17 announcements chapter 16 – online quiz deadline - tonight
Post on 22-Dec-2015
215 views
TRANSCRIPT

17 Announcements
• Chapter 16 – Online Quiz Deadline - Tonight

17Molecular Biology and
Medicine

17 Molecular Biology and Medicine
• Abnormal or Missing Proteins: The Mutant Phenotype
• Mutations and Human Diseases
• Detecting Human Genetic Variations: Screening for Human Diseases
• Cancer: A Disease of Genetic Changes
• Treating Genetic Diseases
• Sequencing the Human Genome

17 Annual Checkup
Suppose you graduate from college, get a good job, have a few kids, and then go into the doctor’s office for an annual checkup.
After getting the results of your blood tests, the doctor tells you that your white blood cell count is 40 times higher than the norm.
Should you be concerned?http://www.kubby.com/kubby.family.hi-res.jpg
Hypothetical situation for family shown

17 Outcome
Subsequent tests reveal that you have a serious bone marrow cancer where white blood cells are proliferating out of control. You have chronic myeloid leukemia.
http://www.kubby.com/kubby.family.hi-res.jpgHypothetical situation for family shown.
http://images.encarta.msn.com/xrefmedia/sharemed/targets/images/pho/35a5c/35A5C297.jpg
Normal: 5,000 WBC/mL
Your #s: 200,000 WBC/mL

17 Proposed Solution
• Your oncologist prescribes aggressive chemotherapy (3 drugs). [Cancer is uncontrolled proliferation of cells.]
• The intent of the prescribed chemotherapy is to kill dividing cells. One downside of the drugs administered is that they will kill any cells that are dividing, including normal, non-cancerous, cells.
• How do the three drugs work? Drug 1: blocks microtubule formation; mitotic spindle
fibers are not assembled Drug 2: drug inserts into DNA helix and interferes
with DNA replication Drug 3: drug inhibits an enzyme involved in the
synthesis of nucleotides, which are needed for DNA

17 Results?
• Serious side effects from the drugs.
• White blood cell count drops from 200,000/mL to 80,000/mL, but still well above the normal level of 5,000/mL.
• Most of the WBCs are not mature, but rather undifferentiated cells from the bone marrow. Chemotherapy drugs have killed your bone marrow cells.
• You’re told that it is likely you will die within months.
• What do you do?http://www.kubby.com/kubby.family.hi-res.jpgHypothetical situation for family shown

17 Thank Scientists
You thank scientists for the long hours they spend in the laboratory to understand the causes of diseases!
http://mmserver.cjp.com/images/image/2661081.jpg
Chronic Myeloid Leukemia Blood Smear
http://www.bekkoame.ne.jp/~take-tomo/WBC/CML/ph1.jpg
Chromosomal translocation between chromosome 9 and 22

17 A Better Understanding
Fusion of parts of two genes: bcr (chr.22) and abl (chr. 9).
http://www.ispub.com/xml/journals/ijgp/vol1n1/taq-fig6.jpg
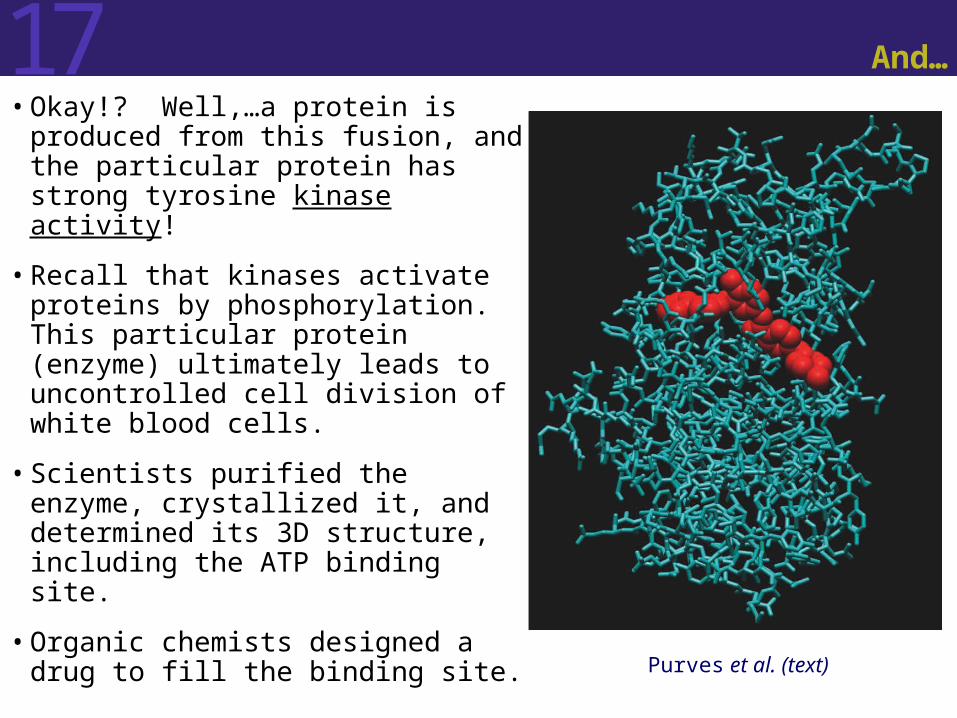
17 And…
• Okay!? Well,…a protein is produced from this fusion, and the particular protein has strong tyrosine kinase activity!
• Recall that kinases activate proteins by phosphorylation. This particular protein (enzyme) ultimately leads to uncontrolled cell division of white blood cells.
• Scientists purified the enzyme, crystallized it, and determined its 3D structure, including the ATP binding site.
• Organic chemists designed a drug to fill the binding site. Purves et al. (text)

17 Outcome
The drug called Gleevec dropped your WBC count to 5400 within weeks. Your cancer is virtually eliminated!
http://www.kubby.com/kubby.family.hi-res.jpgHypothetical situation
Now you can watch your kids grow up…
http://www.firstmed.co.uk/images/uploadedimages/Gleevec.jpg

17 Assortment of Diseases
Many diseases are caused by abnormal or missing proteins. For example: Phenylketonuria (PKU) Sickle-cell Anemia Familial Hypercholesterolemia (FH) Cystic Fibrosis Duchenne Muscular Dystrophy Transmissible Spongiform Encephalopathies
Enzymes, receptors, transport proteins, structural proteins, and nearly all other functional classes of proteins have been implicated in genetic diseases.

17 Phenylketonuria
• Dysfunctional enzymes can cause diseases: A defect in the gene that codes for the enzyme
phenylalanine hydroxylase causes phenylketonuria (PKU).
Clinical Signs
• light skin and hair
• mental retardation
• phenylpyruvic acid in urine
http://www.emedicine.com/derm/images/15571557DER0712-01.jpg

17 Phenylketonuria
Normally, the enzyme, phenylalanine hydroxylase, catalyzes the conversion of dietary phenylalanine to tyrosine in the liver. (Recall that both are amino acids, but with different R groups.)
At one position along the string of amino acids (primary structure) that make up this enzyme, affected individuals have tryptophan instead of arginine. The defective enzyme fails to convert phenylalanine to tyrosine.

