document
TRANSCRIPT
Nature © Macmillan Publishers Ltd 1998
8
letters to nature
NATURE | VOL 394 | 13 AUGUST 1998 697
24. Robinson, K. M. & Lemire, B. D. Flavinylation of succinate: ubiquinone oxidoreductase fromSaccharomyces cervisiae. Meth. Enzymol. 260, 34–51 (1995).
25. Ackrell, B. A. C., Kearney, E. B. & Singer, T. P. Mammalian succinate dehydrogenase. Meth. Enzymol.53, 466–483 (1978).
Acknowledgements. We thank M. Hengartner for strains and for suggestions that facilitated the mappingof mev-1. The wild-type strain was from the C. elegans Genetics Center, which is supported by theNational Center for Research Resources (NCRR). This work was supported by Tokai University School ofMedicine Research Project and by a Grant in Aid for Aging Research from the Ministry of Human andWelfare, Japan, and for Scientific Research from the Ministry of Education, Science, Sports and Culture,Japan.
Correspondence and requests for materials should be addressed to N.I. (e-mail: [email protected]).
ProteinkinaseC regulatesthenuclear localizationof diacylglycerol kinase-zMatthew K. Topham*†‡, Michaeline Bunting*†‡,Guy A. Zimmerman‡, Thomas M. McIntyre‡,Perry J. Blackshear§ & Stephen M. Prescott*†‡* The Huntsman Cancer Institute, † Eccles Program in Human Molecular Biology& Genetics, and ‡ Department of Internal Medicine, University of Utah,Salt Lake City, Utah 84112, USA§ National Institute of Environmental Health Sciences, Research Triangle Park,North Carolina 27709, USA. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Diacylglycerol kinases (DGKs) terminate signalling from diacyl-glycerol by converting it to phosphatidic acid1–8. Diacylglycerolregulates cell growth and differentiation, and its transient accu-mulation in the nucleus may be particularly important in thisregulation9,10. Here we show that a fraction of DGK-z is foundin the nucleus, where it regulates the amount of nuclear diacyl-glycerol. Reducing nuclear diacylglycerol levels by conditionalexpression of DGK-z attenuates cell growth. The nuclear-localiza-tion signal of DGK-z is located in a region that is homologous tothe phosphorylation-site domain of the MARCKS protein. This is,to our knowledge, the first evidence that this domain, which isa major target for protein kinase C, can localize a protein tothe nucleus. Two isoforms of protein kinase C, but not others,regulate the localization of DGK-z. Our results define a cycle inwhich diacylglycerol activates protein kinase C, which thenregulates the metabolism of diacylglycerol by alternating theintracellular location of DGK-z. This may be a general mechanismto control mitogenic signals that depend on nuclear diacylglycerol.
