281155/fulltext01.pdf · million cases and 600,000 deaths (parry, hien et al. 2002). s. typhimurium...
TRANSCRIPT

������������������� ���������� �����
���������� �������������� ������������������� ������� ����������������������������
������ �� ��� �������� �����
������������������������������
����� ����������
���� � ��! "� ���� �#$!��!��%!# $#!$&��'�(�'��'&&')*�!���%"$

����������������������� ������� ���������������������������������������������� ����!������� ��������"�������"�����������#��������$����%���&����!����%������%'&����&��("�������%��������)*�+&�����������,�����������������-!���&*
��������
.�/�������0*�#���*���������%��&������������1���*�2�����������&������%�������*3���� ������������ ���������*����������� �������������� ������������������� ������� ���������������������������$*��4���*� ������*�50�6�$789$�9��:9748798*
�����������&�����������&�!&���������������������&�!�!�,��&�����������!������������;��������������%�����������������!����������&������������!������*� ,��������%���������,&���������&��������&��%����%��&����&����*5�������55�,��������!�����&,��&��������������%%������&�������������������
������������ ���*������������������������� �&�����9%������,�����%%������&�������������������� ���!������������!�%������� ��������� �&�����������!�,�&������%� �&������*�<�������������!!�����&����&���&���������&����!�����,��&���������������������������&������������������!����������%�������������=�����%�������������������&����&�������%�������������������*5������� 5�,���������� ���� ��������������!�&,� �������� ������������������ �*�*�!��
��������������*� ��!���������������!��������&��,���&,����&������������!����������������������������������������&�����������������������*�"��������������,�������������������&��!������&������������������������!��&�������������������%���%���&��!&���2��3����������������*5�������555�,����������%���&���&,����!������������������%�������������������
�&,��� �&���$�>�%��������������&�����������������=����������9���� ���������63������������%���&����%�����*�?���������&����&��������������������������������������%������������,�!��������%�������!�����!��9�������63����*5� ������ 5@� ,�� ������!����� &,� �&�� ��������� �63� ���������� '�� 5@�� 2�0� ��
��%%�����������%�����������%%���������������������������*������������%��&��A��3���!���������������������������������������������������&���,���%��������������%���������������������������������������� ����������63�����������*�2�0� ���������!�,�&����������&�����������%%������������������*�+&��������������������������������������������,��/����%%���������!�,�&������������������%�����������������������2�0*
���� ��������������������������������!�������������!��������������������!����63&��!������������63������������������
�������� ������!����� ����������������"�������� ��������� #����!�"$�%&'!������������� ����!��()*%+',��������!�������
B�0���.�/������#���
5006��4��94#�450�6�$789$�9��:9748798���������������9���:#8�(&����CC��*/�*��C������D��E���������������9���:#8)

I think emotions should rule us. I have good reasons why.
Logic, you see can be faulty. Emotions never lie.
Reality brings us doubts. We know an emotion’s real.
But I want logic to rule us.
It’s just the way I feel.


List of Papers
This thesis is based on the following papers, which are referred to in the text by their Roman numerals.
I Nilsson, A.I., Koskiniemi, S., Eriksson, S., Kugelberg, E., Hin-
ton, J.C., Andersson, D.I. (2005) Bacterial genome size reduc-tion by experimental evolution. Proc. Natl. Acad. Sci., 102(34):12112–6
II Koskiniemi, S., Andersson, D.I., (2009) Variation in spontane-ous deletion rates at different locations of the Salmonella ty-phimurium chromosome. (Manuscript)
III Koskiniemi, S., Andersson, D.I., (2009) Translesion DNA po-lymerases are required for spontaneous deletion formation in Salmonella typhimurium. Proc. Natl. Acad. Sci. 106(25):10248-53
IV Koskiniemi, S., Hughes, D., Andersson, D.I., (2009) Effect of translesion DNA polymerases, endonucleases and RpoS on mu-tation rates in Salmonella typhimurium. (Submitted manuscript)
Reprints were made with permission from the respective publishers.


Contents
Introduction...................................................................................................11 Salmonella enterica..................................................................................11 The bacterial genome ...............................................................................12 The eukaryotic cell as a growth niche ......................................................13 Reductive evolution..................................................................................13 Chromosome organization .......................................................................14 Mechanisms of reductive evolution .........................................................15
Homologous recombination.................................................................15 Illegitimate recombination...................................................................17 Mechanisms of deletion formation ......................................................19 DNA breaks and deletion formation....................................................20
Mutagenesis and DNA repair ...................................................................21 Origins of change.................................................................................21
Fidelity of DNA replication ............................................................22 Sources of DNA damage.................................................................23
Maintaining genome integrity..............................................................24 Base excision repair (BER).............................................................24 Nucleotide excision repair (NER)...................................................25 Methyl directed mismatch repair ....................................................26
Mutators...............................................................................................28 Cellular responses to stress..................................................................29 The adaptive mutation controversy .....................................................29
Present investigations....................................................................................32 Reductive evolution can be a rapid process in laboratory settings...........32 Mechanism of genome reduction .............................................................33 Mechanism of deletion formation ............................................................34 Large variation in deletion rate at different chromosomal locations........36 Fitness effects of large chromosomal deletions .......................................37 Mutation rates are remarkably robust.......................................................38 Concluding remarks .................................................................................39
Future perspectives .......................................................................................41 Acknowledgements.......................................................................................43 References.....................................................................................................45


Abbreviations
DNA Deoxyribonucleic acid NHEJ Non-homologous end joining S. typhimurium Salmonella typhimurium E. coli Escherichia coli BER Base excision repair NER Nucleotide excision repair ORF Open reading frame SSB Single strand binding protein ssDNA Single stranded DNA dsDNA Double stranded DNA Pol III holoenzyme The replicative polymerase with all subunits AP site An apurinic or apyrimidic site in the DNA PFGE Pulsed field gel electrophoresis PCR Polymerase chain reaction Genes recA Major homologous recombination protein recBCD Exonuclease V ruvAB Holliday junction solving helicase recFOR Homologous recombination proteins ligD ATP-dependent DNA ligase dnaE Pol III polymerase subunit dnaQ Pol III editing subunit dnaB Pol III helicase subunit dnaN Pol III beta-clamp subunit dnaQ Pol III clamploader subunit holC Pol III clamploader subunit dinB Translesion DNA polymerase Pol IV polB Translesion DNA polymerase Pol II umuDC Translesion DNA polymerase Pol V samAB Translesion DNA polymerase SamAB tag 3-Methyl adenine DNA glycosylase alkA 3-Methyl adenine DNA glycosylase ung Uracil DNA glycosylase uvrABC NER enzymes mutSHL Methyl directed mismatch repair enzymes

lexA Repressor of the SOS-response rpoS Alternative transcription sigma factor S katEG Catalase enzymes sodC Superoxide dismutase lacI Repressor of the lac-operon lacZ β-galactosidase enzyme

11
Introduction
Evolution is essential for our understanding of biology, from the understand-ing of the beginning of life on earth to modern issues such as the develop-ment of antibiotic resistance. Evolution, i.e. changes in allele frequencies, can occur by a number of processes including mutation, migration, drift and by natural selection. The latter requires in its simplest form two elements, genetic variability in a population and selection among this variation. Muta-tion is one of the major driving forces of evolution as it creates genetic di-versity that allows organisms to adapt to the constantly changing environ-ment through natural selection. The fundamental process of evolution by natural selection was first recognized by Darwin when he described it in his “Origin of the Species” 150 years ago (Darwin 1859). Natural selection acts on individuals with variable genetic traits, selecting those that are most fit for survival in each specific situation. The opposite of natural selection and the second major evolutionary process is chance (i.e. genetic drift). Whereas natural selection fixes the fittest individual for survival in a population, chance will fix any random individual irrespective of fitness. The two key-players of evolution, natural selection and chance, are often battling for precedence in life. In this thesis, I have studied how the forces of evolution form bacterial genomes and what factors affect this process. To this end I have used Salmonella enterica serovar typhimurium as a model organism throughout my work.
Salmonella enterica Salmonella enterica serovar typhimurium, hereafter referred to as S. typhi-murium, is a rod shaped gram-negative bacteria belonging to the class of γ-proteobacteria (McClelland, Sanderson et al. 2001). Unlike its close relative Escherichia coli (E. coli), that is mainly found as a human commensal, Sal-monella enterica never colonizes the human gut without causing infection (Winfield and Groisman 2003). Salmonella enterica serovar typhi is the most virulent of the Salmonella species and rare in that it can only colonize hu-mans. It is the cause of typhoid fever, with an annual global burden of 16 million cases and 600,000 deaths (Parry, Hien et al. 2002).
S. typhimurium on the other hand is not specific to humans and causes a disease similar to typhoid fever in mice (Mastroeni and Sheppard 2004). In

12
humans S. typhimurium causes a milder disease called salmonellosis with symptoms such as diarrhea, fever, and abdominal cramps which develop 12 to 72 hours after infection and may last for up to 7 days (Salyers and Whitt 1994). Infection with Salmonella ssp is through the fecal/oral route, mostly by ingestion of contaminated food or water (Oosterom 1991).
Together with Escherichia coli, Salmonella enterica is one of the two main workhorses of bacterial genetics and with its fully sequenced genome and the extensive knowledge of its regulatory networks, S. typhimurium is one of the most convenient model organisms to use (Neidhardt 1996).
The bacterial genome Genetic material can be acquired by cells via horizontal gene transfer or through amplification of the cellular DNA. In contrast DNA can be lost from cells by deletions (Fig 1). As a consequence bacterial genomes are not fixed but are con-tinuously changing in a population with regard to size and gene content.
Amplification
DeletionHorizontal gene transfer
Forces affecting chromosome composition
Fig. 1
Genome sizes vary to a large extent between bacterial species, from 0.16 kbp for the smallest genome known today to up to 13 kbp for the larger genomes (Casjens 1998; Nakabachi, Yamashita et al. 2006; Schneiker, Perlova et al. 2007). In spite of this size variation, bacterial genomes of all sizes are mostly tightly packed with genes and contain little non-coding genetic material. This raises a multitude of questions. For example how can certain bacteria sustain life when they contain 100 times less genetic material than their lar-ger relatives? From the opposite perspective, if 200 genes are enough to uphold life, why do organisms with larger genomes carry all the additional baggage? The answer to these questions is found in the environment where our microscopic companions reside. In general, intracellular bacteria harbor smaller genomes than their free-living relatives. To understand why, a closer look at the intracellular lifestyle of bacteria is needed.

13
The eukaryotic cell as a growth niche Inside a host cell bacteria living freely in the cell cytoplasm have unlimited access to most nutrients required for growth, thereby relaxing selection to maintain many of the bacterial metabolic genes. Relaxed selection allows these genes to become inactivated by mutation and subsequently lost without affecting cell fitness or the cells chance to overcome extinction by natural selection. Furthermore, intracellular bacteria usually encounter population bottlenecks during transfer from one host to another. By chance any individ-ual cell, irrespective of its fitness in the intracellular environment, might be selected to establish the new population. These population bottlenecks allow deleterious mutations such as deletions to become fixed in the population by chance, which would not have been possible in an environment where natu-ral selection predominates (Andersson and Kurland 1998). The third factor affecting the genomic content of intracellular bacteria is the lack of horizon-tal gene transfer. Inside the eukaryotic cell the individual bacteria are unlikely to receive genetic material from other bacteria, except their closest relatives, which contain more or less identical genetic material. This results in a ratchet where the DNA that is lost cannot be regained as the cell cannot reconstruct the deleted regions through lateral transfer (Moran 1996).
Reductive evolution A small genome size was previously believed to reflect an evolutionary primitive state from which free-living species with larger genomes origi-nated (Wallace and Morowitz 1973). The sequencing of many bacterial ge-nomes in the last decade has proved this hypothesis wrong. Instead, sequenc-ing data implies that the smaller genomes of intracellular pathogens and endosymbionts evolved from larger free-living species (Andersson and Kur-land 1998). It is now believed that low rates of gene influx combined with a mutation bias towards deletions and relaxed selection for certain genes causes a gradual shrinkage in genome size. Furthermore, in organisms with small population sizes, recurrent bottlenecks and low rates of recombination, even slightly deleterious mutations may become fixed in the population. This phenomenon, known as Mullers ratchet, (Muller 1964; Felsenstein 1974) has been extensively studied in the genus Buchnera which includes some of the smallest genomes known to date (Gil, Sabater-Munoz et al. 2002). Two line-ages of the Buchnera species, Buchnera aphidicola (Ap) an endosymbiont of Acyrthosiphon pisum, and Buchnera aphidicola (Sg) an endosymbiont of Schizaphis graminum, have undergone 200-250 million years (My) of endo-symbiotic evolution since their divergence from a free-living gamma-proteobacterial ancestor (Moran and Mira 2001). Reconstruction of the ge-nome of the free-living gamma-proteobacterial ancestor suggests that

