1. punto isoelettrico (pi) 2. peso molecolare (mw)omero.farm.unipi.it/matdidfarm/16/10-elettroforesi...
TRANSCRIPT

PROTEOMA E PROTEOMICA
Le proteine hanno due valori intrinseci che ne determinano le caratteristiche specifiche:
1. Punto isoelettrico (pI)
2. Peso molecolare (Mw)

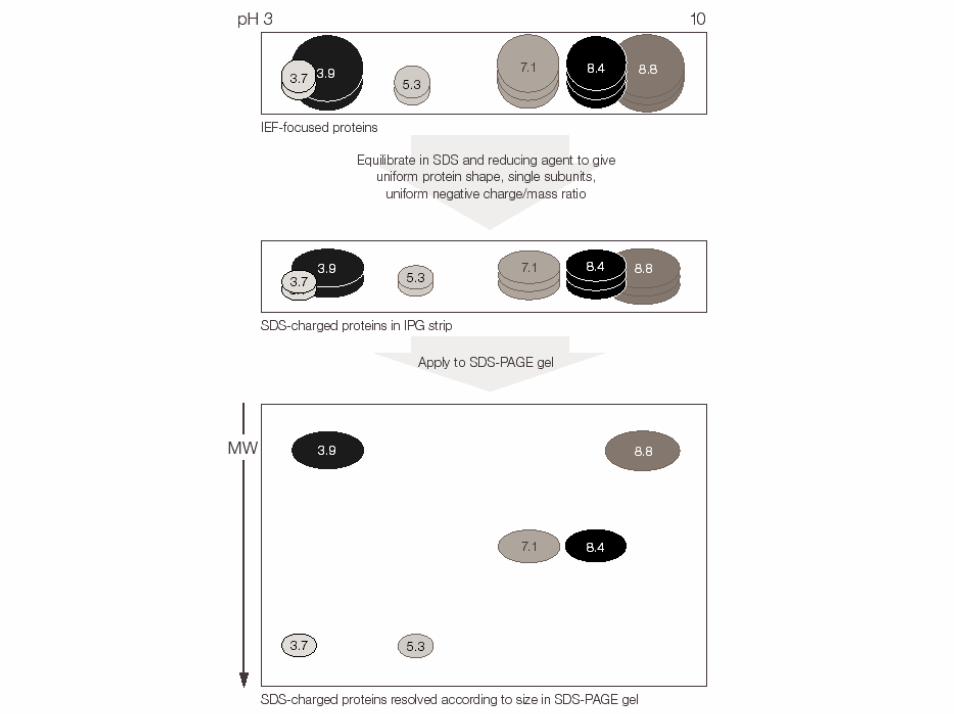
PRINCIPIO DELL’ELETTROFORESI BIDIMENSIONALE
IEF
SDS-PA
GE
Peso molecolare
pH 10.0pH 3.0Grande
Piccolo
-ve (catodo)+ve (anodo)
+ve (anodo)
Carica

PREPARAZIONE DEL CAMPIONE PROTEICO
Una efficace procedura di preparazione del campione deve includere
Solubilizzazione riproducibile di tutte le classi proteiche, incluse quelle idrofobichePrevenzione dell’aggregazione proteica e mantenimento della solubilità durante IEFPrevenzione di modificazioni chimiche ed enzimatiche dovute al processo di estrazioneLa rimozione di macromolecole che possono interferireUna resa proteica adeguata per la valutazione che potrebbe implicare la rimozione di specie proteiche molto abbondanti

PREPARAZIONE DEL CAMPIONE PROTEICO
Le proteine devono essere denaturate e solubili
Questo processo si ottiene tramite trattamenti specifici con:Urea (denaturante)Tioli (riducenti)Detergenti Inibitori di proteasi

UREA
L’urea è l’agente caotropico più utilizzato, ma in casi specifici con problemi di solubilizzazione si può utilizzare la tiourea. Entrambi questi agenti rompono i ponti idrogeno intra e inter –molecolari.
Normalmente si utilizza urea 8M, ma anche miscele di tiourea 2M e urea 5-8M.
DETERGENTI
I detergenti vengono aggiunti per rompere interazioni idrofobiche e incrementare la solubilità proteica al relativo punto isolelettrico. I detergenti devono essere non ionici o zwitterionici, quindi SDS non può essere utilizzato.
Vengono utilizzati CHAPS, CHAPSO o octilglucoside 1-2%

AGENTI RIDUCENTI
Per rompere i ponti disolfuro si utilizzano o ditiotreitolo(DTT) o tributil-fosfina (TBP).Una soluzione di DTT 50mM è sufficiente per la maggior parte delle proteine, ma per casi difficili è consigliato l’uso di TBP
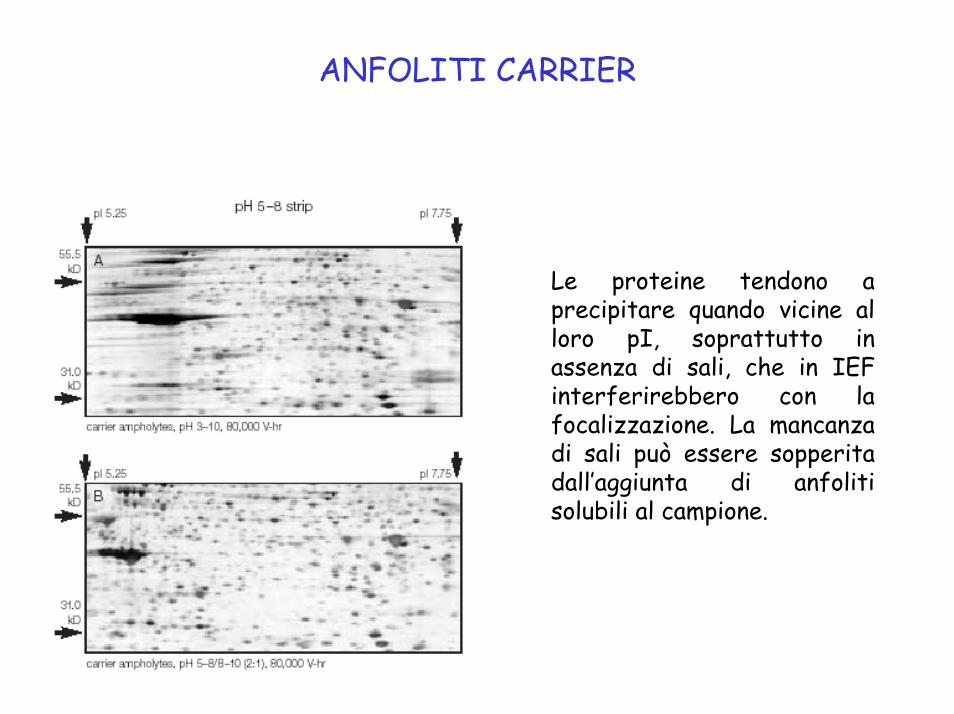
ANFOLITI CARRIER
Le proteine tendono a precipitare quando vicine al loro pI, soprattutto in assenza di sali, che in IEF interferirebbero con la focalizzazione. La mancanza di sali può essere sopperita dall’aggiunta di anfoliti solubili al campione.
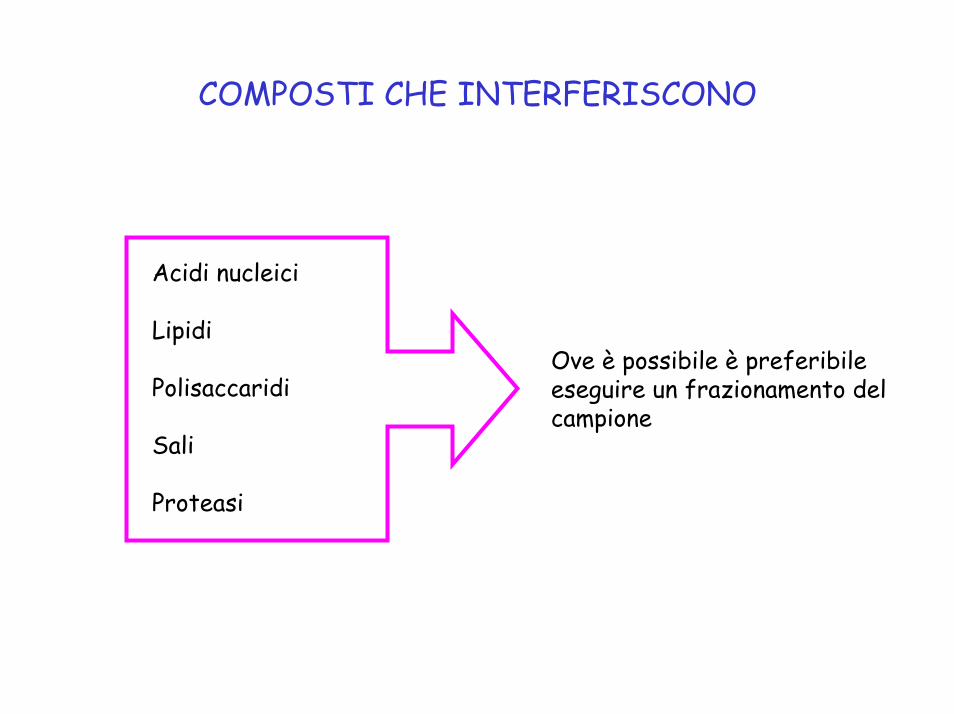
COMPOSTI CHE INTERFERISCONO
Acidi nucleici
Lipidi
Polisaccaridi
Sali
Proteasi
Ove è possibile è preferibile eseguire un frazionamento del campione

