tyrosine phosphatases as receptor tyrosine kinases162453/fulltext01.pdfpersson c. 2003. protein...
TRANSCRIPT

Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 1232
Protein Tyrosine Phosphatases asRegulators of Receptor Tyrosine Kinases
BY
CAMILLA PERSSON
ACTA UNIVERSITATIS UPSALIENSISUPPSALA 2003

2
Dissertation to be publicly examined in Room B42, Biomedical Center, on April 4, 2003, at 13.15, for the Degree of Doctor of Philosophy (Faculty of Medicine) in Molecular Cell Biology. The discussion will be conducted in English. ABSTRACT Persson C. 2003. Protein tyrosine phosphatases as regulators of receptor tyrosine kinases. Acta Universitatis Upsaliensis. Comprehensive Summaries of Uppsala Dissertations from the Faculty of Medicine 1232. 54pp. Uppsala. ISBN 91-554-5556-5
Tyrosine phosphorylation is a crucial mechanism in cellular signaling and regulates proliferation, differentiation, migration and adhesion. The phosphorylation reaction is reversible and is governed by two families of enzymes: protein tyrosine kinases and protein tyrosine phosphatases (PTPs). This thesis investigates the role of PTPs in regulating receptor protein tyrosine kinases (RTKs), and explores a mechanism for regulation of phosphatase activity.
Most receptor tyrosine kinases are activated by ligand induced dimerization, which results in an increase in receptor phosphorylation. Preparations of ligand-stimulated dimeric PDGF β-receptors were shown to be less susceptible to dephosphorylation compared with unstimulated receptors. This revealed that reduced receptor dephosphorylation contributes to ligand-induced increase in RTK phosphorylation.
The receptor-like phosphatase DEP-1 site-selectively dephosphorylates the PDGF β-receptor. One of the most preferred sites is the PLC-γ binding phosphotyrosine pY1021, and the autoregulatory pY857 is one of the least preferred sites. By using chimeric phospho-peptides derived from these two sites as substrate for DEP-1, it was shown that a lysine residue at position +3 acts as a negative determinant for DEP-1 and that an aspartic acid residue at position –1 is a positive determinant.
The modulatory effect of TC-PTP on PDGF β-receptor signaling was explored by using mouse embryonic fibroblasts derived from TC-PTP knockout mice. PDGF β-receptors derived from knockout cells exhibited a higher level of ligand-induced phosphorylation compared to receptors from wildtype cells. The increase was unevenly distributed between different autophosphorylation sites. The PLC-γ binding site, previously implicated in chemotactic response, displayed the largest increase. Consistently, a cell migration assay revealed hyper-responsiveness to PDGF of TC-PTP knockout cells as compared to wildtype cells.
Reversible oxidation of the active site cysteine in PTPs is a mechanism, which have been postulated to regulate phosphatase specific activity. An antibody-based generic method for detection of oxidized PTPs was developed. Using this method it was revealed for the first time that UV-induced inactivation of PTPs involves oxidation of the active site cysteine. Camilla Persson, Ludwig Institute for Cancer Research, Biomedical center, Box 595, SE-751 24 Uppsala, Sweden Camilla Persson 2003 ISSN 0282-7476 ISBN 91-554-5556-5 Printed in Sweden by Eklundshofs Grafiska, Uppsala 2003

3
To my parents

4
This thesis is based on the following papers, referred to in the text by their roman numerals: I. Shimizu A., Persson C., Heldin C-H. and Östman A. (2001). Ligand
stimulation reduces platelet-derived growth factor β-receptor susceptibility to tyrosine dephosphorylation. J. Biol. Chem. 276, 27749-27752
II. Persson C., Engström U., Mowbray SL. and Östman A. (2002). Primary
sequence determinants responsible for site-selective dephosphorylation of the PDGF β-receptor by the receptor-like protein tyrosine phosphatase DEP-1. FEBS Letters 517, 27-31
III. Persson C., Sävenhed C., Tremblay M., Heldin C-H., Rönnstrand L., Östman
A, and Hellberg C. (2003). Deletion of TC-PTP results in site-selective increase in PDGF-induced receptor tyrosine phosphorylation and enhanced chemotactic response. Manuscript
IV. Persson C., Sjöblom T., Engström U., Hellman U., den Hertog J. Heldin C-H.
and Östman A. (2003). UV-induced oxidation of RPTPα revealed by a novel generic method for detection of oxidation-inactivated protein tyrosine phosphatases. Submitted.
Reprints were made with permission from the publisher.

5
TABLE OF CONTENTS PAGE Abbreviations 6 Introduction 7 The receptor tyrosine kinase and protein tyrosine phosphatase 8
enzyme families Receptor tyrosine kinases (RTKs) 8 Protein tyrosine phosphatases (PTPs) 9 Regulation of RTKs and PTPs 12 RTK regulation 12 Ligand binding, receptor dimerization and autophosphorylation 12 Endocytosis, degradation and dephosphorylation 14 PTP regulation 15 Subcellular targeting 15 Proteolytic cleavage 16 Ligand binding 16 Dimerization 17 Phosphorylation 18 Oxidation 18 PTP regulation of RTK signaling 19 Identification of RTK targets for PTPs 19 Site-selective versus general dephosphorylation 23 Molecular basis for substrate recognition 23 The role of PTPs in redox regulation of RTK signaling 25 RTKs and PTPs as drug targets 26 Present investigation 29
Aim 29 Ligand stimulation reduces platelet-derived growth factor β-receptor 29
susceptibility to tyrosine dephosphorylation (paper I) Primary sequence determinants responsible for site-selective 30
dephosphorylation of the PDGF β-receptor by the receptor-like protein tyrosine phosphatase DEP-1 (Paper II)
Deletion of TC-PTP results in site-selective increase in PDGF-induced 31 receptor tyrosine phosphorylation and enhanced chemotactic response (Paper III)
UV-induced oxidation of receptor protein-tyrosine phosphatase α 33 revealed by a novel generic method for detection of oxidation- inactivated protein-tyrosine phosphatases (Paper IV)
Future perspectives 34 Acknowledgements 36 References 37

6
Abbreviations CAM Cell adhesion molecule CSF-1 Colony stimulating factor-1 DEP-1 Density enhanced protein tyrosine phosphatase 1 EGF Epidermal growth factor HGF Hepatocyte growth factor FERM 4.1, ezrin, radixin, moesin FRET Flourescence resonance energy transfer FGF Fibroblast growth factor LAR Leukocyte common antigen-related LMW-PTP Low molecular weight protein tyrosine phosphatase LyPTP Lymphoid protein tyrosine phosphatase MEF Mouse embryonic fibroblast PAE Porcine aortic endothelial PDGF Platelet-derived growth factor PDZ PSD-95, DlgA, ZO-1 PEST Pro, Glu, Ser, Thr PTB Phosphotyrosine-binding PTK Protein tyrosine kinase PTP Protein tyrosine phosphatase RTK Receptor tyrosine kinase SH2 Src homology 2 SHP Src homology containing phosphatase STAT Signal transducer and activator of transcription TC-PTP T cell protein tyrosine phosphatase VEGF Vascular endothelial growth factor

7
Introduction Multicellular organisms are composed of a variety of cell types, each having different assignments depending on which organ or tissue it resides in. In order for the organism to function properly, the behaviour of each cell needs to be tightly regulated. This regulation is based on an elaborate communication system including signaling between cells, between cells and extracellular molecules and between molecules within a cell. A central mechanism in this signaling system is phosphorylation on tyrosine residues in a protein. The phosphorylated protein can span the cell membrane transmitting signals from the environment to the inside of the cell. Alternatively, it resides inside the cell and takes part in a signaling cascade transmitting signals to the cell nucleus. One effect evoked by phosphorylation may be a conformational change, which modulates the enzymatic activity of the protein. Another event induced by tyrosine phosphorylation is the creation of binding sites, to which downstream signaling proteins are recruited and subsequently becomes acivated. Tyrosine phosphorylation controls major cellular events such as proliferation, differentiation, cell adhesion, and migration. Tyrosine phosphorylation is reversible and two families of counteracting enzymes determine the phosphorylation status of specific cellular proteins, i.e. protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs). The signaling role of many PTKs has been thoroughly characterized. Activation of many PTKs by overexpression or mutation is often linked to cancer, which emphasize their importance as positive regulators of cell growth and survival (Blume-Jensen and Hunter, 2001). Aberrant PTK signaling has also been shown to be involved in other diseases such as cardiovascular diseases and developmental defects. The signaling roles of PTPs and their modes of regulation are much less characterized. However, extensive studies are ongoing to elucidate the action of PTPs in cell signaling, and accumulating data suggests that they are regulated enzymes with important roles in the regulation of signal transduction. This thesis will focus on biochemical aspects on the RTK and PTP families, and on modulation of RTK signaling by PTPs. In addition, structural and biochemical aspects on these enzyme families will be put in an applied context by a brief description of RTKs and PTPs as drug targets.

8
The receptor tyrosine kinase and protein tyrosine phosphatase enzyme families Receptor tyrosine kinases (RTKs) Protein tyrosine kinases can be divided into two families: receptor tyrosine kinases (RTKs) and non-receptor tyrosine kinases (nRTKs). Sequencing of the human genome has revealed 90 tyrosine kinases; 58 RTKs and 32 nRTKs (Manning et al., 2002; Robinson et al., 2000). The RTK family can be divided into 20 subfamilies based on the sequence of the kinase domain and the type of domins in the extracellular parts. The prototypes of the major subfamilies include EGFR, PDGFR, FGFR, and insulin receptor. The nRTKs can be subdivided into 10 subfamilies, including the Src and Abl subfamilies. Below the focus will mainly be on receptor tyosine kinases. RTKs are transmembrane enzymes responsible for transducing extracellular signals from peptide growth factors across the cell membrane. They are characterized by extracellular ligand binding domains, a single transmembrane helix and an intracellular portion containing tyrosine kinase activity (Hubbard and Till, 2000). Immunoglobulin-like repeats, fibronectin type III repeats and cysteine rich domains are common extracellular domains. The variability of the extracellular portions provides specificity for ligand binding. The structure of the kinase domain, composed of ~300 amino acid residues, is conserved between tyrosine kinases (Hubbard, 1999). It exhibits a two-domain architecture consisting of an amino-terminal lobe and a larger carboxy-terminal lobe. The cleft formed by the two lobes harbours the reaction where the γ-phosphate from ATP is transferred to a hydroxyl group of the tyrosine in the protein substrate. Most RTKs are monomeric in their inactive state and dimerize upon ligand binding (Schlessinger, 2000). One exception is the insulin receptor family, the members of which are dimerized also in absence of ligand. Ligand binding induces autophosphorylation of tyrosine residues in the so-called activation loop, and subsequently of residues outside the catalytic domain. The phosphorylated non-catalytic tyrosines act as specific binding sites for downstream signaling proteins containing phosphotyrosine binding (PTB) and src homology 2 (SH2) domains (Pawson and Scott, 1997). The specificity of individual SH2 domains is determined by the three to five amino acid residues carboxy-terminal of the phosphotyrosine (Songyang et al., 1993), whereas the PTB domain binding specificity motif is conferred by five to eight amino-acid residues amino-terminal of the phosphotyrosine (van der Geer et al., 1996). Activation of downstream signaling molecules is conferred by different mechanisms. The activation may involve conformational changes induced directly by the binding to the RTK, and it can also be a consequence of tyrosine phosphorylation. The PTP SHP-2

9
and the tyrosine kinase Src are examples of signaling proteins, which undergo conformational changes and subsequent activation upon RTK recruitment. (Hof et al., 1998; Xu et al., 1999). Activation may also involve translocation of the molecule in proximity to its substrate. For instance, phosphatidylinositol 3’-kinase (PI3-kinase), is recruited to RTKs and thereby translocated to the membrane where its substrate phosphatidylinositol-4,5-bisphosphate is located (Cantley, 2002). RTKs can also recruit and phosphorylate transcription factors. For instance phosphorylation of STATs results in dimerization and nuclear translocation of the transcription factor (Darnell, 1997). Another group of molecules recruited to activated RTKs is adaptor molecules e.g. Shc and Grb2. They lack catalytic activity but have the ability to bind several proteins simultaneously and can thus assemble signaling complexes. The variety of downstream signaling molecules thus explains the diversity of biological response, induced by activated RTKs. Protein tyrosine phosphatses (PTPs) PTPs can be divided into three subclasses: dual-specificity PTPs (DSP-PTPs), low molecular weight PTPs (LMW-PTPs) and classical tyrosine-specific PTPs. Dual-specificity PTPs includes VHR, MAPK-PTP, KAP and the cell cycle regulators Cdc25, and are capable of dephosphorylating serine- and threonine residues in addition to tyrosine residues. LMW-PTPs constitute a family of 18 kDa enzymes with specificity primarily towards phosphotyrosine. To date 37, members of the classical tyrosine-specific PTP family have been identified (Andersen et al., 2001b). The classical tyrosine-specific PTPs can be further divided in two subfamilies: cytosolic and receptor-like PTPs (rPTPs). The majority of the receptor-like PTPs contain two catalytic domains: a membrane proximal domian (D1), mainly responsible for catalysis, and a membrane distal domain (D2), containing little or no phosphatase activity. The function of D2 is unclear but it has been suggested to possess a regulatory function (Blanchetot et al., 2002). In addition to the phosphatase domains, rPTPs are composed of a single transmembrane segment and an extracellular domain. The extracellular portion exhibits a broad structural variation. Based on stuctural features of the extracellular domain, rPTPs have been divided into 5 subtypes (Mourey and Dixon, 1994). CD45 is the prototypic type 1 PTP, which is characterized by a heavily glycosylated cysteine-rich domain. Several isoforms exist due to differential splicing. Type 2 PTPs contain immunoglobulin-like domains and fibronectin type III repeats and include LAR, PTPµ and PTPκ. Type 3 PTPs, e.g. DEP-1 and PTPβ, contain fibronectin type III repeats alone. Type 4 includes PTPα and PTPε,

10
which are characterized by short and highly glycosylated extracellular segments. Finally, type 5 members, including PTPγ and PTPζ/β, contain an amino-terminal carbonic anhydrase-like motif. The fact that some structural features of the rPTP extracellular parts, such as immunoglobulin-like domains and fibronectin type III repeats, are similar to cell adhesion molecules, suggests that PTPs might be involved in cell-cell and cell-extracellular matrix interactions (Brady-Kalnay and Tonks, 1995). The diversity of the extracellular domains suggests that they serve as specific ligand binding sites but still only a few ligands have been identified. Cytosolic PTPs contain one single catalytic domain and additional flanking regions with putative roles in regulation of catalytic activity, protein-protein interactions and subcellular targeting (Tonks and Neel, 2001). SHP-1 and SHP-2 are two related PTPs containing SH2 domains, which are involved in both activity regulation and substrate binding. LyPTP and PTP-PEST contain PEST domains, suggested to have a role in substrate targeting. PDZ domains are also involved in substrate binding and are found for instance in PTP-BAS. PTP1B and TC-PTP share high sequence similarity and contain a carboxy-terminal endoplasmic reticulum-targeting domain. Subcellular targeting of PTPs by flanking domains will be discussed further below. Receptor-like and cytosolic PTPs have also been classified into 17 subfamilies based on catalytic domain homology. This classification yields subfamilies that are very similar to the ones based on extracellular features in rPTPs and flanking domains in cytosolic PTPs (Andersen et al., 2001b). Three-dimensional structures, together with kinetic analysis, have provided a clear picture of the catalytic mechanism of PTPs (Barford et al., 1998). The catalytic domains of five cytosolic PTPs (PTP1B, Yop51, SHP-1, SHP-2 and TC-PTP) and four rPTPs (PTPα, PTPµ, LAR and PTP-SL) have been crystallized to date (Barford et al., 1994; Bilwes et al., 1996; Hof et al., 1998; Hoffmann et al., 1997; Iversen et al., 2002; Nam et al., 1999; Stuckey et al., 1994; Szedlacsek et al., 2001; Yang et al., 2000). All members of the classical PTP family contain a highly conserved catalytic signature motif, [V/I]HCSxGxGR[S/T]G, residing at the bottom of an active site cleft. The invariant cysteine residue in the signature motif has a crucial role in catalysis (Guan and Dixon, 1991). The cleft is surrounded by four loops, which contain residues essential for catalysis and substrate recognition. The depth of the cleft confers specificity towards

11
phosphotyrosine since hydrolysis of the shorter phospho-serine and phospho-threonine residues is prevented (Jia et al., 1995). Binding of substrate induces a conformational change of one of the loops, thus closing the cleft and trapping the phosphotyrosine close to the active site. The reaction is performed in two steps and involves formation of a cysteinyl-phosphate intermediate followed by hydrolysis of the intermediate (Pannifer et al., 1998). The cysteinyl-phosphate intermediate is formed through a nucleophile attack on the phosphate by the unprotonated catalytic cysteine residue. An invariant protonated aspartic acid residue in the flexible loop enhances cleavage of the P-O bond by acting as a general acid catalyst. The intermediate is then hydrolysed by a water molecule, which is positioned by an invariant glutamine residue and activated by the aspartic acid now acting as a general base. The crystal structure of several DSP-PTPs has revealed that they possess a shallower active site cleft, which allows access to serine and threonine residues in addition to tyrosine residues (Fauman et al., 1998; Reynolds et al., 1999; Song et al., 2001; Stewart et al., 1999; Yuvaniyama et al., 1996). The overall three-dimensional structure of the catalytic domain of VHR and MAPK-PTP and KAP is similar to that of classical PTPs, whereas Cdc25 family members exhibit a more unique topology. However, all DSP-PTP share the catalytic signature motif HCxxxxxR with classical PTPs and use the same catalytic mechanism (Denu et al., 1996; Fauman et al., 1998). LWM-PTP is completely different from classical PTPs in its structure but still exhibits similar active site architecture (Zhang et al., 1998). The cysteine and arginine residues from the signature motif is conserved and also this PTP family uses the same two-step mechanism for catalysis.

