afitch.sites.luc.eduafitch.sites.luc.edu/electrochemistry files/chapter 2...
TRANSCRIPT

CHAPTER 2: INORGANIC ELECTROCHEMISTRY (l)
A. TRANSITION METAL COMPLEX REDOX RATES
I. Introduction.
In this section we consider "simple" electrochemistry of
inorganic compounds. We will concentrate on systems where the
electron transfer event is rapid and uncomplicated by associated
chemical reactions:
Reaction Classification E=electrochemical step C= chemical step
E, [ 2 . 1 ]
but not:
CE [2. 2 ]
A+ + e = A -+ B EC [ 2 . 3 ]
Or: slow
A+ + e = A [ 2 . 4 ]
where E, refers to a reversible electron transfer event and E~,
refers to a quasi-reversible electron transfer event. By confining
our attention to a rapid, reversible, simple E, reaction we hope to
see which system is amenable to analytical applications based on
DP (differential pulse), SW (square wave), and/or CV (cyclic
voltammetric) analysis.
As an example, in Figure 2.1 (2), we show a plot of a
dimensionless (normalized) current function ~ for a linear sweep
experiment:
[ 2 • 5 ]
The different lines on the plot illustrate the effect of relative

----
•••••••a= 0.5
0.4 ~-----,#--;a~:---I-----i-----t----j
-+--+__~e.-_+_----+_ j • 0.3 ~ ~+-__'¥(E)
•0.2 1----J.-/-l.:---,~-_+-+--_+_----+_-_1•0.1 I--~t+---t+-----T~----+-----t-----j
••-128 o 128 257 385
-n(E-E,I2)' mV (25°)
••2.1 variation of quasi-reversible current function, tP (E), for
different values of Q (0.7, 0.5, 0.3, as indicated) and the
following values of A: I, A = 10; II, A = 1; III, A = 0.1;
•IV, ). = 10.
2• Dashed curve is for a reversible reaction.
1jl(E) = ijnFAC *D 'l2(nFjRT) 112 T)1/2
•o o
). = kOjD'I2(nFjRT)'I2T)'/2.
(From Re f. 2.)
•••

magnitude of the electron transfer rate constant (k,) to the time
scale of the experiment established by the scan rate of the linea~
sweep experiment (v).
= k 50.68 -Y,
'it:. v
where the constant 50.68 arises when we assume 0 = 10,5 cm 2Is, an:::
nF/RT is 38.92 V".
Note from Figure 2.1 that a A of 10 is required to achieve the
full reversible linear sweep scan. The advantage of a reversible
system is that one gets the maximuD peak height that is easil~,
related to the bulk solution concentration. In Table 2.1 variou~
values of A and k" as a function of scan rate are shown. Fro:
Table 2.1 we note that we need electron transfer rate constants 0:
the order of 0.05 cmls or more depending upon the scan rate, whic~
controls the time scale of the experinent.
Reversibility and fast electron transfer are related t:
solvent structural changes and to structural changes expected i~
the complex. We predict that systems involving the leas:
reorganization should be the fastest. In order to have aver',
basic understanding of structural changes in inorganic complexe~
we turn our attention to crystal field theory.
II. Crystal Field theory
, The ease of electron transfer in metal complexes proceeds,
one part, from the spreading of charge over a large volume. Thi~
results in a low charge density which requires little solven:
reorganization around the complex. If, at the same time, littl~

I I I I I I TABLE 2.1: Values of >. as a function of T) and k o •
I A ko cm/s
v(V/s): 0.05 0.5 .2- 50
1.0 4.4x10·2 1.39xl0' 4.4Xl0·' 1. 39
I 1 4.4XI0·3 1.29xIO< 4.4X10 2 1.39xIO·'
0.1 4.4xl0·4 1.29xIO" 4.4xl0·3 1.39xl0·2
~

