1 lessons 5-6 classifying a protein / inside the genome
Post on 20-Dec-2015
218 views
TRANSCRIPT

11
Lessons 5-6Lessons 5-6
Classifying a protein /Classifying a protein /Inside the genomeInside the genome

22
Learning about a proteinLearning about a protein
What does a protein do??What does a protein do?? Post-translational modifications – Post-translational modifications –
phosphorylation, glycosylation, etc.phosphorylation, glycosylation, etc. Identifying patterns, motifsIdentifying patterns, motifs Secondary structureSecondary structure Tertiary/quaternary structureTertiary/quaternary structure Protein-protein interactionsProtein-protein interactions

33
Domains & MotifsDomains & Motifs

44
DomainsDomains
An analysis of known 3-D protein An analysis of known 3-D protein structures reveals that, rather than structures reveals that, rather than being monolithic, many of them being monolithic, many of them contain multiple folding unitscontain multiple folding units. .
Each such folding unit is a domain Each such folding unit is a domain (>50 aa, < 500 aa)(>50 aa, < 500 aa)
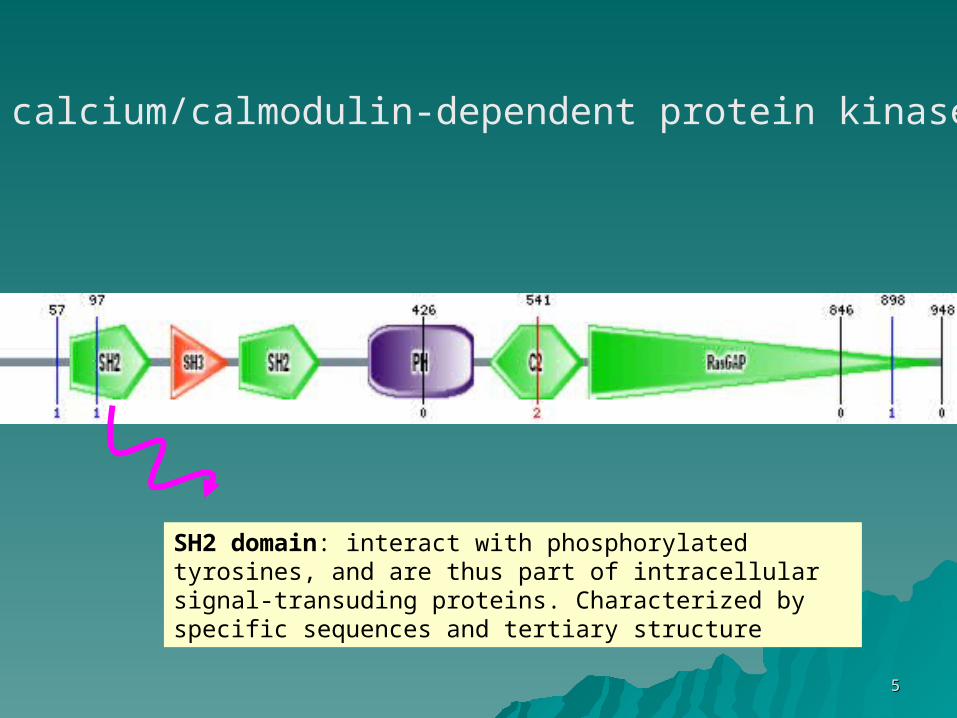
55
calcium/calmodulin-dependent protein kinase
SH2 domain: interact with phosphorylated tyrosines, and are thus part of intracellular signal-transuding proteins. Characterized by specific sequences and tertiary structure

66
What is a motif??What is a motif??
A sequence motifA sequence motif = a certain = a certain sequence that is widespread and sequence that is widespread and conjectured to have biological conjectured to have biological significancesignificance
Examples:Examples:KDELKDEL – ER-lumen retention signal – ER-lumen retention signalPKKKRKVPKKKRKV – an NLS (nuclear – an NLS (nuclear localization signal)localization signal)

77
More loosely defined motifsMore loosely defined motifs
KDEL (usually)KDEL (usually)++
HDEL (rarely) HDEL (rarely) ==
[HK]-D-E-L:[HK]-D-E-L:H H oror K at the first position K at the first position
This is called a pattern (in Biology), or This is called a pattern (in Biology), or a regular expression (in computer a regular expression (in computer science)science)

88
Syntax of a patternSyntax of a pattern
Example:Example: W-x(9,11)-[FYV]-[FYW]-x(6,7)-[GSTNE]W-x(9,11)-[FYV]-[FYW]-x(6,7)-[GSTNE]

99
PatternsPatterns
W-x(9,11)-[FYV]-[FYW]-x(6,7)-[GSTNE]W-x(9,11)-[FYV]-[FYW]-x(6,7)-[GSTNE]
Any amino, between 9-
11 times
F or Y or
V
WOPLASDFGYVWPPPLAWSROPLASDFGYVWPPPLAWSWOPLASDFGYVWPPPLSQQQ

1010
Patterns - syntaxPatterns - syntax The standard IUPAC one-letter codes. The standard IUPAC one-letter codes. ‘‘x’x’ : any amino acid. : any amino acid. ‘‘[]’[]’ : residues allowed at the position. : residues allowed at the position. ‘‘{}’{}’ : residues forbidden at the position. : residues forbidden at the position. ‘‘()’()’ : repetition of a pattern element are indicated in : repetition of a pattern element are indicated in
parenthesis. X(n) or X(n,m) to indicate the number or parenthesis. X(n) or X(n,m) to indicate the number or range of repetition. range of repetition.
‘‘-’-’ : separates each pattern element. : separates each pattern element. ‘‹’‘‹’ : indicated a N-terminal restriction of the pattern. : indicated a N-terminal restriction of the pattern. ‘›’‘›’ : indicated a C-terminal restriction of the pattern. : indicated a C-terminal restriction of the pattern. ‘‘.’.’ : the period ends the pattern. : the period ends the pattern.

1111
Pattern ~ motif ~ signaturePattern ~ motif ~ signature
A A patternpattern (similar to consensus and (similar to consensus and profile) is a way to represent a profile) is a way to represent a conserved sequenceconserved sequence
Whereas a profile and consensus Whereas a profile and consensus usually relate to the entire sequence, usually relate to the entire sequence, a pattern usually relates to a a few a pattern usually relates to a a few tens of amino-acidstens of amino-acids
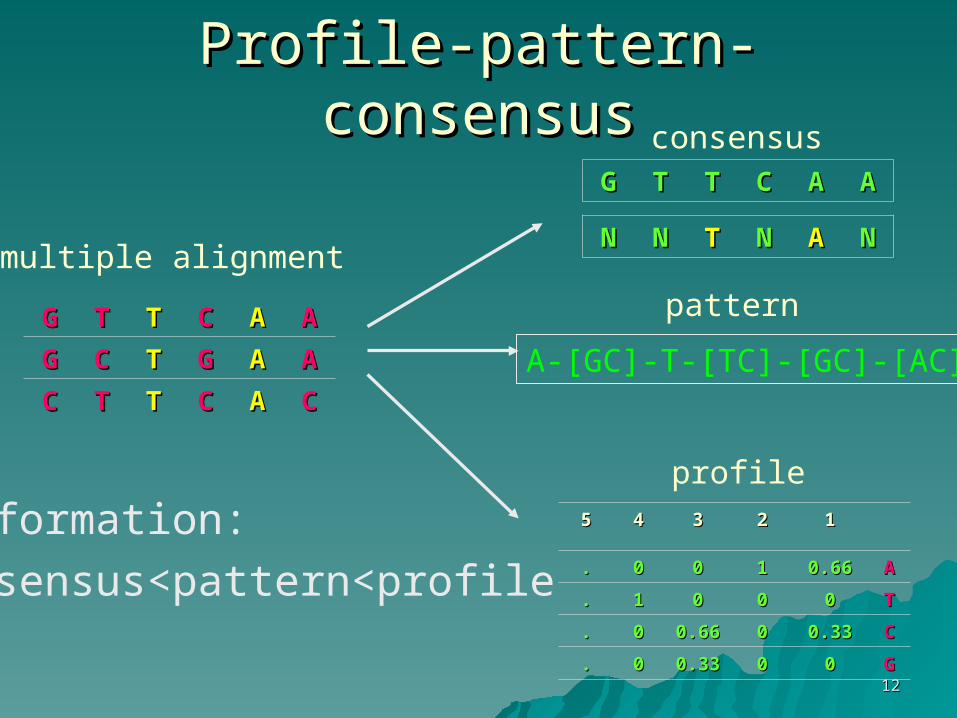
1212
Profile-pattern-consensusProfile-pattern-consensus
AAAACCTTTTGG
AAAAGGTTCCGG
CCAACCTTTTCC
1122334455
AA0.660.66110000..
TT00000011..
CC0.330.33000.660.6600..
GG00000.330.3300..
AAAACCTTTTGG
[AC-]A-[GC]-T-[TC]-[GC]
multiple alignment
consensus
pattern
profile
•Information:
consensus<pattern<profile
NNAANNTTNNNN