Figure 17.1 One Gene, One Enzyme in Humans (Part 1)
nonfunctionalenzyme
normal product if phenylalanine hydroxylase is functional
Product if enzyme is nonfunctional, both phenylalanine and phenylpyruvic acid accumulate. Some eliminated in urine.
Why light skin and hair? Melanin, which is responsible, at least in part, for the pigmentation of our skin and hair, is made from tyrosine. Why mental retardation? Currently unknown.
All because of one amino acid substitution!

17 Sickle-Cell Anemia
• Sickle-cell anemia is caused by a missense mutation that affects the -globin subunit of hemoglobin.
http://www.kacr.or.kr/img/gene_expression/hemoglobin.jpg

17 Sickle-Cell Anemia
• Recall that a missense mutation is one in which a base substitution results in a substitution of one amino acid for another.
Of the 146 amino acids in β-globin subunits, the sixth is changed from a glutamic acid to a valine.
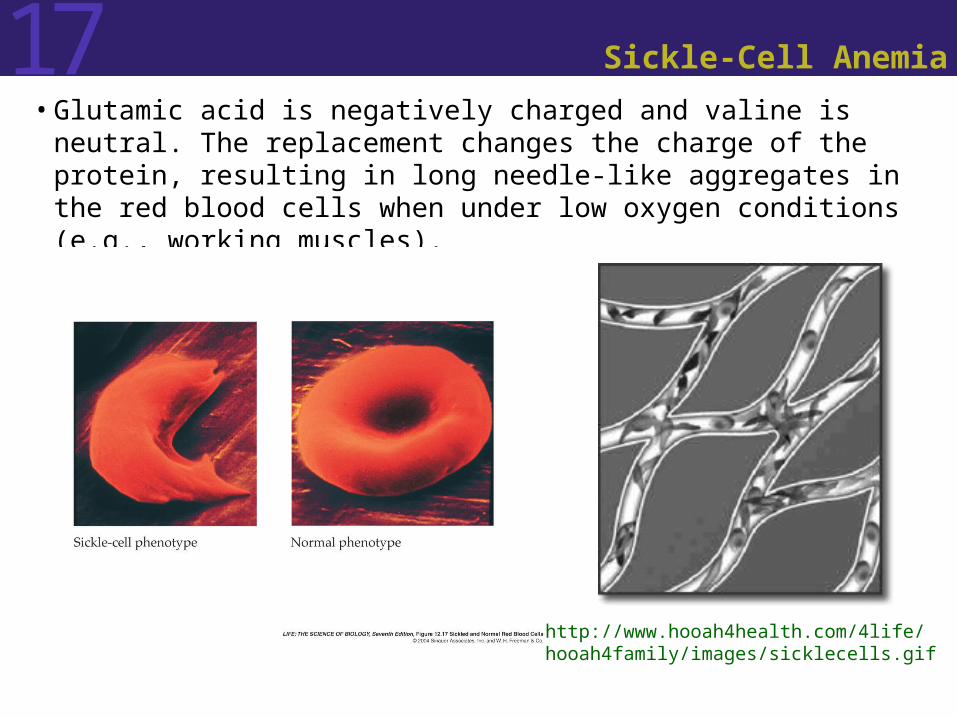
17 Sickle-Cell Anemia
• Glutamic acid is negatively charged and valine is neutral. The replacement changes the charge of the protein, resulting in long needle-like aggregates in the red blood cells when under low oxygen conditions (e.g., working muscles).
http://www.hooah4health.com/4life/hooah4family/images/sicklecells.gif

17 Sickle-Cell Anemia
• The Result: Anemia, a deficiency of
normal red blood cells and an impaired ability of the blood to carry oxygen.
Blocked capillaries, resulting in tissue damage and eventually death by organ failure (e.g., heart attack)
http://health.enotes.com/images/genetic-disorders/gecd_02_img0196.jpg

17 Familial Hypercholesterolemia (FH)
• Some common human genetic disorders show up as altered proteins in cell membranes.
• In familial hypercholesterolemia (FH), there is an altered cell surface receptor protein for the lipid carrier protein LDL (low-density lipoprotein). One person in 500 is born with FH.
http://www.virtualsciencefair.org/2004/thog4n0/public_html/LDL.jpg
http://www.calmainefoods.com/hdl-ldl.jpg

17 Familial Hypercholesterolemia (FH)
• In people with FH, the receptor protein is nonfunctional, so LDL, which carries cholesterol, cannot be transported into cells and therefore it accumulates in the blood to levels several times higher than normal.
• Excess cholesterol in the blood can accumulate on the inner walls of blood vessels
http://160.114.99.91/astrojan/Prot/Ldl.jpg
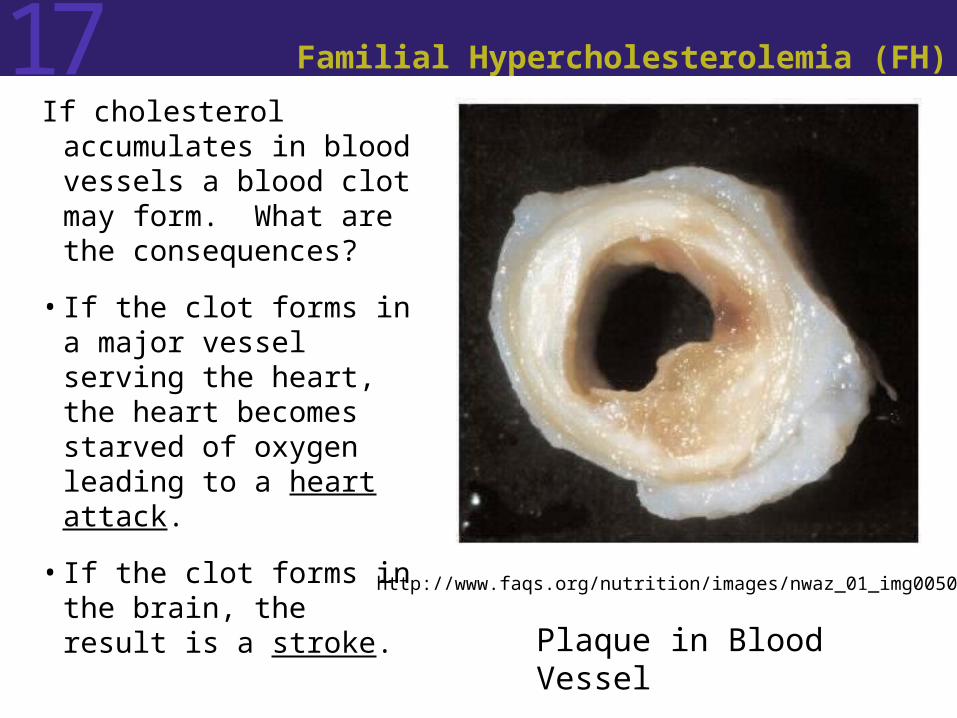
17 Familial Hypercholesterolemia (FH)
If cholesterol accumulates in blood vessels a blood clot may form. What are the consequences?
• If the clot forms in a major vessel serving the heart, the heart becomes starved of oxygen leading to a heart attack.
• If the clot forms in the brain, the result is a stroke.
http://www.faqs.org/nutrition/images/nwaz_01_img0050.jpg
Plaque in Blood Vessel

17 Familial Hypercholesterolemia (FH)
• Recall: In familial hypercholesterolemia (FH), there is an altered cell surface receptor protein for the lipid carrier protein LDL (low-density lipoprotein).
• 840 amino acids make up the receptor; often only one is abnormal in FH.

