Çukurova Ünİversİtesİ fen bİlİmlerİ …erkan tetİk Çukurova Ünİversİtesİ fen...
TRANSCRIPT

ÇUKUROVA ÜNİVERSİTESİ FEN BİLİMLERİ ENSTİTÜSÜ
DOKTORA TEZİ
Erkan TETİK
KARBON NANOTÜPLERİN FİZİKSEL ÖZELLİKLERİ ÜZERİNE KATKILAMANIN ETKİLERİNİN İNCELENMESİ
FİZİK ANABİLİM DALI
ADANA, 2012

ÇUKUROVA ÜNİVERSİTESİ FEN BİLİMLERİ ENSTİTÜSÜ
KARBON NANOTÜPLERİN FİZİKSEL ÖZELLİKLERİ ÜZERİNE
KATKILAMANIN ETKİLERİNİN İNCELENMESİ
Erkan TETİK
DOKTORA TEZİ
FİZİK ANABİLİM DALI Bu Tez 20/01/2012 Tarihinde Aşağıdaki Jüri Üyeleri Tarafından Oybirliği/Oyçokluğu ile Kabul Edilmiştir. ……………….................... ………………………….. ………………………….. Doç.Dr. Faruk KARADAĞ Prof.Dr.Sadullah SAKALLIOĞLU Yrd.Doç.Dr. M. Zeki KURT DANIŞMAN ÜYE ÜYE ...………………............... ……………………….. Prof. Dr. Emirullah MEHMETOV Yrd.Doç. Dr. Muharrem KARAASLAN ÜYE ÜYE Bu Tez Enstitümüz Fizik Anabilim Dalında hazırlanmıştır. Kod No:
Prof. Dr. İlhami YEĞİNGİL Enstitü Müdürü
Bu Çalışma Ç. Ü. Araştırma Projeleri Birimi Tarafından Desteklenmiştir. Proje No: FEF2010D28 Not: Bu tezde kullanılan özgün ve başka kaynaktan yapılan bildirişlerin, çizelge ve fotoğrafların
kaynak gösterilmeden kullanımı, 5846 sayılı Fikir ve Sanat Eserleri Kanunundaki hükümlere tabidir.

I
ÖZ
DOKTORA TEZİ
KARBON NANOTÜPLERİN FİZİKSEL ÖZELLİKLERİ ÜZERİNE KATKILAMANIN ETKİLERİNİN İNCELENMESİ
Erkan TETİK
ÇUKUROVA ÜNİVERSİTESİ FEN BİLİMLERİ ENSTİTÜSÜ
FİZİK ANABİLİM DALI
Danışman :Doç. Dr. Faruk KARADAĞ İkinci Danışman :Doç. Dr. Murat DURANDURDU Yıl: 2012, Sayfa: 169 Jüri :Doç. Dr. Faruk KARADAĞ :Prof. Dr. Sadullah SAKALLIOĞLU :Yrd. Doç. Dr. M. Zeki KURT :Prof. Dr. Emirullah MEHMETOV :Yrd. Doç. Dr. Muharrem KARAASLAN
Bu tez çalışmasının içeriğini üç aşamada hazırladık. Birinci aşamada, tek duvarlı karbon nanotüplerin geometrik yapılarını ve nasıl elde edildiği inceledik. İkinci aşamada, yoğunluk fonksiyoneli teorisi (DFT) ve ab-initio pseudo-potansiyel yöntemini kullanarak, geometrik yapılarına göre seçtiğimiz bazı grafen ve nanotüplerin elektronik bant yapılarını, toplam durum yoğunluklarını ve elektron yoğunluklarını inceledik. Son aşamada ise, seçtiğimiz bir nanotüp üzerine katkılamanın etkileri gözledik.
Çalışmada kullandığımız grafen ve nanotüpler zigzag (6, 0), zigzag (7, 0) chiral (6, 2), chiral (6, 3) ve armchair (7, 7) olarak sıralanmaktadır. İlk olarak bu nanotüpleri elde edebileceğimiz bir program hazırladık. İkinci aşamda grafen ve nanotüpün elektronik özelliklerini elde edip, grafenden nanotüpe döndürmenin etkilerini inceledik. Yaptığımız hesaplamalarda zigzag (6, 0) nanotüpün metalik özellik gösterdiğini bulduk. Chiral vektörünü değiştirerek elde ettiğimiz zigzag (7, 0) nanotüpün yarıiletken özellik gösteren bir malzeme olarak bulduk. Yarıiletken yasak bant aralığını 0,5022 eV olarak elde ettik. Armchair (7, 7) nanotüpün metalik özellik gösteren bir nanotüp olarak elde ettik. Chiral (6, 2) nanotüpün 0,8291 eV yasak bant aralığına sahip bir yarıiletken olduğunu bulduk. Chiral (6, 3) nanotüpü düşük bant aralığına sahip bir sankimetalik (quasimetallic) nanotüp olarak elde ettik. Son aşamada bor ve berilyum atomunun nanotüplerin iletkenlik özelliğini arttırdığını, azot, oksijen ve klor katkılanan nanotüplerin iletkenlik özelliğinin azaldığını ve silisyum katkılanan nanotülerin iletkenlik özelliğinin değişmediğini bulduk.
Anahtar Kelimeler: Karbon Nanotüpler, DFT, Elektronik Özellikler, Elektron
Yoğunluğu, Katkılama

II
ABSTRACT
PhD THESIS
INVESTIGATION OF DOPING EFFECTS ON PHYSICAL PROPERTIES OF CARBON NANOTUBES
Erkan TETİK
ÇUKUROVA UNIVERSITY
INSTITUTE OF NATURAL AND APPLIED SCIENCES DEPARMANT OF PHYSICS
Supervisor :Assoc. Prof. Dr. Faruk KARADAĞ Second Supervisor :Assoc .Prof. Dr. Murat DURANDURDU Yıl: 2012, Sayfa: 169 Jury :Assoc. Prof. Dr. Faruk KARADAĞ :Prof. Dr. Sadullah SAKALLIOĞLU :Asst. Prof. Dr. M. Zeki KURT :Prof. Dr. Emirullah MEHMETOV :Asst. Prof. Dr. Muharrem KARAASLAN
We prepared the contents of this thesis in three stages. The first stage, we
examined the single-walled carbon nanotubes geometric structures and how they were obtained. The second stage, we examined the electronic band structures, total density of states and electron density of some the graphene and nanotubes which we selected according to the geometric structures, using density functional theory (DFT) and ab-initio pseudo-potential method. In the last stage, we observed the doping effects on a nanotube which we selected.
We used the zigzag (6, 0), zigzag (7, 0) chiral (6, 2), chiral (6, 3) and armchair (7, 7) graphene and carbon nanotubes. The first stage, we prepared a program which we will obtain this nanotubes. We obtained the elektronic properties of this graphene and carbon nanotubes and examined the effect of rolling from graphene to nanotubes. In our calculations, we found that zigzag (6, 0) nanotube show matallic behavior. We changed the chiral vector of zigzag (6, 0) and obtained the zigzag (7, 0) nanotube which shows simiconductor behavior and have energy gap and has Eg = 0,5022 eV band gap. We obtained the armchair (7, 7) carbon nanotube which shows matallic behavior. We found that chiral (6, 2) carbon nanotube is a semiconductor and has Eg = 0,8291 eV band gap. We obtained a low band gap for chiral (6, 3) carbon nanotube which is a quasimetallic carbon nanotube. Finally, we found that the bor and beryllium increase the conductivity property of carbon nanotubes. The nitrogen, oxygen and chlorine decrease the conductivity property of carbon nanotubes. The silicon is not changed the conductivity property of nanotube. Anahtar Kelimeler: Carbon Nanotubes, DFT, Electronic Properties, Charge Density, Doping

III
TEŞEKKÜR
Doktora tezimin yönetiminde ve oluşumunda aynı zamanda çalışmalarım
sırasında karşılaştığım sorunların çözümünde her türlü desteğini esirgemeyen, değerli
hocam Doç Dr. Faruk KARADAĞ’a sonsuz saygı ve teşekkürlerimi sunarım. Ayrıca
çalışmalarımdaki desteklerinden dolayı Fizik Bölümü'ndeki tüm öğretim üyelerine
teşekkür ederim.
Tez çalışmam sırasında desteğini ve yardımlarını esirgemeyen, kendisinden
çok şey öğrendiğim ve öğrenmeye devam edeceğim ikinci danışmanım Prof. Dr.
Murat DURANDURDU'ya teşekkürlerimi sunarım.
Tez çalışmam sırasında her türlü bilgi ve desteğini esirgemeyen sayın Prof.
Dr. Emirullah Mehmetov’a çok teşekkür ederim.
Tez çalışmam sırasında yardımlarını esirgemeyen tüm arkadaşlarıma çok
teşekkür ederim.
Böyle yoğun bir çalışma sürecinde beni sonuna kadar destekleyen ve hayatım
boyunca benim için her türlü fedakârlıkları gösteren değerli annem, babam ve
kardeşim Tamer TETİK’e sonsuz teşekkürlerimi sunarım.

IV
İÇİNDEKİLER SAYFA
ÖZ ............................................................................................................................ I
ABSTRACT ............................................................................................................ II TEŞEKKÜR ...........................................................................................................III İÇİNDEKİLER ...................................................................................................... IV
ÇİZELGELER DİZİNİ ......................................................................................... VII ŞEKİLLER DİZİNİ ............................................................................................. VIII SİMGELER VE KISALTMALAR ......................................................................XIV
1. GİRİŞ .................................................................................................................. 1
1.1. Karbon Nanotüplerin Yapısı ve Simetrisi ...................................................... 6
1.1.1. Tek Duvarlı Karbon Nanotüplerin Sınıflandırılması ................................ 6
1.1.3. Öteleme Vektörü: 푇 ...............................................................................11
1.1.4. Simetri Vektörü: 푅 ................................................................................13
1.1.5. Birim Hücre ve Brillouin Bölgeleri ........................................................17
1.2. Karbon Nanotüplerin Elektronik Özellikleri .................................................19
1.2.1. Enerji Dağılımının Zone-Folding Yaklaşımı ..........................................19
1.2.2 Armchair ve Zigzag Nanotüplerin Enerji Dağılımı ..................................21
1.2.3. Chiral Nanotüplerin Enerji Dağılımı ......................................................24
1.2.4. Elektronik Durum Yoğunluğu ve Yasak Bant Aralığı ............................26
1.3. Karbon Nanotüplerde Katkılama İşlemi ........................................................27
2. ÖNCEKİ ÇALIŞMALAR ...................................................................................30
2.1. Karbon Nanotüpler İçin Deneysel Kanıtlar ...................................................30
2.2. Karbon Nanotüplerle İlgili Teorik Çalışmalar ...............................................35
2.3. Karbon Nanotüplerde Katkılama Çalışmaları................................................39
2.3.1. Katkılama İle İlgili Deneysel Çalışmalar................................................40
2.3.2. Katkılama İle İlgili Teorik Çalışmalar ....................................................43
3. MATERYAL VE METOT ..................................................................................48
3.1. Yoğunluk Fonksiyoneli Teorisine Giriş ........................................................48
3.1.1. Kohn-Sham Hesaplamaları ....................................................................48
3.2. Fonksiyonel Kavramı ...................................................................................51
3.3. Kuantum Mekaniği Dalga Fonksiyonu: Tek Elektron ...................................52
3.3.1. İki Elektron ...........................................................................................54

V
3.3.2. İki Elektron İçin Hartree-Fock ...............................................................54
3.3.3. Korelasyon ............................................................................................57
3.3.5. N Elektron .............................................................................................58
3.3.6. Elektronik Yoğunluk .............................................................................60
3.4. Modern Yoğunluk Fonksiyoneli Teorisi .......................................................63
3.4.1. Hohenberg-Kohn Teoremleri .................................................................65
3.4.2. Thomas-Fermi Teorisi ...........................................................................71
3.4.3. Kohn-Sham Denklemleri .......................................................................73
3.4.4. Kohn-Sham Formülasyonu ....................................................................77
3.4.5. Değiş-Tokuş ve Korelasyon Enerjisi ......................................................79
3.4.6. Değişim-Korelasyon Fonksiyonellerinin Genel Analitik Özellikleri .......83
3.5. Yerel Yoğunluk Yaklaşımı (The Local Density Approximation, LDA) .........84
3.5.1. Yerel Yaklaşımlar ..................................................................................85
3.5.2. Yerel Yoğunluk Yaklaşımı ....................................................................86
3.5.3. Düzgün Elektron Gazı ...........................................................................87
3.6. Genelleştirilmiş Gradyent Yaklaşımları ........................................................90
3.7. Pseudo-Potansiyel Yaklaşımı .......................................................................91
3.8. Hesaplama Metotları ....................................................................................92
3.9. Paralel Programlama ....................................................................................93
3.9.1. Siesta İçin Paralel Programlama ............................................................94
4. BULGULAR VE TARTIŞMA ............................................................................96
4.1. Hesaplama Adımları .....................................................................................96
4.2. Karbon Nanotüplerin Geometrik Yapısının Elde Edilmesi ............................97
4.2.1. Hesaplaması Yapılan Nanotüplerin Geometrik Yapıları ....................... 100
4.3. Karbon Nanotüplerin Örgü Sabitinin Hesaplanması .................................... 102
4.4. Karbon Nanotüplerin Elektronik Bant Yapısı ve Durum Yoğunluğu ........... 106
4.5. Karbon Nanotüplerin Yük Yoğunluğu ........................................................ 114
4.6. Karbon Nanotüplerde Katkılamanın Etkilerinin İncelenmesi ...................... 115
4.6.1. Bor Katkılanmış Karbon Nanotüpler .................................................... 118
4.6.2. Berilyum Katkılanmış Karbon Nanotüpler ........................................... 121
4.6.3. Azot Katkılanmış Karbon Nanotüpler .................................................. 124
4.6.4. Silisyum Katkılanmış Karbon Nanotüpler ............................................ 127
4.6.5. Oksijen Katkılanmış Karbon Nanotüpler ............................................. 130
4.6.6. Klor Katkılanmış Karbon Nanotüpler .................................................. 133

VI
5. SONUÇLAR VE ÖNERİLER ........................................................................... 137
5.1. Grafen ve Karbon Nanotüplerin Geometrik Yapıları ................................... 137
5.2. Grafen ve Karbon Nanotüplerin Elektronik Özellikleri ............................... 138
5.2.1. Zigzag (6, 0) Grafen ve Nanotüpün Sonuçları ...................................... 139
5.2.2. Zigzag (7, 0) Grafen ve Nanotüpün Sonuçları ...................................... 139
5.2.3. Armchair (7, 7) Grafen ve Nanotüpün Sonuçları .................................. 140
5.2.4. Chiral (6, 2) Grafen ve Nanotüpün Sonuçları ....................................... 140
5.2.5. Chiral (6, 3) Grafen ve Nanotüpün Sonuçları ....................................... 141
5.3. Karbon Nanotüplerin Katkılama İle İlgili Sonuçları .................................... 141
5.3.1. Zigzag (8, 0) Nanotüpe Bor Katkılama Sonuçları ................................. 142
5.3.2. Zigzag (8, 0) Nanotüpe Berilyum Katkılama Sonuçları ........................ 142
5.3.3. Zigzag (8, 0) Nanotüpe Azot Katkılama Sonuçları ............................... 143
5.3.4. Zigzag (8, 0) Nanotüpe Silisyum Katkılama Sonuçları ......................... 143
5.3.5. Zigzag (8, 0) Nanotüpe Oksijen Katkılama Sonuçları .......................... 144
5.3.6. Zigzag (8, 0) Nanotüpe Klor Katkılama Sonuçları ............................... 144
5.4. Karbon Nanotüple İlgili Teorik Çalışma Önerileri ...................................... 144
KAYNAKLAR ..................................................................................................... 146
ÖZGEÇMİŞ.......................................................................................................... 152
EKLER ................................................................................................................. 153

VII
ÇİZELGELER DİZİNİ SAYFA
Çizelge 1.1. Karbonun farklı formlarının sınıflandırılması........................................ 2
Çizelge 1.2. TDKNT’lerin sınıflandırılması .......................................................... ...8
Çizelge 1.3. KNT’lerin temel parametreleri ........................................................... .16
Çizelge 1.4. Chiral nanotüplerin sınıflandırılması. gcd değeri en büyük ortak
bölendir ............................................................................................. .25
Çizelge 4.1. TDKNT’lerin elde edilen temel parametreleri .................................... 101
Çizelge 4.2. Nanotüp programı ile elde edilen örgü sabitleri .................................. 101
Çizelge 4.3. KNT’lerin optimize edilen örgü Sabitleri ........................................... 104
Çizelge 4.4. Elektronik bant yapısı ve durum yoğunluğu hesaplamaları ................. 113

VIII
ŞEKİLLER DİZİNİ SAYFA
Şekil 1.1. Karbonun bazı allotropları: (a) Elmas, (b) Grafit, (c) Lonsdaleite, (d)
Karbon nanotop (C60) ve (e) Karbon nanotüp ......................................... 1
Şekil 1.2. Karbon atomunun enerji seviyeleri ........................................................ 2
Şekil 1.3. Grafit ve Grafen Yapıları ....................................................................... 6
Şekil 1.4. (a) Zigzag, (b) Armchair ve (c) Chiral TDKNT ..................................... 7
Şekil 1.5. Grafen katmanından zigzag (6,0) TDKNT yapının elde edilmesi ........... 9
Şekil 1.6. Grafen katmanından armchair (4,4) TDKNT yapısının elde edilmesi ..... 9
Şekil 1.7. Grafenin yapısından chiral (4,2) TDCNT yapısının elde edilmesi ........ 10
Şekil 1.8. T, R ve Ch arasındaki vektörel ilişki .................................................... 13
Şekil 1.9. Nanotüpün döndürülmesi. NR = ψτN vektörü silindirik yüzey
üzerinde görülmektedir. Tüp 2π kadar döndürüldükten sonra, NR
vektörü O noktasının eşdeğeri olan C noktasına ulaşır. Fakat C
noktası, o noktasından MT vektörü kadar ayrılır................................... 16
Şekil 1.10. Karbon nanotüpün Birillouin bölgesi K2 vektörüne paralel olan WW'
çizgi parçası ile temsil edilmektedir ..................................................... 18
Şekil 1.11. İki boyut grafitin birim hücresi (a) ve Brillouin bölgesi (b) sırasıyla
eşkenar dörtken ve gölgeli hegzagonal ile gösterilmektedir .................. 20
Şekil 1.12. Metalik enerji bandları için koşul: YK vektörünün uzunluğunun K1
vektörünün uzunluğuna oranı bir tamsayı olduğu zaman, metalik
enerji bandları elde edilir ..................................................................... 21
Şekil 1.13. Armchair (a) ve zigzag (b) nanotüplerin extended Brillouin bölgesi
ve birim hücre bölümleri ...................................................................... 22
Şekil 1.14. Armchair (5, 5) (a), zigzag (9, 0) (b) ve zigzag (10, 0) karbon
nanotüpler için bir boyutlu enerji dağılım grafikleri ............................. 23
Şekil 1.15. Bir boyutlu metalik chiral (9, 6) (a) ve (7, 4) (b) nanotüp için enerji
bant grafikleri. Fermi seviyesi sıfırdadır............................................... 25
Şekil 1.16. İki boyutlu grafen katmanının birim hücresinin bir boyutlu durum
yoğunluğu: chiral (10, 0) yarıiletken (a) ve (9, 0) (b) metalik
nanotüp................................................................................................ 27

IX
Şekil 1.17. B atomu katkılanmış bir karbon nanotüp ............................................. 28
Şekil 2.1. TDKNT’lerin bir demetinin TEM ile üstten görüntüsü. Nanotüp
demetinin çapı 1,4nm ve ortalama tüpler arası mesafesi 1,7nm
civarındadır ......................................................................................... 31
Şekil 2.2. Bu taslak chiral açısını belirlemek için kullanılan H oryantasyonu ile
düzlemler üzerindeki tesadüfi elektron ışını için girişim örneğinin
nasıl olduğunu göstermektedir ............................................................. 33
Şekil 2.3. (17, 3) CNT’nin atomik yapısının herhangi bir dalga vektörü için
normal düzlemi üzerindeki izdüşümü................................................... 33
Şekil 2.4. TDKNT’ün 4,2 K sıcaklığında STM topografik resmi. Karanlık
bölgeler hegzogonları göstermektedirler ve bunlar arasındaki boşluk
2,46 A° civarındadır. Örgü yapısından bu nanotüpün zigzag olmadığı
ve armchair bir yapıya gösterdiği anlaşılmaktadır ................................ 34
Şekil 2.5. Lazer buharlaştırma metodu tarafından hazırlanan TDKNT’ün XRD
yapısı ................................................................................................... 35
Şekil 2.6. KNT’lerin geometrik yapısına göre metalik ve yarı iletken özellik
göstermesi ........................................................................................... 43
Şekil 2.7. (a) Ab initio ile grafenin elektronik bant yapısı, (b) Ab initio ile
zigzag (10, 0) nanotüpün elektronik bant yapısı ve (c) Zigzag (10, 0)
nanotüpün Tight-binding metoduyla elektronik bant yapısı .................. 37
Şekil 2.8. (a) Ab initio ile grafenin elektronik bant yapısı, (b) Ab initio ile
armchair (6, 6) nanotüpün elektronik bant yapısı ve (c) Armchair (6,
6) nanotüpün Tight-binding metoduyla elektronik bant yapısı .............. 38
Şekil 2.9. Katkılama yapılmayan TDKNT’ün TEM görüntüsü (a, b) ve bor
atomu eklenen TDKNT’nin TEM görüntüsü ........................................ 40
Şekil 2.10. Katkılama yapılmayan TDKNT’ün ve %10 ile %15 oranında bor
katkılanan TDKNT’nin optik soğurması .............................................. 41
Şekil 2.11. TDKNT’nin SEM görüntüsü ............................................................... 41
Şekil 2.12. Zigzag (16, 0) nanotüpün elektronik bant yapısı ve durum yoğunluğu
(a), %6,25 bor atomu eklenmiş (16, 0) nanotüpün elektronik bant
yapısı ve durum yoğunluğu (b), %12,5 bor atomu eklenmiş (16, 0)

X
nanotüpün elektronik bant yapısı ve durum yoğunluğu (c) ve %25
bor atomu eklenmiş (16, 0) nanotüpün elektronik bant yapısı ve
durum yoğunluğu (b) ........................................................................... 43
Şekil 2.13. Katkısız (a) ve H-sağurması olan (b) zigzag (8, 0) nanotüpü
elektronik bant yapısı ve durum yoğunluğu .......................................... 44
Şekil 2.14. Bor katkılanmış (a) ve bor katkılanmış (H-sağurma) (b) zigzag (8, 0)
nanotüpü elektronik bant yapısı ve durum yoğunluğu .......................... 45
Şekil 2.15. Azot katkılanmış (a) ve azot katkılanmış (H-sağurma) (b) zigzag (8,
0) nanotüpü elektronik bant yapısı ve durum yoğunluğu ...................... 45
Şekil 3.1. H2 Molekülünün Toplam Enerjisi ........................................................ 48
Şekil 3.2. He atomu için dış ve Kohn-Sham potansiyeli ....................................... 50
Şekil 3.3. Ar atomunun yarıçapa bağlı yoğunluğu ............................................... 60
Şekil 3.4. (a) Bir boyutlu He atomu için HF yaklaşımı, (b) He atomu için HF
ve gerçek grafik ................................................................................... 61
Şekil 3.5. r = 1 + 0,1 cos(20 θ) eğrisi (n=20, eps=0,1) ...................................... 84
Şekil 3.6. Düzgün elektron gazı için değiş-tokuş ve korelasyon enerjileri ............ 88
Şekil 3.7. Pseudo potansiyel, Pseudo ve gerçek dalga fonksiyonları .................... 90
Şekil 4.1. Hesaplaması yapılan karbon nanotüpler. (a) zigzag (6, 0) nanotüp,
(b) chiral (6, 2) nanotüp, (c) armchair (7, 7) nanotüp, (d) chiral (6, 3)
nanotüp ve (e) zigzag (7, 0) nanotüp .................................................. 100
Şekil 4.2. CNT(6, 0) için a (A)-enerji (eV) optimizasyon grafiği ....................... 103
Şekil 4.3. CNT(6, 2) için a (A)-enerji (eV) optimizasyon grafiği ....................... 103
Şekil 4.4. CNT(6, 3) için a (A)-enerji (eV) optimizasyon grafiği ....................... 103
Şekil 4.5. CNT(7, 0) için a (A)-enerji (eV) optimizasyon grafiği ....................... 104
Şekil 4.6. CNT(7, 7) için a (A)-enerji (eV) optimizasyon grafiği ....................... 104
Şekil 4.7. Hegzagonal yapıdaki karbon nanotüplerin Brillouin bölgesi .............. 105
Şekil 4.8. Zigzag (6, 0) grafen (a) ve nanotüpün (b) elektronik bant yapısı ve
durum yoğunluğu .............................................................................. 106
Şekil 4.9. Zigzag (6, 0) grafen (a) ve nanotüpün (b) iki ve üç boyutlu yük
yoğunluğu ......................................................................................... 106

XI
Şekil 4.10. Zigzag (7, 0) grafen (a) ve nanotüpün (b) elektronik bant yapısı ve
durum yoğunluğu .............................................................................. 108
Şekil 4.11. Zigzag (7, 0) grafen (a) ve nanotüpün (b) iki ve üç boyutlu yük
yoğunluğu ......................................................................................... 108
Şekil 4.12. Armchair (7, 7) grafen (a) ve nanotüpün (b) elektronik bant yapısı ve
durum yoğunluğu .............................................................................. 109
Şekil 4.13. Armchair (7, 7) grafen (a) ve nanotüpün (b) iki ve üç boyutlu yük
yoğunluğu ......................................................................................... 109
Şekil 4.14. Chiral (6, 2) grafen (a) ve nanotüpün (b) elektronik bant yapısı ve
durum yoğunluğu .............................................................................. 110
Şekil 4.15. Chiral (6, 2) grafen (a) ve nanotüpün (b) iki ve üç boyutlu yük
yoğunluğu ......................................................................................... 110
Şekil 4.16.Chiral (6, 3) grafen (a) ve nanotüpün (b) elektronik bant yapısı ve
durum yoğunluğu .............................................................................. 112
Şekil 4.17. Chiral (6, 3) grafen (a) ve nanotüpün (b) iki ve üç boyutlu yük
yoğunluğu ......................................................................................... 112
Şekil 4.18. Zigzag (8, 0) TDKNT’nin geometrik yapısı ....................................... 115
Şekil 4.19. Zigzag (8, 0) nanotüpün elektronik bant yapısı ve durum yoğunluğu . 116
Şekil 4.20. %3,125, %6,25, %12,5 ve %25 oranlarında bor eklenmiş (8, 0)
nanotüpün geometrik yapısı ............................................................... 117
Şekil 4.21. %3,125 bor eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu ......................................................................................... 118
Şekil 4.22. %6,25 bor eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu ......................................................................................... 118
Şekil 4.23. %12,5 bor eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu ......................................................................................... 119
Şekil 4.24. %25 bor eklenmiş (8, 0) nanotüpün bant yapısı ve durum yoğunluğu . 119
Şekil 4.25. %3,125, %6,25, %12,5 ve %25 oranlarında berilyum eklenmiş (8, 0)
nanotüpün geometrik yapısı ............................................................... 120
Şekil 4.26. %3,125 berilyum eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu ......................................................................................... 121

XII
Şekil 4.27. %6,25 berilyum eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu ......................................................................................... 121
Şekil 4.28. %12,5 berilyum eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu ......................................................................................... 122
Şekil 4.29. %25 berilyum eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu ......................................................................................... 122
Şekil 4.30. %3,125, %6,25, %12,5 ve %25 oranlarında azot eklenmiş (8, 0)
nanotüpün geometrik yapısı ............................................................... 123
Şekil 4.31. %3,125 azot eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu ......................................................................................... 124
Şekil 4.32. %6,25 azot eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu ......................................................................................... 124
Şekil 4.33. %12,5 azot eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu ......................................................................................... 125
Şekil 4.34. %25 azot eklenmiş (8, 0) nanotüpün bant yapısı ve durum yoğunluğu 125
Şekil 4.35. %3,125, %6,25, %12,5 ve %25 oranlarında silisyum eklenmiş (8, 0)
nanotüpün geometrik yapısı ............................................................... 126
Şekil 4.36. %3,125 silisyum eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu ......................................................................................... 127
Şekil 4.37. %6,25 silisyum eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu ......................................................................................... 127
Şekil 4.38. %12,5 silisyum eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu ......................................................................................... 128
Şekil 4.39. %25 silisyum eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu ......................................................................................... 128
Şekil 4.40. %3,125, %6,25, %12,5 ve %25 oranlarında oksijen eklenmiş (8, 0)
nanotüpün geometrik yapısı ............................................................... 129
Şekil 4.41. %3,125 oksijen eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu ......................................................................................... 130
Şekil 4.42. %6,25 oksijen eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu ......................................................................................... 130

XIII
Şekil 4.43. %12,5 oksijen eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu ......................................................................................... 131
Şekil 4.44. %25 oksijen eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu ......................................................................................... 131
Şekil 4.45. %3,125, %6,25, %12,5 ve %25 oranlarında klor eklenmiş (8, 0)
nanotüpün geometrik yapısı ............................................................... 132
Şekil 4.46. %3,125 klor eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu ......................................................................................... 133
Şekil 4.47. %6,25 klor eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu ......................................................................................... 133
Şekil 4.48. %12,5 klor eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu ......................................................................................... 134
Şekil 4.49. %25 klor eklenmiş (8, 0) nanotüpün bant yapısı ve durum yoğunluğu 134

XIV
SİMGELER VE KISALTMALAR
ab initio : Temel ilkelere dayanan
ABINIT : Yoğunluk fonksiyonel teorisine dayalı olarak pseudo potansiyel yöntem kullanan ab-initio yazılımı
B : Bor
Be : Berilyum
BN : Bor ve Azot
BZ : Brillouin Bölgesi
CASTEP : Yoğunluk fonksiyonel teorisine dayalı olarak pseudo potansiyel yöntem kullanan ab-initio yazılımı
ÇDKNT : Çok duvarlı karbon nanotüp
DFT : Yoğunluk fonksiyoneli teorisi (Density Functional Theory)
DOS : Durum Yoğunluğu (Density of States)
DZP : Düzlem Dalga Baz Setleri
EELS : Elektron Enerji-Kayıp Spektroskopisi (Electron Energy-Loss Spectroscopy, EELS)
εxc : Değiş-tokuş ve korelasyon enerjisi
GGA : Genelleştirilmiş Gradyent Yaklaşımı (Generalized Gradient Approximation
HF : Hatree-Fock
Cl : Klor
KNT : Karbon nanotüp
LDA : Yerel Yoğunluk Yaklaşımı (Local Density Approximation)
MPI : Mesaj Geçirme Arayüzü (Message Passing Interface), bilgisayar bilimlerinde, paralel programlama amacıyla kullanılan bir platformun ismidir
N : Azot
O : Oksijen
OAS : Optik Soğurma Spektroskopisi
SEM : Taramalı elektron mikroskobu (Scanning electron microscope)
SIESTA : Yoğunluk fonksiyonel teorisine dayalı olarak pseudo potansiyel yöntem kullanan ab-initio tabanlı bir yazılım
Si : Silisyum
STM : Taramalı tünelleme mikroskopu (Scanning tunneling microscopy)

XV
TDKNT : Tek duvarlı karbon nanotüp
TEM : Geçirmeli Elektron Mikroskobu (Transmission electron microscopy)
VASP : Yoğunluk fonksiyonel teorisine dayalı olarak pseudo potansiyel yöntem kullanan ab-initio yazılımı
WIEN2K : Yoğunluk fonksiyonel teorisine dayalı olarak pseudo potansiyel yöntem kullanan ab-initio yazılımı
XC : Değiş-tokuş ve Korelasyon

1. GİRİŞ ERKAN TETİK
1
1. GİRİŞ
Üç boyutlu (3B) yarıiletken elmas yapıdan, iki boyutlu (2B) yarı metalik
grafite, bir boyutlu (1B) iletken ve yarıiletken nanotüplere ve sıfır boyutlu (0B)
nanotoplara kadar farklı kararlı yapılara ve birçok ilginç özelliğe sahip olan karbon
elementi, Nanoteknoloji çağının başlamasında önemli bir rol oynamıştır. Hidrojen,
helyum ve oksijenden sonra kütle bakımından evrende en çok bulunan dördüncü
element olan karbon, Şekil 1.1’de gösterildiği gibi pek çok allotropik (elementlerin
bazı özel şartlarda farklı geometrik yapılar göstermesi) formda bulunmaktadır.
Şekil 1.1. Karbonun bazı allotropları: (a) Elmas, (b) Grafit, (c) Lonsdaleite, (d) Karbon nanotop (C60) ve (e) Karbon nanotüp
Karbon atomu altı elektrona sahiptir. Bu elektronlardan dört tanesi dış
kabuktadır ve valans elektronları olarak isimlendirilmektedir. Şekil 1.2’de karbon
atomunun enerji seviye grafiği görülmektedir. Bu şekilde karbon atomunun altı
elektronu oklarla (okun doğrultusu elektronun spinini temsil eder) gösterilmiştir. İlk
iki elektron çekirdeğe yakın olan 1s orbitalinde, sonraki ikisi ise 2s orbitalinde yer

1. GİRİŞ ERKAN TETİK
2
alır. Geriye kalan iki elektron 2p orbitalinde ayrı yerleri işgal ederler. Bu p
orbitallerinin aynı enerjiye sahip olmasından ve elektronların ayrı orbitallerde olmayı
tercih etmesinden kaynaklanmaktadır.
Şekil 1.2. Karbon atomunun enerji seviyeleri
Elektronik hipritleşme kovalent bağlanmanın pek çok türüne izin
vermektedir. Çizelge 1.1’de karbonun farklı formlarının (1s2, 2s2, 2p2 elektronları)
sınıflandırılmasının şematik gösterimi yer almaktadır.
Çizelge 1.1. Karbonun farklı formlarının sınıflandırılması Kristal Yapı Özelliği Elmas Grafit Carbynes Nanotüp
Hibritleşme sp3 sp2 sp1 sp2 Z Kordinatı 4 3 2 3 Bağ Uzunluğu (A0) 1,54 1,42 1,21 1,33 – 1,40 Bağ Enerjisi (eV/mol) 15 25 35 >25
Karbon atomları kendi aralarında bağ yapan elektronların sayısına göre sp1,
sp2 ve sp3 gösterimleriyle ifade edilen üç farklı bağlanma türünü de gösterirler. Bu
gösterimler aynı zamanda bağlanma geometrisini de temsil eder. Karbon elementi,
her üç bağlanma geometrisini gösterebilen tek element olması bakımından istisnai bir
özelliğe sahiptir ve bu özellik karbonun 0B'den 3B'ye kadar farklı yapıda
olabilmesine olanak tanır. Karbonun 1B ve 0B yapıları nanometre düzeyinde
oldukları için, bu sistemlere nanotüpler ve nanotoplar deniyor ki karbon
nanoyapıların aslını toplar ve tüpler oluşturmaktadır. Farklı çap ve boyda, uçları açık
ENERJİ
1
22p 2p 2p x y z

1. GİRİŞ ERKAN TETİK
3
ya da kapalı olabilen Karbon Nanotüpler (KNT) ise Nanoteknolojinin en önemli
konularından biridir.
Karbon nanotüplerin fiziği, 1991 yılında Japonya’daki NEC
laboratuvarlarında Sumio Ijima tarafından çok duvarlı ve iki yıl sonra da tek duvarlı
karbon nanotüplerin keşfedilmesinden bu yana bir araştırma alanı olarak hızlı bir
şekilde gelişmektedir. Elektron mikroskobu uzmanı olan Sumia Iijima, ark
boşalmasıyla elde edilen fulleren yapının Geçirmeli Elektron Mikroskobu (TEM)
görüntüsünde, Çok Duvarlı Karbon Nanotüp (ÇDKNT) olarak isimlendirilen tüp
şeklinde bir yapı gözlemlemiştir. Sonraki araştırmalar sonucunda, grafit elektrotuna
kobalt gibi bazı geçiş metallerin eklenmesi sonucunda Tek Duvarlı Karbon
Nanotüpler (TDKNT) elde edilmiştir. TDKNT’lerin elde edilmesi, karbon
nanotüplerin gelişmesinde büyük bir aşama olmuştur. 1996’da Rice Üniversitesi
Araştırma Grubunun TDKNT oluşturmada daha etkin bir yöntem bulmasıyla, çok
sayıda karbon nanotüp deneylerinin önü açılmış oldu. Arzu edilen nanotüpler 1200
°C fırında karbonun lazer-buharlaştırılmasıyla elde edilmektedir. Daha sonra
Montpellier Üniversitesinden Catherine Journet, Patrick Bernier ve çalışma
arkadaşlarının karbon ark-buharlaşma metoduyla iyonlaşmış karbon plazmasından
TDKNT elde etmişlerdir. ÇDKNT’lerin büyütülmesi için katalizör gerekmezken,
TDKNT’ler ancak katalizör ile büyütülebilmektedir.
Grafen plakasının kıvrılma yönüne göre nanotüpler farklı elektronik ve
mekanik özellikler göstermektedir. Karbon nanotüplerin çapları nanometre, boyları
mikrometre düzeyinde olabilmektedir. Nanotüplerin çapları şimdiye kadar
üretilebilen en ileri yarıiletken aygıtlarınkinden bile çok daha küçüktür. KNT’lerin
yarıiletken teknolojisinde kullanılmaya başlaması yarıiletken fiziğinde çok büyük bir
atılıma vesile olacağı açıktır. Tüpün geometrisine (çapına ve silindir yüzeyinin
kıvrılma yönüne) bağlı olarak nanotüpler metal veya yarıiletken özellik
gösterebiliyorlar. Tüpün elektronik özellikleri, katkı maddesi olmaksızın yalnızca
geometrik parametrelerle ayarlanabiliyor. Bu yarıiletken nanotüplerin yasak enerji
aralığı ise 1 ile 0 eV arasında değiştirilebilmektedir (Reich ve ark., 2004).
Karbon nanotüp fiberler çok geniş yüzey alanına sahiptir. Nanotüp fiberin
kütlesiyle alanı arasındaki oran, normal malzemelere göre çok daha büyüktür.

1. GİRİŞ ERKAN TETİK
4
Karbon nanotüp fiberlerin bu özelliği nanometre düzeyinde süper kapasitörler elde
edilmesine olanak tanır. Ayrıca dikkat çekici mekanik özelliklere sahip olan KNT’ler
hafif ve çok yüksek bir elastik modülüne sahiptirler. KNT’lerin young modülü de
oldukça yüksektir. Küçük çaplı (yaklaşık 1–2 nm) tüplerden oluşturulmuş bir demeti
koparabilmek için uygulanan çekme kuvvetinin büyüklüğü yaklaşık 36 gigapaskal
civarında olmakla birlikte oluşturulan bu demet çok esnek bir yapıya sahiptir. Bu
ilginç mekanik özellikleriyle KNT’ler, malzeme endüstrisini tamamıyla
değiştirebileceğini açıkça ortaya koymuştur. Ayrıca hidrojen depolamaya da olanak
sağlayan geniş yüzey alanı, karbon nanotüpleri potansiyel enerji depolama
malzemesi adayı konumuna getirmektedir.
KNT’lerin teknolojide kullanılması için aşılması gereken sayısız zorluklar
vardır. Örneğin henüz yeterli miktarda ve saflıkta KNT sentez teknikleri
geliştirilememiştir. Ayrıca var olan sentezleme teknikleri de çok pahalıya mâl
olmaktadır. Ancak KNT’lerin fiziksel özellikleri teorik olarak yoğun bir şekilde
çalışılmaktadır. Bu çalışmaların gelecekte yapılacak olan KNT tabanlı projelere ışık
tutacağı açıktır. Teorik çalışmaların büyük bir kısmı paket programlarla yapılan
simülasyonlara dayanmaktadır. Paket programların pek çoğu ise ab initio (temel
prensip) yöntemlerine göre çalışmaktadır.
Atom ve molekül yapıların anlaşılması, dolayısıyla bir sistemin moleküler
özelliklerinin tayin edilebilmesi için Schrödinger denkleminin çözülmesi
gerekmektedir. Fakat çok az sayıda sistem (Hidrojen Atomu, Harmonik Osilatör ve
Kutudaki Parçacık) için Schrödinger denkleminin analitik bir çözümü mümkün
olmaktadır. Çok parçacıklı sistemlerde analitik çözümün mümkün olmamasının ana
nedeni, elektron-elektron ve elektron-çekirdek etkileşmelerinin formülasyonunun
tam olarak yapılamamasıdır. Bir sistemin fiziksel ve kimyasal özellikleri bu
etkileşmelerle doğrudan ilişkilidir. Bu nedenle bu etkileşmelerin modellenmesinde
yaklaşık yöntemlere ihtiyaç duyulmuştur. Born-Oppenheimer (1927)’de çok
parçacıklı sistemler için toplam dalga fonksiyonunun, elektronik dalga fonksiyonu
biçiminde yazılabilir olduğunu öngörmesiyle birlikte yaklaşık çözüm yöntemleri
geliştirilmeye başlanmıştır. Hartree-Fock (1957) teorisinin geliştirilmesi çok parçacık
sistemleri için toplam enerjinin ve enerjiye bağlı olan pek çok fiziksel ve kimyasal

1. GİRİŞ ERKAN TETİK
5
niceliğin, Schrödinger denkleminin yaklaşık çözümüyle elde edilmesini mümkün
hale getirmiştir. Hohenberg ve Kohn (1964), Yoğunluk Fonksiyoneli Teorisi
(Density Functional Theory, DFT) ile sistemin çok elektronlu dalga fonksiyonunu
kullanarak hesaplama yapmak yerine, konum ve zamanın bir fonksiyonu olan
elektron yoğunluğunu kullanarak hesaplama yapma yöntemini geliştirmişlerdir.
Günümüzde kristal yapıların özelliklerinin araştırılmasında ab initio
yöntemlere dayanan SIESTA, ABINIT, VASP, Wien2k, Castep ve Quantun Espresso
gibi pek çok paket program kullanılmaktadır. Bu programlardan bir kısmı ücretli bir
kısmı ise ücretsiz ve açık kaynaklıdır. Son yıllarda bu tür programlar ile oldukça
karmaşık yapılardaki malzemelerin elektronik yapı hesaplarını yapmak oldukça
kolaylaşmış ve hesaplama zamanı da kısalmıştır. Bir taraftan bu hesaplara yönelik
yeni teoriler geliştirilirken, diğer taraftan ise bu programların yazılımları geliştirilmiş
ve güncellenmiştir. Bu tür programların yazılımlarının çoğu DFT temel alınarak
yazılmaktadır.
KNT’lerin şaşırtıcı elektronik, optik ve elastik özellikleri, bu konuda
çalışmayı tercih etmemizin temelini oluşturmaktadır. Çalışmamızda TDKNT’ler
öncelikle, yapısı ve simetrik özellikleri açısından incelenecektir. Sonrasında, başta
elektronik ve optik olmak üzere pek çok özelliği üzerinde çalışmalar yapılacaktır.
TDKNT’lere bor ve nitrojen gibi atomlar katkılandığında, geometrik yapısında,
elektronik ve optik özelliklerinde değişmeler meydana gelmektedir. TDKNT’ler
üzerinde SIESTA paket programı kullanılarak yapılan hesaplamalar doğrultusunda,
nanotüplere, periyodik cetveldeki A grubu elementlerinden yarıçap ve özellik olarak
karbona en yakın olanlar seçilerek katkılama işlemi yapılacaktır. Bu katkılama
işleminden sonra optimizasyon işlemi ve elektronik özelliklerle ilgili hesaplamlar
yeniden yapılacaktır. Elde edilen sonuçlar incelenerek TDKNT’nin fiziksel
özelliklerindeki değişmeler gözlenecektir. Ayrıca katkılama yapılan karbon nanotüp
ile yapılmamış olana nanotüplerin özellikleri, birbirleri arasında karşılaştırmalar
yapılarak incelenecektir.