1313
InterproInterpro
Interpro: a collection of many protein Interpro: a collection of many protein signature databases (Prosite, Pfam, signature databases (Prosite, Pfam, Prints…) integrated into a Prints…) integrated into a hierarchical classifying systemhierarchical classifying system
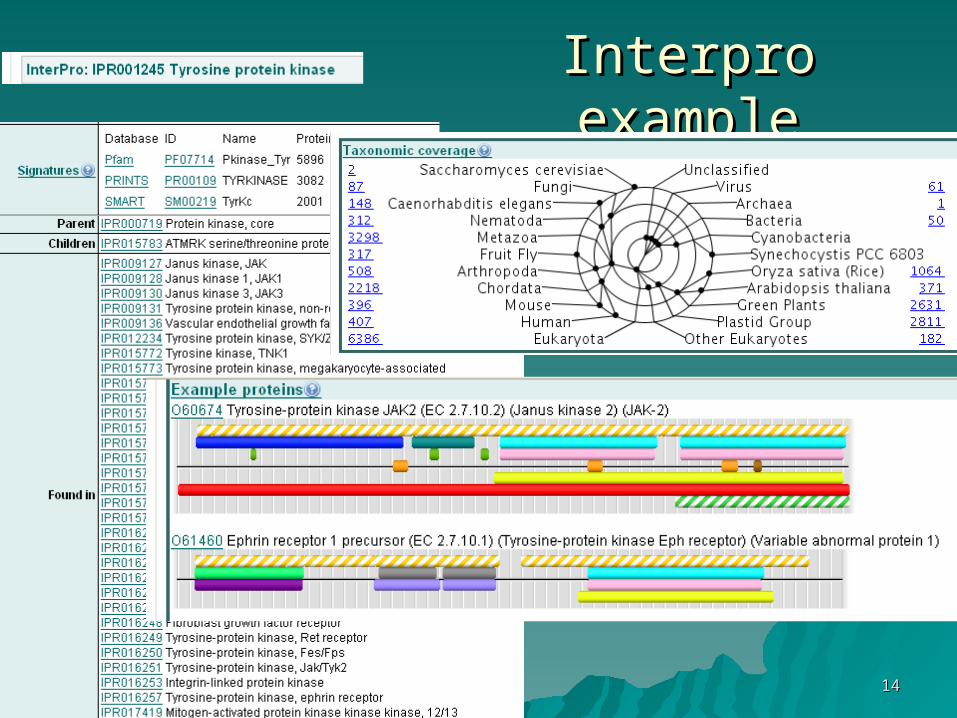
1414
Interpro exampleInterpro example

1515
PTM – Post-PTM – Post-Translational Translational ModificationModification
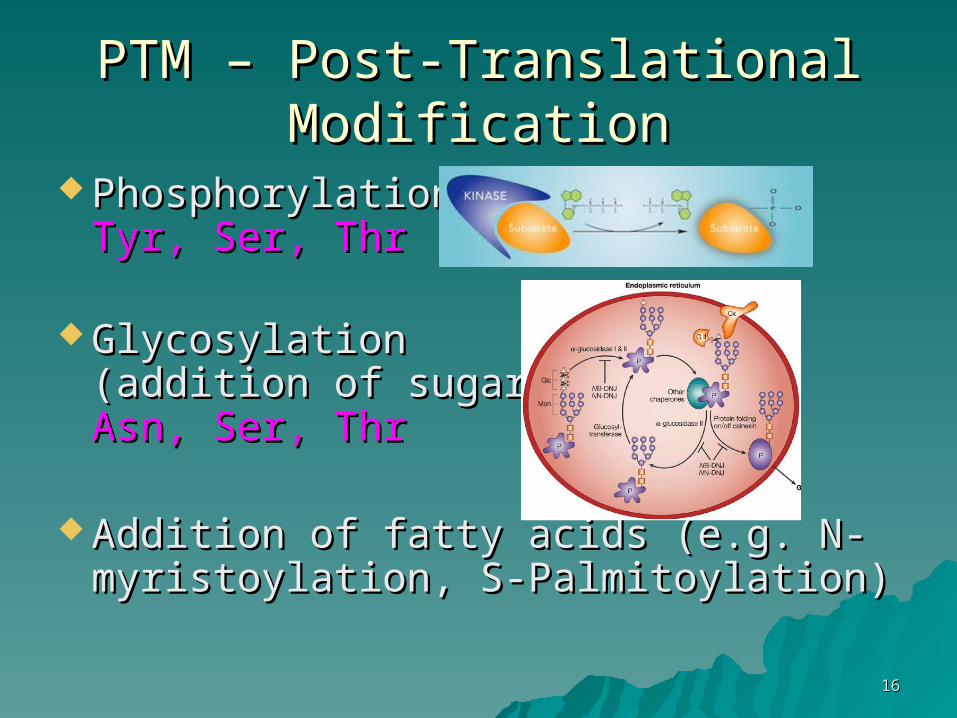
1616
PTM – Post-Translational PTM – Post-Translational ModificationModification
PhosphorylationPhosphorylationTyr, Ser, ThrTyr, Ser, Thr
GlycosylationGlycosylation(addition of sugars)(addition of sugars)Asn, Ser, ThrAsn, Ser, Thr
Addition of fatty acids (e.g. N-Addition of fatty acids (e.g. N-myristoylation, S-Palmitoylation)myristoylation, S-Palmitoylation)

1717
So how to predictSo how to predict
Take into account:Take into account:
1.1. Context (motif):Context (motif):PKC (a kinase) recognizes PKC (a kinase) recognizes X S/T X R/KX S/T X R/KN-Myristoylation at M G X X X S/TN-Myristoylation at M G X X X S/TSeveral times – we don’t know the exact Several times – we don’t know the exact motif!motif!
2.2. ConservationConservationIs the motif found (for instance, in Is the motif found (for instance, in human) also conserved in related human) also conserved in related organisms (for instance, in chimp)?organisms (for instance, in chimp)?

1818
Prediction problemsPrediction problems
Signal for detection is very shortSignal for detection is very short Not enough biological knowledge for Not enough biological knowledge for
characterizing the signalcharacterizing the signal Tertiary structureTertiary structure
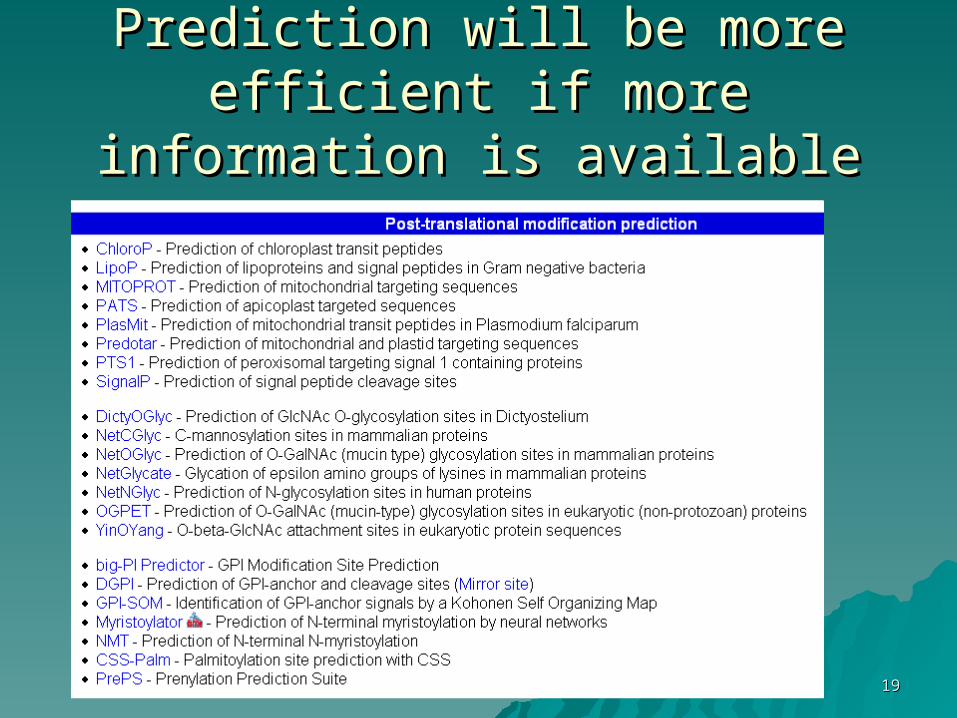
1919
Prediction will be more efficient if Prediction will be more efficient if more information is availablemore information is available

