targeting the subtypes of breast cancer: rethinking investigational drugs
TRANSCRIPT

1. Introduction
2. Luminal A and luminal B breast
cancer
3. New targeted therapies for the
treatment of HER2-positive
breast cancer
4. Ductal triple-negative breast
cancer
5. Conclusion
6. Expert opinion
Review
Targeting the subtypes of breastcancer: rethinking investigationaldrugsGiuseppe Curigliano†, Marzia Locatelli, Luca Fumagalli, Janaina Brollo,Elisabetta Munzone, Franco Nole, Carmen Criscitiello & Aron Goldhirsch†European Institute of Oncology, Division of Medical Oncology, Department of Medicine, Milan,
Italy
Introduction: The choice of adjuvant treatments for women with breast can-
cer is based on several features that take into account the heterogeneity of
the disease. Questions raised during the decision process include the follow-
ing: i) What leads to the use of endocrine therapy? ii) What leads to the use
of anti-HER2 therapy? iii) What justifies the use of chemotherapy?.
Areas covered: Choices of adjuvant treatment are based on parameters
defined by molecular characterization of breast cancer subtypes or by approx-
imations to this classification using traditional clinical--pathological features.
Clinicians should consider cases within the various distinct subpopulation
in order to properly select the most ‘personalized’ adjuvant therapeutic
approach. Sensitivity to chemotherapy and/or targeted agents in subtypes
of breast cancers are predictable based on gene pathway alterations and asso-
ciated gene products. This review covers several clinical data on several inves-
tigational agents for early-stage breast cancer molecular subtypes. We
selected from literature data prospective Phase I, II and III clinical trials of che-
motherapy (weekly or daily schedules), including indicators of activity and
toxicity and data on survival/mortality.
Expert opinion: The future of many investigational therapeutics in breast
cancer is linked to our ability to identify the most druggable target in
each subtype.
Keywords: breast cancer subtypes, early breast cancer, investigational drugs
Expert Opin. Investig. Drugs (2012) 21(2):191-204
1. Introduction
Molecular characterization tools allow the identification of multiple breast cancersubtypes [1-3]. Any subtype is characterized by specific molecular events and canbe also defined by a traditional immunohistochemical approach in breast cancersubtype approximations [4-7]. Molecular subtypes have different risk factors [8,9], nat-ural histories [10-12] and different sensitivity to systemic chemotherapy and targetedagents [13-16].
Clinical decisions in systemic adjuvant therapy for patients with early breast can-cer must address three distinct questions: i) what justifies the use of endocrine ther-apy, ii) what justifies the use of anti-HER2 therapy, and iii) what justifies the use ofchemotherapy? Integration of adjuvant investigational agents should address thesethree major questions.
The availability of next-generation human genomic sequencing tools and prog-ress in sequencing and bio-computational technologies will enable genome-wide investigation of somatic mutations in human breast cancers [17] at diagnosisand during their natural history. These studies have the potential to highlightunderlying mechanisms of metastasis and resistance to drugs. Genome-wide
10.1517/13543784.2012.651456 © 2012 Informa UK, Ltd. ISSN 1354-3784 191All rights reserved: reproduction in whole or in part not permitted
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y M
cMas
ter
Uni
vers
ity o
n 05
/02/
13Fo
r pe
rson
al u
se o
nly.

sequencing studies will, therefore, identify two specifictypes of mutations: the ‘drivers,’ providing a survival- andproliferation-selective advantage, and the ‘passengers,’ neutralto the selection process [18-20]. It will be essential to identify allmolecular pathways that emphasize the heterogeneity andcomplexity of human breast cancer in order to explain mecha-nisms sustaining proliferation hallmarks of cancer and ‘drive’tumor progression and resistance to chemotherapy andtargeted agents.Disease segmentation in subtypes can offer insights to person-
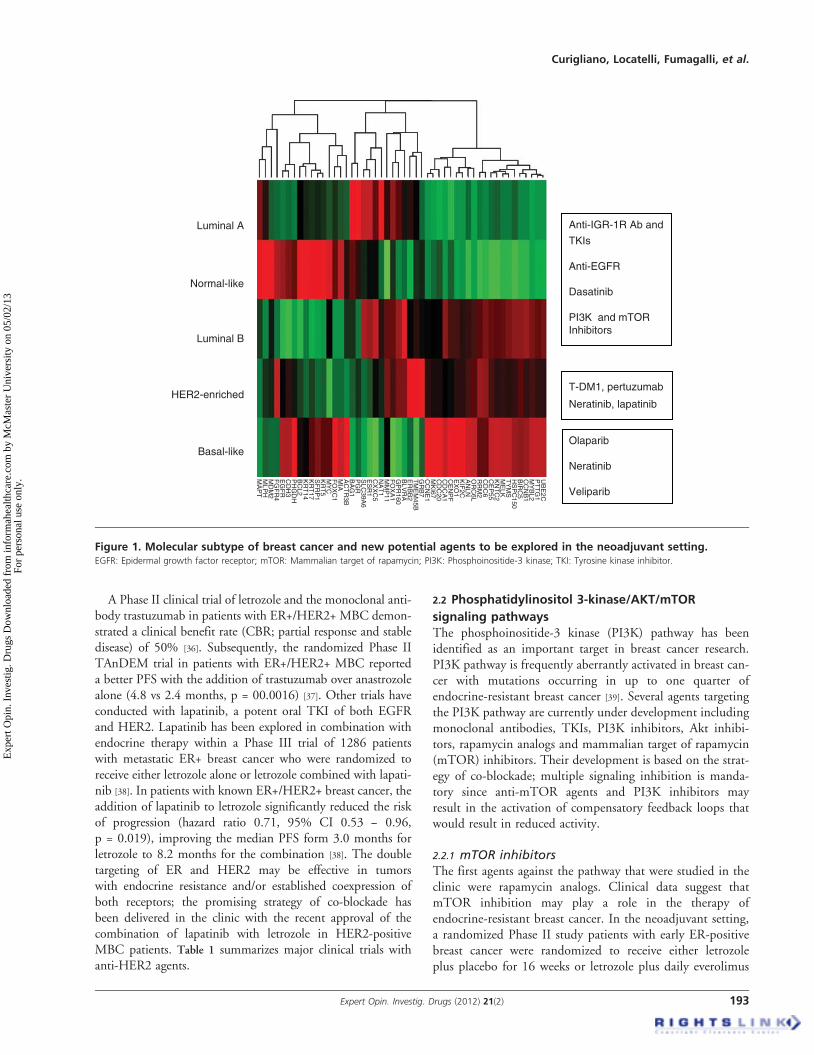
alize treatment [21]. Subtypes defined by clinical--pathological cri-teria are similar to but not identical to intrinsic subtypes andrepresent a convenient approximation. The St. Gallen approachuses immunohistochemical definition of estrogen (ER) and pro-gesterone receptor (PgR), the detection of overexpression and/oramplification of the human epidermal growth factor receptor 2(HER2) oncogene, and Ki-67 labeling index, a marker of cellproliferation, as the means of identifying tumor subtypes [21].Surrogate definition of intrinsic subtype can identify i) luminalA breast cancer, ER and PgR positive with a low Ki-67;ii) luminal B/HER2 negative, ER and PgR positive with highKi-67; iii) luminal B/HER2 positive; iv) HER2 positive, withER and PgR absent and v) ductal triple negative, ER, PgR andHER2 negative. We will overview all new promising moleculesunder investigation in breast cancer subtypes (Figure 1).
2. Luminal A and luminal B breast cancer
Endocrine therapy is the standard of care for hormone receptor-positive breast cancer. Tamoxifen alone for postmenopausal, orovarian function suppression plus tamoxifen as reasonable in pre-menopausal, is the primary option for patient with luminal Abreast cancer [21]. A substantial proportion of patients, despitebeing ER and/or PR positive, are either primarily resistant to
hormone therapies or will develop hormone resistance duringthe course of their disease. Signaling through complex growthfactor receptor pathways, which activate the ER, is emerging asimportant causes of endocrine resistance.
Several new targeted agents in pipeline are actually in develop-ment for targeting signaling pathways in patients with luminal Bbreast cancer. Clinical clue mechanisms to understand resistanceto endocrine therapy can be related to loss of ER expression [22],decrease in ER level over time; gradual loss of E dependence [23]
and upregulation of several transcriptional pathways [24]. Inpatients with endocrine-responsive disease, a ‘real-time’ testingof tumor tissue for genotype sequencing could be ideal. Earlydrug response and development of acquired resistance shouldbe monitored by serial biopsy of the tumor or, noninvasively,by circulating tumor cell analysis [25]. Ongoing clinical trialswith signal transduction inhibitors in combination with hor-monalmanipulation as ameans to overcome endocrine resistancein patients with breast cancer will be reviewed.
2.1 EGFR pathwaySeveral early clinical trials have been conducted with theepidermal growth factor receptor (EGFR) tyrosine kinaseinhibitors (TKIs) gefitinib or erlotinib either alone or in com-bination with endocrine therapy. Results from the monother-apy Phase II studies with gefitinib in patients with advancedbreast cancer were all relatively disappointing [26-28]. Two otherPhase II studies explored the potential benefit for combiningeither gefitinib or erlotinib with an aromatase inhibitor in unse-lected patients with ER-positive advanced breast cancer withvery low clinical efficacy [29,30]. In the setting of neoadjuvanttherapy for ER-positive postmenopausal breast cancer, a ran-domized trial of anastrozole alone or in combination withgefitinib given for 3 months prior to surgery showed noimprovement in tumor response rate or antiproliferative effectas determined by Ki-67 [31]. On the other hand, a preoperativetrial of gefitinib versus gefitinib combined with anastrozole for4 -- 6 weeks prior to surgery in women with ER+ EGFR+ pri-mary breast cancer reported that combined treatment inducedthe greatest reduction in tumor cell proliferation [32]. A -double-blind placebo-controlled Phase II trial of tamoxifenwith or without gefitinib was conducted in 290 patients asfirst-line endocrine therapy in postmenopausal women withER-positive metastatic breast cancer (MBC) [33], with anincrease in progression-free survival (PFS) from 8.8 to10.9 months (hazard ratio 0.84, 95% confidence interval (CI)0.59 -- 1.18, p = 0.31) [33]. A second randomized trial ofgefitinib and anastrozole versus anastrozole alone in a similarfirst-line patient population of women with ER-positiveadvanced breast cancer reported a prolongation of PFSfrom a median of 8.2 months with anastrozole to 14.6 monthswith the combination (hazard ratio 0.55, 95% CI0.32 -- 0.94) [34]. A second randomized Phase II trial with thesame combination of gefitinib and anastrozole did not showany statistically significant benefit [35]. Table 1 summarizesmajor clinical trials with anti-EGFR-targeted agents.