Figure 17.3 Genetic Diseases of Membrane Proteins (Part 1)

Figure 17.3 Genetic Diseases of Membrane Proteins (Part 2)

17 Cystic Fibrosis
• Cystic fibrosis is a genetic disease which results in unusually thick and dry mucus in the respiratory system. This interferes with the normal functioning of the cilia.
http://www.angelfire.com/comics/marfloors/4.png
http://www.emc.maricopa.edu/faculty/farabee/biobk/humrespsys_1.gifNote: cystic fibrosis affects other organs as well (e.g., liver, pancreas, digestive organs)

17 Cystic Fibrosis
• In the respiratory passageways, the thick mucus obstructs the passage of air and also prevents the cilia on the surfaces of the epithelial cells from working efficiently to clear out the bacteria and fungal spores that we take in with every breath.
• The results are recurrent and serous infections as well as liver, pancreatic, and digestive failures causing malnutrition and poor growth.
• People with cystic fibrosis often die in their twenties or thirties.
http://content.answers.com/main/content/wp/en/thumb/3/3e/200px-CFtreatmentvest2.JPG

17 Cystic Fibrosis
• The defect has been traced to a chloride transporter in a membrane protein.
• Normally, an imbalance of Cl– ions (more of them outside than inside) causes cellular water to leave the cell and form moist extracellular mucus.
• In people with cystic fibrosis, the lack of functional transporters changes the normal imbalance.
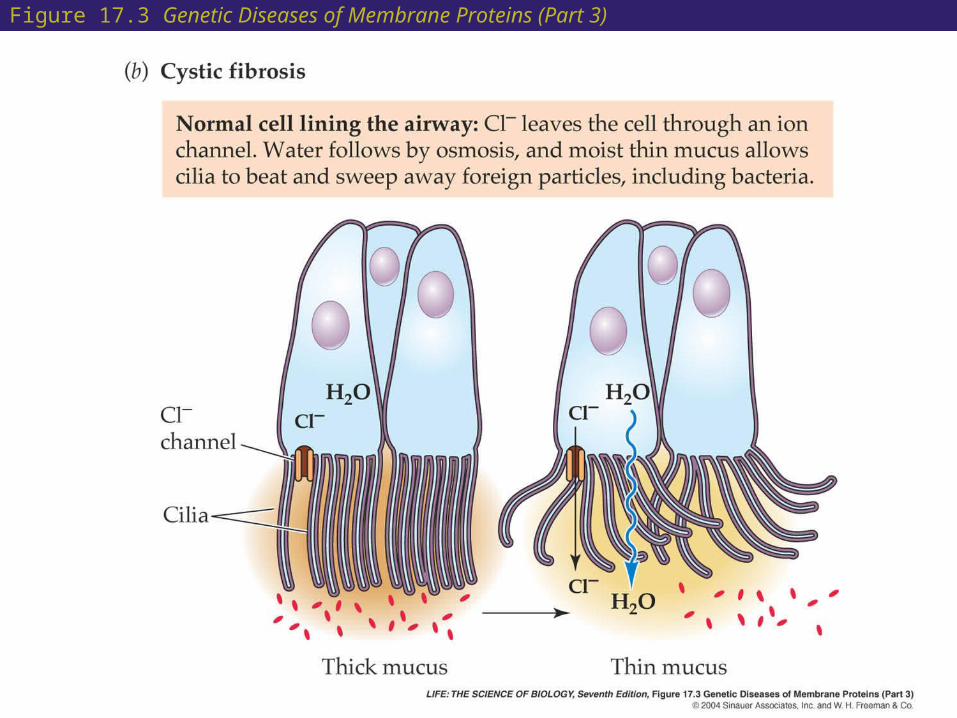
Figure 17.3 Genetic Diseases of Membrane Proteins (Part 3)
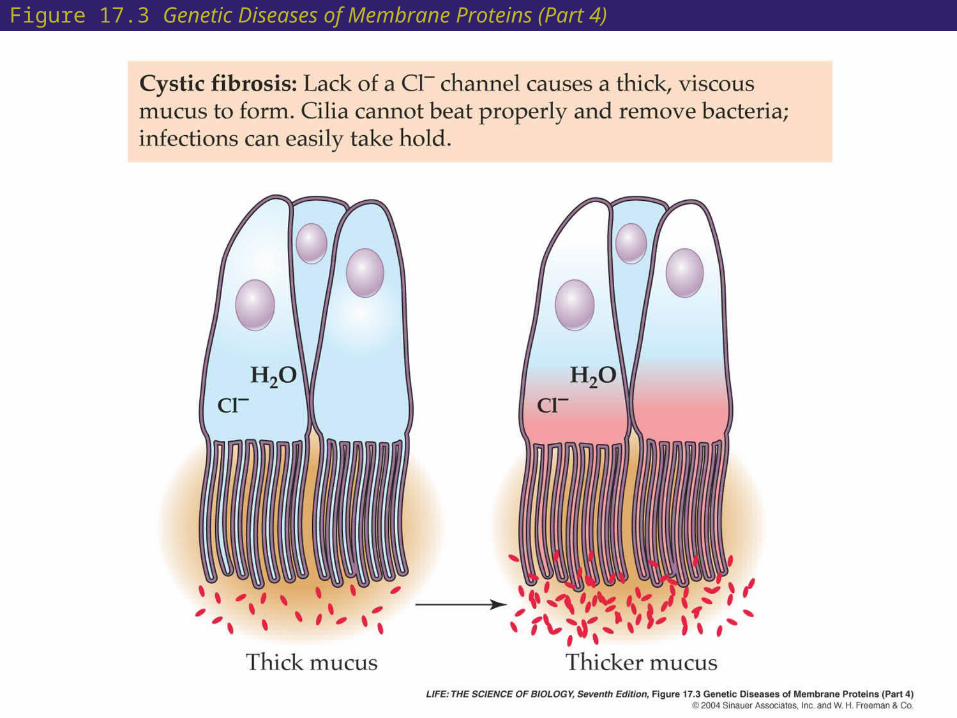
Figure 17.3 Genetic Diseases of Membrane Proteins (Part 4)

17

17 Abnormal or Missing Proteins:The Mutant Phenotype
• Altered structural proteins can cause disease.
• People born with Duchenne’s muscular dystrophy usually die in their twenties, when the muscles that serve their respiratory system fail.
• Dystrophin, which attaches actin to the plasma membrane in muscle cells, is missing or nonfunctional in people with this disease.
• Hemophilia is a genetic disease caused by a lack of one of the coagulation proteins. Affected people risk bleeding to death from even minor cuts.

17 Abnormal or Missing Proteins:The Mutant Phenotype
• Transmissible spongiform encephalopathies (TSEs) are degenerative brain diseases that occur in mammals, including humans.
• The infectious agent causing TSE is a protein.
• Stanley Prusiner purified the protein and called it a proteinaceous infective particle, or prion.

17 Abnormal or Missing Proteins:The Mutant Phenotype
• The mechanism of disease in TSEs: Normal brain cells have a membrane protein
called PrPc. Abnormal TSE-affected brain cells have the
same protein but with an altered shape, PrPsc. In PrPsc the amino acid sequence is the same,
but the shape of the protein has been altered. Insoluble PrPsc accumulates as fibers and
causes cell death.