2020
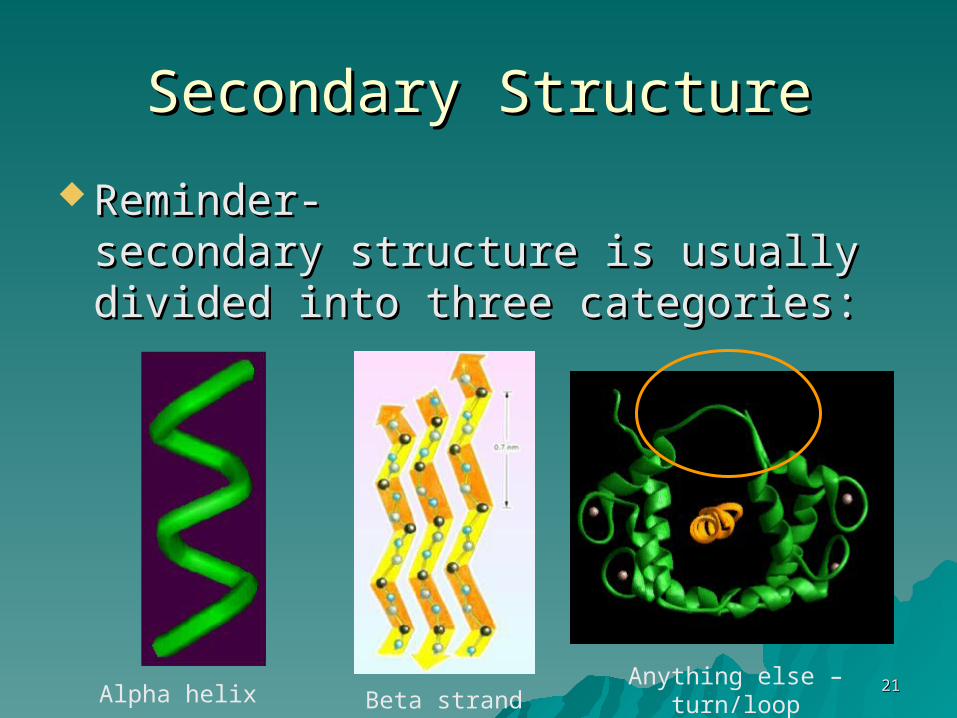
Secondary StructureSecondary Structure
2121
Secondary StructureSecondary Structure
Reminder- Reminder- secondary structure is usually secondary structure is usually divided into three categories:divided into three categories:
Alpha helix Beta strand (sheet)Anything else –
turn/loop

2222
Secondary StructureSecondary Structure
An easier question – what is the An easier question – what is the secondary structure when the 3D secondary structure when the 3D structure is known?structure is known?

2323
DSSPDSSP
DSSPDSSP (Dictionary of Secondary (Dictionary of Secondary Structure of a Protein) – assigns Structure of a Protein) – assigns secondary structure to proteins secondary structure to proteins which have a crystal structurewhich have a crystal structure
H = alpha helix
B = beta bridge (isolated residue)
E = extended beta strand
G = 3-turn helix
I = 5-turn helix
T = hydrogen bonded turn
S = bend

2424
Predicting secondary structure from Predicting secondary structure from primary sequenceprimary sequence

2525
Chou and Fasman (1974)Chou and Fasman (1974)Name P(a) P(b) P(turn)
Alanine 142 83 66Arginine 98 93 95Aspartic Acid 101 54 146Asparagine 67 89 156Cysteine 70 119 119Glutamic Acid 151 037 74Glutamine 111 110 98Glycine 57 75 156Histidine 100 87 95Isoleucine 108 160 47Leucine 121 130 59Lysine 114 74 101Methionine 145 105 60Phenylalanine 113 138 60Proline 57 55 152Serine 77 75 143Threonine 83 119 96Tryptophan 108 137 96Tyrosine 69 147 114Valine 106 170 50
The propensity of an amino acid to be part of a certain secondary structure (e.g. – Proline has a low propensity of being in an alpha helix or beta sheet breaker)

2626
Chou-Fasman predictionChou-Fasman prediction
Look for a series of >4 amino acids which all have Look for a series of >4 amino acids which all have (for instance) alpha helix values >100(for instance) alpha helix values >100
Extend (…)Extend (…) Accept as alpha helix if Accept as alpha helix if
average alpha score > average beta scoreaverage alpha score > average beta score
Ala Pro Tyr Phe Phe Lys Lys His Val Ala Thr
α 142 57 69 113 113 114 114 100 106 142 83
β 83 55 147 138 138 74 74 87 170 83 119

2727
Chou and Fasman (1974)Chou and Fasman (1974)
Success rate of 50%Success rate of 50%

2828
Improvements in the 1980’sImprovements in the 1980’s
Conservation in MSAConservation in MSA Smarter algorithms (e.g. HMM, neural Smarter algorithms (e.g. HMM, neural
networks).networks).

2929
AccuracyAccuracy
Accuracy of prediction seems to hit a Accuracy of prediction seems to hit a ceiling of 70-80% accuracyceiling of 70-80% accuracy
MethodMethodAccuracyAccuracy
Chou & FasmanChou & Fasman50%50%
Adding the MSAAdding the MSA69%69%
MSA+ sophisticated MSA+ sophisticated computationscomputations
70-80%70-80%

3030
Gene OntologyGene Ontology

3131
GOGO
GGeneene O Ontology – a project for ntology – a project for consistent descriptionconsistent description of gene of gene products in products in different databasesdifferent databases. .
Consistent descriptionConsistent description - Common key - Common key definitions. definitions.
Example:Example: ‘protein synthesis’ or ‘protein synthesis’ or ‘translation’‘translation’

3232
GOGO
GO - GO describes proteins in terms of :GO - GO describes proteins in terms of :
biological processbiological process
cellular componentcellular component
molecular functionmolecular function
GO is GO is notnot::
– A sequence database.A sequence database.
– A portal for sequence informationA portal for sequence information
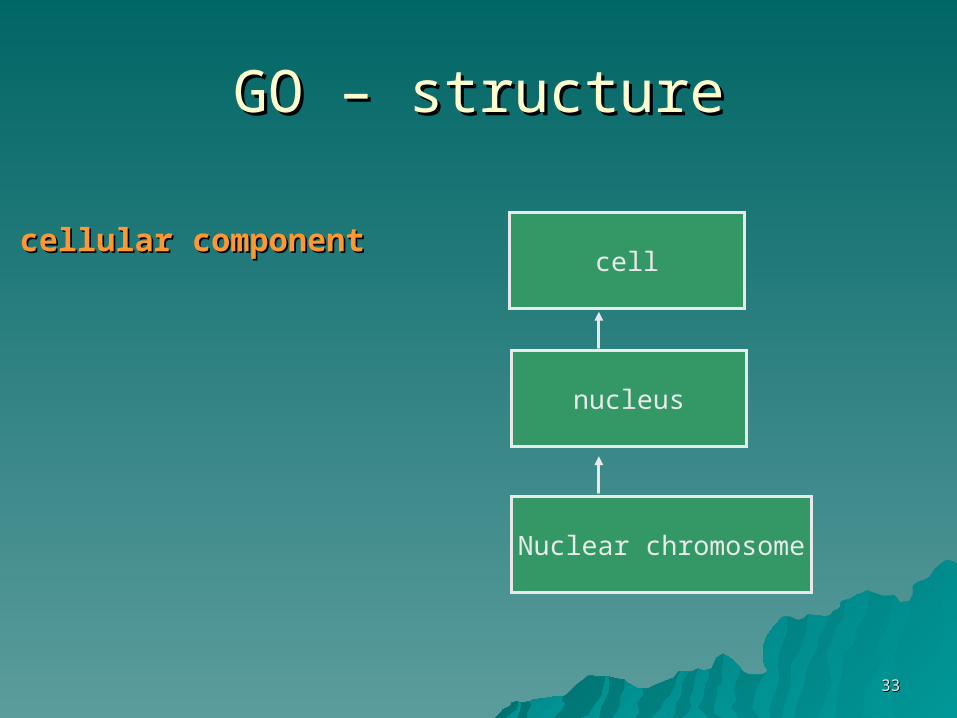
3333
GO – structureGO – structure
nucleus
Nuclear chromosome
cellcellular componentcellular component

3434
GO exampleGO example
Links from the swissprot entry of human protein kinase C alpha
3535
Examples for use of GOExamples for use of GO
Enrichment for a GO category:Enrichment for a GO category:1.1. Do all upregulated genes in a Do all upregulated genes in a
microarray you built belong to the microarray you built belong to the same GO “molecular function” same GO “molecular function” category?category?
2.2. You have predicted a new You have predicted a new transcription factor binding site. Do transcription factor binding site. Do all genes with this site belong to the all genes with this site belong to the same GO biological process?same GO biological process?

3636
Evaluation of prediction Evaluation of prediction methodsmethods

3737
Evaluation of prediction methodsEvaluation of prediction methods
Comparing our results to experimentally Comparing our results to experimentally verified sitesverified sites
Positive (hit)Positive (hit)NegativeNegative
TrueTrueTrue-positiveTrue-positive
True-negativeTrue-negative
FalseFalseFalse-positiveFalse-positive(false alarm)(false alarm)
False-negativeFalse-negative(miss)(miss)
Our prediction gives:
Is t
he
pre
dic
tio
n c
orr
ect
?