1. GİRİŞ ERKAN TETİK
6
1.1. Karbon Nanotüplerin Yapısı ve Simetrisi
Grafitin tek bir katmanı grafen olarak isimlendirilmektedir. Grafen 6 tane
karbon atomundan oluşan bal peteği şeklindeki yapılardır. Şekil 1.3(a)’de grafit ve
şekil 1.3(b)’de grafen yapıları görülmektedir.
(a) (b)
Şekil 1.3. Grafit ve Grafen Yapıları
Grafen katmanı bir levhanın çevresine sarıldığı zaman tek duvarlı karbon
nanotüp (TDKNT) elde edilir. TDKNT’ler yaklaşık olarak 0,7-10 nm çapındadırlar.
TDKNT’lerin düzenli olarak iç içe geçmesiyle oluşan yapı çok duvarlı karbon
nanotüp (ÇDKNT) olarak isimlendirilir. ÇDKNT’ler TDKNT’ler ile benzer uzunluğa
sahipken, çapları dikkate alındığında ÇDKNT’lerin daha büyük bir çapa sahip
olduğu görülür. ÇDKNT’lerin iç ve dış çapı 5 ile 100 nm arasında değişmektedir ve
iç içe geçen TDKNT’lerin birbirleri arasındaki mesafe yaklaşık olarak 0,35 nm
civarındadır. Bu ise grafitin katmanları arasındaki mesafeye yakın bir değerdir. Bu
bakımdan ÇDKNT’ler grafit ile benzer özellikler göstermektedir.
1.1.1. Tek Duvarlı Karbon Nanotüplerin Sınıflandırılması
TDKNT’lerin yapısı, grafen katmanın bir silindir etrafında nasıl sarıldığı ile
doğrudan ilişkilidir. Bu sarılma şekli aynı zamanda TDKNT’lerin fiziksel

1. GİRİŞ ERKAN TETİK
7
özelliklerini de etkilemektedir. Bir TDKNT’nin yapısı hakkındaki ilginç ve önemli
bir gerçek nanotüp eksenine göre bal peteği örgüsündeki altı atomlu karbon (altıgen
yapı) halkasının oryantasyonudur. Bu oryantasyona göre TDKNT’lerin 3 temel
yapısı şekil 1.4’de görülmektedir. Şekil 1.4’den bal peteği örgüsündeki altıgen
yapının doğrultusunun, nanotüpün sarılmasından dolayı oluşan bozulmalar dışında
başka herhangi bir bozulma oluşmaksızın keyfi olarak seçilebileceği görülmektedir.
KNT duvarının temel yapısının bir silindir olduğu gerçeği, farklı özelliklerde pek çok
nanotüpün oluşturulmasına olanak tanımaktadır.
(a)
(c)
(b)
Şekil 1.4. (a) Zigzag, (b) Armchair ve (c) Chiral TDKNT
TDKNT’ler temel simetri sınıflandırmasına göre achiral ve chiral olmak
üzere iki grupta ele alınmaktadır. Achiral nanotüp ayna görüntüsü kendisiyle aynı
yapıya sahip olan bir KNT olarak tanımlanır. Achiral yapının armchair ve zigzag
olmak üzere iki durumu söz konusudur (şekil 1.4(a) ve (b)). Chiral nanotüp sarmal
simetriye sahiptir ve ayna görüntüsü kendisiyle anti-simetriktir. Bu tür yapılar
kimyasal terminolojide chiral olarak nitelendirildiği için, nanotüpler bu isimleri
almıştır. Bunlara ek olarak bazı fiziksel özellikler göz önünde tutularak TDKNT’ler
5 grupta ele alınacaktır. Bu gruplar çizelge 1.2’de verilmektedir. Çizelge 1.2’deki 퐶
chiral vektörü, 휃 ise chiral açısı olarak tanımlanan parametreler sonraki bölümlerde
açıklanacaktır.
1. GİRİŞ ERKAN TETİK
8
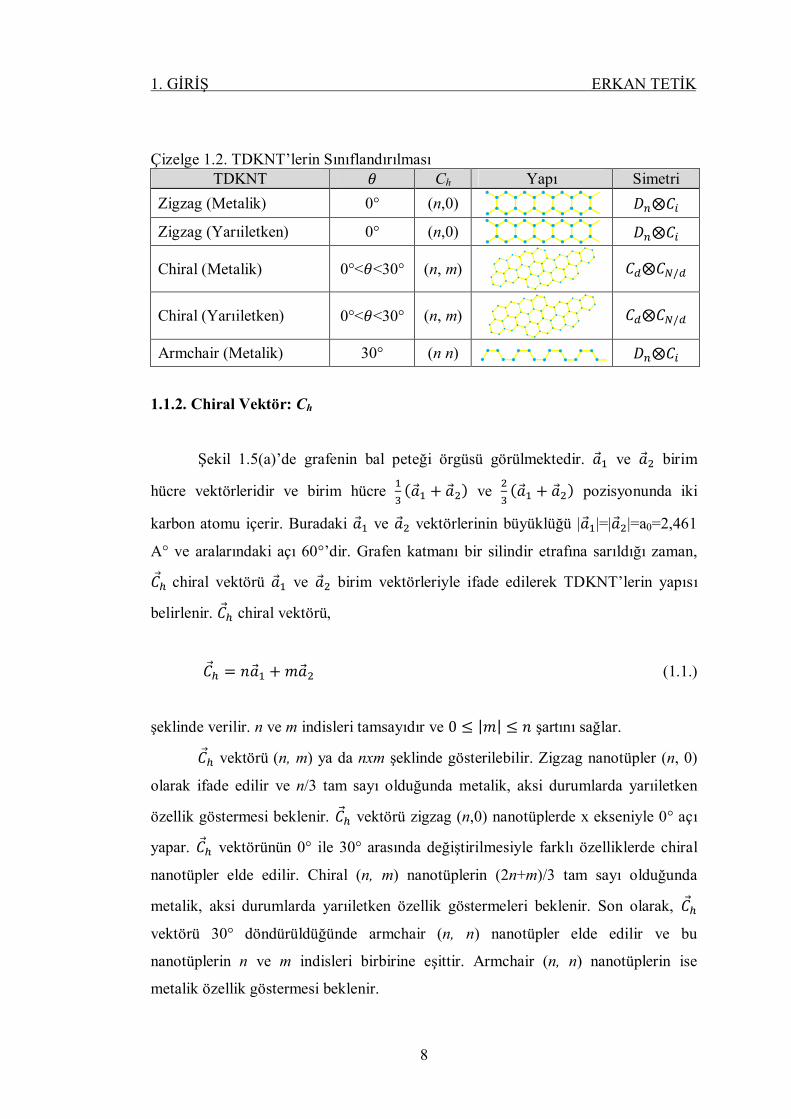
Çizelge 1.2. TDKNT’lerin Sınıflandırılması TDKNT 휃 Ch Yapı Simetri
Zigzag (Metalik) 0° (n,0) 퐷 ⨂퐶
Zigzag (Yarıiletken) 0° (n,0) 퐷 ⨂퐶
Chiral (Metalik) 0°<휃<30° (n, m)
퐶 ⨂퐶 /
Chiral (Yarıiletken) 0°<휃<30° (n, m)
퐶 ⨂퐶 /
Armchair (Metalik) 30° (n n) 퐷 ⨂퐶
1.1.2. Chiral Vektör: Ch
Şekil 1.5(a)’de grafenin bal peteği örgüsü görülmektedir. 푎 ve 푎 birim
hücre vektörleridir ve birim hücre (푎 + 푎 ) ve (푎 + 푎 ) pozisyonunda iki
karbon atomu içerir. Buradaki 푎 ve 푎 vektörlerinin büyüklüğü |푎 |=|푎 |=a0=2,461
A° ve aralarındaki açı 60°’dir. Grafen katmanı bir silindir etrafına sarıldığı zaman,
퐶 chiral vektörü 푎 ve 푎 birim vektörleriyle ifade edilerek TDKNT’lerin yapısı
belirlenir. 퐶 chiral vektörü,
퐶 = 푛푎 + 푚푎 (1.1.)
şeklinde verilir. n ve m indisleri tamsayıdır ve 0 ≤ |푚| ≤ 푛 şartını sağlar.
퐶 vektörü (n, m) ya da nxm şeklinde gösterilebilir. Zigzag nanotüpler (n, 0)
olarak ifade edilir ve n/3 tam sayı olduğunda metalik, aksi durumlarda yarıiletken
özellik göstermesi beklenir. 퐶 vektörü zigzag (n,0) nanotüplerde x ekseniyle 0° açı
yapar. 퐶 vektörünün 0° ile 30° arasında değiştirilmesiyle farklı özelliklerde chiral
nanotüpler elde edilir. Chiral (n, m) nanotüplerin (2n+m)/3 tam sayı olduğunda
metalik, aksi durumlarda yarıiletken özellik göstermeleri beklenir. Son olarak, 퐶
vektörü 30° döndürüldüğünde armchair (n, n) nanotüpler elde edilir ve bu
nanotüplerin n ve m indisleri birbirine eşittir. Armchair (n, n) nanotüplerin ise
metalik özellik göstermesi beklenir.
1. GİRİŞ ERKAN TETİK
9
1
퐶 h 2
푎
푇
퐶
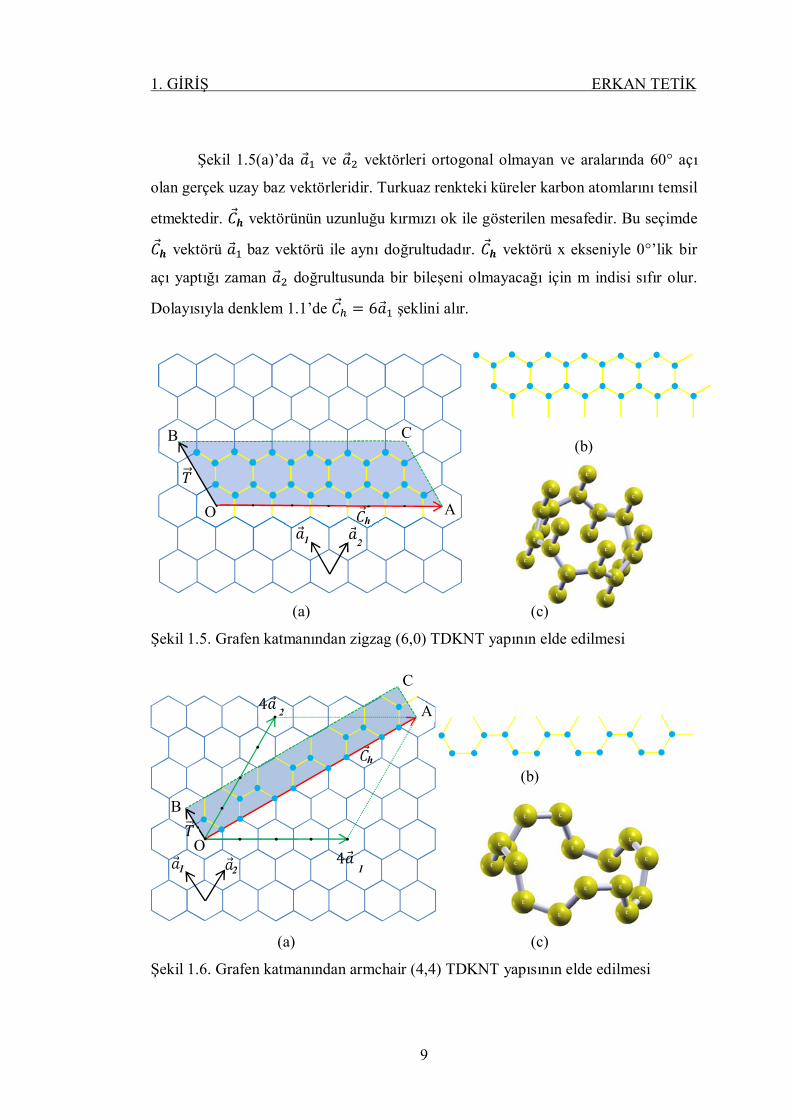
Şekil 1.5(a)’da 푎 ve 푎 vektörleri ortogonal olmayan ve aralarında 60° açı
olan gerçek uzay baz vektörleridir. Turkuaz renkteki küreler karbon atomlarını temsil
etmektedir. 퐶풉 vektörünün uzunluğu kırmızı ok ile gösterilen mesafedir. Bu seçimde
퐶풉 vektörü 푎 baz vektörü ile aynı doğrultudadır. 퐶풉 vektörü x ekseniyle 0°’lik bir
açı yaptığı zaman 푎 doğrultusunda bir bileşeni olmayacağı için m indisi sıfır olur.
Dolayısıyla denklem 1.1’de 퐶 = 6푎 şeklini alır.
(b)
(a) (c)
Şekil 1.5. Grafen katmanından zigzag (6,0) TDKNT yapının elde edilmesi
(b)
(a) (c)
Şekil 1.6. Grafen katmanından armchair (4,4) TDKNT yapısının elde edilmesi
O
h
푎 2푎 1
1
2
푇
A
C
B
O
B
A
C
푎
4푎
4푎

1. GİRİŞ ERKAN TETİK
10
Şekil 1.5(a)’da 24 tane karbon atomu seçilmiştir. Bu seçim 푇 (1, −2) öteleme
ve 푅(1, −1) simetri vektörlerine göre yapılmaktadır. Seçim bölgesindeki (OABC
dörtgeni) bu atomlar grafen örgü üzerinde simetrik olarak kendisini tekrar
etmektedir. Kendisini simetrik olarak tekrar eden ve 24 tane karbon atomundan
oluşan yapı şekil 1.5(b)’de gösterilmektedir. Şekil 1.5(b)’deki yapı bir silindir
etrafında sarıldığında, şekil 1.5(c)’deki nanotüp elde edilir ve bu nanotüp (6,0)
TDKNT yapısıdır.
(a)
(b) (c) (d)
Şekil 1.7. Grafenin yapısından chiral (4,2) TDKNT yapısının elde edilmesi
푇
퐶h
O
A
C
4푎 1
2푎 2

1. GİRİŞ ERKAN TETİK
11
Şekil 1.6(a)’daki grafen katmanı üzerinde, 퐶풉 vektörü x ekseni ile 30°’lik bir
açı yapmaktadır. Bu durumda 퐶풉 = 4푎 + 4푎 şeklinde olur. 푇 (1, −1) öteleme ve
푅(1,0) simetri vektörlerine göre yapılan seçim şekil 1.6(a)’da OACB dikdörtgeni ile
ifade edilmektedir. Seçilen 16 tane karbon atomu Şekil 1.6(b)’de gösterilmektedir.
Şekil 1.6(b)’deki yapı bir silindir etrafında sarıldığında, şekil 1.6(c)’deki yapı elde
edilir. Bu yapı ise armchair (4,4) TDKNT olarak isimlendirilmektedir.
Şekil 1.7(a)’deki grafen katmanı üzerindeki 퐶풉 vektörü x ekseniyle 7,5° bir
açı yapmaktadır. Bu durumda 퐶풉 = 4푎 + 2푎 şeklinde yazılır. 푇 (4, −5) öteleme ve
푅(1, −1) simetri vektörlerine göre yapılan seçim şekil 1.7(a)’da OACB dikdörtgeni
ile ifade edilmektedir. Seçim bölgesinde toplam 56 tane karbon atomu
bulunmaktadır. 56 atomun oluşturduğu yapı ve bu yapının 퐶풉 vektörü Şekil 1.7(b)’de
gösterilmektedir. Şekil 1.7(b)’deki yapı bir silindir etrafında sarıldığında, şekil
1.7(c)’deki yapı elde edilir. Şekil 1.7(d)’deki yapı şekil 1.7(c)’deki yapının farklı bir
görünümüdür. Bu yapı ise chiral (4,2) TDKNT olarak isimlendirilmektedir.
1.1.3. Öteleme Vektörü: 푻
Öteleme vektörü 푇 bir boyutlu KNT’nin birim vektörü olarak tanımlanır. 푇
vektörü nanotüp eksenine paraleldir ve 퐶풉 chiral vektörünün normalidir. 푇 örgü
vektörü, 푎 ve 푎 baz vektörleri olmak üzere,
푇 = 푡 푎 + 푡 푎 (1.2.)
şeklinde yazılır ve burada 푡 ve 푡 tamsayıdır. Nanotüpün çapı dt ve çevre
uzunluğunu L kabul edersek 퐿 = 푑 휋 olur. (1.2) denkleminden,
퐿 = 퐶풉 = 퐶풉. 퐶풉 = 푎√푛 + 푚 + 푚푛 (1.3.)

1. GİRİŞ ERKAN TETİK
12
푎 . 푎 = 푎 . 푎 = 푎 , 푎 . 푎 = (1.4.)
elde edilir. Denklem (1.2), (1.3), (1.4) ve 퐶풉. 푇 = 0 şartını kullanarak,
푡 = , 푡 = − (1.5.)
elde edilir. Burada 푑 , 2푚 + 푛 ve 2푛 + 푚 değerlerinin en büyük ortak bölenleridir.
Aynı zamanda, d’yi n ve m’nin en büyük ortak böleni olarak kabul edersek 푑 ,
푑 = 푛 − 푚 3푑 nin katı değilse 푑푛 − 푚 3푑 nin katı ise 3푑
(1.6.)
şeklinde elde olur. Örneğin 퐶 = (4, 2) ise 푑 = 푑 = 2 olur. Buradan 푇 = (4, −5)
olur. Öteleme vektörünün uzunluğu ise,
푇 = 푇 = √3 (1.7.)
şeklindedir. Çemberin çevre uzunluğu olan L denklem 1.3’de verilmektedir. 퐶 =
(5, 5) armchair nanotüp için, 푑 = 3푑 = 15 ve 푇 = (1, −1) olur. 퐶 = (9,0) zigzag
nanotüp için ise, 푑 = 푑 = 9 ve 푇 = (1, −2) olur.
푎 ve 푎 vektörleri 2 boyutlu grafenin birim hücresini tanımlarken, 퐶 ve 푇
vektörleri 1 boyutlu nanotüpün birim hücresini tanımlar. Nanotüpün birim hücresi
퐶 × 푇 , |푎 × 푎 | ile ifade edilen hegzagonlara bölünür. Birim hücredeki hegzagon
sayısını N ile gösterirsek,
푁 =풉 ×
| × |= ( ) = (1.8.)
şeklinde elde edilir. Her hegzagon 2 karbon atomu içerir. Dolayısıyla nanotüpün her
birim hücresinde 2N karbon atomu bulunur (Saito ve ark., 1998).

1. GİRİŞ ERKAN TETİK
13
1.1.4. Simetri Vektörü: 푹
Karbon atomlarının bir boyutlu nanotüp birim hücresi içerisindeki konum
vektörleri, grup simetri vektörü 푅’nin i kez çarpımı ile yani 푖푅 ile belirlenmektedir.
Burada 푖 bir tamsayıdır (푖 = 1 … 푁). 푖푅 çarpımı birim hücre dışına çıktığı zaman
periyodik sınır koşulları kullanılarak 푇 veya 퐶 ’ın bir integral sayısı tarafından
öteleme boyunca birim hücre içerisinde kalması sağlanır.
Şekil 1.8. 푇, 푅 ve 퐶 arasındaki vektörel ilişki
푅 vektörü nanotüpteki karbon atomlarının koordinatlarını oluşturmak için
kullanılır (Şekil 1.8). 푅 vektörü 푎 ve 푎 vektörleri tarafından,
푅 = 푝푎 + 푞푎 (1.9)
bağıntısı ile ifade edilir. p ve q birleşmeler dışında ortak bölene sahip değildir. 푅
vektörünün 퐶풉 bileşeni yani 퐶풉. 푅 çarpımı 푇 × 푅 ile orantılıdır. 푇 × 푅 ise,
푇 × 푅 = (푡 푞 − 푡 푝)(푎 × 푎 ) (1.10.)
ile ifade edilir. Denklemin sağ tarafındaki (푡 푞 − 푡 푝) tam sayıdır. 푅 vektörünü en
küçük konum vektörü formunda seçmek için (푖 = 1),
퐶풉
푇
푶 Ψ = 2휋/푁 2휋
휏 푅(Ψ|휏)

1. GİRİŞ ERKAN TETİK
14
푡 푞 − 푡 푝 = 1, (0 < 푚푝 − 푛푞 ≤ 푁) (1.11)
olarak alınır. Denklem 1.2’deki 푡 ve 푡 birleşmeler dışında ortak bölene sahip
değilse, p ve q için denklem 1.11’in çözümü belirlenebilir. Denklem 1.11’in sağ
tarafındaki koşul, 푅 vektörünün bir boyutlu nanotüp içerisinde olmasından dolayı
ortaya çıkar. Denklem 1.3, 1.7 ve 1.8 kullanılarak,
0 <.
=풉×
= < 1 (1.12.)
şeklinde elde edilir. Benzer olarak, denklem 1.5 ve 1.8 kullanılarak, bir boyutlu birim
hücre içerisinde 푅 vektöründen ortaya çıkan bir diğer gerekli koşul,
0 <. 풉 =
× = < 1 (1.13.)
olur ve denklem 1.3’den,
0 < 푡 푞 − 푡 푝 ≤ 푁 (1.14.)
koşulu elde edilir. Denklem 1.11’in ilk koşulu denklem 1.13’den belirlenebileceği
için, 푅’nin tanımlanması için bu koşul şart değildir. Nanotüpün birim hücresinin 푖푅
(푖 = 1 … 푁) konum vektörleri ile tüm N noktalarını belirlerken, her i değeri için
푖(푡 푞 − 푡 푝) = 푖 ifadesi kullanılır. 푁푅’nin 퐶 bileşeni her zaman 퐶 = 퐿 eşitliğini
sağladığı gerçeği kullanılarak, 푖푅 vektörleri nanotüp birim hücresi içerisinde N farklı
konum tanımlar ve böylece 퐶 vektörünün doğrultusu boyunca izdüşümleri için
farklı değerlere sahip olur. Bu sebeple, 푖푅 (푖 = 1 … 푁) nanotüpün birim hücresinde
N farklı atom konumu oluşturur.
푅 vektörü, 푇 doğrultusunda 휏 ötelemesiyle birleşmiş bir 휓 açısının nanotüp
ekseni etrafındaki dönmesiyle ilişkilidir ve chiral nanotüpün temel uzay grubu
simetri operatörünü ifade eder ve şekil 4’de görüldüğü gibi 푅 = (휓|휏) şeklinde

1. GİRİŞ ERKAN TETİK
15
yazılır. 퐶 chiral vektörü üzerindeki 푅’nin izdüşümü 퐿/푑 tarafından ölçeklenmiş 휓
açısını verir. 푇 üzerindeki 푅’nin izdüşümü ise KNT’nin bir boyutlu uzay grubunun
temel simetri operatörünü 휏 ötelemesini verir. (푝, 푞) tamsayıları, (휓|휏) simetri
operatörünün (0,0) konumundaki bir atomu hareket ettirdiği zaman ulaşılan
koordinatları ifade eder. (휓|휏) nanotüp için simetri operatörü ise, o zaman
(휓│휏)ퟐ, (휓│휏)ퟑ, … (휓│휏) 퐶 tarafından ifade edilen bir abelyen gurubun farklı
simetri operatörleridir. 푅 × 퐶 ve 푅 × 푇 vektör çarpımları alınarak ve denklem 1.3,
1.8 ve 1.11 kullanılarak 휏 uzunluğunun ve 휓 dönme açısının ifadesi,
휏 =× 풉 = ( )| × | = ( ) (1.15.)
휓 =×
= ( )√
√ = (1.16.)
olarak ifade edilir.
CNT’nin elde edilmesi için 퐶 vektörün şekil 1.5, 1.6 ve 1.7’de görüldüğü
gibi bir silindir etrafında sarılması gerekir. (휓│휏) simetri operatörü O örgü
noktasını, C örgü noktasına taşır ve atom konumlarını belirler. 푁푅 = (휓|휏) vektörü
silindirik yüzeyi gösterir. Tüp etrafındaki 2휋’lik dönmeden sonra, 푁푅 vektörü O
noktasından C noktasına gelir, ancak bu iki nokta birbirinden 푀푇 vektörü kadar
ayrılır. 푁푅,
푁푅 = 퐶풉 + 푀푇 (1.17.)
ile ifade edilir. M ise,
푀 ≡ 푚푝 − 푛푞 (1.18.)

1. GİRİŞ ERKAN TETİK
16
şartını sağlar ve bir tamsayıdır. M, O noktasından C noktasına ulaşmak için
uygulanması gereken 푇 vektörü sayısıdır (Şekil 1.9).
Şekil 1.9. Nanotüpün döndürülmesi. 푁푅 = (휓|휏) vektörü silindirik yüzey üzerinde görülmektedir. Tüp 2휋 kadar döndürüldükten sonra, 푁푅 vektörü O noktasının eşdeğeri olan C noktasına ulaşır. Fakat C noktası, o noktasından 푀푇 vektörü kadar ayrılır.
Çizelge 1.3. KNT’lerin Temel Parametreleri 퐶풉 d dR dt (A°) L/a 푇 T/a N 푅 M
(4, 2) 2 2 4,15 √28 (4, -5) √21 28 (1, -1) 6 (5, 5) 5 15 6,78 √75 (1, -1) 1 10 (1, -0) 5 (9, 0) 9 9 7,05 9 (1, -2) √3 18 (1, -1) 9 (6, 5) 1 1 7,47 √91 (16, -17) √273 182 (1, -1) 11 (7, 4) 1 3 7,55 √93 (5, -6) √31 62 (1, -1) 11 (8, 3) 1 1 7,72 √97 (14, -19) √291 194 (3, -4) 41
(10, 10) 10 30 13,56 √300 (1, -1) 1 20 (1, -0) 10
(n, n) n 3n √3푛푎휋 √3푛 (1, -1) 1 2n (1, -0) n
(n, 0) n n 푛푎휋 n (1, -2) √3 2n (1, -1) n
Çizelge 1.3’de (n, m) indisleri tarafından sınıflandırılan KNT’lerin bazı
önemli parametreleri listelenmektedir. Tablodaki d parametresi, denklem 1.6’da
C hh
C
MT
T R
NR
Oh

1. GİRİŞ ERKAN TETİK
17
verilen dR niceliğine bağlı olarak n ve m indislerinin en büyük ortak bölenini temsil
etmektedir. Tablo 1.3’de dt sayısı ile A° biriminde nanotüpün çapı, 퐿 ile chiral
vektörünün uzunluğu, 푇 öteleme vektörü, 푅 simetri vektörü ve N bir boyutlu
nanotüpün birim hücresindeki hegzagon sayısı verilmektedir.
Çizelge 1.3’ün kullanımını incelemek için şekil 1.7’deki chiral (4, 2)
nanotüpü ele alalım. (4, 2) nanotüp 푇 = (4, −5), 푅 = (1, −1), N=28, d= dR=2 ve
M=6 parametreleri ile verilir ve birim hücresinde 56 tane atoma sahiptir. İkinci bir
örnek olarak, (7, 4) cihral nanotüpü incelersek, ortak bölene sahip olmadığını
görürüz ve d=1 olur. 푛 − 푚 = 3 olduğu için dR=3 olur. Böylece tabloda da
görüldüğü gibi 퐿 = √93푎, 푇 = √31푎, 푁 = 62 ve 푀 = 11 olur. Herhangi bir
nanotüpün elde edilmesindeki temel parametreler çizelge 1.3 verilmiştir. Bu
parametreler n ve m indileri ile 퐶 chiral vektörüne bağlı olarak elde edilmektedir.
1.1.5. Birim Hücre ve Brillouin Bölgeleri
Gerçek uzayda bir nanotüp için birim hücre şekil 1.7(a)’da OACB ile
gösterildiği gibi, 푇 öteleme vektörü ve 퐶 chiral vektörü tarafından oluşturulan
dikdörtgen ile verilmektedir. Bu birim hücrede 2N tane karbon atomu olduğu için, π
bağı ve 휋∗ anti-bağı elektronik enerji bantlarının N çiftinden oluşur. Benzer olarak
fonon dağılım bağıntıları birim hücredeki her karbon atomunun vektörel olarak yer
değiştirmesinin bir sonucu olarak 6N bölümden oluşacaktır.
Çembersel doğrultudaki 퐾 ve nanotüp ekseni boyunca 퐾 ters örgü
vektörlerinin ifadesi
푅 . 퐾 = 2휋훿 (1.19.)
bağıntısından elde edilir. Burada 퐾 ve 퐾 sırasıyla gerçek ve ters uzaydaki örgü
vektörleridir. Nanotüpler bir boyutlu materyaller olduğu için, sadece 퐾 ters örgü

1. GİRİŞ ERKAN TETİK
18
vektörüdür. 퐾 , 퐶 vektörü doğrultusundaki farklı k değerlerini verir. Denklem 1.5,
1.9’i ve aşağıdaki bağıntıları kullanarak,
퐶 . 퐾 = 2휋, 푇. 퐾 = 0 퐶 . 퐾 = 0, 푇. 퐾 = 2휋
(1.20.)
퐾 ve 퐾 ’nin ifadesini,
퐾 = −푡 푏 + 푡 푏 , 퐾 = 푚푏 − 푛푏 (1.21.)
şeklinde elde edilir. Burada 푏 ve 푏 iki boyutlu grafitin ters örgü vektörleridir.
Şekil 1.10. Karbon nanotüpün Birillouin bölgesi 퐾 vektörüne paralel olan WW'
çizgi parçası ile temsil edilmektedir
Şekil 1.10’da, 퐶 = (4, 2) chiral nanotüp için 퐾 ve 퐾 ters örgü vektörleri
görülmektedir. Bu bir boyutlu nanotüpün ilk Brillouin bölgesi olan 푊푊 çizgisi ile
verilmektedir. 푁퐾 = −푡 푏 + 푡 푏 iki boyutlu grafitin ters örgü vektörleriyle
benzer olduğu için 푁퐾 ’den farklı olan iki dalga vektörü birbirine eşittir. 푏 ve 푏
birleşme noktaları dışında ortak bölene sahip olmadıkları için, 푁 − 1 vektör dışında
휇퐾 (burada 휇 = 1, … , 푁 − 1) iki boyutlu grafitin ters örgü vektörleridir. Böylece N
tane dalga vektörü 휇퐾 (burada 휇 = 1, … , 푁 − 1), şekil 1.10’da 푁 = 28 paralel çizgi
parçasıyla gösterildiği gibi N farklı 푘 vektörü verir. Bu 푘 vektörleri 퐶 üzerindeki
푏
Г M
K
퐾
W
W'
푏
퐾

1. GİRİŞ ERKAN TETİK
19
periyodik sınır koşullarıyla ilişkili dalga vektörlerinden ortaya çıkar. Şekil 3.5’deki
tüm paralel çizgi parçalarının uzunluğu bir boyutlu ilk Brillouin bölgesinin uzunluğu
olan 2휋/푇 uzunluğuna eşittir. 푘 vektörlerinin N farklı değeri için, N tane bir boyutlu
enerji bandı ortaya çıkar. 푇’nin öteleme simetrisinden dolayı, sınırsız uzunlukta bir
nanotüp için 퐾 doğrultusunda sürekli dalga vektörüne sahip oluruz. Ancak, 퐿
uzunluğunda bir nanotüp için dalga vektörleri arasındaki mesafe 2휋/퐿
uzunluğundadır. Dalga vektörleri arasındaki bu uzunluk deneysel olarak
gözlenmektedir (Tans ve ark., 1997).
1.2. Karbon Nanotüplerin Elektronik Özellikleri
Karbon nano tüplerin iki bağ çeşidi vardır. En saf haliyle grafit’te bulunan
silindir duvar boyunca 휎 bağları hegzagonal ağı oluşturur. 휋 bağları ise nanotüplerin
yüzeyine diktir. Bu bağlar farklı tüpler arasındaki van-der-Waals etkileşimini
zayıflatmaktan sorumludurlar. İlk bakışta, karbon nanotüplerin (ya da grafitin),
elektronik özellikleri için en önemli unsurun düz σ bağları olduğu düşünülebilir.
Ancak durum böyle değildir. Örneğin bu bağlar, görülebilir enerji aralığında
elektronik taşıma ya da optik emilimde rol almak için Fermi seviyesinden çok
uzaktadırlar. 휋 bağları ise karbon nanotüplerin elektronik yapısının oluşumunda
önemli bir yol oynamaktadır.
1.2.1. Enerji Dağılımının Zone-Folding Yaklaşımı
Tek duvarlı karbon nanotüplerin elektronik yapısı basit olarak iki boyutlu
grafitten elde edilebilir. 퐶 chiral vektörü tarafından nitelendirilen dairesel
doğrultudaki periyodik sınır koşulları kullanılarak, 푇 öteleme vektörünün
doğrultusuyla ilişkili dalga vektörü sınırsız uzunluktaki bir nanotüp için sürekli
kalırken, 퐶 doğrultusu ile bağlantılı dalga vektörünün kuantumlanmış olduğu
görülür. Böylece, enerji bantları bir boyutlu enerji dağılımının bir setinden oluşur.

1. GİRİŞ ERKAN TETİK
20
A B
Г
K
M
Denklem 1.8’deki N değerinden faydalanarak bir boyutlu enerji dağılım
bağıntılarını
퐸 (푘) = 퐸 푘 + 휇퐾 , 휇 = 0, … , 푁 − 1, 푣푒 − < 푘 < (1.22.)
olarak elde ederiz. Bu denklemde 퐸 özdeğerleri 휔 푘 , 푘 ve 푘 ’nin bir
fonksiyonu olarak
퐸 푘 = ∈ ∓
∓ (1.23.)
şeklinde yazarız. Herhangi bir (n, m) nanotüp için, şekil 1.11’de gösterildiği gibi
kesikli çizgiler iki boyutlu Brillouin bölgesindeki bir K noktası boyunca ilerler.
Burada iki boyutlu grafitin 휋 ve 휋∗ enerji bantları simetriye göre dejeneredir. Bir
boyutlu enerji bantları ise sıfır enerji aralığına sahiptir. Dahası, Fermi seviyesindeki
durum yoğunlukları bu karbon nanotüpler için sınırlı bir değere sahiptir ve bu yüzden
onlar metalik özellik gösterirler.
Şekil 1.11. İki boyut grafitin birim hücresi (a) ve Brillouin bölgesi (b) sırasıyla
eşkenar dörtken ve gölgeli hegzagonal ile gösterilmektedir
푎
푎
푏
푏

1. GİRİŞ ERKAN TETİK
21
Ancak kesikli çizgiler K noktasından geçmezse, o zaman valans ve iletim
bantları arasındaki sınırlı bir enerji aralığı ile yarı iletken davranış göstermesi
beklenir. Metalik enerji bandını elde etmek için gerekli koşul şekil 1.12’deki 푌퐾
vektörünün uzunluğunun oranıdır ki bu bir tam sayıdır. Metalik nanotüpler için
vektör koşulu (2푛 + 푚) ya da (푛 − 푚) değerinin 3’ün katları olmasını gerektirir. Bu
durumda (n, n) ile ifade edilen armchair nanotüpler her zaman metalik özellik
gösteriler. (n, 0) zigzag nanotüpler ise sadece n 3’ün katı olduğu durumlarda
metaliktir.
Şekil 1.12. Metalik enerji bandları için koşul: 푌퐾 vektörünün uzunluğunun 퐾 vektörünün uzunluğuna oranı bir tamsayı olduğu zaman, metalik enerji bandları elde edilir
1.2.2 Armchair ve Zigzag Nanotüplerin Enerji Dağılımı
Dağılım bağıntılarının açık ifadelerini elde etmek için, dikkate alınması
gereken en basit yapı yüksek simetriye sahip nanotüplerdir. Şekil 1.13’ü incelersek,
yüksek simetrili (achiral nanotüp) nanotüpler için Brillouin bölgelerini ve birim
hücreleri görebiliriz. Şekil 1.13’de (a) armchair ve (b) zigzag nanotüpleri temsil
etmektedir. (n, n) armchair nanotüpler için enerji özdeğerlerini elde etmek için
kullanılan uygun periyodik sınır koşulları az sayıda izin verilen 푘 , dalga
vektörleriyle sınırlıdır, dairesel doğrultuda,
Г
K
K'
K'
M
M
Y
W
W' 퐾
퐾
1. GİRİŞ ERKAN TETİK
22
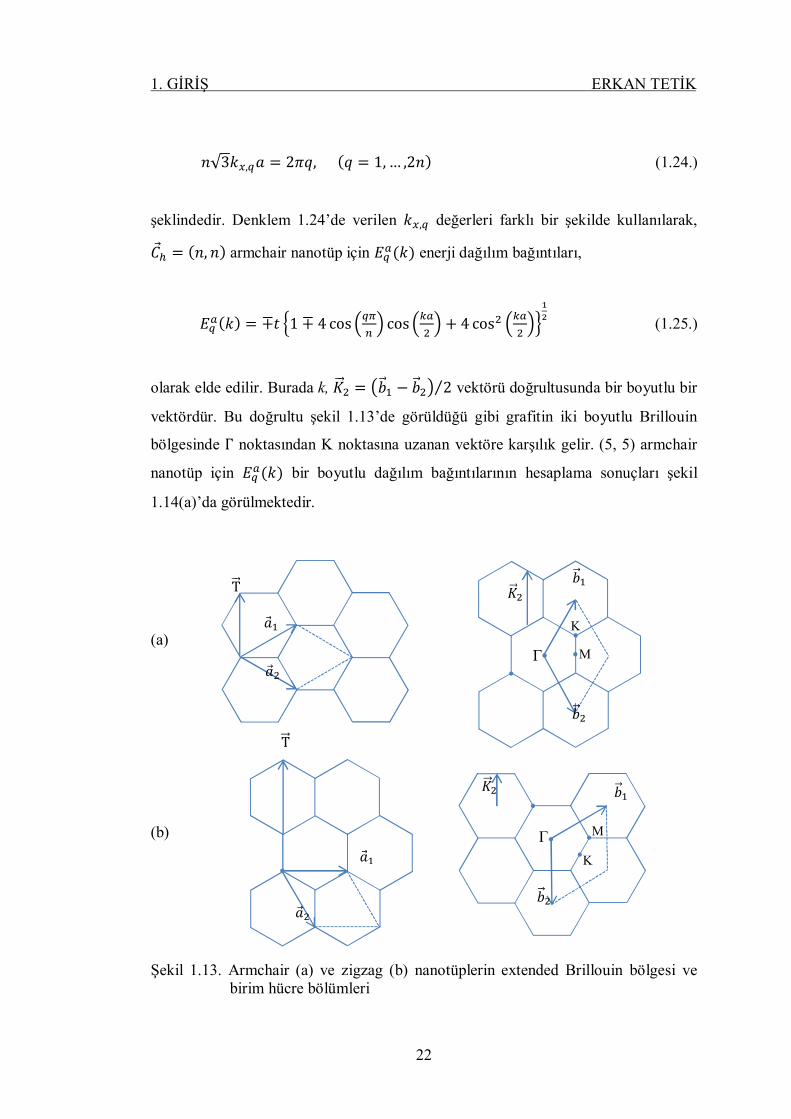
푛√3푘 , 푎 = 2휋푞, (푞 = 1, … ,2푛) (1.24.)
şeklindedir. Denklem 1.24’de verilen 푘 , değerleri farklı bir şekilde kullanılarak,
퐶 = (푛, 푛) armchair nanotüp için 퐸 (푘) enerji dağılım bağıntıları,
퐸 (푘) = ∓푡 1 ∓ 4 cos cos + 4 cos (1.25.)
olarak elde edilir. Burada k, 퐾 = 푏 − 푏 2⁄ vektörü doğrultusunda bir boyutlu bir
vektördür. Bu doğrultu şekil 1.13’de görüldüğü gibi grafitin iki boyutlu Brillouin
bölgesinde Γ noktasından K noktasına uzanan vektöre karşılık gelir. (5, 5) armchair
nanotüp için 퐸 (푘) bir boyutlu dağılım bağıntılarının hesaplama sonuçları şekil
1.14(a)’da görülmektedir.
(a)
(b)
Şekil 1.13. Armchair (a) ve zigzag (b) nanotüplerin extended Brillouin bölgesi ve birim hücre bölümleri
푎 K
Г M
T
K Г M
푎
퐾 푏
푏
T
푎
푎
퐾 푏
푏

1. GİRİŞ ERKAN TETİK
23
(a) (b) (c)
Şekil 1.14. Armchair (5, 5) (a), zigzag (9, 0) (b) ve zigzag (10, 0) (c) karbon nanotüpler için bir boyutlu enerji dağılım grafikleri
Şekil 1.14(a)’de valans bantları için eşit sayıda ve iletim bantları için 6
dağılım bağıntısı görülmektedir. Her durumda, ‘a’ ile gösterilen 2 bant dejenere
değildir ve ‘e’ ile gösterilen 4 bant çift katlı dejeneredir. Tüm armchair nanotüpler
için, enerji bantları sınır bölgesinde büyük bir dejenerelik göstermektedir. Burada
푘푎 = 휋 ve buna göre,
퐸 푘 , , = ∓푡 (1.26.)
şeklinde olur. Gerçek uzay örgü için resim 1.13(a)’ün birim hücresinde 4 karbon
atomu olmasına rağmen, bir grafen katmanının aynı alt örgüsü üzerinde iki karbon
atomu simetrik olarak eşittir. Bu eşitlik Brillouin bölgesinin sınırında enerji
bantlarında bir dejenereliği sebep olur. Armchair nanotüp için şekil 1.14(a)’da valans
ve iletim bandı k noktasında karşı karşıyadır. Karşılaşma Fermi seviyesinde meydana
gelir ve enerji bantları ∓푘 değerleri için simetriktir. Valans ve iletim bandı
arasındaki dejenere noktadan dolayı, (5, 5) armchair nanotüp sıfır bant aralıklı bir
yarı iletken olur. Fakat aynı zamanda sınırlı sıcaklıklarda metalik iletkenlik de
gösterir. Çünkü bölünemeyecek kadar küçük uyarılmalar iletim bandı içindeki
uyarıcı taşıyıcılara ihtiyaç duyarlar. Tüm (n, n) armchair nanotüpler 푘 =

1. GİRİŞ ERKAN TETİK
24
∓2휋 (3푎)⁄ ’da en yüksek valans bandı ve en düşük iletim bandı arasında bir dejenere
banda sahiptir. Bu bantlar Fermi seviyesinde karşı karşıyadır. Böylece tüm armchair
nanotüplerin iki boyutlu grafen katmanına benzer olarak metalik iletkenlik
göstermesi beklenir.
퐶 = (푛, 0) zigzag nanotüpler için enerji bantları periyodik sınır koşulları
yazılarak benzer şekilde elde edilebilir. 푘 ’ye göre,
푛푘 , , 푎 = 2휋푞, (푞 = 1, … , 2푛) (1.27.)
şeklinde (n, 0) zigzag nanotüpün 4n durumları için bir boyutlu dağılım bağıntılarını
elde etmek için,
퐸 (푘) = ∓푡 1 ∓ 4 cos √ cos + 4 cos (1.28.)
olarak yazarız. (9, 0) ve (10, 0) zigzag nanotüpler için 퐸 (푘) bir boyutlu dağılım
bağıntılarının bir boyutlu hesaplama sonuçları sırasıyla şekil 1.15(b) ve 1.15(c)’de
gösterilmektedir. (9, 0) nanotüp için 푘 = 0’da bant aralığı yoktur, oysa (10, 0)
nanotüp bant aralığına sahiptir. Genel olarak (n, 0) zigzag nanotüpler için n 3’ün katı
olduğu zaman 푘 = 0’da enerji bant aralığı sıfır olur. Ancak, n 3’ün katı değilse
푘 = 0’da enerji bant aralığı açıktır.
1.2.3. Chiral Nanotüplerin Enerji Dağılımı
Metalik nanotüplerin bant dejenereliğindeki k değerlerinin armchair ve zigzag
nanotüpler için 푘 = 0 yada 푘 = ∓ 2휋 3푇⁄ olduğunu dikkate almamız gerekir. Bu k
değerleri aynı zamanda yarı iletken zigzag nanotüpler için bant aralığının bulunduğu
yere karşılık gelir. Aynı k değerleri aynı zamanda chiral nanotüplerin genel
durumları için enerji aralıklarının pozisyonlarını göstermektedir.

1. GİRİŞ ERKAN TETİK
25
(a) (b)
Şekil 1.15. Bir boyutlu metalik chiral (9, 6) (a) ve (7, 4) (b) nanotüp için enerji bant grafikleri. Fermi seviyesi sıfırdadır
Şekil 1.15(a) ve (b)’de sırasıyla (9, 6) ve (7, 4) chiral nanotüplerin dağılım
grafikleri görülmektedir. her iki durum için n-m değeri 3’ün katı olduğu için, bu iki
chiral nanotüp metalik özellik göstermektedir. Ayrıca, Fermi seviyesinde çakışan
bantların k değerleri 푘 = 0 ve 푘 = ∓ 2휋 3푇⁄ ’dedir. Chiral nanotüpler için denklem
1.21’de verilen 퐸(푘) bağıntısının daha detaylı analizi, nanotüplerin 3 kategoride
sınıflandırılabileceğini göstermektedir. Bu kategoriler Çizelge 1.4’de
özetlenmektedir (Dresselhaus ve ark., 1994). Çizelge 1.4’de göre nanotüpün
yarıiletken özellik göstermesi için bir durum, metalik özellik göstermesi için iki
durum söz konusudur. Bu durumlar n ve m indislerinin aldığın değerlere göre
belirlenmektedir.
Çizelge 1.4. Chiral nanotüplerin sınıflandırılması. gcd değeri en büyük ortak bölendir.
Özellikler gcd(푛 − 푚, 3) 푑( ) Dejenerelik Yarıiletken 1 d 0 Metal-1 3 d 푘 = 0 için 4 Metal-2 3 3d 푘 = ∓2휋/3푇 için 2

1. GİRİŞ ERKAN TETİK
26
1.2.4. Elektronik Durum Yoğunluğu ve Yasak Bant Aralığı
Tüm metalik nanotüpler için, onların çaplarına ve 퐶 değerine bağımlılığı,
nanotüp ekseni boyunca birim uzunluk başına durum yoğunluğu sabittir ve
푁(퐸 ) =√ | |
(1.29.)
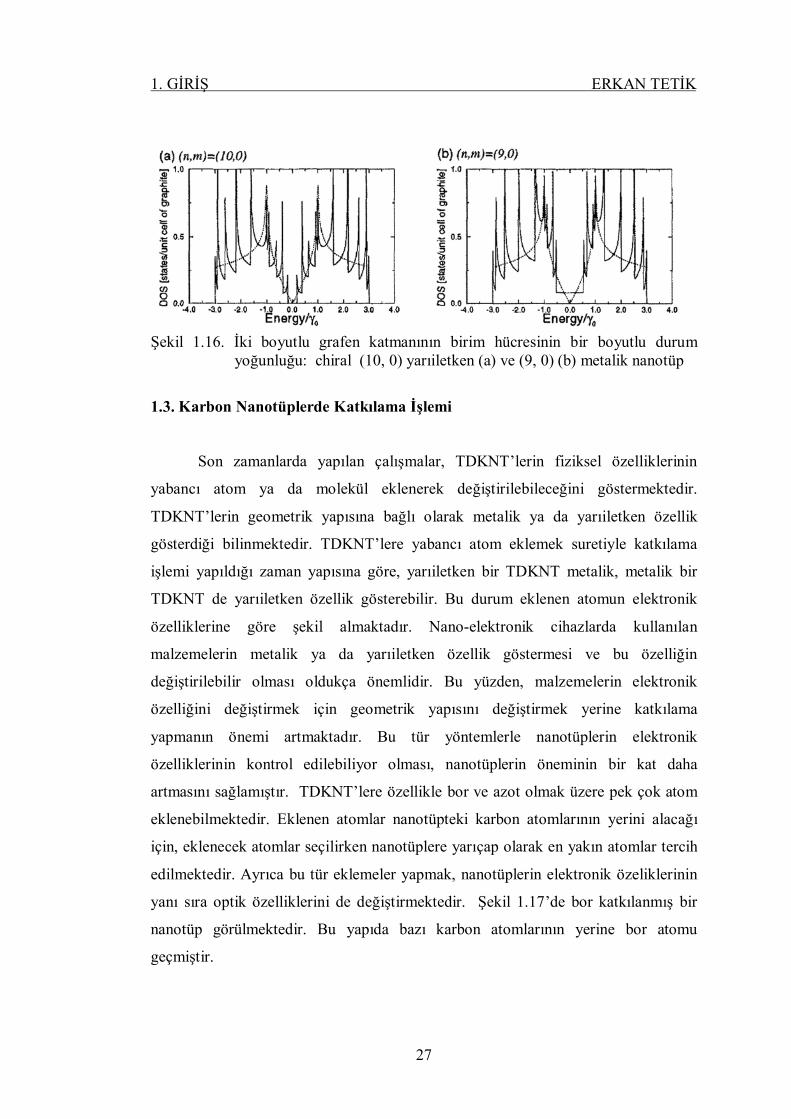
denklemi ile verilir. Burada a grafen katmanının örgü sabitidir. İki tane nanotüpün
enerji dağılımı şekil 1.16’da gösterilmektedir. Bu şekilde (9, 0) metalik ve (10, 0)
yarıiletken nanotüpler karşılaştırılmaktadır. Yarıiletken nanotüplerde Fermi enerjisi
seviyesindeki durum yoğunluğu sıfır değerine sahiptir, fakat metalik nanotüplerde bu
değer sıfır değildir ve aynı zamanda çok küçüktür. Ayrıca, yarıiletken nanotüplerin
yasak bant aralıkları nanotüpün çapı olan 푑 değerine bağlıdır. Yani,
퐸 = | | (1.30.)
denklemine göre nanotüpler chiral açısından bağımsız davranış sergilerler. Burada
푎 = 푎/√3 grafen katmanı üzerindeki en yakın komşu 퐶 − 퐶 bağı uzaklığıdır.
Buna göre yarıiletken nanotüpler chiral açısından bağımsız olarak yalnızca nanotüp
çapına göre karakterize edilirler. Taramalı Tünelleme Spestrokopi (STS) ile yapılan
durum yoğunluğu ölçümleri KNT’lerin elektronik yapıları hakkında önemli bilgiler
vermektedir (Olk ve ark., 1994). Bazı ölçümler nanotüplerin 2/3 civarının
yarıiletken, 1/3 civarının iletken olduğunu doğrulamaktadır. Ayrıca, yarıiletken
nanotüpler üzerinde yapılan ölçümler bant aralıklarının 1/푑 ile orantılı olduğunu
göstermektedir (Mintnire ve ark., 1995). Durum yoğunluğundaki rezonanslar
Taramalı Tünelleme Mikroskopi (STM) ile yapılan çalışmalarla çapları ve chiral
açıları belirlenen yarıiletken ve metalik nanotüpler gözlemlendi. STS sonuçları yasak
bant aralığı ile ilgili teorik modeldeki sonuçları doğrulamaktadır.
1. GİRİŞ ERKAN TETİK
27
Şekil 1.16. İki boyutlu grafen katmanının birim hücresinin bir boyutlu durum yoğunluğu: chiral (10, 0) yarıiletken (a) ve (9, 0) (b) metalik nanotüp
1.3. Karbon Nanotüplerde Katkılama İşlemi
Son zamanlarda yapılan çalışmalar, TDKNT’lerin fiziksel özelliklerinin
yabancı atom ya da molekül eklenerek değiştirilebileceğini göstermektedir.
TDKNT’lerin geometrik yapısına bağlı olarak metalik ya da yarıiletken özellik
gösterdiği bilinmektedir. TDKNT’lere yabancı atom eklemek suretiyle katkılama
işlemi yapıldığı zaman yapısına göre, yarıiletken bir TDKNT metalik, metalik bir
TDKNT de yarıiletken özellik gösterebilir. Bu durum eklenen atomun elektronik
özelliklerine göre şekil almaktadır. Nano-elektronik cihazlarda kullanılan
malzemelerin metalik ya da yarıiletken özellik göstermesi ve bu özelliğin
değiştirilebilir olması oldukça önemlidir. Bu yüzden, malzemelerin elektronik
özelliğini değiştirmek için geometrik yapısını değiştirmek yerine katkılama
yapmanın önemi artmaktadır. Bu tür yöntemlerle nanotüplerin elektronik
özelliklerinin kontrol edilebiliyor olması, nanotüplerin öneminin bir kat daha
artmasını sağlamıştır. TDKNT’lere özellikle bor ve azot olmak üzere pek çok atom
eklenebilmektedir. Eklenen atomlar nanotüpteki karbon atomlarının yerini alacağı
için, eklenecek atomlar seçilirken nanotüplere yarıçap olarak en yakın atomlar tercih
edilmektedir. Ayrıca bu tür eklemeler yapmak, nanotüplerin elektronik özeliklerinin
yanı sıra optik özelliklerini de değiştirmektedir. Şekil 1.17’de bor katkılanmış bir
nanotüp görülmektedir. Bu yapıda bazı karbon atomlarının yerine bor atomu
geçmiştir.

