twolysineresiduesinthebacterialluciferasemobileloop ... · stabilizereactionintermediates ......
TRANSCRIPT

Two Lysine Residues in the Bacterial Luciferase Mobile LoopStabilize Reaction IntermediatesReceived for publication, June 9, 2009, and in revised form, August 22, 2009 Published, JBC Papers in Press, August 26, 2009, DOI 10.1074/jbc.M109.031716
Zachary T. Campbell‡ and Thomas O. Baldwin§1
From the ‡Department of Biochemistry and Molecular Biophysics, University of Arizona, Biological Sciences West,Tucson, Arizona 85721-0088 and the §Department of Biochemistry, University of California at Riverside, Riverside, California 92521
Bacterial luciferase catalyzes the reaction of FMNH2, O2, anda long chain aliphatic aldehyde, yielding FMN, carboxylic acid,and blue-green light. The most conserved contiguous region ofthe primary sequence corresponds to a crystallographically dis-ordered loop adjacent to the active center (Fisher, A. J., Raushel,F. M., Baldwin, T. O., and Rayment, I. (1995) Biochemistry 34,6581–6586; Fisher, A. J., Thompson, T. B., Thoden, J. B., Bald-win, T. O., and Rayment, I. (1996) J. Biol. Chem. 271, 21956–21968). Deletion of themobile loop does not alter the chemistryof the reaction but decreases the total quantum yield of biolu-minescence by 2 orders of magnitude (Sparks, J. M., and Bald-win, T. O. (2001) Biochemistry 40, 15436–15443). In this study,we attempt to localize the loss of activity observed in the loopdeletionmutant to individual residues in themobile loop. Usingalanine mutagenesis, the effects of substitution at 15 of the 29mobile loop residues were examined. Nine of the point mutantshad reduced activity in vivo. Twomutations, K283A andK286A,resulted in a loss in quantum yield comparable with that of theloop deletion mutant. The bioluminescence emission spectrumof both mutants was normal, and both yielded the carboxylicacid chemical product at the same efficiency as the wild-typeenzyme. Substitution of Lys283 with alanine resulted in destabi-lization of intermediate II, whereas mutation of Lys286 had anincrease in exposure of reaction intermediates to a dynamicquencher. Based on amodel of the enzyme-reduced flavin com-plex, the two critical lysine residues are adjacent to the quininoi-dal edge of the isoalloxazine.
The initial step in the luciferase reaction mechanism is thereversible binding of FMNH2 (Fig. 1 andRef. 1). Enzyme-boundFMNH2 subsequently reacts with molecular oxygen yieldingthe 4a,5-dihydro-4a-hydroperoxyflavin or intermediate II (1,2). Intermediate II is unstable at room temperature but is suffi-ciently stable at low temperatures to allow for chromatographicpurification in the presence of long chain alcohols (3, 4). Inter-mediate II reacts with a long chain aliphatic aldehyde to formintermediate IIA (1, 5). The decomposition of intermediate IIAresults in the formation of water, FMN, carboxylic acid, and aphoton of blue-green light (1, 5).Luciferase is a heterodimer composed of two homologous
subunits designated � and � (6). Although the � subunit is
required for activity, the catalytic site resides exclusively on the� subunit (7, 8). The majority of mutations known to result inkinetic defects reside within a single solvent-exposed cleft onthe � subunit (7, 9, 10). Near this cavity is the most stringentlyconserved region of the enzyme, spanning residues 262–290 onthe� subunit (Fig. 2a). This portion of the enzyme correspondsto a protease-labile mobile loop disordered in the two reportedsubstrate-free luciferase structures (10–12). The location of theactive center has recently been experimentally demonstrated(13). The asymmetric unit of crystals of the luciferase-flavincomplex contained� subunits from twonon-symmetry-relatedheterodimers. Neither � subunit contained interpretable elec-tron density for a small segment between residues 283 and 290(13) (Fig. 2b). Unlike the high resolution structure (10), thesequence including residues 262–283 was observed. Despitethis progress in structural characterization of the mobile loop,relatively little progress has been made in assigning functionalroles to residues in the mobile loop.At least two nonexclusive roles are conceivable for the
bacterial luciferase mobile loop. First, it has been suggestedthat the mobile loop might serve to protect reaction inter-mediates from quenching by solvent (14) Second, the mobileloop might afford a docking surface allowing transient com-plex formation between enzyme and a flavin oxidoreductasethat supplies the enzyme with FMNH2. The former hypoth-esis is attractive because the mobile loops of other triose-phosphate isomerase barrel proteins have been shown toserve this role (15). The latter hypothesis is attractivebecause the sequence of the loop region is conserved acrossluminous bacteria, and complex formation would requirestructural specificity. The experiments reported here wereundertaken to investigate these nonexclusive possibilities.A variant has been constructed in which the entire mobile
loop was deleted (14) This deletion results in loss of �8% of theluxA gene. Although the tertiary structure, the yield of the car-boxylic acid, and the affinity for aldehyde, FMN, and FMNH2
were relatively unaltered, the total quantum yield was reducedby 2 orders of magnitude. It was suggested that the cause of thisreductionwas an inability to shield reaction intermediates frombulk solvent (14) This hypothesis was formulated based on theobservation that many triosephosphate isomerase barrelenzymes utilize substrate sensitive mobile loop movements foranalogous lid gating mechanisms (15, 16). To determinewhether the enzymatic properties of the loop deletion mutantwere due to the loss of specific residues, we targeted positionsthroughout the mobile loop for mutagenesis in an attempt to
1 To whom correspondence should be addressed: College of Natural andAgricultural Sciences, 900 University Ave., 302 College Bldg. North, River-side, CA 92521. Tel.: 951-827-3101; Fax: 951-827-4190; E-mail: [email protected].