3838
Method evaluationMethod evaluation
Positive (hit)Positive (hit)NegativeNegative
TrueTrueTrue-positiveTrue-positive
True-negativeTrue-negative
FalseFalseFalse-False-positivepositive
(false alarm)(false alarm)
False-negativeFalse-negative(miss)(miss)
A good method will be one with a high level of A good method will be one with a high level of true-positives and true-negatives, and a low true-positives and true-negatives, and a low level of false-positives and false-negativeslevel of false-positives and false-negatives
Our prediction gives:
Is t
he
pre
dic
tio
n c
orr
ect
?

3939
Calibrating the methodCalibrating the method
All methods have a parameter (or a All methods have a parameter (or a score) that can be calibrated to score) that can be calibrated to improve the accuracy of the method.improve the accuracy of the method.
For example: the E-value cutoff in For example: the E-value cutoff in BLASTBLAST

4040
Calibrating E-value cutoffCalibrating E-value cutoff
Reminder: the lower the E-value, the Reminder: the lower the E-value, the more ‘significant’ the alignment more ‘significant’ the alignment between the query and the hit.between the query and the hit.

4141
Calibrating the E-valueCalibrating the E-value
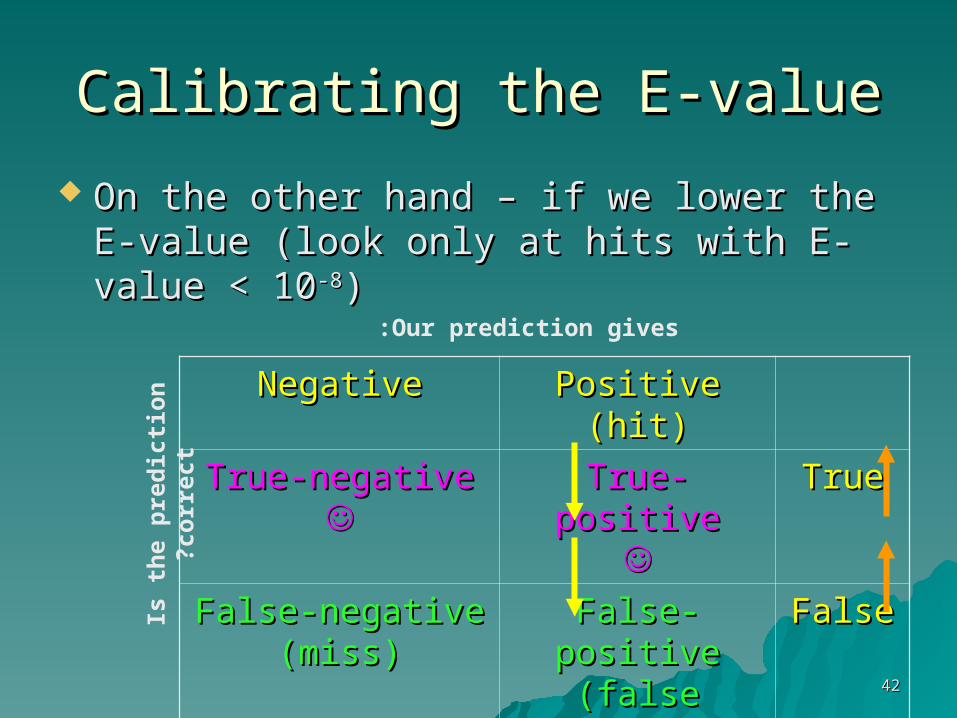
What will happen if we raise the E-value What will happen if we raise the E-value cutoff (for instance – work with all hits with cutoff (for instance – work with all hits with an E-value which is < 10) ?an E-value which is < 10) ?
Positive (hit)Positive (hit)NegativeNegative
TrueTrueTrue-positiveTrue-positive
True-negativeTrue-negative
FalseFalseFalse-positiveFalse-positive(false alarm)(false alarm)
False-negativeFalse-negative(miss)(miss)
Our prediction gives:
Is t
he
pre
dic
tio
n c
orr
ect
?
4242
Calibrating the E-valueCalibrating the E-value
On the other hand – if we lower the E-value On the other hand – if we lower the E-value (look only at hits with E-value < 10(look only at hits with E-value < 10-8-8))
Positive (hit)Positive (hit)NegativeNegative
TrueTrueTrue-positiveTrue-positive
True-negativeTrue-negative
FalseFalseFalse-positiveFalse-positive(false alarm)(false alarm)
False-negativeFalse-negative(miss)(miss)
Our prediction gives:
Is t
he
pre
dic
tio
n c
orr
ect
?

4343
Improving predictionImproving prediction
Trade-off between Trade-off between specificityspecificity and and sensitivitysensitivity

4444
Sensitivity vs. specificitySensitivity vs. specificity
Sensitivity = Sensitivity =
Specificity =Specificity =
True positive
True positive + False negative
Represent all the proteins which are
really phosphorylated
True negative
True negative + False positive
Represent all the proteins which are
really NOT phosphorylated
How good we hit real
phosphorylations
How good we avoid real non-
phosphorylations
4545
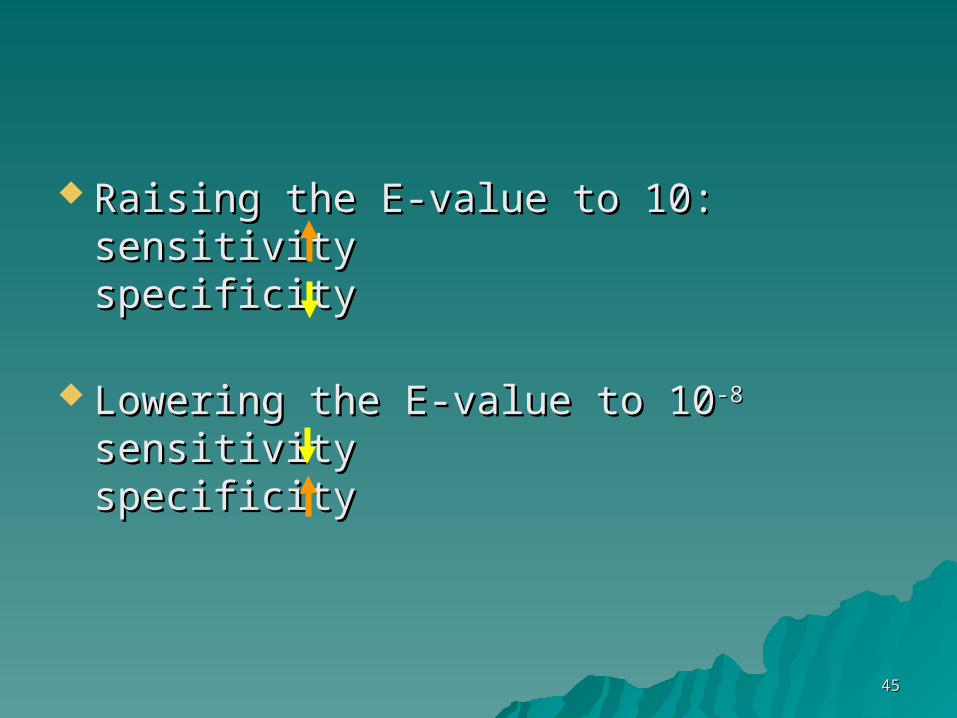
Raising the E-value to 10:Raising the E-value to 10:sensitivitysensitivityspecificityspecificity
Lowering the E-value to 10Lowering the E-value to 10-8-8
sensitivity sensitivity specificityspecificity

4646
Over-predictions: exampleOver-predictions: example
Many PTM-predictors tend to Many PTM-predictors tend to over-over-predictpredict high level of false high level of false positives positives low specificity low specificity
WHY?WHY?
1.1. Tertiary structure! (buried/exposed, Tertiary structure! (buried/exposed, tertiary motifs)tertiary motifs)
2.2. The phosphorylation recognition The phosphorylation recognition mechanism is not completely clear!mechanism is not completely clear!