change in internal bond structure occurs (little lability causir:
bond length changes and/or ligand replacement), the electrc
transfer reaction should be rapid and reversible.
Crystal field theory presumes that the main interactic
between the metal ion and the ligands is electrostatic in nature
Assuming an octahedral complex (6 coordinate), incoming 1 igan::: ~
approach along the x, y and z axis (Figure 2.2), perturbing the c
y2 and the d z2 (Figure 2.3) ra is ing them in energy (Figure 2.4) (J,
Orbitals lying off axis (d ' d ' dyJ are less perturbed in ener::xy xz
and are lowered, resul ting in splitting of the 5 degenerate
orbi tals into 3 degenerate t2~ (d,y' d,z' d\,z) orbitals and
The total energy splitting between orbitals is arbitrarily S'C°
at 10Dq. The absolute value of this energy difference is relat~
to the charge transfer bands observed'spectrochemically in the 4C:
700 nm region. By noting that the sum of the e~ orbitals with t~
lowered t 2g orbitals must equal zero, simple arithmetic thus she.
that the energy is divided between the e g orbitals and the :
orbitals as weighted values of +6Dq and -4Dq, respectively. L,,'
x = the t 2g orbi ta 1 energy and y the e] orb i tal energy:
2x = 3y 2x = 30 - 3x
x + = 10 30 5xY
Y = 10 -x x = 6 so
3y = 30 - 3x Y = 4
As an example, let us examine chromium. Cr has an electro~.
. . f d 5 , 0conf1.gurat1.on 0 3 4s 4p . Notice that the electronic conf igurat:..:

y )' ..r:y
x
: z
//
x x
y
.v xz vz
2.2 Complete set of d orbitals in an octahedral field. The e g
orbitals are shaded and the t2~ orbitals are unshaded. The
torus of the d:2 orbital has been omitted for clarity. (From
Re f. 3.)
- "......!--/? I\j~3 I 2; ....{. 0

I .
t•
•I
I \
2.3 Spatial arrangement of the five d orbitals. (From Ref. 3.)
fie, j

_~__eg
1 1 /
/ /
6Dq/ /
IODq=Ll----I
/
---\-"---\ \ 4Dq \\ 1 \----(2g
2.4 Splitting of the degeneracy of the five d orbitals by an
octahedral ligand field. (From Ref. 3.)

1S written to imply that the 4s orbitals are higher in energy than
the 3d orbitals, therefore electrons removed from Cr come first
from the 4s orbital. The orbital configuration is obtained by
placing the d electrons in the lowest possible orbitals shown in
Figure 2.5. Two examples are diagrammed. That in which the
incoming ligand greatly perturbs the d orbitals (high field ligand)
and that which only weakly perturbs the d orbitals (low field
ligand) .
The total energy of the two states is the crystal fielc
stabilization energy, CFSE. Note that for the low field case, the
splitting between the t 29 and e g orbital levels is less and it is
energetically more favorable to fill the upper e g orbital for the
d 4 complex than to pair up the electrons in the lower t 29 orbital.
The low field complex for Cr2• (d J
) has higher spin than the hig:-.
field (more unpaired electrons).
The energy for the high ligand field Cr 2• complex 1S compute::
from four t 29 electrons minus the energy, P, required to pair u:=
electrons wi thin the same orbit. The energ ies of the varies
electronic configurations can be calculated as shown in Table 2.2.
Similar analysis can be made of other metal ions. We shal~
/work through C0 2+ ]+ (F igure 2. 6) and Fe2
>J+ (Figure 2. 7). Co has tr.·
electronic configuration of 3d 74s", so Co2• is 3d74So and Co]· i::
3d64so. Fe has the electronic configuration of 3d64s 2, so Fe2
• ai.::
Fe]· have the configurations of 3d" and 3d5 • The correspondii.::
electronic configurations are shown in Table 2.3.

- --- --
- --
....
I I I I I
I I FIGURE 2.5
HIGH FIELD LIGAND LOW FIELD LIGAND
I i
i
t6Dq i
i
J.4Dq ~ t- i -- - -
- i -- - t - ~ t J. i
2.5 Diagram of Cr2+ and Cr3+ d orbital spl i tting of in the presence
of a high field and low field 1 igand. The dotted 1 ine
represents the degenerate energy level of the unperturbed
ligands.
I

it
TABLE 2.2
Crystal Field Stabilization Energies in Dq units
a) high field Cr2+ (d 4 ) 4 (-4 Dq) + o (+6Dq) + IP = -16Dq+P
cr)+b) high field (d j) ] (-4Dq) + o (+6Dq) + OP -12Dq
c) low field cr2+ (d 4) ] (-4 Dq) + 1 (+6Dq) + OP = -6Dq
d) low field CrJ + (d j ) ] (-4 Dq) + O(+6Dq) + OP -12Dq
/
,> L·

-- .......
I I I , I I ~ FIGURE 2.6 , HIGH FIELD LIGAND LOW FIELD LIGAND
~ I
t- -
t ~
t ~
t ~
t i
i
~
~
~
t t -- --
t i-- -- ---- t --------- i-- --
t ~ i - - t ~ i ~
I
2.6 Diagram of Co2, and CoJ
• d orbital splitting in the presence of
a high and low field ligand. Dotted line marks the ener~y
level of the five degene=ate orbitals in the absence of
perturbation.

