szerves kÉmia i - tankonyvtar.hu · a "szerves kémia" név berzeliustól (1806)...
TRANSCRIPT

Created by XMLmind XSL-FO Converter.
SZERVES KÉMIA I

Created by XMLmind XSL-FO Converter.
SZERVES KÉMIA I
Publication date 2005 Szerzői jog © 2005 Veszprémi Egyetemi Kiadó

iii Created by XMLmind XSL-FO Converter.
Tartalom
ELŐSZÓ ............................................................................................................................................ vi 1. TÖRTÉNETI ÁTTEKINTÉS ......................................................................................................... 1
1. A szerves kémia kialakulása; "szerves" és "szervetlen" kémia ............................................. 1 2. A szerves kémia klasszikus szerkezetelmélete ...................................................................... 1
2. A SZERVES VEGYÜLETEK SZERKEZETÉNEK ELEKTRONELMÉLETE ........................... 3 1. A szénatom "kitüntetett" helyzete a periódusos rendszerben ................................................ 3 2. A szénatom hibridállapotai ................................................................................................... 4 3. Kémiai kötés a szerves vegyületekben .................................................................................. 5
3.1. A polározott kovalens kötés ...................................................................................... 5 3.2. Elektronaffinitás és elektronegativitás ...................................................................... 6 3.3. Homonukleáris kötések: a C,C-kötések különböző típusai ...................................... 6 3.4. Heteronukleáris kötések ............................................................................................ 9
4. Elektroneltolódási effektusok .............................................................................................. 10 4.1. Induktív effektus ..................................................................................................... 10 4.2. Mezomer effektus ................................................................................................... 11
5. Molekulák közötti kölcsönhatások ...................................................................................... 12 5.1. Molekulakomplexek kialakulása ............................................................................ 12 5.2. Hidrogénhíd-kötés (protonkötés) ............................................................................ 13 5.3. van der Waals-erők ................................................................................................. 13
3. A SZERVES KÉMIAI REAKCIÓK CSOPORTOSÍTÁSA ........................................................ 15 1. Gyökös és ionos reakciók .................................................................................................... 15 2. Szubsztitúció, addíció, elimináció ....................................................................................... 16 3. Elektrofil és nukleofil reakciók ........................................................................................... 16 4. Molekularitás ...................................................................................................................... 17 5. Átmeneti állapot és közti termék ......................................................................................... 17 6. A csoportositás egyéb lehetséges szempontjai .................................................................... 18
4. IZOMÉRIA LEHETŐSÉGEK A SZERVES KÉMIÁBAN ......................................................... 20 1. Szerkezeti izomériák ........................................................................................................... 20 2. Térszerkezeti izomériák (sztereoizomériák) ........................................................................ 21 3. Geometriai izoméria ............................................................................................................ 21 4. Optikai izoméria: szimmetria és optikai aktivitás; kiralitás ................................................ 23 5. Egy aszimmetriacentrumot tartalmazó szerves vegyületek optikai izomériája ................... 25
5.1. A térszerkezet Fischer-féle ábrázolása ................................................................... 25 5.2. Relatív és abszolút konfiguráció ............................................................................. 26 5.3. A Cahn-Ingold-Prelog (C.I.P.) konvenció .............................................................. 27 5.4. Az aszimmetriacentrum reakcióközbeni viselkedése ............................................. 28
6. Két aszimmetriacentrumot tartalmazó szerves vegyületek optikai izomériája .................... 29 6.1. A két aszimmetriacentrum azonos környezetű ....................................................... 29 6.2. A két aszimmetriacentrum nem azonos környezetű ............................................... 30
7. Forgatóképesség és szerkezet .............................................................................................. 31 8. Az optikai forgatóképesség oka .......................................................................................... 32 9. Enantiomerek elválasztása .................................................................................................. 33
9.1. Mechanikus módszer .............................................................................................. 33 9.2. Biológiai módszer ................................................................................................... 33 9.3. Diasztereomer pár képzése ..................................................................................... 33 9.4. Kromatográfiás elválasztás ..................................................................................... 33
10. Aszimmetrikus szintézis .................................................................................................... 33 5. ALKÁNOK .................................................................................................................................. 35
1. Nyíltláncú alkánok; általános áttekintés .............................................................................. 35 1.1. Az alkánok homológ sora ....................................................................................... 35 1.2. A szénatomok rendűsége ........................................................................................ 36 1.3. Az alkánok elnevezése ........................................................................................... 36 1.4. Az alkánok térbeli felépítése .................................................................................. 38 1.5. Az alkánok szerkezeti izomériája ........................................................................... 41 1.6. Az alkánok fizikai tulajdonságai ............................................................................ 41
2. Alkánok kinyerése és előállítása ......................................................................................... 41

SZERVES KÉMIA I
iv Created by XMLmind XSL-FO Converter.
2.1. Előfordulás és kinyerés; kőolaj és földgáz .............................................................. 41 2.2. Egyenesláncú alkánok kinyerése kőolaj-párlatokból .............................................. 43 2.3. Alkánok előállítása szintézissel .............................................................................. 43
3. Az alkánok kémiai tulajdonságai ........................................................................................ 44 3.1. Klórozás .................................................................................................................. 44 3.2. Oxidáció ................................................................................................................. 47 3.3. Hőbomlás ................................................................................................................ 47 3.4. Izomerizálás ............................................................................................................ 47
4. Cikloalkánok ....................................................................................................................... 49 4.1. Egy-gyűrűs rendszerek ........................................................................................... 50 4.2. Többgyűrűs cikloalkánok ....................................................................................... 53
6. ALKÉNEK (olefin-szénhidrogének), A SZÉN-SZÉN KETTŐS KÖTÉS ................................... 56 1. Általános áttekintés ............................................................................................................. 56 2. Alkének előállítása .............................................................................................................. 57
2.1. Nagyipari eljárások ................................................................................................. 57 2.2. Alkének laboratóriumi előállítása ........................................................................... 59
3. Az alkének kémiai tulajdonságai ......................................................................................... 62 3.1. Reakciók halogénekkel ........................................................................................... 63 3.2. H-X típusú molekulák addiciója ............................................................................. 64 3.3. Hidrogénezés .......................................................................................................... 66 3.4. Hidroformilezés ...................................................................................................... 67 3.5. Oxidációs reakciók ................................................................................................. 68 3.6. Polimerizáció .......................................................................................................... 70 3.7. Oligomerizáció ....................................................................................................... 75 3.8. Alkilezés ................................................................................................................. 75 3.9. Izomerizálás (Z,E-izomerizáció és kettős kötés vándorlás) .................................... 76 3.10. Metatézis (alkén-diszpoporcionálódás) ................................................................ 77 3.11. Komplexképzés .................................................................................................... 78
4. Cikloalkének ....................................................................................................................... 79 4.1. Ciklomonoének ....................................................................................................... 79 4.2. Izolált kettős kötéseket tartalmazó ciklodi- és poliének ......................................... 79
7. ALKINEK (ACETILÉN SZÉNHIDROGÉNEK) ÉS KUMULÉNEK. AZ sp SZÉNATOM ÉS A SZÉN-
SZÉN HÁRMAS KÖTÉS ................................................................................................................ 81 1. Alkinek - általános áttekintés .............................................................................................. 81 2. Alkinek előállitása ............................................................................................................... 82
2.1. Acetilén ipari előállitása ......................................................................................... 82 2.2. Acetilén-homológok előállítása (a C,C hármas kötés kialakításának laboratóriumi
módszerei) ..................................................................................................................... 83 3. Az alkinek kémiája .............................................................................................................. 84
3.1. Halogénaddíció ....................................................................................................... 84 3.2. Vinilezés (HX-addíció) ........................................................................................... 84 3.3. Hidrogénezés .......................................................................................................... 85 3.4. Sóképzés ................................................................................................................. 85 3.5. Etinilezés (alkinilezés) ............................................................................................ 86 3.6. Dimerizáció ............................................................................................................ 86 3.7. Ciklooligomerizáció ............................................................................................... 87
4. Kumulált diének és poliének ............................................................................................... 87 8. KONJUGÁLT DIÉNEK ÉS A π-ELEKTRON DELOKALIZÁCIÓ ........................................... 89
1. A π-elektron delokalizáció értelmezése .............................................................................. 89 1.1. A butadién példája - az MO-megközelítés .............................................................. 89 1.2. Az allilkation példája - a VB-megközelítés ............................................................ 91 1.3. A π-elektron delokalizáció termodinamikai és kinetikai hatása ............................. 93
2. A konjugált diének legfontosabb képviselői és előállitásuk ................................................ 94 2.1. Butadién .................................................................................................................. 94 2.2. Izoprén .................................................................................................................... 94
3. A konjugált diének kémiája ................................................................................................ 95 3.1. Hidrogénezés .......................................................................................................... 95 3.2. Halogén- és HX-addíció ......................................................................................... 95 3.3. Diels-Alder-szintézis .............................................................................................. 97 3.4. Polimerizáció .......................................................................................................... 99

SZERVES KÉMIA I
v Created by XMLmind XSL-FO Converter.
4. Gyűrűs diének és poliének ................................................................................................ 102 4.1. Ciklobutadién ....................................................................................................... 102 4.2. Ciklopentadién ...................................................................................................... 103 4.3. Ciklohexadién ....................................................................................................... 105 4.4. Ciklooktatetraén - fluxionális molekulák .............................................................. 106
9. AROMÁS SZÉNHIDROGÉNEK (Arének) ............................................................................... 108 1. Általános áttekintés ........................................................................................................... 108
1.1. Az aromás szénhidrogének csoportosítása és elnevezése ..................................... 108 1.2. Az aromás szénhidrogének fizikai tulajdonságai .................................................. 112
2. Az aromás szénhidrogének molekulaszerkezete ............................................................... 112 2.1. A benzolgyűrű elektronszerkezete - a VB-megközelítés ...................................... 113 2.2. Az aromás kötésrendszer stabilitása - az MO-megközelítés ................................. 114 2.3. Az aromás jelleg kritériumai ................................................................................ 116 2.4. A kondenzált gyűrűs aromás szénhidrogének szerkezete ..................................... 117 2.5. Az izolált gyűrűs aromás szénhidrogének szerkezete ........................................... 118
3. Aromás szénhidrogének előállítása ................................................................................... 119 3.1. Kinyerés kőszénkátrányból ................................................................................... 119 3.2. Aromatizálás és dehidrociklizálás ........................................................................ 119 3.3. Alkil-aromások átalakítása ................................................................................... 120
4. Az aromás szénhidrogének reakciókészsége ..................................................................... 121 4.1. A jellegzetes reakció: az elektrofil szubsztitúció .................................................. 121 4.2. Elektroneltolódások a helyettesített benzolgyűrűben ........................................... 123
5. Az aromás szénhidrogének reakciói .................................................................................. 128 5.1. Az aromás gyűrű szubsztitúciós reakciói .............................................................. 128 5.2. Az aromás gyűrű addíciós reakciói ....................................................................... 130 5.3. Az aromás gyűrű oxidálása ................................................................................... 132 5.4. Az aromás gyűrű komplexképző tulajdonságai .................................................... 133 5.5. Az alifás oldallánc reakciói .................................................................................. 133
6. Nem-benzoid aromás szénhidrogének ............................................................................... 134 7. Polisztirol és származékai ................................................................................................. 135

vi Created by XMLmind XSL-FO Converter.
ELŐSZÓ
Ez a jegyzet egy négy kötetesre tervezett - a szerves kémia alapjait összefoglaló - munka első kötetének anyagát
tartalmazza.
A teljes anyag összesen 29 fejezetből fog állni, ezekből ez az első kötet az 1...9. fejezeteket tartalmazza,
amelyek az általános ismereteket és a szénhidrogének kémiáját tárgyalják. Amennyiben tehát az olvasó a 10.
vagy annál nagyobb fejezetszámra utaló hivatkozással találkozik, ez a II...IV. kötetekben megtalálható fejezetek
valamelyikére vonatkozik. Ezeket az utóbbi köteteket terveink szerint a későbbiekben fokozatosan fogjuk
megjelentetni ebben a digitális formában. Az ismeretanyag tehát ekkor lesz teljes mértékben használható.
Tartalmát tekintve lényegében alapfokú szerves kémia tankönyvről van szó. Az ebben a témakörben megszokott
összeállításokhoz képest azonban néhány szempontból kiegészítettem ezt a klasszikusnak nevezhető
ismeretanyagot. Így a hozzáférhető adatok alapján igyekszem ismertetni azokat a szerves kémiai technológiai
eljárásokat, amelyek ma korszerűeknek tekinthetők, valamint a biológiai szempontból fontosabb vegyületek
esetén több helyen biokémiai vonatkozásokat is megemlítek.
Tekintettel voltam arra, hogy mára egyérteműen az angol vált a kémia nemzetközi kommunikációjának
nyelvévé. Ezért a szakirodalom megértésének megkönnyítése érdekében az angol kémiai szaknyelv azon
kifejezéseit, amelyek a magyar szakkifejezésekből kézenfekvő módon nem következtethetők, a megfelelő
helyeken külön megemlítem. Ugyancsak hivatkozom az angol vegyületelnevezésekre azokban az esetekben,
amikor ezek - a magyartól eltérően - logikusabban követik a nevezéktan szabályait.
Veszprém, 2005. március
Markó László, Veszprémi Egyetem

1 Created by XMLmind XSL-FO Converter.
1. fejezet - TÖRTÉNETI ÁTTEKINTÉS
1. A szerves kémia kialakulása; "szerves" és "szervetlen" kémia
Szerves kémián ma a szénvegyületek kémiáját értjük.
A kémia tudománya a XVIII. század végén kezdett szétválni a ma is megkülönböztetett két ágra: a szerves és
szervetlen kémiára. A "szerves kémia" név Berzeliustól (1806) származik. Az akkori felfogás szerint azokat a
vegyületeket tekintették "szerves"-nek, melyeknek elöállítására csak az élő (állati vagy növényi) szervezetek
"életereje" (vis vitalis) képes. Ennek a felfogásnak a megdöntése Wöhler nevéhez fűződik, aki 1824-ben először
állította elő diciánból az addig kifejezetten növényi eredetűnek tartott oxálsavat, majd 1828-ban az ásványi
eredetűként ismert ammónium-cianátból hőkezeléssel a vizeletben előforduló (tehát "szerves" eredetű)
karbamidot:
Annak ellenére, hogy a szerves és szervetlen kémia közötti elvi különbség Wöhler felfedezése következtében
megszűnt, e két tudományág szétválása továbbra is állandósult. Ennek oka, hogy a molekulát alkotó atomok
kapcsolódási módja e két területen még sokáig nem volt egységesen értelmezhető. A "szervetlen" molekulákat
alkotó atomok kapcsolódására sokáig jól bevált értelmezés - az ionok elektrosztatikus vonzása - a
szénvegyületekre nem volt alkalmazható. A szénvegyületek klasszikus szerkezetelmélete az atomok
összekapcsolódását még a gömbszerű atomok felületén lévő (a vegyértéknek megfelelő számú) "horgocskák"
összeakaszkodásával magyarázta.
Mivel a szerves és szervetlen kémia közötti különbség egyre inkább elmosódik, ez a megkülönböztetés és a
szénvegyületek kémiájának "szerves kémia" elnevezése ma már inkább csak történelmi hagyománynak
tekinthető.
A szerves és szervetlen molekulák atomjainak kapcsolódási elvét a modern kötéselmélet ma már egységes
keretbe foglalja. Mi teszi ennek ellenére ma is jogosulttá a kémia e két ágának megkülönböztetését?
• Elsősorban a szénvegyületek nagy száma: az eddig (2004) a szakirodalomban leírt szénvegyületek száma több
mint 20 millió (a szervetlen és fémorganikus vegyületeké "csak" mintegy 1,5 millió) annak ellenére, hogy a
szerves vegyületek szénatomokon kívül viszonylag kevés másféle atomot tartalmaznak (elsősorban H, O, N,
halogének, P, S). Nagyrészt a szerves kémia fejlődésére vezethető vissza az, hogy a kémiai szakirodalom
volumene - a fejlődés mai fokán - kb. 5-10 évenként megkétszereződik.
• A szénvegyületek körében gyakori az izoméria jelensége; a szerves vegyületek nagy száma tulajdonképpen
erre vezethető vissza. Az izoméria miatt az összegképlet általában nem jellemző a szerves vegyületekre; ezek
egyértelműen csak szerkezeti képletekkel jellemezhetők. (Az Na2SO4 képlet például csak egy szervetlen
vegyületet jelöl; a C2H6O összegképlet azonban egyaránt jelentheti az etil-alkoholt és a dimetil-étert.) Az
izomerek száma a molekulát alkotó atomok számával rohamosan növekszik: C10H12O2 összegképlettel pl. már
több, mint 700 vegyületet ismerünk.
Az előforduló kötéstípusok szerinti megkülönböztetés nem általánosan jogosult. Megállapítható azonban, hogy a
szervetlen vegyületekre inkább az ionos kötés, szerves vegyületekre pedig inkább a kovalens kötés jellemző.
2. A szerves kémia klasszikus szerkezetelmélete
A szerkezetelmélet fejlődése szempontjából döntő jelentőségű volt Kekulé munkássága (1857-től), aki
felismerte, hogy az addig megismert vegyületekben a szénatom mindig négy "vegyértékű" volt, és a szénatomok
egymással is képesek kapcsolódni szénlánc kialakulása közben. Ezt a négy vegyértéket Kekulé még azonos
értékűnek tekintette. Ugyancsak Kekulé ismerte fel elsőként (1865) a benzol gyűrűs szerkezetét, és a gyűrűt
alkotó szénatomok egyenértékűségét.

TÖRTÉNETI ÁTTEKINTÉS
2 Created by XMLmind XSL-FO Converter.
Az atomok egymáshoz kapcsolódásának jelölésére a fejlődés során számos ábrázolási mód alakult ki. A múlt
század hetvenes éveitől kezdve terjedt el a ma is használatos irásmód: az atomok közé a kapcsolódó vegyértékek
számának megfelelő számú kötővonal irása.
A szénvegyületek modern szerkezetelméletének kialakulásához nagyban hozzájárult Le Bel és van't Hoff (1874)
tétele, mely szerint a szénatom négy vegyértéke a "telített" vegyületekben tetraéderes térorientációjú.
A klasszikus szerkezetelmélet alapját tehát a következő tételek képezték:
• a szénatom vegyületeiben mindig négy vegyértékű;
• ez a négy vegyérték egymással egyenértékű;
• a szénatom négy vegyértéke a telített vegyületekben tetraéderes térorientációjú;
• a szénatomok egymással is kapcsolódhatnak, lánc vagy gyűrűs szerkezetű molekulaváz kialakulása közben.

3 Created by XMLmind XSL-FO Converter.
2. fejezet - A SZERVES VEGYÜLETEK SZERKEZETÉNEK ELEKTRONELMÉLETE
1. A szénatom "kitüntetett" helyzete a periódusos rendszerben
A periódusos rendszer elsö három főcsoportjában elhelyezkedő fémek elektropozitív jellege a rendszer közepe
felé haladva csökken. A Na+ képződésekor leszakadó vegyértékelektronnak még csak egységnyi magtöltés
vonzását kell leküzdenie. A két pozitív töltéssel rendelkező Mg2+ ion képződésekor már nagyobb az ionizációs
potenciál. Még nagyobb ez az Al3+ esetében; a "C4+" ion képződése pedig már energetikailag szinte lehetetlen.
A periódusos rendszer jobb oldalán elhelyezkedő nemfémes (savképző) elemek esetében - fordított értelemben -
hasonló a helyzet: az elektronegatív jelleg csökken a rendszer közepe felé haladva. Ennélfogva a C4- ion
képződése is gyakorlatilag kizárható. A szénatom (és hozzá hasonlóan a szilíciumatom) ennek következtében
csak úgy tud nemesgáz-konfigurációt elérni vegyületképzés során, ha homopoláros (kovalens) kötéseket létesít.
A szénatom négyvegyértékűsége tehát a vegyértékhéjban található elektronok számától, a kovalens kötések
kialakítására való hajlam pedig elektroneutralitásából (a periódusos rendszerben elfoglalt közbenső helyzetéből)
adódik. Ez utóbbi magyarázza a szén-szén kötések kialakítására (tehát a lánc- vagy gyűrűképzésre) való
különleges hajlamot.
A szénatomnak a periódusos rendszerben elfoglalt különleges helyzetét az ugyanazon főcsoportba tartozó
szilíciummal való összehasonlítás is mutatja. A szilícium sok hasonlóságot mutat a szénnel, van azonban
közöttük olyan különbség, amely a szénnnek a 14. csoport (a IV. főcsoport) elemei között is különleges
helyzetet biztosít: a szénatom nem rendelkezik alacsony energiaszintű d-pályákkal, ennek következtében nem
csak kötő elektronjainak száma, hanem koordinációs száma is négy. Magános elektronpárt tartalmazó atomok
tehát a szénnel nem tudnak az üres d-pályákat felhasználva úgy térhálós makromolekulákat kialakítani, mint a
szilíciummal. A szénvegyületek molekulái ennek következtében általában diszkrét molekulák (a CO2 gáz, a SiO2
polimer szerkezetű, szilárd kristályos anyag). A szén és a szilícium közötti különbséget jól mutatja a szén-
tetraklorid (CCl4) és a szilícium-tetraklorid (SiCl4) vízzel szembeni viselkedése is. A szilícium koordinációs
száma hat; a koordinatíve telítetlen SiCl4-hoz tehát vízmolekulák tudnak kapcsolódni, aminek hevesen
lejátszódó hidrolízis az eredménye:
H2O + SiCl4 → [H2O) → SiCl4] → (HO)SiCl3 + HCl
H2O + (HO)SiCl3 → (HO)2SiCl2 + HCl stb.
A víz kölcsönhatása az alacsony energiaszintű, üres d-pályával nem rendelkező szénatommal ezzel szemben
energetikailag igen kedvezőtlen, ezért a szilícium-tetrakloriddal analóg szénvegyület, a szén-tetraklorid
hidrolízissel szemben ellenálló.
A szén kötőelektronjainak és koordinációs számának egybeeséséből következik, hogy a szerves vegyületek
többsége nem ionos, kevéssé poláros, diszkrét molekula. Ez a magyarázata a szerves vegyületek jellegzetes
fizikai tulajdonságainak:
• a legtöbb szerves vegyület nem atom- vagy ionrácsban, hanem molekularácsban kristályosodik, a rácsenergia
aránylag csekély, ezért az olvadáspont általában alacsony (< 300 °C);
• a legtöbb szerves vegyület jól oldódik különböző oldószerekben;
• számos szerves vegyület illékony és bomlás nélkül desztillálható;
• a szerves vegyületek zöme az elektromos áramot rosszul vezeti.

A SZERVES VEGYÜLETEK
SZERKEZETÉNEK
ELEKTRONELMÉLETE
4 Created by XMLmind XSL-FO Converter.
2. A szénatom hibridállapotai
Az izolált szénatom az alapállapotában érvényes K 2s22px2py elektronszerkezete alapján két kötőelektronnal
rendelkezne.
Tapasztalat szerint azonban a legtöbb szerves vegyületben a szénatom mind a négy kettes főkvantumszámú
elektronjának felhasználásával létesít kapcsolatot kettő, három vagy négy szomszédos atommal. (A különleges
helyzetet elfoglaló és nem is a szerves vegyületekhez számított szén-monoxiddal és még néhány kivételes
szerves vegyülettel most nem foglalkozunk.)
Négy atommal kapcsolatban álló (négyszomszédos) szénatomhoz a szomszédos négy atom tetraéderes
szimmetria szerint (a tetraéder négy csúcsa irányából) kapcsolódik. Kisérleti tény, hogy amennyiben a négy
kapcsolódó atom azonos minőségű (mint pl. a metánban vagy a szén-tetrakloridban), a négy kapcsolat teljesen
egyenértékű. A tetraéderes szimmetriából és a kapcsolt atomok egyenértékűségéből szükségszerűen következik,
hogy a kapcsolatokat létesítő négy kötőelektronpár azonos energiaszintű molekula pályákon (molekula
orbitálokon, MO) tartózkodik.
Ezt a kvantumkémia nyelvén úgy írjuk le, hogy a szénatom négy vegyértékelektronja négy azonos, egymással
109,5°-os szöget bezáró, a tetraéder négy csúcsa felé irányuló sp3 hibrid atomi pályán (atomi orbitálon, AO)
helyezkedik el és ezen hibrid (s és p jelleget egyaránt tartalmazó) atomi pályák és a négy szomszédos atom
megfelelő atomi pályáinak lineáris kombinációjával kapjuk a kötéseket létesítő lokalizált, hengerszimmetrikus
σ-molekulapályákat. Erre mondjuk azt, hogy a szénatom az ilyen vegyületekben sp3-hibridállapotban van.
Ha a szénatom csak három atommal kapcsolódik (háromszomszédos), ezek az atomok egy síkban, egymáshoz
képest 120°-os szöget bezáró irányokban helyezkednek el és a szénatom ezekkel a szomszédos atomokkal
három σ-kötést alakít ki: az ilyen szénatomot nevezzük sp2 hibridállapotúnak. A szénatom negyedik
kötőelektronja a σ-kötések síkjára merőlegesen orientált p atomi pályán helyezkedik el és valamelyik
szomszédos atom hasonló atomi pályán lévő szabad kötőelektronjával kölcsönhatásba lépve, a két atom között
még egy további (π-) kapcsolatot létesít: kettős kötés (σ,π-kötés) alakul ki. (A kettős kötésról és annak
tulajdonságairól a továbbiakban még részletesen lesz szó.)
A négyszomszédos szénatom esetében tehát négy - azonos szimmetriájú és értékű - σ-kötés alakul ki; a három
atommal kapcsolódó szénatomnál a kapcsolat összetettebb, a kötések nem egyenértékűek (3σ + 1π).
A két atommal kapcsolatban álló (kétszomszédos) szénatom már csak két (egyenértékű) σ-kötést létesít, ez a
két σ-kötés a szénatomon egy egyenes mentén, egymáshoz képest 180°-ra helyezkedik el. Az ilyen szénatomot
nevezzük sp-hibridállapotúnak. A szénatom két fennmaradt kötőelektronja két, egymásra merőleges p atomi
orbitálra kerül és egyik vagy mindkét szomszédos atom megfelelően orientált párosítatlan elektronjaival
kölcsönhatásba lépve egy hármas vagy két kettős kötést alakít ki (lásd később).
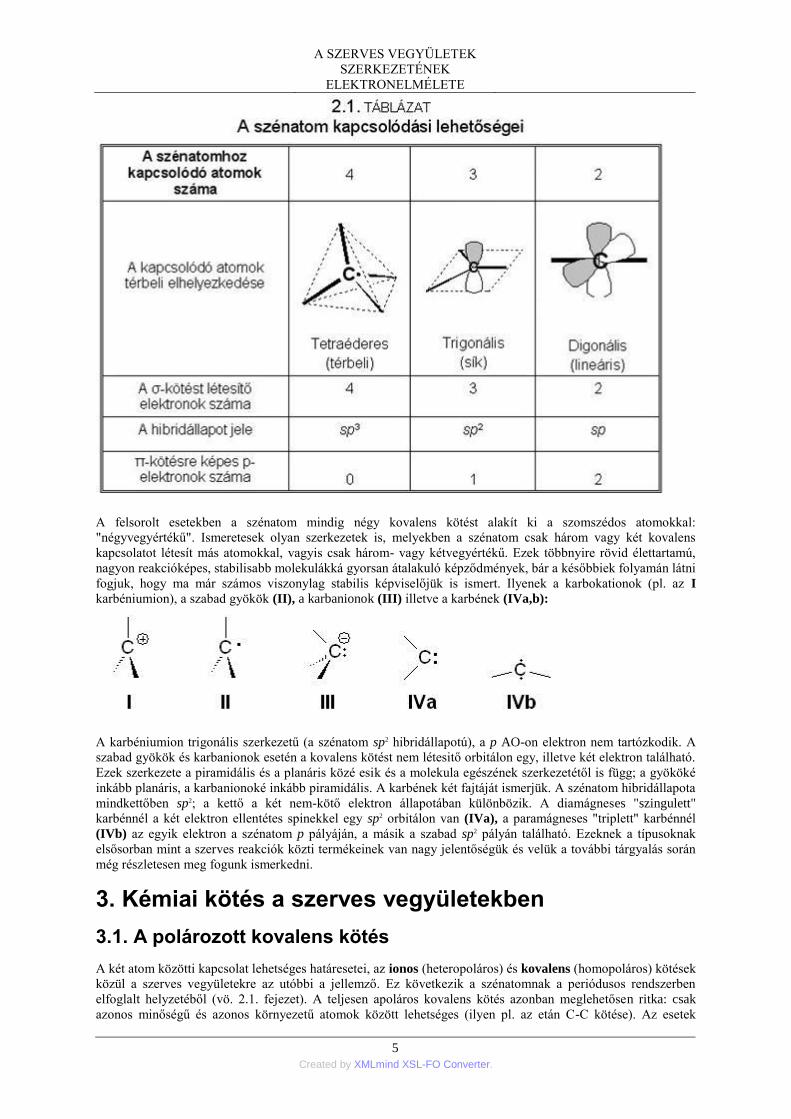
A szénatom alapvető kapcsolódási módjainak legjellemzőbb adatait a 2.1. TÁBLÁZAT szemlélteti.
A SZERVES VEGYÜLETEK
SZERKEZETÉNEK
ELEKTRONELMÉLETE
5 Created by XMLmind XSL-FO Converter.
A felsorolt esetekben a szénatom mindig négy kovalens kötést alakít ki a szomszédos atomokkal:
"négyvegyértékű". Ismeretesek olyan szerkezetek is, melyekben a szénatom csak három vagy két kovalens
kapcsolatot létesít más atomokkal, vagyis csak három- vagy kétvegyértékű. Ezek többnyire rövid élettartamú,
nagyon reakcióképes, stabilisabb molekulákká gyorsan átalakuló képződmények, bár a későbbiek folyamán látni
fogjuk, hogy ma már számos viszonylag stabilis képviselőjük is ismert. Ilyenek a karbokationok (pl. az I
karbéniumion), a szabad gyökök (II), a karbanionok (III) illetve a karbének (IVa,b):
A karbéniumion trigonális szerkezetű (a szénatom sp2 hibridállapotú), a p AO-on elektron nem tartózkodik. A
szabad gyökök és karbanionok esetén a kovalens kötést nem létesitő orbitálon egy, illetve két elektron található.
Ezek szerkezete a piramidális és a planáris közé esik és a molekula egészének szerkezetétől is függ; a gyököké
inkább planáris, a karbanionoké inkább piramidális. A karbének két fajtáját ismerjük. A szénatom hibridállapota
mindkettőben sp2; a kettő a két nem-kötő elektron állapotában különbözik. A diamágneses "szingulett"
karbénnél a két elektron ellentétes spinekkel egy sp2 orbitálon van (IVa), a paramágneses "triplett" karbénnél
(IVb) az egyik elektron a szénatom p pályáján, a másik a szabad sp2 pályán található. Ezeknek a típusoknak
elsősorban mint a szerves reakciók közti termékeinek van nagy jelentőségük és velük a további tárgyalás során
még részletesen meg fogunk ismerkedni.
3. Kémiai kötés a szerves vegyületekben
3.1. A polározott kovalens kötés
A két atom közötti kapcsolat lehetséges határesetei, az ionos (heteropoláros) és kovalens (homopoláros) kötések
közül a szerves vegyületekre az utóbbi a jellemző. Ez következik a szénatomnak a periódusos rendszerben
elfoglalt helyzetéből (vö. 2.1. fejezet). A teljesen apoláros kovalens kötés azonban meglehetősen ritka: csak
azonos minőségű és azonos környezetű atomok között lehetséges (ilyen pl. az etán C-C kötése). Az esetek

A SZERVES VEGYÜLETEK
SZERKEZETÉNEK
ELEKTRONELMÉLETE
6 Created by XMLmind XSL-FO Converter.
túlnyomó részében többé-kevésbé polározott kovalens kötésről van szó. Különösen a nem-azonos atomok
közötti "heteronukleáris" kötésekre jellemző, hogy a kötőelektronpár nem egyenletesen oszlik el a két
kapcsolódó atom erőterében, de azonos minőségű atomok "homonukleáris" kapcsolatában is függ az
elektroneloszlás az illető atomokhoz kapcsolódó egyéb atomok (vagy atomcsoportok) jellegétől. A kötés
polározottságának oka a két kapcsolódó atom eltérő elektronaffinitása és elektronegativitása.
3.2. Elektronaffinitás és elektronegativitás
Két kapcsolódó atom közül az képes erősebben magához szívni a kötőelektronpárt, amelyiknek nagyobb az
elektronaffinitása (vagyis anionná alakulásakor nagyobb energia szabadul fel). Ennek a fogalomnak
értelmezéséhez abból indulhatunk ki, hogy ha a kapcsolódó A és B atomok elektronaffinitása egyenlő, az A,B-
kötésenergia az A,A és B,B kapcsolatok energiáinak számtani középértéke:
EAB = 1/2 (EAA + EBB)
Ha az A és B atomok elektronaffinitása eltérő, a mért kötéserősség ΔAB értékkel nagyobb:
E'AB = 1/2 (EAA + EBB) + ΔAB
Pauling szerint az energiakülönbség a
ΔAB = 23 (xA - xB)2
egyenlettel fejezhető ki, melyben xA és xB a kapcsolódó atomok elektronegativitásai. Az így definiált
elektronegativitás nem azonos az atom elektronaffinitásával, de arányos azzal. (Pauling a legelektronegatívabb
fluor elektronegativitását önkényesen 4,0-nak vette.) Az így jellemzett "elektronszívás" nagyjából fokozatosan
növekszik a magtöltéssel a periódusos rendszer minden periódusában, az egyes oszlopokban lefelé haladva
pedig (a magtöltést árnyékoló elektronburok vastagodása miatt) fokozatosan csökken.
A fontosabb elemek Pauling-féle elektronegativitását a 2.2. TÁBLÁZAT tartalmazza. Ismeretesek más módon
definiált elektronegativitás értékek is.
Az eltérő elektronegativitású atomok közti kapcsolat (pl. a C→Cl kapcsolat; a kötővonalra rajzolt nyíl az
elektroneltolódás irányát jelöli) mindig dipólus-momentummal rendelkezik. A molekulában lévő kötések
dipólusmomentumai részben vagy teljesen kompenzálhatják egymást (a CCl4 eredő dipólusmomentuma például
zérus).
3.3. Homonukleáris kötések: a C,C-kötések különböző típusai
-- A C-C egyes kötés

A SZERVES VEGYÜLETEK
SZERKEZETÉNEK
ELEKTRONELMÉLETE
7 Created by XMLmind XSL-FO Converter.
A C,C egyes kötés ún. σ-kötés, amely a két szénatom azonos fázisú AO-jainak kölcsönhatásával (a)
("átlapolásával") kialakuló hengerszimmetrikus molekulapálya (MO) (b). Ezen - a molekula alapállapotában -
két, ellentett spinű, párosított elektron tartózkodik (az atomi orbitálok "pozitív" és "negatív" fázisviszonyait a
pályák árnyékolásával, illetve annak hiányával szemléltetjük). A kötésben lévő atomok távolsága és a kötés
erőssége a kapcsolódó szénatomok hibrid állapotaitól függően változik (vö. 2.3. és 2.4. TÁBLÁZATOK). A σ-
kötés hengerszimmetrikus jellege lehetővé teszi a szénatomoknak a kötés, mint tengely körüli (korlátozottan)
szabad rotációját (c) (részletesen l. 5.1.4. fejezet):
Az ellenkező fázisú sp3 atomi pályák kombinációja (d) vezet a σ*-gal jelölt lazító molekulapályához (e), amely a
nem gerjesztett molekulában üres:
A kötő és lazító molekulapályák kialakulását az MO-elméletben szokásos szimbolikával az alábbi (az
energiaviszonyokat kvalitatíve tükröző) vázlat szemlélteti arra az esetre, ha mindkét atomi orbitál egy-egy
elektronnal járul hozzá a σ-molekulapálya kialakulásához. Ez erős σ-kötéshez vezet:
Ha a rendelkezésre álló elektronok száma kettőnél több, ezek csak a σ* pályán tudnak elhelyezkedni, ami
(három elektron esetén) a kötés gyengülését, vagy (négy elektron esetén) a kötés teljes megszűnését
eredményezi.
-- A C=C kettős kötés
Ez a kötés két sp2 vagy egy sp2 és egy sp hibrid szénatom között alakulhat ki. Egy hengerszimmetrikus σ-
kötésből és egy ún. π-kötésből áll (σ,π-kötés). A leggyakoribb esetben síktrigonális szimmetriájú sp2
szénatomok kapcsolódnak egymáshoz; erre a legegyszerűbb példa az eténmolekula (CH2=CH2). Az etén C,C és
C,H σ-kötései a szénatomok síktrigonális szimmetriájából adódóan egy síkban helyezkednek el. Mindkét
szénatom rendelkezik egy - a σ-kötések síkjára merőlegesen orientált - atomi p-pályával (a), melyeken egy-egy
elektron tartózkodik. Az azonos fázisú p-pályák kombinálásával alakul ki a két sp2 szénatomra kiterjedő -
molekulapálya (b). Ha ezen két, ellentett spinű elektron tartózkodik (ez a helyzet a molekula alapállapotában), a
két szénatom között egy újabb kötés (π-kötés) alakult ki (c):
Az így kialakult π-kapcsolat orientációja a molekula síkjára merőleges, az atomok kötésvonalára nem
hengerszimmetrikus, hanem a molekula síkjában "csomósíkkal" rendelkezik. Mivel a kapcsolatot létesítő
elektronfelhő kisebb magvonzás alatt áll és könnyebben polározható, mint a σ-kötés elektronfelhője, a π-kötés
gyengébb és reakcióképesebb a σ-kötésnél.
Az ellenkező fázisú atomi p-pályák kombinációjából (d) alakul ki a lazító, π*-gal jelölt molekulapálya (e) amely
az etén alapállapotában (nem gerjesztett állapotában) üres:
Látható, hogy a π*-pálya szimmetriája még alacsonyabb, mint a π-pályáé, két csomósíkkal is rendelkezik. A π-
és π*-pályák kialakulására is megszerkeszthető a σ-pályáknál már megismert vázlat:
A SZERVES VEGYÜLETEK
SZERKEZETÉNEK
ELEKTRONELMÉLETE
8 Created by XMLmind XSL-FO Converter.
Az azonos környezetű szénatomok közötti C-C, C=C és C≡C kötések jellemző adatait a 2.3. TÁBLÁZATBAN
hasonlítjuk össze. Láthatjuk, hogy a kötéshossz a kapcsolódás módjától függően jellemzően változik.
A kötésdisszociációs energia - amely a kovalens kötés homolitikus bontásához (3.1. fejezet) szükséges energiát
jelenti - csak igen áttételesen áll összefüggésben a reakciókészséggel, hiszen a kémiai folyamatok ritkán
igénylik a kémiai kötések ilyen jellegű teljes felbontását. Ezt figyelembevéve is azonban a fenti táblázatból
levonható az a következtetés, hogy a C,C kettős (σ,π)-kötés a szénatomok közötti két elektronpár nagyobb
elektronkoncentrációja következtében
• rövidebb és erősebb a C,C egyes (σ)-kötésnél és ezért magasabb hőmérsékletig stabilis;
• a π-elektronpár viszonylag gyengébb kötöttsége, valamint a π- és π*-pályák könnyebb hozzáférhetősége miatt
reakcióképesebb: elektrofil reagensek hatására sokkal könnyebben polározódik.
Az energiaviszonyokat vázlatosan a 2.1. ÁBRA szemlélteti.
2.1. ÁBRA
A C,C σ,π-kötés a π-elektronpárok nem hengerszimmetrikus eloszlása következtében merev, vagyis
megakadályozza a kapcsolódó atomok rotációját: a kettős kötésben álló atomok és a hozzájuk közvetlenül
kapcsolódó szomszédos atomok megfelelő energia befektetése (a π-kötés megbontása) nélkül nem mozdulnak ki

A SZERVES VEGYÜLETEK
SZERKEZETÉNEK
ELEKTRONELMÉLETE
9 Created by XMLmind XSL-FO Converter.
közös térsíkjukból. A szabad rotáció megszünése a geometriai, vagy Z,E- (cisz, transz)-izomériához vezet (4.3.
fejezet).
-- A C≡C hármas kötés
Az acetilén (etin) hármas kötésében lévő kétszomszédos szénatomok digonális szimmetriában, egymáshoz
viszonyítva 180°-os szögben kapcsolódnak a szomszédos atomokhoz egy-egy σ-kötéssel: az acetilén molekula
tehát lineáris felépítésű. Az ezekre a kötésekre merőleges két - egymásra is merőleges - diszkrét térsíkban
orientált p-elektronok két egymástól független, energetikailag egyenértékű (vö. 2.1. ÁBRA) π-kötést alakítanak
ki ugyanazon két szénatom között (σ,π,π-kötés):
A hármas kötésben lévő szénatomok egymáshoz közelebb helyezkednek el és erősebben is kötődnek, mint az
egyszeres, vagy kétszeres C,C-kapcsolat szénatomjai (vö. 2.3. TÁBLÁZAT).
A C,C hármas kötés elektroneloszlása szimmetrikusabb, mint a C,C kettős kötésé, ezért bizonyos
vonatkozásokban kevésbé reakcióképes (nehezebben polározható) mint a kettős kötés, annak ellenére, hogy π-
elektronpárjai lazábban kötöttek, mint a C,C kettős kötésé (vö. 2.1. ÁBRA)
3.4. Heteronukleáris kötések
Az eddig tárgyalt homonukleáris C,C-kötések nem polározott, vagy csak kismértékben polározott
atomkapcsolatok: a kötő elektronpárok a molekula alapállapotában nagyjából egyenletesen oszlanak meg a
kapcsolódó atomok között. A heteronukleáris kötések a kapcsolódó atomok elektronegativitásai közötti
különbségek arányában mindig polározottak (dipólus jellegűek). Például:
(X a szénnél nagyobb, Y a szénnél kisebb elektronegativitású atomot jelöl.)
Poláros kötés jelenléte a molekulában befolyásolja a molekula elektroneloszlását; az így kialakuló
elektroneltolódási effektusokkal a 2.4. fejezetben foglalkozunk részletesebben.
A szerves vegyületekben gyakrabban előforduló kötéshosszúságokat a 2.4. TÁBLÁZATBAN foglaltuk össze.
A SZERVES VEGYÜLETEK
SZERKEZETÉNEK
ELEKTRONELMÉLETE
10 Created by XMLmind XSL-FO Converter.
4. Elektroneltolódási effektusok
A szerves vegyületek molekuláiban a kémiai kötések általában nem függetlenek egymástól: az elektroneloszlás
a molekulában más lehet, mint ami a molekulában lévő diszkrét kötéseknek megfelelne. Ez a molekulában
kialakuló elektroneltolódási effektusoknak (amelyek tulajdonképpen a molekulán belüli kölcsönhatások közé
sorolhatók) az eredménye. Ha az elektroneltolódás a molekula σ-kötésein jelentkezik, induktív effektusnak (I),
ha a π-kötéseken jelentkezik, nevezzük (az utóbbira használatos kifejezések még: rezonancia, konjugáció). A
molekula alapállapotában kialakuló elektroneltolódásokat szokás sztatikus, a csak valamilyen külső hatásra
kialakulókat pedig dinamikus jelzővel megkülönböztetni. A továbbiakban csak a sztatikus elektroneltolódási
effektusokkal foglalkozunk.
4.1. Induktív effektus
Dipólus jellegű polározott σ-kötés jelenléte a molekulában a vele szomszédos kötés(ek)ben is indukálja a
töltések bizonyos fokú szétválását, vagyis újabb dipólus képződését. Ennek hatására ismét indukálódhat dipólus
a szomszédos σ-kötéseken. Az elektroneltolódás (polározódás) mértéke a hatást kiváltó atomtól vagy
atomcsoporttól távolodva természetesen csökken.
Ha a molekula valamelyik szénatomjához a szénnél nagyobb elektronegativitású ("elektronszívó",
elektronakceptor) X atom vagy atomcsoport kapcsolódik, a kötés C→X értelemben polározódik, a σ-kötések
elektronjai az X irányába tolódnak el és a szénatomon parciális pozitív töltés alakul ki: ezt nevezzük negatív
induktív effektusnak (-I). Ennek hatására a szomszédos kötés is polározódik, de már jóval kisebb mértékben:
az induktív effektus erőssége a funkciós csoporttól távolodva szénatomonként kb. 1/3-ára csökken, tehát a
molekulának csak az X csoporthoz közel eső részére terjed ki. Elektronküldő (elektrondonor) csoport (jelöljük
Y-nal) kapcsolódása esetén a hatás természetesen fordított: pozitív induktív effektus (+I).