4747
Inside the genomeInside the genome

4848
2001: the human genome2001: the human genome

4949
Neck to neck competitionNeck to neck competition
Celera Genomics (private company) Celera Genomics (private company) versus the International Human versus the International Human Genome Sequencing Consortium Genome Sequencing Consortium (public company)(public company)

5050
The highlightsThe highlights
~30,000 genes in the human ~30,000 genes in the human genome genome (today – estimated at 20-25K)(today – estimated at 20-25K)
Oases of genes in empty desertsOases of genes in empty deserts Long-range variation in GC contentLong-range variation in GC content Repetitive elements ruleRepetitive elements rule

5151
How many genes in the genomeHow many genes in the genome??
Ratio of average gene size to Ratio of average gene size to genome size:genome size:100,000100,000
Based on ESTs: Based on ESTs: 35,000-120,00035,000-120,000

5252
Detecting genes in the human Detecting genes in the human genomegenome
Gene finding methods:Gene finding methods: Ab initioAb initio
The challenge: small exons in a sea The challenge: small exons in a sea of intronsof introns
Homology-based Homology-based The problem: will not detect novel The problem: will not detect novel genesgenes

5353
Genscan (ab initio)Genscan (ab initio)
Based on a probabilistic model of a Based on a probabilistic model of a gene structuregene structure
Takes into account:Takes into account:- gene composition – exons/introns- gene composition – exons/introns- GC content- GC content- splice signals- splice signals- promoters- promoters
Goes over all 6 reading framesGoes over all 6 reading frames
Burge and Karlin, 1997, Prediction of complete gene structure in human genomic DNA, J. Mol. Biol. 268
5454
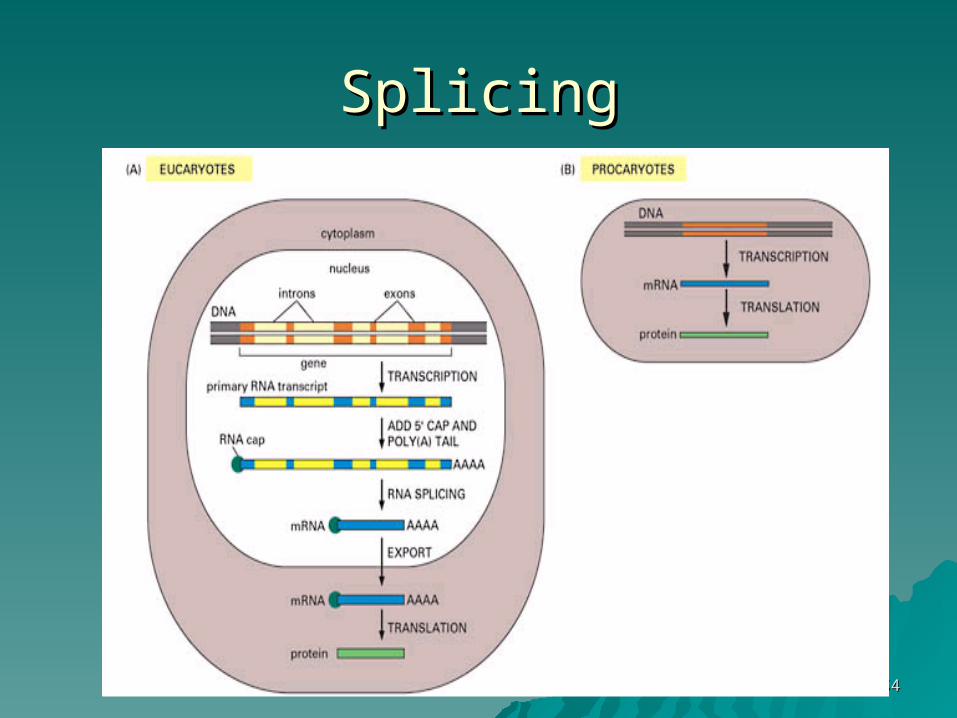
SplicingSplicing
5555
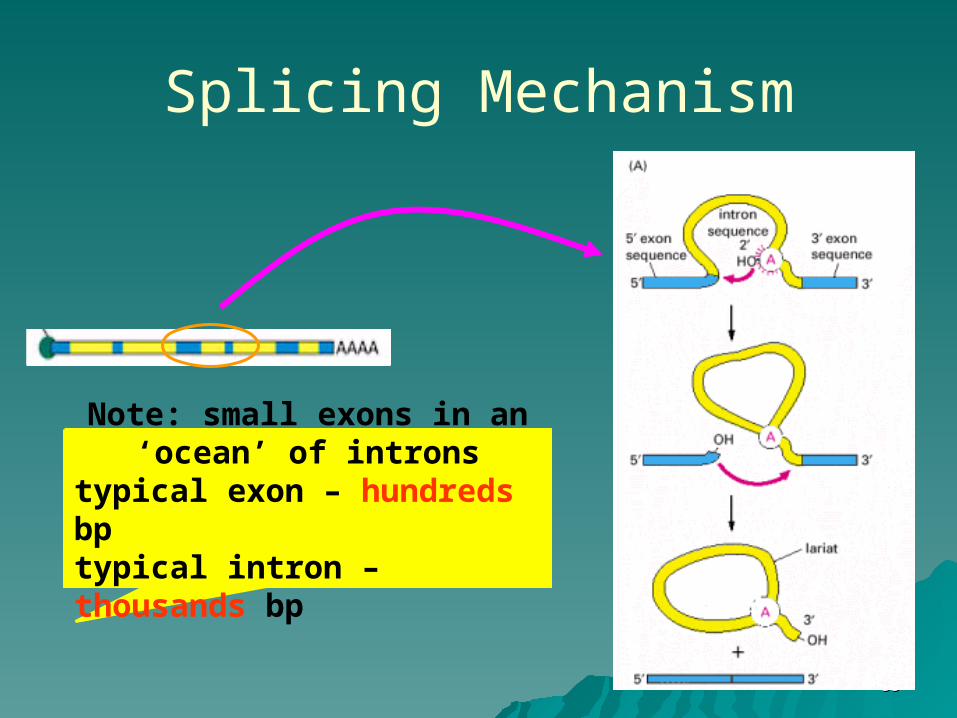
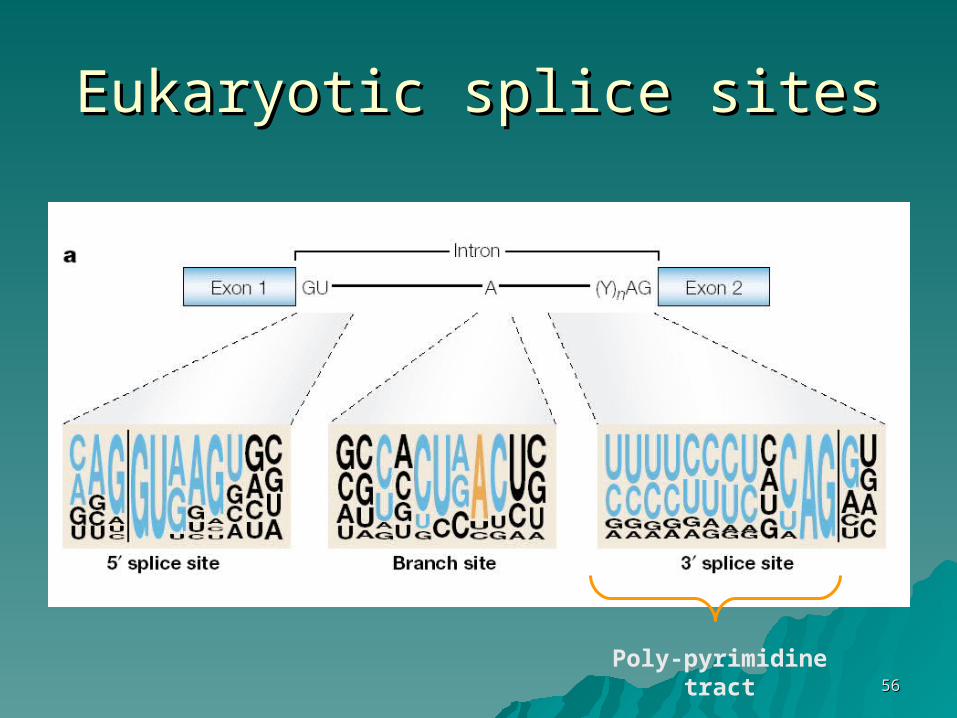
Splicing Mechanism
Note: small exons in an ‘ocean’ of introns
typical exon – hundreds bptypical intron – thousands bp
5656
Eukaryotic splice sitesEukaryotic splice sites
Poly-pyrimidine tract

5757
CpG Islands: another signalCpG Islands: another signal
CpG islands are regions of the CpG islands are regions of the genome with a higher frequency of genome with a higher frequency of CG dinucleotides (not base-pairs!) CG dinucleotides (not base-pairs!) than the rest of the genomethan the rest of the genome
CpG islands often occur near the CpG islands often occur near the beginning of genesbeginning of genes maybe maybe related to the binding of the related to the binding of the TF Sp1TF Sp1

5858
Human genome gene countHuman genome gene count
1.1. Ab initio – GenscanAb initio – Genscan
2.2. Confirmation using Confirmation using ESTsESTs mRNAmRNA Known protein motifs (Pfam) from Known protein motifs (Pfam) from
any organismany organism
3.3. Known genes: Refseq, Swissprot, Known genes: Refseq, Swissprot, TrEMBLTrEMBL