14
Buchnera lost many of its genes through fixation of large deletions soon after entering an endosymbiotic lifestyle (Moran and Mira 2001). Interest-ingly, comparison of the two Bucnera genomes that diverged 50 My ago revealed that no rearrangements, duplications or horizontal gene transfers had occurred from the time of their divergence (Tamas, Klasson et al. 2002). In comparison, free-living relatives belonging to the same gamma-proteobacterial family, such as S.typhimurium and E.coli are highly dynamic and contain several large rearrengements of their chromosomes (Rocha 2008). This evolutionary stasis in Buchnera species over a period of 50 My has been ascribed to the lack of the major homologous recombination protein RecA. As a result of this observation it has been inferred that early gene losses in Buchnera species occurred through a RecA-dependent mechanism, before the two species diverged (Tamas, Klasson et al. 2002).
The tempo and mode of gene loss has been widely debated throughout the literature. Are genes generally lost as large fragments of DNA or are they eradicated by slow erosion of the genetic material? A beautiful illustration of the latter can be seen in the genome of the obligate intracellular parasite Ricketsia prowazekii, which is notable for its high number of pseudogenes. Up to 27% of the R. prowazekii chromosome consists of non-coding genetic material and it has been speculated that the high fraction of pseudogenes represent remnants of ancient genes that are in the process of elimination from the genome. A detailed analysis of the pattern of inactivation in R. prowazekii has revealed that deletions are far more common than insertions and are, on average, much larger in size (Andersson and Andersson 1999). Examples of the former, where genome reduction occurred through large deletions removing several genes at once, are also found. For example an analysis of the total gene content of natural isolates of E. coli shows that large deletions occasionally become fixed within lineages (Ochman and Jones 2000). Likewise, at least 25 long deletions, in one case with the re-moval of as many as 16 ORFs at once, has been identified in Mycobacterium tuberculosis (Kato-Maeda, Rhee et al. 2001). Also for endosymbionts, re-ductive evolution through formation of large deletions has been inferred (Ochman and Moran 2001). In conclusion, the loss of DNA from a genome can occur through either large deletions or through slow erosion, although the relative importance of these two routes will vary among different bacte-rial species (Mira, Ochman et al. 2001).
Chromosome organization In contrast to the size and gene content of bacterial genomes the overall gene order and orientation of the bacterial chromosome remains relatively un-changed, even between different species. For example when the genome of E. coli is compared to that of Salmonella enterica, synonymous mutations

15
are almost saturated in the genome and a difference of more than 10% is seen on the protein level. In comparison, aside from insertions and deletions, the organization of the two genomes is almost identical (Rocha 2008). So what are the forces that preserve the gene order of bacterial chromosomes and why are some changes allowed when others are not? One possible an-swer to these questions is the operon theory, where genes present in an op-eron are kept together for regulatory purposes. Operons could also be main-tained due to horizontal gene transfer, since transfer of just one of the genes in an operon would be non-selective for the recipient if genes in an operon code for different parts of a certain pathway. In contrast, receiving a com-plete operon could enable the recipient to access a new growth niche (Lawrence and Roth 1996)). Another possible explanation for the lack of large chromosomal rearrangements is the symmetry of replication (equal distance from the origin of replication to the terminus for each replichore) (Liu, Liu et al. 2006). If a large deletion spans a portion of one half of the chromosome but not the other, one replichore would be longer than the other, i.e. the two replichores will not finish replication at the same time, possibly resulting in problems with decatenation and segregation (reviewed in (Rocha 2008)).
Mechanisms of reductive evolution Homologous recombination For a long time reductive evolution was thought to occur mainly through homologous recombination between long sequence repeats. A key enzyme in homologous recombination is the RecA protein, which binds and filaments on single stranded (ss) DNA (Story, Weber et al. 1992). As replication in bacterial cells often stalls or stops at different types of lesions or DNA breaks, a repair system designed to retain the genetic information is neces-sary. When a DNA break is encountered, break-processing enzymes target the break and prepare it for homologous recombination. A double stranded (ds) DNA break is rapidly degraded by the RecBCD enzyme complex, which possesses high affinity for dsDNA ends. These enzymes degrade the dsDNA until they reach a recognition site known as chi (χ). At chi the speci-ficity of the enzyme is altered so that only the 5’ DNA strand is degraded. Simultaneously the enzyme recruits RecA to filament on the created 3’ DNA overhang (Fig. 2). Filamented RecA can now bind to the double stranded sister chromatid and search for homology. When homology is reached, RecA mediates strand transfer and recruits factors for polymerization from the 3’ DNA end, which repair the degraded DNA. The junction that remains after the strand exchange event will eventually be resolved by the Holliday junc-tion resolving enzymes RuvABC (Kowalczykowski, Dixon et al. 1994).

16
RecA
RecBRecCRecDchi
chi
DSGap
5’5’3’
3’
5’3’
5’3’
5’3’
5’3’
5’3’
5’3’
5’3’
5’3’
5’3’
Replication restart
Fig. 2
Any single stranded (ss) DNA break is rapidly coated with single strand binding protein, SSB. For RecA to be able to filament on the ssDNA the SSB protein must be removed. To this end, RecFOR enzymes assist in the removal of SSB and in the loading of RecA on the ssDNA. RecA then searches for homology and performs the rest of its task as described for dsDNA breaks, resulting in a repaired ssDNA break (Fig. 3) (Kowalczykowski, Dixon et al. 1994).
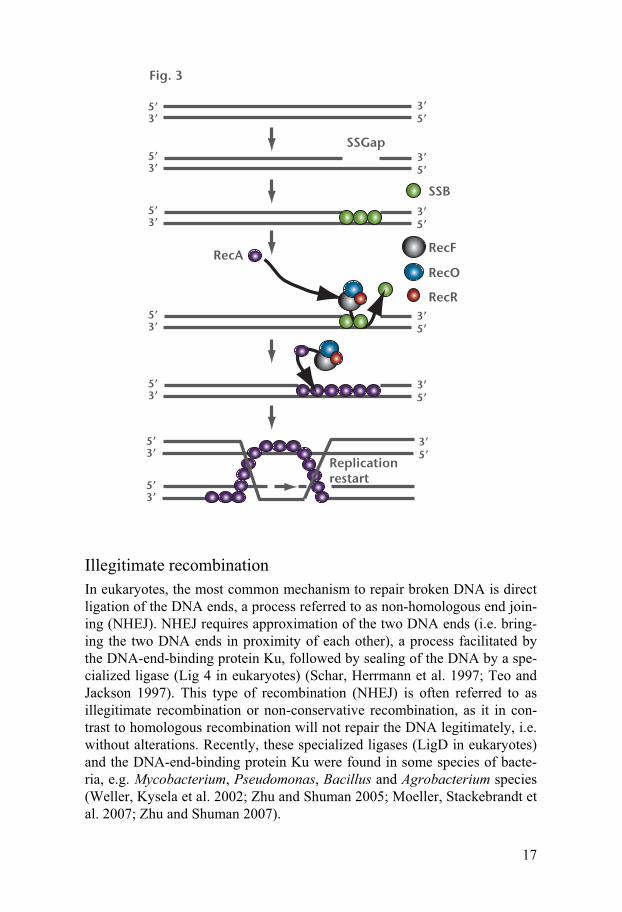
17
SSGap
5’5’3’
3’
5’3’ 5’
3’
5’3’ 5’
3’SSB
5’3’ 5’
3’
RecF
RecORecR
RecA
5’3’ 5’
3’
5’3’
5’3’
5’3’
Replication restart
Fig. 3
Illegitimate recombination In eukaryotes, the most common mechanism to repair broken DNA is direct ligation of the DNA ends, a process referred to as non-homologous end join-ing (NHEJ). NHEJ requires approximation of the two DNA ends (i.e. bring-ing the two DNA ends in proximity of each other), a process facilitated by the DNA-end-binding protein Ku, followed by sealing of the DNA by a spe-cialized ligase (Lig 4 in eukaryotes) (Schar, Herrmann et al. 1997; Teo and Jackson 1997). This type of recombination (NHEJ) is often referred to as illegitimate recombination or non-conservative recombination, as it in con-trast to homologous recombination will not repair the DNA legitimately, i.e. without alterations. Recently, these specialized ligases (LigD in eukaryotes) and the DNA-end-binding protein Ku were found in some species of bacte-ria, e.g. Mycobacterium, Pseudomonas, Bacillus and Agrobacterium species (Weller, Kysela et al. 2002; Zhu and Shuman 2005; Moeller, Stackebrandt et al. 2007; Zhu and Shuman 2007).

18
As many bacteria lack the specialized ligase required for NHEJ, another mechanism for illegitimate recombination in bacteria has been suggested, which involves DNA gyrase (Ikeda, Moriya et al. 1981; Ikeda, Shiraishi et al. 2004). As a replication fork unwinds the DNA, negative supercoiling ahead of the fork must be relieved and as a result positive supercoiling ac-cumulates in the wake of the replication fork. It is essential for the cell to convert this positive supercoiling back to its negative form to condense the DNA and establish the correct balance of constrained and unconstrained superhelicity (Liu and Wang 1987; Cook, Ma et al. 1992). To this end the bacterial cell utilizes topoisomerase II, also known as DNA gyrase. The to-poisomerase reaction is distinguished by a reaction cycle where an active tyrosine residue attacks the phosphodiester bond of a DNA strand, creating a break in the DNA and forming a covalent bond between the 5’ end of the broken DNA and the tyrosine residue of the topoisomerase (Fig. 4) (Wang 2002). Type I topoisomerases will only break one of the DNA strands, thereby allowing the other strand to pass through before resealing the DNA (Champoux 2001).
Fig. 4
However, for type II topoisomerases such as DNA gyrase, the break consists of two staggered cuts where both 5’ ends remain linked to the tyrosine resi-dues of the enzyme. This complex is called the cleavage complex and once it has been formed, the enzyme allows dsDNA to pass through the gap chang-ing the DNA topology before resealing the gap (Fig. 4) (Wang 1998; Wang 2002). The model for gyrase-mediated illegitimate recombination proposed by Ikeda and his coworkers involves two separate gyrase molecules cova-lently bound to DNA. The two DNA gyrases then meet and undergo subunit exchange resulting in the formation of recombinant DNA (Fig. 5) (Ikeda, Shiraishi et al. 2004).

19
Fig. 5
Mechanisms of deletion formation The most studied mechanism of deletion formation is that of non-conservative recombination between long sequence repeats. Several studies have used artificial template on plasmids or on the chromosome to elucidate possible mechanisms of deletion formation (Lovett, Gluckman et al. 1994; Bierne, Seigneur et al. 1997; Bierne, Vilette et al. 1997; Bzymek and Lovett 2001). The slipped mispairing model is conceptually related to the copy-choice model of recombination (Lederberg 1955) which was initially pro-posed to explain frame-shift mutations (Streisinger, Okada et al. 1966). The key feature of this model is the slippage of a growing DNA chain from one sequence repeat to another and the use of the tip of the growing DNA chain as a primer for further DNA synthesis (Fig. 6a) (Weston-Hafer and Berg 1989; Trinh and Sinden 1991; Lovett, Hurley et al. 2002). This type of slip-page is easy to envision as an event occurring inside a replication fork, but studies of transposon excision have revealed that this type of slippage can also occur between distant sites (Egner and Berg 1981; Weston-Hafer and Berg 1989; Gordenin, Malkova et al. 1992). Another model, the break and join model, introduces a break between the repeats by specific or non-specific enzymes. Subsequent processing of the resulting DNA ends by ex-onucleases could then expose the repeats, which could anneal and the result-ing molecule could be repaired by DNA synthesis and ligation (Fig. 6b). The two models make different predictions with regard to the genetic material from which the illegitimate recombination product molecule is formed. The

20
break and join model predicts the deleted DNA to be absent whereas accord-ing to the slipped mispairing model, the deleted DNA is present in the cell. Both of these models require some sequence homology at the deletion end-points and experimental setups have estimated how the length of the repeat affects the rate of non-conservative recombination. For RecA independent events, the rate of recombination is constant at homologies >50bp whereas for RecA dependent events homologies >150bp are required for the highest rates of recombination (Lovett, Hurley et al. 2002). Interestingly, experimen-tal data shows that the formation of large spontaneous deletions on the chromosome of S. typhimurium (Paper I) (Nilsson, Koskiniemi et al. 2005) and E.coli (Mashimo, Kawata et al. 2003) frequently occurs between very short (<10bp) or no repeats. Thus, several processes might be at play in spontaneous deletion formation on the bacterial chromosome.
DNA breaks and deletion formation The mechanism of deletion formation between short or no direct DNA re-peats might still be unsolved but several factors affecting deletion formation have been elucidated both for short-homology independent illegitimate re-combination (gyrase mediated illegitimate recombination) and the short-homology dependent illegitimate recombination described above. Many of these factors can be directed back to one fact, the presence of stalled or bro-ken replication forks in the cell. For gyrase mediated illegitimate recombina-tion, an increase in recombination is seen when cells are treated with oxolinic acid. Oxolinic acid is an antibiotic belonging to the quinolone anti-biotics as it targets DNA gyrase directly. It locks the gyrase enzyme in its open conformation, resulting in an increase in DNA breaks and increased deletion formation (Ikeda, Moriya et al. 1981; Ikeda, Aoki et al. 1982; Shi-