A SZERVES VEGYÜLETEK
SZERKEZETÉNEK
ELEKTRONELMÉLETE
11 Created by XMLmind XSL-FO Converter.
A -I vagy +I hatást kifejtő fontosabb atomokat és csoportokat a 2.5. TÁBLÁZATBAN tüntettük fel. Látható,
hogy a -I effektussal rendelkező elektronszivó csoportok száma nagyobb. Pozitív induktív effektust csak egyes
negatív töltésű ionok, és a kis elektronegativitású elemek (pl. P, Si, Sn, fémek) fejtenek ki.
Az I-effektusok nagyon jól kimutathatók az egyes csoportoknak a karbonsavak saverősségére kifejtett hatása
révén (19.1.3. fejezet). Az aromás gyűrű SE reakciókészségével kapcsolatban szintén kitérünk még az I-
effektusokra (9.4.2. fejezet).
4.2. Mezomer effektus
A mezomer (rezonancia, konjugációs) effektus (M) a molekula szerkezeti adottságai következtében a π-
elektronrendszerben kialakult elektroneltolódást jelenti. Lokalizált és delokalizált π-kötések esetén egyaránt
felléphet.
Az M-effektus kialakulására akkor van lehetőség, ha a hatást kifejtő csoport (vagy atom) közvetlenül
kapcsolódik egy telitetlen rendszerhez. Ha a telítetlen rendszerhez kapcsolódó atom magános elektronpárral
rendelkezik (pl. Cl), a hatást kifejtő csoport elektronsűrűsége csökken, a telítetlen rendszeré nő: +M effektus.
Ha a telítetlen rendszerhez kapcsolódó "kulcs"-atom magános elektronpárral nem rendelkezik és egyidejűleg
nagy elektronegativitású atom(ok)hoz kapcsolódik többszörös kötéssel [pl. -C(=O)H], a heteronukleáris π-
kötésen is fellépő polározódás következében a kulcsatom (példánkban a szénatom) elektronszívóvá válik, s a
telitetlen rendszer elektronsűrűsége csökken: -M effektus.
A mezomer hatás következtében kialakuló elektroneloszlást a VB (valence bond) leírás alapján ún. mezomer
határszerkezetekkel lehet érzékeltetni. Ezek csak erre a célra szolgáló "fiktív" szerkezetek, amelyek azt
kívánják szemléltetni, hogy a molekula tényleges elektroneloszlása a határszerkezetek által leírt szélsőséges
elektroneloszlások közé esik. Közöttük mindig a kétfejű nyíl (←→) jelet fogjuk használni, amelyet gondosan
meg kell különböztetnünk az egyensúlyi folyamatok jelölésére használt kettős nyíltól. Míg ugyanis az utóbbi azt
szimbolizálja, hogy a kémiai egyensúlyok dinamikus jellegűek, azaz az egyensúlyi összetétel valójában a két
ellentétes irányú folyamat azonos sebességének következménye, a mezomer határszerkezetek közé írt kétfejű
nyíl csak logikai szimbólumnak tekintendő és nem jelenti azt, hogy a molekula elektroneloszlása e szélső
állapotok között fluktuál. Valójában a molekula elektroneloszlása (külső hatás távollétében) nem változik és a
határformák közül ahhoz áll legközelebb, amely a legalacsonyabb energiájú.
Szokás az M-effektus fellépését és az elektroneltolódás irányát görbe nyilakkal is jelölni. Példaként bemutatjuk
a vinil-csoporthoz kapcsolódó klóratom +M, és a formil (-CHO) csoport -M hatását:
illetve:

A SZERVES VEGYÜLETEK
SZERKEZETÉNEK
ELEKTRONELMÉLETE
12 Created by XMLmind XSL-FO Converter.
A mezomer és induktív effektusok előjele adott esetben lehet azonos vagy ellenkező; az így kialakuló kombinált
hatásoknak fontos szerepe van például az aromás SE reakcióra vonatkozó írányítási szabályoknál (9.4.2. fejezet).
A +M vagy -M effektust kifejtő atom(csoport)ok fontosabb képviselőit a 2.6. TÁBLÁZAT tartalmazza.
Fontos jellemzője a mezomer effektusnak, hogy konjugált kettős kötéseket tartalmazó molekulákban a π-
elektronok közvetítésével gyakorlatilag korlátlan hosszúságú szénláncon át is kifejtheti hatását (az ilyen
szerkezetű molekulák elektromos vezetőknek tekinthetők). Például:
5. Molekulák közötti kölcsönhatások
A molekulán belül az egyes atomok között kialakuló erős kémiai kötések mellett meg kell emlékeznünk a
szerves vegyületek molekulái között fellépő gyengébb kölcsönhatásokról is. E kölcsönhatások kialakulása
nyilvánvalóan függ a molekulát alkotó atomok minőségétől és kapcsolódásuk jellegétől, tehát a molekula
szerkezete meghatározza a molekulák közötti kölcsönhatások jellegét és erősségét is.
5.1. Molekulakomplexek kialakulása
A molekulakomplex vagy donor-akceptor komplex fogalom tulajdonképpen egy elektrondonor molekula és
egy elektronakceptor molekula asszociátumát jelenti. A donormolekula rendszerint Lewis-bázis, az
akceptormolekula pedig Lewis-sav.
Ezek közül az erősebb kölcsönhatások olyan kovalens kötéseknek tekinthetők, amelyek kötőelektronpárja a
donormolekulából származik (datív kötés). Ilyen típusú kapcsolatok alakulhatnak ki például egyrészről FeCl3,
AlCl3, SbCl5, BF3 stb., másrészről NH3, továbbá oxigén-, nitrogén- vagy kéntartalmú szerves vegyületek között,
50-200 kJ/mol nagyságrendű kötési energiával.
A molekulakomplexek egy gyengébb kötöttségű típusát képviselik a kinhidron (17.3.2. fejezet) és a pikrátok
(24.8.5. fejezet). Ezekben a kapcsolat erőssége rendszerint < 50 kJ/mol. Az ilyen típusú molekulakomplexek
nagy része erősen színes, annak ellenére, hogy az őket felépítő diszkrét molekulák rendszerint színtelenek (a
színtelen hidrokinonból és sárga kinonból kialakuló kinhidron pl. csillogó sötétzöld kristályokat alkot).

A SZERVES VEGYÜLETEK
SZERKEZETÉNEK
ELEKTRONELMÉLETE
13 Created by XMLmind XSL-FO Converter.
5.2. Hidrogénhíd-kötés (protonkötés)
Olyan molekulák között, amelyekben magános elektronpárral rendelkező elektronegatív atomhoz hidrogénatom
kapcsolódik, viszonylag erős kapcsolat tud kialakulni az ún. hidrogénhíd révén. Klasszikus példái ennek a
hidroxilcsoportot (-OH) tartalmazó vegyületek, pl. az alkoholok, melyeknek molekulái hidrogénhidak révén
viszonylag nagy "molekulatömegű" asszociátumokká kapcsolódnak össze:
A legerősebb hidrogénhíd-kötés a KHF2 F-H---F kötése; ennek energiája kb. 115 kJ/mol; az N-H---N
kapcsolatoké kb. 10 kJ/mol. Az O-H---O kapcsolatok erőssége a kettő közötti tartományba esik (a metanolban
például 26 kJ/mol). Az adatokból is látszik, hogy a hidrogénhíd erőssége a kialakításában résztvevő atomok
elektronegativitásával nő.
A hidrogénatom nem szimmetrikusan helyezkedik el a két elektronegatív atom között (a jégben pl. a két O,H
távolság 97 és 179 pm). A kapcsolat főleg elektrosztatikus, de egyben dinamikus is: a hidrogénatom helyzetét
igen gyorsan változtatja, átlagos tartózkodási ideje egy meghatározott helyen csupán 10-12 sec nagyságrendű. Az
ilyen, ún. aktív hidrogénatomok ezért igen könnyen és gyorsan cserélődnek, így pl. deutériummal jelzett
alkoholokhoz vizet adva pillanatszerűleg lejátszódik az (egyensúlyi) H,D csere:
A H-híd kialakulása a két elektronegatív atomot közelebb hozza egymáshoz: az O-H---O távolság mint láttuk
276 pm, míg a két oxigénatom van der Waals sugarainak összege 304 pm.
Két molekula között - azok szerkezetétől függően - több hidrogénhíd-kötés is kialakulhat, ami már viszonylag
erős kapcsolatnak felel meg. Ennek fontos szerepe van például a DNS kettős spirális szerkezetének
kialakulásában (29.2.2. fejezet).
5.3. van der Waals-erők
A molekulák közötti gyenge kölcsönhatásoknak három típusát soroljuk a van der Waals-erők közé: az
orientációs effektust, az indukciós effektust és a diszperziós erőket. Ezek a kölcsönhatások néhány kJ/mol
vonzóerőt jelentenek a molekulák között, de fontos szerepük van pl. a gázok cseppfolyósodásában, a
molekularácsból álló kristályok összetartásában stb.
Az orientációs effektus dipólusmontummal rendelkező molekulák között lép fel. A parciális pozitív és negatív
töltésekkel rendelkező dipólusmolekulák (pl. a metil-klorid):
között megfelelő orientáció esetén elektrosztatikus vonzás tud kialakulni. Például:
Az indukciós effektus egy állandó dipólus és a szomszédos molekulában általa indukált ellentétes értelmű
dipólus kölcsönös vonzásából származik.
Diszperziós erők dipólusmomentummal nem rendelkező molekulák között is hatnak. Ez olyan
kvantummechanikai kölcsönhatás, amely kvalitatíve felfogható az atomokban átmenetileg fellépő dipólusok
kölcsönhatásaként (a pozitív és negatív töltés centruma az atomban időben nem mindig esik egybe). A
diszperziós erők a molekulák között mindig érvényesülnek, függetlenül attól, hogy más van der Waals-erők
fellépnek-e. Legalább egy nagyságrenddel erősebbek az orientációs vagy indukciós erőknél.
A van der Waals-erők hatása a molekulák közti távolság növekedésével rendkivül erősen csökken; általában
csak a részecskenagysággal összemérhető távolságban hatnak. Apoláros jellegű vegyületekben (pl. az

A SZERVES VEGYÜLETEK
SZERKEZETÉNEK
ELEKTRONELMÉLETE
14 Created by XMLmind XSL-FO Converter.
alkánokban) csak a diszperziós erők hatnak. Poláros molekulák között mindhárom típus fellép, de az indukciós
effektus mindig kicsi és itt is a diszperziós effektus az uralkodó.

15 Created by XMLmind XSL-FO Converter.
3. fejezet - A SZERVES KÉMIAI REAKCIÓK CSOPORTOSÍTÁSA
A szerves kémiai reakciókat sok szempont szerint lehet csoportosítani, a szerveskémiai reakciók típusainak
rövid jelölései is mindig több irányú információt tartalmaznak (pl. SN1: monomolekuláris nukleofil
szubsztitúció)
1. Gyökös és ionos reakciók
Ismerünk a szerves kémiában olyan reakciókat is, amelyek során molekulák közvetlenül, egy lépésben reagálnak
egymással. Később tárgyalandó okok miatt azonban a szerves kémiai reakciók zömében a kémiai folyamatok
ionos vagy gyökös jellegű közbenső termékeken keresztül, több lépésben játszódnak le. Ilyen intermedierek
kialakulásához a kovalens kötés felhasadására van szükség.
A kötésfelhasadásnak két fő típusa van, aszerint, hogy a kötőelektronpár szimmetrikusan vagy aszimmetrikusan
oszlik el a keletkező részek között. A lehetőségeket a C-H kötés példáján mutatjuk be.
• Homolitikus kötésfelhasadás ("homolízis") esetén a kapcsolatot létesítő kötőelektronpár egyenletesen oszlik
meg a kialakuló két rész között: elektromos töltés nélküli, párosítatlan elektronnal rendelkező gyökök
keletkeznek, pl.:
• A heterolitikus kötésfelhasadásnak ("heterolízis"-nek) két változata lehet aszerint, hogy a kötőelektronpár
melyik atomon marad vissza:
Az első esetben karbanion (karbeniátion) és hidrogénion, a második esetben karbokation (karbéniumion vagy
- régebbi nevén - karbóniumion) és hidridion képződik.
A hidrogénion képződésével járó folyamatnak ez természetesen erősen leegyszerűsített ábrázolása: szabad
proton kémiai folyamat reakcióelegyében nem fordul elő (a proton elemi részecske!), hanem mindig valamilyen
nukleofil ágenshez kötött alakban (pl. vizes oldatban hidroxónium-ion, H3O+ formájában). A továbbiakban
gyakran fogjuk alkalmazni az egyszerűség kedvéért a H+ jelölést, azonban természetesen annak tudatában, hogy
ez szimbólikusan értendő.
Homolízis, vagy heterolízis természetesen π-kötéseken is bekövetkezhet, például:
Az is nyilvánvaló, hogy a homolízis nagyobb valószínűséggel az apoláros vagy kevéssé poláros kovalens
kötésekre, a heterolízis pedig az erősebben polározott kötésekre jellemző. A polározottság iránya egyúttal
megszabja a keletkező ionok jellegét is.
A reakciót indító kötésfelhasadás típusa, illetve a keletkező részek jellege szerint tehát a kémiai reakciókat két
nagy csoportba sorolhatjuk: gyökös és ionos reakciók.
Gyökös mechanizmusú reakciók

A SZERVES KÉMIAI REAKCIÓK
CSOPORTOSÍTÁSA
16 Created by XMLmind XSL-FO Converter.
A gyökös mechanizmusal lejátszódó reakciók ("gyökös reakciók") homolitikus kötésfelhasadással indulnak.
Jellemzőik:
• apoláros közegben, gyakran gázfázisban játszódnak le;
• a reakciót labilis paramágneses gyök vagy atom indítja;
• fény, oxigén gyakran befolyásolja a reakciót.
Ionos mechanizmusú reakciók
Az ionos mechanizmussal lejátszódó reakciók ("ionos reakciók") heterolitikus kötésfelhasadással indulnak
(esetleg karbokation vagy karbanion képződésével). Jellemzőik:
• poláros közegben (oldószerben) játszódnak le;
• a reakciót pozitív vagy negatív töltésű, a szénvegyület elektronfelhőjét deformáló (polározó) ionok indítják;
• savak vagy lúgok gyakran katalizálják a folyamatot.
2. Szubsztitúció, addíció, elimináció
A reakció eredménye szerint ezt a három fő típust szokás megkülönböztetni; itt a reakció során a
szubsztrátumon felhasadó és kialakuló kötések jellege a döntő, függetlenül a kötésfelhasadás módjától.
• A szubsztitúciós (S) reakció egyszeres kötés hasadásával és új egyszeres kötés kialakulásával játszódik le. A
kilépő atom vagy atomcsoport helyére egy új csoport lép be.
• Az addiciós (A) reakció során egy telítetlen molekulába új csoportok lépnek be. Az eredeti π-kötés
megszűnése közben két új σ-kötés alakul ki.
• Az eliminációs (E) reakció lényegében az addíció megfordítása: két σ-kötés helyébe egy π-kötés lép.
A lehetőségeket szemléletesen mutatja a 3.1. TÁBLÁZAT.
3. Elektrofil és nukleofil reakciók
A reagáló anyagok közül egyiket szubsztrátumnak, a másikat reagensnek tekintve, az ionos reakciókat tovább
csoportosíthatjuk aszerint, hogy a reagens elektronleadásra képes, "nukleofil" jellegű, vagyis a szubsztrátum
elektronszegény (az atommagokhoz közeli) helyein támad (pl. a Lewis-bázisok), vagy elektronfelvételre
hajlamos, "elektrofil" jellegű, tehát a szubsztrátum elektrondús helyein támad (pl. a Lewis-savak). Eszerint
megkülönböztetünk nukleofil és elektrofil reakciókat. (Ezeket a típusokat a rövid jelölésben N, illetve E
indexekkel jellemezzük.)
Ez a felosztás önkényesnek tűnhet, hiszen ha az A + B → AB reakcióban A pl. elektrofilként, akkor B
nyilvánvalóan nukleofilként fog viselkedni és így a reakció típusát aszerint adhatjuk meg, hogy A és B közül
melyiket tekintjük szubsztrátumnak, illetve reagensnek. Ezért itt azt a konvenciót kell követnünk, hogy a
szénatomjával reagáló részecskét tekintjük szubsztrátumnak és a heteroatommal reagálót reagensnek. Pl. az

A SZERVES KÉMIAI REAKCIÓK
CSOPORTOSÍTÁSA
17 Created by XMLmind XSL-FO Converter.
előző táblázatban szereplő etén + bróm reakcióban az etén a szubsztrátum és a bróm (pontosabban a belőle
képződő bróm(I)-kation, Br+) a reagens. Amennyiben mindkét részecskében szénatom a reakciócentrum (tehát
C-C kötés alakul ki) általában a nukleofil partnert tekintjük reagensnek.
Lényeges itt még azt is megjegyeznünk, hogy a rendszerint több, egymást követő (ún. konszekutív)
reakciólépésekből álló folyamatoknál mindig a leglassabb - tehát a bruttó reakció sebességét meghatározó -
elemi lépés jellegét vesszük figyelembe.
4. Molekularitás
Reakciókinetikai szempontból csoportosíthatjuk a reakciókat aszerint, hogy a sebességmeghatározó lépésben
hány molekula változtatja meg kötésállapotát. Monomolekuláris a reakció, ha a sebességmeghatározó lépésben
csak a szubsztrátum vesz részt, bimolekuláris, ha a reagens is résztvesz. Szokás úgy is fogalmazni, hogy a
monomolekuláris reakció esetén csak a szubsztrátum, a bimolekuláris reakció esetén pedig a szubsztrátum és a
reagens koncentrációja szerepel a reakció sebességi egyenletében (vö. az alkil-halogenidek reakcióival, 11.2.
fejezet). A molekularitást a rövid jelölésben a típusjel utáni számmal adjuk meg.
Az eddig tárgyalt szempontok a legfontosabbak egy reakció jellemzése szempontjából. Az erre a célra használt
rövid jelöléseken is mindig ezekre találunk utalást. Példaként megadjuk a fontosabb reakciótípusok rövid
jelöléseit:
5. Átmeneti állapot és közti termék
A kémiai folyamatoknál megkülönböztethetünk egy, vagy több "lépésben" lejátszódó reakciókat. A két típus
közötti különbséget a legegyszerűbben egy "egylépéses" és egy "kétlépéses" reakció ún. energiaprofiljával
szemléltethetjük:

A SZERVES KÉMIAI REAKCIÓK
CSOPORTOSÍTÁSA
18 Created by XMLmind XSL-FO Converter.
I. Egylépéses reakció, "mechanizmus nélküli" reakció, koncertikus reakció. 1: reaktánsok, 2: átmeneti állapot,
3: termékek, ΔE*: aktiválási energia (ΔE‡), ΔE: reakció során felszabaduló energia
II. Példák kétlépéses reakciókra. 1: reaktánsok, 2: az első részreakció átmeneti állapota, 3: közti termékek, 4: a
második részreakció átmeneti állapota, 5: termékek
Az átmeneti állapot és a közti termék között a lényeges különbség az, hogy míg az előbbi energiamaximumot
jelent és élettartama elvileg végtelen kicsi, az utóbbi energiavölgyben fekszik és ennek folytán véges élettartama
van. A közti termék ezért sok esetben kimutatható, vagy akár el is különíthető - stabilitása annál nagyobb, minél
mélyebb az energiavölgy. Egy reakciónak természetesen több, egymást követő közti terméke is lehet.
6. A csoportositás egyéb lehetséges szempontjai
• A reakcióelegy jellege szerint megküiönböztethetünk homogén és heterogén fázisban lejátszódó reakciókat.
Utóbbi típus tovább bontható a heterogén rendszer folyadék/folyadék, gáz/folyadék, gáz/szilárd stb. jellege
szerint. (Folyadék/folyadék heterogénfázisú reakció pl. a benzol nitrálása kevertsavval, 24.8.4. fejezet.)
• A szerveskémiai reakciókat fel lehet osztani katalitikus és nem-katalitikus reakciókra. A katalitikus
reakciók - attól függően, hogy a katalizátor külön fázist képvisel-e a rendszerben, vagy sem - lehetnek
heterogénkatalitikus, illetve homogénkatalitikus reakciók. Külön típusnak tekinthetjük a mikroheterogén
jellegű, enzimek által katalizált biokémiai reakciókat.
• Reakciókinetikai szempontból megkülönböztethetünk egymással párhuzamosan (esetleg összemérhető
sebességgel) lejátszódó ún. konkurens (vagy kompetitív) reakciókat, vagy egymást követő ún. konszekutív
reakciókat. A konkurens reakciók kiindulási anyaga azonos; a konszekutív reakció kiindulási anyaga az előző
reakció terméke.
• Termokémiai szempontból (a reakció hőszinezete szempontjából) beszélhetünk exoterm, endoterm vagy
termoneutrális reakciókról. Utóbbira példa az alkohol oxidáció és dehidrogénezes kombinációja (12.3.3.
fejezet).
• A szerveskémiai reakciókat gyakran nevezzük el a molekulába bevitt csoport vagy a bekövetkezett átalakulás
jellege szerint külön névvel. Ezek a nevek rendszerint nem adnak felvilágosítást a kiindulási anyag(ok)ról, a
lejátszódó folyamat ionos vagy gyökös jellegéről, stb. Ilyen típusú nevek például: halogénezés,, nitrálás,
szulfonálás, diazotálás, hidratálás, hidrogénezés, hidrolízis, oxidáció, redukció, kondenzáció, polimerizáció
stb.
• Végül megemlitjük, hogy a szerves kémiában gyakori a reakcióknak felfedezőjükről való elnevezése: ún.
névvel jelölt reakciók - névreakciók. Példák, melyekkel a tárgyalás során is fogunk találkozni: Friedel-
Crafts-reakció, Cannizzaro-reakció, Koch-szintézis, Grignard-reakciók, Zemplén-féle lebontás, stb. Ezeknek
a neveknek a használata kényelmes, mert feleslegessé teszi az egyes reakciótípusok más szempontok szerinti
körülírását, s egyúttal a tárgyalás tömörségét is elősegiti.
Itt hívjuk fel a figyelmet arra, hogy bizonyos reakciótípusok csak telítetlen rendszerek esetén játszódhatnak le,
pl. a hidrogénezés, hidratálás mindig π-kötés megszűnésével járó addíciós reakciók. A "-lízís" végű nevek, mint

A SZERVES KÉMIAI REAKCIÓK
CSOPORTOSÍTÁSA
19 Created by XMLmind XSL-FO Converter.
pl, hidrogenolízis, hidrolízis, klorolízis, pirolízis viszont egyértelműen σ-kötés(ek)nek a reagens hatására
bekövetkező felszakadására utalnak.
A különféle reakciótípusokra a későbbiek során számos példát fogunk ismertetni.

20 Created by XMLmind XSL-FO Converter.
4. fejezet - IZOMÉRIA LEHETŐSÉGEK A SZERVES KÉMIÁBAN
A bevezető fejezetben már utaltunk arra, hogy a szerves és szervetlen kémia megkülönböztetésének egyik oka
az izoméria [iszosz = egyenlő; merosz = rész (görög)] gyakori fellépése a szerves vegyületek körében.
Az egymással izomer vegyületekre általánosan jellemző, hogy azonos összegképlet (elemi összetétel) mellett
eltérő molekulaszerkezettel rendelkeznek. Ha a molekulát felépítő (azonos számú és minőségű) atomok
kapcsolódási sorrendje vagy a kémiai kötések elrendeződése különbözik, szerkezeti izomériáról beszélünk. Ha
az összegképlet azonossága mellett a molekulát alkotó atomoknak csak a térbeli elrendeződése különböző, az
izomériának egy másik alaptípusáról, a térszerkezeti vagy sztereoizomériáról van szó. Az utóbbin belül
megkülönböztetjük a geometriai és az optikai izomériát.
1. Szerkezeti izomériák
Az egymással szerkezeti izomer viszonyban álló vegyületek fizikai és kémiai tulajdonságai különböznek
egymástól.
A szénatomoknak az a hajlama, hogy tetszés szerint kapcsolódhatnak 1, 2, 3, vagy 4 másik szénatomhoz, már
viszonylag kis molekulákban is lehetővé teszi az atomok eltérő kapcsolódási sorrendjének kialakulását, vagyis
ún. "helyzeti izomerek" fellépését.
Az egyszerű felépítésű alkánok (paraffin-szénhidrogének) körében vizsgálva a kérdést, a négy szénatomot
tartalmazó C4H10 összegképletű molekula atomjai már kétféleképpen kapcsolódhatnak egymáshoz: kétféle bután-
szerkezet ismeretes. Az el nem ágazó "egyenesláncú" butánban (régebben szokás volt ezt normál, vagy n-
butánnak nevezni) a negyedik szénatom a propán egyik láncvégi szénatomjához kapcsolódik. Az elágazó láncú
butánban (amelyet általában izobutánként emlegetünk, de szisztematikus neve 2-metilpropán) a kapcsolódás a
propán-szénlánc középső szénatomján következik be, így alakul ki az elágazó szénlánc:
A homológ vegyületek sorában a szénatomok számának növekedésével rohamosan emelkedik a szerkezeti
izomerek matematikailag levezethető száma. Ezt az alkánokra a következő összeállítás szemlélteti:

IZOMÉRIA LEHETŐSÉGEK A
SZERVES KÉMIÁBAN
21 Created by XMLmind XSL-FO Converter.
Az atomok térigényét és a kötéshosszakat figyelembe véve azonban térbeli okok miatt a lehetséges izomerek
száma - elsősorban a nagyobb szénatomszámú homológok esetén - a fent megadottaknál lényegesen kisebb.
Az azonos funkciós csoportokat a szénlánc különböző helyein tartalmazó vegyületek szintén szerkezeti
izomerek. Ilyenek például a C3H8O összegképletű (normál) propil-alkohol (propan-1-ol) és az izopropil-alkohol
(propan-2-ol):
A gyűrűs vegyületeknél szintén fellép a szerkezeti izomériának ez a formája: azonos számú és minőségű
helyettesítéseket a gyűrűn különböző helyzetekben tartalmazó vegyületek egymásnak izomerjei. Példaként
bemutatjuk a benzol 1,2- (orto-), 1,3- (meta-) és 1,4- (para-) diszubsztituált származékait:
Az azonos összegképletű és láncszerkezetű, de a telítetlen kötés(ek) helyzetében különböző szerkezeteket, mint
pl. az egyenesláncú butén izomerek, szintén a szerkezeti izomerek közé soroljuk.
Az azonos szénatomszámú nyíltláncú alkének és a cikloalkánok szerkezeti izomériája más jellegű. Az egyaránt
C6H12 összegképletű hexének (3 szerkezeti izomer) és a ciklohexán között a molekulát összetartó kémiai kötések
jellegében van különbség. Például:
A később tárgyalandó tautomériát (12.5.1. fejezet) is szokás a szerkezeti izoméria egyik különleges esetének
tekinteni.
2. Térszerkezeti izomériák (sztereoizomériák)
Az idetartozó izomerekre az a jellemző, hogy az összegképleten kívül az atomok kapcsolódási sorrendje és az
atomkapcsolatok jellege is azonos a molekulákban, csak az atomok térbeli elrendeződésében van különbség. Két
fajtája ismeretes ennek az izomériának: a geometriai izoméria (Z,E-izoméria, vagy cisz, transz-izoméria) és az
optikai izoméria.
3. Geometriai izoméria
A butánnak a 4.1. ÁBRÁN bemutatott térbeli formái (konformációi) a C,C-kötések körüli szabad rotáció miatt
már szobahőmérsékleten is könnyen alakulnak át egymásba. Ezek a konformerek egymástól nem különíthetők
el, ezért csak egyféle bután ismert.

IZOMÉRIA LEHETŐSÉGEK A
SZERVES KÉMIÁBAN
22 Created by XMLmind XSL-FO Converter.
4.1. ÁBRA
A bután egymásba könnyen átalakuló konformerei
A but-2-énnek a 4.2 ÁBRÁN bemutatott geometriai izomerjei a molekulákat alkotó atomok minőségében,
számában és egymáshoz való kapcsolódásuk sorrendjében szintén nem különböznek egymástól, csupán az sp2
szénatomokhoz kapcsolódó atomcsoportok térbeli helyzetében van különbség.
4.2. ÁBRA
A but-2-én merev szerkezetű geometriai izomerjei
Azt a térszerkezeti formát, amelyben a két metilcsoport a sík azonos oldalára esik (tehát térbelileg is közelebb
van egymáshoz) (Z)-(vagy cisz)-izomernek, a másik térszerkezeti formát, melyben a két metilcsoport ellentétes
oldalon foglal helyet és így egymástól távolabb kerül, (E)- (vagy transz)-izomernek nevezzük. A két szerkezet
(a két konfiguráció) egymásnak geometriai izomerje. [A dőlt betűs - és zárójelek közé helyezett - (Z) és (E)
jelölés a német "zusammen" (együtt) és "entgegen" (szemben) szavakból ered.]
Nem mindig azonnal ilyen egyértelmű, hogy a (Z) vagy (E) konfigurációt milyen - az sp2-szénatomokhoz
kapcsolódó - csoportok térbeli helyzete alapján adjuk meg. Ilyenkor a kapcsolódó atomoknak vagy
csoportoknak a Cahn-Ingold-Prelog-szabály (4.5.3. fejezet) szerinti rangsorolása értelmében kell eljárni
olymódon, hogy a kettős kötés két szénatomjához kapcsolódó csoportokat mindkét oldalon külön-külön
rangsoroljuk és a kisebb sorszámot kapott ligandumok egymáshoz viszonyított helyzetét vesszük figyelembe.
Például:
Mivel a C,C kettős kötés körüli szabad rotációt a mintegy 270 kJ/mol erősségű π-kötés akadályozza meg,
geometriai izomereket katalizátor nélkül csak ennek megfelelő energia befektetése árán (tehát pl. magas
hőmérsékleten) lehet egymásba átalakítani. A geometriai izomerek fizikai állandói (olvadáspont, forráspont,
égéshő, dipólusmomentum stb.) eltérnek egymástól: a (Z)-but-2-én olvadáspontja például -139 °C, az (E)-
módosulaté -106 °C. Általában az (E)-forma a termodinamikailag stabilisabb. Eltérő fizikai tulajdonságaik
következtében az ilyen izomerpárok el is választhatók egymástól.
Geometriai izoméria felléphet kettős kötést nem tartalmazó, de merev gyűrűs szerkezetekben is. A cikloalkán
gyűrűkben ugyanis a gyűrűt alkotó C,C egyes kötéseknek nincs forgási szabadságuk, s ez a gyűrűhöz
kapcsolódó szubsztituensek egymáshoz viszonyított helyzetét is rögzíti. Ez ugyanolyan típusú izomériára ad
lehetőséget, mint amilyen a C,C kettős kötést tartalmazó rendszerek geometriai izomériája. Azok a gyűrűs
szerkezetek tehát, amelyek csupán a gyűrűhöz kapcsolódó szubsztituenseknek a gyűrű síkjához viszonyított

IZOMÉRIA LEHETŐSÉGEK A
SZERVES KÉMIÁBAN
23 Created by XMLmind XSL-FO Converter.
helyzetében különböznek egymástól, egymásnak ugyanolyan természetű izomerjei, mint pl. a but-2-én cisz- (Z)-
és transz- (E)- izomerjei (azonos szerkezet egymástól térbeli felépítésben különböző formái). Különösen jól
szemléltethető ez a három-, négy- és öttagú cikloalkánoknál, amelyeknél a gyűrűt alkotó szénatomok egy síkban
helyezkednek el. A 4.3. ÁBRA példaként az 1,3-dimetilciklopentán két izomerjét mutatja be.
4.3. ÁBRA
A szerkezeti izomerekhez hasonlóan, a geometriai izomerek kémiai tulajdonságai között is van különbség.
4. Optikai izoméria: szimmetria és optikai aktivitás; kiralitás
Az optikai aktivitás (forgatóképesség) jelenségét Arago fedezte fel (1811), amikor megfigyelte, hogy a
hemiéderes kvarckristályok a lineárisan polározott (rezgéseket a haladás irányára merőlegesen csak egy
térsikban végző) fény rezgési síkját elforgatják. (A hemiéderes enantiomorf alkatú kristályoknak sem
szimmetriasíkjuk, sem szimmetriacentrumuk nincs.) A kétféle hemiéderes kristályszerkezet forgatási iránya
egymással ellentétes, a forgatás nagysága pedig azonos.
Az olyan anyagokat, amelyek a poláros fény rezgési síkját elforgatják, optikailag aktív anyagoknak, magát a
jelenséget optikai aktivitásnak nevezték el. Ezt a jelenséget kezdetben csak a kristályos anyagok szerkezetének
aszimmetriájával hozták összefüggésbe.
1838-ban Biot észlelte, hogy a borkősav vizes oldata is mutat optikai aktivitást; a forgatóképesség tehát nem a
kristályos anyagok kizárólagos tulajdonsága.
Pasteur volt az első, aki 1848 és 1853 között folytatott vizsgálatai során feltételezte, hogy a szerves vegyületek
oldatban mutatott optikai aktivitásának a molekulák aszimmetrikus felépítésével kell összefüggésben lennie.
Csak később - Kekulé (1856-1863) majd Le Bel és van't Hoff (1874) munkássága révén - vált nyilvánvalóvá,
hogy az azonos összetételü molekulák aszimmetrikus térszerkezetét a Iegegyszerűbb és legáltalánosabb esetben
a szénatom tetraéderes vegyértékorientációja teszi lehetővé.
Két azonos összetételű molekula csak akkor teljesen azonos, ha egymással fedésbe hozhatók. Fedésbe nem
hozható, tehát nem azonos molekulák esetén is két lehetőség van: tükörképei vagy nem tükörképei egymásnak.
Az egymással fedésbe nem hozható tükörképi szerkezetek olyan viszonyban vannak egymással, mint a jobb és
bal kéz (a kéz görög nevéből - kheir - származik az ilyen szerkezetek királis elnevezése). Tetraéderes szénatom
esetén a kapcsolódó négy atom vagy atomcsoport ("ligandum") azonos vagy eltérő voltától függően a
következőképpen csökken a szerkezet- szimmetriaelemeinek száma:

IZOMÉRIA LEHETŐSÉGEK A
SZERVES KÉMIÁBAN
24 Created by XMLmind XSL-FO Converter.
Az utolsó, négy különböző csoporttal helyettesített központi szénatom esetén a molekula már nem rendelkezik
szimmetriaelemmel: aszimmetriacentrumot (un. aszimmetrikus szénatomot, jele: C*) tartalmazó királis
vegyület, melynek a következő két, egymással fedésbe nem hozható és optikai aktivitást mutató tükörképi
szerkezete (antipódja) lehetséges:
Az ilyen királis szerkezeteket enantiomer vegyületpároknak nevezzük. Egy királis vegyületnek csak egy
enantiomer párja lehet.
A tükörképével fedésbe nem hozható molekulaszerkezet nem feltétlenül aszimmetrikus, mert egy forgástengelyt
még tartalmazhat (ún. disszimmetrikus molekula). Optikai aktivitása ettől még megmarad. Nem tartalmazhat
azonban a molekula tükrözési sziminetriaelemeket (pl. tükörsík, szimmetriacentrum stb.) mert az ezekkel
rendelkező molekulákra felírható tükörképi szerkezetek megfelelő eltolási vagy elforgatási műveletekkel
fedésbe hozhatók egymással (akirális vegyületek) és ennek megfelelően az ilyen molekulák optikai aktivitást
nem mutatnak.
Az optikai aktivitás jelensége tehát nincs az aszimmetrikus szénatomhoz kötve, feltétele csupán az, hogy a
molekulának ne legyen tükrözési szimmetriaeleme. Tekintettel azonban arra, hogy az ilyen speciális esetek jóval
ritkábbak, itt nem foglalkozunk velük, de néhány vegyülettípus kapcsán erre a jelenségre konkrét példák
formájában még visszatérünk (Pl. 9.1.2. fejezet).
Az optikai izoméria fellépésének leggyakoribb esete tehát az aszimmetriacentrumot tartalmazó vegyületekre
jellemző centrális kiralitás. A központi atomhoz kapcsolódó atomoknak vagy atomcsoportoknak egymáshoz és
az aszimmetriacentrumhoz viszonyított elhelyezkedését a centrum konfigurációjának nevezzük. Az
enantiomerek konfigurációja mindig ellentétes.
A központi atom leggyakrabban aszimmetrikus szénatom, de lehet más tetraéderes koordinációjú atom, pl.
foszforatom is. "Ligandum"-ként magános elektronpár is szerepelhet.
Ha egy királis molekulában az aszimmetriacentrumhoz kapcsolódó csoportok közül bármelyik kettőt
felcseréljük, új, az eredetivel fedésbe nem hozható sztereoizomerhez jutunk. Egy aszimmetrikus szénatom
esetén például a kiindulási vegyület enantiomerjét kapjuk:
Az enantiomer vegyületpár definíciójából következik, hogy az enantiomerekben mind az egyes atomtávolságok,
mind az atomokat összekötő vegyértékek által bezárt szögek is azonosak, ezért nem királis (szimmetrikus)
környezetben fizikai és kémiai tulajdonságaik megegyeznek, s megegyezik a poláros fény elforgatásának
nagysága (szöge) is (a különbség az elforgatás irányában van).
Mindez azonban nem jelenti azt, hogy a két enantiomer között nincs lényeges különbség. Minden olyan
behatással szemben ugyanis, amely királis jellegű, a két enantiomer eltérő módon fog viselkedni. Ilyen királis
jellegű behatások mindenekelőtt a más királis molekulákkal lejátszódó reakciók (itt a reakciósebesség és a
termék is más lehet) és a lineárisan polározott fény (ennek magyarázatát lásd a 4.8. fejezetben), de akár pl. egy
királis oldószer is (amelyben az oldhatóságok lesznek eltérőek). Axiómaként kell azonban elfogadnunk azt,
hogy egy királis molekula enantiomerjei csak egy másik királis molekulával szemben mutatnak eltérő kémiai

IZOMÉRIA LEHETŐSÉGEK A
SZERVES KÉMIÁBAN
25 Created by XMLmind XSL-FO Converter.
viselkedést. Ebből következik pl. az is, hogy enantiomereket csak királis "segédanyag" segítségével lehet
elválasztani egymástól.
5. Egy aszimmetriacentrumot tartalmazó szerves vegyületek optikai izomériája
Egy aszimmetriacentrum esetén mindig csak két enantiomer térszerkezet lehetséges, melyek az eddigiek
alapján a következőkkel jellemezhetők:
• kémiai tulajdonságaik akirális reagensekkel szemben azonosak (eltérőek viszont királis reagensekkel
szemben!);
• fizikai és termodinamikai állandóik azonosak;
• fajlagos forgatóképességük nagysága is azonos, csak iránya ellentétes.
Inaktív anyagokból kiinduló szerveskémiai szintézisekkel előállított, aszimmetrikus szénatomot tartalmazó
vegyületek nem mutatnak optikai aktivitást. Ez érthető, ha meggondoljuk, hogy akirális környezetben az
aszimmetriacentrum kialakulásakor a két aktív módosulat képződésének valószínűsége teljesen azonos. Ezek
optikai aktivitásai tehát a keletkezett ekvimoláris elegyben kompenzálják egymást (intermolekuláris
kompenzáció): optikailag inaktív, ún. racém módosulat (racém elegy, racemát) alakul ki.
A természetben gyakran előforduló optikailag aktív anyagok az élő szervezetekben enzimatikusan lejátszódó ún.
aszimmetrikus szintézisek eredményei (vö. 4.10. fejezet).
Az egy aszimmetriacentrumot tartalmazó vegyületeknek tehát három módosulatuk lehetséges: két optikailag
aktív enantiomer és az optikailag inaktív racém módosulat. Utóbbinak már nem minden fizikai állandója azonos
az aktív módosulatokéval. Az optikailag aktív módosulat forgatási irányát (+)-szal jelöljük, ha az az óramutató
járásával megegyezik és (-)-szal, ha azzal ellentétes. (Régebben a forgatási irány jelölésére a "dexter" (jobb) és
"laevus" (bal) latín szavakból származó d- és l- jelölést használták.)
A racém - racemát elnevezések Pasteurnek a szerves vegyületek kiralitása terén végzett úttörő munkásságára
vezethetők vissza. Pasteur ugyanis alapvető kisérleteit a boroshordók falára kivált (optikailag aktív) d-borkősav
és (optikailag inaktív) szőlősav (20.2.2. fejezet) Na-NH4-sóival végezte. Ő ismerte fel, hogy a szőlősav
valójában a d- és l- borkősav 1:1 arányú elegye. Mivel pedig az ő idejében a szőlősavat racémsavnak nevezték
(a szőlőfürt latin neve racemus), az optikai izomerek 1:1 arányú elegyére a racém elegy elnevezés terjedt el.
5.1. A térszerkezet Fischer-féle ábrázolása
Az aszimmetriacentrumhoz kapcsolódó csoportokat az Emil Fischer által javasolt vetítési szabálynak
megfelelően úgy ábrázoljuk, hogy a szénlánc a vetítéssel kapott képletben függőlegesen és az aszimmetrikus
szénatomhoz képest a papír síkja felé helyezkedjék eI, s a legmagasabb oxidációs fokú funkciós csoportot felül,
a leghosszabb szénláncot pedig alul tartalmazza. A központi szénatomhoz csatlakozó egyéb csoportok helyzetét
(vízszintes irányban) így a térszerkezet egyértelműen meghatározza. Példaképpen bemutatjuk a tejsav egyik
optikai antipódjának a Fischer-féle konvenció szerinti projektív képletét:
A Fischer-féle projektív képletek a papír síkjában végzett forgatással nem hozhatók egymással fedésbe. A
képletet 90°-kal elforgatva (1), vagy két ligandumot megcserélve (2) az ellentétes konfigurációhoz, 180°-os

IZOMÉRIA LEHETŐSÉGEK A
SZERVES KÉMIÁBAN
26 Created by XMLmind XSL-FO Converter.
elforgatással (3) viszont ismét az eredeti konfigurációhoz jutunk (az S, R, D és L jelölések értelmezését lásd a
4.5.2. és 4.5.3. fejezetekben):
5.2. Relatív és abszolút konfiguráció
A tejsav két aktív módosulatáról most még csak annyit tudunk, hogy az egyik jobbra, a másik balra forgató.
Szükséges azonban, hogy a molekula térszerkezete és az anyag forgatási iránya között valamilyen összefüggést
létesítsünk. Végső célként meg kell határoznunk, hogy melyik forgatási irányhoz melyik térszerkezet tartozik,
azaz meg kell ismernünk az optikai izomerek abszolút konfigurációját.
Az abszolút konfiguráció ma már igényes röntgenkrisztallográfiai módszerekkel egyértelműen meghatározható,
azokban az időkben azonban, amikor a biológiai szempontból fontos és optikailag aktív molekulák (aminosavak,
cukrok) kémiája rohamos fejlődésnek indult, erre még nem volt lehetőség. Mivel pedig a forgatási irány és a
térszerkezet között nincs egyszerűen felismerhető összefüggés, akkoriban különböző konvenciók alakultak ki a
konfiguráció és a forgatási irány önkényes egymáshoz rendelésére.
Ezek közül végül a XIX század végén Emil Fischer javaslatára fogadták el azt a konvenciót, amely szerint a
jobbraforgató glicerinaldehidhez az előbbi szabályok értelmében kapott két térszerkezeti képlet közül azt
rendelték, amelyben a hidroxilcsoport a központi szénatomtól jobbra, a hidrogén pedig balra esik. Ezt a
konfigurációt D-vel jelölték, az ellentétes konfiguráció jele pedig L lett.
A D- és L- jelölés konvencionálisan csak a glicerinaldehid megfelelő térszerkezeti formájával
(konfigurációjával) való genetikai összefüggést, másképpen a glicerinaldehidre vonatkoztatott relatív
konfigurációt jelzi. A relatív konfiguráció és a forgatás iránya között nincs egyértelmű összefüggés. Például:
Gyakorlati szempontból még ma is fontos a relatív konfiguráció használata bizonyos vegyületcsoportoknál, mint
pl. a cukrok, aminosavak, stb. A cukrok esetében a relatív konfiguráció megadása speciális konvenció szerint
történik (v.ö. 18.3.2. fejezet). A fehérjékben előforduló α-aminosavak mind azonos térszerkezeti típusba
tartoznak, relatív konfigurációjuk a balraforgató szerinnek a glicerinaldehidre vonatkoztatott konfigurációja
alapján L, függetlenül az R-csoport jellegétől.