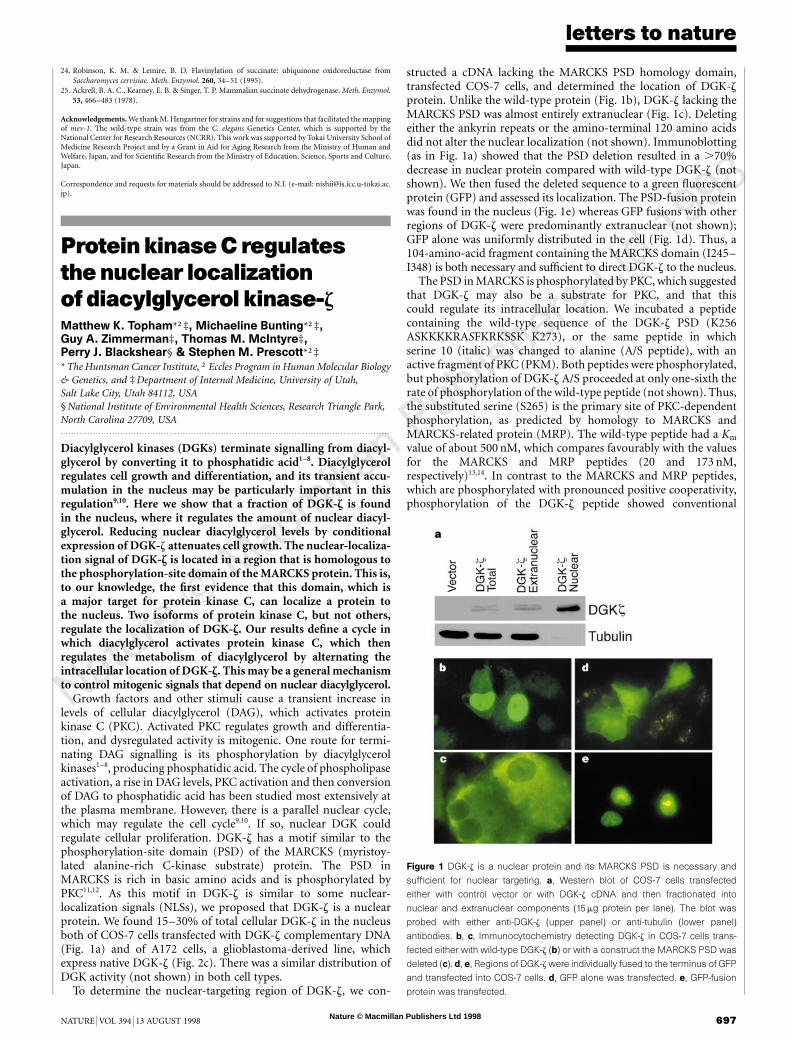
Growth factors and other stimuli cause a transient increase inlevels of cellular diacylglycerol (DAG), which activates proteinkinase C (PKC). Activated PKC regulates growth and differentia-tion, and dysregulated activity is mitogenic. One route for termi-nating DAG signalling is its phosphorylation by diacylglycerolkinases1–8, producing phosphatidic acid. The cycle of phospholipaseactivation, a rise in DAG levels, PKC activation and then conversionof DAG to phosphatidic acid has been studied most extensively atthe plasma membrane. However, there is a parallel nuclear cycle,which may regulate the cell cycle9,10. If so, nuclear DGK couldregulate cellular proliferation. DGK-z has a motif similar to thephosphorylation-site domain (PSD) of the MARCKS (myristoy-lated alanine-rich C-kinase substrate) protein. The PSD inMARCKS is rich in basic amino acids and is phosphorylated byPKC11,12. As this motif in DGK-z is similar to some nuclear-localization signals (NLSs), we proposed that DGK-z is a nuclearprotein. We found 15–30% of total cellular DGK-z in the nucleusboth of COS-7 cells transfected with DGK-z complementary DNA(Fig. 1a) and of A172 cells, a glioblastoma-derived line, whichexpress native DGK-z (Fig. 2c). There was a similar distribution ofDGK activity (not shown) in both cell types.
To determine the nuclear-targeting region of DGK-z, we con-
structed a cDNA lacking the MARCKS PSD homology domain,transfected COS-7 cells, and determined the location of DGK-zprotein. Unlike the wild-type protein (Fig. 1b), DGK-z lacking theMARCKS PSD was almost entirely extranuclear (Fig. 1c). Deletingeither the ankyrin repeats or the amino-terminal 120 amino acidsdid not alter the nuclear localization (not shown). Immunoblotting(as in Fig. 1a) showed that the PSD deletion resulted in a .70%decrease in nuclear protein compared with wild-type DGK-z (notshown). We then fused the deleted sequence to a green fluorescentprotein (GFP) and assessed its localization. The PSD-fusion proteinwas found in the nucleus (Fig. 1e) whereas GFP fusions with otherregions of DGK-z were predominantly extranuclear (not shown);GFP alone was uniformly distributed in the cell (Fig. 1d). Thus, a104-amino-acid fragment containing the MARCKS domain (I245–I348) is both necessary and sufficient to direct DGK-z to the nucleus.
