the spectrum of large adrenal masses: a case series
TRANSCRIPT

Cooke-Barber et al. HCA Healthcare Journal of Medicine (2020) 1:2 https://doi.org/10.36518/2689-0216.1028
83
Case Series
The Spectrum of Large Adrenal Masses: A Case Series Jo Cooke-Barber, MD,1 Sarah E. Hatef, BS, MPH,2 Analise McGreal, BS,3 Andrew Schwemmer, MD,1 Christopher Senkowski, MD1, 3
Abstract
BackgroundAdrenocortical carcinoma (ACC) is a rare malignancy that is challenging to diagnose and has important implications for surgeons who approach this disease. Despite its rarity, it must always be in the differential diagnosis when investigating and treating large adrenal masses. We aim to demonstrate the complexities of this disease through a review of five recent patients at a single tertiary care center.
Methods A series of five patients are described, each of whom presented to a single institution as referrals for “large adrenal mass” in the past sixteen months. Their pre-operative diagnosis, radiographic findings, the operative approach and the pathology results were examined.
ResultsThe first patient had a 12 cm high grade adrenocortical carcinoma. The second patient had pathology consistent with a 9 cm, high grade ACC. The third had a liposarcoma. The fourth patient had a myelolipoma. The fifth was diagnosed with a benign process.
ConclusionAlthough adrenocortical carcinoma is an uncommon cancer, it has significant implications for the patient’s prognosis and ultimately, their treatment algorithm. Therefore, when evaluating large adrenal masses, surgeons must remain vigilant of the possibility of adreno-cortical carcinoma.
Keywordsadrenal cortex neoplasms; adrenocortical carcinoma; pheochromocytoma; adrenal glands; neoplasm metastasis; carcinoma, diagnosis; carcinoma, pathology
Author affiliations are listed
at the end of this article.
Correspondence to:
Analise McGreal
Mercer School of Medicine
1250 East 66th Street
Savannah, GA 31404
(Analise.Elizabeth.Mcgre-
www.hcahealthcarejournal.com
© 2020 HCA Physician Services, Inc. d/b/a Emerald Medical Education
HCA Healthcare Journal of Medicine
IntroductionAdrenocortical carcinoma is a rare form of cancer accounting for about 0.02% of all carci-nomas,1 with just two cases per million people in the world and 200 new cases per year in the United States.2 Nonetheless, when investi-gating a large adrenal mass, especially greater than ten centimeters, adrenocortical carcinoma must be high on the differential diagnosis.
Adrenocortical cancer (ACC) affects women more than men and has a bimodal age distribu-tion with peaks in the first and fourth decades of life.2, 3 It has no geographic pattern and no
confirmed etiology. ACC has been found to be associated with several genetic factors includ-ing insulin-like growth factor overexpression, constitutive activation of the Wnt-b-catenin signaling pathway, as well as TP53 tumor sup-pressor mutations.4 Additionally, studies have shown a germline mutation in TP53 in certain Brazilian populations which correlates with an ACC prevalence in Brazilian children 10-15 times greater than that of the world pediatric popu-lation.5 This association, along with the strong association between ACC and Li-Fraumeni syndrome, offer further evidence for TP53’s role in adrenocortical carcinoma. In addition to

HCA Healthcare Journal of Medicine
84
Li-Fraumeni syndrome, higher rates of ACC are found in those with Beckwith-Widemann syn-drome and Carney complex, implicating genes IGF2 and PRKAR1A in the pathogenesis.4
Diagnosis and determination of prognosis are of utmost importance in ACC, since prompt surgical intervention and complete surgical re-section are the only known curative treatments for the disease (5-year disease free survival), lowering recurrence rates below 30%.6 Sur-gical resection is the mainstay of treatment, followed by adjuvant chemotherapy with mitotane and radiation. Patient prognosis can be predicted with relative accuracy using the European Network for the Study of Adrenal Tumors (ENSAT) Staging Score. (Table 1) The ENSAT Staging Score not only assists clini-cians in giving prognostic information to their patients, but aids in choosing a therapeutic approach as well.6 A retrospective review of 330 patients at MD Anderson Cancer Center in Tex-as found that the factors associated with local recurrence were positive surgical margins and advanced disease stage. In addition, the factors associated with poor survival were functioning tumors, R1/R2 surgical resections, stage III/IV at diagnosis and associated venous thrombo-embolism.7
Table 1. ENSAT Staging Score
Score Components of Staging Score
I T1, N0, M0II T2, N0, M0III T3–T4, N1IV T1–T4, N0–N1, M1
T1, tumor ≤ 5 cm T2, tumor > 5 cm T3, histologically proven tumor invasion of surround-ing tissue T4, tumor invasion of adjacent organs or venous tumor thrombus in vena cava or renal vein. (Venous tumor thrombus is a criterion for poorer prognosis in the ENSAT classification.) N0, negative lymph nodes N1, positive lymph nodes M0, absence of distant metastases M1, presence of distant metastases
Despite the importance of a timely diagno-sis, the ability to diagnose ACC pre-surgically remains limited. ACC commonly presents with steroid hormone excess and rapidly progress-
ing Cushing’s syndrome with or without viril-ization.3 Other clinical presentation patterns in women include androgen-secreting tumors which lead to hirsutism, virilization, dysphonia, male-patterned baldness and amenorrhea. In males, estrogen-secreting tumors cause testic-ular atrophy, which can lead to gynecomastia and baldness. Aldosterone-secreting ACC can cause hypertension and hypokalemia.1
Surprisingly, high levels of steroid precursors can also be found in hormonally inactive ACC possibly simply due to the size of these tumors. These hormonally inactive tumors commonly present with abdominal mass effect involv-ing the kidney causing hematuria and dysuria. Abdominal pain, nausea, vomiting or back pain may also be present. ACC can also invade vasculature such as the left renal vein or the inferior vena cava leading to embolism, throm-bosis or occlusion.