Article highlights.
. Breast cancer is not a single disease. Genetic array toolscan define several subtypes.
. Specific biological processes and distinct gene pathwaysare associated with prognosis and sensitivity tochemotherapy and targeted agents in different subtypesof breast cancers.
. A primary challenge for future drug development inbreast cancer will be to distinguish genes and pathwaysthat ‘drive’ cancer proliferation (drivers) from genes andpathways that have no role in the development ofcancer (passengers).
. The identification of functional pathways that areenriched for mutated genes will select subpopulationof patients who will most likely be sensitive tobiology-driven targeted agents.
. The selection of driver pathways in resistant tumors willpermit to discover a biology-driven platform for newdrug development to overcome resistance.
This box summarizes key points contained in the article.
Targeting the subtypes of breast cancer: rethinking investigational drugs
192 Expert Opin. Investig. Drugs (2012) 21(2)
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y M
cMas
ter
Uni
vers
ity o
n 05
/02/
13Fo
r pe
rson
al u
se o
nly.
A Phase II clinical trial of letrozole and the monoclonal anti-body trastuzumab in patients with ER+/HER2+ MBC demon-strated a clinical benefit rate (CBR; partial response and stabledisease) of 50% [36]. Subsequently, the randomized Phase IITAnDEM trial in patients with ER+/HER2+ MBC reporteda better PFS with the addition of trastuzumab over anastrozolealone (4.8 vs 2.4 months, p = 00.0016) [37]. Other trials haveconducted with lapatinib, a potent oral TKI of both EGFRand HER2. Lapatinib has been explored in combination withendocrine therapy within a Phase III trial of 1286 patientswith metastatic ER+ breast cancer who were randomized toreceive either letrozole alone or letrozole combined with lapati-nib [38]. In patients with known ER+/HER2+ breast cancer, theaddition of lapatinib to letrozole significantly reduced the riskof progression (hazard ratio 0.71, 95% CI 0.53 -- 0.96,p = 0.019), improving the median PFS form 3.0 months forletrozole to 8.2 months for the combination [38]. The doubletargeting of ER and HER2 may be effective in tumorswith endocrine resistance and/or established coexpression ofboth receptors; the promising strategy of co-blockade hasbeen delivered in the clinic with the recent approval of thecombination of lapatinib with letrozole in HER2-positiveMBC patients. Table 1 summarizes major clinical trials withanti-HER2 agents.
2.2 Phosphatidylinositol 3-kinase/AKT/mTOR
signaling pathwaysThe phosphoinositide-3 kinase (PI3K) pathway has beenidentified as an important target in breast cancer research.PI3K pathway is frequently aberrantly activated in breast can-cer with mutations occurring in up to one quarter ofendocrine-resistant breast cancer [39]. Several agents targetingthe PI3K pathway are currently under development includingmonoclonal antibodies, TKIs, PI3K inhibitors, Akt inhibi-tors, rapamycin analogs and mammalian target of rapamycin(mTOR) inhibitors. Their development is based on the strat-egy of co-blockade; multiple signaling inhibition is manda-tory since anti-mTOR agents and PI3K inhibitors mayresult in the activation of compensatory feedback loops thatwould result in reduced activity.
2.2.1 mTOR inhibitorsThe first agents against the pathway that were studied in theclinic were rapamycin analogs. Clinical data suggest thatmTOR inhibition may play a role in the therapy ofendocrine-resistant breast cancer. In the neoadjuvant setting,a randomized Phase II study patients with early ER-positivebreast cancer were randomized to receive either letrozoleplus placebo for 16 weeks or letrozole plus daily everolimus
Anti-IGR-1R Ab and
TKIs
Anti-EGFR
Dasatinib
PI3K and mTORInhibitors
T-DM1, pertuzumab
Neratinib, lapatinib
Olaparib
Neratinib
Veliparib
Luminal A
Luminal B
HER2-enriched
Basal-like
UB
E2C
PT
TG
1M
YB
L2C
CN
B1
BIR
C5
HS
PC
150T
YM
S
KN
TC
2C
EP
55C
DC
6R
RM
2O
RC
6LA
NLN
KIF
2CE
XO
1C
EN
PF
CD
CA
1C
DC
20M
KI67
CC
NE
1G
RB
7T
ME
M45B
ER
BB
2B
LVR
AG
PR
160F
OX
A1
MM
P11
NA
T1
CX
XC
5E
SR
1S
LC39A
6P
GR
BA
G1
AC
TR
3BM
IAF
OX
C1
MY
CK
RT
5S
FR
P1
KR
T17
KR
T14
BC
L2P
HG
DH
CD
H3
EG
FR
FG
FR
4M
DM
2M
LPH
MA
PT
ME
LK
Normal-like
Figure 1. Molecular subtype of breast cancer and new potential agents to be explored in the neoadjuvant setting.EGFR: Epidermal growth factor receptor; mTOR: Mammalian target of rapamycin; PI3K: Phosphoinositide-3 kinase; TKI: Tyrosine kinase inhibitor.
Curigliano, Locatelli, Fumagalli, et al.
Expert Opin. Investig. Drugs (2012) 21(2) 193
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y M
cMas
ter
Uni
vers
ity o
n 05
/02/
13Fo
r pe
rson
al u
se o
nly.
(RAD001), a rapamycin analog. The primary endpoint of thetrial was response rate to the combination therapy [40].Response rate by clinical palpation in the everolimus armwas higher than that with letrozole alone (i.e., placebo;68.1 vs 59.1%). An antiproliferative response, as defined bya reduction in Ki-67 expression to natural logarithm ofpercentage positive Ki-67 of less than 1 at day 15, occurredin 52 (57%) of 91 patients in the everolimus arm and in25 (30%) of 82 patients in the placebo arm (p <. 01) [40]. Inanother trial in the metastatic setting, patients (n = 109)were randomly assigned to receive 75 or 250 mg of temsiroli-mus (monoclonal antibody against mTOR) weekly [41].Patients were evaluated for tumor response, time to tumorprogression, adverse events (AEs) and pharmacokinetics oftemsirolimus. Temsirolimus produced an objective responserate of 9.2% (10 partial responses) in the intent-to-treat pop-ulation. Median time to tumor progression was 12.0 weeks.Efficacy was similar for both dose levels but toxicity wasmore common with the higher dose level [41]. Ridaforolimus(MK-8669) in combination to exemestane is actually underinvestigation in early clinical trials for ER-positive breastcancer progressing to nonsteroidal aromatase inhibitors.
2.2.2 PI3K inhibitorsPan-PI3K inhibitors include GDC-0941 (Genentech, Inc.),which is under investigation in a Phase I -- II clinical trial,in combination with endocrine therapy and mTOR inhibi-tors. The XL147 agent is a potent inhibitor of the class IPI3K family. A Phase I/II randomized study of letrozole andXL147 versus letrozole and XL765 (Exelixis/Sanofi-Aventis),a dual mTOR and PI3K inhibitors, is also planned.
2.3 Histone deacetylaseThe use of demethylating agents or histone deacetylase(HDAC) inhibitors can reactivate expression of a functionalER in cell lines in which ER silencing exists because of pro-moter methylation [42]. HDACs are crucial components ofthe ER transcriptional complex. Preclinically, HDACinhibitors can reverse tamoxifen/aromatase inhibitor resis-tance in hormone receptor-positive breast cancer [42]. In aPhase II trial, patients with ER-positive MBC progressingon endocrine therapy were treated with 400 mg of vorino-stat (Merck & Co., Inc., White House Station, New Jersey)daily for 3 of 4 weeks and 20 mg/day tamoxifen, continu-ously. The objective response rate was 19% and the CBRwas 40%. The median response duration was 10.3 months(CI: 8.1 -- 12.4) [43].
2.4 Insulin-like growth factor receptorInsulin-like growth factor-1 receptor (IGF-1R) is a homodi-meric receptor tyrosine kinase activated by IGF I/II ligand bind-ing, which results in tumor growth and apoptosis blockade [44].IGF-1R antagonists have been shown to interact with both ERpathways. This cross talk between ER suggests that IGF-1Rmay be an attractive treatment target especially for the ‘luminalT
able
1.Endocrinetherapiesco
mbinedwithbiologicaltargetedagents
inestrogenreceptor(ER)-positivebreast
cancer.
Clinicalsetting
Trialphase
No.ofpatients
Intervention
Clinicalendpoints
Ref.
Metastaticbreast
cancer
Phase
IIn=15
Anastrozole
andgefitinib
Response
rate
Noresponse
Nostable
disease
MitaM
etal.
[29]
Phase
IIn=150
Letrozole
andgefitinib
Clinicalbenefit11/20patients
MayerIetal.
[30]
Earlybreast
cancer
Phase
IIrandomized
n=206
Anastrozole
vsgefitinib
plus
anastrozole
Response
rate
61%
anastrozole
vs45%
(combinationarm
)p=0.067
Smithetal.
[31]
Metastaticbreast
cancer
Phase
IIIrandomized
n=207
Anastrozole
vstrastuzumabplus
anastrozole
PFS
=2.4
(anastrozole)vs
4.8
months
(anastrozole
plustrastuzumab)p=0.0016
Kaufm
anBetal.
[37]
Metastaticbreast
cancer
Phase
IIIrandomized
n=219
Letrozole
vsletrozole
pluslapatinib
PFS
=3.0
(letrozole)vs
8.2
months(letrozole
pluslapatinib)
JohnsonSetal.