12
Regulation of RTKs and PTPs RTK regulation Ligand binding, receptor dimerization and autophosphorylation The mechanism of receptor kinase activation has been thoroughly characterized by structural and biochemical studies. Even though dimerization appears as a general activation mechanism for most RTKs, the structural diversity of ligands and receptors suggest various dimerization strategies for different ligand-receptor couples. Three different modes of receptor activation will be discussed using the well-characterized receptors for VEGF, FGF and EGF as examples. VEGF is a homodimeric glycoprotein, which is a member of the cystine-knot growth factor family also including PDGF, NGF and TGF-β (Sun and Davies, 1995). VEGF bind to and induces homodimerization of the receptors KDR and Flt-1. The extracellular portion of VEGF receptors contains seven immunoglobulin domains. The crystal structure of VEGF in complex with immunoglobulin domain 2 of the Flt-1 receptor has provided insights in ligand-induced receptor dimerization (Wiesmann et al., 1997). The antiparallel VEGF dimer exhibits a flat and elongated structure. Flt-1 binds to the poles of the dimer, where it is in contact with both subunits and the complex forms an approximate two-fold symmetry. Domain deletion experiments showed that only domain 2 and 3 are involved in ligand binding. Mutational studies of the Flt-1 receptor suggest that receptor-receptor interactions involving immunoglobulin domains 4 stabilize the dimeric structure (Barleon et al., 1997). The fact that the structures of members of the PDGF receptor family are similar to those of the VEGF receptor family suggests that the same dimerization mechanism also apply for the PDGF receptor family. FGF is a family of at least 23 peptide growth factors (Yamashita et al., 2000). FGF isoforms bind to four related receptors, FGFR1-FGFR4, each having three extracellular immunoglobulin domains (Ornitz et al., 1996). In contrast to VEGF, FGF is a monomeric molecule, which is unable to alone induce receptor dimerization. In addition to FGF, binding of heparin or heparan sulfate proteoglycans is necessary to promote stable receptor dimerization and activation (Spivak-Kroizman et al., 1994). The molecular mechanism underlying FGF receptor activation was elucidated by the crystal structure of FGF2 in complex with immunoglobulin domain 2 and 3 of FGFR1 (Plotnikov et al., 1999). The structure revealed two FGF:FGFR complexes forming a two-fold symmetric dimer. The ligands interact with both domain 2 and 3 of the receptor but in contrast to the dimeric VEGF there is no contact between the two FGFs. The dimeric structure is stabilized by the two FGF molecules, which can interact with both receptor subunits, and by direct receptor-receptor interactions. A positively

13
charged canyon, formed by the inside of the two ligands and domain 2 of the receptor dimer, constitutes a putative heparin-binding site. Co-crystalization of the complex together with heparin revealed extensive interactions between heparin and both FGF and FGFR in a manner that promotes receptor dimerization (Schlessinger et al., 2000). The EGF receptor family comprises four members, EGFR, ErbB-2, ErbB-3 and ErbB-4. The receptors are activated by a number of ligands, including EGF, TGF-α and neuregulins, which are capable of inducing receptor homo- or heterodimerization (Olayioye et al., 2000). The mode of EGFR dimerization has remained unclear until recently when the crystal structure of EGF in complex with the extracellular domains of EGFR was solved (Ogiso et al., 2002). The dimer consists of two EGF:EGFR complexes similarly to the FGF:FGFR. However, each of the monomeric ligands bind only one receptor. The EGFs are localized on opposite sides of the receptor dimer and there are no interactions between the ligands. The complex differs from other receptor -ligand complexes in that the dimerization is mediated exclusively through receptor-receptor interactions. Hence, ligand binding most likely induces a conformational change in the receptors, leading to dimerization of the EGFR monomers. In conclusion, these examples illustrate that receptor dimerization can be achieved through at least three different stabilizing mechanisms: dimeric ligands, accessory ligands and extensive receptor-receptor interactions. Dimerization of RTKs results in autophosphorylation of tyrosines in the intracellular catalytic domains, however the mechanism whereby dimerization and autophosphorylation activate the receptors is not yet fully understood. It is postulated that trans-phosphorylation between the receptors occurs when the cytoplasmic domains are in close proximity to each other. Another mechanism, which has been suggested, is that ligand binding induces a conformational change of the receptor allowing for cis-phosphorylation. Phosphorylation of tyrosines in the activation loop has been shown to be important for activation of several RTKs including insulin receptor, FGFR, VEGFR and PDGFR (Ellis et al., 1986; Fantl et al., 1989; Kendall et al., 1999; Mohammadi et al., 1996a). The crystal structures of the insulin receptor in phosphorylated and unphosphorylated forms showed that phosphorylation induced a conformational change of the activation loop, which allows for access to ATP and protein substrates (Hubbard, 1997; Hubbard et al., 1994). The kinase is thus autoinhibited, with no access to substrate or ATP, in its inactive state. Given that the insulin receptor is constitutively dimerized, autoinhibition may be an important mechanism to prevent ligand-independent activation. Also for the FGF receptor, crystallographic studies have unveiled conformational changes in the activation loop between the phosphorylated and

14
unphosphorylated forms, although the unphosphorylated form is not obstructed to the same extent as the insulin receptor (Mohammadi et al., 1996b). In summary, ligand-induced autophosphorylation of the activation-loop appears to be a central mechanism in stabilizing the kinase in an active state. In the case of the ephrin receptor EphB2 and the PDGF receptor an additional autoregulatory mechanism has been implicated, which is mediated through the cytoplasmic juxtamembrane region. The crystal structure of unphosphorylated EphB2 showed that the juxtamembrane segment forms a helical structure, which distorts the small lobe of the kinase domain and hampers the activation loop from adopting the activated conformation. Phosphorylation of two juxtamembrane tyrosine residues is predicted to relieve the inhibitory interaction with the activation loop (Binns et al., 2000; Wybenga-Groot et al., 2001). Mutational analyses of the juxtamembrane region of the PDGF receptor suggest that a similar mechanism could apply also for this RTK (Baxter et al., 1998; Irusta et al., 2002). Endocytosis, degradation and dephosphorylation One way to attenuate the signal from activated RTKs on the cell surface is through receptor downregulation by endocytosis and subsequent degradation. The mechanism of endocytosis has been well characterized for several RTKs including the EGF, and insulin receptors (Di Guglielmo et al., 1998; Waterman and Yarden, 2001). Activated receptors are internalized via clathrin-coated pits, which then pinch off from the membrane to form coated vesicles. The coated vesicle fuses with the endosome and the receptor is then delivered through the endosomal compartments, either to the lysozome for degradation, or recycled back to the cell membrane. The sorting of the receptor in the endosome for degradation is achieved by ubiquitin tagging of the receptor, a process mediated by the ubiquitin ligase Cbl (Joazeiro et al., 1999). In addition to its ligase activity, Cbl has also been shown to regulate endocytosis of EGF and HGF receptors by acting as an adaptor protein (Petrelli et al., 2002; Soubeyran et al., 2002). Cbl recruits CIN85 and endophilin and forms a complex with ligand stimulated receptors, thus enhancing membrane invagination. Furthermore, ubiquitination of receptor-associated CIN85 by Cbl has been proposed to have a role in endosomal sorting leading to degradation of receptors (Haglund et al., 2002). Evidence that receptor downregulation is important for attenuating RTK signaling is also provided by studies in which inhibition of EGFR downregulation caused enhanced receptor activation and tumorigenesis (Wells et al., 1990; Worthylake et al., 1999). However, endocytosis may also be a way of extending and modulating RTK signaling. Early studies demonstrated increased autophosphorylation activity of internalized insulin and EGF receptors (Khan et al., 1989; Lai et al., 1989). Furthermore, inhibition of endocytosis of the insulin

15
receptor resulted in a selective attenuation of MAP kinase and PI3 kinase signaling, while other downstream signaling pathways were unaffected (Ceresa et al., 1998). Similarly, the pattern of associating downstream signaling molecules differed between internalized and cell surface bound EGF receptors (Burke et al., 2001). Hence, in addition to its role in receptor downregulation, endocytosis might also provide a mechanism for restricting RTK signaling to specific subcellular compartments. An important mechanism for RTK signal termination is the action of PTPs. Treatment of cells with PTP inhibitor results in phosphorylation and activation of RTKs, even in the absence of ligand (Jallal et al., 1992), which emphasizes the tight regulation of RTKs by PTPs. Notably, there is one example that PTP-mediated tyrosine dephosphorylation, rather than receptor internalization, was of major importance for RTK signal attenuation (Chiarugi et al., 2002b). Regulation of RTK signaling by PTPs will be discussed in more detail below. PTP regulation Subcellular targeting Targeting to different subcellular localisations is one way to restrict the action of PTPs. While receptor-like PTPs are restricted to the plasma membrane, different sequences and domains outside the catalytic domains in cytosolic PTPs mediate binding to specific targets in the cell. PTP1B contains a hydrophobic carboxy- terminal tail, which confers localization to the endoplasmic reticulum (Frangioni et al., 1992), hence contributing to limited substrate recognition. Consistent with its targeting domain, PTP1B-catalyzed dephosphorylation of EGFR and PDGFR occurs at specific locations on the surface of the endoplasmic reticulum (Haj et al., 2002). In addition, PTP1B contains an amino-terminal region conferring specific binding to the insulin receptor (Dadke et al., 2000). In PTP-PEST, sequences carboxy-terminal of the catalytic domain bind to paxillin and the paxillin homologue Hic-5, both localized at focal contacts (Nishiya et al., 1999; Shen et al., 2000). PTPH1, PTPBAS, PTPMEG and PTPD1 belong to a family containing FERM domains (Gu and Majerus, 1996; Maekawa et al., 1994; Moller et al., 1994; Zhang et al., 1995). These domains mediate targeting of cytoskeleton associating proteins to cytoskeletal-membrane interfaces (Arpin et al., 1994; Bretscher, 1999). The presence of FERM domains in PTPs indicates targeting to cytoskeleton- and membrane associated proteins, which might be putative substrate for dephosphorylation. Alternative splicing generates two forms of TC-PTP (TC45 and TC48) that differ in the carboxy-terminal motifs. Due to differences in the carboxy-terminal structure, TC45 and TC48 are targeted to the nucleus and endoplasmic reticulum, respectively (Lorenzen et al., 1995).

16
SHP-1 and SHP-2 are two well-characterized PTPs, which contain SH2 domains mediating recruitment to the appropriate tyrosine phosphorylated protein (Feng, 1999; Zhang et al., 2000). The SH2 domains of SHP-1 and SHP-2 have an additional regulatory function. When the PTP is not bound to a substrate, the amino-terminal SH2 domain occupies and inactivates the catalytic site. Upon binding of the carboxy-terminal SH2-domain to a phosphorylated tyrosine substrate, a conformational change disrupts the interaction and activates the PTP (Barford et al., 1998; Hof et al., 1998; Yang et al., 2003). Proteolytic cleavage Several studies indicate that proteolytic cleavage is a mechanism inducing cellular redistribution of PTPs. This process is sometimes associated with changes in phosphatase activity. Calpain-catalyzed cleavage of PTP-1B results in relocalization from membranes to the cytosol concomitant with a two-fold increase in catalytic activity (Frangioni et al., 1993). Proteolytic cleavage has also been demonstrated for receptor-like PTPs. Type 2 rPTPs including LAR and PTPσ are expressed at the cell surface as two subunits due to proteolytic processing during biosynthesis (Aicher et al., 1997; Streuli et al., 1992). Induced proteolysis of LAR and PTPσ at an extracellular juxtamembrane site was shown to result in shedding of the extracellular domain, subsequent internalization, and redistribution of the intracellular domain away from sites of cell-cell contact. Naturally occurring cytoplasmic forms of PTPα and PTPε are generated by calpain cleavage at an intracellular juxtamembrane site (Gil-Henn et al., 2001). The translocation impaired the ability of PTPα to dephosphorylate substrates associated with the membrane, such as Src. Membrane localization of PTPε has also been shown to be crucial for attenuating insulin induced signaling (Andersen et al., 2001a). Ligand binding The structural diversity of the extracellular domain of receptor-like PTPs implicates that it may be a target for binding of regulatory ligands. To date, only a few ligands for rPTPs have been identified. Homophilic interactions of extracellular domains have been demonstrated for PTPµ, PTPκ and PTPδ (Brady-Kalnay et al., 1993; Sap et al., 1994; Wang and Bixby, 1999). Interactions between PTPs and extracellular matrix molecules have also been shown. A laminin-nidogen complex and heparan sulfate proteoglycans are ligands for LAR and PTPσ, respectively (Aricescu et al., 2002; O'Grady et al., 1998). Several ligands for PTPβ/ζ have been identified (Peles et al., 1998). The neuronal recogition molecules contactin and TAG1/axionin, the adhesion molecules Ng-CAM, N-CAM and Nr-CAM, and the extracellular matrix protein tenascin have been

17
shown to bind to different parts of the extracellular domain of PTPβ/ζ (Milev et al., 1994; Milev et al., 1996; Peles et al., 1995). In addition, a soluble ligand of PTPβ/ζ , the heparin-binding pleiothrophin, has been identified (Maeda et al., 1996). However, a modulating effect of ligand binding on PTP activity has been detected only in two cases. Pleiothrophin have been shown to inactivate PTPβ/ζ (Meng et al., 2000). The pleiothrophin-induced inactivation was associated with increased phosphorylation of β-catenin, a candidate PTPβ/ζ substrate shown to interact with and to be dephosphorylated by the phosphatase. In another case of regulatory ligand binding, PTP activation was demonstrated. In this study an extracellular matrix preparation, Matrigel, was shown to up-regulate the activity of DEP-1 through interactions with the extracellular domain (Sörby et al., 2001). Dimerization Dimerization is a suggested inhibitory regulatory mechanism for rPTPs. The crystal structure of the membrane proximal catalytic domain of PTPα revealed that the PTP forms dimers (Bilwes et al., 1996). In the dimeric structure, an amino-terminal wedge of one subunit is inserted into the catalytic site of the opposing subunit, hence blocking access to substrate. Also, disulfide-bonded full-length dimeric PTPα, generated by introduction of cysteines in the extracellular juxtamembrane region, exhibits a reduced ability to dephosphorylate and activate Src (Jiang et al., 1999). CD45, an rPTP implicated to have a positive role in T cell receptor signaling, may also be regulated by dimerization. Expression of a chimeric protein consisting of extracellular EGFR and intracellular CD45 in a CD45 deficient cell line resulted in EGF-mediated inhibition of T cell receptor signaling (Desai et al., 1993). Two acidic amino acid residues in the inhibitory wedge in PTPα are conserved in rPTPs, including CD45. Mutation in this wedge in the EGFR/CD45 chimera diminished the EGF-mediated inhibition of T cell receptor signaling (Majeti et al., 1998). Consistently, knock-in mice bearing a point mutation in the putative inhibitory wedge of CD45, develop lymphoproliferation and autoimmunity (Majeti et al., 2000). Moreover, dimeric forms of both PTPα and CD45 have been identified by cross-linking of cell surface proteins (Jiang et al., 2000; Takeda et al., 1992). In addition to homo-dimerization, hetero-dimerization of PTPs has been proposed as a regulatory mechanism. The rPTP-like proteins IA-2 and IA2β contain single inactive PTP domains and have been shown to form heterodimers with PTPα and PTPε (Gross et al., 2002). Dimerization with PTPα resulted in down-regulation of PTP activity. Furthermore, heterodimerization between domain 1 of one PTP and domain 2 of another PTP has been demonstrated for several PTPs, including PTPα, PTPµ, PTPε, PTPδ,

18
PTPσ, LAR and CD45 (Gross et al., 2002). In the case of PTPδ domain 2 binding to PTPσ domain 1, inhibition of PTPσ has been demonstrated (Wallace et al., 1998). The dimerization-mediated regulatory mechanism may not apply to all phosphatases. PTPµ was shown to crystallize as dimers but no inhibitory interactions between the active site and the inhibitory wegde could be seen (Hoffmann et al., 1997). Moreover, the crystal structure of LAR suggests that steric hindrance prevents dimerization (Nam et al., 1999). Phosphorylation Phosphorylation of PTPs can modulate phosphatase activity in both a positive and a negative manner. Tyrosine phosphorylation of PTP1B upon association with EGF- and insulin receptor has been shown to increase phosphatase activity (Dadke et al., 2001; Liu and Chernoff, 1997). Similarly, LMW-PTP displays upregulated catalytic activity upon tyrosine phosphorylation by Src in response to PDGF stimulation (Bucciantini et al., 1999). Phosphorylation on serine residues has been shown to positively regulate activity of PTPα due to both an increase in specific activity and to a decrease of the inhibitory binding to Grb2 (Zheng and Shalloway, 2001). CD45 is another PTP which displays increased PTP activity upon phosphorylation of serine residues (Wang et al., 1999). Serine phosphorylation may also negatively regulate PTP activity. For instance, serine phosphorylation of PTP-PEST results in a decrease in catalytic activity (Garton and Tonks, 1994). Oxidation The conserved catalytic cysteine in PTPs has a low pKa due to charge interactions with the microenvorinment. The cysteine residue thus exists at physiological pH as a thiolate anion, which is vulnerable to oxidation (Denu and Dixon, 1998; Denu et al., 1995; Lohse et al., 1997; Zhang and Dixon, 1993). The cysteine can be reversibly oxidized to sulfenic acid or further irreversibly oxidized to sulfinic or sulfonic acid (Denu and Tanner, 1998; Huyer et al., 1997). One of the first evidence of oxidation-mediated inactivation of PTPs was demonstrated upon H2O2-treatment of PTP1B, LAR and VHR in vitro. Chemical modification and mutational analysis showed that the catalytic cysteine was the target for oxidation to sulfenic acid (Denu and Tanner, 1998). Inhibition of catalytic activity and oxidation of the catalytic cysteine in response to H2O2-treatment in vitro have also been demonstrated for PTP1B (Lee et al., 1998). Furthermore, mass spectrometric analysis revealed oxidation-induced glutathionylation