Figure 17.4 Prion Proteins

17 Abnormal or Missing Proteins:The Mutant Phenotype
• Human diseases that can be traced to a single altered protein have a 1 percent frequency in the total population.
• Most diseases are multifactorial, caused by many genes and proteins interacting with one another and with the environment.
• Estimates suggest that up to 60 percent of all people have diseases that are genetically influenced.

17 Abnormal or Missing Proteins:The Mutant Phenotype
• Human genetic diseases have several patterns of inheritance:
Autosomal recessive pattern
Autosomal dominant pattern
X-linked recessive pattern
Chromosomal abnormalities

17 Abnormal or Missing Proteins:The Mutant Phenotype
• Autosomal recessive pattern: PKU, sickle-cell anemia, and cystic fibrosis are
autosomal recessive genetic diseases. People who are homozygous for the mutant
allele are affected. Those who are heterozygous may have less of
the normal gene product, but they have enough to have a normal phenotype.

17 Abnormal or Missing Proteins:The Mutant Phenotype
• Autosomal dominant pattern: With this pattern of inheritance, the presence of
just one mutant allele is enough to produce the clinical phenotype.
An example in humans is familial hyper-cholesterolemia. Having half the receptors for LDL is inadequate to prevent accumulation of cholesterol.

17 Abnormal or Missing Proteins:The Mutant Phenotype
• X-linked recessive pattern: Hemophilia is inherited as X-linked recessive
diseases. Sons inherit this condition from their mothers,
because the mutant allele is located on the X chromosome.
If a daughter with an unaffected father inherits a mutant allele from her mother, she will be a heterozygous carrier.
Until recently, few affected males lived to reproduce, so the most common pattern of inheritance has been from carrier mother to son.

17 Abnormal or Missing Proteins:The Mutant Phenotype
• Chromosomal abnormalities include loss or gain of one or more chromosomes, loss or gain of a piece of a chromosome, or transfer of a piece from one chromosome to another.
• Some abnormalities are inherited. Some are the result of nondisjunction during meiosis (or early mitosis).
• About 90 percent of human zygotes that have one X chromosome and no Y (Turner Syndrome) fail to survive beyond 4 months of gestation.
• A common cause of mental retardation is fragile-X syndrome.

Figure 17.5 A Fragile-X Chromosome at Metaphase

17 Mutations and Human Diseases
• The isolation and description of human mutant genes relies on mRNA, chromosome deletions, and DNA markers.
• Some gene mutations associated with human diseases are easy to clone. Hemoglobin abnormalities are an example.
• Finding the troublesome gene is much more difficult when the molecular causes are unknown.

17 Mutations and Human Diseases
• Sickle-cell anemia is caused by a single amino-acid defect in the -globin subunits of hemoglobin.
• It was possible to find the exact cause of sickle-cell anemia because the protein involved was known.
• Once -globin mRNA was isolated, cDNA copies were made and used to probe a human DNA library to find the -globin gene.

17 Mutations and Human Diseases
• In the case of Duchenne’s muscular dystrophy, chromosome deletions helped to identify the gene and the protein defect associated with the disease.
• Several boys with the disease were found to have a small deletion in their X chromosome.
• Comparison of the affected chromosomes with normal X chromosomes made possible the isolation of the gene.

Figure 17.6 Strategies for Isolating Human Genes

17 Mutations and Human Diseases
• An approach called positional cloning can be used when no candidate protein or deletion is known for a gene.
• Reference points for positional cloning are genetic markers on the DNA.
• Restriction enzymes are used to cut DNA molecules at specific recognition sequences.
• These become genetic markers called RFLPs (restriction fragment length polymorphisms).
• RFLPs have been found at more than 1000 sites for the human genome.
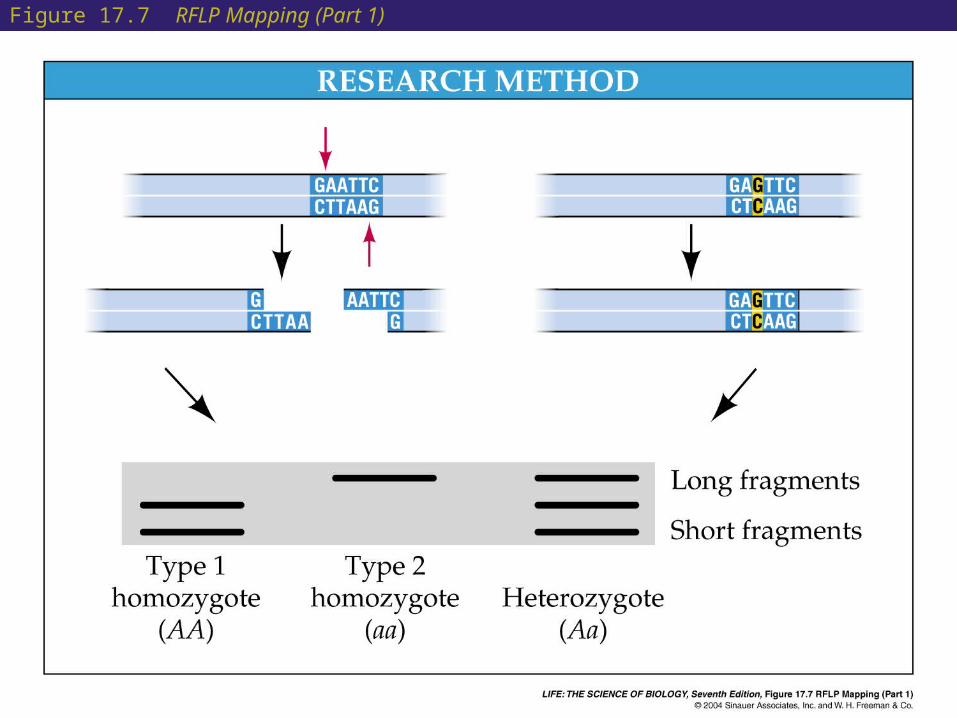
Figure 17.7 RFLP Mapping (Part 1)
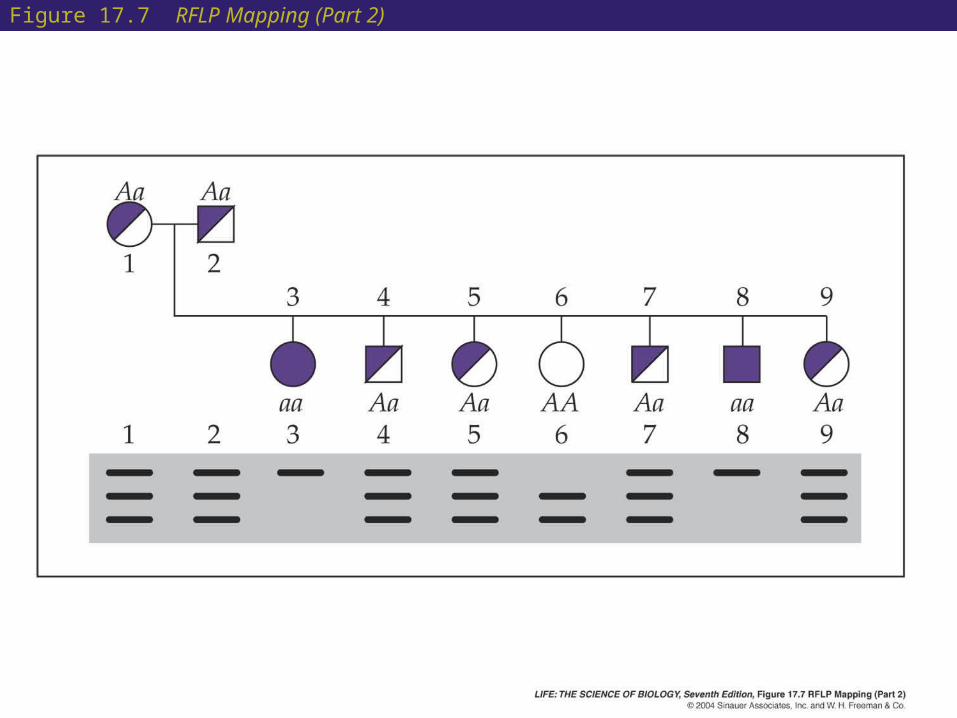
Figure 17.7 RFLP Mapping (Part 2)