[38]
Metastaticbreast
cancer
Randomizedphase
IIn=150
Tamoxifenvs
tamoxifenplusgefitinib
PFS
=8.8
(tamoxifen)vs
10.9
months
(tamoxifenplusgefitinib)
OsborneCK
[33]
Randomizedphase
IIn=206
Anastrozole
plusgefitinib
vsanastrozole
PFS
=14.6
(anastrozole
plusgefitinib)vs
8.2
months(anastrozole)
CristofanilliM
[34]
Targeting the subtypes of breast cancer: rethinking investigational drugs
194 Expert Opin. Investig. Drugs (2012) 21(2)
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y M
cMas
ter
Uni
vers
ity o
n 05
/02/
13Fo
r pe
rson
al u
se o
nly.

B’ breast cancers. This is supported by in vitro experimentsshowing a synergistic effect when co-targeting the IGF-1Rreceptor along with antiestrogen agent [45]. Moreover, growthof tamoxifen-resistant MCF-7 cells declines when anti-IGF-1Rantibody is added to the cells [46]. Several monoclonal antibodiesand TKIs are in early clinical development in the treatment ofbreast cancer. Phase II randomized trials are currently ongoingfor patients progressing to nonsteroidal aromatase inhibitorsand randomized to exemestane versus exemestane with figitu-mumab (Pfizer, Inc.), a fully humanized anti-IGF-1R antibody.In a recent randomized Phase II study, the investigational agentAMG 479 (Amgen, Inc.), a fully human monoclonal antibodyagainst the IGF-1R, failed to revert resistance to hormonal ther-apy in patients with endocrine therapy-resistant, ER-positiveMBC [47]. Indeed, the drug showed a trend toward worse PFSand objective response in a Phase II trial [47]. When AMG479 was paired with exemestane or fulvestrant, patients in theexperimental arm had a median PFS of 3.9 months, comparedwith 5.7 months for patients on exemestane or fulvestrant alone(hazard ratio, 1.17; p =. 435). In this study, AMG 479 in com-bination with either fulvestrant or exemestane does not appear todelay or reverse resistance to hormonal therapy in this popula-tion of patients with prior endocrine therapy-resistant hormonereceptor-positive MBC. Other trials are currently ongoing inhormone-resistant breast cancer patents using TKIs targetingthe IGF-1R pathway.
2.5 Src family tyrosine kinaseSrc is specifically involved in coordinating signaling from the ste-roid receptors, including the ER and androgen receptor (AR).Multiple studies have shown cross talk between ER/AR andSrc, with ER/AR activation leading to activation of Src, and sub-sequent Src-mediated cell proliferation [48]. Blocking the interac-tion between ER/AR and Src leads to inhibition of downstreamcellular pathways and cessation of cell growth [48]. Several studieshave shown associations between resistance to endocrine therapyand both increased levels of Src activity and an increasingly inva-sive and aggressive tumor phenotype [49,50]. Given this data, spe-cifically targeting Src may overcome endocrine resistance inhormonally driven cancers. Several inhibitors of Src have beendeveloped. One of the best studied is dasatinib (Sprycel,BMS354825; Bristol-Myers Squibb Oncology). Dasatinib is apotent oral small-molecule inhibitor of the Src tyrosine kinase.Another agent, bosutinib (SKI-606, Wyeth), is an oral dual-selective competitive inhibitor of both Src (IC50 = 1.0 nmol/l)and Abl tyrosine kinases, with moderate inhibition of the Axltyrosine kinase, Eph receptors and Ste20 family kinases [51].Mul-tiple other agents with activity against Src, including saracatinib(AZD0530; AstraZeneca) and XL999 (Exelixis), are in preclini-cal or early-phase clinical development. A Phase II monotherapystudy was open to patients with both ER-positive and/or HER2-positive disease. Of the response-evaluable population from bothsubtypes, a response rate of 4% was seen, with a CBR of 8% inthe HER2+ cohort, and 16% in the ER+ cohort. Interestingly,all benefit was seen in patients with ER+ tumors [52]. Another
Phase II randomized trial was designed for patients withER-positive MBC progressing to nonsteroidal aromatase inhibi-tors. Patients are randomized to exemestane plus dasatinib versusexemestane plus placebo. The accrual to this trial has just beencompleted. Progesterone receptors also interact with c-Src. PRcontains an SH3 domain interaction motif in the N-terminusthat mediates interaction with Src tyrosine kinases and othersignaling molecules. This interaction mediates rapid progester-one activation of Src/MAP K signaling pathways and defines amolecularmechanism for some of the rapid non-genomic actionsof progesterone. Interactions of PR with c-Src or proteinkinases downstream of c-Src are required for breast cancer cellproliferation [53].
3. New targeted therapies for the treatmentof HER2-positive breast cancer
Many breast cancer patients with HER2 overexpression donot respond to initial therapy with trastuzumab (Herceptin,Roche), and a vast majority of these develop resistance tothis monoclonal antibody. Several molecular mechanismsleading to the development of trastuzumab resistance havebeen described, including circulating HER2 extracellulardomain [54], loss of PTEN [55], activation of alternative path-ways (e.g., IGFR) [56], receptor--antibody interaction block [57]
or innate modulation of the immunological response [58].Identification of upregulated pathways may lead to develop-ment on new therapeutic targets that potentially overcomeresistance to trastuzumab. Several agents are currently underdevelopment to overcome trastuzumab resistance.
3.1 Trastuzumab-DM1Genentech, Inc., in collaboration with Roche, has recentlydeveloped trastuzumab-DM1 (a maytansine conjugated to tras-tuzumab, RG-3502, T-DM1), which is active on HER2 overex-pressing breast cancer and also on trastuzumab-refractorytumors. Maytansinoids are very potent anticancer agents origi-nally isolated from plant families: Celastraceae, Rhamnaceaeand Euphorbiaceae and later from microorganism producingantibiotics (Actinosynnema pretiosum) [59,60]. They are 19-mem-bered microcyclic lactams related to amsamycin. The maytansi-noid DM1 is 100- to 1000-fold more potent that anticanceragents in clinical use [59,60]. The maytansine DM1 bindsto microtubules in a manner similar to Vinca alkaloids, but is20- to 100-fold more potent than vincristine in blockingmitosis [59]. Therefore, the maytansinoid DM1 was conjugatedto the humanized HER2 antibody trastuzumab (Tmab, whichis a protein) using --S-S- (disulfide)-containing linkers(Tmab-SPDT-DM1, Tmab-SPP-DM1, Tmab-SSNPPDM1,Tmab-SSNPP-DM4) (Figure 2).
The first-in-human Phase I, multicenter, open-label, dose-escalation study of single-agent T-DM1 in patients withHER2-positive MBC, who had previously received atrastuzumab-containing chemotherapy regimen, demonstratedthat, at the maximum-tolerated dose (MTD) of 3.6 mg/kg every
Curigliano, Locatelli, Fumagalli, et al.
Expert Opin. Investig. Drugs (2012) 21(2) 195
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y M
cMas
ter
Uni
vers
ity o
n 05
/02/
13Fo
r pe
rson
al u
se o
nly.

3 weeks, T-DM1 was safe and had considerable clinicalactivity. The CBR (RR plus stable disease (SD) at 6 months)among 15 patients treated at MTD was 73%, including fiveobjective responses [61]. Phase II studies of T-DM1 in patientswith HER2-positive MBC who progressed while receivingHER2--directed therapy (trastuzumab or lapatinib), or whowere previously treated with several lines of chemotherapy,have demonstrated an objective response rate, by independentassessment, of 25.9% (95% CI 18.4 -- 34.4%). Median dura-tion of response was not reached as a result of insufficient events(lower limit of 95% CI 6.2 months), and median PFS time was4.6 months (95% CI 3.9 -- 8.6 months) [62]. Several randomizedclinical trials are actually ongoing in metastatic HER2-positivebreast cancer patients. An open-label, Phase III trial (EMILIA)will compare the safety and efficacy of T-DM1 with that ofcapecitabine in combination with lapatinib in patients withHER2-positive MBC previously treated with a trastuzumab-based therapy [63]. Another first-line trial (MARIANNE) is cur-rently ongoing for the treatment of MBC [64]. This randomized,three-arm, multicenter study will evaluate the efficacy and safetyof trastuzumab-DM1 with pertuzumab or T-DM1 with pertu-zumab--placebo, versus the combination of trastuzumab plustaxane (docetaxel or paclitaxel) in patients with HER2-positiveprogressive or recurrent locally advanced or previously untreatedMBC. Patients will be randomized to one of three treatmentarms (arms A, B or C). Arm A will be open-label, whereasarms B and C will be blinded [64].
3.2 PertuzumabPertuzumab is a novel recombinant humanized monoclonalantibody directed against the highly conserved dimerizationdomain of HER2, and as such, it inhibits HER2 homo- andheterodimerization. Pertuzumab-mediated blockage of HER2dimerization inhibits HER family downstream signaling (i.e.,the Akt cell survival pathway and the mitogen-activated proteinkinase pathway) [65]. In a Phase II randomized trial
investigating the efficacy and safety of pertuzumab in patientswith HER2-positive MBC, the only measurable therapeuticbenefit observed was a stable disease of a relatively short dura-tion [66]. The idea that the combination of pertuzumab andtrastuzumab might be a clinically meaningful therapy inMBC came from the single-arm Phase II trial of trastuzumabplus pertuzumab, which demonstrated that the combinationwas well tolerated and active in patients with HER2-positiveMBC who had progressed during trastuzumab therapy [67]. Inthis trial, the objective response rate was 24.2%, and theCBR was 50%. Cardiac dysfunction was minimal, and nopatients withdrew as a result of cardiac-related AEs. Recentlythe combination of pertuzumab and trastuzumab has beentested in patients with HER2-positive first diagnosed earlybreast cancer. The NEOSPHERE study (Neoadjuvant Studyof Pertuzumab and Herceptin in an Early Regimen Evaluation)is a randomized multicenter Phase III study, which was con-ducted in 417 women with newly diagnosed HER2-positiveearly, inflammatory or locally advanced breast cancer whonever received trastuzumab. Prior to surgery (neoadjuvant treat-ment), these women were randomized to four study arms. Theprimary endpoint was pathological complete response (pCR)and the results were i) Arm A: pCR of 29% for trastuzumaband docetaxel; ii) arm B: pCR of 45.8% for trastuzumab, per-tuzumab and docetaxel; iii) arm C: pCR of 16.8% for trastuzu-mab and pertuzumab; and iv) arm D: pCR of 24 % forpertuzumab and docetaxel [68]. The findings of the NEO-SPHERE study suggested that this new approach was effectivefor early HER2-positive breast cancer and suggest a potentialapplication of the double targeting approach.