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 284, NO. 47, pp. 32827–32834, November 20, 2009© 2009 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
NOVEMBER 20, 2009 • VOLUME 284 • NUMBER 47 JOURNAL OF BIOLOGICAL CHEMISTRY 32827
by guest on September 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

disrupt the “seal” of the active center provided by the mobileloop.An additional focus of the present study was to define the
possible role of themobile loop in facilitating the transient asso-ciation of accessory proteins. Luciferase requires reduced flavinto catalyze the bioluminescence reaction (17). Maintaining aconstant level of reduced flavin may be difficult during aerobicgrowth, because free reduced flavin readily reacts with molec-ular oxygen (18). It has been proposed that a complex is formedbetween luciferase and flavin-oxidoreductase enzymes inwhich FMNH2 is transferred directly from the reductase to theactive site of luciferase (19, 20). The transfer model impliesmolecular specificity in the transient complex between the twoenzymes. Bacterial luciferase is highly active in a variety ofrecombinant bacteria such asEscherichia coli (21, 22). The geneencoding the bioluminescence-supporting oxidoreductase inE. coli, fre, was recently identified and characterized (23) Theloop is a promising potential docking platform between lucif-erase and the oxidoreductase based on both stringent sequenceconservation and proximity to the active center (Fig. 2, a and b,and Ref. 13, 24, and 25). If a given residue within the loop facil-itates flavin transfer, bioluminescence may be impaired inassays utilizing enzymatic reduction of FMN but display nor-mal levels of activity when supplied with excess catalyticallyreduced FMN. Therefore, activity measurements wereobtained for point mutants throughout the mobile loop usingactivity measurements in vivo and by a second method wherethe flavin is reduced chemically and provided by free diffusionin vitro.
EXPERIMENTAL PROCEDURES
Chemicals—All of the chemicals were obtained from Sigma-Aldrich unless otherwise noted and were of reagent grade orhigher. The cells were cultured in standard Luria-Bertani brothor agar supplemented with 100 �g/ml ampicillin.Vector Construction and Mutagenesis—Using a high fidelity
polymerase (Pfu Turbo; Stratagene), Vibrio harveyi lux AB wasamplified from pJHD500 plasmid using the nucleotide primers5�-gagcccctcgagcgagtgatatttg (sense) and 5�-ccatatgaaattcg-gaaacttccttc (antisense) (26) All DNA mutagenesis andsequencing primers were obtained from IDT DNA. Amplifica-tion with this pair of primers led to incorporation of an NdeI
site and an XhoI site downstream.After digestionwith both restrictionendonucleases (New England Bio-labs), fragments were gel-purified(Qiaex gel purification system) andligated into a prepared pET21-bvector (Novagen). The vectorresulted in the addition of a series ofsix histidine residues onto the C ter-minus of the LuxB gene. Sequencingof the entire insertwas used to verifyfidelity (Arizona Research Labora-tories Sequencing Facility, Univer-sity of Arizona). The mutants weregenerated from pZCH2 using site-directed mutagenesis (27). These
clones were verified by local sequencing at the site ofmutation.Luciferase Purification—Luciferase protein was expressed
from pZCH2 in a BL21 (�DE3) cell line after growth to an A600of 0.5 (Stratagene). Expression was initiated by the addition ofisopropyl �-D-thiogalactopyranoside to 1 mM. Expression wascontinued for �6.5 h at 25 °C with constant agitation. Clarifiedlysate was applied to a custom nickel affinity column (Amer-sham Biosciences) and purified to �90% purity assessed bySDS-PAGE analysis (28). Purified protein was dialyzed exten-sively into buffer containing 100 mM Na�/K� phosphate and100 mM NaCl, pH 7.0.Determination of Protein Concentration—Concentrations of
luciferase and the individual � and � subunits were determinedusing extinction coefficients at 280 nm of 1.136, 1.41, and 0.71(mg/ml)�1 cm�1, respectively (29).Western Blotting—A small volume of each culture was with-
drawn and normalized for cell density at 600 nm. The cells werecollected by centrifugation and lysed in an equal volume of 200mM Tris-HCl, 200 mM dithiothreitol, 4% SDS, 0.2% bromphe-nol blue, and 25% glycerol. The samples were then boiled for 5min and briefly centrifuged. Subsequent electrophoresis wasconducted using 12.5% SDS-PAGE gels prior to transfer onto anitrocellulose membrane (28, 30). The membrane was incu-bated overnight at 4 °C in blocking buffer (1% powdered milkwith 0.05% Tween 20). The membrane was washed repeatedlyin phosphate-buffered saline containing 0.05% Tween 20.Luciferase was detected with a polyclonal anti-serum (1:5000dilution in blocking buffer) followed by exposure to a Cy dye-conjugated anti-rabbit secondary antibody (Odyssey IRDye).Unbound secondary antibody was removed by repeated wash-ing with phosphate-buffered saline. Fluorescence excitationwas carried out using an Odyssey Li-COR IR imager at 680 nm.Emission was detected at 700 nm and automatically correctedfor background.Activity Assays—Specific activities were determined by the
flavin injection assay (31). Enzyme was incubated in 1.0 ml of100 mM Na�/K� phosphate containing 0.5 mg/ml bovineserum albumin and 0.001% aldehyde. The reactions were initi-ated with the rapid injection of 1.0 ml of 50 �M photo-reducedFMNH2. Reduced flavin was obtained by photo-reductionusingwhite fluorescent light in the presence of EDTA (14). Peak
FIGURE 1. Simplified chemical mechanism depicting bacterial bioluminescence. The isoalloxazine ring ofthe flavin is oxygenated at position C(4a) prior to formation of the aldehyde hemiacetal (2, 55). The resolutionof this tetrahedral intermediate leads to the population of an excited state emitter and ultimately lightemission.
Mutagenesis of the Luciferase Mobile Loop
32828 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 284 • NUMBER 47 • NOVEMBER 20, 2009
by guest on September 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from
luminescence was recorded using a custom bench top lumi-nometer (32). Aldehyde dependence was measured using theflavin injection assay varying the aldehyde incubated withenzyme. The rate of luminescence decay (k � 0.69/t1⁄2) wasdetermined from the elapsed time for the light intensity todecay by 50% (t1⁄2) (33). Total quantum yield was determinedbased on the equation I(t) � Io2�t/t
(1/2). The yield was calcu-lated based on integration over the first 100 s of the reactionbased on observed values of Io and t1⁄2. Activitymeasurements invivoweremade on 1.0-ml aliquots. Light emissionwas recordedfollowing the rapid injection of 1.0 ml of 0.1% decanal (v/v inwater). Peak luminescence was reached �5 s after injection.
Dissociation constants for flavin were determined by the dithion-ite method as described (34) Intermediate II was formed andassayed as described (35).Kd values for aldehydewere determinedby the using the standard flavin injectionmethod (31).Bioluminescent Emission Spectra—Samples contained 1 nm
V. harveyi Frp, 2 �M FMN, 1 mM NADPH, 80 �M n-decyl alde-hyde suspension, and 10 �M protein solution in 100 mM
Na�/K� phosphate, 100 mM NaCl, pH 7.0. The spectra wereacquired on Cary Eclipse fluorimeter lacking excitation. Theinstrument detected relative light intensity as a function ofwavelength using a wavelength dispersion for emission bandpass fixed at 5 nm. The scan speed was �500 nm/min.
