tesi triennale
TRANSCRIPT

UNIVERSITA’ DI ROMA “LA SAPIENZA”
FACOLTA’ DI SCIENZE MATEMATICHE FISICHE E NATURALI
Corso di Laurea triennale in Scienze Biologiche Curricolo generale
ELABORATO FINALE
Ruolo della vitamina B6 nelle Encefalopatie Epilettiche Neonatali Candidato: Alessandra Cicalini 1131336
Relatore interno: Prof. Roberto Contestabile
(Dipartimento di Scienze Biochimiche)
Dott. Martino L. di Salvo (Dipartimento di Scienze Biochimiche)
Anno Accademico 2011 – 2012

INTRODUZIONE
Le Encefalopatie Epilettiche Neonatali (NEE) sono sindromi convulsive che
si manifestano nei neonati già nelle prime ore di vita, causate da disfunzioni
di varia natura a carico del sistema nervoso centrale e generalmente curate
mediante somministrazione di farmaci anticonvulsivanti.
In qualche raro caso sono state riscontrate NEE refrattarie alle cure con i
convenzionali farmaci, le quali hanno tuttavia risposto bene al trattamento
con la vitamina B6 ed in modo speciale ai vitameri piridossina (PN) e
piridossal-5'-fosfato (PLP).
Queste particolari NEE dipendenti dalla vitamina B6 (NEE-B6 responsive)
sono risultate associabili principalmente ad una carenza di PLP nel corpo.
I pazienti affetti da NEE presentano inoltre fenotipi caratteristici, indici dei
metabolismi alterati, strumenti grazie ai quali è stato possibile risalire agli
enzimi mutati e comprendere il ruolo del PLP in queste sindromi.
Nella prima parte di questa tesi verranno descritte struttura e funzioni della
vitamina B6 con particolare attenzione al piridossal-5'-fosfato, il suo ruolo
come cofattore enzimatico e la sua reattività. Successivamente l'attenzione
verrà portata sugli enzimi della via di recupero del PLP, per illustrarne,
anche qui, struttura e funzione, correlate all'assorbimento ed al
metabolismo del cofattore. Nella parte centrale della tesi verranno
affrontate le possibili cause della deficienza del PLP e la conseguente
alterazione dei metabolismi, con particolare attenzione a quelli correlati
con la funzionalità cerebrale e l'epilessia.
Nella parte conclusiva verranno infine trattate le Encefalopatie Epilettiche
Neonatali ed il coinvolgimento del PLP in queste malattie.
Lo scopo finale della tesi è quello di esporre con chiarezza il ruolo
interpretato dalla vitamina B6, in particolar modo dal cofattore piridossal-
5'-fosfato, nelle NEE-B6 responsive, mettendo in luce le possibili cause della
deficienza del cofattore e delineando i possibili livelli di indagine
affrontabili nello studio della problematica.

INDICE
1. PIRIDOSSAL-5'-FOSFATO: LA FORMA BIOLOGICAMENTE
ATTIVA DELLA VITAMINA B6 ……………………………………… pag. 1
1.1 Vitamina B6: struttura ……………………………………………………………. pag. 1
1.2 Vitamina B6: funzione ……………………………………………………………. pag. 2
1.3 Il cofattore piridossal-5'-fosfato (PLP) e la sua reattività ………………………... pag. 3
1.4 Vitamina B6: biosintesi …………………………………………………………... pag. 6
1.5 Vitamina B6: assorbimento ……………………………………………………….. pag. 7
2. ENZIMI DELLA VIA DI RECUPERO DEL PLP …………………….. pag. 10
2.1 Generalità ………………………………………………………………………… pag. 10
2.2 Struttura e funzione dell'enzima piridossal chinasi (PLK) ………………………. pag. 11
2.3 Struttura e funzione dell'enzima PNP ossidasi (PNPOx) ………………………… pag. 16
2.4 Regolazione dell'espressione del salvage pathway e controllo dei livelli di PLP … pag. 19
2.5 Tossicità del PLP …………………………………………………………………. pag. 21
3. DEFICIENZA DI PLP: CAUSE ED EFFETTI DELLA CARENZA
DI PLP SUL METABOLISMO …………………………………………. pag. 22
3.1 Malfunzionamento degli enzimi del salvage pathway …………………………… pag. 22
3.2 Sequestro del PLP da parte di intermedi di vie metaboliche mutate …………….. pag. 26
3.3 Effetto dei farmaci sull’ uptake della vitamina B6 ………………………………. pag. 27
3.4 Ruolo degli enzimi PLP-dipendenti nel metabolismo ed alterazione delle
vie metaboliche dovuta a carenza del PLP ………………………………………. pag. 28
3.5 Enzimi PLP-dipendenti nel metabolismo dei neurotrasmettitori e funzione
cerebrale …………………………………………………………………..…….. pag. 30
3.6 L'epilessia e gli enzimi PLP-dipendenti ………………………………………….. pag. 32
4. COINVOLGIMENTO DEL PLP NELLE ENCEFALOPATIE
EPILETTICHE NEONATALI (NEE) …………………………………... pag. 35
4.1 Riepilogo introduttivo ……………………………………………………………. pag. 35
4.2 Introduzione alle Encefalopatie Epilettiche Neonatali (NEE) …………………… pag. 36
4.3 NEE PLP-dipendenti ……………………………………………………………... pag. 37
4.4 NEE PN-dipendenti (PDE) ……………………………………………………….. pag. 41
CONCLUSIONI E POSSIBILI LIVELLI DI INDAGINE ……………….. pag. 44
RINGRAZIAMENTI …………………………………………………..…... pag. 48
ELENCO ABBREVIAZIONI …………………………………………..…. pag. 49
INDICE FIGURE …………………………………………………………… pag. 50
BIBLIOGRAFIA ……………………………………………………………. pag. 51

1
1. PIRIDOSSAL-5'-FOSFATO: LA FORMA
BIOLOGICAMENTE ATTIVA DELLA VITAMINA B6
1.1 Vitamina B6: struttura
Il termine vitamina B6 è stato spesso usato per indicare la piridossina.
Oggi ha acquisito un carattere più generico riferendosi all'insieme di sei composti o vitameri,
presenti in tutti gli organismi, che condividono la struttura base 2-metil-3-idrossipiridina e
differiscono per la natura dei diversi sostituenti sul C4 e C5 .
I vitameri B6 qui di seguito riportati sono: la piridossina (PN), il piridossale (PL), la
piridossamina (PM) ed i loro rispettivi derivati fosforilati sul C5; la piridossin-5'-fosfato
(PNP), il piridossal-5'-fosfato (PLP) e la piridossamin-5'-fosfato (PMP). (Figura 1)
FIGURA 1. Struttura dei vitameri B6
(PMP)

2
1.2 Vitamina B6: funzione
Il piridosal-5'-fosfato (PLP) è considerato la forma biologicamente attiva della vitamina B6
poiché, oltre a svolgere svariate funzioni in molteplici processi cellulari, assume il ruolo
predominante di cofattore enzimatico.
Attua infatti oltre 140 differenti reazioni enzimatiche, le quali rappresentano circa il 4% di
tutte le attività catalitiche conosciute e sono eseguite da enzimi appartenenti a cinque delle
sei classi enzimatiche fin ora catalogate.
La vitamina B6 svolge inoltre altre funzioni importanti come dimostrato da studi recenti
effettuati su molti organismi:
È stato dimostrato, sia nelle piante che nell'uomo, il ruolo della vitamina nel
“quenching” delle specie reattive dell'ossigeno (ROS) e nella protezione da molecole
citotossiche, che incrementa la resistenza agli stress biotici e abiotici. (Bilski P. et al.
2000; Büyükokuroglu ME 2007)
Vi sono prove che nel corpo umano i vitameri possano agire come fattori
antiossidanti. (Bilski P. et al. 2000)
Studi passati hanno evidenziato il ruolo del PLP e della piridossina come regolatori
del trasporto ionico di membrana (Lambrecht G. et al. 2002; Salhany JM et al.
1987; Dakshinamurti K. et al. 1998)
Sempre il PLP e la PN sono stati identificati come ligandi di recettori steroidei (Oka
T. 2001) e come regolatori di fattori di trascrizione. (Huq M. D. et al. 2007)
Il PLP lega il dominio D1 della proteina CD4, una proteina transmembrana di 55
kDa presente su Linfociti T, implicata nella trasduzione del segnale di
riconoscimento antigenico. (Salhany J.M. et al. 1993)

3
1.3 Il cofattore piridossal-5'-fosfato (PLP) e la sua reattività
La capacità di effettuare un così grande numero di reazioni il PLP lo deve alla sua elevata
reattività, dovuta al gruppo aldeidico in posizione C4, il quale può reagire con:
Ammino-gruppi, tramite reazioni di condensazione per formare una base di Schiff.
Idrazine, tramite reazioni di condensazione.
Idrossilammine sostituite, tramite reazioni di condensazione.
Composti sulfidrilici
Il PLP è infatti in grado di catalizzare anche in assenza di enzimi, in soluzione, numerose
reazioni. Tuttavia la velocità delle reazioni catalizzate è inferiore rispetto a quando si trova
legato come cofattore agli enzimi. (Snell E.E. 1985)
Questo viene spiegato dal fatto che, nella tasca enzimatica, le proprietà catalitiche del
cofattore vengono modulate dalle catene polipeptidiche che lo circondano, le quali da una
parte aumentano la velocità di reazione e dall'altra gli conferiscono un più ampio spettro di
possibilità catalitiche.
I passaggi iniziali del meccanismo d'azione del PLP, in tutte le reazioni enzimatiche PLP-
dipendenti, sono essenzialmente gli stessi. (Figura 2)
Il cofattore è sempre covalentemente legato, in forma di aldimmina, al gruppo ε-amminico di
un residuo di lisina, nella tasca dell'enzima PLP-dipendente, formando la cosiddetta
"aldimmina interna". Il gruppo -NH2 del substrato effettua poi un attacco nucleofilo al C4' del
PLP, con formazione di una diammina geminale intermedia. In questa struttura intermedia
entrambi i gruppi -NH2, dell'enzima e del substrato, sono legati al C4', il quale assume una
geometria tetraedrica. Successivamente il legame con il residuo di lisina viene scisso,
generando, per condensazione del PLP con il substrato, quella che viene chiamata un'
"aldimmina esterna" (Base di Schiff).
Sia nell'aldimina interna che in quella esterna, invece, il C4' ha una geometria planare.
(Metzler et al. 1954)

4
Aldimina interna
FIGURA 2. Meccanismo di reazione del PLP; formazione della base di Schiff
A questo punto i legami del Cα del substrato vengono indeboliti dall'effetto di attrazione
elettronica, esercitato dall'anello aromatico del PLP e dall'azoto piridinico protonato,
portando alla rottura eterolitica di uno di questi.
Le successive fasi della reazione si diversificano a seconda del tipo di substrato e di quale
dei legami del Cα del substrato viene scisso.
Il legame che viene rotto dipende anche dalla conformazione e dalla natura dei residui
amminoacidici della tasca enzimatica, poiché sono questi a modulare l'attività catalitica del
cofattore stabilizzando un legame piuttosto che un altro.
Tuttavia la rottura può essere di tre tipi: l’eliminazione di CO2, la deprotonazione,
l’eliminazione della catena laterale.
Comunque la perdita di uno dei sostituenti del Cα del substrato, per taglio eterolitico, porta
sempre alla formazione di un carbanione (chiamato anche carbanione α), con una coppia di
elettroni libera, altamente instabile.
Il PLP si comporta però come una “trappola elettronica”, grazie all'estesa coniugazione degli
elettroni π dell'anello piridinico, delocalizzando per risonanza la carica negativa ed il
carbanione viene immediatamente stabilizzato formando un intermedio chinonoide.. (Nelson
D.L. and COX M.M 2010) (Figura 3)
Aldimina esterna (base di Schiff)
Diamina geminale 1
Aldimina interna Diamina geminale 2

5
FIGURA 3. Struttura di risonanza del
carbanione con formazione di un intermedio
chinonoide.
A questo punto seguono, a seconda dell'enzima coinvolto nella catalisi, molteplici vie, che si
ramificano in una serie di passaggi e portano al distacco di prodotti finali differenti. A
seconda del legame rotto precedentemente possono presentarsi diverse reazioni:
1) Reazioni che procedono dopo l’eliminazione di CO2 dal Cα:
α-decarbossilazione, α-decarbossilazione seguita da transaminazione
2) Reazioni che procedono dopo la deprotonazione del Cα:
racemizzazione, transaminazione, β-decarbossilazione, β-eliminazione,
β-sintesi, γ-eliminazione, γ-sintesi
3) Reazioni che procedono dopo l’eliminazione della catena laterale:
α-sintesi, scissione aldolica
Per esempio le reazioni centrali del metabolismo degli amminoacidi, catalizzate dagli enzimi
PLP-dipendenti, sono le più note sin dagli anni '40.
In questo caso il cofattore è coinvolto principalmente in diversi tipi di reazione a livello degli
atomi di carbonio α,β,γ (da C2 a C4) degli amminoacidi. Le reazioni riguardanti l’atomo di
carbonio α comprendono racemizzazioni, decarbossilazioni e transamminazioni (Hayashi H
1995; Eliot AC et al. 2004).
In ognuna di queste reazioni il PLP agisce sempre scindendo uno dei legami del carbonio α
del substrato, attraverso la rimozione di un protone o di un gruppo carbossilico.

6
1.4 Vitamina B6: biosintesi
Solamente le piante, gli Eubatteri, gli Archaea ed alcuni funghi sono in grado di sintetizzare
de novo la vitamina B6 e sono due le vie di sintesi fin ora conosciute.
La prima via, utilizzata da Escherichia coli e altri membri della divisione γ dei proteobatteri,
viene definita “DXP-dependente” e richiede per la sintesi di PNP sei enzimi, riportati nella
figuura sottostante. (Figura 4)
Gli altri microrganismi, inclusi gli Archea, e la maggior parte degli Eubatteri, i funghi e le
piante, invece, utilizzano una seconda via biosintetica definita “DXP-independente”. Questa
via è catalizzata solo da due enzimi (Pdx1-Pdx2), che agiscono insieme per convertire il
ribosio 5-fosfato (R5P) e la gliceraldeide 3-fosfato (G3P) direttamente in PLP. Questo
complesso enzimatico viene chiamato PLP sintasi. (Figura 4)
Anche alcuni batteri patogeni sono in grado di sintetizzare la vitamina (Neisseria meningitis,
Salmonella typhimurium, Vibrio cholerae, Yersina pestis ed altri), quindi progettare farmaci
specifici potrebbe avere interessanti fini applicativi. (Fitzpatrick et al., 2010)
FIGURA 4.
Vie biosintetiche della vitamina
B6.
Via De novo DXP-dipendente
(presente in alcuni eubatteri):
GapB, d-eritrosio-4-fosfato
deidrogenasi; PdxB, eritronato-4-
fosfato deidrogenasi; PdxF/SerC,
fosfoserina aminotransferasi;
PdxA, 4-idrossitreonina-4-fosfato
deidrogenasi; DPXS, 1-deossi-d-
xilulosio-5-fosfato sintasi; PNP
sintasi, dal gene PdxJ. Via De
novo DXP-indipendente (presente
negli altri eubatteri, funghi ,piante
ed Archaea): complesso PLP
sintasi: dominio sintasico dal gene
Pdx1; dominio glutaminasico dal
gene Pdx2. Salvage pathway
(presenti in tutti gkli organismi
inclusi I mammiferi): PLK,
piridossal chinasi dal gene PdxK e
piridossal chinasi 2 dal gene PdxY;
PNPOx, piridossina-5’-fosfato
ossidasi dal gene PdxH.