3.3 LapatinibIntegration of lapatinib in early breast cancer has been investi-gated in the Neoadjuvant Lapatinib and/or Trastuzumab Treat-ment Optimisation Study (Neo-ALTTO study). This is arandomized, open-label multicenter Phase III study comparingthe efficacy of neoadjuvant lapatinib plus paclitaxel, versus trastu-zumab plus paclitaxel, versus concomitant lapatinib and trastuzu-mab plus paclitaxel given as neoadjuvant treatment in HER2/ErbB2 overexpressing and/or amplified primary breast cancer.Patients have been randomized to receive either lapatinib1500 mg/day, trastuzumab 4 mg/kg i.v. load followed by2 mg/kg i.v. weekly or lapatinib 1000 mg/day with trastuzumab4 mg/kg i.v. load followed by 2 mg/kg i.v. weekly for a total of6 weeks. After this biological window, patients on monotherapyarms continued on the same targeted therapy plus weekly pacli-taxel 80 mg/m2 for a further 12 weeks, up to definitive surgery.In the combination arm, patients received lapatinib 750 mg/day in combination with trastuzumab 2 mg/kg i.v. plus weeklypaclitaxel 80 mg/m2 i.v. for a further 12 weeks, up to definitivesurgery. After surgery, patients received three courses of adjuvantchemotherapy with fluorouracil, epirubicin and cyclophospha-mide followed by the same targeted therapy as in the biologicalwindow of the neoadjuvant setting for a further 34 weeks (inthe combination arm, lapatinib dose will be 1000 mg/day in
Trastuzumab
DM1MCC
Figure 2. T-DM1: maytansinoid DM1 was conjugated to the
humanized HER2 antibody trastuzumab.
Targeting the subtypes of breast cancer: rethinking investigational drugs
196 Expert Opin. Investig. Drugs (2012) 21(2)
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y M
cMas
ter
Uni
vers
ity o
n 05
/02/
13Fo
r pe
rson
al u
se o
nly.

combination with trastuzumab). The planned total duration ofthe anti-HER2 therapy was 1 year. Results of this study havebeen presented during the SanAntonio 2010Breast Cancer Sym-posium [69]. In this study 455 patients have been randomized.The primary endpoint of pCR at the time of surgery in theITT population was statistically significantly higher in subjectsreceiving lapatinib plus trastuzumab with paclitaxel comparedwith those receiving trastuzumab with paclitaxel. The pCR inthe lapatinib plus trastuzumab group was 51.32% comparedwith 29.53% in the trastuzumab group; unstratified binomialp-value was p < 0.0001. No difference in pCR was observedbetween lapatinib with paclitaxel and trastuzumabwith paclitaxel(24.68 vs 29.53%). When adjusting for the stratification factors(tumor size, clinical nodal status, ER/PgR status, candidate forconservative surgery), the pCR remained higher in the lapatinibplus trastuzumab group compared with trastuzumab. The ORwas 2.62 (97.5% CI 1.50, 4.58; stratified log rank p < 0.001).When considering the secondary endpoint of locoregional pCR(extended definition of pCR including regional lymph nodestatus of pN0), the rate was also higher in the lapatinib plus tras-tuzumab group compared with trastuzumab (43.66 vs 26.53%).No difference in locoregional pCR was observed between lapati-nib and trastuzumab (18.12 vs 26.53%). In the ITT population,94.2, 90.8 and 85.9% of subjects reported AEs in the lapatinib,lapatinib plus trastuzumab and trastuzumab groups, respectively.More subjects in the lapatinib and lapatinib plus trastuzumabgroups reported serious adverse events (SAEs) compared withtrastuzumab (39 (25.3%) and 34 (22.4%) subjects, respectively,vs 9 (6.0%)). The most frequently reported SAEs in thelapatinib-containing treatment groups were hypertransaminase-mia, diarrhea and hyperbilirubinemia. No clinically significantcardiac dysfunction occurred and there were no deaths duringthe neoadjuvant treatment phase.
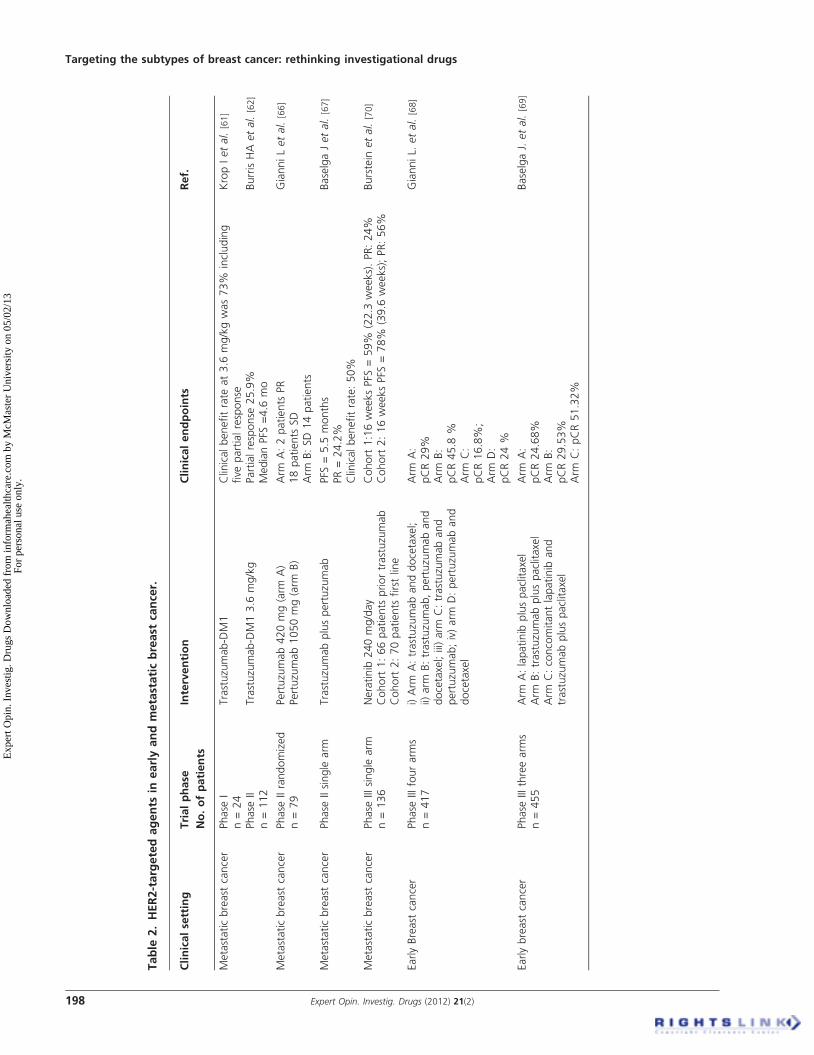
3.4 NeratinibNeratinib, is an orally available pan-ErbB TKI, differing inthat it inhibits HER4 as well as HER1/EGFR andHER2 [70]. The efficacy and safety of neratinib were evaluatedin a trial including two cohorts of patients with advancedErbB2-positive breast cancer, and those with and those with-out prior trastuzumab treatment, in an open-label, multicen-ter, Phase II trial. The 16-week PFS rates were 59% forpatients with prior trastuzumab treatment and 78% forpatients with no prior trastuzumab treatment. Median PFSwas 22.3 and 39.6 weeks, respectively. Objective responserates were 24% among patients with prior trastuzumab treat-ment and 56% in the trastuzumab-naive cohort [70]. Emergingagents for treatment of HER2-resistant tumors are reportedin Table 2.
4. Ductal triple-negative breast cancer
Numerous transcriptional pathways are under investigation todetermine how best to target therapies to specific mutations ormolecular events in basal-like breast cancers. Each one of these
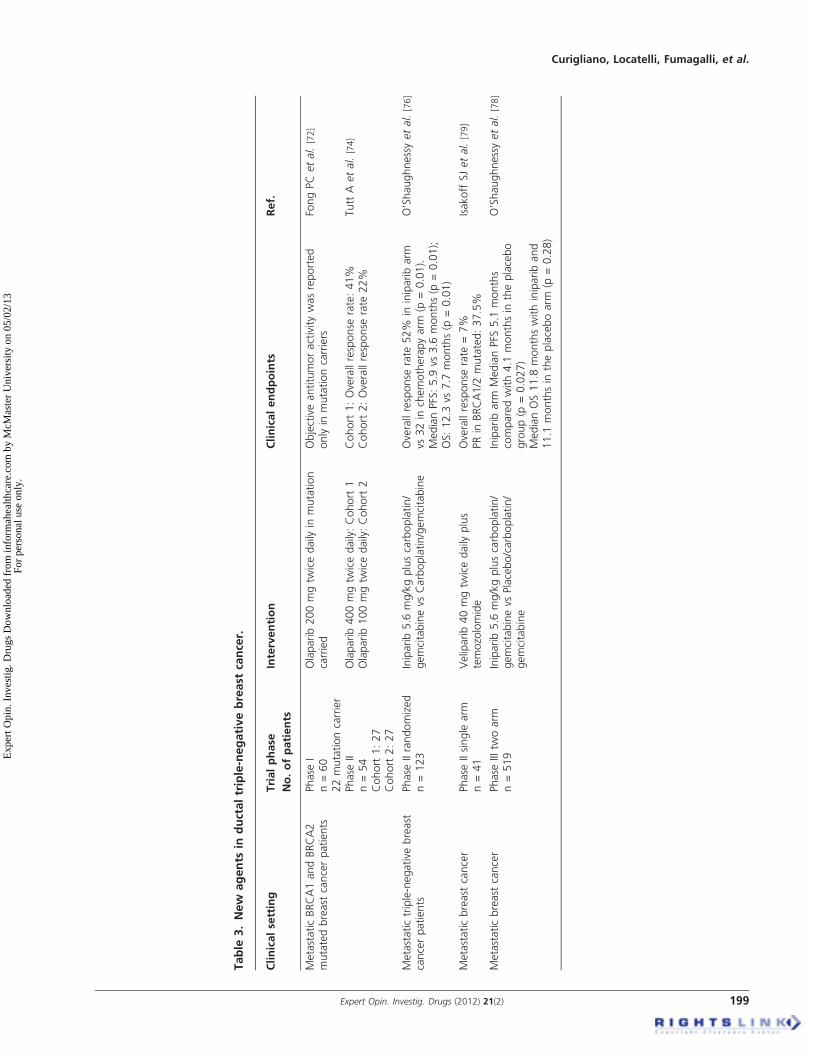
pathways will require careful investigation to assess howimportant therapeutic interventions along this pathway willbe. Table 3 summarizes clinical data of new agents in ductaltriple-negative breast cancer (TNBC).