IZOMÉRIA LEHETŐSÉGEK A
SZERVES KÉMIÁBAN
27 Created by XMLmind XSL-FO Converter.
Az optikailag aktív anyagok abszolút térszerkezetének későbbi meghatározása során kiderült, hogy a
glicerinaldehidnek a Fischer-féle konvenció szerint D-vel jelzett szerkezete a valóságban is a jobbraforgató
módosulathoz tartozik. Ez a szerencsés véletlen azt eredményezte, hogy mindazon vegyületek esetében,
amelyek a glicerinaldehid valamelyik módosulatával - a konfigurációt meg nem változtató kémiai átalakítás
révén - tényleges genetikai kapcsolatba hozhatók, az így megállapított relatív konfiguráció azonos az abszolút
konfigurációval. A tejsav esetében tehát a balraforgató módosulathoz ténylegesen a D-vel jelölt szerkezet
tartozik.
Ez az egyezés a Fischer-féle és az abszolút konfiguráció között az előbbiek folytán az α-aminosavak esetében is
fennáll.
5.3. A Cahn-Ingold-Prelog (C.I.P.) konvenció
Fischer konvenciójának döntő szerepe volt abban, hogy a XIX-XX század fordulóján már ismert (viszonylag
egyszerű felépítésű) természetes szerves vegyületek egyértelmű sztereokémiai rendszerezése lehetővé vált. A
glicerinaldehidtől erősebben eltérő vagy bonyolultabb molekulák konfigurációjának jellemzésére azonban ez a
módszer már nem lehetett alkalmas. Mivel pedig a fejlődés során egyre több olyan vegyület abszolút
konfigurációja vált ismertté, amely a glicerinaldehiddel semmiféle genetikai kapcsolatba nem volt hozható,
szükségessé vált egy új jelölésmód kialakítása.
A C.I.P. konvenció szerint egy Cabde tipusú molekulában a központi atomhoz kapcsolódó atomokat,
atomcsoportokat (ligandumokat) részletesen kidolgozott szabályok szerint, elsősorban a csökkenő rendszám
sorrendjében 1-től 4-ig rangsoroljuk. A tetraédert ezután a 4. jelű csoporttal ellentétes oldalról nézve
megállapítjuk, hogy az 1-2-3 sorrend az óramutató járásával megegyező-e vagy ellentétes. Az abszolút
konfigurációt az első esetben (R) [rectus = jobb (latin)]), a második esetben (S) (sinister = bal) - zárójelek közé
tett - dőlt betűkkel jelöljük.
A csökkenő rendszám szerinti rangsorolás jól bemutatható például a bróm-klór-ecetsavon:
A rangsorolás főbb szabályai a következők: először a központi atomhoz közvetlenül kapcsolódó (az első
övezetbe tartozó) atomokat rangsoroljuk. Ha ez nem dönthető el egyértelműen, az ezekhez kapcsolódó atomok
rendszámait vesszük figyelembe (második övezet), s ezt mindaddig folytatjuk, amig a rangsorolás egyértelművé
nem válik. A szénatomokhoz kapcsolódó ligandumok számát mindig négyre egészítjük ki: kettős vagy hármas
kötéssel kapcsolódó atom kétszer, illetve háromszor számít. Egy övezeten belül a kapcsolódó atom rendszáma
az elsődleges, utána az azonos atomok száma a döntő (pl. -CHO > -CH2OH, de -CH2SH > -CHO). Példaképpen
a glicerinaldehid ligandumainak rangsorolását mutatjuk be:
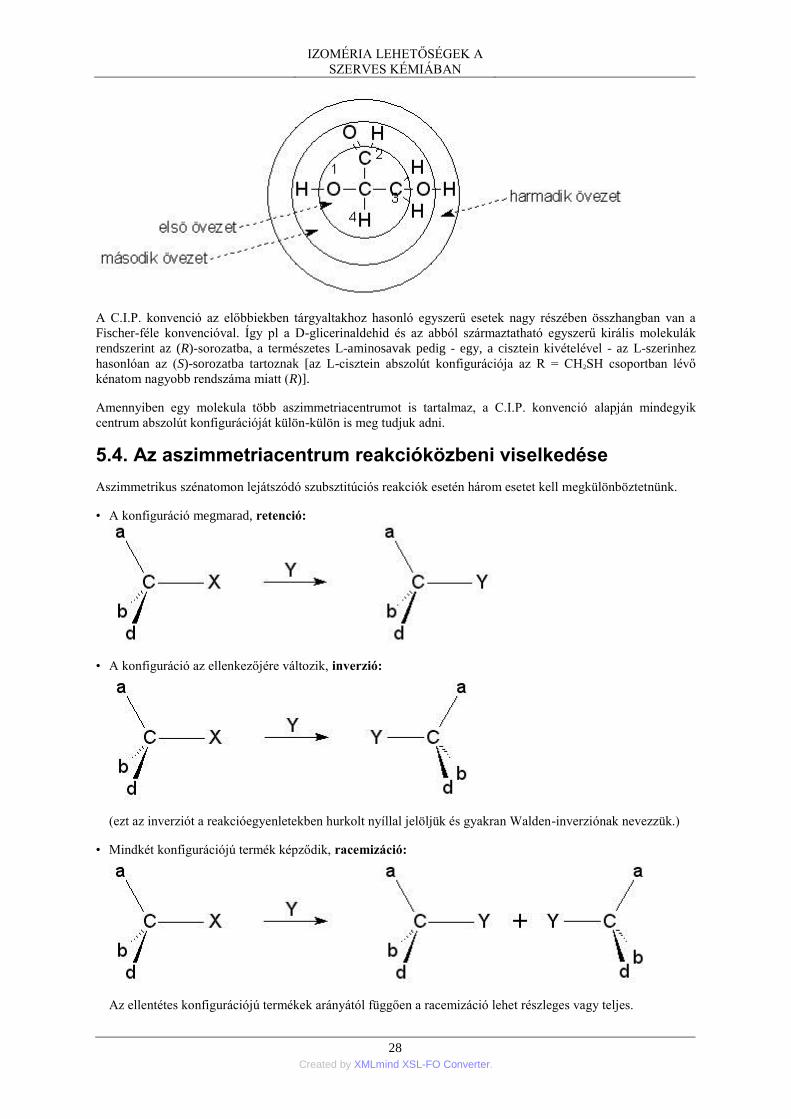
IZOMÉRIA LEHETŐSÉGEK A
SZERVES KÉMIÁBAN
28 Created by XMLmind XSL-FO Converter.
A C.I.P. konvenció az előbbiekben tárgyaltakhoz hasonló egyszerű esetek nagy részében összhangban van a
Fischer-féle konvencióval. Így pl a D-glicerinaldehid és az abból származtatható egyszerű királis molekulák
rendszerint az (R)-sorozatba, a természetes L-aminosavak pedig - egy, a cisztein kivételével - az L-szerinhez
hasonlóan az (S)-sorozatba tartoznak [az L-cisztein abszolút konfigurációja az R = CH2SH csoportban lévő
kénatom nagyobb rendszáma miatt (R)].
Amennyiben egy molekula több aszimmetriacentrumot is tartalmaz, a C.I.P. konvenció alapján mindegyik
centrum abszolút konfigurációját külön-külön is meg tudjuk adni.
5.4. Az aszimmetriacentrum reakcióközbeni viselkedése
Aszimmetrikus szénatomon lejátszódó szubsztitúciós reakciók esetén három esetet kell megkülönböztetnünk.
• A konfiguráció megmarad, retenció:
• A konfiguráció az ellenkezőjére változik, inverzió:
(ezt az inverziót a reakcióegyenletekben hurkolt nyíllal jelöljük és gyakran Walden-inverziónak nevezzük.)
• Mindkét konfigurációjú termék képződik, racemizáció:
Az ellentétes konfigurációjú termékek arányától függően a racemizáció lehet részleges vagy teljes.

IZOMÉRIA LEHETŐSÉGEK A
SZERVES KÉMIÁBAN
29 Created by XMLmind XSL-FO Converter.
A szerves vegyületek kémiájának részletes tárgyalása során mindhárom esetre fogunk konkrét példákat találni.
6. Két aszimmetriacentrumot tartalmazó szerves vegyületek optikai izomériája
Ha egy molekulában két aszimmetriacentrum van, ezek környezete (szerkezete, vagyis a hozzájuk kapcsolódó
ligandumok minősége) lehet azonos vagy különböző. Ezenkívül mindegyik centrum rendelkezhet kétféle [(R)
vagy (S)] konfigurációval.
6.1. A két aszimmetriacentrum azonos környezetű
A két azonos környezetű aszimmmetrikus szénatomot tartalmazó vegyületek klasszikus példája a borkősav.
Egyik enantiomerjének szerkezetét bemutatjuk a Fischer-féle projekcióban (A), a tényleges térbeli szerkezetet
érzékeltető ábrázolásban (B) és az azt (jobbról nézve) egyszerűsítve kifejező, ún. Newman-féle ábrázolásban (C,
vö. 5.1.4. fejezet):
A két C* környezete azonos és ebben az esetben konfigurációjuk is azonos: a C.I.P. szabályok értelmében
mindkét szénatom (R)-konfigurációjú. Az ábrázolt vegyület tehát a (2R,3R)-borkősav (I). Ennek enantiomer
párja értelemszerűen a (2S,3S)-borkősav (I'), de lehetségesek a (2R,3S) (II) és (2S,3R) (II') szerkezetek is,
amelyek szintén tükörképei egymásnak. Az összefüggéseket egyszerűsítve így ábrázolhatjuk:
I-ben és I'-ben a konfigurációk azonossága folytán a két centrum forgatóképessége összeadódik. Az (R,R)-
borkősav, amelynek konvencionális relatív konfigurációja D, jobbraforgató (+), az (S,S)-izomer (L) a
balraforgató (-) aktív módosulat. II és II'-ben az azonos környezetű (tehát külön-külön azonos mértékű forgatást
okozó) aszimmetriacentrumok konfigurációja ellentétes; ezek egymás hatását intramolekulárisan kompenzálják.
Az ilyen, intramolekuláris kompenzáció miatt inaktív módosulatokat mezo-módosulatoknak nevezzük.
Könnyen meggyőződhetünk arról, hogy ezek az utóbbi szerkezetek egymással fedésbe is hozhatók, azonosak,
vagyis II és II' ugyanazon inaktív módosulatot jelentik. A következő ábrázolásból jól látszik, hogy a mezo-
módosulat szimmetriacentrummal (i) rendelkezik, s hogy a két szerkezet a C*-C* tengely körüli 180°-os
elforgatással fedésbe hozható egymással:

IZOMÉRIA LEHETŐSÉGEK A
SZERVES KÉMIÁBAN
30 Created by XMLmind XSL-FO Converter.
Ugyanígy belátható az is, hogy a C*-C* tengely körül csak az egyik aszimmetriacentrumot elforgatva 180°-kal,
a molekulán belül jelentkezik két tükörképi szerkezet, vagyis a fedő-állásnak megfelelő konformációban a
molekulának a C*-C* kötésirányra merőlegesen szimmetriasíkja van.
Két azonos környezetű aszimmetriacentrumot tartalmazó vegyületek esetében tehát összesen négy módosulat
lehetséges: két aktív [(R,R)- és (S,S)-], a mezo [(R,S) = (S,R)] és a racém módosulat, mely utóbbi a két aktív
forma 50-50%-os elegye. Az aktiv formák egymásnak enantiomerjei, és a mezo-formával mindegyikük
diasztereomer viszonyban van.
Diasztereomer vegyületpárt alkotnak az olyan, több aszimmetriacentrummal rendelkező optikai izomerek,
amelyek a teljes molekulát tekintve nincsenek tükörképi viszonyban egymással, csak részlegesen tükörképei
egymásnak. A borkősavnak például az I és II vagy az I és II' módosulatai diasztereomer viszonyban vannak
egymással, mert az egyik aszimmetriacentrumuk azonos, a másik ellentétes (tükörképi) konfigurációjú.
A diasztereomer párok fogalmának meghatározásából következik, hogy ezek nem-azonos, egymással fedésbe
nem hozható, de ugyanakkor nem is tükörképi szerkezetek. Ebből adódóan a molekulán belüli atomtávolságok
sem mind azonosak, ezért nem azonosak a diasztereomerek
• kémiai tulajdonságai;
• fizikai állandói;
• optikai forgatás irányai és mértékei sem.
A diasztereomer vegyületpárok tehát már egyszerű fizikai módszerekkel is elválaszthatók egymástól (aminek -
mint azt a 4.9. fejezetben látni fogjuk - nagy jelentősége van az enantiomerek elválasztása szempontjából).
A diasztereoméria fogalmán belül meg szokás különböztetni az epiméria fogalmát (v.ö. 18.3.2. fejezet): az
epimerek olyan - több aszimmetriacentrummal rendelkező - vegyületek, amelyek egymástól csak egy szénatom
konfigurációjában különböznek. A borkősav előbb tárgyalt diasztereomerjei tehát egyúttal epimerek is, de ezt a
kifejezést inkább csak a kettőnél több aszimmetriacentrummal rendelkező molekuláknál szokás használni. Több
aszimmetriacentrumos aktív vegyület egy centrumának inverzióját epimerizációnak nevezzük. Így például egy
három aszimmetrikus szénatommal (A, B és C) rendelkező molekula esetében az alábbi epimerizációs
folyamatok játszódhatnak le:
A-B-C → A'-B-C, vagy
A-B-C → A-B'-C, vagy
A-B-C → A-B-C'
6.2. A két aszimmetriacentrum nem azonos környezetű
A két azonos centrumot tartalmazó vegyületek esetén - mint láttuk - a mezo-forma fellépése csökkenti a
lehetséges diasztereomer szerkezetek számát. Más a helyzet, ha a két centrum a molekulában nem azonos
környezetű.
A két eltérő környezetű centrumot A-val illetve B-vel jelölve a viszonyok legegyszerűbb ábrázolása a
következő:
Az A és B centrumok eltérő környezete (és ebből adódóan különböző mértékű forgatása) következtében a IV és
IV' szerkezetek itt már nem egy közös mezo-módosulatnak felelnek meg, hanem szintén külön-külön létező
enantiomerek. Példaképpen vizsgáljuk meg a 3-Cl-butan-2-ol sztereoizomerjeit. (Az egyszerűség kedvéért a
Fischer-féle képleteket használjuk, de megadjuk a C.I.P. konvenciónak megfelelő konfigurációjelöléseket is.)

IZOMÉRIA LEHETŐSÉGEK A
SZERVES KÉMIÁBAN
31 Created by XMLmind XSL-FO Converter.
A III-III' illetve IV-IV' molekulák enantiomer párok; a III-IV, III-IV', III'-IV és III'-IV' molekulák
páronként diasztereomer viszonyban vannak egymással. A vegyület szintézisekor a III és III' vegyületek -
amelyek azonos nagyságú, de ellentétes irányú forgatással rendelkező enantiomerek - azonos valószínűséggel
keletkeznek és racém módosulatot alkotnak. Ugyanez érvényes külön a IV és IV' enantiomerekre is, de a két
enantiomerpár mennyisége (mivel ezek egymással diasztereomer viszonyban vannak) általában eltérő. Az
enantiomerek tehát páronként külön képeznek racém módosulatokat és a teljes szintetikus termék optikailag
inaktív lesz.
Azon vegyületek esetében tehát, amelyeknek molekulájában két nem azonos környezetű aszimmetriacentrum
van, négy optikailag aktív izomer és két racém módosulat (vagyis összesen hat módosulat) lehetséges.
Megemlitjtük, hogy az III és III' típusú molekulákat (amelyek tehát a két aszimmetrikus szénatomon egy-egy
azonos szubsztituenst (itt hidrogént) hordoznak és az azonos szubsztituensek a Fischer-féle projekcióban
ellentétes oldalra esnek) összefoglaló névvel treo, a IV és IV' típusúakat (ahol ezek a szubsztituensek az azonos
oldalra kerülnek) eritro konfigurációjú vegyületeknek nevezzük.
A kettőnél több aszimmetriacentrumot tartalmazó vegyületekben minden újabb aszimmetriacentrum elvben
megkétszerezi a lehetséges optikailag aktív izomerek számát: n aszimmetrikus szénatomhoz ezért általában 2n
számú enantiomer tartozik (pl. szénhidrátok, 18. fejezet). Többgyűrűs vegyületeknél azonban a merev szerkezet
csökkentheti a lehetséges enantiomerek számát (pl. gyűrűs terpének, 10.2.1. fejezet).
7. Forgatóképesség és szerkezet
Az elforgatás nagyságának és irányának meghatározására használt polariméterek többsége a Na D-vonalnak
(589 nm) megfelelő monokromatikus fénnyel működik. Ennek megfelelően az optikailag aktív vegyületekre
jellemző fizikai adatként az ezen a hullámhosszon mért, ún. fajlagos forgatóképességet adjuk meg.
A forgatóképesség a lineárisan polározott fény hullámhosszával változik, ezen a jelenségen alapulnak az optikai
rotációs diszperzió és a cirkuláris dikroizmus elnevezésű fizikai-kémiai mérési eljárások, amelyekkel azonban
ezen jegyzet keretében nem fogalkozunk. Ezzel kapcsolatban általánosságban annyit érdemes megjegyezni,
hogy a legtöbb szerves vegyület forgatóképessége a méréshez használt fény hullámhosszának csökkenésével nő.
A fajlagos forgatóképesség meghatározása:
Ha egy anyag forgatóképességét etanolos oldatban 20 °C-on mérjük és c = 15 g aktív anyag/100 ml oldat
koncentrációnál l = 2 dm rétegvastagságú küvettában α = 9,6° forgatást mértünk az óramutató járásával
megegyező irányban, akkor az
összefüggésből meghatározható az anyag [α] fajlagos forgatóképessége:
[α] = + 32°
Mivel a fajlagos forgatóképesség az anyagi minőségen kivül a hőmérséklettől, a fény hullámhosszától és
sokszor nagy mértékben az alkalmazott oldószer minőségétől is függ, a forgatóképesség egzakt megadásakor
mindezekre utalni kell. Meg szokás adni továbbá annak az oldatnak a töménységét is, amelyben a fajlagos
forgatóképességet meghatározták:

IZOMÉRIA LEHETŐSÉGEK A
SZERVES KÉMIÁBAN
32 Created by XMLmind XSL-FO Converter.
Az optikailag aktív vegyületek fajlagos forgatóképességével kapcsolatban gyakorlati szempontból is fontos
törvényszerűség, hogy a D-vonalnál mért [α] értéke általában annál nagyobb, minél közelebb van az
aszimmetriacentrumhoz a molekulának valamilyen, a láthatóban, vagy az ultraibolyában abszorbeáló π-
elektronrendszere. Ez lehet kettőskötés, aromás gyűrű, stb., vagyis valamilyen, úgynevezett kromofor csoport,
amelynek hatása annál kifejezettebb, mennél kiterjedtebb ez a π-elektronrendszer. Ez utóbbi abból adódik, hogy
a konjugált kettős kötések számának növekedésével a kromofor fényabszorpciója az ultraibolyából egyre inkább
a látható tartomány felé tolódik el (9.1.2. fejezet), a fajlagos forgatóképesség pedig az abszorpciós maximum
közelében a legnagyobb. A viszonyokat az alábbi néhány vegyület tiszta állapotban mért forgatóképességének
összehasonlítása érzékelteti:
A legnagyobb fajlagos forgatóképességet azok a molekulák mutatják, amelyekben maga a kromofor
aszimmetrikus (vagy disszimmetrikus), vö. hexahelicén (9.1.2. fejezet).
8. Az optikai forgatóképesség oka
A 4.5. fejezetben említettük, hogy az enantiomerek kémiai viselkedése nem-királis reagensekkel szemben
azonos, de királis reagensekkel szemben eltérő. Mivel ennek az elvnek a fizikai behatások esetében is
érvényesülnie kell, a lineárisan polározott fény síkjának a két enantiomer által ellenkező irányba való elforgatása
csak akkor értelmezhető, ha a lineárisan polározott fény valamilyen szempontból királis fizikai jelenségnek
tekinthető. Valóban ez a helyzet, amint az az alábbi magyarázatból kiderül.
A lineárisan polározott fény valójában két, azonos szögsebességű, egymással ellentétes irányba (jobbra, illetve
balra) cirkulárisan polározott fénysugár eredője. A két összetevő úgy képzelhető el, mint egy jobbmenetes és
egy balmenetes csavarvonal, a csavarvonalak pedig királis alakzatok (gondoljunk arra, hogy a jobb és
balmenetes csavarok egymásnak tükörképei, de pl. a jobbmenetes csavarra a balmenetes anya nem illeszthető
rá). A két azonos szögsebességű és amplitudójú, ellentétes "menetű" cirkulárisan polározott fénysugarak tehát
"enantiomer" viszonyban vannak egymással. A kettő eredője a forgás minden fázisában ugyanazon sikba esik
mindaddig, amíg ez a fénynyaláb izotróp (nem-királis, optikailag inaktív) közegen halad át, mert ebben a két
cirkulárisan polározott komponens haladási sebessége azonos annak folytán, hogy kölcsönhatásuk a közeget
alkotó molekulákkal azonos mértékű.
Ha azonban ez a két cirkulárisan polározott fénysugár olyan közegen halad át, amelyben valamilyen királis
vegyület egyik enantiomerje feleslegben van jelen, a két komponens haladási sebessége eltérő lesz, mivel
kölcsönhatásuk mértéke ezekkel a királis molekulákkal nem lesz azonos. Ennek eredményeként az egyik
komponens kismértékben "lemarad" a másikhoz képest és ezért a kettő eredőjeként jelentkező lineárisan
polározott fény rezgési síkja a kezdeti állapothoz képest elfordul; jobbra (+), ha a jobbmenetes komponens
sebességcsökkenése a kisebb, és fordítva.
A kiralitással kapcsolatban megismert fogalmak segítségével a helyzet a következőképpen is értelmezhető: a
cirkulárisan polározott fény "enantiomerjei" a királis anyagon áthaladva azzal "diasztereomer" viszonyba
kerülnek. Az ilyen "diasztereomerek" viselkedése közötti eltérés a két fénysugár haladási sebességének
különbségében mutatkozik meg.

IZOMÉRIA LEHETŐSÉGEK A
SZERVES KÉMIÁBAN
33 Created by XMLmind XSL-FO Converter.
9. Enantiomerek elválasztása
A szerves kémiai szintézisek során általában (eltekintve az ún. aszimmetrikus szintézisektől, 4.10. fejezet) az
enantiomerek azonos mennyiségben keletkeznek. Az így keletkezett racém elegynek enantiomerekre való
szétválasztását rezolválásnak nevezzük.
A rezolválásra ma már számos módszer ismert. A mechanikus eljárás kivételével mindegyikre érvényes az,
hogy az elválasztás csak valamilyen másik királis (optikailag aktív) anyag közreműködésével valósítható meg.
Az elválasztás hatásfoka a kapott enantiomer(ek) optikai tisztaságával jellemezhető:
9.1. Mechanikus módszer
Ha az enantiomerek külön-külön kristályosodnak (ún. konglomerátumot alkotnak), a szépen fejlett enantiomorf
hemiéderes kristályok kiválogathatók a kristályhalmazból. Ez a módszer azonban ritkán alkalmazható, egyrészt,
mert viszonylag kevés racém elegy enantiomer komponensei kristályosodnak külön-külön, másrészt pedig
megfelelő méretű kristályok is csak kivételesen képződnek.
Az egyik enantiomer további tisztítására viszont alkalmazható az átkristályosítás abban az esetben, ha az
elegyben a másik enantiomerből már csak kevés (szennyezésként) van jelen. Ha ezt az oldatot hűtjük, először a
nagyobb koncentrációban jelenlévő enantiomer kezd tisztán kikristályosodni.
9.2. Biológiai módszer
Az enantiomerek oldatából az egyik komponenst megfelelő mikroorganizmussal elemésztetjük. Hátránya, hogy
ez a komponens elvész.
9.3. Diasztereomer pár képzése
A gyakorlatban rendszerint ezt a módszert alkalmazzák, mert ez a legáltalánosabban használható. Valamilyen
optikailag aktív reagens hozzáadásával diasztereomer sókat, komplexeket, vagy vegyületpárokat képezhetünk az
elválasztandó anyag jellegétől függően. Lényeges, hogy a diasztereomerekből az eredeti enantiomerek
racemizálódás nélkül regenerálhatók legyenek. Ha egy A aszimmetria-centrummal rendelkező vegyület racém
formáját (A,A') feloldás után B centrumú optikailag aktív vegyülettel reagáltatjuk, a keletkező AB és A'B
vegyületek már nem enantiomer, hanem diasztereomer viszonyban lesznek egymással, tehát pl. frakcionált
kristályosítással elkülöníthetők. Regenerálás után többé-kevésbé tiszta enantiomer(eke)t kapunk:
9.4. Kromatográfiás elválasztás
A rezolválás megoldható a racém elegynek optikailag aktív adszorbensen (pl. cellulózon) végzett
kromatografálásával is. Ezt az teszi lehetővé, hogy az enantiomerek az adszorbens királis molekuláihoz eltérő
módon illeszkednek, s így az adszorbensen különböző mértékben kötődnek meg [lényegében itt is (labilis)
"diasztereomer" párok keletkeznek)].
10. Aszimmetrikus szintézis

IZOMÉRIA LEHETŐSÉGEK A
SZERVES KÉMIÁBAN
34 Created by XMLmind XSL-FO Converter.
Amennyiben a reakció során új aszimmetrikus szénatom alakul ki a molekulában, mindig számolnunk kell
mindkét konfigurációjú termék képződésével. A gyakorlat szempontjából két lényeges esetet kell
megkülönböztetnünk:
1. a molekula már tartalmazott (egy vagy több) aszimmetrikus szénatomot,
2. az új aszimmetrikus szénatom az első a molekulában.
Az 1) esetben diasztereomerek képződnek: A → AB + AB', melyek minden szempontból eltérő tulajdonságú
vegyületek lévén, nem azonos sebességgel, tehát eltérő mennyiségben fognak keletkezni. Az ilyen típusú
átalakítások során ezért gyakran igen jó szelektivitás érhető el az egyik vagy a másik diasztereomer javára.
A 2) eset ezzel szemben, ha teljesen akirális körülmények között dolgozunk, szükségszerűen racém elegy
képződéséhez vezet. Ilyenkor az aszimmetrikus szintézis csak úgy valósítható meg, ha valamilyen módon
kiralitást viszünk a rendszerbe, pl. királis katalizátor (enzim, királis komplex) vagy királis reagens segitségével.
Ez az aszimmetrikus komponens mintegy indukálja a termék aszimmetriáját ("aszimmetrikus indukció").

35 Created by XMLmind XSL-FO Converter.
5. fejezet - ALKÁNOK
(Telített szénhidrogének, "paraffinok")
Az alkánok olyan szerves vegyületek, melyeknek molekulái csak σ-kötésekkel összekapcsolt szén- és
hidrogénatomokból épülnek fel, C,C kettős-, vagy hármaskötéseket nem tartalmaznak. Ezeknek a "telített"
szénhidrogéneknek szénlánca tehát kizárólag sp3 hibridizációjú "négyszomszédos" (négy szén és/vagy
hidrogénatomhoz kapcsolódó) szénatomokból áll.
A "telített" jelző azt kívánja kifejezni, hogy a molekula nem tartalmaz olyan "telítetlen" C,C kapcsolatot - vagyis
π-kötést - amely a szénlánc elszakadása nélkül még további atomokat vehet fel ("telítődhet").
Még ma is igen elterjedten használják ezekre a vegyületekre a régi, hagyományos, "paraffinok" elnevezést. Ez a
név arra utal, hogy az alkánok a szerves kémiában használatos reagensek nagyrészével szemben nem, vagy csak
igen kevéssé reakcióképesek; ezt a nevet eredetileg a latin "parum affinis" (alig vonzódó) kifejezésből képezték.
-- A molekulaszerkezet ábrázolása
A legegyszerűbb alkánban, a metánban (CH4) az egyetlen szénatomhoz négy hidrogénatom kapcsolódik,
tetraéderes elrendezésben. Ha ebben a szerkezetben a kapcsolódó atomok térbeli helyzetét az atomszimbólumok
segitségével jelöljük, a molekula térszerkezeti képletét kapjuk [5.1. ÁBRA a)]. A gyakorlatban ennek síkba
vetített változatát, az ún. szerkezeti képletet szokás inkább használni [5.1. ÁBRA; b), c)]; ez az írásmód a vetítés
már tárgyalt szabályainak ismeretében (4.5. fejezet) mindig egyértelmű képet ad az atomok kapcsolódási
sorrendjéről és egymáshoz viszonyított helyzetéről.
5.1. ÁBRA
Az összes atomkapcsolatoknak (különösen a C,H-kapcsolatoknak) ilyen részletes jelölése azonban sokszor csak
zavarólag hat; ezért általában a célszerűség határozza meg, hogy milyen egyszerűsitést alkalmazunk a
képletirásban. Ilyen pl. a gyakran alkalmazott d) szerinti megoldás, de a következő fejezetben ismertetünk egy
még radikálisabb egyszerűsítést, amelyet majd elsősorban nagyobb molekulák esetében tudunk és fogunk
használni.
1. Nyíltláncú alkánok; általános áttekintés
Nyíltláncúaknak nevezzük azokat az alkánokat, amelyekben a szénatomok lánca nem zárt, azaz nem alkot
gyűrűs szerkezeteket. Az alkánok tárgyalását ezzel a csoporttal kezdjük, a zártláncú, gyűrűs cikloalkánokról az
5.4. fejezetben lesz szó.
1.1. Az alkánok homológ sora

ALKÁNOK
36 Created by XMLmind XSL-FO Converter.
A két szénatomot tartalmazó telített nyíltláncú szénhidrogén, az etán összegképlete C2H6. Szerkezete úgy
fogható fel, hogy a metán egy hidrogénatomja helyébe egy három hidrogénatomhoz kapcsolódó szénatom lépett
(a különbség tehát -CH2-):
Az etántól ismét csak egy -CH2- (metilén) csoporttal különböző, három szénatomos propán szénláncán már
csak a "láncvégi" szénatomokat fedi három-három hidrogénatom; a "láncmenti" szénatomhoz csak két
hidrogénatom tud kapcsolódni:
A szénatomok számát fokozatosan tovább növelve a nyíltláncú alkánoknak olyan sorozatát kapjuk, melynek
minden tagja (egyébként azonos típusú szénváz-szerkezet mellett) csak egy -CH2- csoporttal nagyobb, mint az
előtte álló tag. Az ilyen vegyületsorozatokat homológ soroknak nevezzük [homos = azonos (görög)]. A
homológ sorok tagjainak összegképletére általános összefüggés adható meg: a nyíltláncú alkánok (paraffin
szénhidrogének) homológ sorának általános képlete például CnH2n+2 (ahol n = 1,2,3,...).
Az egymással homológ (egy sorozatba tartozó) vegyületek kémiai és fizikai tulajdonságai is közel állnak
egymáshoz; a homológ sorok fogalmának bevezetése ennélfogva egyszerűsítést jelent a szerves vegyületek
rendszerezésében.
1.2. A szénatomok rendűsége
A szénláncot alkotó sp3 szénatomok láncbeli helyzete különböző lehet; ezt a szénatom rendűségével
jellemezzük. A láncvégi (csak egy szénatommal szomszédos) szénatomot primer, a láncmentit szekunder, az
egyszeres láncelágazásnál állót tercier; a kétszeres láncelágazásnál állót pedig kvaterner szénatomnak
nevezzük. Ennek megfelelően a különböző rendű szénatomokhoz kapcsolódó hidrogénatomok száma
sorrendben: 3, 2, 1, illetve 0. Egy nyolc szénatomos alkán (oktán) izomer alábbi képletében (baloldali szerkezet)
a szénatomok rendűségét római számokkal jelöltük:
A már megismert szerkezetábrázolás mellett a jobboldali vonalas ábra ugyanennek az alkánnak a - már a
bevezetésben is említett - lehető legegyszerűbb, de mégis egyértelmű szerkezeti leírása. A vonalak csak a C,C-
kötéseket jelzik, az elágazásoknál, a töréseknél és a vonalak végeinél vannak a szénatomok, az ezekhez
kapcsolódó hidrogének száma pedig a szénatom négyvegyértékűségéből kikövetkeztethető. Később látni fogjuk,
hogy ez az ábrázolásmód igen bonyolult molekulákra is alkalmazható olymódon, hogy az egyéb (ún. hetero-
)atomokat az ezeknek megfelelő betűszimbólumokkal jelöljük.
1.3. Az alkánok elnevezése
A szerves vegyületek elnevezésére általában használhatunk
• triviális nevet, amely nem szisztematikusan levezetett, de megszokás alapján a gyakorlatban elterjedt,
többnyire rövid elnevezés, vagy
• racionális, (szisztematikus) nevet, amely a vegyület molekulaszerkezetéről is felvilágosítást ad.
A molekulaszerkezetet legtökéletesebben a IUPAC nevezéktannal tudjuk megadni.
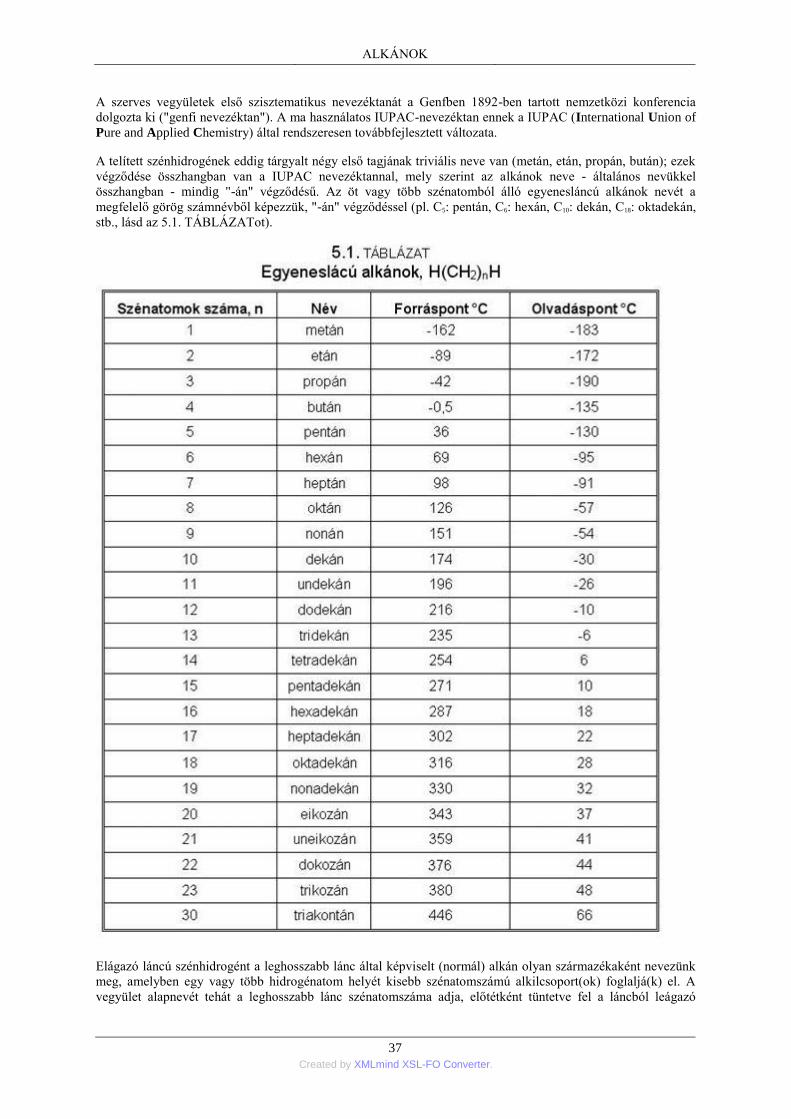
ALKÁNOK
37 Created by XMLmind XSL-FO Converter.
A szerves vegyületek első szisztematikus nevezéktanát a Genfben 1892-ben tartott nemzetközi konferencia
dolgozta ki ("genfi nevezéktan"). A ma használatos IUPAC-nevezéktan ennek a IUPAC (International Union of
Pure and Applied Chemistry) által rendszeresen továbbfejlesztett változata.
A telített szénhidrogének eddig tárgyalt négy első tagjának triviális neve van (metán, etán, propán, bután); ezek
végződése összhangban van a IUPAC nevezéktannal, mely szerint az alkánok neve - általános nevükkel
összhangban - mindig "-án" végződésű. Az öt vagy több szénatomból álló egyenesláncú alkánok nevét a
megfelelő görög számnévből képezzük, "-án" végződéssel (pl. C5: pentán, C6: hexán, C10: dekán, C18: oktadekán,
stb., lásd az 5.1. TÁBLÁZATot).
Elágazó láncú szénhidrogént a leghosszabb lánc által képviselt (normál) alkán olyan származékaként nevezünk
meg, amelyben egy vagy több hidrogénatom helyét kisebb szénatomszámú alkilcsoport(ok) foglaljá(k) el. A
vegyület alapnevét tehát a leghosszabb lánc szénatomszáma adja, előtétként tüntetve fel a láncból leágazó
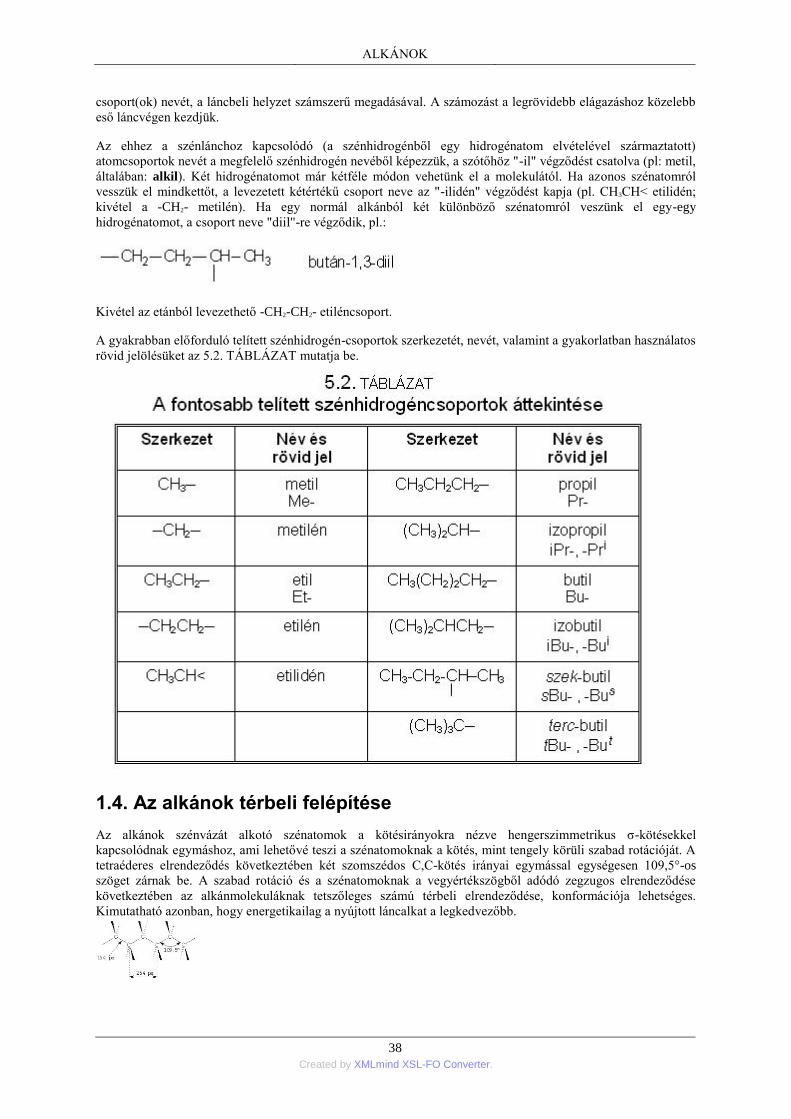
ALKÁNOK
38 Created by XMLmind XSL-FO Converter.
csoport(ok) nevét, a láncbeli helyzet számszerű megadásával. A számozást a legrövidebb elágazáshoz közelebb
eső láncvégen kezdjük.
Az ehhez a szénlánchoz kapcsolódó (a szénhidrogénből egy hidrogénatom elvételével származtatott)
atomcsoportok nevét a megfelelő szénhidrogén nevéből képezzük, a szótőhöz "-il" végződést csatolva (pl: metil,
általában: alkil). Két hidrogénatomot már kétféle módon vehetünk el a molekulától. Ha azonos szénatomról
vesszük el mindkettőt, a levezetett kétértékű csoport neve az "-ilidén" végződést kapja (pl. CH3CH< etilidén;
kivétel a -CH2- metilén). Ha egy normál alkánból két különböző szénatomról veszünk el egy-egy
hidrogénatomot, a csoport neve "diil"-re végződik, pl.:
Kivétel az etánból levezethető -CH2-CH2- etiléncsoport.
A gyakrabban előforduló telített szénhidrogén-csoportok szerkezetét, nevét, valamint a gyakorlatban használatos
rövid jelölésüket az 5.2. TÁBLÁZAT mutatja be.
1.4. Az alkánok térbeli felépítése
Az alkánok szénvázát alkotó szénatomok a kötésirányokra nézve hengerszimmetrikus σ-kötésekkel
kapcsolódnak egymáshoz, ami lehetővé teszi a szénatomoknak a kötés, mint tengely körüli szabad rotációját. A
tetraéderes elrendeződés következtében két szomszédos C,C-kötés irányai egymással egységesen 109,5°-os
szöget zárnak be. A szabad rotáció és a szénatomoknak a vegyértékszögből adódó zegzugos elrendeződése
következtében az alkánmolekuláknak tetszőleges számú térbeli elrendeződése, konformációja lehetséges.
Kimutatható azonban, hogy energetikailag a nyújtott láncalkat a legkedvezőbb.
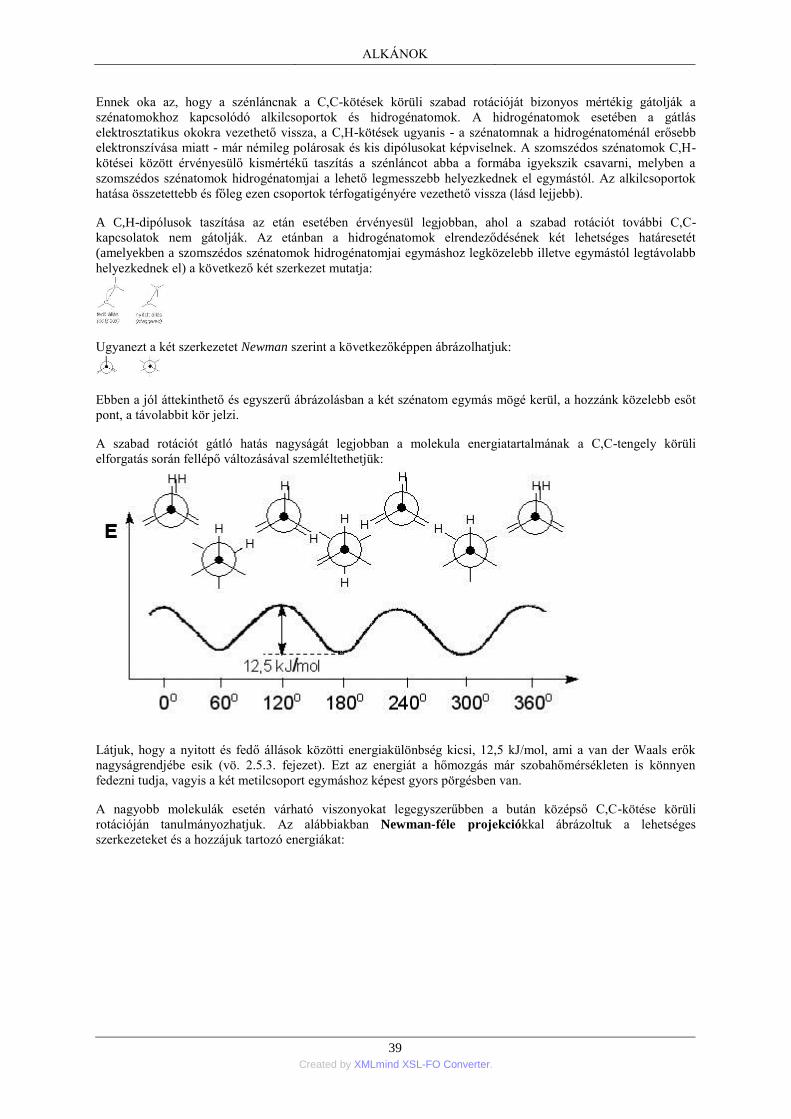
ALKÁNOK
39 Created by XMLmind XSL-FO Converter.
Ennek oka az, hogy a szénláncnak a C,C-kötések körüli szabad rotációját bizonyos mértékig gátolják a
szénatomokhoz kapcsolódó alkilcsoportok és hidrogénatomok. A hidrogénatomok esetében a gátlás
elektrosztatikus okokra vezethető vissza, a C,H-kötések ugyanis - a szénatomnak a hidrogénatoménál erősebb
elektronszívása miatt - már némileg polárosak és kis dipólusokat képviselnek. A szomszédos szénatomok C,H-
kötései között érvényesülő kismértékű taszítás a szénláncot abba a formába igyekszik csavarni, melyben a
szomszédos szénatomok hidrogénatomjai a lehető legmesszebb helyezkednek el egymástól. Az alkilcsoportok
hatása összetettebb és főleg ezen csoportok térfogatigényére vezethető vissza (lásd lejjebb).
A C,H-dipólusok taszítása az etán esetében érvényesül legjobban, ahol a szabad rotációt további C,C-
kapcsolatok nem gátolják. Az etánban a hidrogénatomok elrendeződésének két lehetséges határesetét
(amelyekben a szomszédos szénatomok hidrogénatomjai egymáshoz legközelebb illetve egymástól legtávolabb
helyezkednek el) a következő két szerkezet mutatja:
Ugyanezt a két szerkezetet Newman szerint a következőképpen ábrázolhatjuk:
Ebben a jól áttekinthető és egyszerű ábrázolásban a két szénatom egymás mögé kerül, a hozzánk közelebb esőt
pont, a távolabbit kör jelzi.
A szabad rotációt gátló hatás nagyságát legjobban a molekula energiatartalmának a C,C-tengely körüli
elforgatás során fellépő változásával szemléltethetjük:
Látjuk, hogy a nyitott és fedő állások közötti energiakülönbség kicsi, 12,5 kJ/mol, ami a van der Waals erők
nagyságrendjébe esik (vö. 2.5.3. fejezet). Ezt az energiát a hőmozgás már szobahőmérsékleten is könnyen
fedezni tudja, vagyis a két metilcsoport egymáshoz képest gyors pörgésben van.
A nagyobb molekulák esetén várható viszonyokat legegyszerűbben a bután középső C,C-kötése körüli
rotációján tanulmányozhatjuk. Az alábbiakban Newman-féle projekciókkal ábrázoltuk a lehetséges
szerkezeteket és a hozzájuk tartozó energiákat:
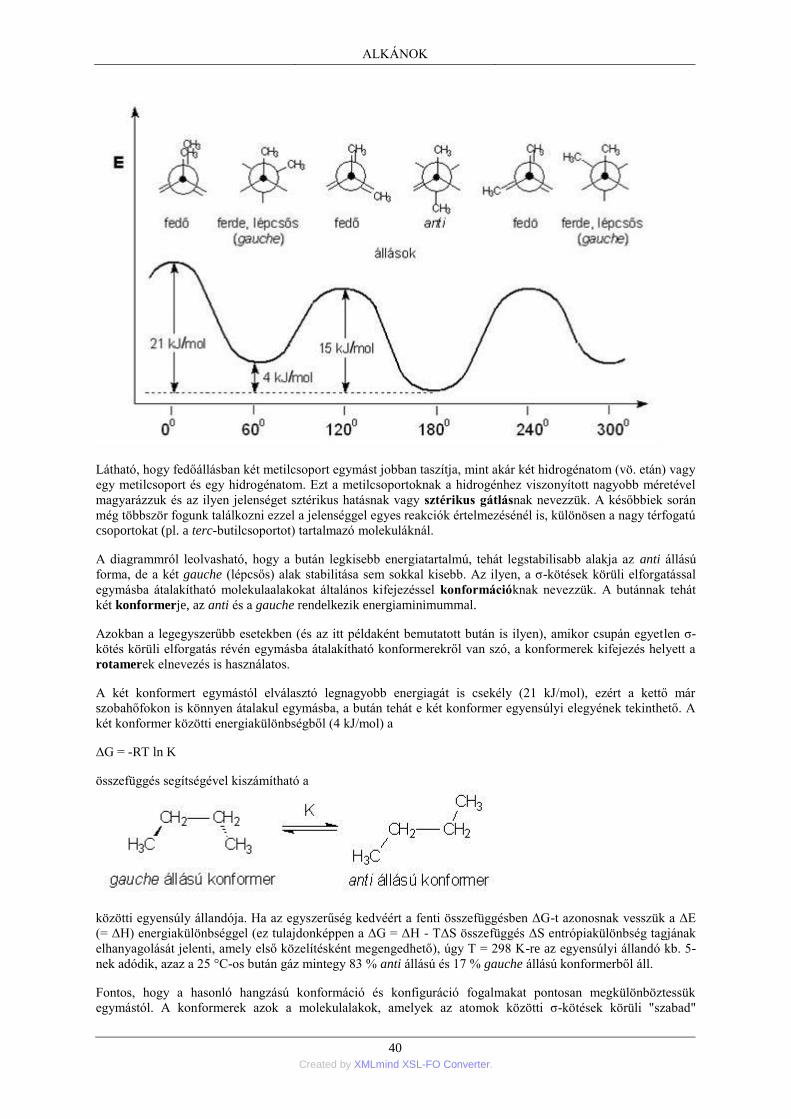
ALKÁNOK
40 Created by XMLmind XSL-FO Converter.
Látható, hogy fedőállásban két metilcsoport egymást jobban taszítja, mint akár két hidrogénatom (vö. etán) vagy
egy metilcsoport és egy hidrogénatom. Ezt a metilcsoportoknak a hidrogénhez viszonyított nagyobb méretével
magyarázzuk és az ilyen jelenséget sztérikus hatásnak vagy sztérikus gátlásnak nevezzük. A későbbiek során
még többször fogunk találkozni ezzel a jelenséggel egyes reakciók értelmezésénél is, különösen a nagy térfogatú
csoportokat (pl. a terc-butilcsoportot) tartalmazó molekuláknál.
A diagrammról leolvasható, hogy a bután legkisebb energiatartalmú, tehát legstabilisabb alakja az anti állású
forma, de a két gauche (lépcsős) alak stabilitása sem sokkal kisebb. Az ilyen, a σ-kötések körüli elforgatással
egymásba átalakítható molekulaalakokat általános kifejezéssel konformációknak nevezzük. A butánnak tehát
két konformerje, az anti és a gauche rendelkezik energiaminimummal.
Azokban a legegyszerűbb esetekben (és az itt példaként bemutatott bután is ilyen), amikor csupán egyetlen σ-
kötés körüli elforgatás révén egymásba átalakítható konformerekről van szó, a konformerek kifejezés helyett a
rotamerek elnevezés is használatos.
A két konformert egymástól elválasztó legnagyobb energiagát is csekély (21 kJ/mol), ezért a kettő már
szobahőfokon is könnyen átalakul egymásba, a bután tehát e két konformer egyensúlyi elegyének tekinthető. A
két konformer közötti energiakülönbségből (4 kJ/mol) a
ΔG = -RT ln K
összefüggés segítségével kiszámítható a
közötti egyensúly állandója. Ha az egyszerűség kedvéért a fenti összefüggésben ΔG-t azonosnak vesszük a ΔE
(= ΔH) energiakülönbséggel (ez tulajdonképpen a ΔG = ΔH - TΔS összefüggés ΔS entrópiakülönbség tagjának
elhanyagolását jelenti, amely első közelítésként megengedhető), úgy T = 298 K-re az egyensúlyi állandó kb. 5-
nek adódik, azaz a 25 °C-os bután gáz mintegy 83 % anti állású és 17 % gauche állású konformerből áll.
Fontos, hogy a hasonló hangzású konformáció és konfiguráció fogalmakat pontosan megkülönböztessük
egymástól. A konformerek azok a molekulalakok, amelyek az atomok közötti σ-kötések körüli "szabad"