The PSD in MARCKS is phosphorylated by PKC, which suggestedthat DGK-z may also be a substrate for PKC, and that thiscould regulate its intracellular location. We incubated a peptidecontaining the wild-type sequence of the DGK-z PSD (K256ASKKKKRASFKRKSSK K273), or the same peptide in whichserine 10 (italic) was changed to alanine (A/S peptide), with anactive fragment of PKC (PKM). Both peptides were phosphorylated,but phosphorylation of DGK-z A/S proceeded at only one-sixth therate of phosphorylation of the wild-type peptide (not shown). Thus,the substituted serine (S265) is the primary site of PKC-dependentphosphorylation, as predicted by homology to MARCKS andMARCKS-related protein (MRP). The wild-type peptide had a Km
value of about 500 nM, which compares favourably with the valuesfor the MARCKS and MRP peptides (20 and 173 nM,respectively)13,14. In contrast to the MARCKS and MRP peptides,which are phosphorylated with pronounced positive cooperativity,phosphorylation of the DGK-z peptide showed conventional
Figure 1 DGK-z is a nuclear protein and its MARCKS PSD is necessary and
sufficient for nuclear targeting. a, Western blot of COS-7 cells transfected
either with control vector or with DGK-z cDNA and then fractionated into
nuclear and extranuclear components (15 mg protein per lane). The blot was
probed with either anti-DGK-z (upper panel) or anti-tubulin (lower panel)
antibodies. b, c, Immunocytochemistry detecting DGK-z in COS-7 cells trans-
fected either with wild-type DGK-z (b) or with a construct the MARCKS PSD was
deleted (c). d, e, Regions of DGK-z were individually fused to the terminus of GFP
and transfected into COS-7 cells. d, GFP alone was transfected. e, GFP-fusion
protein was transfected.

Nature © Macmillan Publishers Ltd 1998
8
letters to nature
698 NATURE | VOL 394 | 13 AUGUST 1998
Michaelis–Menten kinetics, with a Hill constant of 0.7 (data notshown). The DGK A/S peptide possibly had biphasic kinetics,indicating that it was phosphorylated at one or two other, loweraffinity, sites. As predicted from studies of the MARCKS tetra-alapeptide14, the DGK-z A/S peptide inhibited PKM when histone IIISwas the substrate (K i,800 nM).
To test whether this phosphorylation regulates nuclear localiza-tion of DGK-z, we expressed in COS-7 cells the full-length DGK-zwith PKC-a or PKC-g, which both phosphorylate MARCKS15 andare expressed in A172 cells16. PKC-a (Fig. 2a) and PKC-g (notshown) both reduced the fraction of DGK-z in the nucleus by 30 to50%. We further tested whether the nuclear localization of DGK-zwas regulated in the same way in A172 cells by downregulating thesePKC isozymes by prolonged treatment with phorbol 12-myristate13-acetate (PMA), and then determining the location of DGK-z.This procedure resulted in complete loss of these PKC isozymes(Fig. 2b), and the localization of DGK-z to the nucleus was
enhanced (Fig. 2c), as was nuclear DGK activity (not shown).Growth factors and other stimuli cause turnover of phosphati-
dylinositol-4,5-biphosphate and a rapid increase in DAG levels incell nuclei17–20, which can regulate growth and differentiation. AsPKC regulates the amount of DGK-z in the nucleus, we next askedwhether this DGK activity could attenuate the accumulation ofnuclear DAG and modulate cell growth. In A172 cells treated withepidermal growth factor (EGF), nuclear DAG levels rose two- tothreefold above baseline by 10 minutes after treatment, whereastotal cellular levels were unchanged (not shown). To determine thefunction of DGK activity in the nucleus, we compared nuclear DAGlevels in control A172 cells with levels in cells with high nuclearDGK-z levels (cells exposed to PMA; Fig. 2c). When the A172 cellswere treated with EGF for 10 minutes, nuclear DAG in cells notpretreated with PMA reached 241% (637%) of that in unstimu-lated cells, but nuclear DAG in cells that had been pretreated withPMA (that is, cells with high nuclear DGK-z levels) reached only
Con
trol
PM
A
Con
trol
PM
A
DGK-ζ
Total Nuclear
PKC-α PKC-γ
Con
trol
PM
A
Con
trol
PM
A
PKC
Vec
tor
tota
l
Vec
tor
nucl
ear
PK
C-α
tota
l
PK
C-α
nuc
lear
DGK-ζ Fol
d in
crea
se in
DA
G
PMAControl0
1
2
3
*
* TotalNuclear
Dou
blin
g tim
e (h
ours
)
Control GFP DGK0
20
40
60
80 ControlInducedControlInduced
G0/G1
28.5 ± 3.2
25.5 ± 0.8
24.7 ± 4.2
28.1 ± 2.2
32.7 ± 1.9
27.2 ± 2.7
37.4 ± 0.2
35.1 ± 0.3
S G2/M
47.2 ± 1.9
38.0 ± 3.9
36.9 ± 2.0
36.8 ± 1.3
WT
∆ATP
∆NLS
Vector
Per cent in:
a
b
c
d
e
f
Figure 2 PKC controls nuclear localization of DGK-z, and overexpression of DGK-
z regulates nuclear DAG and the cell cycle. a, Immunoblot (for DGK-z) of total
cellular homogenates or nuclear fractions (10 mg protein per lane) from COS-7
cells into which DGK-z had been transfected with control vector or with PKC-a.