5959
Human genome gene countHuman genome gene count
31,000 genes31,000 genes 1.5% of the genome: coding1.5% of the genome: coding 33% - transcribed into genes33% - transcribed into genes
6060
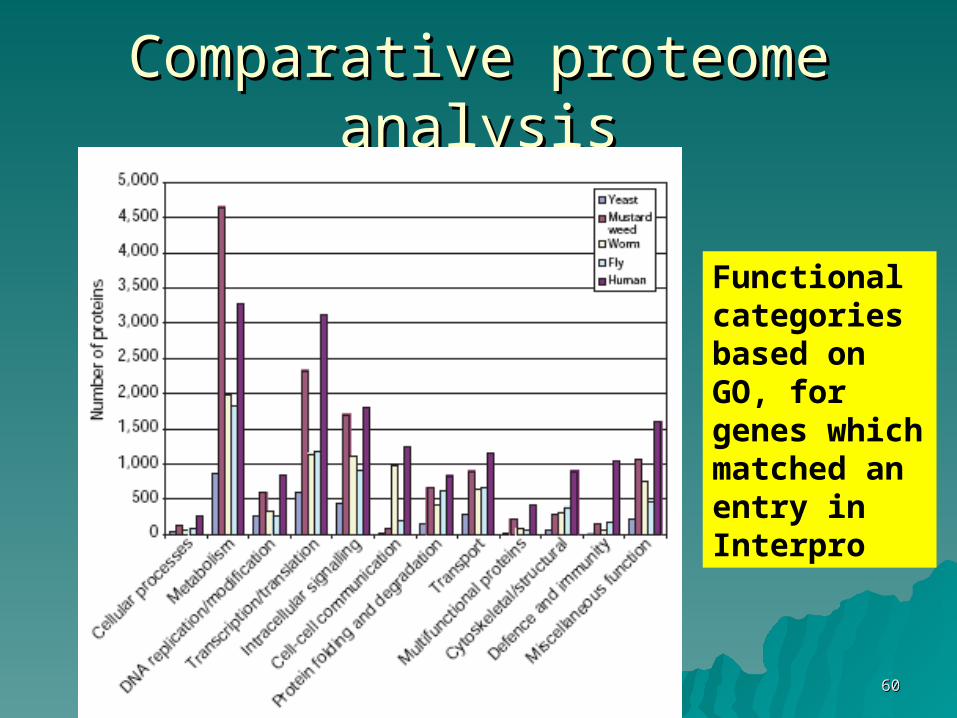
Comparative proteome analysisComparative proteome analysis
Functional categories based on GO, for genes which matched an entry in Interpro

6161
Comparative proteome analysisComparative proteome analysis
Humans have more proteins involved Humans have more proteins involved in cytoskeleton, immune defense, in cytoskeleton, immune defense, and and transcriptiontranscription
6262
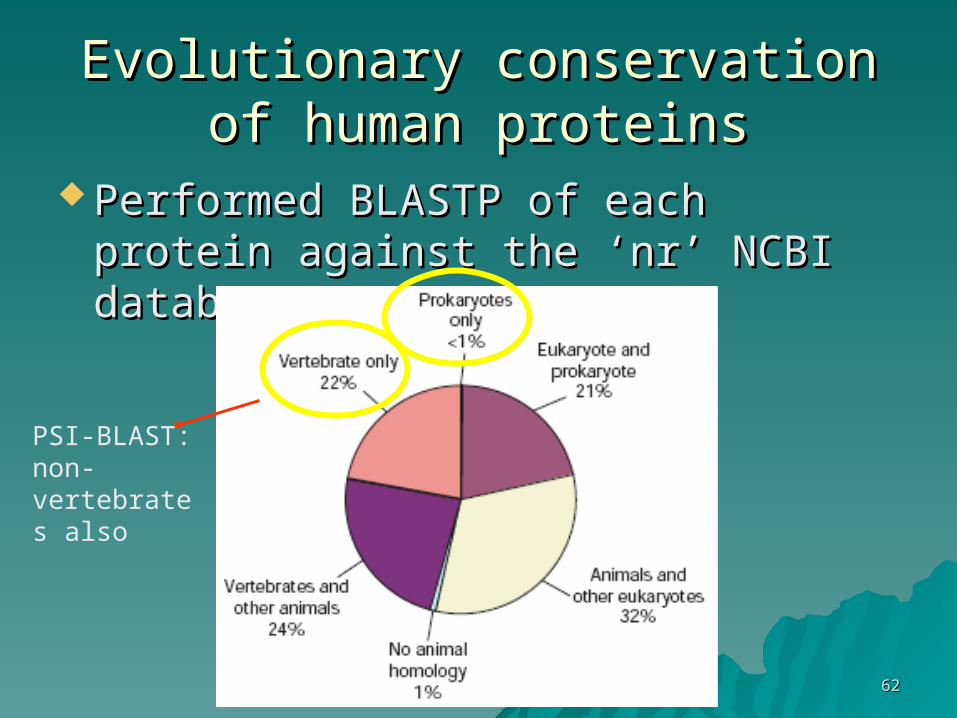
Evolutionary conservation of Evolutionary conservation of human proteinshuman proteins
Performed BLASTP of each protein Performed BLASTP of each protein against the ‘nr’ NCBI databaseagainst the ‘nr’ NCBI database
PSI-BLAST: non-vertebrates also

6363
Horizontal (lateral) gene transferHorizontal (lateral) gene transfer
Lateral Gene Transfer (LGT) is any process in which an organism transfers genetic material to another organism that is not its offspring

6464
Mechanisms:
Transformation
Transduction (phages/viruses)
Conjugation

6565
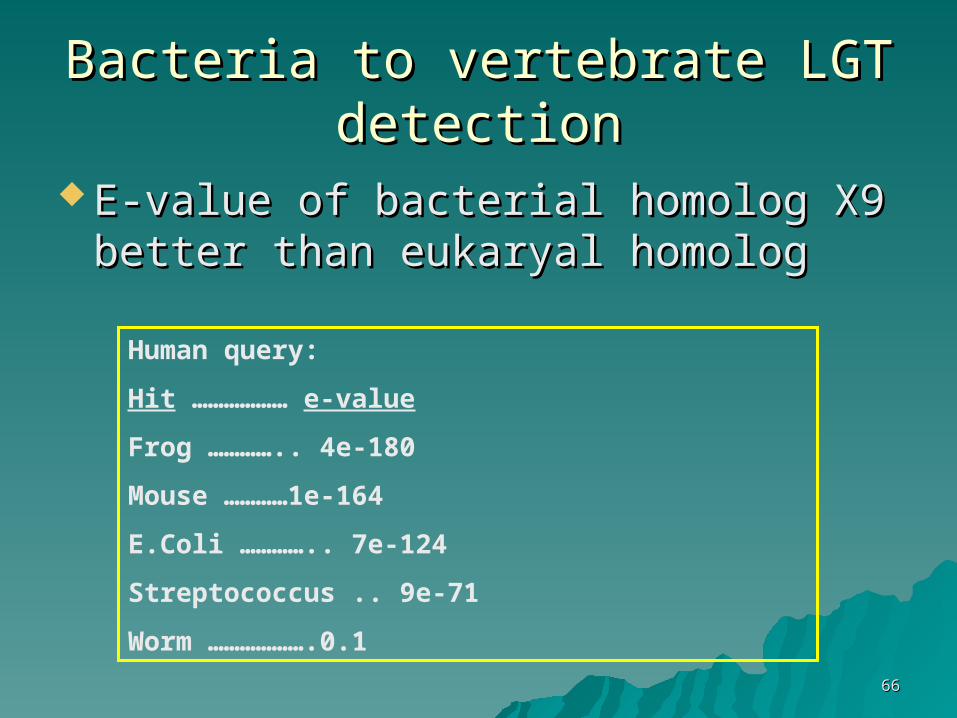
Bacteria to vertebrate LGT criteriaBacteria to vertebrate LGT criteria
Homologs in bacteriaHomologs in bacteria Homologs in vertebrates (detected in Homologs in vertebrates (detected in
PSI-BLAST)PSI-BLAST) NoNo significant homologs in non- significant homologs in non-
vertebratesvertebrates
6666
Bacteria to vertebrate LGT Bacteria to vertebrate LGT detectiondetection
E-value of bacterial homolog X9 E-value of bacterial homolog X9 better than eukaryal homologbetter than eukaryal homolog
Human query:
Hit ……………… e-value
Frog ………….. 4e-180
Mouse …………1e-164
E.Coli ………….. 7e-124
Streptococcus .. 9e-71
Worm ……………….0.1
6767
Bacteria to vertebrate LGTBacteria to vertebrate LGT
vertebratesBacteria Non-vertebrates

6868
Bacteria to vertebrate LGTBacteria to vertebrate LGT
Genes with a role in metabolism of Genes with a role in metabolism of xenobiotics or stress responsexenobiotics or stress response
Selective advantage for these transfers. Selective advantage for these transfers. May be highly important immune geneMay be highly important immune gene

6969

7070
Bacteria to vertebrate LGT??Bacteria to vertebrate LGT??
Hundreds of sequenced bacterial Hundreds of sequenced bacterial genome vs. handful of eukaryotesgenome vs. handful of eukaryotes
Gene finding in bacteria is much Gene finding in bacteria is much easier than in eukaryoteseasier than in eukaryotes
On the practical side: rigid On the practical side: rigid mechanical barriers to LGT in mechanical barriers to LGT in eukaryotes (nucleus, germ line)eukaryotes (nucleus, germ line)

7171
Repetitive elements in the human Repetitive elements in the human genomegenome

7272
The C-value paradoxThe C-value paradox
Genome size does not correlate with Genome size does not correlate with organism complexityorganism complexity
YeastYeastHumanHumanRiceRiceAmoebaAmoeba
Genome Genome sizesize
12 million12 million3 billion3 billion4.3 billion4.3 billion67 billion67 billion
Number of Number of genesgenes
6,2756,27520-25,00020-25,000~30,000~30,000??