Although ACC can present in several ways, it is often asymptomatic and studies have demonstrated that up to 15% are discovered incidentally.3 Li et al. studied 1,941 patients with adrenal incidentaloma (AI) and found that only 1.62% of them were diagnosed with ACC.8 For this reason, the disease is often discovered in advanced stages. Thus, 20-30% of patients with adrenocortical carcinoma already have dis-tant metastases at the time of diagnosis and the 5-year survival for this disease is low, rang-ing between 16-40%.9 Improving the ability to diagnose ACC preoperatively could potentially increase survival. The purpose of this case se-ries is to identify common issues in diagnosing adrenocortical carcinoma and to offer insight into choosing the best surgical approach.
The diagnosis of ACC relies on careful investi-gation of clinical, biological and imaging fea-tures before surgery and pathological exam-ination. The ENSAT Staging Score suggests a preoperative hormonal workup for suspected ACC. In particular, the assessment of basal cortisol, ACTH, dehydroepiandrostenedione sulfate, 17-hydroxyprogesterone, testosterone, androstenedione and estradiol, as well as a dexamethasone suppression test and urinary free cortisol are recommended.3
The risk for ACC increases with tumor size, with the index of suspicion increasing for tu-
Cooke-Barber et al. (2020) 1:2. https://doi.org/10.36518/2689-0216.1028
85
mors > 4 cm (sensitivity, 97%; specificity, 52%) and > 6 cm (sensitivity, 91%; specificity, 80%).10 An abdominal computed tomography (CT) scan is mandatory in suspicion of ACC, studies have established a threshold of ≤ 10 Hounsfield Unit (HU) in unenhanced CT for the diagnosis of benign lesions. If the basal density is > 10 HU, a contrast media washout will be helpful in differentiating a benign lesion from ACC; an absolute washout > 50% suggests a benign lesion.11-14 If CT imaging cannot adequately char-acterize the adrenal mass, three major charac-teristics of an MRI will be helpful in diagnosing ACC: the presence of isointense to hypointense signal on T1-weighted images, a hyperintense signal on T2-weighted images and a heteroge-neous signal drop on chemical shift.15
Although a percutaneous adrenal biopsy was widely used in the past, with the advances in imaging modalities and the associated compli-cations with biopsy, it is less frequently used at present.16 Moreover, it is difficult to differenti-ate an adrenal adenoma from ACC with a fine needle aspiration (FNA) biopsy.17 The clinical indications for a needle biopsy are the presence of an adrenal mass in an oncologic patient for staging or characterization of a primary lesion when the differentiation of benign from ma-lignant lesions using imaging findings is not resolved.17
MethodsFive patients are presented from Memorial Health University Medical Center. All referrals for “large adrenal lesion” over a 16-month peri-od were selected for inclusion in this case se-ries. All patients received pre-operative imag-
ing, which is reviewed. The surgical approaches for each case are discussed. The primary outcome was presence or absence of pathology consistent with adrenocortical carcinoma.
ResultsCase 1