PREFRAZIONAMENTO
Estrazione differenziale
Frazionamento subcellulare
Cromatografia
IEF preparativa

ELETTROFORESI BI-DIMENSIONALE
PRIMA DIMENSIONE
Focalizzazione isoelettrica

Focalizzazione isoelettrica
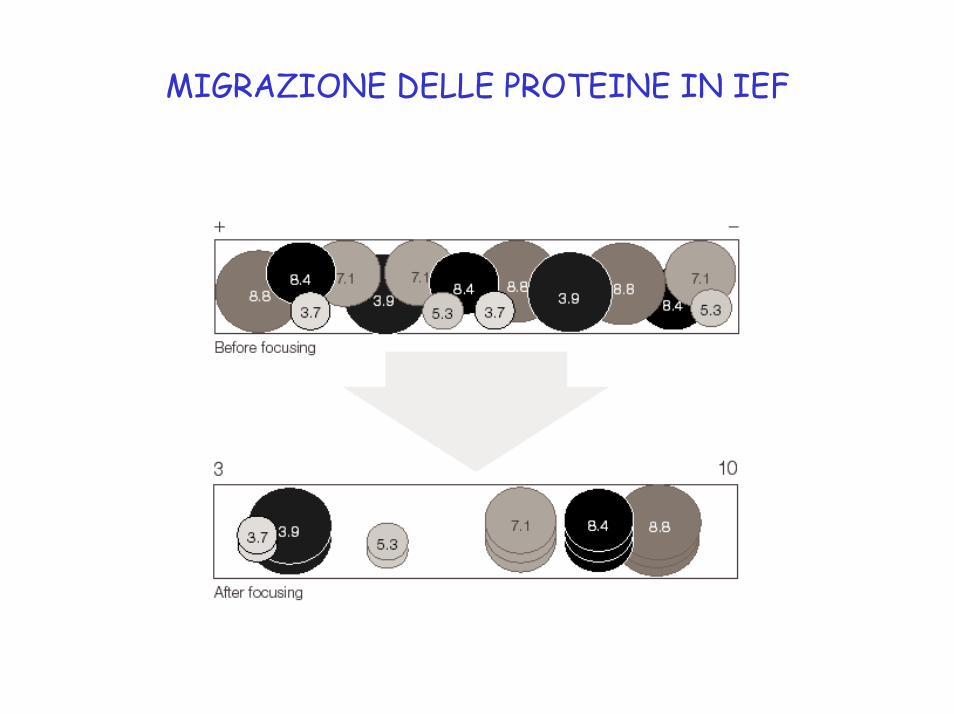
MIGRAZIONE DELLE PROTEINE IN IEF
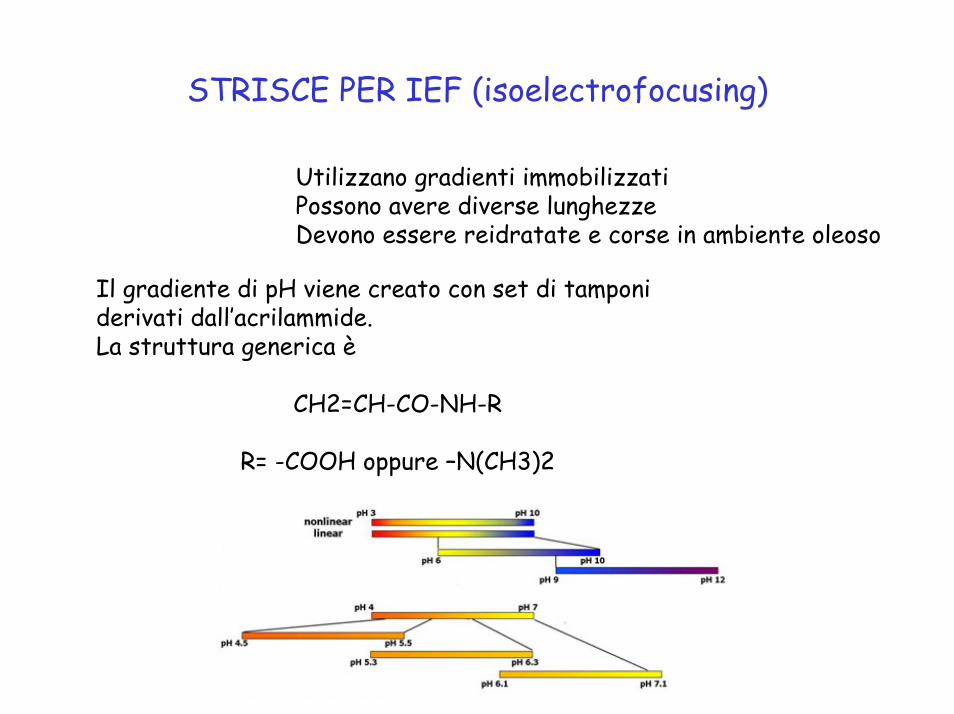
STRISCE PER IEF (isoelectrofocusing)
Utilizzano gradienti immobilizzatiPossono avere diverse lunghezzeDevono essere reidratate e corse in ambiente oleoso
Il gradiente di pH viene creato con set di tamponi derivati dall’acrilammide. La struttura generica è
CH2=CH-CO-NH-R
R= -COOH oppure –N(CH3)2