4.1 Poly-(adenosine diphosphate (ADP)--ribose)
polymerases pathwayDNA lesions such as single-strand breaks (SSBs) and double-strand breaks (DSBs) are common by products of normal cellularmetabolism and may also result from exposure to harmful envi-ronmental agents. Briefly four DNA repair mechanisms areresponsible for repairing these lesions: i) base-excision repair(BER), ii) nucleotide-excision repair (NER), iii) mismatch repair(MMR) and iv) recombinational repair (with homologousrecombination and nonhomologous end joining (NHEJ)) [71].When SSBs occur, they are repaired using the intact complemen-tary strand as a template by BER, NER and MMR. A key com-ponent of the BER pathway, PARP1, is the most importantmember of the poly-(adenosine diphosphate (ADP)--ribose)polymerase (PARP) family of enzymes [72,73]. PARP inhibitionleads to accumulation of DNA SSBs and subsequent DNADSBs at replication forks. These breaks normally are repairedvia the homologous recombination double-strandedDNA repairpathway, major components of which are the tumor-suppressorproteins BRCA1 and BRCA2 [73]. PARP1 is upregulated differ-entially in primary breast cancers, including ERnegative, PRneg-ative, HER2 negative (ductal triple negative). Preclinical studiesof in vitro activity of PARP inhibitors demonstrated inhibitionof tumor cell growth only if the cell was BRCA deficient [73].Inhibition of PARP to kill tumor cells selectively, therefore, is anovel approach to cancer therapy.
4.1.1 OlaparibOlaparib, a novel, orally active PARP inhibitor, induced syn-thetic lethality in BRCA-deficient cells. A proof-on-concept trialin BRCA-mutated patients assessed the efficacy, safety and toler-ability of olaparib alone in women with advanced breast can-cer [74]. Patients had been given a median of three previouschemotherapy regimens (range 1 -- 5 in cohort 1, and 2 -- 4 incohort 2). Response rate was 11 (41%) of 27 patients (95% CI25 -- 59) in the cohort assigned to 400 mg twice daily, and six(22%) of 27 (11 -- 41) in the cohort assigned to 100 mg twicedaily [74]. The results of this study provide positive proof of con-cept for PARP inhibition in BRCA-deficient breast cancers andshow a favorable therapeutic index for a novel targeted treatmentstrategy in patients with tumors that have genetic loss of functionof BRCA1-associated or BRCA2-associated DNA repair.Phase I studies are currently ongoing combining cisplatin andolaparib. Agents such as platinum salts bind to DNA directlyand result in the formation of DNA-platinum adducts and, con-sequently, intrastrand and interstrand DNA cross-links thatimpede cell division. As a consequence, cisplatinmay be an effec-tive treatment for patients with hereditary BRCA1-mutatedbreast cancers. Because sporadic TNBC and BRCA1-associatedbreast cancer share features suggesting common pathogenesis, a
Curigliano, Locatelli, Fumagalli, et al.
Expert Opin. Investig. Drugs (2012) 21(2) 197
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y M
cMas
ter
Uni
vers
ity o
n 05
/02/
13Fo
r pe
rson
al u
se o
nly.
Table
2.HER2-targetedagents
inearlyandmetastaticbreast
cancer.
Clinicalsetting
Trialphase
No.ofpatients
Intervention
Clinicalendpoints
Ref.
Metastaticbreast
cancer
Phase
In=24
Trastuzumab-DM1
Clinicalbenefitrate
at3.6
mg/kgwas73%
including
five
partialresponse
KropIetal.
[61]
Phase
IIn=112
Trastuzumab-DM13.6
mg/kg
Partialresponse
25.9%
MedianPFS
=4.6
mo
BurrisHA
etal.
[62]
Metastaticbreast
cancer
Phase
IIrandomized
n=79
Pertuzumab420mg(arm
A)
Pertuzumab1050mg(arm
B)
Arm
A:2patients
PR
18patients
SD
Arm
B:SD14patients
GianniLetal.
[66]
Metastaticbreast
cancer
Phase
IIsingle
arm
Trastuzumabpluspertuzumab
PFS
=5.5
months
PR=24.2%
Clinicalbenefitrate:50%
BaselgaJetal.
[67]
Metastaticbreast
cancer
Phase
IIIsingle
arm
n=136
Neratinib
240mg/day
Cohort1:66patients
priortrastuzumab
Cohort2:70patients
firstline
Cohort1:16weeksPFS
=59%
(22.3
weeks).PR:24%
Cohort2:16weeksPFS
=78%
(39.6
weeks);PR:56%
Burstein
etal.
[70]
EarlyBreast
cancer
Phase
IIIfourarm
sn=417
i)Arm
A:trastuzumabanddocetaxel;
ii)arm
B:trastuzumab,pertuzumaband
docetaxel;iii)arm
C:trastuzumaband
pertuzumab;iv)arm
D:pertuzumaband
docetaxel
Arm
A:
pCR29%
Arm
B:
pCR45.8
%Arm
C:
pCR16.8%
;Arm
D:
pCR24%
GianniL.
etal.
[68]
Earlybreast
cancer
Phase
IIIthreearm
sn=455
Arm
A:lapatinib
pluspaclitaxel
Arm
B:trastuzumabpluspaclitaxel
Arm
C:concomitantlapatinib
and
trastuzumabpluspaclitaxel
Arm
A:
pCR24.68%
Arm
B:
pCR29.53%
Arm
C:pCR51.32%
BaselgaJ.etal.
[69]
Targeting the subtypes of breast cancer: rethinking investigational drugs
198 Expert Opin. Investig. Drugs (2012) 21(2)
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y M
cMas
ter
Uni
vers
ity o
n 05
/02/
13Fo
r pe
rson
al u
se o
nly.
Table
3.New
agents
inductaltriple-negativebreast
cancer.
Clinicalsetting
Trialphase
No.ofpatients
Intervention
Clinicalendpoints
Ref.
MetastaticBRCA1andBRCA2
mutatedbreast
cancerpatients
Phase
In=60
22mutationcarrier
Olaparib200mgtw
icedaily
inmutation
carried
Objectiveantitumoractivitywasreported
onlyin
mutationcarriers
FongPCetal.
[72]
Phase
IIn=54
Cohort
1:27
Cohort
2:27
Olaparib400mgtw
icedaily:Cohort1
Olaparib100mgtw
icedaily:Cohort2
Cohort1:Overallresponse
rate:41%
Cohort2:Overallresponse
rate
22%
TuttA
etal.
[74]
Metastatictriple-negative
breast
cancerpatients
Phase
IIrandomized
n=123
Iniparib5.6
mg/kgpluscarboplatin/
gemcitabinevs
Carboplatin/gemcitabine
Overallresponse
rate
52%
ininiparibarm
vs32in
chemotherapyarm
(p=0.01).
MedianPFS:5.9
vs3.6
months(p
=0.01);
OS:12.3
vs7.7
months(p
=0.01)
O’Shaughnessyetal.
[76]
Metastaticbreast
cancer
Phase
IIsingle
arm
n=41
Veliparib40mgtw
icedaily
plus
temozolomide
Overallresponse
rate
=7%
PRin
BRCA1/2
mutated:37.5%
IsakoffSJetal.
[79]
Metastaticbreast
cancer
Phase
IIItw
oarm
n=519
Iniparib5.6
mg/kgpluscarboplatin/
gemcitabinevs
Placebo/carboplatin/
gemcitabine
Iniparibarm
MedianPFS
5.1
months
comparedwith4.1
monthsin
theplacebo
group(p
=0.027)
MedianOS11.8
monthswithinipariband
11.1
monthsin
theplaceboarm
(p=0.28)
O’Shaughnessyetal.
[78]
Curigliano, Locatelli, Fumagalli, et al.
Expert Opin. Investig. Drugs (2012) 21(2) 199
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y M
cMas
ter
Uni
vers
ity o
n 05
/02/
13Fo
r pe
rson
al u
se o
nly.

neoadjuvant trial of cisplatin in TNBC was conducted [75]. Six(22%) of twenty-eight patients achieved pathologic completeresponses, including both patients with BRCA1 germline muta-tions; 18 (64%) patients had a clinical complete or partialresponse in the BRCA1 mutation group. These backgrounddata suggest that combination of PARP inhibitors and cisplatincan be potentially very active.