The cells were treated with PMA for 30min before collecting. b, We treated
A172 cells with 60ng ml−1 PMA or vehicle for 24 h, and then immunoblotted
total cellular homogenates (10 mg per lane) using anti-PKC-a or anti-PKC-g
antibodies. c, Immunoblot for DGK-z in A172 cells treated with PMA as in b (10 mg
per lane total and 20 mg per lane nuclear). d, A172 cells were treated with PMA as
in b and then with EGF (40 ng per ml) for 10 min. Nuclear and total cellular lipids
were extracted and DAG/phosphate levels determined. Values are fold-increase
over basal levels (n ¼ 4). Asterisk indicates significance (P , 0:05). e, Doubling
times of 293 cells stably expressing either vector, GFP, or DGK-z with or without
inducing agent (1 mM muristerone A). The values presented are the averages
from two experiments. f, COS-7 cells were transfected with either control vector,
wild-type DGK-z (WT), kinase-dead DGK-z (DATP), or a MARCKS PSD mutant
(DNLS) and then cell-cycle analysis was performed by flow cytometry. Mean
values from two experiments are shown.
Figure 3 Introduction of negative charge into the MARCKS PSD excludes it from
the nucleus. a–d, COS-7 cells were transfected with GFP fusions containing
either the wild-type NLS (a) or constructs in which serines were altered to
alanines (b), aspartates (c) or asparagines (d). The cells were then examined by
fluorescence microscopy. The sequences are shown with altered amino acids in
red and the MARCKS PSD in green.
Figure 4 Phosphorylation by PKC-a causes nuclear exclusion of DGK-z. a, b, A
GFP construct containing wild-type DGK-z NLS (Fig. 3a) was transfected into
COS-7 cells together with control vector (a) or PKC-a (b). c, A GFP–NLS mutant
with all serines changed to alanines (Fig. 3b) was transfected together with PKC-
a. d, Western blot, using an anti-DGK-z antibody, of nuclear fractions (10 mg
protein per lane) into which control vector or PKC-a was transfected together
with full-length wild-type (WT) DGK-z, or a full-length mutant DGK-z construct with
all serines of the MARCKS PSD changed to asparagines (Mut). Controls (not
shown) showed similar expression levels of total DGK-z.

Nature © Macmillan Publishers Ltd 1998
8
letters to nature
NATURE | VOL 394 | 13 AUGUST 1998 699
130% (672%) of unstimulated cells (Fig. 2d, P , 0:05). Thissupported the hypothesis that DGK-z in the nucleus regulates theconcentration of DAG there. To verify these observations, wegenerated a stable cell line with inducible overexpression of DGK-z. When the cells were induced, nuclear DAG levels fell by 38%; incontrol cells nuclear DAG levels were unchanged with induction (notshown). As DAG levels may be an important determinant ofcell growth9, we tested the effect of DGK-z on doubling time; over-expression increased doubling time from 33.5 (63.5) to 77.0 (63.0)hours (Fig. 2e, P , 0:05). Similar results were obtained with anotherDGK-z-overexpressing clone, but not with cell lines stably transfectedeither with vector without insert (no change with induction) or withan inducible GFP (1.2-fold increase in doubling time with induction).
As a separate test of whether nuclear targeting, and DGK activity,were essential for the effects of DGK-z on the cell cycle, wetransfected cDNAs encoding wild-type DGK-z, a kinase-deadmutant (DATP, G355 → D355), or a mutant that did not localizeto the nucleus (DNLS; see below) along with GFP as a reporter. Thecells were collected at 48 hours, stained with propidium iodide, andthen sorted by flow cytometry to identify transfected cells (GFP-positive). The cell-cycle profile of the transfected cells show thatoverexpression of wild-type DGK-z, but not of the kinase-dead orNLS mutants, induced an accumulation of the cells in the G0/G1phases of the cell cycle (Fig. 2f). In another experiment, wesynchronized the cells with nocodazole and saw a qualitativelysimilar, but more marked, accumulation in cells in G0/G1 (notshown). We conclude that DGK-z regulates cell growth and thatboth its enzymatic activity and its nuclear localization are essential.