CORSA ELETTROFORETICA
Fino a 4 mg di proteine totali per strip
Il campione viene comunemente pre-adsorbito sulle strip durante la loro reidratazione, permettendo anche alle HMW proteins di entrare nel gel. Questo processo dura almeno 11 ore, dopodichè si può passare alla focalizzazione
Durante la corsa elettroforetica la conduttività del gel cambia dipendentemente dal numero di specie cariche, diminuendo mano a mano che le varie specie si focalizzano

ELETTROFORESI BI-DIMENSIONALE
SECONDA DIMENSIONE
SDS-PAGE
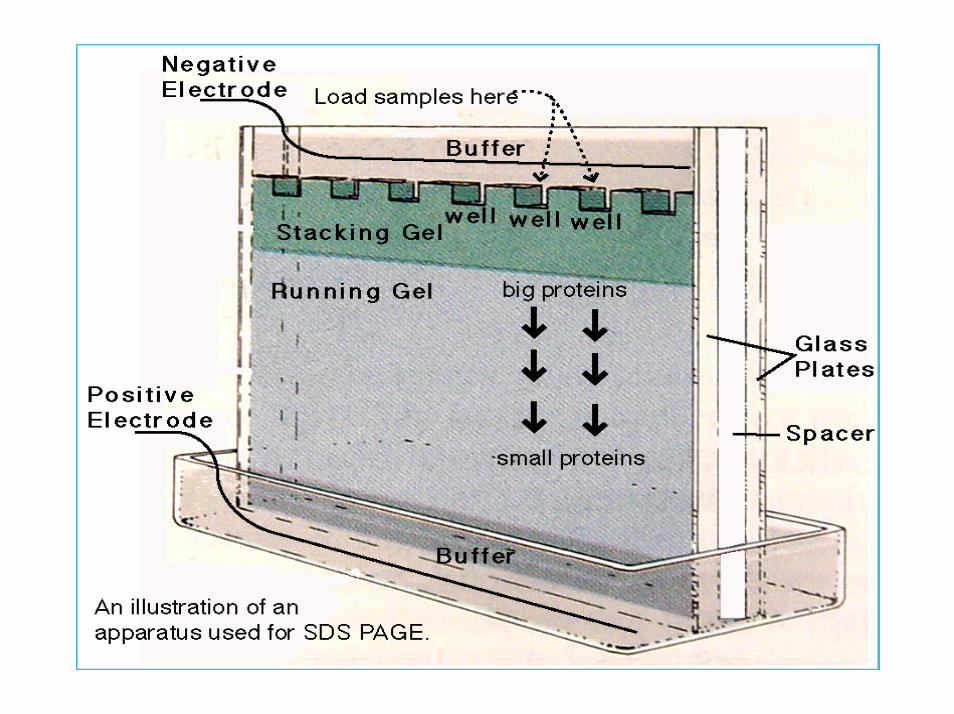

SECONDA DIMENSIONE
La transizione dalla prima alla seconda dimensione coinvolge l’equilibrazione della striscia focalizzata in tampone contenente SDS, la deposizione di essa sul gel di corsa e l’inglobamento in un gel di agarosio.
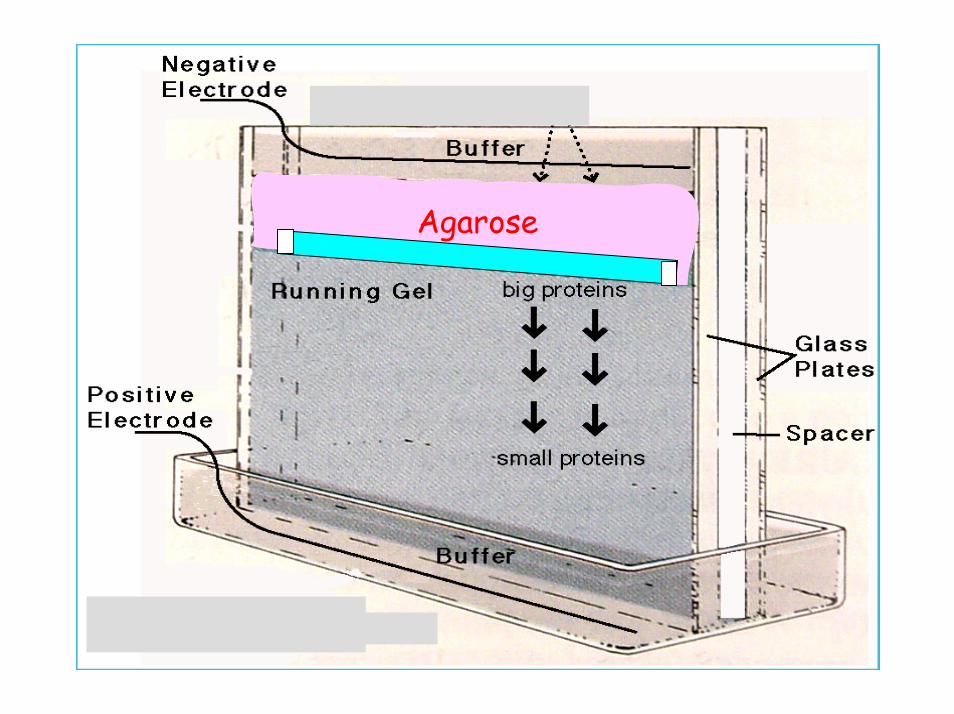
Agarose

SDS-PAGE
E’ esattamente uguale all’SDS-PAGE mono-dimensionale. Per avere un quadro generale si utilizza solitamente un gradiente (es. 8-16%). Una volta individuato il PM della regione di interesse, si passa a gel a percentuale uniforme che mostra una maggiore definizione.Nel caso si debbano paragonare due campioni è opportuno utilizzare due gel identici con corsa parallela.

COLORAZIONE DELLE PROTEINE
Una volta separato il campione in elettroforesi è possibile continuare con un Western blot, nel caso si cerchi qualcosa di specifico, come modificazioni proteiche post-traduzionali. Normalmente si procede alla colorazione del gel.

CHE COLORAZIONE EFFETTUARE?
La scelta della colorazione dipenderà soprattutto dalla quantità di proteine caricate.
Le colorazioni non devono interferire con i processi successivi e, in ordine di sensibilità, sono le seguenti:
Blu CoomassieArgentoColoranti fluorescenti

CHE COLORAZIONE EFFETTUARE?
I gel sono o preparativi o analitici. Se lo scopo del gel è quello di identificare proteine poco abbondanti, si eseguirà un grosso caricamento con una colorazione finale molto sensibile
Se la procedura viene utilizzata per ottenere un antigene, le proteine non devono essere fissate al gel, quindi la colorazione con argento verrà esclusa
In ogni caso ogni colorazione ha le proprie limitazioni

CHE COLORAZIONE EFFETTUARE?
La sensibilità raggiungibile con la colorazione è determinata
dalla quantità di colorante che si lega alla proteina
dalla differenza di colorazione tra il campione e il background del gel (signal-to-noise-ratio)
Ogni colorazione interagisce diversamente con le proteine e non esiste una colorazione che sia in grado di colorare tutte le proteine allo stesso modo. Sembra però che tutti i coloranti interagiscano meglio con aminoacidi basici

CHE COLORAZIONE EFFETTUARE?
Premettendo che sia meglio effettuare colorazioni diverse dello stesso campione, il colorante che sembra riuscire a dare il più ampio spettro di colorazione è il Blu Coomassie colloidale.
E’ possibile effettuare colorazioni successive dello stesso gel (fluorescente, Coomassie e infine argento)