1. GİRİŞ ERKAN TETİK
28
Şekil 1.17. Bor atomu katkılanmış bir karbon nanotüp
Son yıllarda, p-tipi ve n-tipi nanotüpler yapmak için bor ve azot atomlarının
nanotüplere yer değiştirme ile katkılama yapılması yoğun olarak çalışılmaktadır.
Bazı deneysel çalışmalarda bor ve azot atomları ile KNT’lere yer değiştirme yoluyla
katkılama yapılmasına rağmen, nanotüplere katkılama ile ilgili deneysel çalışmaların
çoğunluğu alkali metaller ile yapılmaktadır (Zhou ve ark., 1994). Bu işlem yer
değiştirme ile katkılama olarak dikkate alınmasa da, nanotüplerde metalik
iletkenliğin artmasına yol açtığı tahmin edilmektedir (Gal’pern ve ark., 1993). Kristal
yapı içerisindeki grafen katmanları arasına yerleşen alkali metaller grafitin orta
kısımlarına bir katman olarak ilave edilmiş olur. Bu alkali metal katmanları grafit
katmanlarına elektronlarını verirler, böylece elektrik iletkenliğine büyük miktarlarda
artış meydana gelir. KNT’lerin kapalı silindirik yapısından dolayı, büyük alkali metal
iyonları mükemmel nanotüplere kolayca giriş yolu bulamaz. Ayrıca ÇDKNT’lerin
katmanları arasında bu iyonlar istedikleri yeri bulması oldukça zordur. Bu yüzden,
alkali metaller neredeyse sadece yapısal kusurlarının olduğu yerlerde yer bulabilirler.
İyonlar nanotüpün daha dış yüzeylerine girdikleri zaman, nanotüp duvarlarında
büyük boyutta yarıklar açarlar. Bu etki alkali metallerin katkılandığı karbon
nanotüplerle ilgili mikroskobik çalışmalarda görülebilir.
TDKNT’lere alkali metal ve holojen katkılamak için, kristal grafite yapılan
katkılamaya benzer şekilde iki bölgeli bir yapı oluşturuldu (Dresselhaus ve ark.,

1. GİRİŞ ERKAN TETİK
29
1981). Bu çalışmalarda tipik sıcaklık alkali metaller için yaklaşık 120C ve
nanotüpler için 160C olarak alınmıştır. Bu çalışmalarda Rb, K ve Br2 gibi alkali
metaller kullanılmıştır. Ölçümler K ve Br2 katkılanan nanotüplerin elektriksel
iletkenliklerinde ciddi bir artış olduğunu doğrulamaktadır.
Dış tarafında 5 karbon katmanı, orta bölgesinde 6 bor-azot katmanı ve iç
kısımda 3 karbon katmanında oluşan 12 nm çapındaki bir ÇDKNT, N2 atmosferdeki
karbon elektrodu ve HfB2 elektrodu kullanarak ark boşaltma metodu tarafından
sentezlenebilmektedir (Suenaga ve ark., 1997). Sentezelenen nanotüplerin elektronik
özellikleri hakkında bilgiler elde edilmektedir.
Teorik çalışmalarda nanotüp duvarındaki karbon atomlarının bulunduğu
konumların yerine katkılama yapılacak atomlar yerleştirilmektedir. Atomlar istenilen
konuma, istenildiği kadar yerleştirilebilir. İstenilen yapı elde edildikten sonra
hesaplamalar yapılır. Bu yapılarda herhangi bir yapısal kusur olmadığından deneysel
verilerden yaklaşık % 30 ile % 40 arasında farklı sonuçlar verir. Bu farklılık
hesaplanan kristalin diğer özelliklerine göre de değişmektedir.

2. ÖNCEKİ ÇALIŞMALAR ERKAN TETİK
30
2. ÖNCEKİ ÇALIŞMALAR
Nanoteknoloji, zamanımızın en önemli teknolojik gelişmesi halini almaya
başlamıştır. 1974 yılında Tokyo Üniversitesinde Norio Taniguchi tarafından ortaya
atılan Nanoteknoloji mevcut teknolojilerin daha ileri düzeyde duyarlılık ve
küçültülmesine dayalı olarak hızla gelişen bir teknolojidir. Karbon nanotüpler
(KNT), dikkat çekici özellikleri ile Nanoteknoloji içerisinde en önemli rollerden
birini oynamaktadır. Bu önem, KNT’lerin nano boyuttaki yapısal özelliklerinden
kaynaklanmaktadır (Charles ve ark., 2003).
KNT’lerin keşfi, 1991 yılında Japonya’nın NEC Laboratuarlarında Sumio
Iijima ve grubu tarafında iç içe çok duvarlı karbon nanotüplerin (ÇDKNT) elektron
mikroskobu ile gözlenmesiyle gerçekleşmiştir. Bu keşifle beraber KNT’ler bir
araştırma alanı olarak hızla gelişmektedir (Iijima, 1991). Bu keşiften iki yıl sonra
S.Ijima ve T.Ichihashi tarafından tek duvarlı karbon nanotüpler (TDKNT)
sentezlenerek elde edilmiştir (Iijima ve Ichihashi, 1993). Daha sonra, nanotüplerle
ilgili deneysel ve teorik pek çok çalışma yapılmış ve yapılmaya devam etmektedir.
KNT’lerle ilgili yoğun çalışılmasının en önemli sebeplerinden biri farklı
geometrilerinin bulunması ve bu geometrilerde farklı elektronik özellikler
gösterebilmeleridir. Temel amaç nanotüpleri nano-elektronik aletlerde
kullanabilmektir. Ayrıca KNT’lere yabancı atom eklenerek de elektronik özellikleri
değiştirilebilmektedir. Katkılama olarak bilinen bu yöntem de yoğun olarak
çalışılmaktadır.
Çalışmamızda KNT’leri tercih etmemizin en önemli sebeplerinden biri sebebi
farklı geometrik yapıları ve bu farklı geometrik yapılarda farklı elektronik özellikler
göstermeleridir. Ayrıca geometrik olarak karbon atomuna yakın olan pek çok atomun
katkılanabilmesidir.
2.1. Karbon Nanotüpler İçin Deneysel Kanıtlar
TDKNT’lerin varlığı yüksel çözünürlüklü iletim elektron mikroskopu (High
resulution transmission electron microscopy, TEM) ve taramalı tünelleme
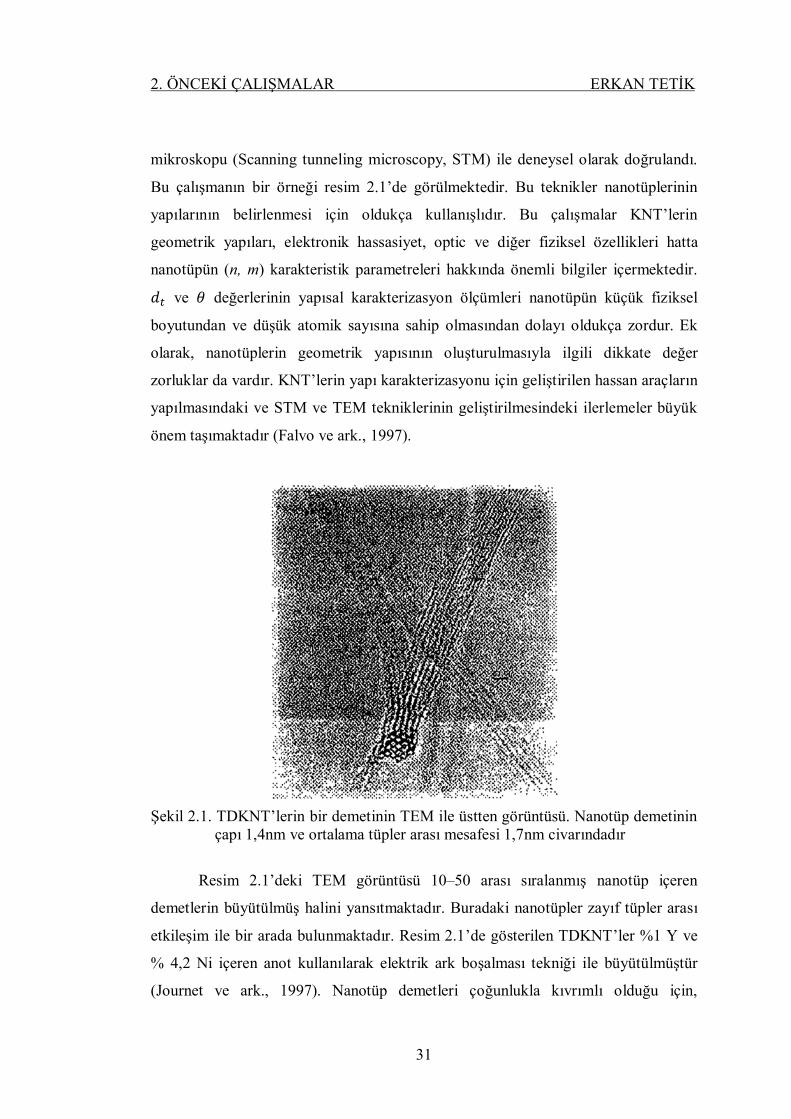
2. ÖNCEKİ ÇALIŞMALAR ERKAN TETİK
31
mikroskopu (Scanning tunneling microscopy, STM) ile deneysel olarak doğrulandı.
Bu çalışmanın bir örneği resim 2.1’de görülmektedir. Bu teknikler nanotüplerinin
yapılarının belirlenmesi için oldukça kullanışlıdır. Bu çalışmalar KNT’lerin
geometrik yapıları, elektronik hassasiyet, optic ve diğer fiziksel özellikleri hatta
nanotüpün (n, m) karakteristik parametreleri hakkında önemli bilgiler içermektedir.
푑 ve 휃 değerlerinin yapısal karakterizasyon ölçümleri nanotüpün küçük fiziksel
boyutundan ve düşük atomik sayısına sahip olmasından dolayı oldukça zordur. Ek
olarak, nanotüplerin geometrik yapısının oluşturulmasıyla ilgili dikkate değer
zorluklar da vardır. KNT’lerin yapı karakterizasyonu için geliştirilen hassan araçların
yapılmasındaki ve STM ve TEM tekniklerinin geliştirilmesindeki ilerlemeler büyük
önem taşımaktadır (Falvo ve ark., 1997).
Şekil 2.1. TDKNT’lerin bir demetinin TEM ile üstten görüntüsü. Nanotüp demetinin çapı 1,4nm ve ortalama tüpler arası mesafesi 1,7nm civarındadır
Resim 2.1’deki TEM görüntüsü 10–50 arası sıralanmış nanotüp içeren
demetlerin büyütülmüş halini yansıtmaktadır. Buradaki nanotüpler zayıf tüpler arası
etkileşim ile bir arada bulunmaktadır. Resim 2.1’de gösterilen TDKNT’ler %1 Y ve
% 4,2 Ni içeren anot kullanılarak elektrik ark boşalması tekniği ile büyütülmüştür
(Journet ve ark., 1997). Nanotüp demetleri çoğunlukla kıvrımlı olduğu için,

2. ÖNCEKİ ÇALIŞMALAR ERKAN TETİK
32
nanotüpün bazı parçalarının elektron ışınıyla paralel oryantasyonu ile tüpün sonunda
TEM ile belirlenebilir. Demetler tipik olarak 5-20 nm çapındadır ve 1,7 nm tüpler
arası mesafeye sahip üçgen örgü şeklinde gösterilmektedir (52, 55). Bu nanotüplerin
periyodik paketlenmesi bu şekilde nanotüp demetlerinin birleşiminden elde edilen
elektron kırınım örnekleri tarafından doğrulanmaktadır. Nanotüp demetlerinin benzer
iki boyutlu yapısı ilk olarak lazer buharlaştırma tekniği kullanılarak rapor edilmiştir
(Thess ve ark., 1996).
Çapı nm civarında olan KNT’lerden çok zayıf elektron saçılması (atom
numarası Z=6) ve TEM araçlarının 100-200 KeV elektron ışını tarafından hasarı için
onların yüksek hassasiyeti elektron kırınım çalışmalarındaki başarıyı
zorlaştırmaktadır. Düşük elektron ışını kullanmak için alınan önlemlerle, TDKNT
(Cowley ve ark., 1997) ve ÇDKNT (Kiang ve ark., 1995) hakkında elektron kırınım
örneklerinin ve örgü resimlerinin TEM ölçümlerini elde etmek mümkün olmaktadır.
Ancak, bu TEM deneylerinin hala yetersizlikleri vardır. Yine de, TEM sonuçları
TDKNT’nin duvarlarının bir grafen katmanının yerel yapısına sahip olduğunu
göstermektedir (Iijima ve Ichihashi, 1993).
Şekil 2.2. Bu taslak chiral açısını belirlemek için kullanılan H oryantasyonu ile
düzlemler üzerindeki tesadüfi elektron ışını için girişim örneğinin nasıl olduğunu göstermektedir
Şekil 2.2’de, nanotüpün chiral açısını değiştirmek gibi oryantasyonları
ölçmek için elektron kırınım tekniklerinin nasıl kullanıldığı görülmektedir. Ayrıca

2. ÖNCEKİ ÇALIŞMALAR ERKAN TETİK
33
TEM ÇDKNT’lerde gözlemler yaparak pek çok yapı kusurunu karakterize etmek
için yaygın bir şekilde kullanılmaktadır (Endo ve ark., 1993). Çapları 2 nm’den
büyük olana nanotüpler pek çok yapısal kusur göstermek eğiliminde olmakla birlikte,
çok küçük çaplı nanotüplerde daha az yapısal kusur görülmektedir ve bunlar daha
çok mükemmele yakındır.
TDKNT’ler hakkındaki TEM elektron kırınım örnekleri üzerindeki
çalışmalar, (n, m) nanotüp için beklenen kırınım örneklerini veren bilgisayar
simülasyonları kullanılarak açıklanmaktadır (Lambin ve ark., 1997). Bu bilgisayar
simülasyonları X-ışını kırınım deneylerinden DNA molekülünün yapısının
açıklamaları ile bağlantılı olarak 1952 yılında Cochran, Crick ve Vand (Cochran ve
ark., 1952) tarafından ortaya atılan bir analiz temel alınarak oluşturulmuştur.
Herhangi bir nanotüpün 1 boyutlu birim hücresindeki tüm atomlar için yapı faktörü
hesaplanarak, beklenen kırınım kusurları bulunabilmektedir. Şekil 2.3’de (17, 3)
TDKNT için bir kırınım örneği gösterilmektedir.
Şekil 2.3. (17, 3) CNT’nin atomik yapısının herhangi bir dalga vektörü için normal düzlemi üzerindeki izdüşümü
Günümüzde, deneysel olarak çalışanlar yapılan simülasyon çalışmalarıyla,
elde ettikleri gözlemleri karşılaştırıp, sonuçlara göre çalışmalarını sürdürmektedirler.
Aslında, bilgisayar simülasyonları 1 boyutlu birim hücre içinde atomik bölgeler için
gerçek bir uzay yapısı içinde gözlenen kırınım örneklerini dönüştürme işleminde
henüz yeteri kadar başarılı değildirler. Bu alandaki çalışmalar da yoğun bir şekilde
devam etmektedir.

2. ÖNCEKİ ÇALIŞMALAR ERKAN TETİK
34
Şekil 2.4. TDKNT’ün 4,2 K sıcaklığında STM topografik resmi. Karanlık bölgeler hegzogonları göstermektedirler ve bunlar arasındaki boşluk 2,46 퐴° civarındadır. Örgü yapısından bu nanotüpün zigzag olmadığı ve armchair bir yapıya gösterdiği anlaşılmaktadır
Nanotüp içindeki karbon atomlarının periyodik dizilimi ve konumları
hakkında detaylı bilgiler STM ile yapılan çalışmalarda elde edilmektedir ve böyle
deneylerler düşük sıcaklıklarda TDKNT’ler üzerinde başarılı bir şekilde
yapılabilmektedir. Yaklaşık 1 nm civarında çapa sahip bir TDKNT’nin STM ile elde
edilen topografik örneği şekil 2.4’de görülmektedir. Bu çalışma 4,2 K civarında
yapılmıştır. 1 boyutlu durum yoğunluklarını vermek için 푑 , 휗 ve taramalı tünelleme
spektroskopisi (scanning tunneling spectroscopy, STS) için birleştirilmiş STM
ölçümleri nanotüplerin elektronik yapı çalışmaları için önemli bir teknik
sağlamaktadır (Heremans ve ark., 1994). STM ile yapılan örnekler nanotüpün yüzey
eğriliği için doğrudan bir kanıt teşkil etmektedir (Sattler, 1995). Tüp çapı STM ile
ilgili yapılana çalışmalarda sürekli olarak ölçülmektedir. Bu verilerden chiral açısı
belirlenerek nanotüp eksenine göre karbon atomlarının pozisyonları
belirlenebilmektedir. Ayrıca STM ile en yakın komşu karbon atomu mesafesinin
ölçülmesi de mümkündür ve bu değer grafit için ölçülen değerin aynısı olan 1,42 퐴°
civarındadır (Sattler, 1995).
KNT’lerin yapısının belirlenmesi ve kullanılması için kullanılan tekniklerdeki
gelişmelerin devam edeceği beklenmektedir ve bu teknikler nanotüpler üzerindeki
çalışmaların artması için sürekli olarak gelişmektedir. Dahası gelecekte KNT’lerin
farklı geometrik yapılarından, mükemmel elektronik ve mekanik özelliklerinden
dolayı nanoyapıların elde edilmesi için etkin bir şekilde kullanılması beklenmektedir.

2. ÖNCEKİ ÇALIŞMALAR ERKAN TETİK
35
Şekil 2.5. Lazer buharlaştırma metodu tarafından hazırlanan TDKNT’ün XRD yapısı
Teorik çalışmalarda TDKNT’lerin iki boyutlu formunun van der Waals
etkileşimi boyunca zayıf bir şekilde etkileştiği şeklinde davranmaktadır (Saito ve
ark., 1992). Bu hesaplama (6, 6) nanotüpün 2 boyutlu yapısının stabilizasyonu olarak
dikkate alınmaktadır ve P6/mcc uzay grubu ile üçgensel örgünün olduğu
konfigürasyonun en dengeli konfigürasyon olduğu görülmektedir. Ayrıca bu
çalışmalarda en yakın komşu karbon atomlarının 3 boyutlu grafitin dizilimi olan
ABAB oryantasyonuna sahip olduğu ortaya çıkmaktadır. X-ışını kırınımı teknikleri
kullanılarak TDKNT’lerin 2 boyuttaki örgü yapıları üzerinde yapılan son
zamanlardaki deneysel çalışmalar, 3 boyutlu yapının 1,38∓0,02 nm çapına ve 0,315
nm tüpler arası mesafeye sahip nanotüpler için 17 퐴° örgü sabiti ile üçgensel örgüye
sahip olduğunu doğrulamaktadır (Şekil 2.5).
2.2. Karbon Nanotüplerle İlgili Teorik Çalışmalar
KNT’ler keşfedildiğinden bu yana teorik olarak da yoğun bir şekilde
çalışılmaktadır. Teorik çalışmalardan yapısal, elektronik ve optik özellikleri
hakkında çok önemli bilgiler elde edilmiştir. Yapısal olarak pek çok farklı formda
bulunması ve fiziksel olarak da hem metalik hem de yarı iletken özellik
göstermesinden dolayı teorik çalışma alanı oldukça genişlemiştir. İlk teorik
çalışmalar KNT’lerin yapısal özellikleri hakkında yapılmıştır. KNT’lerin hangi
yapısal durumlarda metalik hangi durumlarda yarı iletken olduğu incelenmiştir. Şekil

2. ÖNCEKİ ÇALIŞMALAR ERKAN TETİK
36
2.6’da KNT’lerin geometrik yapısına göre nasıl metalik ve yarı iletken özellik
sergilediği gösterilmektedir. Bu tüpler sırasıyla açık ve kapalı dairelerle belirtilmiştir.
Şekil 2.6’dan açık olarak söyleyebiliriz ki, karbon nanotüplerin üçte biri metalik
geriye kalan üçte ikisi yarıiletkendir.
Şekil 2.6. KNT’lerin geometrik yapısına göre metalik ve yarı iletken özellik göstermesi
KNT’ler grafen katmanının silindirik olarak katlanmasıyla elde edildiği için,
nanotüplerle ilgili çalışmalar grafenin geometrik ve elektronik özelliklerini de
kapsamaktadır. Yapılan ilk teorik çalışmalarda grafenin ve nanotüpün elektronik
özellikleri ve birbiri arasındaki ilişkiler incelenmiştir. Daha sonra bilgisayar donanım
sistemlerinin de gelişmesiyle birlikte, süper hücreli yapı olarak isimlendirilen birden
fazla birim hücreden oluşan yapılar çalışılmaya başlanmıştır. Süper hücreli
nanotüplerin özellikleri birim hücredeki nanotüplerden farklılıklar göstermektedir.
Örneğin bu KNT’lerin yasak bant aralıklarında küçülmeler meydana gelmektedir.
Süper hücreli yapılarda nanotüpler arası uzaklığın değişmesi de nanotüpün elektornik
özelliklerini etkilemektedir. Nanotüpler arası mesafenin arttırılıp ya da azaltılmasıyla
nanotüp metalik yapıdan yarılektken yapıya geçebilmektedir. KNT’lerin çapına
bağlılığını dikkate alırsak, zigzag (19, 0) gibi büyük çaplı nanotüplerin dikkat çekici
(0,0) (1,0) (2,0) (3,0) (4,0) (5,0) (6,0) (7,0) (8,0) (9,0) (10,0)
(1,1) (2,1) (3,1) (4,1) (5,1) (6,1) (7,1) (8,1) (9,1)
(2,2) (3,2) (4,2) (5,2) (6,2) (7,2) (8,2) (9,2)
(3,3) (4,3) (5,3) (6,3) (7,3) (8,3)
(4,4) (5,4) (6,4) (7,4) (8,4)
(5,5) (6,5) (7,5)
(6,6) (7,6) Metal Yarıiletken Armchair
Zigzag

2. ÖNCEKİ ÇALIŞMALAR ERKAN TETİK
37
farklılıklar oluşmaktadır. Bu nanotüplerin bant yapısında eğriliğin etkisi daha açık
olarak görülmektedir.
Teorik çalışmalarda öncelikle nanotüpün geometrik özellikleri elde
edilmektedir. Bu özellikler kullanılacak programa göre genellikle nanotüpün örgü
vektörleri ve atomik konumlarıdır. Nanotüpün örgü vektörleri analitik olarak
çözülerek ya da bu çözümlerin kullanıldığı bir program ile ilde edilebilir. Daha sonra
hesaplama yapmak için kullanılacak programın özelliklerine göre hesaplama yöntemi
ile ilgili bilgiler girilir. Örneğin DFT hesaplamalarında bu parametreler LDA ya da
GGA hesaplama yönteminin seçilmesi, baz setlerinin belirlenmesi, Pseudo-
Potansiyel dosyası, atom sayısı, kesilim enerjisi gibi sıralanabilir. Bu parametreler
hesaplanacak özelliğe göre değişiklik göstermektedir. Girdi dosyası hazırlandıktan
sonra hesaplama başlatılır ve hesaplama bittikten sonra sonuçlar analiz edilir.
Şekil 2.7. (a) Ab initio ile grafenin elektronik bant yapısı, (b) Ab initio ile zigzag (10,
0) nanotüpün elektronik bant yapısı ve (c) Zigzag (10, 0) nanotüpün Tight-binding metoduyla elektronik bant yapısı
Şekil 2.7 ve 2.8’de S. Reich ve C. Thomsen (2002) tarafından SIESTA paket
programı kullanılarak yapılan hesaplamalardan elde edilen grafikler görülmektedir.
Bu çalışmada farklı yapılardaki TDKNT’ler elektronik özelliklerine göre izole ve
süper hücre olarak incelenmiştir. Şekil 2.7 (a)’da (10, 0) grafenin bant yapısı

2. ÖNCEKİ ÇALIŞMALAR ERKAN TETİK
38
görülmektedir. Bu yapı (10, 0) nanotüpün bir silindir etrafında katlanmadan önceki
halidir. Şekil (2.7) (b)’de ise (10, 0) nanotüp yani şekil 2.7 (a)’daki grafen yapının bir
silindir etrafında sarılmış halinin elektronik bant yapısı vardır. Çalışmada grafen ve
nanotüplerin elektronik özelliklerinin yanı sıra döndürmenin etkileri gözlenmiştir.
Şekilden de görüldüğü gibi iletim bantlarının valans bantlarına göre döndürmeden
daha fazla etkilendiği görülmektedir. Şekil 2.7 (c)’de ise (10, 0) nanotüpün
elektronik bant yapısı tight-binding yöntemi kullanılarak elde edilmiştir. Şekildeki
boş ve dolu siyah daireler dejenere ve dejenere olmayan bantları göstermektedir.
Şekil 2.8. (a) Ab initio ile grafenin elektronik bant yapısı, (b) Ab initio ile armchair
(6, 6) nanotüpün elektronik bant yapısı ve (c) Armchair (6, 6) nanotüpün Tight-binding metoduyla elektronik bant yapısı.
Eğriliğin bant yapısı üzerindeki etkilerini daha iyi incelemek için şekil 2.8’de
armchair (6, 6) nanotüp için benzer hesaplamalar yapılmıştır. Bu çalışmada da önce
grafenin daha sonra nanotüpün elektronik bant yapısı elde edilip karşılaştırmalar
yapılmıştır. Daha sonra tight-binding yöntemi ile hesaplanan bant yapısı
incelenmiştir. Ayrıca S. Reich ve C. Thomsen (2002) tarafından yapılan bu
çalışmada nanotüplerin süper hücreli yapılarıyla da hesaplamalar yapılarak sonuçlar
analiz edilmiştir. İlk çalışamda zigzag (10, 0) nannotüpün yarıiletken özellik
gösterdiği bulunmuştur. İkinci çalışmada ise armchair (6, 6) nanotüpün metalik

2. ÖNCEKİ ÇALIŞMALAR ERKAN TETİK
39
özellik gösterdiği bulunmuştur. Bu çalışma iki boyutta yapılmıştır ve elektronik bant
yapısı Γ ile π/a arasında seçilmiştir. Ayrıca hesaplaması yapılan grafen ve
nanotüplerin iletim ve valans bantlarının Γ ie M noktalarındaki enerkileri elde
edilmiştir. Ayrıca bu çalışmada benzer hesaplamalar çok hücreleri KNT’ler için de
yapılmıştır. İzole nanotüplerle çok hücreli nanotüpler arasındaki farklar incelenerek
analiz edilmiştir. Çalışmanın son kısmında ise KNT’lerin elektronik özelliklerinin
çapa bağlılığı araştırlımıştır.
Nanotüplerle ilgili yapılan teorik hesaplamalar nanotüplerin elektronik
özelliklerinin yanı sıra optik, fonon ve elastik özellikleri de çalışılmaktadır. Bu
çalışmalarda da çoğunlukla benzer yöntem ve paket programlar kullanılmaktadır.
2.3. Karbon Nanotüplerde Katkılama Çalışmaları
KNT’ler keşfedildikten sonra üzerinde çalışılan en büyük konu chirality
açısının ve çapının değiştirilerek farklı türlerinin elde edilmesiydi. Geometrik
yapının değişmesiyle elde edilen farklı nanotüpler elektronik özellikleri bakımından
farklılık göstermesi oldukça önemli bir özellikti. Bu özellik pek çok nano-elektronik
cihazda kullanılabileceği düşünülmektedir. Nano-elektronik cihazlarda kullanılan
malzemelerin metalik ya da yarıiletken özellik göstermesi ve bu özelliğin
değiştirilebilir olması oldukça önemlidir. Nanotüplerin geometrik yapısının
değiştirilerek elektronik özelliğinin değiştirilmesi üzerine pek çok çalışma yapılmış
ve yapılmaya devam etmektedir. Bunlara alternatif olarak, nanotüplere yabancı atom
ekleyerek elektronik özelliğinin değiştirilmesi de oldukça önemli bir yöntemdir.
KNT’lerde katkılama ile ilgili de pek çok çalışma yürütülmektedir. Katkılama için
tercih edilen atomlar genellikle karbon atomuna yakın olan bor ve azot atomlarıdır.
Katkılama işleminde katkılanacak atomun karbon atomuna geometrik olarak yakın
bir atom olması önemlidir. Aksi halde yapı bozularak ve örgü kusuru oluşabilir.
Bunun dışında silikon, oksijen ve alkali metallerden olan lityum gibi atomlarla da
katkılama çalışmaları yapılmaktadır. Katkılama yapılırken, eklenen yabancı atomun
elektronik özellikleri de önemlidir. Çünkü katkılanan nanotüpün özellikleri, eklenen
atomun özelliklerine doğru değişim gösterecektir.

2. ÖNCEKİ ÇALIŞMALAR ERKAN TETİK
40
2.3.1. Katkılama İle İlgili Deneysel Çalışmalar
Katkılama ile ilgili deneysel çalışmalar genellikle bor, azot ve lityum gibi
atomlarla yapılmaktadır. Bu çalışmalar çoğunlukla STM ve TEM gibi
mikroskoplarla yapılmaktadır. Ayrıca Elektron Enerji-Kayıp Spektroskopisi
(Electron Energy-Loss Spectroscopy, EELS) ve Optik Soğurma Spektroskopisi
(OAS) kullanılarak yapılan çalışmalar da yürütülmektedir. Borowiak-Palen ve
arkadaşları (2004) tarafından yapılan bir çalışmada TDKNT’lere bor atomu
katkılandığı bir çalışma yapılmıştır. Katkılama sonucu elde edilen yapının elektronik
ve optik özellikleri incelenmiştir. Çalışmada %10 ve %15 olmak üzere farklı bor
konsantrasyonları kullanılmıştır. Ayrıca bor eklenmiş TDKNT’lerin elektronik ve
optik özelliklerinin analizi için, ince film hazırlanmıştır. Bor eklenen örneklerde
kimyasal etkinin daha detaylı analiz edilmesi için EELS ve OAS kullanılarak bor
eklenen yapının elektronik özellikleri analiz edilmiştir.
Şekil 2.9’da çalışmada elde edilen sonuçların mikroskobik görüntüsü yer
almaktadır. Şekil 2.9(a) ve (b)’de yabancı atom eklenmemiş TDKNT, şekil 2.9(c) ve
(d) ise bor atomu eklenmiş TDKNT’nin TEM görüntüsü yer almaktadır. Bu
görüntülerden katkılama işleminin geometrik yapı üzerindeki etkileri
gözlenmektedir.
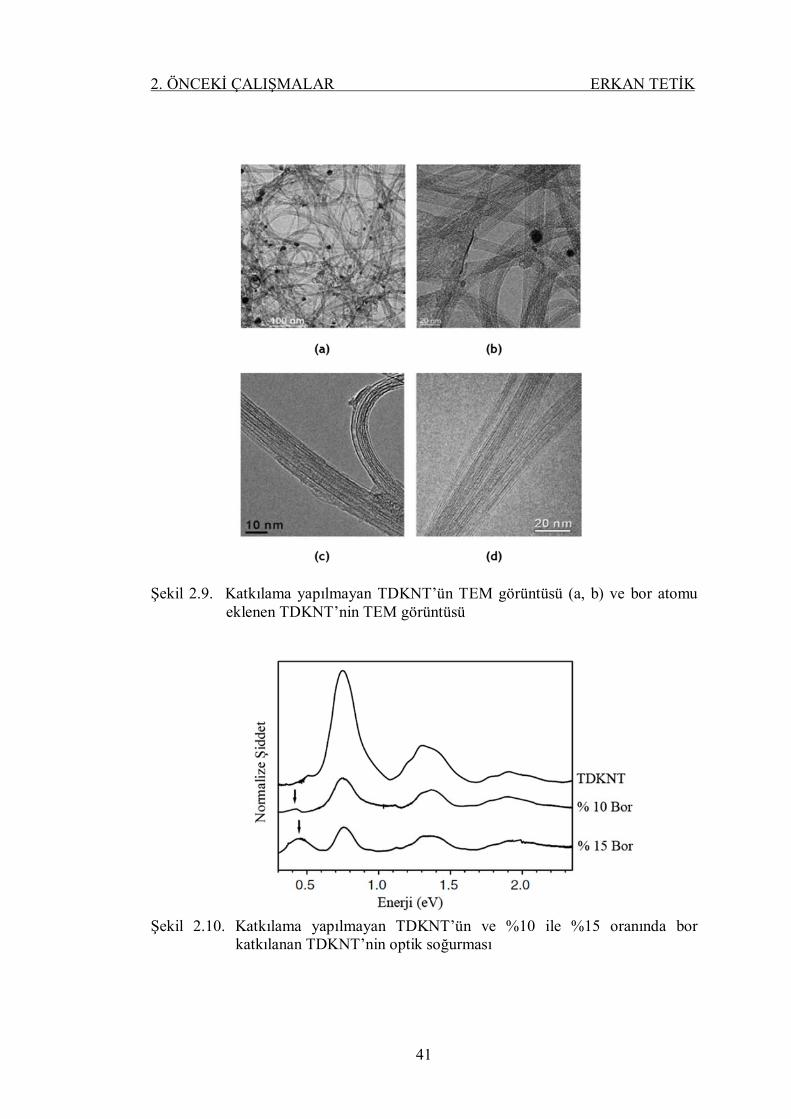
2. ÖNCEKİ ÇALIŞMALAR ERKAN TETİK
41
Şekil 2.9. Katkılama yapılmayan TDKNT’ün TEM görüntüsü (a, b) ve bor atomu
eklenen TDKNT’nin TEM görüntüsü
Şekil 2.10. Katkılama yapılmayan TDKNT’ün ve %10 ile %15 oranında bor katkılanan TDKNT’nin optik soğurması

2. ÖNCEKİ ÇALIŞMALAR ERKAN TETİK
42
Şekil 2.10’da ise bor eklenen TDKNT’nin optik soğurma grafiği
görülmektedir. Bu grafiğe göre bor eklenen TDKNT’lerin optik ölçümleri ilginç
sonuçlar ortaya çıkarır. Grafikte pik pozisyonları ve izin verilen optik geçişler 0,3 ve
2,35 eV enerji aralığında görülmektedir. Tipik optik geçişler 0,75 eV değerinde
gözlenmektedir. Katkılama yapılan nanotüplerde 0,4 eV civarında yeni bir pik ortaya
çıkmaktadır. Bu yeni pik valans bandından akseptör bandına optik uyarılmalara işaret
etmektedir.
Şekil 2.11. TDKNT’nin SEM görüntüsü
Elektronik ve optik özellikler dışında elastik özellikler, elektriksel iletkenlik
ve iletim özellikler ile ilgili yapılan çalışmalar da dikkat çekmektedir. Barberio ve
arkadaşları (2009) sodyum (Na), potasyum (K), Sezyum (Cs) ve lityum (Li) gibi
alkali atomlar katkılanmış TDKNT’lerle ilgili yaptıkları çalışmalarda iletim
özellikleri araştırmışlardır. Bu çalışmada TDKNT’nin elde edilen görüntüsü şekil
2.11’de görülmektedir. Bu resim taramalı elektron mikroskobu (Scanning electron
microscope, SEM) kullanılarak elde edilmiştir. Daha sonra bu nanotüpe alkali
atomları eklenerek sonuçlar alınmış ve analiz edilmiştir. Çalışmada KNT’nin
katkılama işlemine maruz kalma süresinin bir fonksiyonu olarak rezistansın değeri
ölçülmüştür. Elektriksel özelliklerin alkali atomunun eklendiği nanotüp yapılarda,
güçlü bir şekilde değiştiği gözlenmiştir. Eklenen alkali metalin türüne göre de

2. ÖNCEKİ ÇALIŞMALAR ERKAN TETİK
43
elektriksel iletkenlik farklılıklar göstermektedir. Tüm alkali türlerinde rezistansın
artış gösterdiği görülmüştür.
2.3.2. Katkılama İle İlgili Teorik Çalışmalar
KNT’ler farklı elektronik, optik ve mekanik özellikleri ile dikkat
çekmektedir. Bu özelliklerin KNT’ye yabancı atom eklenerek değiştirilebiliyor
olması ayrı bir önem taşımaktadır. Ancak KNT’lerin geleceğin teknolojisinde
kullanılması için aşılması gereken birçok zorluk vardır. KNT sentezleme teknikleri
hala yetersizdir. Ayrıca var olan sentezleme teknikleri de çok pahalıya mâl
olmaktadır. Dolayısıyla yeterli hassaslıkta katkılama yapmak oldukça zor ve külfetli
bir iştir. Ancak yabancı atom eklenen KNT’lerin fiziksel özellikleri teorik olarak
yoğun bir şekilde çalışılmaktadır. Çalışmaların çoğunluğu teorik olarak
yürütülmektedir. Bu çalışmalar KNT’lere yabancı atom ekleme ile ilgili deneysel
çalışmalara ışık tutmaktadır.
Teorik çalışmalarda KNT’lere eklenen yabancı atomlar çoğunlukla B, N, BN,
Si ve Li atomlarıdır. Bu çalışmaların büyük bir çoğunluğu YFT tabanlı paket
programlar kullanılarak yapılmaktadır. Seçilen bazı nanotüplere belirli oranlarda
yabancı atom eklenerek yani karbon atomlarının yerine seçilen yapancı atol
yerleştirilerek yeni bir yapı oluşturulmaktadır. Bu yeni yapı ab initio yaklaşımına
göre optimize edilerek ideal hale getirilir. Elde edilen son yapının kullanılan
programın özelliklerine göre, elektronik, optik ve mekanik gibi pek çok fiziksel
özelliği elde edilebilir. Teorik çalışmalarda elde edilen bu sonuçlar analiz
edilmektedir.
Wirtz ve Rubio (2009) bor eklenmiş KNT’ler üzerinde katkılamanın etkilerini
incelemişlerdir. Hesaplamalarda Abinit paket programını kullanmışlardır. Yabancı
atom eklenmiş KNT’lerin elektronik bant yapısını ve durum yoğunluğunu elde
etmişlerdir. Hesaplamalarda zigzag (16, 0) nanotüp kullanmışlardır. İlk olarak
katkılama yapılmamış KNT üzerinde hesaplama yapıp daha sonra belirli oranlarda
yabancı atom eklenen zigzag (16, 0) nanotüp üzerinde çalışmışlardır. Zigzag (16, 0)
nanotüpe eklenen bor atomu yüzdeleri %6,25, %12,5 ve %25 şeklindedir. Zigzag

2. ÖNCEKİ ÇALIŞMALAR ERKAN TETİK
44
(16, 0) nanotüpün ve katkılama yapılana nanotüplerin elektronik bant yapıları ve
durum yoğunlukları şekil 2.12’de görülmektedir.
Şekil 2.12. Zigzag (16, 0) nanotüpün elektronik bant yapısı ve durum yoğunluğu (a),
%6,25 bor atomu eklenmiş (16, 0) nanotüpün elektronik bant yapısı ve durum yoğunluğu (b), %12,5 bor atomu eklenmiş (16, 0) nanotüpün elektronik bant yapısı ve durum yoğunluğu (c) ve %25 bor atomu eklenmiş (16, 0) nanotüpün elektronik bant yapısı ve durum yoğunluğu (b)
Şekil 2.12(a)’daki grafikte yabancı atom eklenmemiş zigzag (16, 0)
nanotüpün yasak bant aralığı 0,5978 eV olarak elde edilmiştir. %6,25 bor atomu
eklenmiş şekil 2.12(b)’deki nanotüpün bant arağının biraz daraldığını görüyoruz.
Ayrıca katkılama yapılan bu nanotüpün Γ noktasındaki valans bantlarının Fermi
enerji seviyesinin üzerine çıktığı görülmektedir. Bor atomu ekleme yüzdesi arttıkça
Fermi enerji seviyesinin üzerine çıkan valans bantlarının sayısı da artmaktadır. Bu
durum şekil 2.12(c) ve (d) grafiklerinde görülmektedir. Aynı zamanda DOS
grafiklerindeki gri alanlar, katkılama sonrası Fermi seviyesinin altında kalan valans
bantlarını göstermektedir. Bu gri alanlardan ne kadar valans bandının Fermi
seviyesinin üzerine çıktığını daha net olarak görebiliriz. Bu zigzag (16, 0) nanotüpün

2. ÖNCEKİ ÇALIŞMALAR ERKAN TETİK
45
iletkenlik özelliğinin artması anlamına gelir. Çünkü valans bantlarının Fermi enerji
seviyesinin üzerine çıkmasıyla birlikte serbest elektronların sayısı da artar. Bu da
iletkenliğin artması anlamına gelir.
Zhou ve arkadaşları (2006) katkılama ile ilgili daha kapsamlı ve daha farklı
bir çalışma yapmışlardır. Çalışmalarında TDKNT’lere bor ve azot atomlarını
katkılamışlardır. Ayrıca bu katkılama işlemine hidrojen soğrulmasını da ekleyerek
elektronik özelliklerdeki farklılıkları incelemişlerdir. Hesaplamalarda zigzag (8, 0)
nanotüpü kullanmışlardır. Hesaplama sonucu elde edilen grafikler şekil 2.13, 2.14 ve
2.15’de verilmiştir. Her bir şekilde H-soğurma ve H-soğurma olmayan olmak üzere
iki ayrı grafik vardır.
Şekil 2.13. Katkısız (a) ve H-sağurması olan (b) zigzag (8, 0) nanotüpü elektronik
bant yapısı ve durum yoğunluğu
Şekil 2.14. Bor katkılanmış (a) ve bor katkılanmış (H-sağurma) (b) zigzag (8, 0)
nanotüpü elektronik bant yapısı ve durum yoğunluğu

2. ÖNCEKİ ÇALIŞMALAR ERKAN TETİK
46
Şekil 2.15. Azot katkılanmış (a) ve azot katkılanmış (H-sağurma) (b) zigzag (8, 0)
nanotüpü elektronik bant yapısı ve durum yoğunluğu
Şekil 2.14’deki grafikten de görüldüğü gibi bor katkılanan nanotüpte valans
bantları Fermi seviyesinin üzerine çıkmıştır. Yani serbest elektronların sayısı atmış
ve nanotüpün iletkenlik özelliğini arttırmıştır. H-soğurmasının olduğu durumda ise
katkılama sonrası Fermi seviyesinin üzerine çıkan valans elektronları yeniden Fermi
seviyesinin altına inmiştir. Şekil 2.15’deki grafikte ise zigzag (8, 0) nanotüpe azot
atomu katkılanmıştır. Katkılama sonrasında nanotüpün iletim bantlarının Fermi
seviyesinin altına indiğini görüyoruz. Dolayısıyla nanotüpteki serbest elektronların
sayısı azalmaktadır ve nanotüp yalıtkan duruma geçmektedir. Azot katkılana nanotüp
H-soğurma durumumda ise katkılama sonrası Fermi seviyesinin altına inen iletim
bantları yeniden Fermi seviyesinin üzerine çıkmıştır. Sonuç olarak, zigzag
nanotüplerde bor atomu katkılamak hidrojen atomik soğurma enerjisini
arttırmaktadır. Azot atomu katkılama ise hidrojen atomik soğurma enerjisini
azaltmaktadır.
Biz yapacağımız çalışmamızın ilk aşamasında grafen ve TDKNT’lerin
elektronik bant yapısını, durum yoğunluğunu ve elektron yoğunluğunu
inceleyeceğiz. Daha sonra granfende nanotüpe döndürmenin etkilerinin elektronik
özellikler açısından değerlendireceğiz. Çalışmamızda kullanacağımız nanotüplerin
bazılarını, daha önce yapılmış çalışmalarla karşılaştırmak için, üzerinde çalışılmış
nanotüplerden seçeceğiz. Bazılarını üzerinde daha önce çalışılmamış nanotüpleden
seçeceğiz. Çalışmamızın ikinci aşamasında ise KNT’lere katkılamanın etkilerini

2. ÖNCEKİ ÇALIŞMALAR ERKAN TETİK
47
inceleyeceğiz. KNT’lere karbon atomuna geometrik olarak yakın özellikte olan ve
periyodik cetveldeki A gurubunda seçtiğimiz bazı atomları katkılayacağız.
Dolayısıyla KNT’lere bazıları daha önce katkılanmamış olan birçok farklı atom
katkılayarak, katkılama ile ilgili kapsamlı bir çalışma yapmış olacağız.