19
of the cysteine (Barrett et al., 1999a; Barrett et al., 1999b). This finding supports the notion that glutathionylation is a mechanism to protect the unstable sulfenic acid intermediates from further oxidation to the irreversible sulfinic and sulfonic acid forms. In addition, in vivo-oxidation and inactivation of several PTPs including PTP1B, LMW PTP, SHP-1 and SHP-2 have been demonstrated in response to H2O2-treatment of cells (Chiarugi et al., 2001; Cunnick et al., 1998; Lee et al., 1998; Meng et al., 2002). The oxidation-induced reversible inactivation of LMW PTP was proposed to occur by formation of an intramolecular disulfide bond between the catalytic cysteine and a nearby residing cysteine. Similarly, the cell cycle regulating dual specificity PTPs Cdc25A, Cdc25B, Cdc 25C and KAP have been implicated to form oxidation-induced catalytic site disulfide bonds (Fauman et al., 1998; Reynolds et al., 1999; Savitsky and Finkel, 2002; Song et al., 2001). Moreover, a mechanism of PTP inactivation, which involves oxidation-induced stabilization of inhibitory PTPα dimers, has been proposed (Blanchetot et al., 2002). The stabilization of the dimeric structure was suggested to involve conformational changes of the membrane distal catalytic domain due to oxidation. The physiological relevance of oxidation as a regulatory mechanism is emphasized by the fact that growth factor-stimulation induces H2O2 production, which is crucial for downstream signaling. The issue of PTP inactivation upon growth factor stimulation will be discussed further below. PTP regulation of RTK signaling PTPs may exert a regulatory effect on signal transduction on several levels. They can modulate signaling by dephosphorylating membrane receptors such as RTKs and ion channels. They can also modulate signaling cascades by dephosphorylating adaptor molecules, intracellular kinases and transcription factors. Below the focus will be mainly on the regulation of RTKs by PTPs. Identification of RTK targets for PTPs Various approaches have been undertaken to reveal physiological targets for PTP mediated dephosphorylation, including gene knockout studies, antisense mediated suppression and substrate trapping. RTKs identified to associate with or be modulated by PTPs are summarized in Table 1. In many cases different methods identify the same substrate, increasing the confidence

20
of the individual findings, but strikingly few specific PTP-RTK couples have been identified. Instead many PTP appears able to dephosphorylate several RTKs, and almost each RTK is dephosphorylated by more than one PTP. This might be explained by the experimental setup. In a physiological context, a combination of different spatial or temporal expression pattern and subcellular localization of PTPs and RTKs, as well as intrinsic structural substrate specificity determinants in PTPs may confer more stringent substrate specificity. Most PTPs have been associated with a negative role in RTK signaling (Östman and Böhmer, 2001). However, some PTPs have been shown to act as positive regulators of signaling. SHP-2, which is closely related to SHP-1, can function as a signal enhancer. A decreased response to growth factors and defects in insulin signaling have been demonstrated in fibroblasts derived from mice with a targeted mutation in SHP-2 (Oh et al., 1999; Shi et al., 1998). One of the strongest evidence for a biological function of PTPs as negative regulators of RTKs is provided by gene knockout studies. PTP1B knockout mice exhibit increased autophosphorylation of the insulin receptor, enhanced sensitivity to insulin, and are resistant to high fat diet induced obesity. This indicates that the insulin receptor is a physiological substrate for PTP1B (Elchebly et al., 1999; Klaman et al., 2000). A comparison of mouse embryonic fibroblast (MEF) cell lines derived from PTP1B knockout mice with fibroblasts derived from wildtype mice revealed that PDGFR and EGFR from knockout cells exhibited enhanced phosphorylation in response to ligand stimulation (Haj et al., 2003). Using the same strategy, another study demonstrated increased IGF-1-induced phosphorylation of IGF-1 receptors in PTP1B knockout fibroblasts. In addition, PTP1B deficient cells displayed enhanced IGF-1 mediated suppression of apoptosis and motility (Buckley et al., 2002). Another way to identify substrates for PTPs is by the use of substrate trapping mutants, in which the catalytic site cysteine or aspartic acid residue is changed to a serine or alanine residue, respectively (Flint et al., 1997). These mutants have lost their enzymatic activity and form a stable complex with their substrate. Substrate-trapping mutant forms of PTP1B have been shown to associate with the insulin receptor and EGF receptor (Bandyopadhyay et al., 1997; Flint et al., 1997; Walchli et al., 2000). Additional PTP-RTK couples which have been identified in this manner is DEP-1 in association with the HGF receptor Met, and TC-PTP associating with the EGF receptor (Flint et al., 1997; Palka et al., 2003; Tiganis et al., 1998).

21
A useful approach to identify possible substrates is through antisense mediated suppression of PTPs. Antisense mediated suppression of PTP1B has been shown to increase insulin receptor signaling (Clampit et al., 2003). Experiments suppressing the rPTP LAR resulted in an increased ligand-induced autophosphorylation of the insulin, HGF, and EGF receptors (Kulas et al., 1996; Kulas et al., 1995). The opposite approach, PTP overexpression, has also been used. The notion that PTP1B act as a negative regulator of insulin signaling is supported by the fact that PTP1B overexpression in cells inhibits insulin-induced receptor autophosphorylation (Kenner et al., 1996). Further evidence that the insulin receptor is a substrate for LAR is provided by the fact that overexpression of LAR reduces insulin-induced insulin-receptor phosphorylation (Zhang et al., 1996). Two naturally occurring mutations of murine SHP-1, the motheaten (me/me) and viable motheaten (me-v/me-v), cause severe hematopoietic abnormalities and have provided insights in SHP-1 regulation of hematopoietic signaling pathways (Shultz et al., 1997). Genetic complementation studies suggest that SHP-1 negatively regulates signaling from the stem cell factor receptor c-Kit (Lorenz et al., 1996; Paulson et al., 1996). Other RTKs that have been implicated as substrates for SHP-1 from studies using these SHP-1 mutant mice are CSF-1R and Ros (Berg et al., 1999; Chen et al., 1996; Keilhack et al., 2001; Umeda et al., 1999). In a recent study, an in-gel PTP assay was used to find PTPs associating with RTKs. PTP activity was found to be associated with PDGFR, EGFR, and c-Kit immunoprecipitated from cell lysates. Immunodepletion identified PTP-PEST, SHP-2, PTP-1B and TC-PTP to be the PTPs associating with the PDGFR, whereas PTP-PEST and SHP-1 were the main PTPs interacting with EGFR and c-Kit, respectively (Markova et al., 2003).
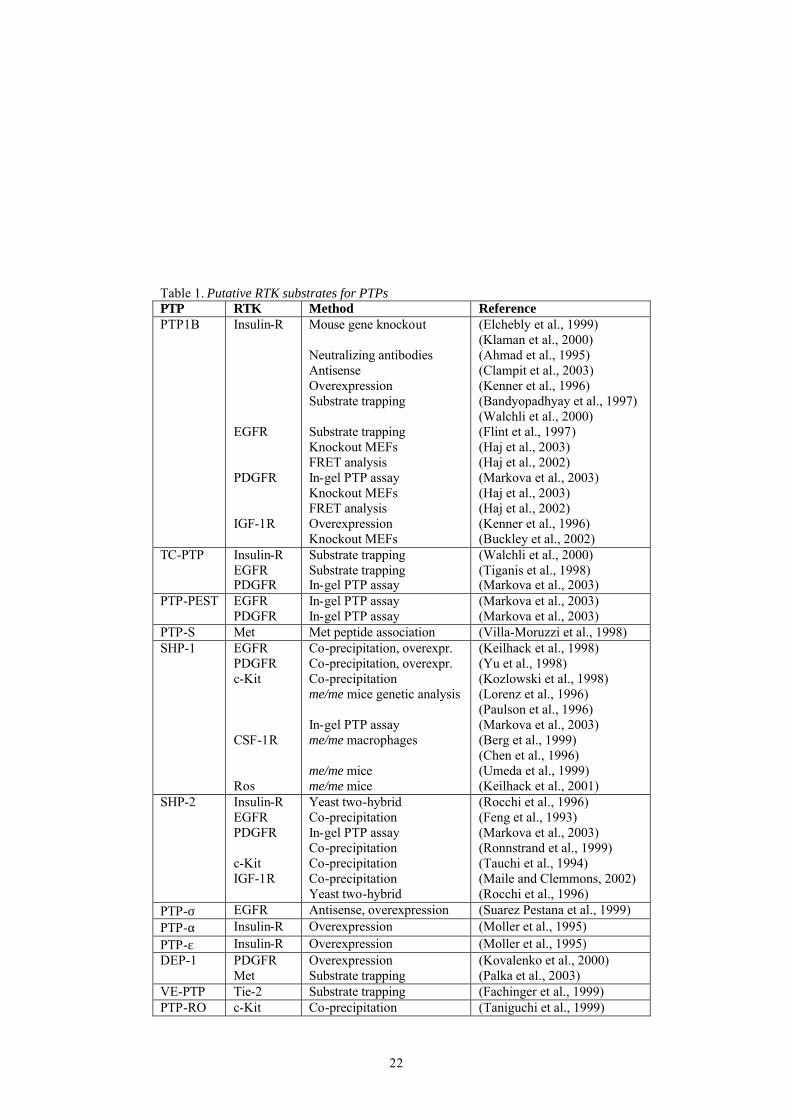
22
Table 1. Putative RTK substrates for PTPs PTP RTK Method Reference PTP1B Insulin-R
EGFR PDGFR IGF-1R
Mouse gene knockout Neutralizing antibodies Antisense Overexpression Substrate trapping Substrate trapping Knockout MEFs FRET analysis In-gel PTP assay Knockout MEFs FRET analysis Overexpression Knockout MEFs
(Elchebly et al., 1999) (Klaman et al., 2000) (Ahmad et al., 1995) (Clampit et al., 2003) (Kenner et al., 1996) (Bandyopadhyay et al., 1997) (Walchli et al., 2000) (Flint et al., 1997) (Haj et al., 2003) (Haj et al., 2002) (Markova et al., 2003) (Haj et al., 2003) (Haj et al., 2002) (Kenner et al., 1996) (Buckley et al., 2002)
TC-PTP Insulin-R EGFR PDGFR
Substrate trapping Substrate trapping In-gel PTP assay
(Walchli et al., 2000) (Tiganis et al., 1998) (Markova et al., 2003)
PTP-PEST EGFR PDGFR
In-gel PTP assay In-gel PTP assay
(Markova et al., 2003) (Markova et al., 2003)
PTP-S Met Met peptide association (Villa-Moruzzi et al., 1998) SHP-1 EGFR
PDGFR c-Kit CSF-1R Ros
Co-precipitation, overexpr. Co-precipitation, overexpr. Co-precipitation me/me mice genetic analysis In-gel PTP assay me/me macrophages me/me mice me/me mice
(Keilhack et al., 1998) (Yu et al., 1998) (Kozlowski et al., 1998) (Lorenz et al., 1996) (Paulson et al., 1996) (Markova et al., 2003) (Berg et al., 1999) (Chen et al., 1996) (Umeda et al., 1999) (Keilhack et al., 2001)
SHP-2 Insulin-R EGFR PDGFR c-Kit IGF-1R
Yeast two-hybrid Co-precipitation In-gel PTP assay Co-precipitation Co-precipitation Co-precipitation Yeast two-hybrid
(Rocchi et al., 1996) (Feng et al., 1993) (Markova et al., 2003) (Ronnstrand et al., 1999) (Tauchi et al., 1994) (Maile and Clemmons, 2002) (Rocchi et al., 1996)
PTP-σ EGFR Antisense, overexpression (Suarez Pestana et al., 1999) PTP-α Insulin-R Overexpression (Moller et al., 1995) PTP-ε Insulin-R Overexpression (Moller et al., 1995) DEP-1 PDGFR
Met Overexpression Substrate trapping
(Kovalenko et al., 2000) (Palka et al., 2003)
VE-PTP Tie-2 Substrate trapping (Fachinger et al., 1999) PTP-RO c-Kit Co-precipitation (Taniguchi et al., 1999)
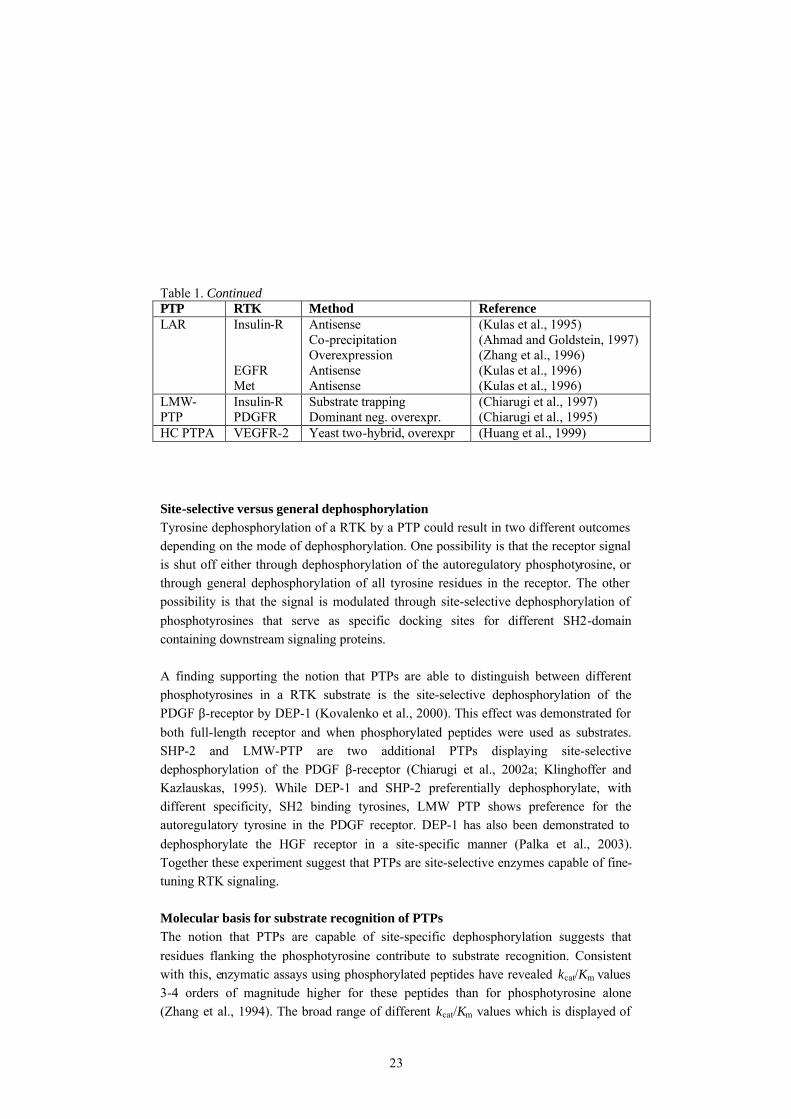
23
Table 1. Continued PTP RTK Method Reference LAR Insulin-R
EGFR Met
Antisense Co-precipitation Overexpression Antisense Antisense
(Kulas et al., 1995) (Ahmad and Goldstein, 1997) (Zhang et al., 1996) (Kulas et al., 1996) (Kulas et al., 1996)
LMW-PTP
Insulin-R PDGFR
Substrate trapping Dominant neg. overexpr.
(Chiarugi et al., 1997) (Chiarugi et al., 1995)
HC PTPA VEGFR-2 Yeast two-hybrid, overexpr (Huang et al., 1999) Site-selective versus general dephosphorylation Tyrosine dephosphorylation of a RTK by a PTP could result in two different outcomes depending on the mode of dephosphorylation. One possibility is that the receptor signal is shut off either through dephosphorylation of the autoregulatory phosphotyrosine, or through general dephosphorylation of all tyrosine residues in the receptor. The other possibility is that the signal is modulated through site-selective dephosphorylation of phosphotyrosines that serve as specific docking sites for different SH2-domain containing downstream signaling proteins. A finding supporting the notion that PTPs are able to distinguish between different phosphotyrosines in a RTK substrate is the site-selective dephosphorylation of the PDGF β-receptor by DEP-1 (Kovalenko et al., 2000). This effect was demonstrated for both full-length receptor and when phosphorylated peptides were used as substrates. SHP-2 and LMW-PTP are two additional PTPs displaying site-selective dephosphorylation of the PDGF β-receptor (Chiarugi et al., 2002a; Klinghoffer and Kazlauskas, 1995). While DEP-1 and SHP-2 preferentially dephosphorylate, with different specificity, SH2 binding tyrosines, LMW PTP shows preference for the autoregulatory tyrosine in the PDGF receptor. DEP-1 has also been demonstrated to dephosphorylate the HGF receptor in a site-specific manner (Palka et al., 2003). Together these experiment suggest that PTPs are site-selective enzymes capable of fine-tuning RTK signaling. Molecular basis for substrate recognition of PTPs The notion that PTPs are capable of site-specific dephosphorylation suggests that residues flanking the phosphotyrosine contribute to substrate recognition. Consistent with this, enzymatic assays using phosphorylated peptides have revealed kcat/Km values 3-4 orders of magnitude higher for these peptides than for phosphotyrosine alone (Zhang et al., 1994). The broad range of different kcat/Km values which is displayed of