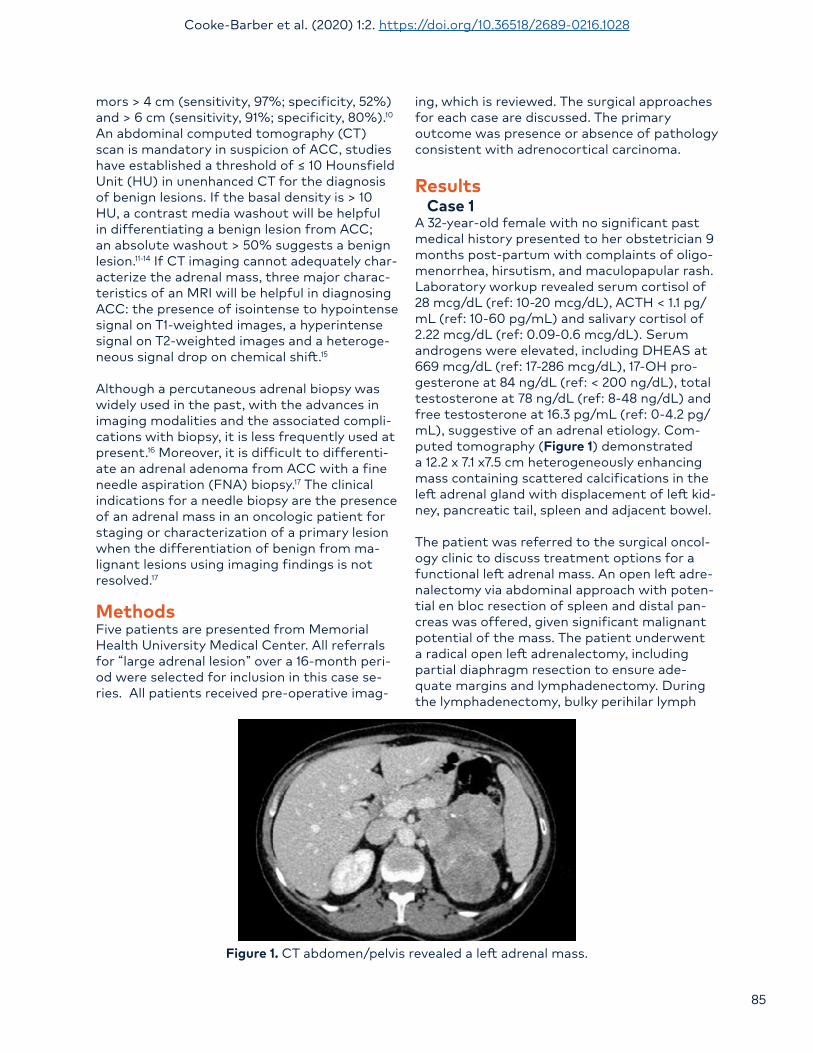
A 32-year-old female with no significant past medical history presented to her obstetrician 9 months post-partum with complaints of oligo-menorrhea, hirsutism, and maculopapular rash. Laboratory workup revealed serum cortisol of 28 mcg/dL (ref: 10-20 mcg/dL), ACTH < 1.1 pg/mL (ref: 10-60 pg/mL) and salivary cortisol of 2.22 mcg/dL (ref: 0.09-0.6 mcg/dL). Serum androgens were elevated, including DHEAS at 669 mcg/dL (ref: 17-286 mcg/dL), 17-OH pro-gesterone at 84 ng/dL (ref: < 200 ng/dL), total testosterone at 78 ng/dL (ref: 8-48 ng/dL) and free testosterone at 16.3 pg/mL (ref: 0-4.2 pg/mL), suggestive of an adrenal etiology. Com-puted tomography (Figure 1) demonstrated a 12.2 x 7.1 x7.5 cm heterogeneously enhancing mass containing scattered calcifications in the left adrenal gland with displacement of left kid-ney, pancreatic tail, spleen and adjacent bowel.
The patient was referred to the surgical oncol-ogy clinic to discuss treatment options for a functional left adrenal mass. An open left adre-nalectomy via abdominal approach with poten-tial en bloc resection of spleen and distal pan-creas was offered, given significant malignant potential of the mass. The patient underwent a radical open left adrenalectomy, including partial diaphragm resection to ensure ade-quate margins and lymphadenectomy. During the lymphadenectomy, bulky perihilar lymph
Figure 1. CT abdomen/pelvis revealed a left adrenal mass.
HCA Healthcare Journal of Medicine
86
nodes required resection of renal vessels and necessitated a left nephrectomy. Pathology revealed a high-grade adrenocortical carcinoma with positive nodes. The kidney was uninvolved and diaphragm margins were negative. Resec-tion of the spleen and distal pancreas was not required. At time of resection, the tumor was Stage III (T4, N1, M0).
Surveillance imaging demonstrated hepatic metastases 3 months after her surgery and an area concerning for local recurrence along the left retroperitoneum. Cytoreductive ablation was performed for the hepatic lesions. Addi-tionally, she was referred to medical oncology and underwent adjuvant chemotherapy with mitotane, etoposide, doxorubicin and cispla-tin, which yielded minimal results. Second line treatment was subsequently undertaken with four cycles of pembrolizumab. Patient expired from disease at 8 months from resection.
Case 2 A 72-year-old female presented to her pri-mary care physician for complaints of vague abdominal pain, weight loss, headaches, sore throat, voice changes, leg swelling, back pain, skin changes, nausea and diarrhea. Esoph-agogastroduodenoscopy and colonoscopy revealed esophageal candidiasis and gastritis. The patient’s past medical history was perti-nent for thyroid disease, HTN, anemia, arthri-tis, glaucoma, uterine and breast cancer, and tobacco use. The patient’s past surgical history was pertinent for a hysterectomy, hernia repair, laparoscopic gastric banding and left mastecto-my. Her family history was positive for cancer, diabetes, heart disease and hypertension. The patient’s physical exam demonstrated tender-
ness in the left upper quadrant. Outside hospi-tal non-contrasted CT imaging revealed a large mass in the left adrenal gland.