CHE COLORAZIONE EFFETTUARE?
Sypro Ruby
Silver staining

2D ELETTROFORESI DELLE PROTEINE DI C. elegans
3 1pI 0
kDa
43
30
20
14

COLLEGAMENTO A BANCHE DATI

COMPARAZIONE CON 2D IN BANCA DATI
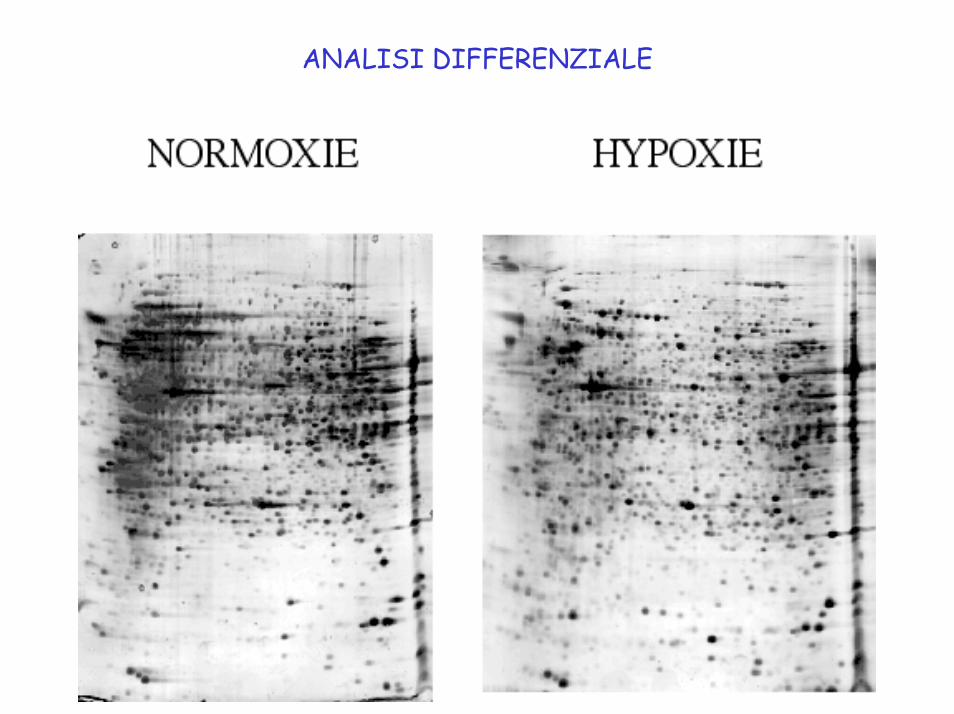
ANALISI DIFFERENZIALE

Coomassie Blue Dyes- commonly used- does not interfere with subsequent protein identification- inexpensive- sensitivity well below silver and fluorescent dyes
Silver stain- sensitivity 10-50 times greater than CB- ability to detect 1 ng of protein- silver diammine/silver nitrate- relatively expensive (reagents/waste disposal)- high background
Fluorescent Stains and Dyes- accurately determine changes in protein expression- greater sensitivity than silver stain- DIGE- cost
General detection methods
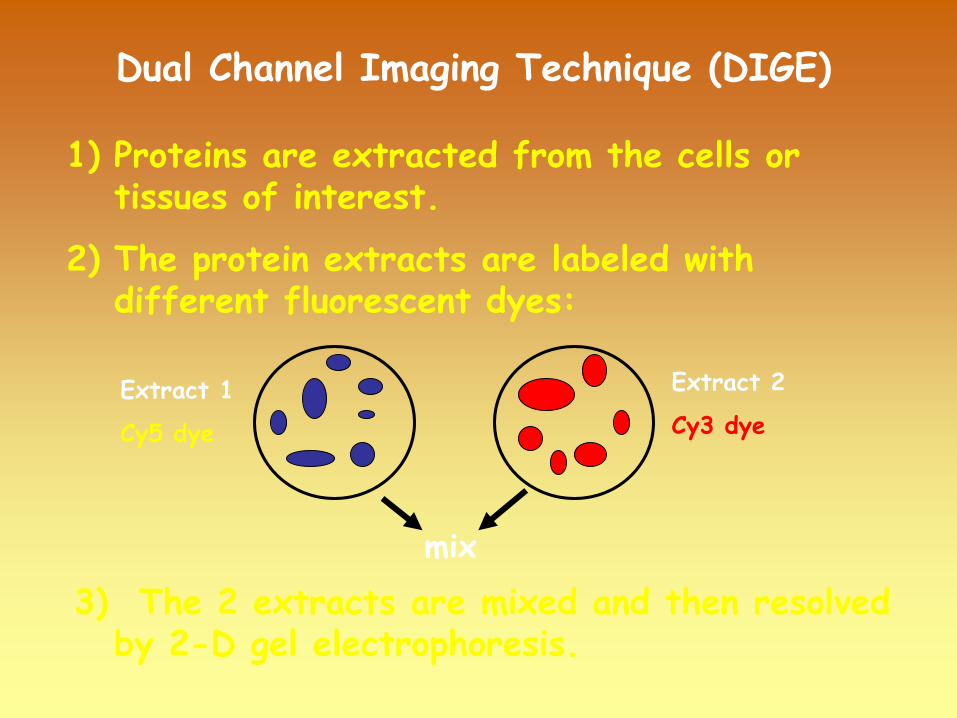
1)
Proteins are extracted from the cells or tissues of interest.
2)
The protein extracts are labeled with different fluorescent dyes:
3)
The 2 extracts are mixed and then resolved by 2-D gel electrophoresis.
Extract 1
Cy5 dye
Extract 2
Cy3 dye
mix
Dual
Channel
Imaging
Technique
(DIGE)
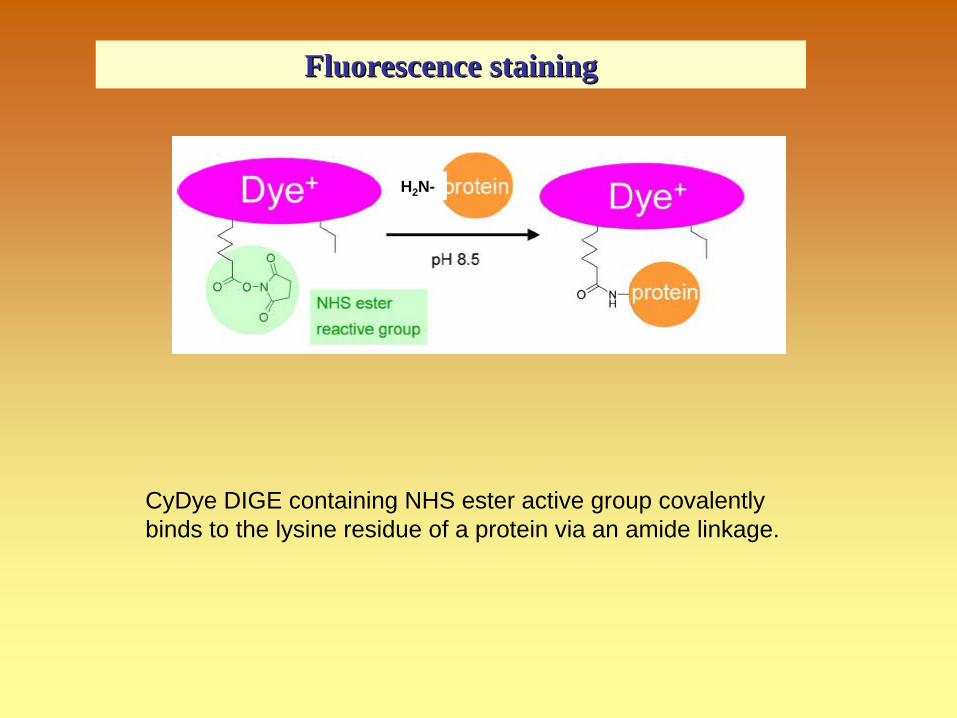
CyDye DIGE containing NHS ester active group covalently binds to the lysine residue of a protein via an amide linkage.
H2 N-
FluorescenceFluorescence stainingstaining