24
PTPs for peptides derived from different phoshorylation sites further reinforce the importance of the primary sequence surrounding the phosphotyrosine as specificity determinants. The substrate specificity of PTP1B towards phosphorylated peptides has been well characterized using various biochemical methods such as library affinity-selection and alanine scanning in combination with dephosphorylation assays (Huyer et al., 1998; Pellegrini et al., 1998; Vetter et al., 2000; Zhang et al., 1993). The results from these different approaches are similar; in summary, PTP1B show a preference for acidic or aromatic residues amino-terminal of the phosphotyrosine, in particular at position –1. Basic residues were disliked in any position both amino-terminal and carboxy- terminal of the phosphotyrosine (Vetter et al., 2000). A preference for acidic residues and a non-preference for basic residues in proximal amino-terminal positions, relative to the phosphotyrosine, have also been observed for TC-PTP and SHP-1 (Ruzzene et al., 1993; Wang et al., 2002). Two substrate-trapping mutant forms of PTPs have been crystallized in complex with phosphorylated peptide substrates; PTP1B with peptides derived from the insulin and EGF receptor, and SHP-1 together with peptides derived from the signal regulatory protein SIRP-α (Jia et al., 1995; Salmeen et al., 2000; Yang et al., 2000). These structures reveal interactions between side-chains of residues in the loops surrounding the catalytic cleft and residues adjacent to the phosphotyrosine, consistant with the notion that these residues are specificity determinants. PTP1B has also been co-crystallized in complex with peptides containing aromatic residues in position –1 (Sarmiento et al., 2000). These studies have provided structural basis for plasticity of PTP1B in substrate recognition. Depending on the amino acid sequence amino-terminal of the phosphotyrosine in the substrate, PTP1B can adopt different conformations conferred by a single arginine residue, explaining its ability to accommodate both aromatic and acidic residues in position –1. Sequence alignment has shown that some of the substrate interacting residues are conserved among PTPs and may thus be involved in catalysis and substrate binding mechanisms common to most PTPs, whereas several of the interacting residues exhibit variability and possibly act as specificity determinants (Andersen et al., 2001b). Combined biochemical and structural studies further support the importance of specific side-chain residue interactions for substrate recognition. A glycine residue in PTP1B forms a pocket close to the active site that allows binding to a broad range of peptide substrates, whereas the corresponding residue in PTPα is a bulkier glutamine, restricting substrate binding by steric hindrance. Replacement of the glycine residue with a

25
glutamine residue in PTP1B and vice verse in PTPα, reversed the substrate specificity of the PTPs when assayed for catalytic efficiency using different phosphopeptides (Peters et al., 2000). A catalytic domain switching experiment provides another example supporting the notion that the catalytic domain contains intrinsic substrate specificity. The catalytic domain in SHP-1 was replaced with the catalytic domain of the closely related SHP-2 known to dephosphorylate the EGFR. The PTP chimera, like native SHP-1, showed no activity towards the EGF recepter even though the association ability with the receptor was preserved (Tenev et al., 1997). The structure of SHP-1 in complex with SIRP-α peptides shows substrate interactions in regions not conserved in SHP-2, which could explain the observed SHP-1 specificity (Andersen et al., 2001b). The role of PTPs in redox regulation of RTK signaling Several studies have shown that stimulation of cells of with various growth factors such as PDGF, EGF and insulin induces production of H2O2 (Bae et al., 1997; May and de Haen, 1979; Sundaresan et al., 1995). The increase in H2O2 concentration is crucial for phosphorylation and activation of downstream signaling proteins. Also, exogenously added H2O2 induces tyrosine phosphorylation and activation of signaling pathways. Given the vulnerability of the PTP active site cysteine for oxidation, it is likely that the increase in tyrosine phosphorylation is a consequence of oxidation-induced inactivation of phosphatases. Indeed, accumulating studies demonstrate oxidation of PTPs upon ligand-stimulation of cells. Reduced sensitivity to incorporation of radiolabelled iodoacetc acid has been used as readout for active site cysteine oxidation. Stimulation of cells with EGF resulted in a decrease in iodoacetic acid bound to PTP1B (Lee et al., 1998). The maximal decrease was apparent after 10 minutes and the incorporation returned to baseline after 40 minutes. Similar findings have been demonstrated for LMW-PTP upon stimulation of cells with PDGF (Chiarugi et al., 2001). The reversible oxidation was concurrent with a reversible inhibition of PTP activity. Formation of an intramolecular disulfide bond was proposed as a mechanism for protecting the catalytic site cysteine in LMW-PTP from irreversible oxidation. Inactivation of PTP1B has also been demonstrated following stimulation of cells with insulin (Mahadev et al., 2001). PTP inactivation and insulin-induced tyrosine phosphorylation of the insulin receptor was suppressed when cells were pretreated with the H2O2 scavenger catalase. Recently, a modified in-gel PTP assay was developed to identify oxidized PTPs (Meng et al., 2002). Upon growth factor stimulation, cells were lysed in presence of alkylating agent irreversibly inactivating reduced PTPs. Oxidized PTPs, which do not become alkylated, were then reactivated by

26
addition of reducing agent and visualized by in-gel PTP assay. Using this method, SHP-2 was identified as a phosphatase undergoing oxidation upon PDGF-stimulation of cells. Furthermore, it was revealed that the oxidation was dependent on association of SHP-2 with the PDGF β-receptor. In addition to ligand stimulation and H2O2 treatment, activation of RTKs also occurs upon other stimuli independently of ligand binding. UV-irradiation and stimulation of G-protein coupled receptosr are examples of events that induce RTK trans-activation (Weiss et al., 1997). Interestingly, both these types of stimuli induces H2O2 production in cells (Rittie and Fisher, 2002; Ushio-Fukai et al., 1999). Reversible oxidation of PTPs may have a role in these processes. Consistent with this idea, UV-induced EGFR phosphorylation was linked with a reduced rate in dephosphorylation of the receptor (Knebel et al., 1996). Furthermore, inactivation of SHP-1, PTPα, PTPσ and DEP-1 has been demonstrated upon UV irradiation of cells (Gross et al., 1999). RTKs and PTPs as drug targets Since aberrant receptor tyrosine kinase signaling has been implicated in a variety of diseases, including cancer, cardiovascular disease and fibrosis, they have become attractive drug targets. The well-characterized mechanism of RTK activation and the availability of various structures of intracellular and extracellular domains, have enabled several different approaches in the development of RTK targeted drugs. The different strategies undertaken include development of compounds, which either target the extracellular domain, or the kinase domain, thus inhibiting ligand binding and receptor activation, respectively. Below are described some RTK directed inhibitors, which have shown promising result in recent clinical trials. One way of preventing ligand induced activation of RTK is the use of monoclonal antibodies directed to the extracellular domain (Fan and Mendelsohn, 1998). Herceptin is a monoclonal antibody against the EGFR family member HER2, which has proven successful for treatment of HER2 overexpressing breast cancer (Pegram et al., 1998). The antibody development was based on genetic characerization of breast cancer tumors, which revealed HER2 gene amplification. Low molecular weight compounds that specifically bind to the active site of the kinase have also been developed. Despite the high degree of sequence conservation of the ATP binding site of tyrosine kinases, several compounds that relatively selectively compete with ATP binding to different RTKs have been developed (Shawver et al., 2002).

27
STI571 is a selective inhibitor of the PDGFR, c-Kit and the non-receptor tyrosine kinase Abl (Capdeville et al., 2002). A crystal structure of STI571 in compex with Abl showed that selectivity is conferred by the specific conformation of the unphosphorylated activation loop (Schindler et al., 2000). The drug has shown to be effective in treatment of chronic myeloid leukemia (CML), which is caused by expression of the fusion protein Bcr-Abl (Druker et al., 2001). In addition, it has successfully been used in treatment of gastrointestinal stromal tumors (GIST) which are caused by gain-of-function mutatuins in c-Kit (van Oosterom et al., 2001). STI571 has now gained FDA approval for use in therapy of CML and GIST. The drug has also shown promising results in treatments of chronic myelomonocytic leukemia and dermatofibrosarcoma protuberans where it targets constitutively activated PDGF receptors (Apperley et al., 2002; Rubin et al., 2002). Another example of an RTK kinase inhibitor is the EGFR inhibitor ZD-1839, which displays anti-tumor effect on cells co-expressing EGFR and its ligand TGFα (Ciardiello et al., 2000). In clinical trials ZD-1839 has shown some therapeutic effects in patients with non-small-cell lung cancer (Herbst et al., 2002). The accumulating data that PTPs are important regulators of signal transduction have resulted in a view of them as promising drug targets. The discovery that PTP1B knockout mice exhibit increased insulin receptor autophosphorylation and enhanced sensitivity to insulin has established PTP1B as a therapeutic target for diabetes and obesity. This PTP is the most exploited phosphatase as drug target, and has been subject of numerous screenings and structural studies in search for specific inhibitors. Several inhibitors targeting the catalytic site of PTP1B have been developed. Since the phosphotyrosine-binding site exhibits a highly conserved nature, it has been critical to consider the sourrounding variable sites to achieve specificity. One approach has been to incorporate nonhydrolyzable analogues of phosphotyrosine into peptides, which are optimized for PTP1B recognition (Burke and Zhang, 1998). However, due to their susceptibility for proteolysis, peptides are not optimal for development of effective drugs. Hence, the focus has mainly been on the development of non-peptide low molecular weight drugs. Bis-(para-phosphoenyl) methane (BPPM) has been identified as a high affinity low molecular weight substrate for PTP1B (Montserat et al., 1996). The crystal structure of PTP1B in complex with this compound revealed an additional aryl phosphate binding site, which is not conserved among PTPs, adjacent to the phophotyrosine binding site (Puius et al., 1997). This feature enables design of specific high affinity PTP1B inhibitors, which bind both these sites simultaneously. To date structural studies and library screening have yielded numerous selective low molecular

28
weight PTP1B inhibitors (Andersen et al., 2002; Larsen et al., 2002; Shen et al., 2001). Some of them have also been shown to lower blood glucose levels in diabetic mouse models (Malamas et al., 2000). Other putative drug targets are CD45 and PTPα. CD45 has been suggested to have a positive regulatory role on T cell receptor signaling (Byth et al., 1996). Inhibition of CD45 could thus be a useful treatment against autoimmunity diseases and transplant rejection. Indeed, a monoclonal antibody against CD45 has been shown to prevent renal allograft rejection in mice (Lazarovits et al., 1996). PTPα activates Src family kinases by dephosphorylating the inhibitory carboxy-terminal phosphotyrosine and primary embryonic fibroblasts from PTPα knockout mice displays reduced Src and Fyn activities (Ponniah et al., 1999; Su et al., 1999). Hence, a PTPα inhibitor might have therapeutic effects in tumors with elevated Src kinase activity. Another PTP target for tumor therapy is the Cdc25 family of dual specificity PTPs. They play important roles in regulation of cell cycle by removing inhibitory phosphates on tyrosine and threonine residues of cyclin-dependent kinases. Cdc25B is overexpressed in more than 30% of primary breast tumors and may thus be a potential oncogene (Galaktionov et al., 1995). A significant number of studies have aimed at developing Cdc25 inhibitors as anticancer drugs (Eckstein, 2000).

29
Present investigation Aim The aim of this thesis was to investigate mechanisms whereby PTPs control cellular signaling mediated by RTKs. The purpose was also to explore the role of oxidation as a regulatory mechanism of PTP activity. Ligand stimulation reduces platelet-derived growth factor β-receptor susceptibility to tyrosine dephosphorylation (paper I) Most RTKs are activated by ligand-induced dimerization and subsequent autophosphorylation. The aim of this study was to investigate whether the increase in tyrosine phosphorylation of RTKs that follows ligand stimulation involves decreased sensitivity to the action of PTPs by using the PDGF β-receptor as a prototype RTK. Activation of PDGF receptors is mediated by PDGF-induced dimerization and phosphorylation. Tyrosine phosphorylated unstimulated and ligand-stimulated forms of PDGF β-receptors were prepared by treating PAE cells, stably transfected with PDGF β-receptor, with pervanadate only, PDGF only, or both pervanadate and PDGF-BB. The cell-surface proteins were subjected to cross-linking and the existence of PDGF β-receptor dimers was examined. Dimeric receptors were generated only after stimulation with PDGF-BB, either alone or in combination with pervanadate. SDS-PAGE and immunoblotting revealed that all three preparations contained similar amounts of tyrosine phosphorylation. Two-dimensional phospho-peptide mapping showed that the tyrosine phosphorylation pattern was similar in the different receptor preparations. Furthermore, the pattern of associating SH2-domain containing proteins was similar for unstimulated receptors and receptors stimulated with PDGF-BB. Immunoblotting also confirmed similar amounts of co-precipitating PI3-kinase. The only deviation was a 120 kDa protein associating solely receptors from cells treated with PDGF. To investigate the susceptibility to dephosphorylation of the different PDGF β-receptor preparations, WGA-sepharose immobilized receptors were incubated with recombinant forms of the phosphatases PTP1B and DEP-1. After elution and immuno-precipitation, the phosphotyrosine content was analyzed by immunoblotting. Incubation with PTPs resulted in a dose-dependent decrease of phosphotyrosine in all receptor preparations. However, the ligand-stimulated receptors required 10-100 fold higher amount of PTP to be dephosphorylated to the same level as the unstimulated receptors. This ligand-dependent effect was also seen when PDGF β-receptors in intact cell membranes were incubated with either recombinant DEP-1 or membrane preparations from untransfected PAE cells.

30
In conclusion, ligand stimulated receptors, immobilized on WGA sepharose or in intact cell membranes, were less sensitive to dephosphorylation compared to the unstimulated receptors, both when recombinant PTP or cell membranes where used as source of phosphatase activity. Hence, the increase in RTK tyrosine phosphorylation upon ligand binding is likely to involve a decreased sensitivity to the action of phosphatases. Since cross-linking experiment showed that PDGF β-receptors existed as dimers only in the ligand stimulated cells, the decreased sensitivity may thus be due to dimeric RTKs being intrinsically poorer substrates for PTPs. Another explanation is that PTP activity is regulated upon ligand stimulation, e.g. by a phosphatase inhibitory protein associating with ligand stimulated receptors, or a phosphatase activating protein associating with unstimulated receptors. Primary sequence determinants responsible for site-selective dephosphorylation of the PDGF β-receptor by the receptor-like protein tyrosine phosphatase DEP-1 (Paper II) DEP-1 has previously been shown to site-selectively dephosphorylate the PDGF β-receptor (Kovalenko et al., 2000). The aim of this study was to further investigate the contribution of primary sequences surrounding the phosphotyrosine to the observed site-selectivity. Experiments in which DEP-1 was shown to site-selectively dephosphorylate the PDGF β-receptor revealed that one of the preferred sites was pY1021, whereas pY857 and pY562 were identified as poorer substrates (Kovalenko et al., 2000). In this study, kinetic analyses of wild type and mutated decapeptides corresponding to these sites were performed. Dephosphorylation of wildtype pY1021 peptide and pY857 peptide by recombinant DEP-1 revealed similar kcat values, however the Km for the pY1021 peptide was ten-fold lower than for the pY857 peptide. Hence, the difference in dephosphorylation efficiency is most likely due to a difference in affinity between the peptides. The arginine and lysine in positions -4 and +3 relative to the phosphotyrosine in pY857 were presumed to act as negative determinants since positively charged residues in the same positions also occurred in the poorly dephosphorylated peptide pY562. Consistently, DEP-1 dephosphorylation efficiency was reversed for chimeric peptides, pY1021/pY857, which contained substitutions in positions -4 and +3. Similarly, the pY562 peptide was improved as a DEP-1 substrate when substituted with amino acid residues from the pY1021 peptide in these positions. The introduction of alanine residues in position -4 and +3 in the pY857 peptide resulted in a decrease in Km to nearly the value of the pY857/pY1021 chimeric peptide. This suggests that the