21
mizu, Yamaguchi et al. 1995). For short-homology dependent illegitimate recombination, one process affecting deletion formation is DNA replication. A wide variety of mutants with impaired processivity of replication were shown to have an increased deletion rate. For example, mutations in the normal replicative polymerase Pol III (dnaE) (Bierne, Vilette et al. 1997; Saveson and Lovett 1997) and other enzymes of the Pol III holoenzyme; dnaQ (the editing subunit), dnaB (the helicase), dnaN (the beta-clamp) and dnaX and holC (clamp loader complex) increased deletion rates (Saveson and Lovett 1997). Moreover, mutants lacking one of two helicases, rep or uvrD (DNA helicase II) have a decreased speed of replication and more ille-gitimate recombination (Shiraishi, Imai et al. 2005; Shiraishi, Imai et al. 2006). In eukaryotes, special DNA conformations, known as non-B DNA, have been associated with deletion endpoints (Bacolla, Jaworski et al. 2004). Specific sequence motifs, such as inverted repeat structures, have been shown to undergo structural changes during replication, resulting in such non-B DNA conformations, e.g hairpins or cruciforms (Bowater and Wells 2001; Majumdar and Patel 2002). Such structures could function as blockage for the replication fork, resulting in stalling and breakage of the fork, which could explain why non-B DNA is associated with deletion endpoints in eu-karyotes. Finally, the presence of stalled replication of forks and DNA breaks has been shown more directly to result in deletion formation (Kong and Masker 1994; Bierne, Ehrlich et al. 1997; Michel, Ehrlich et al. 1997). How slow processivity or stalling of the replication fork could mediate dele-tion between sites that are hundreds of kilobases apart is however still un-known.
Mutagenesis and DNA repair Mutation plays a fundamental role in evolution but it is also a potential threat to the viability and survival of cells. An inactivating mutation in an essential gene results in cell death, and for unicellular organisms such as bacteria, the loss of that individual from the population. As a consequence all character-ized species harbor mechanisms to limit and prevent the accumulation of too many mutations and a resulting mutational meltdown.
Origins of change Spontaneous genomic mutation rates are rather similar among unicellular microbes. A survey conducted by Jan Drake (1991) compared mutation rates in bacteria, phage and fungi with very different genome sizes and found that the mutation frequencies were similar for all species examined, approxi-mately 0.003 mutations / genome / generation (Drake 1991). This would

22
suggest that the rate of mutation and the rate of repair have been optimized during evolution.
Fidelity of DNA replication DNA replication is the largest contributor to maintaining genomic stability. Faithful replication of genetic material allows for unaltered inheritance of essential characteristics from parent to offspring. Nevertheless, every time a genome is duplicated, there is a possibility of base misincorporation. Even the most accurate DNA polymerases make errors as they replicate the ge-nome and these mistakes form part of the mutation frequencies observed in all organisms. The replicative DNA polymerase III (Pol III) normally repli-cates the bacterial chromosome in S. typhimurium and E. coli. Pol III is a complex multicomponent enzyme where the core holoenzyme consists of 10 different subunits. The holoenzyme includes both proofreading and exonu-clease activities which limit the amount of errors produced (reviewed in (Nohmi 2006)). Factors that cause incorporation of the wrong base include damaged or modified bases. In S. typhimurium and E. coli specialized poly-merases exist to replicate past various types of DNA lesions and damaged bases. These polymerases are known as the translesion DNA polymerases, and they are capable of replication past lesions that the normal replicative polymerase Pol III cannot pass (Ohmori, Friedberg et al. 2001). In this proc-ess the translesion DNA polymerases produce errors in their wake, which has led to the suggestion that the majority of mutations are the result of er-ror-prone replication by the translesion DNA polymerases (Wagner and Nohmi 2000; Nohmi 2006). Five DNA polymerases are found in S. typhi-murium and E. coli; the normal replicative polymerase Pol III, the major gap-filling polymerases Pol I and Pol II and the translesion DNA poly-merases Pol IV and Pol V. Despite its proofreading activity Pol II is also counted as a translesion DNA polymerase as it is involved in error-prone translesion synthesis across various damages in the template DNA (reviewed in (Nohmi 2006)). In Salmonella a sixth polymerase SamAB, a homologue of Pol V, is encoded on the virulence plasmid pSLT (Nohmi, Hakura et al. 1991). The translesion DNA polymerases are each specialized in bypassing different types of lesions although a high degree of redundancy can be seen (reviewed in (Nohmi 2006)). The current model of translesion synthesis proposes that the replicative DNA polymerase Pol III stalls at damaged bases and is replaced by one of the SOS induced polymerases to enable bypass of the lesion. However, recent findings suggest that the normal replicative po-lymerase Pol III itself has the ability to bypass several lesions in an error-prone manner (Fujii and Fuchs 2004; Kokubo, Yamada et al. 2005). So how are these polymerases coordinated when multiple polymerases possess the ability to bypass the same lesions? The beta-subunit of Pol III holoenzyme (HE), hereafter referred to as the beta-clamp, appears to have a key role in coordinating polymerase switching as it interacts with all the polymerases at

23
the same site (Lopez de Saro, Georgescu et al. 2003; Burnouf, Olieric et al. 2004). Interestingly, a hierarchy appears to exist (Pol III > Pol I > Pol II > Pol IV = Pol V, in S. typhimurium) among the polymerases in their propen-sity to gain access to the beta-clamp of the replication fork (reviewed in (Nohmi 2006)). As the beta-clamp is a homodimer two polymerases can bind to the replication fork simultaneously, one for leading strand synthesis and the other for lagging strand synthesis (Indiani, McInerney et al. 2005). Recent evidence however, suggests that three polymerases are bound to the fork simultaneously, where the third polymerase is held in reserve (McInerney, Johnson et al. 2007). This illustrates the replication fork as a highly dynamic complex that is able to frequently switch between poly-merases with little effort.
Sources of DNA damage DNA can be damaged both by endogenous and external factors. An example of the former is spontaneous decomposition of DNA in the form of depurina-tion or depyrimidination. This occurs when the N-glycosyl bond of the de-oxyribnonucleotides is exposed to hydrolytic attack, resulting in a apurinic /apyrimidic (AP) site. An AP site is non-coding and the passing polymerase could, in theory, insert any base at that position. However, the AP sites are difficult to pass and in E. coli adenine (A) is inserted with high frequency opposite the AP site as the A/AP pair is thermodynamically more stable than others. Insertion of an A base opposite the AP site results in GC to TA or AT to TA transversions (Loeb and Preston 1986). Transversions are mutations that change a purine to a pyrimidine and vice versa and result in more dra-matic structural changes, thought to be toxic. In contrast, a purine to purine or a pyrimidine to pyrimidine exchange are called transitions and considered less deleterious than transversions. Deamination is another type of spontane-ous DNA damage, that occurs when the exocyclic amino group of the deoxy-ribonucleotides is hydrolyzed. Mutations caused by deamination of cytosine, 5-methylcytosine, adenine and guanine are mostly transitions and in the case of guanine, the correct base is inserted (Kreutzer and Essigmann 1998).
With the exception of endogenous DNA damage bacterial cells are con-tinuously exposed to various damaging agents from the environment, such as UV-light, acidic or alkaline pH, temperature shifts and ionizing radiation. External damaging agents will result in a variety of base damage, the most studied being the formation of pyrimidine dimers caused by exposure to UV-light. Bacteria living in an aerobic environment need protection from oxida-tive damage due to the production of hydroxyl free radicals (OH.) that can deprotonate any DNA position causing a wide variety of base damage. The best-characterized hydroxyl-radical induced DNA lesions are 8-oxoguanosine (8-oxo-dG) and cis-thymine glycol. Thymine glycol is toxic in dsDNA as it blocks the replication fork and is mutagenic in ssDNA where it causes AT to GC transitions. 8-oxo-dG on the other hand, can mispair with

24
adenine during replication resulting in GC to TA transversions (Cheng, Ca-hill et al. 1992).
Maintaining genome integrity Only a limited subset of DNA damage is repaired by direct reversal. Instead, most DNA damage is repaired by excision of the damaged or inappropriate base followed by replacement with the correct one. To this end bacterial cells possess a cellular response, referred to as excision repair, which can be further divided into subclasses according to the mechanism of action.
Base excision repair (BER) The excision of damaged bases is initiated by the action of DNA glycosy-lases, which catalyze the hydrolysis of the N-glycosylic bonds linking bases to the phosphate backbone of DNA. Excision repair initiated by DNA glyco-sylases is referred to as base excision repair (BER), as the excised elements consist of free bases (Fig. 7a) (Duncan, Hamilton et al. 1976). Removal of the base however, results in another form of DNA damage, an apurinic / apyrimidic (AP) site. The repair of AP-sites require another set of enzymes known as AP-endonucleases which will target the 3’ or 5’ phosphodiester-bonds next to the AP-site, nicking the phosphate backbone of the DNA. Sub-sequently, exonucleases can enter the DNA at the nick and remove the AP-site from the phosphate backbone, resulting in repair by replication and sub-sequent ligation (Lindahl 1979). Each DNA glycosylase acts on very specific damaged bases, e.g. Tag and AlkA only recognize and excise 3-methyladenine whereas Ung removes uracil residues that are incorporated instead of thymine residues (Lindahl, Ljungquist et al. 1977; Yamamoto, Katsuki et al. 1978; Steinum and Seeberg 1986). As a consequence E. coli and S. typhimurium possess a wide variety of DNA glycosylases all with their own specificity for different damaged bases.

25
O
O
P O-O
O
N
O
O
P O-O
O
H2C
N
O
NH2
H2C
O
O
P O-O
O
H2C N
N
NH2
N
N
N
NH
O
O
Guanine
Cytosine
Uracil
O
O
P O-O
O
H2C N
N
NH2
N
N
Adenine
O
Uracil-DNA glycosylase
O
O
P O-O
O
N
O
O
P O-O
O
H2C
N
O
NH2
H2C
O
O
P O-O
O
H2C N
N
NH2
N
N
Guanine
Cytosine
O
O
P O-O
O
H2C N
N
NH2
N
N
Adenine
O
N
NH
O
O
Uracil
OH
H
+
Base excission repairFig. 7a
O
O
P O-O
O
N
O
O
P O-O
O
H2C
N
O
NH2
H2C
O
O
P O-O
O
H2C N
N
NH2
N
N
N
NH
O
O
Guanine
Cytosine
Thymine
O
O
P O-O
O
H2C N
N
NH2
N
N
Adenine
O
UvrC endonuclease
O
O
P O-O
O
N
O
OH
P O-O
O
H2C
N
O
NH2
H2C
O
O
P O-O
O
H2C N
N
NH2
N
N
Guanine
Cytosine
O
O
P O-O
O
H2C N
N
NH2
N
N
Adenine
O
OH
Nucleoide excission repair
HC
Fig. 7b
Nucleotide excision repair (NER) Like base excision repair, nucleotide excision repair (NER) is a multistep process that results in the formation of a gap in the DNA duplex that needs to be repaired by DNA polymerases and DNA ligase. In contrast to BER, NER excises the damaged bases, e.g. pyrimidine dimers or photoproducts, as intact nucleotides, or parts of oligonucleotide fragments (Grossman, Caron et al. 1988). The excision itself produces a gap in the DNA duplex that can be repaired by replication and ligation (Fig. 7b). A fundamental property of NER is the ability of the repair machinery to locate and recognize damaged bases. In E. coli and S. typhimurium two proteins collaborate to mediate this task. The UvrA protein in its dimeric form (UvrA2) binds DNA containing various forms of base damage (Grossman, Caron et al. 1988). However, these DNA-UvrA2 complexes are very unstable and it has been suggested that the role of UvrA in NER is that of a “molecular matchmaker” as it re-cruits UvrB to the site of damage before dissociating from the DNA (Lin and Sancar 1992). UvrB-DNA complexes are, on the contrary, highly stable. The third factor involved is the damage specific endonuclease, UvrC, which pos-sesses high affinity for the UvrB-DNA complexes. The UvrC protein is con-stitutively expressed at low levels and possesses the endonuclease activity required to incise the DNA (Orren and Sancar 1989). When over-expressed from a plasmid in the absence of DNA damage UvrC has been shown to

26
attack undamaged DNA (Branum, Reardon et al. 2001). UvrC nicks the DNA on both sides of the damaged base and an oligonucleotide fragment is removed. The small ssDNA gap created can then be repaired by DNA poly-merases and DNA ligase (Fig. 7c).
TT
TT
UvrAUvrA
UvrB
TT
UvrC
UvrC
TT
UvrC
TT
UvrC
TT
TT
DNA helicase II
DNA polymerase I
Fig. 7c