We then further tested whether the phosphorylation of DGK-z byPKC, and subsequent intracellular relocalization of DGK-z, ismediated at the MARCKS domain. We transfected cells with aplasmid encoding GFP fused to a 27-amino-acid sequence thatcontained the MARCKS PSD of DGK-z. Like the 104-amino-acidfragment (see above), this region directed GFP to the nucleus (seebelow). Changing the basic amino acids at either end of theMARCKS-homology domain abolished the nuclear localization ofGFP. As the GFP-fusion proteins could have passively diffused intothe nucleus, the nuclear localization may have resulted from acharge interaction there. To address this, we mutated to alanine thebasic amino acids of the MARCKS PSD of full-length DGK-z(DNLS, A256 ASAAAAAASFAAASSA A273), transfected it intoCOS-7 cells, and immunoblotted subcellular fractions. TheseK=R → A mutations resulted in .90% reduction in levels of nuclearprotein (not shown). Thus, the MARCKS-homology domain ofDGK-z is its predominant nuclear-localization signal.
To verify that phosphorylation of this region relocalizes DGK-z,we transfected GFP constructs containing either the native 27-amino-acid sequence, or one in which the serines had been changedto aspartates, which mimics phosphorylation21. The S → D GFPfusion protein was extranuclear, whereas an uncharged, structurallysimilar construct (S → N) was nuclear, as was an S → A construct(Fig. 3). Thus, phosphorylation of the serine residues in theMARCKS-homology region of DGK-z regulates its intracellularlocation—the unphosphorylated form is in the nucleus whereas thephosphorylated form is extranuclear. As these constructs were nottruly phosphorylated, we transfected cells with cDNAs encodingone of eight PKC isoforms (a, bI, bII, g, d, e, m or z) with GFP-reporter constructs containing wild-type DGK-z NLS or the samesequence with serines changed to alanines. In agreement with Fig.2a, when the native NLS–GFP fusion was with PKC-a or PKC-g, itwas redistributed from the nucleus to the cytoplasm (Fig. 4a–c). Incontrast, the GFP construct in which the NLS serines had beenreplaced with alanines was nuclear, verifying that the phosphoryla-tion occurred at the MARCKS domain. Expression of the nativeNLS–GFP with the other isoforms of human PKC did not influencethe location (not shown). To ensure that the GFP-reporter con-structs reflected the behaviour of the native protein, we expressed
PKC-a with a full-length construct of DGK-z in which the serinesin the MARCKS PSD had been mutated to asparagines (K256ANKKKKRANFKRKNNK K273) so that this region could nolonger be phosphorylated. Unlike the wild-type protein, theamount of this construct in the nucleus was not affected byexpression of PKC (Fig. 4d).
Lipid signalling pathways regulate cell growth and differentiation,and transient accumulation of DAG, particularly in the nucleus, isimportant for this regulation18,19,22,23. Our results show that thissignal is turned off by DGK-z. This requires DGK-z to be active andin the nucleus, a localization that is dynamically regulated byphosphorylation of the MARCKS-homology domain by some,but not all, isoforms of PKC. M. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Methods
cDNA constructs. We cloned DGK-z cDNA into pcDNA1/Amp orpcDNA3.1/Zeo plasmids (Invitrogen). To delete the MARCKS domain, thiscDNA was digested with BamHI and then religated. We constructed MARCKS-domain mutants of full-length DGK-z by incorporating a new EagI site(guanine was altered to cytosine at nucleotide 919) and then ligating annealedoligonucleotides with desired mutations into the BamHI/EagI-digested cDNA.The DATP mutant was made by a guanine-to-adenosine change at nucleotide1152 using a Muta-Gene kit (Bio-Rad). Coding regions of DGK-z were fused tothe carboxy terminus of pEGFP (Clontech) using the following restriction sites:EcoRI/BamHI (amino acids 2–245), BamHI/BamHI (amino acids 245–348),ApaI/XbaI (amino acids 333 to stop codon), and SacI/XbaI (amino acids 685 tostop codon). To generate the GFP–NLS constructs, forward and reverseoligonucleotides were synthesized, annealed, and ligated into pEGFP vectors.Transfections. Transfections were performed and cells collected as described7.In the PKC co-transfection experiments, 500 ng of PKC-a, bI, bII, g, d, e, m or z