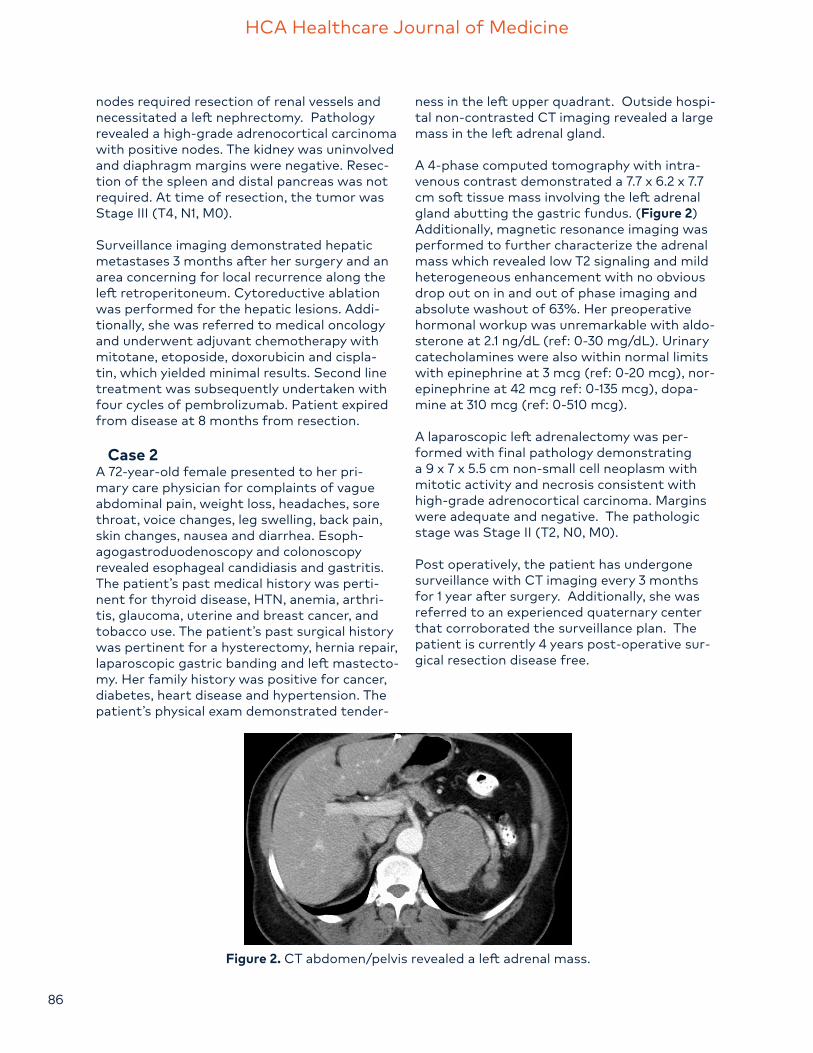
A 4-phase computed tomography with intra-venous contrast demonstrated a 7.7 x 6.2 x 7.7 cm soft tissue mass involving the left adrenal gland abutting the gastric fundus. (Figure 2) Additionally, magnetic resonance imaging was performed to further characterize the adrenal mass which revealed low T2 signaling and mild heterogeneous enhancement with no obvious drop out on in and out of phase imaging and absolute washout of 63%. Her preoperative hormonal workup was unremarkable with aldo-sterone at 2.1 ng/dL (ref: 0-30 mg/dL). Urinary catecholamines were also within normal limits with epinephrine at 3 mcg (ref: 0-20 mcg), nor-epinephrine at 42 mcg ref: 0-135 mcg), dopa-mine at 310 mcg (ref: 0-510 mcg).
A laparoscopic left adrenalectomy was per-formed with final pathology demonstrating a 9 x 7 x 5.5 cm non-small cell neoplasm with mitotic activity and necrosis consistent with high-grade adrenocortical carcinoma. Margins were adequate and negative. The pathologic stage was Stage II (T2, N0, M0).
Post operatively, the patient has undergone surveillance with CT imaging every 3 months for 1 year after surgery. Additionally, she was referred to an experienced quaternary center that corroborated the surveillance plan. The patient is currently 4 years post-operative sur-gical resection disease free.
Figure 2. CT abdomen/pelvis revealed a left adrenal mass.

Cooke-Barber et al. (2020) 1:2. https://doi.org/10.36518/2689-0216.1028
87
Case 3An 87-year-old female presented with symptoms of abdominal discomfort, weight loss and an abdominal mass that was discovered on self-examination. Past medical history was significant for atrial fibrillation and breast cancer. Family history included a son and granddaughter with melanoma. There was no history of trauma, tobacco or alcohol use and no symptoms of functioning adrenal tumor.
Computed tomography (Figure 3) was performed revealing a mixed density partially enhancing mass within the left upper quadrant adjacent to the kidney with cystic and solid components with calcifications measuring 17.6 x 11.1 x 26.0 cm. The mass displaces the spleen anteriorly and the left kidney, aorta and bowel are displaced to the right hemiabdomen.
Given the patient’s age and comorbidities, image guided core needle biopsy was offered. However, secondary to ongoing pain the decision was made with the patient to proceed with surgical resection. Preoperative biochemical evaluation was within normal limits.
After standard preoperative assessment and medical optimization, a radical resection of the retroperitoneal mass was performed. This included a distal pancreatectomy, splenectomy, left nephrectomy and left adrenalectomy, which were performed to resect the mass en bloc. Pathology demonstrated a grade 3
dedifferentiated liposarcoma with R1 resection margins. The pathologic stage was Stage III (T4, N1, M0). Patient received adjuvant radiation therapy due to the high grade of the sarcoma. Post operative surveillance imaging at 18 months revealed a 4 cm lesion in her resection bed which also had PET avidity suggesting recurrence. She subsequently underwent an endoscopic ultrasound guided FNA of the mass that confirmed recurrence.
Case 4A 41-year-old female with multiple symptoms, including abdominal pain, flushing, sweating and weight loss, presented to her primary care physician. Abdominal examination was unremarkable. An abdominal ultrasound was performed revealing a 7 x 6 x 7 cm, solid, echo-genic mass between the liver and right kidney. This prompted a CT abdomen/pelvis with IV contrast (Figure 4), which demonstrated a 6 x 7 cm lesion of the right adrenal gland with mild internal soft tissue density.
A right adrenal resection was performed lap-aroscopically with removal of a necrotic and hemorrhagic tumor. Pathology showed a 6 cm mass with mildly elevated increase in dopa-mine/epinephrine. Pathology demonstrated a myelolipoma.
Adrenal myelolipoma is a rare benign tumor typically with lipid tissue containing a bone marrow component. Expanded use of imaging is leading to increased detection of myelolipo-mas. Generally, they are considered non-func-
Figure 3. CT abdomen/pelvis revealed a left adrenal mass.