31
improvement is most likely due to removal of the positively charged residues, rather than the specific introduction of residues from the pY1021 sequence. Substitutions of single amino acid residues in the pY857 peptide, with residues from the pY1021 peptide, revealed that the lysine in position +3 was most responsible for the low affinity of the pY857 peptide. Furthermore, substitution of asparagine for aspartic acid in position -1 in the pY857 peptide resulted in a notable increase in affinity. To investigate whether the non-preference by DEP-1 for positively charged residues were exclusive for position +3, lysines were also introduced in other positions carboxy-terminal of the phosphotyrosine in the pY1021 peptide. This revealed that a lysine in position +2 also resulted in a substantial decrease in affinity. In conlusion, basic amino acid residues in position +2 and +3 act as negative determinants and acidic residues in position -1 act as positive determinants in DEP-1 substrate selection. Peptides containing acidic residues amino-terminal of the phosphotyrosine are preferred substrates for other PTPs including TC-PTP and PTP1B (Huyer et al., 1998; Ruzzene et al., 1993). PTP1B also exhibits a strong non-preference for basic amino acids both in amino-terminal and carboxy-terminal positions (Vetter et al., 2000). In contrast, LMW-PTP exhibits a reversed site-selectivity towards PDGF β-derived peptides compared with DEP-1 (Bucciantini et al., 1999). Previously solved structures of PTPs in complex with phosphopeptides have revealed specific side-chain interactions between residues in the catalytic domian and residues from position -4 throughout position +1 in the peptide (Jia et al., 1995; Salmeen et al., 2000; Yang et al., 2000). Our study suggests that additional interactions further carboxy-terminal of the phosphotyrosine are important for substrate recognition. This is consistent with the notion that a unique combination of determinants in the PTP active site confers specific substrate recognition and dephosphorylation in a site-selective manner. Deletion of TC-PTP results in site-selective increase in PDGF -induced receptor tyrosine phosphorylation and enhanced chemotactic response (Paper III) Overexpression of a truncated active form of TC-PTP has been shown to reduce tyrosine phosphorylation of several proteins in PDGF-stimulated cells and TC-PTP was recently shown to associate with the PDGF β-receptor (Cool et al., 1990; Markova et al., 2003). The aim of this study was to investigate the regulatory role, with emphasis on site-selective effects, of TC-PTP on PDGF β-receptor signaling. The approach was to use embryonic fibroblasts (MEFs) derived from TC-PTP knockout mice and littermate wildtype mice. PDGF β-receptors from TC-PTP knockout fibroblast exhibited a fivefold increase in tyrosine phosphorylation upon stimulation with PDGF-DD. The phosphorylation pattern of the PDGF β-receptors was further analyzed by

32
using site-specific phosphotyrosine antibodies. The increase in phosphorylation of the receptors from knockout cells was distributed unevenly between the different phosphotyrosine sites. Y1021 showed more than average increase of phosphorylation whereas Y579 and Y751 showed less than average increase of phosphorylation. The PDGF β-receptor phosphorylation in response to PDGF-DD was also examined in mouse embryonic fibroblasts derived from PTP1B and PTPε knockout mice. The increase in receptor phosphorylation in PTP1B knockout cells was smaller than what was obseved in TC-PTP knockout cells. Furthermore, PDGF β-receptors from PTP1B knockout cells displayed a different distribution of the increase in tyrosine phosphorylation as compared to receptors from TC-PTP knockout cells. In PTPε knockout cells, there was no increase in tyrosine phosphorylation at any sites in the receptors. This suggests that the observed increase in receptor phosphorylation in TC-PTP knockout cells is due to the loss of TC-PTP-mediated dephosphorylation of the PDGF β-receptor. To further investigate whether the different distribution of phosphorylation increase between individual sites was caused by the loss of TC-PTP, an in vitro dephosphorylation assay was performed. Recombinant TC-PTP was incubated with WGA immobilized phosphorylated PDGF β-receptors, and the remaining amount of phosphotyrosine was analyzed by immunoblotting with phosphotyrosine specific antibodies. The sites pY1021 and pY751 were efficiently dephosphorylated by TC-PTP whereas pY771 was a poor TC-PTP substrate. Hence, this indicates that Y1021 is directly dephosphorylated by TC-PTP in vivo, whereas the hyperphosphorylation of Y771 in TC-PTP knockout cells cannot be explained by a direct effect of TC-PTP removal. The fact that pY751 is highly dephosphorylated in vitro but very little affected by TC-PTP loss in vivo, might be explained by the existence of other PTPs with high preference for this site, which compensate for the loss of TC-PTP. Furthermore, we used PDGF β-receptor derived phosphopeptides as substrates for TC-PTP to evaluate the contribution of the primary sequence surrounding the phosphotyrosine to substrate specificity. TC-PTP dephosphorylated peptides containing pY1021, pY751 and pY771 with similar efficiency, indicating that the observed site-selectivity is not due to the amino acid residues surrounding the phosphotyrosine. To study the effects of TC-PTP removal on PDGF β-receptor induced signaling, a cell migration assay was performed. This assay revealed an increase in migration towards PDGF of TC-PTP knockout cells as compared to wild type cells. This is in accordance with the observed hyperphosphorylation of the PLC-γ binding site pY1021 in knockout

33
cells, since PLC-γ has been linked to PDGF-induced chemotaxis (Kundra et al., 1994; Rönnstrand et al., 1999). In conclusion, these findings suggest that TC-PTP dephosphorylates the PDGF β-receptor in a site-selective manner. Furthermore, the site-selective dephosphorylation was consistent with TC-PTP mediated modulation of a biological response to PDGF β-receptor signaling. UV-induced oxidation of RPTPα revealed by a novel generic method for detection of oxidation-inactivated protein tyrosine phosphatases (paper IV) Accumulating data indicate that the specific activity of protein tyrosine phosphatases (PTPs) is regulated by transient and reversible oxidation of the conserved active site cysteine residue following growth factor-induced H2O2 production. The aim of this study was to further explore the role of oxidation as a regulatory mechanism of PTPs.
In order to facilitate studies of this PTP regulatory mechanism, we developed an antibody-based method for detection of oxidized, inactivated PTPs. Affinity purified antibodies (oxPTP) were produced from an antiserum raised against a peptide corresponding to the catalytic site signature motif, VHCSAG, containing sulfonic acid, which is an irreversibly oxidized form of cysteine. In vitro analysis confirmed that the antibody recognized pervanadate oxidized, but not native, forms of recombinant DEP-1 in immunoblotting. Furthermore, the antibody did not recognize a mutant form of DEP-1, in which the active site cysteine residue was changed to a serine residue, confirming the specificity of the antibody for the active site. In addition to DEP-1, we showed that the antibody also recognized oxidized TC-PTP, PTPα and SHP-2, indicating that the oxPTP antibody can be used as a generic tool to detect several oxidized PTPs. To be able to use the antibody to distinguish between reduced and reversibly oxidized PTPs in the cell, the reduced forms were alkylated with iodoacetic acid and thereby protected from oxidation. Subsequently, reversibly oxidized PTPs, which do not react with iodoacetic acid, were further oxidized to sulfonic acid that is recognized by the antibody. Using this strategy we could detect oxidized PTPα in cell lysates upon H2O2-stimulation of cells. The antibody was also used to confirm previous findings that SHP-2 is oxidized upon PDGF stimulation of cells. Furthermore, using the oxPTP antibody, we demonstrated for the first time oxidation of a PTP upon UV-irradiation of cells. Earlier studies have demonstrated UV-induced inactivation of PTPs (Gross et al., 1999). Our finding identifies oxidation of the active site as the mechanism underlying this inactivation.

34
Different methods have been used in assays to detect oxidized PTPs including activity assays and monitoring of alkylation susceptibility of the catalytic cysteine. The advantage of our method is that it detects the increase of oxidized PTPs rather than the decrease of reduced PTPs, which should improve sensitivity when the fraction of oxidized PTP is small. Future perspectives We have shown that ligand-stimulated PDGF β-receptors were less susceptible to dephosphorylation compared to unstimulated receptors. This suggests a previously unrecognized mechanism of ligand induced RTK activation in addition to increase in kinase activity. Future studies should aim at defining the mechanism underlying this finding. A possible candidate with a role in negative regulation of phosphatase activity is the 120 kDa protein associating only with the PDGF-stimulated receptor preparation. The hypothesis of inactivation of PTPs would be consistent with previous findings showing PTP inactivation due to oxidation upon ligand induced production of H2O2 (Chiarugi et al., 2001; Lee et al., 1998; Mahadev et al., 2001; Meng et al., 2002). Whether this mechanism is involved in the current findings remains to be investigated. We have identified possible substrate specificity determinants for DEP-1 in the primary sequence surrounding the phosphotyrosine. A crystal structure of DEP-1 in complex with the preferred peptide substrate would provide important information about the molecular interactions involved in substrate-recognition. In addition, identification and characterisation of higher affinity DEP-1 substrates is warranted. Further understanding of the molecular basis for substrate recognition would be valuable for structure-based design of PTP inhibitors. Our finding that also TC-PTP site-selectively dephosphorylates the PDGF β-receptor supports the notion that PTPs are site-selective enzymes fine-tuning the biological response elicited by RTKs. Further studies would include additional analysis of effects on downstream signaling pathways emanating from the PDGF β-receptor, as well as identification of other PTPs with site-selective effects on RTK dephosphorylation. It will also be interesting to investigate if increased responsiveness to PDGF is a phenotype of TC-PTP knockout mice. The establishment of a novel method for antibody detection of oxidized PTPs creates new possibilities to explore PTP oxidation. Further studies using the antibody should aim at investigating UV-induced PTP oxidation under more physiological conditions,

35
for instance in UV-irradiated skin. The possibility of using the antibody in immunohistochemistry and immunopreciptiation should also be explored. The antibody-based method will be useful in further characterization of oxidative signaling. Constitutive generation of H2O2 has been observed in cell lines transformed with oncogenic Ras (Irani et al., 1997). Moreover overexpression of the ROS generating oxidase Mox1 has been shown to induce transformation (Suh et al., 1999). It has also been demonstrated that ligand-induced H2O2 production is decreased in dense growth-inhibited fibroblasts compared to sparse cell cultures (Pani et al., 2000). Hence, it is interesting to examine the role of PTP oxidative regulation in the context of density dependent growth arrest and cell transformation. Finally, it will be interesting to explore if this antibody-based strategy also is applicable for other enzyme families containing an oxidation sensitive catalytic cysteine within a conserved region, e.g. caspases (Hampton and Orrenius, 1997).

36
Acknowledgements This study was carried out at the Ludwig Institute for Cancer Research. I have encountered lots of people who have made these years fun and exiting. Thank you all! In particular I would like to express my gratitude to Arne Östman, my supervisor, for guiding me through the world of phosphatases with a never-ending enthusiasm, and for creating an inspireing atmosphere in the lab. Carl-Henrik Heldin for sharing your immense scientific knowledge and for running the institute in a way that makes it a friendly and enjoyable place to be at. Past and present members of the GR-lab: Jill, Tobias, Kristian, Carina, Gudrun, Daniel, Masao, Patrick, Olli, Maria, Marina and Akira for being such skilled, helpful, and generous people. It has been fun working together with you! Ulf Hellman for eagerly analyzing all my proteins, maintaining my bike, and never giving up on convincing me that clay-pot chicken is better than Stinas. Ulla Engström for skilful synthesis of a huge amount of peptides and Christer Wernstedt for mass spec analyses. Sherry Mowbray for sharing your expertize in enzyme kinetics and structural biology and Ulrika Magnusson for setting up piles of crystalization screens. All past and present PhD students of the Ludwig Institute: Ninna, Sofia, Anders, Johan, Greger, Katerina, Ann-Sofi, Kaisa, Maria, Åsa, Iwona, Inga, Kaska, and Marcin for being great companions during the “PhD-journey”. Aino, Anita, Aive, Susanne and Lotti for sharing skills and laughs. Eva Grönros for your tremendous willingness to help and give advice. Bengt Ejdesjö, Uffe Hedberg, Lasse Herrmansson, Inger Schönquist, Gullbritt Pettersson, Eva Hallin and Ingegärd Shiller for invaluable help with a great deal of things. My friends outside the lab: Karin, Anette, Cilla, Christina, Malin, Camilla, Josefin, Jenny, Karro, Annika and Lena for lots of fun; holliday trips, parties, sports and telephone shats. You are all great friends! Mats for continuous support and feeding with crepes and chocolate. My parents for your endless encouragement and support in life.

37
References Ahmad, F., and Goldstein, B. J. (1997). Functional association between the insulin
receptor and the transmembrane protein-tyrosine phosphatase LAR in intact cells, J Biol Chem 272, 448-57.
Ahmad, F., Li, P. M., Meyerovitch, J., and Goldstein, B. J. (1995). Osmotic loading of neutralizing antibodies demonstrates a role for protein-tyrosine phosphatase 1B in negative regulation of the insulin action pathway, J Biol Chem 270, 20503-8.
Aicher, B., Lerch, M. M., Müller, T., Schilling, J., and Ullrich, A. (1997). Cellular redistribution of protein tyrosine phosphatases LAR and PTPsigma by inducible proteolytic processing, J Cell Biol 138, 681-696.
Andersen, H. S., Olsen, O. H., Iversen, L. F., Sorensen, A. L., Mortensen, S. B., Christensen, M. S., Branner, S., Hansen, T. K., Lau, J. F., Jeppesen, L. , et al. (2002). Discovery and SAR of a novel selective and orally bioavailable nonpeptide classical competitive inhibitor class of protein-tyrosine phosphatase 1B, J Med Chem 45, 4443-59.
Andersen, J. N., Elson, A., Lammers, R., Romer, J., Clausen, J. T., Moller, K. B., and Moller, N. P. (2001a). Comparative study of protein tyrosine phosphatase-epsilon isoforms: membrane localization confers specificity in cellular signalling, Biochem J 354, 581-90.
Andersen, J. N., Mortensen, O. H., Peters, G. H., Drake, P. G., Iversen, L. F., Olsen, O. H., Jansen, P. G., Andersen, H. S., Tonks, N. K., and Moller, N. P. (2001b). Structural and Evolutionary Relationships among Protein Tyrosine Phosphatase Domains, Mol Cell Biol 21, 7117-7136.
Apperley, J. F., Gardembas, M., Melo, J. V., Russell-Jones, R., Bain, B. J., Baxter, E. J., Chase, A., Chessells, J. M., Colombat, M., Dearden, C. E., et al. (2002). Response to imatinib mesylate in patients with chronic myeloproliferative diseases with rearrangements of the platelet-derived growth factor receptor beta, N Engl J Med 347, 481-7.
Aricescu, A. R., McKinnell, I. W., Halfter, W., and Stoker, A. W. (2002). Heparan sulfate proteoglycans are ligands for receptor protein tyrosine phosphatase sigma, Mol Cell Biol 22, 1881-92.
Arpin, M., Algrain, M., and Louvard, D. (1994). Membrane-actin microfilament connections: an increasing diversity of players related to band 4.1, Curr Opin Cell Biol 6, 136-141.
Bae, Y. S., Kang, S. W., Seo, M. S., Baines, I. C., Tekle, E., Chock, P. B., and Rhee, S. G. (1997). Epidermal growth gactor (EGF)-induced generation of hydrogen peroxide. Role in EGF receptor-mediated tyrosine phosphorylation, J Biol Chem 272, 217-221.
Bandyopadhyay, D., Kusari, A., Kenner, K. A., Liu, F., Chernoff, J., Gustafson, T. A., and Kusari, J. (1997). Protein-tyrosine phosphatase 1B complexes with the insulin receptor in vivo and is tyrosine-phosphorylated in the presence of insulin, J Biol Chem 272, 1639-1645.
Barford, D., Das, A. K., and Egloff, M. P. (1998). The structure and mechanism of protein phosphatases: Insights into catalysis and regulation, Annu Rev Biophys Biomol Struct 27, 133-164.
Barford, D., Flint, A. J., and Tonks, N. K. (1994). Crystal structure of human protein tyrosine phosphatase 1B, Science 263, 1397-1404.

38
Barleon, B., Totzke, F., Herzog, C., Blanke, S., Kremmer, E., Siemeister, G., Marmé, D., and Martiny-Baron, G. (1997). Mapping of the sites for ligand binding and receptor dimerization at the extracellular domain of the vascular endothelial growth factor receptor FLT-1, J Biol Chem 272, 10382-10388.
Barrett, W. C., DeGnore, J. P., Keng, Y. F., Zhang, Z. Y., Yim, M. B., and Chock, P. B. (1999a). Roles of superoxide radical anion in signal transduction mediated by reversible regulation of protein-tyrosine phosphatase 1B, J Biol Chem 274, 34543-34546.
Barrett, W. C., DeGnore, J. P., Konig, S., Fales, H. M., Keng, Y. F., Zhang, Z. Y., Yim, M. B., and Chock, P. B. (1999b). Regulation of PTP1B via glutathionylation of the active site cysteine 215, Biochem 38, 6699-6705.
Baxter, R. M., Secrist, J. P., Vaillancourt, R. R., and Kazlauskas, A. (1998). Full activation of the platelet-derived growth factor beta-receptor kinase involves multiple events, J Biol Chem 273, 17050-17055.
Berg, K. L., Siminovitch, K. A., and Stanley, E. R. (1999). SHP-1 regulation of p62(DOK) tyrosine phosphorylation in macrophages, J Biol Chem 274, 35855-65.
Bilwes, A. M., den Hertog, J., Hunter, T., and Noel, J. P. (1996). Structural basis for inhibition of receptor protein-tyrosine phosphatase-α by dimerization, Nature 382, 555-559.
Binns, K. L., Taylor, P. P., Sicheri, F., Pawson, T., and Holland, S. J. (2000). Phosphorylation of tyrosine residues in the kinase domain and juxtamembrane region regulates the biological and catalytic activities of Eph receptors, Mol Cell Biol 20, 4791-805.
Blanchetot, C., Tertoolen, L. G., and den Hertog, J. (2002). Regulation of receptor protein-tyrosine phosphatase alpha by oxidative stress, Embo J 21, 493-503.
Blume-Jensen, P., and Hunter, T. (2001). Oncogenic kinase signalling, Nature 411, 355-365.
Brady-Kalnay, S. M., Flint, A. J., and Tonks, N. K. (1993). Homophilic binding of PTPµ, a receptor-type protein tyrosine phosphatase, can mediate cell-cell aggregation, J Cell Biol 122, 961-972.
Brady-Kalnay, S. M., and Tonks, N. K. (1995). Protein tyrosine phosphatases as adhesion receptors, Curr Opin Cell Biol 7, 650-657.
Bretscher, A. (1999). Regulation of cortical structure by the ezrin-radixin-moesin protein family, Curr Opin Cell Biol 11, 109-16.
Bucciantini, M., Chiarugi, P., Cirri, P., Taddei, L., Stefani, M., Raugei, G., Nordlund, P., and Ramponi, G. (1999). The low Mr phosphotyrosine protein phosphatase behaves differently when phosphorylated at Tyr131 or Tyr132 by Src kinase, FEBS Lett 456, 73-78.
Buckley, D. A., Cheng, A., Kiely, P. A., Tremblay, M. L., and O'Connor, R. (2002). Regulation of insulin-like growth factor type I (IGF-I) receptor kinase activity by protein tyrosine phosphatase 1B (PTP-1B) and enhanced IGF-I- mediated suppression of apoptosis and motility in PTP-1B-deficient fibroblasts, Mol Cell Biol 22, 1998-2010.
Burke, P., Schooler, K., and Wiley, H. S. (2001). Regulation of epidermal growth factor receptor signaling by endocytosis and intracellular trafficking, Mol Biol Cell 12, 1897-910.
Burke, T. R., Jr., and Zhang, Z. Y. (1998). Protein-tyrosine phosphatases: structure, mechanism, and inhibitor discovery, Biopolymers 47, 225-41.