3. MATERYAL VE METOT ERKAN TETİK
48
3. MATERYAL VE METOT
3.1. Yoğunluk Fonksiyoneli Teorisine Giriş
Yoğunluk Fonksiyoneli Teorisi (YFT) elektronik yapı hesaplamalarında
önemli bir yer tutmaktadır. Doğruluk ve hesaplama maliyeti arasında faydalı bir
denge sağladığı ve geleneksel ab initio metotlarından daha büyük sistemler için
kullanılabildiği için son zamanlarda yoğun olarak kullanılmaktadır. Geleneksel dalga
fonksiyonu metotları küçük sistemler üzerinde yüksek doğrulukta sonuçlar elde
etmek için kullanılıp, elde edilen sonuçlar YFT vasıtasıyla daha büyük sistemlere
uygulanabilmektedir. YFT Schrödinger denklemini çözmenin farklı bir yolu ya da
deneysel sonuçları değerlendirmenin basit bir metodu değildir. YFT yer ve zamanın
bir fonksiyonu olan elektron yoğunluğunu temel bir değişken olarak alan tamamen
farklı bir teoridir.
3.1.1. Kohn-Sham Hesaplamaları
YFT’nin ne olduğu ve niçin bu kadar kullanışlı olduğu hakkında bilgi vermek
için basit bir örnek olan hidrojen molekülünü ele alalım. Bu işlem için Born-
Oppenheimer yaklaşımından faydalanacağız ve elektronlar için temel-durum
kuantum mekaniksel problem çözümünü yapacağız. Schrödinger denklemi,
− ∑ ∇, +| |
+ ∑ 휐 ş(r ), Ψ(r , r ) = 퐸Ψ(r , r ) (3.1.)
şeklindedir. Burada i indeksi sadece iki elektron içindir ve dış potansiyel,
휐 ş(r) = − −| |
(3.2.)
burada Z = 1 her çekirdek üzerindeki yük, 푧 bağ ekseni boyunca birim vektör ve R
çekirdekler arası mesafedir. Hesaplamalarda atomik birim kullanacağımız için,
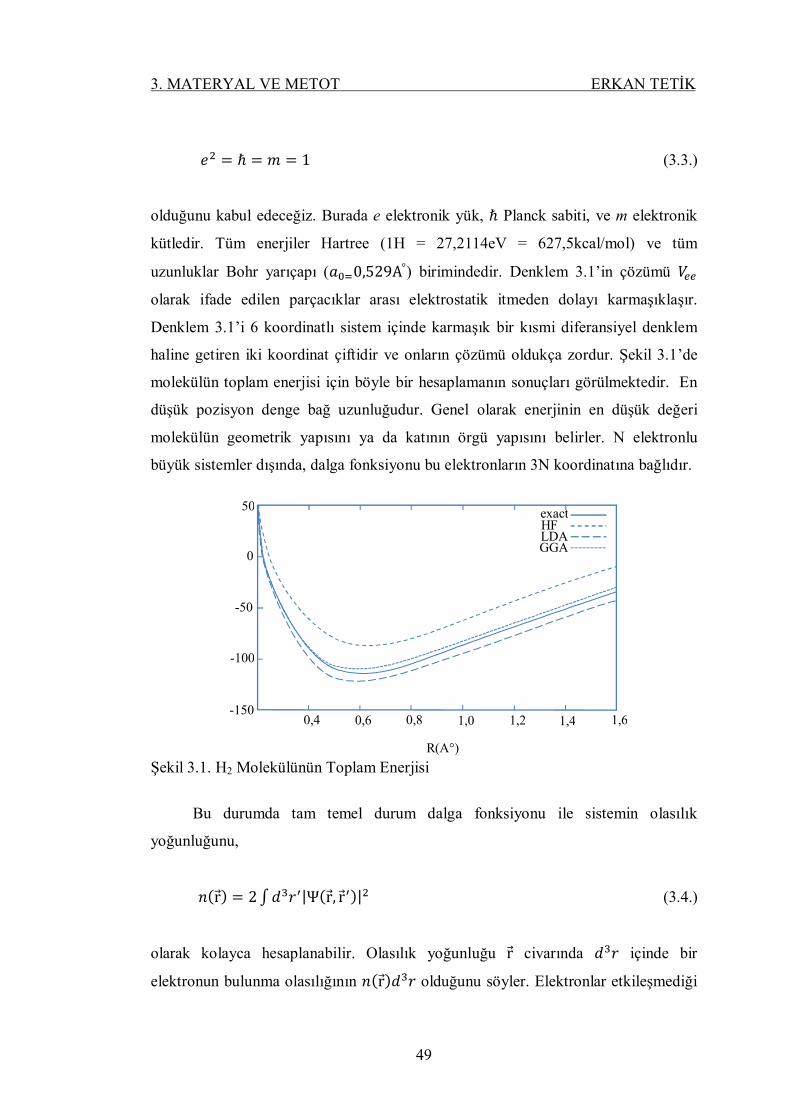
3. MATERYAL VE METOT ERKAN TETİK
49
푒 = ℏ = 푚 = 1 (3.3.)
olduğunu kabul edeceğiz. Burada e elektronik yük, ℏ Planck sabiti, ve m elektronik
kütledir. Tüm enerjiler Hartree (1H = 27,2114eV = 627,5kcal/mol) ve tüm
uzunluklar Bohr yarıçapı (푎 0,529A°) birimindedir. Denklem 3.1’in çözümü 푉
olarak ifade edilen parçacıklar arası elektrostatik itmeden dolayı karmaşıklaşır.
Denklem 3.1’i 6 koordinatlı sistem içinde karmaşık bir kısmi diferansiyel denklem
haline getiren iki koordinat çiftidir ve onların çözümü oldukça zordur. Şekil 3.1’de
molekülün toplam enerjisi için böyle bir hesaplamanın sonuçları görülmektedir. En
düşük pozisyon denge bağ uzunluğudur. Genel olarak enerjinin en düşük değeri
molekülün geometrik yapısını ya da katının örgü yapısını belirler. N elektronlu
büyük sistemler dışında, dalga fonksiyonu bu elektronların 3N koordinatına bağlıdır.
Şekil 3.1. H2 Molekülünün Toplam Enerjisi
Bu durumda tam temel durum dalga fonksiyonu ile sistemin olasılık
yoğunluğunu,
푛(r) = 2 ∫ 푑 푟 |Ψ(r, r )| (3.4.)
olarak kolayca hesaplanabilir. Olasılık yoğunluğu r civarında 푑 푟 içinde bir
elektronun bulunma olasılığının 푛(r)푑 푟 olduğunu söyler. Elektronlar etkileşmediği
LDA
-100
-150
-50
0
50
0,4 0,6 0,8 1,0 1,2 1,4 1,6
exact HF GGA
R(A°)
E(H )+1/R
-E(2E) (kcal/mol)
2

3. MATERYAL VE METOT ERKAN TETİK
50
için, onların koordinatları çözülmüş değildir ve dalga fonksiyonu orbitaller olarak
isimlendirilen tek elektronlu dalga fonksiyonlarının bir ürünüdür. Yani,
− ∇ + 휐 (r) 휙 {r} = 휀 휙 {r} (3.5.)
şeklindedir. Burada Φ(r , r ) = 휙 (r )휙 (r ) eşittir. Bu durumda sistem sadece 3
koordinata sahip olduğu için denklemin çözümü basitleşir. Etkileşmeyen sistemi
gerçek sistemin taklidi olarak alırsak, çözmek için çok daha kolay bir denkleme sahip
olacağız. Bu durumda enerjiyi minimize etmek istersek, efektif potansiyelin bir
ürünü olan Hartree-Fock denklemlerine ulaşırız. Yani,
휐 (r) = 휐 ş(r) + ∫ 푑 푟 | |
(3.6.)
şeklinde olur. Buradaki dış potansiyel ikinci elektronun etkisini taklit eder. Denklem
3.5’deki potansiyel ise elektronik yoğunluğa bağlıdır. Bu yüzden bu denklem setinin
öz uyumlu terimidir. Potansiyel için ilk tahmin yapılır, daha sonra özdeğer problemi
çözülür, yoğunluk hesaplanır ve yeni bir potansiyel bulunur. Bu adımlar sonraki
döngüdeki sonuç ile bir önceki döngüdeki sonuç arasındaki fark en düşük değerinin
alana kadar kendini tekrar eder. Denklemlerdeki bu tür setler genellikle öz uyumlu
alan denklemleri olarak isimlendirilir. Şekil 3.1’de Hartree-Fock denkleminin
sonuçları görülmektedir. En düşük enerjinin olduğu değer en doğru sonucu verir.
Kohn-Sham hesaplamalarında temel adım buna benzerdir fakat mantık biraz
farklıdır. Etkileşmeyen elektron çifti fiziksel sisteme göre aynı 푛(r) yoğunluğuna
sahiptir. YFT kullanılan Kohn-Sham sisteminde yoğunluğa bağlı E toplam enerjisi
bilinirse, 휐 (r) potansiyeli türetilebilir. Yoğunluk üzerindeki enerjinin bilinmeyen
bağımlılığı için basit bir yaklaşım tüm elektronik sistemine uygulanabilir ve hayali
etkileşmeyen elektronlar için öz uyumlu potansiyel ve enerji tahmin edilebilir.

3. MATERYAL VE METOT ERKAN TETİK
51
3.2. Fonksiyonel Kavramı
Bir fonksiyon bir sayıyı başka bir sayıyla eşleştirir. Bir fonksiyonel bir sayıya
bir fonksiyon atar. Örneğin 0 ≤ 휃 ≤ 2휋 aralığındaki tüm 푟(휃) fonksiyonlarını
dikkate alalım. Bu fonksiyonlar periyodiktir yani 푟(휃 + 2휋) = 푟(휃) denklemini
sağlar. Bu tür fonksiyonlar şekil 3.2’deki gibi kendilerini aynı yoldan geri tekrar
etmeyen eğrilerin iki boyuttaki şekilleri olarak tanımlanırlar. Bu tür eğriler için,
eğrinin uzunluğu olarak P çevre uzunluğunu ve eğri tarafından kapalı bölge olarak A
bölgesini tanımlayabiliriz. Bunlar 푟(휃) değerinin fonksiyonelleridir.
Şekil 3.2. He atomu için dış ve Kohn-Sham potansiyeli
Şekil 3.2’de verilen eğri için,
푟(휃) =( ) ( )
(3.7.)
olarak yazılır. P ve A’nın tek iyi tanımlanmış bir değeri vardır. Bu fonksiyonel
bağımlılığını göstermek için 푃[푟] ve 퐴[푟] şeklinde yazarız. Bağıntıyı açık olarak
bilmiyoruz ama onun var olduğunu biliyoruz yani her sınırlandırılmış eğri bir çevre
-6
-8
-4
0
0 0,2 0,4 0,6 0,8
r 1,0
-2
2
4
yoğunluk
Kohn-Sham Potensiyeli
He Atomu
-2/r

3. MATERYAL VE METOT ERKAN TETİK
52
uzunluğuna ve bir alana sahiptir. 푑휃 açısındaki çok küçük değişikliklerden ve açının
tüm aralıklar üzerinden gelen katkıyı dikkate alalım. 푟 cos(푑휃) ≈ 푟 ve
푟(휃) sin(푑휃) ≈ 푟푑휃 bağıntılarından,
퐴[푟] = ∫ 푟푑휃푟 = ∫ 푑휃 푟 (휃) (3.8.)
olarak yazılır. Böylece bu fonksiyonel eğri tarafından sınırlandırılmış bölgede, bir
sayı için bir argümanın gerçek bir fonksiyonu ile eşleştir. Benzer olarak, çevre
uzunluğundaki küçük değişiklikler,
훿푃 = 푑푟 + 푟 (휃)푑휃 (3.9.)
kutupsal koordinatlarda çizgi parçalarıdır. 푟 = 푟(휃) için, 푑푟 = 푑휃(푑푟/푑휃) yazarsak,
푃[푟] = ∫ 푑휃 푟 (휃) + (푑푟/푑휃) (3.10.)
şeklinde türetilir. Her iki durumda, 푟(휃) değerini bilirsek, fonksiyoneli
hesaplayabiliriz. Bölge 푟(휃) değerinin yerel fonksiyonelidir ve,
퐴[푟] = ∫ 푑휃 푓 푟(휃) (3.11.)
formunda yazılabilir. Burada 푓(푟) r’nin bir fonksiyonudur ve bölge için, 푓 = 푟 /2
olarak yazılır.
3.3. Kuantum Mekaniği Dalga Fonksiyonu: Tek Elektron
Temel kuantum mekaniğinde bir boyutta tek elektron için Rayleigh-Ritz
varyasyonel ilkesi,
퐸 = min 휙 퐻 휙 , ∫ 푑푥|휙(푥)| = 1 (3.12.)

3. MATERYAL VE METOT ERKAN TETİK
53
şeklindedir. Bu prensip enerjinin beklenen değerini hesaplamak için herhangi bir
normalize dalga fonksiyonu kullanabileceğimizi söyler. Örneğin, bir boyutlu
problem için aynı dalga fonksiyonunu kullanabiliriz ve temel-durum enerjisinin bir
üst sınırını elde edebiliriz. Bu prensip için,
푇[휙] = ∫ 푑푥휙∗(푥) − 휙(푥) = ∫ 푑푥|휙(푥)| (3.13.)
bu fonksiyoneli yazabiliriz. Buradan herhangi bir normalize dalga fonksiyonu için
denklem 3.9’den hesaplanabilen tek bir T sayısı vardır. Benzer olarak, basit bir
foksiyonel olarak potansiyel enerji,
푇[휙] = ∫ 푑푥 푉(푥) |휙(푥)| (3.14.)
olarak yazılır. Problem hangi dalga fonksiyonuna uygulanırsa uygulansın, verilen
dalga fonksiyonun kinetik enerjisi her zaman aynıdır. Kinetik enerji evrensel bir
fonksiyoneldir yani fiziksel problem ne olursa olsun, verilen dalga fonksiyonu
üzerine aynı operatör uygulanabilir. Fakat potansiyel enerji her problem için farklıdır
yani evrensel değildir. Bu işlem 퐹(푥, 푦) = (푥 /2) − 푥푦 denkleminin x üzerinden en
küçük değerini bulmaya benzerdir. İlk terim y’den bağımsızdır, bu yüzden her x için
bir değer listesi oluşturabiliriz ve aynı listeyi y‘nin en düşük değerini bulmak için
kullanabiliriz. Bu durumda varyasyonel prensip,
퐸 = min{푇[휙] + 푉[휙]}, ∫ 푑푥 |휙(푥)| = 1 (3.15.)
olarak yazılabilir. Buradan tüm olası normalize dalga fonksiyonlarının enerjisini
hesaplamanız ve bu değerlerin en düşük olanını seçmemiz gerekiyor.

3. MATERYAL VE METOT ERKAN TETİK
54
3.3.1. İki Elektron
Sistemde bir parçacıktan daha fazlası olduğu zaman, Hamiltonyeni ve dalga
fonksiyonu her parçacık için bir koordinat içerir. Dahası, elektronlar aşağı ve yukarı
olmak üzere iki spin durumuna sahiptir ve bu yüzden dalga fonksiyonu uzaysal ve
spin koordinatlarının her iki fonksiyonuna da sahiptir. Çok elektronlu kuantum
mekaniğinin temel prensibi herhangi iki koordinat setinin değişikliği altında dalga
fonksiyonunun antisimetrik olarak kalmasıdır. Bunu iki elektron için uygularsak,
dalga fonksiyonu,
Ψ(r , σ , r , σ ) = −Ψ(r , σ , r , σ ) (3.16.)
ile ifade edilir. Bu durumda sistem temel durumda, dalga fonksiyonun spin kısmı
antisimetrik ve uzaysal kısmı simetrik olacaktır,
Ψ(r , σ , r , σ ) = Ψ(r , r )휒 (σ , σ ) (3.17.)
burada uzaysal kısım uzaysal koordinatların değişimi altında simetriktir ve spin
kısmı 휒 (σ , σ ) =√
(| ↑↓ ⟩ − | ↑↓ ⟩) şeklinde antisimetriktir.
3.3.2. İki Elektron İçin Hartree-Fock
Bir elektrondan daha fazlası için, Hamiltonyan operatörü bütüm koordinatlara
bağlıdır. Kinetik enerji ve dış potansiyel,
푇 = − + 푉 ş = 휐 ş(푥 ) + 휐 ş(푥 ) (3.18.)
şeklinde yazırlır. Elektron-elektron itme operatörü iki cisim operatörüdür ve her
terim eşzamanlı olarak iki koordinata bağlıdır. Üç boyutta iki elektron arasındaki
etkileşim 1/|r − r | şeklindedir. Bir boyutta 1/|r − r | homojendir ancak oldukça

3. MATERYAL VE METOT ERKAN TETİK
55
güçlü bir çekime sahiptir ve bu yüzden bu terimi, 훿(푥 − 푥 ) olarak kullanmayı tercih
edeceğiz. 훿(푥 − 푥 ), 1/|r − r | ile aynı özelliklere sahiptir fakat bu terim daha
zayıftır. Genel problem için bir boyutta He atomunu dikkate alalım. Bu durumda
Hamiltonyan,
푇 = − + − 푍훿(푥 ) − 푍훿(푥 ) + 훿(푥 − 푥 ) (3.19.)
olarak yazarız. Bu denklemin tam çözümünü bulmak için, bu Hamiltonyan ile
Schrödinger denklemini çözmek isteyebiliriz, ancak varyasyon ilkesini kullanarak
yaklaşık çözüm bulmak daha kolaydır yani,
퐸 = min Ψ 퐻 Ψ , ∫ 푑푥 푑푥 |Ψ(푥 , 푥 )| = 1 (3.20.)
olur ve elektron-elektron itmesini ihmal edersek diferansiyel denklem,
Ψ(푥 , 푥 ) = 휙(푥 ) 휙(푥 ) (3.21.)
şeklinde olur. Burada her iki orbital de aynıdır ve bir elektron problemi için
uygundur bu durumda,
휙(푥) = √푍 exp(−푍푟) (3.22.)
şeklide yazılır. Koordinatlardaki bu durum, sadece bir elektron çözmeye ihtiyaç
duyacağımız için, büyük sistemleri incelemeyi mümkün kılar. Ancak bu yaklaşım
çok eksiktir. Kinetik ve potansiyel enerjinin katkısı tek elektronlu sistemde iki kat
daha büyüktür bu durumda,
푇 = 2 , 푉 ş = 2(−푍 ), 푉 = 0 (3.23.)
olur ve bu yüzden toplam enerji,

3. MATERYAL VE METOT ERKAN TETİK
56
퐸 = 2휖 = −푍 (3.24.)
olur ve He (Z=2) için enerjiyi 퐸 = −4 olarak elde ederiz. Bu değer problemin doğru
taban durumu enerjisinden düşüktür, çünkü Hamiltonian’deki enerjinin yazılmasında
eksikler var. Daha doğru tahmin için, varyosyonel prensip üzerinde biraz daha
düzenleme yapalım. Dalga fonksiyonumuzun 푉 kısmının beklenen değerinde
düzenleme yaparsak,
푉 = ∫ 푑푥 ∫ 푑푥 |휙(푥)| |휙(푥 )| 훿(푥 − 푥 ) (3.25.)
푉 = 푍 ∫ 푑푥 exp(−4푍|푥|) = (3.26.)
denklemlerini elde ederiz ve He (Z=2) için, 푉 = +1 olur ve temel durum enerjisini
ise 퐸 = −4 + 1 = −3 olarak elde ederiz. Burada 푉 = 0 problemi için orbitalin
uzunluk ölçeğini seçtik yani, 훼 = 푍 kabul ettik. Enerjinin minimize değeri için,
푇 = 2 , 푉 ş = −2푍훼, 푉 = 훼/2 (3.27.)
yazarız ve toplam enerjiyi 훼 değerinin bir fonksiyonu olarak,
퐸(훼) = 훼 − 2훼 푍 − (3.28.)
şeklinde elde ederiz. Bu minimize işlemine göre 훼 = 푍 − 1/4 olarak buluruz ve
böylece,
퐸 = 훼 = − 푍 − = −3.0625 (3.29.)

3. MATERYAL VE METOT ERKAN TETİK
57
olarak elde ederiz. Bu durumda bir Hartree’nin 1/16 kadar daha düşün bir enerji elde
ettik, bu 1,7 eV yani 40 kcal/mol civarı olduğu için çok uygun olmayabilir. Kimyasal
doğruluk bu değeri 1 ya da 2 kcal/mol civarı olmasını gerektirir.
Bu problem için en iyi çözüm en düşük enerjiyi üreten orbitali bulmaktır.
Bunu orbitaldeki varyasyonel parametrelerle yapabiliriz ve onların her biri için
enerjiyi minimize edebiliriz. Orbitalin bir fonksiyoneli olarak enerji,
퐸[휙] = 2푇 [휙] + 2푉 ş[휙] + [ ] , 푈[휙] = 2 ∫ 푑푥 |휙(푥)| (3.30.)
olarak yazarız. 푇 ve 푉 ş tek elektron fonksiyonelleridir ve 푈 Hartree enerjisinin bir
boyuttaki eş değeridir. Orbitalin bu fonksiyonelini basit olarak minimize edersek,
− − 푍훿(푥) + 휐 (푥) 휙(푥) = 휀 휙(푥) (3.31.)
Burada 휐 (푥) değeri,
휐 (푥) =( )
= ∫ 푑푥 푛(푥 ) 훿(푥 − 푥 ) = 푛(푥) (3.32.)
denklemi ile ifade edilir ve bu Hartree potansiyelidir. Bu denklem aynı zamanda
klasik veya elektrostatik potansiyel olarak bilinir. Bu denklem bu problem için
Hartree-Fock denklemidir.
3.3.3. Korelasyon
Hartree-Fock denkleminin çözümü tam değildir, çünkü doğru dalga
fonksiyonu iki orbitalin ürünü değildir fakat her ikisi için de eş zamanlı olarak
oldukça karmaşık bir fonksiyondur. Doğru dalga fonksiyonu tam Schrödünger
denklemi için uygundur ve aynı zamanda verilen dış potansiyel için temel durum
enerjisini minimize eder. Geleneksel kuantum kimyasında, korelasyon enerjisi

3. MATERYAL VE METOT ERKAN TETİK
58
Hartree-Fock enerjisi ve tam temel durum enerjisi arasındaki fark olarak tanımlanır,
yani,
퐸 = 퐸 − 퐸 (3.33.)
şeklinde yazılır. Korelasyon enerjileri sistemin toplam enerjisinin çok küçük bir
kısmını oluşturmasına karşın oldukça önemlidir. Genellikle 20-40 mH/electron
civarındadırlar.
3.3.5. N Elektron
Bu noktada, N elektronlu elektronik sistem için notasyonumuzu
tanımlayabiliriz. İlk olarak, N elektron için dalga fonksiyonu 3N uzaysal ve N spin
koordinatının bir fonksiyonudur. Dalga fonksiyonunu normalize edersek,
∫ 푑푥 … ∫ 푑푥 |Ψ(푥 , … , 푥 )| = 1 (3.34.)
burada ∫ 푑푥 her iki spin üzerinden toplam ve tüm uzay üzerinden integrali ifade eder.
Antisimetri prensibi,
Ψ 푥 , … , 푥 , … , 푥 , … = −Ψ 푥 , … , 푥 , … , 푥 , … (3.35.)
ile ifade edilir. Elektronik yoğunlu ise,
푛(r) = 푁 ∑ ∫ 푑푥 … ∫ 푑푥 |Ψ(r, 휎, 푥 , … , 푥 )| (3.36.)
şeklinde tanımlanır. 푛(r)푑 푟, r vektörü etrafında bir 푑 푟 bölgesinde herhangi bir
elektronu bulmak için kullanılan olasılık yoğunluğudur. Bu yoğunluk normalizedir,
yani,
∫ 푑 푟 푛(r) = 푁 (3.37.)

3. MATERYAL VE METOT ERKAN TETİK
59
eşitliğini sağlar. Bir ve iki parçacıklı operatörler özerinden toplam alırsak,
푇 = − ∑ ∇ (3.38.)
푉 ş = ∑ 휐 ş(r풊) (3.39.)
푉 = ∑풊 풋
(3.40.)
denklemlerini elde ederiz. Schrödinger denklemini,
푇 + 푉 + 푉 ş Ψ(푥 , … , 푥 ) = EΨ(푥 , … , 푥 ) (3.41.)
olarak yazar ve minimizasyon işlemini tüm normalize antisimetirk dalga
fonksiyonları üzerinden yazdığımız zaman, varyasyonel prensibe göre temel durum
enerjisini,
퐸 = min Ψ 푇 + 푉 + 푉 ş Ψ (3.42.)
şeklidne yazabiliriz. Etkileşmeyen parçacıların özel bir durumuna göre, Ψ yerine Φ
dalga fonksiyonunu alırsak, tek Slater determinantını,
Φ(푥 , … , 푥 ) =휙 (푥 ) ⋯ 휙 (푥 )
⋮ ⋮휙 (푥 ) ⋯ 휙 (푥 )
(3.43.)
şeklinde yazabiliriz. Spin-bağımsız bir dış potansiyeldeki aşağı ve yukarı parçacıların
sayısı eşit olan sistemler için, tüm orbitaller 휙 (푥) = 휙 (퐫)|휎⟩ denekleminin bir
ürünü olarak yazılabilir ve her uzaysal orbital iki kez görülür. Bu durumda, Hartree-
Fock enerjisi,

3. MATERYAL VE METOT ERKAN TETİK
60
퐸 = min Ψ 푇 + 푉 + 푉 ş Ψ (3.44.)
ile ifade edilir. Slater determinantı için Coulomb etkileşimi determinantın
antisimetrik doğasının ürünüdür ve buradaki iki katkı vardır,
Ψ 푉 Ψ = 푈[Ψ] + 퐸 [Ψ] (3.45.)
Bunlardan ilki doğrudan, Coulomb, elektrostatik yada klasik katkı olarak
isimlendirilir. İkincisi ise Fock ya da değiş-tokuş integrali, yani,
퐸 [휙 ] = − ∑ ∑ ∫ 푑 푟, ∫ 푑 푟∗ ( ) ∗ ( )
| | (3.46.)
şeklindedir. Bu teorik olarak Pauli-dışarlama ilkesinin bir etkisidir.
3.3.6. Elektronik Yoğunluk
Tüm atomik yoğunluklar orijinde çekirdekle r’nin bir fonksiyonu olarak
çizildiği zaman, tam olarak ispatlanmamış olmasına karşın, düzenli olarak azaldığı
görülmektedir. Çekirdeğin yakınlarında, dış potansiyel Schrödinger denkleminde
baskındır ve yoğunlukta piklerin oluşmasını sağlar. Piklerin boyu çekirdeğin yükü ile
orantılıdır yani,
= −2푍 푛(0) (3.47.)
şeklindedir. Bu Kato tarafından ortaya atılan bir pik koşuludur. Küresel yoğunluklar
4휋푟 faz-uzayı faktörü tarafından çizilmektedir. Bu eğri altındaki bölgelerin N
olduğu anlamına gelir. Ayrıca farklı elektronik kabuklar kolayca görülebilir.

3. MATERYAL VE METOT ERKAN TETİK
61
Şekil 3.3. Ar atomunun yarıçapa bağlı yoğunluğu
Örnek olarak Ar atomunu inceleyelim. Şekil 3.3’de Ar atomunun yarıçapa
bağlı yoğunluğu görülmektedir. Bu yoğunluk 18 elektrondan oluşmaktadır. 2
elektron içeren 푟 = 0,13, 4 elektron içeren 푟 = 0,25, 10 elektron içeren 푟 = 0,722
ve 12 elektron içeren 푟 = 1,13’e kadar olan kısımların integralini bulabiliriz. Bunlar
sırasıyla 2 tane 1s, 2 tane 2s, 6 tane 2p ve 2 tane 3s elektronuna karşılık gelmektedir.
Yarıçapa bağlı yoğunluktaki pikler ve çukurluklar bu kabukları temsil etmektedir.
Dalga fonksiyonundaki bir koordinat çekirdekten uzak mesafelerde alındığı
zaman, N-elektronlu taban durumu dalga fonksiyonu (푁 − 1)-elektronlu dalga
fonksiyonunun yoğunluk zamanının karekökünün bir sonucu olarak azalır. Bu
yoğunluğun karekökünün Schrödinger benzeri bir denklemle tanımlandığı anlamına
gelir. Özdeğerler iki sistem arasındaki enerjilerde farklılık gösterir yani,
푛(푟) = 퐴푟 exp(−α 푟) (푟 → ∞) (3.48.)
şeklindedir ve burada α = √2퐼 ve,
퐼 = 퐸(푁 − 1) − 퐸(푁) (3.49.)
r
4π r n(r)
10
5
0
25
0,5 1,0 1,5 2,0 2,5 3,0 0
Ar Atomu 2 15
20
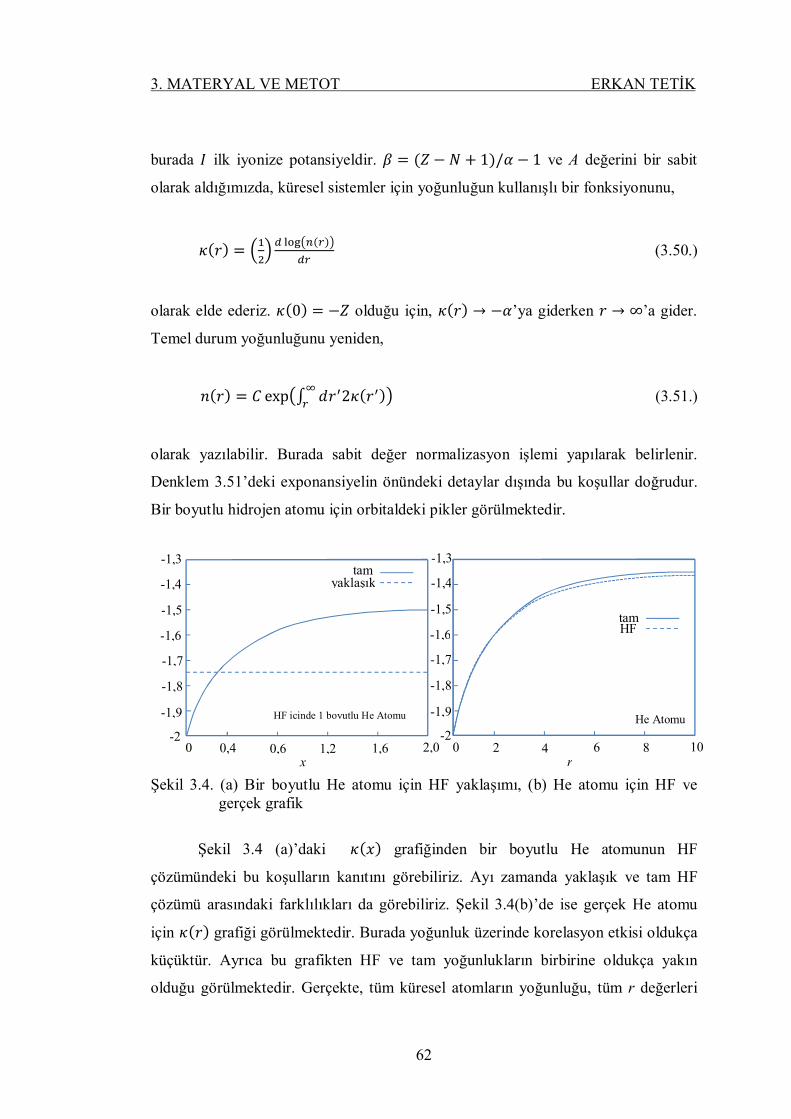
3. MATERYAL VE METOT ERKAN TETİK
62
-1,8 -1,9
-2
-1,6
0 0,4 0,6 1,2 1,6 x
2,0
-1,7
HF içinde 1 boyutlu He Atomu
0 2 4 6 8
r 10
He Atomu
-1,5 -1,4 -1,3
-1,8 -1,9
-2
-1,6 -1,7
-1,5 -1,4 -1,3 κ tam
yaklaşık
tam HF
burada 퐼 ilk iyonize potansiyeldir. 훽 = (푍 − 푁 + 1)/훼 − 1 ve A değerini bir sabit
olarak aldığımızda, küresel sistemler için yoğunluğun kullanışlı bir fonksiyonunu,
휅(푟) = ( ) (3.50.)
olarak elde ederiz. 휅(0) = −푍 olduğu için, 휅(푟) → −훼’ya giderken 푟 → ∞’a gider.
Temel durum yoğunluğunu yeniden,
푛(푟) = 퐶 exp ∫ 푑푟 2휅(푟 ) (3.51.)
olarak yazılabilir. Burada sabit değer normalizasyon işlemi yapılarak belirlenir.
Denklem 3.51’deki exponansiyelin önündeki detaylar dışında bu koşullar doğrudur.
Bir boyutlu hidrojen atomu için orbitaldeki pikler görülmektedir.
Şekil 3.4. (a) Bir boyutlu He atomu için HF yaklaşımı, (b) He atomu için HF ve gerçek grafik
Şekil 3.4 (a)’daki 휅(푥) grafiğinden bir boyutlu He atomunun HF
çözümündeki bu koşulların kanıtını görebiliriz. Ayı zamanda yaklaşık ve tam HF
çözümü arasındaki farklılıkları da görebiliriz. Şekil 3.4(b)’de ise gerçek He atomu
için 휅(푟) grafiği görülmektedir. Burada yoğunluk üzerinde korelasyon etkisi oldukça
küçüktür. Ayrıca bu grafikten HF ve tam yoğunlukların birbirine oldukça yakın
olduğu görülmektedir. Gerçekte, tüm küresel atomların yoğunluğu, tüm r değerleri

3. MATERYAL VE METOT ERKAN TETİK
63
için düzenli olarak azaldığı bilinmektedir, fakat henüz hiç kimse bu gerçeği
ispatlayamamıştır.
3.4. Modern Yoğunluk Fonksiyoneli Teorisi
Yoğunluk Fonksiyonel Teorisinin temelleri Hohenberg ve Kohn (1964)
tarafından atılmıştır. Hohenberg ve Kohn yapmış oldukları çalışmada, geleneksel
yöntemlerle katı veya molekülün enerjisini hesaplarken kullanılan çok parçacıklı
dalga fonksiyonlarını temel değişken olarak almanın problemi oldukça
güçleştirdiğini öne sürerek, dalga fonksiyonunun yerine yer ve zamanın bir
fonksiyonu olan elektron yoğunluğunu temel değişken olarak almışlardır. Yani
YFT’de temel değişken olarak, çok-parçacık dalga fonksiyonu yerine, tek parçacık
yoğunluğu kullanılır. Yoğunluk sadece üç tane uzaysal koordinatın (dalga
fonksiyonunun ya da N tane koordinatın değil) fonksiyonu olduğundan, DFT çok
büyük sistemleri hesaplamada kolaylık sağlar. Buradaki temel amaç elektron
yoğunluk fonksiyonları aracılığıyla elektron korelasyonunu modellemektir.
Çok parçacıklı bir sistemde r noktasına yerleşmiş 푉 ş(푟) olarak tanımlanan
bir potansiyele sahip N tane elektron olduğunu kabul edelim. Schrödinger denklemi
kullanılarak bu sistemin özellikleri hesaplanabilir. N elektronlu sistem için
Hamiltonyen,
퐻 = ∑ ( ℏ ∇ ) + ∑ 푉 ş(푟 ) + ∑
(3.52.)
olarak yazılır. Bu denklem N tane tek elektron Schrödinger denklemine
indirgendiğinde,
− ∇ + 푉 ş(푟) Ψ (푟) = 휀 Ψ (푟) (3.53.)
şeklinde olur. Buradaki Ψ (푟)’ler tek elektron dalga fonksiyonları ve 푉(푟) tek
elektronun tüm etkileşimlerini içeren terimidir yani,

3. MATERYAL VE METOT ERKAN TETİK
64
푉(푟) = 푉 ş(푟) + 푉 (푟) + 푉 (푟) (3.54.)
denklemi ile ifade edilir. Bu denklemdeki, potansiyeldeki ilk terim iyonlarla olan
etkileşimi, ikinci terim diğer elektronlarla olan etkileşimi, üçüncü terim ise değiş-
tokuş ve korelasyon etkileşimini gösterir.
Hohenberg ve Kohn 1964 yılında homojen olmayan elektron gazının taban
durumunu bulmak için YFT’yi geliştirmişlerdir (Hohenberg ve Kohn, 1964). Böyle
bir sistem için parçacık yoğunluğu,
휌(푟) = 푁 ∫|Ψ (푟 , 푟 , … , 푟 )| 푑푟 … 푟 (3.55.)
ile verilir. Burada 0 sistemin taban durumu dalga fonksiyonudur. Hohenberg ve
Kohn sistemin taban durum enerjisinin yoğunluğun fonksiyoneli olarak
verilebileceğini ve enerji fonksiyonelinin, iyonlarla etkileşim ile ilgili olan terimi
dışındaki kısmının (퐹[휌]) evrensel olduğu gösterilmiştir. 푉 ş(푟) ile belirlenen
yoğunluk sistemi tek olarak betimlenmektedir. Ayrıca 퐹[휌] minimum değerini
sadece taban durumu yoğunluğunda alır. Kohn ve Sham, Hohenberg ve Kohn
teoremlerini kullanarak bugün Kohn-Sham denklemleri olarak bilinen ve enerji
fonksiyonelini minimum yapan yoğunluğun denklemlerini,
( ') ( )( ) ( ) ' ( ) ( ) ( )' XC dış
r rE r T r dr dr E r r V r drr r
(3.56.)
olarak ifade etmişlerdir. Yoğunluk fonksiyonelinin,
( ')' ( ) ( )'etk XC dış
rV dr V r V rr r
(3.57.)
şeklinde tanımlanması ve

3. MATERYAL VE METOT ERKAN TETİK
65
N
ii rr
1
2)()( (3.58.)
ile verilen yoğunluğa göre minimize edilmesiyle
)()()(21 2 rrrV iietki
(3.59.)
denklemi elde edilir. Bu denklem öz uyumlu çözülmektedir. Bunun için verilen
başlangıç yoğunluğundan etkV hesaplanır. etkV , Denk. 3.59’da yazılarak i ’ler elde
edilir. Daha sonra bulunan i ’ler, Denk. 3.58’da kullanılarak yeni yoğunluk elde
edilir. Hesaplamalar tamamlandığında elde edilen yoğunluk Denk. 3.56’de yazılarak
sistemin taban durum enerjisi elde edilir.
Yöntemin kesinliğini bozan etken denklem 3.56’de verilen XCE terimidir. Bu
terimin formu bilinmediğinden, yoğunluğun fonksiyoneli olarak yazmak zordur.
Bunun için ise iki yaklaşım kullanılmaktadır. Bir tanesi yerel yoğunluk yaklaşımı
(local density approximation, LDA) diğeri ise genelleştirilmiş gradiyent yaklaşımıdır
(generalized gradient approximation, GGA).
3.4.1. Hohenberg-Kohn Teoremleri
Hohenberg ve Kohn (1964)'de, temel değişken olan elektron yoğunluğunu ele
alarak, toplam enerjiyi yük yoğunluğunun bir fonksiyoneli olarak gösterilebileceğini
kanıtladılar. Bu toplam enerji fonksiyonunun minimum değeri sistemin temel durum
enerjisine karşılık gelmektedir. Buna ek olarak, bu minimum değere neden olan yük
yoğunluğu tek parçacık temel durum yoğunluğudur.
Herhangi bir kuantum mekaniksel problemin çözümü dalga
fonksiyonunun belirlenmesine bağlıdır. Dalga fonksiyonu bilinebilen tüm bilgilere
geçiş sağladığı için merkezi bir niceliktir. Katı gibi geniş bir sistem için, dalga
fonksiyonunun belirlenmesinin bazı zorlukları vardır. Dalga fonksiyonu çok

3. MATERYAL VE METOT ERKAN TETİK
66
karmaşık bir niceliktir. Deneysel olarak ölçülemez ve N tane elektronun her biri için
3 uzaysal ve bir spin değişkeni olmak üzere N4 değişkene bağlıdır. Katıhal
sistemlerinin çoğu, çok sayıda elektron ve iyonlar içerdiği için, herhangi bir dalga
fonksiyonunu hesaplamak oldukça zor ve zaman alıcı bir işlemdir. Bu çözümü
zorlaştırmakla kalmaz aynı zamanda sistem hakkında tanımlayıcı olmayı da karmaşık
hale getirmektedir. Diğer taraftan elektron yoğunluğunun değişken fonksiyon olduğu
temel durum enerjisi için varyasyon prensibini geliştirmek mümkündür. Bu yoğunluk
fonksiyonu )(r , yalnızca üç uzaysal koordinata bağlıdır.
YFT’de, ana değişken olan )(r yoğunluğunu seçmek, herhangi bir atomik
veya moleküler sistemin Hamiltonyen operatörünün, çekirdek yükler )( kZ , uzaydaki
çekirdeğin konumu )( kR
ve elektronların sayısı )(N tarafından tanımlanmasıyla
olmaktadır. M çekirdek ve N elektrondan oluşan bir sistem için, atomik birimlerde
)1( eme temel hamiltonyen
nuclel HHH (3.60.)
şeklinde verilir. Bu denklemdeki 퐻 ve 퐻 terimleri
N
i
N
i
M
k
N
i
N
ij ijik
kiel rr
ZH
1 1 1 1
2 121
(3.61.)
M
k
M
k
M
kl kl
lkk
knucl R
ZZM
H1 1
2121 (3.62.)
olarak ifade edilir. Çekirdek ve elektronların kütleleri arasındaki farklar göz önüne
alınırsa Hamiltonyen basitleştirilebilir. Bütün çekirdeklerin en hafifi olan proton bir
elektrondan yaklaşık olarak 1800 kez daha ağırdır. Böylece, çekirdek elektronlarla
kıyaslandığında çok daha yavaş hareket eder. Bu nedenle, elektronlar sabit çekirdek
alanında hareket ediyor olarak düşünülebilir. Bu Born-Oppenheimer yaklaşımı olarak

3. MATERYAL VE METOT ERKAN TETİK
67
bilinir. Çekirdek uzayda hareketsiz ise, kinetik enerjileri sıfırdır ve çekirdek-çekirdek
itmelerinden dolayı potansiyel enerjileri sadece bir sabittir. Sonuç olarak denklem
3.68’deki Hamiltonyen, 퐻 elektronik Hamiltonyene indirgenir. Böylece sistemin
çözümü sadece elektronik dalga fonksiyonu olur. Bu durumda öz değer problemi
elelelel EH (3.63.)
şeklinde verilir. Bu yaklaşımdan sonra sistemin toplam enerjisi
nucleltot EEE
(3.64.)
denklemi ile ifade edilir. Burada nuclE , Denk. 4.70’deki ikinci terimdir ve bir sabit
olarak ele alınır. Sonuç olarak herhangi bir atomik veya moleküler sistem için toplam
Hamiltonyen, elektronların sayısı )(N , çekirdek konumları )( kR
ve çekirdek yükleri
)( kZ ile tanımlanabilir. Biz yalnızca temel durumun dejenere olmadığı durumlarla
ilgileneceğiz. Diğer taraftan Hamiltonyeni elektron yoğunluğu açısından
gösterebiliriz. Hamiltonyen ile ilişkilendirilmiş üç önemli özellik vardır.
1) Elektron yoğunluğu )(r , sistemdeki parçacıkların toplam sayısına normalize
edilir.
Nrrd )( (3.65.)
2) Doğal olarak )(r , yalnızca iyon merkezlerinde )( kR
maksimuma sahiptir.
3) )(r nükleer koordinatlarda )( kR
nükleer yükler )( kZ hakkında bilgi içerir.
)0(2)(0
krkk
Zrr k
(3.66.)

3. MATERYAL VE METOT ERKAN TETİK
68
burada kr , k indisi ile belirtilen iyon göbeğinden radyal uzaklıktır ve )( kr aynı
iyon çevresindeki yük yoğunluğunun küresel ortalamasıdır. Bu nedenle )(r , özel
hamiltonyen sistemin tanımlanabildiği tek değişken olarak seçilebilir ve tüm
moleküler özelliklerin tanımlanabilmesi için )(r yeterli olur. Elektron yoğunluğu
)(r , tüm sistemin fiziksel özellikleri hakkında bilgi almak için kullanılabilir. Temel
durumda )(r ,
nN xdxddsxxxNr ...),...,,()( 21
2
21
* ( ) ( )el elr r
(3.67.)
olarak tanımlanır. İlk Hohenberg-Kohn teoremi sistemin tüm özelliklerini ve
Hamiltonyen operatörünü belirleyen elektron yoğunluğunu sağlamaktadır.
Hohenberg ve Kohn (1964)’de makalelerinde çok basit bir ispat verdiler. Bir )(rV
dış potansiyeli etkisi altındaki bir elektron gazını dikkate aldılar. Hamiltonyeni
aşağıdaki gibi verdiler,
UVTH (3.68.)
bu denklemde,
rdrrT )()(
21 * (3.69.)
rdrrrVV )()()( * (3.70.)
)()()()(21 ** rrrr
rrrdrdU
(3.71.)

3. MATERYAL VE METOT ERKAN TETİK
69
olarak tanımlanır. Denk. 3.67’de tanımlandığı gibi )(r , )(rV ’nin bir
fonksiyonelidir. Sonra aynı yük yoğunluğu )(r ’ye neden olan )(rV ve )(rV iki
dış potansiyeli dikkate aldılar. Bu düşünce, yoğunluk formülünün denklem 3.67’de
verilen bir dalga fonksiyonundan nasıl inşa edilmesi gerektiğini göstermektedir.
Temel durum ve sonraki ilk sonuçlar dejenere olmayan elektron sistemi ile
ilişkilendirilmiş ' sağlıyor. Orijinal Hohenberg- Kohn ispatının dejenere olmayan
temel durumlarına getirilen kısıtlama daha sonra kaldırılacaktır. Potansiyeller en az
bir sabitle birbirinden farklı olmasaydı, farklı Schrödinger denklemlerini
sağlamadıkları için ,' ’ye eşit olmazdı. )(rV dış potansiyeline karşılık gelen
temel durum enerjisi
HHE
VVH (3.72.)
olarak yazılabilir. Burada HE , gibi nicelikler ile karakterize edilen sisteme ait
niceliklerdir ve HE, gibi nicelikler ise aittir. Hamiltonyenler sadece dış
potansiyellerde farklı olduğu için
rdrrVrVEE )()()( (3.73.)
olarak yazılır. VE , ve VE, niceliklerdeki değişme, aşağıdaki aynı işlemle bulunur.
rdrrVrVEE )()()( (3.74.)
denklem 3.73 ve 3.74’nin toplamı bir tutarsızlığa neden olur.
EEEE (3.75.)

3. MATERYAL VE METOT ERKAN TETİK
70
Bu yüzden aynı temel durum elektron yoğunluğunu sağlayan iki farklı potansiyel
olmayabilir. Ayrıca )(rV , küçük bir sabitle toplanan elektron yoğunluğu )(r
vasıtası ile belirlenir.
İkinci teorem, varyasyonel ilkesi (variational theorem) olarak adlandırılır.
Varyasyonel ilkesi, kinetik ve elektron-elektron etkileşim enerjisi olan tüm
parçacıkların temel durum enerjisi )(r ’nin tek bir fonksiyoneli olduğu gerçeğinden
ortaya çıkar. Bu nedenle, temel durum toplam enerji E, temel durum yük yoğunluğu
)(r terimine bağlı olarak iki kısma ayrılabilir.
FrdrrVE )()( (3.76.)
burada
UTF (3.77.)
olarak verilir. Bu ifadede ki F Hohenberg-Kohn fonksiyoneli olarak adlandırılır
ve elektronların sayısından )(N , çekirdek koordinatlarından )( kR
ve çekirdek yükü
)( kZ ’den bağımsızdır. Yani F , parçacıkların herhangi bir sayısından ve herhangi
bir dış potansiyel için geçerli olan evrensel bir fonksiyoneldir.
Hohenberg-Kohn fonksiyoneli F , giriş yoğunluğu sadece doğru temel
durum yoğunluğu ise en düşük enerjiyi tanımlayan sistemin temel varyasyonel
ilkesini ortaya çıkarır. Doğru temel durum enerjisini bulmak için değişken prensip
kullanılabilir. Bu nedenle denklem 3.76’deki ifade bir değişken problem olarak ele
alınabilir. Varyasyonel ilkesinin uygulanabilir olmasında bazı kısıtlamalar vardır. İlk
olarak bu metot sistemin en düşük enerji durumunu gösterdiği için temel duruma
sınırlandırılıyor. İkincisi, deneme yoğunluğu pozitif olmalı ve denklem 3.65’de
verildiği gibi parçacıkların sayısı N ’yi integre etmelidir.