HCA Healthcare Journal of Medicine
88
tional and do not require surgical excision. Additionally, they can be regarded as an excep-tion to the mandatory metabolic workup for a newly detected adrenal mass,18, 19 but rarely they can be functional. Given the patient’s presenting symptoms, laparoscopic surgical excision was warranted, in accordance with the accepted indications for surgical excision of a myelolipoma, including symptomatic tumor > 4 cm, metabolically active tumor and suspicion of malignancy on imaging.20
Case 5A 79-year-old female presented to the emer-gency department with shortness of breath and chest pain. Initially, she was treated for presumed acute congestive heart failure and pulmonary edema. However, the patient did not improve despite appropriate therapy. This prompted CT imaging of the chest which revealed a partially visualized incidentaloma superior to the left kidney. An MRI of the abdo-
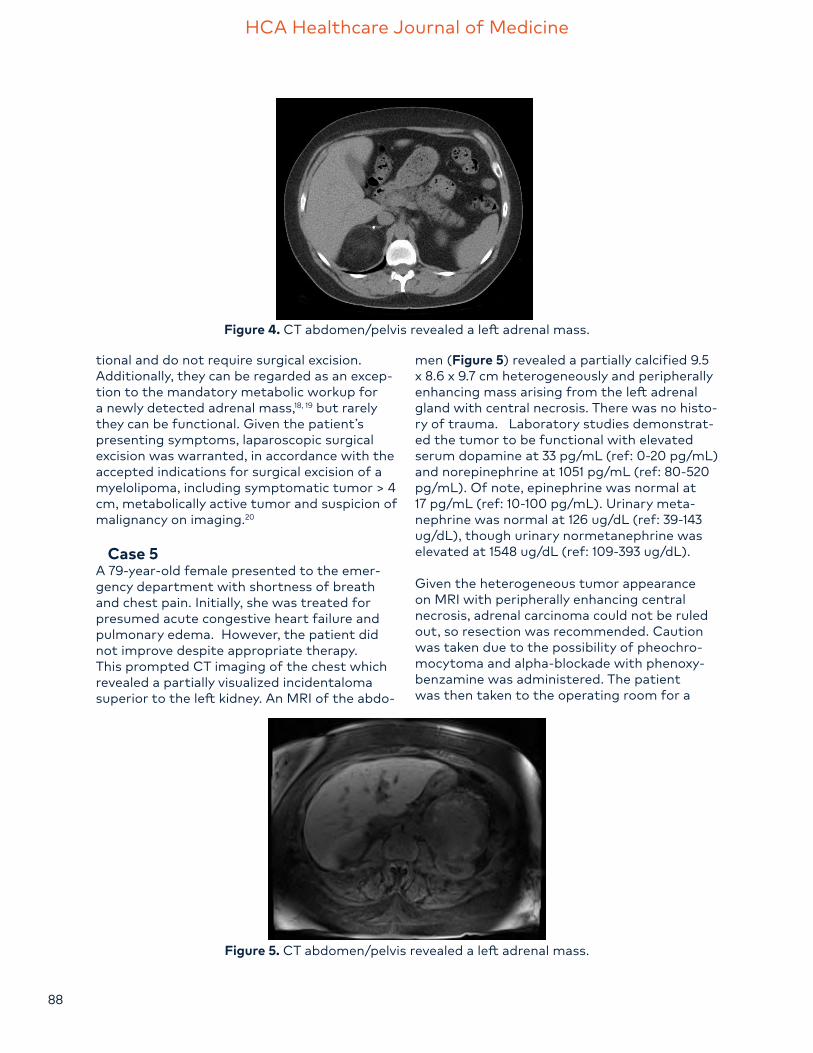
men (Figure 5) revealed a partially calcified 9.5 x 8.6 x 9.7 cm heterogeneously and peripherally enhancing mass arising from the left adrenal gland with central necrosis. There was no histo-ry of trauma. Laboratory studies demonstrat-ed the tumor to be functional with elevated serum dopamine at 33 pg/mL (ref: 0-20 pg/mL) and norepinephrine at 1051 pg/mL (ref: 80-520 pg/mL). Of note, epinephrine was normal at 17 pg/mL (ref: 10-100 pg/mL). Urinary meta-nephrine was normal at 126 ug/dL (ref: 39-143 ug/dL), though urinary normetanephrine was elevated at 1548 ug/dL (ref: 109-393 ug/dL).
Given the heterogeneous tumor appearance on MRI with peripherally enhancing central necrosis, adrenal carcinoma could not be ruled out, so resection was recommended. Caution was taken due to the possibility of pheochro-mocytoma and alpha-blockade with phenoxy-benzamine was administered. The patient was then taken to the operating room for a
Figure 4. CT abdomen/pelvis revealed a left adrenal mass.
Figure 5. CT abdomen/pelvis revealed a left adrenal mass.

Cooke-Barber et al. (2020) 1:2. https://doi.org/10.36518/2689-0216.1028
89
left radical adrenalectomy. Given the tumor’s encroachment on the left renal artery, the diminutive size of the left kidney, the patient’s age and comorbidities, and a functioning right kidney; a left nephrectomy was also performed. The lesion measured 14 x 13 x 14 cm. Pathology indicated the mass to be a benign hemorrhagic adrenal cyst with focal myelolipomatous meta-plasia. The patient tolerated the surgery well and was discharged home. Ultimately, she died 40 days later due to complications of pre-exist-ing COPD and chronic kidney disease.
DiscussionThe diagnosis of adrenocortical carcinoma is difficult to make based upon pre-operative imaging alone. These cases demonstrate the importance of a comprehensive history and physical exam in determining the nature of a mass. Pertinent positives, including hyper-tension, headaches, hirsutism, etc., on review of symptoms, may reveal a functional tumor. When an adrenal mass is seen it is imperative to characterize the mass with cross-sectional imaging and obtain labs.