39
Byth, K. F., Conroy, L. A., Howlett, S., Smith, A. J., May, J., Alexander, D. R., and Holmes, N. (1996). CD45-null transgenic mice reveal a positive regulatory role for CD45 in early thymocyte development, in the selection of CD4+CD8+ thymocytes, and B cell maturation, J Exp Med 183, 1707-18.
Cantley, L. C. (2002). The phosphoinositide 3-kinase pathway, Science 296, 1655-7. Capdeville, R., Buchdunger, E., Zimmermann, J., and Matter, A. (2002). Glivec
(STI571, imatinib), a rationally developed, targeted anticancer drug, Nat Rev Drug Discov 1, 493-502.
Ceresa, B. P., Kao, A. W., Santeler, S. R., and Pessin, J. E. (1998). Inhibition of clathrin-mediated endocytosis selectively attenuates specific insulin receptor signal transduction pathways, Mol Cell Biol 18, 3862-70.
Chen, H. E., Chang, S., Trub, T., and Neel, B. G. (1996). Regulation of colony-stimulating factor 1 receptor signaling by the SH2 domain-containing tyrosine phosphatase SHPTP1, Mol Cell Biol 16, 3685-97.
Chiarugi, P., Cirri, P., Marra, F., Raugei, G., Camici, G., Manao, G., and Ramponi, G. (1997). LMW-PTP is a negative regulator of insulin-mediated mitotic and metabolic signalling, Biochem Biophys Res Commun 238, 676-82.
Chiarugi, P., Cirri, P., Raugei, G., Camici, G., Dolfi, F., Berti, A., and Ramponi, G. (1995). PDGF receptor as a specific in vivo target for low M(r) phosphotyrosine protein phosphatase, FEBS Lett 372, 49-53.
Chiarugi, P., Cirri, P., Taddei, M. L., Giannoni, E., Fiaschi, T., Buricchi, F., Camici, G., Raugei, G., and Ramponi, G. (2002a). Insight into the role of low molecular weight phosphotyrosine phosphatase (LMW-PTP) on platelet-derived growth factor receptor (PDGF- r) signaling. LMW-PTP controls PDGF-r kinase activity through TYR-857 dephosphorylation, J Biol Chem 277, 37331-8.
Chiarugi, P., Cirri, P., Taddei, M. L., Talini, D., Doria, L., Fiaschi, T., Buricchi, F., Giannoni, E., Camici, G., Raugei, G., and Ramponi, G. (2002b). New perspectives in PDGF receptor downregulation: the main role of phosphotyrosine phosphatases, J Cell Sci 115, 2219-32.
Chiarugi, P., Fiaschi, T., Taddei, M. L., Talini, D., Giannoni, E., Raugei, G., and Ramponi, G. (2001). Two vicinal cysteines confer a peculiar redox regulation to low molecular weight protein tyrosine phosphatase in response to platelet- derived growth factor receptor stimulation, J Biol Chem 276, 33478-87.
Ciardiello, F., Caputo, R., Bianco, R., Damiano, V., Pomatico, G., De Placido, S., Bianco, A. R., and Tortora, G. (2000). Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor- selective tyrosine kinase inhibitor, Clin Cancer Res 6, 2053-63.
Clampit, J. E., Meuth, J. L., Smith, H. T., Reilly, R. M., Jirousek, M. R., Trevillyan, J. M., and Rondinone, C. M. (2003). Reduction of protein-tyrosine phosphatase-1B increases insulin signaling in FAO hepatoma cells, Biochem Biophys Res Commun 300, 261-7.
Cool, D. E., Tonks, N. K., Charbonneau, H., Fischer, E. H., and Krebs, E. G. (1990). Expression of a human T-cell protein-tyrosine-phosphatase in baby hamster kidney cells, Proc Natl Acad Sci U S A 87, 7280-4.
Cunnick, J. M., Dorsey, J. F., Mei, L., and Wu, J. (1998). Reversible regulation of SHP-1 tyrosine phosphatase activity by oxidation, Biochem Mol Biol Int 45, 887-894.

40
Dadke, S., Kusari, A., and Kusari, J. (2001). Phosphorylation and activation of protein tyrosine phosphatase (PTP) 1B by insulin receptor, Mol Cell Biochem 221, 147-54.
Dadke, S., Kusari, J., and Chernoff, J. (2000). Down-regulation of insulin signaling by protein-tyrosine phosphatase 1B is mediated by an N-terminal binding region, J Biol Chem 275, 23642-23647.
Darnell, J. E., Jr. (1997). STATs and gene regulation, Science 277, 1630-1635. Denu, J. M., and Dixon, J. E. (1998). Protein tyrosine phosphatases: mechanisms of
catalysis and regulation, Curr Opin Chem Biol 2, 633-41. Denu, J. M., Lohse, D. L., Vijayalakshmi, J., Saper, M. A., and Dixon, J. E. (1996).
Visualization of intermediate and transition-state structures in protein-tyrosine phosphatase catalysis, Proc Natl Acad Sci U S A 93, 2493-8.
Denu, J. M., and Tanner, K. G. (1998). Specific and reversible inactivation of protein tyrosine phosphatases by hydrogen peroxide: Evidence for a sulfenic acid intermediate and implications for redox regulation, Biochemistry 37, 5633-5642.
Denu, J. M., Zhou, G., Guo, Y., and Dixon, J. E. (1995). The catalytic role of aspartic acid-92 in a human dual-specific protein- tyrosine-phosphatase, Biochemistry 34, 3396-403.
Desai, D. M., Sap, J., Schlessinger, J., and Weiss, A. (1993). Ligand-mediated negative regulation of a chimeric transmembrane receptor tyrosine phosphatase, Cell 73, 541-554.
Di Guglielmo, G. M., Drake, P. G., Baass, P. C., Authier, F., Posner, B. I., and Bergeron, J. J. (1998). Insulin receptor internalization and signalling, Mol Cell Biochem 182, 59-63.
Druker, B. J., Talpaz, M., Resta, D. J., Peng, B., Buchdunger, E., Ford, J. M., Lydon, N. B., Kantarjian, H., Capdeville, R., Ohno-Jones, S., and Sawyers, C. L. (2001). Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia, N Engl J Med 344, 1031-1037.
Eckstein, J. W. (2000). Cdc25 as a potential target of anticancer agents, Invest New Drugs 18, 149-56.
Elchebly, M., Payette, P., Michaliszyn, E., Cromlish, W., Collins, S., Loy, A. L., Normandin, D., Cheng, A., Himms-Hagen, J., Chan, C. C., et al. (1999). Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene, Science 283, 1544-1548.
Ellis, L., Clauser, E., Morgan, D. O., Edery, M., Roth, R. A., and Rutter, W. J. (1986). Replacement of insulin receptor tyrosine residues 1162 and 1163 compromises insulin-stimulated kinase activity and uptake of 2-deoxyglucose, Cell 45, 721-732.
Fachinger, G., Deutsch, U., and Risau, W. (1999). Functional interaction of vascular endothelial-protein-tyrosine phosphatase with the angiopoietin receptor Tie-2, Oncogene 18, 5948-53.
Fan, Z., and Mendelsohn, J. (1998). Therapeutic application of anti-growth factor receptor antibodies, Curr Opin Oncol 10, 67-73.
Fantl, W. J., Escobedo, J. A., and Williams, L. T. (1989). Mutations of the platelet-derived growth factor receptor that cause a loss of ligand-induced conformational change, subtle changes in kinase activity, and impaired ability to stimulate DNA synthesis, Mol Cell Biol 9, 4473-4478.
Fauman, E. B., Cogswell, J. P., Lovejoy, B., Rocque, W. J., Holmes, W., Montana, V. G., Piwnica-Worms, H., Rink, M. J., and Saper, M. A. (1998). Crystal structure of

41
the catalytic domain of the human cell cycle control phosphatase, Cdc25A, Cell 93, 617-25.
Feng, G. S. (1999). Shp-2 tyrosine phosphatase: signaling one cell or many, Exp Cell Res 253, 47-54.
Feng, G. S., Hui, C. C., and Pawson, T. (1993). SH2-containing phosphotyrosine phosphatase as a target of protein- tyrosine kinases, Science 259, 1607-11.
Flint, A. J., Tiganis, T., Barford, D., and Tonks, N. K. (1997). Development of "substrate-trapping" mutants to identify physiological substrates of protein tyrosine phosphatases, Proc Natl Acad Sci USA 94, 1680-1685.
Frangioni, J. V., Beahm, P. H., Shifrin, V., Jost, C. A., and Neel, B. G. (1992). The nontransmembrane tyrosine phosphatase PTP-1B localizes to the endoplasmic reticulum via its 35 amino acid C-terminal sequence, Cell 68, 545-560.
Frangioni, J. V., Oda, A., Smith, M., Salzman, E. W., and Neel, B. G. (1993). Calpain-catalyzed cleavage and subcellular relocation of protein phosphotyrosine phosphatase 1B (PTP-1B) in human platelets, EMBO J 12, 4843-4856.
Galaktionov, K., Lee, A. K., Eckstein, J., Draetta, G., Meckler, J., Loda, M., and Beach, D. (1995). CDC25 phosphatases as potential human oncogenes, Science 269, 1575-7.
Garton, A. J., and Tonks, N. K. (1994). PTP-PEST: a protein tyrosine phosphatase regulated by serine phosphorylation, EMBO J 13, 3763-3771.
Gil-Henn, H., Volohonsky, G., and Elson, A. (2001). Regulation of protein-tyrosine phosphatases alpha and epsilon by calpain-mediated proteolytic cleavage, J Biol Chem 276, 31772-9.
Gross, S., Blanchetot, C., Schepens, J., Albet, S., Lammers, R., den Hertog, J., and Hendriks, W. (2002). Multimerization of the protein-tyrosine phosphatase (PTP)-like insulin- dependent diabetes mellitus autoantigens IA-2 and IA-2beta with receptor PTPs (RPTPs). Inhibition of RPTPalpha enzymatic activity, J Biol Chem 277, 48139-45.
Gross, S., Knebel, A., Tenev, T., Neininger, A., Gaestel, M., Herrlich, P., and Böhmer, F. D. (1999). Inactivation of protein-tyrosine phosphatases as mechanism of UV-induced signal transduction, J Biol Chem 274, 26378-26386.
Gu, M., and Majerus, P. W. (1996). The properties of the protein tyrosine phosphatase PTPMEG, J Biol Chem 271, 27751-9.
Guan, K. L., and Dixon, J. E. (1991). Evidence for protein-tyrosine-phosphatase catalysis proceeding via a cysteine-phosphate intermediate, J Biol Chem 266, 17026-30.
Haglund, K., Shimokawa, N., Szymkiewicz, I., and Dikic, I. (2002). Cbl-directed monoubiquitination of CIN85 is involved in regulation of ligand-induced degradation of EGF receptors, Proc Natl Acad Sci USA 99, 12191-12196.
Haj, F. G., Markova, B., Klaman, L. D., Bohmer, F. D., and Neel, B. G. (2003). Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatase-1B, J Biol Chem 278, 739-44.
Haj, F. G., Verveer, P. J., Squire, A., Neel, B. G., and Bastiaens, P. I. (2002). Imaging sites of receptor dephosphorylation by PTP1B on the surface of the endoplasmic reticulum, Science 295, 1708-11.
Hampton, M. B., and Orrenius, S. (1997). Dual regulation of caspase activity by hydrogen peroxide: implications for apoptosis, FEBS Lett 414, 552-6.

42
Herbst, R. S., Maddox, A. M., Rothenberg, M. L., Small, E. J., Rubin, E. H., Baselga, J., Rojo, F., Hong, W. K., Swaisland, H., Averbuch, S. D., et al. (2002). Selective oral epidermal growth factor receptor tyrosine kinase inhibitor ZD1839 is generally well-tolerated and has activity in non- small-cell lung cancer and other solid tumors: results of a phase I trial, J Clin Oncol 20, 3815-25.
Hof, P., Pluskey, S., Dhe-Paganon, S., Eck, M. J., and Shoelson, S. E. (1998). Crystal structure of the tyrosine phosphatase SHP-2, Cell 92, 441-50.
Hoffmann, K. M., Tonks, N. K., and Barford, D. (1997). The crystal structure of domain 1 of receptor protein-tyrosine phosphatase mu, J Biol Chem 272, 27505-27508.
Huang, L., Sankar, S., Lin, C., Kontos, C. D., Schroff, A. D., Cha, E. H., Feng, S. M., Li, S. F., Yu, Z., Van Etten, R. L., et al. (1999). HCPTPA, a protein tyrosine phosphatase that regulates vascular endothelial growth factor receptor-mediated signal transduction and biological activity, J Biol Chem 274, 38183-8.
Hubbard, S. R. (1997). Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog, EMBO J 16, 5572-5581.
Hubbard, S. R. (1999). Structural analysis of receptor tyrosine kinases, Prog Biophys Mol Biol 71, 343-58.
Hubbard, S. R., and Till, J. H. (2000). Protein tyrosine kinase structure and function, Annu Rev Biochem 69, 373-398.
Hubbard, S. R., Wei, L., Ellis, L., and Hendrickson, W. A. (1994). Crystal structure of the tyrosine kinase domain of the human insulin receptor, Nature 372, 746-754.
Huyer, G., Kelly, J., Moffat, J., Zamboni, R., Jia, Z., Gresser, M. J., and Ramachandran, C. (1998). Affinity selection from peptide libraries to determine substrate specificity of protein tyrosine phosphatases, Anal Biochem 258, 19-30.
Huyer, G., Liu, S., Kelly, J., Moffat, J., Payette, P., Kennedy, B., Tsaprailis, G., Gresser, M. J., and Ramachandran, C. (1997). Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate, J Biol Chem 272, 843-51.
Irani, K., Xia, Y., Zweier, J. L., Sollott, S. J., Der, C. J., Fearon, E. R., Sundaresan, M., Finkel, T., and Goldschmidt-Clermont, P. J. (1997). Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts, Science 275, 1649-52.
Irusta, P. M., Luo, Y., Bakht, O., Lai, C. C., Smith, S. O., and DiMaio, D. (2002). Definition of an inhibitory juxtamembrane WW-like domain in the platelet-derived growth factor beta receptor, J Biol Chem 277, 38627-34.
Iversen, L. F., Moller, K. B., Pedersen, A. K., Peters, G. H., Petersen, A. S., Andersen, H. S., Branner, S., Mortensen, S. B., and Moller, N. P. (2002). Structure determination of T cell protein-tyrosine phosphatase, J Biol Chem 277, 19982-90.
Jallal, B., Schlessinger, J., and Ullrich, A. (1992). Tyrosine phosphatase inhibition permits analysis of signal transduction complexes in p185HER2/neu-overexpressing human tumor cells, J Biol Chem 267, 4357-63.
Jia, Z., Barford, D., Flint, A. J., and Tonks, N. K. (1995). Structural basis for phosphotyrosine peptide recognition by protein tyrosine phosphatase 1B, Science 268, 1754-1758.
Jiang, G., den Hertog, J., and Hunter, T. (2000). Receptor-like protein tyrosine phosphatase α homodimerizes on the cell surface, Mol Cell Biol 20, 5917-5929.
Jiang, G., den Hertog, J., Su, J., Noel, J., Sap, J., and Hunter, T. (1999). Dimerization inhibits the activity of receptor-like protein-tyrosine phosphatase-α, Nature 401, 606-610.