FIGURE 2.7
HIGH FIELD LIGAND LOW FIELD LIGAND
i -- i-- i-- i--
---- i --------- i-- -- i -- i-- i ~ i
i ~ i- - i ~ i ~
i ~ i ~
2.7 Diagram of Fe2+ and Fe). d orbital spl i tting in the presence 0:
a high and low field ligand. Dotted 1 ine marks the energ~·
level of the five degenerate orbitals in the absence 0:
perturbation.
./ --~I~ ,.
L
30

I I
III. Ease of oxidation/Reduction from CFSE theory
I t-ie may make some inferences from Table 2.3. First, let's
I
::::nsider the reduction/oxidation of chromium (CrN3+). In the
I ~~esence of a high field ligand there is little major change in the
electronic configuration, so, in the absence of other
I considerations, we might expect that there is little structural
change in the complex, thus facilitating rapid electron transfer
reactions. This, of course, presumes that the CFSE energy shown
I ~n Table 2.3 is of a similar order of magnitude for. the divalent
and trivalent complexes (Dq similar). In fact, the Dq values are
I I always quite a lot larger for the trivalent complex due to
electrostatic considerations. This suggests that trivalent
complexes should be more stable to sUbstitution reactions as
I compared to divalent complexes. Figure 2.8 (4) confirms these
I
expectations by showing that the on/off rate of inner sphere water
I molecules is much slower for trivalent complexes than for divalent
complexes. Thus, reduction of a trivalent complex to a divalent
I complex should always be checked for lability and attack of the
reduced complex. Such attack would convert our simple E reaction
to an EC reaction (see equation 2.3).
I This analysis holds up even more when looking at a complex in
which the ligand produces a weak interacting field with Cr (see
I I Table 2.3). Here we see that a change in orbital configuration
accompanies a change in the redox state. Similar changes in the
orbital state are noted for the oxidation/reduction of Co high
I field complexes, as compared to Fe or Ru high field complexes.
I -1.7
./ I
I

TABLE 2.3
Table of Electronic Configurations and CFSE
For Several Metal Ions
electronic #d e High Field Low Field
Cr configuration
Jd5 4s'4p(J Electronic Config Energy Electronic Config ~
cr2' Jd4 4So Jpo d4 4t 2g -lGDq+P ) 1t g e g -6Dq
'-"-.)
"1-' Cr]' 3d)4SoJpO d) l t,u -120q )
t,u -12Dq
Co Jd 74S'
Cot. 3d74S° d l b 1t 2tl e g -180q+JP 5 2
t'Q ell -BD:}~2P
Co]' 3do4S° db t h 2g -24Dq+JP 4 ,
t 29 e g -4cqtP
Fe 3do4 S°
Fe" 3do4 S° d6 °t 29 -240q+JP 4 2t 29 e g -4D:J+P
Fe)' 3d5 4so d5 t 5 y -200q+2P ) 2t 2g e g OOq
Ru 55'4d7
Ru2 ' 5so4d6 dO 6t
29
R,,/' 5s0 4d5 d5 5t 2g

\_,,)
0~
'--
Na+ K+Cs+ u+ ~!' ~Rb~
Be2 + Mg2+ Ca2+ s,-2 + Ba2 +
i" f
c~+R 3+ Fe3 + 3+ Ti3 +
3 Yb3+_ 03+- Gd3+ A1 3+ a In + "I~ ,-, u /V3+
Ru2+ V2 + Ni2+ Co2+ Fe2+ Cu2 + c,-2 +
" 1 Mn 2 +~ ,
Pt2+ Pd2 + Zn2+ Cd2 + Hi+
10- 6 10- 4 10- 2 100 102 104 106 loS 1010
---... -.:\
1'\.[\ 2.8 Characteristic rate constants (s") for sUbstitution of inner~
sphere water molecules on various metal ions. (From Ref. 4.),:./\.