3. MATERYAL VE METOT ERKAN TETİK
71
Herhangi bir deneme yoğunluğu ~ kendi Hamiltonyeni H~ ve kendi dalga
fonksiyonu ~ tanımlıyor. Dolayısıyla bu dalga fonksiyonu gerçek dış potansiyel
)(rV ’den türetilen Hamiltonyen için deneme dalga fonksiyonu olarak alınabilir.
3.4.2. Thomas-Fermi Teorisi
Thomas-Fermi Teorisini ilk YFT teorisi olarak değerlendirebiliriz. Bu teori
1927 yılında, L. Thomas ve E. Fermi tarafından öne sürülen yarı klasik bir
yaklaşımdır (Thomas, 1927; Fermi, 1928). Thomas-Fermi teorisinde başlangıç
noktası çok-elektron dalga fonksiyonu değil elektron yoğunluğuydu. Birkaç yıl sonra
ise Dirac (1930) bu teoriye değişim terimini ekledi. Thomas ve Fermi atomun kinetik
enerjisini elektron yoğunluğunun fonksiyoneli olarak kabul edip bir atomun
enerjisini hesaplamışlar ve bunu iyon-elektron ve elektron-elektron etkileşimlerine
ait klasik ifadelerle birleştirmişlerdir. Bu iki etkileşmeyi elektron yoğunluğu
cinsinden ifade edip, sistemin toplam enerjisi minimize etmişlerdir.
Bu yaklaşımda elektronlar bağımsız parçacıklar olarak düşünülür. Sistemin
toplam enerjisini oluşturan terimlerden biri elektron-elektron etkileşim enerjisidir ve
sadece elektrostatik enerjiden kaynaklanır (Jones ve Gunnarsson, 1989) yani,
'
')'()(
2)(
2
rdrdrr
rrerEes
(3.78.)
şeklinde ifade edilir. )(rt , )(r yoğunluklu etkileşmeyen elektronlar sisteminin
kinetik enerji yoğunluk fonksiyonelidir. Homojen elektron gazı için kinetik enerji
yoğunluk fonksiyoneli,
kdmkrt
Fkk
2)2(
2)(22
3 (3.79.)
şeklinde verilir. Fermi dalga vektörü

3. MATERYAL VE METOT ERKAN TETİK
72
3/12 )(3 rkF
(3.80.)
eşitliği ile verilir. Buradan,
3/222 )3(10
3
mC k (3.81.)
olmak üzere kinetik enerji fonksiyoneli
rdrCrT k 3/5)()( (3.82.)
şeklinde verilir. Toplam enerjideki son terim ise iyon-elektron arasındaki
elektrostatik çekim enerjisidir. Dolayısıyla elektron yoğunluğunun fonksiyoneli
olarak sistemin toplam enerjisi
rdrvrrCrdrdrr
rrerE kTF
)()()('
')'()(
2)( 3/5
2
(3.83.)
şeklinde yazılır. Burada
M
RrZrv
1)(
(3.84.)
iyonlardan kaynaklanan statik Coulomb potansiyelidir. Elektronların sayısının sabit
olduğu varsayıldığında
Nrdr )( (3.85.)

3. MATERYAL VE METOT ERKAN TETİK
73
ile verilen yoğunluğa göre )(rE fonksiyoneli varyasyon metodu kullanılarak
minimize edilebilir yani,
0))(()( NrdrrE 3.86.)
varyasyonu alındığında bilinen Thomas-Fermi denklemi
0)()(35'
')'( 3/22 rVrCrd
rrre extk
(3.87.)
şeklinde elde edilir. Dirac (1930), Thomas-Fermi teorisine bir değişim terimi ekledi.
Thomas-Fermi-Dirac teorisinde elektron yoğunluğunun fonksiyoneli olarak enerji
rdrCrErE eTFD )(
43)()( 3/4 3.88.)
şeklinde yazılır (Lieb, 1981). Bu denklemin sağ tarafındaki ikinci terim Dirac
değişim terimi olarak adlandırılan değiş-tokuş enerjisidir ve eC pozitif bir sabittir.
Thomas-Fermi teorisi önemli bir ilk adım olmasına rağmen, doğruluğu
sınırlıdır. Çünkü Hartree-Fock teorisinde öngörülen bir atomun değiş-tokuş enerjisi
dikkate alınmamıştır. Değiş-tokuş enerjisi 1928’de Dirac tarafından eklenmiş ama
yinede teori pek çok uygulamada tam doğru sonuçlar vermemiştir. Bunun en önemli
nedeni kinetik enerjinin temsilinden kaynaklanmakta olup, ayrıca değiş-tokuş
enerjisinin ve elektron korelasyonu’nun tamamen ihmal edilmiş olmasındandır.
3.4.3. Kohn-Sham Denklemleri
Kohn-Sham 1965 yılında, çok elektron sistemleri için değişim ve korelasyon
etkilerini de içeren, Hartree-Fock denklemlerine benzer öz-uyumlu denklemler için
bir formülasyon oluşturdular. Bu formülasyonda reel ve etkileşen elektronlar sistemi,

3. MATERYAL VE METOT ERKAN TETİK
74
etkileşmeyen hayali bir sisteme dönüştürülerek elektronlar etkin bir potansiyelde
hareket ettirilir. Bu potansiyel “Kohn-Sham tek-parçacık potansiyeli” olarak
isimlendirilir.
Yoğunluk fonksiyoneli teorisi, dalga fonksiyonlarını kullanmadan yalnızca
temel durum elektron yoğunluğunu kullanarak çok elektron sistemlerinin
özelliklerini belirtmeyi amaçlar. N elektron içeren relativistik olmayan Schrödinger
denklemi için elektronik Hamiltonyen,
ee
N
i
N
i
N
ji jiidııi VVT
rrrVH ˆˆˆ1
21)(
21ˆ
1 1
2
(3.89.)
olarak ifade edilmektedir. Burada )( idıı rV i. elektrona etkiyen dış potansiyeldir.
Atomlar, moleküller ve katılar için )( idıı rV , R konumunda Z yüklü çekirdeklerin
Coulomb potansiyelidir yani,
RrZrV
iidıı )( (3.90.)
olarak yazılır. Schrödinger denklemi * ile soldan çarpılır ve herbir terim uzaysal
)( ir ve spin )( i koordinatları üzerinden integre edilip, ’nin normalize olduğu
kabul edilirse, sistemin enerjisi
21
21
2122 ),()(),(21 rdrd
rrrrPrdrVrdrrE dıırrr
(3.91.)
şeklinde bulunur. Burada ),( rr indirgenmiş tek elektron yoğunluk matrisidir ve
),( 212 rrP çift yoğunluğu,

3. MATERYAL VE METOT ERKAN TETİK
75
NNN rdrdrrrNNrrPN
...),...,,(...2
)1(),( 3
2
...2211212
1
(3.92.)
olarak verilir. Yoğunluk matrisi ve çift yoğunluğu
2212111 ),(
12),()( rdrrP
Nrrr
(3.93.)
ile ilişkilidir. Denk. 3.88’deki ilk terim elektronların kinetik enerjisini
rdrrT rrr
,(21 2 . (3.94.)
ikinci terim elektron-çekirdek etkileşim enerjisini
rdrrVV dıı )()( (3.95.)
ve üçüncü terim elektron-elektron etkileşim enerjisini
2121
2,12 )(rdrd
rrrrP
Vee
(3.96.)
gösterir. Bu son terim klasik Coulomb itmelerini ve kuantum mekaniksel değişim-
korelasyon etkilerini içerir. Klasik ve kuantum mekaniksel kısımlarının ayrılması,
çift yoğunluğunun aşağıdaki şekilde yeniden yazılmasıyla yapılabilir.
),()()(21),( 2121212 rrhrrrrP XC
(3.97.)
Buradaki ),( 21 rrhXC , 1r
konumundaki bir elektronun değişim-korelasyon holudur.
Denk. 3.94 Denk. 3.93’de yerine yazılırsa,

3. MATERYAL VE METOT ERKAN TETİK
76
)(c
XCee EUV (3.98.)
olarak yazılabilir. Burada U , klasik Coulomb itme enerjisi
2121
21 )()(21 rdrd
rrrrU
(3.99.)
şeklinde yazılır. )(cXCE konvansiyonel değişim-korelasyon enerjisi (Scuseria ve
Staroverov, 2005) ise
2121
211)( ),()(21 rdrd
rrrrhr
E XCcXC
(3.100.)
şeklinde verilir. Şimdi Denk. 3.88 yeniden
)(c
XCEUVTE (3.101.)
olarak yazılabilir. V ve U )(r ’nin açık fonksiyoneli olduğuna T ve )(cXCE ’nin ise
olmadığına dikkat edilmelidir. T ve )(cXCE , ),( rr
yoğunluk matrisinin ve ),( 212 rrP
çift fonksiyonun bilinmesini gerektirdiği için )(r ’den belirlenemeyeceği izlenimi
doğar. Hohenberg-Kohn teoremleri statik dış potansiyel )(rVdıı
’de bulunan gerçek
çok elektron sisteminin temel durum enerjisinin yoğunluğun fonksiyoneli olduğunu
iddia ettiği için bu öngörü doğru değildir. Bu durumda enerji
FrdrrVE dıı )()( (3.102.)

3. MATERYAL VE METOT ERKAN TETİK
77
şeklinde ifade edilir. Burada F , kinetik enerji ve elektron-elektron etkileşim
terimlerini içerir. Bu durumda
)(cXCEUTF (3.102.)
şeklinde yazılır. Hohenberg-Kohn teoremleri sadece F fonksiyonelinin varlığını
garanti eder, fakat F ’nin açık formu bilinmemektedir ( U hariç) ve yaklaşım
yapılmalıdır. Elektronların sayısı sabit olduğunda herhangi iki sistemin hamiltoniyen
operatörleri arasındaki fark )(rVdıı
ile belirlenir. Bundan dolayı F evrensel bir
fonksiyoneldir.
3.4.4. Kohn-Sham Formülasyonu
Denklem 3.99’daki genel Hamiltoniyendeki eeV terimi bir elektron-elektron
çiftlenim sabiti ile ayarlanmıştır. değerleri 0 ile 1 arasındadır. ’nın herbir
değeri farklı evrensel yoğunluk fonksiyoneline karşılık gelir (Levy, 1979). Evrensel
yoğunluk fonksiyoneli
min,min, ˆˆ eeVTF (3.104.)
olarak ifade edilir. Burada min, , eeVT ˆˆ beklenen değerini minimize eden ve
)(r yoğunluğunu üreten N-elektron dalga fonksiyonudur. Reel sistemler için,
FF 1 olsun diye 1 ’dir. 0 değeri, )(rVdıı
dış potansiyelde hareket
eden etkileşmeyen bir elektronlar sistemine karşılık gelir. Etkileşmeyen elektronlar
sistemi için Schrödinger denklemini çözmek mümkündür. 0min,0 çözümü, tek
parçacık denklemlerinden elde edilen tek elektron i dalga fonksiyonlarının tek bir
Slater determinantıdır.

3. MATERYAL VE METOT ERKAN TETİK
78
)()()(21 2 rrrV iiidıı
(3.105.)
Etkileşen bu sistem için evrensel yoğunluk fonksiyoneli,
N
iiisTF
1
20 2
1 (3.106.)
olarak verilir. Burada elektron yoğunluğu
N
ii rr
1
2)()( (3.107.)
ile verilir. sT orbitaller cinsinden yazılabilir ve bundan dolayı )(r ’nin bir
fonksiyonelidir.
Kohn-Sham varsayımına göre, )(r temel durum yoğunluklu herhangi bir
real (etkileşen) sistem için daima aynı )(r temel durumlu etkileşmeyen bir sistem
vardır. Bu durumda
XCs EUTF (3.108.)
olarak yazılabilir. Burada sT , etkileşmeyen sistemin kinetik enerjisidir. XCE
ise DFT değiş-tokuş ve korelasyon enerjisidir ve formal olarak
)(cXCsXC ETTE (3.109.)
şeklinde tanımlanır. 0)( rE varyasyon ilkesinin uygulanmasıyla Kohn-Sham
formülasyonu

3. MATERYAL VE METOT ERKAN TETİK
79
XCsdıı EUTrdrVrE )()( (3.110.)
olarak verilir. ijii | ortonormallik şartı ile N tane Hartree tipi tek elektron
denklemi
)()()()()(21 2 rrrvrd
rrrrV iiiXCdıı
(3.111.)
şeklinde elde edilir. Burada )(ri
Kohn-Sham orbitalleri, i Kohn-Sham orbital
enerjisi ve )(rv XC ise değişim-korelasyon potansiyelidir. Yani
)(
)(r
Erv XC
XC
(3.112.)
Yoğunluk ile ilgili XCE ’nin fonksiyonel türevidir. )(ri
orbitalleri Kohn-Sham
dalga fonksiyonu olarak adlandırılan bir min Slater determinantını oluştururlar.
Kohn-Sham denklemleri olarak bilinen (3.107), (3.111) ve (3.112) denklemleri
formal olarak tam ve kesindirler. Yalnızca XCE bilinmeyen terimini içerirler.
Kohn-Sham DFT’de yaklaşıklığı yapılan XCE terimidir; konvensiyonel değiş-
tokuş ve korelasyon terimi )(cXCE değildir.
3.4.5. Değiş-Tokuş ve Korelasyon Enerjisi
Denklem 3.106 ile verilen değiş-tokuş ve korelasyon enerjisinin formal
tanımıyla yaklaşık yoğunluk fonksiyonellerini oluşturmak çok elverişli değildir. Ama
XCE için çok iyi formüller vardır. Hellmann(1937)-Feynman(1939) teoremiyle

3. MATERYAL VE METOT ERKAN TETİK
80
min,min, ˆ
eeVF (3.113.)
şeklinde verilir. Burada F denklem 3.104 ile verilir. (3.113) denkleminde, tüm
’larda )(r ’yi sabit tutarak üzerinden 0’dan 1’e kadar integrali alınırsa,
UEFFdF
XC
01
1
0
(3.114.)
şeklinde olur. Bu yöntem “adyabatik integrasyon” olarak adlandırılır (Scuseria ve
Staroverov, 2005). Bu son denklemde FF 1 için sırasıyla (3.103) ve (3.105)
denklemleri kullanıldı. 3.113 ve 3.114 denklemleri birleştirildiğinde adyabatik
bağlantı formülleri
1
0
1
0
min,min, ˆ
dEUdVE XCeeXC (3.115.)
şeklinde elde edilir (Harris ve Jones, 1974; Langreth ve Perdew, 1975, 1977;
Gunnarsson ve Lundqvist, 1976). Denklem 3.94’daki çift fonksiyonu ),( 212 rrP ve
değiş-tokuş ve korelasyon hol ),( 21 rrhXC tanımları genelleştirilerek ve 3.97, 3.98 ve
3.99 denklemleri kullanılarak 3.105 denklemi
21
21
2111
0
),()(21 rdrd
rrrrhrdE XC
XC
(3.116.)
şeklinde yeniden yazılabilir. Bu denklemdeki )( 1r ’de elektron yoğunluğu sabit
olduğu için indisi yoktur. üzerinden integrasyon uzay koordinatları üzerinden
integrasyona dönüştürülerek

3. MATERYAL VE METOT ERKAN TETİK
81
1
02121 ),(),( drrhrrh XCXC (3.117.)
şeklinde tanımlanır ve 3.116 denklemi
2121
211 ),()(21 rdrd
rrrrhr
E XCXC
(3.118.)
şeklinde yeniden yazılabilir. 3.118 denklemi 3.100 denklemine benzerdir. Şimdi
121, rrurr değişken değişimi yapılır ve u’nun açısal koordinatları üzerinden
integral alınır. Bu
2
0 0
sin),(41),( uuXCuXC durrhdurh (3.119.)
şeklinde ortalama değiş-tokuş ve korelasyon holu verir. Bu son ifade kullanılarak
3.118 denklemi
duu
urhurdrE XCXC
0
2 ),(4)(21
(3.120.)
olarak yazılabilir. Bu denklem birçok yoğunluk fonksiyoneli yaklaşımları için
başlangıç noktasıdır.
Kohn-Sham DFT’de XCE değiş-tokuş ve korelasyon fonksiyoneli
genellikle
CXXC EEE (3.121.)
şeklinde değiş-tokuş ve korelasyon kısımlarına ayrılır. Değişim enerjisi,

3. MATERYAL VE METOT ERKAN TETİK
82
UVE eeX minmin ˆ (3.122.)
ile tanımlanır. Burada min Kohn-Sham determinantıdır. Korelasyon enerjisi ise
formal olarak
minminminmin ˆˆ eeeeXXCC VVEEE (3.123.)
şeklinde tanımlanır. Burada min etkileşen dalga fonksiyonudur. min
gibi tek
determinant dalga fonksiyonu için çift yoğunluğu (Scuseria ve Staroverov, 2005).
),(),(),(),(21)()(
21),( 1221122121212 rrrrrrrrrrrrP
(3.124.)
olarak verilir. Spini dengelenmiş sistemler için 3.97 ve 3.122 denklemlerinde 3.124
denkleminin yerine yazılmasıyla değişim enerjisi için
21
21
221,(
41 rdrd
rrrr
EX
(3.125.)
şeklinde bir ifade elde edilir. Burada ),( 21 rr Kohn-Sham tek elektron yoğunluk
matrisidir. Bu yoğunluk
)()(),( 21
121 rrrrN
iii
(3.126.)
şeklinde verilir. 3.125 denklemi, tam olarak Hatree-Fock (HF) teorisindeki değişim
terimine benzerdir. Fakat 3.126 denklemindeki )(ri
’ler Hatree-Fock orbitalleri
değil, Kohn-Sham orbitalleridir. Orbitallerdeki bu iki setler birbirinden farklıdır.
Çünkü bunlar farklı denklemlerin çözümlerinden elde edilirler. Bu yüzden genellikle

3. MATERYAL VE METOT ERKAN TETİK
83
HFX
KSX EE ilkesi geçerlidir. 3.125 denklemindeki değişim fonksiyonelinin elektron
yoğunluğuna bağlılığı açık değildir. Bu nedenle yaklaşık değişim fonksiyonellerine
gereksinim vardır.
3.4.6. Değişim-Korelasyon Fonksiyonellerinin Genel Analitik Özellikleri
Herhangi bir elektron yoğunluğu için korelasyon enerjisi asla pozitif değilken,
değişim enerjisi daima negatiftir.
0,0 CX EE (3.127.)
Lieb ve Oxford (1981), Coulomb sistemlerinde elektronların değiş-tokuş ve
korelasyon enerjisinin
rdrCEE LOXCX)(,, 3/4 (3.128.)
şeklinde sınırlı bir aralıkta olması gerektiğini göstermişlerdir. Burada
68.144,1 LOC ’dir. Tek elektron yoğunlukları )(1 r için XE enerjisi,
0)(0, 11 UEX (3.129.)
şeklinde Coulomb öz-itme enerjisini yok etmelidir. Burada CE ,
00,1 CE (3.130.)
şeklinde tamamen yok olmalıdır. Düzenli elektron yoğunlukları için ,XCE ,
düzgün elektron gazının değiş-tokuş ve korelasyon formüllerine
,, LSDAXCXC EE , )(r =sabit ise (3.131.)

3. MATERYAL VE METOT ERKAN TETİK
84
şeklinde indirgenmelidir (Scuseria ve Staroverov, 2005). Yoğunluk
fonksiyonellerinin bilinen kesin özelliklerinin birçoğu, koordinat ölçekli
dönüşümlerini içerir. Bu ilişkilerin çoğu Levy ve çalışma arkadaşları (Levy ve
Perdew, 1985, 1993; Yang ve Levy, 1990a,b; Levy, 1989, 1991; Görling ve Levy,
1992) tarafından türetilmiştir. Yoğunluğun düzgün ölçeği
)()( 3 rr (3.132.)
şeklinde tanımlanır. Değiş-tokuş ve korelasyon fonksiyonelleri için koordinat ölçekli
sınırlamalar Levy (1995) tarafından yeniden incelenmiştir. Bu sınırlamaların en
önemlileri
C
XX
E
EE
lim
,
(3.133.)
şeklinde verilir. Spini dengelenmiş ve spini polarize sistemlerin değişim fonksiyonel
yaklaşımı spin ölçekli ilişkiyle birbirlerine
2221, XXX EEE (3.134.)
şeklinde bağlıdır (Oliver ve Perdew, 1979). Burada 2/,2/ XX EE ’dir.
3.5. Yerel Yoğunluk Yaklaşımı (The Local Density Approximation, LDA)
Yerel yoğunluk yaklaşımı (LDA) değiş-tokuş ve korelasyon fonksiyoneli
XCE sabit yoğunluklu (düzgün elektron gazı) elektron sistemindeki çok elektron
etkileşimlerine ait bilinen sonuçları kullanır. Yaklaşımın hem hesaplama kolaylığı
hem de şaşırtıcı derecede doğru sonuçlar verdiği görülmüştür. LDA yaklaşımında,

3. MATERYAL VE METOT ERKAN TETİK
85
bir molekül veya katıdaki her bir noktanın belirli bir elektron yoğunluğuna sahip
olduğu kabul edilir ve her noktadaki elektronun, çevresindeki aynı yoğunluklu öteki
elektronlarla aynı çok cisim etkileşmeye maruz kaldığı varsayılır. O zaman tüm
moleküllerin veya bir katı maddenin toplam değiş-tokuş ve korelasyon fonksiyoneli,
bütün hacim elemanları üzerinden alınacak katkıların integrali olarak verilir.
3.5.1. Yerel Yaklaşımlar
Yerel yaklaşımın, fonksiyoneli incelemede niçin en önemli aşama olduğunu
göstermek için 푟(휃) eğrisinin çevre uzunluğunu inceleyelim. Bu problem için
öncelikle eğrinin daha basit bir formunu dikkate alalım,
푟(휃) = 푟 (1 + 휀 cos(푛휃)) (3.135.)
burada 푛 periyodikliği korumak için 푛 = 1,2,3, … şeklinde tamsayı olarak seçilir.
Burada 휀 bir daireden meydana gelen büyük sapmaların nasıl olduğunu belirlerken, n
açı ile yarıçapın nasıl hızlı bir şekilde değiştiğinin bir ölçüsüdür. Şekil 3.5’de
푛 = 20 ve 휀 = 0,1 için bu durum gösterilmektedir.
Şekil 3.5. 푟 = 1 + 0,1 cos(20 휃) eğrisi (n=20, eps=0,1)
푟’nin yerel fonksiyoneli ile çevre uzunluğuna bir yaklaşım yapmak istersek,
푃 = ∫ 푑휃 푓(푟) (3.136.)

3. MATERYAL VE METOT ERKAN TETİK
86
olarak yazarız. Buradaki 푓 fonksiyonunu belirlemek için dairenin çevre uzunluğunu
2휋푟 olarak kabul ediyoruz. Yaklaşımın 푟 değeri 휃 açısından bağımsız olduğu için,
yaklaşımımız tam olabilir. Bu durumda fonksiyonelin her zaman doğru olmasını
istersek, 푓 = 푟 olarak seçmemiz gerekir. Böylece,
푃 = ∫ 푑휃 푟(휃) (3.137.)
denklemini elde ederiz ve bu denklem çevre uzunluğu için yerel yaklaşım olur.
3.5.2. Yerel Yoğunluk Yaklaşımı
Yerel yoğunluk yaklaşımı kurmak için ilk olarak aynı-spin elektronlarının
etkileşmeyen kinetik enerjisi için,
푇 [푛] = ∫ 푑푥 푡 푛(푥) (3.138.)
yazabiliriz. Boyut analizine göre 푡 kinetik enerji yoğunluğu 푛 ile orantılı olmalıdır.
Buna göre,
푡 (푛) = 푎 푛 (3.139.)
olur. Burada 푎 = 휋 /6 olarak dikkate alacağız. Bu noktada Fermi dalga vektörü
olarak isimlendirilen bir sistemi inceleyelim. Bu dalga vektörü sistemdeki en yüksek
işgal edilen orbitalin dalga vektörüdür. N elektronlu bir sistemi temel aldığımız için,
푘 = = 푛휋 (1 boyutlu polarize) (3.140.)
ve
푇 [푛] = ∫ 푑푥 푛(푥)휏 푛(푥) , 휏 (푛) = (3.141.)

3. MATERYAL VE METOT ERKAN TETİK
87
olarak yazarız. Böylece, 3 boyutlu Coulomb- etkileşim problemi için,
퐸 [푛] = ∫ 푑 푟 푓 푛(r) (3.142.)
denklemini yazabiliriz. 푓(푛), 푛’nin bir fonksiyonudur. Bu fonksiyonu belirlemek
için, düzgün elektron gazını inceleyeceğiz.
3.5.3. Düzgün Elektron Gazı
Yerel yaklaşım, uzayın sınırsız bir bölgesine yerleşen elektronlar gibi uniform
elektronik bir sistemin özel durumları için tamdır. Böyle bir sistemin kinetik ve
değiş-tokuş enerjileri, Kohn-Sham dalga fonksiyonları düzlem dalgaların basit Slater
Determinantları olduğu için kolayca değerlendirilebilir. Korelasyon enerjisi ise
Monte Carlo hesaplamalarından çıkartılabilir. Periyodik sınır koşullarını dikkate
alırsak, durumlar momentumun katları olur ve en düşük işgal edilen seviyeler
momentum uzayında bir küre olarak şekillenir. Bu kürenin yarıçapı ise Fermi dalga
vektörüdür ve bu vektör,
푘 = (3휋 푛) / (3.143.)
olarak verilir. Burada farklı kuvvet ve katsayılar farklı boyutlardan gelir. Boyut
analizi 1 boyutta kinetik enerji için aynı formlar üretir. Fakat uniform gaz için farklı
bir sabit elde etmek için,
푇 [푛] = ∫ 푑 푟 푘 (r)푛(r) = 퐴 ∫ 푑 푟 푛 / (r) (3.144.)
denklemini yazarız. Burada 퐴 = (3휋 ) / = 2,871 olarak elde edilir. Düzgün
gazın değiş-tokuş enerjisi için, Coulomb etkileşiminin ters uzunluğunun boyutlarına
sahip olduğunu dikkate alalım. Böylece onun enerji yoğunluğunun 푘 ile orantılı
olması gerektiği sonucuna ulaşırız. Bu durumda,

3. MATERYAL VE METOT ERKAN TETİK
88
퐸 [푛] = 퐴 ∫ 푑 푟 푛 / (r) (3.145.)
şeklinde olur. Düzlem-dalga orbitallerinin Slater determinantı için Fock integralinin
değeri uniform gazın elektronu vasıtasıyla değiş-dokuş enerjisini,
휀 (푛) = (3.146.)
olarak elde ederiz. Denklem 3.145’deki 퐴 değeri, 퐴 = − = −0,738
olarak elde edilir. Korelasyon uniform gazın fiziksel olarak temel-durum dalga
fonksiyonuna bağlı olduğu için daha karmaşıktır. Yoğunluğun bir diğer faydalı
ölçümü Wigner-Seitz yarıçapıdır yani,
푟 = ( ) / = , (3.147.)
olarak yazarız. Bu elektronların toplam yoğunluğunu tüm kürelerin hacmine
eşitlemek için elektron etrafındaki bir kürenin yarıçapıdır. Böylece, 푟 → 0 yüksek
yoğunluk limiti, 푟 → ∞ ise düşük yoğunluk limiti olur. Buna göre,
퐸 [푛] = ∫ 푑 푟 푛(r) 휀 푟 (r) (3.148.)
olarak yazarız. Burada 휀 uniform gazın korelasyon enerjisidir. Bu korelasyon
değiş-tokuş enerjisi üzerinden bir artış olarak,
휀 (푟 ) = 퐹 (푟 ) 휀 (푟 ) (3.149.)
şeklinde ifade edilir ve bu artış faktörü şekil 3.6’da görülmektedir. Şekil 3.6’daki
eğride pek çok önemli özellik vardır:

3. MATERYAL VE METOT ERKAN TETİK
89
1) 푟 = 0 iken (sonsuz yoğunluk), exchange korelesyonun üzerindedir ve 퐹 = 1’e
eşittir.
2) 푟 → 0’a giderken (yüksek yoğunluk), 1’e doğru keskin bir pik vardır. Bu sonsuz
bir sistemdeki Coulomb etkileşiminin uzun-aralıklı olmasından dolayıdır. Bu
durumda
휀 (푟 ) → 0,0311 ln푟 − 0,047 − 0,009푟 ln푟 − 0,017푟 (푟 → 0) (3.150.)
şeklinde olur.
3) 푟 limiti azaldığında (düşük yoğunluk),
휀 (푟 ) → − + − ⋯ (푟 → ∞) (3.151.)
şeklinde olur. Buradaki sabitler 푑 = 0,896 ve 푑 = 1,325 şeklindedir. 푑 sabiti ilk
olarak bu sistem için Wigner kristalinden Wigner tarafından oluşturulmuştur. Bu
korelasyonun değiş-tokuş enerjisi kadar büyük olduğu anlamına gelir ve buna göre
de 퐹 (푟 → ∞) = 1,896 olur.
Şekil 3.6. Düzgün elektron gazı için değiş-tokuş ve korelasyon enerjileri
r
0,25
-0,3 1 2 3 4 5 6 0
Homojen Gaz 0,20
0,15
0,10
0,05
0
C X

3. MATERYAL VE METOT ERKAN TETİK
90
3.6. Genelleştirilmiş Gradyent Yaklaşımları
Yerel yoğunluk yaklaşımındam bir adım daha ileri gidilerek genelleştirilmiş
gradyan yaklaşımının (GGA) oluşturulmuştur. Bu yaklaşım yerel yoğunluk
yaklaşımına ek olarak, her noktada elektronik yük yoğunluğunun (휌) yanı sıra bu
yoğunluğun |∇푛(r)| olarak ifade edilen gradyanının da hesaplanması gerektiği
fikrine dayanır. Bu yaklaşımın genel şekli,
퐸 [푛] = ∫ 퐹 [푛(r), |∇푛(r)|]푑푟 (3.152.)
şeklindedir. Bu denklemdeki 퐹 fonksiyonelinin yapısını pek çok bilim adamı
tarafından çalışılmıştır. Bu çalışmalara örnek olarak Becke (1988), Perdew ve ark.
(1996), Perdew ve Wang (1992), Perdew ve ark. (1992), Perdew-Burke ve Ernzerhof
(1996) gösterilebilir. GGA’da değiş-tokuş korelasyon enerjisi bir 퐹 fonksiyoneli ile
yerel yoğunluk yaklaşımı üzerine eklenerek,
퐸 [푛] = ∫ 푛(r) 휀 퐹 (푟 , 푠) 푑푟 (3.153.)
olarak genişletildi. Denklemdeki 푠(푟) = |∇ | boyutsuz yoğunluk gradyentidir.
Ayrıca 푘 = 4푘 /휋 ve 푘 = (3휋 푛) / şeklindedir. PBE’de 퐹 (푠) değiş-tokuş
terimi,
퐹 (푠) = 1 + 푘 − , 휇 = 훽 ( ) (3.154.)
şeklindedir. Bu denklemlerdeki 푘 = 0,804 ve β≅0,066725 olur. Düzeltilen kısım
olan 퐹 ise,
퐹 [푛(r), |∇푛(r)|] = ∫ 푛(r) 휀 푛(r) + 퐻 (푟 , 푡) 푑푟 (3.155.)

3. MATERYAL VE METOT ERKAN TETİK
91
denklemi ile ifade edilir. Bu denklemdeki 퐻 (푟 , 푡) ise,
퐻 (푟 , 푡) = 훾 ln 1 + 푡 (3.156.)
퐴 =/
, 훾 = (3.157.)
olarak tanımlanır ve ayrıca buradaki 푡 = ∇ ise diğer bir boyutsuz yoğunluk
gradyentidir.
3.7. Pseudo-Potansiyel Yaklaşımı
Pseudo-potansiyel, verilen bir yarıçap, kor yarıçap )( cr olarak alınarak
gerçek potansiyel gibi üretilir. Benzer şekilde her bir pseudo-dalga fonksiyonu
şekil.3.7’de gösterildiği gibi cr kesme yarıçapının ötesinde uygun gerçek dalga
fonksiyonuna uymalıdır. Ayrıca kor bölgesinin dışında elde edilen yük yoğunlukları
gerçek yük yoğunluğuna özdeş olmalıdır. Bu yüzden kor bölgesi üzerinde gerçek ve
pseudo-dalga fonksiyonlarının genliklerinin karesinin integrali özdeş olmalıdır. Bu
şart norm-koruma olarak bilinir. Bu tür yerel ve yerel olmayan pseudo-
potansiyellerin çeşitli atomik ortamlardaki iyon korlarından dolayı saçılmayı
tanımlayabildiği bilinir.
Şekil 3.7. Pseudo potansiyel, Pseudo ve gerçek dalga fonksiyonları
r
-Z/r
c r V pseudo
Ψ gerçek
Ψ pseudo

3. MATERYAL VE METOT ERKAN TETİK
92
Pseudo-potansiyeller, bir ab-initio yöntem kullanarak üretilirler. Pseudo-
dalga fonksiyonları, yoğunluk fonksiyonel teorisini kullanarak izole edilmiş bir atom
için hesaplanır. Sonra valans dalga fonksiyonları, norm-koruma sınırlamasına
uyarken titreşimleri kaldırmak için kor bölgesinde değiştirilir. Ondan sonra
Scrödinger denklemi, pseudo-fonksiyonları üretecek olan pseudo-potansiyelleri
bulmak için tersine çevrilir. Bu yöntem, geniş çapta değişken sistemler arasında
transfer edilebilen bir pseudo-potansiyel üretir. Bu, belirli bir atomik ortamı
tanımlamak için üretilen yarı-ampirik potansiyeller ile çelişir ve farklı ortamlara
kolay bir şekilde transfer edilemezler.
3.8. Hesaplama Metotları
Kuantum Fiziğinin oluşmaya başlaması günümüzden yaklaşık yüz yıl önceye
rastlar ve bu güne kadarki evrimi sonucu birçok alt dalı oluşmuştur. Oluşumunun ilk
yıllarında, kuramı oluşturan fizikçiler birçok zorluğa da işaret etmişlerdir. Bu
zorlukların temelinde fiziğin de en temel hatta tek problemi olan çok parçacıklı
sistemler için Schrodinger denkleminin çözümü yatmaktadır. Denklemin çözümü
olan ve sistemi tanımlayan dalga fonksiyonu, sistemin tüm serbestlik derecelerinin
fonksiyonudur ve Hamiltoniyen de sistemin serbestlik derecesi arttıkça oldukça
karmaşık hale gelir. Tüm bu koşullar altında temel denklem olan Schrodinger
denkleminin çok parçacıklı bir sistem için çözümü imkânsızlaşır. Bu nedenle bazı
yaklaşımlar yaparak denklemi çözülebilir hale getirmek kaçınılmazdır.
Yaklaşık yöntemlerle denklemi çözülebilir hale getirdikten sonra,
uygulanacak yapının durumu da çözüme ulaşmayı etkilemektedir. Karmaşık
yapılarda sonuca ulaşmayı kolaylaştırmak için çeşitli programlar geliştirilmektedir.
DFT temelli bu programlar temel olarak sistemi optimize edip toplam enerjiyi
hesaplamaktadırlar. Bunların başında Siesta, Abinit, Quantum Espresso, Wien2k ve
Gaussian gibi programlar gelmektedir. Derinleşen araştırmalar neticesinde artan
atom sayısı nedeniyle bilgisayar programları oldukça hızlı bir gelişim grafiği
izlemiştir. 90’ların başında Hamiltonians doğrusal ölçekli hesaplamalarının
algoritmalarla çözülmesinde önemli bir artış gözlemlenmiştir. Farklı yöntemlerin bu

3. MATERYAL VE METOT ERKAN TETİK
93
hesaplamalar için denenmesinin ardından yeni bir problem ortaya çıkmıştır. DFT
hesaplamalarının tamamı için yapılacak ölçeklendirme çalışmaları nasıl mümkün
kılınabilir? Zira o sıralar Kohn-Stam Hamiltonian hesaplamaları için bile henüz
ölçeklendirme çalışmaları istenen sonuçları vermemiştir. SIESTA yöntemi,
Sankey’in (1995) atomik orbitalleri 3B gridleme yöntemiyle gözlemlemesi ve
hesaplamalarını yoğunluk ve temel hal fonksiyonlarına dayandırmasıyla
kullanılmaya başlanmıştır. Gridleme sayesinde geniş aralıklar için geçerli olan
elektrostatik kuvvetlerle doğal yoldan başa çıkılmıştır. Böylelikle lokalizasyon
iyileştirilmiştir (lokalizasyon, doğrusal ölçeklendirmenin temel gereksinimidir). Bu
yöntem pek çok doğrusal ölçeklendirme çözümlemesi için geçerli olup, 1996 yılında
yazılan SIESTA (internet, 2011; Ordejon ve ark, 1996; Soler ve ark, 2002) programı
sayesinde DFT lineer ölçeklendirmesi mümkün kılınmıştır.
SIESTA yöntemi gibi periyodik bağlanma koşullarını üç boyutlu uzayda
içermektedir. 0 B ‘de moleküller, noktasal bozulmalar; 1 B’de zincirler, tüpler ve
doğrusal bozulmalar, 2B’de yüzeyler, ara yüzler ve düzlemsel bozulmalar ve 3 B’de
süper hücreler bulunmaktadır. Bu çözümlemeyi yapan tek algoritma bağlanma
koşullarını içeren Hartree takımı tarafından kullanılan Fourier dönüşümüdür ve
ölçeklendirme N’e karşılık logN şeklindedir. Böylelikle SIESTA yöntemi daha kolay
lineer ölçeklendirilebilir ve bağlılık koşulları 0D, 1D, 2D ve 3D hesaplamaları için
daha kolay bir hal almaktadır. Dirichlet bağlanma koşulları tekli gruplar ya da
moleküller için kullanılmaktadır. Burada gruplar tekrarlanmamıştır. Ancak
elektrostatik potansiyel kutu bağlantılarının yük yoğunluklarının düşük momentlerine
göre hesaplanır. Çoklu gridleme yöntemi sayesinde 3D PBC ve hibrit BC beraber
kullanılabilir. Böylelikle tekrarlamayan tüp ve tabakalar elde edilir.
3.9. Paralel Programlama
MPI (Mesaj Geçirme Arayüzü) başta dağıtık bellekli sistemler olmak üzere
paralel algoritma yazmaya yarayan bir paralel programlama kütüphanesidir. Bir
programlama dili değildir, paralel programlama için kullanılır. Paralel programlama
bir işin tek bir bilgisayarda değil de birçok bilgisayara dağıtılıp sonuç olarak geriye

3. MATERYAL VE METOT ERKAN TETİK
94
dönen değerlerin ana bilgisayarda toparlanıp işlemlerin daha hızlı
gerçekleştirilmesini sağlar. MPI kütüphanelerinin C, C++ ve Fortran programlama
dilleri için sürümleri bulunmaktadır. MPI, “mesaj geçme" mantığına dayalıdır yani
farklı işlemciler, birbirleriyle iletiler sayesinde haberleşirler. MPI ile oluşturulan
küme bilgisayarların bellek ve işlemci sayısı oldukça yüksektir. Böylelikle atomik
boyutta karmaşık yapıların hesaplanması mümkün olmaktadır.
C ve Fortran gibi programlama dillerinde yazılmış olan Siesta, Abinit,
Kuantum Espresso, Wien2k ve Gaussian gibi paket programlar MPI kütüphanesini
desteklemektedir. Bu paket programlar atomik boyutta hesaplamalar yaptığı için,
atom sayısı arttıkça hesaplama zamanı artmaktadır. Hatta bazı durumlarda karmaşık
bir yapının hesaplanması tek bir bilgisayarda yapılamamaktadır. Bu hesaplamalar
için yüksek bellek ve işlemci gerekmektedir. Dolayısıyla karmaşık sistemlerin
hesaplanabilmesi için bu paket programların MPI kütüphanelerine uygun olarak
yazılması standart haline gelmiştir.
3.9.1. Siesta İçin Paralel Programlama
SIESTA kodlamaları yardımıyla lineer ölçeklendirme çözümleri, 3B grid
uygulamaları ve köşegenleştirme uygulanmaları yapılabilmektedir. Lineer
ölçeklendirme çözümlemeleri Si kristalleri için 2001 yılında 288’den 524 atoma
kadar ve 256 düğüm noktasının bulunduğu bir yapı baz alınarak paralel bir sistemde
hesaplama yapılmıştır. Veriler 2001 yılında SGI Altix içerisine kaydedilmiştir ve
kullanılmaktadır.
Köşegenleştirmedeki ölçeklendirme aynı ölçüde başarılı olmamıştır.
ScalPACK kullanılarak blok döngü dağılımının yörünge başına düşen düğüm
sayısıyla çakışması hedeflenmiştir. Bu yöntemin etkili kullanımındaki en önemli
adım İngiliz HPCx şirketinde Hein tarafından elde edilen başarıdır. Burada 1B
dağılım yerine 2B dağılım yapılmıştır. 2B uygulamada ScalPACK uygulaması
normalde gerekenden daha az yönlendirmeye ihtiyaç duyar. Pilin proteini için
yapılan gerçekçi bir hesaplamaya bakacak olursak 944 atom, iki taraflı polar dağılım,
2’ye kadar ulaşan hız faktörü, devam edildikçe 1,82, 1,67 ve 1,53 gibi hız faktörü

3. MATERYAL VE METOT ERKAN TETİK
95
değerlerine, 8 den 16’ya, 16’dan 32’ye, 32’den 64’e ve 64’ten 128’e ulaşan işlemci
sayısına ulaşıldığı görülmüştür. Cambridge HPCF süper bilgisayarlarında averaj
zaman aralığında (10 SCF adımında) 25 dakika süre harcandığı ve 32 işlemci
kullanıldığı gözlemlenmiştir. Yüksek kalitede irtibat sağlanamadığı durumlarda
işlemci sayısı 8-16 arasında kısıtlanacaktır. Paralel hale getirme stratejilerinden biri
de etki alanı dahilindeki çözümlemelerin optimizasyonuna ek olarak, evrensel
irtibatın en aza indirilmesidir. Bu durum grafik-teorisi analizinden de görülmektedir.

4. BULGULAR VE TARTIŞMA ERKAN TETİK
96
4. BULGULAR VE TARTIŞMA
4.1. Hesaplama Adımları
Nanotüplerin geometrik yapıları üzerinde çalışarak elde edilen verilerle,
nanotüplerin elektronik bant yapıları, durum yoğunlukları yoğunluk fonksiyoneli
teorisi baz alınarak yazılmış paket programlar kullanarak hesaplanmaktadır.
Hesaplamalarda ab-initio kod olarak SIESTA paket programı, değişim ve korelasyon
terimleri için yaklaşım olarak yerel yoğunluk yaklaşımı (LDA) kullanıldı. Elektronik
bant yapıları ve toplam durum yoğunlukları Troullier-Martins norm-koruyucu
pseudo-potansiyeli kullanılarak hesaplanmıştır. Karbon atomlarının elektronik
dağılımında 1s çekirdek elektronlarını, 2s ve 2p ise valans elektronlarını temsil
etmektedir. Valans elektronları elektronik dalga fonksiyonları için düzlem dalga baz
setleri (DZP) kullanılmıştır. Kohn-Sham denklemlerinin çözümleri ise “conjugate
gradient minimization method” (Payne ve ark., 1992) kullanılarak yapılmıştır. Hem
pseudo-potansiyellerin üretiminde hem de bant yapısı hesaplarında değiş-tokuş ve
korelasyon etkileri, yerel yoğunluk yaklaşımı altında Ceperley-Alder (Ceperley ve
ark., 1980) fonksiyonelleri kullanılarak hesaba katılmıştır. Bu parametrelere
nanotüpün örgü vektörü ve atomik konumları eklenerek öncelikle optimizasyon
işlemi yapılmıştır. Optimizasyon işlemi stres tensörleri 0,04 eV/A°3 değerinin altına
düşene kadar yapılmıştır. Optimizasyon işlemiyle minimize teorik örgü sabiti ve
atomik konumları elde edilir. Bu programdan elde edilen parametreler ve atomik
konumlar kullanılarak nanotüplerin elektronik bant yapısı ve durum yoğunlukları
hesaplandı.
Çalışmanın ikinci aşamasında seçilen bir nanotüpe, karbon atomuna
geometrik özellikler bakımından yakın olan A grubu atomlarından bazıları seçilerek
katkılama işlemi yapılmıştır. Katkılama işlemi seçilen her atom için ayrı ayrı
%3,125, %6,25, %12,5 ve %25 oranlarında yapıldı. Katkılama ile elde edilen yeni
yapıların elektronik bant yapısı ve durum yoğunlukları yeniden hesaplandı.
Hesaplanan yeni sonuçlar birbirleri ile karşılaştırılarak katkılamanın etkileri
incelendi.

4. BULGULAR VE TARTIŞMA ERKAN TETİK
97
4.2. Karbon Nanotüplerin Geometrik Yapısının Elde Edilmesi
Karbon nanotüplerin geometrik yapıları m ve n indislerinin aldığı değerlere
göre farklılıklar göstermektedir. Bu yapı değişikliği nanotüplerin elektronik
özelliklerini de değiştirmektedir. Nanotüplerle ilgili çalışmalarda öncelikle
çalışılacak nanotüpün örgü sabitleri ve atomik koordinatları belirlenmelidir. Bunun
için öncelikle n ve m indisleri belirlenir (Örneğin (7, 4) karbon nanotüp seçilebilir).
Bu indisler kullanılarak indislerin en büyük ortak böleni hesaplanır. Bu hesaplanan
en büyük ortak böleni 푛 olarak kabul edelirse,
푛 = 푛 − 푚 3푛 nin katı değilse 푛푛 − 푚 3푛 nin katı ise 3푛 (4.1.)
formülünü kullanarak 푛 değerini hesaplanır. 푛 değeri, 2푚 + 푛 ve 2푛 + 푚
değerlerinin en büyük ortak bölenleridir. İki karbon atomu arasındaki uzaklığı
푎 = 1,42 A olarak kabul edilir. n ve m indisleri ve a değerleri kullanılarak,
퐿 = 푎√푛 + 푚 + 푚푛 (4.2.)
formülünden nanotüpün çevre uzunluğu olan 퐿 hesaplanır. Nanotüpün çapı 푑 = 퐿/휋
ve yarıçapı 푟 = 푑 /2 formülleri ile hesaplanır. Öteleme vektörü 푇 = (푛 , 푛 ),
푛 = (2푚 + 푛)/푛 ve 푛 = −(2푛 + 푚)/푛 formülleri ile elde edilir. 푇 öteleme
vektörünün uzunluğu,
푇 = √3 (4.3.)
formülü hesaplanır. Nanotüpün birim hücresindeki hegzagon sayısı,
푁 = ( ) (4.4.)