17 Mutations and Human Diseases
• The RFLP band patterns are inherited in a Mendelian fashion and can be followed through a pedigree.
• Marker types and pedigrees are compared. If a marker corresponds to a phenotype, the gene that causes the phenotype must be near the marker.
• The neighborhood around the RFLP can be screened for other RFLPs. If one is linked directly, a DNA fragment from the region can be used to identify a cDNA sequence.
• The gene in affected and unaffected people is compared to determine the genetic difference responsible for the disease.

17 Mutations and Human Diseases
• Many diseases, such as sickle cell anemia, are caused by point mutations that alter a protein’s function, usually by affecting its three-dimensional structure.
• Some mutations lead to shortened protein chains, with loss of function.
• In some people with cystic fibrosis, a codon that would normally code for an amino acid near the beginning of a long protein has a nonsense mutation that changes it to a stop codon.
• Protein translation stops at the stop codon, and a very short, nonfunctional peptide results.

17 Mutations and Human Diseases
• Some base pairs are more prone to mutation than others:
When cytosine is methylated it can lose its amino group and becomes thymine. Regions of DNA with methylated cytosine are prone to mutation and are called hot spots.
• Larger mutations may involve many base pairs of DNA:
Duchenne’s muscular dystrophy varies in severity depending on how much of the dystrophin gene is deleted.
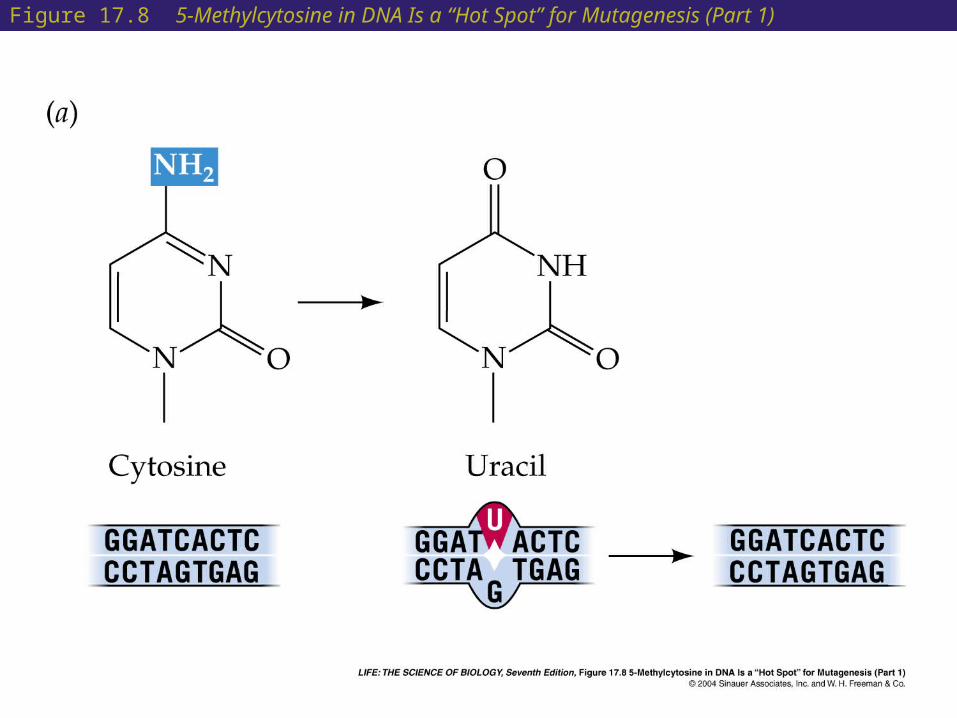
Figure 17.8 5-Methylcytosine in DNA Is a “Hot Spot” for Mutagenesis (Part 1)

Figure 17.8 5-Methylcytosine in DNA Is a “Hot Spot” for Mutagenesis (Part 2)

17 Mutations and Human Diseases
• Some diseases are cause by expanding triplet repeats.
In fragile-X syndrome, the gene FMR1 has a repeated triplet sequence: CGG.
In normal people, this triplet is repeated 6 to 54 times. In those affected with fragile-X, CGG is repeated 200 to 1,300 times.
More than 200 repeats leads to methylation of the CGG units and inactivation of FMR1.
Other diseases that involve expanding triplet repeats are myotonic dystrophy and Huntington’s disease.

Figure 17.9 The CGG Repeat in the Fragile-X Gene Expands with Each Generation

17 Mutations and Human Diseases
• In mammals, the DNA from fathers and mothers is expressed differently, a phenomenon known as genomic imprinting.
• Just after mammal egg fertilization, there are two haploid pronuclei—one from the sperm, and one from the egg.
• It is possible to make a mouse zygote with two male or two female pronuclei, but these fail to develop beyond the diploid cells.

17 Mutations and Human Diseases
• A small deletion in chromosome 15 produces completely different results depending on whether the deletion is in the chromosome from the mother or the father.
• If the mutated gene comes from the father, the child is short and obese, with small hands and feet (Prader-Willi syndrome).
• If the mutated gene comes from the mother, the child is thin with a wide mouth and prominent jaw (Angelman syndrome).

17 Detecting Human Genetic Variations:Screening for Human Diseases
• Genetic screening is used to identify people who have, are predisposed to, or are carriers of, genetic diseases.
• Screening can be done at several times in life:
Prenatal screening of an embryo or fetus
Newborn screening
Screening of asymptomatic people whose relatives have genetic diseases

17 Detecting Human Genetic Variations:Screening for Human Diseases
• Screening for PKU is legally required in many countries, including the United States.
• When babies homozygous for this disease consume protein, phenylalanine enters the blood and within days the accumulation causes brain damage.
• If PKU is detected early, a diet low in phenylalanine will prevent the damage.
• In the screening test, auxotrophic bacteria are used to detect the presence of phenylalanine in the blood. The bacteria require phenylalanine for growth.

17 Detecting Human Genetic Variations:Screening for Human Diseases
• DNA testing is the most accurate way to test for an abnormal gene.
• This works best if just a few different allelic forms of the disease gene exist.

17 Detecting Human Genetic Variations:Screening for Human Diseases
• Preimplantation screening: PCR allows testing of DNA from a single cell.
• If both parents are heterozygous for a recessive gene, such as the gene for cystic fibrosis, a single cell from an 8-cell zygote can be tested in the laboratory for presence of the disease.
• Postimplantation screening, such as chorionic villus sampling (tenth week of pregnancy) and amniocentesis (thirteenth to seventeenth week) are more common forms of prenatal genetic testing.