Recall that the e" orbital is most directly in the path of the
oncoming ligands and so we might expect large differences in bond
d4lengths in going from the to the d 3 complexes of Cr when in the
low field system. Consequently, we might also expect that the rate
of electron transfer should be low. Marcus theory (5, 6, 7, 8)
predicts that the rate of a self exchange reaction, k,,:
k" A + A = A + A" [ 2 • 7 J
should relate to the rate of electron transfer at an electrode,
heterogeneous electron transfer, k o :
[2.8:
via the relationship:
k" == Zel ( ~,,_ ) '/2
Zsoin
where Zel and Z'OII1 are the collisional frequency factors generall::o
taken to be 103 to 104 cm/s and 10~' M·'s·'. The frequency factors
tell you how many times the reactants collide at the electrodE
surface or together in solution before an electron transfer even:
occurs. Zsoln is estimated from the thermal velocity of the react in:::
molecules:
ZSOln - (kT/27Tm) 1/2 [2.1C
where k is the gas constant, T is the temperature, and m is t~~
effective mass of molecules (9). From equation [2.9J we note tha:
the rate of electron transfer will mirror the self exchans::
constant. Table 2.4 (4) and Appendix 8.1 show some data for rate~
of self exchange for several metal complexes. Note that the sel:

I I I I I I Some Outer-Sphere Electron-Exchange Reactions , Rate Difference in M-L
Reacting pair Electron configuration (L mol-I S-I at 2SOC) bond lengths (A) , I
[Fe(bipy)3]2 + 13 +
[Mn(CN)6]~-/3
[Mo(CN)s]~-/3
[W(CN)S]"-/3[IrCI6P-/2[Os(bipy) ]2+/3+
[Fe(CN)6]~-/3
[Ru(en)3]H /3.+
[Ru(NH3)6]2+f3+ [Ru(H20)6]2+/3+
[Fe(H20)6]2+f3+
[MnO~J2-/I
[Coen3J2+/3+ }
[CO(NH3)6]2+f3+
[CO(~O~)3]4-/3
" " t~g/ t~g
t~g/tig
t~g/ tig .
t~g/ tig
tt/dg
t~g / t~g
t 4 2/ 3 22geg t 2geg
"
-lOS 4 x Hr' 4 x 1()3
20C 4 >1()3
-10- 4
0.00 ± 0.01
Very small
0.04 ± 0.01 0.09 ± 0.02 0.14 ± 0.02
0.18 :±: 0.02
aNot octahedral, but the change in electronic configuration occurs in a nonbonding orbital.

exchange rate of the cobalt complexes is a good deal slower tha~
that of the Fe and Ru complexes which involve only a t~ electron~
(orbitals out of the path of the incoming ligand).
From Table 2.4 we can also observe the effect of moving do~~
the periodic table. Note that even though Fe and Ru are in tt~
same group, their electronic configurations In the presence of ~
low field ligand (water) are different. Fe behaves as a low fiel:
complex with electrons in the e g orbitals while Ru behaves as a hig.
field complex with electrons conf ined to the t?_ orbitals. This':'" ,~
because size and electron cloud density effects change as one mOVE~
down the periodic chart. The l~lger d orbitals of Ru are mo~~
greatly perturbed by the incoming ligand and greater stabilizatic~
results. The magnitude of Dq increases down the periodic char~
Complexes of the 2nd and 3rd row transition series are almos~
exclusively low spin (high field) in nature (3). Because Fe hc.'
a low field configuration with electron density in the e g orbitals
the bond length changes in the oxidation/reduction reaction a~·:
greater and, therefore, the self exchange rates are much slo~E~
than for the aquo complex of ruthenium.
IV. Summary - reversibility in Metal Complexes
We look for reversible electron transfer events in met:::.
complexes lower in the periodic chart and in which t~ electrons a=,
removed exclusively (Fe, Os, Ru).
B. TRANSITION METAL REDOX POTENTIALS
Having looked at the complexes and gained some rough feel fc~
their reversibility, can we also get a feel for the absolu:·

~ I I
e~ergetics required to cause the electron transfer to occur? That
:s, can we get a feel for the redox potentials? The answer is yes.
Let's consider some redox potentials for CoJ~'. We can first
, ,
~rite the Nernst equation for simple reduction of trivalent Co to
divalent Co:
E = EO - RT ln [Co2,] [2.11] nF [Coo.]
Next we note the complex formation reactions for both the di- and
tri-valent complexes:
CoJ + + 6L := CoL/· Kill
:= r <;:0 L,~~""l [2.12] [Co'""] [Lf
C0 2+ + 6L := coLt 1\1 = [COL)~_ [2.13]
[Co""] [L]"
-w'here and K I are the formation constants reactions [2.12] andKill
[2.13]. By combining equations [2.11-2.13] and separating out
constants:
E = { EO - RT ln K II } - RT ln ~~ [2.14] nF ~I [ C0 4'·]
The term in the {} on the right hand side of equation [2,14] is the
formal potential, EO', for the Co complex. Note that when the
trivalent species is more strongly complexed (stabilized) the
potential will shift negative from the formal potential of the
simple cation. These trends are observed for the cobalt metal
complexes as shown in Table 2.5. Dq is a measure of the strength
of the ligand in creating the ligand field, as is If', a ranking
of ligand strength. In general ligands follow the order of:

