heparin9. Intranasal prostacyclin
For each of the following patients not currently on anyof the drugs on the list above, select the most appropri-ate drug (more than one may be correct). Each item canbe used once, more than once, or not at all.
A. A 65-year-old woman with repeated carotidtransient ischaemic attacks (TIAs) who has a < 70%stenosis of the internal carotid artery and intimalplaque seen on carotid doppler.
B. A 40-year-old man newly admitted with an acutepulmonary embolism 14 days after fracturing hisfemur.
C. A 59-year-old patient admitted to the coronary careunit with unstable angina on no treatment.
D. A 72-year-old man with atrial fibrillation and asmall stroke, which resolved completely within 5days, whose echocardiogram shows left atrialthrombus.
E. An 88-year-old man with early dementia and inatrial fibrillation who is to be discharged to livealone having been admitted after a fall.
A
B
Fig. 69 Thoracic computed tomographic scans before (a) andafter (b) an intravenous contrast agent.

6
283
Objective structured clinical examination(OSCE)
This is a 5 minute station with a normal volunteer on acouch.Examiner: This 35-year-old man has been found to have
a mediastinal mass and lymphoma is suspected. Pleaseexamine him.
After you have examined him:Examiner: What features of any lymph nodes found
on examination would help your differentialdiagnosis?
Self-assessment: questions

Haematology6
284
Multiple choice answers
1. a. False. This is true in vitro but hypocalcaemiasevere enough to have the same effect in vivowould be incompatible with life.
b. True. Because hepatocellular dysfunction impairsthe synthesis of vitamin K-dependent clottingfactors.
c. True. This is used as a measure of heparinisation.d. False. Vitamin K reverses the action of warfarin;
protamine reverses heparin action.e. False. FDPs are raised by massive intravascular
fibrin formation, as in disseminated intravascularcoagulation (DIC), and may be increased inthrombotic conditions but are not sensitive orspecific enough to be a useful diagnostic test.Measurement of D-dimer is sensitive enough todetect venous thrombosis but is not specific.
2. a. False. A purpuric rash or easy bruising on thelimbs or trunk are more likely first symptoms.
b. True. The appearance of retinal haemorrhagesindicates that the patient is at risk ofhaemorrhage.
c. True.d. False. Corticosteroids are given to some patients
with idiopathic thrombocytopenic purpura butthrombocytopenia is often not steroid responsive.
e. False. The goal is to prevent bleeding, not tonormalise the blood count.
3. a. False.b. True. Thalassaemia is one of the causes of
microcytosis.c. True.d. False. Anticonvulsants may cause macrocytosis.e. False. Haemolysis increases the reticulocyte count
which, since reticulocytes are large, causesmacrocytosis.
4. a. True. In Hodgkin’s disease, thelymphadenopathy is often confined to a singlesite, most commonly the neck, at presentation.
b. True.c. True.d. False. Alcohol-related pain is typical of
Hodgkin’s disease.e. False. There is no evidence for this in clinical
trials to date.
5. a. False. A minor risk. It is when the count fallsbelow 0.5 × 109 cells/l and particularly 109 cells/lthat the risk becomes major.
b. False. Mostly neutrophil release from themarginating pool. The left shift (or band forms) isthe proportion of new neutrophils from themarrow.
c. False. Immediate intravenous broad-spectrumantibiotics are indicated.
d. True. Especially caused by the drugs zidovudineand ganciclovir.
e. True. Neutropenia occurs in 1:10 000 patientstreated with carbimazole for thyrotoxicosis.
Case history answers
Case history 1
1. Whilst anticoagulation is indicated to preventcerebral embolism in patients with atrial fibrillation,you must not initiate it unless you are sure it will besafe. Age, in itself, is not a contraindication, butthere are aspects of the history which suggest thereare other contraindications. There are strong hintsthat he may be abusing alcohol. The confusion isworrying because someone who is having falls andbecomes confused could have a subduralhaematoma, which is an absolute contraindication toanticoagulation. You should ask about:
● his alcohol intake● dyspepsia or history of blood loss● his likelihood of taking tablets and attending for
anticoagulant monitoring reliably.
2. You should measure his prothrombin time beforestarting anticoagulants; autoanticoagulation iscommon and would affect your choice of loadingdose. It would also increase your anxiety about thepossibility of alcohol abuse (prolonging the PT bycausing hepatocellular damage). You should alsocheck the haemoglobin concentration; anaemiawould be a relative contraindication toanticoagulation or should, at least, be investigatedbefore anticoagulation.
3. These might include
● alcohol abuse● dementia (if it would interfere with compliance)● difficulty attending for anticoagulant monitoring● inability of wife or carer to supervise treatment● history of GI bleeding or anaemia● history of cerebral haemorrhage● concomitant drug therapy affecting the stability of
warfarin levels● falls.
Self-assessment: answers