7
1.5 Vitamina B6: assorbimento
Gli animali non sono in grado di sintetizzare de novo la vitamina B6 e la devono pertanto
acquisire dalla dieta mediante assorbimento intestinale:
Dal cibo di origine animale l'uomo assimila la vitamina in parte sotto forma di PMP ma
principalmente sotto forma di PLP, che si trova associato agli enzimi PLP-dipendenti.
In particolar modo ricava il PLP dalla degradazione della glicogeno fosforilasi presente
in abbondante quantità nel muscolo e quindi nella carne. (McCormick D. B. 1989)
Dal cibo di origine vegetale la vitamina B6 viene assunta principalmente sotto forma di
PN e PN-5'-β-glucoside (Figura 5) anche se in quest'ultima forma sembra essere
assimilata con più difficoltà poiché richiede l'intervento di glucosidasi intestinali.
(Mackey et al 2003)
FIGURA 5. Piridossina-5'-β glucoside
É stato dimostrato che anche il latte umano presenta grandi quantità di vitamina B6
sotto forma di PLP e PL.(Morrison and Driskell 1985)

8
Tuttavia, anche non disponendo di enzimi per la sua sintesi, gli animali possiedono gli
enzimi in grado di interconvertire tra loro le forme defosforilate e fosforilate della vitamina
(PL chinasi e varie fosfatasi) ed un' ossidasi in grado di convertire poi le forme fosforilate in
PLP (PNP ossidasi; PNPOx). (Mccormick D. B., Chen H. 1999; Jang, Y. M. 2003)
(Figura 6)
FIGURA 6. Interconversione tra le forme fosforilate e defosforilate della vitamina B6 ed enzimi
della via di recupero del PLP
Tutti questi enzimi prendono il nome di “enzimi della via di recupero” del PLP ed il loro
funzionamento è indispensabile per l'assorbimento di tutti i vitameri e per permettere che
raggiungano i tessuti.
Infatti a livello intestinale le forme fosforilate (PLP, PMP, PNP), che non riuscirebbero a
passare la membrana delle cellule ed essere così assorbite, vengono defosforilate a PN, PL e
PM da ecto-enzimi tessuto specifici rilasciati nel lume dell'intestino (fosfatasi intestinali; IP).
Le forme defosforilate, sia quelle attivamente formate dalle fosfatasi sia quelle direttamente
ottenute dal cibo, vengono così assorbite dall'intestino tenue mediante un sistema mediato da
carrier e riversate nel circolo sanguigno. (Said H. M. 2004)
A questo punto il sistema portale epatico trasporta i vitameri, attraverso la vena portale
epatica, fino al fegato dove la PL kinasi li rifosforila a PLP, PNP, PMP e la PNPOx converte
poi PNP e PMP in PLP.
Circa il 60% della vitamina che viene riversato nel circolo sistemico e raggiunge i tessuti è
sotto forma di PLP legato alla Lys190 all'albumina. La restante percentuale circola sotto
forma di PL, PM, e PN. (Surtees R. A. et al. 2006)

9
Una volta raggiunti i tessuti i vitameri defosforilati passano dai letti capillari al liquido
interstiziale ed entrano nelle cellule, mentre il PLP deve essere defosforilato a PL.
A mediare questo passaggio vi sono, in tutti i tessuti, delle fosfatasi alcaline tessuto-non-
specifiche (TNAP) associate alla membrana delle cellule endoteliali dei vasi. (Clayton P.T.
2006) Studi effettuati nel 2006 da diversi laboratori hanno dimostrato anche l'esistenza di
fosfatasi tessuto-specifiche espresse nelle cellule germinali e nella placenta implicate nella
stessa funzione.
Di particolare interesse in questa trattazione è evidenziare che anche a livello della barriera
emato-encefalica sono presenti le TNAP e che infatti il PL è il vitamero maggiormente
acquisito dai neuroni. Il PL, formato a livello del plesso coroideo, viene trasportato
attraverso il fluido cerebrospinale da un meccanismo di trasporto attivo ed attraversa la
membrana plasmatica dei neuroni. (Surtees R. A. et al. 2006)
All'interno delle cellule le forme defosforilate vengono nuovamente fosforilate dalla PL
kinasi e la PNPOx converte ancora i vitameri PNP e PMP nella forma biologicamente attiva
della vitamina: il PLP. Quest'ultimo viene così ceduto agli apo-enzimi PLP-dipendenti.
Tutto il ciclo di assorbimento è qui di seguito riassunto in figura 7.
FIGURA 7. Ciclo di assorbimento e trasporto della vitamina B6

10
2. ENZIMI DELLA VIA DI RECUPERO DEL PLP
2.1 Generalità
FIGURA 8. Enzimi della via di recupero o “salvage pathway” del PLP
Gli enzimi della via di recupero o “salvage pathway” (McCormick DB et al. 1999; Jang
YM et al. 2003) del PLP (Figura 8) svolgono un ruolo centrale in tutto il metabolismo della
vitamina B6. Oltre a permettere l'assorbimento dei vitameri ed il loro trasporto ai tessuti
(come trattato nel paragrafo precedente), sono indispensabili anche per la formazione
intracellulare del PLP, per regolarne il livello, evitarne la tossicità e per la formazione degli
oloenzimi PLP-dipendenti, poiché sembrano svolgere un ruolo attivo nel cedere il PLP
direttamente agli apoenzimi.
FMNH2 FMN
PNPOx

11
2.2 Struttura e funzione dell'enzima piridossal chinasi (PLK)
L'enzima piridossal chinasi (PLK) è stato purificato e caratterizzato da vari organismi:
cervello di mucca, cervello di pecora, fegato di maiale, fegato di pecora, eritrociti umani,
Escherichia coli ed Arabidopsis thaliana.
Mostra un peso molecolare che varia da 80 KDa, come gli enzimi del cervello di pecora e di
mucca, a 64 KDa, come l’enzima del fegato di maiale, a 70 KDa per la PLK di Arabidopsis
thaliana. (Karawya e Fonda 1978; Churchich e Wu 1981; Tagaya et al. 1989; Hirakawa-
Sakurai et al. 1993; Lainè-cessac e Allain 1996; Lum et al. 2002)
Nella maggior parte degli organismi la PLK viene codificata del gene pdxK, che nell'uomo è
collocato sul cromosoma 21q22.3.
Tuttavia, già uno studio del 1998 di Yang et al. dimostrò, in E. coli, la presenza di un
secondo gene che codifica per una PLK: il gene pdxY, che ha una significativa omologia di
sequenza con il già noto gene pdxK.
Con l'utilizzo di mutanti di E. coli bloccati nella biosintesi de novo del PLP e con il gene
pdxK inattivato, dimostrarono che la PLK codificata dal gene pdxY utilizza in modo
specifico solo il PL. Infatti le cellule erano in grado di crescere su PL ma non su PN o PM.
Gli autori hanno così concluso che negli eucarioti esiste un solo gene, pdxK, che codifica per
la chinasi, a cui è stato dato il nome di PLK1, la quale catalizza la reazione di fosforilazione
di tutti e tre i vitameri. La gran parte dei procarioti, invece, possiede entrambi i geni, pdxK e
pdxY ed il secondo enzima, chiamato PLK2, catalizza la reazione di fosforilazione solo del
PL..
La PLK catalizza l'addizione del fosfato dall’ATP al gruppo alcolico in posizione 5' dei
vitameri PL, PM, PN formando PLP, PMP, PNP rispettivamente (Figura 9). Le costanti
cinetiche della PLK umana (hPLK) sono state determinate e riportano i seguenti valori: Km
PL 24 µM, Km MgATP 190 M, kcat 29 min-1
. (Gandhi et al. 2009)

12
Figura 9. Reazione catalizzata dalla PLK.
In questo tipo di reazione entrambi i substrati (ATP e vitamero) si trovano legati all’enzima
formando un complesso ternario. Il complesso ternario si può generare mediante il legame
dei substrati in una sequenza casuale o con un ordine specifico. Sia nell'enzima di pecora che
di E. coli il meccanismo segue un criterio random, cioè i due substrati si legano all’enzima in
ordine casuale. (di Salvo et al. 2004)
L'analisi cristallografica della struttura della PLK espressa dal gene pdxK di molti organismi,
come quella di E. coli (ePLK1) o quella umana (hPLK), ha mostrato che la proteina è
un'omodimero nel quale ciascun monomero presenta un sito attivo, formato esclusivamente
dai residui del monomero in cui si trova. I due siti attivi si presentano simmetrici nella
struttura dimerica e legano una molecola di ATP, il substrato (PL o PM o PN) ed ioni
metallici (Mg2+
e K+ o Na
+). (Safo et al. 2006; Li M.H. et al. 2002) (figura 10)
Questi studi hanno messo in evidenza anche la presenza alcune caratteristiche che sono
comuni ai membri della superfamiglia delle ribochinasi, alla quale infatti questo enzima
appartiene:
1. La presenza di un “coperchio” nel sito attivo la cui funzione è quella di
coprire il substrato e i gruppi fosfato dell’ATP.
2. Un core centrale costituito da foglietti-β centrali circondati da α-eliche.
(Zhang et al. 2004)

13
Il confronto tra la ePLK1 e la hPLK dimostra questa struttura altamente conservata nella
quale il core differisce solo per il numero di α-eliche e β-foglietti: nella ePLK1 ciascun
monomero è costituito da 9 foglietti-β ed 8 α-eliche, nella hPLK 12 foglietti-β e 9 α-eliche.
(Cao et al. 2006) (Figura 10-11)
Figura 10.
Struttura
cristallina del
dimero della
ePLK1.
Figura 11. Diagramma che rappresenta
la struttura dimerica dell’hPLK, in rosso
sono mostrate le α-eliche, in azzurro i
foglietti-β e in grigio i due foglietti-β non
presenti in altre PLK.

14
In ciascun sito attivo l’ATP si lega sulla superficie della tasca mentre il substrato è collocato
in profondità, in direzione opposta e frontalmente al fosfato in γ dell’ATP.
I residui della tasca enzimatica che interagiscono con il substrato formano legami idrogeno e
interazioni idrofobiche e sono altamente conservati tra le varie PLK. (Cao et al. 2006)
La parte adenosinica dell’ATP forma sia legami di van der Waals che legami idrogeno con i
residui della proteina che lo circondano.
I tre gruppi fosforici dell’ATP formano anch’essi legami idrogeno con alcuni residui della
proteina e con ioni Mg2+
, K+ o Na
+.
Il fosfato in γ dell’ATP è orientato in profondità verso il core interno della proteina. Questo
gruppo fosforico interagisce con i residui Asp125 e Glu162 e si pensa che questi residui
siano importanti nello stabilizzare il fosfato in γ e proteggerlo da una eventuale idrolisi
prematura che comprometterebbe la buona riuscita della catalisi. (Figura 12)
FIGURA 12. Diagramma schematico che mostra le interazioni idrofobiche (linee tratteggiate
spesse) e i legami idrogeno (linee tratteggiate sottili) tra l’Mg2+
ATP e i residui della ePLK1
In particolare i fosfati dell'ATP contattano mediante legami idrogeno un loop-P, costituito
dal motivo conservato GTGD (in ePLK: Gly230, Thr231, Gly232 e Asp233). I residui del
motivo GTGD, ad eccezione di Thr 231, sono i più conservati nella superfamiglia delle
ribochinasi e formano un “buco anionico” che aiuta a neutralizzare le cariche negative ed a
stabilizzare lo stato di transizione durante il trasferimento del gruppo fosfato dall’ATP al
substrato (Safo et al. 2006; di Salvo et al. 2011)

15
Il residuo di Asp di questo motivo GTGD, in particolare, è il residuo più conservato nel
corso dell’evoluzione e sembra svolgere un ruolo importante nella catalisi, sia nella hPLK
(Asp235), che nella ePLK1 (Asp233). Stabilisce un legame idrogeno con il gruppo ossidrile
in C5' del substrato e l'ipotesi è che ne medi la deprotonazione. Il meccanismo proposto è
quello del trasferimento del protone dal 5'-OH del substrato al gruppo carbossilico dell'Asp
con formazione di una carica negativa sull' O5' in grado di attuare un attacco nucleofilo
diretto al g-fosfato dell'ATP che viene così trasferito al substrasto. (Gandhi et al. 2009;
Sigrell et al. 1998) (Figura 13)
Inoltre i fosfati β e γ dell'ATP sono stabilizzati da legami con gli ioni Mg2+
e K+ (o Na
+).
FIGURA 13. Meccanismo di reazione proposto per la PLK
Bisogna sottolineare che i metalli, sia cationi monovalenti che bivalenti, sono necessari per
la funzione di molte chinasi in quanto forniscono forze guida per il legame con l’ATP e la
catalisi. Nel caso della ePLK1di E. coli, Mg2+
e K+ sono richiesti per l’attività enzimatica (di
Salvo et al. 2011), mentre nella hPLK sono i metalli Zn2+
e K+ ad essere necessari
(McCormik et al. 1961); in uno studio più recente sull’hPLK, di Salvo et al. (2004) hanno
evidenziato che in condizioni fisiologiche, a pH 7.3, la catalisi richiede Mg2+, mentre Zn2+
addirittura inibisce la reazione. Lo Zn2+
favorisce la reazione solo in condizioni non
fisiologiche (pH 6) e con concentrazioni non fisiologiche di substrato. Studi del 2007 hanno
evidenziato inoltre che nonostante altri ioni monovalenti come il Na+ possano stimolare di
molto l'attività enzimatica rispetto al K+ esso resta lo ione che maggiormente si trova legato
all'enzima in condizioni fisiologiche. (Musayev et al. 2007)

16
Analisi più dettagliate sulla ePLK1 hanno anche rilevato la presenza del tipico “coperchio”
composto di due loop che protegge il sito attivo.
Nella struttura cristallografica, il primo, loop I (residui da 129 a 137), è flessibile, si trova in
una parziale posizione aperta in uno dei monomeri, è mancante nell’altro monomero e non
interagisce con i substrati. Il loop II (residui da 55 a 64), invece, è collocato tra i foglietti β2
e β3, interagisce, attraverso i residui Pro58 e His59, con la posizione 3' e il 4' del PL. È
questo secondo loop che oltre ad avere interazioni con il substrato, è anche strategicamente
collocato in modo da coprire il sito attivo dal solvente (Safo et al., 2006). (Figura 10)
Nella hPLK è stata osservata la presenza di una larga regione a potenziale negativo che si
trova su un lato della superficie dell'enzima e si pensa che possa interagire con gli enzimi
PLP-dipendenti. Inoltre è anche presente una cavità carica negativamente che potrebbe
favorire l’attrazione di substrati con carica positiva, come l’anello piridinico della vitamina
B6, o una carica positiva non netta, come nell’anello adeninico dell’ATP. (Cao et al. 2006)
2.3 Struttura e funzione dell'enzima PNP ossidasi (PNPOx)
La piridossina/piridossamina 5'-fosfato ossidasi (PNPOx) è una flavoproteina: un enzima
dipendente dal coenzima idrosolubile flavina mononucleotide (FMN) (Figura 14), derivato,
quest'ultimo, dalla vitamina riboflavina (B12).
FIGURA 14. Struttura del coenzima flavin
mononucleotide (FMN). L'anello isoallosazinico si riduce
accettando due equivalenti riducenti sotto forma di uno o
due atomi di idrogeno (1 elettrone + 1 protone per ogni
atomo) per volta, formando FMNH2. Il ribitolo originario
della riboflavina è fosforilato.
L'enzima PNPOx viene codificato dal gene pdxH (OMIM 6032870), che nell'uomo è
localizzato sul cromosoma 17q21.2 ed è stato purificato e cartterizzato da vari organismi;
pecora , ratto, maiale, coniglio, insetto, E. coli e l'uomo. (Musayev F.N. 2003; Di Salvo M.
1998; Huang S.H. 2009; di Salvo M.L. 2003; Choi J.D. 1983)

17
La reazione catalizzata da questa ossidasi (Figura 15) consiste nel trasferimento diretto
dell'idrogeno, dal gruppo idrossilico in posizione 4' del PNP o dall'amino gruppo sempre in
posizione 4' del PMP, all'anello isoallosazinico del FMN ossidato. Si formano così PLP e
FMNH2 ridotto. Il coenzima viene poi rigenerato grazie alla cessione di due elettroni
all'ossigeno con formazione quindi di perossido di idrogeno. (Di Salvo M. L. et al. 2002)
FIGURA 15. Reazione catalizzata dalla PNPOx
L'enzima ha una lenta reattività con valori di costante catalitica che vanno da 0.2 sec-1
a 0.8
sec-1
e una Km in un intervallo di valori nel micromolare molto basso per entrambi i substrati
(Km PNPOx umana per PMP e PNP ~ 1.0 mM). (Zhao G. et al. 1995; Di Salvo M. L. et al.
2003; Musayev F. N. et al. 2003)
Il prodotto di reazione, il PLP, ha invece un'ottima affinità per l'enzima, migliore di quella
dei substrati. Questa caratteristica avvalora l'ipotesi, discussa nel paragrafo precedente, che il
prodotto di reazione possa avere una funzione inibitoria sull'enzima. (Zhao G. et al. 1995,
Kazarinoff M. N. et al. 1975; Choi S. Y. Et al. 1987)
La struttura cristallografica della PNPOx è stata ottenuta per la prima volta dal gruppo di
ricerca di Salvo et al. Successivamente sono stati effettuati studi cristallografici anche
sull'enzima umano (Musayev F.N. 2003) e tutti mostrano una struttura omodimerica, la
quale possiede due siti catalitici di legame per l'FMN. Ogni monomero proteico è formato da
due domini α/β-barile, i due siti catalitici si collocano all'interfaccia tra le due subunità del
dimero e ciascuno lega il cofattore FMN ed il substrato PNP o PMP.