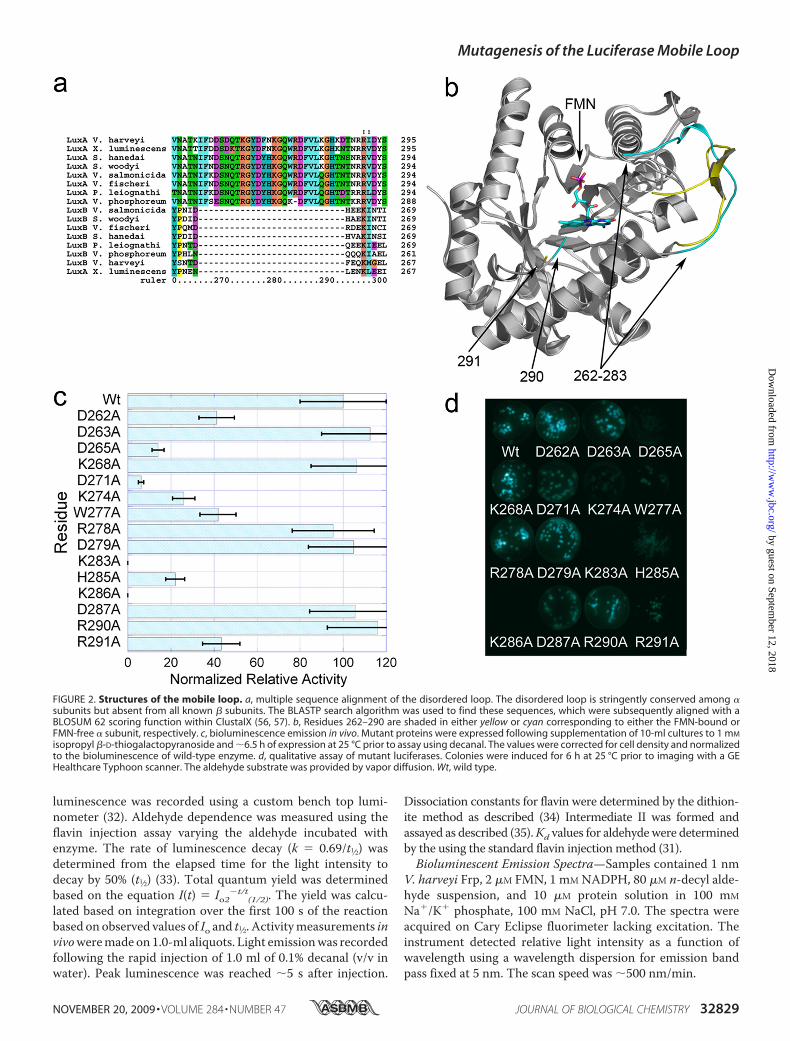
FIGURE 2. Structures of the mobile loop. a, multiple sequence alignment of the disordered loop. The disordered loop is stringently conserved among �subunits but absent from all known � subunits. The BLASTP search algorithm was used to find these sequences, which were subsequently aligned with aBLOSUM 62 scoring function within ClustalX (56, 57). b, Residues 262–290 are shaded in either yellow or cyan corresponding to either the FMN-bound orFMN-free � subunit, respectively. c, bioluminescence emission in vivo. Mutant proteins were expressed following supplementation of 10-ml cultures to 1 mM
isopropyl �-D-thiogalactopyranoside and �6.5 h of expression at 25 °C prior to assay using decanal. The values were corrected for cell density and normalizedto the bioluminescence of wild-type enzyme. d, qualitative assay of mutant luciferases. Colonies were induced for 6 h at 25 °C prior to imaging with a GEHealthcare Typhoon scanner. The aldehyde substrate was provided by vapor diffusion. Wt, wild type.
Mutagenesis of the Luciferase Mobile Loop
NOVEMBER 20, 2009 • VOLUME 284 • NUMBER 47 JOURNAL OF BIOLOGICAL CHEMISTRY 32829
by guest on September 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from
Collisional Quenching—The experiments were conducted at20 °C essentially as described (36). Decay rates and initial veloc-ities were determined from samples containing �1 �M lucifer-ase and an appropriate concentration of KCl tomaintain a con-stant ionic strength of 250 mM.DecanoicAcid Production—Carboxylic acid analysiswas per-
formed as described with minor modifications (14). Gas chro-matography was used to quantify the yield of decanoate follow-ing a single enzymatic turnover. 1.0 ml of photo-reducedFMNH2 was injected into 3.0-ml samples containing 50 �M
protein and �10 �M sonicated decanal (31). 1.0-ml aliquotswere withdrawn and supplemented with 100 �l of a 200 �g/mldodecanoic acid solution as internal standard. The sample wasacidified with 50 �l of 4 M H2SO4. The acid was removed withthe addition of 1.0ml of CH2Cl2. This mixture was vortexed for1min vigorously prior to removal of the organic phase. The acidwas then derivitized with the addition of 0.4ml ofN-methyl-N-trifluoroacetamide and 10 min of incubation at 37 °C. 1 �l ofeach sample was loaded onto a 5988A gas chromatography/mass spectrometry. It was found that the decanoic acid had aretention time of 3.61 min, and the dodecanoic acid standardhad a retention time of 9.29 min. The total area under eachcurvewas integrated for both peaks. The values for the decanoicacid yield are given relative to the quantity of the dodecanoicacid internal standard.