4.1.2 IniparibIniparib (previously BSI 201) (4-iodo-3-nitrobenzamide) is adrug that acts as an irreversible inhibitor of PARP1 (hence, itis a PARP inhibitor) and possibly other enzymes throughcovalent modification [76]. An open-label, Phase II study tocompare the efficacy and safety of gemcitabine and carbopla-tin with or without iniparib, a small molecule with PARP-inhibitory activity, in patients with metastatic TNBC, wasconducted. A total of 123 patients were randomly assignedto receive gemcitabine (1000 mg/m2 of body surface area)and carboplatin (at a dose equivalent to an area under theconcentration-time curve of 2) on days 1 and 8 -- with orwithout iniparib (at a dose of 5.6 mg/kg of body weight) ondays 1, 4, 8 and 11 -- every 21 days. Primary endpoints werethe rate of clinical benefit (i.e., the rate of objective response(complete or partial response) plus the rate of stable diseasefor ‡ 6 months) and safety. Additional endpoints includedthe rate of objective response, PFS and overall survival [76].The addition of iniparib to gemcitabine and carboplatinimproved the rate of clinical benefit from 34 to 56%(p = 0.01) and the rate of overall response from 32 to 52%(p = 0.02). The addition of iniparib also prolonged themedian PFS from 3.6 to 5.9 months (hazard ratio for progres-sion, 0.59; p = 0.01) and the median overall survival from7.7 to 12.3 months (hazard ratio for death, 0.57;p = 0.01) [76]. Another large randomized trial included519 women with metastatic TNBC. Patients were random-ized to receive a standard chemotherapy regimen (gemcitabineand carboplatin) on days 1 and 8 of each 21-day cycle, with orwithout iniparib 5.6 mg/kg, which was administered on days1, 4, 8 and 11 of each 21-day cycle [77]. Patients in the studyhad received up to two previous lines of chemotherapy in ametastatic setting. The co-primary endpoints were overall sur-vival and PFS. Recently final data of this study have been pre-sented at ASCO 2011 meeting. Authors announced that thetrial did not meet the prespecified criteria for significancefor co-primary endpoints of overall survival and PFS [78].The Phase III trial involved 519 women with metastaticTNBC, treated with two or fewer metastatic regimens. Allpatients received gemcitabine and carboplatin and were ran-domized to iniparib or placebo. Patients who progressed onplacebo could cross over to the iniparib arm, and 96% ofpatients in the placebo arm subsequently crossed over. Thetrial had co-primary endpoints of overall survival and PFS,and the study would be considered positive if either endpointfavored iniparib. Secondary endpoints included objectiveresponse rate, safety and tolerability. The study population
had a median age of 53 -- 54, and all had good performancestatus. O’Shaughnessy reported that 56 -- 57% of patientswere receiving first-line therapy for metastatic disease, and43 -- 44% were being treated in second or third line. Patientsin the placebo group had a median disease-free interval of15 months compared with 12 months for the iniparib group.The rates and types of treatment-emergent AEs were similarbetween the two groups, suggesting that iniparib did notadd appreciably to the anticipated toxicity of the chemother-apy. When the trial ended, the iniparib arm had a medianPFS of 5.1 months compared with 4.1 months in the placebogroup. The associated probability value (p = 0.027) did notmeet the prespecified definition of p = 0.01. Median overallsurvival was 11.8 months with iniparib and 11.1 months inthe placebo arm (p = 0.28). Analysis of secondary endpointsshowed an objective response rate of 34% in the iniparibgroup and 30% in the placebo arm. The two groups had sim-ilar rates of complete and partial responses. An exploratoryanalysis of the primary endpoints by treatment history showedthat patients receiving first-line metastatic therapy had similaroverall survival and PFS. By contrast, patients in second- andthird-line therapy had a median PFS of 4.2 months with ini-parib and 2.5 months with placebo, a difference that trans-lated into a hazard ratio of 0.57 in favor of iniparib. Medianoverall survival was 10.8 months in the iniparib arm and8.1 months in the placebo arm, representing a 35% reductionin the hazard [78].
4.1.3 VeliparibVeliparib (ABT-888) is a potent inhibitor of both PARP-1and PARP-2 [78]. Preclinical studies showed that temozolo-mide potentiation by PARP inhibition occurs in TNBC [79].A single-arm Phase II trial of veliparib in combination withtemozolomide was conducted in patients who had receivedat least one prior regimen for MBC [79]. Patients received veli-parib (40 mg twice daily on days 1 -- 7) and oral temozolo-mide (150 mg/m2/day on days 1 -- 5) every 28 days;temozolomide was increased to 200 mg/m2 as tolerated. Theprimary endpoint was objective response rate and secondaryendpoints were PFS, CBR and safety and tolerability. Ofthe 41 patients, 23 had TNBC. Objective response was 7%(one complete response and two partial responses; 95% CI2 -- 20%). The rate of stable disease at 16 weeks was 10%(four patients). When response among all BRCA1/BRCA2 mutation carriers was determined, objective responsewas 37.5% (one complete response and two partial responses)and CBR was 62.5% (n = 5) [79].
5. Conclusion
The ‘wiring diagrams’ of breast cancer subtypes define thatthe signaling circuitry describing the intercommunicationbetween various pathways should be charted in far greaterdetail and clarity, in order to better understand ‘drivers’ and‘passengers.’ We continue to foresee breast cancer research as
Targeting the subtypes of breast cancer: rethinking investigational drugs
200 Expert Opin. Investig. Drugs (2012) 21(2)
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y M
cMas
ter
Uni
vers
ity o
n 05
/02/
13Fo
r pe
rson
al u
se o
nly.

an increasingly ‘computational’ science, in which in silicomodels should predict underlying pathways that sustain can-cer progression and proliferation. The selection of patientsfor targeted therapy remains a challenge, because we lack reli-able biomarkers to predict activity for most of the targetedagents. Traditional methodologies applied for drug develop-ment may be inappropriate for new targeted agents. Resis-tance to many of traditional and new drugs is a majorclinical challenge. The use of high-throughput technologieswill help us to understand the molecular biology of signalingpathways as the roads of the ‘genomic landscape’ of breastcancer. The number of potential driver genes is large, even ifmore limited is the number of ‘driver’ pathways. Patientselection, rational combination therapies, surrogate markersidentification and tumor tissue banking will be key areasof research.
6. Expert opinion
Clinical research in the field of breast cancer should be appliedfor the future to specific subtypes of breast cancer. Trials of thepast did not follow such concept of heterogeneity, using extrap-olation of data from retrospective analyses, frequently difficultand imprecise. Future research should achieve the goal to recog-nizing the diversity of targets in each subtype of breast cancer,taking advantage from molecular characterization tools. Newprospective trials will specifically address the questions of tar-geting multiple pathways in each breast cancer subtype, to max-imize response to treatment and minimize the toxicity. Recentlarge-scale tumor sequencing studies, including wide genomeanalysis studies, have identified a number of mutations thatmight be involved in breast cancer tumorigenesis. Analysis ofthe frequency of specific mutations across different tumorshas been able to identify some, but not all of the mutated genesthat contribute to tumor initiation and progression. One reasonfor this is that other functionally important genes are likely tobe mutated more rarely and only in specific contexts. Thus,for example, mutation in one member of a collection of func-tionally related genes may result in the same net effect, and/or mutations in certain genes may be observed less frequentlyif they play functional roles in later stages of tumor develop-ment, such as metastasis. The biggest challenge for the futurewill be to apply a network reconstruction and coexpressionmodule identification-based approach to identify functionally
related gene modules targeted by somatic mutations in cancer.The ultimate goal of this approach is to identify network ofpathways and potential cross talks within pathways. Dual ormultiple targeting in order to shut down ‘driver’ pathwayswill be the future of breast cancer treatment within several sub-types. This method was applied to available breast cancersequence data and identified several pathways as targets ofrare driver mutations in breast. These mutations do not appearto alter genes that play a central role in these pathways, butrather contribute to a more refined shaping or ‘tuning’ of thefunctioning of these pathways in such a way as to result inthe inhibition of their tumor-suppressive signaling arms andthereby conserve or enhance tumor-promoting processes. Webelieve a gene network reconstruction strategy-based approachcan successfully identify cancer driver mutations throughenrichment of mutations within modules. We should high-light a few important caveats in the field. Next-generationsequencing technologies used to reconstruct genetic networkscan be altered to generate networks of different sizes, or toreflect different coexpression relationships, depending on theinvestigator requirements and/or sample size and likelihoodthat module enrichment will be observed in different-sizedmodules. Specifically genome remodeling during cancerprogression or upon resistance to therapies can upregulatepathways due to downregulation of current driver path-ways. Additionally, this approach probably does not captureall of the secondary driver mutations, which may requireeither additional complementary systems biology appro-aches or larger sample sizes to capture other mutation-enriched coexpression modules. Overall, we believe that thisapproach shows tremendous promise for the identification ofrare tumorigenic driver mutations, which is a crucial task forthe upcoming large-scale cancer resequencing projects, as it isthese more private mutations that may be driving intratumorheterogeneity, inter-patient heterogeneity and ultimately alter-ing response to therapeutic intervention. The future of manyinvestigational therapeutics in breast cancer is, therefore,linked to our ability to identify the most druggable target ineach subtype.
Declaration of interest
The authors state no conflict of interest and have received nopayment in preparation of this manuscript.
Curigliano, Locatelli, Fumagalli, et al.
Expert Opin. Investig. Drugs (2012) 21(2) 201
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y M
cMas
ter
Uni
vers
ity o
n 05
/02/
13Fo
r pe
rson
al u
se o
nly.

BibliographyPapers of special note have been highlighted as
either of interest (�) or of considerable interest(��) to readers.
1. Perou CM, Sorlie T, Eisen MB, et al.
Molecular portraits of human breast
tumours. Nature 2000;21:747-52... This is a seminal paper that describes
molecular breast cancer subtypes.
2. Prat A, Perou CM. Deconstructing the
molecular portraits of breast cancer.
Mol Oncol 2011;5:5-23
3. Parker JS, Mullins M, Cheang MCU,
et al. Supervised risk predictor of breast
cancer based on intrinsic subtypes.
J Clin Oncol 2009;27:1160-7
4. Nielsen TO, Hsu FD, Jensen K, et al.
Immunohistochemical and clinical
characterization of the basal--like subtype
of invasive breast carcinoma.