18
Inoltre vari studi strutturali e funzionali dell'enzima effettuati su E. coli hanno mostrato la
presenza di un secondo sito, non catalitico, di legame per il PLP, che si trova su ogni
subunità del dimero. Anche se in questo sito il PLP è legato con forza all'enzima sembra che
sia proprio questa molecola ad essere trasferita agli apo-enzimi. Questo sito non catalitico
potrebbe dunque giocare un ruolo importante nel regolare i livelli di PLP libero. (Musayev
F.N. et al. 2003; Yang E.S. et al. 2000) (figura 16)
FIGURA 16. Struttura della PNPOx di E. coli
L’FMN si colloca in una profonda fenditura all'interfaccia tra le due sub-unità dell'enzima e
prende contatto, mediante legami sia idrogeno che di natura idrofobica, con residui molto
conservati di entrambe le sub-unità della proteina. (Musayev F.N. et al. 2003)
Il substrato PNP o PMP, così come il prodotto di reazione PLP, si legano frontali e paralleli
all'FMN, ma con il loro gruppo fosfato che punta verso l'esterno della tasca, orientato quindi
in verso opposto a quello del FMN che punta verso il fondo della cavità. Il cofattore ed il
substrato instaurano legami deboli di tipo van der Waals ed il C4' del PLP substrato e l' N5
del FMN vengono a trovarsi ad una distanza di 3,4 Å, ottimale per il trasferimento
dell'idrogeno. (Di Salvo M.L. et al. 2011)

19
Vengono proposti due possibili meccanismi catalitici qui sotto riportati in figura 17.
Il primo mostra un trasferimento diretto dell'idrogeno dal C4' del substrato al N5 del FMN
(B1). Il secondo implica il coinvolgimento di una base del sito attivo che media la rimozione
del protone dal C4' del substrato ed attacca poi il cofattore formando con esso un complesso
covalente. La successiva rottura del legame tra i due reagenti genera PLP e FMNH2.(B2)
FIGURA 17. Due meccanismi catalitici proposti per la PNPOx
2.4 Regolazione dell'espressione del salvage pathway e controllo
dei livelli di PLP
Gli enzimi della via di recupero sono espressi in tutte le cellule, ma la quantità dei trascritti
(mRNA) e la funzionalità di questi enzimi sono altamente regolate in maniera tessuto-
specifica.
I livelli di regolazione su questi enzimi sono due principalmente: una regolazione a livello
trascrizionale per controllare la loro concentrazione intracellulare ed una post traduzionale
per regolarne l'attività.

20
Nel primo caso notiamo che vi è una trascrizione differenziale nei diversi tessuti. La PLK,
per esempio, è espressa in tutti i tessuti ma in diverse isoforme. (Fang,X. Et al. 2004)
La PNPOx, invece, viene espressa principalmente nel fegato, nel muscolo scheletrico , nei
reni e nel cervello, in particolar modo nella corteccia cerebrale. (Kang J. H. et al. 2004)
Per quanto riguarda il secondo tipo di regolazione studi effettuati su E. Coli hanno
dimostrato che la PNPOx e la PLK sono in grado di reagire con i propri prodotti di reazione i
quali mediano quindi un'inibizione a feedback negativo inibendone l'attività. (Safo M. K. et
al. 2006; Zhao G., Winkler M. E. 1995)
Questo tipo di controllo sull'espressione e l'attività degli enzimi della via di recupero è
effettuato dalle cellule per mantenere bassi i livelli PLP libero nel citoplasma poiché esso è
un'aldeide altamente reattiva.
Inoltre il controllo viene effettuato principalmente sulla PLK e la PNPOx che sono gli
enzimi chiave del “salvage pathway”.
Infatti questi enzimi determinano la formazione del PLP ma sembrano concorrere anche a
non lasciarlo circolare liberamente nel citoplasma, trattenendolo dopo la sua formazione e
cedendolo direttamente agli apo-enzimi. Numerosi studi recenti effettuati appunto sulla PLK
e sulla PNPOx suggeriscono l'esistenza di diversi meccanismi di scambio del PLP con gli
apo-enzimi. La maggior parte di questi studi sembra però avvalorare l'ipotesi che la PLK,
così come la PNPOx, interagisca con gli enzimi PLP dipendenti mediante un'interazione
proteina-proteina e trasferisca poi il PLP a questi apo-enzimi. Tuttavia ancora non sono ben
noti i passaggi specifici di questo meccanismo di rilascio né come la PLK, l’enzima
donatore, possa riconoscere ed interagire in modo specifico con le dozzine di apo-enzimi
accettori. (Kim, Y. T. et al. 1988 ; Yang E. S. et al. 2000 ; Cheung P. Y. et al. 2003; di
Salvo et al., 2011)
Altri meccanismi che concorrono a mantenere bassa la concentrazione intracellulare di PLP
libero sono: la sua defosforilazione da parte delle fosfatasi e la conversione in acido
piridossico (Figura 18) da parte di ossidasi specifiche per le aldeidi o da parte di
deidrogenasi NAD-dipendenti. (Stanulovic M. et al. 1976)

21
FIGURA 18. Acido piridossico
La concentrazione del PLP libero nelle cellule eucariotiche viene mantenuta bassa da questi
meccanismi regolatori ad un valore intorno a 1 µM. (Li T. K. Et al. 1974)
Sia dosi alte, superiori a 200 mg/day, che valori di assunzione al di sotto di 1-2 mg/day,
possono causare danni ingenti al metabolismo e a livello del sistema nervoso centrale come
riportato da diversi studi. (Chung J. Y et al. 2008 ; Morra M. et al. 1983)
2.5 Tossicità del PLP
Livelli eccessivi di PLP nel corpo, dovuti ad un'errata regolazione del pathway di recupero
oppure all'assunzione di massicce dosi del vitamero, hanno effetti collaterali deleteri,
principalmente a carico del sistema nervoso. (Clayton, 2006)
Già tra gli anni '70 e '90, studi effettuati in diversi laboratori, dimostrarono che dosi eccessive di
PLP, dovute alla sovraespressione di PNPOx e PLK, causavano una degenerazione della
neurotrasmissione GABAergica. Sembra infatti che il PLP vada ad agire direttamente sui
recettori GABAergici o diminuisca l'uptake del neurotrasmettitore da parte dei neuroni.
(Ishioka N. et al. 1995; Ebadi M. et al. 1979)
Salazar et al. (2001), studiando gli effetti di farmaci GABAergici e antagonisti dei recettori del
glutammato, osservarono che l’iniezione intracranica diretta di PLP nel cervello, quindi una dose
eccessiva in loco, induceva convulsioni in mammiferi adulti, andando ad interagire con un
residuo di lisina cruciale collocato in un loop extracellulare dei recettori.
Ancora, Scott et al. (2008) e Gdynia et al. (2008) hanno ipotizzato che elevati livelli di vitamina
B6 nel sangue potrebbero essere la causa di polineuropatie sensomotorie, le quali sembrano infatti
recedere appena viene interrotta la somministrazione di vitamina (Foca, 1985).
In uno studio condotto negli anni '90 da Morra et al. sono state rilevate anche neuropatie
permanenti indotte da alti livelli di PLP.
Inoltre, come già discusso in precedenza, alte dosi di PLP sembrano influenzare anche l'attività
catalitica degli stessi enzimi della via di recupero, inibendoli.

22
3. DEFICIENZA DI PLP: CAUSE ED EFFETTI DELLA
CARENZA DI PLP SUL METABOLISMO
L'alterazione dei livelli intra- ed extracellulari di PLP può essere molto dannosa per
l'organismo: l'eccesso di PLP nel corpo, come affermato in precedenza, induce effetti tossici
per l'organismo, ma anche la deficienza di questo cofattore è molto pericolosa, poiché porta
al malfunzionamento degli enzimi PLP-dipendenti ed all'alterazione di tutti i metabolismi ad
essi associati.
I meccanismi che portano ad una deficienza di vitamina B6 e di conseguenza ad un
incremento nella richiesta di PLP o PN possono essere molteplici:
Mutazioni nei geni che codificano gli enzimi del salvage pathway del PLP.
Alterazioni di metabolismi che portano all’accumulo di piccole molecole
che reagiscono con il PLP inattivandolo.
Farmaci che reagiscono con il PLP oppure interferiscono con il
funzionamento degli enzimi del salvage pathway.
Malattie, oppure terapie ad esse associate, che portano ad un malassorbimento dei
vitameri B6 o alla loro perdita: per esempio la celiachia che si pensa porti ad un
malassorbimento dei vitameri B6 o la dialisi renale, che porta ad un aumento della
perdita dei vitameri B6 dalla circolazione.
3.1 Malfunzionamento degli enzimi del salvage pathway
Il mancato funzionamento degli enzimi della via di recupero del PLP, dovuto ad una
mutazione dei geni che li codificano o alla loro inibizione, porta una diminuzione di
disponibilità di questo cofattore per gli enzimi PLP-dipendenti.
La deficienza del PLP può essere così conseguenza del mancato contributo necessario per il
suo assorbimento e trasporto, della sua mancata sintesi intracellulare o dell'impossibilità di
essere ceduto agli apo-enzimi; tutte azioni, queste, svolte dagli enzimi della via di recupero.
L’enzima PLK, per esempio, svolge un ruolo chiave nella formazione del PLP. É
responsabile della fosforilazione dei vitameri PL, PN, PM a PLP, PNP e PMP

23
rispettivamente. Attraverso questa reazione permette sia la fosforilazione a livello epatico
delle forme defosforilate assimilate dalla dieta, che vengono così trasportate dall'albumina
fino ai tessuti, sia la fosforilazione intracellulare diretta del PL a PLP.
In Figura 19 è riportato il probabile effetto del malfunzionamento della PLK
sull'assorbimento ed il trasporto dei vitameri dove si nota che: non solo il cofattore attivo
non può essere formato a livello epatico, con conseguente totale carenza di PLP legato
all'albumina nel sangue, ma anche le forme defosforilate che raggiungono i tessuti non
possono essere convertite in cofattore attivo.
Ciò che ne risulta è l'impossibilità di sintesi ed accumulo di PLP nel corpo: deficienza
plasmatica ed una conseguente, forte, deficienza intracellulare che non permettere il
funzionamento degli enzimi PLP-dipendenti.
FIGURA 19. Effetto del malfunzionamento della PLK sull'assorbimento ed il trasporto della
vitamina B6.
É stato dimostrato, inoltre, che molti farmaci, così come alcune sostanze naturali, possono
avere un'azione inibitoria sulla PLK: caffeina, teobromina, teofillina, gincotossine, levodopa,
cicloserina e molte altre. (Kastner U. et al. 2007; Laine-Cessac P. et al. 1997; Ebadi M.
1982)
L'alterazione dell’attività PL chinasica sembra essere un punto nodale in molte patologie e
molti farmaci sono oggi disponibili per modularne l’attività.
Lo studio effettuato da Gandhi et al., 2009 su un mutante artificiale, mostra come mutazioni
relative al residuo di Asp235 della hPLK portino ad un decremento notevole dell'attività
catalitica dell'enzima.

24
Nel 2006, Adams J. B. et al., analizzando lo spettro clinico di bambini affetti da autismo,
hanno proposto proprio un'associazione tra le alte concentrazioni di vitameri defosforilati e
bassi livelli di PLP nel corpo con un malfunzionamento della PLK.
Tuttavia mutazioni a carico dei geni per la PLK, che ne impediscono l'attività catalitica in
maniera costitutiva, sono molto probabilmente mortali per l'organismo e per tale motivo non
sono state mai riportate in letteratura.
La PNPOx, d'altro canto, è necessaria per permettere la formazione del PLP dalle forme
fosforilate PMP e PNP e cosa non meno importante sembra essere implicata, più della PLK,
nella cessione diretta del cofattore agli apoenzimi.
A livello epatico ed intracellulare nei tessuti, l'azione dell'ossidasi produce e rende
disponibile il PLP per gli enzimi PLP-dipendenti e per il trasporto.
Il probabile effetto del malfunzionamento della PNPOx sull'assorbimento ed il trasporto
risulta nella diminuzione di PLP sia nella circolazione sistemica legato all'albumina (poiché
il solo PLP circolante è stato prodotto dall'azione della PLK epatica a partire dal PL) che al
livello dei tessuti. Inoltre i tessuti risentono maggiormente dell'azione della PNPOx nel
cedere il cofattore agli apoenzimi. (Figura 20)
FIGURA 20. Effetto del malfunzionamento della PNPOx sull'assorbimento ed il trasporto della
vitamina B6.
Il risultato complessivo, in questo caso, è una deficienza di PLP disponibile a carico di tutti i
tessuti del corpo con conseguente malfunzionamento degli enzimi PLP-dipendenti.
Uno studio effettuato da Musayev FN et al. 2009 su mutanti PNPOx ha mostrato

25
l'importanza di alcuni residui amminoacidici dell'enzima ed il decremento dell'attività
catalitica associata.
Mutazioni a carico dei geni per la PNPOx, che ne impediscono l'adeguata attività catalitica,
risultano in gravi disfunzioni generalizzate nei metabolismi che si servono di enzimi PLP-
dipendenti.
Inoltre, poiché il buon funzionamento dell'enzima dipende dalla presenza del suo cofattore
FMN, l'attività della PNPOx e la conseguente disponibilità di PLP sono correlate anche al
metabolismo della riboflavina. ( Bowling F. G. 2011)
Tra le patologie da deficienza di PLP, correlate al malfunzionamento di questo enzima, si
riscontrano le Encefalopatie Epilettiche Neonatali PLP-dipendenti (NEE PLP-dipendenti) di
cui si parlerà in seguito.
Anche il buon funzionamento delle fosfatasi è importante per il corretto assorbimento della
vitamina. Infatti permettono alle forme fosforilate PLP, PMP, PNP, assunte con il cibo, di
attraversare in forma di PL, PM, PN il tessuto intestinale e di essere trasportate fino al
fegato. Anche a livello dei tessuti periferici, così come a livello dell’encefalo, permettono
alla vitamina circolante, in maggior parte sotto forma di PLP, di essere defosforilata a PL e
passare dalla circolazione alle cellule dei tessuti.
Il probabile effetto del malfunzionamento delle fosfatasi risulta in un ridotto assorbimento
intestinale e tessutale della vitamina, con conseguente deficienza intracellulare del PLP che
si accumula invece nel plasma. (Figura 21)
FIGURA 21. Effetto del malfunzionamento delle fosfatasi sull'assorbimento ed il trasporto della
vitamina B6.