27
Methyl directed mismatch repair
One of the most important contributors to mismatched base pairs in the bac-terial cell is replication. In this case the correct base is located on the paren-tal strand of the newly replicated DNA and to maintain fidelity the newly
synthesized strand must be repaired. To this end the cell possesses a mechanism to separate between pa-rental and newly synthesized DNA, DNA methylation. After replication, the newly synthesized DNA strand is rapidly methylated by Dam-methylase at GATC consensus se-quences. The short window of time between replication and methylation allows the cell to distinguish between newly synthesized DNA and parental DNA. During this time the MutS enzyme of the methyl directed mis-match repair system scans the newly synthesized DNA for errors. When MutS recognizes a mispaired base pair it recruits the MutL and MutH proteins to form a complex, which is believed to facilitate bending of the DNA. The bent DNA brings the near-est GATC sequence into contact with the MutH protein which nicks the unmethylated DNA strand at the GATC sequence. Thus, the state of methylation determines the specificity of repair (Modrich 1991). DNA heli-case II then unwinds the newly syn-thesized DNA strand between the GATC site and the mismatch. As the
GATC site can be located either 3’ or 5’ to the mismatch, the excised DNA will be degraded by the proper exonuclease depending on the direction (Exo VII and RecJ for 5’ and Exo I for 3’) (Grilley, Griffith et al. 1993). When the mismatched DNA has been removed, DNA polymerase can re-synthesize the remaining gap to complete the repair process (Fig. 8).
MutS
MutL
MutH
DNA helicase II
Exo IRecJExo X
Fig. 8

28
Mutators Strains with an increased mutation rate compared to the standard (i.e. the median rate for the population) rate of mutation of a particular species, are referred to as mutators. A mutator phenotype can be generated in several ways, but the most common route is inactivation of one of the cellular repair pathways. Inactivating mutations in, for example, mutS, mutH or mutL have been shown to increase mutation rates approximately 200-fold in E. coli (Schaaper and Dunn 1991). Mutators are a source of genetic diversity in a population and it has been assumed that a subpopulation of mutators is al-ways present. However, in the absence of variable environments and strong novel selection, mutators do not generally benefit from their high mutation rate. As beneficial mutations only arise at very low frequencies (Denamur and Matic 2006), most mutations accumulating in these cells are detrimental. If the selection pressure would change however, the mutators have a higher chance of carrying or acquiring a mutation that is beneficial in the new selec-tive environment. It is well documented that mutators have a selective ad-vantage over non-mutators during strong selection and in variable environ-ments (Mao, Lane et al. 1997; Miller, Suthar et al. 1999; Giraud, Matic et al. 2001). For example during the evolution of 12 lineages of E. coli in minimal M9 media supplemented with glucose, mutators arose in 3 of those lineages after 10000 generations (Cooper, Rozen et al. 2003). Furthermore, constitu-tive mutS- mutators were shown to initially have an advantage compared to the wild type in colonizing the mouse gut. However, when the two strains had undergone 400 generations of preadaptation in the mice gut, the mutator no longer possessed an advantage in colonizing as the beneficial mutations had already been acquired by both wild type and mutS- strains (Giraud, Matic et al. 2001). Selection of constitutive mutators and their role in adapta-tion has been well documented in natural isolates where mutators are fre-quently found among both pathogenic and commensal bacteria (reviewed in (Giraud, Radman et al. 2001)). Mutator bacteria succeed in short-term adap-tation as they can accumulate adaptive mutations faster than wild type bacte-ria, allowing them to take over the growth niche. After selection is relieved however, the mutators will have accumulated mutations that may be neutral in the present environment but may decrease their fitness upon a shift to a new environment. However, mutator bacteria are not necessarily doomed. Mutator phenotypes can be reversed either by back mutation or by replacing the mutated gene by means of horizontal gene transfer (Denamur, Lecointre et al. 2000). Alternatively, the mutators could acquire suppressor mutations of the mutator phenotype, thereby preventing further accumulation of disad-vantageous mutations (Trobner and Piechocki 1984).

29
Cellular responses to stress When bacterial cells encounter stressful situations such as nutrient starva-tion, low pH, oxidative stress, or the consequences of such stress i.e. DNA damage, response systems in the cell are activated to protect the cell from damage.
Upon DNA damage a response system in the cell, known as the SOS-response, is activated (George, Devoret et al. 1974; Radman 1975). Dam-aged DNA is rapidly processed and filamentation of RecA on ssDNA medi-ates proteolytic cleavage of the LexA repressor of the SOS-response (Moreau 1985). Induction of the LexA regulon alters the expression of ap-proximately 40 genes under LexA control. Among these genes are the tran-slesion DNA polymerases (Fernandez De Henestrosa, Ogi et al. 2000; Quil-lardet, Rouffaud et al. 2003). Under non-induced conditions these poly-merases are present at relatively low levels but their expression is increased 10-20 fold during the SOS-response (Delmas and Matic 2006). It is assumed that this increase facilitates replication across various types of DNA lesions including the DNA damage that may have activated the stress response to begin with. LexA also regulates the expression of the UvrA and UvrB pro-teins of the nucleotide excision repair pathway described above, which can then remove and repair any modified nucleotides causing the stress.
Also, conditions such as oxidative stress, nutrient starvation or low pH, induce a cellular stress response that is regulated by the alternative transcrip-tion sigma factor RpoS. Induction of Rpos changes the expression of more than 300 genes to varying degrees (Patten, Kirchhof et al. 2004; Weber, Po-len et al. 2005). Among these genes, many proteins involved in the protec-tion and repair of DNA are found, e.g. one of the TLS DNA polymerases Pol IV (dinB) and the catalase genes katE and katG, which protect against the formation of oxygen free radicals by catalysing the formation of water and oxygen from hydrogen peroxide (Loewen 1984; Loewen, Triggs et al. 1985; Layton and Foster 2003). The sodC gene is also up-regulated during RpoS induction and is involved in protection against superoxide, as the gene prod-uct superoxide dismutase breaks down superoxide ions (Fridovich 1989; Imlay and Imlay 1996).
The adaptive mutation controversy A key to survival for microorganisms such as bacteria is their ability to adapt to a rapidly changing environment. Adaptation requires a phenotypic change, which will bestow the individual with this new phenotype with higher fitness in the changed environment. Such phenotypic change can be the result of altered regulation of a certain pathway but for adaptation to be hereditary a genetic change is required, i.e. a mutation. The classical neo-Darwinian view of evolution postulates that mutations occur randomly and at a rate that is

30
essentially independent of selection. The fact that at least some spontaneous mutations arise before and independently of selection was demonstrated early by the classic experiments of Luria and Delbruck (1943) and Lederberg (1952) (Luria and Delbruck 1943; Lederberg and Lederberg 1952). Luria and Delbruck studied the origin of phage-resistant mutants that appeared when a culture of E. coli was plated in the presence of bacteriophage T1. Differences in the number of mutants which arose in independent cultures led them to conclude that the mutations were rare spontaneous events and occurred prior to selection (Luria and Delbruck 1943). However, the lethal selections they used made it impossible to detect mutations inducible by selective conditions. Several researchers (Shapiro 1984; Cairns, Overbaugh et al. 1988; Hall 1990; Hall 1990) pointed out this deficiency and described genetic systems where selective conditions seemed to increase the mutation rate (Shapiro 1984; Cairns, Overbaugh et al. 1988; Hall 1990; Hall 1990). The Cairns system has been analyzed in greatest detail. In this system Lac- mutant cells, (Lac- due to of a frameshift mutation in the lacI region of a lacIZ gene fusion), incubated on lactose-containing medium accumulated mutations, allowing them to grow on lactose, over several days (Cairns, Overbaugh et al. 1988) (Cairns and Foster 1991). From these observations several models have been presented, suggesting that starvation for lactose induces a stress which in turn increases the mutation rate of the cells, result-ing in the increased number of Lac+ revertants observed after several days. One specific model system suggests that the increased mutagenesis requires induction of the RpoS regulon and over-expression of the translesion DNA polymerase Pol IV (dinB), found under the control of LexA and RpoS. The model suggests that a subpopulation of cells under stress will enter a hyper-mutable state and that these cells will accumulate mutations at a high rate until a beneficial mutation restoring growth occurs. For cells to enter the hypermutable state two factors are required simultaneously: induction of the general RpoS stress response by any stress inducing factor and induction of Pol IV expression by spurious SOS-induction. Induction of Pol IV expres-sion by two pathways simultaneously would then increase the mutation rate of the cell by an error-prone break repair process (Fig. 9a) (Ponder, Fonville et al. 2005). It is easy to interpret the observation of increasing Lac+ cells at later time points as a global or localized increase in mutation rate. If, how-ever, even a subset of the population grows, new opportunities for mutation are provided and any pre-existing variant with the smallest ability to grow, can improve. In fact, for many of the systems where stress induced mutagenesis have been proposed, the evidence that exists favors models where a subpopulation of cells grow under selection and a stepwise im-provement occurs during the selection period (Cairns, Overbaugh et al. 1988; Taddei, Halliday et al. 1997; Andersson, Slechta et al. 1998; Wrande, Roth et al. 2008). Thus, an alternative explanation for the accumulation of mutants on lactose in the lac-system suggests that if a subpopulation of the

31
plated cells carry a pre-existing duplication of the leaky lac-gene, that sub-population can grow slowly on the lactose medium. Further amplification of this pre-existing duplication increases growth on lactose further. The in-creased copy number of the leaky lac-gene increases the number of target genes for which a beneficial -1 frameshift mutation can revert the lac-gene back to wild type function. After reversion there is no selection to retain the amplification and the cells will revert back to one copy of lac (Fig. 9b) (re-viewed in (Roth, Kugelberg et al. 2006)).
lacI lacZ
-1 frameshift Growth on lactose(+)
+
n+++
n+++
0 frameshift
+++
Fig. 9a-1 frameshift DSB
Fig. 9b
SOS-induction -> Pol IVHomologous recombination
RpoS-induction
Stress
Pol IV
Stress-induced point mutations
High fidelity repair

32
Present investigations
Reductive evolution can be a rapid process in laboratory settings Obligate parasites and endosymbionts possess the smallest bacterial ge-nomes seen today, but they are believed to have originated from free-living ancestors with larger genomes (Gil, Sabater-Munoz et al. 2002). The tempo and mode of reductive evolution has been widely debated throughout the literature. An attempt to calculate some approximate rate of early genome reduction in Buchnera species established that the initial gene loss must have occurred faster than one gene / 5-10 million years (My), a number that was calculated for the last 50 My of separate evolution for two lineages of Buchnera species (Tamas, Klasson et al. 2002).
In paper I we set out to study genome reduction in a laboratory setting and set up an experimental system where we could partly mimic the intracel-lular lifestyle of endosymbiotic and parasitic bacteria. By growing cells on rich medium plates, hematin agar, we mimicked the nutrient rich environ-ment bacteria would encounter inside host cells. As population bottlenecks are associated with fixation of deleterious mutations (e.g. deletions) transfer-ring cells with one-cell bottlenecks could mimic the extensive genetic drift the intracellular bacteria typically encounter inside host. The lack of horizon-tal gene transfer inside a host represents a ratchet that only allows the reduc-tive process of endosymbiontic genomes to go in one direction. To simulate this a single colony was always picked during passaging to minimalize hori-zontal gene transfer among the bacteria.
Fig. 10

33
The evolution experiment was performed using two strains with either a low wild type or high (mutS-) mutation rate. 60 lineages of S.typhimurium LT2 wild type and the mutS- mutant each were grown on hematin agar plates. Every day one random colony from each lineage was re-streaked onto a new plate (Fig. 10). This was repeated for 300 and 60 cycles, for wild type and mutS- mutant respectively, resulting in a total of ~7500 and ~1500 genera-tions of growth. Cells were continuously (every 30 cycles) checked for chromosomal rearrangements by PFGE but for the wild type strain no rear-rangements could be seen even after 300 cycles, ~7500 generations. Using the mod-gal assay the deletion rate of the mutS- mutant was determined to be 50-fold higher than that of the wild type. The deletions found in the mutS- strain varied from 173bp to 1,2 kbp in size and from the four deletions found in the mutS- after ~1500 generations of growth, an initial rate of gene loss could be calculated. Assuming a generation time of one day, the initial rate of gene loss could be calculated to 2,5bp /cell /generation for the mutS- mu-tant and 0,05bp /cell /generation for a wt strain. These rates most likely rep-resent the maximum rate of genome reduction, as subsequent deletions probably would occur at a lower rate due to the reduction in amount of de-letable DNA. However, on an evolutionary time scale these data indicate that genome reduction could occur during a very short time, approximately 1Mbp in 50,000 years if a generation time of one day is assumed. This is an ex-tremely high rate of gene-loss compared to the estimation of the initial rate of genome reduction in the Buchnera species, 1 gene / 50 My. These calcula-tions are obviously associated with a considerable level of uncertainty since many factors such as the size of the bottleneck and the degree of horizontal gene transfer will affect the rate of DNA loss. However, for the Buchnera species there is reason to believe that reductive evolution initially occurred at high rates, to slow down as the genomes shrunk and the amount of non-essential genes decreased (Tamas 2002).
Mechanism of genome reduction How genome reduction occurs has been a subject of debate during the last decades (Moran and Mira 2001). Are genes lost in big pieces of DNA or slowly eroded by smaller deletions? According to our results in paper I ge-nome reduction can initially occur through large genomic deletions, up to 200kb at the time. Furthermore genes appearing to be essential in gene knock-out studies (Knuth, Niesalla et al. 2004), can be lost in these large deletions with no impairment of growth. It has been proposed that genome reduction during evolution of the intracellular parasites and endosymbionts was mainly carried out through RecA- dependent homologous recombina-tion. This was used as an argument for the evolutionary stasis in B.aphdicola the last 50million years as these strains long were the only sequenced bacte-