Formal Potential,
for Co 3' Complexes
Reaction
Co 3+ + e = Co 2,
Co (ox) / +e = Co (ox) 34
Co (phen) /+ + e = Co (phen) 32+
Co(bpy)/+ + e = Co(bpy)/+
3 2Co(NH3)6 + + e = Co(NH3)6 +
co(en)/+ + e = Co(en)32+
3Co (CN) / + e = Co (CN) 5
TABLE 2.5
CFSE, and f, ligand strength factors
E C (vs NHE) D9(3+) IkJ-mol- 1 .f
1.808
0.57 .99
0.37 to 0.42 1. 3~
0.31 to 0.37 1. 3 =
0.1 278 1. 2::
-0.26 278 1. 2::
-0.83 401 1. 7

~here ox is oxalate, en is ethylenedia~ine, bpy is bipyridine, phen
is phenanthroline. Similar trends can be cODpiled from Appendix
B.2.
From the EO' values for Co we see that in the absence of any
ligand and for the oxalate complex, the divalent state is preferred
over the trivalent complex. This might be expected from the
relative second and third ionization potentials of cobalt which
increase indicating the greater difficulty in removing a second or
third electron from the atom. As the ligand strength increases to
ammonia, and cyano complexes, the trivalent complex can be
stabilized in solution (oxidation potentials shift negative). The
chelate complexes of bipyridine and phenanthroline do not follow
the sequence perfectly due to the unique structural effects of the
chelating ligand.
The reason the trivalent complex can be favored is related to
the large energy gain from the complex in the trivalent state (from
-18Dq to -24Dq for the high field complexes) .
C. ANALYTICAL APPLICATIONS: STRIPPING ANALYSIS
The aquated complexes of the metal ions lie between high and
low field complexes. That is, we can not assume that both the
divalent and trivalent complex of the aquated specie should be
particularly well stabilized in a similar electronic configuration
involving only a transfer of a t;~ electron. Thus we might infer
•••••••
••, •
that the reversible reduction of the aquated metal ions would be
poor and not a good candidate for electrochemistry as an analytical
method. Tables 2.6 (3) and 2.7 (6) show the data for aquo
()1
.

TABLE 2.6:
Electron configuration and CFSE for Aqua Complexes
Of Some Metal Ions
Ion Electrons 10Dg/em.,
Cr3 +
3 1760t 2Q
Cr2 • t 2g3 e g
, 1400
Mn 3 + t 2g3 e g
, 2100
Mn2 + t 2g
3e g2 750
Fe 3 + t 2g
3e g2 1400
Fe 2 +
4 2 1000t 2g e g
Co 3 +
6t 2g
Co 2 + t 2g
5e g2 1000

I I
. .Calculated values of .1G:~ Ao and AI for some Inorgamc self-exchange reactions In aqueous solution t
L1G*o A. IJI A.I~ 1e oReduced form r .'A r/A kcal rno\-I kcal mol-I kcal mol-I .dG*· d L1G~bSI' cole
Co(HP)~" 3.56 3.40 3.6 26.3 48.4 22.3 14.3
Fe(HP)~" 3.59 3.43 3.6 26.1 48.4 22.2 14.2
Mn(Hp)~.j. 3.66 3.46 3.6 25.7 75.2 28.8 19.8
Cr(Hp)~~ 3.58 3.40 3.6 26.2 60.4 25.3 ~21.4
V(HP)~' 3.56 3.41 3.7 26.2 12.0 13.3 17.6
Ti(/lP)~~ 3.56 3.45 3.6 26.1 22.8 15.8 17.7
(CO'IIW 11 0 4 0 r 5.0 5.0 12.7 18.3 11.0 20.0 16.7
/Ru.,O(CH)COOMpY)J l~ 7.0 7.0 0 9.0 I a 2.3 4.0
'-I I