Clin Cancer Res 2004;10:5367-74
5. Blows FM, Driver KE, Schmidt MK,
et al. Subtyping of breast cancer by
immunohistochemistry to investigate a
relationship between subtype and short
and long term survival: a collaborative
analysis of data for 10,159 cases from
12 studies. PLoS Med 2010;7:e1000279
6. Hugh J, Hanson J, Cheang MC, et al.
Breast cancer subtypes and response to
docetaxel in node-positive breast cancer:
use of an immunohistochemical
definition in the BCIRG 001 trial.
J Clin Oncol 2009;27:1168-76
7. Cheang MCU, Chia SK, Voduc D, et al.
Ki67 index, HER2 status, and prognosis
of patients with luminal B breast cancer.
J Natl Cancer Inst 2009;101:736-50
8. Millikan RC, Newman B, Tse CK, et al.
Epidemiology of basal-like breast cancer.
Breast Cancer Res Treat
2008;109:123-39
9. Phipps AI, Chlebowski RT, Prentice R,
et al. Body size, physical activity, and
risk of triple negative and estrogen
receptor-positive breast cancer.
Cancer Epidemiol Biomarkers Prev
2011;20:454-63
10. Phipps AI, Buist DS, Malone KE, et al.
Reproductive history and risk of three
breast cancer subtypes defined by three
biomarkers. Cancer Causes Control
2011;22:399-405
11. Liedtke C, Mazouni C, Hess KR, et al.
Response to neoadjuvant therapy and
long-term survival in patients with
triple-negative breast cancer.
J Clin Oncol 2008;26:1275-81
12. Dignam JJ, Dukic V, Anderson SJ, et al.
Hazard of recurrence and adjuvant
treatment effects over time in lymph
node-negative breast cancer.
Breast Cancer Res Treat
2009;116:595-602
13. Aebi S, Sun Z, Braun D, et al.
Differential efficacy of three cycles of
CMF followed by tamoxifen in patients
with ER-positive and ER-negative
tumors: long-term follow up on IBCSG
Trial IX. Ann Oncol
2011. [Epub ahead of print]
14. Albain KS, Barlow WE, Shak S, et al.
Prognostic and predictive value of the
21-gene recurrence score assay in
postmenopausal women with
node-positive, oestrogen receptor-positive
breast cancer on chemotherapy:
a retrospective analysis of a randomised
trial. Lancet Oncol 2010;11:55-65
15. Nguyen PL, Taghian AG, Katz MS,
et al. Breast cancer subtype approximated
by estrogen receptor, progesterone
receptor, and HER-2 is associated with
local and distant recurrence after
breast-conserving therapy. J Clin Oncol
2008;26:2373-8
16. Sotiriou C, Pusztai L. Gene-expression
signatures in breast cancer. N Engl
J Med 2009;360:790-800
17. Ding L, Ellis MJ, Li S, et al. Genome
remodelling in a basal-like breast cancer
metastasis and xenograft. Nature
2010;464(7291):999-1005.. This is an important manuscript to
understand mechanisms of
genome remodeling.
18. Vogelstein B, Kinzler KW. Cancer genes
and the pathways they control. Nat Med
2004;10(8):789-99
19. Parmigiani G, Boca S, Lin J, et al.
Design and analysis issues in
genome-wide somatic mutation studies of
cancer. Genomics 2009;93(1):17:1-8. The manuscript provides important
highlights on statistical design of
genome-wide analysis studies.
20. Hanahan D, Weinberg RA. Hallmarks of
Cancer: the next generation. Cell
2011;144(5):646-74... Seminal papers in the field of
cancer pathways.
21. Goldhirsch A, Wood WC, Coates AS,
et al. Panel members. Strategies for
subtypes--dealing with the diversity of
breast cancer: highlights of the St. Gallen
International Expert Consensus on the
Primary Therapy of Early Breast Cancer
2011. Ann Oncol 2011;22(8):1736-47
22. Curigliano G, Bagnardi V, Viale G, et al.
Should liver metastases of breast cancer
be biopsied to improve treatment choice?
Ann Oncol 2011. [Epub ahead of print
23. Kuukasjarvi T, Kononen J, Helin H,
et al. Loss of estrogen receptor in
recurrent breast cancer is associated with
poor response to endocrine therapy.
J Clin Oncol 1996;14:2584-9
24. Martin LA, Farmer I, Johnston SR, et al.
Enhanced estrogen receptor (ER) alpha,
ERBB2, and MAPK signal transduction
pathways operate during the adaptation
of MCF-7 cells to long term estrogen
deprivation. J Biol Chem
2003;278:30458-68
25. Haber DA, Gray NS, Baselga J. The
evolving war on cancer. Cell
2011;145(1):19-24
26. Albain K, Elledge R, Gradishar WJ,
et al. Open-label phase II multicenter
trial of ZD1839 (Iressa) in patients with
advanced breast cancer. Breast Cancer
Res Treat 2002;76:A20
27. Baselga J, Albanelli J, Ruiz A, et al.
Phase II and tumor pharmacodynamic
study of gefitinib in patients with
advanced breast cancer. J Clin Oncol
2005;23:5323-33
28. Robertson JFR, Gutteridge E,
Cheung KL, Cheung Eet al. Gefitinib
(ZD1839) is active in acquired tamoxifen
(TAM) resistant oestrogen receptor (ER)-
positive and ER-negative breast cancer:
results from a phase II study. Proc Annu
Meet Am Soc Clin Oncol 2003;22:7
29. Mita M, Bono J, Mita A. A phase II and
biologic correlative study investigating
anastrozole (A) in combination with
gefitinib (G) in postmenopausal patients
with estrogen receptor positive (ER)
metastatic breast carcinoma (MBC) who
have previously failed hormonal therapy.
Breast Cancer Res Treat
2005;94:abstract 1117
30. Mayer I, Ganja N, Shyr Y, et al.
A phase II trial of letrozole plus erlotinib
in post-menopausal women with
hormone-sensitive metastatic breast
Targeting the subtypes of breast cancer: rethinking investigational drugs
202 Expert Opin. Investig. Drugs (2012) 21(2)
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y M
cMas
ter
Uni
vers
ity o
n 05
/02/
13Fo
r pe
rson
al u
se o
nly.

cancer: preliminary results of toxicities
and correlative studies. Breast Cancer
Res Treat 2006;100:abstract 4052
31. Smith IE, Walsh G, Skene A, et al.
A phase II placebo-controlled trial of neo
adjuvant anastrozole alone or with
gefitinib in early breast cancer.
J Clin Oncol 2007;25:3816-22
32. Polychronis A, Sinnet HD,
Hadjiminas D, et al. Pre-operative
gefitinib versus gefitinib and anastrozole
in postmenopausal patients with
oestrogen-receptor positive and epidermal
growth factor receptor positive primary
breast cancer: a double blind
placebo-controlled phase II randomised
trial. Lancet Oncol 2005;6:383-91
33. Osborne CK, Dirix L, Mackey J, et al.
Randomized Phase II study of gefitinib
(IRESSA) or placebo in combination
with tamoxifen in patients with hormone
receptor positive metastatic breast cancer.
Breast Cancer Res Treat
2007;106(Suppl):abstract 2067
34. Cristofanilli M, Valero V, Mangalik A,
et al. Phase II, randomized trial to
compare anastrozole combined with
gefitinib or placebo in postmenopausal
women with hormone receptor-positive
metastatic breast cancer. Clin Cancer Res
2010;16(6):1904-14
35. Mauriac L, Cameron D, Dirix L, et al.
Results of randomised phase II trial
combining Iressa (gefitinib) and
Arimidex in women with
advanced breast cancer. EORTC
protocol 10021. Cancer Res
2009;69(Supp l2):abstract 6133
36. Marcom PK, Isaacs C, Harris L, et al.
The combination of letrozole and
trastuzumab as first or second-line
biological therapy produces durable
responses in a subset of HER2 positive
and ER positive advanced breast cancers.
Breast Cancer Res Treat 2007;102:43-9
37. Kaufman B, Mackey JR, Clemens MR,
et al. Trastuzumab plus anastrozole
versus anastrozole alone for the treatment
of postmenopausal women with human
epidermal growth factor receptor
2-positive, hormone receptor-positive
metastatic breast cancer: results form the
randomized TAnDEM study.
J Clin Oncol 2009;27:5529-37
38. Johnston S, Pippen J Jr, Pivot X, et al.
Lapatinib combined with letrozole versus
letrozole and placebo as first-line therapy
for postmenopausal
hormone-receptor-positive metastatic
breast cancer. J Clin Oncol
2009;27:5538-46
39. Stemke-Hale K, Gonzalez-Angulo AM,
Lluch A, et al. An integrative genomic
and proteomic analysis of PIK3CA,
PTEN, and AKT mutations in breast
cancer. Cancer Res 2008;68:6084-91
40. Baselga J, Semiglazov V, van Dam P,
et al. Phase II randomized study of
neoadjuvant everolimus plus letrozole
compared with placebo plus letrozole in
patients with estrogen receptor-positive
breast cancer. J Clin Oncol
2009;27:2630-7
41. Chan S, Scheulen ME, Johnston S, et al.
Phase II study of temsirolimus (CCI-
779), a novel inhibitor of mTOR, in
heavily pretreated patients with locally
advanced or metastatic breast cancer.
J Clin Oncol 2005;23(23):5314-22
42. Ferguson AT, Lapidus RG, Baylin SB,
et al. Demethylation of the estrogen
receptor gene in estrogen
receptor-negative breast cancer cells can
reactivate estrogen receptor gene
expression. Cancer Res 1995;55:2279-83
43. Munster PN, Thurn KT, Thomas S,
et al. A phase II study of the histone
deacetylase inhibitor vorinostat combined
with tamoxifen for the treatment of
patients with hormone therapy-resistant
breast cancer. Br J Cancer
2011;104(12):1828-35
44. Fagan DH, Yee D. Crosstalk between
IGF1R and estrogen receptor signaling in
breast cancer. J Mammary Gland
Biol Neoplasia 2008;13:423-9
45. Chakraborty AK, Welsh A,
Digiovanna MP. Co-targeting the
insulin-like growth factor I receptor
enhances growth-inhibitory and
pro-apoptotic effects of anti-estrogens in
human breast cancer cell lines.