34
ria lacking RecA (Tamas, Klasson et al. 2002). However, analysis of the endpoints for the deletions analyzed in paper I indicated little (<15bp) or no homology at the deletion endpoints. Actually, 5/6 deletions found could not have been formed through RecA dependent homologous recombination, as this requires at least 25bp of homology (Shen and Huang 1986). These re-sults suggest that many large spontaneous deletions are not formed through RecA mediated homologous recombination, implying that RecA plays a less important role in reductive evolution than previously thought.
Mechanism of deletion formation If large spontaneous deletions are not formed between long direct repeats, how are they generated? As S. typhimurium does not appear to posses the enzymes required for NHEJ, another mechanism must be involved in the formation of these deletions. Genes involved in DNA repair and replication have been shown to influence deletion formation and the presence of DNA breaks, or stalled replication forks have been shown to increase deletion formation in several systems (Kong and Masker 1994; Bierne, Ehrlich et al. 1997; Bierne, Vilette et al. 1997; Michel, Ehrlich et al. 1997; Saveson and Lovett 1997). In paper III we set out to study the mechanism of spontane-ous deletion formation on the bacterial chromosome. Our aim was to inves-tigate the role of four translesion DNA polymerases found in S.typhimurium, in spontaneous deletion formation. We studied spontaneous deletion forma-tion in a specific region of the S. typhimurium chromosome. In the moaA-gal region of the S.typhimurium chromosome two operons, moaA and gal are separated by approximately 40 kb. The moaA gene encodes a protein in-volved in molybdate biosynthesis. Molybdate is a co-factor of nitrate reduc-tase and inactivation of nitrate reductase turns cells chlorate resistant as they no longer convert chlorate to the toxic chlorite under anaerobic growth con-ditions (Miller and Amy 1983). Also, any inactivating mutation in any of the gal genes, makes cells unable to grow on galactose, which can be seen as white colony appearance on MacConkey agar plates containing galactose. Finally we included a kanamycin resistance gene between the two operons. By selecting for cells that are chlorate resistant and screening for white kanamycin sensitive colonies, we could measure the deletion rate (Fig. 11) (Nilsson, Koskiniemi et al. 2005). We found that 90% of all spontaneous deletions required the four translesion DNA polymerases, Pol II, Pol IV, Pol V and SamAB for their formation. Conversely, increased expression of these polymerases, via SOS induction or artificial overproduction from an arabi-nose inducible plasmid increased the rate of deletion formation about 10- to 30-fold. To our knowledge this is the first direct evidence provided showing that deletion formation is dependent on the translesion DNA polymerases. Previous reports suggest an involvement of the normal replicative Pol III in

35
deletion formation between short and long tandem repeats (Trinh and Sinden 1993; Lovett and Feschenko 1996) and in illegitimate recombination (Dutra, Sutera et al. 2007). Furthermore, an important role for the translesion poly-merases, in particular Pol IV, has been shown for frameshift mutations (Kim, Maenhaut-Michel et al. 1997) and in deletion formation between long tan-dem repeats, but the mechanism of frameshift formation and RecA depedent homologous recombination is different from that observed for large deletions with no or little homology at deletion endpoints (Lovett, Hurley et al. 2002; Tippin, Pham et al. 2004).
mod kan gal837339 848198
Chlorate resistance
White appearance on MacConkey agar plates
Kanamycin sensitivity
Fig. 11
Inactivation results in
So how would these translesion DNA polymerases contribute to deletion formation in the cell? One possible mechanism is by interference at the rep-lication fork. Since the translesion DNA polymerases have a lower proces-sivity at the replication fork compared to the normal replicative polymerase Pol III (Wagner, Fujii et al. 2000), an excess of these polymerases at the fork could result in frequent replication pausing. To test this hypothesis we used the TUNEL assay to measure the number of DNA breaks in the cell. The TUNEL assay uses terminal deoxyribonucleotidyltransferase, an enzyme that adds on fluorescent nucleotides to free 3’OH DNA ends and as a conse-quence the relative increase in DNA breaks can be measured as an increase in fluorescence. We found that cells with a constitutively de-repressed LexA-regulon had a 10-fold increase in DNA breaks. The increase in breaks was not mediated by the translesion DNA polymerases but by three endonu-cleases, UvrBC and Cho, under LexA-control. Earlier reports confirm that over-expression of UvrC, in the absence of DNA damage resulted in an in-crease in DNA breaks (Branum, Reardon et al. 2001). Interestingly, removal of the three endonucleases under LexA-control resulted in a concomitant reduction in deletion rate. This indicates that two separate factors are limit-ing deletion formation, the number of DNA breaks and the amount of tran-slesion DNA polymerases in the cell. In conclusion we suggest that the tran-slesion DNA polymerases do not increase spontaneous deletion formation by

36
an increase in DNA breaks, but possibly use the breaks as template for ex-tension of misaligned DNA ends thereby allowing deletion formation be-tween sites with little or no homology.
Large variation in deletion rate at different chromosomal locations Even though bacterial chromosomes are highly dynamic in their nature the overall gene organization of the chromosome remains relatively unaltered between different species. For example, comparison of the genomes of two closely related bacteria, E. coli and S. typhimurium, revealed that while syn-onymous substitutions were nearly saturated between these two genomes and amino acid changes occurred up to 10%, the organization of the chromo-some remained almost the same (Rocha 2008). One of the factors affecting chromosomal rearrangements is the presence of supercoiling and other macro domains in the chromosome. The presence of supercoiling domains has been shown to confer mechanistic constraints on chromosomal rear-rangements, such as deletions, creating boundaries across which rearrange-ments rarely occur (Charlebois and St Jean 1995). In paper II we con-structed a genetic tool, the “deletometer”, to study how chromosomal loca-tion will affect large chromosomal rearrangements in the form of deletions. The deletometer can be used to study spontaneous chromosomal deletion rates at various chromosomal locations using the same genetic screen. The deletometer consists of a transposable backbone (Tn10dtet) carrying the lac operon, the moaA gene and a chloramphenicol resistance gene. The gene product of the moaA gene is involved in the molybdate biosynthesis pathway and loss of this gene renders cells chlorate resistant. Cells lacking the lac operon will no longer be able to grow on lactose, which can be seen as white colony appearance on MacConkey agar plates containing lactose. Finally, cells lacking a chloramphenicol gene are not able to grow on plates contain-ing chloramphenicol. Using these selections and screens the deletometer could be used to study deletion rates at various chromosomal locations, by plating cells on MacConkey agar plates containing lactose and screening for chlorate resistant white colonies that were sensitive to chloramphenicol.
We measured deletion rates at 12 chromosomal locations using the dele-tometer and found that the spontaneous deletion rate varied 44-fold between different chromosomal locations. As the rate of deletion formation is likely to appear higher when the deletometer is surrounded by more deletable ma-terial, we corrected for this effect by normalizing the deletion rate at each location to the experimentally defined deletable region. The deletable re-gions were mapped as the largest deletion found at each location using pulsed field gel electrophoresis (PFGE) and PCR. After this normalization,

37
deletion rates varied more than a 100-fold between different chromosomal locations. The highest deletion rates were found with three individual dele-tometers all located around 2 Mbp of the S.typhiumurium chromosome, im-plying a potential hotspot for deletion formation in this region. This could be expected if some of the deletometers were flanked by perfect direct repeats, such as IS-elements. However, no longer (>25bp) direct repeats were found in the regions with higher deletion rates than those with lower deletion rates. Differences in the rate of deletion formation at different chromosomal loca-tions could affect the trajectory of reductive evolution. As deletions are gen-erally considered deleterious, they will be fixed in a population by chance due to high drift and small population bottlenecks. However, if deletion rates at one chromosomal location are 100-fold higher as compared to other re-gions, the chance of fixing deletions in this region is likely to increase, af-fecting the rate of reductive evolution in that specific region.
Fitness effects of large chromosomal deletions Chromosomal rearrangements, such as large deletions are prone to alter the highly conserved organization of the chromosome. Bacterial chromosomes are selected for specific gene order and orientation and large rearrangements in the genome organization are often deleterious for the cells (Rocha 2008). Using the deletions found with the deletometer, in paper II we set out to study how different large deletions at various chromosomal locations would affect cellular fitness. Fitness was measured as growth rate during exponen-tial growth in rich LB media and poorer M9-media supplemented with glyc-erol. The isolated deletions ranged from 1-200 kbp in size but no negative correlation between fitness (measured as exponential growth rate) and dele-tion size could be seen. Instead the gene content of the deletion seemed to affect fitness more than the actual size. Surprisingly, some of the deletions, ranging from 18- to 38 kbp in size, increased the exponential growth rate of the cells with up to 10%. Why removal of certain genetic elements would increase the growth rate of the cells is at present unclear but one explanation could be that loss of certain genetic elements that are unused in both rich and minimal growth media, reduces energetic costs of DNA, RNA and protein synthesis, freeing resources for synthesis of other required gene products. Another alternative explanation could be that the deleted regions contain sequences that posses a direct inhibitory effect on growth at these conditions, as has been observed for anti-virulence genes in bacterial pathogens (Maurelli 2007).

38
Mutation rates are remarkably robust The classical neo-Darwinian view of evolution postulates that mutations arise randomly and at a rate that is independent of selection. It has however been suggested that bacteria can change both the rate and specificity of mu-tation formation in response to various stresses. One specific model-system proposes that this putative stress-induced mutagenesis requires both induc-tion of the error-prone DNA polymerase IV under control of the DNA dam-age-inducible LexA regulon (SOS response) and an increase in levels of the starvation-induced sigma-factor RpoS (Galhardo, Hastings et al. 2007). In paper IV we have examined this idea by determining whether a simultane-ous induction of the LexA-mediated SOS response and an increase in RpoS levels will cause an increase in mutation rates. We have studied the impact of these two different stress responses on four different types of mutations: resistance to the antibiotics nalidixic acid and rifampicin caused by amino acid changes in gyrA and rpoB, respectively, resistance to chlorate caused by any inactivating mutation in the chlA-G genes and reversion to Lac+ caused by -1 frameshift mutations in the lacI gene of the the lacIZ gene fusion.
We observed that constitutive de-repression of the LexA regulon by a lexA(def) mutation increased mutation rates for all types of mutations. How-ever, the increase was due to the translesion DNA polymerases for only one type of mutations. For rifampicin- and nalidixic acid resistance mutations and Lac+ reversion no decrease in mutation rates was seen in a strain lacking all four translesion DNA polymerases in a lexA(def) background. In contrast, for chlorate resistance mutations the mutation rate was decreased to wild type levels when inactivating all four translesion DNA polymerases in a lexA(def) background. Chlorate resistance can be acquired by a wide variety of mutations in a large number of genes whereas rifampicin and nalidixic acid resistance mutations are acquired through a limited subset of basepair substitutions in specific genes. Our results imply that the translesion DNA polymerases do contribute to the increased mutagenesis seen when de-repressing the LexA-regulon, but only to some types of mutations. For other types of mutations, such as base pair substitutions, the translesion DNA po-lymerases do not contribute considerably to mutagenesis, at least not at the polymerase levels conferred when the LexA regulon is de-repressed by a lexA(def) mutation. In contrast to the translesion DNA polymerases, three endonucleases found under LexA-control, decreased mutation rates to wild type levels for all types of mutations in a lexA(def) background. In paper III we showed that these endonucleases under LexA-control, increase the amount of ssDNA gaps in the cell when overexpressed constitutively in a lexA(def) mutant. These results indicate that an increase in DNA breaks/gaps increases the mutation rate, through any polymerase at the fork in an error-prone gap repair process. Error-prone break repair has been described ear-lier, but as a mechanism where the translesion DNA polymerase Pol IV re-

39
pairs DNA breaks in an error-prone manner, resulting in an increased mutagenesis in the cell (Ponder, Fonville et al. 2005). Here we show that the DNA breaks per se appear to increase mutagenesis, with or without the tran-slesion DNA polymerases.
With regard to the potential involvement of RpoS in mutagenesis, neither an increase in RpoS levels conferred by artificial over-expression from a plasmid nor long-term stationary phase incubation or slow growth caused an increase in any of the four mutation rates measured, alone or in combination with over-expression of the translesion DNA polymerases. In conclusion, mutation rates are remarkably robust and no combination of growth condi-tions, induction of translesion polymerases or increased RpoS expression could confer an increase in mutation rates higher than the moderate increase caused by de-repression of the LexA regulon alone.
Concluding remarks Bacteria frequently encounter rapidly changing environments and their abil-ity to adapt is the key for their survival. An understanding of the mecha-nisms and rates of genetic change is necessary if we want to gain insight into the future of our microscopic companions. Microorganisms are masters of adaptation, and to co-exist we must find ways to keep up, for example by limiting their rate of adaptation to antibiotics. In this thesis my work has focused on the changes in the bacterial chromosome and which factors affect this change.
It is fascinating to see how fast selection, or the lack of selection, can alter the composition of a bacterial genome. In paper I we showed that during conditions of high genetic drift, genome reduction could occur at very high rates. Furthermore, in paper II we show that large spontaneous deletions can actually increase fitness of the cells in a specific environment. Thus, it is possible that reductive genome evolution can be driven by selection as well. This is an example of where genetic drift (i.e. chance) and natural selection could result in essentially the same outcome, i.e. gene loss. Yet another ex-ample of how little we know about the forces affecting chromosome compo-sition is seen in studies where the aim has been to establish the minimal gene set required for life (Koonin 2000; Gil, Silva et al. 2004; Knuth, Niesalla et al. 2004; Glass, Assad-Garcia et al. 2006). In paper I we find that many genes found to be essential in such knock-out studies (Knuth, Niesalla et al. 2004), were easily disposable in large deletions, with no obvious affects on bacterial fitness.
Even though selection is often the factor deciding which changes are fixed in a population and which are not, the rates of mutation will play an important role in environments where purifying selection is low. In paper II we find that the rate of deletion formation can vary at least 100-fold depend-