The DIGE technology platform
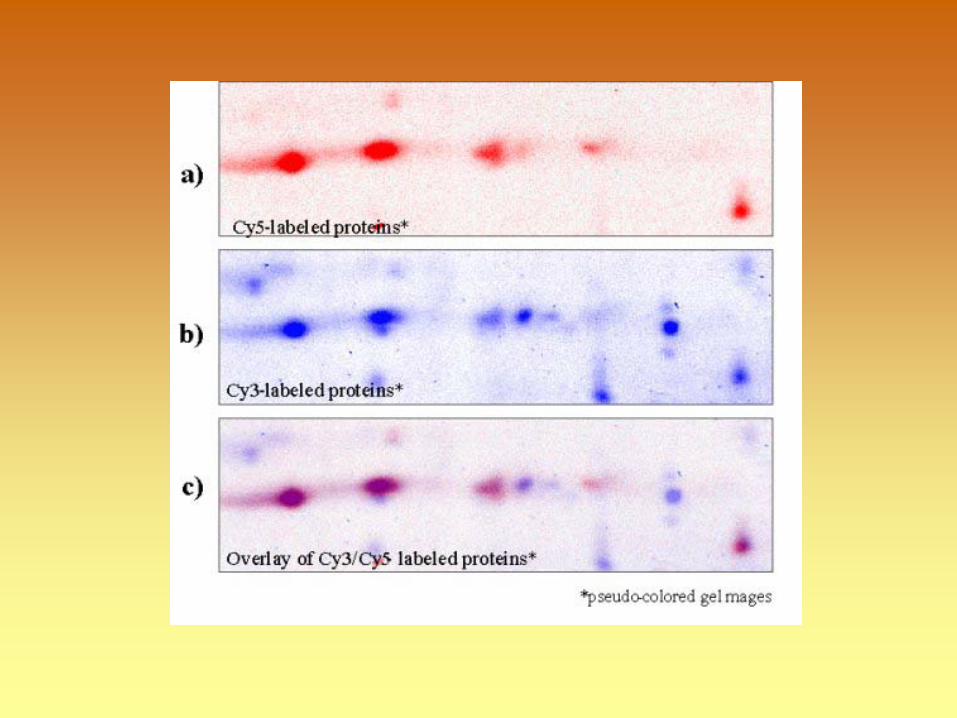

E-Cy5 E-Cy3
Excise spots; elute; digest, extract peptides; MS analyze, Protein identification

PSHA_1_0658
IEF (pH 4-7)
PSHA_1_2360
PSHA_1_0871PSHA_1_0021
PSHA_1_0849
PSHA_2_0363PSHA_2_0259PSHA_2_0420
PSHA_1_2262
PSHA_1_0328
PSHA_1_1875
PSHA_1_0255
PSHA_1_2758
PSHA_1_1059
PSHA_2_0557
PSHA_1_1740
PSHA_2_0150 PSHA_1_2771
PSHA_1_0341PSHA_1_2145
PSHA_1_3038 PSHA_2_0144
PSHA_1_0416
PSHA_1_0689
PSHA_2_0370
PSHA_1_0119PSHA_1_3009
PSHA_1_0870
PSHA_2_0474
PSHA_1_0595
PSHA_1_3001PSHA_1_2400
SDS
PAG
E-
PSHA_1_0658
PSHA_1_2360
PSHA_1_0871PSHA_1_0021
PSHA_1_0849
PSHA_2_0363PSHA_2_0259PSHA_2_0420
PSHA_1_2262
PSHA_1_0328
PSHA_1_1875
PSHA_1_0255
PSHA_1_2758
PSHA_1_1059
PSHA_2_0557
PSHA_1_1740
PSHA_2_0150 PSHA_1_2771
PSHA_1_0341PSHA_1_2145
PSHA_1_3038 PSHA_2_0144
PSHA_1_0416
PSHA_1_0689
PSHA_2_0370
PSHA_1_0119PSHA_1_3009
PSHA_1_0870
PSHA_2_0474
PSHA_1_0595
PSHA_1_3001PSHA_1_2400
PSHA_1_0658
PSHA_1_2360
PSHA_1_0871PSHA_1_0021
PSHA_1_0849
PSHA_2_0363PSHA_2_0259PSHA_2_0420
PSHA_1_2262
PSHA_1_0328
PSHA_1_1875
PSHA_1_0255
PSHA_1_2758
PSHA_1_1059
PSHA_2_0557
PSHA_1_1740
PSHA_2_0150 PSHA_1_2771
PSHA_1_0341PSHA_1_2145
PSHA_1_3038 PSHA_2_0144
PSHA_1_0416
PSHA_1_0689
PSHA_2_0370
PSHA_1_0119PSHA_1_3009
PSHA_1_0870
PSHA_2_0474
PSHA_1_0595
PSHA_1_3001PSHA_1_2400
PSHA_1_0658
PSHA_1_2360
PSHA_1_0871PSHA_1_0021
PSHA_1_0849
PSHA_2_0363PSHA_2_0259PSHA_2_0420
PSHA_1_2262
PSHA_1_0328
PSHA_1_1875
PSHA_1_0255
PSHA_1_2758
PSHA_1_1059
PSHA_2_0557
PSHA_1_1740
PSHA_2_0150 PSHA_1_2771
PSHA_1_0341PSHA_1_2145
PSHA_1_3038 PSHA_2_0144
PSHA_1_0416
PSHA_1_0689
PSHA _2_0370
PSHA_1_0119PSHA_1_3009
PSHA_1_0870
PSHA_2_0474
PSHA_1_0595
PSHA_1_3001PSHA_1_2400
Dual
Channel
Imaging
Technique
Green label
Proteome
A; Red label
= Proteome
B
Identification of differently expressed proteins by Dual
Channel
Imaging
Technique
(DIGE)

PROTEOMA E PROTEOMICA
"The analysis of the entire PROTEin content expressed by a genOME, or by a cell or tissue type.“ Wasinger VC et al, Electrophoresis 16 (1995)
“PROTEOMICS is the systematic analysis and documentation of proteins in biological samples with focus on state-related expression of proteins.”