In patients with adrenocortical carcinoma, CT is more likely to reveal a tumor greater than 6 cm in diameter, reduced contrast washout on contrast CT, calcifications, heterogeneity, central low attenuation and poorly-defined margins.12 A contrast CT showing greater than 10 Hounsfield Units (HU), as mentioned earlier, has been considered a predictor for malignancy, although some have suggested this threshold be changed to 13 HU.11
Lab work can aid in preoperative diagnosis and includes a full endocrine workup, including studies to rule out hypercortisolism, adrenocor-ticotropic hormone measurement and aldoste-rone-renin ratio. Additional tests to consider include dehydroepiandrosterone sulfate level, which is often elevated in malignant adrenal tumors, and plasma metanephrine and norme-tanephrine measurement, which can rule out pheochromocytoma.21
Despite the recommendations in place for diagnosing ACC based on imaging and labs, the diagnosis is frequently missed preoperatively, and improvement is needed in this field.22 One area in which further research must be done
is in patient selection for preoperative biopsy, since imaging characterization has become quite sensitive and specific.11, 17 When an adrenal cancer is suspected, FNA biopsy is relatively contraindicated due to the risk of seeding the tumor.16 Therefore, when malignancy is a dif-ferential diagnosis, resection without biopsy is often the preferred treatment.
Several new modalities have been suggested for improving the capability to diagnose ACC preoperatively. New imaging methods, such as FDG-PET scan or PET using C-labeled metomi-date (MTO), have shown some utility in diag-nosing cortical tumors.23, 24 FDG-PET has been shown to diagnose ACCs with very high sensi-tivity and specificity, and therefore, is becoming more widely used. However, the efficacy of its routine use is unclear.23, 24 Several molecular markers for ACC are currently being explored. MiRNAs 483-5p and 483-3p are generally over-expressed in ACC, and overexpression of miR-483, miR-503 and miR-1275 are associated with decreased survival.25, 26 Detection of aberrant methylation patterns and abnormal gene ex-pression may also provide new methods of di-agnosing adrenocortical carcinoma.4, 26 As these methods become validated and more widely available for clinical use, clinicians may be able to diagnose ACC more efficiently, improving survival length and cure rates for patients with the disease.
The surgical goal in treatment of ACC is com-plete resection with negative margins, which offers the best and possibly the only chance at survival. In fact, a study of 3,982 patients by Bilimoria et al. showed that patients with margin-negative resections have a 44.2-month greater median survival than patients who undergo margin-positive resections.27 Although laparoscopic adrenalectomy (LA) is generally the best option for benign adrenal tumors, it has been shown to increase the likelihood of having positive margins compared to open adrenalectomy when used on patients with ACC, thus decreasing patients’ chances for a cure.28 Studies have found lower overall survival in patients with stage II and greater ACC who undergo a laparoscopic approach, along with a higher risk of peritoneal carcinomatosis at the time of recurrence, which decreases the likeli-hood of a successful salvage surgery.28-31

HCA Healthcare Journal of Medicine
90
Adrenal artery embolization is a minimal-ly invasive procedure that can be used as an alternative or adjunct to surgery. Of the varied indications for this procedure, it is most often performed for oncologic palliation, such as pain relief, reduction of tumor bulk or preoperative reduction of tumor bulk. Emergency emboli-zation for the hemostasis of ruptured tumors with retroperitoneal hemorrhage is an addition-al application.32 This procedure is not routinely used in the management of ACC but can be an adjunct with surgery in massive hemorrhagic tumors.
Due to the discouraging prognosis, Autorino et al. argues that open resection of ACC is the best approach, with laparoscopic resection being saved for a select few cases.33 On the other hand, Vanbrugghe et al. supports the use of laparoscopic adrenalectomy, especially for patients with smaller tumors and no evidence of invasion. This group argues that the extent of the resection is the important variable, and that if a complete resection with negative margins can be achieved using a laparoscopic approach, this is a viable option.34 Case 2 in our series could be regarded as support for this opinion. In that case, a laparoscopic approach was attempted despite imaging consistent with malignancy, and the intervention success-fully achieved negative margins. The patient has shown no sign of recurrence since 2014. Furthermore, the exclusion of a laparoscopic approach as an option rests on the assumption that the ACC will be diagnosed and staged with invasive disease ruled out. Therefore, in cases that ultimately do not turn out to be ACC, the open approach is likely unnecessary. This again highlights the importance of improv-ing methods of preoperative diagnosis.
In our series we describe five cases:
Case one was highly suspicious preoperative-ly for ACC and was approached aggressively. In most instances the kidney can be salvaged; however, complete lymphadenectomy is crucial and in this case, resulted in nephrectomy, due to the necessary resection of nodes around hilar vessels.
Case two was managed as a nonfunctional adrenal mass at 7 cm and thus, was approached laparoscopically with oncologic principles to
resect en bloc or convert to open which was not required.
Case three represents an aggressive sarcoma. A preoperative biopsy with radiation may have been a suitable alternative, although the radi-ation field would have been quite large. It was believed on imaging to represent bleeding into a tumor, causing the pain and; therefore, a sur-gical option was advised.
Case four represented a symptomatic myelo-lipoma causing abdominal pain, sweating and weight loss. The tumor was excised via laparo-scopic resection.