40
ing on the chromosomal region. Differences like this are likely to affect the trajectory of reductive evolution, as the probability of fixing frequent dele-tions is likely to increase.
Another aspect of adaptation is how it occurs mechanistically. How are large chromosomal rearrangements formed in the cell and what are the sources of mutation? In paper I we observed that most large chromosomal deletions were not generated by homologous recombination, an important finding since homologous recombination has been suggested as the major path to genome reduction (Tamas, Klasson et al. 2002). In paper III we follow up this finding and show that large spontaneous deletions require the translesion DNA polymerases and DNA gaps for their formation. The impor-tance of stalled replication forks and DNA gaps in deletion formation has been established previously (Kong and Masker 1994; Bierne, Ehrlich et al. 1997), but the forces acting on the stalled or broken forks to mediate dele-tions have not been identified. The same two factors, DNA breaks and tran-slesion DNA polymerases have been described to induce an error-prone break repair process that will repair the DNA, but with errors (Ponder, Fon-ville et al. 2005; Lovett 2006). In paper IV we found that mutation rates increased with increased numbers of DNA breaks, but even though the tran-slesion DNA polymerases did contribute to mutagenesis, they were only responsible for a subset of mutations. In conclusion, restart of a stalled or collapsed replication fork per se seems to increase mutagenesis in the form of basepair substitution, whereas deletions require the translesion DNA po-lymerases at the restarting fork. It will be interesting to see how and why restart of a replication fork results in increased mutagenesis.

41
Future perspectives
From the results of this thesis several interesting follow-up projects have emerged.
At first it will be interesting to determine why deletion rates vary more than a 100-fold between different chromosomal locations. One explanation could be the presence of supercoiling domains that affect the rate of recombination and deletion formation in the cells. To test this the level of supercoiling of the cells could be altered. Generally the DNA of E. coli cells is more negatively super-coiled than that of its close relative S. typhimurium. It is possible to change the supercoiling of S. typhimurium to that of E. coli and see how that would affect the rates of deletion formation (Champion and Higgins 2007). Will the rates change uniformly over the chromosome, or will only some regions be af-fected? Another possible explanation is that regions with highly transcribed genes have higher rates of deletion formation as the DNA in these regions is more accessible for DNA damaging agents that could generate DNA breaks. To test this, the deletometer could be transferred to a gene known to have low expression but that is in close proximity to the original region with high dele-tion rate. If the hypothesis is true, the deletion rate should be lower at this new location. The third possible reason is the presence of DNA structures such as hairpins or cruciforms in the DNA. Cruciforms and hairpins can function as roadblocks for the replication fork and the presence of such cruciforms could therefore increase deletion rate locally. This hypothesis could be tested in a mutant lacking the hairpin specific endo- and exonuclease SbcCD. SbcCD cleaves and degrades these structures resulting in more DNA gaps, which have been shown increase the frequency of deletion formation. Mutants lacking this enzyme should have lower deletion rates in regions where cruciforms are in-fluencing deletion formation.
Secondly it will be interesting to determine why some large chromosomal deletions increase cellular fitness. Initially, we need to find out the exact endpoints of these deletions. Possibly the deletions contain genes that are highly expressed but not required for growth in that specific environment. Alternatively, the region could contain specific DNA sequences that inhibit growth as seen for anti-virulence genes in pathogens. It will also be impor-tant to find out if all the deletions that increase exponential growth rate also out-compete the wild type during the whole growth cycle.

42
Thirdly, we still do not know which types of deletion endpoints are gener-ated in the large spontaneous chromosomal deletions. It would be interesting to compare the deletion endpoints in cells with and without the four transle-sion DNA polymerases present. This could give us clues to whether the tran-slesion DNA polymerases contribute to short-homology dependent illegiti-mate recombination or short-homology independent illegitimate recombina-tion, or both.
Finally, it would be interesting to find out why stalled and broken replication forks increase the formation of deletions and mutations. Are restarted repli-cation forks somehow different from normal replicative replication forks and if they are, how are they different?

43
Acknowledgements
I am not a master with words nor someone who is known to extensively show my gratitude, so for all of you that have helped me through the years, here are my thanks to you. First of all I would like to thank my supervisor Dan Andersson without whom this thesis would never have come to be. Thank you for all the inspi-ration and guidance along my path to becoming an independent researcher, I do not think anyone else could have done a better job. I am deeply grateful for the time I have spent under your supervision. I would also like to thank my co-supervisor Diarmaid Hughes for all of your critical and constructive opinions on whatever the project. Together you and Dan added up to the perfect balance of enthusiasm and structure. You are greatly appreciated. I would also like to thank my co-authors and other nice scientific people that I have had the opportunity to meet over the years. Special thanks to John Roth for all the great suggestions about my projects and for communicating one of the PNAS papers. And the people in John's lab, Drew, Amiko, Se-marhy, Doug, Jacob and Eric, it has been a pleasure to get to know you. Thanks to Jim Sawitzke for many tips and tricks about linear transforma-tions. And to Kelly Hughes, Sue Lovett, Stanley Malloy, Paul Rainey, Mar-tin Marinus and many more for inspiring me to become a better researcher and for all good comments on my projects over the years. I am grateful to all the past members of the DA-lab: To Annika for taking care of me when I first started and for all the support since then. You were my lab supervisor, but you soon became one of my dearest friends. Wille, who has brought fun and energy to my world of science. You are out-standing in intensity and those who have not met you do not know what hard work really is. Elisabeth, I counted the days to you're return to Stockholm as your presence always made the lab brighter. Sophie, the one who knew eve-rything, you were and still are a role-model in science for me. Cissi, who always had great suggestions and time to answer stupid questions. Therese, for all the great times in the gym and for discussions on every perspective of life. And finally to Caroline who became one of my best friends from the

44
first day in the lab and have remained by my side for all these years, you are my safety vent. Thanks also to all the present members of the DA-lab, without whom I don’t think I would have survived. You feed me and help me to remain sane, but most of all, the lab is a better place with you people in it. Special thanks to Anna who has supported me like no one else. You are truly one of a kind and I am sure that nothing I have done during this time would have been the same without you. Maria, for the joyful singing and the atmosphere you bring to the lab. Thank you for being you. Linus for always making me feel brilliant and creative even when I’m not. You are the hub that our lab circles around. Chris, for all the fun times in and outside of the lab and for being a great friend. You bring joy to my world. Peter, who began as my student and soon became the master. Thank you for all the mind ravelling discussions and for being special. To Song who is outstanding and scientifically way past the rest of us already. It has been a pleasure to get to know you. To Jocke for all the great scientific discussions in and outside of the lab, you are my problem solver. And to the newcomers; Amira, Hervé, Marlen, Erik and Hava, with hopes of good times to come and wishing you the best of luck in the future. I would also like to thank the people of B9:3, D7:3 and many others at IM-BIM for great times and fun Iphab pubs and Imbim days. And the secretar-ies especially Barbro, Marianne and Rehné for your help with all the paper work. Special thanks to Chris and Anna for helping me with the language as I wrote this thesis and to Erik or helping me with the cover. Special thanks to Lena for taking care of me when I first arrived in Uppsala and for you're continued support throughout all the years that followed. I would also like to thank Jojjo for being my friend and for supporting me over the years. You are the greatest. And last but not least, I would like to thank my family. My parents for sup-porting me every step of the way. To know you are always there for me is what keeps me going. My sister for inspiring me to get into the world of science in the first place. You are my dearest friend and the one I turn to when troubled. Thank you for being there. My brother and Janette for sup-plying me with my own safe haven. Thank you for providing a home full of joy and atmosphere when needed. And finally to my Tomas without whom I would never have lasted this long. I love you.

45
References
Andersson, D. I., E. S. Slechta and J. R. Roth (1998). "Evidence that gene amplification underlies adaptive mutability of the bacterial lac operon." Science 282(5391): 1133-5. Andersson, J. O. and S. G. Andersson (1999). "Insights into the evolutionary process of genome degradation." Curr Opin Genet Dev 9(6): 664-71. Andersson, S. G. and C. G. Kurland (1998). "Reductive evolution of resident genomes." Trends Microbiol 6(7): 263-8. Bacolla, A., A. Jaworski, J. E. Larson, J. P. Jakupciak, N. Chuzhanova, S. S. Abeysinghe, C. D. O'Connell, D. N. Cooper and R. D. Wells (2004). "Breakpoints of gross deletions coincide with non-B DNA conformations." Proc Natl Acad Sci U S A 101(39): 14162-7. Bierne, H., S. D. Ehrlich and B. Michel (1997). "Deletions at stalled replica-tion forks occur by two different pathways." Embo J 16(11): 3332-40. Bierne, H., M. Seigneur, S. D. Ehrlich and B. Michel (1997). "uvrD muta-tions enhance tandem repeat deletion in the Escherichia coli chromosome via SOS induction of the RecF recombination pathway." Mol Microbiol 26(3): 557-67. Bierne, H., D. Vilette, S. D. Ehrlich and B. Michel (1997). "Isolation of a dnaE mutation which enhances RecA-independent homologous recombina-tion in the Escherichia coli chromosome." Mol Microbiol 24(6): 1225-34. Bowater, R. P. and R. D. Wells (2001). "The intrinsically unstable life of DNA triplet repeats associated with human hereditary disorders." Prog Nu-cleic Acid Res Mol Biol 66: 159-202. Branum, M. E., J. T. Reardon and A. Sancar (2001). "DNA repair excision nuclease attacks undamaged DNA. A potential source of spontaneous muta-tions." J Biol Chem 276(27): 25421-6. Burnouf, D. Y., V. Olieric, J. Wagner, S. Fujii, J. Reinbolt, R. P. Fuchs and P. Dumas (2004). "Structural and biochemical analysis of sliding clamp/ligand interactions suggest a competition between replicative and translesion DNA polymerases." J Mol Biol 335(5): 1187-97.

46
Bzymek, M. and S. T. Lovett (2001). "Instability of repetitive DNA se-quences: the role of replication in multiple mechanisms." Proc Natl Acad Sci U S A 98(15): 8319-25. Cairns, J. and P. L. Foster (1991). "Adaptive reversion of a frameshift muta-tion in Escherichia coli." Genetics 128(4): 695-701. Cairns, J., J. Overbaugh and S. Miller (1988). "The origin of mutants." Na-ture 335(6186): 142-5. Casjens, S. (1998). "The diverse and dynamic structure of bacterial ge-nomes." Annu Rev Genet 32: 339-77. Champion, K. and N. P. Higgins (2007). "Growth rate toxicity phenotypes and homeostatic supercoil control differentiate Escherichia coli from Sal-monella enterica serovar Typhimurium." J Bacteriol 189(16): 5839-49. Champoux, J. J. (2001). "DNA topoisomerases: structure, function, and mechanism." Annu Rev Biochem 70: 369-413. Charlebois, R. L. and A. St Jean (1995). "Supercoiling and map stability in the bacterial chromosome." J Mol Evol 41(1): 15-23. Cheng, K. C., D. S. Cahill, H. Kasai, S. Nishimura and L. A. Loeb (1992). "8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G----T and A----C substitutions." J Biol Chem 267(1): 166-72. Cook, D. N., D. Ma, N. G. Pon and J. E. Hearst (1992). "Dynamics of DNA supercoiling by transcription in Escherichia coli." Proc Natl Acad Sci U S A 89(22): 10603-7. Cooper, T. F., D. E. Rozen and R. E. Lenski (2003). "Parallel changes in gene expression after 20,000 generations of evolution in Escherichiacoli." Proc Natl Acad Sci U S A 100(3): 1072-7. Darwin, C. (1859). On the Origin of Species by Means of Natural Selection, or the Preservation of Favoured Races in the Struggle for Life. London, John Murray Albemarle street. Delmas, S. and I. Matic (2006). "Interplay between replication and recombi-nation in Escherichia coli: impact of the alternative DNA polymerases." Proc Natl Acad Sci U S A 103(12): 4564-9. Denamur, E., G. Lecointre, P. Darlu, O. Tenaillon, C. Acquaviva, C. Sayada, I. Sunjevaric, R. Rothstein, J. Elion, F. Taddei, M. Radman and I. Matic