PROTEOMA E PROTEOMICA
L’approccio genomico non considera le possibili
modificazioni post-traduzionali
che tutte le proteine degli organismi superiori posseggono

APPROCCIO PROTEOMICO
Ricerca proteine in un lisato cellulare o in estratto tissutale la cui espressione differenziale è determinata da fattori come
1. Processi patologici2. Delezioni o over-espressioni geniche3. Trattamenti farmacologici4. Stimolazioni chimiche e fisiche5. Rimozione di nutrienti o di ossigeno

SCOPO DELL’APPROCCIO PROTEOMICO
Valutazione di marcatori diagnostici in fluidi corporei
Studi di farmacologia quali identificazione e/o validazione
di nuovi target proteici per trattamenti farmacologici e
“drug profiling”
Studi di tossicità
Glicosilazioni, fosforilazioni o processamenti proteici
Analisi tissutali in situazioni patologiche

TECNOLOGIA LEGATA ALLA PROTEOMICA
Elettroforesi bidimensionale
Rilevazione degli “spot” proteici
Analisi di immagine
Excisione degli “spot” proteici
Digestione enzimatica
Spettrometria di massa
Bioinformatica

ANALISI DEL GEL
L’acquisizione dell’immagine dipende dal sistema di colorazione
Per colorazioni in luce diretta si possono utilizzare
DensitometroTelecamera
Per colorazioni in fluorescenza o radioattive
Scanner a fluorescenzaPhosphoimagerTelecamera su transilluminatore

COME IDENTIFICARE NUOVI SPOT?
Classica degradazione di Edman
Rimozione chimica di un residuo aminoacidico alla volta e analisi dello stesso. Metodo lento che necessita di grosse quantità proteiche
Spettrometria di massa
“Fingerprint” proteico e sequenza ex novo.Metodo veloce che necessita di piccole quantità proteiche

SPETTROMETRIA DI MASSA
E’ una metodica che
Misura accuratamente la massa proteica
Attraverso sequenze peptidiche identifica proteine note e sequenzia
proteine ignote
Identifica modificazioni post-traduzionali

EXCISIONE DEGLI SPOT PROTEICI
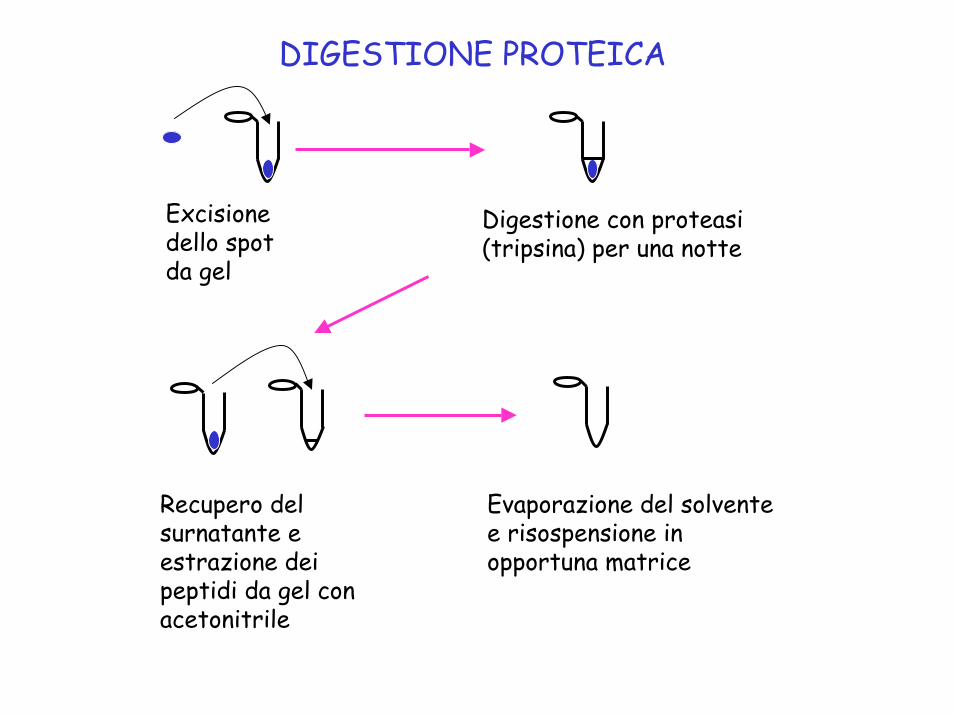
DIGESTIONE PROTEICA
Excisione dello spot da gel
Digestione con proteasi(tripsina) per una notte
Recupero del surnatante e estrazione deipeptidi da gel con acetonitrile
Evaporazione del solvente e risospensione in opportuna matrice

PEPTIDE MASS FINGERPRINT
La tripsina digerisce la sequenza proteica dopo un residuo di lisina (K) e di arginina (R). Il peptide
YAEKGLQQPVRAYMESEKSEEIDLPK
verrà quindi digerito in corrispondenza delle frecce
I 4 peptidi derivati hanno una massa ben definita:
YAEK 609.31GLQQPVR 797.45AYMESEK 857.38SEEIDLPK 957.54
Queste 4 masse definiscono esattamente il peptide della sequenza precedente

COME RILEVARE I PEPTIDI?
Dopo la digestione il campione deve essere ionizzato tramite:
Protonazione: M + H+ = MH+
Cationizzazione: M + Cat+ = MCat+
Deprotonazione: MH = M- + H+
Rilascio di elettroni: M = M+ + e-
Cattura di elettroni: M + e- = M-

COME RILEVARE I PEPTIDI?
Dopo la ionizzazione il campione viene fatto “volare” in
condizioni opportune.
Applicando lo stesso impulso a tutti i peptidi ionizzati, questi
percorreranno uno stesso tratto con un tempo relativo alla
loro massa. Questo tempo viene definito “tempo di volo”
(Tof)