Breast Cancer Res Treat
2010;120:327-35
46. Weroha SJ, Haluska P. IGF-1 receptor
inhibitors in clinical trials----early lessons.
J Mammary Gland Biol Neoplasia
2008;13:471-83
47. Kaufman PA, Ferrero JM, Bourgeois H,
et al. A randomized, double-blind,
placebo-controlled, phase 2 study of
AMG 479 with exemestane (E) or
fulvestrant (F) in postmenopausal women
with hormone-receptor positive (HR+)
metastatic (M) or locally advanced (LA)
breast cancer (BC). Cancer Research
2010;70:76s
48. Migliaccio A, Di Domenico M,
Castoria G, et al. Tyrosine kinase/p21ras/
MAP-kinase pathway activation by
estradiol-receptor complex in
MCF-7 cells. EMBO J
1996;15:1292-300
49. Hiscox S, Morgan L, Green TP, et al.
Elevated Src activity promotes cellular
invasion and motility in tamoxifen
resistant breast cancer cells. Breast Cancer
Res Treat 2006;97:263-74
50. Riggins RB, Thomas KS, Ta HQ, et al.
Physical and functional interactions
between Cas and c-Src induce tamoxifen
resistance of breast cancer cells through
pathways involving epidermal growth
factor receptor and signal transducer and
activator of transcription 5b. Cancer Res
2006;66:7007-15
51. Bantscheff M, Eberhard D, Abraham Y,
et al. Quantitative chemical proteomics
reveals mechanisms of action of clinical
ABL kinase inhibitors. Nat Biotechnol
2007;25:1035-44
52. Mayer EL, Baurain JF, Sparano J, et al.
A phase 2 trial of dasatinib in patients
with advanced HER2-positive and/or
hormone receptor-positive breast cancer.
Clin Cancer Res 2011 Nov 1;
17(21):6897-904
53. Edwards DP, Wardell SE,
Boonyaratanakornkit V. Progesterone
receptor interacting coregulatory proteins
and cross talk with cell signaling
pathways. J Steroid Biochem Mol Biol
2002;83(1-5):173-86
54. Zabrecky JR, Lam T, McKenzie SJ, et al.
The extracellular domain of p185/neu is
released from the surface of human
breast carcinoma cells, SK-BR-3.
J Biol Chem 1991;266:1716-20
55. Nagata Y, Lan KH, Zhou X, et al.
PTEN activation contributes to tumor
inhibition by trastuzumab, and loss of
PTEN predicts trastuzumab resistance in
patients. Cancer Cell 2004;6:117-27
56. Lu Y, Zi X, Zhao Y, et al. Insulin-like
growth factor-I receptor signaling and
resistance to trastuzumab (Herceptin).
J Natl Cancer Inst 2001;93:1852-7
57. Nagy P, Friedlander E, Tanner M, et al.
Decreased accessibility and lack of
activation of ErbB2 in JIMT-1, a
herceptin-resistant, MUC4-expressing
Curigliano, Locatelli, Fumagalli, et al.
Expert Opin. Investig. Drugs (2012) 21(2) 203
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y M
cMas
ter
Uni
vers
ity o
n 05
/02/
13Fo
r pe
rson
al u
se o
nly.

breast cancer cell line. Cancer Res
2005;65:473-82
58. Musolino A, Naldi N, Bortesi B, et al.
Immunoglobulin G fragment C receptor
polymorphisms and clinical efficacy of
trastuzumab-based therapy in patients
with HER-2/neu-positive metastatic
breast cancer. J Clin Oncol
2008;26:1789-96
59. Tin-Wein Y, Bai L, Clade D, et al. The
biosynthetic gene cluster of the
maytansinoid antitumor agent
ansamitocin from Actinosynnema
pretiosum. PNAS 2002;12:7968-73
60. Tadayoni M, Bourret LA, Liu C, et al.
Eradication of large colon tumor
xenografts by targeted delivery of
maytansinoids. PNAS 1996;93:8618-23
61. Krop IE, Beeram M, Modi S et al.
Phase I study of trastuzumab-DM1, an
HER2 antibody-drug conjugate, given
every 3 weeks to patients with
HER2-positive metastatic breast cancer.
J Clin Oncol 2010;28:2698-704
62 Burris HA 3rd, Rugo HS, Vukelja SJ,
et al. Phase II study of the antibody drug
conjugate trastuzumab-DM1 for the
treatment of human epidermal growth
factor receptor 2 (HER2)-positive breast
cancer after prior HER2-directed
therapy. J Clin Oncol
2011;29(4):398-405
63. ClinicalTrials.gov. An Open-Label Study
of Trastuzumab-MCC-DM1 (TDM1)
vs. Capecitabine Lapatinib in Patients
With HER2-Positive Locally Advanced
or Metastatic Breast Cancer (EMILIA).
Available from: http:/clinicaltrials.gov/
show/NCT00829166, [Accessed 28 May
2011]
64. ClinicalTrials.gov. A Study of
Trastuzumab-DM1 Plus Pertuzumab
Versus Trastuzumab [Herceptin] Plus a
Taxane in Patients With Metastatic
Breast Cancer (MARIANNE). Available
from: http://clinicaltrials.gov/ct2/show/
NCT01120184, [Accessed 28 May 2011]
65. Adams CW, Allison DE, Flagella K,
et al. Humanization of a recombinant
monoclonal antibody to produce a
therapeutic HER dimerization inhibitor,
pertuzumab.
Cancer Immunol Immunother
2006;55:717-27
66. Gianni L, Llado A, Bianchi G, et al.
Open-label, phase II, multicenter,
randomized study of the efficacy and
safety of two dose levels of Pertuzumab,
a human epidermal growth factor
receptor 2 dimerization inhibitor, in
patients with human epidermal growth
factor receptor 2-negative metastatic
breast cancer. J Clin Oncol
2010;28(7):1131-7
67. Baselga J, Gelmon KA, Verma S, et al.
Phase II trial of pertuzumab and
trastuzumab in patients with human
epidermal growth factor receptor
2-positive metastatic breast cancer that
progressed during prior trastuzumab
therapy. J Clin Oncol 2010;28:1138-44
68. Gianni L, Pienkowski T, Im Y-H, et al.
Neoadjuvant pertuzumab (P) and
trastuzumab (H): antitumor and safety
analysis of a randomized phase II study
(NeoSphere)” [abstract S3-2].
San Antonio. Breast Cancer Symposium;
2010
69. Baselga J, Bradbury I, Eidtmann H, et al.
First results of the NeoALTTO trial
(BIG 01-06/ EGF 106903): A phase III,
randomized, open label, neoadjuvant
study of lapatinib, trastuzumab, and their
combination plus paclitaxel in women
with HER2-positive primary breast
cancer [abstract S3-3]. 33rd Annual
San Antonio Breast Cancer Symposium;
10 December 2010
70. Burstein HJ, Sun Y, Dirix LY, et al.
Neratinib, an irreversible ErbB receptor
tyrosine kinase inhibitor, in patients with
advanced ErbB2-positive breast cancer.
J Clin Oncol 2010;28(8):1301-7
71. Underhill C, Toulmonde M,
Bonnefoi H. A review of PARP
inhibitors: From bench to bedside.
Ann Oncol 2011;22:268-79
72. Fong PC, Boss DS, Yap TA, et al.
Inhibition of poly(ADPribose)
polymerase in tumors from
BRCA mutation carriers. N Engl J Med
2009;361:123-34
73. Farmer H, McCabe N, Lord CJ, et al.
Targeting the DNA repair defect in
BRCA mutant cells as a therapeutic
strategy. Nature 2005:434;917-21
74. Tutt A, Robson M, Garber JE, et al.
Oral poly(ADP-ribose) polymerase
inhibitor olaparib in patients with
BRCA1 or BRCA2 mutations and
advanced breast cancer:
a proof-of-concept trial. Lancet
2010;376(9737):235-44
75. Silver DP, Richardson AL, Eklund AC,
et al. Efficacy of neoadjuvant Cisplatin in
triple-negative breast cancer.
J Clin Oncol 2010;28(7):1145-53
76. O’Shaughnessy J, Osborne C, Pippen JE,
et al. Iniparib plus chemotherapy in
metastatic triple-negative breast cancer.
N Engl J Med 2011;364(3):205-14
77. ClinicalTrials.gov. Phase 3, multi-center,
open-label, randomized study of
gemcitabine/carboplatin, with or without
iniparib, in patients with ER-, PR-, and
HER2-negative metastatic breast cancer.
Available from: http://clinicaltrials.gov/
ct2/show/NCT01130259, [Accessed
28 May 2011
78. O’Shaughnessy J, Schwartzberg LS,
Danso MA, et al. A randomized
phase III study of iniparib (BSI-201) in
combination with gemcitabine/
carboplatin (G/C) in metastatic
triple-negative breast cancer (TNBC).
J Clin Oncol
2011;29(Suppl):abstract 1007
79. Isakoff SJ, Overmoyer B, Tung NM,
et al. A phase II trial of the PARP
inhibitor veliparib (ABT888) and
temozolomide for metastatic breast
cancer [abstract 1019]. J Clin Oncol
2009;28 (15S, Part I):118s
AffiliationGiuseppe Curigliano† MD PhD,
Marzia Locatelli, Luca Fumagalli, Janaina Brollo,
Elisabetta Munzone, Franco Nole,
Carmen Criscitiello & Aron Goldhirsch†Author for correspondence
Division of Medical Oncology,
Istituto Europeo di Oncologia,
Via Ripamonti 435,
20141 Milano, Italia
Tel: +39 02 57489788; Fax: +39 02 57489581;
E-mail: [email protected]
Targeting the subtypes of breast cancer: rethinking investigational drugs
204 Expert Opin. Investig. Drugs (2012) 21(2)
Exp
ert O
pin.
Inv
estig
. Dru
gs D
ownl
oade
d fr
om in
form
ahea
lthca
re.c
om b
y M
cMas
ter
Uni
vers
ity o
n 05
/02/
13Fo
r pe
rson
al u
se o
nly.