47
(2000). "Evolutionary implications of the frequent horizontal transfer of mismatch repair genes." Cell 103(5): 711-21. Denamur, E. and I. Matic (2006). "Evolution of mutation rates in bacteria." Mol Microbiol 60(4): 820-7. Drake, J. W. (1991). "A constant rate of spontaneous mutation in DNA-based microbes." Proc Natl Acad Sci U S A 88(16): 7160-4. Duncan, J., L. Hamilton and E. C. Friedberg (1976). "Enzymatic degradation of uracil-containing DNA. II. Evidence for N-glycosidase and nuclease ac-tivities in unfractionated extracts of Bacillus subtilis." J Virol 19(2): 338-45. Dutra, B. E., V. A. Sutera, Jr. and S. T. Lovett (2007). "RecA-independent recombination is efficient but limited by exonucleases." Proc Natl Acad Sci U S A 104(1): 216-21. Egner, C. and D. E. Berg (1981). "Excision of transposon Tn5 is dependent on the inverted repeats but not on the transposase function of Tn5." Proc Natl Acad Sci U S A 78(1): 459-63. Felsenstein, J. (1974). "The evolutionary advantage of recombination." Ge-netics 78(2): 737-56. Fernandez De Henestrosa, A. R., T. Ogi, S. Aoyagi, D. Chafin, J. J. Hayes, H. Ohmori and R. Woodgate (2000). "Identification of additional genes be-longing to the LexA regulon in Escherichia coli." Mol Microbiol 35(6): 1560-72. Fridovich, I. (1989). "Superoxide dismutases. An adaptation to a paramag-netic gas." J Biol Chem 264(14): 7761-4. Fujii, S. and R. P. Fuchs (2004). "Defining the position of the switches be-tween replicative and bypass DNA polymerases." Embo J 23(21): 4342-52. Galhardo, R. S., P. J. Hastings and S. M. Rosenberg (2007). "Mutation as a stress response and the regulation of evolvability." Crit Rev Biochem Mol Biol 42(5): 399-435. George, J., R. Devoret and M. Radman (1974). "Indirect ultraviolet-reactivation of phage lambda." Proc Natl Acad Sci U S A 71(1): 144-7. Gil, R., B. Sabater-Munoz, A. Latorre, F. J. Silva and A. Moya (2002). "Ex-treme genome reduction in Buchnera spp.: toward the minimal genome needed for symbiotic life." Proc Natl Acad Sci U S A 99(7): 4454-8.

48
Gil, R., F. J. Silva, J. Pereto and A. Moya (2004). "Determination of the core of a minimal bacterial gene set." Microbiol Mol Biol Rev 68(3): 518-37. Giraud, A., I. Matic, O. Tenaillon, A. Clara, M. Radman, M. Fons and F. Taddei (2001). "Costs and benefits of high mutation rates: adaptive evolu-tion of bacteria in the mouse gut." Science 291(5513): 2606-8. Giraud, A., M. Radman, I. Matic and F. Taddei (2001). "The rise and fall of mutator bacteria." Curr Opin Microbiol 4(5): 582-5. Glass, J. I., N. Assad-Garcia, N. Alperovich, S. Yooseph, M. R. Lewis, M. Maruf, C. A. Hutchison, 3rd, H. O. Smith and J. C. Venter (2006). "Essential genes of a minimal bacterium." Proc Natl Acad Sci U S A 103(2): 425-30. Gordenin, D. A., A. L. Malkova, A. Peterzen, V. N. Kulikov, Y. I. Pavlov, E. Perkins and M. A. Resnick (1992). "Transposon Tn5 excision in yeast: influence of DNA polymerases alpha, delta, and epsilon and repair genes." Proc Natl Acad Sci U S A 89(9): 3785-9. Grilley, M., J. Griffith and P. Modrich (1993). "Bidirectional excision in methyl-directed mismatch repair." J Biol Chem 268(16): 11830-7. Grossman, L., P. R. Caron, S. J. Mazur and E. Y. Oh (1988). "Repair of DNA-containing pyrimidine dimers." Faseb J 2(11): 2696-701. Hall, B. G. (1990). "Directed evolution of a bacterial operon." Bioessays 12(11): 551-8. Hall, B. G. (1990). "Spontaneous point mutations that occur more often when advantageous than when neutral." Genetics 126(1): 5-16. Ikeda, H., K. Aoki and A. Naito (1982). "Illegitimate recombination medi-ated in vitro by DNA gyrase of Escherichia coli: structure of recombinant DNA molecules." Proc Natl Acad Sci U S A 79(12): 3724-8. Ikeda, H., K. Moriya and T. Matsumoto (1981). "In vitro study of illegiti-mate recombination: involvement of DNA gyrase." Cold Spring Harb Symp Quant Biol 45 Pt 1: 399-408. Ikeda, H., K. Shiraishi and Y. Ogata (2004). "Illegitimate recombination mediated by double-strand break and end-joining in Escherichia coli." Adv Biophys 38: 3-20. Imlay, K. R. and J. A. Imlay (1996). "Cloning and analysis of sodC, encod-ing the copper-zinc superoxide dismutase of Escherichia coli." J Bacteriol 178(9): 2564-71.

49
Indiani, C., P. McInerney, R. Georgescu, M. F. Goodman and M. O'Donnell (2005). "A sliding-clamp toolbelt binds high- and low-fidelity DNA poly-merases simultaneously." Mol Cell 19(6): 805-15. Kato-Maeda, M., J. T. Rhee, T. R. Gingeras, H. Salamon, J. Drenkow, N. Smittipat and P. M. Small (2001). "Comparing genomes within the species Mycobacterium tuberculosis." Genome Res 11(4): 547-54. Kim, S. R., G. Maenhaut-Michel, M. Yamada, Y. Yamamoto, K. Matsui, T. Sofuni, T. Nohmi and H. Ohmori (1997). "Multiple pathways for SOS-induced mutagenesis in Escherichia coli: an overexpression of dinB/dinP results in strongly enhancing mutagenesis in the absence of any exogenous treatment to damage DNA." Proc Natl Acad Sci U S A 94(25): 13792-7. Knuth, K., H. Niesalla, C. J. Hueck and T. M. Fuchs (2004). "Large-scale identification of essential Salmonella genes by trapping lethal insertions." Mol Microbiol 51(6): 1729-44. Kokubo, K., M. Yamada, Y. Kanke and T. Nohmi (2005). "Roles of replica-tive and specialized DNA polymerases in frameshift mutagenesis: mutability of Salmonella typhimurium strains lacking one or all of SOS-inducible DNA polymerases to 26 chemicals." DNA Repair (Amst) 4(10): 1160-71. Kong, D. and W. Masker (1994). "Deletion between direct repeats in T7 DNA stimulated by double-strand breaks." J Bacteriol 176(19): 5904-11. Koonin, E. V. (2000). "How many genes can make a cell: the minimal-gene-set concept." Annu Rev Genomics Hum Genet 1: 99-116. Kowalczykowski, S. C., D. A. Dixon, A. K. Eggleston, S. D. Lauder and W. M. Rehrauer (1994). "Biochemistry of homologous recombination in Es-cherichia coli." Microbiol Rev 58(3): 401-65. Kreutzer, D. A. and J. M. Essigmann (1998). "Oxidized, deaminated cytosi-nes are a source of C --> T transitions in vivo." Proc Natl Acad Sci U S A 95(7): 3578-82. Lawrence, J. G. and J. R. Roth (1996). "Selfish operons: horizontal transfer may drive the evolution of gene clusters." Genetics 143(4): 1843-60. Layton, J. C. and P. L. Foster (2003). "Error-prone DNA polymerase IV is controlled by the stress-response sigma factor, RpoS, in Escherichia coli." Mol Microbiol 50(2): 549-61. Lederberg, J. (1955). "Recombination mechanisms in bacteria." J Cell Physiol Suppl 45(Suppl. 2): 75-107.

50
Lederberg, J. and E. M. Lederberg (1952). "Replica plating and indirect selection of bacterial mutants." J Bacteriol 63(3): 399-406. Lin, J. J. and A. Sancar (1992). "(A)BC excinuclease: the Escherichia coli nucleotide excision repair enzyme." Mol Microbiol 6(16): 2219-24. Lindahl, T. (1979). "DNA glycosylases, endonucleases for apurinic/apyrimidinic sites, and base excision-repair." Prog Nucleic Acid Res Mol Biol 22: 135-92. Lindahl, T., S. Ljungquist, W. Siegert, B. Nyberg and B. Sperens (1977). "DNA N-glycosidases: properties of uracil-DNA glycosidase from Es-cherichia coli." J Biol Chem 252(10): 3286-94. Liu, G. R., W. Q. Liu, R. N. Johnston, K. E. Sanderson, S. X. Li and S. L. Liu (2006). "Genome plasticity and ori-ter rebalancing in Salmonella typhi." Mol Biol Evol 23(2): 365-71. Liu, L. F. and J. C. Wang (1987). "Supercoiling of the DNA template during transcription." Proc Natl Acad Sci U S A 84(20): 7024-7. Loeb, L. A. and B. D. Preston (1986). "Mutagenesis by apurinic/apyrimidinic sites." Annu Rev Genet 20: 201-30. Loewen, P. C. (1984). "Isolation of catalase-deficient Escherichia coli mu-tants and genetic mapping of katE, a locus that affects catalase activity." J Bacteriol 157(2): 622-6. Loewen, P. C., B. L. Triggs, C. S. George and B. E. Hrabarchuk (1985). "Genetic mapping of katG, a locus that affects synthesis of the bifunctional catalase-peroxidase hydroperoxidase I in Escherichia coli." J Bacteriol 162(2): 661-7. Lopez de Saro, F. J., R. E. Georgescu, M. F. Goodman and M. O'Donnell (2003). "Competitive processivity-clamp usage by DNA polymerases during DNA replication and repair." Embo J 22(23): 6408-18. Lovett, S. T. (2006). "Replication arrest-stimulated recombination: Depend-ence on the RecA paralog, RadA/Sms and translesion polymerase, DinB." DNA Repair (Amst) 5(12): 1421-7. Lovett, S. T. and V. V. Feschenko (1996). "Stabilization of diverged tandem repeats by mismatch repair: evidence for deletion formation via a mis-aligned replication intermediate." Proc Natl Acad Sci U S A 93(14): 7120-4. Lovett, S. T., T. J. Gluckman, P. J. Simon, V. A. Sutera, Jr. and P. T. Drap-kin (1994). "Recombination between repeats in Escherichia coli by a recA-

51
independent, proximity-sensitive mechanism." Mol Gen Genet 245(3): 294-300. Lovett, S. T., R. L. Hurley, V. A. Sutera, Jr., R. H. Aubuchon and M. A. Lebedeva (2002). "Crossing over between regions of limited homology in Escherichia coli. RecA-dependent and RecA-independent pathways." Genet-ics 160(3): 851-9. Luria, S. E. and M. Delbruck (1943). "Mutations of Bacteria from Virus Sensitivity to Virus Resistance." Genetics 28(6): 491-511. Majumdar, A. and D. J. Patel (2002). "Identifying hydrogen bond alignments in multistranded DNA architectures by NMR." Acc Chem Res 35(1): 1-11. Mao, E. F., L. Lane, J. Lee and J. H. Miller (1997). "Proliferation of muta-tors in A cell population." J Bacteriol 179(2): 417-22. Mashimo, K., M. Kawata and K. Yamamoto (2003). "Roles of the RecJ and RecQ proteins in spontaneous formation of deletion mutations in the Es-cherichia coli K12 endogenous tonB gene." Mutagenesis 18(4): 355-63. Mastroeni, P. and M. Sheppard (2004). "Salmonella infections in the mouse model: host resistance factors and in vivo dynamics of bacterial spread and distribution in the tissues." Microbes Infect 6(4): 398-405. Maurelli, A. T. (2007). "Black holes, antivirulence genes, and gene inactiva-tion in the evolution of bacterial pathogens." FEMS Microbiol Lett 267(1): 1-8. McClelland, M., K. E. Sanderson, J. Spieth, S. W. Clifton, P. Latreille, L. Courtney, S. Porwollik, J. Ali, M. Dante, F. Du, S. Hou, D. Layman, S. Leo-nard, C. Nguyen, K. Scott, A. Holmes, N. Grewal, E. Mulvaney, E. Ryan, H. Sun, L. Florea, W. Miller, T. Stoneking, M. Nhan, R. Waterston and R. K. Wilson (2001). "Complete genome sequence of Salmonella enterica serovar Typhimurium LT2." Nature 413(6858): 852-6. McInerney, P., A. Johnson, F. Katz and M. O'Donnell (2007). "Characteri-zation of a triple DNA polymerase replisome." Mol Cell 27(4): 527-38. Michel, B., S. D. Ehrlich and M. Uzest (1997). "DNA double-strand breaks caused by replication arrest." Embo J 16(2): 430-8. Miller, J. B. and N. K. Amy (1983). "Molybdenum cofactor in chlorate-resistant and nitrate reductase-deficient insertion mutants of Escherichia coli." J Bacteriol 155(2): 793-801.