17 Detecting Human Genetic Variations:Screening for Human Diseases
• Screening for allele-specific cleavage differences: This method uses a restriction enzyme that can
recognize either the sequence at the mutation or the original sequence that is altered by that mutation.
In sickle alleles, for example, there is also a change at a restriction site, making the DNA sequence unrecognizable by the restriction enzyme.
When the enzyme fails to make the cut in the mutant gene, gel electrophoresis detects a larger-than-normal DNA fragment.

Figure 17.11 DNA Testing by Allele-Specific Cleavage

17 Detecting Human Genetic Variations:Screening for Human Diseases
• Screening for allele-specific oligonucleotide hybridization:
This method is easier and faster than allele-specific cleavage.
Oligonucleotides can be made in the laboratory that will hybridize to the denatured DNA sequences for either normal or sickle -globin.
Usually a probe of at least a dozen bases is needed to form a stable double helix with the target DNA.
If the probe is radioactively or fluorescently labeled, hybridization is readily detected.

Figure 17.12 DNA Testing by Allele-Specific Oligonucleotide Hybridization (Part 1)
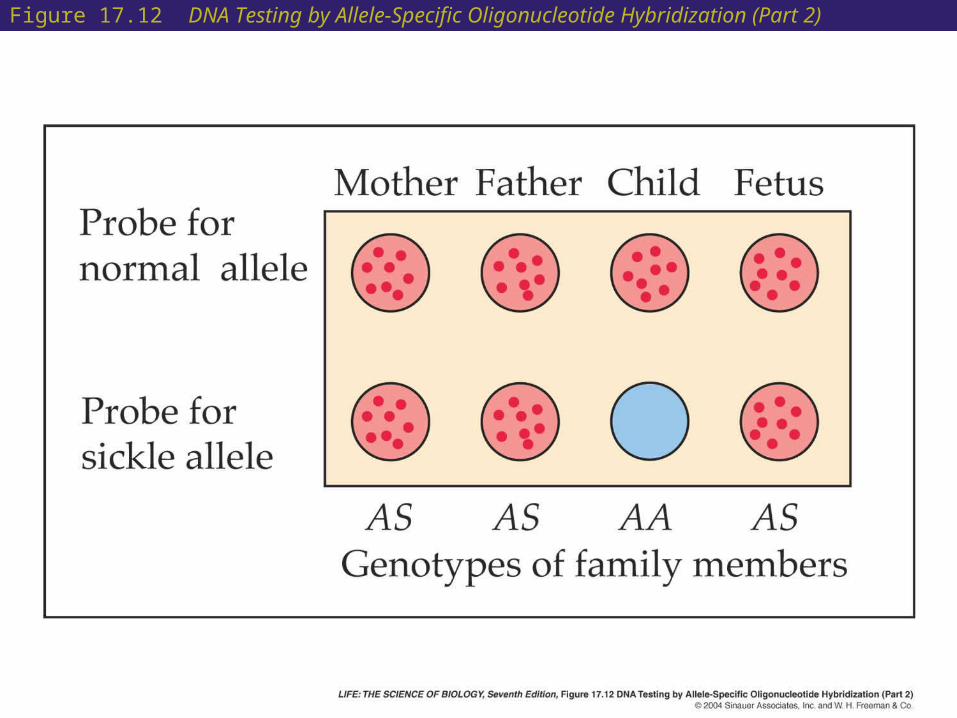
Figure 17.12 DNA Testing by Allele-Specific Oligonucleotide Hybridization (Part 2)

17 Cancer: A Disease of Genetic Changes
• Cancer is caused primarily by genetic changes and is more common in later life.
• Cancer is more frequent than in the past, in part due to longer life spans.

17 Cancer: A Disease of Genetic Changes
• Cancer cells have lost control over appropriate cell division.
• Cancer cells divide more or less continuously, not responding to growth factor or hormonal control.
• They form tumors, which often contain millions of cells by the time they are detected.
• A benign tumor resembles the tissue it comes from and remains localized.
• Malignant tumors do not look like parent tissues and often have irregular structures.

Figure 17.13 A Cancer Cell with Its Normal Neighbors

17 Cancer: A Disease of Genetic Changes
• Spreading of cancer to surrounding tissue and other body parts is called metastasis.
• The malignant tumor secretes chemical signals that cause blood vessels to grow into it. This is called angiogenesis.
• Next, the cells of the tumor secrete enzymes that digest and disintegrate surrounding tissues.
• Then they erode blood vessels. Some cells gain the ability to divide free from the tumor.
• These enter the bloodstream or lymphatic system. A few of these survive and form additional tumors.

17 Cancer: A Disease of Genetic Changes
• About 85 percent of all human tumors are carcinomas, which form from epithelial cells.
• Some skin cancers are of this type, as are lung, breast, colon, and liver cancers.
• Sarcomas are cancers of tissues such as bone, blood vessels, and muscle.
• Leukemias and lymphomas affect the cells that give rise to blood cells.

17 Cancer: A Disease of Genetic Changes
• Viruses cause at least five types of human cancer.
• Hepatitis B virus is associated with liver cancers, although it does not cause cancer by itself and its role is unclear.
• Papillomaviruses, spread via sexual transmission, cause genital and anal warts that can often develop into tumors. These viruses can cause cancer on their own, without mutations in the host tissue.

17 Cancer: A Disease of Genetic Changes
• Most cancer is caused by mutations, usually in the cells of older people.
• These mutations occur usually in the somatic cells.
• Spontaneous mutations arise because of changes in the nucleotides, damaging DNA.
• Carcinogens can cause mutations that lead to cancer.
• Tobacco smoke, meat preservatives, ultraviolet light from the sun, and ionizing radiation are common carcinogens.

17 Cancer: A Disease of Genetic Changes
• Many carcinogens are naturally occurring agents, including chemicals naturally present in food.
• Most carcinogens damage DNA by shifting one base to another.
• Cells that divide often, such as epithelial and bone marrow cells, are especially susceptible to genetic damage because they spend less time on DNA repair.
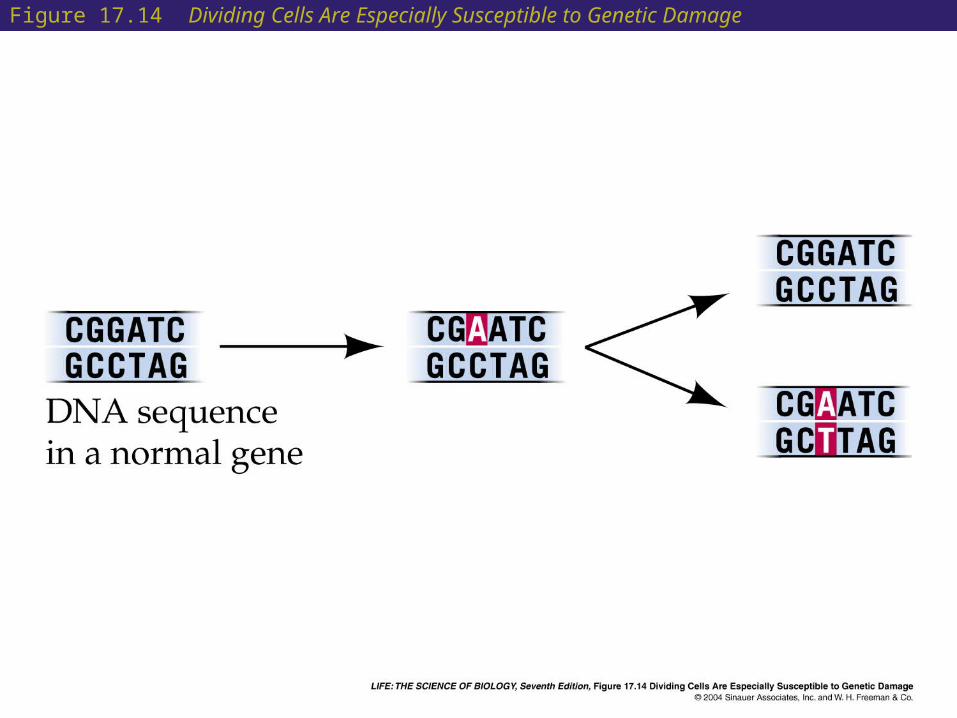
Figure 17.14 Dividing Cells Are Especially Susceptible to Genetic Damage