(in pcDNA3/Amp from J. Metherall) or control vector were combined with500 ng of either full-length DGK-z or a GFP construct. In the GFP experimentswith PKC, the transfection medium was removed at 5 h and replaced with60 ng ml−1 PMA or ethanol vehicle in DMEM medium for another 18 h. In theexperiments with full-length DGK-z and PKC constructs, the transfectionmedium was removed at 5 h and replaced with DMEM for another 18 h. At thatpoint, PMA was added for 30 min and the cells were collected.Immunoblotting and immunocytochemistry. An anti-peptide antibody wasgenerated in rabbits7 and was affinity-purified. Immunoblots were performedas described7. Anti-PKC antibodies (Transduction Laboratories) were used atthe recommended dilutions. Bands on some blots were quantified by densito-metry using Kodak 1D image-analysis software. Immunocytochemistry wasperformed as described24. Cells transfected with GFP alone were washed andfixed as above. A172 cells (from ATCC) were stained as above except that theincubation times with anti-DGK-z and goat anti-rabbit antibodies were 90 and45 min at room temperature.Stable transfects. Cell lines that could be induced to overexpress DGK-z orGFP were made using the ecdysone system (Invitrogen). Full-length DGK-zcDNA was cloned into pIND(SP1). This construct or pIND(SP1) withoutinsert was transfected into EcR 293 cells (Invitrogen), which were then selectedin 400 mg ml−1 G418 (Life Technologies) and 400 mg ml−1 zeocin (Invitrogen).After induction with muristerone A, colonies were screened for DGK-z byimmunocytochemistry, or for GFP by fluorescent phase-contrast microscopy.Appropriate colonies were replated and selected twice more until ahomogenous, inducible population was verified.Nuclear isolation. COS-7 and A172 nuclear isolation was done as described25,typically from two 35-mm wells. Immunoblots of cell homogenates were thencarried out or DAG levels were determined. To assess the purity of these nuclearisolates, immunoblots for tubulin (anti-tubulin antibody Clontech) were per-formed or lipid phosphate levels were determined. Nuclear lipid phosphate wastypically 2–5% of total lipid phosphate, which concurred with previous reports26.DAGassay. Stable cell lines treated with inducing agent (1 mM muristerone A)or control vehicle, or A172 cells treated with PMA or control vehicle, weregrown to 80% confluence. For total cellular DAG, cells from one 35-mm wellwere collected in 2 ml methanol. Nuclei were collected using two 35-mm wellsand the final nuclear pellet was brought into 2 ml methanol. Lipid extractionswere done as described27. DAG levels were determined using the DAG assay

Nature © Macmillan Publishers Ltd 1998
8
letters to nature
700 NATURE | VOL 394 | 13 AUGUST 1998
system (Amersham), and phosphate levels also were determined from the lipidextracts28.Synthesis, purification and phosphorylation of peptides. Peptides weresynthesized and phosphorylated by mixed rat brain active PKC isozymes(PKM) for 10 min at 30 8C (refs 13, 14). When the inhibitory effects of the DGKA/S peptide (0–100 mM) were assayed, histone IIIS (final concentration20 mg ml−1) was used as a substrate for PKM. This reaction was linear for atleast 40 min; we used 10 and 15 min time points to evaluate inhibitory effects ofthe peptide29.
Received 27 April; accepted 28 May 1998.
1. Goto, K. & Kondo, H. Molecular cloning and expression of a 90-kDa diacylglycerol kinase thatpredominantly localizes in neurons. Proc. Natl Acad. Sci. USA 90, 7598–7602 (1993).
2. Kai, M., Sakane, F., Imai, S.-i., Wada, I. & Kanoh, M. Molecular cloning of a diacylglycerol kinaseisozyme predominantly expressed in human retina with a truncated and inactive enzyme expressionin most other human cells. J. Biol. Chem. 269, 18492–18498 (1994).
3. Sakane, F., Yamada, K., Kanoh, H., Yokoyama, C. & Tanabe, T. Porcine diacylglycerol kinase sequencehas zinc finger and E-F hand motifs. Nature 344, 345–348 (1990).
4. Houssa, B. et al. Cloning of a novel human diacylglycerolkinase (DGKu) containing three cysteine-rich domains, a proline-rich region and a pleckstrin homology domain with overlapping ras-associating domain. J. Biol. Chem. 272, 10422–10428 (1997).