RESULTS
Point mutants were generated using site-directed mutagen-esis at 15 of the 29 mobile loop residues between positions 262and 291. This collection represents all of the charged residues inthemobile loop plus the singlemobile loop tryptophan residingat position 277. Activity measurements, normalized for celldensity, were determined in vivo 6.5 h post-induction (Fig. 2, cand d). Nine mutants had reduced levels of activity in vivo. Toexclude the possibility that reduced activity was due to changesin protein synthesis and/or folding, the mutants were analyzedby Western blots (data not shown). The level of expression forall of the mutants was the same as for the wild-type control. Todetermine relative activity using a nonenzymatic method for
providing reduced flavin, mutants with decreased activity invivo were purified and subjected to detailed kinetic analysis(Table 1). All of the mutants had specific activities in vitrowithin a factor of 2 of the activity obtained in vivo.The turnover number of luciferases from different biolumi-
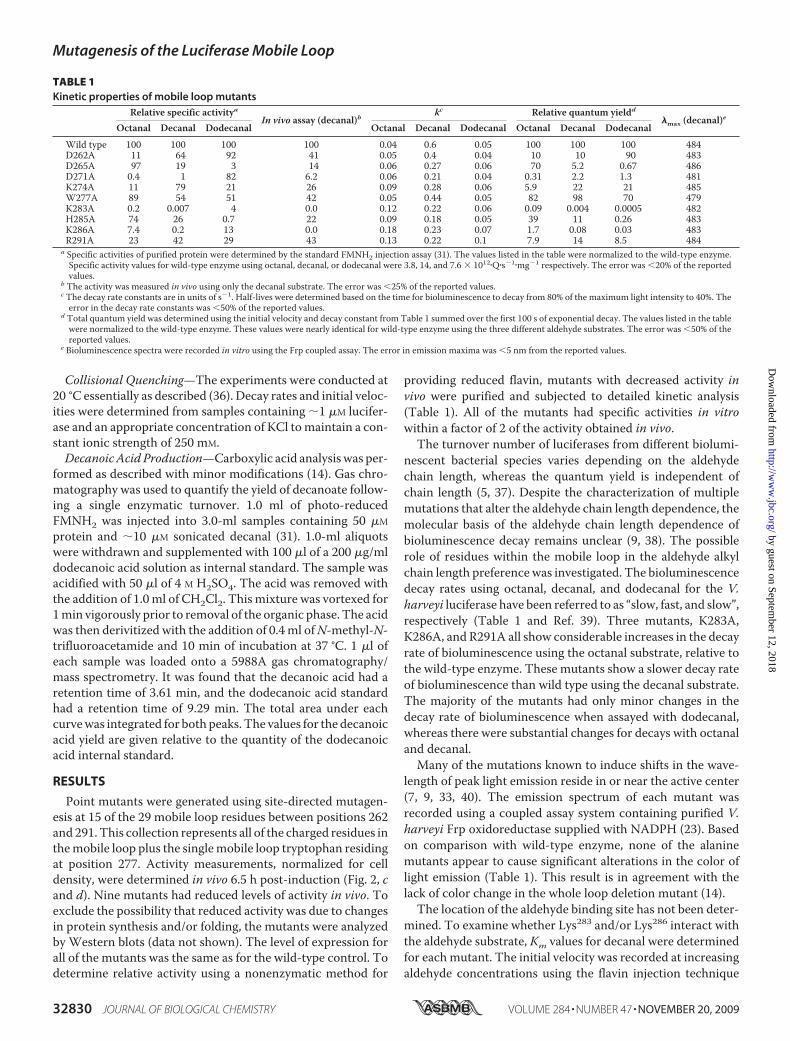
nescent bacterial species varies depending on the aldehydechain length, whereas the quantum yield is independent ofchain length (5, 37). Despite the characterization of multiplemutations that alter the aldehyde chain length dependence, themolecular basis of the aldehyde chain length dependence ofbioluminescence decay remains unclear (9, 38). The possiblerole of residues within the mobile loop in the aldehyde alkylchain length preference was investigated. The bioluminescencedecay rates using octanal, decanal, and dodecanal for the V.harveyi luciferase have been referred to as “slow, fast, and slow”,respectively (Table 1 and Ref. 39). Three mutants, K283A,K286A, and R291A all show considerable increases in the decayrate of bioluminescence using the octanal substrate, relative tothe wild-type enzyme. These mutants show a slower decay rateof bioluminescence than wild type using the decanal substrate.The majority of the mutants had only minor changes in thedecay rate of bioluminescence when assayed with dodecanal,whereas there were substantial changes for decays with octanaland decanal.Many of the mutations known to induce shifts in the wave-
length of peak light emission reside in or near the active center(7, 9, 33, 40). The emission spectrum of each mutant wasrecorded using a coupled assay system containing purified V.harveyi Frp oxidoreductase supplied with NADPH (23). Basedon comparison with wild-type enzyme, none of the alaninemutants appear to cause significant alterations in the color oflight emission (Table 1). This result is in agreement with thelack of color change in the whole loop deletion mutant (14).The location of the aldehyde binding site has not been deter-
mined. To examine whether Lys283 and/or Lys286 interact withthe aldehyde substrate, Km values for decanal were determinedfor each mutant. The initial velocity was recorded at increasingaldehyde concentrations using the flavin injection technique
TABLE 1Kinetic properties of mobile loop mutants
Relative specific activityaIn vivo assay (decanal)b
kc Relative quantum yieldd�max (decanal)eOctanal Decanal Dodecanal Octanal Decanal Dodecanal Octanal Decanal Dodecanal
Wild type 100 100 100 100 0.04 0.6 0.05 100 100 100 484D262A 11 64 92 41 0.05 0.4 0.04 10 10 90 483D265A 97 19 3 14 0.06 0.27 0.06 70 5.2 0.67 486D271A 0.4 1 82 6.2 0.06 0.21 0.04 0.31 2.2 1.3 481K274A 11 79 21 26 0.09 0.28 0.06 5.9 22 21 485W277A 89 54 51 42 0.05 0.44 0.05 82 98 70 479K283A 0.2 0.007 4 0.0 0.12 0.22 0.06 0.09 0.004 0.0005 482H285A 74 26 0.7 22 0.09 0.18 0.05 39 11 0.26 483K286A 7.4 0.2 13 0.0 0.18 0.23 0.07 1.7 0.08 0.03 483R291A 23 42 29 43 0.13 0.22 0.1 7.9 14 8.5 484
a Specific activities of purified protein were determined by the standard FMNH2 injection assay (31). The values listed in the table were normalized to the wild-type enzyme.Specific activity values for wild-type enzyme using octanal, decanal, or dodecanal were 3.8, 14, and 7.6 � 1012�Q�s�1�mg�1 respectively. The error was �20% of the reportedvalues.
b The activity was measured in vivo using only the decanal substrate. The error was �25% of the reported values.c The decay rate constants are in units of s�1. Half-lives were determined based on the time for bioluminescence to decay from 80% of the maximum light intensity to 40%. Theerror in the decay rate constants was �50% of the reported values.
d Total quantum yield was determined using the initial velocity and decay constant from Table 1 summed over the first 100 s of exponential decay. The values listed in the tablewere normalized to the wild-type enzyme. These values were nearly identical for wild-type enzyme using the three different aldehyde substrates. The error was �50% of thereported values.
e Bioluminescence spectra were recorded in vitro using the Frp coupled assay. The error in emission maxima was �5 nm from the reported values.