17 Cancer: A Disease of Genetic Changes
• Oncogenes stimulate cell division but are “turned off” in normal undividing cells.
• They have been identified as the genes carried by cancer-causing viruses.
• Some oncogenes control apoptosis. If mutated, apoptosis is prevented, and cells continue dividing.
• Some oncogenes can be activated by point mutations, others by chromosome changes, and others by gene amplification.
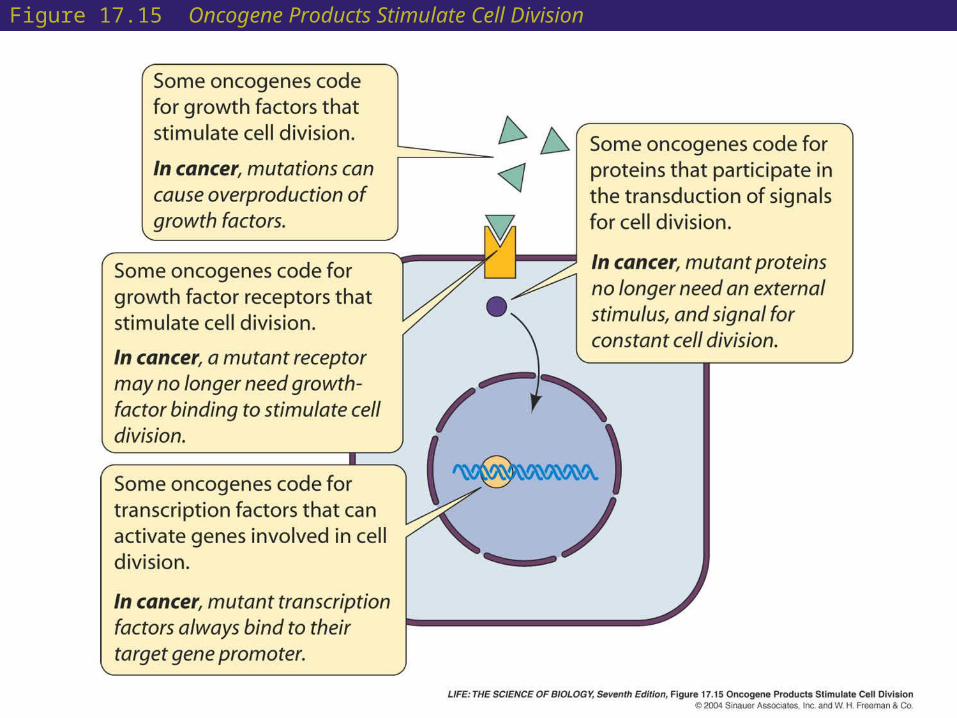
Figure 17.15 Oncogene Products Stimulate Cell Division

17 Cancer: A Disease of Genetic Changes
• About 10 percent of cancers are caused by defective tumor suppressor genes. These are inherited cancers, which usually appear in the form of multiple tumors and earlier in life than noninherited cancer.
• When functioning normally, tumor suppressor genes prevent cell division.
• People with inherited cancer are born with one mutant allele for the gene; just one mutational event is then needed to cause the disease.

Figure 17.16 The “Two-Hit” Hypothesis for Cancer

17 Cancer: A Disease of Genetic Changes
• An inherited form of breast cancer demonstrates the effect of tumor suppressor genes.
• The 9 per cent of women with one mutant BRCA1 allele have a 60 percent chance of having breast cancer by age 50 and an 82 percent chance by age 70.
• Women who inherit two normal alleles have a 2–7 percent chance.

17 Cancer: A Disease of Genetic Changes
• Tumor suppressor genes are normally involved in vital cell functions.
• Rb encodes a protein that inactivates transcription during the G1 phase of the cell cycle.
• When the Rb protein is inactivated by mutation, the cell cycle moves forward independently of growth factors and retinoblastoma can result.
• Another tumor suppressor gene is p53. The protein product of this gene stops cells during G1 of the cell cycle.
• Mutations in the p53 gene are associated with many cancers, including lung and colon cancer.

17 Cancer: A Disease of Genetic Changes
• A sequence of events must occur before a normal cell becomes malignant.
• At least three tumor suppressor genes and one oncogene must be mutated in sequence for an epithelial cell in the colon to become metastatic.
• Although the likelihood of this happening to any given cell is small, the colon has millions of cells that divide constantly in the presence of carcinogens.

Figure 17.18 (a) Multiple Mutations Transform a Normal Colon Epithelial Cell into a Cancer Cell

Figure 17.18 (b) Multiple Mutations Transform a Normal Colon Epithelial Cell into a Cancer Cell

17 Treating Genetic Diseases
• To treat genetic disease, physicians must diagnose the disease correctly, know the molecular mechanisms of the disease, and be able to intervene early, before the disease causes damage.
• Physicians are now applying the knowledge of pathogenesis at the molecular level to treat genetic diseases.

17 Treating Genetic Diseases
• One treatment approach is to modify the phenotype:
Restrict the substrate of a deficient enzyme
Use metabolic inhibitors
Supply the missing protein

17 Treating Genetic Diseases
• Restricting the substrate: The mutation that causes PKU results in an
enzyme that is unable to break down phenylalanine.
Treatment involves restricting intake of the substrate for this enzyme.

17 Treating Genetic Diseases
• Metabolic inhibitors: Cholesterol synthesis by the liver can be
lowered by metabolic inhibitors such as mevinolin.
This blocks the patient’s own cholesterol synthesis and helps those with familial hypercholesterolemia.
Metabolic inhibitors also form the basis for chemotherapy, or treatment with drugs that kill rapidly dividing cells.

Figure 17.19 Strategies for Killing Cancer Cells

17 Treating Genetic Diseases
• Supplying the missing protein: Hemophilia A can be treated by supplying the
protein that people with hemophilia fail to produce, a clotting factor protein.
This protein is now produced in a pure form using biotechnology.

17 Treating Genetic Diseases
• Gene therapy involves inserting a new gene into a patient’s cells.
• Different methods are being tried to get cells to take up and incorporate the new DNA.
• Cells have been removed from patients, genetically modified ex vivo, and then reintroduced into the same patient.