43
Joazeiro, C. A., Wing, S. S., Huang, H., Leverson, J. D., Hunter, T., and Liu, Y. C. (1999). The tyrosine kinase negative regulator c-Cbl as a RING-type, E2-dependent ubiquitin-protein ligase, Science 286, 309-312.
Keilhack, H., Muller, M., Bohmer, S. A., Frank, C., Weidner, K. M., Birchmeier, W., Ligensa, T., Berndt, A., Kosmehl, H., Gunther, B., et al. (2001). Negative regulation of Ros receptor tyrosine kinase signaling. An epithelial function of the SH2 domain protein tyrosine phosphatase SHP- 1, J Cell Biol 152, 325-34.
Keilhack, H., Tenev, T., Nyakatura, E., Godovac-Zimmermann, J., Nielsen, L., Seedorf, K., and Bohmer, F. D. (1998). Phosphotyrosine 1173 mediates binding of the protein-tyrosine phosphatase SHP-1 to the epidermal growth factor receptor and attenuation of receptor signaling, J Biol Chem 273, 24839-46.
Kendall, R. L., Rutledge, R. Z., Mao, X., Tebben, A. J., Hungate, R. W., and Thomas, K. A. (1999). Vascular endothelial growth factor receptor KDR tyrosine kinase activity is increased by autophosphorylation of two activation loop tyrosine residues, J Biol Chem 274, 6453-60.
Kenner, K. A., Anyanwu, E., Olefsky, J. M., Kusari, J., Marynen, P., Cassiman, J. J., and van den Berghe, H. (1996). Protein-tyrosine phosphatase 1B is a negative regulator of insulin- and insulin-like growth factor-I-stimulated signaling, J Biol Chem 271, 19810-19816.
Khan, M. N., Baquiran, G., Brule, C., Burgess, J., Foster, B., Bergeron, J. J., and Posner, B. I. (1989). Internalization and activation of the rat liver insulin receptor kinase in vivo, J Biol Chem 264, 12931-40.
Klaman, L. D., Boss, O., Peroni, O. D., Kim, J. K., Martino, J. L., Zabolotny, J. M., Moghal, N., Lubkin, M., Kim, Y. B., Sharpe, A. H., et al. (2000). Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice, Mol Cell Biol 20, 5479-89.
Klinghoffer, R. A., and Kazlauskas, A. (1995). Identification of a putative Syp substrate, the PDGF beta receptor, J Biol Chem 270, 22208-17.
Knebel, A., Rahmsdorf, H. J., Ullrich, A., and Herrlich, P. (1996). Dephosphorylation of receptor tyrosine kinases as target of regulation by radiation, oxidants or alkylating agents, EMBO J 15, 5314-5325.
Kovalenko, M., Denner, K., Sandstrom, J., Persson, C., Gross, S., Jandt, E., Vilella, R., Bohmer, F., and Ostman, A. (2000). Site-selective dephosphorylation of the platelet-derived growth factor beta-receptor by the receptor-like protein-tyrosine phosphatase DEP-1, J Biol Chem 275, 16219-26.
Kozlowski, M., Larose, L., Lee, F., Le, D. M., Rottapel, R., and Siminovitch, K. A. (1998). SHP-1 binds and negatively modulates the c-Kit receptor by interaction with tyrosine 569 in the c-Kit juxtamembrane domain, Mol Cell Biol 18, 2089-99.
Kulas, D. T., Goldstein, B. J., and Mooney, R. A. (1996). The transmembrane protein-tyrosine phosphatase LAR modulates signaling by multiple receptor tyrosine kinases, J Biol Chem 271, 748-754.
Kulas, D. T., Zhang, W.-R., Goldstein, B. J., Furlanetto, R. W., and Mooney, R. A. (1995). Insulin receptor signaling is augmented by antisense inhibition of the protein tyrosine phosphatase LAR, J Biol Chem 270, 2435-2438.
Kundra, V., Escobedo, J. A., Kazlauskas, A., Kim, H. K., Rhee, S. G., Williams, L. T., and Zetter, B. R. (1994). Regulation of chemotaxis by the platelet-derived growth factor receptor-β, Nature 367, 474-476.

44
Lai, W. H., Cameron, P. H., Doherty II, J.-J., Posner, B. I., and Bergeron, J. J. M. (1989). Ligand-mediated autophosphorylation activity of the epidermal growth factor receptor during internalization, J Cell Biol 109, 2751-2760.
Larsen, S. D., Barf, T., Liljebris, C., May, P. D., Ogg, D., O'Sullivan, T. J., Palazuk, B. J., Schostarez, H. J., Stevens, F. C., and Bleasdale, J. E. (2002). Synthesis and biological activity of a novel class of small molecular weight peptidomimetic competitive inhibitors of protein tyrosine phosphatase 1B, J Med Chem 45, 598-622.
Lazarovits, A. I., Poppema, S., Zhang, Z., Khandaker, M., Le Feuvre, C. E., Singhal, S. K., Garcia, B. M., Ogasa, N., Jevnikar, A. M., White, M. H., et al. (1996). Prevention and reversal of renal allograft rejection by antibody against CD45RB, Nature 380, 717-20.
Lee, S. R., Kwon, K. S., Kim, S. R., and Rhee, S. G. (1998). Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor, J Biol Chem 273, 15366-15372.
Liu, F., and Chernoff, J. (1997). Protein tyrosine phosphatase 1B interacts with and is tyrosine phosphorylated by the epidermal growth factor receptor, Biochem J 327, 139-145.
Lohse, D. L., Denu, J. M., Santoro, N., and Dixon, J. E. (1997). Roles of aspartic acid-181 and serine-222 in intermediate formation and hydrolysis of the mammalian protein-tyrosine-phosphatase PTP1, Biochemistry 36, 4568-75.
Lorenz, U., Bergemann, A. D., Steinberg, H. N., Flanagan, J. G., Li, X., Galli, S. J., and Neel, B. G. (1996). Genetic analysis reveals cell type-specific regulation of receptor tyrosine kinase c-Kit by the protein tyrosine phosphatase SHP1, J Exp Med 184, 1111-26.
Lorenzen, J. A., Dadabay, C. Y., and Fischer, E. H. (1995). COOH-terminal sequence motifs target the T cell protein tyrosine phosphatase to the ER and nucleus, J Cell Biol 131, 631-43.
Maeda, N., Nishiwaki, T., Shintani, T., Hamanaka, H., and Noda, M. (1996). 6B4 proteoglycan/phosphacan, an extracellular variant of receptor-like protein-tyrosine phosphatase ζ/RPTPβ, binds pleiotrophin/heparin-binding growth-associated molecule (HB-GAM), J Biol Chem 271, 21446-21452.
Maekawa, K., Imagawa, N., Nagamatsu, M., and Harada, S. (1994). Molecular cloning of a novel protein-tyrosine phosphatase containing a membrane-binding domain and GLGF repeats, FEBS Lett 337, 200-6.
Mahadev, K., Zilbering, A., Zhu, L., and Goldstein, B. J. (2001). Insulin-stimulated hydrogen peroxide reversibly inhibits protein-tyrosine phosphatase 1b in vivo and enhances the early insulin action cascade, J Biol Chem 276, 21938-21942.
Maile, L. A., and Clemmons, D. R. (2002). Regulation of insulin-like growth factor I receptor dephosphorylation by SHPS-1 and the tyrosine phosphatase SHP-2, J Biol Chem 277, 8955-60.
Majeti, R., Bilwes, A. M., Noel, J. P., Hunter, T., and Weiss, A. (1998). Dimerization-induced inhibition of receptor protein tyrosine phosphatase function through an inhibitory wedge, Science 279, 88-91.
Majeti, R., Xu, Z., Parslow, T. G., Olson, J. L., Daikh, D. I., Killeen, N., and Weiss, A. (2000). An inactivating point mutation in the inhibitory wedge of CD45 causes lymphoproliferation and autoimmunity, Cell 103, 1059-1070.

45
Malamas, M. S., Sredy, J., Moxham, C., Katz, A., Xu, W., McDevitt, R., Adebayo, F. O., Sawicki, D. R., Seestaller, L., Sullivan, D., and Taylor, J. R. (2000). Novel benzofuran and benzothiophene biphenyls as inhibitors of protein tyrosine phosphatase 1B with antihyperglycemic properties, J Med Chem 43, 1293-310.
Manning, G., Whyte, D. B., Martinez, R., Hunter, T., and Sudarsanam, S. (2002). The protein kinase complement of the human genome, Science 298, 1912-34.
Markova, B., Herrlich, P., Rönnstrand, L., and Böhmer, F.-D. (2003). Identification of protein tyrosine phosphatases associating with the PDGF receptor, Biochemistry in press.
May, J. M., and de Haen, C. (1979). Insulin-stimulated intracellular hydrogen peroxide production in rat epididymal fat cells, J Biol Chem 254, 2214-20.
Meng, K., Rodriguez-Peña, A., Dimitrov, T., Chen, W., Yamin, M., Noda, M., and Deuel, T. F. (2000). Pleiotrophin signals increased tyrosine phosphorylation of β-catenin through inactivation of the intrinsic catalytic activity of the receptor-type protein tyrosine phosphatase β/ζ , Proc Natl Acad Sci USA 97, 2603-2608.
Meng, T. C., Fukada, T., and Tonks, N. K. (2002). Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo, Mol Cell 9, 387-99.
Milev, P., Friedlander, D. R., Sakurai, T., Karthikeyan, L., Flad, M., Margolis, R. K., Grumet, M., and Margolis, R. U. (1994). Interactions of the chondroitin sulfate proteoglycan phosphacan, the extracellular domain of a receptor-type protein tyrosine phosphatase, with neurons, glia, and neural cell adhesion molecules, J Cell Biol 127, 1703-1715.
Milev, P., Maurel, P., Häring, M., Margolis, R. K., and Margolis, R. U. (1996). TAG-1/axonin-1 Is a high-affinity ligand of neurocan, phosphacan/protein-tyrosine phosphatase-ζ/β, and N-CAM, J Biol Chem 271, 15716-15723.
Mohammadi, M., Dikic, I., Sorokin, A., Burgess, W. H., Jaye, M., and Schlessinger, J. (1996a). Identification of six novel autophosphorylation sites on fibroblast growth factor receptor 1 and elucidation of their importance in receptor activation and signal transduction, Mol Cell Biol 16, 977-989.
Mohammadi, M., Schlessinger, J., and Hubbard, S. R. (1996b). Structure of the FGF receptor tyrosine kinase domain reveals a novel autoinhibitory mechanism, Cell 86, 577-587.
Moller, N. P., Moller, K. B., Lammers, R., Kharitonenkov, A., Hoppe, E., Wiberg, F. C., Sures, I., and Ullrich, A. (1995). Selective down-regulation of the insulin receptor signal by protein- tyrosine phosphatases alpha and epsilon, J Biol Chem 270, 23126-31.
Moller, N. P., Moller, K. B., Lammers, R., Kharitonenkov, A., Sures, I., and Ullrich, A. (1994). Src kinase associates with a member of a distinct subfamily of protein- tyrosine phosphatases containing an ezrin-like domain, Proc Natl Acad Sci U S A 91, 7477-81.
Montserat, J., Chen, L., Lawrence, D. S., and Zhang, Z. Y. (1996). Potent low molecular weight substrates for protein-tyrosine phosphatase, J Biol Chem 271, 7868-72.
Mourey, R. J., and Dixon, J. E. (1994). Protein tyrosine phosphatases: Characterization of extracellular and intracellular domains, Curr Opin Genet Dev 4, 31-39.
Nam, H. J., Poy, F., Krueger, N. X., Saito, H., and Frederick, C. A. (1999). Crystal structure of the tandem phosphatase domains of RPTP LAR, Cell 97, 449-457.

46
Nishiya, N., Iwabuchi, Y., Shibanuma, M., Cote, J. F., Tremblay, M. L., and Nose, K. (1999). Hic-5, a paxillin homologue, binds to the protein-tyrosine phosphatase PEST (PTP-PEST) through its LIM 3 domain, J Biol Chem 274, 9847-9853.
Ogiso, H., Ishitani, R., Nureki, O., Fukai, S., Yamanaka, M., Kim, J. H., Saito, K., Sakamoto, A., Inoue, M., Shirouzu, M., and Yokoyama, S. (2002). Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains, Cell 110, 775-87.
O'Grady, P., Thai, T. C., and Saito, H. (1998). The laminin-nidogen complex is a ligand for a specific splice isoform of the transmembrane protein tyrosine phosphatase LAR, J Cell Biol 141, 1675-1684.
Oh, E. S., Gu, H., Saxton, T. M., Timms, J. F., Hausdorff, S., Frevert, E. U., Kahn, B. B., Pawson, T., Neel, B. G., and Thomas, S. M. (1999). Regulation of early events in integrin signaling by protein tyrosine phosphatase SHP-2, Mol Cell Biol 19, 3205-15.
Olayioye, M. A., Neve, R. M., Lane, H. A., and Hynes, N. E. (2000). The ErbB signaling network: receptor heterodimerization in development and cancer, EMBO J 19, 3159-3167.
Ornitz, D. M., Xu, J., Colvin, J. S., McEwen, D. G., MacArthur, C. A., Coulier, F., Gao, G., and Goldfarb, M. (1996). Receptor specificity of the fibroblast growth factor family, J Biol Chem 271, 15292-7.
Palka, H. L., Park, M., and Tonks, N. K. (2003). Hepatocyte Growth Factor Receptor Tyrosine Kinase Met Is a Substrate of the Receptor Protein-tyrosine Phosphatase DEP-1, J Biol Chem 278, 5728-35.
Pani, G., Colavitti, R., Bedogni, B., Anzevino, R., Borrello, S., and Galeotti, T. (2000). A redox signaling mechanism for density-dependent inhibition of cell growth, J Biol Chem 275, 38891-9.
Pannifer, A. D., Flint, A. J., Tonks, N. K., and Barford, D. (1998). Visualization of the cysteinyl-phosphate intermediate of a protein- tyrosine phosphatase by x-ray crystallography, J Biol Chem 273, 10454-62.
Paulson, R. F., Vesely, S., Siminovitch, K. A., and Bernstein, A. (1996). Signalling by the W/Kit receptor tyrosine kinase is negatively regulated in vivo by the protein tyrosine phosphatase Shp1, Nat Genet 13, 309-15.
Pawson, T., and Scott, J. D. (1997). Signaling through scaffold, anchoring, and adaptor proteins, Science 278, 2075-2080.
Pegram, M. D., Lipton, A., Hayes, D. F., Weber, B. L., Baselga, J. M., Tripathy, D., Baly, D., Baughman, S. A., Twaddell, T., Glaspy, J. A., and Slamon, D. J. (1998). Phase II study of receptor-enhanced chemosensitivity using recombinant humanized anti-p185HER2/neu monoclonal antibody plus cisplatin in patients with HER2/neu-overexpressing metastatic breast cancer refractory to chemotherapy treatment, J Clin Oncol 16, 2659-71.
Peles, E., Nativ, M., Campbell, P. L., Sakurai, T., Martinez, R., Lev, S., Clary, D. O., Schilling, J., Barnea, G., Plowman, G. D., et al. (1995). The carbonic anhydrase domain of receptor tyrosine phosphatase β is a functional ligand for the axonal cell recognition molecule contactin, Cell 82, 251-260.
Peles, E., Schlessinger, J., and Grumet, M. (1998). Multi-ligand interactions with receptor-like protein tyrosine phosphatase beta: implications for intercellular signaling, Trends Biochem Sci 23, 121-124.

47
Pellegrini, M. C., Liang, H., Mandiyan, S., Wang, K., Yuryev, A., Vlattas, I., Sytwu, T., Li, Y. C., and Wennogle, L. P. (1998). Mapping the subsite preferences of protein tyrosine phosphatase PTP-1B using combinatorial chemistry approaches, Biochemistry 37, 15598-606.
Peters, G. H., Iversen, L. F., Branner, S., Andersen, H. S., Mortensen, S. B., Olsen, O. H., Moller, K. B., and Moller, N. P. (2000). Residue 259 is a key determinant of substrate specificity of protein- tyrosine phosphatases 1B and alpha, J Biol Chem 275, 18201-9.
Petrelli, A., Gilestro, G. F., Lanzardo, S., Comoglio, P. M., Migone, N., and Giordano, S. (2002). The endophilin-CIN85-Cbl complex mediates ligand-dependent downregulation of c-Met, Nature 416, 187-90.
Plotnikov, A. N., Schlessinger, J., Hubbard, S. R., and Mohammad, M. (1999). Structural basis for FGF receptor dimerization and activation, Cell 98, 641–650.
Ponniah, S., Wang, D. Z., Lim, K. L., and Pallen, C. J. (1999). Targeted disruption of the tyrosine phosphatase PTPalpha leads to constitutive downregulation of the kinases Src and Fyn, Curr Biol 9, 535-8.
Puius, Y. A., Zhao, Y., Sullivan, M., Lawrence, D. S., Almo, S. C., and Zhang, Z. Y. (1997). Identification of a second aryl phosphate-binding site in protein- tyrosine phosphatase 1B: a paradigm for inhibitor design, Proc Natl Acad Sci U S A 94, 13420-5.
Reynolds, R. A., Yem, A. W., Wolfe, C. L., Deibel, M. R., Jr., Chidester, C. G., and Watenpaugh, K. D. (1999). Crystal structure of the catalytic subunit of Cdc25B required for G2/M phase transition of the cell cycle, J Mol Biol 293, 559-68.
Rittie, L., and Fisher, G. J. (2002). UV-light-induced signal cascades and skin aging, Ageing Res Rev 1, 705-20.
Robinson, D. R., Wu, Y. M., and Lin, S. F. (2000). The protein tyrosine kinase family of the human genome, Oncogene 19, 5548-57.
Rocchi, S., Tartare-Deckert, S., Sawka-Verhelle, D., Gamha, A., and van Obberghen, E. (1996). Interaction of SH2-containing protein tyrosine phosphatase 2 with the insulin receptor and the insulin-like growth factor-I receptor: studies of the domains involved using the yeast two-hybrid system, Endocrinology 137, 4944-52.
Ronnstrand, L., Arvidsson, A. K., Kallin, A., Rorsman, C., Hellman, U., Engstrom, U., Wernstedt, C., and Heldin, C. H. (1999). SHP-2 binds to Tyr763 and Tyr1009 in the PDGF beta-receptor and mediates PDGF-induced activation of the Ras/MAP kinase pathway and chemotaxis, Oncogene 18, 3696-702.
Rubin, B. P., Schuetze, S. M., Eary, J. F., Norwood, T. H., Mirza, S., Conrad, E. U., and Bruckner, J. D. (2002). Molecular targeting of platelet-derived growth factor B by imatinib mesylate in a patient with metastatic dermatofibrosarcoma protuberans., J Clin Oncol 20, 3586-3591.
Ruzzene, M., Donella-Deana, A., Marin, O., Perich, J. W., Ruzza, P., Borin, G., Calderan, A., and Pinna, L. A. (1993). Specificity of T-cell protein tyrosine phosphatase toward phosphorylated synthetic peptides, Eur J Biochem 211, 289-95.
Rönnstrand, L., Siegbahn, A., Rorsman, C., Johnell, M., Hansen, K., and Heldin, C.-H. (1999). Overactivation of phospholipase C-γ1 renders platelet-derived growth factor β-receptor-expressing cells independent of the phosphatidylinositol 3-kinase pathway for chemotaxis, J Biol Chem 274, 22089-22094.