Mutagenesis of the Luciferase Mobile Loop
32830 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 284 • NUMBER 47 • NOVEMBER 20, 2009
by guest on September 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from
(31). A version of theMichaelis-Menten equation was fit to thedata to include the effects of substrate inhibition at high con-centrations (14) Wild-type enzyme had a Km of 8 �M, whereasK286A and K283A bind decanal withMichaelis constants of 10and 10.5 �M, respectively. Therefore, neither of these muta-tions appears to significantly impede binding of decanal.In the structure of the luciferase-FMN complex, the segment
of the mobile loop containing Lys286 and Lys283 is near theflavin phosphate group. Therefore, the dissociation constant(Kd) for FMNH2 was determined for K283A and K286A. In thisassay, the concentration of FMNH2was varied, and the reactionwas initiated with the rapid injection of air-equilibrated alde-hyde (34). The initial velocity was determined at different con-centrations of reduced flavin and used to perform a nonlinearleast squares fitting analysis (14). The Kd we obtained for wild-type enzymewas 0.9�M, in good agreement with themicromo-lar value obtained in prior measurements (14). K283A andK286A bind reduced flavin more weakly, with dissociationequilibrium constants of 1.1 and 6.8 �M, respectively.To determine whether the low levels of light emitted by
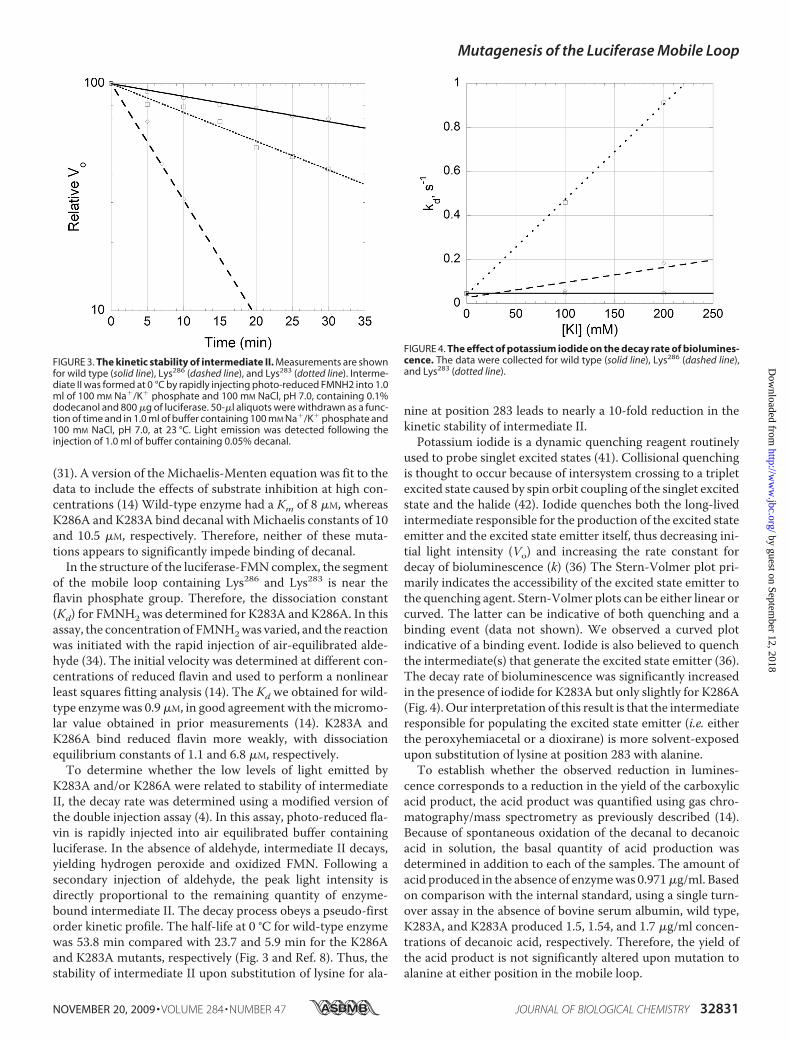
K283A and/or K286A were related to stability of intermediateII, the decay rate was determined using a modified version ofthe double injection assay (4). In this assay, photo-reduced fla-vin is rapidly injected into air equilibrated buffer containingluciferase. In the absence of aldehyde, intermediate II decays,yielding hydrogen peroxide and oxidized FMN. Following asecondary injection of aldehyde, the peak light intensity isdirectly proportional to the remaining quantity of enzyme-bound intermediate II. The decay process obeys a pseudo-firstorder kinetic profile. The half-life at 0 °C for wild-type enzymewas 53.8 min compared with 23.7 and 5.9 min for the K286Aand K283A mutants, respectively (Fig. 3 and Ref. 8). Thus, thestability of intermediate II upon substitution of lysine for ala-
nine at position 283 leads to nearly a 10-fold reduction in thekinetic stability of intermediate II.Potassium iodide is a dynamic quenching reagent routinely
used to probe singlet excited states (41). Collisional quenchingis thought to occur because of intersystem crossing to a tripletexcited state caused by spin orbit coupling of the singlet excitedstate and the halide (42). Iodide quenches both the long-livedintermediate responsible for the production of the excited stateemitter and the excited state emitter itself, thus decreasing ini-tial light intensity (Vo) and increasing the rate constant fordecay of bioluminescence (k) (36) The Stern-Volmer plot pri-marily indicates the accessibility of the excited state emitter tothe quenching agent. Stern-Volmer plots can be either linear orcurved. The latter can be indicative of both quenching and abinding event (data not shown). We observed a curved plotindicative of a binding event. Iodide is also believed to quenchthe intermediate(s) that generate the excited state emitter (36).The decay rate of bioluminescence was significantly increasedin the presence of iodide for K283A but only slightly for K286A(Fig. 4). Our interpretation of this result is that the intermediateresponsible for populating the excited state emitter (i.e. eitherthe peroxyhemiacetal or a dioxirane) is more solvent-exposedupon substitution of lysine at position 283 with alanine.To establish whether the observed reduction in lumines-
cence corresponds to a reduction in the yield of the carboxylicacid product, the acid product was quantified using gas chro-matography/mass spectrometry as previously described (14).Because of spontaneous oxidation of the decanal to decanoicacid in solution, the basal quantity of acid production wasdetermined in addition to each of the samples. The amount ofacid produced in the absence of enzymewas 0.971�g/ml. Basedon comparison with the internal standard, using a single turn-over assay in the absence of bovine serum albumin, wild type,K283A, and K283A produced 1.5, 1.54, and 1.7 �g/ml concen-trations of decanoic acid, respectively. Therefore, the yield ofthe acid product is not significantly altered upon mutation toalanine at either position in the mobile loop.