4. Advice would include:
● avoid alcohol● avoid non-steroidal analgesics and aspirin● report any excessive or unusual bleeding
immediately● attend regularly for anticoagulant monitoring.
You should give the patient and his wife an informationsheet about anticoagulants, listing drugs to be avoided.
Case history 2
1. a. True. This is always appropriate in febrileneutropenia, even in the absence of chest signs orsymptoms.
b. True. Very important.c. False. Unnecessary if done that morning, which it
will have been to see whether platelettransfusions were required.
d. False. You must inspect his anal area as he has asymptom there, but a formal rectal examination isnot appropriate as it may cause bacteraemia.
e. False. Not appropriate; i.v. therapy required withbroader coverage.
2. a. True.b. False. He is not that ill and you would take him
out of protective isolation. In fact, even if he wereso ill and requiring ventilation, neutropenicleukaemic patients do so badly in ITU that it isused rarely.
c. True. Always true in this group of patients.Especially mouth, chest, skin and rectal area (p. 268).
d. True. Candidaemia or invasive aspergillosis arenow more likely. This group of patients withpersistent fever during neutropenia have a 30%mortality, mostly as a result of fungal infection.
e. True. It is always worth repeating blood cultures,even though he is on antibiotics, as these patientsget breakthrough bacteraemia.
3. a. True. Now you have a complex problem with twopossible sites of infection.
b. True. If one was not done that day. A CT scan ofhis chest is a much better investigation andshould be done within the next 24 hours.
c. False. Not useful in febrile neutropenia (unlikeAIDS), although pneumocystis pneumonia is adiagnostic possibility because fludarabinetherapy ‘paralyses’ T cells.
d. False. As the cough is not productive, of no benefit.e. False. His lesion is almost certainly not a pressure
sore but a developing infection called icthymagangrenosum (Pseudomonas aeruginosa).
4. He probably has two focal infections: one in hislungs and the other perirectally. The perirectalinfection is likely to be caused by Pseudomonasaeruginosa, other bacteria including anaerobes,Aspergillus or mucormycosis. His lung disease maybe caused by any of these or other bacteria orpossibly Pneumocystis. He needs large doses ofantifungals (amphotericin), a CT scan of the lung,which is helpful diagnostically, bronchoscopy, withlavage, to obtain material for microscopy andculture, and a biopsy of his rectal lesion, all as soonas possible.
Data interpretation answers
1. Patient A. This is a fairly typical picture ofmegaloblastic anaemia caused by vitamin B12 orfolate deficiency with severe macrocytosis,leucopenia and thrombocytopenia; hypersegmentedneutrophils on the blood film would confirm thediagnosis, as would bone marrow aspirationalthough this is not done as a routine. Haematinicsshould be measured.
Patient B. This is a moderately severe microcyticanaemia (the differential diagnosis is given on p. 260). Iron deficiency is the most likely cause. Theleucocytosis may be caused by infection or inflamma-tion but could signify acute or subacute blood loss, apossible cause for the iron deficiency.
Patient C. There is erythrocytosis, thrombocytosisand leucocytosis, suggestive of polycythaemia vera.The red cell mass is likely to be increased andsplenomegaly may be present.
Patient D. There is a normocytic anaemia with nor-mal white cell and platelet counts. This would be typ-ical of the anaemia of chronic disease (e.g. renalfailure) but could also be caused by a mixed deficien-cy. This would be suggested by an increased: red celldistribution width (RDW) and a ‘dimorphic’ bloodfilm. You should examine and investigate the patientfor an underlying disease.
Patient E. Here, there is a normochromic anaemiawith mild thrombocytopenia, but the striking abnor-mality is the marked leucocytosis; this is typical of achronic leukaemia. A differential white cell count andblood film would be crucial.
Patient F. This shows moderate pancytopenia andcould be seen in a patient with acute leukaemia,hypoplastic anaemia or after chemotherapy (see p. 262 for a fuller list of causes).
2. Prolongation of the prothrombin time may becaused by warfarin, consumption of clotting factorsas in DIC, vitamin K deficiency or deficiency of thevitamin K-dependent clotting factors owing to liverdisease. At 48 hours after a massive paracetamol
6
285
Self-assessment: answers