26
Anche le mutazioni a carico dei geni che codificano per le fosfatasi, risultano in una
deficienza di PLP intracellulare e nella conseguente alterazione dei metabolismi che si
servono di enzimi PLP-dipendenti.
Il ruolo indispensabile giocato dalle fosfatasi alcaline tessuto non specifiche (TNAP), nell’
uptake della vitamina B6 a livello cellulare, è evidente nell’insorgenza dell’ Ipofosfatasia.
Quest’ultima è una rara sindrome genetica, risultante da mutazioni nel gene che codifica
un’isoenzima della fosfatasi alcaline tessuto non specifiche (ALPL; OMIM 171760) ed è
caratterizzata dall’incremento della concentrazione plasmatica di PLP e di vanillattato,
indicatore, quest’ultimo, del mal funzionamento degli enzimi PLP-dipendenti decarbossilasi
di amminoacidi aromatici. (di Salvo et al. 2012; Iqbal S. J. et al. 1998; Litmanovitz et al.
2002)
3.2 Sequestro del PLP da parte di intermedi di vie metaboliche
mutate
Un'altra possibile causa della deficienza di piridossal-5'-fosfato nel corpo è da ricercare
nell'interazione del vitamero con prodotti chimici derivati da altre vie metaboliche.
Sono state riportate alcune patologie, causate dall'alterazione di vie metaboliche, nelle quali
alcuni enzimi mutati (e quindi inattivi) determinano l’accumulo di intermedi di reazione
instabili e particolarmente reattivi, ai quali il PLP può legarsi ed essere così sequestrato
dall'ambiente intracellulare.
In questi casi agli effetti sintomatici della patologia si sommano anche quelli dovuti alla
deficienza del PLP.
Due esempi sono rappresentati dall’ Iperprolinemia di tipo II (Walker V. et al. 2000) e da
quelle sindromi classificate come PDE: epilessie dipendenti da piridossina. (Mills P. B et al.
2006)
Entrambe queste patologie sono caratterizzate dall’avere metabolismi che presentano enzimi
mutati e dall’accumulo di intermedi di reazione capaci di sequestrare il PLP dalla soluzione.
Esse sono implicate nell’insorgenza di encefalopatie, come conseguenza della deficienza di
PLP nel tessuto nervoso.
Le PDE verranno discusse in maggiore dettaglio nel capitolo conclusivo della tesi trattando
delle Encefalopatie Epilettiche Neonatali.
27
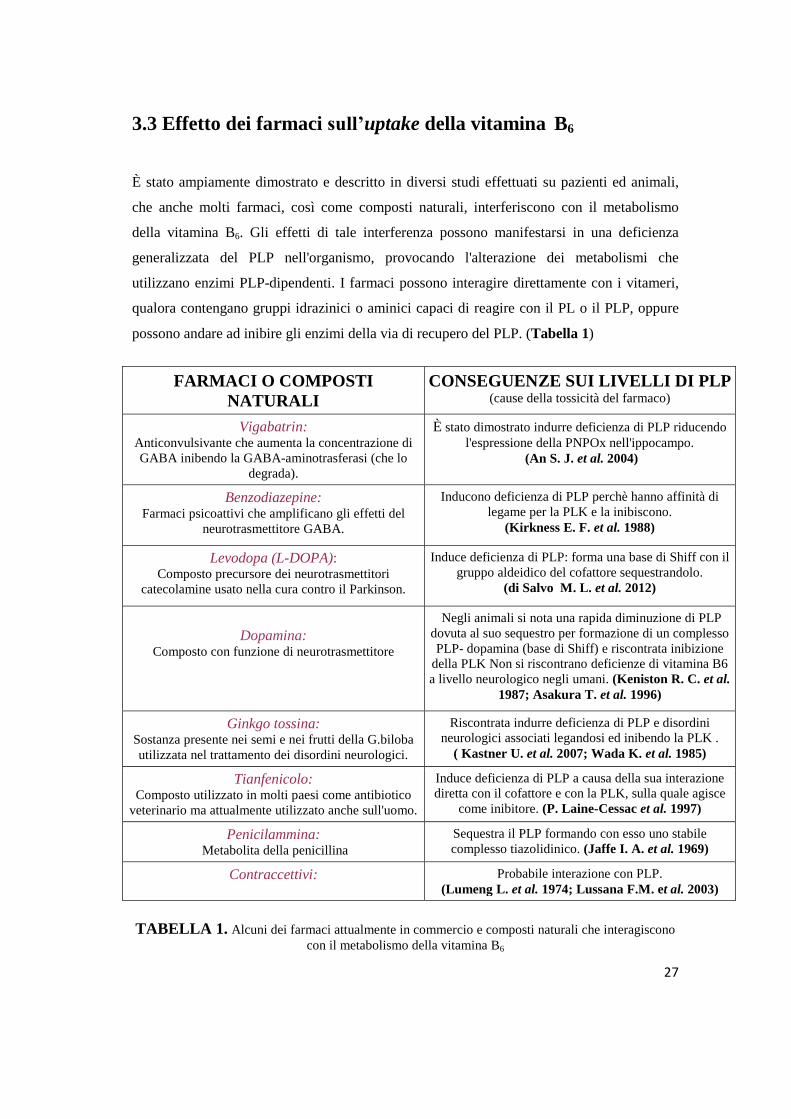
3.3 Effetto dei farmaci sull’uptake della vitamina B6
È stato ampiamente dimostrato e descritto in diversi studi effettuati su pazienti ed animali,
che anche molti farmaci, così come composti naturali, interferiscono con il metabolismo
della vitamina B6. Gli effetti di tale interferenza possono manifestarsi in una deficienza
generalizzata del PLP nell'organismo, provocando l'alterazione dei metabolismi che
utilizzano enzimi PLP-dipendenti. I farmaci possono interagire direttamente con i vitameri,
qualora contengano gruppi idrazinici o aminici capaci di reagire con il PL o il PLP, oppure
possono andare ad inibire gli enzimi della via di recupero del PLP. (Tabella 1)
FARMACI O COMPOSTI
NATURALI
CONSEGUENZE SUI LIVELLI DI PLP (cause della tossicità del farmaco)
Vigabatrin: Anticonvulsivante che aumenta la concentrazione di
GABA inibendo la GABA-aminotrasferasi (che lo
degrada).
È stato dimostrato indurre deficienza di PLP riducendo
l'espressione della PNPOx nell'ippocampo.
(An S. J. et al. 2004)
Benzodiazepine: Farmaci psicoattivi che amplificano gli effetti del
neurotrasmettitore GABA.
Inducono deficienza di PLP perchè hanno affinità di
legame per la PLK e la inibiscono.
(Kirkness E. F. et al. 1988)
Levodopa (L-DOPA): Composto precursore dei neurotrasmettitori
catecolamine usato nella cura contro il Parkinson.
Induce deficienza di PLP: forma una base di Shiff con il
gruppo aldeidico del cofattore sequestrandolo.
(di Salvo M. L. et al. 2012)
Dopamina: Composto con funzione di neurotrasmettitore
Negli animali si nota una rapida diminuzione di PLP
dovuta al suo sequestro per formazione di un complesso
PLP- dopamina (base di Shiff) e riscontrata inibizione
della PLK Non si riscontrano deficienze di vitamina B6
a livello neurologico negli umani. (Keniston R. C. et al.
1987; Asakura T. et al. 1996)
Ginkgo tossina: Sostanza presente nei semi e nei frutti della G.biloba
utilizzata nel trattamento dei disordini neurologici.
Riscontrata indurre deficienza di PLP e disordini
neurologici associati legandosi ed inibendo la PLK .
( Kastner U. et al. 2007; Wada K. et al. 1985)
Tianfenicolo: Composto utilizzato in molti paesi come antibiotico
veterinario ma attualmente utilizzato anche sull'uomo.
Induce deficienza di PLP a causa della sua interazione
diretta con il cofattore e con la PLK, sulla quale agisce
come inibitore. (P. Laine-Cessac et al. 1997)
Penicilammina: Metabolita della penicillina
Sequestra il PLP formando con esso uno stabile
complesso tiazolidinico. (Jaffe I. A. et al. 1969)
Contraccettivi: Probabile interazione con PLP.
(Lumeng L. et al. 1974; Lussana F.M. et al. 2003)
TABELLA 1. Alcuni dei farmaci attualmente in commercio e composti naturali che interagiscono
con il metabolismo della vitamina B6

28
3.4 Ruolo degli enzimi PLP-dipendenti nel metabolismo ed
alterazione delle vie metaboliche dovuta a carenza del PLP
Le conseguenze della deficienza di piridossal-5'-fosfato nel corpo possono essere molto
gravi. Infatti questo cofattore enzimatico è una molecola centrale nel metabolismo degli
esseri viventi poiché, attraverso gli enzimi che lo richiedono, prende parte ad importanti
processi cellulari quali:
- Rifornimento delle unità monocarboniose
- Sintesi dei composti tetrapirrolici, come per esempio l’eme
- Metabolismo degli amminozuccheri
- Sintesi e degradazione di ammine biogene
- Sintesi del DNA/RNA
- Produzione di neurotrasmettitori
- Sintesi, interconversione e degradazione degli amminoacidi
(Percudani R. and A. Peracchi 2009)
Numerose sono le vie metaboliche che presentano enzimi PLP-dipendenti e qui di seguito, in
tabella 2, vengono riportati alcuni di questi enzimi, i metabolismi ad essi correlati e gli
effetti dell'alterazione dovuti alla carenza del cofattore o alla mutazione dei geni che li
codificano.
I valori alterati degli intermedi delle vie metaboliche, vengono utilizzati oggi come marcatori
dell’ insufficiente apporto di vitamina B6 nel corpo ed in particolare della deficienza di PLP,
per la diagnosi di molte malattie.

29
ENZIMI PLP-DIP. COIVOLTI
IN VARI METABOLISMI
EFFETTI CARENZA PLP/
MARCATORI
Serina idrossimetil-transferasi:
Catalizza la conversione reversibile di serina e
tetraidrofolato in glicina e N5-N
10- metilen-
tetraidrofolato. Quest’ultimo viene ridotto da
una reduttasi a N5-metil-THF implicato a sua
volta nel riciclo di omocisteina a metionina.
Accumulo e quindi elevata concentrazione di omocisteina
nel plasma e nelle urine.
Cistationina beta-sintasi:
Catalizza la condensazione della serina con
l'omocisteina producendo cistationina nella via
della transulfurazione del ciclo della metionina.
Accumulo e quindi elevata concentrazione di omocisteina
nel plasma e nelle urine ed in questo caso si parla di
Omocistinuria o iper-omocisteinemia: malattia
multisistemica, che coinvolge occhi, scheletro, sistema
nervoso ed apparato vascolare.
Cistationina gamma-liasi:
Catalizza la rottura non idrolitica della
molecola della cistationina con formazione di
cisteina e acido α-chetobutirrico.
Accumulo e quindi elevata concentrazione di cistationina
nelle urine
Kinureninasi e kinurenina
aminotransferasi:
Implicate nella via ossidativa della kinurenina,
un prodotto della via di degradazione del
triptofano, dalla quale vengono prodotti
intermedi implicati nella regolazione del
sistema nervoso centrale.
Accumulo e quindi elevata concentrazione di acido
xanturenico nelle urine implicato nell’insorgenza di
disordini neurodegenerativi.
Treonina deidratasi:
Enzima epatico che catalizza la deaminazione
non ossidativa della treonina.
Accumulo e quindi elevata concentrazione di treonina nel
CSF e plasma.
Serina deidratasi:
Enzima epatico che catalizza la deaminazione
non ossidativa della serina.
Accumulo e quindi elevata concentrazione di serina nel
CSF e plasma
Δ-Aminolevulinato- δ-ALA sintasi:
Catalizza la condensazione di Succinil-CoA e
glicina per produrre acido Δ-aminolevulinico,
CoA e CO2. L'aminolevulinato è un precursore
dei composti tetrapirrolici come l'eme.
Accumulo di Fe nel fegato e bassi livelli nel muscolo.
Insorgenza di sindromi definite anemia ipocromica,
microcitica e sieroblastica.
TABELLA 2. Enzimi PLP-dipendenti coinvolti in vari metabolismi cellulari e gli effetti del loro
malfunzionamento.

30
3.5 Enzimi PLP-dipendenti nel metabolismo dei
neurotrasmettitori e funzione cerebrale
Il piridossal-5'-fosfato è correlato al buon funzionamento del sistema nervoso centrale,
poiché è coinvolto anche nel metabolismo dei neurotrasmettitori.
Infatti, l’alterazione, sia per eccesso che per difetto, del livello di PLP nell’organismo
sembra essere correlata con l’insorgenza di gravi patologie del sistema nervoso centrale
come il morbo di Parkinson, l’Alzheimer, l’epilessia ed altre.
Il sistema nervoso è costituito da una complessa rete di cellule, neuroni e cellule gliali,
capaci di ricevere ed integrare le informazioni provenienti dall'esterno. Il trasferimento delle
informazioni e la comunicazione tra queste cellule dipendono dai segnali elettrici, o
potenziali di azione, che corrono lungo i neuroni e dalle molecole segnale, neurotrasmettitori
e neuromodulatori, che vengono rilasciati dai neuroni presinaptici sui recettori di quelli
postsinaptici.
I neurotrasmettitori vengono sintetizzati nel corpo cellulare o nelle terminazioni della cellula
nervosa, possono essere composti chimici di varia natura e molti di questi sono sintetizzati
direttamente o indirettamente da enzimi PLP-dipendenti (Schema 1):
. Dopamina
. Epinefrina Catecolamine Amine Biogene o Monoamine
. Norepinefrina (monoamine derivate (molecole derivate da aminoacidi,
. Serotonina da Tirosina) caratterzzate da un solo gruppo
. Istamina amminico)
. Glicina
. L-Glutammato
. GABA Aminoacidi e loro derivati
. D-Serina
. D-aspartato
SCHEMA 1. Neurotrasmettitori sintetizzati da enzimi PLP-dipendenti.

31
Qui di seguito, in tabella 3, vengono riassunti molti degli enzimi PLP-dipendenti coinvolti
nel metabolismo dei neurotrasmettitori.
ENZIMI PLP-DIP. NEL
METABOLISMO DEI
NEUROTRASMETTITORI
CATALISI
FUNZIONE CEREBRALE
Decarbossilasi degli L-
aminoacidi aromatici (AAAD)
:
Sintesi dopamina e
serotonina.
La dopamina è il precursore di
noradrenalina ed adrenalina. Hanno effetti
sia eccitatori che inibitori sul SNC.
Carenze determinano sindromi
neurodegenerative.
Aminotransferasi
dell'aminoacido a catena
ramificata 2-oxoglutarato
(BCAT):
Sintesi di L-glutammato.
Il glutammato è il principale
neurotrasmettitore eccitatorio nel SNC. É
importante nei processi cognitivi, di
apprendimento e memoria.
Glutammato Decarbossilasi:
Conversione di glutammato in
GABA
É il principale neurotrasmettitore inibitorio
nel SNC. Carenze sono associate a
sindromi epilettiche e neurodegenerative.
GABA Transaminasi:
Degradazione GABA,
conversione in glutammato
Un eccesso di neurotrasmettitore inibitorio
induce ritardo di risposta sinaptica.
Serina Idossimetiltransferasi:
Sintesi della glicina dalla L-
serina
La glicina è un neurotrasmettitore sia
inibitorio che eccitatorio del SNC cruciale
per la regolazione dei motoneuroni
Glicina Decarbossilasi:
Catabolismo della glicina. Un eccesso di neurotrasmettitore inibitorio
induce ritardo di risposta sinaptica.
Racemasi della L-serina:
Racemizzazione della L-
serina in D-serina
La D-serina è un neurotrasmettitore
richiesto per la plasticità neuronale, la
memoria e l'apprendimento.
Istidina Decarbossilasi:
Sintesi di istamina dalla
decarbossilazione
dell'istidina.
L' Istamina è un neurotrasmettitore
coinvolto nella memoria
TABELLA 3. Enzimi PLP-dipendenti coinvolti nel metabolismo dei neurotrasmettitori
È ovvio che inadeguati livelli di PLP inducano un decremento dell'attività degli enzimi PLP-
dipendenti, di conseguenza un significante decremento dei livelli del neurotrasmettitore
associato e lo sviluppo quindi di patologie.
Molti disordini neurologici sono oggi associati alla deficienza di PLP: Alzheimer, Parkinson,
disfunzioni di apprendimento, ansietà, deficit mentali, schizofrenia e sindromi convulsive di
varia natura. (Adams J. B. et al. 2006; Nakagawa E. et al. 1997; Sandyk R et al. 1990)

32
3.6 L'epilessia e gli enzimi PLP-dipendenti
L'epilessia è una patologia caratterizzata dalla comparsa ricorrente di alterazioni transitorie
della funzione cerebrale, di intensità variabile (crisi epilettiche), causate da un'attività
elettrica eccessiva ed incontrollata di un gruppo di cellule nervose. Generalmente le crisi
hanno una durata breve e si possono manifestare come convulsioni (contrazioni e spasmi
muscolari), perdita della coscienza e/o movimenti incontrollati. (Bear MF et al. 2005)
L'epilessia può avere cause genetiche (“epilessia idiopatica”) o insorgere come conseguenza
di malattie o altri fattori (“epilessia sintomatica”): traumi cerebrali, tumori, lesioni infettive o
ferite prenatali. (Jallon P. et al. 2001)
Le epilessie idiopatiche umane riscontrate fino ad oggi sono state associate principalmente a
mutazioni nei geni codificanti canali ionici: voltaggio-dipendenti che generano il potenziale
d'azione e voltaggio-indipendenti (recettori).
Tuttavia anche una de-regolazione nella produzione dei neurotrasmettitori, dovuta a
mutazioni a carico di enzimi implicati nel loro metabolismo, può essere la causa di crisi
convulsive.
Infatti la sincronizzazione dell'attività dei neuroni è essenziale per l'elaborazione dei segnali
ed una sua alterazione, generalmente associata proprio ad uno sbilanciamento tra produzione
di neurotrasmettitore inibitorio ed eccitatorio, può generare l'epilessia.
Ma come può essere tutto questo collegato alla vitamina B6 ed agli enzimi PLP-dipendenti?
Il sistema nervoso centrale (SNC) dei mammiferi utilizza principalmente, per la trasmissione
e l'integrazione dei segnali nervosi, due neurotrasmettitori, tra loro antagonisti: l' L-
glutammato, il quale ha un effetto eccitatorio sulle sinapsi e l'acido γ-amminobutirrico
(GABA) con funzione inibitoria. (Figura 22)
FIGURA 22. Principali neurotrasmettitori del SNC: L-glutammato ed acido γ-amminobutirrico
(GABA)
L-GLUTAMMATO
GABA

33
I neuroni glutammatergici (che sintetizzano ed utilizzano L-glutammato) e quelli
GABAergici (che sintetizzano GABA) sono infatti ubiquitari a livello cerebrale ma la quota
predominante si ritrova nella corteccia cerebrale, nell'ippocampo e nel cervelletto e sono
pertanto implicati in processi come il controllo della funzione motoria, l'apprendimento, la
memoria, la percezione localizzata delle sensazioni e del dolore.
Bassi o anormali livelli di questi neurotrasmettitori sono infatti stati rilevati in occasione di
sindromi epilettiche, invecchiamento cerebrale e malattie neurodegenerative come l'
Alzheimer, il Parkinson, l' Huntington e la sindrome di Tourette. (Andre V. M. et al. 2010;
Leckman J. F. et al. 2010; Rupsingh R. et al. 2011; Blandini F. et al. 1996; Butterworth
J. Et al. 1983)
Il metabolismo di questi due neurotrasmettitori è strettamente associato alla funzionalità di
enzimi PLP-dipendenti ed inoltre sono collegati tra loro dalla via dello shunt del GABA,
costituita da tre enzimi, due dei quali sono proprio PLP-dipendenti. (Figura 23)
FIGURA 23. Via dello shunt del GABA.
L' L-glutammato è sintetizzato a partire da amminoacidi come la leucina e la valina,
attraverso una reazione di transaminazione, effettuata da enzimi PLP-dipendenti: le
amminotrasferasi di amminoacidi a catena ramificata (BCAT).