ALKÁNOK
41 Created by XMLmind XSL-FO Converter.
rotációval egymásba átforgathatók (pl. a bután anti és gauche alakjai). Ezzel szemben az eltérő konfigurációjú
molekulák olyan izomerek, amelyek csak valamilyen kovalens kötés elszakítása révén alakíthatók át egymásba
(pl. a but-2-én (E)- és (Z)- izomerjei a C,C π-kötés felszakításával).
1.5. Az alkánok szerkezeti izomériája
Az alkánok sorában a C4H10 butántól kezdve lehetőség van a szerkezeti izoméria fellépésére (részletesen lásd
4.1. fejezet). Az elágazó láncú izomereket általában megkülönböztethetjük az izo- előtéttel, de az n- előtétet a
normál, vagyis egyenes szénláncú vegyületeknél nem használjuk - pl. a pentán név önmagában mindig a normál
pentánt jelenti. Bonyolultabb esetekben a szerkezetek egyértelmű jellemzésére a IUPAC nevezéktant kell
alkalmazni.
1.6. Az alkánok fizikai tulajdonságai
-- Halmazállapot
Az alkánok fizikai tulajdonságait a molekulák közötti erőhatások határozzák meg. A molekulákat általában a
hőmozgással konkuráló, igen kis hatótávolságú van der Waals-erők kapcsolják össze. A molekulák között ható
erők kicsinységéből és kis hatótávolságából következik, hogy csak akkor képesek a szomszédos molekulákat
viszonylag rendezett halmazokká asszociálni, ha a molekulák elég nagy felületen érintkezhetnek egymással,
vagyis a kapcsolódási pontok száma elég nagy a hőmozgás ellensúlyozásához. Ebből következik, hogy az
olvadáspont és forráspont függ a molekula nagyságától és alakjától, vagyis
• az egyforma láncszerkezetű szénhidrogének homológ sorában az oIvadáspont és forráspont a szénlánc
hosszával fokozatosan emelkedik. Szobahőmérsékleten a butánnál hosszabb egyenesláncú alkánok már
folyadékok, a hexadekántól (C16) kezdve pedig szilárd halmazállapotúak (5.1. TÁBLÁZAT);
• az elágazó láncú alkánok olvadás- és forráspontja alacsonyabb, mint az egyenesláncú izomereké.
Olvadáspont szempontjából kivételt képeznek a teljesen merev, szimmetrikus felépitésü szerkezetek: a
neopentán (2,2-dimetilpropán) olvadáspontja például csak alig valamivel alacsonyabb a forráspontjánál.
Ugyanezt a jelenséget tükrözi az is, hogy a merev szerkezetű metán és etán olvadáspontja magasabb, mint a
"hajlékonyabb" propáné (5.1. TÁBLÁZAT).
-- Oldhatóság
A szénhidrogének általában apoláros oldószerekben jól oldódó, "lipofil" (zsírkedvelő, zsíroldó) vegyületek.
Hidrogénhíd-kötés kialakítására képes, ("protonaktiv", protikus), erősen poláros oldószerekben azonban
gyakorlatilag oldhatatlanok, "hidrofóbok" (víztaszítók).
Az oldhatóságot az határozza meg, hogy a vegyület molekulái milyen mértékben képesek az adott oldószer
molekuláival asszociálódni. Erősen poláros protikus oldószerek (pl. víz) az alkánokat azért nem oldják, mert az
oldószermolekulák asszociációja olyan erős, hogy halmazrendszerükbe a csak kismértékben polározódó
szénhidrogének nem tudnak behatolni. Ezek az oldószerek tehát nem szolvatálják a szénhidrogén-molekulákat.
Az alkánok a különböző oldószerekben azok apoláros jellegének arányában (elsősorban tehát
szénhidrogénekben) oldódnak. Oldódnak továbbá a hasonlóan apoláros halogénezett szénhidrogénekben
(kloroform, szén-tetraklorid). A poláros vegyületek közül elegyednek azokkal, amelyek hidrogénhíd-kötés
kialakítására nem képesek (pl. aceton), vagy amelyekben a szénhidrogén jelleg dominál (pl. magasabb
alkoholok).
2. Alkánok kinyerése és előállítása
2.1. Előfordulás és kinyerés; kőolaj és földgáz
Alkánok nagy mennyiségben állnak rendelkezésre a természetben a földgázban és kőolajban, de minden esetben
egymáshoz fizikai és kémiai tulajdonságaikban közelálló vegyületek elegyeinek formájában. A homológ sor
első tagjai (C1-C5) megfelelő módszerekkel tisztán elkülöníthetők a természetes előfordulásokból; a nagyobb
molekulatömegű komponensek tiszta állapotban való előállítása azonban gyakran csak szintetikus úton,
laboratóriumi módszerekkel lehetséges.

ALKÁNOK
42 Created by XMLmind XSL-FO Converter.
-- A földgáz
A nyers földgáz - előfordulási helyétől függően - 70-90 % (esetenként közel 100 %) metánt, 1-7 % etánt, 0-3 %
propánt és 0-4 % magasabb (C4-C6) telitett szénhidrogént tartalmaz. Nem-szénhidrogén szennyezésként a
legtöbb földgáz tartalmaz dinitrogént, szén-dioxidot és kén-hidrogént is.
A kőolajjal együtt előforduló ("nedves") földgáz általában nagyobb arányban tartalmaz szénhidrogén-
homológokat, míg egyes önálló ("száraz") földgáz előfordulásokra tekintélyes mennyiségű nem-szénhidrogén
alkotórész jelenléte lehet jellemző.
A világ földgáztermelése évente jelenleg kb. 2600 milliárd m3 (2003-as adat). Ennek nagy részét
tüzelőanyagként hasznosítják, de a vegyipar (a petrolkémiai ipar) is egyre nagyobb mennyiséget dolgoz fel. A
földgáz metántartalmából többek között szintézisgázt (CO + H2), hidrogént, acetilént, finom eloszlású kormot
állítanak elő.
-- A kőolaj
Az kőolaj (ásványolaj) többnyire sötét színű, sűrűn folyó anyag, amelynek összetétele az előfordulási helytől
függően változik. Megkülönböztetünk paraffinbázisú, intermedierbázisú és nafténbázisú kőolajat (utóbbi főleg
telitett gyűrűs szénhidrogéneket, ún. nafténeket tartalmaz). Egyes kőolajokban viszonylag nagy mennyiségű
(30-40 %) aromás szénhidrogén is található.
A különböző kőolajok átlagos elemi összetétele a következő határok között mozog: 81-87 % C, 10-14 % H, 0-7
% O, 0-6 % S, 0-1 % N. Kis mennyiségben más elemeket (így különböző fémeket) is tartalmazhatnak. A világ
évi kőolaj termelése 2003-ban kb. 3800 milliárd tonna volt.
A kőolaj-feldolgozás első lépése a víz és a kőzettörmelék elkülönitése ülepítéssel. Az ezt követő gáztalanitás (a
20 °C-nál alacsonyabb hőmérsékleten forró komponensek elválasztása) során tekintélyes mennyiségű propán-
bután gázt nyernek. Az így "stabilizált" kőolaj atmoszférikus desztillációjakor a következő frakciókat szokás
elkülöníteni:
• 60-200 °C között forró párlat: nyers benzin
• 180-280 °C között forró párlat: petróleum
• 260-360 °C között forró párlat: gázolaj
• 360 °C felett forró párlat: kenő- és paraffinolaj
• párlási maradék: petróleumaszfalt (gudron)
Az egyes frakciókat a könnyen gyantásodó alkatrészek eltávolítása céljából további tisztításnak vetik alá
("raffinálás"), majd újbóli desztillálással további frakciókra bontják. A kenő- és paraffinolaj-frakció illetve a
párlási maradék továbbfeldolgozására vákuumdesztillációt vagy vákuum-vízgőz-desztillációt szokás alkalmazni.
A kőolaj-feldolgozás főbb termékei:
• motorhajtó anyagok (benzin, gázolaj);
• különböző kenőolajok (orsó- gép-, motorolaj, stb.), szilárd paraffin;
• bitumen.
Előállításuk részletesebb ismertetése az kőolajipari technológia feladata.
A motorhajtó anyagok közül a benzinek kompressziótűrésének jellemzésére használják az ún. oktánszámot.
Oktánszámon annak a (normál) heptán - izooktán (2,2,4-trimetilpentán) elegynek a százalékos izooktán
tartalmát értjük, melynek kompressziótűrése azonos a vizsgált benzinével. Az izooktán oktánszáma tehát 100, a
heptáné 0. Az egyenesláncú szénhidrogének ugyanis a motorban öngyulladásra hajlamosak (a levegő -
szénhidrogén elegy a komprimálás közben felmelegedve gyújtás nélkül is berobban), vagyis rossz a
kompressziótűrésük. A gázelegy idő előtti gyulladása a motor káros "kopogását" okozza. Üzemanyagként tehát
elsősorban a nagy oktánszámú, főleg elágazóláncú szénhidrogéneket, vagy nagy oktánszámú adalékokat (pl.
terc-butil-metiléter) tartalmazó benzinek alkalmasak. A benzinek oktánszámát régebben ún. kopogásgátló

ALKÁNOK
43 Created by XMLmind XSL-FO Converter.
adalékokkal (pl. tetraetil-ólom) is javították, az ólomtartalmú benzinek felhasználását azonban
környezetvédelmi okokból mára már betiltották.
A kőolajtermékek között legértékesebb motorhajtó benzin mennyiségének növelésére szolgáló különböző
eljárások közül a magasabb szénhidrogéneket tartalmazó párlatok katalitikus krakkolását, az aromás
szénhidrogének arányát növelő reformálást, az izobutánnak C3-C4 alkénekkel végzett alkilezését (alkilát benzin),
valamint az egyenesláncú alkánoknak elágazó láncúvá való izomerizálását említjük meg. (Néhány ilyen
módszer kémiájával a későbbiekben meg fogunk ismerkedni.)
A Diesel-motorok működése, a benzinmotorokkal ellentétben, az üzemanyag öngyulladására épül, vagyis a
főleg egyenesláncú szénhidrogéneket tartalmazó gázolaj (Diesel-olaj) tekinthető jobb üzemanyagnak. A
gázolajok jellemzésére használatos ún. cetánszám olyan (normál) cetán - α-metilnaftalin elegy százalékos
cetántartalmát jelenti, melynek gyulladási tulajdonságai a vizsgált olajéval azonosak.
2.2. Egyenesláncú alkánok kinyerése kőolaj-párlatokból
-- Molekulaszűrővel
A molekulaszűrők (molekulasziták) zeolitszerü, mesterséges, kristályos szerkezetü porózus alumínium-
szilikátok, melyeknek pórusnyílása nagyon egyenletes (a pórusátmérő áItalában 4-500 pm). A metánt és etánt
mindkét típus, a propántól C14-ig terjedő normál-alkánokat csak az 500 pm-es molekulaszűrő abszorbeálja. Az
elválasztást az teszi lehetővé, hogy az elágazó láncú és aromás szénhidrogéneket sem a 400 pm, sem az 500 pm
pórusátmérőjű molekulaszűrő nem abszorbeálja (a pórusokban nem férnek el). Az adszorbeálódott egyenesláncú
alkánokat a molekulaszitából melegítéssel lehet visszanyerni.
Mivel az egyenesláncú alkánok kémiai feldolgozás szempontjából értékesebbek, mint az izoalkánok, a
molekulaszitákat alkalmazó eljárás gyakorlati szempontból is jelentős.
-- Karbamidos adduktképzéssel
A karbamid az egyenesláncú alkánokkal (és általában a négynél nagyobb szénatomszámú egyenesláncú
szénhidrogén-származékokkal) kristályos adduktot, molekulavegyületet (ún. zárványvegyületet, "klatrátot")
képez, mely szűréssel vagy centrifugálással elválasztható az olajtól. Ennek mosás utáni vizes elbontásával a
normál alkánok elegye kinyerhető. Elágazó láncú alkánokkal, naftén vagy aromás szénhidrogénekkel a
karbamid szilárd terméket nem képez. Az egyenes szénláncú vegyületek jelenlétében a karbamid hexagonális
kristályrácsban kristályosodik és ezek a vegyületek ennek a "csatornáiban" helyezkednek el; elágazó láncok
ezekbe a csatornákba nem tudnak beilleszkedni. Az ilyen klatrátok egy szénatomra szárnitott képződéshője kb. 6
kJ/mol.
2.3. Alkánok előállítása szintézissel
A kőolajfeldolgozás nagyobb molekulatömegű termékei mindig szénhidrogének elegyei. Bonyolultabb
szerkezetű egyedi szénhidrogének előállítása laboratóriumi módszerekkel történik az erre alkalmas, funkciós
csoportokkal rendelkező származékokból. Ennek legegyszerűbb módja valamely - a kívánt szénláncot már
tartalmazó - vegyület funkciós csoportjának kicserélése hidrogénre:
R-Y → R-H
Általában alkoholokból szokás kiindulni, a hidroxilcsoportot valamilyen közbenső funkciós csoporton [-X, -
MgX (X = halogén)], vagy C,C kettős kötésen keresztül cserélve ki hidrogénre. A szokásos módszerekről itt
csak vázlatos áttekintést adunk; ezeket részletesebben a megfelelő funkciós csoportot tartalmazó vegyületeknél
fogjuk tárgyalni. Néhány lehetőség vázlatosan:

ALKÁNOK
44 Created by XMLmind XSL-FO Converter.
3. Az alkánok kémiai tulajdonságai
Az alkánok általában kémiailag kevéssé reakcióképes vegyületek. Ennek oka C,C- és C,H-kötéseik egyenletes
elektroneloszlása, apoláros karaktere. Apoláros természetükből következően az alkánok kíméletes körülmények
között elektronszívó vagy elektrondús reakciópartnerekkel általában nem reagálnak (savaknak, lúgoknak,
oxidálószereknek ellenállnak). Ugyancsak apoláros jellegükből következik, hogy kémiai reakcióik nagyrésze
gyökös mechanizmussal lejátszódó láncreakció. Kémiai reakciót ugyanis elsősorban azok a reakciópartnerek
képesek kiváltani, amelyek elegendő energiával rendelkeznek ahhoz, hogy az alkánmolekulák atomkapcsolatait
- még az erős C,H-kapcsolatokat is - homolitikusan szakítsák fel (ilyenek többek között a labilis paramágneses
atomok, mint pl. a klóratom). Az alkánok gyökös reakcióinak rossz szelektivitása is arra vezethető vissza, hogy
a különböző helyzetű szénatomok reakciókészségében csekély különbségek vannak ugyan, de a molekula
támadhatósága nagyjából mindenütt egyforma, azaz az alkánmolekulának nincsenek speciálisan támadható
pontjai (funkciós csoportjai).
Az alkánok - a rájuk elsősorban jellemző gyökös reakciók mellett - megfelelő katalizátorok jelenlétében ionos
mechanizmussal, viszonylag enyhe körülmények között lejátszódó reakciókra is hajlamosak (pl.
izomerizálódás). Reakcióikat a következő csoportositásban tárgyaljuk:
3.1. Klórozás
Az alkánok klórozása erősen exoterm folyamat. A metán klórozásának reakcióhője ΔH = -104 kJ/mol; a reakció
egyes részlépéseire a következőkben megadott reakcióhő értékek is konkrétan a metán klórozására vonakoznak.
Alkánok klórgázzal csak akkor reagálnak, ha a klórgáz molekulái energiafelvétellel "aktiválódnak" (A), vagyis
szétesnek párosítatlan elektront tartalmazó energiadús szabad klóratomokra:
A képződött szabad klóratomok egy része az elegyben klórmolekulákká rekombinálódik, másik része az alkán
molekuláival ütközve - sikeres ütközés esetén - arról hidrogénatomot szakit le, paramágneses alkilgyök
keletkezése közben (L):
Ez a lépés gyengén endoterm, ΔH = + 12 kJ/mol. Ha a reakcióképes labilis alkilgyök a párosítatlan elektront
hordozó szénatomjánál klórmolekulával ütközik, alkil-klorid mellett új szabad klóratom keletkezik (L'), amely
újabb (L) reakcióban tud résztvenni és ílymódon exoterm "láncreakció" indulhat meg:
Ez az a lépés, amely jelentős energianyereséggel jár, ΔH = - 116 kJ/mol.

ALKÁNOK
45 Created by XMLmind XSL-FO Converter.
Az (L) és (L') ún. láncvivő reakciók mindaddig ismétlődnek, amíg a reakciópartnerek egyike el nem fogy.
Lánczáró reakció akkor következik be, ha vagy a klóratomok rekombinálódnak, vagy két másik paramágneses
partner sikeres ütközés révén összekapcsolódik (Z vagy Z'):
A lánczárás feltétele, hogy a paramágneses gyökök kombinálódásakor felszabaduló energiát a rendszer ütközés
révén le tudja adni pl. a reaktor falának vagy valamilyen szennyező anyagnak, molekulának.
Az alkánok ilyen közvetlen halogénezésének csak a klórszármazékok előállítása szempontjából van gyakorlati
jelentősége. A közvetlen fluorozás ugyanis olyan hevesen játszódik le, hogy a termék lángtünemény közben
rögtön el is bomlik; a bróm közvetlen belépése a molekulába már csak rossz hatásfokkal megy, a jód pedig már
egyáltalán nem reagál az alkánokkal. A reakciósorozat kritikus részlépése az (L) láncvivő reakció, amelynek
endoterm (energiaigényes) jellege a Cl → Br → I sorban nő és ezzel együtt nő az ezen lépéshez szükséges
aktiválási energia nagysága is.
Az aktiválási (A) folyamat energiaszükségletének fedezése szerint a klórozás három típusát különböztetjük meg:
--Termikus klórozás
Megvalósításához 300-400 °C szükséges. Ez az iparban leginkább alkalmazott eljárás (pl. metán, pentánok vagy
táblás paraffin klórozására).
--Fotokémiai klórozás
A molekuláris klór Cl,Cl-kötésének termikus elszakításához 240 kJ/mol energia szükséges, fotolízis esetén
ennek a 495 nm hullámhosszuságú fény felel meg. Később tárgyalandó okok (10.4. fejezet) miatt azonban
fotokémiai aktiválásnál ehhez ennél nagyobb energia, illetve kisebb, 375 nm hullámhosszuságú fény szükséges.
A metán és etán klórozása már napfény hatására is robbanásszerűen mehet végbe; megfelelő körülmények
alkalmazásával azonban az alkánok fotokémiai klórozása irányított folyamattá alakítható. A túlklórozódás
elkerülése végett általában a sztöchiometrikusnál kevesebb klórt szokás alkalmazni.
--Katalitikus (iniciátoros) klórozás
Általában peroxidok bomlásakor képződő gyökökkel indítják a láncreakciót. Gyökforrás lehet pl. valamilyen
diacil-peroxid:
Helyesebb a peroxidot iniciátornak nevezni és nem katalizátornak, mert bomlásával csak elindítja a gyökös
láncfolyamatot, de nem katalizálja azt.
--Termékösszetétel
Az alkánok gyökös szubsztitúciós reakciói közül a klórozás az egyik legjobban tanulmányozott. Ezért ennek a
reakciótipusnak kapcsán tárgyaljuk a telitett szénhidrogének szubsztitúciós reakcióinak legfontosabb
törvényszerűségeit. Megjegyezzük, hogy az alább ismertetendő szabályok - főbb vonásaikban - a telitett
szénhidrogén-csoportokat (molekularészeket) tartalmazó bonyolultabb molekulák ezen csoportjaira is
érvényesek.
Az alkánok szubsztitúciója során felmerülő legfontosabb kérdés a keletkező termékek szerkezete. A
tapasztalat ugyanis azt mutatja, hogy a szubsztitúció nem teljesen statisztikusan következik be, hanem a tercier
szénatomokhoz kapcsolódó hidrogénatom könnyebben cserélődik ki a szekunder szénatomhoz kapcsolódó
hidrogénatomnál és a primer szénatomhoz kapcsolódó hidrogénatom szubsztituálható legkevésbé. Ennek oka a
C,H-kötések erőssége közötti különbségben keresendő:
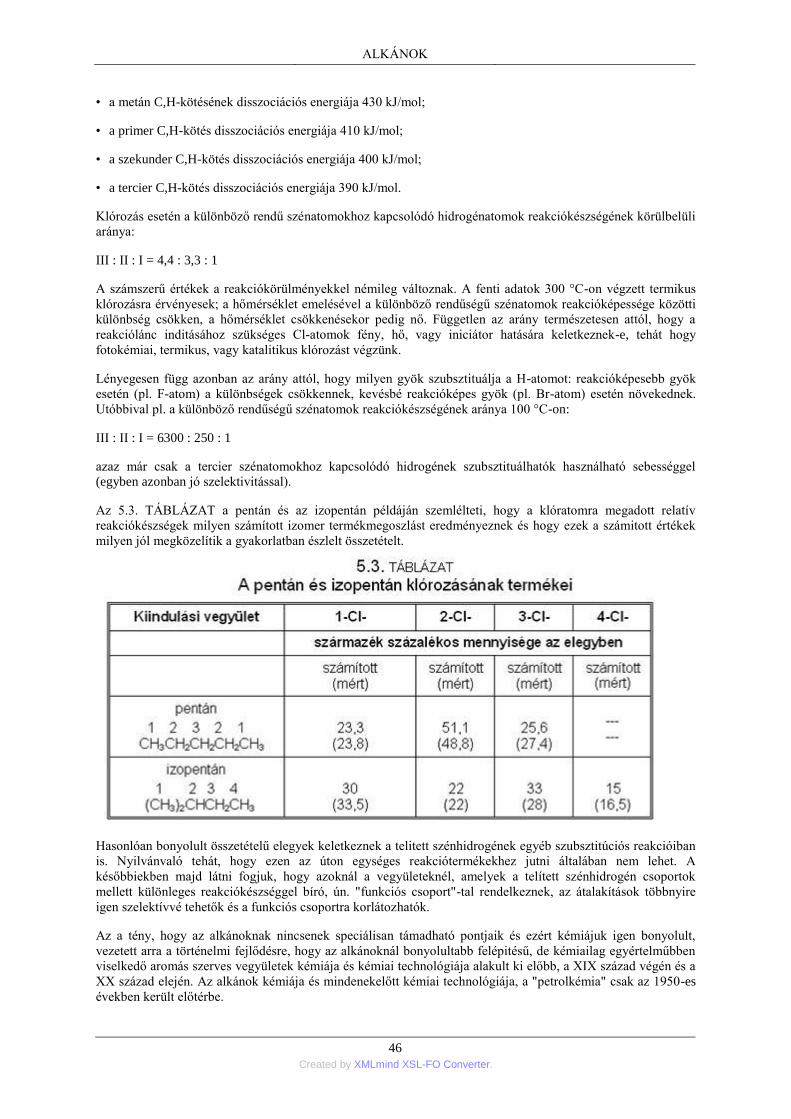
ALKÁNOK
46 Created by XMLmind XSL-FO Converter.
• a metán C,H-kötésének disszociációs energiája 430 kJ/mol;
• a primer C,H-kötés disszociációs energiája 410 kJ/mol;
• a szekunder C,H-kötés disszociációs energiája 400 kJ/mol;
• a tercier C,H-kötés disszociációs energiája 390 kJ/mol.
Klórozás esetén a különböző rendű szénatomokhoz kapcsolódó hidrogénatomok reakciókészségének körülbelüli
aránya:
III : II : I = 4,4 : 3,3 : 1
A számszerű értékek a reakciókörülményekkel némileg változnak. A fenti adatok 300 °C-on végzett termikus
klórozásra érvényesek; a hőmérséklet emelésével a különböző rendűségű szénatomok reakcióképessége közötti
különbség csökken, a hőmérséklet csökkenésekor pedig nő. Független az arány természetesen attól, hogy a
reakciólánc inditásához szükséges Cl-atomok fény, hő, vagy iniciátor hatására keletkeznek-e, tehát hogy
fotokémiai, termikus, vagy katalitikus klórozást végzünk.
Lényegesen függ azonban az arány attól, hogy milyen gyök szubsztituálja a H-atomot: reakcióképesebb gyök
esetén (pl. F-atom) a különbségek csökkennek, kevésbé reakcióképes gyök (pl. Br-atom) esetén növekednek.
Utóbbival pl. a különböző rendűségű szénatomok reakciókészségének aránya 100 °C-on:
III : II : I = 6300 : 250 : 1
azaz már csak a tercier szénatomokhoz kapcsolódó hidrogének szubsztituálhatók használható sebességgel
(egyben azonban jó szelektivitással).
Az 5.3. TÁBLÁZAT a pentán és az izopentán példáján szemlélteti, hogy a klóratomra megadott relatív
reakciókészségek milyen számított izomer termékmegoszlást eredményeznek és hogy ezek a számitott értékek
milyen jól megközelítik a gyakorlatban észlelt összetételt.
Hasonlóan bonyolult összetételű elegyek keletkeznek a telitett szénhidrogének egyéb szubsztitúciós reakcióiban
is. Nyilvánvaló tehát, hogy ezen az úton egységes reakciótermékekhez jutni általában nem lehet. A
későbbiekben majd látni fogjuk, hogy azoknál a vegyületeknél, amelyek a telített szénhidrogén csoportok
mellett különleges reakciókészséggel bíró, ún. "funkciós csoport"-tal rendelkeznek, az átalakítások többnyire
igen szelektívvé tehetők és a funkciós csoportra korlátozhatók.
Az a tény, hogy az alkánoknak nincsenek speciálisan támadható pontjaik és ezért kémiájuk igen bonyolult,
vezetett arra a történelmi fejlődésre, hogy az alkánoknál bonyolultabb felépitésű, de kémiailag egyértelműbben
viselkedő aromás szerves vegyületek kémiája és kémiai technológiája alakult ki előbb, a XIX század végén és a
XX század elején. Az alkánok kémiája és mindenekelőtt kémiai technológiája, a "petrolkémia" csak az 1950-es
években került előtérbe.

ALKÁNOK
47 Created by XMLmind XSL-FO Converter.
3.2. Oxidáció
A paramágneses molekuláris oxigén kémiai reakciókban gyökként viselkedik: viszonylag alacsony
hőmérsékleten megtámadja az alkánokat. Az oxigénnel szembeni reakciókészség a hőmérséklet emelkedésével
rohamosan növekszik, a szénhidrogének tűzveszélyes anyagok. Bizonyos hőmérsékleten a szénhidrogének
meggyulladnak és elégnek (tüzelés, robbanómotorok). A telített szénhidrogének tökéletes égésének általános
reakcióegyenlete:
CnH2n+2 + (3n + 1)/2 O2 → n CO2 + (n + 1) H2O
Alacsony hőmérsékleten az oxidáció termékei különböző oxigéntartalmú vegyületek. A termék összetétele még
bonyolultabb, mint a klórozásnál; a reakció kevésbé szelektív. A termékek főleg karbonsavak,
karbonilvegyületek és alkoholok.
A reakciót indító alkilgyök képződésének energiaszükséglete elég tekintélyes, ezért az aktiválás aránylag lassú
folyamat:
A keletkezett alkilgyök már számottevő aktiválási energia nélkül reagál tovább:
Az alkil-hidroperoxidok könnyen bomlanak tovább, főleg karbonilvegyületek és karbonsavak keletkezése
közben (vö. 14.3.3. fejezet).
3.3. Hőbomlás
A telitett szénhidrogének magas hőmérsékleten (500-1000 °C) a C,C- és C,H-kötések homolitikus felszakadása
következtében bomlást szenvednek, melynek során kisebb molekulatömegű szénhidrogének bonyolult
összetételű elegye képződik. A hőbomlási folyamatok kémiáját részletesebben a telítetlen szénhidrogéneknél
fogjuk tárgyalni.
3.4. Izomerizálás
A telített szénhidrogénekben a C,C-kötések igen stabilisak és általában csak magasabb hőmérsékleten szakadnak
el (lásd hőbomlás). A telített szénhidrogéneknél, vagy az ilyen csoportot tartalmazó bonyolultabb szerves
vegyületeknél (amelyeket később fogunk tárgyalni) ezért a kémiai átalakítások során a vegyület szénvázával
mint a molekula változatlan "gerincével" szokás számolni.
Ismeretesek azonban olyan katalizátorok, amelyeknek hatására a telített szénhidrogének szénváza már enyhe
reakciókörülmények között is átrendeződik: izomerizálódás következik be. Ilyen hatású katalizátorok az
alumínium-klorid (100 °C-on), valamint az alumínium-szilikátok (500 °C-on). Így pl. bután AlCl3, HCl és kis
mennyiségű (< 0,1 %) telítetlen szénhidrogén (alkén, 6. fejezet), pl. butén jelenlétében izobutánná
izomerizálható:
A telített szénhidrogének izomerizációja egyensúlyi reakció, amelynél a katalizátor természetesen csak a
termodinamikai jellemzők által meghatározott eqyensúlyi összetétel eléréséig tud hatást kifejteni. Nagyobb

ALKÁNOK
48 Created by XMLmind XSL-FO Converter.
molekulatömegű szénhidrogéneknél már nem csak két, hanem több izomer is képződhet és a reakciótermék
bonyolult összetételű.
Az izomerizáció - az eddig tárgyalt reakcióktól eltérően - nem gyökös, hanem ionos közbenső termékeken át
játszódik le. A folyamat induló lépése az alumínium-kloridból és hidrogén-kloridból képződő komplex sav
reakciója a kis mennyiségben jelen lévő telítetlen szénhidrogénnel, melynek során karbokation
(karbéniumion) alakul ki:
Alumínium-szilikát katalizátor esetén a karbokation kialakulásához szükséges protont a katalizátor felületén
elhelyezkedő SiO4 tetraéderek egyik oxigénatomjához kapcsolódó - erősen savas jellegű - hidrogénatom
szolgáltatja.
A karbokation úgy stabilizálódik, hogy egy telített szénhidrogén molekulából hidridiont von el és ezzel újabb
karbokationt alakit ki:
A karbéniumionok jellemző tulajdonsága, hogy molekulán belüli hidridion vagy alkanidion (alkil-anion)
vándorlással - az egyensúlyi koncentráció eléréséig - átalakulhatnak egymásba (vagyis átrendeződhetnek).
Például:
Az izomerizáció befejező lépéseként az izobutil-kation a nagy feleslegben jelenlévő telitett szénhidrogénből
hidridiont vonhat el, és új karbokation kialakítása (vagyis újabb reakciósorozat inditása) közben izobután
formájában stabilizálódik:
A láncreakció mindaddig folytatódik, amig az izomer szénhidrogének egyensúlyi elegye ki nem alakul.
Általánosan érvényes szabály, hogy a karbokationok stabilitása annál nagyobb, minél nagyobb a pozitív
töltést hordozó szénatom rendűsége. Ez a megfigyelés a "karbénium-szénatom"-hoz kapcsolódó alkilcsoportok
töltés-delokalizáló tulajdonságával értelmezhető. Minél több primer vagy szekunder alkilcsoport kapcsolódik a
karbénium szénatomhoz, annál egyenletesebb elektroneloszlású (vagyis annál stabilisabb) szerkezet alakul ki.
Ezt a hatást legjobban a terc-butil kationban a karbénium-szénatomhoz kapcsolódó három metilcsoport
biztositja. Ez esetben a pozitív töltés eloszlása egyenletesebb:

ALKÁNOK
49 Created by XMLmind XSL-FO Converter.
Az alkilcsoportok ezen töltés-delokalizáló tulajdonsága nem valamilyen pozitív induktív effektus
következménye (a 2.4. fejezetben leírtak értelmében az alkilcsoportoknak sem pozitív, sem negatív induktív
effektusuk nem lehet), hanem az ún. σ-konjugáció (hiperkonjugáció) eredménye (lásd lejjebb).
Az alkánok izomerizációja a legújabb eredmények szerint igen erős savakkal (ún. "szupersavakkal" - ezzel a
névvel illetik a tömény kénsavnál erősebb savakat) alkének távollétében is könnyen lejátszódik. Ilyenkor a
láncreakciót indító karbéniumion az alkánból hidridion elvonás révén alakul ki:
Hangsúlyozni kell, hogy a szénváz könnyű átalakulása, izomerizálódása csak az elektronhiányos
karbokationok tulajdonsága. A telített szénhidrogénekből szubsztitúciós reakciók közben (pl, klórozás,
oxidáció) kialakuló alkilgyökök (amelyek párosítatlan elektront tartalmaznak ugyan, de nem elektrondeficites
molekulatöredékek) átrendeződésre nem hajlamosak. Ez az oka annak, hogy a szénhidrogének reakcióik
során általában megtartják az eredeti szénvázat.
--A σ-konjugáció (hiperkonjugáció)
A σ-konjugáció (régebbi nevén: hiperkonjugáció) lényegét legjobban a karbénium-szénatomhoz kapcsolódó
metilcsoport hatásán szemléltethetjük. Az sp2 hibridizációjú, tehát planáris karbénium-szénatom üres p-pályája
és a metilcsoport egyik C,H-kötésének σ-molekulapályája ugyanis - megfelelő konformáció esetén - fedő
állásba kerülnek (5.1. fejezet), ami némi töltés-delokalizációt tesz lehetővé:
Hatását tekintve a σ-konjugáció azt a látszatot kelti, mintha a metilcsoportnak pozitív induktív effektusa volna,
mivel csökkenti a pozitív töltést az sp2 szénatomon. A régebbi szakirodalomban ezért elterjedten találkozunk
azzal a megfogalmazással, hogy az alkilcsoportok pozitív induktív effektussal rendelkeznek.
Ugyanez a kölcsönhatás tud fellépni a karbénium-szénatom üres p-pályája és egy C,C σ-kötés között is. A σ-
konjugáció tehát akkor is jelentkezik, ha hosszabb szénláncú alkilcsoportok kapcsolódnak a karbénium-
szénatomhoz. Ez azt jelenti, hogy minden alkilcsoport rendelkezik ezzel a látszólagos pozitív induktív
effektussal.
4. Cikloalkánok
A cikloalkánokban a molekula vázát alkotó sp3 szénlánc (vagy annak egy része) gyűrűt, "ciklust" alkot. Ezek a
vegyületek, melyeket cikloparaffinoknak vagy naftén-szénhidrogéneknek is szokás nevezni, lényegében a
nyíltláncú alkánokhoz hasonló természetű vegyületek.
A szénatomok gyűrűvé kapcsolódása természetesen merevíti a molekulát: a vázalkotó szénatomok mozgási
lehetősége kisebb, mint a nyílt szénláncban. Az atomok erősebb rendezettségének megfelelően a cikloalkánok
molekulái szorosabban illeszkednek egymáshoz (vagyis olvadás- és forráspontjuk magasabb, sűrűségük
nagyobb), mint az analóg nyíltláncú szénhidrogéneké:
Mint látjuk, az egyszerű (egy-gyűrűs) cikloalkánok elnevezésének alapelvei azonosak a nyíltláncú
szénhidrogéneknél tárgyaltakkal, azzal a különbséggel, hogy a gyűrűs jelleget az alapnév elé tett "ciklo" szóval
jelezzük és csak a gyűrűt alkotó szénatomok láncát tekintjük alapszénhidrogénnek. A gyűrűhöz kapcsolódó
szénlánc(ok) ("oldalláncok") helyzetét számozással adjuk meg: a gyűrű szénatomjait úgy számozzuk meg, hogy
mindig a rövidebb oldallánc helyét jelölő szám legyen a kisebb.