Figure 17.20 Gene Therapy: The Ex Vivo Approach (Part 1)

Figure 17.20 Gene Therapy: The Ex Vivo Approach (Part 2)

17 Treating Genetic Diseases
• In people with hemophilia, skin cells have been modified and reintroduced.
• Skin cells from their arms were removed and transfected with a plasmid containing a normal allele for the clotting protein.
• The cells were then reintroduced into the patients, where they produced adequate protein for normal clotting.

17 Treating Genetic Diseases
• Attempts also have been made to insert genes directly into cells, the in vivo approach.
• Tumor suppressor genes have been put in vectors and targeted at tumors.
• Vectors carrying functional alleles of the tumor suppressor genes that are mutated in lung cancer, as well as vectors expressing antisense RNAs against oncogene mRNAs, have been introduced in this way with some clinical success.

17 Sequencing the Human Genome
• In 1986 Renato Dulbecco, who won a Nobel prize for his work on cancer-causing viruses, suggested that determining the normal sequence of human DNA could be a boon to cancer research.
• The Human Genome Project is an internationally funded program to determine the sequences of the human genome.
• Private industry launched its own sequencing effort in the 1990s.

17 Sequencing the Human Genome
• Chromosomes can be sorted by a machine based on their different sizes.
• The DNA of a chromosome is too long to be sequenced directly.
• The DNA must be fragmented using restriction enzymes into sections of about 700 base pairs, and these fragments can be mapped.
• Then all the millions of fragments must be put back together like the pieces of a jigsaw puzzle—a formidable challenge.

17 Sequencing the Human Genome
• Hierarchical sequencing: Restriction enzymes recognizing 8–12 base
pair sequences are used to generate a small number of relatively large DNA fragments.
These large fragments are added to a vector called a bacterial artificial chromosome (BAC) and inserted into bacteria to create a gene library.
The fragments of this library are arranged in the proper sequences by using the marker sequences.

17 Sequencing the Human Genome
• Shotgun sequencing: Human DNA is randomly broken into
fragments that are 500–800 base pairs long. Each fragment is sequenced. A computer then finds and uses overlapping
sequences shared by fragments to align them. The entire 180-million-base-pair fruit fly
genome was sequenced by the shotgun method.
This method is much faster than the hierarchical approach.

Figure 17.21 Two Approaches to Sequencing DNA

17 Sequencing the Human Genome
• The human genome sequence was completed by 2003.
• Of the 3.2 billion base pairs, less than 2 percent are coding regions, containing a total of 30,000–35,000 genes.
• The average gene has 27,000 base pairs and has many introns.
• Over 50 percent of the genome is made up of highly repetitive sequences.
• Almost all (99.9%) of the genome is the same in all people. Over 2 million single-nucleotide polymorphisms (SNPs) have been mapped.
• Genes are not evenly distributed over the genome.

Figure 17.22 The Human Genome

17 Sequencing the Human Genome
• Gene sequencing has many applications.
• Many organisms have gene sequences in common with humans. These homologs have helped identify human genes.
• Mapping technologies make isolation of genes easier and allow disease-causing genes to be identified.
• Better drug treatments based on determining genetic variations in drug metabolism (pharmacogenomics) may be developed.

17 Sequencing the Human Genome
• Differential gene expression can be studied using DNA chips.
• The Cancer Genome Anatomy Project is seeking to make a “fingerprint” of a tumor at each stage of its development.
• “Genome prospecting” is the search for genes that might predispose a population to certain conditions.
• Knowledge of the human genome may lead to new approaches to medical care based on the individual’s genetic predisposition and potential.
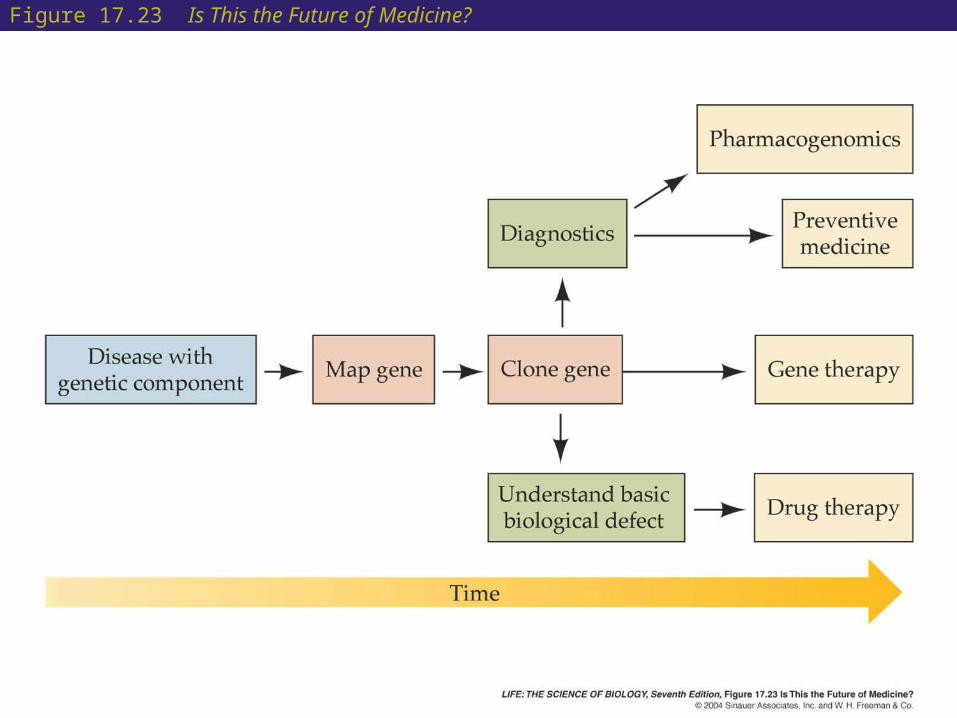
Figure 17.23 Is This the Future of Medicine?

17 Sequencing the Human Genome
• How should genetic information be used?
• Many people are uninterested in their genetic makeup unless they or a close relative are known to have a genetic disease.
• There is fear that insurance companies may try to use the information for health insurance exclusions.
• Many concerns have been raised about commercialization of people’s DNA sequences.
• The question of who will profit from the project has not yet been resolved.

17 Sequencing the Human Genome
• Humans have about one-third as many genes as predicted based on the number of proteins found in human cells.
• Many genes encode more than one protein by processes such as alternative splicing and posttranslational modifications.
• The sum total of the proteins produced by an organism is called its proteome.
• The one-gene, one-polypeptide relationship that was once a central theme of biology has been laid to rest by genomics.

17 Sequencing the Human Genome
• The proteome can be analyzed using two-dimensional gel electrophoresis or mass spectrometry.
• The field of proteomics seeks to describe the phenotypes of expressed proteins.

Figure 17.24 Proteomics (Part 1)

Figure 17.24 Proteomics (Part 2)

17 Sequencing the Human Genome
• Proteomics has been used in conjunction with DNA chip technology to compare brain proteins in chimpanzees and humans.
• 12,000 DNA sequences from the cortex of human and chimpanzee brains were tested for expression as mRNA.
• Only 1.4 percent showed differences between the two species.
• Proteomics, however, showed that the kinds of proteins produced by the two species differed by 7.4 percent, probably due to alternate splicing.
• Control of gene expression may be the key to human evolution.