Case five demonstrated benign pathology with false elevation of normetanephrine. It is possi-ble this false elevation occurred as a medication side effect. The patient was taking a beta-2 agonist inhaler, which has been shown to cause false elevation of normetanephrines in the lit-erature.35 Regardless, these lesions can undergo angioembolization if classic imaging character-istics suggest angiomyolipoma. Transarterial embolization of angiomyolipoma demonstrates low rates of mortality and serious complica-tions.36
ConclusionIn this case series we have highlighted the chal-lenges and complexities of preoperatively diag-nosing Adrenocortical Carcinoma and choosing the best operative approach. Further research is needed to improve preoperative diagnosis of ACC with imaging and laboratory assessment. This could potentially be accomplished through a randomized, prospective trial separating patients by pre-operative imaging modality and laboratory studies, but would require a well-or-ganized, multi-center study given its overall low incidence in any given population.
Conflicts of InterestThe authors declare they have no conflicts of interest.
Drs. Jo Cooke-Barber, Andrew Schwemmer and Christopher Senkowski are employees of Memorial Health University Medical Center, a hospital affiliated with the journal’s publisher.
This research was supported (in whole or in

Cooke-Barber et al. (2020) 1:2. https://doi.org/10.36518/2689-0216.1028
91
part) by HCA Healthcare and/or an HCA Healthcare affiliated entity. The views expressed in this publication represent those of the author(s) and do not necessarily represent the official views of HCA Healthcare or any of its affiliated entities.
Author Affiliations1. Memorial Health University Medical Center2. Ohio State University School of Medicine3. Mercer University School of Medicine
References1. Clark OH, Duh Q-Yang. Textbook of Endocrine
Surgery. Philadelphia: W.B. Saunders; 1997.2. Allolio B, Fassnacht M. Adrenocortical carcino-
ma: Clinical update. J Clin Endocrinol Metab. 2006;91(6):2027-2037. https://doi.org/10.1210/jc.2005-2639
3. Libé R. Adrenocortical carcinoma (ACC): di-agnosis, prognosis, and treatment. Front Cell Dev Biol. 2015;3:45. https://doi.org/10.3389/fcell.2015.00045
4. Szyszka P, Grossman AB, Diaz-Cano S, Sworczak K, Dworakowska D. Molecular pathways of human adrenocortical carcinoma—translating cell signalling knowledge into diag-nostic and treatment options. Endokrynol Pol. 2016;67(4):427-450. https://doi.org/10.5603/EP.a2016.0054
5. Ribeiro RC, Sandrini F, Figueiredo B, et al. An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc Natl Acad Sci U S A. 2001;98(16):9330-9335. https://doi.org/10.1073/pnas.161479898
6. Jouinot A, Bertherat J. MANAGEMENT OF ENDOCRINE DISEASE: Adrenocortical car-cinoma: differentiating the good from the poor prognosis tumors. Eur J Endocrinol. 2018;178(5):R215-R230. https://doi.org/10.1530/EJE-18-0027
7. Ayala-Ramirez M, Jasim S, Feng L, et al. Ad-renocortical carcinoma: clinical outcomes and prognosis of 330 patients at a tertiary care center. Eur J Endocrinol. 2013;169(6):891-899. https://doi.org/10.1530/EJE-13-0519
8. Li L, Yang G, Zhao L, et al. Baseline Demo-graphic and Clinical Characteristics of Pa-tients with Adrenal Incidentaloma from a Single Center in China: A Survey. Int J Endocrinol. 2017;2017:3093290. https://doi.org/10.1155/2017/3093290
9. Creemers SG, Hofland LJ, Korpershoek E, et al. Future directions in the diagnosis and medical treatment of adrenocortical carcinoma. Endocr Relat Cancer. 2016;23(1):R43-R69. https://doi.
org/10.1530/ERC-15-045210. Sturgeon C, Shen WT, Clark OH, Duh QY, Kebe-
bew E. Risk assessment in 457 adrenal cortical carcinomas: how much does tumor size predict the likelihood of malignancy? J Am Coll Surg. 2006;202(3):423-430. https://doi.org/10.1016/j.jamcollsurg.2005.11.005
11. Ilias I, Sahdev A, Reznek RH, Grossman AB, Pa-cak K. The optimal imaging of adrenal tumours: a comparison of different methods. Endocr Relat Cancer. 2007;14(3):587-599. https://doi.org/10.1677/ERC-07-0045
12. Zhang HM, Perrier ND, Grubbs EG, et al. CT features and quantification of the character-istics of adrenocortical carcinomas on unen-hanced and contrast-enhanced studies. Clin Ra-diol. 2012;67(1):38-46. https://doi.org/10.1016/j.crad.2011.03.023
13. Young WF Jr. Conventional imaging in adreno-cortical carcinoma: update and perspectives. Horm Cancer. 2011;2(6):341-347. https://doi.org/10.1007/s12672-011-0089-z
14. Ierardi AM, Petrillo M, Patella F, et al. Interven-tional radiology of the adrenal glands: current status. Gland Surg. 2018;7(2):147-165. https://doi.org/10.21037/gs.2018.01.04
15. Elsayes KM, Mukundan G, Narra VR, et al. Adre-nal masses: mr imaging features with patho-logic correlation. Radiographics. 2004;24 Suppl 1:S73-S86. https://doi.org/10.1148/rg.24si045514
16. Berruti A, Baudin E, Gelderblom H, et al. Adre-nal cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23 Suppl 7:vii131-vii138. https://doi.org/10.1093/annonc/mds231
17. Terzolo M, Stigliano A, Chiodini I, et al. AME position statement on adrenal incidentaloma. Eur J Endocrinol. 2011;164(6):851-870. https://doi.org/10.1530/EJE-10-1147
18. NIH state-of-the-science statement on man-agement of the clinically inapparent adrenal mass (“incidentaloma”). NIH Consens State Sci Statements. 2002;19(2):1-25.