ALKÁNOK
50 Created by XMLmind XSL-FO Converter.
4.1. Egy-gyűrűs rendszerek
-- Kémiai viselkedés és szerkezet
Bár a cikloalkánok általában ugyanolyan indifferens (kevéssé reakcióképes) vegyületek, mint a nyíltláncú
alkánok, a homológ sor első tagja, a ciklopropán, sőt bizonyos vonatkozásban még a ciklobután is "telítetlen"
jellegű: az alkénekhez hasonlóan addíciós reakciókban reagál.
Ez a "telítetlenség"-ből adódó reakciókészség a gyűrűtagszám növekedésével (tehát a vegyértékszögnek a
tetraédereshez való közeledésével) fokozatosan csökken: a ciklopropán 80 °C-on, a ciklobután pedig csak 200
°C-on hidrogéneződik ugyanolyan sebeséggel, mint (a nagyon formálisan "kéttagú gyűrűs vegyületként" is
felfogható) etén szobahőmérsékleten.
A fenti reakciókban megnyilvánuló addíciós készséget a klasszikus Baeyer-féle feszültségelmélet az egy
térsíkba eső C,C-kapcsolatoknak a tetraéderes vegyértékiránytól eltérő irányából adódó feszültségnek
tulajdonította. Az elmélet szerint a molekula ebből a feszített állapotból szabadulni igyekszik, és ez okozza a
fokozott reakciókészséget.
A három- és négytagú cikloalkán-gyűrűk látszólagos telítetlenségét és ennek a gyűrűtagszámmal való változását
ma már kvantumkémiai alapon értelmezzük. A gyűrűt felépítő sp3 szénatomok AO-jainak orientációja ugyanis
kisebb-nagyobb mértékben eltér a kialakult gyűrű geometriájából adódó kapcsolódási irányoktól. Ennek két
következménye van:
• egyrészt a σ-kötéseket kialakító orbitálok nem tudnak olyan mértékben átlapolni, mint a kevésbé merev
nyíltláncú szerkezetekben, az átfedés tehát annál kisebb, minél nagyobb az előbb említett irányok közötti
szögeltérés;
• másrészt az így kialakult - már eleve gyengébb - σ-kötés részben kikerül a szénatomokon lévő
szubsztituensek árnyékoló hatása alól és ezáltal egy külső reakciópartner számára könnyebben
hozzáférhetővé, támadhatóbbá válik:
Az egyes C,C-kötéseknek az átfedés csökkenéséből adódó gyengülése a következő: a ciklopropán gyűrűben 40
kJ/mol, a ciklobutánban 27 kJ/mol, az alább tárgyalandó ciklopentánban pedig már csak 5 kJ/mol.
A ciklopentán kémiai viselkedésében már teljesen a nyíltláncú alkánokra hasonlít, a szabályos ötszög szárai
ugyanis 108°-os szöget zárnak be egymással (a tetraéderes vegyértékirány 109,5°). Az öttagú gyűrűt alkotó sp3
szénatomok tehát csaknem pontosan a normális kapcsolódási irányaiknak megfelelően és majdnem a C,C-
egyeskötés normális erősségével kapcsolódnak egymáshoz. Ezért a ciklopentán hidrogénezéssel szemben magas
hőmérsékleten is indifferens és a kloroformos brómoldatot sem színteleníti el.

ALKÁNOK
51 Created by XMLmind XSL-FO Converter.
A ciklopentán gyűrű azonban - annak ellenére, hogy az említett szögeltérés minimális - mégsem teljesen
planáris. Ennek oka az, hogy a szénatomok síkbeli elrendeződése esetén a hidrogénatomok a lehető legközelebb
kerülnének egymáshoz (ugyanúgy, mint az etán energiamaximummal rendelkező fedő állásában), s ez
feszültséget okoz a molekulában. Mivel viszont a molekula általában azt az alakot igyekszik felvenni, amely az
összes alkotó atomok elrendezése szempontjából energiaminimumot képvisel, a gyűrűt alkotó szénatomok kissé
kitérnek az egysíkú elrendezésből, annak ellenére, hogy ezáltal a kötésirányok alkotta szögek a gyűrűben kissé
csökkennek, s az ebből adódó feszültség némileg megnő.
-- A ciklohexán szerkezete
A ciklohexán kémiai viselkedés szempontjából szintén teljesen alkánjellegű: katalitikus hidrogénezéssel,
molekuláris brómmal, oxidálószerekkel szemben ugyanolyan indifferens, mint a hexán. A hattagú gyűrűt alkotó
sp3 szénatomok ugyanis a tetraéderes vegyértékszögeknek megfelelő irányokból kapcsolódnak össze. Ez
természetesen csak úgy lehetséges, hogy a gyűrű nem egy síkban helyezkedik el.
A ciklohexánnak több konformációja (térbeli alakja) létezik, ezek közül a legfontosabb - és legstabilisabb - a
szék konformáció (vagy szék forma). Ebben a konformációban a szomszédos szénatomok egymáshoz
viszonyított helyzete az etán nyitott állásának felel meg. Tulajdonképpen két székforma létezik, melyek a C,C-
kötések körüli elfordulások útján, 45 kJ/mol energia befektetése révén egymásba át tudnak alakulni (5.2.
ÁBRA).
5.2. ÁBRA
A ciklohexángyűrű székformájában mindegyik szénatomon az egyik szubsztituens közelítőleg merőleges a
gyűrű átlagos síkjára, "axiális" helyzetű (a), a másik pedig megközelítőleg radiálisan fekszik a gyűrű síkjában,
"ekvatoriális" helyzetű (e). Amikor a molekula a hőmozgás hatására másik székformába megy át, a
szubsztituensek kapcsolódási iránya is megváltozik: az axiális kapcsolat ekvatoriálissá válik és fordítva (lásd
5.2. ÁBRA). Mivel a gyűrűt alkotó szénatomokhoz kapcsolódó atomok zsúfoltsága (egymásra gyakorolt taszító
hatása) az axiális helyzetben nagyobb, a ciklohexángyűrűhöz kapcsolódó szubsztituensek - ha erre lehetőség van
- elsősorban ekvatoriális helyzetet foglalnak el (vö. a glükopiranóz térszerkezetével, 18.3.3. fejezet).
A metilciklohexánban például az a szék-konformáció az uralkodó, melyben a metilcsoport ekvatoriális helyzetű:
a másik lehetséges székforma már nagyobb energiájú. Nagyterjedelmű szubsztituens, amely axiális helyzetben
el sem fér (pl. a terc-butilcsoport), már teljesen egy adott konformációba merevíti a gyűrűt.
A ciklohexán szék konformációja mellett meg kell még különböztetnünk a kád és csavart kád konformációkat,
melyeket az alábbi rajzok szemléltetnek:
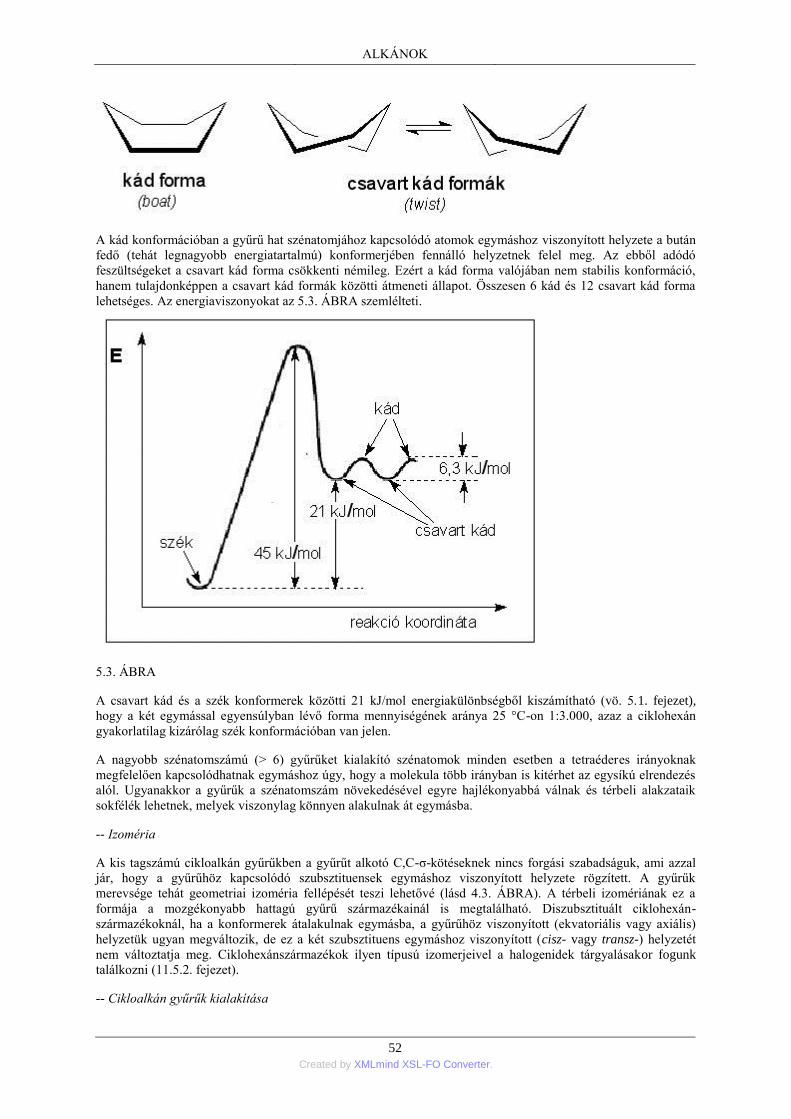
ALKÁNOK
52 Created by XMLmind XSL-FO Converter.
A kád konformációban a gyűrű hat szénatomjához kapcsolódó atomok egymáshoz viszonyított helyzete a bután
fedő (tehát legnagyobb energiatartalmú) konformerjében fennálló helyzetnek felel meg. Az ebből adódó
feszültségeket a csavart kád forma csökkenti némileg. Ezért a kád forma valójában nem stabilis konformáció,
hanem tulajdonképpen a csavart kád formák közötti átmeneti állapot. Összesen 6 kád és 12 csavart kád forma
lehetséges. Az energiaviszonyokat az 5.3. ÁBRA szemlélteti.
5.3. ÁBRA
A csavart kád és a szék konformerek közötti 21 kJ/mol energiakülönbségből kiszámítható (vö. 5.1. fejezet),
hogy a két egymással egyensúlyban lévő forma mennyiségének aránya 25 °C-on 1:3.000, azaz a ciklohexán
gyakorlatilag kizárólag szék konformációban van jelen.
A nagyobb szénatomszámú (> 6) gyűrűket kialakító szénatomok minden esetben a tetraéderes irányoknak
megfelelően kapcsolódhatnak egymáshoz úgy, hogy a molekula több irányban is kitérhet az egysíkú elrendezés
alól. Ugyanakkor a gyűrűk a szénatomszám növekedésével egyre hajlékonyabbá válnak és térbeli alakzataik
sokfélék lehetnek, melyek viszonylag könnyen alakulnak át egymásba.
-- Izoméria
A kis tagszámú cikloalkán gyűrűkben a gyűrűt alkotó C,C-σ-kötéseknek nincs forgási szabadságuk, ami azzal
jár, hogy a gyűrűhöz kapcsolódó szubsztituensek egymáshoz viszonyított helyzete rögzített. A gyűrűk
merevsége tehát geometriai izoméria fellépését teszi lehetővé (lásd 4.3. ÁBRA). A térbeli izomériának ez a
formája a mozgékonyabb hattagú gyűrű származékainál is megtalálható. Diszubsztituált ciklohexán-
származékoknál, ha a konformerek átalakulnak egymásba, a gyűrűhöz viszonyított (ekvatoriális vagy axiális)
helyzetük ugyan megváltozik, de ez a két szubsztituens egymáshoz viszonyított (cisz- vagy transz-) helyzetét
nem változtatja meg. Ciklohexánszármazékok ilyen típusú izomerjeivel a halogenidek tárgyalásakor fogunk
találkozni (11.5.2. fejezet).
-- Cikloalkán gyűrűk kialakítása

ALKÁNOK
53 Created by XMLmind XSL-FO Converter.
A kis tagszámú gyűrűk előállításának legáltalánosabb módja α,ω-dihaloalkánokból indul ki (vö. alkének
előállítása vicinális dihalogenidekből, 6.2.2. fejezet). Ez a módszer elsősorban ciklopropán előállítására
alkalmas. A propán klórozásával nyert izomerelegyből elkülönített 1,3-diklór-propánt fémcinkkel kezelik,
amikor - 70 %-os hozammal - intramolekuláris folyamatban ciklopropán képződik:
A gyűrűtagszám növelésekor egyre inkább előtérbe lép az intermolekuláris folyamat, azaz a molekulák
összekapcsolódása és ezzel a hosszabb láncok kialakulásával járó mellékreakció. A halogénatomok láncbeli
távolságának növekedésével ugyanis rohamosan csökken annak a valószínűsége, hogy ugyanazon szénlánc két
vége kapcsolódjék össze. Így pl. 1,4-dibróm-butánból már csak 7 %-os hozammal keletkezik ciklobután:
Az intermolekuláris kapcsolódásnak - vagyis a polimer láncok kialakulásának - valószínűsége az oldat
hígításával csökkenthető, mert ezáltal az egymással összekapcsolódni képes α,ω-dihalogenidek messzebb
kerülnek egymástól.
Ciklohexán előállitása. Az egy-gyűrűs cikloparaffinok közül legnagyobb ipari jelentősége a ciklohexánnak van.
Előállitása benzol katalitikus hidrogénezésével történik:
Ciklohexánszármazékok előállítására különösen alkalmas a Diels-Alder-szintézis. Butadién és
maleinsavanhidrid cikloaddíciójával például - katalitikus hidrogénezés után - ciklohexán-1,2-
dikarbonsavanhidrid nyerhető (ezt a reakciót a 8.3.3. fejezetben ismertetjük részletesen):
Közepes szénatomszámú gyűrűk származékainak laboratóriumi előállítására jól bevált módszer az α,ω-
dihaloalkánok reagáltatása nátrium-etilát jelenlétében malonészterrel, amely egy szénatommal járul hozzá a
gyűrű kialakitásához:
4.2. Többgyűrűs cikloalkánok
Nagy szénatomszámú egyszerű cikloalkán gyűrűk viszonylag ritkán fordulnak elő a természetes eredetű szerves
vegyületekben. Annál gyakoribbak azonban a szerkezetet merevítő keresztkötéseket tartalmazó többgyűrűs
szénhidrogén-vázak. Ezek a policiklusos szerkezetek leggyakrabban öt- és hattagú gyűrűkből épülnek fel.
-- Elnevezés
Többgyűrűs vegyületek szisztematikus elnevezésekor - különösen bonyolultabb esetekben - problémát jelenthet,
hogy hány gyűrűsnek kell tekintenünk a vegyületet. Ennek eldöntésére a következő szabályt kell alkalmazni:

ALKÁNOK
54 Created by XMLmind XSL-FO Converter.
egy többgyűrűs vegyület mindig annyi "gyűrűs", ahány atomkapcsolat felhasítása szükséges a szerkezet
nyíltláncúvá alakításához.
A többgyűrűs szénhidrogének szisztematikus nevezéktana az alapnév megállapításakor mindig a gyűrűrendszert
alkotó összes atomok számából indul ki, előtéttel adva meg a szerkezetben foglalt gyűrűk számát: pl. biciklo,
triciklo, stb. A név egyértelműségét a szögletes zárójelben megadott három szám biztosítja: ezek a két "hídfő"-
szénatomot összekapcsoló szénatomok számát adják meg, csökkenő nagyság szerinti sorrendben. Kétgyűrűs
vegyületek esetén például:
A példákon a gyűrűt alkotó szénatomok szokásos számozását is feltüntettük. A szögletes zárójelet szorosan,
kötőjelek nélkül illesztjük a névbe.
A biciklo[4,4,0]dekán tulajdonképpen a naftalin teljesen telített származéka, "perhidro"- vagy inkább
dekahidronaftalin. Az ebből levezetett félig-meddig triviális név a dekalin.
A biciklo[2,2,1]heptánt szokás norbornánnak is nevezni. A "nor" előtét arra utal, hogy a vegyület váza
lényegében azonos a névadó alapvegyületével (jelen esetben a bornánéval), de annál egy (vagy több)
szénatommal kevesebbet tartalmaz. A nor-származék jelentheti az oldalláncaitól teljesen megfosztott vegyületet
is (mint pl. esetünkben). Ez a nevezéktan tehát - mint látjuk - nem teljesen egyértelmű. Hasonló meggondolás
alapján szokás az alapvegyületnél egy szénatommal többet tartalmazó származékot "homo"-előtéttel jelölni
(olyan esetben is, ha homológ sorról nem beszélhetünk).
Három vagy többgyűrűs molekuláknál a bonyolult szisztematikus elnevezések helyett gyakran használnak rövid
triviális neveket is (pl. adamantán).
-- Szerkezet
A többgyűrűs szerkezetek mindig merevebbek (többnyire stabilisabbak, vagyis kevésbé reakcióképesek is),
mint egy-gyűrűs analogonjaik, mert a keresztkötés tovább csökkenti a váz amúgyis korlátozott hajlékonyságát.
A biciklo[2,2,1]heptánban (norbornánban) például a ciklohexánváz az egyébként nem stabilis kádformában
rögzítődik azáltal, hogy átellenes szénatomjait a híd-szénatom egymáshoz kapcsolja. A biciklo[4,4,0]dekánban
(dekalinban) a tíz szénatom egy keresztkötés révén két hatos gyűrűt alakít ki. Ennek az utóbbi vegyületnek két
izomerje ismert:

ALKÁNOK
55 Created by XMLmind XSL-FO Converter.
A cisz- és transz-dekalin nem konformerjei egymásnak, hanem tulajdonképpen egy diszubsztituált
ciklohexánszármazék cisz- illetve transz-izomerjeiként foghatók fel. Az ilyen izomerek már csak
kötésfelhasítással (pl. alumínium-bromidos kezeléssel) alakithatók át egymásba.
A policiklusos telített szénhidrogének közül elméleti szempontból érdekes a háromgyűrűs adamantán, mely
egyes ásványolajokban is előfordul. A háromgyűrűs szerkezet négy szék-konformációjú ciklohexángyűrű
teljesen szimmetrikus és feszülésmentes "kondenzálásával" képzelhető el. A molekulában a szénatomok
elhelyezkedése a gyémántrácsban találhatóhoz hasonlít; innen ered a neve és ezzel magyarázható rendkivüli
stabilitása. A nagyobb tagszámú gyűrűk merevitésének egy másik lehetséges formája az ún. spirán-szerkezet,
melyben két gyűrűnek egyetlen atomja közös. A közös kvaterner szénatom tetraéderes vegyértékorientációja a
két gyűrű síkját egymásra merőleges helyzetben rögziti.

56 Created by XMLmind XSL-FO Converter.
6. fejezet - ALKÉNEK (olefin-szénhidrogének), A SZÉN-SZÉN KETTŐS KÖTÉS
Ezek a vegyületek az ún. telítetlen szénhidrogének közé tartoznak, szénláncaik ugyanis olyan szénatompár(oka)t
is tartalmaznak, mely(ek)ben két egymással szomszédos sp2 szénatom kétszeres (σ,π) kötéssel kapcsolódik
egymáshoz (2.3. fejezet). A kétszeres C,C-kapcsolat - viszonylag nagy elektronkoncentrációja következtében -
sokkal könnyebben polározható s ezért reakcióképesebb, mint az egyszeres C,C-kapcsolat. A C,C kettőskötés
tehát az alkének szelektíven reagáló, jellegzetes "funkciós csoportja".
Ebben a fejezetben elsősorban az egy C,C kettős kötést tartalmazó alkénekkel (régebbi nevükön
monoolefinekkel) foglalkozunk, de röviden megemlítünk néhány - egymástól a molekulán belül távol eső két,
vagy három kettős kötést tartalmazó - diént, illetve triént is, mivel ezek kémiája nem különbözik lényegesen az
alkénekétől. Az egyszerű alkénektől több szempontból is jellegzetesen eltérő ún. kumulált és konjugált diéneket
a 7.4., illetve a 8. fejezetben fogjuk tárgyalni.
1. Általános áttekintés
-- A homológ sor
Az alkének homológ sorának tagjai ugyanúgy vezethetők le a sorozat legegyszerűbb tagjából, az eténből, mint
az alkán-sorozat tagjai a metánból. Az etén (C2H4) szerkezeti képletében egy hidrogénatomot metilcsoporttal
helyettesítve, a háromszénatomos alkén, a propén (C3H6) szerkezetéhez jutunk:
A propénben mind a három szénatom különböző mértékben van fedve hidrogénatomokkal. A sorozatban a
további helyettesítés három egymással izomer négyszénatomos szerkezethez, buténekhez (C4H8) vezet. Ezek
közül az egyiknek két térszerkezeti (geometriai izomer) változata van:
Az azonos felépítésű alkének (pl. a kettős kötést a láncvégi szénatomon - "terminális" helyzetben - tartalmazó 1-
alkének vagy másként α-olefinek) az alkánokhoz hasonlóan homológ sort alkotnak, melynek általános
összegképlete CnH2n.
-- Az alkének elnevezése
C,C kettős kötés jelenlétét a molekulában a IUPAC nevezéktan a név "-én" végződésével jelöli. Az elágazó
szénláncú alkének nevét a C,C kettős kötést tartalmazó leghosszabb lánc szénatomszáma, a kettős kötés helyét
pedig a lánc végéhez közelebb eső szénatom sorszáma határozza meg. (A számozást általában úgy kezdjük,
hogy a kettős kötések helyét jelölő számok minél kisebbek legyenek). Ha telítetlen kötés és elágazás együtt

ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
57 Created by XMLmind XSL-FO Converter.
fordul elő a láncon, a kötés helyét jelölő szám legyen a kisebb. Példaként szolgáljon a pentén öt szerkezeti
izomerjének elnevezése:
A két legegyszerűbb alként még ma is gyakran említik régebbi, "-ilén" szóvégződésű triviális nevükön: etén -
etilén; propén - propilén.
A telítetlen szénhidrogének nevét bizonyos esetekben az eténre, mint alapvegyületre is visszavezethetjük. Pl.:
Az alkénekből levezethető "alkenil"-csoportok közül a kisebb szénatomszámúaknak külön triviális neveik
használatosak:
-- Fizikai tulajdonságok
Az alkének forráspontja általában néhány fokkal alacsonyabb, sűrűségük valamivel nagyobb, mint a megfelelő
alkánoké. Törésmutatójuk - a kettőskötés π-elektronpárjának lazább kötöttsége miatt - lényegesen és jellemzően
nagyobb, mint az alkánoké.
-- Geometriai izoméria
A kettős kötést nem-terminális helyzetben tartalmazó ún. "belső alkének"-re jellemző a Z,E- (cisz, transz)-
izoméria fellépése. A sztereoizomériának ezzel a fajtájával már részletesen foglalkoztunk a 4.3. fejezetben.
2. Alkének előállítása
Az egyszerűbb alkének a petrolkémiai ipar legfontosabb nyersanyagai, melyeket nagy mennyiségben
használnak fel vegyipari alapanyagok, különböző polimerek és motorhajtó anyagok előállítására. Mivel a
kőolajban és földgázban nem fordulnak elő, a legfontosabb kis molekulatömegű alkének előállítására nagyipari
eljárásokat dolgoztak ki. Nagyobb tisztaságú vagy bonyolultabb összetételű alkének előállitására laboratóriumi
módszereket használnak.
2.1. Nagyipari eljárások
-- Hőbontás

ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
58 Created by XMLmind XSL-FO Converter.
Az ipari alkének legnagyobbrészt telitett szénhidrogének hőbontásából (krakkolásából, vagy pirolíziséből)
származnak. Mindkettő során a telített szénláncok kisebb szénláncú telített és telítetlen szénhidrogénekre
tördelődnek szét, hidrogén keletkezése közben. A lejátszódó folyamatok kémiai lényege, hogy C,C- és C,H-
kötések hasadnak fel magas hőmérsékleten.
A krakkolás lehet tisztán termikus vagy katalitikus. A csupán hőhatásra bekövetkezett bomlást nevezzük
termikus krakkolásnak (hőbontásnak). Különbséget szokás tenni krakkolás és pirolízis között: a szénhidrogének
650 °C alatti hőmérsékleten végzett hőbontását nevezzük krakkolásnak, 650 °C felett pedig pirolízisnek.
A termikus hőbontás jellemzői:
• Gyökös mechanizmussal lejátszódó folyamat, mert a C,C- és C,H-kötések homolitikus hasításának
energiaszükséglete gázfázisban csak mintegy 1/3 része a heterolitikus bomlásénak. Gyökös jellegéből
következően a termikus hőbontás részben láncreakció is. Pl.:
• A szénhidrogéneknek a termikus krakkolással szembeni stabilitása a szénlánc nagyságával fokozatosan
csökken, vagyis nagyobb szénhidrogén molekulák már viszonylag alacsonyabb hőmérsékleten is
krakkolódnak. Ez a szabály az alkénekre is érvényes. Az alkalmazott hőmérséklet emelkedésével tehát a
képződött szénhidrogénelegy átlagos molekulatömege egyre kisebb lesz: 650-850 °C között már csak C2-C3
alkének (etén és propén), 850 °C felett pedig metán, szén és hidrogén lesznek a termék fő alkotói.
• Katalizátorok távollétében - különösen nagyobb szénatomszám esetén - a C,C-kötések hasadnak fel nagyobb
valószínűséggel, mert ezeknek kötésdisszociációs energiája átlagosan 40-60 kJ/mol értékkel kisebb a C,H-
kötésekénél. A kisebb mennyiségben keletkező hidrogénatomok azonban már kisebb aktiválási energiával
(20-40 kJ/mol) képesek újabb C,H-kötéseket hasítani:
A 650 °C alatti hőmérsékleten végrehajtott termikus krakkolási folyamatoknak - minthogy ezek általában
folyékony szénhidrogének bonyolult elegyét szolgáltatják - elsősorban a motorhajtó anyagok előállításában volt
régebben jelentőségük. Mára azonban ezeket az eljárásokat a kőolajiparban a katalitikus krakkolási eljárások
teljesen kiszorították. A 650 °C felett lejátszódó termikus pirolízist viszont egyedi alkéneknek - főleg eténnek és
propénnek - kifejezetten kémiai továbbfeldolgozás céljából való nagyipari előállitására ma is igen nagy
méretekben alkalmazzák.
Az így előállított etén (etilén) a szerves vegyipar legnagyobb mennyiségben gyártott egyedi terméke.
Legfontosabb felhasználási területei a polimerizáció (polietén-polietilén), a sztirolgyártás (polisztirol), az
etilénglikol előállítás (fagyálló folyadék és poliészter típusú műanyagok), valamint az ecetsav szintézis.
Érdekességként megemlítjük, hogy az etén növényi hormon is: gyorsítja a növények fejlődését és a gyümölcsök
érését.
A katalitikus krakkolást rendszerint alumínium-szilikát típusú katalizátorokon (elsősorban zeolitokon) végzik
400-500 °C hőmérsékleten, motorhajtó anyagok előállítása céljából. Mivel itt izomerizáció, hidrogénátvitel és
gyűrűzáródás is fellép, a folyékony termék csak igen kevés alként tartalmaz, elsősorban (nagyobbrészt erősen
elágazó láncú) alkánokból és aromás szénhidrogénekből áll.
A katalitikus krakkolás végrehajtására rendkivül sok ipari eljárás alakult ki, melyeknek ismertetése a kőolajipari
és petrolkémiai szakkönyvek feladata.
-- Dehidrogénezés
Alkének előállítása alkánok dehidrogénezésével fontos ipari eljárás. Megkülönböztetünk termikus és katalitikus
dehidrogénezést.
Mivel a C,C- és C,H-kötések energiájából következően gázfázisban a dehidrogénezés nagyobb
energiabefektetést igényel, mint a szénlánc szakítása, katalizátor alkalmazása nélkül, tisztán termikus hatásra

ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
59 Created by XMLmind XSL-FO Converter.
nagyobb valószínűsége van az alkán láncszakadásának, mint alkénné való dehidrogéneződésének. A
láncszakadásra való hajlam - mint már emlitettük - a szénatomszámmal nő. Ezért termikus dehidrogénezéssel
csak az etán alakítható át egyértelműen a megfelelő alkénné, eténné (800-900 °C-on). Propán termikus
bontásakor - a C,C-kötés könnyebb hasadása miatt - már etén a főtermék, mert itt már lehetőség van stabilis
kisebb szénatomszámú telített szénhidrogén (metán) keletkezésére is.
Mivel a láncszakadási folyamat a szénhidrogén molekulatömegének növekedésével egyre gyorsabb lesz, C4 és
annál nagyobb szénatomszámú alkének ipari előállítására már csak a katalitikus dehidrogénezés alkalmas.
Katalizátorként alumínium-oxidra felvitt platinát használnak 400-600 °C hőmérsékleten, kis nyomású hidrogén
jelenlétében. A dehidrogénezés során természetesen mindig az izomer alkének elegye képződik.
A katalizátor szerepe itt az, hogy a C,H-kötések hasadását (vagyis a dehidrogénezési reakciót) gyorsítsa és
ezáltal olyan kis reakcióidők alkalmazását tegye lehetövé, ami alatt a C,C-kötés(ek)nek az alkalmazott magas
hőmérsékleten bekövetkező hasadása (tehát a krakkolódás) még jelentéktelen.
2.2. Alkének laboratóriumi előállítása
A C,C kettős kötés laboratóriumi kialakításának általános elve az, hogy a megfelelő telített szénhidrogén
funkciós származékának X csoportját vagy csoportjait hasítjuk ki a molekulából H-X illetve X-X formában. A
jellemző reakció tehát az elimináció (E).
-- Alkoholok dehidratálása
Alkoholokból erősen savanyú katalizátorok képesek vizet kihasítani. Ilyenek:
• alumínium-oxid 300-400 °C-on
• kénsav, foszforsav 100-200 °C-on
Például:
A reakció megfordítható: megfelelő körülmények között alkénekből alkoholok állíthatók elő (lásd 6.3.2. és
12.2.2. fejezetek). A dehidratálási reakció törvényszerűségei a következőkben foglalhatók össze:
• A különböző rendű alkoholok dehidratálódási készsége a rendűséggel nő. A stabilisabb karbéniumion
kialakítására képes (magasabb-rendű) alkoholok könnyebben (már hígabb vagy gyengébb sav hatására és
alacsonyabb hőmérsékleten is) dehidratálódnak. A reakció-készségi sorrend tehát: III > II > I rendű alkohol.
• A hidroxilcsoportot hordozó szénatomhoz képest szomszédos helyzetű szénatomok közül elsősorban arról
hasad le hidrogén a vízkilépés során, amelyen már eleve kevesebb volt (Zajcev-szabály):
A Zajcev-szabály értelmében tehát a termodinamikailag stabilisabb alkén (vö. 6.3.9. fejezet) a főtermék. A
szabály azonban nem kizárólagos érvényű: ha a vízkihasadásra többféle lehetőség van, mindig izomer alkének
elegye keletkezik.

ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
60 Created by XMLmind XSL-FO Converter.
A könnyen dehidratálható tercier alkoholok esetén az alkénképződés mechanizmusa monomolekuláris
elimináció (E1):
A kénsav szerepe elsősorban az, hogy az alkohol OH csoportját protonálva oxóniumiont alakít ki, amelyből
vízkilépéssel könnyen képződik karbéniumion. Később még látni fogjuk, hogy alkoholból protonálás nélkül
karbéniumion csak igen nehezen alakulna ki az OH csoport ún. "rossz kilépő (távozó) csoport" jellege miatt
(6.3.2. fejezet), ugyanakkor az OH2+ már jobb "kilépő csoport" (leaving group).
A reakció "oldalágában" gyors egyensúlyi - nukleofil szubsztitúciós - reakcióban keletkező kénsav-félészter a
dehidratáláshoz alkalmazott reakciókörülmények között csak átmenetileg jelenik meg (a hidrogénszulfát anion
könnyen távozik a félészterből, mert az OH2+-hoz hasonlóan jó kilépő csoport). A viszonylag magas hőmérséklet
az entrópianövekedéssel járó alkén + víz képződésnek kedvez: egy alkohol molekulából így két molekula
keletkezik.
A nehezen dehidratálható primer alkoholok bimolekuláris eliminációs mechanizmussal (E2) veszítenek vizet
kénsav hatására. A protonált alkoholból vagy a HSO4- anion, vagy egy másik alkohol molekula szakítja le az
egyik β-helyzetű hidrogént proton formájában:
Alacsonyabb hőmérsékleten, alkohol felesleg jelenlétében - különösen elsőrendű alkoholok esetén -
mellékreakcióban éterek is keletkeznek. Pl.:
Ez a szubsztitúciós mellékreakció primer alkohol esetén SN2 mechanizmussal játszódik le, az alkohol kiszorítja a
molekulából a protonált OH csoportot:
Mellékreakcióként lehetőség van arra is, hogy a protonfelvétellel majd vízvesztéssel kialakult karbéniumion
átrendeződésével a szénlánc eredeti szerkezete megváltozzék (vö. 5.3.4. fejezet).
-- Alkil-halogenidek dehidrohalogénezése
Alkil-halogenidekből halogén-hidrogén kihasításával alkén keletkezik:

ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
61 Created by XMLmind XSL-FO Converter.
Ez a reakció is megfordítható: alkének halogén-hidrogén-addiciójával alkil-halogenid állítható elő (lásd 6.3.2. és
11.2.2. fejezetek). Lúgos közegben a HX protonjának megkötése folytán a reakció az alkénképződés irányába
tolható el. Célszerűen homogén oldatban, alkoholos lúggal dolgozunk. A reakcióképességi sorrendek a
halogénatom minősége és láncbeli helyzete szerint a következők:
I > Br > Cl, illetve III > II > I rendű halogenid
A terc-butil-jodid már forró vízbe csepegtetve is kvantitatíve alakul át izobuténné. A szekunder, vagy még
inkább a primer halogenidek csak viszonylag erős bázisok (alkálilúgok, alkáli-alkoholátok, tercier aminok)
hatására veszítenek halogén-hidrogént és különbözik a reakciók mechanizmusa is. Alkoholos közegben a
reakció kétirányú:
Tercier alkil-halogenidekből, amelyekből igen könnyen alakul ki a tercier karbéniumion, SN1 reakcióban éter,
E1 reakcióban a Zajcev-szabálynak megfelelő összetételben izomer alkének képződnek (vizes lúg hatására a
halogén helyébe hidroxilcsoport lép be; vö. az alkil-halogenidek hidrolízisével, 11.3. fejezet). Az eliminációs
mechanizmusnak itt az a lényege, hogy a bázis a karbénium szénatommal szomszédos, a σ-konjugáció miatt
fellazult egyik hidrogént szakítja le (ehhez sokszor már gyenge bázis, pl. víz is elegendő - lásd terc-butil-jodid
HI-vesztése):
Primer és szekunder származékok esetében, amelyek erős bázist igényelnek és amelyekből a megfelelő
karbéniumion kialakulása nehezebb, az elimináció bimolekuláris mechanizmus (E2) szerint játszódik le, ezt a
folyamatot éterképződéssel járó SN2 reakció kíséri. Ilyenkor a mechanizmus azért bimolekuláris, mert az erős
bázis előbb szakítja le a halogénnel szubsztituált szénatomról a hidrogént, mint ahogy a karbéniumion
kialakulhatna:

ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
62 Created by XMLmind XSL-FO Converter.
Az E2 mechanizmusú elimináció sztereoszelektív: a bázis azt a hidrogént támadja meg és szakítja le
hidrogénion formájában, amely az alkil-halogenid stabilis konformációjánál a halogénatomhoz képest anti
állásban van. 2-Brómpentán esetében ezért a főtermék az (E)-pent-2-én:
Az (E) izomer túlsúlyát okozó sztereokémiai viszonyokat az átmeneti állapotok Newman szerinti ábrázolásával
tudjuk legjobban érzékeltetni. A két belső alkénhez vezető út két különböző konformációjú átmeneti állapotát az
alábbi ábrák mutatják:
A két konformer közül az alkil csoportok anti állása miatt az A stabilisabb, mint a B, és mivel ez a domináns
forma, az elimináció valószínűsége ebből nagyobb. A bázis pedig azért támadja meg elsősorban a halogénhez
képest anti helyzetű hidrogént, mert
• egyrészt a negatív töltésű bázis így van a legtávolabb a parciális negatív töltésű halogénatomtól,
• másrészt az átmeneti állapot elektronpályái ebben az esetben mennek át legkönnyebben az alkén π-kötésének
elektronpályájába:
-- Vicinális dihalo-alkánok dehalogénezése
Az alkilláncon egymással szomszédos - vicinális - helyzetben [vicinus = szomszédos (latin)] kapcsolódó
halogénatomok (rendszerint brómatomok) leszakításával egyértelműen a két halogént-vesztő szénatom között
alakul ki C,C kettős kötés (tehát nem izomer alkének elegye keletkezik). A dehalogénezés vagy fém
nátriummal, vagy fém cinkkel (az utóbbival ecetsavas közegben) oldható meg:
Ez a reakció a preparatív munkában a brómaddicióval átmenetileg megvédett kettős kötés egyértelmű
regenerálására használható.
-- Wittig-féle alkénszintézis
Ezen az úton tetszésszerinti szerkezetű alkének állíthatók elő foszfónium-ilidekből és karbonilvegyületekből
kiindulva. Ezt a módszert azonban csak a foszforvegyületekkel kapcsolatban fogjuk részletesen tárgyalni
(25.2.2. fejezet).
3. Az alkének kémiai tulajdonságai
A C,C kettős kötés az alkének jellegzetes funkciós csoportja, ezért a továbbiakban elsősorban ennek reakcióival
foglalkozunk részletesen. Ez a kettős kötés "telítetlen" jellegű, azaz a C,C π-kötés felszakadásával járó addíciós
reakciókra képes. Ezen addíciós reakciók során a C,C σ-kötés érintetlen marad.
Az alkének telitett szénláncának tulajdonságai általában megegyeznek az alkánok kémiai tulajdonságaival.
Kivételt képez az allil-helyzet: a kettős kötésben lévő szénatom melletti szénatom C,C-, vagy C,H-kapcsolata.
Az allil-helyzetű kötések ugyanis (a konjugált diéneknél később részletesen tárgyalandó okok miatt) az

ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
63 Created by XMLmind XSL-FO Converter.
átlagosnál könnyebben hasíthatók. Az allil-helyzetű hidrogénatom ezért különösen reakcióképes, könnyen
szubsztituálható és a szénlánc is itt szakad el legkönnyebben. Ezen a helyen tehát szubsztitúciós reakciók
játszódhatnak le.
A következőkben mindkét reakciótípussal részletesen foglalkozunk.
3.1. Reakciók halogénekkel
-- Halogének addíciója vízmentes közegben
Vízmentes közegben a C,C kettős kötés általában könnyen köt meg brómot vicinális dibrómalkán keletkezése
közben. A kloroformos (vagy jégecetes) híg brómoldatot C,C kettős kötést tartalmazó vegyületek elszíntelenítik.
Molekuláris klór ís hasonlóan kötődik meg, de jód gyakorlatilag már nem addicionálódik a kettős kötésre. A
bruttó folyamat eredményeként mindig vicinális dihalogenid keletkezik:
A brómaddició általában kvantitative játszódik le, ezért a C,C kettős kötés mennyiségi meghatározására is
alkalmas.
Vízmentes poláros oldószerekben molekuláris halogénnel általában ionos mechanizmus szerint, anti-addíció
játszódik le. Az addíció tehát sztereoszelektív - a két lehetséges (anti és fedő) konformer (vö. bután
konformerek, 5.1.4. fejezet) közül csak az egyik keletkezik:
A halogénmolekula az alkén elektrondús kettős kötésével ütközve (I) polarizálódik: az alkén kettőskötése π-
komplex átmeneti keletkezése közben "megfogja" a brómmolekula δ+ brómatomját (II). Ezt követi a
brómmolekula heterolízise: a képződött bróm(I)kationból és az alkén sp2 szénatomjaiból gyűrűs bromóniumion
(III) alakul ki, amely a szénatomok másik oldalán elektrondús partnerek (bromid- vagy bármilyen egyéb anion)
által támadhatóvá válik, anti-addíció játszódik le. Ez a mechanizmus magyarázza azt a megfigyelést, hogy pl. a
kloridionok jelenlétében végzett brómaddició során vegyes (Br,Cl) addíciós termék is keletkezik.
A halogénaddíció anti sztereoszelektivitása által meghatározott termékszerkezet a nyíltláncú alkéneknél a C,C-
kötés körüli szabad rotáció miatt esetleg nem, vagy csak közvetve ismerhető fel. A gyűrűs alkéneknél azonban
az anti addíció egyértelműen meghatározza a termék transz térszerkezetét (6.4.1. fejezet).
Az alkén addíciós készségét (a kettős kötés polározhatóságát), illetve az addíciós reakció sebességét
• a kettősen kötött szénatom(ok)hoz kapcsolódó alkilcsoportok növelik (a szomszédos C,H-kötések σ-
konjugáció révén részben delokalizálják a bromóniumion pozitív töltését és ezzel növelik stabilitását).
A hex-1-én brómaddiciója jégecetes közegben például tízszer gyorsabban játszódik le, mint az etén esetében.
2-Metilbut-1-énnel ez az arány már 300-szoros. Nagyon elágazó alkilcsoportok az ezeknél fellépő sztérikus

ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
64 Created by XMLmind XSL-FO Converter.
gátlás miatt azonban már csak kisebb hatást képesek kifejteni: az erős σ-konjugációs effektussal rendelkező
terc-butil-csoportot tartalmazó 3,3-dimetilbut-1-én brómaddiciójának sebessége az eténhez viszonyítva pl.
csak hétszeres.
• A kettős kötésben álló szénatom(ok)hoz kapcsolódó elektronszívó csoportok (pl. karboxilcsoport,
halogének) csökkentik az addíciós reakció sebességét (negatív induktív effektus, -I). Az akrilsav pl. sokkal
lassabban vesz feI brómot, mint az etén.
-- Hipoklórossav addíciója
Vizes oldatban a molekuláris klór (vagyis a hipoklórossav) reakciója alkénekkel, amelynek során
végeredményként egy klóratom és egy OH-csoport addicionálódik a C,C kettős kötésre, tulajdonképpen a
vegyes halogénaddíció (lásd előbb) speciális eseteként fogható fel. A reakció (a brómaddicióhoz hasonlóan) a
molekuláris klór és az alkén π-kötése közötti kölcsönhatással indul, majd az ebből a π-komplexből kialakuló
kloróniumion reagál vízzel, mint nagy feleslegben jelenlévő nukleofil reagenssel. A termék végül
protonvesztéssel "halohidrin" (β-helyen halogénezett alkohol) formájában stabilizálódik:
Ez a reakció is sztereoszelektíven anti-addíción át játszódik le vagyis a termék gyűrűs alkén esetén itt is transz
konfigurációjú lesz.
-- Halogének reakciója gázfázisban
Magasabb hőmérsékleten (>400 °C) a halogének (klór, bróm) alkénekkel nem addícióval, hanem az alkén allil-
helyzetű hidrogénatomjának szubsztitúciójával reagálnak, gyökös mechanizmusú reakcióban. Pl. propén
gázfázisú klórozása allil-kloridot eredményez:
Hasonlóan gyökös mechanizmussal, de szobahőmérsékleten játszódik le az allil-helyzetű hidrogénatom
helyettesítése brómmal N-bróm-szukcinimid hatására, peroxid katalizátor jelenlétében. (Ezt a reakciót
részletesebben a telítetlen halogénszármazékokkal kapcsolatban fogjuk tárgyalni, 11.6.2. fejezet.)
3.2. H-X típusú molekulák addiciója
-- Halogén-hidrogének ionos addiciója
A C,C kettős kötés H-X típusú, protonleadásra képes molekulákat is addicionál. Az addíciót a H+ támadása
indítja, tehát elektrofil addícióról (AE) van szó. Minthogy az addíció sebessége arányos a H+ koncentrációjával,
gyakorlatilag csak az ásványi savak addicionálódnak; a szerves savak disszociációja ehhez túl gyenge.
Az addícióra érvényes ún. Markovnyikov-szabály szerint a sav protonja a kettős kötés kevésbé szubsztituált
(tehát több hidrogénatomhoz kapcsolódó) szénatomjához kapcsolódik. Általában tehát a magasabbrendű
szénatomon szubsztituált származékok keletkeznek. Ez a szabály összhangban van a karbokationok
stabilitásáról tanultakkal: a H+ belépésekor elsősorban a magasabbrendű karbokation keletkezik. Pl.:
A reakció tehát regioszelektív: a két lehetséges szerkezeti izomer közül csak az egyiket szolgáltatja.
Halogén-hidrogének vízben, vagy poláros szerves oldószerekben (éter, kloroform, jégecet) a Markovnyikov-
szabálynak megfelelően, a fenti reakcióegyenlet szerint (X = Cl, Br, I) addicionálódnak a C,C kettős kötésre. A
halogén-hidrogének reakciókészségének sorrendje: HI > HBr >> HCl. Ez a savak erősségének, tehát a
hidrogénion-koncentrációk nagyságának sorrendjével azonos és egyben annak következménye is. (Az etén
sósavat már csak alumínium-klorid katalizátor jelenlétében vesz fel gyakorlatilag használható sebességgel.)

ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
65 Created by XMLmind XSL-FO Converter.
A halogén-hidrogén addiciója megfelelően megválasztott reakciókörülmények között reverzibilis: lásd az alkil-
halogenidek E1 vagy E2 eliminációs mechanizmusát (alkének előállítása, 6.2.2. fejezet).
-- A hidrogén-bromid gyökös, "anti-Markovnyikov" addiciója
A hidrogén-bromid már kis mennyiségben jelenlévő peroxidok (illetve fény vagy levegő, mely utóbbinak
oxigéntartalma az oldószert megtámadva hoz létre peroxidokat) hatására a Markovnyikov-szabállyal ellentétes
értelemben addicionálódik. A hidrogén-bromidból ugyanis a peroxidokból képződő gyökök hatására atomos
bróm keletkezik és az addíció, az L és L' láncvivő lépéseken keresztül, igen gyors gyökös láncmechanizmussal
megy végbe. A láncreakciót a brómatom támadása indítja:
A brómatom belépésének helyét (az addíció regioszelektivitását) két tényező határozza meg:
• Az alkilgyökök stabilitása. Mivel ezek stabilitása (hasonlóan a karbokationokéhoz) a primer < szekunder <
tercier sorrendben nő, a brómatom a több hidrogént hordozó szénatomhoz fog kapcsolódni, mert így
keletkezik a magasabb rendű, stabilisabb gyök.
• A brómatom nagy térigénye ugyancsak a könnyebben hozzáférhető láncvégi szénatom megtámadásának
kedvez.
A láncreakció feltétele, hogy mindkét részlépés energiafelszabadulással járjon. Ez a hidrogén-halogenidek közül
csak a HBr esetében áll fenn, ezért a HCl és a HI esetében ilyen értelmű addició nem játszódhat le. Hasonlóan
"anti-Markovnyikov" szerint addicionálhatók viszont a C,C kettős kötésre az S-H kötést tartalmazó molekulák
(H2S, RSH) is.
-- Kénsavas hidratálás
Alkének vizes kénsav hatására addiciós reakcióban alkoholokká hidratálhatók. A reakció mechanizmusa
megegyezik az alkoholok kénsavas dehidratálásának (6.2.2. fejezet) mechanizmusával. Ez a tény az ún.
mikroszkópikus reverzibilitás elvének megnyilvánulása, vagyis annak az elvnek, hogy egy reverzibilis
reakciónak oda és vissza egyaránt minden részlépése azonos kell, hogy legyen (vagyis nem lehet szó valamiféle
"körfolyamat"-ról).
Az alkohol dehidratálási reakció megfordítását elsősorban a hőmérséklet csökkentése teszi lehetővé:
A reakciót híg kénsavval végezve a hidratálás a fő reakció, mert a hidrogén-szulfát anionnal a nagy feleslegben
jelenlévő víz molekulái sikeresen konkurálnak.
A látszólag teljesen analóg halogén-hidrogén addícióknál alkohol nem képződik annak ellenére, hogy ezeket a
savakat is alkalmazhatják vizes oldatban. Ezt azzal magyarázzuk, hogy a Cl-, Br- és I- ionok sokkal erősebb
nukleofil reagensek a HOSO3- anionnál - nagyobb nukleofil erővel rendelkeznek - és ezért ezekkel a
vízmolekula, amely viszonylag gyenge nukleofil reagens, már nem tud versenyezni.

ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
66 Created by XMLmind XSL-FO Converter.
A hidrogén-szulfát anion kis nukleofil erejét két tényezőre vezetjük vissza:
• a támadó atom, tehát az oxigénatom magános elektronpárjai viszonylag nehezen polározhatók, az oxigénatom
ún, merev (kemény, "hard") bázis;
• negatív töltése (ami egyébként minden reagens nukleofil erejét növeli) sok atomon oszlik meg, erősen
delokalizálódott és ezért nem olyan hatásos.
A hidrogénszulfát anion kis nukleofil ereje nyilvánvalóan csak más megfogalmazása annak, az alkoholokból
való alkénelőállitás során már megismert tulajdonságának, miszerint ez az anion "jó távozó csoport".
Általánosan megfogalmazva jó távozó csoportok az erős savak anionjai (ún. konjugált bázisai), rossz távozó
csoportok pedig értelemszerűen a gyenge savak anionjai (konjugált bázisai). Ezért jó távozó csoport pl. a H2O
(mert a H3O+ erős sav) és rossz távozó csoport az OH- (mert a H2O igen gyenge "sav" - hétköznapi értelemben
nem is sav, hanem semleges molekula).
Az alkének kénsavas hidratálása fontos ipari reakció: a pirolízis termékek C2-C4 alkénjeiből a Markovnyikov-
szabálynak megfelelő szerkezetű alkoholok állíthatók elő. A reakcióképességi sorrend: izobutén > butének >
propén > etén (a kapott alkoholok: terc-butil-alkohol, szek-butil-alkohol, izopropil-alkohol, etil-alkohol).
3.3. Hidrogénezés
Alkének molekuláris hidrogénnel, katalizátorok jelenlétében alkánokká hidrogénezhetők:
A reakció reverzibilis: alacsony hőmérséklet és nagy H2 nyomás a hidrogénezésnek, magas hőmérséklet és kis
H2 nyomás a dehidrogénezésnek kedvez.
A C,C kettős kötés naszcensz hidrogénnel vagy hidridionok leadására képes komplex fém-hidridekkel (amelyek
a karbonil csoport (>C=O) redukálására alkalmasak) nem telíthető: a hidrogénezést minden esetben molekuláris
hidrogénnel, megfelelő katalizátor jelenlétében kell végezni. Egy izolált α-helyzetű C,C kettős kötés
hidrogénezésekor általában kereken 127 kJ/mol hő szabadul fel. A katalitikus hidrogénezés során
szénvázátrendeződés nem következik be.
A hidrogénezési reakciók túlnyomó részét heterogén fázisban jelenlevő szilárd katalizátorokkal hajtják végre, de
egyre nagyobb jelentőségre tesz szert a telítetlen rendszerek homogénkatalitikus hidrogénezése is.
-- Heterogénkatalitikus hidrogénezés
Katalizátorként hordozóra felvitt vagy hordozó nélküli átmeneti-fémeket (Pd, Pt, Ni, stb.) vagy átmenetifém-
oxidokat szokás alkalmazni. A leggyakrabban használt hidrogénező katalizátorok:
• Laboratóriumban
Palládium-csontszén: (10-50-szeres mennyiségű csontszénre gyengén alkálikus közegben redukáló
anyaggal, pl. formaldehiddel lecsapott finom eloszlású fémpalládium): szobahőmérsékleten és atmoszférikus
nyomáson az izolált C,C kettős kötés, de a konjugált diének és cikloalkének hidrogénfelvételét is katalizálja.
Platina-csontszén: hasonló módon készül, mínt a palládium-csontszén, de annál aktívabb, az aromás C,C-
kötések hidrogénezését is katalizálja.
Raney-nikkel: nikkel-alumínium ötvözetből az alumíniumnak vizes lúgoldattal történő kioldásával készült,
hordozó nélküli finom eloszlású "pirofóros" fémnikkel. Általában magasabb hőmérsékleten (20-200 °C) és
nyomáson (1-100 bar) végzett hidrogénezésre használatos.
• Iparban
Az iparban a katalizátor szelektivitása és aktivitása mellett igen fontos szerepe van mechanikai ellenálló-
képességének is.

ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
67 Created by XMLmind XSL-FO Converter.
Hordozóra (horzsakőre vagy alumínium-oxidra) felvitt nikkel katalizátorok: nikkelsóból a hordozó
felületén redukcióval állítják elő a finom eloszlásu fémnikkelt. Tablettázott formában használják 100-200 °C-
on és 100-200 bar nyomáson.
Oxid-katalizátorok: keverék-katalizátorok (pl. CuO + Cr2O3), melyek bizonyos vegyületeket (pl. aldehideket
alkoholokká) szelektíven képesek hidrogénezni. Ezek 150-250 °C-on és 200-300 bar nyomáson aktívak.
A heterogénkatalitikus hidrogénezés mechanizmusa: az alkalmazott katalizátorok "kemiszorbeálják" a telítetlen
vegyületet és a hidrogént; a katalizátorfémből és a molekuláris hidrogénből fém-hidrid képződik (vagyis a H-H
kötés felszakad), az alkén pedig lapjával helyezkedve a katalizátor felületére, az átmeneti fémmel π-komplexet
képez. A két hidrogénatom átadása az alkénre fokozatosan játszódik le. A bonyolult folyamat igen erősen
leegyszerűsített vázlata:
A két H a kettős kötésnek azonos oldalára lép be: sztereospecifikus syn-addíció [szün = együtt (görög)]
játszódik le (megfelelő szerkezetű alkénszármazékból pl. eritro-dihidro termék képződik; vö. 4.6.2. és 6.3.5.
fejezetek).
-- Homogénkatalitikus hidrogénezés
Számos átmenetifém-komplex képes aktiválni a molekuláris hidrogént homogén fázisban is (azaz a katalizátor
oldódik a reakcióelegyben). A katalizátorkomplex központi atomjához koordinálódott alkénmolekula a
komplexben hidrid-formában kötött hidrogént veszi fel; a hidrogénezés tehát a központi atom koordinációs
övezetében játszódik le.
Homogén hidrogénező katalizátorok elsősorban a kobalt, ródium, irídium és ruténium foszfin és/vagy karbonil
ligandumokat tartalmazó komplexei között találhatók [pl. Co2(CO)8, RhCl(PPh3)3].
A homogénkatalitikus hidrogénezést (és általában a fémorganikus katalizátorok hatásmechanizmusát)
részletesebben az átmenetifém-organikus vegyületek kapcsán, a 26.13.3. fejezetben fogjuk tárgyalni, ahol majd
kitérünk a katalitikus hidrogénezés néhány elméleti kérdésére is.
3.4. Hidroformilezés
A hidroformilezés (oxo-szintézis) az alkének egyik - ipari szempontból is fontos - jellegzetes reakciója. Az
alként kobalt- vagy ródiumtartalmú katalizátor jelenlétében reagáltatják nagy nyomáson és magas
hőmérsékleten szén-monoxid és hidrogén elegyével. A reakció főterméke az alkénnél eggyel nagyobb
szénatomszámú aldehid izomerek elegye:
A reakció tehát - bár formailag H-X addíciónak fogható fel - nem követi a Markovnyikov szabályt, az addíció
nem regioszelektív. Ennek oka az ionos H-X addícióktól teljesen eltérő, fémorganikus mechanizmus, amelyet -
egyszerűsített formában - kobalt, mint katalizátor esetére mutatunk be.
A tulajdonképpeni katalizátor a kobaltból, vagy kobalt vegyületekből a reakció körülményei között
oktakarbonil-dikobalton keresztül keletkező komplex hidrid, mely a reakcióelegyben oldva, mint homogén
katalizátor fejti ki hatását:

ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
68 Created by XMLmind XSL-FO Converter.
Az utolsó lépésben felszabaduló HCo(CO)4 visszakerül a reakciósorozat elejére és ezzel a homogénkatalitikus
ciklus bezárul. A fémorganikus mechanizmusból következik, hogy a H és -CHO (formil) csoport a kettőskötés
azonos oldalára lép be, az addíció tehát a katalitikus hidrogénezéshez hasonlóan sztereospecifikusan syn.
Újabban a kobalt mellett előtérbe kerültek a ródiumtartalmú katalizátorok. A rendkívül drága ródium
alkalmazását az egyszerűbb technológia és a jobb szelektivitás indokolják.
3.5. Oxidációs reakciók
-- A C=C kettős kötés oxidációja
Oxidálószerek (krómsav, ózon, permanganát stb.) könnyen addicionálódnak a C,C kettős kötésre. Minthogy ezt
az addíciót sok esetben követi láncszakadásra vezető további oxigénfelvétel, a szénlánc a telítetlen kötésnél
általában könnyen elszakítható.
Jégecetben oldott krómsav vagy ózon a C,C kettős kötést tartalmazó szénláncot a kettős kötés helyén simán
elszakítja. A keletkezett termékekből következtetni lehet arra, hogy a szénláncban hol volt a kettős kötés.
Néhány évtizeddel ezelőtt ezeket a reakciókat elterjedten használták telítetlen vegyületek szerkezetének
felderítésére, ma már azonban ezeknek a módszereknek a műszeres analitikai eljárások fejlódése folytán nincs
analitikai jelentőségük, inkább csak egyes speciális esetekben van preparatív értékük.
Szódaalkálikus kálium-permanganáttal kiméletesebben lehet oxidálni. Ha az oxidálószert nem alkalmazzuk
feleslegben, a kettős kötés csak hidroxilálódik vicinális diol keletkezése közben anélkül, hogy további oxidálás
folytán láncszakadás következnék be. Az alkoholos hidroxil-csoportok syn helyzetben épülnek be a molekulába,
mert a permanganát gyűrűs szerkezetű közbenső termék kialakításával kapcsolódik a kettősen kötött
szénatomokra: syn-dihidroxilálás. A reakció sztöchiometriája és részletes mechanizmusa igen bonyolult, a
folyamat vázlatosan:
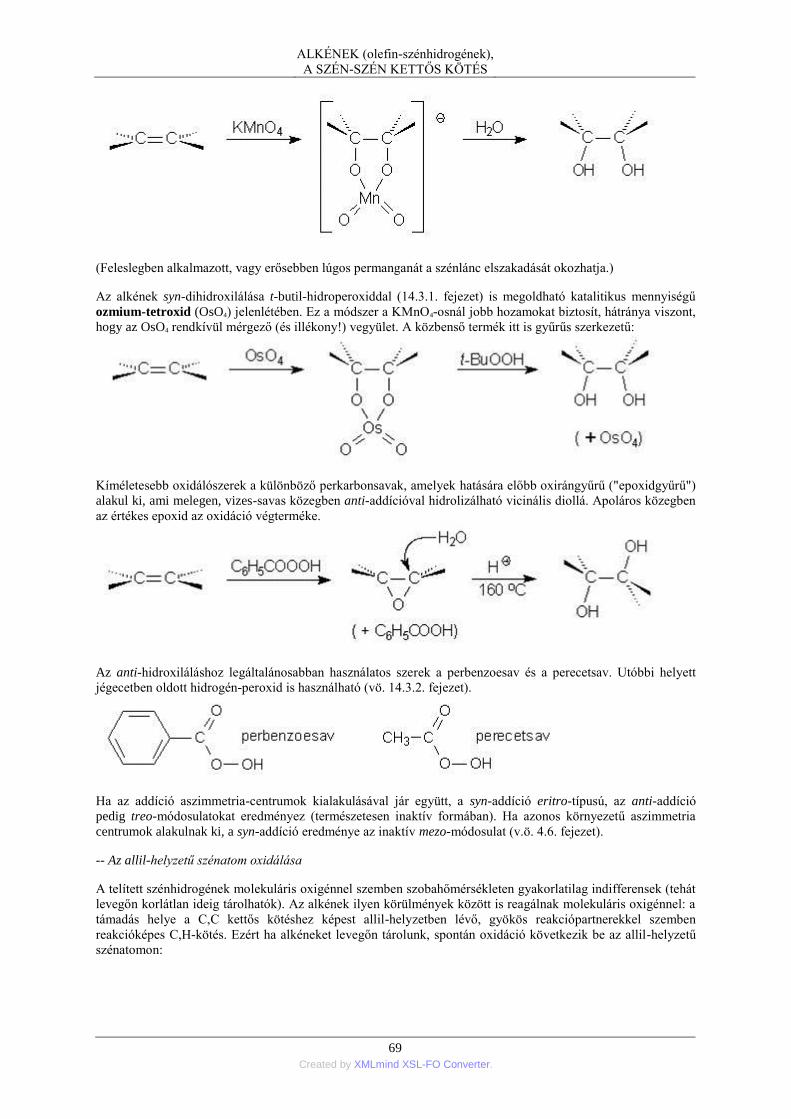
ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
69 Created by XMLmind XSL-FO Converter.
(Feleslegben alkalmazott, vagy erősebben lúgos permanganát a szénlánc elszakadását okozhatja.)
Az alkének syn-dihidroxilálása t-butil-hidroperoxiddal (14.3.1. fejezet) is megoldható katalitikus mennyiségű
ozmium-tetroxid (OsO4) jelenlétében. Ez a módszer a KMnO4-osnál jobb hozamokat biztosít, hátránya viszont,
hogy az OsO4 rendkívül mérgező (és illékony!) vegyület. A közbenső termék itt is gyűrűs szerkezetű:
Kíméletesebb oxidálószerek a különböző perkarbonsavak, amelyek hatására előbb oxirángyűrű ("epoxidgyűrű")
alakul ki, ami melegen, vizes-savas közegben anti-addícióval hidrolizálható vicinális diollá. Apoláros közegben
az értékes epoxid az oxidáció végterméke.
Az anti-hidroxiláláshoz legáltalánosabban használatos szerek a perbenzoesav és a perecetsav. Utóbbi helyett
jégecetben oldott hidrogén-peroxid is használható (vö. 14.3.2. fejezet).
Ha az addíció aszimmetria-centrumok kialakulásával jár együtt, a syn-addíció eritro-típusú, az anti-addíció
pedig treo-módosulatokat eredményez (természetesen inaktív formában). Ha azonos környezetű aszimmetria
centrumok alakulnak ki, a syn-addíció eredménye az inaktív mezo-módosulat (v.ö. 4.6. fejezet).
-- Az allil-helyzetű szénatom oxidálása
A telített szénhidrogének molekuláris oxigénnel szemben szobahőmérsékleten gyakorlatilag indifferensek (tehát
levegőn korlátlan ideig tárolhatók). Az alkének ilyen körülmények között is reagálnak molekuláris oxigénnel: a
támadás helye a C,C kettős kötéshez képest allil-helyzetben lévő, gyökös reakciópartnerekkel szemben
reakcióképes C,H-kötés. Ezért ha alkéneket levegőn tárolunk, spontán oxidáció következik be az allil-helyzetű
szénatomon:

ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
70 Created by XMLmind XSL-FO Converter.
A képződött alkil-hidroperoxid bomlása gyökös polimerizációt indíthat be, ami gyantásodáshoz vezet. Ezért
kettős kötéseket tartalmazó anyagokhoz oxidációt gátló inhibítorokat (pl. hidrokinon, aminok, alkil-fenolok)
szokás adni, hogy a gyantásodást megakadályozzák. Az inhibítor úgy akadályozza meg a polimer
továbbnövekedését, hogy a párosítatlan elektron átvételével önmaga stabilis, tehát új polimerizációs lánc
inditására már nem eléggé reakcióképes gyökké alakul át (vö. 13.3.7. fejezet).
Az alkének kettős kötésének, vagy allil-helyzetü C,H-kötésének oxidálására az utóbbi években számos
katalitikus ipari eljárás alakult ki, melyek a szerves vegyipar számára alapvető fontosságú kiindulási anyagokat
szolgáltatnak. Ilyen eljárások például az etén katalitikus oxidálása etilén-oxiddá vagy acetaldehiddé, a propén
katalitikus oxidálása akril-nitrillé, stb. Ezekkel az eljárásokkal az egyes oxidációs termékek tárgyalásakor
fogunk részletesebben foglalkozni.
3.6. Polimerizáció
Az alkének polimerizációja a kiindulási alkén (a monomer) sorozatos önaddíciója: a monomerből megfelelő
iniciátorral kialakított instabilis származék a kettős kötések felszakadása révén sorozatosan reagál további
monomermolekulákkal, miközben a növekvő molekula instabilis (tehát növekedésre képes) marad mindaddig,
amíg valamilyen lánczáró lépésben nem stabilizálódik. Az így képződött nagy molekulatömegű polimer
szerkezetileg sok azonos kisebb részből álló molekulának tekinthető. A láncnövekedés fokozatosan megy végbe:
két monomer-molekulából előbb dimer, majd ebből egy harmadik monomermolekulával trimer keletkezik és a
folyamat elvben addig folytatódik, míg a monomer el nem fogy.
Adott monomerből láncreakcióban keletkező polimer szerkezetét egyszerűen jelölhetjük a következő módon:
A polimer lánc egyik végéhez a láncot indító iniciátor (helytelenül: katalizátor) kapcsolódik, másik végén pedig
a lánczáró lépés jellegétől függően C,C kettős kötést, telitett szénatomot, vagy más, az iniciátorból származó
funkciós csoportot találunk.
Az alkének polimerizációjának különböző típusait a láncot inditó iniciátor (illetve a láncnövekedési reakció
mechanizmusának) jellege szerint a következőképpen csoportosíthatjuk:
Az alkének polimerizációja az addíciós, vagy láncpolimerizáció tipikus példája. Az ilyen típusú
polimerizációk közös jellemzője, hogy egy aktiválási (iniciálási) lépés után kialakult aktív centrumon megy
végbe a láncnövekedés. A polimerizáció sebessége az iniciátor (és a monomer) koncentrációjától függ és a

ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
71 Created by XMLmind XSL-FO Converter.
polimerizációs fok (a polimer molekulatömege) az iniciátor koncentrációjával szabályozható. Könnyen
belátható, hogy minél nagyobb az iniciátor koncentrációja, annál több lánc indul növekedésnek és a monomer
elfogyása után annál kisebb lesz a polimer molekulák átlagos molekulatömege.
A polimerizációk másik típusával, a lépcsős polimerizációval, a tereftálsav kapcsán, a 19.9.4. fejezetben fogunk
megismerkedni.
-- Gyökös polimerizáció
Ez az alkének polimerizálásának legáltalánosabban alkalmazott formája. A reakciót valamilyen gyök indítja el
néhány alkénmolekula π-kötésének homolitikus felbontásával. Az így keletkezett alkilgyök reagál egy másik
alkénmolekulával, mivel pedig az ennek során kialakuló dimer hasonlóan gyökös természetű, a reakciósorozat
mindaddig folytatódik, amig valamilyen lánczáró lépés (vagy a monomer elfogyása) le nem állítja a folyamatot:
Lánczárás bekövetkezhet:
vagy valamilyen R' gyökkel való kapcsolódás révén:
(Az R' lehet egy másik növekvő polimergyök is; ez esetben dimerizációval záródik a láncnövekedés.)
A gyökös polimerizációt inditó iniciátorok lehetnek:
• Könnyen bomló, gyököt szolgáltató szerves molekulák, többnyire peroxidok, pl. dibenzoil-peroxid:
vagy azo-bisz(izobutiro-nitril):
• Kis mennyiségben hozzáadott molekuláris oxigén indítja például az etén polimerizálását a nagynyomású
polietén (polietilén, PE) előállitásakor:
(Az etén polimerjét a IUPAC nevezéktan szerint természetesen polieténnek kell nevezni, a szakmai
közbeszédben azonban többnyire ma is az alkén polimerek eredeti, régi nevét - polietilén, polipropilén,
poli(izobutilén), általánosságban poliolefinek - használják.) Ezt figyelembevéve a továbbiakban ezeket a nem-
szisztematikus elnevezéseket is megemlítjük.
• Esetenként kivánatos, hogy a polimerizáció alacsony hőmérsékleten (kb. 0 °C) menjen végbe (igy
keletkezhetnek nagyon hosszú szénláncú polimer molekulák). Ilyen esetben iniciátorként redox-rendszerek
használatosak.
Pl. hidrogén-peroxid vas(II)sóval bontva hidroxilgyököt szolgáltat (ún. Fenton reagens, műkaucsuk
előállitásában használatos iniciátor):
• Magas hőmérsékleten (500-800 °C) külön hozzáadott iniciátorok nélkül is lejátszódik az alkének
polimerizációja.

ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
72 Created by XMLmind XSL-FO Converter.
Magas hőmérsékleten a szénhidrogének krakkolódási folyamatai szolgáltatnak iniciátorként ható gyököket
(vö. 6.2.1. fejezet). Ez a spontán polimerizáció (és az azt követő további dehidrogéneződési, aromatizálódási
folyamatok) okozzák például a szénhidrogén-krakkolásnál és pirolizisnél fellépő kokszképződést is.
-- Kationos polimerizáció
A láncnövekedési reakciót az iniciátor (pl. protont szolgáltató sav, bór-trifluorid, alumínium-klorid, stb.),
hatására keletkezett karbokation indítja el és ennek a karbokationnak a szénlánca növekedik a pozitív töltés
lépésenkénti átrendeződésével mindaddig, míg be nem következik valamilyen lánczáró lépés.
Kationos mechanizmussal polimerizálható például az izobutén. Bór-trifluoridos iniciálás esetén a reakció
megindításához nyomnyi mennyiségű víz szükséges, mely a bór-trifluoriddal reagálva protont szolgáltat:
Mivel a komplex sav anionja kovalens kötés kialakítására nem képes, az izobuténből protonaddicióval
keletkezett monomer karbokation nem a sav anionjával, hanem a nagy feleslegben jelenlévő alkénnel reagál,
dimer karbokation képződése közben. Ezen reakció ismétlődésével makromolekulához vezető láncreakció indul
meg. Lánczáró lépés protonledobással következhet be:
A képződött poli(izobutén) [poli(izobutilén)] átlagos molekulatömege elérheti a 200.000-et, vagyis
polimerizációs foka (az egy makromolekulában átlagosan egymáshoz kapcsolódott monomermolekulák száma)
maximálisan 3500. Ez a gumiszerű műanyag telített lévén nem vulkanizálható (lásd 8.3.4. fejezet); az izobutént
2 % izoprénnel együtt polimerizálva (kopolimerizálva) azonban kiváló tulajdonságú butilkaucsuk nyerhető,
melynek vulkanizált terméke kémiailag nagyon ellenálló és gyakorlatilag nem öregszik.
A fenti kationos mechanizmus szerint játszódik le sok más alkén savval iniciált (sokszor nem kívánatos)
polimerizálódása is.
-- Anionos polimerizáció
Anionosan azok a C,C kettős kötést tartalmazó monomerek polimerizálhatók, melyek a kettős kötés mellett
valamilyen erősen elektronszívó (vagy az elektronokat delokalizálni képes) "Z" csoportot (pl. -CN, -Cl, -fenil)
tartalmaznak. Az iniciáló anion (A-) lehet szervetlen, pl. NH2- a kálium-amid disszociációjából, vagy karbanion,
pl. butil-lítium disszociációjából:
A folyamat vázlatos mechanizmusa:
-- Koordinációs polimerizáció
Alkének és diének polimerizálására egyaránt alkalmasak a. különböző redukálószerekből (rendszerint fém-
alkilekből) és átmeneti-fémsókból képződő katalizátorrendszerek is (pl. a Ziegler-féle katalizátorok). A hatást a
keletkező komplex átmenetifém-organikus vegyület fejti ki. Ezeknél a polimerizációknál jogosan beszélhetünk
katalizátorokról, mert az átmenetifém-komplex nem csak elindítja a láncnövekedést, hanem az mindvégig az
átmeneti-fématom koordinációs övezetében játszódik is le.

ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
73 Created by XMLmind XSL-FO Converter.
A trietil-alumíniumból és titán-tetrakloridból képződő klasszikus, ún. Ziegler-katalizátor az etént
atmoszférikus nyomáson és szobahőmérsékleten képes polimerizálni (vö. 26.13.3. fejezet). A Natta által
kidolgozott propén polimerizálás eredeti katalizátora trietil-alumínium mellett titán-trikloridot tartalmaz.
Ezekkel a fémorganikus komplex katalizátor-rendszerekkel a vinil típusú alkének polimerizációja
sztereospecifikusan is végrehajtható.
A reakciósorozat vázlatos mechanizmusa:
Az utóbbi években ezeket a katalizátorrendszereket sok szempontból (elsősorban a definiált szerkezetű
katalizátorok irányába) tovább fejlesztették és ma már igen sok változatuk ismert, sőt iparilag alkalmazott is.
Ezekkel vázlatosan a 26.13.3. fejezetben foglalkozunk.
-- A vinil típusú alkének polimerjeinek sztereokémiája
A vinilcsoportot tartalmazó alkén monomerek polimerizációjával előállított makromolekulák általában ún. fej-
láb polimerek, azaz a polimerizáció regioszelektív:
A fej-fej és láb-láb kapcsolódás ritka:
A makromolekulás anyagok kristályszerkezetét (és ezen keresztül mechanikai tulajdonságaikat) azonban a
polimerizáció (általában kedvező) regioszelektivitásán kívül döntő módon befolyásolja annak
sztereoszelektivitása is, azaz az R-csoportok térbeli helyzete a polimer láncban.
A makromolekulát láncalakban a térben kihúzva szembetünik, hogy az R szubsztituensek a láncot alkotó
szénatomok által meghatározott sík egyik vagy másik oldalára kerülhetnek:
Amennyiben a polimerizációt sztereospecifitással nem rendelkező katalizátorral (pl. valamilyen gyökös iniciátor
segitségével) hajtják végre, az R csoportok térbeli elhelyezkedését csupán statisztikai törvények határozzák meg
és a makromolekulás lánc térbeli felépítése a fenti ábrán láthatóhoz lesz hasonló. Az ilyen polimereket nevezik
ataktikusnak, utalva arra, hogy az R csoportot hordozó és azonos térbeli felépitésü szénatomok a láncban
szabálytalan térközökben követik egymást. Az ilyen polimer - mivel annak minden egyes molekulája más térbeli
felépitésű - nem kristályos, amorf (az ataktikus polipropén (polipropilén) pl. alacsony olvadáspontú,
gyakorlatilag nem hasznosítható anyag).
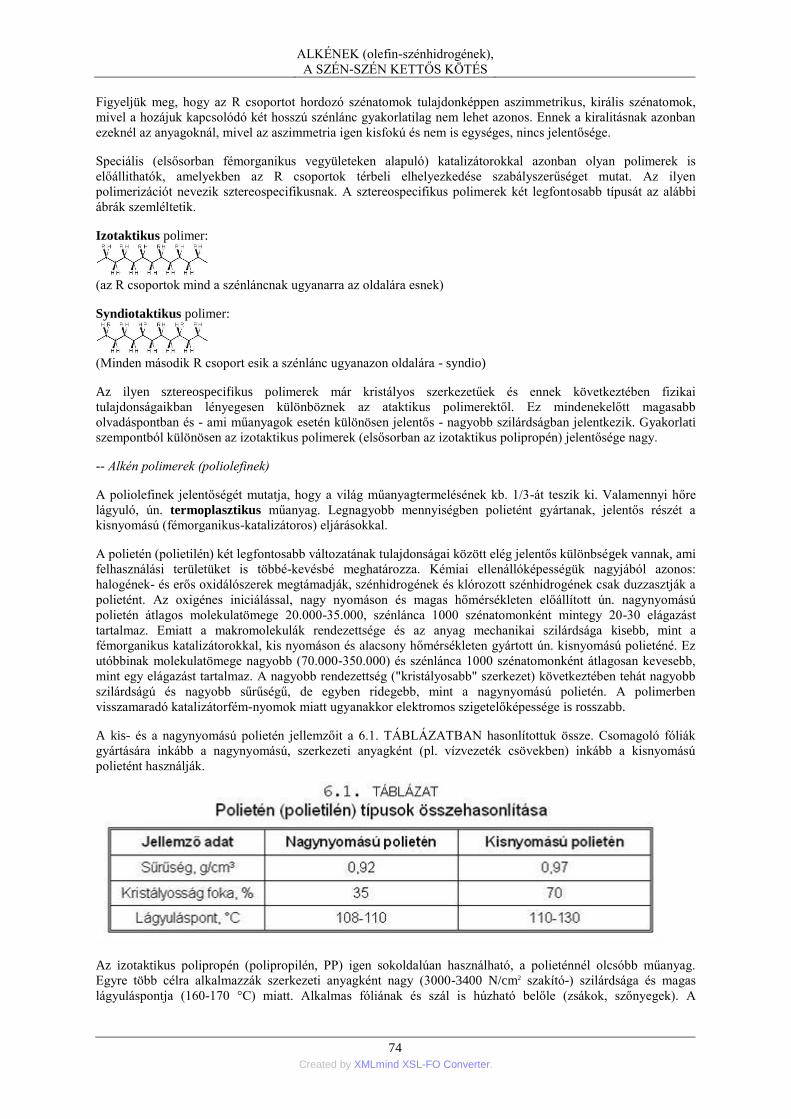
ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
74 Created by XMLmind XSL-FO Converter.
Figyeljük meg, hogy az R csoportot hordozó szénatomok tulajdonképpen aszimmetrikus, királis szénatomok,
mivel a hozájuk kapcsolódó két hosszú szénlánc gyakorlatilag nem lehet azonos. Ennek a kiralitásnak azonban
ezeknél az anyagoknál, mivel az aszimmetria igen kisfokú és nem is egységes, nincs jelentősége.
Speciális (elsősorban fémorganikus vegyületeken alapuló) katalizátorokkal azonban olyan polimerek is
előállithatók, amelyekben az R csoportok térbeli elhelyezkedése szabályszerűséget mutat. Az ilyen
polimerizációt nevezik sztereospecifikusnak. A sztereospecifikus polimerek két legfontosabb típusát az alábbi
ábrák szemléltetik.
Izotaktikus polimer:
(az R csoportok mind a szénláncnak ugyanarra az oldalára esnek)
Syndiotaktikus polimer:
(Minden második R csoport esik a szénlánc ugyanazon oldalára - syndio)
Az ilyen sztereospecifikus polimerek már kristályos szerkezetűek és ennek következtében fizikai
tulajdonságaikban lényegesen különböznek az ataktikus polimerektől. Ez mindenekelőtt magasabb
olvadáspontban és - ami műanyagok esetén különösen jelentős - nagyobb szilárdságban jelentkezik. Gyakorlati
szempontból különösen az izotaktikus polimerek (elsősorban az izotaktikus polipropén) jelentősége nagy.
-- Alkén polimerek (poliolefinek)
A poliolefinek jelentőségét mutatja, hogy a világ műanyagtermelésének kb. 1/3-át teszik ki. Valamennyi hőre
lágyuló, ún. termoplasztikus műanyag. Legnagyobb mennyiségben polietént gyártanak, jelentős részét a
kisnyomású (fémorganikus-katalizátoros) eljárásokkal.
A polietén (polietilén) két legfontosabb változatának tulajdonságai között elég jelentős különbségek vannak, ami
felhasználási területüket is többé-kevésbé meghatározza. Kémiai ellenállóképességük nagyjából azonos:
halogének- és erős oxidálószerek megtámadják, szénhidrogének és klórozott szénhidrogének csak duzzasztják a
polietént. Az oxigénes iniciálással, nagy nyomáson és magas hőmérsékleten előállított ún. nagynyomású
polietén átlagos molekulatömege 20.000-35.000, szénlánca 1000 szénatomonként mintegy 20-30 elágazást
tartalmaz. Emiatt a makromolekulák rendezettsége és az anyag mechanikai szilárdsága kisebb, mint a
fémorganikus katalizátorokkal, kis nyomáson és alacsony hőmérsékleten gyártott ún. kisnyomású polieténé. Ez
utóbbinak molekulatömege nagyobb (70.000-350.000) és szénlánca 1000 szénatomonként átlagosan kevesebb,
mint egy elágazást tartalmaz. A nagyobb rendezettség ("kristályosabb" szerkezet) következtében tehát nagyobb
szilárdságú és nagyobb sűrűségű, de egyben ridegebb, mint a nagynyomású polietén. A polimerben
visszamaradó katalizátorfém-nyomok miatt ugyanakkor elektromos szigetelőképessége is rosszabb.
A kis- és a nagynyomású polietén jellemzőit a 6.1. TÁBLÁZATBAN hasonlítottuk össze. Csomagoló fóliák
gyártására inkább a nagynyomású, szerkezeti anyagként (pl. vízvezeték csövekben) inkább a kisnyomású
polietént használják.
Az izotaktikus polipropén (polipropilén, PP) igen sokoldalúan használható, a polieténnél olcsóbb műanyag.
Egyre több célra alkalmazzák szerkezeti anyagként nagy (3000-3400 N/cm2 szakító-) szilárdsága és magas
lágyuláspontja (160-170 °C) miatt. Alkalmas fóliának és szál is húzható belőle (zsákok, szőnyegek). A

ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
75 Created by XMLmind XSL-FO Converter.
poli(izobutén) nagy részét butilkaucsukká dolgozzák fel, de használják saválló és szigetelő burkolások
készítésére is. A polisztirollal a 9.7. fejezetben fogunk foglalkozni.
3.7. Oligomerizáció
Oligomerizációnak nevezzük a polimerizációnak azt a formáját, amikor csak néhány monomermolekula
kapcsolódik össze. Az összekapcsolódó molekulák számától függően beszélünk dimerizációról, trimerizációról,
tetramerizációról, stb. A keletkező termék a dimer, trimer, illetve tetramer.
Az oligomerizáció mechanizmusa és az iniciálás módja lényegében azonos a polimerizációval kapcsolatban már
megismertekkel. A lényeges különbség a polimerizációval szemben az, hogy a lánczáró reakciók sebességét a
hőmérséklet emelésével megnövelik a láncnövekedési reakció sebességéhez képest. A reakciólánc ilyenkor már
néhány (2, 3, 4, stb.) monomer-molekula egymáshoz kapcsolódása után megszakad és az intermedier
stabilizálódik.
Gyakorlati szempontból fontos oligomerizáció az izobutén dimerizálása kénsav katalizátorral. Az izobutén
kationos polimerizációjához hasonlóan ez is karbokationos mechanizmussal lejátszódó reakció:
A keletkező két izomer oktén elegyét katalitikus hidrogénezéssel 2,2,4-trimetilpentánná, közhasználatú nevén
"izooktán"-ná alakitják, amely kedvező tulajdonságú (100-as oktánszámú) motorbenzin komponens.
Nagyipari jelentőségű, fémorganikus katalizátorokkal végzett oligomerizálásra példaként megemlítjük az etén
oligomerizálását nikkel-foszfin komplex katalizátoron, amelynek során 80-120 °C-on és 70-140 bar nyomáson
lineáris α-alkének keletkeznek (SHOP eljárás = Shell higher olefins process):
A reakció itt is az átmenetifém (Ni) koordinációs övezetében játszódik le, lényegében ahhoz hasonló
mechanizmussal, mint amilyent a koordinációs polimerizációval kapcsolatban a Ti-ra már vázoltunk. A
keletkező α-alkének egyenes láncúak, ami felhasználásuknál környezetvédelmi szempontból előnyös (vö.
szintetikus mosószerek, 15.6.3. fejezet).
3.8. Alkilezés
Alkilezésnek azt a reakciótípust nevezzük, amelynek során valamely molekula egyik hidrogénatomját
alkilcsoportra cseréljük ki.
Ennek a sokféle változatban ismert reakciónak itt csak azt a megoldását tárgyaljuk, melyben az alkilezett
molekula alkán, az alkilezőszer pedig alkén. Példa erre az izobután alkilezése buténnel:

ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
76 Created by XMLmind XSL-FO Converter.
Ezt a reakciót is protonleadásra hajlamos vegyületek (kénsav, HF, AlCl3 + HCl) katalizálják, mechanizmusa
ionos. Az átmenetileg keletkező karbokationok átrendeződésével magyarázható, hogy a termék mindig izomerek
elegyéből áll.
Iniciálás:
Alkilezési láncreakció:
Gyakorlatilag is használható sebességgel csak a tercier szénatommal rendelkező telített szénhidrogéneket lehet
alkénekkel alkilezni, mert az alkalmazott reakciókörülmények között csak ilyen alkánokból tud könnyen
karbéniumion keletkezni. Izobutánnak propén-butén elegyekkel való alkilezése nagyipari eljárás (lásd
alkilátbenzin, 5.2.1. fejezet)
3.9. Izomerizálás (Z,E-izomerizáció és kettős kötés vándorlás)
Láttuk, hogy protonleadásra képes H-X típusú molekuláknak C,C kettős kötésre való addicionálódásakor
karbokation keletkezik (6.3.2. fejezet). Ez a karbokation egyrészt H- és R- vándorlással átrendeződésekre képes
(5.3.4. fejezet), másrészt kialakulásával megszűnik a C,C π-kötés és így lehetővé válik a C,C σ-kötés körüli
szabad rotáció. Mindezek a folyamatok azt eredményezik, hogy H+ ionok jelenlétében az alkéneknek számos
izomerizációs reakciója tud lejátszódni. Ezek a reakciók egyensúlyi folyamatok.
A C,C σ-kötés körüli szabad rotációval értelmezhető az alkének H+ hatására lejátszódó Z,E-izomerizációja:
A Z,E-izomerizációnál a kialkuló egyensúlyi elegyben sztérikus okok miatt általában azok az alkének lesznek
jelen nagyobb mennyiségben, amelyekben a nagyobb térigényű szubsztituensek egymástól távolabb
helyezkednek el.
Az alkének izomerizációjának másik fajtája az, amely a C,C kettős kötés láncbeli helyzetének megváltozásával
jár együtt:
A reakció mechanizmusa:

ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
77 Created by XMLmind XSL-FO Converter.
A különböző karbokationok eltérő stabilitása (tercier > szekunder >> primer) az oka annak, hogy a pozitív töltés
• egyenes láncú karbokationoknál a lánc vége felől a közepe felé,
• elágazó láncú karbokationoknál a tercier szénatomra tolódik el.
A kettős kötés vándorlás is általában gyors reakció, ezért az izomer alkének egyensúlyi elegye hamar kialakul.
Ennek az elegynek az összetételét az alkének termodinamikai stabilitása határozza meg. Ez az sp2
szénatomokhoz kapcsolódó alkilcsoportok számával növekszik, azonos C,C-kötés elrendeződés esetén pedig a
Z,E-izomerizációra jellemző sztérikus hatás érvényesül. Sok alkén-reakciónál ezek miatt a gyors izomerizációk
miatt még egységes kiindulási anyag esetén is olyan lehet a végtermék szerkezete, míntha az izomer alkének
elegyét reagáltattuk volna.
Mind a két típusú kettős kötés izomerizálódás nem-savas jellegű katalizátorok (pl. átmenetifém-komplexek)
hatására is lejátszódik.
Néhány egyeneslácú alkén izomer egyensúlyi koncentrációinak 190 °C-ra vonatkozó értékeit a 6.2.
TÁBLÁZAT mutatja be. A hőmérséklet emelkedésével a terminális alkén aránya általában nő, a belső alkéneké
csökken. A butén izomerek elegye szobahőmérsékleten pl. csupán 3% but-1-ént tartalmaz.
3.10. Metatézis (alkén-diszpoporcionálódás)
Molibdén- vagy volfrámtartalmú katalizátorok hatására játszódik le az alkének metatézise, melynek bruttó
reakcióegyenlete:
A reakciónak mind heterogénkatalitikus (Mo/Al2O3, 300 °C), mind homogénkatalitikus (W- és Mo-komplexek
oldatban, 20 °C) változata ismert. Ezen termoneutrális egyensúlyi reakció végtermékének összetétele azonos a
statisztikusan várhatóval (50% R,R', 25-25% R,R, illetve R',R' alkén). A reakció fém-karbén és
metallaciklobután komplex intermediereken át játszódik le:
Indító lépés:

ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
78 Created by XMLmind XSL-FO Converter.
Katalitikus ciklus:
3.11. Komplexképzés
Az alkének különböző átmenetifém-vegyületekkel - a kettős kötés felhasadása nélkül - komplexeket képeznek,
melyekből az alkén sok esetben vissza is nyerhető. Egyes alkén-komplexeknek gyakorlati jelentőségük is van
például alkéntartalmú elegyek szétválasztásában, vagy alkének kémiai átalakításában.
E komplexek összetétele igen változatos lehet. Egy átmeneti-fématom általában egy alkénmolekulát köt meg, de
igen sok ettől eltérő összetételű alkénkomplex is ismeretes. Ezeket a vegyületeket sokszor nem is lehet tiszta
állapotban eIkülöniteni, ez azonban sok esetben nem akadályozza gyakorlati felhasználásukat.
Ismeretesek Cu, Ag, Pd, Pt, Co, Fe, stb. tartalmú alkénkomplexek, melyek közül például a palládium
eténkomplexe az etén katalitikus oxidálásával történő acetaldehid-előállításban játszik fontos szerepet.
Legrégebben ismert a Zeise által 1827-ben előállított kálium-etén-trikloro-platinát(II) K[(C2H4)PtCl3] ("Zeise-
féle só"). Ma már egyértelműen tisztázott, hogy az eténmolekula az etén-platina irányra merőlegesen
kapcsolódik a platinaatomhoz:
Jelenlegi felfogásunk szerint ez a szerkezeti elv a legtöbb átmenetifém-alkén komplexre jellemző.
Az alkén-platina kötés elektronszerkezete a következő: egyrészt a C,C kettős kötés π molekulaorbitálja
átfedésbe kerül a platina egyik üres (megfelelően orientált) dsp2 hibrid atomi orbitáljával, és így egy -
szimmetriáját tekintve - σ-kötéshez hasonló részkötés alakul ki, melynek két elektronját az alkén szolgáltatja, ez
az elektrondonor partner. A negatív töltésnek a fémen ezáltal előálló felhalmozódásának ellensúlyozására
viszont a platina egyik betöltött dp hibridorbitáljáról kerül át elektron - átfedés révén - az etén lazitó π*
molekulaorbitáljára. Ez a "viszont-koordináció" (back-donation) egy π-szimmetriájú részkötést alakít ki:

ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
79 Created by XMLmind XSL-FO Converter.
Az alkén és a fém közötti kötés az alábbi mezomer határszerkezetekkel is jellemezhető:
Az ilyen típusú komplex vegyületeket, melyekben a kötés a központi fématom és a koordinálódott ligandum
között az utóbbi π-molekulapályáinak felhasználásával jön létre, π-komplexeknek nevezzük.
A π-komplexekben kötött alkének bizonyos mértékig "aktivált" állapotban vannak jelen. A viszont-
koordinálódás folytán ugyanis az alkén lazító π* molekulaorbitáljára is van elektronátmenet, ezért az alkén
eredeti C,C π-kötése gyengül. Lényeges továbbá, hogy a π-komplexben kötött alkén sp2-szénatomjain az
elektronkoncentráció csökken, s így az alkén ezeken a helyeken nukleofil reagensek által is támadhatóvá válik
(vö. 26.13.2. fejezet).
4. Cikloalkének
A cikloalkének elnevezéséről annyit kell tudnunk, hogy a kettős kötés helyzetét jelölő szám lesz a legkisebb,
ehhez igazodik azután a gyűrűhöz kapcsolódó szénláncok számozása. Pl: 3-metil-5-etil-ciklohex-1-én:
4.1. Ciklomonoének
Az egy C,C kettős kötést tartalmazó telítetlen aliciklusos szénhidrogének (cikloalkének) kémiai viselkedése
hasonló a nyíltláncú alkénekéhez. Kis tagszámú gyűrűkben azonban az sp2 szénatomok 120°-os (a tetraéderes
109,5°-nál nagyobb) vegyértékszöge által okozott még erősebb feszültségnövekedés a vegyület
reakciókészségét a stabilitás rovására rendkívüli mértékben megnöveli. Ezért például a ciklopropén ismeretes
ugyan, de már szobahőmérsékleten robbanásszerű hevességgel bomlik. A C,C kettős kötés még a ciklobutén
szénvázát is annyira feszültté teszi, hogy a molekula sokkal reakcióképesebb (labilisabb) a nyíltláncú
buténeknél.
A ciklopentén és ciklohexén kémiai viselkedése már közelebb áll a nyíltláncú penténekéhez, illetve
hexénekéhez. Szénvázuk merevsége következtében azonban hajlamosabbak a szénváz feszültségét csökentő
polimerizációra.
A ciklohexén ciklohexanol dehidratálásával nyerhető, tömény kénsavas kezeléssel, vagy Al2O3 kontakton,
magasabb hőmérsékleten:
A ciklohexén brómmal a már ismert anti-addíciós mechanizmus szerint reagál és transz-1,2-dibrómciklohexán
keletkezik:
A halogénaddíció anti-sztereoszelektivitása - amelynek mechanisztikus hátterét a 6.3.1. fejezetben részletesen
tárgyaltuk - itt, a cikloalkéneknél, amelyeknél a C,C-kötés körüli szabad rotáció nem lehetséges, tehát
egyértelműen transz-dihaloalkánt eredményez.
4.2. Izolált kettős kötéseket tartalmazó ciklodi- és poliének
Az izolált kettős kötéseket tartalmazó di- és poliénekben a kettős kötéseket legalább egy sp3 szénatom választja
el egymástól. Ennek következtében egymással kölcsönhatásba nem léphetnek és egymástól függetlenül
reagálnak, vagyis tulajdonságaik lényegében azonosak az alkének kémiai tulajdonságaival. Részletesebben ezért
ezekkel a vegyületekkel a nyíltláncú alkének keretében nem is foglalkoztunk.