34
La glutammato decarbossilasi (GAD), della quale esistono due isoforme, è un enzima PLP-
dipendente che catalizza la conversione dell'amminoacido L-glutammato in GABA. (Wei
and Wu 2008)
Anche la GABA amminotransferasi (GABA-AT) è un'enzima PLP-dipendente, ubiquitario,
che catalizza la conversione del GABA in semialdeide succinica attraverso una tipica
reazione di transaminazione, bimolecolare di tipo “ping- pong”, reversibile, che presenta
come substrati l' α-chetoglutarato ed il GABA e come prodotti la semialdeide succinica e
l'Lglutammato.
La semialdeide succinica deidrogenasi (SSADH) è l'ultimo enzima della via che catalizza la
conversione della semialdeide succinica in succinato, il quale entra poi nel ciclo di Krebs.
La buona funzionalità di questi enzimi PLP-dipendenti permette la regolazione della sintesi
del GABA e dell' L-glutammato ed è quindi fondamentale per mantenere il delicato
equilibrio tra eccitazione ed inibizione sinaptica.
Nel momento in cui, nell' encefalo, la concentrazione del GABA diminuisce del 30% rispetto
al livello normale e quella dell' L-glutammato aumenta, insorgono le convulsioni, a causa
dell'elevato grado di eccitazione neuronale, provocato dal rilascio massiccio del
neurotrasmettitore eccitatorio. (Sherwin A.L. 1999; Karlsson et al. 1974)
Tale situazione può verificarsi proprio a causa di una deficienza di PLP a livello encefalico,
la quale induce un malfunzionamento degli enzimi della via dello shunt del GABA.
L'alterata funzionalità di GAD, infatti, che è l'enzima e monte dello shunt del GABA, sembra
essere coinvolta nella genesi di attacchi epilettici, in quanto la sua down-regulation provoca
un abbassamento dei livelli di GABA ed un eccessivo accumulo di glutammato, sbilanciando
il delicato equilibrio tra eccitazione ed inibizione sinaptica. (Nanavati and Silverman 1989)
Anche un' up-regulation della GABA-AT, la quale funzione è quella di distruggere una
molecola di neurotrasmettitore inibitorio per produrne una eccitatoria, può generare gli stessi
effetti, ma quest' aumento di attività non sembra poter essere associabile alla deficienza di
PLP.

35
4. COINVOLGIMENTO DEL PLP NELLE
ENCEFALOPATIE EPILETTICHE NEONATALI (NEE)
4.1 Riepilogo introduttivo
Il piridosal-5'-fosfato (PLP) è considerato la forma biologicamente attiva della vitamina B6
poiché, oltre a svolgere svariate funzioni in molteplici processi cellulari, è il cofattore
enzimatico di oltre 160 enzimi.
Come conseguenza del ruolo cruciale svolto dagli enzimi PLP-dipendenti nella fisiologia
dell’organismo, è facile intuire che alterazioni dei livelli di piridossal-5'-fosfato nel corpo
determinano lo sviluppo di patologie.
Patogenesi associate alla deficienza di PLP possono derivare da molteplici fattori: alterata
regolazione sul pathway di recupero e quindi uno squilibrio tra biosintesi e degradazione del
cofattore, inefficiente assorbimento e trasporto ai tessuti periferici, mutazioni a carico degli
enzimi della via di recupero del PLP, mutazioni a carico di enzimi PLP-dipendenti o fattori
esterni come farmaci, malattie, terapie ed intermedi di vie metaboliche mutate che possono
interferire con l'uptake del cofattore.
Tuttavia, nonostante l'ampio background di cause, le patologie legate ad alterazioni nei
livelli di PLP si manifestano soprattutto come gravi disfunzioni neurologiche a carico del
sistema nervoso centrale, come la malattia di Parkinson, di Alzheimer, l’epilessia o la
schizofrenia.
Questo accade perché, come già detto precedentemente, molti degli enzimi PLP-dipendenti
sono coinvolti nel metabolismo di neurotrasmettitori e sembra che questi enzimi siano
particolarmente sensibili anche a modeste diminuzioni dei livelli di PLP, competendo tra
loro per quello disponibile. (Rahman MK et al. 1982)
Allo scopo di definire tale legame tra il PLP e la funzione cerebrale abbiamo discusso come,
il malfunzionamento degli enzimi PLP-dipendenti, dello shunt del neurotrasmettitore GABA,
possa essere direttamente implicato nella generazione delle sindromi epilettiche.

36
Inoltre, ciò che la precedente trattazione ha voluto mettere in evidenza è l'ipotesi secondo la
quale, poiché la maggior parte della vitamina B6 viene acquisita dai neuroni in forma
defosforilata (PL), molte delle sindromi epilettiche e neurodegenerative PLP-dipendenti
potrebbero essere attribuibili al mal funzionamento degli enzimi della via di recupero della
vitamina, responsabili della sintesi in loco del cofattore PLP attivo. (Li T. K. Et al. 1974)
Proprio a dimostrazione di tale ipotesi verrà qui trattata, nella parte conclusiva della tesi, una
rara sindrome epilettica associata alla carenza della vitamina B6, oggetto recente di studi da
parte di diversi laboratori nel mondo: l'Encefalopatia Epilettica Neonatale.
4.2 Introduzione alle Encefalopatie Epilettiche Neonatali
Le Encefalopatie Epilettiche Neonatali (NEE) sono sindromi convulsive che si manifestano
nei neonati già nelle prime ore di vita e sono generalmente causate da disfunzioni di varia
natura a carico del sistema nervoso centrale: infiammazioni, anomalie strutturali
dell'encefalo, malattie cerebrovascolari e metaboliche. (Ghatge MS et al. 2012)
In qualche raro caso sono state riscontrate NEE refrattarie alle cure con i convenzionali
anticonvulsivanti, le quali hanno tuttavia risposto bene al trattamento con la vitamina B6 ed
in modo speciale ai vitameri piridossina (PN) e piridossal-5'-fosfato (PLP).
Queste particolari NEE dipendenti dalla vitamina B6 risultano associate principalmente ad
una carenza di PLP nel corpo, dovuta a diverse cause. In rare situazioni la deficienza può
essere causata dalla mancata assimilazione della vitamina dalla dieta. Nella maggior parte
dei casi si sono evidenziate mutazioni a carico di enzimi ubiquitari, coinvolti nel riciclo e
nella sintesi della forma attiva della vitamina, come l'enzima piridossina 5'-fosfato ossidasi
(PNPOx). Oppure si sono riscontrate mutazioni di enzimi coinvolti in diversi altri
metabolismi cellulari, che comportano l'accumulo di intermedi capaci di interagire e
sequestrare il PLP, riducendone la disponibilità. (Mills P.B. et al. 2005; Gospe S. M 2010)
I pazienti affetti da NEE dipendenti dalla vitamina B6 riportano inoltre fenotipi caratteristici,
che sono indici dei metabolismi alterati e grazie ai quali è stato possibile risalire agli enzimi
mutati e comprendere il ruolo del PLP in queste sindromi.

37
In base ai tipi di metabolismi alterati, al fenotipo risultante ed alla risposta al PN piuttosto
che al PLP sono stati identificati due sottogruppi di NEE B6-dipendenti:
NEE che rispondono/dipendenti dal PLP (NEE PLP-responsive)
NEE che rispondono/dipendenti dal PN (NEE PN-responsive o PDE)
Tutt'oggi questi due sottogruppi non sono stati ancora ben delineati, poiché alcuni casi
riportano fenotipi e risposte ai trattamenti non sempre associabili a questa suddivisione.
4.3 NEE PLP-dipendenti
Le più severe forme di Encefalopatie Epilettiche Neonatali sono state riscontrate in pazienti
con mutazioni nel gene che codifica la PNPOx e queste sindromi convulsive sono state
pertanto definite anche NEE da deficienza di PNPOx (PNPOx deficiency).
Inoltre, in questi casi, i pazienti risultano rispondere solo alla somministrazione di PLP o PL
esogeno, con un miglioramento progressivo delle crisi fino alla loro completa cessazione,
motivo per il quale a questi disordini è stato dato il nome più specifico di NEE PLP-
dipendenti. (Ghatge MS et al. 2012)
Molti dei pazienti che presentano queste mutazioni alla PNPOx nascono prematuramente e
quelli che sopravvivono manifestano generalmente ritardi mentali e richiedono la
somministrazione di vitamin B6 esogena, sotto forma di PLP o PL, per tutta la vita.
È interessante inoltre osservare che questa particolare e rara sindrome convulsiva si è
manifestata principalmente in pazienti di origine o discendenza turca o asiatica, come
riportano i referti clinici osservati. (Mills P.B. et al. 2005)
Le caratteristiche cliniche riscontrate nei pazienti con NEE PLP-dipendenti generalmente
includono: convulsioni, ipoglicemia, acidosi lattica, anemia, incremento di lattato nel sangue,
elettroencefalogramma con caratteristico pattern di soppressione a scoppio, asfissia ed un
basso indice di Apgar. (Clayton P.T. 2006) Inoltre presentano caratteristici valori alterati di
alcune specie chimiche nel fluido cerebrospinale (CSF), nel plasma e nelle urine.
Nel CSF si riscontrano alte concentrazioni di 3-metoxitirosina (3-O-metil-DOPA), L-DOPA,
glicina, treonina, istidina e basse concentrazioni di acido omovanillico (HVA), acido 5-
idrossiindolacetico (5-HIAA), arginina. Nelle urine vi è alta concentrazione di vanillattato

38
(VLA), cistationina ed acido xanturenico. Nel plasma inoltre si riscontra un’elevata
concentrazione di omocisteina. (Mills P.B. et al. 2005)
Come discusso nel capitolo precedente, tutte queste caratteristiche cliniche e biochimiche,
che si accompagnano alla sindrome convulsiva, sono indici di metabolismi alterati.
Studi effettuati su pazienti affetti da questa sindrome hanno dimostrato che questi
metabolismi alterati possiedono enzimi PLP-dipendenti ed in assenza del cofattore non
possono svolgere la loro attività catalitica, alterando le concentrazioni dei metaboliti.
Nelle tabelle 2 e 3 esposte precedentemente nei capitoli 3.4 e 3.5 sono mostrati anche gli
enzimi PLP-dipendenti implicati direttamente nella produzione delle alterazioni riscontrate
nelle NEE PLP-dipendenti.
In particolare si nota che molti degli intermedi metabolici che si accumulano derivano dal
malfunzionamento di enzimi PLP-dipendenti presenti in abbondanza nei neuroni, come la
decarbossilasi degli L-aminoacidi aromatici (AAAD), che mediano la sintesi di dopamina e
serotonina: accumulo di metaboliti intermedi nella sintesi dell’ L-DOPA (3-metoxitirosina
nel CSF, vanillattato VLA nelle urine), decremento di metaboliti secondari della dopamina
(acido omovanillico HVA) e della serotonina (l’acido 3-idrossiindolacetico 5-HIAA).
Allo scopo di identificare, in queste NEE PLP-dipendenti, le cause della carenza di
piridossal-5’-fosfato, che induce il malfunzionamento degli enzimi PLP-dipendenti, il
gruppo di ricerca universitario di Mills P.B. si è interrogato sulla responsività dei pazienti
alla somministrazione della vitamina esogena. (Mills P.B. et al. 2005)
Osservando che le crisi, così come i valori alterati dei metaboliti, tornano alla normalità solo
grazie alla somministrazione di PLP o PL e che a livello cellulare solo i vitameri defosforilati
possono esser assorbiti, i ricercatori hanno ipotizzato un difetto nell’enzima richiesto per la
sintesi intracellulare di PLP a partire da PMP o PNP.
L’enzima in questione non può che essere la PNP ossidasi (PNPOx) del salvage pathway del
PLP, poiché essa è richiesta per la conversione intracellulare di PMP e PNP a PLP. Mentre la
conversione di PL in PLP è effettuata direttamente dalla PLK e non richiede la PNPOx.
Quindi, nello studio effettuato da Mills P.B et al. 2005, venne sequenziato il gene PNPOx in
pazienti con NEE PLP-dipendenti e vennero identificate ed analizzate tre delle sette
mutazioni autosomiche recessive a perdita di funzione della PNPOx oggi riconosciute
responsabili della sua inattività e dell’insorgenza di tale sindrome. Le sette mutazioni
includono: mutazione missenso in omozigosi (mutanti R95C, R95H e R229W), una

39
mutazione di un codone di stop (X262Q), una mutazione non senso (p.A174X), la mutazione
di un sito di splicing (IVS31g>a) ed una mutazione frameshift (c.del246T). Ciascuna delle
sette mutazioni riscontrate corrispondono ad una nulla o particolarmente ridotta attività della
PNPOx. (Mills P.B et al. 2005; Hoffmann G. F. et al. 2007; Khayat M. et al. 2008; Bagci
S. et al. 2008; Ruiz A. et al. 2008; Musayev F. N et al. 2009)
Il gene umano per la PNPOx (OMIM 6032870) è situato sul cromosoma 17q21.2 ed ha
un’estensione di ~7.5 kb. Contiene sette esoni e produce un trascritto mRNA che codifica
261 amminoacidi. La proteina ha una struttura omodimerica, ciascuna subunità del dimero
possiede un sito catalitico che lega il cofattore FMN ed il substrato PNP o PMP e catalizza
l’ossidazione del PMP e PNP a PLP. (Mills P.B. et al. 2005)
Lo studio più recente dei mutanti della PNPOx umana, ed in particolar modo l'analisi
cristallografica del mutante R229W (mutazione autosomica missenso in omozigosi) di
questa, effettuate dal gruppo di ricerca di Musayev F.N., di Salvo M.L., Contestabile R. et
al. 2009, ha permesso l'identificazione dei residui amminoacidici implicati direttamente o
indirettamente nella catalisi e nella corretta attività dell'enzima. Qui di seguito prenderò in
analisi proprio lo studio del mutante R229W.
In genetica la mutazione missenso è definita come una mutazione puntiforme nella quale un
singolo nucleotide è mutato ed il codone che ne risulta codifica per un amminoacido
differente rispetto al codone del wild-tipe.
Nel mutante R229W la mutazione missenso causa la sostituzione del residuo Arg229, nel
sito catalitico, con un residuo di triptofano, la quale induce gravi effetti sulla struttura del sito
attivo ed un decremento dell’attività catalitica dell’enzima di ben oltre il 70%.
Nel sito catalitico del wild-tipe l’FMN si colloca in una profonda fenditura all'interfaccia tra
le due sub-unità dell'enzima e prende contatto, mediante legami sia idrogeno che di natura
idrofobica, con residui molto conservati di entrambe le sub-unità della proteina. Il substrato
PNP o PMP, così come il prodotto di reazione PLP, si legano frontalmente e parallelamente
all'FMN, ma con il loro gruppo fosfato che punta verso l'esterno della tasca, orientato quindi
in verso opposto a quello del FMN che punta verso il fondo della cavità. Il cofattore ed il
substrato instaurano legami deboli di tipo van der Waals ed il C4' del PLP e l' N5 del FMN
vengono a trovarsi ad una distanza di ~3,4 Å, ottimale per il trasferimento dell'idrogeno.
(Musayev F.N. et al. 2003)

40
FIGURA 24. Struttura del sito attivo
della hPLK, che mostra la distanza di ~3,4 Å tra
il C4' del PLP e l' N5 del FMN nel sito catalitico
della PNPOx
Il risultato della sostituzione dell’ Arg229 con un residuo ingombrante come il triptofano, nel
sito di legame dell’FMN, porta alla perdita di legami idrogeno e ponti salini tra la tasca
dell’enzima e questo cofattore, riducendone l’affinità di legame. Impedisce anche la
formazione di alcuni legami idrogeno critici tra il substrato (PNP) ed i residui His227 e
Arg225. Questi due legami idrogeno si sono rivelati molto importanti per permettere il
posizionamento dell’anello piridinico del substrato in corretto orientamento parallelo con
l’anello isoallosazinico del cofattore. Infatti i reagenti, nel mutante, perdono l'orientazione
appropriato per reagire, non si ritrovano più posizionati parallelamente ed i gruppi interessati
nella reazione di trasferimento dell’idrogeno, il C4’ del PNP e l’N5 dell’FMN sono separati
da ~4,5 Å, rispetto ai ~3,4 Å nel wild-tipe. (Figura 25)
FIGURA 25. Sovrapposizione tra il sito
attivo del mutante PNPOx umano R229W
(azzurro) e il sito attivo della PNPOx umana
wild-type.
La perdita dei legami idrogeno tra la tasca enzimatica e l’anello piridinico del substrato porta
quindi ad un decremento dell’efficienza catalitica del mutante rispetto al wild-tipe.