4. BULGULAR VE TARTIŞMA ERKAN TETİK
98
formülü kullanılarak hesaplanır. Simetri vektörü 푅 = (푛 , 푛 ) hesaplamak için sınır
koşulları kullanılır. Buradaki 푛 , 푛 tamsayıdır. Öteleme vektörü ve n ile m ilgili
aşağıdaki,
푛 푛 − 푛 푛 = 1, (0 < 푚푛 − 푛푛 ≤ 푁) (4.5.)
denklemleri kullanılarak 푛 , 푛 değerleri belirlenir. 푅 vektörü, 푇 doğrultusunda 휏
ötelemesiyle birleşmiş bir 휓 açısının nanotüp ekseni etrafındaki dönmesiyle
ilişkilidir ve chiral nanotüpün temel uzay grubu simetri operatörünü ifade eder.
Nanotüplerle ilgili son parametre olan M değerini elde etmek için,
푀 = 푚푛 − 푛푛 (4.6.)
formülünü kullanırız. M, nanotüpün alt noktasından üst noktasına ulaşmak için
uygulanması gereken 푇 vektörü sayısıdır. Temel parametreler elde edildikten sonra
seçilen nanotüpün atomlarının koordinatları hesaplanır. Atomik koordinatları
hesaplamak için gerekli olan formüller,
푟 = 푎 푛 + 푛 + 푛 푛 (4.7.)
푐 = 푎√푛 + 푚 + 푛푚 (4.8.)
푡 = √3푐/푛 (4.9.)
푞 = 푎 tan (√3푚/(2푛 + 푚)) (4.10.)
푞 = 푎 tan (√3푛 /(2푛 + 푛 )) (4.11.)
푞 = 푞 − 푞 (4.12.)

4. BULGULAR VE TARTIŞMA ERKAN TETİK
99
푞 = 2 (3,14)/푁 (4.13.)
푞 = 푎 푠푖푛(3,14/6 − 푞 )/2푐 (3,14) (4.14.)
ℎ = |푡| /|푠푖푛(푞 )| (4.15.)
ℎ = 푎 sin ((3,14/6) − 푞 ) (4.16.)
formülleri kullanılarak nanotüpteki her bir atomun konumu belirlenir. Bu
formüllerdeki 푟 simetri vektörünün uzunluğu, 푐 chiral vektörünün uzunluğu, 푡
öteleme vektörünün uzunluğu, 푞 chiral vektörü için chiral açısı, 푞 simetri vektörü
için chiral açısı, 푞 simetri ve chiral vektörü arasındaki açıdır. Atomik konum
hesaplanırken ilk atomu ‘A atomu’ ve ikinci atomu ‘B atomu’ olarak kabul edersek,
diğer atomlar bu atomlardan türetilebilir. Bu durumda 푞 ilk atom olan A atomu için
nanotüp döndürülürken oluşan ilk açının periyodudur. 푞 ise A ve B atomları
arasındaki açısal farktır. Son olarak ℎ ve ℎ parametreleri A ve B atomları
arasındaki delta z değerini temsil eder. Bu aşamadan sonra referans atomu olan A
atomunun koordinatları,
푥 = 푟 푐표푠 (|푖| 푞 ) (4.17.)
푦 = 푟 푠푖푛 (|푖| 푞 ) (4.18.)
푧 = ( |푖||푟| − |푘| ℎ )푠푖푛 (푞 ) (4.19.)
formülleri ile türetilir. İkinci referans atomu olan B atomunun koordinatları ise,
푥 = 푟 푐표푠 (|푖| 푞 + 푞 ) (4.18.)
푦 = 푟 푠푖푛 (|푖| 푞 + 푞 ) (4.19.)

4. BULGULAR VE TARTIŞMA ERKAN TETİK
100
푧 = ( |푖||푟| − |푘| ℎ )푠푖 푛(푞 ) − ℎ (4.20.)
formülleri ile türetilir. A ve B atomu hegzagon oluşturmaktadır. A ve B atomunun
oluşturduğu hegzagonlardan diğer karbon atomlarının koordinatları türetilir.
Denklem (4.1)’den (4.20)’ya kadar oluşan denklem dizisi kullanılarak
herhangi bir nanotüpün temel parametreleri hesaplanabilir. Üzerinde çalışılacak
nanotüpün elde edilen temel parametreleri de diğer özelliklerin hesaplanması için
kullanılır. Nanotüplerin geometrik açıdan pek çok farklı türü olduğunu düşünürsek,
herhangi bir nanotüpl için bu denklemleri çözmek uzun süreceğinden bu denklemleri
bir programlama dili kullanarak yazmak daha kolay, doğru ve hızlı bir süreç
sağlayacaktır. Ek 1’de bu denklemlerin Fortran programlama dili kullanılarak
yazılmış bir programı bulunmaktadır. Ek 1’deki kodlar herhangi bir fortran
derleyicisi kullanılarak derlenebilir. Derlendikten sonra çalıştırılan program
kullanıcıdan n ve m indis değerlerini isteyecektir. Kullanıcı tarafından bu değerler
girildikten sonra program nanotüpün temel parametreleri ve atomik konumları
hesaplar.
Ek 1’deki nanotüp programı kullanılarak elde edilen atomik konumlar ‘.xyz’
formatındadır. Elde edilen bu çıktı dosyası herhangi bir atomik yapı görüntüleme
programı tarafından görüntülenebilir. Burada elde edilen ‘.xyz’ dosyası siesta
programının atomik konum yapısına göre düzenlenerek, siesta input dosyasına
eklenmiştir. Hazırlanan siesta input dosyası siesta programı kullanılarak
hesaplanmıştır.
4.2.1. Hesaplaması Yapılan Nanotüplerin Geometrik Yapıları
Hesaplanacak nanotüpler, karbon nanotüplerin geometrik ve elektronik
özellikleri dikkate alınarak seçilmiştir. Bu seçimde Zigzag (Metalik), zigzag
(Yarıiletken), chiral (Metalik), chiral (Yarıiletken) ve armchair (Metalik) olmak
üzere 5 grup oluşturulmuştur. Bu gruplarda için pek çok nanotüp seçilebilir.
Çalışmamızda nanotüpler zigzag (6, 0), zigzag (7, 0) chiral (6, 2), chiral (6, 3) ve
armchair (7, 7) olarak seçilmiştir. Seçilen nanotüplerin Ek 1’deki program

4. BULGULAR VE TARTIŞMA ERKAN TETİK
101
kullanılarak temel parametreleri ve atomik konumları hesaplanmıştır. Elde edilen
atomik konumlar xcrysden (internet, 2011) atomik konum görüntüleme programı
yardımıyla görüntüleri elde edilmiştir. Elde edilen görüntüler ise şekil 4.1’de
gösterilmektedir.
Şekil 4.1. Hesaplaması yapılan karbon nanotüpler. (a) zigzag (6, 0) nanotüp, (b)
chiral (6, 2) nanotüp, (c) armchair (7, 7) nanotüp, (d) chiral (6, 3) nanotüp ve (e) zigzag (7, 0) nanotüp
Hesaplaması yapılan nanotüplerde sırasıyla 24, 28, 104, 84 ve 28 atom
bulunmaktadır. Ayrıca bu nanotüplerle ilgili ek 1’deki program kullanılarak elde
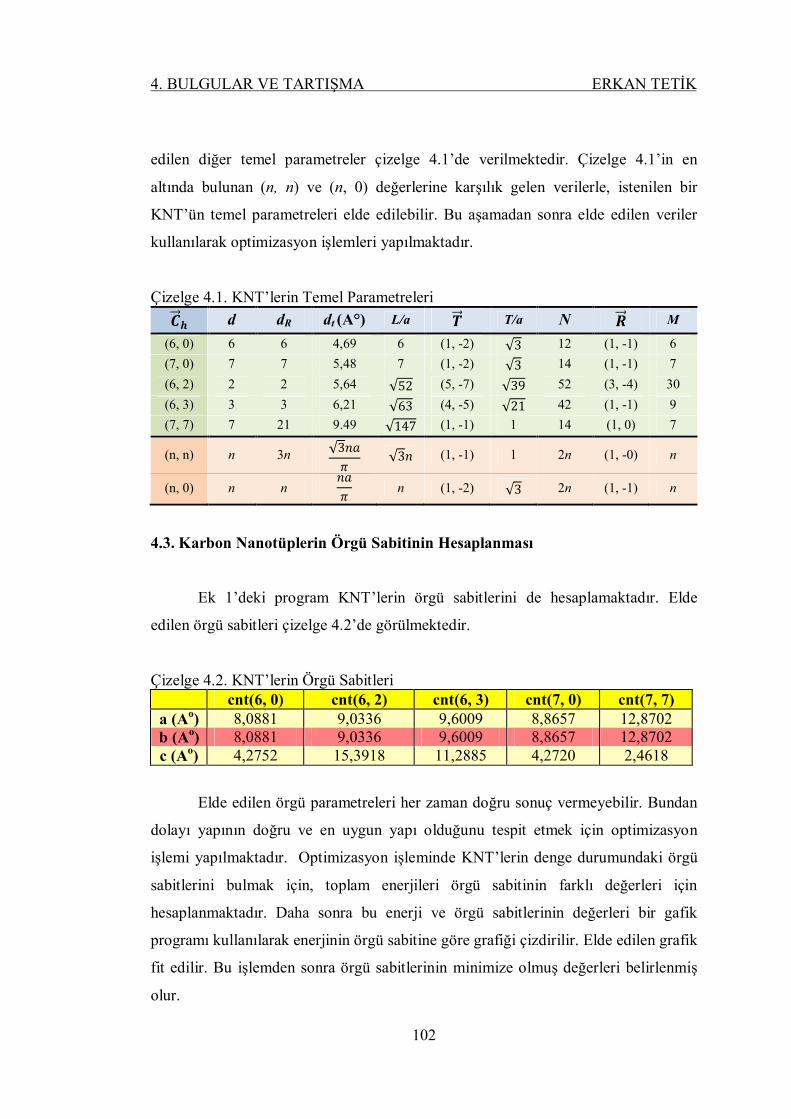
4. BULGULAR VE TARTIŞMA ERKAN TETİK
102
edilen diğer temel parametreler çizelge 4.1’de verilmektedir. Çizelge 4.1’in en
altında bulunan (n, n) ve (n, 0) değerlerine karşılık gelen verilerle, istenilen bir
KNT’ün temel parametreleri elde edilebilir. Bu aşamadan sonra elde edilen veriler
kullanılarak optimizasyon işlemleri yapılmaktadır.
Çizelge 4.1. KNT’lerin Temel Parametreleri 푪풉 d dR dt (A°) L/a 푻 T/a N 푹 M
(6, 0) 6 6 4,69 6 (1, -2) √3 12 (1, -1) 6 (7, 0) 7 7 5,48 7 (1, -2) √3 14 (1, -1) 7 (6, 2) 2 2 5,64 √52 (5, -7) √39 52 (3, -4) 30 (6, 3) 3 3 6,21 √63 (4, -5) √21 42 (1, -1) 9 (7, 7) 7 21 9.49 √147 (1, -1) 1 14 (1, 0) 7
(n, n) n 3n √3푛푎휋 √3푛 (1, -1) 1 2n (1, -0) n
(n, 0) n n 푛푎휋 n (1, -2) √3 2n (1, -1) n
4.3. Karbon Nanotüplerin Örgü Sabitinin Hesaplanması
Ek 1’deki program KNT’lerin örgü sabitlerini de hesaplamaktadır. Elde
edilen örgü sabitleri çizelge 4.2’de görülmektedir.
Çizelge 4.2. KNT’lerin Örgü Sabitleri cnt(6, 0) cnt(6, 2) cnt(6, 3) cnt(7, 0) cnt(7, 7)
a (Ao) 8,0881 9,0336 9,6009 8,8657 12,8702 b (Ao) 8,0881 9,0336 9,6009 8,8657 12,8702 c (Ao) 4,2752 15,3918 11,2885 4,2720 2,4618
Elde edilen örgü parametreleri her zaman doğru sonuç vermeyebilir. Bundan
dolayı yapının doğru ve en uygun yapı olduğunu tespit etmek için optimizasyon
işlemi yapılmaktadır. Optimizasyon işleminde KNT’lerin denge durumundaki örgü
sabitlerini bulmak için, toplam enerjileri örgü sabitinin farklı değerleri için
hesaplanmaktadır. Daha sonra bu enerji ve örgü sabitlerinin değerleri bir gafik
programı kullanılarak enerjinin örgü sabitine göre grafiği çizdirilir. Elde edilen grafik
fit edilir. Bu işlemden sonra örgü sabitlerinin minimize olmuş değerleri belirlenmiş
olur.

4. BULGULAR VE TARTIŞMA ERKAN TETİK
103
Özdeş küreleri, aralarında boşluklar en aza indirilecek şekilde ve düzenli bir
tarzda yığmanın iki yolu vardır. Bunlardan biri yüzey merkezli kübik (fcc) yapı öteki
ise hegzagonal sıkı (hcp) yapıdır. Bu yapıların her ikisi de sıkı paket yapılardır.
Düzlemde, her küre altı küreye değecek şekilde bir sıkı paket tabakası oluşturur.
Böyle bir tabaka, hcp yapının taban düzlemi veya fcc yapının (111) düzlemi olur. Bu
tabakanın üstüne ikinci bir tabaka, her küre tabandaki üç küreye değecek şekilde
yerleşir. Bunun üstüne de üçüncü tabaka iki değişik şekilde yerleşebilir. Bunlar;
1) Üçüncü tabakadaki küreler; ikinci tabaka kurulurken işgal edilmemiş olan
birinci tabakadaki boşlukların üstüne gelecek şekilde yerleşir. Bunun sonunda
yüzey merkezli kübik yapı oluşur.
2) Üçüncü tabakadaki atomlar, birinci tabakadaki atomların üzerine gelecek
şekilde yerleşir. bu durumda ise, hegzagonal sıkı paket yapı oluşur.
Hegzagonal sıkı paket yapı, hegzagonal ilkel hücreye sahiptir ve bazında iki
atom bulunur (Dikici, 1993). Hegzagon yapıların optimizasyon işlemi c/a ve a
değerlerine göre yapılır. KNT’ler hegzagonal yapıda oldukları için hem c/a hem de a
parametresi için ayrı ayrı otimizasyon işlemi yapıldı. Bu işlem yapılırken önce c/a
parametresinin farklı değerleri için siesta paket programı kullanılarak hesaplama
yapıldı. Hesaplamalarda diğer tüm parametreler sabit tutularak, sadece c/a oranı sabit
aralıklarla artırıldı. Daha sonra bu c/a değerlerin karşılık geldiği toplam enerji
değerlerinin grafiği çizildi. Çizilen grafik fit edilerek c/a parametresinin minimum
enerji değerindeki değeri elde edildi. Daha sonra elde edilen bu c/a değeri
kullanılarak a parametresi farklı değerler için optimize edildi. Yine c/a’ya benzer
şekilde a’nın enerjiye karşı grafiği çizildi. Grafik fit edildikten sonra a değerinin
minimum enerji değeri elde edilmiş oldu. Optimizasyon sonuçlarının grafikleri şekil
4.2, 4.3, 4.4, 4.5 ve 4.6’da görülmektedir. Grafiklerde a (A)’ın değerine karşılık
gelen toplam enerji (eV) değerleri, daha doğru sonuç için iki aşamalı olarak
çizilmiştir. İkinci grafikte yıldız işareti ile gösterilen nokta minimum enerji değerine
karşılık gelen a (eV) örgü parametresinin değeridir. Böylelikle hesaplamaların ilk
basamağı olan optimizasyon ve optimizasyon işleminin sonuçlarının analiz edilmesi
tamanlanmış oldu.
4. BULGULAR VE TARTIŞMA ERKAN TETİK
104
Şekil 4.2. CNT(6, 0) için a (A)-enerji (eV) optimizasyon grafiği
Şekil 4.3. CNT(6, 2) için a (A)-enerji (eV) optimizasyon grafiği
Şekil 4.4. CNT(6, 3) için a (A)-enerji (eV) optimizasyon grafiği

4. BULGULAR VE TARTIŞMA ERKAN TETİK
105
Şekil 4.5. CNT(7, 0) için a (A)-enerji (eV) optimizasyon grafiği
Şekil 4.6. CNT(7, 7) için a (A)-enerji (eV) optimizasyon grafiği
Optimizasyon ve hesaplama işlemleri CNT’lerin izole durumları için
yapılmıştır. Optimizasyon işlemi sonucunda elde edilen c/a ve a parametrelerinin
optimize değerleri çizelge 4.2’de görülmektedir. KNT’lerin diğer fiziksel özellikleri
de bu optimize değerler kullanılarak hesaplandı.
Çizelge 4.2. KNT’lerin Örgü Sabitleri cnt(6, 0) cnt(6, 2) cnt(6, 3) cnt(7, 0) cnt(7, 7)
c/a 0,528579024 1,703839001 1,175775188 0,1708 0,098472 a (A) 8,0831 9,0466 9,6029 24,9973 24,9609

4. BULGULAR VE TARTIŞMA ERKAN TETİK
106
4.4. Karbon Nanotüplerin Elektronik Bant Yapısı ve Durum Yoğunluğu
Hegzagonal yapıdaki karbon nanotüplerin Brillouin bölgesindeki yüksek
simetri noktaları şekil 4.7’de gösterilmektedir. Optimizasyon işlemini yaptığımız
KNT’lerin elektronik özelliklerini elde etmek için, birinci Brillouin bölgesinin
yüksek simetri noktaları k-uzayında A (0, 0, 1/2), (0, 0, 0), M (1/2, 0, 0), K
(0,33333, 0,33333, 0), (0, 0, 0), A (0, 0, 1/2) olarak seçilmiştir. KNT’leri yapısal
ve fiziksel özelliklerine göre 5 guruba ayırmıştık. Bu guruptaki her bir nanotüp için
bu yüksek simetri noktalarını kullanarak, enerji bant grafiklerini, yasak bant
aralıklarını, durum yoğunluğunu ve yük yoğunluğunu elde ettik. Enerji bant ve
durum yoğunluğu grafiklerini iki boyutlu, yük yoğunluğu grafiklerini hem iki hem de
üç boyutlu olarak XY düzlemine göre elde ettik.
Şekil 4.7. Hegzagonal yapıdaki karbon nanotüplerin Brillouin bölgesi
KNT’ler grafen katmanından elde edildiği için, KNT’lerin elektronik
özellikleri incelenirken grafenin de elektronik özelliklerini dikkate almak faydalı
olacaktır. Bu yüzden hesaplamalarımızda öncelikle KNT’yi oluşturan grafen katmanı
üzerinde hesaplamalar yaptık. Grafen katmanı için de enerji bant grafiğini, yasak
bant aralığını, durum yoğunluğunu ve yük yoğunluğunu elde ettik. Daha sonra bu
grafenden elde edilen nanotüpün hesaplama sonuçlarıyla karşılaştırmalar yaptık.
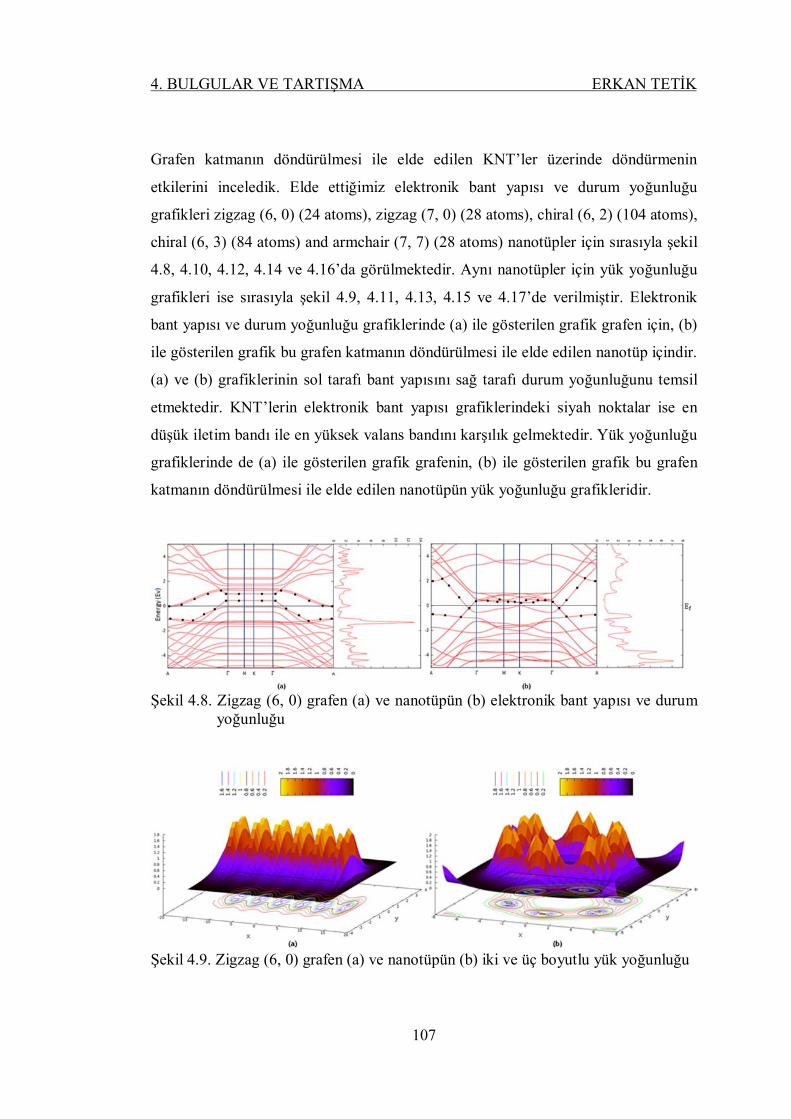
4. BULGULAR VE TARTIŞMA ERKAN TETİK
107
Grafen katmanın döndürülmesi ile elde edilen KNT’ler üzerinde döndürmenin
etkilerini inceledik. Elde ettiğimiz elektronik bant yapısı ve durum yoğunluğu
grafikleri zigzag (6, 0) (24 atoms), zigzag (7, 0) (28 atoms), chiral (6, 2) (104 atoms),
chiral (6, 3) (84 atoms) and armchair (7, 7) (28 atoms) nanotüpler için sırasıyla şekil
4.8, 4.10, 4.12, 4.14 ve 4.16’da görülmektedir. Aynı nanotüpler için yük yoğunluğu
grafikleri ise sırasıyla şekil 4.9, 4.11, 4.13, 4.15 ve 4.17’de verilmiştir. Elektronik
bant yapısı ve durum yoğunluğu grafiklerinde (a) ile gösterilen grafik grafen için, (b)
ile gösterilen grafik bu grafen katmanın döndürülmesi ile elde edilen nanotüp içindir.
(a) ve (b) grafiklerinin sol tarafı bant yapısını sağ tarafı durum yoğunluğunu temsil
etmektedir. KNT’lerin elektronik bant yapısı grafiklerindeki siyah noktalar ise en
düşük iletim bandı ile en yüksek valans bandını karşılık gelmektedir. Yük yoğunluğu
grafiklerinde de (a) ile gösterilen grafik grafenin, (b) ile gösterilen grafik bu grafen
katmanın döndürülmesi ile elde edilen nanotüpün yük yoğunluğu grafikleridir.
Şekil 4.8. Zigzag (6, 0) grafen (a) ve nanotüpün (b) elektronik bant yapısı ve durum
yoğunluğu
Şekil 4.9. Zigzag (6, 0) grafen (a) ve nanotüpün (b) iki ve üç boyutlu yük yoğunluğu

4. BULGULAR VE TARTIŞMA ERKAN TETİK
108
Şekil 4.8’da zigzag (6, 0) grafen ve nanotüpün elektronik bant yapıları
görülmektedir. Grafenin birim hücresi baz alınarak yapılan hesaplamalarda, grafenin
yarımetal özellik gösterdiği görülmektedir. Ancak grafenin Fermi yüzeyi 6 farklı
noktadan oluşmaktadır. Grafendeki bu özel Fermi yüzeyinden dolayı, KNT’ler bazı
durumlarda metalik bazı durumlarda yarı iletken özellik göstermektedir. Grafenin
birim hücresinde iki tane karbon atomu bulunmaktadır. Grafendeki atomları x-ekseni
boyunca 24 atoma kadar periyodik olarak arttırırsak, 24 atomlu süper örgüye sahip
bir grafen elde ederiz. Şekil 4.8(a)’daki grafik bu süper hücreli grafenin bant
yapısıdır. Bu 24 atomlu grafen bir silindir etrafında sarıldığında oluşan KNT’nin
elektronik bant yapısı ise şekil 4.8(b)’de görülmektedir. Grafen ve nanotüpün bant
yapıları genel olarak birbirine yakın görünse de ikisi arasında farklılıklar vardır.
KNT’nin valans ve iletim bantlarının her ikisi de grafen katmanının
döndürülmesinden etkilenir. Fakat iletim bantları nanotüpün eğriliğinden daha fazla
etkilendiği görülmektedir. Grafenin bant yapısında bazı valans bantları Fermi
seviyesinin üzerine çıkmıştır. Bunun sebebi grafene x-ekseni boyunca eklenen
atomlardır. Bu şekilde eklenen atomlar grafenin yapısında örgü kusuru oluşmasına
neden olmuştur. Grafen ve nanotüpün durum yoğunluklarından da görüldüğü gibi,
grafene göre nanotüpün fermi enerji seviyesinde bantlar daha fazla yoğundur. Bu da
nanotüpe metalik özellik kazandırmaktadır. Zigzag (6, 0) nanotüp için 퐶 = 14,8945
A° ve çapı 4,74106 A° olarak elde edilmiştir. İletim bandının minimum enerji değeri
A-Г simetri noktaları arasında ve Г noktasına daha yakın olmak üzere -0,1556 eV,
valans bandının maksimum enerji değeri ise K-Г simetri noktaları arasında ve Г
noktasına daha yakın olmak üzere 0,4101 eV değerindedir. Elektronik bant yapısı
grafiğindeki enerji değerlerinden de nanotüpün metalik özellik gösterdiği
görülmektedir. Yapılan deneysel çalışmalarda zigzag (6, 0) nanotüpün metalik
özellik gösterdiği ortaya çıkmıştır. Buradaki çalışmamız da deneysel çalışmalarla
uyum içindedir.

4. BULGULAR VE TARTIŞMA ERKAN TETİK
109
Şekil 4.10. Zigzag (7, 0) grafen (a) ve nanotüpün (b) elektronik bant yapısı ve durum
yoğunluğu
Şekil 4.11. Zigzag (7, 0) grafen (a) ve nanotüpün (b) iki ve üç boyutlu yük
yoğunluğu
Nanotüplerin grafen yapıdan silindirik yapıya dödürmenin KNT’lerin bant
yapısına etkilerini incelemek için benzer bir hesaplamayı zigzag (7, 0) grafen ve
nanotüp için yaptık. Şekil 4.10’da zigzag (7, 0) grafen (a) ve nanotüpün (b)
elektronik bant yapısı ve durum yoğunluğu yer almaktadır. Zigzag (7, 0) grafeni elde
etmek için zigzag (6, 0) grafene x-ekseni boyunca 4 atom daha ekledik. Bu durumda
zigzag (7, 0) nanotüp için 퐶 = 17,3374 A° ve çap 5,51868 A° oldu. Yine grafen ve
nanotüpün bant yapıları birbirine benzemektedir. Grafenin süper hücre yapısından
dolayı bazı valans bantlarının bu yapıda da Fermi enerji seviyesinin üzerine çıktığı
görülmektedir. Grafen yapıdan nanotüp elde edildiğinde ise bantlar Fermi
seviyesinin altına inmektedir. Zigzag (6, 0) grafene göre zigzag (7, 0) grafenin
elektronik bant yapısında önemli bir değişiklik yoktur. Ancak zigzag (7, 0)
nanotüpün bant yapısı tamamen değişmiştir. Şekil 4.10 (b)’deki elektronik bant
yapısı ve durum yoğunluğu grafiklerinden, zigzag (7, 0) nanotüpün yarıiletken
özellik gösterdiği görülmektedir. Bu da 퐶 değerinin diğer bir deyişle nanotüpün

4. BULGULAR VE TARTIŞMA ERKAN TETİK
110
geometrik yapısının değişmesi ile elektronik özelliğinin de değiştiğini
göstermektedir. Bu nanotüp için yarıiletken yasak bant aralığını E = 0,5022 eV
olarak elde ettik.
Şekil 4.12. Armchair (7, 7) grafen (a) ve nanotüpün (b) elektronik bant yapısı ve
durum yoğunluğu
Şekil 4.13. Armchair (7, 7) grafen (a) ve nanotüpün (b) iki ve üç boyutlu yük
yoğunluğu
Şekil 4.12’de ise geometrik olarak farklı bir yapı olan, armchair (7, 7) grafen
(a) ve KNT’nin (b) elektronik bant yapısı ve durum yoğunluğu görülmektedir.
Armchair (7, 7) grafen yapıyı elde etmek için 퐶 vektörünü, zigzag (7, 0) grafene
göre 30 döndürdük. KNT’yi ise yine bu grafenin bir silindir etrafında sararak
oluşturduk. Yeni yapının chiral vektörünü 29,9179 A° ve çapını 9,52316 A° olarak
elde ettik. Chiral vektöründeki bu değişiklik hem grafenin hem de nanotüpün
elektronik özelliklerini değiştirdiğini gözledik. (6, 0) ve (7, 0) grafenin Г-M-K-Г
simetri noktalarındaki yasak bant aralığı sırasıyla 0,5355 eV ve 0,2275 eV
civarındadır. Fakat armchair (7, 7) grafen böyle bir bant aralığına sahip değildir.
Fermi seviyesindeki bant yoğunluğundan dolayı armchair (7, 7) grafenin metalik
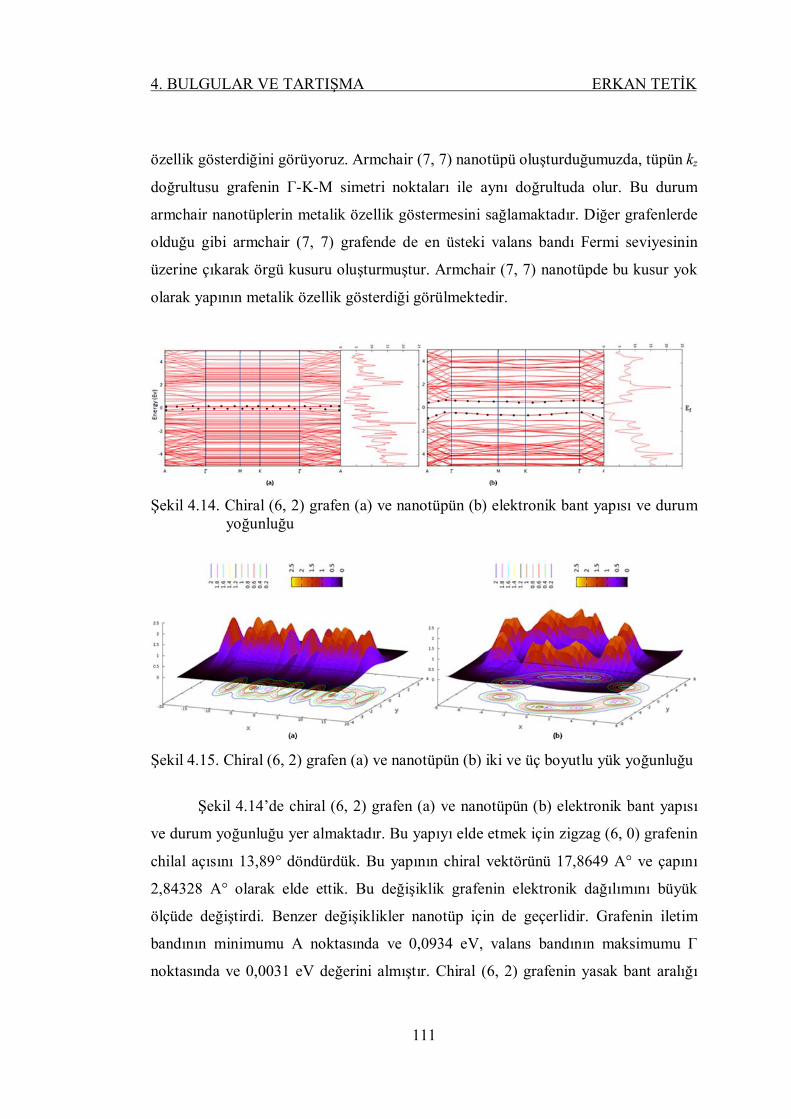
4. BULGULAR VE TARTIŞMA ERKAN TETİK
111
özellik gösterdiğini görüyoruz. Armchair (7, 7) nanotüpü oluşturduğumuzda, tüpün kz
doğrultusu grafenin Г-K-M simetri noktaları ile aynı doğrultuda olur. Bu durum
armchair nanotüplerin metalik özellik göstermesini sağlamaktadır. Diğer grafenlerde
olduğu gibi armchair (7, 7) grafende de en üsteki valans bandı Fermi seviyesinin
üzerine çıkarak örgü kusuru oluşturmuştur. Armchair (7, 7) nanotüpde bu kusur yok
olarak yapının metalik özellik gösterdiği görülmektedir.
Şekil 4.14. Chiral (6, 2) grafen (a) ve nanotüpün (b) elektronik bant yapısı ve durum
yoğunluğu
Şekil 4.15. Chiral (6, 2) grafen (a) ve nanotüpün (b) iki ve üç boyutlu yük yoğunluğu
Şekil 4.14’de chiral (6, 2) grafen (a) ve nanotüpün (b) elektronik bant yapısı
ve durum yoğunluğu yer almaktadır. Bu yapıyı elde etmek için zigzag (6, 0) grafenin
chilal açısını 13,89° döndürdük. Bu yapının chiral vektörünü 17,8649 A° ve çapını
2,84328 A° olarak elde ettik. Bu değişiklik grafenin elektronik dağılımını büyük
ölçüde değiştirdi. Benzer değişiklikler nanotüp için de geçerlidir. Grafenin iletim
bandının minimumu A noktasında ve 0,0934 eV, valans bandının maksimumu Г
noktasında ve 0,0031 eV değerini almıştır. Chiral (6, 2) grafenin yasak bant aralığı

4. BULGULAR VE TARTIŞMA ERKAN TETİK
112
0.0965 eV civarındadır. Dolayısıyla chiral (6, 2) grafeni düşük yasak bant aralığına
sahip bir yarıiletken olarak elde ettik. Ayrıca chiral (6, 2) grafende diğer süper
hücreli grafende oluşan örgü kusurları bulunmamaktadır. Bu da chiral (6, 2) grafeni
süper hücre yapmak için atom sayısını arttırıken seçilen doğrultunun, x ve y
ekseninin bileşke vektörüne yakın doğrultuda seçilmesinden kaynaklanmaktadır.
Chiral (6, 2) nanotüpün elektronik bant yapısına bakarsak, diğer nanotüplere göre A
noktasındaki bant enerjilerinin Fermi seviyesine daha yakın olduğunu görürüz. Bu
durumun nedeni yük yoğunluğu grafiklerinden daha iyi görülmektedir ve bir sonraki
bölümde açıklanacaktır. KNT’nin iletim bandının minimumu A noktasında ve 0,4091
eV, valans bandının maksimumu Г noktasında ve -0,42 eV değerindedir. Dolayısıyla
chiral (6, 2) nanotüp yarıiletkendir ve yasak bant aralığı E = 0,8291 eV olarak elde
edilmiştir. Chiral (6, 2) nanotüpün diğer nanotüplere göre oldukça büyük bir dolaylı
yasak bant aralığına sahip olduğunu söyleyebiliriz.
Son olarak şekil 4.16’da yer alan chiral (6, 3) grafen (a) ve nanotüpün (b)
elektronik dağılımını ve durum yoğunluğunu hesapladık. Bu yapı chiral (6, 2) ile
benzer bir geometrik yapıya sahiptir. Sadece chiral vektörünün döndürme açısı olan
chiral açısı 19.11° olarak döndürülerek elde edilen chiral (6, 3) yapının chiral açısı
farklılık göstermektedir. Bu farklılık chiral (6, 3) atom sayısını da chiral (6, 2)
göre değiştirmiştir. Bu değişiklik grafen ve nanotüpün elektronik yapısına
yansımıştır. İlk olarak grefeni dikkate alırsak, grafenin dolaylı yasak bant
aralığına sahip olduğunu ve bant aralığının da E = 0,0516 eV civarında
olduğunu görüyoruz. Bu değer (6, 2) grafenden daha düşüktür. Ayrıca (6, 3)
grafende en üstteki valans bandı Fermi seviyesini aşarak örgü kusuru meydana
getirmiştir. Chiral (6, 3) nanotüpü incelediğimizde yasak bant aralığının
E = 0,0488 eV civarında olduğunu görüyoruz. İletim bandının minimum değeri
A-Г noktaları arasında, Г noktasına daha yakındır ve 0,0237 eV, valans bandının
maksimum değeri ise A-Г noktaları arasında, Г noktasına daha yakındır ve -0,0251
eV civarındadır. Bu durumda chiral (6, 3) nanotüpün düşük bant aralığını sahip
bir sankimetalik (quasimetallic) nanotüp olduğunu söyleyebiliriz. Genel anlamda,
nanotüplerin valans ve iletim bantlarının fermi seviyesine göre asimetrik olduğu

4. BULGULAR VE TARTIŞMA ERKAN TETİK
113
gözlenmiştir. Bu durumun iki önemli sebebi vardır. Birincisi, grafenin elektronik
dağılımı valans ve iletim bantları için birbirinden farklı olmasındandır. İkincisi, daha
yüksek bantların nanotüpteki eğrilikten dolayı Fermi seviyesine doğru hareket
etmesinden kaynaklanmaktadır.
Şekil 4.16. Chiral (6, 3) grafen (a) ve nanotüpün (b) elektronik bant yapısı ve durum
yoğunluğu
Şekil 4.17. Chiral (6, 3) grafen (a) ve nanotüpün (b) iki ve üç boyutlu yük yoğunluğu
KNT’ler için elektronik bant yapısı ve durum yoğunluğu hesaplamaları
zigzag (6, 0), zigzag (7, 0 ), armchair (7, 7), Chiral (6, 2) ve chiral (6, 3) grafen ve
nanotüpler için yapıldı. Hesaplama sonucunda grafen ve nanotüpün sonuçları
karşılaştırılarak elektronik yapıdaki değişikliklerden döndürmenin etkileri gözlendi.
Bu hesaplamalardan elde veriler ve bu yapıların atom sayıları çizelge 4.3’de
özetlenmiştir.
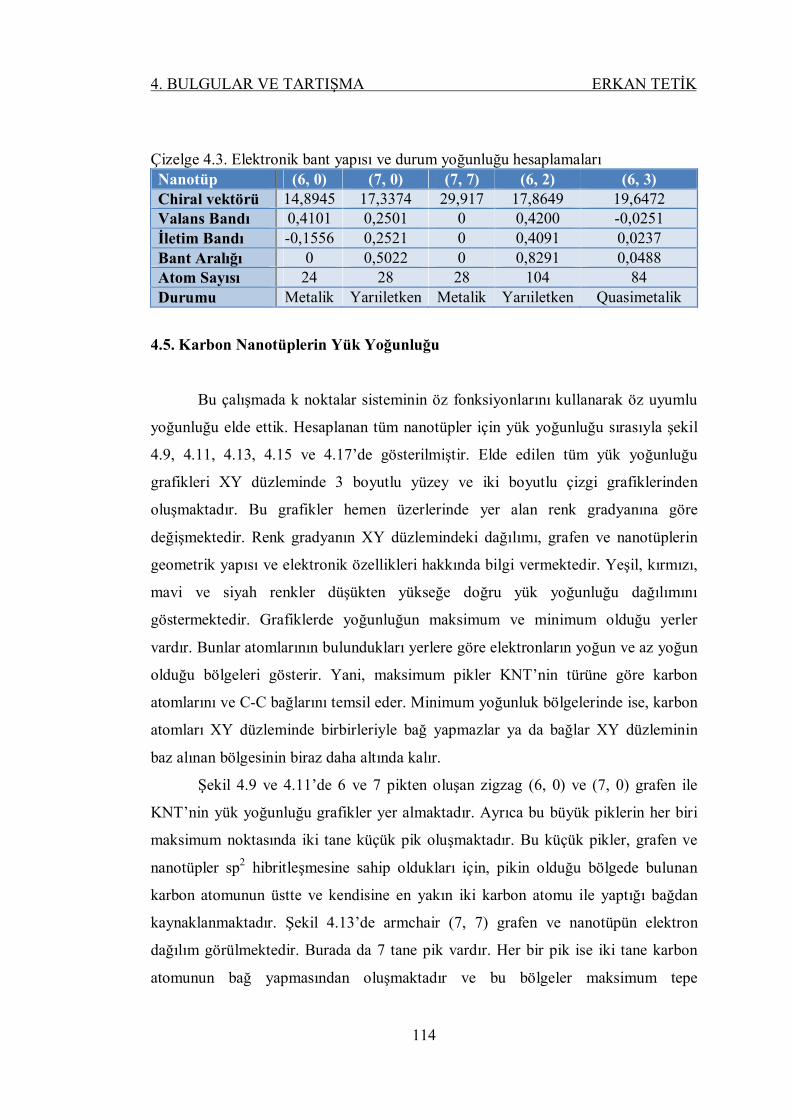
4. BULGULAR VE TARTIŞMA ERKAN TETİK
114
Çizelge 4.3. Elektronik bant yapısı ve durum yoğunluğu hesaplamaları Nanotüp (6, 0) (7, 0) (7, 7) (6, 2) (6, 3) Chiral vektörü 14,8945 17,3374 29,917 17,8649 19,6472 Valans Bandı 0,4101 0,2501 0 0,4200 -0,0251 İletim Bandı -0,1556 0,2521 0 0,4091 0,0237 Bant Aralığı 0 0,5022 0 0,8291 0,0488 Atom Sayısı 24 28 28 104 84 Durumu Metalik Yarıiletken Metalik Yarıiletken Quasimetalik
4.5. Karbon Nanotüplerin Yük Yoğunluğu
Bu çalışmada k noktalar sisteminin öz fonksiyonlarını kullanarak öz uyumlu
yoğunluğu elde ettik. Hesaplanan tüm nanotüpler için yük yoğunluğu sırasıyla şekil
4.9, 4.11, 4.13, 4.15 ve 4.17’de gösterilmiştir. Elde edilen tüm yük yoğunluğu
grafikleri XY düzleminde 3 boyutlu yüzey ve iki boyutlu çizgi grafiklerinden
oluşmaktadır. Bu grafikler hemen üzerlerinde yer alan renk gradyanına göre
değişmektedir. Renk gradyanın XY düzlemindeki dağılımı, grafen ve nanotüplerin
geometrik yapısı ve elektronik özellikleri hakkında bilgi vermektedir. Yeşil, kırmızı,
mavi ve siyah renkler düşükten yükseğe doğru yük yoğunluğu dağılımını
göstermektedir. Grafiklerde yoğunluğun maksimum ve minimum olduğu yerler
vardır. Bunlar atomlarının bulundukları yerlere göre elektronların yoğun ve az yoğun
olduğu bölgeleri gösterir. Yani, maksimum pikler KNT’nin türüne göre karbon
atomlarını ve C-C bağlarını temsil eder. Minimum yoğunluk bölgelerinde ise, karbon
atomları XY düzleminde birbirleriyle bağ yapmazlar ya da bağlar XY düzleminin
baz alınan bölgesinin biraz daha altında kalır.
Şekil 4.9 ve 4.11’de 6 ve 7 pikten oluşan zigzag (6, 0) ve (7, 0) grafen ile
KNT’nin yük yoğunluğu grafikler yer almaktadır. Ayrıca bu büyük piklerin her biri
maksimum noktasında iki tane küçük pik oluşmaktadır. Bu küçük pikler, grafen ve
nanotüpler sp2 hibritleşmesine sahip oldukları için, pikin olduğu bölgede bulunan
karbon atomunun üstte ve kendisine en yakın iki karbon atomu ile yaptığı bağdan
kaynaklanmaktadır. Şekil 4.13’de armchair (7, 7) grafen ve nanotüpün elektron
dağılım görülmektedir. Burada da 7 tane pik vardır. Her bir pik ise iki tane karbon
atomunun bağ yapmasından oluşmaktadır ve bu bölgeler maksimum tepe

4. BULGULAR VE TARTIŞMA ERKAN TETİK
115
noktalarıdır. Büyük pikler arasında kalan bölgelerde de bağ yapan karbon atomları
bulunmaktadır ama bu atomlar seçilen XY düzleminin altında kaldığından dolayı
elektron yoğunluğu görülmemektedir. Renk gradyanına baktığımız armchair (7, 7)
nanotüpün zigzag nanotüplere göre daha yüksek bir yük yoğunluğuna sahip
olduğunu görürüz. Bu durum armchair nanotüplerin yüksekliğinin küçük olmasından
kaynaklanmaktadır. Chiral nanotüplerin yük yoğunlukları ise şekil 4.15 ve 4.17’de
yer almaktadır. Bu nanotüpler geometrik yapılarındaki eğrilikten dolayı zigzag ve
armchair nanotüplere göre yüzeylerinde daha fazla elektron barındırırlar ve daha
yüksek bir yük yoğunluğuna sahiptirler. Chiral (6, 2) grafen (6, 3) grafene göre
yüzeyinde daha fazla yoğunluk gözlenmektedir. Zigzag ve armchair grafenlerin
yapısında örgü kusuru oluştuğu görülmektedir. Ancak chiral grafenlerde örgü kusuru
ya hiç görülmemekte ya da çok az oluşmaktadır. Bu durumun chiral yapıların
yüzeyindeki elektron yoğunluğunun fazla olmasından kaynaklandığını söyleyebiliriz.
4.6. Karbon Nanotüplerde Katkılamanın Etkilerinin İncelenmesi
TDKNT’lerin elektronik özelliklerinin tüpün 퐶 açısına ve çapına hassas bir
şekilde bağlıdır. KNT’leri nano-elektronik cihazlarda bir eleman olarak
kullanabilmek için, tüpün yarıçapını ve 퐶 açısını kontrol edebilen bir yol bulmak
gerekmektedir. Bunu yapmak ise oldukça zor ve zahmetli işleri beraberinde getirir.
Bu işleme alternatif olarak KNT’lere yabancı atom eklemek yani katkılama işlemi
yaparak, elektronik özelliklerde değişiklik yapma yöntemi de kullanılabilir. Nano-
elektronik cihazlarda kullanılan malzemelerin metalik ya da yarıiletken özellik
göstermesi ve bu özelliğin değiştirilebilir olması oldukça önemlidir. Bu yüzden,
malzemelerin elektronik özelliğini değiştirmek için geometrik yapısını değiştirmek
yerine katkılama yapmanın önemi artmaktadır. Nanotüplere bor ve azot gibi atomlar
katkılamak onların elektronik özelliklerini kontrol etme imkânı tanımaktadır. Ancak
katkılama yapılacak atom seçilirken de dikkat edilmelidir. Örneğin katkılama
yapılacak atom karbon atomunun çapına yakın bir atom olmalıdır. Ayrıca katkılama
işlemi nanotüpün orijinal yapısını bozmayacak şekilde yapılmalıdır. Yani C-C

4. BULGULAR VE TARTIŞMA ERKAN TETİK
116
arasındaki bağ uzunluğunun 1,42 A civarında olduğunu düşünürsek eklenen atomlar
bu mesafeyi çok fazla değiştirmemelidir.
TDKNT geometrik yapısına göre metalik ya da yarıiletken özellik gösterdiği
yaptığımız hesaplama sonuçlarından gördük. TDKNT’lere yabancı atom ekleyerek
katkılama işlemi yapıldığı zaman, yarıiletken bir TDKNT metalik, metalik bir
TDKNT de yarıiletken özellik gösterebilir. Bu durum eklenen atomun elektronik
özelliklerine ve ekleme miktarına göre değişiklik göstermektedir. KNT’ye yabancı
atom eklemek, bunun dışında nanotüpün elektriksel ve optik gibi pek çok özelliğini
etkilemektedir.
Şekil 4.18. Zigzag (8, 0) TDKNT’nin geometrik yapısı
Nanotüplerin elektronik yapısına katkılamanın etkilerini incelemek için
yarıiletken zigzag (8, 0) nanotüp üzerinde hesaplamalar yaptık. Öncelikle herhangi
bir yabancı atom eklemeden (8, 0) nanotüpü optimize ettik. Optimize edilen yapı
şekil 4.18’de görülmektedir. Daha sonra optimize yapının elektronik bant yapısını ve
durum yoğunluğunu elde ettik. Bu yapının bant yapısı ve durum yoğunluğu grafiği
şekil 4.19’de görülmektedir. Zigzag (8, 0) nanotüpün yabancı atom eklenmemiş
yapısının ve yabancı atom eklenen diğer tüm yapılarının optimizasyon sonuçları ek
2’de verilmiştir.
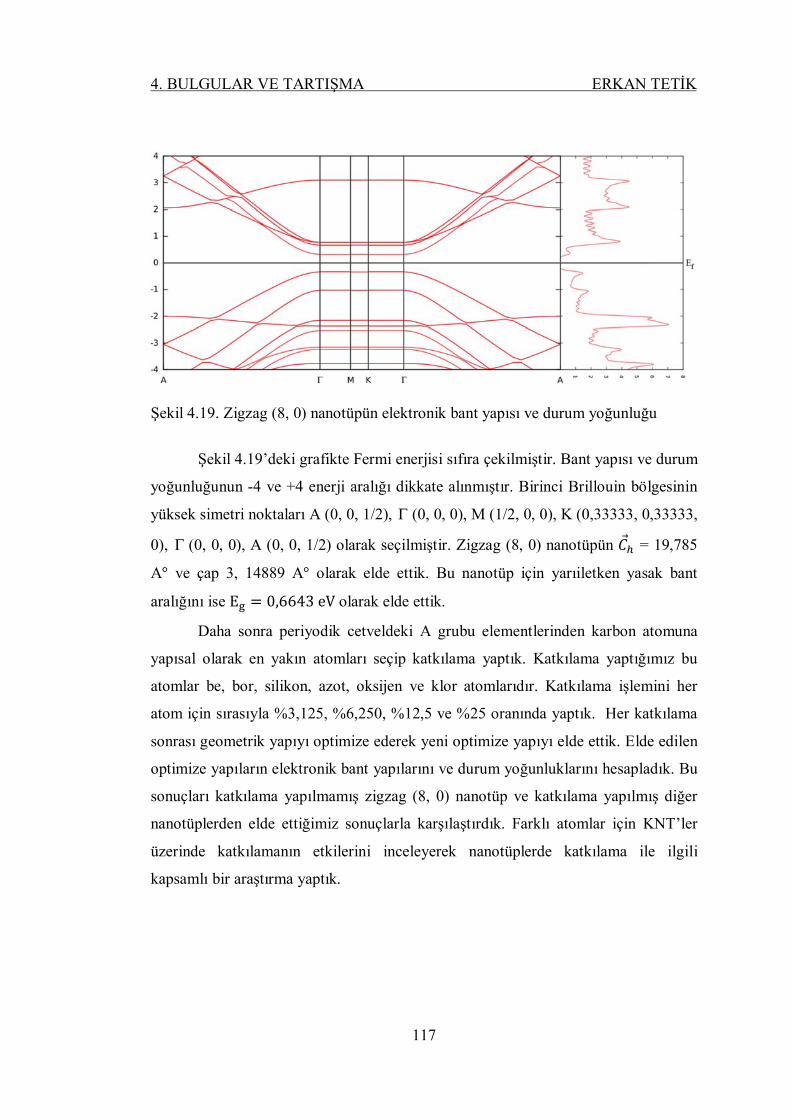
4. BULGULAR VE TARTIŞMA ERKAN TETİK
117
Şekil 4.19. Zigzag (8, 0) nanotüpün elektronik bant yapısı ve durum yoğunluğu
Şekil 4.19’deki grafikte Fermi enerjisi sıfıra çekilmiştir. Bant yapısı ve durum
yoğunluğunun -4 ve +4 enerji aralığı dikkate alınmıştır. Birinci Brillouin bölgesinin
yüksek simetri noktaları A (0, 0, 1/2), (0, 0, 0), M (1/2, 0, 0), K (0,33333, 0,33333,
0), (0, 0, 0), A (0, 0, 1/2) olarak seçilmiştir. Zigzag (8, 0) nanotüpün 퐶 = 19,785
A° ve çap 3, 14889 A° olarak elde ettik. Bu nanotüp için yarıiletken yasak bant
aralığını ise E = 0,6643 eV olarak elde ettik.
Daha sonra periyodik cetveldeki A grubu elementlerinden karbon atomuna
yapısal olarak en yakın atomları seçip katkılama yaptık. Katkılama yaptığımız bu
atomlar be, bor, silikon, azot, oksijen ve klor atomlarıdır. Katkılama işlemini her
atom için sırasıyla %3,125, %6,250, %12,5 ve %25 oranında yaptık. Her katkılama
sonrası geometrik yapıyı optimize ederek yeni optimize yapıyı elde ettik. Elde edilen
optimize yapıların elektronik bant yapılarını ve durum yoğunluklarını hesapladık. Bu
sonuçları katkılama yapılmamış zigzag (8, 0) nanotüp ve katkılama yapılmış diğer
nanotüplerden elde ettiğimiz sonuçlarla karşılaştırdık. Farklı atomlar için KNT’ler
üzerinde katkılamanın etkilerini inceleyerek nanotüplerde katkılama ile ilgili
kapsamlı bir araştırma yaptık.