ALKÉNEK (olefin-szénhidrogének),
A SZÉN-SZÉN KETTŐS KÖTÉS
80 Created by XMLmind XSL-FO Converter.
Itt, a gyűrűs alkének között viszont megemlítjük a butadiénből (lásd 8. fejezet) átmenetifém-organikus homogén
katalizátorokkal megoldható ciklodimerizációval és ciklotrimerizációval előállítható ciklookta-1,5-diént
(COD), illetve ciklododeka-1,5,9-triént (CDT). Az előbbi igen kedvelt komponense az átmenetifém-organikus
π-komplexeknek (vö. 6.3.11. fejezet), az utóbbi viszont a speciális tulajdonságokkal rendelkező Nylon-12 (PA-
12) poliamid típusú műanyag alapanyaga (vö. 21.6.4. és 26.13.3. fejezetek).

81 Created by XMLmind XSL-FO Converter.
7. fejezet - ALKINEK (ACETILÉN SZÉNHIDROGÉNEK) ÉS KUMULÉNEK. AZ sp SZÉNATOM ÉS A SZÉN-SZÉN HÁRMAS KÖTÉS
Az sp szénatom két vegyülettípusban fordul elő a szénhidrogének között: az alkinekben (acetilénekben) és a
kumulénekben. Az előbbiek két, egymáshoz σ,π,π-hármas kötéssel kapcsolódó sp szénatomot tartalmaznak, az
utóbbiakban pedig legalább két (esetleg több) közvetlenül egymás mellett elhelyezkedő (ún. kumulált) σ,π-
kettős kötés van. A két típus szerkezetét vázlatosan a 7.1. TÁBLÁZAT mutatja be; a C,C σ-kötésre merőleges
síkok a π-elektronok térbeli elhelyezkedését szemléltetik vázlatosan.
1. Alkinek - általános áttekintés
Mint arról a bevezetésben már szó volt, az alkinekre az jellemző, hogy szénláncuk két, egymáshoz hármas
kötéssel kapcsolódó szénatomot tartalmaz. A legegyszerűbb ilyen típusú szénhidrogén az etin (acetilén),
melyben az egymással hármas kötést létesítő két szénatomhoz csak egy-egy hidrogénatom kapcsolódik. A
nagyobb szénatomszámú alkinekben a hármas kötésben lévő szénatomok egyikéhez vagy mindkettőhöz
kapcsolódhat (elvben tetszőleges) szénlánc:
A hármas kötést azonos helyzetben tartalmazó alkinek - az alkánokhoz és alkénekhez hasonlóan - homológ sort
alkotnak CnH2n-2 általános képlettel. Az 1-alkinek homológ sorának első tagja az acetilén.
--Elnevezés
A IUPAC-nevezéktan a C,C hármas kötés jelenlétét a szénláncban "-in" szóvégződéssel jelöli. A IUPAC
elnevezésekre vonatkozó eddig megismert szabályok az alkinekre is érvényesek. Példák:

ALKINEK (ACETILÉN
SZÉNHIDROGÉNEK) ÉS
KUMULÉNEK. AZ sp
SZÉNATOM ÉS A SZÉN-SZÉN
HÁRMAS KÖTÉS
82 Created by XMLmind XSL-FO Converter.
A legegyszerűbb (két szénatomos) alkin hivatalos IUPAC neve tehát etin, ezt azonban ma még csak ritkán
használják. Sőt szokás az egyszerűbb alkineket az acetilén alkil-szubsztituált származékaiként is elnevezni.
Például:
A továbbiakban általában a IUPAC nevezéktant fogjuk használni, de az alapvegyületre, az acetilénre,
megtartjuk a régi és nagyon begyökeresedett nevet.
A leggyakrabban előforduló alkinilcsoport nevét az alapvegyület szisztematikus nevéből képezzük:
--Fizikai tulajdonságok
Az alkinek olvadás- és forráspontja (merevebb szerkezetük folytán) általában magasabb, mint az azonos
szénatomszámú alkánoké vagy alkéneké. Az 1-alkinek homológ sorának első három tagja gáz, az 1-hexadecin-
től kezdve szilárd halmazállapotúak.
Az acetilén (a szénhidrogének többi tagjával ellentétben) endoterm képződéshőjű vegyület, vagyis elemeire való
szétesése hőfejlődéssel jár:
C2H2 → 2 C + H2 ΔH = - 230 kJ/mol
Emiatt az igen jelentős hőfejlődés miatt az acetilén rendkívül instabilis, spontán bomlása már kis
energiaközlésre is beindulhat és robbanássá fejlődik. Így pl. önmagában nem is komprimálható. Ezért az
acetilént acélbombában kovafölddel felitatott acetonban oldva szokás tárolni vagy szállítani (disszugáz). Egy
térfogat aceton ugyanis 12 bar nyomáson 300 térfogat acetilént képes oldani (az 50%-os oldat még stabilis,
robbanásra nem hajlamos). Nagy égéshője (1300 kJ/mol) következtében az oxigénnel keverten elégetett acetilén
igen forró lángot hoz létre; az acetilén hegesztőlámpa lángjának hőmérséklete kb. 3000 °C.
2. Alkinek előállitása
2.1. Acetilén ipari előállitása
A XX század első felében az acetilén volt a szerves vegyipar egyik legfontosabb nyersanyaga. Mint alább látni
fogjuk, az acetilén előállítása azonban - bármelyik megoldást válasszuk is - nagyon energiaigényes folyamat. Az
acetilén tehát a többi egyszerű szénhidrogénhez képest drága, ezért ipari nyersanyagként ma már csak néhány
eljárásban használják. A későbbiek során még látni fogjuk, hogy a legtöbb, valamikor még acetilén-bázisú
fontos szerves alapvegyület (acetaldehid, vinil-klorid, vinil-acetát, akrilo-nitril) előállításánál mostanra már
általában más, olcsóbb nyersanyagokra (etén, propén) tértek át (v.ö. 11.6.1., 16.2.4. és 21.8.3. fejezetek).
-- Metánból
Az acetilén előállítása metánból az erősen endoterm
2 CH4 → C2H2 + 3 H2 ΔH = + 380 kJ/mol
reakció szerint 1500 °C feletti hőmérsekleten oldható meg.
Ennek termodinamikai okai vannak. A hőmérséklet növelésével ugyanis az acetilénnek a képződési
szabadenergiával jellemezhető relatív stabilitása nő (bár mindvégig az instabilitás tartományában marad), míg az
alkánoké és alkéneké (mint általában minden szerves vegyületé) csökken és egy bizonyos hőmérséklet felett
ezek is elemeikhez képest termodinamikailag instabilissá válnak. Olyan hőmérsékletet kell tehát elérni,
amelynél az acetilén termodinamikai instabilitása már kisebb, mint a kiindulási szénhidrogéné. Ez a hőmérséklet
metán esetében minimálisan 1200 °C.

ALKINEK (ACETILÉN
SZÉNHIDROGÉNEK) ÉS
KUMULÉNEK. AZ sp
SZÉNATOM ÉS A SZÉN-SZÉN
HÁRMAS KÖTÉS
83 Created by XMLmind XSL-FO Converter.
A metánnak acetilénné való alakításához szükséges magas hőmérséklet elérhető külső fűtéssel (pl. regeneratív
megoldással), ívfény alkalmazásával (3500-4000 °C), vagy a metán egy részének elégetésével ("parciális
oxidáció"). Mindhárom megoldásnál igen lényeges, hogy a metán csak nagyon rövid ideig tartózkodjék a magas
hőmérsékletű térben, addig, amennyi szükséges az intermedier bomlástermékként fellépő acetilén képződéséhez,
de az ezt követő gyors lehűtés már megakadályozza az ezen a hőmérsékleten szintén instabilis acetilén
elbomlását.
Ma az acetilén ipari előállitási módszerei közül a metán parciális oxidációja a legelterjedtebb. Ennek
energiatermelő reakciója:
2 CH4 + O2 → 2 CO + 4 H2
A teljes elégéshez szükségesnél kevesebb oxigénnel kevert metán tartózkodási ideje az 1500-1600 °C-os
reaktortérben (lángban) kb. 0,01 sec. A kilépő gázt, amely 8-9% acetilént tartalmaz, víz befecskendezésével
hűtik le 150 °C-ra. Az acetilént nyomás alatt oldószerrel mossák ki a gázelegyből. Melléktermékként értékes
szintézisgáz (CO + 2 H2) keletkezik.
-- Kalcium-karbidból
Ez a klasszikus megoldás. Ennél az eljárásnál égetett meszet és kokszot olvasztanak össze az ívfény
hőmérsékletén (2500-3000 °C), majd az így kapott kalcium-karbidból (helyesebben: kalcium-acetilidből)
állítják elő vízzel az acetilént:
CaO + 3 C → CaC2 + CO
CaC2 + 2 H2O → C2H2 + Ca(OH)2
Az így nyert (egyébként szagtalan) acetilén arzin, foszfin és kén-hidrogén szennyezései miatt fokhagymaszagú
gáz.
2.2. Acetilén-homológok előállítása (a C,C hármas kötés kialakításának laboratóriumi módszerei)
-- Dihalo-alkánokból
Alkánok vicinális dihalogénszármazékai (amelyek a megfelelő alkénekből nyerhetők halogénaddicióval)
viszonylag magas hőmérsékleten tömény alkoholos alkálilúggal kezelve alkinszármazékokká alakulnak át. Híg
alkoholos lúgoldat csak egy halogén-hidrogén molekulát hasít ki a dihalogenidből, mert a keletkező vinil-
halogenid lúgokkal szemben meglehetősen ellenálló.
1,2-Dibróm-propánból 30%-os alkoholos lúggal propin keletkezik. Melléktermékként csak kevés allén
képződik, mert lúgos közegben ez a vegyület aránylag gyorsan átrendeződik a stabilisabb propinné:
--Fém-alkinidek alkilezése
Nátrium-, vagy főleg magnézium-alkinidek (acetilidek) alkilezése a nagyobb szénatomszámú alkinek
előállításának legcélszerűbb módja:

ALKINEK (ACETILÉN
SZÉNHIDROGÉNEK) ÉS
KUMULÉNEK. AZ sp
SZÉNATOM ÉS A SZÉN-SZÉN
HÁRMAS KÖTÉS
84 Created by XMLmind XSL-FO Converter.
3. Az alkinek kémiája
Az alkinek kémiai tulajdonságait elsősorban legfontosabb képviselőjük, az acetilén példáján ismertetjük. Az
acetilén igen reakcióképes vegyület, reakcióit a következő csoportosításban tárgyaljuk:
A nagy elektronsűrűségű C,C hármas kötést elektrofil reagensek könnyen megtámadják és ennek folytán az
acetilén elektrofil addíciós reakciókra hajlamos. Ugyanakkor az sp-szénatomhoz kapcsolódó hidrogénatom
gyengén savas karakterű, mert a különböző hibridállapotú szénatomok elektronegativitása az sp3 < sp2 < sp
sorrendben növekszik. Ennek oka az s-karakter növekedése a p-karakter rovására: az s elektronok ugyanis az
atommaghoz közelebb helyezkednek el és így a C,H kötés polározottsága a C → H irányban megnő.
Az alkinek ennélfogva addíciós és szubsztitúciós reakciókra egyaránt hajlamosak. Addíciókészségük -
valószinűleg a π-elektronrendszer szimmetrikusabb szerkezete miatt - gyengébb, mint az alkéneké (az addíciós
reakciókban tehát legtöbbször katalizátorokat kell alkalmazni). A hármas kötés két π-kötése egyébként
egymástól gyakorlatilag függetlenül addicionál: az addíció első lépésében keletkező eténszármazék többnyire
elkülönithető a reakcióelegyből.
3.1. Halogénaddíció
Halogéneket a C,C hármas kötés ugyanolyan - egymást követő - anti-addíciók útján vesz fel, mint a C,C kettős
kötés, de valamivel kisebb sebességgel (az első lépés a lassabb):
3.2. Vinilezés (HX-addíció)
HX típusú (protont szolgáltató) molekuláknak a C,C hármas kötésre való addicionálódása a Markovnyikov-
szabálynak megfelelően játszódik le:
Ez a reakció a vinil-származékok előállításának egyik legcélszerűbb laboratóriumi módszere.
-- Szervetlen és szerves savak addíciója
A reakció végrehajtásához általában katalizátorra (legtöbbször a sav higany- vagy cinksójára) van szükség.
Például:
Hasonló módon reagál a hidrogén-cianid is, sósavas ammónium-kloridos réz(I)-klorid oldat jelenlétében:
A reakció rézkomplexeken keresztül, bonyolult mechanizmussal játszódik le.

ALKINEK (ACETILÉN
SZÉNHIDROGÉNEK) ÉS
KUMULÉNEK. AZ sp
SZÉNATOM ÉS A SZÉN-SZÉN
HÁRMAS KÖTÉS
85 Created by XMLmind XSL-FO Converter.
-- Viz addíciója
A reakció híg kénsavas oldatban csak higany(II)-szulfát katalizátor jelenlétében játszódik le. A reakció első
lépésében keletkező vinil-alkohol nem stabilis vegyület és igen gyorsan átrendeződik acetaldehiddé:
Ez az átrendeződés általában jellemző az "enol"-os kettős kötésben lévő szénatomhoz kapcsolódó
hidroxilcsoportra. Az enolok tulajdonságairól később részletesen lesz szó a 12.5.1. fejezetben.
A reakcióban mindig karbonilvegyület keletkezik: propinból kiindulva pl. aceton a termék. A folyamat
mechanizmusa bonyolult és még nem teljesen tisztázott, de feltételezhető, hogy higanyorganikus intermedierek
is fellépnek.
-- Szén-monoxid és alkoholok addíciója
Szén-monoxid is reagál acetilénnel, tetrakarbonil-nikkel katalizátor jelenlétében (Reppe-féle karbonilezés).
Alkohol jelenlétében akrilsav-észter keletkezik:
A reakció csak formailag tartozik ebbe a csoportba, valójában a hidroformilezéséhez hasonló bonyolult
fémorganikus mechanizmussal, π-komplex intermediereken keresztül játszódik le.
3.3. Hidrogénezés
Platina vagy nikkel katalizátor jelenlétében végzett hidrogénezéskor az alkinek hármas kötése telítődik:
Palládium vagy vas katalizátort alkalmazva részlegesen is telíthető a C,C hármas kötés. Magasabb alkinekből
ilyenkor mindig (Z)-konfigurációjú alkének képződnek, mert a H2 addiciójának sztereokémiája itt is syn, mint az
alkének esetében (6.3.3. fejezet). Itt a termék (Z)-konfigurációja - a keletkezett C,C kettős kötés merevsége
folytán - könnyen felismerhető módon bizonyítja a hidrogénaddíció sztereoszelektivitását.
Az acetilén részleges hidrogénezésének gyakorlati jelentősége van például a pirolízissel nyert etén
acetiléntartalmának (szennyezésének) eltávolításában.
3.4. Sóképzés
Mint már említettük, az sp szénatomhoz kapcsolódó hidrogénatom már kis mértékben savanyú jellegű, "aktív".
Maga az acetilén fémnátriummal (folyékony ammóniában) valamint réz és ezüst ammóniás vizes oldataival
(lúgos pH-nál) sóképzés közben reagál. Ezeket az ionos vegyületeket acetilideknek vagy alkinideknek
nevezzük.
A könnyűfémek acetilidjei - mint nagyon gyenge savak sói - vízzel azonnal hidrolizálnak (vö. acetilén
előállítása kalcium-karbidból):

ALKINEK (ACETILÉN
SZÉNHIDROGÉNEK) ÉS
KUMULÉNEK. AZ sp
SZÉNATOM ÉS A SZÉN-SZÉN
HÁRMAS KÖTÉS
86 Created by XMLmind XSL-FO Converter.
A nehézfémek acetilidjei vízzel nem, csak ásványi savakkal bonthatók meg. A réz és ezüst acetilidjei száraz
állapotban robbanékonyak (disszugáz-palackon ezért tilos vörösréz-szerelvény alkalmazása).
Grignard-reagensekből (pl. metil-magnézium-jodidból) az acetilén, mint erősebb sav, felszabadítja a telített
szénhidrogént:
Ez a reakció általánosságban felhasználható alkinil-magnézium-halogenidek, s ezeken keresztül hosszabb
szénláncú alkinek előállitására (vö. 7.2. fejezet).
3.5. Etinilezés (alkinilezés)
Az acetilén (általában az 1-alkinek) katalizátor jelenlétében alkinolok képződése közben reagálnak
karbonilvegyületekkel (aldehidekkel vagy ketonokkal). Lúgra nem érzékeny karbonilvegyületek reakciója
glikoléterben oldott kálium-hidroxiddal katalizálható. Például:
A karbonilcsoportot a lúg által deprotonált acetilénből kialakult alkinid ion támadja meg; a reakció
tulajdonképpen a karbonilvegyületek nukleofil addíciós (AN) reakcióinak egyik esete (16.3.1. fejezet):
Ezt a reakciót alkalmazza az ipar az 1,4-butindiol előállítására, amelyet 1,4-butándiollá hidrogéneznek:
Az 1,4-butándiol az oldószerként igen értékes tetrahidrofurán (THF) gyártásának nyersanyaga (14.1.4. és 28.2.3.
fejezetek). Ez a reakció egyike azon keveseknek, amelyeknél az acetilént még ma is ipari méretekben
alkalmazzák nyersanyagként.
3.6. Dimerizáció
Az acetilén gyengén savas C,H kötése réz(I)-kloridot és ammónium-kloridot tartalmazó sósavas oldatban
addicionálódik egy másik acetilén molekula C,C hármas kötésére:
A vinilacetilén sósav addícióval kloroprénné alakítható:

ALKINEK (ACETILÉN
SZÉNHIDROGÉNEK) ÉS
KUMULÉNEK. AZ sp
SZÉNATOM ÉS A SZÉN-SZÉN
HÁRMAS KÖTÉS
87 Created by XMLmind XSL-FO Converter.
A kloroprén polimerizálva értékes elasztomert, neoprént szolgáltat (8.3.4. fejezet). Ipari előállítása ma azonban
már nem acetilénből, hanem butadiénből indul ki (11.6.1. fejezet).
3.7. Ciklooligomerizáció
Az acetilén ciklooligomerizációját első ízben Berthelot észlelte 1868-ban: acetilént izzó vascsövön átvezetve
benzol keletkezését mutatta ki.
Megfelelő átmenetifém-organikus katalizátorok jelenlétében alkinekből már szobahőmérsékleten is képződnek
gyűrűs oligomerek. A főtermékek általában benzolszármazékok, de a termék összetételét a katalizátor és a
reakciókörülmények megválasztásával befolyásolni lehet. Így pl. kobaltorganikus komplexek hatására alkinek
elsősorban trimerizálódnak és benzolszármazékok keletkeznek:
Ugyanakkor acetilénből komplex nikkel-cianid katalizátor hatására a más úton igen nehezen hozzáférhető
ciklooktatetraén (8.4.4. fejezet) képződik főtermékként:
4. Kumulált diének és poliének
A kumulált kettős kötéseket tartalmazó vegyületekben két, egymásra merőlegesen orientált π-kötés három
szénatomot kapcsol össze egymással lineáris és merev szerkezetté: két sp2 szénatom kapcsolódik egy sp
szénatomon keresztül (lásd 7.1. TÁBLÁZAT). Az orientációs síkok egymásra merőleges állásából következik,
hogy a π-kötések egymástól lényegében függetlenül reagálnak.
A kumulált kötésrendszerű vegyületek legegyszerűbb képviselője az allén (propadién). Előállítható allil-
bromidból brómaddíció utáni hidrogén-bromid és bróm-kihasítással:

ALKINEK (ACETILÉN
SZÉNHIDROGÉNEK) ÉS
KUMULÉNEK. AZ sp
SZÉNATOM ÉS A SZÉN-SZÉN
HÁRMAS KÖTÉS
88 Created by XMLmind XSL-FO Converter.
Az acetilén-kötés stabilisabb, mint a kumulált kettőskötés-rendszer: az allén fémnátrium hatására acetilén-
származékká, Na-metil-acetiliddé alakul át:
A kettőnél több szomszédos (kumulált) helyzetű C,C kettős kötést tartalmazó allén-homológokat kumuléneknek
nevezzük. A legegyszerűbb kumulén a butatrién: CH2=C=C=CH2. A kumulének a bennük lévö sp szénatom(ok)
miatt lineáris (botalakú) molekulák, amelyekben a π-elektronok az egyes C,C kettős kötésekben felváltva
egymásra merőlegesen helyezkednek el (vö. a 7.1. TÁBLÁZAT ábrájával). Sztereokémiai szempontból érdekes
következménye ennek, hogy a kumulált kettős kötések számától függően a kettős kötéses szénlánc végéhez
kapcsolódó szubsztituensek vagy egymásra merőlegesen, vagy ugyanabba a síkba esnek. Az allén és a butatrién
térszerkezete jól szemlélteti ezt:

89 Created by XMLmind XSL-FO Converter.
8. fejezet - KONJUGÁLT DIÉNEK ÉS A π-ELEKTRON DELOKALIZÁCIÓ
Konjugált kettős kötéses rendszereknek nevezzük azokat a szerkezeteket, amelyekben az egyes (σ) kötések és a
kettős (σ,π) kötések felváltva követik egymást a szénatomok láncában. Ebben a fejezetben ennek a
molekulacsoportnak legegyszerűbb képviselőivel, a két, konjugált helyzetű C,C kettős kötést tartalmazó
szénhidrogénekkel, a konjugált diénekkel fogunk foglalkozni.
Látni fogjuk, hogy a konjugált diének kémiai tulajdonságai sok tekintetben alapvetően eltérnek az egy kettős
kötést tartalmazó alkének tulajdonságaitól (6. fejezet). Ennek oka az ún. π-elektron delokalizáció. A konjugált
diének előállításának és kémiájának tárgyalása előtt ezért még meg kell ismerkednünk ezzel a jelenséggel.
1. A π-elektron delokalizáció értelmezése
A konjugált diének legegyszerűbb (és egyben legfontosabb) képviselője a buta-1,3-dién (a továbbiakban
egyszerűen csak butadién) π-kötései nem függetlenek egymástól, hanem többé-kevésbé összefüggő kötés-
rendszerként viselkednek. Ezt mutatja például, hogy
• a butadiént szobahőfokon kloroformos oldatban brómozva egy molekula bróm felvételével nagyobbrészt 1,4-
dibrómbut-2-én keletkezik, amely mellett a "szabályos" reakcióban várt 3,4-dibrómbut-1-én csak
melléktermék:
• Butadiént katalitikusan butánná hidrogénezve nem 2×127, hanem csak 239 kJ/mol hidrogénezési hő szabadul
fel. Ez arra utal, hogy a konjugált π-elektronpárok 15 kJ/mol-lal energiaszegényebbek (stabilisabbak), mint
két izolált π-elektronpár.
Mindkét jelenséget azzal értelmezzük, hogy a kettős kötéseket létrehozó π-elektronpárok delokalizációra
képesek: a butadiénnek (a hidrogénezési hő által mutatott) energiaszegényebb jellegét a butadién két konjugált
π-elektronpárjának négy szénatomra kiterjedő delokalizációjával, az 1,4- és 1,2-brómaddició jelenségét pedig a
brómozás közti termékeként fellépő allil-kation egyetlen π-elektronpárjának három szénatomra való
delokalizációjával magyarázzuk. A következőkben külön-külön vizsgáljuk meg ezt a kétféle delokalizációt.
1.1. A butadién példája - az MO-megközelítés
A butadiénben és a hozzá hasonló konjugált rendszerekben a kvantumkémiai molekulapálya (MO, molecular
orbital) tárgyalásmód értelmében a π-elektronpárok nem két-két szomszédos szénatomon lokalizálva, hanem
valamennyi egymással szomszédos sp2 szénatomra elosztva, delokalizálva helyezkednek el a molekulában. A
delokalizációt az teszi lehetővé, hogy az sp2 szénatomok egy síkban helyezkednek el, s így pl. a butadiénben
nemcsak az 1. és 2., valamint a 3. és 4. szénatomok p-orbitáljai fednek át egymással páronként, hanem a 2. és 3.
szénatomok p-pályái között is kölcsönhatás jöhet létre:
Mivel a butadién mindkét kötő π-elektronpárja közösen oszlik el a négy szénatom erőterében, két - egyenként
két-két szénatomra lokalizált - molekulapálya (molekulaorbitál, MO) helyett a négy szénatomhoz közösen
tartozó ún. négycentrumos delokalizált molekulapályák alakulnak ki. Négy centrummal rendelkező
delokalizált rendszer esetén négy delokalizált MO lehetséges, melyek közül kettő kötő, kettő pedig lazító MO. A
butadién négy π-elektronja alapállapotban a két kötő MO-on helyezkedik el. Ennek a két kötő molekulapályának
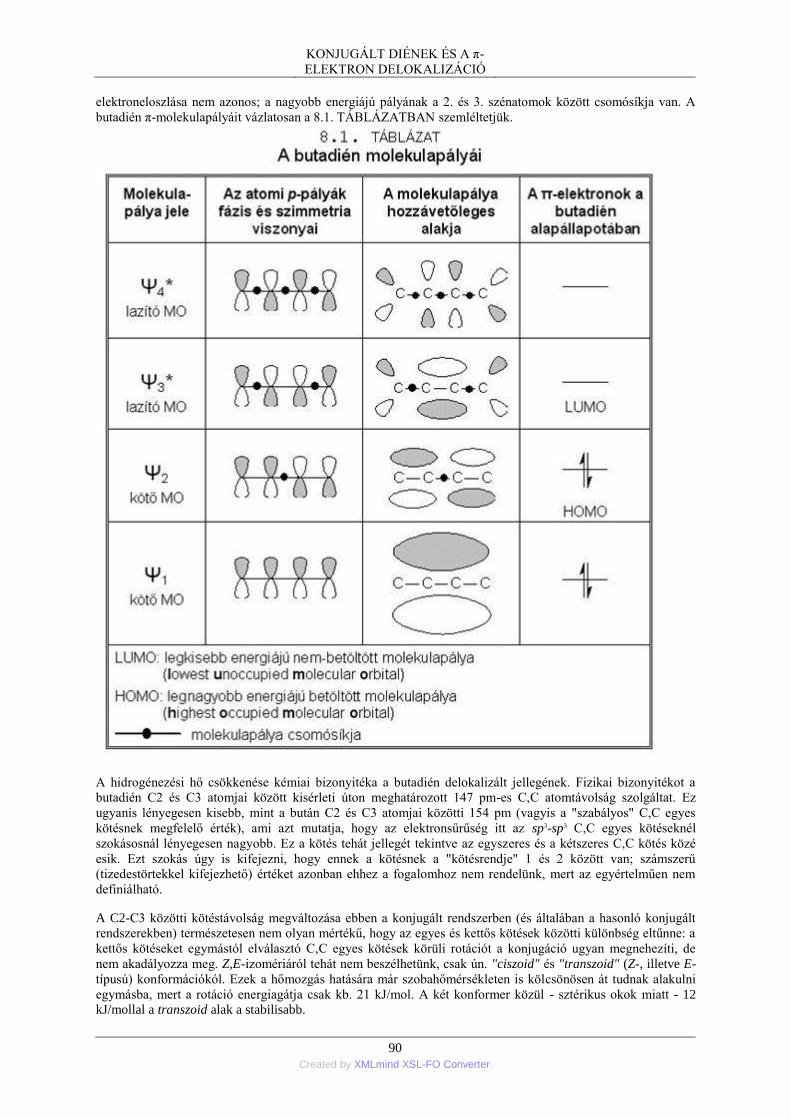
KONJUGÁLT DIÉNEK ÉS A π-
ELEKTRON DELOKALIZÁCIÓ
90 Created by XMLmind XSL-FO Converter.
elektroneloszlása nem azonos; a nagyobb energiájú pályának a 2. és 3. szénatomok között csomósíkja van. A
butadién π-molekulapályáit vázlatosan a 8.1. TÁBLÁZATBAN szemléltetjük.
A hidrogénezési hő csökkenése kémiai bizonyitéka a butadién delokalizált jellegének. Fizikai bizonyitékot a
butadién C2 és C3 atomjai között kisérleti úton meghatározott 147 pm-es C,C atomtávolság szolgáltat. Ez
ugyanis lényegesen kisebb, mint a bután C2 és C3 atomjai közötti 154 pm (vagyis a "szabályos" C,C egyes
kötésnek megfelelő érték), ami azt mutatja, hogy az elektronsűrűség itt az sp3-sp3 C,C egyes kötéseknél
szokásosnál lényegesen nagyobb. Ez a kötés tehát jellegét tekintve az egyszeres és a kétszeres C,C kötés közé
esik. Ezt szokás úgy is kifejezni, hogy ennek a kötésnek a "kötésrendje" 1 és 2 között van; számszerű
(tizedestörtekkel kifejezhető) értéket azonban ehhez a fogalomhoz nem rendelünk, mert az egyértelműen nem
definiálható.
A C2-C3 közötti kötéstávolság megváltozása ebben a konjugált rendszerben (és általában a hasonló konjugált
rendszerekben) természetesen nem olyan mértékű, hogy az egyes és kettős kötések közötti különbség eltűnne: a
kettős kötéseket egymástól elválasztó C,C egyes kötések körüli rotációt a konjugáció ugyan megnehezíti, de
nem akadályozza meg. Z,E-izomériáról tehát nem beszélhetünk, csak ún. "ciszoid" és "transzoid" (Z-, illetve E-
típusú) konformációkól. Ezek a hőmozgás hatására már szobahőmérsékleten is kölcsönösen át tudnak alakulni
egymásba, mert a rotáció energiagátja csak kb. 21 kJ/mol. A két konformer közül - sztérikus okok miatt - 12
kJ/mollal a transzoid alak a stabilisabb.

KONJUGÁLT DIÉNEK ÉS A π-
ELEKTRON DELOKALIZÁCIÓ
91 Created by XMLmind XSL-FO Converter.
Az elektronok delokalizációja csökkenti azok energiáját, ami végeredményben az egész molekula
szempontjából energiacsökkenést, vagyis növekvő stabilitást jelent. A stabilizáló hatás mértékét az ún.
delokalizációs energia fejezi ki. A butadién esetében kísérletileg egyszerűen megmérhető energiáról van szó,
melynek közelítő értéke - mint láttuk - 254-239 = 15 kJ/mol. Általános szabály az, hogy minél több centrumú
delokalizált MO-ok tudnak kialakulni (vagyis minél kiterjedtebb a konjugáció), annál nagyobb a delokalizációs
energia és a stabilitás növekedés. Különösen jelentős a stabilizálódás a később tárgyalandó gyűrűsen konjugált
(aromás) rendszerekben.
A centrumok számának növekedésével, vagyis a delokalizáció kiterjedésével a molekula fényabszorpciója az
ibolyántúli tartományból a látható fény tartományába tolódik el: az anyag színessé válik. Az
elektrondelokalizáció és a fényabszorpció összefüggésével a karotinoidok tárgyalásakor (10.4. fejezet)
foglalkozunk részletesebben.
A többcentrumos molekulaorbitálokkal rendelkező delokalizált rendszerek jellemzésére jól használható a
következő jelölésmód: megadjuk a konjugációban résztvevő atomok, vagyis a centrumok számát és a megadott
centrumú MO-okon elhelyezkedő (vagyis delokalizált) π-elektronok számát. A butadién például eszerint egy
(4C,4π) rendszer. Semleges molekuláknál a két jelzőszám azonos. Ha a centrumok száma a nagyobb, pozitív, ha
a delokalizált π-elektronok száma a nagyobb, negatív töltésű ionokról van szó. Például a később tárgyalandó
allil kation (3C,2π)+, vagy a ciklopentadienid anion (5C,6π)- rendszer.
1.2. Az allilkation példája - a VB-megközelítés
A butadiénnél jelentkező π-elektron delokalizációt két érvvel, a kisebb hidrogénezési hővel és a C,C-távolságok
megváltozásával támasztottuk alá. Vizsgáljuk meg ezek után, mivel tudjuk értelmezni a butadién talán
legváratlanabb kémiai viselkedését, az 1,4-addíciós termék képződését a Br2-addíció során. Ehhez meg kell
értenünk a Br2 addíció mechanizmusát.
Tudjuk, hogy az alkénes (σ,π) kettőskötés hatására a Br2 molekula polározódik és a kettős kötést tulajdonképpen
a Br(I) kation támadja meg (6.3.1. fejezet). Ez a Br+ a butadién delokalizált elektronrendszerét a szélső
szénatomok egyikénél fogja megtámadni. A támadásnak ezt a helyét sztérikus okok is elősegítik, de főként az
indokolja, hogy az így keletkező karbéniumion három megmaradó sp2 szénatomja ezáltal szomszédos helyzetbe
kerül és ezért ezek molekulapályáin a C,Br σ-kötés kialakulása után megmaradó két π-elektron továbbra is
delokalizálódni tud, tehát alacsonyabb energiaállapotban (stabilisabb állapotban) maradhat. A butadiénből így
kialakuló (3C,2π)+ allilkationban a π-elektronok delokalizációját az atomszimbólumok fölé húzott vonallal
szemléltetve:
A bromidion ezután az allilkationt a szélső szénatomok bármelyikén megtámadhatja. Így érthető az 1,2- és 1,4-
diszubsztituált termékek egymás melletti képződése:
Az allilkation elektroneloszlását jól szemlélteti az MO-elmélet szerinti leírása. Eszerint három, háromcentrumos
molekulapálya alakul ki (8.2. TÁBLÁZAT).

KONJUGÁLT DIÉNEK ÉS A π-
ELEKTRON DELOKALIZÁCIÓ
92 Created by XMLmind XSL-FO Converter.
A ψ1 molekulaorbitál energianívója a delokalizáció miatt jóval alacsonyabb az egyszerű π-kötésénél, innen van
az allilkation nagy stabilitása. A ψ2 orbitálon az allilgyökben (•C3H5) egy, az allilanionban (C3H5-) két elektron
foglal helyet. Ennek az orbitálnak nem-kötő (tehát sem nem kötő, sem nem lazító) jellege miatt azonban ezek az
elektronok ezen molekulák stabilitását nem befolyásolják, ezért mindkét utóbbi részecske is kitüntetett
stabilitású.
Az előbbiek alapján most már érthetővé válik, hogy miért szubsztituálhatók könnyen az alkének allil-helyzetű
hidrogénatomjai, akár halogénezésről (6.3.1. fejezet), akár oxidációról (6.3.5. fejezet) legyen szó. Ennek oka
nyilvánvalóan az, hogy ha ez a hidrogén homolitikusan leszakad, háromcentrumos delokalizált allilgyök tud
kialakulni. Ez pedig a delokalizációs energia arányában stabilisabb, mint az egyszerű alkilgyökök és ezért
viszonylag könnyen keletkezik. Az allilhelyzetű C,H-kötés disszociációs energiája csak 370 kJ/mol (vö. 5.3.1.
fejezet).
A delokalizált rendszerek π-elektroneloszlását eddig az MO-elmélet alapján tárgyaltuk. A kérdés másik
megközelitési lehetőségét a vegyértékkötés (VB, valence bond) elméletből kifejlődött rezonanciaelmélet
jelenti. E módszer alkalmazását az alábbiakban az allilkation példáján fogjuk bemutatni.
A rezonanciaelmélet szerint a molekula tényleges elektroneloszlása hipotetikus határszerkezetek
kombinációjaként ("rezonanciahibridjeként") jellemezhető. Az angolszász irodalomban elterjedt rezonancia
kifejezés helyett mi azonban erre a jelenségre a továbbiakban inkább a német szakirodalomban használt
mezoméria kifejezést [mesz = középső, közbenső (görög)] fogjuk használni annak érzékeltetésére, hogy a
molekula tényleges elektroneloszlása "közbenső állapot" a lehetséges klasszikus vegyértékkötésű szerkezetekkel
ábrázolt mezomer határszerkezetek között. Igen fontos ugyanis annak megértése, hogy nem arról van szó,
mintha a molekula a határszerkezetekben "lokalizált" elektronszerkezetű alakok között oszcillálna ("rezonálna").
A mezomer határszerkezeteket tehát élesen meg kell különböztetnünk az egymással kémiai egyensúlyban lévő
konkrét molekuláktól. Ezt a célt szolgálja az is, hogy a mezomer határszerkezeteket kétfejű nyíllal és nem az
egyensúlyi folyamatoknál használt kettős nyíllal "kötjük össze":
Az allilkation alább példaképpen bemutatott mezomer határszerkezetei - bár fizikai realitással a fentiek
értelmében nem rendelkeznek - azt mégis jól mutatják, hogy a pozitív töltés a delokalizált rendszer szélső
szénatomjain jelentkezhet:

KONJUGÁLT DIÉNEK ÉS A π-
ELEKTRON DELOKALIZÁCIÓ
93 Created by XMLmind XSL-FO Converter.
A rezonanciaelmélet szerint a molekula tényleges állapota mindig energiaszegényebb (stabilisabb), mint a
felírható határszerkezetek bármelyike. Az energiakülönbség az ún. rezonanciaenergia vagy mezomériaenergia,
amely értelemszerűen megfelel az MO-elmélet által definiált delokalizációs energiának.
Habár az MO-megközelítés a korszerűbb (és egyben elméleti számításoknak is alapjául szolgálhat), mégis a VB-
megközelítésnek a hétköznapi használatban, didaktikai szempontból három előnyös oldala is van az MO-
megközelítéssel szemben:
• a π-elektron delokalizáció szemléltetésére a szokásos és könnyen áttekinthető szerkezeti képleteket használja;
• a delokalizációs energia nagyságára egyszerű kvalitatív becslést tesz lehetővé: a molekula stabilitásának
növekedése ugyanis várhatóan annál nagyobb lesz, minél hasonlóbbak energiatartalom szempontjából a
felírható mezomer határszerkezetek;
• lehetővé teszi a molekula tényleges elektronszerkezetének kvalitatív megbecsülését is: ez ugyanis a kisebb
energiatartalmú határszerkezethez kell, hogy közelebb álljon.
Az utóbbi két előnyt jól szemlélteti a butadién és az allilkation mezomer határszerkezeteinek összehasonlítása:
A butadiénre felírható három mezomer határszerkezet között nyilvánvalóan igen nagy különbségek vannak, az
"ikerionos" szerkezetek energiatartalma sokkal nagyobb, mint a nem ionizált kovalens szerkezeté. Ez egyrészt
azt jelenti, hogy a delokalizációs energia csekély kell hogy legyen. Valóban, a hidrogénezési hők alapján ez
mindössze 15 kJ/mol, ami kb. a C,C σ-kötések körüli rotáció energiagátjának felel meg és messze van a
kovalens C,C-, vagy C,H-kötések többszáz kJ/mol nagyságrendű energiájától. Másrészt azt is jelenti, hogy a
butadién elektronszerkezetét alapállapotban a középső szerkezet jól megközelíti, a két π-elektronpár
túlnyomórészt az 1. és 2., illetve a 3. és 4. szénatom között helyezkedik el.
Ugyanakkor az allilkation két határszerkezete gyakorlatilag azonos, vagyis nagy delokalizációs energiát
várhatunk és a pozitív töltés lényegében egyenlő mértékben fog megoszlani a két szélső szénatom között. Az
allilkation π-elektronpárja mondhatni "tökéletesen" delokalizált állapotban van. Ennek fizikai bizonyítéka, hogy
a két C,C-kötés azonos hosszúságú, kémiai bizonyítéka pedig a butadién párhuzamosan lejátszódó 1,2 és 1,4
brómaddíciója.
1.3. A π-elektron delokalizáció termodinamikai és kinetikai hatása
Végül még egy látszólagos ellentmondásra kell kitérnünk, nevezetesen arra, hogy a butadién
energiaszegényebb (stabilisabb), mintha a két kettős kötése független volna egymástól, ugyanakkor mégis
reakcióképesebb, pl. brómmal szemben, mint az alkének. Konjugált diének és alkének elegyét brómozva az
előbbiek sokkal gyorsabban lépnek reakcióba. Ennek a kettősségnek magyarázata a következő.
A butadién két π-elektronpárjának delokalizációja révén nyert stabilizációs energia a π-elektronrendszer
egészére érvényes, azaz azt jelenti, hogy a ψ1 és ψ2 molekulapályák együttes energiája kisebb, mint két izolált
C,C π-kötésé. Az energianyereség ezen belül azonban csak a csomósík nélküli ψ1 pályánál jelentkezik, a ψ2
pálya energiaszintje (a C2 és C3 közötti csomósík és az itt jelentkező lazító kölcsönhatás miatt) valójában már
magasabban fekszik, mint egy izolált π-pályáé. Grafikusan vázolva az energiaviszonyokat:
A bruttó energianyereség (vagyis a butadién nagyobb termodinamikai stabilitása) abból adódik, hogy ΔE(π-ψ1)
> ΔE(ψ2-π), ami azt jelenti, hogy E(ψ1 + ψ2) < E(2π). Ugyanakkor a butadién nagyobb reakciókészsége pedig
annak a következménye, hogy E(ψ2) > E(π). A Br+ ugyanis nyilvánvalóan a nagyobb energiatartalmu ψ2
(HOMO) pályán lévő elektronpárral fog kapcsolatba lépni, ez a reakció tehát gyorsabb lesz, mint a Br+ és egy
izolált π-elektronpár között lejátszódó folyamat.