complexes.
From Table 2.6 again note the consistently greater stability
(large 10Dq values) of the trivalent complex. More importantly,
note that only the reduction of Fe"" to Fe2' involves solely t;o;
electrons, thus we might predict that the remaining aquated
complexes would be sluggish at an electrode and not amenable to the
assumption of reversibility at the electrode surface, hindering the
analysis of currents in sweep methods.
This is true. We can beat this problem by taking another
tack. Many metal ions form Hg analgams \·;hen reduced to their
metallic state. The amalgam formation depends upon the metallic
solubility of the compound in liquid Hg. Since Hg is large and
polarizable we would expect similarly large and polarizable metals
to be soluble within Hg. Table 2.8 shows the solubility of various
metals in mercury (10). Those metals grouped on the left-hand side
have larger solubilities than those on the right-hand side. In
general, those with larger solubilities fall to the right of the
periodic chart and almost all are in the 2nd and Jrd row of
transition metal series. Nearly all those ions exhibit ing low
solubility in Hg, are first row transition metals, which will be
smaller and less polarizable.
Metal ions which can be determined analytically by
t. 3· 3+ ·2.preconcentration in Hg are: Bi J. , C U , Ga , Ge 4+ , I n , N 1 ,
The metals are determined via a method
termed anodic stripping voltammetry (ASV). A potential is applied
to the. electrode surface which reduces the metals resul ting in

I I I I I I I Metal
In
Th~ I
Cd
Zn
Sn
Pb
Bi
TABLE 2.8
SolUbility of Metals in Mercury
Solubility (wt%)
68.3
42.4
5
5.6
1.3
1.2
1.2
Metal
Cu
Mn
Sb
Ni
Co
Solubility (wt%)
8xlO- J
6.6xlO- J
3.8xlO·4
2.1X1O- J
3xlO·4

metal-mercury amalgam formation. This process concentrates the
metal ions in the small mercury drop (Figure 2.\.n ( 2) • After a
loading period, the electrode potential is swept positive, causing
the oxidation of the metals and their removal from the mercury
drop. The resulting anodic stripping current is large due to the
preceding concentration period (Figure 2.S; (10)) Listed in Table
2.9 are some typical detection limits for Anodic Stripping
Voltammetry (ASV) utilizing either Differential Pulse or Linear
Sweep techniques (see Introduction) (10) and also typical detection
1 imits (11) for spectroscopic methods 0 f anal ys is, where AAS is
flame atomic absorption spectrosccpic, GFAAS is graphite furnace
atomic absorption specroscopy and ICPAES is inductively coupled
plasma atomic emission spectroscopy.
Note that GFAA has the lowest detection limits, but is useful
for analysis of single components only. ICPAES is a multicomponen~
spectroscopic technique but does not have the detection limits of
ASV with DP detection. ASV can analyze for several components
simul taneously (Figure 2 ./~ .
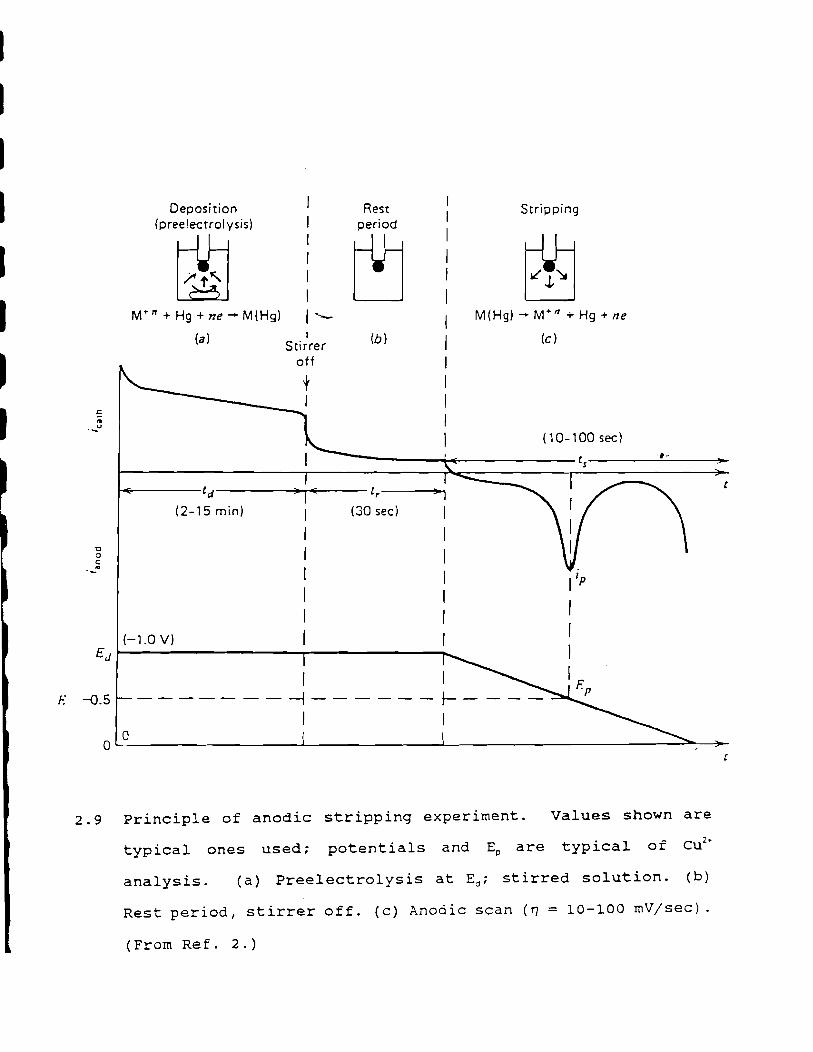
Stripping
M(Hg) -. M+ 11 + Hg + ne
(e)
(10-100 sec)
------k------- 's------=·t~---_;~_
t
2.9 Principle of anodic stripping experiment. Values shown are
typical ones used; potentials and Ep are typical of cu2
•
analysis. (a) Preelectrolysis at E,,; stirred solution. (b)
Rest period, stirrer off. (c) Anodic scan (ry = 10-100 mV/sec).
(From Re f. 2.)