FIGURE 3. The kinetic stability of intermediate II. Measurements are shownfor wild type (solid line), Lys286 (dashed line), and Lys283 (dotted line). Interme-diate II was formed at 0 °C by rapidly injecting photo-reduced FMNH2 into 1.0ml of 100 mM Na�/K� phosphate and 100 mM NaCl, pH 7.0, containing 0.1%dodecanol and 800 �g of luciferase. 50-�l aliquots were withdrawn as a func-tion of time and in 1.0 ml of buffer containing 100 mM Na�/K� phosphate and100 mM NaCl, pH 7.0, at 23 °C. Light emission was detected following theinjection of 1.0 ml of buffer containing 0.05% decanal.
FIGURE 4. The effect of potassium iodide on the decay rate of biolumines-cence. The data were collected for wild type (solid line), Lys286 (dashed line),and Lys283 (dotted line).
Mutagenesis of the Luciferase Mobile Loop
NOVEMBER 20, 2009 • VOLUME 284 • NUMBER 47 JOURNAL OF BIOLOGICAL CHEMISTRY 32831
by guest on September 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

In the luciferase-FMN complex, two basic side chains residewithin an appropriate distance and orientation to coordinatethe phosphate group of the flavin, Arg125 and Arg107 (13).Mutation of Arg107 to glutamate effectively inactivates lucifer-ase and greatly reduces the affinity of the enzyme for substrate(43, 44). To examine whether Lys283 or Lys286 directly contactsthe phosphate group of the flavin, both positions were mutatedto glutamate. Bothmutants appeared to havewild-type levels ofactivity in vivo, in stark contrast to the corresponding alaninemutants (data not shown).
DISCUSSION
The mobile loop from bacterial luciferase was subjected toalanine mutagenesis for two major reasons. First, the region ofthe protein containing the mobile loop is vital for catalysis inmany enzymes with the triosephosphate isomerase barrel fold.The canonical triosephosphate isomerase barrel consists of an8-fold repeat of parallel �-strands forming an inner barrel sur-rounded by eight �-helices on the exterior of the protein (45).The triosephosphate isomerase barrel is present in �10% ofknown structures (16). The approximate location of the activecenter near the C-terminal ends of � strands appears to beconserved, suggesting divergent evolution from a commonancestor (15). The major source of variation between triose-phosphate isomerase barrel enzymes are variable length loopregions connecting secondary structural elements (16). Thearchetypical triosephosphate isomerase barrel, triose phos-phate isomerase, catalyzes the conversion of the ketose,dihydroxyacetone phosphate, into the aldose, glyceralde-hyde-3-phosphate. The enediol intermediate is susceptibleto loss of phosphate by �-elimination (46). To avoid this sidereaction, triosephosphate isomerase uses a mobile loop as alid to sequester the active site following ligand binding (47)Similar lid gating mechanisms are common, making thosewith uncommon properties particularly interesting (48)Luciferase is unusual because of its distinct reaction chem-istry and an unusually long mobile loop consisting of �30residues. The first aim of this study was to determinewhether any labile intermediates might be protected by thebacterial luciferase mobile loop.We found that mutation of either of the mobile loop resi-
dues Lys283 or Lys286 to alanine led to a substantial decreasein the stability of intermediate II. This was more pronouncedfor the K283A mutant. The kinetic stability of intermediateII is decreased upon substitution at a number of active centerresidues including His44, Asp113, Cys106, and Val173 (40,49–51). Because of the distance from the isoalloxazine to themobile loop, the finding that mutations in the mobile loopcan alter the stability of intermediate II is surprising but notwithout precedent. In a prior study, two mutations in themobile loop (G275P and F261D) were found to result in adecreased stability of intermediate II (52). Unlike the lysinemutants we described, the G275P and F261D were unable toeffectively oxidize the aldehyde substrate to form the acidproduct (52). This finding is interesting given the extremephysical separation between the flavin distal portion of themobile loop and the experimentally determined active cen-ter (13). The G275P and F261D mutations may have altered
the dynamics of the loop in such a way that luciferase wasunable to effectively adopt a closed conformation of themobile loop (52).Based on dynamic quenching analysis, we found that the
K286A mutation resulted in an inability to protect the long-lived intermediate responsible for the production of the excitedstate emitter. Following reaction of the aldehyde substrate withintermediate II, it is unclear how the singlet excited state ispopulated. We favor the formation of a dioxirane intermediateprior to population of the excited state (5, 53). According to thismechanism the excited state emitter forms following homolyticcleavage of the peroxide bond. Potentially, there are multiplecauses for an increase in the decay rate of bioluminescence inthe presence of a dynamic quenching agent, such as a defect inthe conversion of the intermediate IIA into the dioxirane.Indeed, the effect could be as simple as solvent-mediated dep-rotonation of N-5 and elimination of the peroxy group or theperacid. Additional NMR studies utilizing isotopically labeledflavins may allow discrimination between these hypotheses.The second question we sought to examine in this work was
whether the mobile loop may play a role in complex formationbetween luciferase and an NAD(P)H-dependent oxidoreduc-tase (30). A complex has been proposed involving luciferase andoxidoreductase in which FMNH2 is transferred directly fromthe reductase to the luciferase (19, 20, 54). Transfer obviates theproblem of reaction of reduced flavin withmolecular oxygen insolution. Implicit in this model is the requirement of molecularspecificity in the transient complex. The features of this com-plex have yet to be described. We were interested in screeningfor sites in the mobile loop responsible for providing the oxi-doreductase binding interface. The expectation was thatmutants defective in flavin transfer because of a docking defectshould display high levels of bioluminescence when assayedwith an exogenous source of flavin in vitro. Therefore, we com-pared normalized activity measurements both in vivo and invitro. We did not observe a significant difference between thetwo measurements, suggesting that either (i) the oxidoreduc-tase interface requires a substantial number of residues and isinsensitive to a singlemutation, (ii) we selected the wrong set ofresidues, or (iii) there is no complex between luciferase and theoxidoreductase.Concurrent with this work, we investigated the source of
reduced flavin during bioluminescence in E. coli (23). In thisstudy, the oxidoreductase enzyme (fre) from a nonbiolumines-cent bacterium (E. coli) was found to be as competent in pro-viding the reduced flavin substrate in vivo or in vitro as theendogenous enzyme in V. harveyi. If direct flavin transferoccurs between proteins, specificitymust exist in the encountercomplex between a given reductase and luciferase to ensureproper positioning of the oxidoreductase over the active site.Given the lack of sequence similarity between the oxidoreduc-tase from E. coli (Fre) and the endogenous enzyme inV. harveyi(Frp), it is unlikely that a complex is formed between enzymes.Further, the E. coli reductase did not physically interact withluciferase based on a pulldown assay (23). Therefore, it was notsurprising that following mutagenesis of the mobile loop, alarge discrepancy in activity caused by defective flavin transferwas not observed.