7373
Repetitive elementsRepetitive elements
The C-value mystery was partially The C-value mystery was partially resolved when it was found that resolved when it was found that large portions of genomes contain large portions of genomes contain repetitive elementsrepetitive elements

7474
Repeats in the human genomeRepeats in the human genome
~50% of the human genome (~1% ~50% of the human genome (~1% coding):coding):
1.1. Transposon derived (=interspersed Transposon derived (=interspersed repeats) (45% of the genome)repeats) (45% of the genome)
2.2. Retrotransposed cellular genesRetrotransposed cellular genes
3.3. Sequence repeats (A)Sequence repeats (A)nn, (CG), (CG)nn, etc., etc.
4.4. Segmental duplicationsSegmental duplications
7575
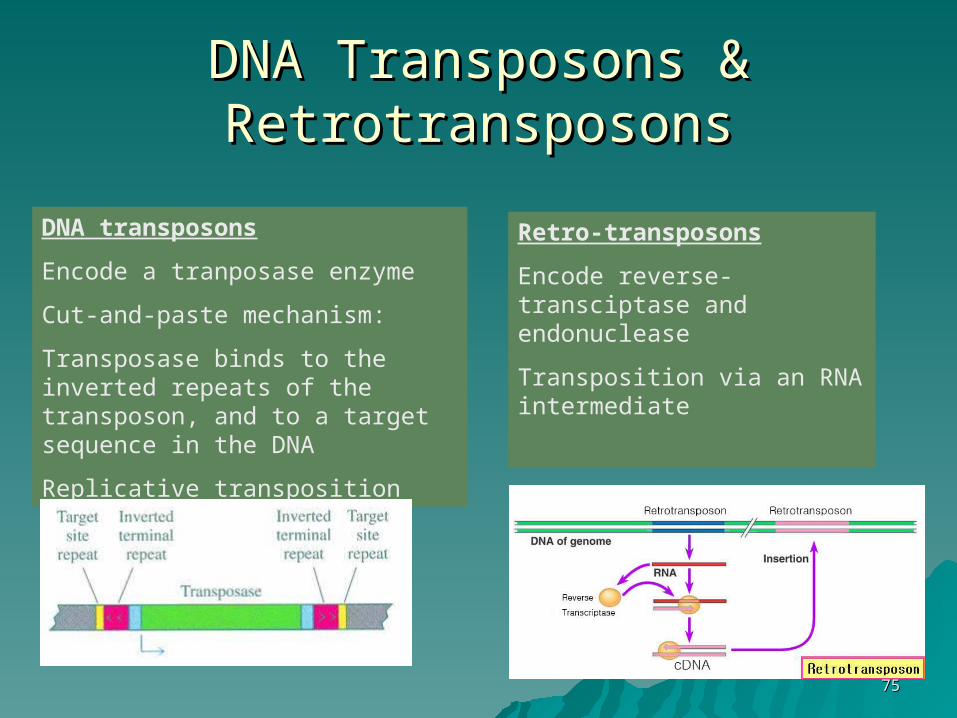
DNADNA Transposons & Transposons & RetrotransposonsRetrotransposons
DNA transposons
Encode a tranposase enzyme
Cut-and-paste mechanism:
Transposase binds to the inverted repeats of the transposon, and to a target sequence in the DNA
Replicative transposition
Retro-transposons
Encode reverse-transciptase and endonuclease
Transposition via an RNA intermediate

7676
Transposable elements in the Transposable elements in the human genomehuman genome
Retrotransposons
*
*
* Non-LTR retrotransposons
** LTR transposon
**

7777
LINEs and SINEsLINEs and SINEs
Highly successful elements in Highly successful elements in eukaryoteseukaryotes
SINEs are freeriders on the backs of SINEs are freeriders on the backs of LINEs – LINEs – encode no proteinsencode no proteins

7878
Determining the age of Determining the age of transposable elementstransposable elements
For each family, a For each family, a consensus consensus sequencesequence was built ===> the was built ===> the ancestral sequenceancestral sequence
Compute the divergence (%) of each Compute the divergence (%) of each sequence from the ancestorsequence from the ancestor
Convert sequence divergence to Convert sequence divergence to actual agesactual ages

7979
Age of transposable elementsAge of transposable elements
Most transposable elements date Most transposable elements date back to the emergence of placental back to the emergence of placental mammals (low disposal rate of mammals (low disposal rate of transposons)transposons)
DNA transposons in the human DNA transposons in the human genome are dead (high divergence genome are dead (high divergence from ancestor)!from ancestor)!

8080
Where are the transposons Where are the transposons locatedlocated??
LINEs LINEs AT-rich regions (less genes) AT-rich regions (less genes) SINEs (MIR, Alu) SINEs (MIR, Alu)
GC-rich areas …… ?? … they use the GC-rich areas …… ?? … they use the LINE machinery …….??LINE machinery …….??

8181
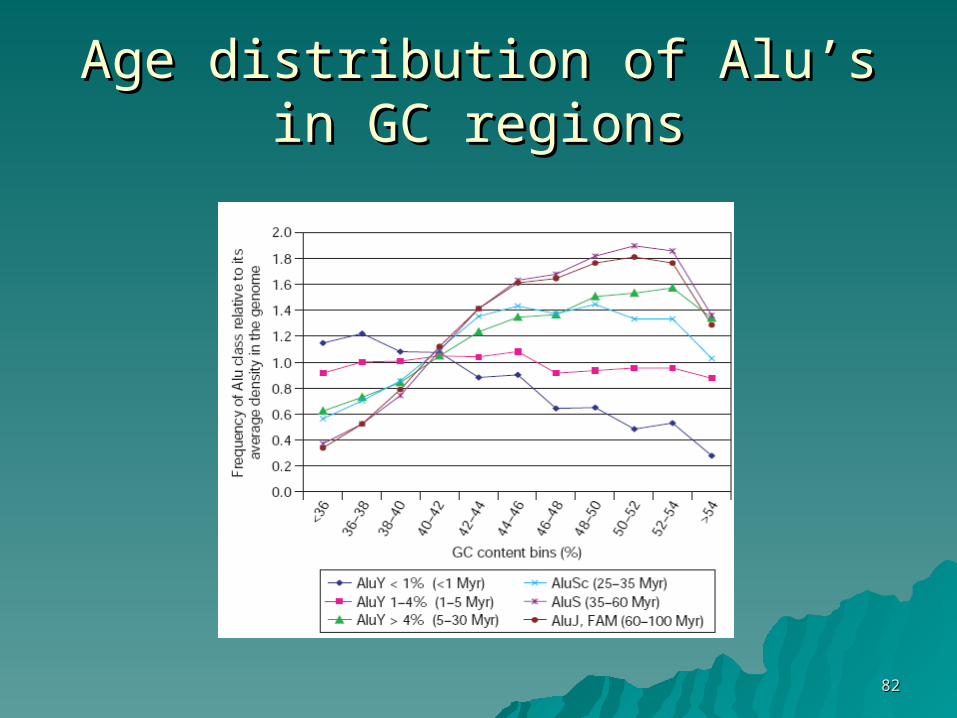
Why are there SINEs in GC-rich Why are there SINEs in GC-rich regionsregions??
1.1. SINEs target GC rich regionsSINEs target GC rich regions
2.2. Evolutionary advantage for SINEs Evolutionary advantage for SINEs that ‘land’ in a GC-rich regionthat ‘land’ in a GC-rich region
How do we resolve between the How do we resolve between the two options?two options?
8282
Age distribution of Alu’s in GC Age distribution of Alu’s in GC regionsregions

8383
SINEs in GC-rich regionsSINEs in GC-rich regions
1.1. High rate of random loss in AT-rich High rate of random loss in AT-rich regionsregions
2.2. Negative selection against Alu in Negative selection against Alu in AT-richAT-rich
3.3. Positive selection (evolutionary Positive selection (evolutionary advantage) for Alu in GC richadvantage) for Alu in GC rich
Comparison with LINEs
Alus correlate with actively transcribed genes