41
4.4 NEE PN-dipendenti (PDE)
Altre forme di Encefalopatie Epilettiche Neonatali sono state osservate in pazienti con
mutazioni al gene ALDH7A1, che codifica per l’enzima ubiquitario semialdeide α-
aminoadipica deidrogenasi, più comunemente chiamata antiquitina.
In questi casi i pazienti rispondono anche alla somministrazione di PN esogeno, con un
miglioramento progressivo delle crisi fino alla loro completa cessazione, motivo per il quale
a questi disordini è stato dato il nome più specifico di NEE PN-responsive o PDE (epilessie
dipendenti da piridossina). (Stockler, S. et al. 2011)
I riscontri clinici dei pazienti con NEE PN-dipendenti riportano convulsioni, gastroenteriti
ricorrenti, encefalopatie e moderati ritardi dello sviluppo, ma raramente presentano stress
fetale e nascita prematura, che invece caratterizzano le NEE PLP-dipendenti.
Anche questi bambini hanno bisogno di essere trattati con dosi farmacologiche di piridossina
ed è necessaria una terapia di lunga durata per tutta la vita per prevenire le convulsioni.
Inoltre i neonati non riportano elevate concentrazioni di treonina, di glicina o alterate
concentrazioni di metaboliti associati al malfunzionamento delle decarbossilasi degli
amminoacidi aromatici, ma sono invece caratterizzati dalla presenza, nel plasma e nel CSF,
di elevate concentrazioni di acido pipecolico e di semialdeide α-aminoadipica (quest’ultima
anche nelle urine). (Ghatge MS et al. 2012)
Nel 2004 Clayton P.T., osservando i valori alterati dei metaboliti che si accompagnano alla
sindrome convulsiva, sviluppò l’ipotesi della mutazione all’antiquitina sulla base di due
osservazioni: si conosceva già, nell’iperprolinemia di tipo II, la capacità del PLP di reagire
ed essere sequestrato, mediante una reazione di condensazione, dall’acido Δ1-pirrolin-5-
carbossilico (P5C) ed i pazienti affetti da NEE PL-dipendenti riportavano concentrazioni
elevate di acido pipecolico, un intermedio di reazione nella via di degradazione della lisina
cerebrale.
La via di degradazione cerebrale della lisina produce acido pipecolico, il quale viene
ossidato ad acido L-Δ1-piperidin-6-carbossilico (P6C), che è strutturalmente simile al P5C e
particolarmente reattivo. Questo intermedio instabile però generalmente, per eliminazione di
una molecola d’acqua, si converte spontaneamente nella semialdeide α-aminoadipica (α-
AASA), substrato della reazione catalizzata dall’antiquitina.

42
Quando l’antiquitina è mutata non catalizza più l’ossidazione NAD-dipendente della
semialdeide α-aminoadipica ad acido α-aminoadipico e l’equilibrio della reazione precedente
si sposta accumulando P6C nel citoplasma, il quale può reagire mediante condensazione di
Knoevenagel con il PLP e sequestrarlo dall’ambiente intracellulare. (Figura 26)
FIGURA 26.
Interazione tra gli
intermedi della via di
degradazione della lisina
ed il PLP. Reazione
dell’antiquitina e
condensazione di
Knoevenagel tra P6C e
PLP.
Negli anni successivi Mills, P. B. et al. 2006 sequenziarono il gene dell’antiquitina in
individui affetti da NEE PL-dipendenti e vennero trovate mutazioni sia in omozigosi che in
eterozigosi a carico di questo enzima. Recentemente Sharer et al. ne hanno riportato
un’analisi complessiva che comprende mutazioni missenso, non senso, siti di splicing e
piccole delezioni.

43
Queste NEE PL-dipendenti generate da una mutazione nel gene per l’antiquitina sono
pertanto dovute ad una deficienza di PLP, indotta dal sequestro del cofattore, da parte di un
intermedio instabile, derivato da una via metabolica mutata. Questa deficienza di piridossal-
5’-fosfato intracellulare che ne risulta influisce poi, molto probabilmente, su enzimi PLP-
dipendenti particolarmente sensibili anche a modeste diminuzioni del cofattore, come quelli
implicati nell’insorgenza dell’epilessia (BCAT, GAD o GABA-AT).
La somministrazione di PN esogeno porta ad un incremento del PLP intracellulare
sintetizzato dagli enzimi del suo salvage pathway che viene direttamente ceduto agli
apoenzimi tramite interazione proteina-proteina. Molto probabilmente anche la
somministrazione diretta del cofattore attivo può esser in grado di far cessare le crisi ma con
una minor efficienza, poiché esso, libero in soluzione, potrebbe essere sequestrato dal P6C.
Sorprendentemente è stato dimostrato che molti dei casi di NEE PN-dipendenti, che
riportano la mutazione del gene dell’antiquitina, rispondono anche alla somministrazione
dell’acido folinico (5-formil tetrraidrofolato) (Figura 27), in maniera variabile rispetto al
trattamento con la piridossina. (Gallagher, R. C. et al. 2009)
In questi casi i soggetti hanno mostrato la presenza di due composti non identificati
nel liquido cerebrospinale, motivo per il quale è parere di Ghatge MS et al. 2012 che la
risposta all’acido folinico, pur essendo associata a casi di NEE PLP-dipendenti potrebbe
essere relativa ad una diversa condizione patologica non ancora dimostrata.
FIGURA 27. Acido folinico (5-formil
tetrraidrofolato)
Tuttavia la classificazione delle casistiche associabili alla NEE PN-dipendenti è più
complessa e meno definita di quella delle NEE PLP-dipendenti. Sono infatti conosciute
diverse altre patologie, caratterizzate da manifestazioni convulsive ed encefalopatie neonatali
che non presentano mutazione all’antiquitina ma rispondono al trattamento con piridossina e
potrebbero quindi ricadere sotto questa denominazione: l’ipofosfatasia e l’iperprolinemia di
tipo II. (Walker V. et al. 2000; Litmanovitz et al. 2002)

44
CONCLUSIONI E POSSIBILI LIVELLI DI INDAGINE
Le Encefalopatie Epilettiche Neonatali sono delle rare sindromi neurologiche correlate alla
deficienza di piridossal-5’-fosfato che causa malfunzionamento di enzimi PLP-dipendenti,
soprattutto implicati nella sintesi dei neurotrasmettitori. Nella maggior parte dei casi la causa
della deficienza ha origine da una mutazione nella PNPOx appartenente al salvage pathway
del PLP (NEE PLP-dipendenti). In questo caso i pazienti rispondono solo al trattamento con
il PL o il PLP, poiché a livello intracellulare è bloccata la via di sintesi del cofattore a partire
da PMP e PNP. In altri casi la causa della deficienza è una mutazione del gene che codifica
l’antiquitina (NEE PN-dipendenti), che genera un intermedio della via di degradazione della
lisina, molto reattivo, capace di sequestrare il PLP dalla soluzione. In questo caso i pazienti
rispondono bene al trattamento con la PN e molto probabilmente anche con il PLP.
Le due forme di NEE possono essere diagnosticate, distinte l’una dall’altra e dalle altre
sindromi neurologiche, in base ai fenotipi ed alla risposta al PN piuttosto che al PL/PLP,
come riportato in tabella 4.
TABELLA 4. Comparazione degli aspetti clinici e metabolici tra NEE PLP-dipendenti e NEE PN-
dipendenti.

45
Tuttavia, molte altre sindromi potrebbero ricadere sotto la denominazione di NEE PN/PLP-
dipendenti, essendo caratterizzate da convulsioni neonatali che rispondono al trattamento con
la vitamina B6. D’altro canto anche alcuni casi clinici riportati appartenere alle NEE
PN/PLP-dipendenti potrebbero non essere proprio associabili a tale suddivisione, avendo
riportato concentrazioni di metaboliti e responsività anomale.
Questo accade perché i metabolismi alterati, in grado di produrre intermedi di reazione
instabili, capaci di legare il PLP, possono essere molti. Anche se l’effetto della deficienza va
ad influire sempre sugli enzimi PLP-dipendenti, le cause e le sindromi neurodegenerative
associate possono variare pur avendo simile risposta alla vitamina B6.
Ad esempio, patologie come l’iperprolinemia di tipo II e l’ipofosfatasia, potrebbero essere
comprese nella denominazione NEE PN-responsive, poiché sono entrambe caratterizzate da
manifestazioni convulsive neonatali, encefalopatie, rispondono bene al trattamento con la
piridossina e sono dovute ad una deficienza di PLP. Tuttavia la causa della deficienza è
diversa. Nel caso dell’iperprolinemia la mutazione nel gene ALDH4A1 genera un intermedio
instabile (Δ1-pirrolin-5-carbossilico; P5C), che sequestra il PLP, similmente a quanto accade
nelle PDE con mutazione all’antiquitina (ALDH7A1). Nell’ipofosfatasia è mutato il gene
che codifica un’isoenzima della fosfatasi alcaline tessuto non specifiche (ALPL; OMIM
171760), che impedisce alla vitamina di raggiungere i tessuti. (Walker V. et al. 2000;
Litmanovitz et al. 2002)
D’altro canto alcune sindromi, dovute alla mutazione di enzimi PLP dipendenti come
l’anemia PN-responsive, l’omocistinuria o la deficienza da ornitina δ-aminotransferasi, non
manifestano attacchi convulsivi ed encefalopatie, ma sono caratterizzate dall’avere una
risposta alla somministrazione di piridossina. In alcuni di questi casi non si è ancora capito in
che modo essa agisca. (Herrmann W et al. 2007; Clayton P.T 2006)
Altri disordini neurologici ben conosciuti come l’autismo, l’Alzheimer, la sindrome di
Down, la schizofrenia, il Parkinson etc. sono stati associati alla deficienza di PLP ed i
pazienti sembrano rispondere bene al trattamento con la vitamina B6 (in alcuni casi con PN
ed in altri con PL/PLP). (Adams J. B. et al. 2006; Nakagawa E. et al. 1997; Sandyk R et
al. 1990)

46
Per comprendere meglio la difficoltà di classificazione dei casi di Encefalopatie Epilettiche
Neonatali ed il coinvolgimento della vitamina B6 nella loro genesi, è opportuno prendere in
considerazione un dato importante; il PLP svolge numerose altre funzioni nel metabolismo
cellulare ed alcune di queste potrebbero concorrere alla formazione del fenotipo della
patologia quali: il ruolo del PLP e della piridossina come regolatori del trasporto ionico di
membrana, come regolatori di fattori di trascrizione e ligandi di recettori steroidei.
Per affrontare una problematica che ha un così ampio raggio di cause e coinvolge, a livello
biochimico e genetico, una fitta rete di metabolismi, è indispensabile, riuscire a definire una
scala di livelli di indagine che aiuti il ricercatore ad avere una visione più completa ed
ordinata del fenomeno.
Livelli di indagine:
1) Analisi degli enzimi del Salvage pathway del piridossal-5‘-fosfato:
In primo luogo, un’analisi attenta degli enzimi che permettono la sintesi e
l’assorbimento della vitamina B6, risulta indispensabile qualora ci si trovi di fronte ad
una sindrome che risponde ad una soimministrazione esogena della vitamina. In base ai
dati fin ora raccolti ed all’analisi cliniche dei pazienti è possibile intuire se vi sia il
malfunzionamento di uno di questi enzimi.
Poco si conosce ancora delle fosfatasi intestinali implicate nel primo assorbimento della
vitamina, di cui sarebbe utile conoscere struttura ed effetti del malfunzionamento.
Inoltre sarebbe interessante approfondire l’analisi sul ruolo svolto dagli enzimi della via
di recupero nel regolare i livelli di PLP intracellulare.
2) Analisi di possibili metabolismi alterati in grado di accumulare intermedi di
reazione che sequestrano il PLP:
L’analisi delle concentrazioni alterate di alcuni metaboliti può essere indice della
presenza di sostanze che interagiscono con il PLP e lo sequestrano.
3) Enzimi che usano il PLP come cofattore e ruolo della piridossina:
Alcune malattie sono causate dal malfunzionamento o dalla mutazione di enzimi PLP-
dipendenti ed in alcuni di questi casi si è verificata una risposta marcata alla
piridossina. Un altro livello di indagine potrebbe proprio esser quello di ricercare ed

47
identificare il meccanismo attraverso il quale la piridossina riesce a trattare queste
sindromi causate da mutazioni di enzimi PLP-dipendenti.
4) Il PLP e la PN come regolatori di fattori di trascrizione:
Il ruolo di regolatori di fattore di trascrizione, potrebbe essere la causa di alcuni aspetti
fenotipici della malattia, che risultano di ignota origine.
5) Ruolo del PLP e la PN nel trasporto di ioni e come fattori di legame ai recettori
steroidei:
Anche questo ruolo, potrebbe essere la causa di alcuni aspetti fenotipici della malattia,
che risultano di ignota origine. Infatti, soprattutto a livello cerebrale, questi vitameri
potrebbero avere una più complessa modulazione della trasmissione nervosa, che
potrebbe spiegare alcuni aspetti fenotipici caratteristici dei pazienti.
6) Ruolo dell'acido folinico:
Anche l’indagine più approfondita sull’azione di questo composto potrebbe far luce su
molti parametri irregolari riscontrati nelle sindromi neurologiche PN-dipendenti, come i
picchi cromatografici riferiti a sostanze sconosciute nelle analisi del CSF in casi di
PDE.
7) Indagine genetica:
La ricerca troverebbe completezza con un’analisi delle mutazioni ai geni ritenuti
responsabili della sindrome. Sarebbe interessante analizzare il differene impatto che le
mutazioni in omozigosi ed in eterozigosi hanno sulla gravità della malattia, ossia
analizzare se le mutazioni causano solo una parziale o ridotta attività degli enzimi
mutati oppure il completo knockout.
Inoltre un’analisi della derivazione genetica delle mutazioni potrebbe far luce
sull’incidenza e le motivazioni di certe mutazioni rispetto ad altre.
Un esempio interssante potrebbe esser quello di comprendere perché le mutazioni alla
PNPOx si sono riscontrate principalmente in pazienti con un a qualche origine asiatica
o turca.