SPETTROMETRIA DI MASSA
generazione di ioni in fase gassosa:
EI - Electron impact Ionisation (1919 A.J. Dempster)
CI - Chemical Ionisation
FAB - Fast Atom Bombardment (1981 M. Barber)
MALDI - Matrix Assisted Laser Desorption Ionisation (1988, K. Tanaka et al.)
ESI - ElectroSpray Ionisation (1985 J. Fenn)
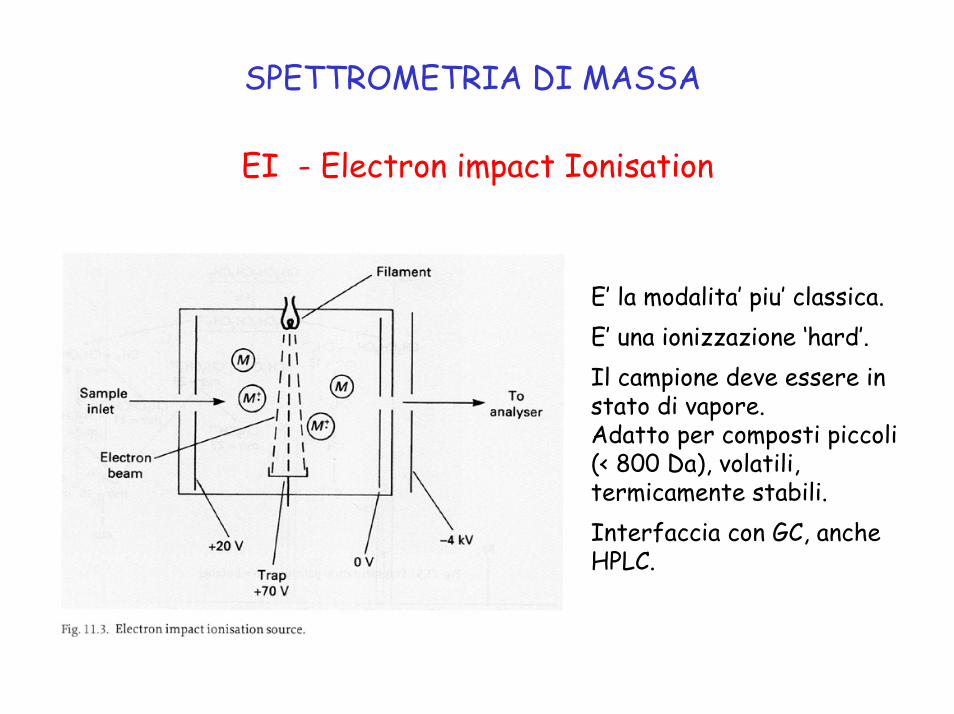
SPETTROMETRIA DI MASSA
EI - Electron impact Ionisation
E’ la modalita’ piu’ classica.E’ una ionizzazione ‘hard’.Il campione deve essere in stato di vapore.Adatto per composti piccoli (< 800 Da), volatili, termicamente stabili.Interfaccia con GC, anche HPLC.
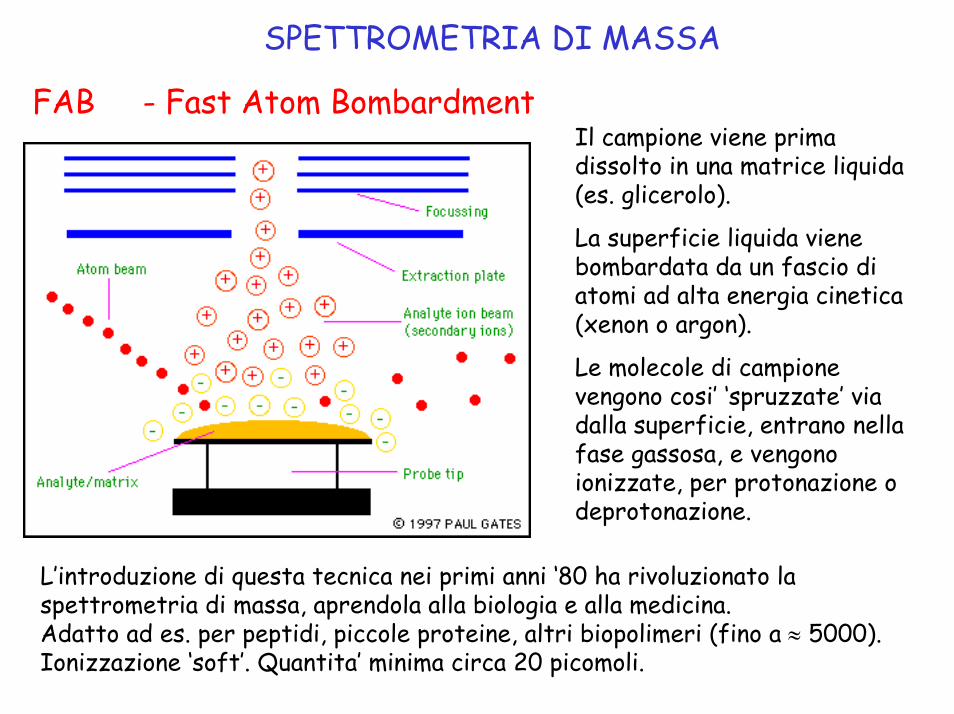
SPETTROMETRIA DI MASSA
FAB - Fast Atom BombardmentIl campione viene prima dissolto in una matrice liquida (es. glicerolo).
La superficie liquida viene bombardata da un fascio di atomi ad alta energia cinetica (xenon o argon).
Le molecole di campione vengono cosi’ ‘spruzzate’ via dalla superficie, entrano nella fase gassosa, e vengono ionizzate, per protonazione odeprotonazione.
L’introduzione di questa tecnica nei primi anni ‘80 ha rivoluzionato la spettrometria di massa, aprendola alla biologia e alla medicina. Adatto ad es. per peptidi, piccole proteine, altri biopolimeri (fino a ≈ 5000).Ionizzazione ‘soft’. Quantita’ minima circa 20 picomoli.
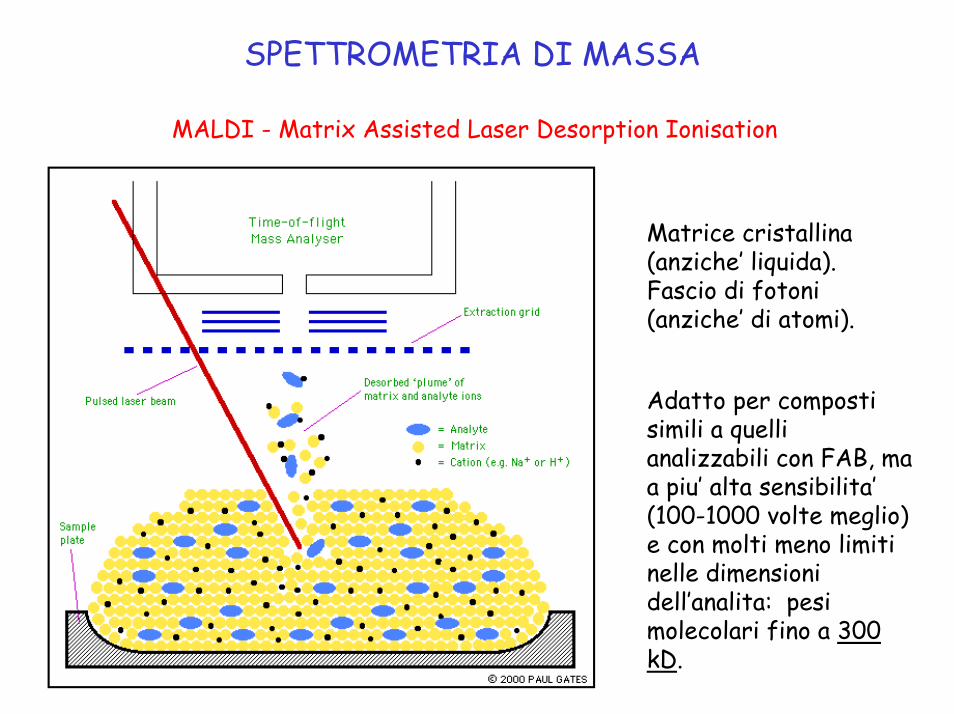
SPETTROMETRIA DI MASSA
MALDI - Matrix Assisted Laser Desorption Ionisation
Matrice cristallina (anziche’ liquida).Fascio di fotoni (anziche’ di atomi).
Adatto per composti simili a quelli analizzabili con FAB, ma a piu’ alta sensibilita’(100-1000 volte meglio) e con molti meno limiti nelle dimensioni dell’analita: pesi molecolari fino a 300 kD.
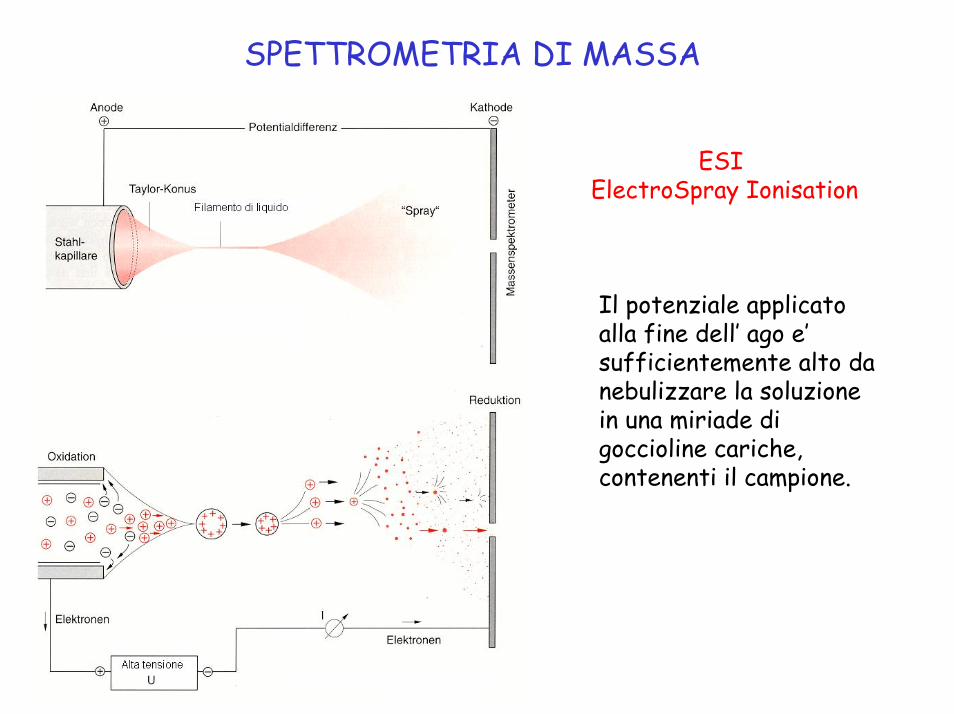
SPETTROMETRIA DI MASSA
ESIElectroSpray Ionisation
Il potenziale applicato alla fine dell’ ago e’ sufficientemente alto da nebulizzare la soluzione in una miriade di goccioline cariche, contenenti il campione.