5. Klauck, T. M., Xu, X., Mousseau, B. & Jaken, S. Cloning and characterization of a glucocorticoid-induced diacylglycerol kinase. J. Biol. Chem. 271, 19871–19788 (1996).
6. Sakane, F., Imai, S. I., Kai, M., Wada, I. & Kanoh, H. Molecular cloning of a novel diacylglycerol kinaseisozyme with a pleckstrin homology domain and a C-terminal tail similar to those of the EPH familyof protein tyrosine kinases. J. Biol. Chem. 271, 8394–8401 (1996).
7. Bunting, M., Tang, W., Zimmerman, G. A., McIntyre, T. M. & Prescott, S. M. Molecular cloning andcharacterization of a novel human diacylglycerol kinase z. J. Biol. Chem. 271, 10230–10236 (1996).
8. Tang, W., Bunting, M., Zimmerman, G. A., McIntyre, T. M. & Prescott, S. M. Molecular cloning of anovel human diacylglycerol kinase highly selective for arachidonate-containing substrates. J. Biol.Chem. 271, 10237–10241 (1996).
9. Jackowski, S. Cell cycle regulation of membrane phospholipid metabolism. J. Biol. Chem. 271, 20219–20222 (1996).
10. Olson, E. N., Burgess, R. & Staudinger, J. Protein kinase C as a transducer of nuclear signals. CellGrowth Differ. 4, 699–705 (1993).
11. Aderem, A. The MARCKS brothers: a family of protein kinase C substrates. Cell 71, 713–716 (1992).12. Blackshear, P. J. The MARCKS family of cellular protein kinase C substrates. J. Biol. Chem. 268, 1501–
1504 (1993).13. Verghese, G. M. et al. Protein kinase C-mediated phosphorylation and calmodulin binding of
recombinant MARCKS and MARCKS-related protein (MRP). J. Biol. Chem. 269, 9361–9367(1994).
14. Graff, J. M., Rajan, R. R., Randall, R. R., Nairn, A. C. & Blackshear, P. J. Protein kinase C substrate andinhibitor characteristics of peptides from the myristoylated alanine-rich C kinase substrate(MARCKS) protein phosphorylation site domain. J. Biol. Chem. 266, 14390–14398 (1991).
15. Herget, T., Oehrlein, S. A., Pappin, D. J. C., Rozengurt, E. & Parker, P. J. The myristoylated alanine-richC-kinase substrate (MARCKS) is sequentially phosphorylated by conventional, novel and atypicalisotypes of protein kinase C. Eur. J. Biochem. 233, 448–457 (1995).
16. Xiao, H., Goldthwait, D. A. & Mapstone, T. The identification of four protein kinase C isoforms inhuman glioblastoma cell lines: PKC alpha, gamma, epsilon, and zeta. J. Neurosurg. 81, 734–740 (1994).
17. Divecha, N., Banfic, H. & Irvine, R. F. Inositides and the nucleus and inositides in the nucleus. Cell 74,405–407 (1993).
18. Divecha, N., Banfic, H. & Irvine, R. F. The polyphosphoinositide cycle exists in the nuclei of Swiss 3T3cells under the control of a receptor (for IGF-1) in the plasma membrane, and stimulation of the cycleincreases nuclear diacylglycerol and apparently induces translocation of protein kinase C to thenucleus. EMBO J. 10, 3207–3214 (1991).
19. Banfic, H., Zizak, M., Divecha, N. & Irvine, R. F. Nuclear diacylglycerol is increased during cellproliferation in vivo. Biochem. J. 290, 633–636 (1993).
20. Leach, K. L. et al. a-Thrombin stimulates nuclear diglyceride levels and differential nuclearlocalization of protein kinase C isozymes in IIC9 cells. J. Biol. Chem. 267, 21816–21822 (1992).
21. Swierczynski, S. L. & Blackshear, P. J. Myristoylation-dependent and electrostatic interactions exertindependent effects on the membrane association of the myristoylated alanine-rich protein kinase Csubstrate protein in intact cells. J. Biol. Chem. 271, 23424–23430 (1996).
22. Sun, B., Murray, N. R. & Fields, A. P. A role for nuclear phosphatidylinositol-specific phospholipase Cin the G2/M phase transition. J. Biol. Chem. 272, 26313–26317 (1997).
23. Divecha, N., Rhee, S.-G., Letcher, A. J. & Irvine, R. F. Phosphoinositide signalling in rat liver nuclei:phosphoinositidase C isoform b1 is specifically, but not predominantly, located in the nucleus.Biochem. J. 289, 617–620 (1993).