52
Miller, J. H., A. Suthar, J. Tai, A. Yeung, C. Truong and J. L. Stewart (1999). "Direct selection for mutators in Escherichia coli." J Bacteriol 181(5): 1576-84. Mira, A., H. Ochman and N. A. Moran (2001). "Deletional bias and the evolution of bacterial genomes." Trends Genet 17(10): 589-96. Modrich, P. (1991). "Mechanisms and biological effects of mismatch re-pair." Annu Rev Genet 25: 229-53. Moeller, R., E. Stackebrandt, G. Reitz, T. Berger, P. Rettberg, A. J. Doherty, G. Horneck and W. L. Nicholson (2007). "Role of DNA repair by nonho-mologous-end joining in Bacillus subtilis spore resistance to extreme dry-ness, mono- and polychromatic UV, and ionizing radiation." J Bacteriol 189(8): 3306-11. Moran, N. A. (1996). "Accelerated evolution and Muller's rachet in endo-symbiotic bacteria." Proc Natl Acad Sci U S A 93(7): 2873-8. Moran, N. A. and A. Mira (2001). "The process of genome shrinkage in the obligate symbiont Buchnera aphidicola." Genome Biol 2(12): RE-SEARCH0054. Moreau, P. L. (1985). "Role of Escherichia coli RecA protein in SOS induc-tion and post-replication repair." Biochimie 67(3-4): 353-6. Muller, H. J. (1964). "The Relation of Recombination to Mutational Ad-vance." Mutat Res 106: 2-9. Nakabachi, A., A. Yamashita, H. Toh, H. Ishikawa, H. E. Dunbar, N. A. Moran and M. Hattori (2006). "The 160-kilobase genome of the bacterial endosymbiont Carsonella." Science 314(5797): 267. Neidhardt (1996). Escherichia coli and Salmonella: Cellular and molecular biology. Washington D.C., ASM press. Nilsson, A. I., S. Koskiniemi, S. Eriksson, E. Kugelberg, J. C. Hinton and D. I. Andersson (2005). "Bacterial genome size reduction by experimental evo-lution." Proc Natl Acad Sci U S A 102(34): 12112-6. Nohmi, T. (2006). "Environmental stress and lesion-bypass DNA poly-merases." Annu Rev Microbiol 60: 231-53. Nohmi, T., A. Hakura, Y. Nakai, M. Watanabe, S. Y. Murayama and T. So-funi (1991). "Salmonella typhimurium has two homologous but different umuDC operons: cloning of a new umuDC-like operon (samAB) present in a

53
60-megadalton cryptic plasmid of S. typhimurium." J Bacteriol 173(3): 1051-63. Ochman, H. and I. B. Jones (2000). "Evolutionary dynamics of full genome content in Escherichia coli." Embo J 19(24): 6637-43. Ochman, H. and N. A. Moran (2001). "Genes lost and genes found: evolu-tion of bacterial pathogenesis and symbiosis." Science 292(5519): 1096-9. Ohmori, H., E. C. Friedberg, R. P. Fuchs, M. F. Goodman, F. Hanaoka, D. Hinkle, T. A. Kunkel, C. W. Lawrence, Z. Livneh, T. Nohmi, L. Prakash, S. Prakash, T. Todo, G. C. Walker, Z. Wang and R. Woodgate (2001). "The Y-family of DNA polymerases." Mol Cell 8(1): 7-8. Oosterom, J. (1991). "Epidemiological studies and proposed preventive measures in the fight against human salmonellosis." Int J Food Microbiol 12(1): 41-51. Orren, D. K. and A. Sancar (1989). "The (A)BC excinuclease of Escherichia coli has only the UvrB and UvrC subunits in the incision complex." Proc Natl Acad Sci U S A 86(14): 5237-41. Parry, C. M., T. T. Hien, G. Dougan, N. J. White and J. J. Farrar (2002). "Typhoid fever." N Engl J Med 347(22): 1770-82. Patten, C. L., M. G. Kirchhof, M. R. Schertzberg, R. A. Morton and H. E. Schellhorn (2004). "Microarray analysis of RpoS-mediated gene expression in Escherichia coli K-12." Mol Genet Genomics 272(5): 580-91. Ponder, R. G., N. C. Fonville and S. M. Rosenberg (2005). "A switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation." Mol Cell 19(6): 791-804. Quillardet, P., M. A. Rouffaud and P. Bouige (2003). "DNA array analysis of gene expression in response to UV irradiation in Escherichia coli." Res Microbiol 154(8): 559-72. Radman, M. (1975). "SOS repair hypothesis: phenomenology of an inducible DNA repair which is accompanied by mutagenesis." Basic Life Sci 5A: 355-67. Rocha, E. P. (2008). "The organization of the bacterial genome." Annu Rev Genet 42: 211-33. Roth, J. R., E. Kugelberg, A. B. Reams, E. Kofoid and D. I. Andersson (2006). "Origin of mutations under selection: the adaptive mutation contro-versy." Annu Rev Microbiol 60: 477-501.

54
Salyers and Whitt (1994). Bacterial pathogenesis. Washington D.C., ASM press. Saveson, C. J. and S. T. Lovett (1997). "Enhanced deletion formation by aberrant DNA replication in Escherichia coli." Genetics 146(2): 457-70. Schaaper, R. M. and R. L. Dunn (1991). "Spontaneous mutation in the Es-cherichia coli lacI gene." Genetics 129(2): 317-26. Schar, P., G. Herrmann, G. Daly and T. Lindahl (1997). "A newly identified DNA ligase of Saccharomyces cerevisiae involved in RAD52-independent repair of DNA double-strand breaks." Genes Dev 11(15): 1912-24. Schneiker, S., O. Perlova, O. Kaiser, K. Gerth, A. Alici, M. O. Altmeyer, D. Bartels, T. Bekel, S. Beyer, E. Bode, H. B. Bode, C. J. Bolten, J. V. Choud-huri, S. Doss, Y. A. Elnakady, B. Frank, L. Gaigalat, A. Goesmann, C. Groeger, F. Gross, L. Jelsbak, L. Jelsbak, J. Kalinowski, C. Kegler, T. Knauber, S. Konietzny, M. Kopp, L. Krause, D. Krug, B. Linke, T. Mah-mud, R. Martinez-Arias, A. C. McHardy, M. Merai, F. Meyer, S. Mormann, J. Munoz-Dorado, J. Perez, S. Pradella, S. Rachid, G. Raddatz, F. Rosenau, C. Ruckert, F. Sasse, M. Scharfe, S. C. Schuster, G. Suen, A. Treuner-Lange, G. J. Velicer, F. J. Vorholter, K. J. Weissman, R. D. Welch, S. C. Wenzel, D. E. Whitworth, S. Wilhelm, C. Wittmann, H. Blocker, A. Puhler and R. Mul-ler (2007). "Complete genome sequence of the myxobacterium Sorangium cellulosum." Nat Biotechnol 25(11): 1281-9. Shapiro, J. A. (1984). "Observations on the formation of clones containing araB-lacZ cistron fusions." Mol Gen Genet 194(1-2): 79-90. Shen, P. and H. V. Huang (1986). "Homologous recombination in Es-cherichia coli: dependence on substrate length and homology." Genetics 112(3): 441-57. Shimizu, H., H. Yamaguchi and H. Ikeda (1995). "Molecular analysis of lambda bio transducing phage produced by oxolinic acid-induced illegiti-mate recombination in vivo." Genetics 140(3): 889-96. Shiraishi, K., Y. Imai, S. Yoshizaki and H. Ikeda (2005). "Rep helicase sup-presses short-homology-dependent illegitimate recombination in Escherichia coli." Genes Cells 10(11): 1015-23. Shiraishi, K., Y. Imai, S. Yoshizaki, T. Tadaki, Y. Ogata and H. Ikeda (2006). "The role of UvrD in RecET-mediated illegitimate recombination in Escherichia coli." Genes Genet Syst 81(4): 291-7.

55
Steinum, A. L. and E. Seeberg (1986). "Nucleotide sequence of the tag gene from Escherichia coli." Nucleic Acids Res 14(9): 3763-72. Story, R. M., I. T. Weber and T. A. Steitz (1992). "The structure of the E. coli recA protein monomer and polymer." Nature 355(6358): 318-25. Streisinger, G., Y. Okada, J. Emrich, J. Newton, A. Tsugita, E. Terzaghi and M. Inouye (1966). "Frameshift mutations and the genetic code. This paper is dedicated to Professor Theodosius Dobzhansky on the occasion of his 66th birthday." Cold Spring Harb Symp Quant Biol 31: 77-84. Taddei, F., J. A. Halliday, I. Matic and M. Radman (1997). "Genetic analy-sis of mutagenesis in aging Escherichia coli colonies." Mol Gen Genet 256(3): 277-81. Tamas, I., L. Klasson, B. Canback, A. K. Naslund, A. S. Eriksson, J. J. Wernegreen, J. P. Sandstrom, N. A. Moran and S. G. Andersson (2002). "50 million years of genomic stasis in endosymbiotic bacteria." Science 296(5577): 2376-9. Teo, S. H. and S. P. Jackson (1997). "Identification of Saccharomyces cere-visiae DNA ligase IV: involvement in DNA double-strand break repair." Embo J 16(15): 4788-95. Tippin, B., P. Pham and M. F. Goodman (2004). "Error-prone replication for better or worse." Trends Microbiol 12(6): 288-95. Trinh, T. Q. and R. R. Sinden (1991). "Preferential DNA secondary struc-ture mutagenesis in the lagging strand of replication in E. coli." Nature 352(6335): 544-7. Trinh, T. Q. and R. R. Sinden (1993). "The influence of primary and secon-dary DNA structure in deletion and duplication between direct repeats in Escherichia coli." Genetics 134(2): 409-22. Trobner, W. and R. Piechocki (1984). "Selection against hypermutability in Escherichia coli during long term evolution." Mol Gen Genet 198(1): 177-8. Wagner, J., S. Fujii, P. Gruz, T. Nohmi and R. P. Fuchs (2000). "The beta clamp targets DNA polymerase IV to DNA and strongly increases its proces-sivity." EMBO Rep 1(6): 484-8. Wagner, J. and T. Nohmi (2000). "Escherichia coli DNA polymerase IV mutator activity: genetic requirements and mutational specificity." J Bacte-riol 182(16): 4587-95.

56
Wallace, D. C. and H. J. Morowitz (1973). "Genome size and evolution." Chromosoma 40(2): 121-6. Wang, J. C. (1998). "Moving one DNA double helix through another by a type II DNA topoisomerase: the story of a simple molecular machine." Q Rev Biophys 31(2): 107-44. Wang, J. C. (2002). "Cellular roles of DNA topoisomerases: a molecular perspective." Nat Rev Mol Cell Biol 3(6): 430-40. Weber, H., T. Polen, J. Heuveling, V. F. Wendisch and R. Hengge (2005). "Genome-wide analysis of the general stress response network in Es-cherichia coli: sigmaS-dependent genes, promoters, and sigma factor selec-tivity." J Bacteriol 187(5): 1591-603. Weller, G. R., B. Kysela, R. Roy, L. M. Tonkin, E. Scanlan, M. Della, S. K. Devine, J. P. Day, A. Wilkinson, F. d'Adda di Fagagna, K. M. Devine, R. P. Bowater, P. A. Jeggo, S. P. Jackson and A. J. Doherty (2002). "Identification of a DNA nonhomologous end-joining complex in bacteria." Science 297(5587): 1686-9. Weston-Hafer, K. and D. E. Berg (1989). "Palindromy and the location of deletion endpoints in Escherichia coli." Genetics 121(4): 651-8. Winfield, M. D. and E. A. Groisman (2003). "Role of nonhost environments in the lifestyles of Salmonella and Escherichia coli." Appl Environ Micro-biol 69(7): 3687-94. Wrande, M., J. R. Roth and D. Hughes (2008). "Accumulation of mutants in "aging" bacterial colonies is due to growth under selection, not stress-induced mutagenesis." Proc Natl Acad Sci U S A 105(33): 11863-8. Yamamoto, Y., M. Katsuki, M. Sekiguchi and N. Otsuji (1978). "Es-cherichia coli gene that controls sensitivity to alkylating agents." J Bacteriol 135(1): 144-52. Zhu, H. and S. Shuman (2005). "A primer-dependent polymerase function of pseudomonas aeruginosa ATP-dependent DNA ligase (LigD)." J Biol Chem 280(1): 418-27. Zhu, H. and S. Shuman (2007). "Characterization of Agrobacterium tumefa-ciens DNA ligases C and D." Nucleic Acids Res 35(11): 3631-45.


���� +�*������� +,����������������� �������������� ������������������� ������� ����������������������������
�)���' -�� ���� �� ��� .��&��� �� ��)���
� )������� )��������� ��� ��� .��&��� �� ��)���/ +,,����+�*�����/ � &�&���� � �&��� �� � �&(�� �� ,�,���0 � ��1��,�� �� ��� ��,���� )��������� ��� 2�,� �� �3�� �1�)���������� �(�����/ 1��� ��� �&��� ����� � )���(&��)������������� ����&4� ��� ����� �4��� 5�,������*��&���� �� +,,���� ����������� ��� ��� .��&��� ����)���0 67��� �� 8��&���/ "���/ ��� ����� 1�� ,&(����)&�)�� ��� ���� 95�,������*� �&���� �� +,,��������������� ��� ��� .��&��� �� ��)���:0;
����(&���' ,&(�������0&&0��&��'�(�'��'&&')*�!���%"$
������������������� ���������� �����