ESI ElectroSpray Ionisation
Quando il solvente evapora, la concentrazione di carica alla superficie della gocciolina aumenta fino a superare la tensione superficiale →esplosione ‘coulombica’ . Si formano ioni dell’ analita privi di solvente.
Le cariche sono statisticamente distribuite tra i siti disponibili dell’analita, portando spesso alla formazione di ioni a cariche multiple.
La tecnica ESI e’ adatta per composti simili a quelli sottoponibili a MALDI, con sensibilita’ ed estensione di dimensioni molecolari un po’ minori.
ESI si adatta all’interfacciamento con HPLC e elettroforesi capillare
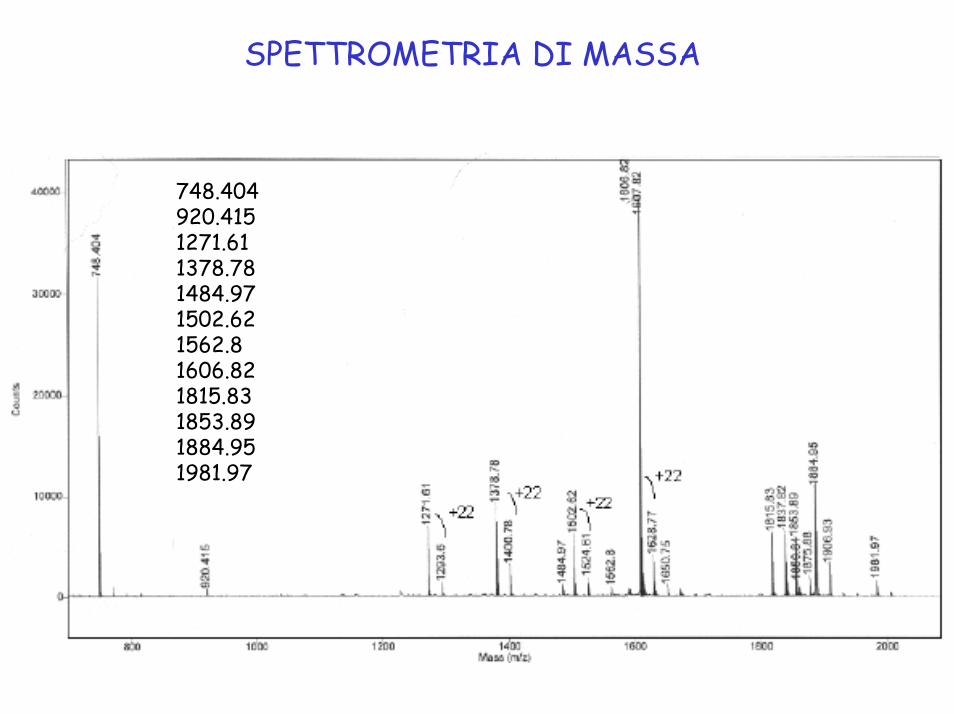
SPETTROMETRIA DI MASSA
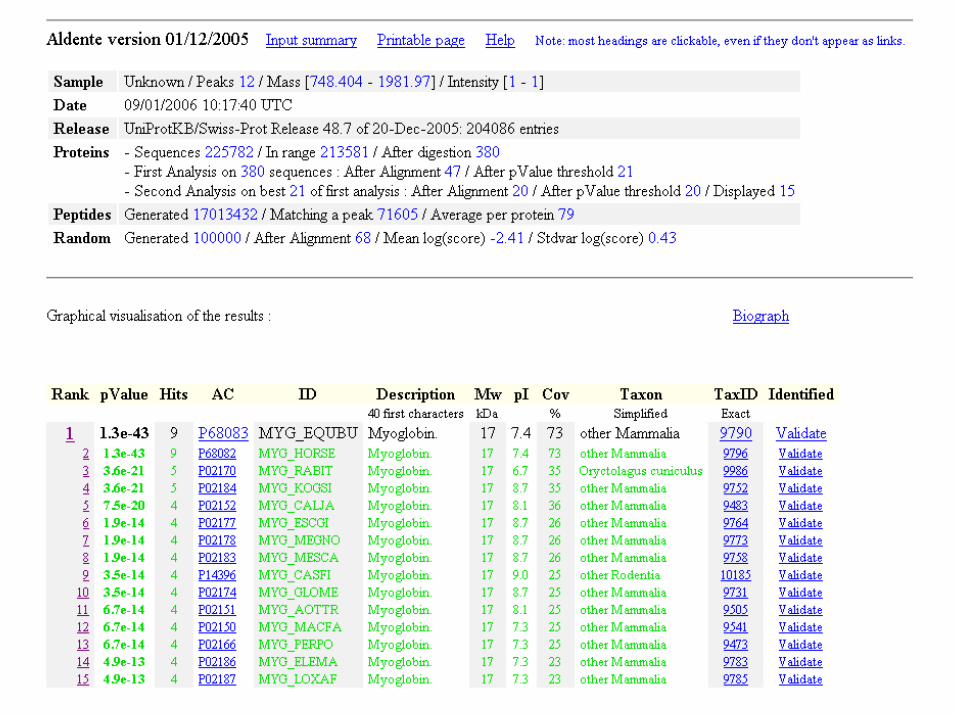
748.404 920.415 1271.61 1378.78 1484.97 1502.62 1562.8 1606.82 1815.83 1853.89 1884.95 1981.97
748.404 920.415 1271.61 1378.78 1484.97 1502.62 1562.8 1606.82 1815.83 1853.89 1884.95 1981.97

Modificazioni post-trasduzionali di proteine
Nominal Mass Common Modifications and Residueschange (Da)-42 ornithine replaces Arg-30 homoserine replaces Met-18 loss of water, (crosslink, dehydration, pyroGlu, replaces Glu)- 2 disulfide formation, dehydroalanine replaces Ala- 1 amide group replaces acid1 acid group replaces amide, citrulline replaces Arg14 methylene group (methylation, homolog, etc)16 oxygen atom (hydroxylation, Met sulfoxide formation, etc.)28 formyl, 2 methyl groups, ethyl32 addition of 2 oxygen atoms (dihydroxyl addition)35 & 37 chlorine atom42 acetyl, trimethyl group44 carboxyl addition56 t-butyl64 + isotopes selenocysteine replaces serine74 glyceryl group78 & 80 bromine atom80 phosphoryl, sulfonyl groups126 iodine atom132 pentosyl176 glucuronosyl210 myristoyl226 biotinyl229 pyridoxyl454 coenzyme A541 ADP-ribosyl617 heme784 FAD

TECNICHE MS E POTENZIALITA’

MALDI-Tof