Table 2.9 Detection Limits (DL) in Anodic Stripping Voltamrnetry (ASV)
DP = Differential Pulse, LS = Linear Swee~
and Spectroscopy AAS = Atomic Absorption Spectroscopy
GFAA = Graphite Furnace Atomic Absorption Spect~:
ICPAES = Inductively Coupled Plasma Atomic Emission Spe:'
Anodic ion ng/ml
DP
Bi
Cd 0.005
Cu 0.005
Ga 0.4
In 0.1
Pb 0.01
Rh
Sn 2
Tl 0.01
Zn 0.04
stripping (ppb)
LS
.01
0.01
0.01
0.02
10
0.04
0.04
Spectroscopy
AAS GFAAS ICPAES
1 10~ 2
2 0.1
10 2
20 0.1 30
2 5x10-5 2

I I I I
<! :J.-to-
Z lJJ c:: a:: :::> u
Zn
Cd
+0.25 0 -0.25 -0.50 -0.75 -1.00 -1.25
POTENTIAL (V vs Ag/AgCU
2.10 Current-sampled polarogram (top) and anodic stripping
vol tammogram (bottom) of 2.5 ppm cu;', Zn 2., and 5 ppm Pb2., Cd2•
in 0.1 M sodium acetate.
rs~' Ie

CHAPTER 2: PROBLEMS
2.1 a) Work out the electron configuration for OS2' and OS3' i;:
the presence of CN- and in the presence of Cl-.
B) Would you expect the sel f exchange rate for OS2o(3. to be
rapid as a CN or as a Cl complex? Justify your answer.
2.2 a) Work out the electron con f igura t ion for Cu 2' and Cu" ir.
the presence of CN- and in the presence of Cl-.
b) Would you expect the self exchange rate for CU 2•
11 - to be
rapid as a CN or as a Cl co~plex? Justify your answer.
2.3 Would you expect a peak splitting .tiL of 59 mV in cyclic
voltammetry for RU(en)/- at a scan rate of 5,000, 500, 50, c:::
• .~ 25 Vis? Assume 0 IS 5x10· cm /s. Assume no iR error.
2.4 Buttrey and Anson (12) ion exchanged Co(bpy)/' into a Nafio:
ion exchange polymer modifying a Pt electrode. As they varie=
the scan rate they found differential changes in the pea:-:
heights for the 3+/2+ vs the 2+/1+ peaks. The current of one
of the redox couples was dependent upon physical diffusion 0:
the complex within the Nafion to the electrode surface. The
second redox couple was found to be dependent upon the rate
of self-exchange of electrons between couples immobilized i:
the Nafion. Which couple was which?

I I I
2.5 You are performing cycl ic vol tammetry of Cr (bpy) /. and
Ru (bpy) t in an aqueous med ia with a small amount
phenanthroline present at a fairly slow scan rate (5 mV/s).
Sketch the cyclic voltammograms that you might expect to see
for the two different couples. Justify your sketches.
2.6 a) Jorgensen (13) has estimated 10Dq values from the
formula:
10Dq = f"gano X g,en'
If Co(II) and Co(III) have g values of 9 and 18.2,
respectively, compute the 10Dq values for Co(phen)6;' and
Co(phen)6 J" respectively.
b) Using these values make some predictions as to the EO
val ue of the Co (phen) 53.;;. coupl e as compared to the EO val ue of
the straight C03~' reduction at +1.8 V vs NHE.
2.7 (From Bard and Faulkner (2).)
a) An analysis for lead at the HMDE gives rise to a peak
current of 1 ~A under conditions in which the deposition time
is held constant at 5 min and s'tleep rate is 5 OmVIs. What
currents would be observed for sweep rates of 25 and 100 mV/s?
You may consider the peak current in the linear sweep to be
roughly described by equation (14) in Chap. 1.
b) The same solution gives a peak current of 25~A at a 100
A thick mercury film electrode (MFE) on glassy carbon when the
deposition time is 1 min, the electrode rotation rate is 2000