24. Ding, L. et al. Alternative splicing of the human diacylglycerol kinase zeta gene in muscle. Proc. NatlAcad. Sci. USA 94, 5519–5524 (1997).
25. Payrastre, B. et al. A differential location of phosphoinositide kinases, diacylglycerol kinase, andphospholipase C in the nuclear matrix. J. Biol. Chem. 267, 5078–5084 (1992).
26. York, J. & Majerus, P. W. Nuclear phosphatidylinositols decrease during S-phase of the cell cycle inHeLa cells. J. Biol. Chem. 269, 7847–7850 (1994).
27. Bligh, E. G. & Dyer, W. J. A rapid method of total lipid extraction and purification. Can. J. Biochem.Physiol. 37, 911–917 (1959).
28. Whatley, R. E. et al. Growth-dependent changes in arachidonic acid release from endothelial cells aremediated by protein kinase C and changes in diacylglycerol. J. Biol. Chem. 268, 16130–16138 (1993).
29. Halsey, D. L., Girard, P. R., Kuo, J. F. & Blackshear, P. J. Protein kinase C in fibroblasts. Characteristicsof its membrane association during growth and after exposure to phorbol esters and other mitogens.J. Biol. Chem. 262, 2234–2243 (1987).
Acknowledgements. We thank E. Kennington, M. Flynn, J. White and H. Rust for technical assistance,M. McAdams for peptide synthesis, and V. Bennett and A. Thorburn for discussions. This work wassupported by a grant from the National Cancer Institute. The core facilities at the University of Utah(DNA Sequencing and Peptide/DNA User Facility) were supported by grants from the National CancerInstitute. P.J.B. was an Investigator of the Howard Hughes Medical Institute when this work wasperformed, and M.K.T. is a Howard Hughes Medical Institute Physician Postdoctoral Fellow.
Correspondence and requests for materials should be addressed to S.M.P. (e-mail: [email protected]).
DNA-dependentproteinkinaseactsupstreamofp53 inresponse toDNAdamageRichard A. Woo*, Kevin G. McLure*, Susan P. Lees-Miller†,Derrick E. Rancourt‡ & Patrick W. K. Lee*
Departments of * Microbiology and Infectious Diseases, † Biological Sciences, and‡ Biochemistry and Molecular Biology, University of Calgary, Calgary,Alberta T2N 4N1, Canada. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
The tumour suppressor p53 becomes activated as a transcriptionfactor in response to DNA damage1–3, but the mechanism for thisactivation is unclear. A good candidate for an upstream activatorof p53 is the DNA-dependent protein kinase (DNA-PK) thatdepends on the presence of DNA breaks for its activity4–6. Herewe investigate the link between DNA damage and the activation ofDNA-PK and of p53. To determine whether DNA-PK is anupstream mediator of the p53 DNA-damage response, we analyseda severe combined-immunodeficiency (SCID) mouse cell line,SCGR11 (refs 7, 8), and the human glioma cell line M059J (ref.9). Both cell lines lack any detectable DNA-PK activity. We findthat p53 is incapable of binding to DNA in the absence of DNA-PK, that DNA-PK is necessary but not sufficient for activation of
Figure 1 The effect of DNA damage on p53 DNA-binding activity in nuclear
extracts from DNA-PK-deficient SCGR11 cells. a, BALB/c 3T3 (lanes 1–5) or
SCGR11 (lanes 6–10) cells were exposed to different DNA-damaging agents
(mock-treated, CTRL; hydrogen peroxide, HP; actinomycin D, AD; mitomycin C,
MMC; g-irradiation, IR). Nuclear extracts were then prepared and assayed for p53
DNA-binding by EMSA. All reactions contained pAb421 unless otherwise stated.
‘Control’ lanes are as follows: lane 11 is identical to lane 3, except pAb421 was not
used; lane 12 contained the same nuclear extract as lane 3, except that a
nonspecific DNA oligonucleotide probe (NB in ref. 23)wasused in the placeof the
p53 consensus binding oligonucleotide probe. b, BALB/c 3T3 or SCGR11 cells
were mock-treated (CTRL) or DNA-damaged as in a and then labelled with 35S-
methionine for 2 h. Nuclear extracts were immunoprecipitatedwith either anti-p53
monoclonal antibodies or control antibody (NS1).