48
RINGRAZIAMENTI
Ringrazio prima di tutto i docenti, che con pazienza ed estrema disponibilità, mi
hanno seguito ed incoraggiato nella scrittura di questa tesi: il Prof. Roberto
Contestabile ed il Dott. Martino Luigi di Salvo.
Un particolare ringraziamento va alla mia famiglia, alle mie nonne, alle persone a
me più vicine ed al mio collega di università, per avermi supportato e sopportato in
questo periodo così pieno ed intenso. Li ringrazio per la pazienza, l’aiuto materiale e
l’affetto dimostratomi.

49
ELENCO ABBREVIAZIONI:
3-O-metil-DOPA: 3-metoxitirosina
5-HIAA: acido 5-idrossiindolacetico
AAAD: decarbossilasi degli L-amminoacidi aromatici
ALDH7A1: gene che codifica per l’enzima antiquitina (α-aminoadipica deidrogenasi)
ANTIQUITINA: semialdeide α-aminoadipica deidrogenasi
BCAT: aminotransferasi di amminoacidi a catena ramificata
FMN: flavina mononucleotide
CSF: fluido cerebrospinale
GABA: acido γ-amminobutirrico
GABA-AT: acido γ-amminobutirrico amminotransferasi
GAD: glutammato decarbossilasi
HVA: acido omovanillico
IP: fosfatasi intestinali
L-DOPA: L-dopamina
NEE: Encefalopatie Epilettiche Neonatali
P5C: acido Δ1-pirrolin-5-carbossilico
P6C: acido L-Δ1-piperidin-6-carbossilico
PDE: epilessie dipendenti da piridossina
PL: piridossale
PLK: piridossal chinasi
PLP: piridossal-5’-fosfato
PM: piridossamina
PMP: piridossamina-5’-fosfato
PN: piridossina
PNP: piridossina-5’-fosfato
PNPOx: piridossina-5’-fosfato ossidasi
SNC: sistema nervosa centrale
SSADH: semialdeide succinica deidrogenasi
TNAP: fosfatasi alcaline tessuto non-specifiche
VLA: vanillattato
α-AASA: semialdeide α-aminoadipica

50
INDICE FIGURE E TABELLE
FIGURA 1. Struttura dei vitameri B6.
FIGURA 2. Meccanismo di reazione del PLP; formazione della base di schiff .
FIGURA 3. Struttura di risonanza del carbanione con formazione di un intermedio chinonoide.
FIGURA 4. Vie biosintetiche della vitamina B6.
FIGURA 5. Piridossina-5’-β- glucoside.
FIGURA 6. Interconversione tra le forme fosforilate e defosforilate della vitamina B6 ed enzimi della via di recupero del
PLP.
FIGURA 7. Ciclo di assorbimento e trasporto della vitamina B6.
FIGURA 8. Enzimi della via di recupero o “salvage pathway” del PLP.
FIGURA 9. Reazione catalizzata dalla PLK.
FIGURA 10. Struttura cristallina del dimero della ePLK.
FIGURA 11. Diagramma della struttura dimerica della hPLK.
FIGURA 12. Diagramma schematico delle interazioni idrofobiche ed i legami idrogeno tra Mg2+ , ATP e residui della
ePLK.
FIGURA 13. Meccanismo di reazione proposto per la PLK.
FIGURA 14. Struttura del coenzima flavin mononucleotide (FMN).
FIGURA 15. Reazione catalizzata dalla PNPOx.
FIGURA 16. Struttura della PNPOx di E. coli.
FIGURA 17. Due meccanismi proposti per la PNPOx.
FIGURA 18. Acido piridossico
FIGURA 19. Effetto del malfunzionamento della PLK sull’assorbimento ed il trasporto della vitamina B6.
FIGURA 20. Effetto del malfunzionamento della PNPOx sull’assorbimento ed il trasporto della vitamina B6.
FIGURA 21. Effetto del malfunzionamento delle fosfatasi sull’assorbimento ed il trasporto della vitamina B6.
FIGURA 22. Principali neurotrasmettitori del SNC: L-glutammato ed acido γ-amminobutirrico (GABA).
FIGURA 23. Via dello shunt del GABA.
FIGURA 24. Struttura del sito attivo della hPLK.
FIGURA 25. Sovrapposizione tra il sito attivo del mutante PNPOx umano R229W e quello del wild-type.
FIGURA 26. Interazione tra gli intermedi della via di degradazione della lisina ed il PLP.
SCHEMA 1. Neurotrasmettitori sintetizzati da enzimi PLP-dipendenti.
FIGURA 27. Acido folinico (5-formil tetraidrofolato)
TABELLA 1. Alcuni dei farmaci attualmente in commercio e composti naturali che interagiscono con il metabolismo
della vitamina B6.
TABELLA 2. Enzimi PLP-dipendenti coinvolti in vari metabolismi cellulari e gli effetti del loro malfunzionamento.
TABELLA 3. Enzimi PLP-dipendenti coinvolti nel metabolismo dei neurotrasmettitori.
TABELLA 4. Comparazione degli aspetti clinici e metabolici tra NEE PLP-dipendenti e NEE PN-dipendenti.

51
BIBLIOGRAFIA
Abbott R., Pye I. And Nahorski S. : Csf And Plasma Gaba Levels In Parkinson's Disease.
J. Neurol. Neurosurg. Psychiatry. 45, 253-256 (1982)
Adams, J. B., George, F. And Audhya, T: Abnormally High Plasma Levels Of Vitamin B6 In Children With Autism Not Taking Supplements Compared To Controls Not Taking Supplements.
J. Altern. Complement. Med. 12, 59-63. (2006)
An SJ, Park SK, Hwang IK, et al ; Vigabatrin inhibits pyridoxine- 5 -phosphate oxidase, not pyridoxal kinase in the hippocampus of seizure prone gerbils.
Neurochem Int 44: 133–137. (2004)
Andre V. M., Cepeda C. And Levine M. S. : Dopamine And Glutamate In Huntington's Disease: A Balancing
Act. Cns Neurosci. Ther. 16, 163-178. Miscellanea on Encephalopathies – A Second Look (2010)
Aoyagi T., Wada T., Nagai M., Kojima F., Harada S., Takeuchi T., Takahashi H.,
Hirokawa K. And Tsumita T. :
Increased Gamma-Aminobutyrate Aminotransferase Activity In Brain Of Patients With Alzheimer's Disease. Chem. Pharm. Bull. (Tokyo). 38, 1748-1749 (1990)
Bagci S., J. Zschocke, G. F. Hoffmann, T. Bast, J. Klepper, A. Muller, A. Heep, P. Bartmann and A. R. Franz: Pyridoxal phosphate-dependent neonatal epileptic encephalopathy.
Arch Dis Child Fetal Neonatal Ed, 93(2), F151-2 (2008)
Bear M.F., Connors B.W., Paradiso M.A.; i ritmi del cervello.
In “Neuroscienze” (Esplorando il cervello). Mei V.Ed.Masson, Mi, cap. 19: 264-655 (2005)
Bilski P, LiMY, Ehrenshaft M, Daub ME, Chignell CF: Vitamin B6 (pyridoxine) and its derivatives are efficient singlet oxygen quenchers and potential fungal antioxidants.
Photochem Photobiol 71: 129–134. (2000)
Blandini F., Porter R. H. And Greenamyre J. T. : Glutamate And Parkinson's Disease.
Mol. Neurobiol. 12, 73-94.(1996)
Bowling F. G.: Pyridoxine supply in human development
Seminars in Cell & Developmental Biology 22 611–618 (2011)
Butterworth J., Yates C. M. And Simpson J. : Phosphate-Activated Glutaminase In Relation To Huntington's Disease And Agonal State.
J. Neurochem. 41, 440-447 (1983)
Büyükokuroglu ME, Gepdiremen A, Tastekin A, Ors R.: Pyridoxine may protect the cerebellar granular cells against glutamate-induced toxicity.
Int J Vitam Nutr Res;77(5):336–40. (2007)

52
Cao P, Gong Y, Tang L, Leung YC, Jiang T : Crystal structure of human pyridoxal kinase.
J. Struct. Biol. 154: 327-332 (2006)
Cheung P. Y., Fong C. C., Ng K. T., Lam W. C., Leung Y. C., Tsang C. W., Yang M. And Wong M. S. : Interaction Between Pyridoxal Kinase And Pyridoxal-5- Phosphate-Dependent Enzymes.
J. Biochem. 134, 731-738 (2003)
Choi J.D., M. Bowers-Komro, M.D. Davis, D.E. Edmondson, D.B. McCormick; Kinetic properties of pyridoxamine (pyridoxine)-5'-phosphate oxidase from rabbit liver.
The Journal of biological chemistry 258 840-845. (1983)
Choi S. Y., Churchich J. E., Zaiden E. And Kwok F. :
Brain Pyridoxine-5- Phosphate Oxidase. Modulation Of Its Catalytic Activity By Reaction With
Pyridoxal 5-Phosphate And Analogs. J. Biol. Chem. 262, 12013-12017 (1987)
Chung J. Y., Choi J. H., Hwang C. Y. And Youn H. Y. Pyridoxine Induced Neuropathy By Subcutaneous Administration In Dogs.
J. Vet. Sci. 9, 127-131 (2008)
Churchich JE, Wu C : Brain pirydoxal kinase. Mechanism of substrate addiction, binding of ATP, and rotational mobility of the inhibitor pyridoxaloxime.
J. Biol. Chem.. 256: 780-784 (1981)
Clayton P.T. : B6-responsive disorders: a model of vitamin dependency
J. Inherit. Metab. Dis., 29 , pp. 317–326 (2006)
Dakshinamurti K. , K. J. Lal and P. K. Ganguly: Hypertension, calcium channel and pyridoxine (vitamin B6).
Mol Cell Biochem, 188(1-2), 137-48 (1998)
Di Salvo M. L., Ko T. P., Musayev F. N., Raboni S., Schirch V. And Safo M. K. :
Active Site Structure And Stereospecificity Of Escherichia Coli Pyridoxine-5'- Phosphate Oxidase. J. Mol. Biol. 315, 385-397. (2002)
Di Salvo M. L., Safo M. K., Musayev F. N., Bossa F. And Schirch V. : Structure And Mechanism Of Escherichia Coli Pyridoxine 5'-Phosphate Oxidase.
Biochim. Biophys. Acta. 1647, 76-82 (2003)
Di Salvo M. L., Safo MK, Contestabile R: Biomedical aspects of pyridoxal-5'-phosphate availability.
Front Biosci (Elite Ed.) Jan 1;4:897-913. (2012)
Di Salvo M., E. Yang, G. Zhao, M.E. Winkler, V. Schirch: Expression, purification, and characterization of recombinant Escherichia coli pyridoxine 5'-phosphate oxidase.
Protein expression and purification 13 349-356 (1998)
Di Salvo ML, Contestabile R, Safo MK : Vitamin B6 salvage enzymes: mechanism, structure and regulation.
BBA. doi: 10.1016/j.bbapap.2010.12.006 (2011)

53
di Salvo ML, Hunt S, Schirch V :
Expression, purification, and kinetic constants for human and Escherichia coli pyridoxal kinases. Prot. Express. Purif. 36: 300-306 (2004)
Ebadi M. , C.F. Gessert, A. Al-Sayegh: Drug-pyridoxal phosphate interactions.
Quarterly reviews on drug metabolism and drug interactions 4289-331. (1982)
Ebadi M. and B. Klangkalya: On the mechanism of pyridoxal phosphate-related convulsions as implicated in enhanced transport of GABA.
Neuropharmacology, 18(3), 301-7 (1979)
Eliot AC, Kirsch JF : Pyridoxal phosphate enzymes: mechanistic, structural and evolutionarily considerations.
Annu. Rev. Biochem. 73: 383-415 (2004)
Fang,X., Zhou Z. M., Lu L., Yin L. L., Li J. M., Zhen Y., Wang H. And Sha J. H. : Expression Of A Novel Pyridoxal Kinase Mrna Splice Variant, Pkh-T, In Human Testis.
Asian J. Androl. 6, 83-91 (2004)
Fitzpatrick TB, Moccand C, Roux C ; Vitamin B6 biosynthesis: charting the mechanistic landscape.
Chem. Bio. Chem. 11: 1185-1193 (2010)
Foca FJ : Motor and sensory neuropathy secondary to excessive pyridoxine ingestion.
Arch. Phys. Med. Rehabil. 66: 634-636 (1985)
Fu TF, di Salvo ML, Schirch V : Distribution of B6 vitamers in Escherichia coli as determined by enzymatic assay.
Anal. Biochem. 298: 314-321 (2001)
Gallagher, R. C., Van Hove, J. L., Scharer, G., Hyland, K., Plecko, B., Waters, P. J., Mercimek-Mahmutoglu, S., Stockler-Ipsiroglu, S., Salomons, G. S., Rosenberg, E. H., Struys, E. A. And Jakobs, C.:
Folinic Acid-Responsive Seizures Are Identical To Pyridoxine-Dependent Epilepsy. Ann. Neurol. 65, 550-556. (2009)
Gandhi AK, Ghatge MS, Musayev FN, Sease A, Aboagye SO, di Salvo ML, Schirch V, Safo MK : Kinetic and structural studies of the role of the active site residue Asp 235 of human pyridoxal kinase.
Biochem. Biophys. Res. Comm. 381: 12-15 (2009)
Gdynia HJ, Muller T, Sperfeld AD, Kuhnlein P, Otto J, Kassubek J, Ludolph AC : Severe sensorimotor neuropathy after intake of highest dosages of vitamin B6.
Neuromuscul. Disord. 18: 156-158 (2008)
Ghatge MS, Di Salvo ML, Contestabile R, Eseonu DN, Karve S, Schirch V, Safo MK: Molecular defects of vitamin B6 metabolism associated with Neonatal Epileptic Encephalopathy.
Miscellanea on Encephalopathies- second look (2012)
Gospe S. M., Jr.: Neonatal vitamin-responsive epileptic encephalopathies.
Chang Gung Med J, 33(1), 1-12 (2010)

54
Hayashi H. : Pyridoxal enzymes: mechanistic diversity and uniformity.
J. Biochem. 118: 463-473 (1995)
Herrmann W., Lorenzl S. And Obeid R. :
Review Of The Role Of Hyperhomocysteinemia And B-Vitamin Deficiency In Neurological And
Psychiatric Disorders--Current Evidence And Preliminary Recommendations. Fortschr Neurol. Psychiatr. 75, 515-527. (2007)
Herrmann, W., Lorenzl, S. And Obeid, R. :
Review Of The Role Of Hyperhomocysteinemia And B-Vitamin Deficiency In Neurological And Psychiatric Disorders--Current Evidence And
Preliminary Recommendations. Fortschr Neurol. Psychiatr. 75, 515-527. (2007)
Hirakawa-Sakurai T, Okhawa K, Matsuda M : Purification and properties of pyridoxal kinase from bovine brain.
Mol. Cell. Biochem. 119: 203-207 (1993)
Hoffmann G. F., B. Schmitt, M. Windfuhr, N. Wagner, H. Strehl, S. Bagci, A. R. Franz, P. B. Mills, P. T. Clayton, M. R.
Baumgartner, B. Steinmann, T. Bast, N. I. Wolf and J. Zschocke:
Pyridoxal 5'-phosphate may be curative in early-onset epileptic encephalopathy. J Inherit Metab Dis, 30(1), 96-9 (2007)
Huang S.H. , R.J. Shi, J.Y. Zhang, Z. Wang, L.Q. Huang.
Cloning and characterization of a pyridoxine 5'-phosphate oxidase from silkworm, Bombyx mori. Insect molecular biology 18 365-371. (2009)
Huq M. D. , N. P. Tsai, Y. P. Lin, L. Higgins and L. N. Wei: Vitamin B6 conjugation to nuclear corepressor RIP140 and its role in gene regulation.
Nat Chem Biol, 3(3), 161-5 (2007)
Iqbal S. J., A. Brain, T. M. Reynolds, M. Penny and S. Holland: Relationship between serum alkaline phosphatase and pyridoxal-5'-phosphate levels in hypophosphatasia.
Clin Sci (Lond), 94(2), 203-6 (1998)
Ishioka N., J. Sato, J. Nakamura, T. Ohkubo, A. Takeda and S. Kurioka: In vivo modification of GABAA receptor with a high dose of pyridoxal phosphate induces tonic-clonic convulsion in immature mice.
Neurochem Int, 26(4), 369-73 (1995)
Jallon P., Loiseau P., Loiseau J.; Newly diagnosed unprovoked epileptic seizures: presentation at diagnosis in CAROLE study. Cordination active du reseau observatoire longitudinal
de l'epilepsie. Epilepsia 42: 464-475 (2001)
Jang Y. M., Kim D. W., Kang T. C., Won M. H., Baek N. I., Moon B. J., Choi S. Y. , Kwon O. S. : Human Pyridoxal Phosphatase. Molecular Cloning, Functional Expression, And Tissue Distribution.
J. Biol. Chem. 278, 50040-50046. (2003)
Kang J. H., Hong M. L., Kim D. W., Park J., Kang T. C., Won M. H., Baek N. I., Moon B. J., Choi S. Y. And Kwon O. S. : Genomic Organization, Tissue Distribution And Deletion Mutation Of Human Pyridoxine 5'-Phosphate Oxidase.
Eur. J. Biochem. 271, 2452-2461. (2004)
Karawya E, Fonda ML : Purification, assay, and kinetic properties of sheep liver pyridoxal kinase.
Anal. Biochem. 90: 525-533 (1978)