rpm, and the sweep rate is 50mV/s. What currents would t"
observed for sweep rates of 25 and 100 mV/s under otherwis
unchanged conditions? (The peak current in a MFE is (10):
where CR is the concentration of the metal in the MFE .
mol/cm3 , e is the thickness of the HME.
c) Why does the current follow a direct v dependence for t~"
MFE in (b), but a v 1a dependence for the HMDE in (a)? (Hint.
Refer to technique oriented textbooks like Bard and Faulkne~
(2), Kissinger and Heineman (10) or Reiger (14).
d) Compare this situation to the one observed for
deposition time of 1 min, a sHeep rate of 50mV/s and
rotation rate of 4000 rpm? (C~ is related to the depositic
time and the rotation rate of the electrode. The rotatic
rate sets the diffusion layer, and hence current, for moveme~~
of the ion from the bulk solution to the mercury electrode
The limiting current of a rotating disk electrode is (14):
= 0 62 nFAC 0 2/3 -1/6 1;2l' L· Oulk Ox 1/ W
where, in this case, 1/ is the kinematic viscosi ty of tr.-::
solution and w is the rotation rate of the electrode in rad!~
(rad=27THz) . A typical kinematic viscosity is lO~ m~s. T:--.
amount of charge deposited is Q = nFN where N is the numbe:
of moles deposited, and q is the integrated current fidt.)
e) Suppose the film thickness were varied by the use c:
different concentrations of the mercuric ion in the analyte
What effect would one see on the peak current under otherwis"
2~_

I I ~
constant conditions?
2.8 Films of Pb02 can be deposited oxidatively on Sn02 ( 15) .
Suggest an analytical determination of Pb based on this
phenomena.
2.9 Your electrode area is 0.05 cm< and your bulk solution
concentration of co(en)/· 5 mM in 0.01 M NaCl (K. :::: 11.85xl0··
n·'cm·'). Assuming that the solution res istance measured at a
disk electrode is p/4a can you attribute peak splitting in
cyclic voltammetry at 500 mV/s to slow electron transfer
kinetics?
(Hint: you will need to compute the extent of iR error at the
peak current, you may wish to refer to Bard and Faulkner (2)
or Reiger (14) for more detail on iR error.)

LITERATURE CITED
1. For an early review see: H. Taube, Electron Transfe~
Reactions of Complex Ions in Solution, Academic Press, 1970.
2. Bard, A. J. and Faulkner, L. R. 1980, Electrochemical Methods,
Wiley and Sons, p. 225.
3. Huheey, J. E. 1978, Inorg. Chem., Chap. 9.
4. Cotton, F. A. and Wilkinson, G. 1988, Advanced Inorganic:
Chemistry, 5th Ed., Chap. 29.
5. Marcus, R. A. Electrochim. Acta, 1968, 13, 995.
6. Eberson, L., Electron-Transfer Reactions in Organic Chemistr;
7. Hush, N. S., Electrochim. Acta., 1968, 113, 13, 1005.
8. Kojima and A. J. Bard, J. Am. Chern. Soc., 1985, 97, 6317.
9. Marcus, R. A., J. Chem. Phys., 1965, 28: 962.
10. Heineman, W. R., Mark, H. B., Jr., J. A. Wise, and D. A.
Roston, in Laboratory Techniques in Electroanalytica~
Chemistry, 1984, Marcel Dekker.
11. Skoog, D.A., Principles of Instrumental Analysis, 3rd Ed.,
1985, Saunders.

12. Buttry,
685.
D. A. and Anson, F. C. J. Amer. Chern. Soc., 1983, 105,
13. Jorgensen, C. K., Oxidation
1969, Springer, N.Y.
Numbers and Oxidation States,
14. Rieger, P.H., Electrochemistry, 1987, Prentice Hall.
15. Laitinen,
1352.
H. A. and Watkins, N. H. Anal. Chern., 1975, 47,
-" ,-,"