8484
Are Alus functionalAre Alus functional????
SINEs are transcribed under stressSINEs are transcribed under stress SINE RNAs may bind a protein kinase SINE RNAs may bind a protein kinase
promote translation under stress promote translation under stress
Need to be in regions which are highly Need to be in regions which are highly transcribedtranscribed
Role in alternative splicingRole in alternative splicing

8585
Repeats in the human genomeRepeats in the human genome
~50% of the human genome (~1% ~50% of the human genome (~1% coding):coding):
1.1. Transposon derived (=interspersed Transposon derived (=interspersed repeats) (45% of the genome)repeats) (45% of the genome)
2.2. Retrotransposed cellular genesRetrotransposed cellular genes
3.3. Sequence repeats (A)Sequence repeats (A)nn, (CG), (CG)nn, etc., etc.
4.4. Segmental duplicationsSegmental duplications

8686
Segment duplicationsSegment duplications
1077 segmental duplications detected1077 segmental duplications detected Several genes in the duplicated regions Several genes in the duplicated regions
associated with diseases (may be related associated with diseases (may be related to homologous recombination)to homologous recombination)
Most are recent duplications (conservation Most are recent duplications (conservation of entire segment, versus conservation of of entire segment, versus conservation of coding sequences only)coding sequences only)

8787

8888
Genome-wide studiesGenome-wide studies

8989
Sequenced genomesSequenced genomesAssembled and annotated eukaryote genomes in Ensembl

9090

9191
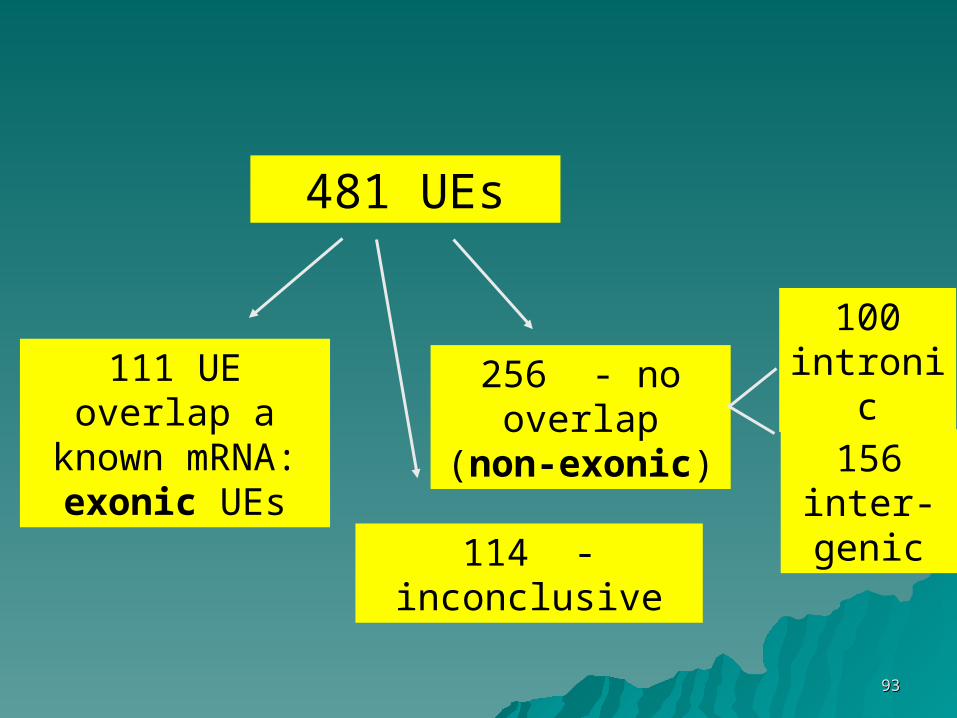
481 segments > 200 bp absolutely 481 segments > 200 bp absolutely conserved (100% identity) between conserved (100% identity) between human, rat and mousehuman, rat and mouse

9292
Comparison with a neutral Comparison with a neutral substitution ratesubstitution rate
Compare the substitution rate in a Compare the substitution rate in a any 1Mb regionany 1Mb region
Probability of 10Probability of 10-22 -22 of obtaining of obtaining 11 ultranconserved element (UE) by ultranconserved element (UE) by chancechance
9393
481 UEs
111 UE overlap a known
mRNA: exonic UEs
256 - no overlap (non-
exonic)
114 - inconclusive
100 intronic
156 inter-genic
9494
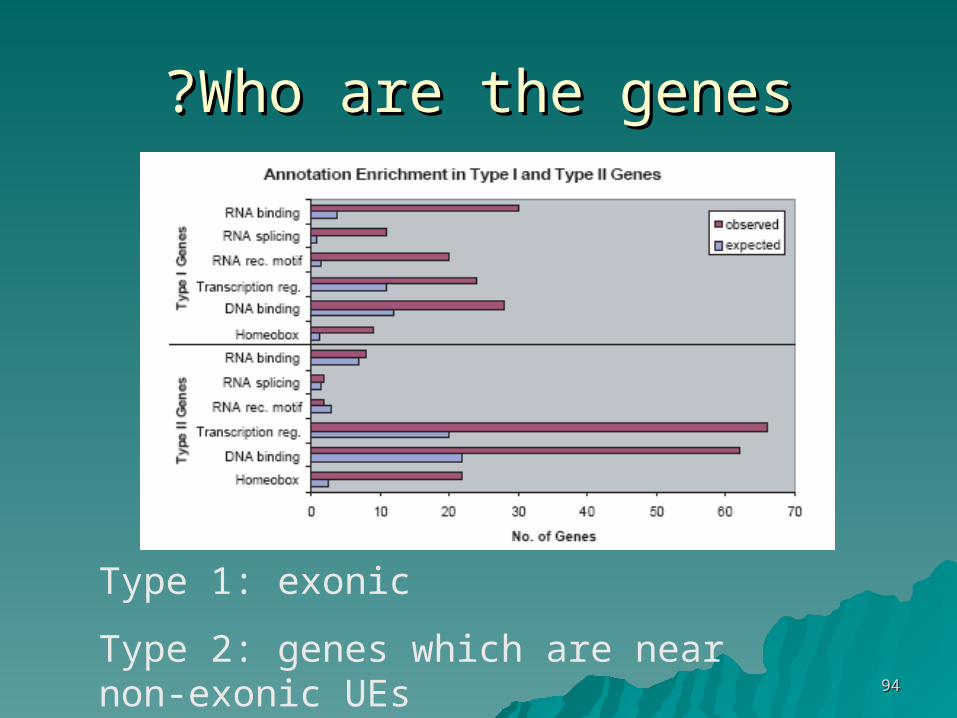
Who are the genesWho are the genes??
Type 1: exonic
Type 2: genes which are near non-exonic UEs

9595
Type 1:Type 1:enrichment for:enrichment for:- RNA binding and splicing regulation- RNA binding and splicing regulation- RRM motif (RNA recognition)- RRM motif (RNA recognition)
Type 2:Type 2:enrichment for:enrichment for:- Transcription regulation, DNA - Transcription regulation, DNA bindingbinding- DNA binding motifs- DNA binding motifs

9696
Intergenic UEsIntergenic UEs
Genes which flank intergenic UEs are Genes which flank intergenic UEs are enriched for early developmental enriched for early developmental genesgenes
Are UEs distal enhancers of these Are UEs distal enhancers of these genes?genes?

9797
Gene enhancerGene enhancer
A short region of DNA, usually quite A short region of DNA, usually quite distant from a gene (due to distant from a gene (due to chromatin complex folding), which chromatin complex folding), which binds an activatorbinds an activator
An activator recruits transcription An activator recruits transcription factors to the genefactors to the gene

9898
Experimental studies of UEsExperimental studies of UEs
Some UEs cluster within regions enriched for genes encoding developmentally important transcription factors
Within these loci, a special pattern of histone methylation (bivalent domains)
Silence the developmental genes when unnecessary
Suggest that the DNA pattern affects the histone methylation
Cell, Vol 125, 315-326, 21 April 2006

9999
Experimental studies of UEsExperimental studies of UEs
Tested 167 UEs (both mouse-human UEs and fish-human UEs) for enhancer activity: cloned before a reporter gene to test their activity
45% functioned as enhancers

100100
A bioinformatic successA bioinformatic success
Ultraconservation can predict highly Ultraconservation can predict highly important function!important function!

101101
BUT …

102102
PLoS Biol. 2007 Sep;5(9):e234
Chose 4 UEs which are near specific genes:
genes which show a specific phenotype when knocked-out
Performed complete deletion of these UEs
… the mice were viable and did not show any different phenotype

103103
ConclusionsConclusions……
Ultraconservation can be indicative Ultraconservation can be indicative of important functionof important function
…… And sometimes not:And sometimes not:
- gene redundancy- gene redundancy- long-range phenotypes- long-range phenotypes- laboratories cannot mimic life- laboratories cannot mimic life