Mutagenesis of the Luciferase Mobile Loop
32832 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 284 • NUMBER 47 • NOVEMBER 20, 2009
by guest on September 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

In addition to examination of the aforementioned aspects ofthe mobile loop, several observations regarding the structureof this region of the enzyme have beenmade. Structural data onthemobile loopwere unavailable until the recent description ofthe luciferase-FMN crystal structure (13). The lysine mutantsreported here were near or within a disordered region betweenresidues 283 and 290 in this structure. This disordered region ofthemobile loop is adjacent to the phosphate group of the flavin.The mobile loop may interact with the anion site directly viaionic interaction or indirectly through contacts to the residuesthat bind the anion. Direct interaction is conceivable based onthe compositional bias toward charged residues in the region.The approximate distance separating the mobile loop from theflavin-binding site is also compatible with a direct interaction(13). The K286A mutant had an increase in the Kd for FMNH2to 6.8 �M. We therefore tested whether either lysine residuedirectly interacted with the anion-binding site for the 5� phos-phate group of FMNH2. Nonconservative mutation at eitherposition to glutamate resulted in a highly active enzyme. Itappears that neither lysine residue is involved in directly bind-ing the anion of the flavin. Thus, we were unable to find clearevidence supporting a direct interaction between either lysineresidue and the 5� phosphate group of FMNH2. Based on theproperties of the alanine mutants relative to substitutions withglutamate, it appears that the size of the residue at either loca-tion is of greater importance for catalytic function than chargestate. Additional mutagenesis experiments probing the rela-tionship between activity and residue size are required to testthis model.
To characterize the dynamics of the loop region, a recentinvestigation simulated the dynamics of the luciferase� subunitusing replica exchange molecular dynamics.2 The authorsdescribed a model for the enzyme:FMNH2 complex by incor-porating decades of experimental results with detailed networkanalysis of a vast collection of loop conformations. At the timeof this study, only two point mutants had been described in theloop region (52). To examine the agreement of the presentstudy with this proposed complex, a heat map was devisedusing the severity of mutation on in vivo activity in relation tothe site of the mutation in the bound model (Fig. 5). The twomost severemutants were extremely close to the quinoidal faceof the isoalloxazine. This result agrees with our proposal thatexclusion of solvent from the active center requires moderatelysized residues at both positions 283 and 286.In summary, we were able to identify residues in the mobile
loop involved in excluding solvent following substrate binding.Our results build upon previous work where the mobile loopwas removed entirely from bacterial luciferase (14). Deletion ofthe loop significantly decreases the total quantum yield andcauses binding of reduced flavin to be slightly weaker.We wereable to produce and characterize two alanine point mutantswith qualitatively similar kinetic defects. Neither position isuniversally conserved across luminous bacterial species. How-ever, the relative size of the residue is conserved. We believethat positions 283 and 286 serve a critical function as compo-nents of the active center lid that prevents entry of solvent intothe active center.
Acknowledgment—We thank Dr. Miriam Ziegler for detailed analy-sis and stylistic suggestions in preparing this manuscript.
REFERENCES1. Eberhard, A., and Hastings, J. W. (1972) Biochem. Biophys. Res. Commun.
47, 348–3532. Balny, C., and Hastings, J. W. (1975) Biochemistry 14, 4719–47233. Hastings, J. W., Balny, C., Peuch, C. L., and Douzou, P. (1973) Proc. Natl.
Acad. Sci. U.S.A. 70, 3468–34724. Tu, S. C. (1982) J. Biol. Chem. 257, 3719–37255. Baldwin, T.O., andZiegler,M.M. (1992) inChemistry andBiochemistry of
Flavoenzymes (Muller, F., ed) pp. 467–530, CRC Press, Boca Raton, FL6. Friedland, J., and Hastings, J. W. (1967) Proc. Natl. Acad. Sci. U.S.A. 58,
2336–23427. Cline, T. W., and Hastings, J. W. (1972) Biochemistry 11, 3359–33708. Nicoli,M. Z.,Meighen, E. A., andHastings, J.W. (1974) J. Biol. Chem. 249,
2385–23929. Chen, L. H., and Baldwin, T. O. (1989) Biochemistry 28, 2684–268910. Fisher, A. J., Thompson, T. B., Thoden, J. B., Baldwin, T. O., and Rayment,
I. (1996) J. Biol. Chem. 271, 21956–2196811. Holzman, T. F., and Baldwin, T. O. (1980) Proc. Natl. Acad. Sci. U.S.A. 77,
6363–636712. Fisher, A. J., Raushel, F. M., Baldwin, T. O., and Rayment, I. (1995) Bio-
chemistry 34, 6581–658613. Campbell, Z. T., Weichsel, A., Montfort, W. R., and Baldwin, T. O. (2009)
Biochemistry 48, 6085–609414. Sparks, J. M., and Baldwin, T. O. (2001) Biochemistry 40, 15436–1544315. Farber, G. K., and Petsko, G. A. (1990) Trends Biochem. Sci. 15, 228–23416. Wierenga, R. K. (2001) FEBS Lett. 492, 193–198
2 Z. T. Campbell, T. O. Baldwin, and O. Migashita, manuscript submitted forpublication.
FIGURE 5. The location of mutated residues relative to the proposedluciferase-FMNH2 complex2. a, the � subunit shown in a cartoon with themobile loop shaded in yellow. The location of the flavin is shown in stickand shaded in green by atom type. b, heat map of activity loss upon sub-stitution with alanine. Activity loss was reported based upon the measure-ments obtained in vivo. Severity of each mutation is indicated by shadeaccording to the key.