4. BULGULAR VE TARTIŞMA ERKAN TETİK
118
4.6.1. Bor Katkılanmış Karbon Nanotüpler
KNT’lere yabancı atom katkılama ile ilgili ilk hesaplamalarımızı bor atomu
ile gerçekleştirdik. Bor atomu periyodik cetvelde 5A gurubunda ve 2. periyottadır.
Atom numarası 5 ve atomik çapı 85 pm değerini alır. Ayrıca bor atomu rombohedral
yapıda olup uzay grubu 166’dır. Bor yarımetaller ailesindendir. KNT’ye eklenen bor
atomları yabancı atom eklenme yüzdelerimize (%3,125, %6,250, %12,5 ve %25)
göre, sırasıyla 1, 2, 4 ve 8 tanedir. Bor eklenen nanotüpler eklenme sırasına göre
şekil 4.20’de verilmiştir. Şekil 4.20’deki katkılama yapılan her nanotüp için
katkılama sonrası yeniden optimizasyon işlemi yapılmıştır. İdeal yapı elde edildikten
sonra bu yapıların elektronik özellikleri hesaplanmıştır.
Şekil 4.20. %3,125, %6,25, %12,5 ve %25 oranlarında bor eklenmiş (8, 0) nanotüpün geometrik yapısı

4. BULGULAR VE TARTIŞMA ERKAN TETİK
119
Bor atomu periyodik cetvelde bulunduğu konum itibarıyla karbon atomuna
yakın bir atomdur. Çapı da karbon atomuna yakın olduğundan, KNT’lere en çok
katkılama yapılan yabancı atomlardan biridir diyebiliriz. Katkılama yapılan
atomların hesaplama sonuçları katkılama sırasına göre, şekil 4.21, 4.22, 4.23 ve
4.24’de verilmiştir. Bu şekillerde yabancı atom eklenen KNT’lerin elektronik bant
yapıları ve durum yoğunlukları vardır. Durum yoğunluğu grafiklerindeki gri alanlar
Fermi enerji seviyesinin altında kalan valans bantlarını temsil etmektedir. Mavi
alanlar ise katkılama sonucunda valans bantlarının ne kadarlık bir kısmının Fermi
enerji seviyesinin üzerine çıktığını göstermektedir.
Şekil 4.21. %3,125 bor eklenmiş (8, 0) nanotüpün bant yapısı ve durum yoğunluğu
Şekil 4.22. %6,25 bor eklenmiş (8, 0) nanotüpün bant yapısı ve durum yoğunluğu
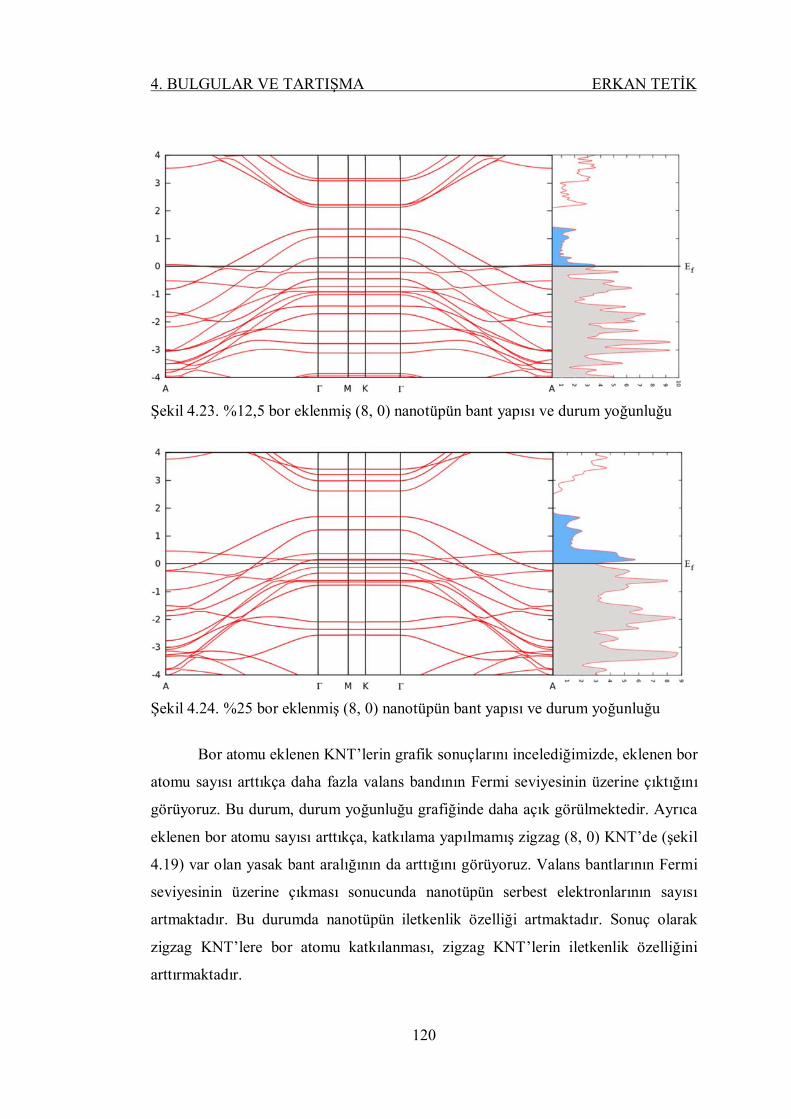
4. BULGULAR VE TARTIŞMA ERKAN TETİK
120
Şekil 4.23. %12,5 bor eklenmiş (8, 0) nanotüpün bant yapısı ve durum yoğunluğu
Şekil 4.24. %25 bor eklenmiş (8, 0) nanotüpün bant yapısı ve durum yoğunluğu
Bor atomu eklenen KNT’lerin grafik sonuçlarını incelediğimizde, eklenen bor
atomu sayısı arttıkça daha fazla valans bandının Fermi seviyesinin üzerine çıktığını
görüyoruz. Bu durum, durum yoğunluğu grafiğinde daha açık görülmektedir. Ayrıca
eklenen bor atomu sayısı arttıkça, katkılama yapılmamış zigzag (8, 0) KNT’de (şekil
4.19) var olan yasak bant aralığının da arttığını görüyoruz. Valans bantlarının Fermi
seviyesinin üzerine çıkması sonucunda nanotüpün serbest elektronlarının sayısı
artmaktadır. Bu durumda nanotüpün iletkenlik özelliği artmaktadır. Sonuç olarak
zigzag KNT’lere bor atomu katkılanması, zigzag KNT’lerin iletkenlik özelliğini
arttırmaktadır.

4. BULGULAR VE TARTIŞMA ERKAN TETİK
121
4.6.2. Berilyum Katkılanmış Karbon Nanotüpler
Berilyum atomu periyodik cetvelde 2A gurubunda ve 2. periyottadır. Atom
numarası 4 ve atomik çapı 90 pm değerini almaktadır. Ayrıca berilyum atomu
hegzagonal yapıda olup uzay grubu 194’dür. Berilyum toprak alkali metal
ailesindendir. KNT’ye eklenen berilyum atomları yabancı atom eklenme
yüzdelerimize (%3,125, %6,250, %12,5 ve %25) göre, sırasıyla 1, 2, 4 ve 8 tanedir.
Berilyum eklenen nanotüpler eklenme sırasına göre şekil 4.25’de verilmiştir. Şekil
4.25’deki katkılama yapılan her nanotüp optimize edilmiştir. İdeal yapı elde
edildikten sonra bu yapıların elektronik özellikleri hesaplanmıştır.
Şekil 4.25. %3,125, %6,25, %12,5 ve %25 oranlarında berilyum eklenmiş (8, 0)
nanotüpün geometrik yapısı

4. BULGULAR VE TARTIŞMA ERKAN TETİK
122
Şekil 4.26. %3,125 berilyum eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu
Şekil 4.27. %6,25 berilyum eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu
Berilyum atomu periyodik cetvelde bulunduğu konum itibarıyla karbon
atomuna uzak durumdadır. Katkılama yapılan berilyum atomlarının hesaplama
sonuçları katkılama sırasına göre, şekil 4.26, 4.27, 4.28 ve 4.29’de verilmiştir. Bu
şekillerde berilyum eklenen KNT’lerin elektronik bant yapıları ve durum
yoğunlukları verilmiştir. Durum yoğunluğu grafiklerindeki gri alanlar, bor katkılanan
nanotüplerin grafiklerinde olduğu gibi, Fermi enerji seviyesinin altında kalan valans
bantlarını temsil etmektedir. Mavi alanlar ise katkılama sonucunda valans bantlarının
ne kadarlık bir kısmının Fermi enerji seviyesinin üzerine çıktığını göstermektedir.
4. BULGULAR VE TARTIŞMA ERKAN TETİK
123
Şekil 4.28. %12,5 berilyum eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu
Şekil 4.29. %25 berilyum eklenmiş (8, 0) nanotüpün bant yapısı ve durum yoğunluğu
Berilyum atomu eklenen KNT’lerin grafik sonuçlarını incelediğimizde,
eklenen berilyum atomu sayısı arttıkça daha fazla valans bandının Fermi seviyesinin
üzerine çıktığını görüyoruz. Berilyum atomu katkılanan nanotüpleri bor atomu
eklenen nanotüplerle kıyaslarsak, berilyum atomu katkılanan nanotüplerde daha fazla
valans bandı Fermi enerjisinin seviyesinin üzerine çıkmıştır. Dolayısıyla berilyum
katkılanan nanotüplerde serbest elektronların sayısı daha fazladır. Bu nanotüplerde
iletkenlik bor katkıllanan nanotüplere göre daha fazla artmaktadır. Sonuç olarak,
berilyum katkılanan KNT’ler bor katkılananlara göre daha fazla iletken özellik
gösterir.

4. BULGULAR VE TARTIŞMA ERKAN TETİK
124
4.6.3. Azot Katkılanmış Karbon Nanotüpler
Azot atomu periyodik cetvelde 5A gurubunda ve 2. periyottadır. Atom
numarası 7 ve atomik çapı 65 pm değerini alır. Ayrıca bor atomu hegzagonal yapıda
olup uzay grubu 194’dür. Azot ametal ailesindendir. KNT’ye eklenen azot atomları
yabancı atom eklenme yüzdelerimize (%3,125, %6,250, %12,5 ve %25) göre,
sırasıyla 1, 2, 4 ve 8 tanedir. Azot eklenen nanotüpler eklenme sırasına göre şekil
4.30’da verilmiştir. Şekil 4.30’daki katkılama yapılan her nanotüp optimize edilip,
ideal yapı elde edildikten sonra elektronik özellikleri hesaplanmıştır.
Şekil 4.30. %3,125, %6,25, %12,5 ve %25 oranlarında azot eklenmiş (8, 0)
nanotüpün geometrik yapısı
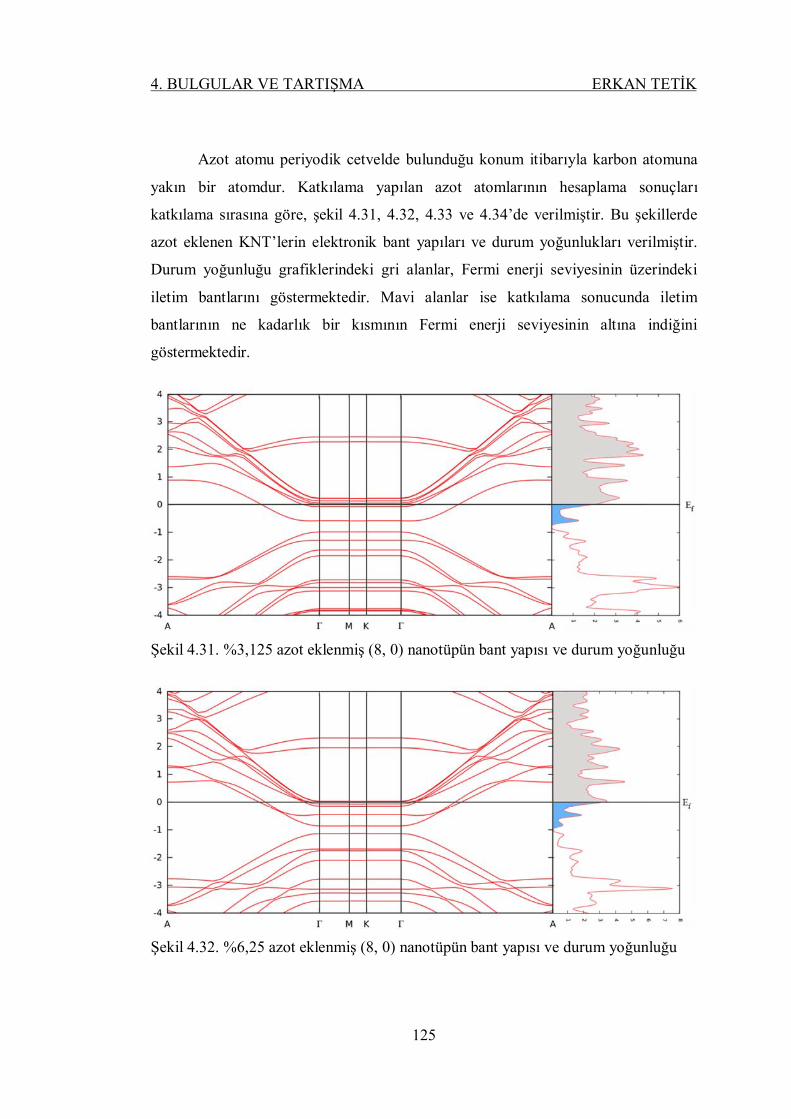
4. BULGULAR VE TARTIŞMA ERKAN TETİK
125
Azot atomu periyodik cetvelde bulunduğu konum itibarıyla karbon atomuna
yakın bir atomdur. Katkılama yapılan azot atomlarının hesaplama sonuçları
katkılama sırasına göre, şekil 4.31, 4.32, 4.33 ve 4.34’de verilmiştir. Bu şekillerde
azot eklenen KNT’lerin elektronik bant yapıları ve durum yoğunlukları verilmiştir.
Durum yoğunluğu grafiklerindeki gri alanlar, Fermi enerji seviyesinin üzerindeki
iletim bantlarını göstermektedir. Mavi alanlar ise katkılama sonucunda iletim
bantlarının ne kadarlık bir kısmının Fermi enerji seviyesinin altına indiğini
göstermektedir.
Şekil 4.31. %3,125 azot eklenmiş (8, 0) nanotüpün bant yapısı ve durum yoğunluğu
Şekil 4.32. %6,25 azot eklenmiş (8, 0) nanotüpün bant yapısı ve durum yoğunluğu
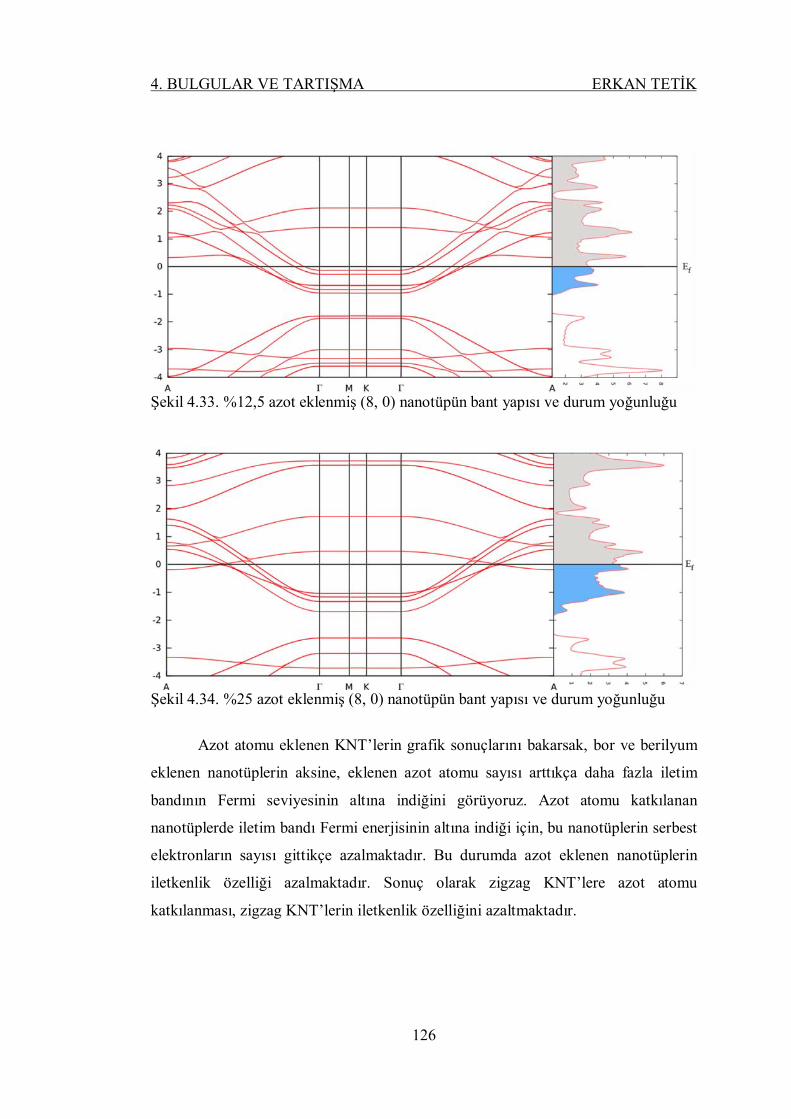
4. BULGULAR VE TARTIŞMA ERKAN TETİK
126
Şekil 4.33. %12,5 azot eklenmiş (8, 0) nanotüpün bant yapısı ve durum yoğunluğu
Şekil 4.34. %25 azot eklenmiş (8, 0) nanotüpün bant yapısı ve durum yoğunluğu
Azot atomu eklenen KNT’lerin grafik sonuçlarını bakarsak, bor ve berilyum
eklenen nanotüplerin aksine, eklenen azot atomu sayısı arttıkça daha fazla iletim
bandının Fermi seviyesinin altına indiğini görüyoruz. Azot atomu katkılanan
nanotüplerde iletim bandı Fermi enerjisinin altına indiği için, bu nanotüplerin serbest
elektronların sayısı gittikçe azalmaktadır. Bu durumda azot eklenen nanotüplerin
iletkenlik özelliği azalmaktadır. Sonuç olarak zigzag KNT’lere azot atomu
katkılanması, zigzag KNT’lerin iletkenlik özelliğini azaltmaktadır.

4. BULGULAR VE TARTIŞMA ERKAN TETİK
127
4.6.4. Silisyum Katkılanmış Karbon Nanotüpler
Silisyum atomu periyodik cetvelde 4A gurubunda ve 3. periyottadır. Atom
numarası 14 ve atomik çapı 110 pm değerini alır. Ayrıca bor atomu yüzey merkezli
kübik yapıda olup uzay grubu 227’dir. Silisyum yarımetal ailesindendir. KNT’ye
eklenen silisyum atomları yabancı atom eklenme yüzdelerimize (%3,125, %6,250,
%12,5 ve %25) göre, sırasıyla 1, 2, 4 ve 8 tanedir. Silisyum eklenen nanotüpler
eklenme sırasına göre şekil 4.30’da verilmiştir. Şekil 4.35’deki katkılama yapılan her
nanotüp optimize edilip, ideal yapı elde edildikten sonra elektronik özellikleri
hesaplanmıştır.
Şekil 4.35. %3,125, %6,25, %12,5 ve %25 oranlarında silisyum eklenmiş (8, 0)
nanotüpün geometrik yapısı

4. BULGULAR VE TARTIŞMA ERKAN TETİK
128
Silisyum atomu periyodik cetvelde bulunduğu konum itibarıyla karbon
atomuna ile aynı guruptadır. Fakat karbon atonumdan bir alt periyottadır. Dolayısıyla
atomil çapı karbon atomundan biraz daha büyüktür. Katkılama yapılan silisyum
atomlarının hesaplama sonuçları katkılama sırasına göre, şekil 4.36, 4.37, 4.38 ve
4.39’de verilmiştir. Bu şekillerde silisyum eklenen KNT’lerin elektronik bant
yapıları ve durum yoğunlukları verilmiştir. Durum yoğunluğu grafiklerindeki önceki
hesaplamalarda var olan gri ve mavi alanlar yoktur.
Şekil 4.36. %3,125 silisyum eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu
Şekil 4.37. %6,25 silisyum eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu
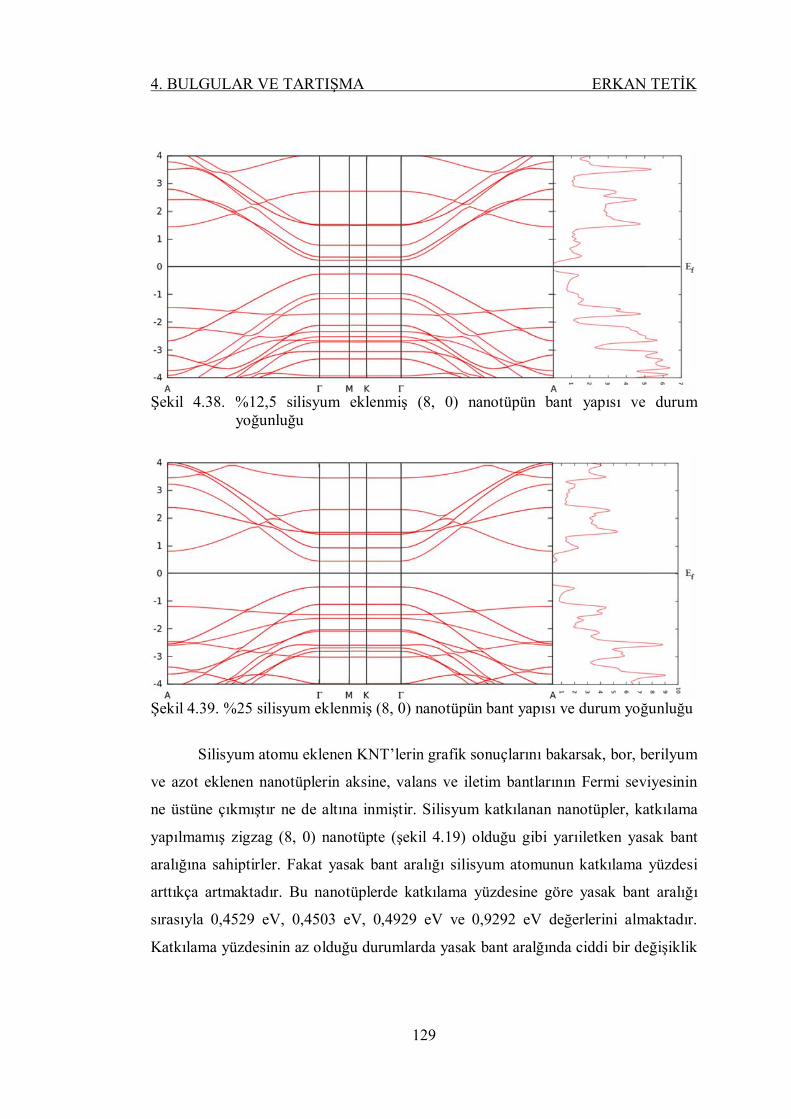
4. BULGULAR VE TARTIŞMA ERKAN TETİK
129
Şekil 4.38. %12,5 silisyum eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu
Şekil 4.39. %25 silisyum eklenmiş (8, 0) nanotüpün bant yapısı ve durum yoğunluğu
Silisyum atomu eklenen KNT’lerin grafik sonuçlarını bakarsak, bor, berilyum
ve azot eklenen nanotüplerin aksine, valans ve iletim bantlarının Fermi seviyesinin
ne üstüne çıkmıştır ne de altına inmiştir. Silisyum katkılanan nanotüpler, katkılama
yapılmamış zigzag (8, 0) nanotüpte (şekil 4.19) olduğu gibi yarıiletken yasak bant
aralığına sahiptirler. Fakat yasak bant aralığı silisyum atomunun katkılama yüzdesi
arttıkça artmaktadır. Bu nanotüplerde katkılama yüzdesine göre yasak bant aralığı
sırasıyla 0,4529 eV, 0,4503 eV, 0,4929 eV ve 0,9292 eV değerlerini almaktadır.
Katkılama yüzdesinin az olduğu durumlarda yasak bant aralğında ciddi bir değişiklik

4. BULGULAR VE TARTIŞMA ERKAN TETİK
130
olmamıştır. Ancak katkılama yüzdesi arttığında silisyum atomu baskın olduğunu
göstermiş ve yasak bant aralığını arttırmıştır.
4.6.5. Oksijen Katkılanmış Karbon Nanotüpler
Oksijen atomu periyodik cetvelde 6A gurubunda ve 2. periyottadır. Atom
numarası 8 ve atomik çapı 60 pm değerini almaktadır. Ayrıca oksijen atomu
monoklinik yapıda olup uzay grubu 12’dür. Oksijen atomu ametaldir. KNT’ye
eklenen oksijen atomları yabancı atom eklenme yüzdelerimize (%3,125, %6,250,
%12,5 ve %25) göre, sırasıyla 1, 2, 4 ve 8 tanedir. Oksijen eklenen nanotüpler
eklenme sırasına göre şekil 4.40’da verilmiştir. Şekil 4.40’daki nanotüpler optimize
edilip, ideal yapı elde edildikten sonra elektronik özellikleri hesaplanmıştır.
Şekil 4.40. %3,125, %6,25, %12,5 ve %25 oranlarında oksijen eklenmiş (8, 0)
nanotüpün geometrik yapısı

4. BULGULAR VE TARTIŞMA ERKAN TETİK
131
Oksijen atomu periyodik cetvelde bulunduğu konum itibarıyla karbon
atomundan biraz uzaktır ve atomik çapı karbon atomundan küçüktür. Katkılama
yapılan oksijen atomlarının hesaplama sonuçları katkılama sırasına göre, şekil 4.41,
4.42, 4.43 ve 4.44’de verilmiştir. Bu şekillerde oksijen eklenen KNT’lerin elektronik
bant yapıları ve durum yoğunlukları yer almaktadır. Durum yoğunluğu
grafiklerindeki gri alanlar, Fermi enerji seviyesinin üzerindeki iletim bantlarını, mavi
alanlar ise katkılama sonucunda iletim bantlarının ne kadarlık bir kısmının Fermi
enerji seviyesinin altına indiğini göstermektedir.
Şekil 4.41. %3,125 oksijen eklenmiş (8, 0) nanotüpün bant yapısı ve durum
yoğunluğu
Şekil 4.42. %6,25 oksijen eklenmiş (8, 0) nanotüpün bant yapısı ve durum yoğunluğu
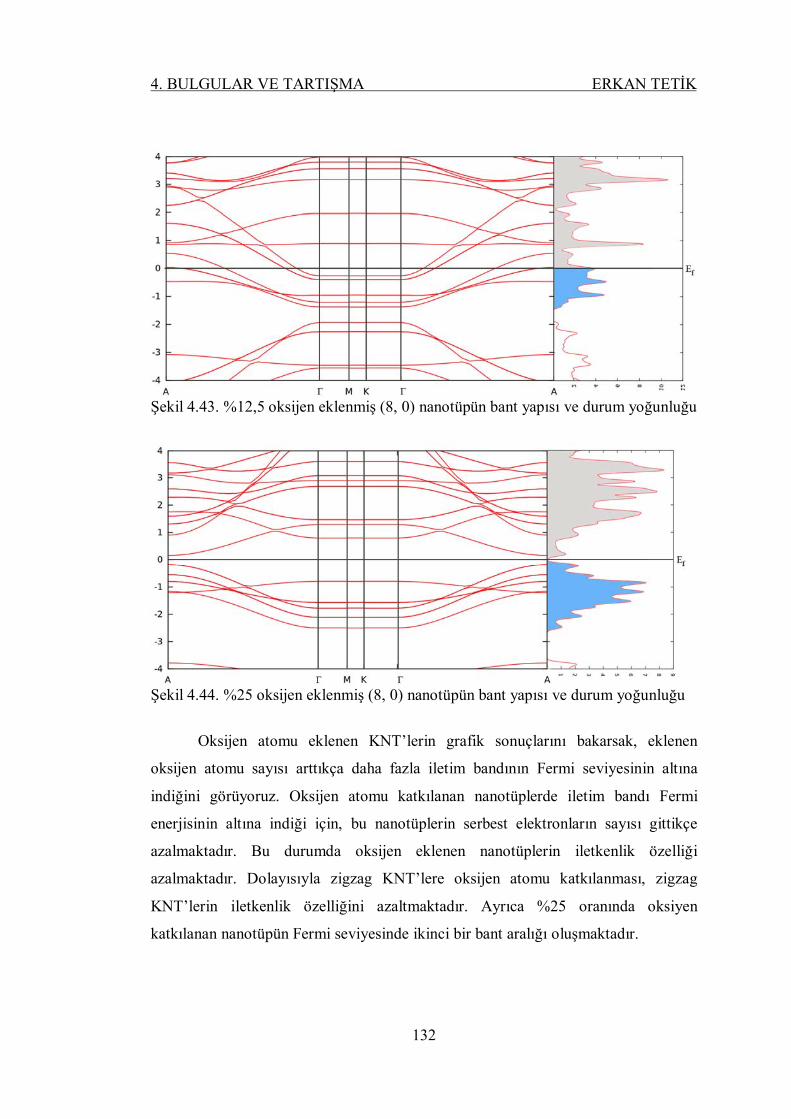
4. BULGULAR VE TARTIŞMA ERKAN TETİK
132
Şekil 4.43. %12,5 oksijen eklenmiş (8, 0) nanotüpün bant yapısı ve durum yoğunluğu
Şekil 4.44. %25 oksijen eklenmiş (8, 0) nanotüpün bant yapısı ve durum yoğunluğu
Oksijen atomu eklenen KNT’lerin grafik sonuçlarını bakarsak, eklenen
oksijen atomu sayısı arttıkça daha fazla iletim bandının Fermi seviyesinin altına
indiğini görüyoruz. Oksijen atomu katkılanan nanotüplerde iletim bandı Fermi
enerjisinin altına indiği için, bu nanotüplerin serbest elektronların sayısı gittikçe
azalmaktadır. Bu durumda oksijen eklenen nanotüplerin iletkenlik özelliği
azalmaktadır. Dolayısıyla zigzag KNT’lere oksijen atomu katkılanması, zigzag
KNT’lerin iletkenlik özelliğini azaltmaktadır. Ayrıca %25 oranında oksiyen
katkılanan nanotüpün Fermi seviyesinde ikinci bir bant aralığı oluşmaktadır.

4. BULGULAR VE TARTIŞMA ERKAN TETİK
133
4.6.6. Klor Katkılanmış Karbon Nanotüpler
Klor atomu periyodik cetvelde 7A gurubunda ve 3. periyottadır. Atom
numarası 17 ve atomik çapı 100 pm değerini almaktadır. Ayrıca klor atomu
ortorombik yapıda olup uzay grubu 64’dür. Klor atomu halojendir. KNT’ye eklenen
klor atomları yabancı atom eklenme yüzdelerimize (%3,125, %6,250, %12,5 ve
%25) göre, sırasıyla 1, 2, 4 ve 8 tanedir. Klor eklenen nanotüpler eklenme sırasına
göre şekil 4.45’de verilmiştir. Şekil 4.45’deki katkılama yapılan her nanotüp
optimize edilip, ideal yapı elde edildikten sonra elektronik özellikleri hesaplanmıştır.
Şekil 4.45. %3,125, %6,25, %12,5 ve %25 oranlarında klor eklenmiş (8, 0)
nanotüpün geometrik yapısı
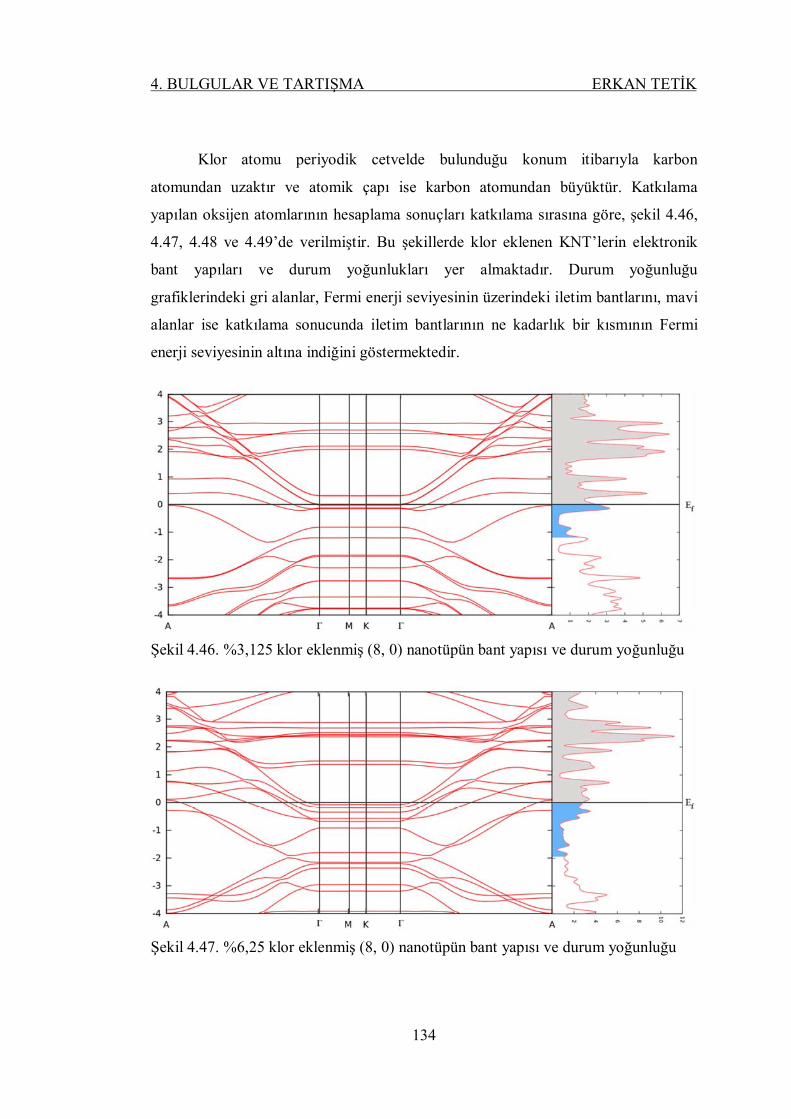
4. BULGULAR VE TARTIŞMA ERKAN TETİK
134
Klor atomu periyodik cetvelde bulunduğu konum itibarıyla karbon
atomundan uzaktır ve atomik çapı ise karbon atomundan büyüktür. Katkılama
yapılan oksijen atomlarının hesaplama sonuçları katkılama sırasına göre, şekil 4.46,
4.47, 4.48 ve 4.49’de verilmiştir. Bu şekillerde klor eklenen KNT’lerin elektronik
bant yapıları ve durum yoğunlukları yer almaktadır. Durum yoğunluğu
grafiklerindeki gri alanlar, Fermi enerji seviyesinin üzerindeki iletim bantlarını, mavi
alanlar ise katkılama sonucunda iletim bantlarının ne kadarlık bir kısmının Fermi
enerji seviyesinin altına indiğini göstermektedir.
Şekil 4.46. %3,125 klor eklenmiş (8, 0) nanotüpün bant yapısı ve durum yoğunluğu
Şekil 4.47. %6,25 klor eklenmiş (8, 0) nanotüpün bant yapısı ve durum yoğunluğu

4. BULGULAR VE TARTIŞMA ERKAN TETİK
135
Şekil 4.48. %12,5 klor eklenmiş (8, 0) nanotüpün bant yapısı ve durum yoğunluğu
Şekil 4.49. %25 klor eklenmiş (8, 0) nanotüpün bant yapısı ve durum yoğunluğu
Klor atomu eklenen KNT’lerin grafik sonuçlarını bakarsak, eklenen klor
atomu sayısı arttıkça daha fazla iletim bandının Fermi seviyesinin altına indiğini
görüyoruz. Klor atomu katkılanan nanotüplerde iletim bandı Fermi enerjisinin altına
indiği için, bu nanotüplerin serbest elektronların sayısı gittikçe azalmaktadır. Bu
durumda klor eklenen nanotüplerin iletkenlik özelliği azalmaktadır. Dolayısıyla
zigzag KNT’lere klor atomu katkılanması, zigzag KNT’lerin iletkenlik özelliğini
azaltmaktadır. Ayrıca klor katkılanan nanotüplerde, azot ve oksijen katkılanan
nanotüplere göre daha fazla valans bandının Fermi enerji seviyesinin altına indiği
görülmetedir. Dolayısıyla nanotüplere klor katkılanması iletkenlik özelliğinin hızlı

4. BULGULAR VE TARTIŞMA ERKAN TETİK
136
bir şekilde azalmasına neden olmaktadır. Hatta katkılama yüzdesi arttıkça elektronik
yapı hesaplamarında kullandığımız -4, +4 enerji aralığının 0 ile -4 bölümün
tamamında Fermi seviyesinin altına inmiş iletim bantları yer almaktadır. Bu durum
%25 klor atomu katkılama sonuçlarının yer aldığı şekil 4.49’daki durum yoğunluğu
grafiğindeki mavi bölge olarak görülmektedir.

5. SONUÇLAR VE ÖNERİLER ERKAN TETİK
137
5. SONUÇLAR VE ÖNERİLER
Karbon nanotüplerin fiziği, 1991 yılında ÇDKNT ve iki yıl sonra da
TDKNT’lerin keşfedilmesiyle başlamış ve önemli bir araştırma alanı olarak hızlı bir
şekilde gelişmektedir. TDKNT’lerin elde edilmesi, karbon nanotüplerin gelişmesinde
büyük bir aşama olmuştur. Çalışmamızda nanotüpleri oluşturan grafen katmanının ve
TDKNT’lerin fiziksel özelliklerini inceledik. Çalışmamızı üç aşamalı olarak
gerçekleştirdik. Birinci aşamada grafen ve TDKNT’lerin geometrik yapılarını ve bu
geometrik yapının nasıl elde edildiğini araştırdık. İkinci aşamada, geometrik
yapılarını elde ettiğimiz bazı grafen ve TDKNT’lerin elektronik özelliklerini
hesapladık. Son aşamada ise seçtiğimiz bir TDKNT’ye periyodik cetveldeki bazı A
grubu atomları katkıladık. Nanotüpe katkılamak için seçtiğimiz atomları belirli
oranlarda ekleyerek, katkılanan atomun nanotüpün fiziksel özellikleri üzerindeki
etkilerini inceledik. Hesaplamalarımızı yoğunluk fonksiyonel teorisini temel alan
SIESTA paket programı ile gerçekleştirdik.
5.1. Grafen ve Karbon Nanotüplerin Geometrik Yapıları
TDKNT’lerin yapısı, grafen katmanın bir silindir etrafında nasıl sarıldığı ile
doğrudan ilişkilidir. Bir TDKNT’nin yapısı hakkındaki ilginç ve önemli bir gerçek
nanotüp eksenine göre bal peteği örgüsündeki altı atomlu karbon (altıgen yapı)
halkasının oryantasyonudur. Bu oryantasyona göre chiral, armchair ve zigzag
nanotüpler olarak isimlendirdiğimiz üç temel yapı ortaya çıkmaktadır.
TDKNT’lerdeki atom sayısı değiştikçe farklı geometriklere sahip farklı nanotüpler
ortaya çıkmaktadır. Dolayısıyla TDKNT’lerin n ve m indislerine göre (6, 3), (18,8),
(12,1) ve (14, 0) gibi per çok türü ortaya çıkmaktadır. Burada önemli olan
istediğimiz geometrik yapıdaki nanotüpü nasıl elde edebileceğimizdir. Çalışmamızın
ilk aşaması bu geometrik yapıların elde edilmesine dayanmaktadır. Bunun için n ve
m indislerini bildiğimiz bir TDKNT’nin çap, öteleme ve simetri vektörü gibi temel
parametreleri hesaplayabilmemiz ve bu değerlerden de nanotüpteki atomik
konumları türetebilmemiz gerekmektedir. Bu aşamada bu temel parametrelerin ve

5. SONUÇLAR VE ÖNERİLER ERKAN TETİK
138
atomik konumların elde etmek için bir program hazırladık. Program yazmak için
fortran programlama dilini kullandık. Programı yazmak için R. Saito ve arkadaşları
(1998) tarafından yazılan “Physical Properties of Carbon Nanotubes” isimli kitapdan
yararlandık. Program n ve m indisleri bilinen TDKNT’lerin temel parametrelerini ve
atomik konumlarını hesaplamaktadır. Atomik konumlar “.xyz” formatında bir çıktı
dosyasına kaydedilmektedir. Programın kodları EK 1’de verilmiştir.
Sonraki aşamalarda bu program farklı şekillerde geliştirilerek farklı bilgiler
elde edilebilir. Örneğin “.xyz” formatına ek olarak, farklı formatlarda atomik konum
dosyalarının çıktısı verecek şekilde geliştirilebilir. TDKNT’lerin dışında ZnO, MgO,
SbI3 gibi farklı yapıların da çıktısını verecek bir program haline getirilebilir. Hatta
gurup teorisi ile ilgili bir çalışma programa entegre edilerek, elde edilen yapıların
uzay grubu belirlenebilir. Programdan elde edilen çıktıları görüntülemek için,
programa bir molekül görüntüleyici grafik arayüzü eklenebilir.
5.2. Grafen ve Karbon Nanotüplerin Elektronik Özellikleri
Artık istediğimiz geometrik yapıdaki TDKNT’yi elde edebiliyor olmamız
çalışmamızın ikinci aşamasına geçmemize olanak tanımaktadır. Çalışmamazın ikinci
aşaması grafen ve TDKNT’lerin elektronik özelliklerinin araştırılmasına
dayanmaktadır. Öncelikle elektronik özelliklerini araştıracağımız TDKNT’leri
belirledik. Belirlediğimiz nanotüpler zigzag (6, 0), zigzag (7, 0) chiral (6, 2), chiral
(6, 3) ve armchair (7, 7) olarak sıralanmaktadır. Bu yapılar şekil 4.1’de
görülmektedir. Bu yapıları belirlerken nanotüplerin üç temel yapısı olan zigzag,
chiral ve armchair durumları dikkate aldık.
Öncelikle EK 1’de verilen program yardımıyla istediğimiz yapıları elde ettik.
Bu bölümle ilgili hesaplamalarımız grafen ve nanotüp yapılardan oluşmaktadır.
Üzerinde çalıştığımız grafenler nanotüplerin silindir şeklini almamış halleridir. Elde
ettiğimiz yapıları SIESTA programını kullanarak optimize ettik. Daha sonra bu
optimize yapıların elektronik özelliklerini hesapladık. Seçtiğimiz grafen ve bu
grafenin bir silindir etrafında sarılmasıyla elde edilen nanotüpün elektronik
özellikleri inceleyerek döndürmenin etkilerini araştırdık.