Haematology6
286
overdose is about the time when hepatocellulardamage becomes apparent, and the prothrombintime is quite a sensitive test for this. The likelydiagnosis is acute hepatocellular necrosis caused byparacetamol.
3. An extremely high ESR is usually caused bymultiple myeloma, giant cell arteritis orchronic/severe infection/inflammation. This patientalso has a moderate normochromic anaemia.Multiple myeloma is a likely diagnosis. Renal failureand hypercalcaemia are common in myeloma. Youshould measure plasma urea, creatinine andcalcium. The definitive diagnosis is made by plasmaand urine immunoelectrophoresis, bone marrowexamination and skeletal survey.
Picture answers
1. a. Figure 68 shows multiple ‘punched-out’ lesions,in keeping with multiple myeloma or anothercause of osteolytic metastases.
b. Hypercalcaemia is a common cause of acute renalfailure in multiple myeloma. Volume depletion isan important and reversible effect ofhypercalcaemia. ‘Myeloma kidney’ may causerenal failure without hypercalcaemia. This isbecause of tubular damage by the paraprotein,secondary hyperuricaemia, amyloidosis andinfection.
c. With the history given, you should immediatelymeasure serum calcium. You should also arrangeserum and urine immunoelectrophoresis toidentify a paraprotein but that is not anemergency investigation.
d. You should assess the patient’s volume status,using a central venous pressure line if necessary,and give saline to increase urinary calciumexcretion. Corticosteroid and/or bisphosphonatetherapy may be needed as second-line treatmentfor hypercalcaemia.
2. a. There is a large soft tissue attenuation masswithin the mediastinum abutting the trachea andaortic arch and compressing the superior venacava (which cannot be seen). This is almostcertainly a large lymph node mass.
b. Although you are not told that she has cervicallymphadenopathy, the scenario makes it verylikely that she is describing alcohol-related lymphnode pain, which is very specific to Hodgkin’sdisease. The combination with mediastinallymphadenopathy makes the diagnosis ofHodgkin’s by far the most likely diagnosis. Othercauses of a mediastinal soft tissue mass includenon-Hodgkin’s lymphoma (not associated with
alcohol-related pain) or sarcoidosis (the secondmost likely possibility in this case). Unlikelycauses are tuberculosis, retrosternal thyroid(unlikely), thymic tumour, dermoid or lungcancer nodes (especially small cell).
Viva answers
1. Aplastic anaemia usually affects the red cells, whitecells and platelets. Patients are anaemic,thrombocytopenic and neutropenic. The anaemiamay be symptomatic; it may precipitate symptomsof coronary, cerebrovascular or peripheral vascularischaemia and may cause heart failure.Thrombocytopenia may cause purpura, bruisingand – in severe cases – mucosal or GI bleeding orcerebral haemorrhage. Neutropenia causes mouthulceration, perineal infection and susceptibility toopportunistic infection. Anaemia is treated withperiodic blood transfusions. Platelet transfusions aregiven if the patient is severely thrombocytopenicand at risk of haemorrhage. Granulocytetransfusions are relatively ineffective and so thetreatment of neutropenia is prophylaxis againstinfection and aggressive treatment of the earliestsigns of it.
2. The choice of antiplatelet or anticoagulation therapydepends upon the pathophysiology of the disease(see p. 275).
Extended matched answers
EMQ 1
A. 3. The dry tap suggests myelofibrosis andsplenomegaly is characteristic.
B. 1. This patient has ‘B’ symptoms and pathologicallymphadenopathy; the fact that it is in adjacent areas(on only one side of the diaphragm) suggestsHodgkin’s disease.
C. 4. The Philadelphia chromosome is pathognomonicof chronic granulocytic leukaemia
D. 6. Either thalassaemia trait or chronic disease couldalso give a microcytic anaemia, but the clinicaldetails strongly favour iron deficiency
E. 13. Of course there are many causes of renal failure,but the heading makes it clear that you should bemaking a haematological diagnosis; myeloma is thehaematological disease most closely associated withrenal failure.
EMQ 2
A. 5. TIAs are caused by platelet emboli so aspirin isthe treatment of choice.

6
287
B. 8. Fibrinolytic therapy is of no proven value, and maycause him to bleed into the fracture site. He willcertainly need to take warfarin but must be treatedwith heparin first. Clinical trials consistently showlow-molecular-weight heparin to be superior tounfractionated heparin so that should be your choice.
C. 8, 5. Either aspirin or heparin would be correct, butlow-molecular-weight heparin is the better answer.
D. 6 or 4. Unlike carotid TIAs, arterial thrombo-embolism from cardiac clot should be treated withwarfarin (or sinthrone, although it is less commonlyused). Depending on your perception of the dangerof recurrence, you might give heparin, but medium-to-long term warfarin is the mainstay of preventingrecurrence.
E. 5. Warfarin is indicated for preventing cerebralembolism in atrial fibrillation unless there arecontraindications. Age, falls and concern about thispatient’s ability to care for himself are relativecontraindications. Depending on likely compliance,you might prescribe aspirin.
Response to OSCE
Do not waffle on about ‘taking a history’ because yourinstruction is clear and do not attempt to perform a‘general’ physical examination. You must use yourknowledge to focus the examination on examining:
● all possible sites of lymphadenopathy: theepitrochlear region of the elbow, neck, axillae, andgroins
● the abdomen for para-aortic nodes (by deeppalpation), hepatomegaly and splenomegaly
● the conjunctivae for anaemia.
It is reasonable to examine the chest if you have time,but make less of a play of this than the parts of the ex-amination listed above because chest examination maywell be normal, and these other features will be muchmore informative, whether negative or positive.
The character and distribution of the nodes are bothimportant; see page 257.
Self-assessment: answers