48
Salmeen, A., Andersen, J. N., Myers, M. P., Tonks, N. K., and Barford, D. (2000). Molecular basis for the dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B, Mol Cell 6, 1401-12.
Sap, J., Jiang, Y.-P., Friedlander, D., Grumet, M., and Schlessinger, J. (1994). Receptor tyrosine phosphatase R-PTP-κ mediates homophilic binding, Mol Cell Biol 14, 1-9.
Sarmiento, M., Puius, Y. A., Vetter, S. W., Keng, Y. F., Wu, L., Zhao, Y., Lawrence, D. S., Almo, S. C., and Zhang, Z. Y. (2000). Structural basis of plasticity in protein tyrosine phosphatase 1B substrate recognition, Biochemistry 39, 8171-9.
Savitsky, P. A., and Finkel, T. (2002). Redox regulation of Cdc25C, J Biol Chem 277, 20535-40.
Schindler, T., Bornmann, W., Pellicena, P., Miller, W. T., Clarkson, B., and Kuriyan, J. (2000). Structural mechanism for STI-571 inhibition of abelson tyrosine kinase, Science 289, 1938-42.
Schlessinger, J. (2000). Cell signaling by receptor tyrosine kinases, Cell 103, 211-225. Schlessinger, J., Plotnikov, A. N., Ibrahimi, O. A., Eliseenkova, A. V., Yeh, B. K.,
Yayon, A., Linhardt, R. J., and Mohammadi, M. (2000). Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization, Mol Cell 6, 743-750.
Shawver, L. K., Slamon, D., and Ullrich, A. (2002). Smart drugs: tyrosine kinase inhibitors in cancer therapy, Cancer Cell 1, 117-23.
Shen, K., Keng, Y. F., Wu, L., Guo, X. L., Lawrence, D. S., and Zhang, Z. Y. (2001). Acquisition of a specific and potent PTP1B inhibitor from a novel combinatorial library and screening procedure, J Biol Chem 276, 47311-9.
Shen, Y., Lyons, P., Cooley, M., Davidson, D., Veillette, A., Salgia, R., Griffin, J. D., and Schaller, M. D. (2000). The noncatalytic domain of protein-tyrosine phosphatase-PEST targets paxillin for dephosphorylation in vivo, J Biol Chem 275, 1405-13.
Shi, Z. Q., Lu, W., and Feng, G. S. (1998). The Shp-2 tyrosine phosphatase has opposite effects in mediating the activation of extracellular signal-regulated and c-Jun NH2-terminal mitogen-activated protein kinases, J Biol Chem 273, 4904-8.
Shultz, L. D., Rajan, T. V., and Greiner, D. L. (1997). Severe defects in immunity and hematopoiesis caused by SHP-1 protein- tyrosine-phosphatase deficiency, Trends Biotechnol 15, 302-7.
Song, H., Hanlon, N., Brown, N. R., Noble, M. E., Johnson, L. N., and Barford, D. (2001). Phosphoprotein-protein interactions revealed by the crystal structure of kinase-associated phosphatase in complex with phosphoCDK2, Mol Cell 7, 615-26.
Songyang, Z., Shoelson, S. E., Chaudhuri, M., Gish, G., Pawson, T., Haser, W. G., King, F., Roberts, T., Ratnofsky, S., Lechleider, R. J., et al. (1993). SH2 domains recognize specific phosphopeptide sequences, Cell 72, 767-778.
Soubeyran, P., Kowanetz, K., Szymkiewicz, I., Langdon, W. Y., and Dikic, I. (2002). Cbl-CIN85-endophilin complex mediates ligand-induced downregulation of EGF receptors, Nature 416, 183-187.
Spivak-Kroizman, T., Lemmon, M. A., Dikic, I., Ladbury, J. E., Pinchasi, D., Huang, J., Jaye, M., Crumley, G., Schlessinger, J., and Lax, I. (1994). Heparin-induced oligomerization of FGF molecules is responsible for FGF receptor dimerization, activation, and cell proliferation, Cell 79, 1015-1024.

49
Stewart, A. E., Dowd, S., Keyse, S. M., and McDonald, N. Q. (1999). Crystal structure of the MAPK phosphatase Pyst1 catalytic domain and implications for regulated activation, Nat Struct Biol 6, 174-81.
Streuli, M., Krueger, N. X., Ariniello, P. D., Tang, M., Munro, J. M., Blattler, W. A., Adler, D. A., Disteche, C. M., and Saito, H. (1992). Expression of the receptor-linked protein tyrosine phosphatase LAR: proteolytic cleavage and shedding of the CAM-like extracellular region, EMBO J 11, 897-907.
Stuckey, J. A., Schubert, H. L., Fauman, E. B., Zhang, Z. Y., Dixon, J. E., and Saper, M. A. (1994). Crystal structure of Yersinia protein tyrosine phosphatase at 2.5 A and the complex with tungstate, Nature 370, 571-575.
Su, J., Muranjan, M., and Sap, J. (1999). Receptor protein tyrosine phosphatase alpha activates Src-family kinases and controls integrin-mediated responses in fibroblasts, Curr Biol 9, 505-11.
Suarez Pestana, E., Tenev, T., Gross, S., Stoyanov, B., Ogata, M., and Bohmer, F. D. (1999). The transmembrane protein tyrosine phosphatase RPTPsigma modulates signaling of the epidermal growth factor receptor in A431 cells, Oncogene 18, 4069-79.
Suh, Y. A., Arnold, R. S., Lassegue, B., Shi, J., Xu, X., Sorescu, D., Chung, A. B., Griendling, K. K., and Lambeth, J. D. (1999). Cell transformation by the superoxide-generating oxidase Mox1, Nature 401, 79-82.
Sun, P. D., and Davies, D. R. (1995). The cystine-knot growth-factor superfamily, Annu Rev Biophys Bipmol Struct 24, 269-291.
Sundaresan, M., Yu, Z. X., Ferrans, V. J., Irani, K., and Finkel, T. (1995). Requirement for generation of H2O2 for platelet-derived growth factor signal transduction, Science 270, 296-299.
Szedlacsek, S. E., Aricescu, A. R., Fulga, T. A., Renault, L., and Scheidig, A. J. (2001). Crystal structure of PTP-SL/PTPBR7 catalytic domain: implications for MAP kinase regulation, J Mol Biol 311, 557-68.
Sörby, M., Sandström, J., and Östman, A. (2001). An extracellular ligand increases the specific activity of the receptor-like protein tyrosine phosphatase DEP-1, Oncogene 20, 5219-5224.
Takeda, A., Wu, J. J., and Maizel, A. L. (1992). Evidence for monomeric and dimeric forms of CD45 associated with a 30- kDa phosphorylated protein, J Biol Chem 267, 16651-9.
Taniguchi, Y., London, R., Schinkmann, K., Jiang, S., and Avraham, H. (1999). The receptor protein tyrosine phosphatase, PTP-RO, is upregulated during megakaryocyte differentiation and Is associated with the c-Kit receptor, Blood 94, 539-49.
Tauchi, T., Feng, G. S., Marshall, M. S., Shen, R., Mantel, C., Pawson, T., and Broxmeyer, H. E. (1994). The ubiquitously expressed Syp phosphatase interacts with c-kit and Grb2 in hematopoietic cells, J Biol Chem 269, 25206-11.
Tenev, T., Keilhack, H., Tomic, S., Stoyanov, B., Stein-Gerlach, M., Lammers, R., Krivtsov, A. V., Ullrich, A., and Bohmer, F. D. (1997). Both SH2 domains are involved in interaction of SHP-1 with the epidermal growth factor receptor but cannot confer receptor-directed activity to SHP-1/SHP-2 chimera, J Biol Chem 272, 5966-73.

50
Tiganis, T., Bennett, A. M., Ravichandran, K. S., and Tonks, N. K. (1998). Epidermal growth factor receptor and the adaptor protein p52Shc are specific substrates of T-cell protein tyrosine phosphatase, Mol Cell Biol 18, 1622-1634.
Tonks, N. K., and Neel, B. G. (2001). Combinatorial control of the specificity of protein tyrosine phosphatases, Curr Opin Cell Biol 13, 182-195.
Umeda, S., Beamer, W. G., Takagi, K., Naito, M., Hayashi, S., Yonemitsu, H., Yi, T., and Shultz, L. D. (1999). Deficiency of SHP-1 protein-tyrosine phosphatase activity results in heightened osteoclast function and decreased bone density, Am J Pathol 155, 223-33.
Ushio-Fukai, M., Alexander, R. W., Akers, M., Yin, Q., Fujio, Y., Walsh, K., and Griendling, K. K. (1999). Reactive oxygen species mediate the activation of Akt/protein kinase B by angiotensin II in vascular smooth muscle cells, J Biol Chem 274, 22699-704.
Walchli, S., Curchod, M. L., Gobert, R. P., Arkinstall, S., and Hooft van Huijsduijnen, R. (2000). Identification of tyrosine phosphatases that dephosphorylate the insulin receptor. A brute force approach based on "substrate-trapping" mutants, J Biol Chem 275, 9792-6.
Wallace, M. J., Fladd, C., Batt, J., and Rotin, D. (1998). The second catalytic domain of protein tyrosine phosphatase delta (PTP delta) binds to and inhibits the first catalytic domain of PTP sigma, Mol Cell Biol 18, 2608-2616.
van der Geer, P., Wiley, S., Gish, G. D., Lai, V. K., Stephens, R., White, M. F., Kaplan, D., and Pawson, T. (1996). Identification of residues that control specific binding of the Shc phosphotyrosine-binding domain to phosphotyrosine sites, Proc Natl Acad Sci U S A 93, 963-8.
van Oosterom, A. T., Judson, I., Verweij, J., Stroobants, S., Donato di Paola, E., Dimitrijevic, S., Martens, M., Webb, A., Sciot, R., Van Glabbeke, M., et al. (2001). Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study, Lancet 358, 1421-1423.
Wang, J., and Bixby, J. L. (1999). Receptor tyrosine phosphatase-delta is a homophilic, neurite-promoting cell adhesion molecular for CNS neurons, Mol Cell Neurosci 14, 370-384.
Wang, P., Fu, H., Snavley, D. F., Freitas, M. A., and Pei, D. (2002). Screening combinatorial libraries by mass spectrometry. 2. Identification of optimal substrates of protein tyrosine phosphatase SHP-1, Biochemistry 41, 6202-10.
Wang, Y., Guo, W., Liang, L., and Esselman, W. J. (1999). Phosphorylation of CD45 by casein kinase 2. Modulation of activity and mutational analysis, J Biol Chem 274, 7454-61.
Waterman, H., and Yarden, Y. (2001). Molecular mechanisms underlying endocytosis and sorting of ErbB receptor tyrosine kinases, FEBS Lett 490, 142-52.
Weiss, F. U., Daub, H., and Ullrich, A. (1997). Novel mechanisms of RTK signal generation, Curr Opin Genet Dev 7, 80-6.
Wells, A., Welsh, J. B., Lazar, C. S., Wiley, H. S., Gill, G. N., and Rosenfeld, M. G. (1990). Ligand-induced transformation by a noninternalizing epidermal growth factor receptor, Science 247, 962-964.
Vetter, S. W., Keng, Y. F., Lawrence, D. S., and Zhang, Z. Y. (2000). Assessment of protein-tyrosine phosphatase 1B substrate specificity using "inverse alanine scanning", J Biol Chem 275, 2265-8.

51
Wiesmann, C., Fuh, G., Christinger, H. W., Eigenbrot, C., Wells, J. A., and de Vos, A. M. (1997). Crystal structure at 1.7 Å resolution of VEGF in complex with domain 2 of the Flt-1 receptor, Cell 91, 695-704.
Villa-Moruzzi, E., Puntoni, F., Bardelli, A., Vigna, E., De Rosa, S., and Comoglio, P. M. (1998). Protein tyrosine phosphatase PTP-S binds to the juxtamembrane region of the hepatocyte growth factor receptor Met, Biochem J 336, 235-9.
Worthylake, R., Opresko, L. K., and Wiley, H. S. (1999). ErbB-2 amplification inhibits down-regulation and induces constitutive activation of both ErbB-2 and epidermal growth factor receptors, J Biol Chem 274, 8865-74.
Wybenga-Groot, L. E., Baskin, B., Ong, S. H., Tong, J., Pawson, T., and Sicheri, F. (2001). Structural basis for autoinhibition of the EphB2 receptor tyrosine kinase by the unphosphorylated juxtamembrane region, Cell 106, 745-757.
Xu, W., Doshi, A., Lei, M., Eck, M. J., and Harrison, S. C. (1999). Crystal structures of c-Src reveal features of its autoinhibitory mechanism, Mol Cell 3, 629-38.
Yamashita, T., Yoshioka, M., and Itoh, N. (2000). Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain, Biochem Biophys Res Commun 277, 494-8.
Yang, J., Cheng, Z., Niu, T., Liang, X., Zhao, Z. J., and Zhou, G. W. (2000). Structural basis for substrate specificity of protein-tyrosine phosphatase SHP-1, J Biol Chem 275, 4066-71.
Yang, J., Liu, L., He, D., Song, X., Liang, X., Zhao, Z. J., and Zhou, G. W. (2003). Crystal Structure of Human Protein-tyrosine Phosphatase SHP-1, J Biol Chem 278, 6516-20.
Yu, Z., Su, L., Hoglinger, O., Jaramillo, M. L., Banville, D., and Shen, S. H. (1998). SHP-1 associates with both platelet-derived growth factor receptor and the p85 subunit of phosphatidylinositol 3-kinase, J Biol Chem 273, 3687-94.
Yuvaniyama, J., Denu, J. M., Dixon, J. E., and Saper, M. A. (1996). Crystal structure of the dual specificity protein phosphatase VHR, Science 272, 1328-31.
Zhang, J., Somani, A. K., and Siminovitch, K. A. (2000). Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signalling, Semin Immunol 12, 361-78.
Zhang, M., Stauffacher, C. V., Lin, D., and Van Etten, R. L. (1998). Crystal structure of a human low molecular weight phosphotyrosyl phosphatase. Implications for substrate specificity, J Biol Chem 273, 21714-20.
Zhang, S. H., Eckberg, W. R., Yang, Q., Samatar, A. A., and Tonks, N. K. (1995). Biochemical characterization of a human band 4.1-related protein- tyrosine phosphatase, PTPH1, J Biol Chem 270, 20067-72.
Zhang, W. R., Li, P. M., Oswald, M. A., and Goldstein, B. J. (1996). Modulation of insulin signal transduction by eutopic overexpression of the receptor-type protein-tyrosine phosphatase LAR, Mol Endocrinol 10, 575-84.
Zhang, Z. Y., and Dixon, J. E. (1993). Active site labeling of the Yersinia protein tyrosine phosphatase: the determination of the pKa of the active site cysteine and the function of the conserved histidine 402, Biochemistry 32, 9340-5.
Zhang, Z. Y., Maclean, D., McNamara, D. J., Sawyer, T. K., and Dixon, J. E. (1994). Protein tyrosine phosphatase substrate specificity: size and phosphotyrosine positioning requirements in peptide substrates, Biochemistry 33, 2285-90.

52
Zhang, Z. Y., Thieme-Sefler, A. M., Maclean, D., McNamara, D. J., Dobrusin, E. M., Sawyer, T. K., and Dixon, J. E. (1993). Substrate specificity of the protein tyrosine phosphatases, Proc Natl Acad Sci U S A 90, 4446-50.
Zheng, X. M., and Shalloway, D. (2001). Two mechanisms activate PTPalpha during mitosis, Embo J 20, 6037-49.
Östman, A., and Böhmer, F.-D. (2001). Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatases, TICB 11, 258-266.