5. SONUÇLAR VE ÖNERİLER ERKAN TETİK
139
5.2.1. Zigzag (6, 0) Grafen ve Nanotüpün Sonuçları
Grafenin birim hücresinde iki tane karbon atomu bulunmaktadır. Grafendeki
atomları x-ekseni boyunca 24 atoma kadar periyodik olarak arttırıldığında, 24 atomlu
süper örgüye sahip bir grafen elde edilir. Bu 24 atomlu grafen bir silindir etrafında
sarıldığında zigzag (6, 0) TDKNT oluşur. Hesaplamalarımız sonucunda zigzag (6, 0)
nanotüp için 퐶 = 14,8945 A° ve çapı 4,74106 A° olarak elde ettik. İletim bandının
minimum enerji değeri A-Г simetri noktaları arasında ve Г noktasına daha yakın
olmak üzere -0,1556 eV, valans bandının maksimum enerji değeri ise K-Г simetri
noktaları arasında ve Г noktasına daha yakın olmak üzere 0,4101 eV olarak bulduk.
Zigzag (6, 0) nanotüpün elektronik bant yapısı ve durum yoğunluğu grafiğini
incelediğimizde, valans ve iletim bantlarının her ikisi de grafen katmanının
döndürülmesinden etkilenmektedir. Fakat iletim bantları nanotüpün eğriliğinden
daha fazla etkilendiği görülmektedir. Bu durum da deneysel verilerle uyum
içersindedir. Grafenin bant yapısında bazı valans bantları Fermi seviyesinin üzerine
çıktığını gördük. Bunun sebebi grafene x-ekseni boyunca eklenen atomlardır. Bu
şekilde eklenen atomlar grafenin yapısında örgü kusuru oluşmasına neden olmuştur.
Çalışmamızda zigzag (6, 0) nanotüpü metalik olarak elde ettik. Yapılan deneysel
çalışmalarda zigzag (6, 0) nanotüpün metalik özellik gösterdiği ortaya çıkmıştır.
Buradaki çalışmamız da deneysel çalışmalarla uyum içindedir.
5.2.2. Zigzag (7, 0) Grafen ve Nanotüpün Sonuçları
Zigzag (7, 0) grafeni elde edebilmek için, zigzag (6, 0) grafene x-ekseni
boyunca 4 atom daha ekledik. Bu durumda zigzag (7, 0) grafen için 퐶 = 17,3374 A°
ve çap 5,51868 A° olarak hesapladık. Zigzag (7, 0) grafenin süper hücre yapısından
dolayı, zigzag (6, 0) grafende olduğu gibi bazı valans bantlarının bu yapıda da Fermi
enerji seviyesinin üzerine çıktığı gördük. Yani zigzag (7, 0) grafende de örgü
kusurları oluşmaktadır. Grafen yapılardan nanotüpler elde edildiğinde bu örgü
kusurlarının yok olduğunu gördük. Elektronik bant yapısı ve durum yoğunluğu
grafiklerinden, zigzag (7, 0) nanotüpün yarıiletken özellik gösteren bir malzeme

5. SONUÇLAR VE ÖNERİLER ERKAN TETİK
140
olarak bulduk. Yarıiletken yasak bant aralığını ise E = 0,5022 eV olarak elde ettik.
Dolayısyla 퐶 değerinin değişmesiyle nanotüplerin elektronik özelliğinin de
değiştiğini gözlemledik.
5.2.3. Armchair (7, 7) Grafen ve Nanotüpün Sonuçları
Armchair (7, 7) grafen yapıyı elde etmek için 퐶 vektörünü, zigzag (7, 0)
grafene göre 30 değiştirdik. Armchair (7, 7) TDKNT’yi ise yine bu grafenin bir
silindir etrafında sararak oluşturduk. Yeni yapının chiral vektörünü 29,9179 A° ve
çapını 9,52316 A° olarak elde ettik. Elde ettiğimiz bant yapısı ve durum yoğunluğu
grafiklerinden, Fermi enerji seviyesinde Г noktasında nanotüpün valans ve iletim
bantları çakıştığını gördük. Dolayısıyla armchair (7, 7) nanotüpün metalik özellik
gösteren bir TDKNT olarak elde ettik.
5.2.4. Chiral (6, 2) Grafen ve Nanotüpün Sonuçları
Chiral (6, 2) grafeni elde edebilmek için zigzag (6, 0) grafenin chilal açısını
13,89° döndürdük. Bu yapının chiral vektörünü 17,8649 A° ve çapını 2,84328 A°
olarak elde ettik. Bu nanotüpün birim hücresinde 104 atom vardır. Grafenin iletim
bandının minimumu A noktasında ve 0,0934 eV, valans bandının maksimumu Г
noktasında ve 0,0031 eV değerini almıştır. Chiral (6, 2) grafenin yasak bant aralığını
0.0965 eV olarak elde ettik. Dolayısıyla chiral (6, 2) grafeni çok küçük yasak bant
aralığına sahip bir yarıiletken olarak elde ettik. Ayrıca chiral (6, 2) grafende diğer
süper hücreli grafende oluşan örgü kusurları oluşmadığını gördük. Bu da chiral (6, 2)
grafeni süper hücre yapmak için atom sayısını arttırıken seçilen doğrultunun, x ve y
ekseninin bileşke vektörüne yakın doğrultuda seçilmesinden kaynaklandığını
dşünüyoruz. Bu KNT’nin iletim bandının minimumu A noktasında ve 0,4091 eV,
valans bandının maksimumu Г noktasında ve -0,42 eV değerindedir. Dolayısıyla
chiral (6, 2) nanotüp yarıiletkendir ve yasak bant aralığı E = 0,8291 eV olarak elde

5. SONUÇLAR VE ÖNERİLER ERKAN TETİK
141
ettik. Chiral (6, 2) nanotüpün diğer nanotüplere göre oldukça büyük bir dolaylı yasak
bant aralığına sahip olduğunu söyleyebiliriz.
5.2.5. Chiral (6, 3) Grafen ve Nanotüpün Sonuçları
Son olarak chiral (6, 3) grafen ve nanotüp ile ilgili hesaplamalar yaptık. Bu
yapı chiral (6, 2) ile benzer bir geometrik yapıya sahiptir. Sadece chiral vektörünün
döndürme açısı olan chiral açısı farklılık göstermektedir. Chiral (6, 3) grafenin chiral
açısı 19.11° olarak döndürülmüştür. Bu farklılık chiral (6, 3) atom sayısını da chiral
(6, 2) göre değiştirmiştir. Chiral (6, 3) nanotüp 84 atoma sahiptir. Chiral (6, 3)
grafenin dolaylı yasak bant aralığına sahip olduğunu ve bant aralığının da
E = 0,0516 eV civarında olduğunu görüyoruz. Bu değer (6, 2) grafenden daha
düşüktür. Chiral (6, 3) nanotüpü incelediğimizde yasak bant aralığının
E = 0,0488 eV olarak elde ettik. Bu yapı için ietim bandının minimum değeri A-Г
noktaları arasında, Г noktasına daha yakındır ve 0,0237 eV, valans bandının
maksimum değeri ise A-Г noktaları arasında, Г noktasına daha yakındır ve -0,0251
eV civarında olduğunu görüyoruz. Bu durumda chiral (6, 3) nanotüpün düşük
bant aralığını sahip bir sankimetalik (quasimetallic) nanotüp olduğunu
söyleyebiliriz.
5.3. Karbon Nanotüplerin Katkılama İle İlgili Sonuçları
Yaptığımız elektronik yapı hesaplamarında TDKNT’lerin elektronik
özelliklerinin tüpün 퐶 açısına ve çapına hassas bir şekilde bulduk. TDKNT’leri
nano-elektronik cihazlarda bir eleman olarak kullanabilmek için, tüpün yarıçapını ve
퐶 açısını kontrol edebilen bir yol bulmak gerekmektedir. Bunu geometrik yapıyı
değiştirerek yapmak oldukça zor ve zahmetli işleri beraberinde getirir. Bu işleme
alternatif olarak KNT’lere yabancı atom ekleyerek yani katkılama işlemi yaparak,
elektronik özelliklerde değişiklik yapma yöntemi de kullanılabilir. Bu konuyu
incelemek için seçtiğimiz bir TDKNT üzerinde katkılama çalışması yaptık. Bu

5. SONUÇLAR VE ÖNERİLER ERKAN TETİK
142
çalışmada yarıiletken zigzag (8, 0) nanotüpü kullandık. Öncelikle herhangi bir
yabancı atom eklemeden (8, 0) nanotüpü optimize ettik. Daha sonra optimize yapının
elektronik bant yapısı ve durum yoğunluğu grafikleri elde ettik. Hesaplama
sonuçlarımızda, zigzag (8, 0) nanotüpün 퐶 = 19,785 A° ve çap 3, 14889 A° olarak
bulduk. Bu nanotüp için yasak bant aralığını ise E = 0,6643 eV olarak elde ettik.
Dolayısıyla zigzag (8, 0) nanotüp yarıiletken özellik göstermektedir. Daha sonra
periyodik cetveldeki A grubu elementlerinden karbon atomuna yapısal olarak en
yakın atomları seçip katkılama yaptık. Katkılama yaptığımız bu atomlar berilyum,
bor, silikon, azot, oksijen ve klor atomlarıdır. Katkılama işlemini her atom için
sırasıyla %3,125, %6,250, %12,5 ve %25 oranında yaptık.
5.3.1. Zigzag (8, 0) Nanotüpe Bor Katkılama Sonuçları
Bor atomu periyodik cetvelde bulunduğu konum itibarıyla karbon atomuna
yakın bir atomdur. Çapı da karbon atomuna yakın olduğundan, KNT’lere en çok
katkılama yapılan yabancı atomlardan biridir diyebiliriz. Bor atomu eklenen
KNT’lerin sonuçlarını incelediğimizde, eklenen bor atomu sayısı arttıkça daha fazla
valans bandının Fermi seviyesinin üzerine çıktığını görüyoruz. Ayrıca eklenen bor
atomu sayısı arttıkça, katkılama yapılmamış zigzag (8, 0) KNT’de var olan yasak
bant aralığının da arttığını görüyoruz. Valans bantlarının Fermi seviyesinin üzerine
çıkması sonucunda nanotüpün serbest elektronlarının sayısı artmaktadır. Bu durumda
nanotüpün iletkenlik özelliği artmaktadır. Sonuç olarak zigzag KNT’lere bor atomu
katkılanması, zigzag KNT’lerin iletkenlik özelliğini arttırmaktadır.
5.3.2. Zigzag (8, 0) Nanotüpe Berilyum Katkılama Sonuçları
Berilyum atomu periyodik cetvelde bulunduğu konum itibarıyla karbon
atomuna uzak bir konumdadır. Berilyum toprak alkali metal ailesindendir ve bu
özelliğini katkılama yapılan nanotüpte baskın olarak göstermiştir. Berilyum atomu
eklenen KNT’lerin sonuçlarını incelediğimizde, eklenen berilyum atomu sayısı
arttıkça bor katkılanan nanotüplere göre daha fazla valans bandının Fermi

5. SONUÇLAR VE ÖNERİLER ERKAN TETİK
143
seviyesinin üzerine çıktığını görüyoruz. Dolayısıyla berilyum katkılanan
nanotüplerde serbest elektronların sayısı daha fazladır. Bu nanotüplerde iletkenlik
bor katkıllanan nanotüplere göre daha fazla artmaktadır. Sonuç olarak, berilyum
katkılanan KNT’ler bor katkılananlara göre daha fazla iletken özellik gösterir.
5.3.3. Zigzag (8, 0) Nanotüpe Azot Katkılama Sonuçları
Azot atomu periyodik cetvelde 5A gurubunda ve 2. periyottadır. KNT’lere
sık katkılanan bir atomdur. Azot atomu eklenen KNT’lerin sonuçlarını göre, bor ve
berilyum eklenen nanotüplerin aksine, eklenen azot atomu sayısı arttıkça daha fazla
iletim bandının Fermi seviyesinin altına indiğini görüyoruz. Azot atomu katkılanan
nanotüplerde iletim bandı Fermi enerjisinin altına indiği için, bu nanotüplerin serbest
elektronların sayısı gittikçe azalmaktadır. Bu durumda azot eklenen nanotüplerin
iletkenlik özelliği azalmaktadır.
5.3.4. Zigzag (8, 0) Nanotüpe Silisyum Katkılama Sonuçları
Silisyum atomu karbon atomu ile aynı guruptadır. Silisyum atomu eklenen
KNT’lerin sonuçlarını bakarsak, bor, berilyum ve azot eklenen nanotüplerin aksine,
valans ve iletim bantlarının Fermi seviyesinin ne üstüne çıkmıştır ne de altına
inmiştir. Silisyum katkılanan nanotüpler, katkılama yapılmamış zigzag (8, 0)
nanotüpte olduğu gibi yarıiletken yasak bant aralığına sahiptirler. Fakat yasak bant
aralığı silisyum atomunun katkılama yüzdesi arttıkça artmaktadır. Bu nanotüplerde
katkılama yüzdesine göre yasak bant aralığı sırasıyla 0,4529 eV, 0,4503 eV, 0,4929
eV ve 0,9292 eV değerlerini almaktadır. Katkılama yüzdesinin az olduğu durumlarda
yasak bant aralğında ciddi bir değişiklik olmamıştır. Ancak katkılama yüzdesi
arttığında silisyum atomu baskın olduğunu göstermiş ve yasak bant aralığını
arttırmıştır.

5. SONUÇLAR VE ÖNERİLER ERKAN TETİK
144
5.3.5. Zigzag (8, 0) Nanotüpe Oksijen Katkılama Sonuçları
Oksijen atomu periyodik cetvelde bulunduğu konum itibarıyla karbon
atomundan biraz uzaktır ve atomik çapı karbon atomundan küçüktür. Oksijen atomu
eklenen KNT’lerden elde ettiğimiz sonuçlara göre, eklenen oksijen atomu sayısı
arttıkça daha fazla iletim bandının Fermi seviyesinin altına indiğini görüyoruz. Fermi
seviyesinin altına inen iletim bantların sayısı, azot katkılanan nanotüplerden daha
fazladır. Oksijen atomu katkılanan nanotüplerde iletim bandı Fermi enerjisinin altına
indiği için, bu nanotüplerin serbest elektronların sayısı gittikçe azalmaktadır. Bu
durumda oksijen eklenen nanotüplerin iletkenlik özelliği azot etkenen nanotüplere
göre daha fazla azalmaktadır. Ayrıca %25 oranında oksiyen katkılanan nanotüpün
Fermi seviyesinde ikinci bir bant aralığı oluşmaktadır.
5.3.6. Zigzag (8, 0) Nanotüpe Klor Katkılama Sonuçları
Halojen olan klor atomu periyodik çetvelde 7A grubundadır ve konum
itibarıyla karbon atomundan uzaktadır. Klor atomu eklenen KNT’lerde, eklenen klor
atomu sayısı arttıkça daha fazla iletim bandının Fermi seviyesinin altına indiğini
görüyoruz. Klor atomu katkılanan nanotüplerde iletim bandı Fermi enerjisinin altına
indiği için, bu nanotüplerde de serbest elektronların sayısı gittikçe azalmaktadır. Bu
durumda klor eklenen nanotüplerde de iletkenlik özelliği azalmaktadır. Bu
KNT’lerde, katkılama yüzdesi arttıkça elektronik yapı hesaplamarında kullandığımız
-4, +4 enerji aralığının 0 ile -4 bölümün tamamında Fermi seviyesinin altına inmiş
iletim bantları yer almaktadır. Dolayısyla klor katkıladığımızda, hızlı bir şekilde
iletim bantlarının Fermi seviyesinin altına indiğini görüyoruz.
5.4. Karbon Nanotüple İlgili Teorik Çalışma Önerileri
Yaptığımız çalışmanın ikinci kısmında grafen ve TDKNT’lerin TDKNT’lerin
elektronik özelliklerini elektronik inceledik. Üçüncü kısmında ise TDKNT’lere
katkılamanın elektronik özellikler üzerine etkilerini kapsamlı bir şekilde inceledik.

5. SONUÇLAR VE ÖNERİLER ERKAN TETİK
145
TDKNT’lerin elektronik özelliklerinin yanı sıra optik ve mekanik gibi özellikleri de
büyük önem taşımaktadır. Örneğin karbon nanotüp fiberler çok geniş yüzey alanına
sahiptir. Nanotüp fiberin kütlesiyle alanı arasındaki oran, normal malzemelere göre
çok daha büyüktür. Karbon nanotüp fiberlerin bu özelliği nanometre düzeyinde süper
kapasitörler elde edilmesine olanak tanır. Ayrıca KNT’ler hafiftir ve çok yüksek bir
elastik modülüne sahiptirler. İlginç mekanik özellikleriyle KNT’ler, malzeme
endüstrisini tamamıyla değiştirebileceği düşünülmektedir. Bundan sonraki
çalışmalarda TDKNT’lerin optik ve mekanik özellikleri üzerine teorik çalışmalar
yapılabilir. Bu çalışmalar gelecekte yapılacak olan KNT tabanlı projelere önemli
ölçüde fayda sağlayacaktır.
KNT’ler geniş bir yüzey alanına sahiptir. Bu özelliğin onlara hidrojen
depolama özelliği kazandırdığı düşünülmektedir. Karbon nanotüpler bu özelliği ile
potansiyel enerji depolama malzemesi adayı konumundadır. Dolayısıyla KNT’lerle
ilgili hidrojen depolama çalışmaları da önemli bir araştırma konusudur.
Bu çalışmada KNT’lere yabancı atom katkılamanın sadece elektronik
özelliklere etkisi üzerinde çalıştık. Sonraki çalışmalarda, katkılamanın optik ve
elastik özellikler özerindeki etkileri de çalışılabilir.

146
KAYNAKLAR
BECKE, A. D., 1988. Density-functional exchange-energy approximation with
correct asymptotic behavior. Phys. Rev. A, 38: 3098-3100.
BARBERIO M., BARONE, P., BONANNO, A., CAMARCA, M., MASCIARI, E.,
OLIVA, A., and XU, F., 2009. Transport properties of alkali-doped single-
wall carbon nanotube mats. Superlattices and Microstructures, 46: 369-373.
BORN, M., and OPPENHEIMER , R., 1927. Zur Quantentheorie der Molekeln. Ann.
Physik, 84: 457-484.
BOROWIAK-PALEN, E., PICHLER, T., GRAFF, A., KALENCZUK, R.J.,
KNUPFER, M., and FINK, J., 2004. Synthesis ve electronic properties of B-
doped single wall carbon nanotubes. Carbon, 42: 1123–1126.
CEPERLEY, D M., ALDER, M. J., 1980. Ground State of The Electron Gas by A
Stochastic Method. Phys. Rev. Lett., 45: 566-569.
CHARLES, P., and FRANK, J., 2003. Introduction of Nanotechnology. A-Wiley
Interscience Publication, United States of America.
COCHRAM, W., CRICK, F. H. C., and VAND, V., 1952. The structure of synthetic
polypeptides. I. The transform of atoms on a helix. Acta Cryst., 5: 581-586.
COWLEY, J. M., NIKOLAEV, P., THESS, A., and SMALLEY, R. E., 1997.
Electron nano-diffraction study of carbon single-walled nanotube ropes.
Chem. Phys. Lett., 265: 379-384.
DIRAC, P. A. M., 1930. Note on Exchange Phenomena in The Thomas-Fermi Atom.
Proc. Cambridge Philos. Soc., 26: 376-385.
DİKİCİ, M., 1993. Katıhal Fiziğine Giriş. Ondokuz Mayıs Üniversitesi Yayınları,
Samsun, 276s.
DRESSELHAUS, M. S., and DRESSELHAUS, G., 1981. Intercalation Compounds
of Graphite. Advances in Phys. 30: 139-326.
DRESSELHAUS, M. S., JISHI, R. A., DRESSELHAUS, G., INOMATA, D.,
NAKAO, K., and SAITO, R., 1994. Group theoretical concepts for carbon
nanotubes. Molecular Materials, 4: 27-40.

147
ENDO, M., Takeuchi, K., Kobori, K., Takahashi, K., Kroto, H., ve Sarkar, A., 1995.
Pyrolytic carbon nanotubes from vapor-grown carbon fibers. Carbon, 33:
873-881.
FALVO, M. R., CLARY, G. J., TAYLOR II, R. M., CHI, V., BROOKS, F. P.,
WASHBURN, S., and SUPERFINE, R., 1997. Bending and buckling of
carbon nanotubes under large strain. Nature, 389: 582-584.
FERMI, E., 1928. A Statistical Method for The Determining of Some Properties of
The Atoms. II. Application to The Periodic System of The Elements. Z.
Phys., 48: 73-79.
FEYNMAN, R. P., 1939. Forces in Molecules. Phys. Rev., 56: 340-343.
GAL’PERN, E. G., STANKEVICH, I. V., CHISTYKOV, A. L., and CHERNOZA-
TONSKII, L. A., 1993. Carbon nanotubes with metal inside: electron
structure of tubelenes [Li@C24]n and [K@C36]n. Chem. Phys. Lett. 214:
345-348.
HARRIS, J., and JONES, R. O., 1974. The Surface Energy of a Bounded Electron
Gas. J. Phys. F, 4: 1170-1186.
HATREE, D. R., and FOCK, V., 1957. Represetation of The Exchange Terms in
Fock’s Equation by A Quasi-Potantiel. Phys. Rew., 107: 1631-1639.
HELLMANN, H., 1937. Einführung in die Quantenchemie (Introduction to Quantum
Chemistry). Deuticke, Leipzig and Wien, 350s.
HEREMANS, C. H., HEREMANS, J. P., and MATER, J., 1994. Scanning tunneling
spectroscopy of carbon nanotubes. Res., 9: 259-262.
GORLING, A., and LEVY, M., 1992. Requirements for Correlation Energy Density
Functionals from Coordinate Transformations. Rev. A, 45: 1509-1517.
GUNNARSSON, O., and LUNDQVIST, B. I., 1976. Exchange and Correlation in
Atoms, Molecules, and Solids by The Spin-Density-Functional Formalism.
Phys. Rev. B, 13: 4274-4298.
HOHENBERG, P., and KOHN, W., 1964. Inhomogeneous Electron Gas. Phys. Rev.,
136: A1133-A1138.
IIJIMA, S., 1991. Helical Microtubules of Graphitic Carbon. Letters to Nature, 354:
56-58.

148
IIJIMA, S., and ICHIHASHI, T., 1993. Single-shell Carbon Nanotubes of 1-nm
Diameter, Letters to Nature, 363: 603-605.
INTERNET: 2011, The Spanish Initiative for Electronic Simulations with Thousands
of Atoms. http://www.uam.es/siesta.
INTERNET: 2011, X-Window Crystalline Structures and Densities (XCrySDen).
http://www.xcrysden.org.
JONES, R. O., and GUNNARSSON, O., 1989. The Density Functional Formalism,
its Applications and Prospects. Rev. Mod. Phys., 61: 689-746.
JOURNET, C., MASER, W. K., BERNIER, P., LOISEAU, A., LAMY DE LA
CHAPELLE, M., LEFRANT, S., DENIARD, P., LEE, R., and FISCHER, J.
E., 1997. Large-scale production of single-walled carbon nanotubes by the
electric-arc technique. Nature, 388: 756- 758.
KIANG, C. H., GODDARD, W. A., BEYERS, R., and BETHUNE, D. S., 1995.
Carbon Nanotubes With Single-Layer Walls. Carbon, 33: 903-914.
LAMBIN, P. and LUCAS, A. A., 1997. Quantitative theory of diffraction by carbon
nanotube. Phys. Rev. B, 56: 3571-3573.
LANGERTH, D. C., and PERDEW, J. P., 1975. The Exchange-Correlation Energy
of a Metallic Surface. Soid State Commun., 17: 1425-1429.
LANGERTH, D. C., and PERDEW, J. P., 1977. The Exchange-Correlation Energy
of a Metallic Surface: Wave-Vector Analysis. Phys. Rev. B, 15: 2884-2901.
LEVY, M., 1979. Universal Variational Functionals of Electron Densities, First-
Order Density Matrices, and Natural Spin-Orbitals and Solution of Te v-
Representability Problem. Proc. Natl. Acad. Sci. Of USA, 76:, 6062-6065.
LEVY, M., and PERDEW, J. P., 1985. Hellmann-Feynman, Virial, and Scaling
Requisites for The Exact Universal Density Functionals. Shape of The
Correlation Potential and Diamagnetic Susceptibility for Atoms. Phys. Rev.
A, 32: 2010-2021.
LEVY, M., 1989. Asymptotic Coordinate Scaling Bound For Exchange - Correlation
Energy in Density - Functional Theory. Int. J. Quantum Chem. Symp., 23:
617-623.

149
LEVY, M., 1991. Density-Functional Exchange Correlation Through Coordinate
Scaling in Adiabatic Connection and Correlation hole. Phys. Rev. A, 43:
4637-4646.
LEVY, M., and PERDEW, J. P., 1993. Tight Bound and Convexity Constraint on
The Exchange-Correlation Energy Functional in The Low-Density Limit, and
Other Formal Test of Generalized-Gradient Approximations. Phys. Rev. B,
48: 11638-11645.
LEVY, M., 1995. Density-Functional Theory (E. K. Gross, R. M. Dreizler Editörler).
Plenum, New York, 696s.
LIEB, E. H., 1981. Thomas-Fermi and Related Theories of Atoms and Molecules.
Rev. Mod. Phys., 53: 603-641.
MINTNIRE, J. W., and WHITE, C. T., 1995. Electronic and structural properties of
carbon nanotubes. Carbon, 33: 893-902.
OLIVER, G. L., and PERDEW, J. P., 1979. Spin-Density Gradient Expansion for
The Kinetic Energy. Phys. Rev. A, 20: 397-403.
OLK, C. H., HEREMANS, J. P., and MATER, J., 1994. Scanning tunneling
spectroscopy of carbon nanotubes. Res, 9: 259-262.
ORDEJON, P., ARTACHO, E., and SOLER, J. M., 1996. Self-Consistent Order-N
Density Functional Calculations for Very Large Systems. Phys. Rev. B
(Rapid Comm.), 223: 411-415.
PAYNE, M. C., TETER, M. P., ALLAN, D. C., ARIAS, T. A., and
JOANNOPOULOS, J. D., 1992. Iterative Minimization Techniques for ab
initio Total-Enerji Calculation: Moleculer Dynamics and Conjugate
Gradients. Rev. Mod. Phys., 64: 1045-1097.
PERDEW, J. P. and WANG, Y., 1992. Accurate and simple analytic representation
of the electron-gas correlation energy. Phys. Rev. B, 45: 13244-13249.
PERDEW, J. P., SHEVAY, J. A., VOSKO, S. H., JACKSON, K. A., PEDERSON,
M. R., SINGH, D. J. and FIOLHAIS, C., 1992. Atoms, molecules, solids, and
surfaces: Applications of the generalized gradient approximation for
exchange and correlation, Phys. Rev. B, 46: 6671-6687.

150
PERDEW, J. P., BURKE, K. and WANG, Y., 1996. Generalized gradient
approximation for the exchange-correlation hole of a many-electron system.
Phys. Rev. B, 54: 16533-16539.
PERDEW, J. P., BURKE, K. and ERNZENHOF M., 1996. Generalized Gradient
Approximation Made Simple. Phys. Rev. Lett., 77: 3865-3868.
REICH, S., ve THOMSEN, C., 2002. Electronic band structure of isolated and
bundled carbon nanotubes. Phys. Rev. B, 65: 155411- 155422.
REICH, S., THOMSEN, C., and MAULTZSCH, J., 2004. Carbon Nanotubes. Betz-
Druck GmbII, Darmstadt, 211s.
SAITO, R., DRESSELHAUS, G., and DRESSELHAUS, M. S., 1998. Physical
Properties of Carbon Nanotubes. Imperial College Press, London, 274s.
SAITO, R., FUJITA, M., DRESSELHAUS, G., and DRESSELHAUS, M. S., 1992.
Electronic Structure of Graphene Tubules Based on C60. Phys. Rev. B, 46:
1804-1811.
SATTLER K., 1995. Scanning Tunneling Microscopy of Carbon Nanotubes and
Nanocones. Carbon, 33, 915-920.
SCUSERIA, G. E., and STAROVEROV, V. N., 2005. Progress in The Development
of Exchange-Correlation Functionals in: Theory and Applications of
Computational Chemistry: The First 40 Years (C. E. Dykstra, G. Frenking, K.
S. Kim and G. E. Scuseria Editörler), Elsevier, Amsterdam, 1336s.
SOLER, J. M., ARTACHO, E., GALE, J. D., GARCIA, A., JUNQUERA, J.,
ORDEJON, P., and PORTAL D. S., 2002. The SIESTA Method for ab initio
Order-N Materials Simulation. J. Phys. Condens. Matter, 14: 2745-2779.
SUENAGA, K., COLLIEX, C., DEMONCY, N., LOISEAU, A., PASCARD, H., and
WILLAIME, F., 1997. Synthesis of Nanoparticles and Nanotubes with Well-
Separated Layers of Boron Nitride and Carbon. Science 278: 653-658.
TANS, S. J., DEVORET, M. H., DAI, H., THESS, A., SMALLEY, R. E.,
GEERLIGS, L. J., and DEKKER, C., 1997. Individual single-wall carbon
nanotubes as quantum wires. Nature 386: 474-477.
THESS, A., LEE, R., NIKOLAEV, P., DAI, H., PETIT, P., ROBERT, J., XU, C.,
LEE, Y. H., KIM, S. G., RINZLER, A. G., COLBERT, D. T., SCUSERIA,

151
G. E., TOMRINET, D., FISCHER, J. E., and SMALLEY, R. E., 1996.
Crystalline Ropes of Metallic Carbon Nanotubes. Science, 273: 483-487.
WIRTZ., L., and RUBIO, A., 2004. Band structure of boron doped carbon
nanotubes AIP Conference Proceedings - Electronic Properties of Novel
Materials, 685: 402-405.
YANG, H. Ou,, and LEVY, M., 1990a. Nonuniform Coordinate Scaling
Requirements in Density-Functional Theory. Phys. Rev. A, 42: 155-160.
YANG, H. Ou,, and LEVY, M., 1990b. Nonuniform Coordinate Scaling
Requirements for Exchange-Correlation Energy. Phys. Rev. A, 42: 651-652.
ZHOU , 0., FLEMING, R. M., MURPHY, D. W., CHEN, C. H., HADDON, R. C.,
RAMIREZ, A. P., and GLARURN, S. H., 1994. Defects in Carbon
Nanostructures. Science, 263: 1744-1747.
ZHOU, Z., XUEPING, G., YAN, J., and SONG, D., 2006. Doping effects of B and
N on hydrogen adsorption in single-walled carbon nanotubes through density
functional calculations. Carbon, 44: 939–947.

152
ÖZGEÇMİŞ
1983 yılında Uşak’da dünyaya geldi. İlkokulu Fatih İlköğretim okulunda ve
ortaokulu Mehmet Sadık Boz İlköğretim Okulunda tamamladı. 1999 yılında Uşak
Atatürk Lisesinde mezun olarak lise öğrenimimi tamamladı. 1999 yılında
Gaziosmanpaşa Üniversitesi Fen-Edebiyat Fakültesinde üniversite öğrenimine
başladı. Üniversite öğrenimi esnasında bilgisayar programlama konusunda da eğitim
alarak yazılım konusunda kendini geliştirdi. 2003 yılında üniversiteden mezun oldu.
Mezun olduktan sonra Eşme Çok Programlı Devlet Lisesinde bir yıl matematik
öğretmeni olarak çalıştı. Daha sonra öğretmenliği bırakıp Konya teknoloji geliştirme
merkezinde yazılım uzmanı olarak çalışmaya başladı. Burada PHP, MySQL, C++,
JAVA ve C# gibi programlama dillerinde kendini geliştirdi. Çalışırken aynı zamanda
Selçuk Üniversitesi’nde tezsiz yüksek lisans öğrenimini tamamladı. 2006 yılında
teknoloji geliştirme merkezindeki işini bırakarak askerlik görevine başladı. 2007
yılında askerliğini tamamladıktan sonra Çukurova Üniversitesi Fen-Edebiyat
Fakültesi Fizik Bölümünde lisansüstü doktoraya başladı ve halen devam etmektedir.

153
EKLER
EK 1
Karbon Nanotüpler İçin Parametre Hesabı
Aşağıdaki kodlar fortran programlama dilinde yazılmıştır. Programı herhangi
bir fortran derleyici kullanarak derleyebilirsiniz. Program Linux ve Windows işletim
sistemlerinde kullanılabilir.
PROGRAM nanotup C IMPLICIT REAL*8(a-h,o-z) PARAMETER (nk=20000,aa=1.42) DIMENSION x(nk),y(nk),z(nk) DIMENSION iic(nk),ic(nk,3),iz(nk,3) C WRITE(*,*) 'n ve m degerlerini giriniz. Ornek: 10 5' READ(*,*) n,m WRITE(*,*) 'Birim hucre sayısını giriniz.' READ(*,*) nu CALL UNITCEL(n,m,np,nq,ndr) C OPEN(61,file='unitube.d') READ(61,*) nn READ(61,*) t DO 10 i=1,nn READ(61,*) j,x(i),y(i),z(i) 10 CONTINUE CLOSE(61,status='keep') C WRITE(*,*) 'Toplam Atom Sayisi = ',nu*nn IF(nu*nn.GT.nk) Stop 'nk yi daha buyuk bir degerle degistirin.' C ii=nn DO 20 i=1,nn DO 21 jj=0,nu-1 iii=ii+i+jj*ii z(iii)=z(i)+dfloat(jj+1)*t x(iii)=x(i) y(iii)=y(i) 21 CONTINUE 20 CONTINUE C OPEN(60,FILE='nanotup.xyz') WRITE(60,*) nu*nn

154
WRITE(60,*) ' ' DO 100 i=1,nu*nn WRITE (60,700) x(i),y(i),z(i) 100 CONTINUE 700 FORMAT('C ',3F10.5) CLOSE(60) C STOP END C ********************************************************************** SUBROUTINE GEN11(n,m,np,nq,ndr) C C C dimension nnp(1000), nnq(1000) C itest=1 itest1=0 C nd=igcm(n,m) IF(mod((n-m),3*nd).EQ.0) THEN ndr=3*nd ELSE ndr=nd ENDIF IF(itest.EQ.1) WRITE(*,21) n,m 21 FORMAT(' C_h = (',I3,',',I3,')',' : CHIRAL VEKTOR') IF(itest.EQ.1) WRITE(*,22) nd 22 FORMAT(' d = ',I3) IF(itest.EQ.1) WRITE(*,23) ndr 23 FORMAT(' d_R = ',I3) C a=sqrt(3.0)*1.42 eps=1.0E-5 C L l2=n*n+m*m+n*m If(l2.LE.0) stop 'l2.LE.0' l=int(sqrt(dfloat(l2))+eps) IF((l2-l**2).EQ.0) THEN IF(itest.EQ.1) WRITE(*,31) l 31 FORMAT(' L/a = ',I4) ELSE

155
IF(itest.EQ.1) WRITE(*,32) l2 32 FORMAT(' L/a = karekok',I6) ENDIF dt=a*sqrt(dfloat(l2))/3.1415926525 rt= dt*0.5 IF(itest.EQ.1) WRITE(*,33) dt,rt 33 FORMAT(' d_t = ',F10.5,' A r_t = ',F10.5,' A : CAP ', & '& YARICAP') C T nr=(2*m+n)/ndr ns=-(2*n+m)/ndr IF(itest.EQ.1) WRITE(*,41) nr,ns 41 FORMAT(' T = (',I3,',',I3,')') nt2=3*l2/ndr/ndr nt=int(sqrt(dfloat(nt2))+eps) IF((nt2-nt**2).EQ.0) THEN IF(itest.EQ.1) WRITE(*,42) nt 42 FORMAT(' T/a = ',I3) ELSE IF(itest.EQ.1) WRITE(*,43) nt2 43 FORMAT(' T/a = karekok',I6) ENDIF C N nn=2*l2/ndr IF(itest.EQ.1) WRITE(*,51) nn 51 FORMAT(' N = ',I4) C R ichk=0 IF(nr.EQ.0) THEN n60=1 ELSE n60=nr ENDIF C C itest2=0 itest2=1 C DO 61 np=-abs(n60),abs(n60) DO 62 nq=-abs(ns),abs(ns) j2=nr*nq-ns*np IF(j2.EQ.1) THEN j1=m*np-n*nq IF(itest2.EQ.1) WRITE(*,68) n,m,nr,ns,np,nq,j1,j2 68 FORMAT('n, m, nr, ns, np, nq, j1, j2 = ',6I4,2I6) IF((j1.GT.0).AND.(j1.LT.nn)) THEN ichk=ichk+1

156
nnp(ichk)=np nnq(ichk)=nq ENDIF ENDIF 62 CONTINUE 61 CONTINUE C IF(ichk.EQ.0) THEN WRITE(*,*) 'n, m, nr, ns = ', n,m,nr,ns STOP 'p ve q degerlerinde hata!!' ENDIF C itest3=1 C IF(ichk.GE.2) THEN IF(itest3.EQ.1) THEN WRITE(*,*) 'n, m, nr, ns = ',n,m,nr,ns WRITE(*,*) 'ichk = ',ichk,' ndr = ',ndr WRITE(*,67) (nnp(i),nnq(i),(m*nnp(i)-n*nnq(i)),nn,i=1,ichk) STOP 'p ve q degerlerinde hata!!' ENDIF C IF((nr.NE.0).AND.(ns.NE.0)) THEN C IF(itest1.EQ.1) THEN DO 77 i=1,ichk IF((m*nnp(i)-n*nnq(i)).LT.nn) GOTO 777 77 CONTINUE ENDIF C WRITE(*,*) 'n, m, nr, ns = ',n,m,nr,ns WRITE(*,*) 'ichk = ',ichk,' ndr = ',ndr WRITE(*,67) (nnp(i),nnq(i),(m*nnp(i)-n*nnq(i)),nn,i=1,ichk) 67 FORMAT(' (',I2,',',I2,') mp-nq = ',I3,' N = ',I3) STOP 'p ve q degerlerinde hata!!' ENDIF ENDIF 777 CONTINUE C IF(itest.EQ.1) WRITE(*,63) nnp(1),nnq(1) 63 FORMAT(' R = (',I3,',',I3,')') C M = mp - nq mmm=m*nnp(1)-n*nnq(1) WRITE(*,69) mmm 69 FORMAT(' M = ',I3) C

157
IF(itest.EQ.1) THEN np=nnp(1) nq=nnq(1) ENDIF 2 CONTINUE RETURN 1 CONTINUE STOP END C C en buyuk ortak bolen hesabi C integer function igcm(ii,jj) i=abs(ii) j=abs(jj) IF(j.GT.1) THEN iw=j j=i i=iw ENDIF IF(j.EQ.0) THEN igcm=i RETURN ENDIF 10 CONTINUE ir=mod(i,j) IF(ir.EQ.0) THEN igcm=j RETURN ELSE i=j j=ir ENDIF GOTO 10 END C ********************************************************************** SUBROUTINE UNITCEL(n,m,np,nq,ndr) C C (n,m) nanotupun birim hucredeki atomik kordinatlari C C

158
IMPLICIT REAL*8(a-h,o-z) C C acc = C-C arasindaki bag uzunlugu C PARAMETER(acc=1.42d0) PARAMETER(nk=20000) DIMENSION x(nk),y(nk),z(nk) C C CALL gen11(n,m,np,nq,ndr) C C a = birim vektor uzunlugu C pi = 3.141592 sq3=1.732 sq3=sqrt(3.0d0) pi=4.0d0*atan(1.0d0) a=sqrt(3.0d0)*acc C C r=|R|, c=|C_h|, t=|T| C r=a*sqrt(dfloat(np*np+nq*nq+np*nq)) c=a*sqrt(dfloat(n*n+m*m+n*m)) t=sqrt(3.0d0)*c/dfloat(ndr) WRITE(*,*) ' t = ',t, dfloat(n*n+m*m+n*m) C WRITE(*,190) c/2.0d0/pi C 1 C C nn: birim hucredeki hegzagon sayisi nn = 2*(n**2+m**2+n*m)/ndr C C WRITE(*,*) ' N = ',nn*2 C C rs: tupun yaricapi C IF(2*nn.GT.nk) stop 'nk cok kucuk.' C rs=c/(2.0d0*pi) C C q1: C_h icin chiral acisi C q2: R icin chiral acisi C q3: C_h ve R arasindaki aci C q1=atan((sq3*dfloat(m))/dfloat(2*n+m)) q2=atan((sq3*dfloat(nq))/dfloat(2*np+nq)) q3=q1-q2 C

159
C C q4=2.0d0*pi/dfloat(nn) q5=acc*cos((pi/6.0d0)-q1)/c*2.0d0*pi C C h1=abs(t)/abs(sin(q3)) h2=acc*sin((pi/6.0d0)-q1) C WRITE(*,*) 'q1: =',q1*180.0d0/pi,' : CHIRAL ACISI' C WRITE(*,*) 'q2: =',q2*180.0d0/pi C WRITE(*,*) 'q4: =',q4*180.0d0/pi C WRITE(*,*) 'q5: =',q5*180.0d0/pi C C C C ii=0 DO 100 i=0,nn-1 x1=0 y1=0 z1=0 k=int(dfloat(i)*abs(r)/h1) x1=rs*cos(dfloat(i)*q4) y1=rs*sin(dfloat(i)*q4) z1=(dfloat(i)*abs(r)-dfloat(k)*h1)*sin(q3) kk2=abs(int(z1/t))+1 C C C IF(z1.GT.t-0.02) THEN z1=z1-t*dfloat(kk2) ENDIF IF(z1.LT.-0.02) THEN z1=z1+t*dfloat(kk2) ENDIF ii=ii+1 x(ii)=x1 y(ii)=y1 z(ii)=z1 C WRITE(*,*) "z(ii) = ",z(ii) C C C z3=(dfloat(i)*abs(r)-dfloat(k)*h1)*sin(q3)-h2 ii=ii+1

160
C C C IF((z3.GE.-0.02).AND.(z3.LE.t-0.02)) THEN C x2=rs*cos(dfloat(i)*q4+q5) y2=rs*sin(dfloat(i)*q4+q5) z2=(dfloat(i)*abs(r)-dfloat(k)*h1)*sin(q3)-h2 x(ii)=x2 y(ii)=y2 z(ii)=z2 ELSE C x2=rs*cos(dfloat(i)*q4+q5) y2=rs*sin(dfloat(i)*q4+q5) z2=(dfloat(i)*abs(r)-dfloat(k+1)*h1)*sin(q3)-h2 kk=abs(int(z2/t))+1 IF(z2.GT.t-0.02) THEN z2=z2-t*dfloat(kk) ENDIF IF(z2.LT.-0.02) THEN z2=z2+t*dfloat(kk) ENDIF x(ii)=x2 y(ii)=y2 z(ii)=z2 ENDIF 100 CONTINUE C C C OPEN(60,file='unitup.xyz') WRITE(60,*) 2*nn WRITE(60,*) ' ' DO i=1,nn*2 WRITE(60,117) x(i),y(i),z(i) ENDDO 117 FORMAT('C',3F10.5) CLOSE(60) C C C OPEN(60,file='unitube.d') WRITE(60,*) 2*nn WRITE(60,118) t,acc 118 FORMAT(2F25.5)
161
DO i=1,nn*2 WRITE(60,116) i,x(i),y(i),z(i) ENDDO 116 FORMAT(I5,3F25.20) CLOSE(60) RETURN STOP END
EK 2
Yabancı Atom Eklenmiş Karbon Nanotüplerin Optimizas Sonuçları
Şekil 1. CNT(8, 0) için a (A)-enerji (eV) optimizasyon grafiği
Şekil 2. %3,125 bor katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon
grafiği
162
Şekil 3. %6,25 bor katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon grafiği
Şekil 4. %12,5 bor katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon grafiği
Şekil 5. %25 bor katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon grafiği

163
Şekil 6. %3,125 berilyum katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon
grafiği
Şekil 7. %6,25 berilyum katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon
grafiği
Şekil 8. %12,5 berilyum katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon
grafiği
164
Şekil 9. %25 berilyum katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon
grafiği
Şekil 10. %3,125 azot katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon
grafiği
Şekil 11. %6,25 azot katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon
grafiği

165
Şekil 12. %12,5 azot katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon
grafiği
Şekil 13. %25 azot katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon grafiği
Şekil 14. %3,125 silisyum katkılanmış CNT(8, 0) için a (A)-enerji (eV)
optimizasyon grafiği

166
Şekil 15. %6,25 silisyum katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon
grafiği
Şekil 16. %12,5 silisyum katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon
grafiği
Şekil 17. %25 silisyum katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon
grafiği

167
Şekil 18. %3,125 oksijen katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon
grafiği
Şekil 19. %6,25 oksijen katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon
grafiği
Şekil 20. %12,5 oksijen katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon
grafiği

168
Şekil 21. %25 oksijen katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon
grafiği
Şekil 22. %3,125 klor katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon
grafiği
Şekil 23. %6,25 klor katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon
grafiği

169
Şekil 24. %12,5 klor katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon
grafiği
Şekil 25. %25 klor katkılanmış CNT(8, 0) için a (A)-enerji (eV) optimizasyon grafiği