Mutagenesis of the Luciferase Mobile Loop
NOVEMBER 20, 2009 • VOLUME 284 • NUMBER 47 JOURNAL OF BIOLOGICAL CHEMISTRY 32833
by guest on September 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

17. McElroy, W. D., Hastings, J. W., Sonnenfeld, V., and Coulombre, J. (1953)Science 118, 385–386
18. Gibson, Q. H., and Hastings, J. W. (1962) Biochem. J. 83, 368–37719. Lei, B., and Tu, S. C. (1998) Biochemistry 37, 14623–1462920. Jeffers, C. E., Nichols, J. C., and Tu, S. C. (2003) Biochemistry 42, 529–53421. Liu, Y., Golden, S. S., Kondo, T., Ishiura, M., and Johnson, C. H. (1995) J.
Bacteriol 177, 2080–208622. Gordon, S., Chung, G., and Andrew, P. (1998)Mycobacteria Protocols, pp.
235–244, Humana Press, Totowa, NJ23. Campbell, Z. T., and Baldwin, T. O. (2009) J. Biol. Chem. 284, 8322–832824. Cohn, D. H., Mileham, A. J., Simon, M. I., Nealson, K. H., Rausch, S. K.,
Bonam, D., and Baldwin, T. O. (1985) J. Biol. Chem. 260, 6139–614625. Baldwin, T. O., Devine, J. H., Heckel, R. C., Lin, J. W., and Shadel, G. S.
(1989) J. Biolumin. Chemilumin. 4, 326–34126. Devine, J. H., Shadel, G. S., and Baldwin, T. O. (1989) Proc. Natl. Acad. Sci.
U.S.A. 86, 5688–569227. Zheng, L., Baumann, U., and Reymond, J. L. (2004) Nucleic Acids Res. 32,
e11528. Schagger, H., and von Jagow, G. (1987) Anal. Biochem. 166, 368–37929. Sinclair, J. F., Waddle, J. J., Waddill, E. F., and Baldwin, T. O. (1993) Bio-
chemistry 32, 5036–504430. Tu, S. C. (2001) Antioxid. Redox Signal. 3, 881–89731. Hastings, J.W., Baldwin, T. O., andNicoli, M. Z. (1978)Methods Enzymol.
57, 135–15232. Mitchell, G. W., and Hastings, J. W. (1971) Anal. Biochem. 39, 243–25033. Lin, L. Y., Sulea, T., Szittner, R., Kor, C., Purisima, E.O., andMeighen, E. A.
(2002) Biochemistry 41, 9938–994534. Tu, S., and Hastings, J. W. (1975) Biochemistry 14, 4310–431635. Tu, S. C. (1979) Biochemistry 18, 5940–594536. Sirokman, G., Wilson, T., and Hastings, J. W. (1995) Biochemistry 34,
13074–1308137. Meighen, E. A., and Bartlet, I. (1980) J. Biol. Chem. 255, 11181–1118738. Hosseinkhani, S., Szittner, R., and Meighen, E. A. (2005) Biochem. J. 385,
575–580
39. Hastings, J.W.,Weber, K., Friedland, J., Eberhard, A.,Mitchell, G.W., andGunsalus, A. (1969) Biochemistry 8, 4681–4689
40. Lin, L. Y., Szittner, R., Friedman, R., and Meighen, E. A. (2004) Biochem-istry 43, 3183–3194
41. Lakowicz, J. R., and Masters, B. R. (2008) J. Biomed. Opt. 13, 215–22042. Kasha, M. (1952) J. Chem. Phys. 20, 71–7443. Lin, L. Y., Sulea, T., Szittner, R., Vassilyev, V., Purisima, E. O., and
Meighen, E. A. (2001) Protein Sci. 10, 1563–157144. Moore, C., Lei, B., andTu, S. C. (1999)Arch. Biochem. Biophys. 370, 45–5045. Banner, D. W., Bloomer, A. C., Petsko, G. A., Phillips, D. C., Pogson, C. I.,
Wilson, I. A., Corran, P. H., Furth, A. J., Milman, J. D., Offord, R. E.,Priddle, J. D., and Waley, S. G. (1975) Nature 255, 609–614
46. Sterner, R., and Schmid, F. X. (2004) Science 304, 1916–191747. Sampson, N. S., and Knowles, J. R. (1992) Biochemistry 31, 8488–849448. Sullivan, S. M., and Holyoak, T. (2008) Proc. Natl. Acad. Sci. U.S.A. 105,
13829–1383449. Abu-Soud, H. M., Clark, A. C., Francisco, W. A., Baldwin, T. O., and
Raushel, F. M. 5(1993) J. Biol. Chem. 268, 7699–770650. Huang, S., and Tu, S. C. (1998) Biochemistry 37, 861451. Cline, T. W., and Hastings, J. W. (1974) J. Biol. Chem. 249, 4668–466952. Low, J. C., and Tu, S. C. (2002) Biochemistry 41, 1724–173153. Raushel, F.M., and Baldwin, T.O. (1989)Biochem. Biophys. Res. Commun.
164, 1137–114254. Tu, S. C., Lei, B., Liu, M., Tang, C. K., and Jeffers, C. (2000) J. Nutr. 130,
331S–332S55. Francisco, W. A., Abu-Soud, H. M., DelMonte, A. J., Singleton, D. A.,
Baldwin, T. O., and Raushel, F. M. (1998) Biochemistry 37, 2596–260656. Altschul, S. F., Gish,W.,Miller,W.,Myers, E.W., and Lipman, D. J. (1990)
J. Mol. Biol. 215, 403–41057. Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan,
P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R.,Thompson, J. D., Gibson, T. J., and Higgins, D. G. (2007) Bioinformatics23, 2947–2948
Mutagenesis of the Luciferase Mobile Loop
32834 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 284 • NUMBER 47 • NOVEMBER 20, 2009
by guest on September 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from

Zachary T. Campbell and Thomas O. BaldwinIntermediates
Two Lysine Residues in the Bacterial Luciferase Mobile Loop Stabilize Reaction
doi: 10.1074/jbc.M109.031716 originally published online August 26, 20092009, 284:32827-32834.J. Biol. Chem.
10.1074/jbc.M109.031716Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/284/47/32827.full.html#ref-list-1
This article cites 55 references, 17 of which can be accessed free at
by guest on September 12, 2018
http://ww
w.jbc.org/
Dow
nloaded from