55
Kastner U. C. Hallmen, M. Wiese, E. Leistner, C. Drewke: The human pyridoxal kinase, a plausible target for ginkgotoxin from Ginkgo biloba,
The FEBS journal 274 1036-1045. (2007)
Kazarinoff M. N. And Mccormick D. B. : Rabbit Liver Pyridoxamine (Pyridoxine) 5'-Phosphate Oxidase. Purification And Properties.
J. Biol. Chem. 250, 3436-3442 (1975)
Keniston R. C. , S. Cabellon, Jr. and K. S. Yarbrough: Pyridoxal 5'-phosphate as an antidote for cyanide, spermine, gentamicin, and dopamine toxicity: an in vivo rat study.
Toxicol Appl Pharmacol, 88(3), 433-41 (1987)
Khayat M., S. H. Korman, P. Frankel, Z. Weintraub, S. Hershckowitz, V. F. Sheffer, M. Ben Elisha, R. A. Wevers and T. C.
Falik-Zaccai:
PNPO deficiency: an under diagnosed inborn error of pyridoxine metabolism. Mol Genet Metab, 94(4), 431-4 (2008)
Kim Y. T., Kwok F. And Churchich J. E. : Interactions Of Pyridoxal Kinase And Aspartate Aminotransferase Emission Anisotropy And Compartmentation Studies.
J. Biol. Chem. 263, 13712-13717 (1988)
Kirkness E. F. and A. J. Turner: Characterization of a cytosolic protein (P36) isolated from pig brain by benzodiazepine- affinity chromatography.
J Neurochem, 50(2), 356-65 (1988)
Lainè-Cessac P, Allain P : Kinetic studies of the effects of K+, Na+ and Li+ on the catalytic activity of human erythrocyte pyridoxal kinase.
Enzyme protein 49: 291-304 (1996)
Lainè-Cessac P, Caillux A, Allain P : Mechanisms of the inhibition of human erythrocyte pyridoxal kinase by drugs.
Biochem. Pharmacol. 54: 863-870 (1997)
Laine-Cessac P., A. Cailleux, P. Allain: Mechanisms of the inhibition of human erythrocyte pyridoxal kinase by drugs,
Biochemical pharmacology 54863-870. (1997)
Laine-Cessac, A. Cailleux and P. Allain: Mechanisms of the inhibition of human erythrocyte pyridoxal kinase by drugs.
Biochem Pharmacol, 54(8), 863-70 (1997)
Lambrecht G. , K. Braun, M. Damer, M. Ganso, C. Hildebrandt, H. Ullmann, M. U. Kassack and P. Nickel: Structure-activity relationships of suramin and pyridoxal-5'-phosphate derivatives as P2 receptor antagonists.
Curr Pharm Des, 8(26), 2371-99 (2002)
Leckman J. F., Bloch M. H., Smith M. E., Larabi D. And Hampson M. : Neurobiological Substrates Of Tourette's Disorder.
J. Child Adolesc. Psychopharmacol. 20, 237-247. (2010)
Li M.H., F. Kwok, W.R. Chang, C.K. Lau, J.P. Zhang, S.O. Liu, Y.C. Leung, T. Jiang, D.C. Liang : Crystal structure of brain pyridoxal kinase, a novel member of the ribokinase superfamily,
J. Biol. Chem. 27746385–46390. (2002)

56
Li T. K., L. Lumeng and R. L. Veitch: Regulation of pyridoxal 5'-phosphate metabolism in liver.
Biochem Biophys Res Commun, 61(2), 677-84 (1974)
Litmanovitz, O. Reish, T. Dolfin, S. Arnon, R. Regev, G. Grinshpan, M. Yamazaki and K. Ozono: Glu274Lys/Gly309Arg mutation of the tissue-nonspecific alkaline phosphatase gene in neonatal hypophosphatasia associated with convulsions.
J Inherit Metab Dis, 25(1), 35-40 (2002)
Lum HK, Kwok F, Lo SC : Cloning and characterization of Arabidopsis thaliana pyridoxal kinase.
Planta 215: 870-879 (2002)
Mackey A. D., Lieu, S. O., Carman, C. And Gregory, J. F. :
Hydrolytic Activity Toward Pyridoxine-5'-Beta-D-Glucoside In Rat Intestinal Mucosa Is Not Increased By Vitamin B-6 Deficiency: Effect Of Basal
Diet Composition And Pyridoxine Intake. J. Nutr. 133, 1362-1367 (2003)
Mccormick D. B., Chen H. : Update On Interconversions Of Vitamin B-6 With Its Coenzyme.
J. Nutr. 129, 325-327 (1999)
McCormick DB, Gregory ME, Snell EE : Pyridoxal-phosphokinases: I. Assay, distribution, purification, and properties.
J. Biol. Chem. 236: 2076-2084 (1961)
Mccormick, D. B. : Two Interconnected B Vitamins: Riboflavin And Pyridoxine.
Physiol. Rev. 69, 1170-1198 (1989)
Metzler, D. E., Ikawa, M., Snell, E. E.: “A general mechanism for vitamin B6-catalyzed reactions”
J. Am. Chem. Soc. 76, 648-652. (1954)
Mills P. B., E. Struys, C. Jakobs, B. Plecko, P. Baxter, M. Baumgartner, M. A. Willemsen, H. Omran, U. Tacke, B.
Uhlenberg, B. Weschke and P. T. Clayton:
Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat Med, 12(3), 307-9 (2006)
Mills P.B., R.A. Surtees, M.P. Champion, C.E. Beesley, N. Dalton, P.J. Scambler, S.J. Heales, A.
Briddon, I. Scheimberg, G.F. Hoffmann, J. Zschocke, P.T. Clayton:
Neonatal epileptic encephalopathy caused by mutations in the PNPO gene encoding pyridox(am)ine 5'-phosphate oxidase. Human molecular genetics 14 1077-1086. (2005)
Mills, P. B., Struys, E., Jakobs, C., Plecko, B., Baxter, P., Baumgartner, M., Willemsen, M.
A., Omran, H., Tacke, U., Uhlenberg, B., Weschke, B. And Clayton, P. T.:
Mutations In Antiquitin In Individuals With Pyridoxine-Dependent Seizures. Nat. Med. 12, 307-309. (2006)
Morra M., Philipszoon H. D., D'andrea G., Cananzi A. R., L'erario R. And Milone F. F. :
Sensory And Motor Neuropathy Caused By Excessive Ingestion Of Vitamin B6: A Case Report. Funct. Neurol. 8, 429-432 (1993)
Morrison, L. A. And Driskell, J. A. : Quantities Of B6 Vitamers In Human Milk By High-Performance Liquid Chromatography. Influence Of Maternal Vitamin B6 Status.
J. Chromatogr. 337, 249-258 (1985)

57
Mousain-Bosc M., M. Roche, A. Polge, D. Pradal-Prat, J. Rapin, J.P. Bali: Improvement of neurobehavioral disorders in children supplemented with magnesium-vitamin B6.
I. Attention deficit hyperactivity disorders, Magnes Res 19 46-52. (2006)
Musayev F. N., Di Salvo M. L., Ko T. P., Schirch V. And Safo M. K. : Structure And Properties Of Recombinant Human Pyridoxine 5'-Phosphate Oxidase.
Protein Sci. 12, 1455-1463 (2003)
Musayev F. N., M. L. Di Salvo, M. A. Saavedra, R. Contestabile, M. S. Ghatge, A. Haynes, V. Schirch and M. K. Safo: Molecular basis of reduced pyridoxine 5'-phosphate oxidase catalytic activity in neonatal epileptic encephalopathy disorder. J
Biol Chem, 284(45), 30949-56 (2009)
Musayev FN, di Salvo ML, Ko TP, Gandhi AK, Goswami A, Schirch V, Safo MK : Crystal structure of human pyridoxal kinase: structural of M+ and M2+ activation.
Protein science 16: 2184-2194 (2007)
Nakagawa, E., Tanaka, T., Ohno, M., Yamano, T. And Shimada, M: Efficacy Of Pyridoxal Phosphate In Treating An Adult With Intractable Status Epilepticus.
Neurology. 48, 1468-1469 (1997)
Nanavati S.M., Silverman R.B.: Design of potential anticonvulsant agents: mechanistic classification of GABA aminotransferase inactivators.
J. Med. Chem. 32: 2413-2421 (1989)
Nelson D.L., Cox M.M. : I principi di biochimica di Lehninger.
Zanichelli Ed., Bologna, pp. 682-684
Nishino N., Fujiwara H., Noguchi-Kuno S. A. And Tanaka C. : Gabaa Receptor But Not Muscarinic Receptor Density Was Decreased In The Brain Of Patients With Parkinson's Disease.
Jpn. J. Pharmacol. 48, 331-339 (1988)
Nogovitsina O.R., E.V. Levitina: Effect of MAGNE-B6 on the clinical and biochemical manifestations of the syndrome of attention deficit and hyperactivity in children.
Eksperimental'naia i klinicheskaia farmakologiia 69 74-77. (2006)
Oka T. : Modulation of gene expression by vitamin B6.
Nutr Res Rev, 14(2), 257-66 (2001)
Pearl P. L. And Gibson K. M. : Clinical Aspects Of The Disorders Of Gaba Metabolism In Children.
Curr. Opin. Neurol. 17, 107-113 (2004)
Percudani R. and A. Peracchi: The B6 database: a tool for the description and classification of vitamin B6-dependent enzymatic activities and of the corresponding protein families.
BMC Bioinformatics, 10, 273 (2009)
Rahman M. K., Nagatsu T., Sakurai T., Hori S., Abe M. And Matsuda M. :
Effect Of Pyridoxal Phosphate Deficiency On Aromatic L-Amino Acid Decarboxylase Activity With L-Dopa And L-5-Hydroxytryptophan As
Substrates In Rats. Jpn. J. Pharmacol. 32, 803-811 (1982)

58
Rajesh R. , A.S. Girija: Pyridoxine-dependent seizures: a review,
Indian pediatrics 40 633-638. (2003)
Ruiz A., J. Garcia-Villoria, A. Ormazabal, J. Zschocke, M. Fiol, A. Navarro-Sastre, R. Artuch, M. A. Vilaseca and A. Ribes: A new fatal case of pyridox(am)ine 5'-phosphate oxidase (PNPO) deficiency.
Mol Genet Metab, 93(2), 216-8 (2008)
Rupsingh R., Borrie M., Smith M., Wells J. L. And Bartha R. : Reduced Hippocampal Glutamate In Alzheimer Disease.
Neurobiol. Aging. 32, 802-810. (2011)
Safo M. K., Musayev F. N., Di Salvo M. L., Hunt S., Claude J. B. And Schirch V. : Crystal Structre Of Pyridoxal Kinase From The Escherichia Coli Pdxk Gene: Implications For The Classification Of Pyridoxal Kinases.
J. Bacteriol. 188, 4542- 4552. (2006)
Said H. M. : Recent Advances In Carrier-Mediated Intestinal Absorption Of Water-Soluble Vitamins.
Annu. Rev. Physiol. 66, 419-446. (2004)
Salazar P, Tapia R : Seizure induced by intracerebral administration of pyridoxal-5'-phosphate: effect of GABAergic drugs and glutamate receptor antagonist.
Neuropharmacology 41: 546-553 (2001)
Salhany J. M. , P. B. Rauenbuehler and R. L. Sloan: Characterization of pyridoxal 5'-phosphate affinity labeling of band 3 protein. Evidence for allosterically interacting transport inhibitory subdomains.
J Biol Chem, 262(33), 15965-73 (1987)
Salhany JM, SchopferLM: Pyridoxal 5-phosphate binds specifically to soluble CD4 protein, the HIV-1 receptor.
J Biol Chem 268:7643–7645.(1993)
Sandyk, R. And Pardeshi, R.: Pyridoxine Improves Drug-Induced Parkinsonism And Psychosis In A Schizophrenic Patient.
Int. J. Neurosci. 52, 225-232 (1990)
Scharer, G., Brocker, C., Vasiliou, V., Creadon-Swindell, G., Gallagher, R. C., Spector, E. And Van Hove, J. L.: The Genotypic And Phenotypic Spectrum Of Pyridoxine-Dependent Epilepsy Due To Mutations In Aldh7a1.
J. Inherit. Metab. Dis. 33, 571-581. (2010)
Schrag, A. : Psychiatric Aspects Of Parkinson's Disease--An Update.
J. Neurol. 251, 795-804. (2004)
Scott K, Zeris S, Kothari MJ : Elevated B6 levels and peripheral neuropathies.
Electromyogr. Clin. Neurophysiol. 48: 219-223 (2008)
Sherwin A.L.: Neuroactive Amino Acid in focally epileptic human brain: a review.
Neurochem. Res. 24: 13871395 (1999)

59
Sigrell JA, Cameron AD, Jones TA, Mowbray SL :
Structure of Escherichia coli ribokinase in complex with ribose and dinucleotide determined to 1.8 Å resolution: insights into e new family of kinase
structures. Structure 6: 183-193 (1998)
Snell EE : In transaminases.
Christen P, Metzler DE Eds, Wiley-Inter-science. NY. Pp 19-30 (1985)
Stanulovic M., Jeremic V., Leskovac V. And Chaykin S. : New Pathway Of Conversion Of Pyridoxal To 4-Pyridoxic Acid.
Enzyme. 21, 357-369 (1976)
Stockler, S., Plecko, B., Gospe, S. M.,Jr, Coulter-Mackie, M., Connolly, M., Van Karnebeek, C., Mercimek-Mahmutoglu, S., Hartmann, H.,
Scharer, G., Struijs, E., Tein, I., Jakobs, C., Clayton, P. And Van Hove, J. L.:
Pyridoxine Dependent Epilepsy And Antiquitin Deficiency Clinical And Molecular Characteristics And
Recommendations For Diagnosis, Treatment And Follow-Up. Mol. Genet. Metab. /J.Ymgme.2011.05.014 (2011)
Surtees R. A., Mills, P. B., And Clayton, P. :
Inborn Errors Affecting Vitamin B6 Metabolism. Future Medicine. 1, 615-620. (2006)
Tagaya M, Yamano K, Fukui T :
Kinetic studies of pyridoxal kinase from pig liver: slow binding inhibition by adenosine tetraphosphopyridoxal. Biochemistry 28: 4670-4675 (1989)
Walker V., G. A. Mills, S. A. Peters and W. L. Merton:
Fits, pyridoxine, and hyperprolinaemia type II.
Arch Dis Child, 82(3), 236-7 (2000)
Wei J., Wu J.Y.;
Post-translational regulation of L-glutamic acid decarboxylase in the brain Neurochem. Res.33: 1459-1465 (2008)
Yang E. S. And Schirch V. :
Tight Binding Of Pyridoxal 5'-Phosphate To Recombinant Escherichia Coli Pyridoxine 5'-Phosphate Oxidase. Arch. Biochem. Biophys. 377, 109-114. (2000)
Yang Y, Tsui HCT, Man TK, Winkler ME : Identification and function of the PdxY gene, which encodes a novel pyridoxal kinase involved in the salvage pathway of pyridoxal 5'-phosphate
biosynthesis in Escherichia coli K-12.
J. Bacteriol. 180: 1814-1821 (1998)
Zhang Y, Dougherty M, Downs DM, Ealick SE :
Crystal structure of an aminoimidazole riboside kinase from Salmonella enterica: implications for evolution of the ribokinase superfamily. Structure 12: 1809-1821 (2004)
Zhao G. And Winkler M. E. :
Kinetic Limitation And Cellular Amount Of Pyridoxine (Pyridoxamine) 5'-Phosphate Oxidase Of Escherichia Coli K-12. J. Bacteriol. 177, 883-891 (1995)