19. Zeiger MA, Thompson GB, Duh QY, et al. American Association of Clinical Endocrinolo-gists and American Association of Endocrine Surgeons Medical Guidelines for the Manage-ment of Adrenal Incidentalomas: executive summary of recommendations. Endocr Pract. 2009;15(5):450-453. https://doi.org/10.4158/EP.15.5.450
20. Shenoy VG, Thota A, Shankar R, Desai MG. Adrenal myelolipoma: Controversies in its management. Indian J Urol. 2015;31(2):94-101. https://doi.org/10.4103/0970-1591.152807
21. Héron E, Chatellier G, Billaud E, Foos E, Plouin PF. The urinary metanephrine-to-creatinine ratio for the diagnosis of pheochromocytoma. Ann Intern Med. 1996;125(4):300-303. https://doi.org/10.7326/0003-4819-125-4-199608150-

HCA Healthcare Journal of Medicine
92
0000822. Ozsari L, Kutahyaliouglu M, Elsayes EM, et al.
Preexisting adrenal masses in patients with adrenocortical carcinoma: clinical and radio-logical factors contributing to delayed diagno-sis. Endocrine. 2016;51(2):351-359. https://doi.org/10.1007/s12020-015-0694-7
23. Boland GW, Dwamena BA, Jagtiani Sangwaiya M, et al. Characterization of adrenal masses by using FDG PET: a systematic review and meta-analysis of diagnostic test performance. Radiology. 2011;259(1):117-126. https://doi.org/10.1148/radiol.11100569
24. Takeuchi S, Balachandran A, Habra MA, et al. Impact of ¹⁸F-FDG PET/CT on the manage-ment of adrenocortical carcinoma: analysis of 106 patients. Eur J Nucl Med Mol Imaging. 2014;41(11):2066-2073. https://doi.org/10.1007/s00259-014-2834-3
25. Soon PS, Tacon LJ, Gill AJ, et al. miR-195 and miR-483-5p Identified as Predictors of Poor Prognosis in Adrenocortical Cancer. Clin Can-cer Res. 2009;15(24):7684-7692. https://doi.org/10.1158/1078-0432.CCR-09-1587
26. Özata DM, Caramuta S, Velázquez-Fernández D, et al. The role of microRNA deregulation in the pathogenesis of adrenocortical carcino-ma. Endocr Relat Cancer. 2011;18(6):643-655. Published 2011 Oct 27. https://doi.org/10.1530/ERC-11-0082
27. Bilimoria KY, Shen WT, Elaraj D, et al. Adreno-cortical carcinoma in the United States: treat-ment utilization and prognostic factors. Cancer. 2008;113(11):3130-3136. https://doi.org/10.1002/cncr.23886
28. Huynh KT, Lee DY, Lau BJ, Flaherty DC, Lee J, Goldfarb M. Impact of Laparoscopic Adre-nalectomy on Overall Survival in Patients with Nonmetastatic Adrenocortical Carcinoma. J Am Coll Surg. 2016;223(3):485-492. https://doi.org/10.1016/j.jamcollsurg.2016.05.015
29. Payabyab EC, Balasubramaniam S, Edgerly M, et al. Adrenocortical Cancer: A Molecularly Complex Disease Where Surgery Matters. Clin Cancer Res. 2016;22(20):4989-5000. https://doi.org/10.1158/1078-0432.CCR-16-1570
30. Leboulleux S, Deandreis D, Al Ghuzlan A, et al. Adrenocortical carcinoma: is the surgical approach a risk factor of peritoneal carcinoma-tosis?. Eur J Endocrinol. 2010;162(6):1147-1153. https://doi.org/10.1530/EJE-09-1096
31. Gonzalez RJ, Shapiro S, Sarlis N, et al. Laparo-scopic resection of adrenal cortical carcinoma: a cautionary note. Surgery. 2005;138(6):1078-1086. https://doi.org/10.1016/j.surg.2005.09.012
32. Fowler AM, Burda JF, Kim SK. Adrenal ar-tery embolization: anatomy, indications, and technical considerations. AJR Am J Roentgenol. 2013;201(1):190-201. https://doi.org/10.2214/AJR.12.9507
33. Autorino R, Bove P, De Sio M, et al. Open Ver-sus Laparoscopic Adrenalectomy for Adreno-cortical Carcinoma: A Meta-analysis of Surgical
and Oncological Outcomes. Ann Surg Oncol. 2016;23(4):1195-1202. https://doi.org/10.1245/s10434-015-4900-x
34. Vanbrugghe C, Lowery AJ, Golffier C, Taieb D, Sebag F. Adrenocortical carcinoma surgery-sur-gical extent and approach. Langenbecks Arch Surg. 2016;401(7):991-997. https://doi.org/10.1007/s00423-016-1462-8
35. Pacak K, Del Rivero J. Pheochromocytoma imaging. In: Feingold KR, Anawalt B, Boyce A, et al., ed., Endotext [Internet]. South Dart-mouth, MA: MDText.com, Inc.; 2013. https://doi.org/10.1530/endoabs.32.S14.2
36. Murray TE, Doyle F, Lee M. Transarterial Em-bolization of Angiomyolipoma: A Systematic Review. J Urol. 2015;194(3):635-639. https://doi.org/10.1016/j.juro.2015.04.081