KONJUGÁLT DIÉNEK ÉS A π-
ELEKTRON DELOKALIZÁCIÓ
94 Created by XMLmind XSL-FO Converter.
Úgy is fogalmazhatunk, hogy a ΔE[(ψ1 + ψ2) - (2π)] energiaváltozás a π-elektron delokalizáció
termodinamikai, a ΔE(ψ2-π) energiaváltozás pedig a π-elektron delokalizáció kinetikai hatásának
magyarázata.
2. A konjugált diének legfontosabb képviselői és előállitásuk
2.1. Butadién
A butadién -4 °C-on cseppfolyósodó gáz. Fontos nagyipari termék, melynek több, mint 90%-át műkaucsukká
dolgozzák fel. Előállítására számos eljárást dolgoztak ki.
-- Benzinpirolízis
A butadién szükséglet legnagyobb részét ma már az etén és propén előállítása céljából nagy méretekben végzett
benzinpirolizis (6.2.1. fejezet) során nyert C4-frakció butadién tartalma fedezi. A C4 frakció mennyisége a
feldolgozott benzinre vonatkoztatva 8-10%, butadién tartalma pedig 40-50%. A butadiént ebből az elegyből
poláros oldószerek (aceto-nitril, dimetil-formamid) jelenlétében végzett extraktív desztillációval különítik el.
-- Bután-butén elegyek dehidrogénezése
A kőolajfeldolgozás során nyert propán-bután frakció bután tartalmából, vagy a krakkgázokból elkülönített
butén elegyből katalitikus dehidrogénezéssel (oxid-katalizátorok, 600-700 °C) állítanak elő butadiént:
-- Acetaldehidből
Az első, ipari méretekben is megvalósított eljárás szerint az acetaldehid lúgos dimerizálásával nyert "aldol"
(16.3.2. fejezet) hidrogénezésével kapott bután-1,3-diolt alakították butadiénné foszforsavas dehidratálással:
Ennek a módszernek ma már nincs gyakorlati jelentősége.
2.2. Izoprén
Az izoprén a butadién homológja: 2-metilbuta-1,3-dién. A természetes kaucsuk "épitőköve" (a kaucsuk száraz
hevítésekor izoprén keletkezik). A természetes kaucsuk mellett más természetes eredetű szerves vegyületek
között is gyakoriak az olyanok, amelyek szénváza az izoprén szénvázát tartalmazó egységekből épül fel (pl.
terpének, szteroidok, stb.) Az ilyen izoprenoidokkal külön fejezetben fogunk foglalkozni (10. fejezet).
Az izoprén a szintetikus kaucsukok egyik fontos komponense, ipari előállítására ezért több módszer is
ismeretes.
-- C5 szénhidrogén frakciókból
A butadiénhez hasonlóan egyrészt előállítható az izoprén a megfelelő C5 szénhidrogén elegyek katalitikus
dehidrogénezésével (Cr2O3/Al2O3 katalizátor, 600 °C):

KONJUGÁLT DIÉNEK ÉS A π-
ELEKTRON DELOKALIZÁCIÓ
95 Created by XMLmind XSL-FO Converter.
másrészt kinyerhető a benzinpirolízis C5 frakciójából dimetil-formamidos (vagy N-metil-pirrolidines) extraktiv
desztillációval. A rohamosan növekvő benzinpirolízis-kapacitások folytán ma már ez az izoprén legfontosabb
forrása.
-- Izobuténből és formaldehidből
Némi ipari jelentősége van még az izobuténből és formaldehidből erősen savas közegben (ún. Prins-reakcióban)
keletkező 1,3-dioxánszármazék (vö. 28.5.1. fejezet) termikus bontásának izoprénné:
--Acetonból és acetilénből
Ma már csak ipartörténeti érdekesége van annak az eljárásnak, amely az aceton etinilezésével (7.3.5. fejezet)
kapott alkinol részleges hidrogénezése, majd az így nyert alkenol dehidratálásával állított elő izoprént:
3. A konjugált diének kémiája
3.1. Hidrogénezés
A konjugált diének katalitikus hidrogénezéssel a reakciókörülményektől és az alkalmazott katalizátortól függően
részlegesen (alkénekké) vagy teljesen (alkánokká) telíthetők. Az utóbbi esetben a "szabályos" kb. 250 kJ/mol-
nál (a pontos érték függ a kettős kötések molekulán belüli helyzetétől) kisebb hidrogénezési hő szabadul fel (vö.
8.1.fejezet).
A diének részleges hidrogénezése alkénekké fontos ipari eljárás: így távolítják el pl. a benzinpirolízis
termékéből kinyert butének mellől a nem kívánatos butadiénszennyezést.
3.2. Halogén- és HX-addíció
A konjugált diének halogénaddícióját már részletesen tárgyaltuk a π-elektron delokalizáció értelmezésével
kapcsolatban (8.1. fejezet). Itt most még a reakció hőmérsékletének az 1,2- és 1,4- termékek arányára gyakorolt
hatását kívánjuk megvizsgálni.
A kísérletek szerint alacsony hőmérsékleten (-80 °C) elsősorban 1,2-, magasabb hőmérsékleten (+40 °C) viszont
főleg 1,4-addíciós termék keletkezik. Megfigyelték ezen kívül, hogy +40 °C-on a két dibróm-butén könnyen

KONJUGÁLT DIÉNEK ÉS A π-
ELEKTRON DELOKALIZÁCIÓ
96 Created by XMLmind XSL-FO Converter.
átalakul egymásba, pontosabban bármelyikből kiindulva ugyanazt az összetételű egyensúlyi reakcióelegyet
kapják. Ez az izomerizáció -80 °C-on nem játszódik le mérhető sebességgel:
Ez a rendszer az egyik iskolapéldája a kinetikusan vagy termodinamikusan szabályozott termékösszetételnek. A
fenti észlelések alapján ugyanis az alábbi következtetéseket lehet levonni:
• az 1,2-addiciós termék képződésének sebessége a nagyobb;
• termodinamikai szempontból az 1,4-addiciós termék a stabilisabb.
Alacsony hőmérsékleten, amikor minden reakció sebessége kicsi, csak a legkisebb aktiválási energiát igénylő
reakció (a viszonylag leggyorsabb folyamat) lesz számottevő, a termék összetételében az ennek során képződő
termék (esetünkben az 1,2-addukt) fog dominálni. Az 1,2-addíciós termék tehát az ún. kinetikailag
szabályozott reakciótermék.
Magasabb hőmérsékleten már valamennyi lehetséges reakció sebessége számottevő és a rendelkezésre álló idő
alatt beállhat a termodinamikai egyensúly, vagyis a termékek képződési szabadentalpiái által meghatározott
termékösszetétel. Ez esetben a termékelegyben a legstabilisabb vegyület fog dominálni, ami ebben az esetben az
1,4-addukt. Ez utóbbi tehát az ún. termodinamikailag szabályozott termék.
Az energiaviszonyokat vázlatosan a 8.1. ÁBRA szemlélteti.
8.1. ÁBRA
Az 1,2-termék nagyobb képződési sebességét azzal magyarázhatjuk, hogy a butadién hatására lejátszódó Br2 →
Br+ + Br- heterolízis során képződő Br-, a Br+ közelében tartózkodik, tehát nagyobb valószínűséggel tud a C2
atommal reagálni, mint a C4 atommal. Az 1,4-termék nagyobb stabilitása pedig részben a brómatomok sztérikus
igényére, részben a rajtuk kialakuló parciális negatív töltések taszítására vezethető vissza.

KONJUGÁLT DIÉNEK ÉS A π-
ELEKTRON DELOKALIZÁCIÓ
97 Created by XMLmind XSL-FO Converter.
A halogénekhez hasonlóan a HX-molekulák is 1,4- és 1,2-helyzetben addicionálódnak a konjugált diénekre.
Például:
3.3. Diels-Alder-szintézis
A konjugált diének ún. dienofil partnerekkel reagálva 1,4-cikloaddícióval hatszénatomos telítetlen gyűrűt
(ciklohexéngyűrűt) alakítanak ki. A dienofil partner rendszerint olyan alkén- vagy alkinszármazék, amely a
telítetlen C,C-kötés mellett elektronszívó csoportot tartalmaz (pl. akrilsav, akrilo-nitril, maleinsav-anhidrid,
stb.). Maguk a telítetlen szénhidrogének csak erélyesebb körülmények között reagálnak. A diének közül viszont
az izoprén (vagy még inkább a dimetilbutadién) könnyebben reagál, mint a butadién. A dienofil
reakciókészségét tehát elektronszívó csoport (-I effektus), a dién reakciókészségét pedig elektrondonor csoport
(σ-konjugáció, vagy +I effektus) jelenléte növeli. Tágabb értelmezésben tehát úgy is fogalmazhatunk, hogy a
dienofil reaktáns elektrofil, a dién reaktáns pedig nukleofil reakciópartner. A reaktánsok elektrondonor, illetve
elektronakceptor jellegének hatását a Diels-Alder-szintézishez szükséges reakciókörülményekre jól szemlélteti
az alábbi két példa:
Ezt a ciklohexán-származékok szintézise szempontjából igen értékes reakciót gyakran használják konjugált
diének szelektív elválasztására is hasonló, de nem-konjugált alkének mellől. A dién ugyanis a termékből
elválasztott (és esetleg tovább tisztított) adduktumból többnyire regenerálható (a reakció reverzibilis).
Az adduktumok térszerkezetére a ciklopentadién tárgyalásakor fogunk részletesebben kitérni.
A Diels-Alder-szintézis olyan koncertikus reakció (vö. 3.5. fejezet), amelynek során hat kötőelektron
rendeződik át egyidejűleg:
Ezt figyelembevéve meglepő az, hogy a reakció - megfelelő szubsztrátumok esetén - milyen könnyen és
szelektíven játszódik le. Ennek okát az ún Woodward-Hoffmann szabályok értelmében a reagáló
molekulapályák kedvező szimmetriaviszonyaiban (és térbeli elrendeződésében) találjuk meg. Ezen szabályok
lényegét egyszerűsített formában a következőkben világítjuk meg.

KONJUGÁLT DIÉNEK ÉS A π-
ELEKTRON DELOKALIZÁCIÓ
98 Created by XMLmind XSL-FO Converter.
Az MO-elméletnek a reakciókészség értelmezése tekintetében továbbfejlesztett formája, az FMO-elmélet
(frontier molecular orbitals - szélső molekulapályák) szerint két reaktáns közeledése során elsőnek az egyik
reaktáns HOMO-ja és a másik reaktáns LUMO-ja (vagy fordítva) lép kölcsönhatásba egymással, így a reakció
sebességét és irányát elsősorban ezek a kölcsönhatások szabják meg.
A molekulapályák energiaviszonyait tekintve érthető, hogy főleg a legnagyobb energiájú (tehát a
"legreakcióképesebb" elektronokat tartalmazó) betöltött pálya és a legkisebb energiájú (tehát legkönnyebben
elérhető) betöltetlen pálya határozza meg egy molekula reakcióképességét. Egy HOMO és egy LUMO
kölcsönhatása stabilis kémiai kötést tud létrehozni, mert így van biztosítva a kialakuló új molekulapálya
kötőszintjének feltöltése két elektronnal és az, hogy a hozzá tartozó lazító szint üres marad:
Két HOMO kombinációja pl. nem-kötő állapothoz vezetne, mert az ezeken lévő, összesen négy elektron a lazító
és a kötő pálya egyidejű feltöltését eredményezné.
Az előbbiekben láttuk, hogy a Diels-Alder-cikloaddicióban a dién a nukleofil és az alkén az elektrofil partner,
logikus tehát, ha a dién HOMO és az alkén LUMO pályáját vesszük szemügyre. (Az FMO elmélet szerint
általánosságban is definiálhatunk úgy, hogy az a nukleofil reagens, amely a HOMO-jával és az az elektrofil,
amely a LUMO-jával reagál.) A 8.2. ÁBRÁBÓL látható, hogy a reakcióhoz szükséges átmeneti állapot
szerkezetét figyelembevéve a két MO szimmetriája azonos (a papír síkjában lévő tükörsíkra nézve mindkettő
antiszimmetrikus, "A" ), e tekintetben tehát adott a kötő molekulapályák kialakulásának feltétele.
8.2. ÁBRA
Ugyanerre az eredményre (vagyis hogy a kölcsönhatásba lépő molekulapályák szimmetriaviszonyai
megfelelőek a reakció lejátszódásához) jutunk egyébként akkor is, ha a butadién ψ3*, LUMO és az etén π,
HOMO pályákat vesszük figyelembe. A különbség az lesz, hogy ez esetben mindkét pálya a papír síkjára nézve
szimmetrikus, "S".
A kedvező szimmetria-tulajdonságokon túlmenően elősegíti a reakciót még az is, hogy a butadién ciszoid
konformációja esetén a kérdéses molekulapályák az átfedéshez kedvező térbeli helyzetben is vannak.
Az FMO-elmélet alapján érthetővé válik az a régóta ismert kísérleti tény is, hogy a rendkívül könnyen
lejátszódó Diels-Alder-cikloaddícióval ellentétben az alkének ciklodimerizációját ciklobután származékokká
termikus koncertikus reakcióban (azaz kizárólag melegítéssel, egy lépésben) nem lehet megvalósítani. Ez a
negatív tapasztalat ugyanis összhangban van azzal, hogy két alkén (pl. két etén molekula) HOMO és LUMO
orbitáljainak szimmetriája nem azonos (8.3. ÁBRA).
8.3. ÁBRA

KONJUGÁLT DIÉNEK ÉS A π-
ELEKTRON DELOKALIZÁCIÓ
99 Created by XMLmind XSL-FO Converter.
Tisztában kell lennünk azonban azzal, hogy ez a reakció nem termodinamikailag lehetetlen, hanem csak
koncertikus folyamatban kinetikailag erősen gátolt. Ilyen és hasonló, ún. szimmetria-tiltott reakciók
megvalósíthatók, ha akár a HOMO vagy a LUMO megváltozik (pl. fotokémiai aktiválás hatására), vagy akár
más - többlépéses - mechanizmusra, tehát "kerülőútra" van lehetőség.
Az elmondottak alapján nem véletlen, hogy a szerves kémiai reakciók nagy többsége többlépéses
"kerülőutakon" át játszódik le: ionos vagy gyökös jellegű közti termékek lépnek fel és a reakcióknak többé-
kevésbé bonyolult mechanizmusuk van. Így pl. az alkének Br2 vagy HX addíciója koncertikus reakcióban
szimmetria-tiltott folyamat volna. Ezért ez a két - a valóságban mégis igen könnyen végbemenő reakció - a Br+,
illetve a H+ támadásával indul, majd az ennek során kialakult karbokation és az anion reakciójával végződik.
Mindkét részlépés külön-külön csak igen egyszerű szimmetriájú atomi-, vagy molekula-orbitálok részvételét
igényli, amelyeknél szimmetriaprobléma nem lép fel. Ennek alapján az is érthetővé válik, hogy miért olyan sok
az olyan szerves kémiai reakció, amelyet a H+ katalizál: ennek a részecskének LUMO pályája egy olyan üres,
gömbszimmetrikus atomi s-orbitál, melynek szimmetriája bármely kötő molekulaorbitállal vagy magános
elektronpárt tartalmazó atomi orbitállal kompatibilis.
A Br2 vagy HX addició tehát ionos intermediereken át lejátszódó kerülőt választ, mert a reagensek vagy már
erősen polárosak (HX), vagy könnyen polározhatók (Br2). A H2 molekula addíciója (alkén-hidrogénezés)
esetében ezek a feltételek nem állnak fenn és ezért a koncertikusan itt is szimmetria-tiltott reakciót a katalizátor
teszi lehetővé, megint csak többlépéses mechanizmus révén (részletesen lásd 26.13.3. fejezet).
3.4. Polimerizáció
A konjugált diének gyökös iniciátorok (fémnátrium, peroxidok, redox-rendszerek), vagy fémorganikus
katalizátorok hatására rugalmas természetű makromolekulás láncokká - elasztomerekké - polimerizálódnak. A
kapcsolódás módja a monomer konjugált szerkezete következtében háromféle lehet (8.4. ÁBRA):
• 1,2-,
• (Z)-cisz-1,4
• (E)-transz-1,4
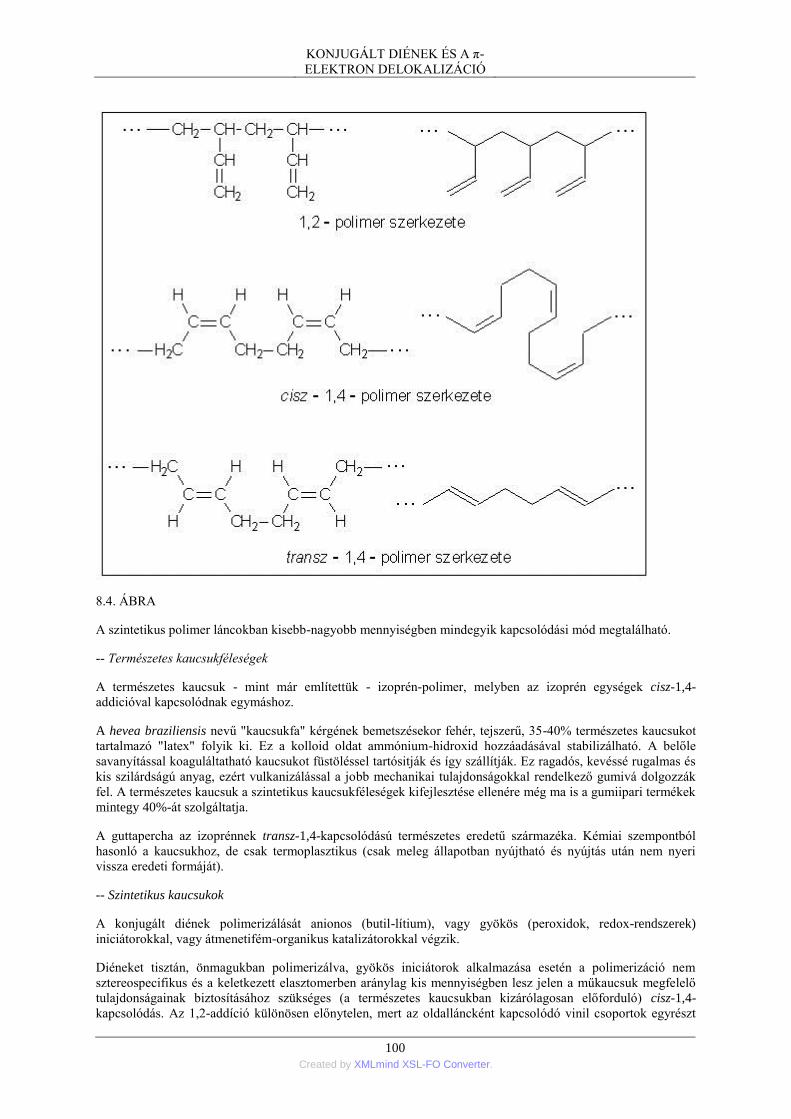
KONJUGÁLT DIÉNEK ÉS A π-
ELEKTRON DELOKALIZÁCIÓ
100 Created by XMLmind XSL-FO Converter.
8.4. ÁBRA
A szintetikus polimer láncokban kisebb-nagyobb mennyiségben mindegyik kapcsolódási mód megtalálható.
-- Természetes kaucsukféleségek
A természetes kaucsuk - mint már említettük - izoprén-polimer, melyben az izoprén egységek cisz-1,4-
addicióval kapcsolódnak egymáshoz.
A hevea braziliensis nevű "kaucsukfa" kérgének bemetszésekor fehér, tejszerű, 35-40% természetes kaucsukot
tartalmazó "latex" folyik ki. Ez a kolloid oldat ammónium-hidroxid hozzáadásával stabilizálható. A belőle
savanyítással koaguláltatható kaucsukot füstöléssel tartósitják és így szállítják. Ez ragadós, kevéssé rugalmas és
kis szilárdságú anyag, ezért vulkanizálással a jobb mechanikai tulajdonságokkal rendelkező gumivá dolgozzák
fel. A természetes kaucsuk a szintetikus kaucsukféleségek kifejlesztése ellenére még ma is a gumiipari termékek
mintegy 40%-át szolgáltatja.
A guttapercha az izoprénnek transz-1,4-kapcsolódású természetes eredetű származéka. Kémiai szempontból
hasonló a kaucsukhoz, de csak termoplasztikus (csak meleg állapotban nyújtható és nyújtás után nem nyeri
vissza eredeti formáját).
-- Szintetikus kaucsukok
A konjugált diének polimerizálását anionos (butil-lítium), vagy gyökös (peroxidok, redox-rendszerek)
iniciátorokkal, vagy átmenetifém-organikus katalizátorokkal végzik.
Diéneket tisztán, önmagukban polimerizálva, gyökös iniciátorok alkalmazása esetén a polimerizáció nem
sztereospecifikus és a keletkezett elasztomerben aránylag kis mennyiségben lesz jelen a műkaucsuk megfelelő
tulajdonságainak biztosításához szükséges (a természetes kaucsukban kizárólagosan előforduló) cisz-1,4-
kapcsolódás. Az 1,2-addíció különösen előnytelen, mert az oldalláncként kapcsolódó vinil csoportok egyrészt
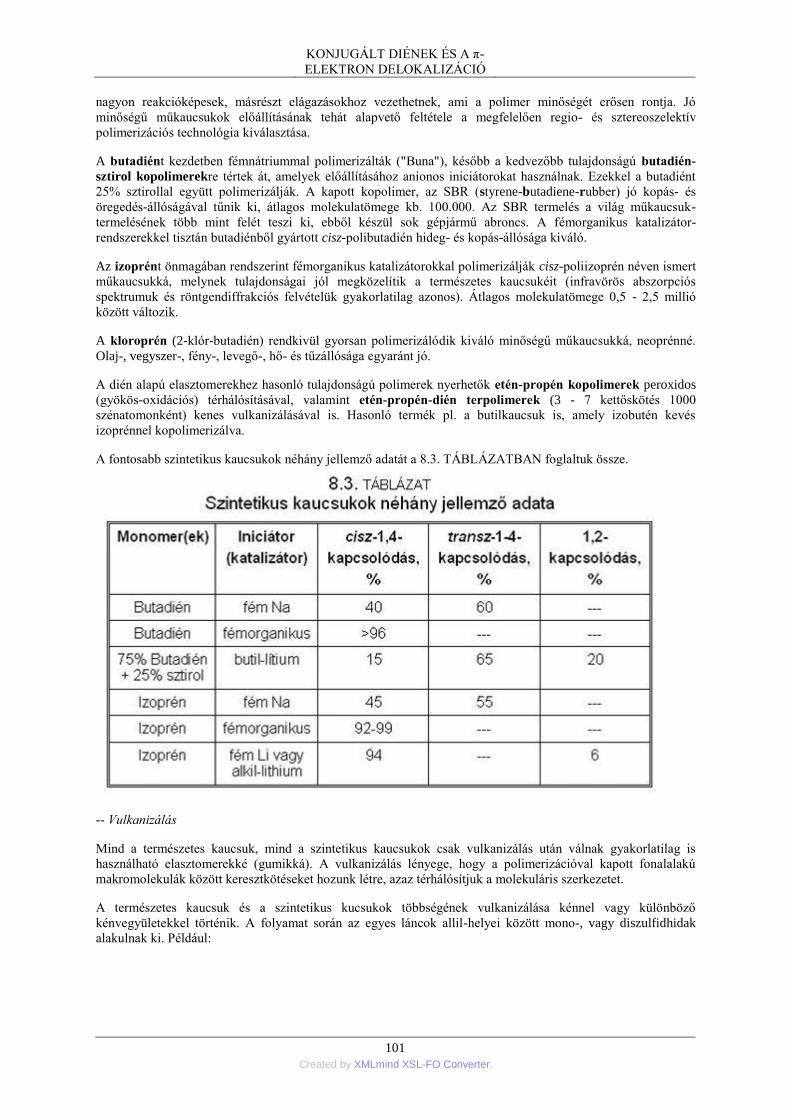
KONJUGÁLT DIÉNEK ÉS A π-
ELEKTRON DELOKALIZÁCIÓ
101 Created by XMLmind XSL-FO Converter.
nagyon reakcióképesek, másrészt elágazásokhoz vezethetnek, ami a polimer minőségét erősen rontja. Jó
minőségű műkaucsukok előállításának tehát alapvető feltétele a megfelelően regio- és sztereoszelektív
polimerizációs technológia kiválasztása.
A butadiént kezdetben fémnátriummal polimerizálták ("Buna"), később a kedvezőbb tulajdonságú butadién-
sztirol kopolimerekre tértek át, amelyek előállításához anionos iniciátorokat használnak. Ezekkel a butadiént
25% sztirollal együtt polimerizálják. A kapott kopolimer, az SBR (styrene-butadiene-rubber) jó kopás- és
öregedés-állóságával tűnik ki, átlagos molekulatömege kb. 100.000. Az SBR termelés a világ műkaucsuk-
termelésének több mint felét teszi ki, ebből készül sok gépjármű abroncs. A fémorganikus katalizátor-
rendszerekkel tisztán butadiénből gyártott cisz-polibutadién hideg- és kopás-állósága kiváló.
Az izoprént önmagában rendszerint fémorganikus katalizátorokkal polimerizálják cisz-poliizoprén néven ismert
műkaucsukká, melynek tulajdonságai jól megközelítik a természetes kaucsukéit (infravörös abszorpciós
spektrumuk és röntgendiffrakciós felvételük gyakorlatilag azonos). Átlagos molekulatömege 0,5 - 2,5 millió
között változik.
A kloroprén (2-klór-butadién) rendkivül gyorsan polimerizálódik kiváló minőségű műkaucsukká, neoprénné.
Olaj-, vegyszer-, fény-, levegő-, hő- és tűzállósága egyaránt jó.
A dién alapú elasztomerekhez hasonló tulajdonságú polimerek nyerhetők etén-propén kopolimerek peroxidos
(gyökös-oxidációs) térhálósításával, valamint etén-propén-dién terpolimerek (3 - 7 kettőskötés 1000
szénatomonként) kenes vulkanizálásával is. Hasonló termék pl. a butilkaucsuk is, amely izobutén kevés
izoprénnel kopolimerizálva.
A fontosabb szintetikus kaucsukok néhány jellemző adatát a 8.3. TÁBLÁZATBAN foglaltuk össze.
-- Vulkanizálás
Mind a természetes kaucsuk, mind a szintetikus kaucsukok csak vulkanizálás után válnak gyakorlatilag is
használható elasztomerekké (gumikká). A vulkanizálás lényege, hogy a polimerizációval kapott fonalalakú
makromolekulák között keresztkötéseket hozunk létre, azaz térhálósítjuk a molekuláris szerkezetet.
A természetes kaucsuk és a szintetikus kucsukok többségének vulkanizálása kénnel vagy különböző
kénvegyületekkel történik. A folyamat során az egyes láncok allil-helyei között mono-, vagy diszulfidhidak
alakulnak ki. Például:

KONJUGÁLT DIÉNEK ÉS A π-
ELEKTRON DELOKALIZÁCIÓ
102 Created by XMLmind XSL-FO Converter.
A lágy és rugalmas gumi általában 1-3% kén bevitelével készül, a térhálónak ugyanis igen lazának (kevés
keresztkötést tartalmazónak) kell lennie. 25...32% kén beépitésével keménygumi (ebonit) keletkezik. A
vulkanizálást elemi kénnel végzik, rendszerint valamilyen gyorsító (akcelerátor) jelenlétében, amely a
vulkanizálószer és a kaucsuk kölcsönhatását segíti elő. Ilyen lehet pl. a 2-szulfanil-benz[4,5]tiazol (régi nevén
merkapto-benztiazol) (I) vagy az N,N,N,N-tetrametil-tiurám-diszulfid (TMTD) (II). Ezekből könnyen
keletkeznek szabad gyökök, amelyek felszakítják az S8 gyűrűt és ezzel iniciálják a gyökös mechanizmusú
vulkanizálást.
A neoprént ZnO-dal vulkanizálják, itt a keresztkötéseket a néhány 1,2-polimer egység között kialakuló éteres
oxigénhidak biztosítják:
A kész gumitermék törékennyé válásának megakadályozására öregedésgátlókat (antioxidánsokat, pl. aromás
aminokat) adagolnak a kaucsukhoz. Az autoabroncsokhoz felhasznált gumi mindig tartalmaz még más
adalékanyagokat is, így jelentős mennyiségű (≈30%) kormot, továbbá lágyítókat stb.
4. Gyűrűs diének és poliének
4.1. Ciklobutadién
Kettős kötések konjugációjára formálisan már a négytagú gyűrűben is van lehetőség. A ciklobutadién azonban
szobahőfokon nem stabilis, mert egyrészt - mint azt később látni fogjuk - négy π-elektront tartalmazó konjugált
rendszer esetében a gyűrűzárás a nyíltláncú szerkezethez (a butadiénhez) képest az MO-ok energiáját
megnöveli, másrészt a 120°-os vegyértékszöget "igénylő" sp2 szénatomok atomi orbitáljainak átlapolása a 90°-
os négyzetes elrendeződésben gyenge.

KONJUGÁLT DIÉNEK ÉS A π-
ELEKTRON DELOKALIZÁCIÓ
103 Created by XMLmind XSL-FO Converter.
Az extrém alacsony hőmérsékleteken speciális módszerekkel előállított ciklobutadién már -200 °C-on Diels-
Alder szerint dimerizál, amennyiben az egyik molekula diénként, a másik dienofilként viselkedik (a két
sztereoizomer termék magyarázatát lásd a következő fejezetben):
Stabilizálhatók azonban a ciklobutadién, vagy annak származékai mint ligandumok különböző átmenetifém-
komplexekben, pl.:
A ciklobutadién a teljesen konjugált gyűrűs poliének legegyszerűbb képviselője. A benzol formailag
tulajdonképpen a clklobutadién "vinilóg"-jának (egy -CH=CH- csoporttal megnövelt láncú származékának)
tekinthető. Az aromás benzolgyűrűhöz való formai hasonlósága miatt ezért a ciklobutadién tulajdonságai
elméleti szempontból érdekesek.
4.2. Ciklopentadién
A legegyszerűbb stabilis konjugált gyűrűs dién a ciklopenta-1,3-dién (ciklopentadién), amely kisebb
mennyiségben a kőszénkátrány előpárlatában, gazdaságosan kinyerhető mennyiségben (15-25%) a
benzinpirolízis termékének C5-frakciójában fordul elő.
-- Dimerizáció
A ciklopentadién a diénekre jellemző tulajdonságokban - szerkezeti hasonlósága miatt - közel áll a butadiénhez,
de annál reakcióképesebb: levegőn gyorsan oxidálódik és polimerizálódik, mert gyűrűje a konjugált rendszer
merevsége miatt meglehetősen feszült és ez alól a molekula kitérni igyekszik. Konjugált kettőskötés-
rendszerének reakciói közül a legfontosabb, hogy szobahőmérsékleten Diels-Alder-reakcióban spontán
dimerizálódik diciklopentadiénné. Ez a reakció - mint általában a Diels-Alder-reakciók - reverzibilis, a
diciklopentadién hevítve két molekula ciklopentadiénre esik szét:
A diciklopentadiénnek két térszerkezeti formája lehetséges, amelyeket endo-, illetve exo- előnevekkel
különböztetünk meg egymástól [endo = belül, exo = kívül (görög)].
Ezeket a fogalmakat hidas biciklusos molekulák térszerkezetének jelölésére használják: az endo helyzetű
szubsztituens az, amely a kádszerűen meghajlított hatos gyűrű belső oldalához közelebb esik. A
diciklopentadiénnél szubsztituensként a harmadik gyűrű szerepel. Általában az endo-módosulat az elsődleges
(tehát a kinetikailag kontrollált) főtermék, mely azután átrendeződhet a termodinamikailag stabilisabb exo-
formába.
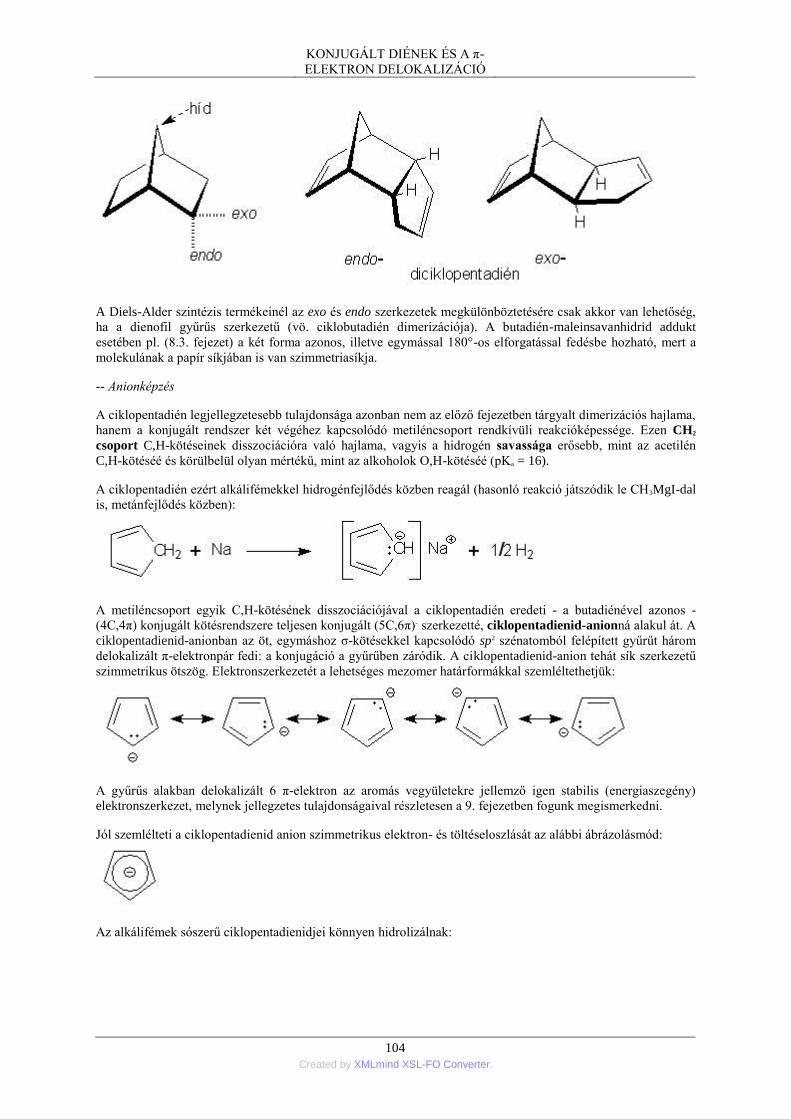
KONJUGÁLT DIÉNEK ÉS A π-
ELEKTRON DELOKALIZÁCIÓ
104 Created by XMLmind XSL-FO Converter.
A Diels-Alder szintézis termékeinél az exo és endo szerkezetek megkülönböztetésére csak akkor van lehetőség,
ha a dienofil gyűrűs szerkezetű (vö. ciklobutadién dimerizációja). A butadién-maleinsavanhidrid addukt
esetében pl. (8.3. fejezet) a két forma azonos, illetve egymással 180°-os elforgatással fedésbe hozható, mert a
molekulának a papír síkjában is van szimmetriasíkja.
-- Anionképzés
A ciklopentadién legjellegzetesebb tulajdonsága azonban nem az előző fejezetben tárgyalt dimerizációs hajlama,
hanem a konjugált rendszer két végéhez kapcsolódó metiléncsoport rendkívüli reakcióképessége. Ezen CH2
csoport C,H-kötéseinek disszociációra való hajlama, vagyis a hidrogén savassága erősebb, mint az acetilén
C,H-kötéséé és körülbelül olyan mértékű, mint az alkoholok O,H-kötéséé (pKa = 16).
A ciklopentadién ezért alkálifémekkel hidrogénfejlődés közben reagál (hasonló reakció játszódik le CH3MgI-dal
is, metánfejlődés közben):
A metiléncsoport egyik C,H-kötésének disszociációjával a ciklopentadién eredeti - a butadiénével azonos -
(4C,4π) konjugált kötésrendszere teljesen konjugált (5C,6π)- szerkezetté, ciklopentadienid-anionná alakul át. A
ciklopentadienid-anionban az öt, egymáshoz σ-kötésekkel kapcsolódó sp2 szénatomból felépített gyűrűt három
delokalizált π-elektronpár fedi: a konjugáció a gyűrűben záródik. A ciklopentadienid-anion tehát sík szerkezetű
szimmetrikus ötszög. Elektronszerkezetét a lehetséges mezomer határformákkal szemléltethetjük:
A gyűrűs alakban delokalizált 6 π-elektron az aromás vegyületekre jellemző igen stabilis (energiaszegény)
elektronszerkezet, melynek jellegzetes tulajdonságaival részletesen a 9. fejezetben fogunk megismerkedni.
Jól szemlélteti a ciklopentadienid anion szimmetrikus elektron- és töltéseloszlását az alábbi ábrázolásmód:
Az alkálifémek sószerű ciklopentadienidjei könnyen hidrolizálnak:

KONJUGÁLT DIÉNEK ÉS A π-
ELEKTRON DELOKALIZÁCIÓ
105 Created by XMLmind XSL-FO Converter.
Nagy mértékben stabilizálódik a ciklopentadienid-anion akkor, ha az alkén-π-komplexekben már megismert
módon (6.3.11. fejezet) átmenetifémhez kapcsolódik. Az ilyen fém-π-ciklopentadienil-komplexeket és
származékaikat általános névvel metallocéneknek nevezzük. Legfontosabb és legismertebb képviselőjük a
bisz(η5-ciklopentadienil)vas vagy ferrocén [Fe(C5H5)2], de ismert pl. a nikkelocén, kobaltocén is.
A szisztematikus névben szereplő η (olvasd: éta vagy hapto) és a hozzárendelt szám itt azt jelképezi, hogy a
ligandumként szereplő szerves molekula hány atomja kapcsolódik a központi fématomhoz (esetünkben mind az
öt szénatom).
A ferrocén termikusan és kémiailag egyaránt igen stabilis kristályos vegyület; a fémorganikus vegyületek
többségétől eltérően lúgokra, savakra, levegőre nem érzékeny. Szerkezete:
A két ciklopentadienid-anion gyűrű tehát lapjával fordul a központi fématom (ion) felé. Az ilyen szerkezetű
komplexeket találó hasonlattal szendvicskomplexeknek is szokás nevezni. A koordinatív kapcsolat a fématom
üres d-orbitáljainak és a ciklopentadienid-gyűrűk delokalizált π-MO-jainak átfedése révén jön létre. A baloldali
ábrázolásmódban feltüntetett töltések teljesen formális jelöléseknek tekinthetők (csak a vegyület kialakulásának
folyamatát kívánják érzékeltetni); a pozitív és negatív töltések a molekulán belül kiegyenlítődnek (a ferrocén
apoláros, szénhidrogénekben oldódó vegyület).
A ferrocén és a hasonló átmenetifém-ciklopentadienid-komplexek tehát tipikus π-komplexek, melyek kémiai és
fizikai tulajdonságaikban alapvetően különböznek az alkálifémek ionos szerkezetű, hidrolízisre érzékeny
ciklopentadienidjeitől. (A ferrocén pl. számos - az aromás szénhidrogénekre jellemző - szubsztitúciós reakcióra
hajlamos.) Az átmenetifém-organikus vegyületekre részletesebben a 26.13. fejezetben fogunk kitérni.
-- Kondenzáció ketonokkal
Lúgos közegben a ciklopentadién ketonokkal konjugált szerkezetű gyűrűs szénhidrogénekké, fulvénekké
kondenzálódik (a metiléncsoport C,H-kötése addicionálódik a karbonil (>C=O) csoportra):
4.3. Ciklohexadién
A két konjugált telítetlen kötést tartalmazó ciklohexa-1,3-diénben az egyszerű (4C,4π) delokalizált rendszer nem
stabilis. Ehhez az egységhez ugyanis - hatos gyűrűben - csak feszültség árán kapcsolódhat két sp3 szénatom. Ha

KONJUGÁLT DIÉNEK ÉS A π-
ELEKTRON DELOKALIZÁCIÓ
106 Created by XMLmind XSL-FO Converter.
ugyanezen két szénatom pl. dehidrogéneződés révén sp2-vé alakul át, stabilis, aromás jellegű rendszer
kialakulására van lehetőség (lásd 9. fejezet):
Ez a dehidrogéneződés a gyűrűfeszültség megszűnése és a gyűrűben záródó konjugáció lehetősége
következtében ezért kis mértékben exoterm (ΔH = -24 kJ/mol). Ez igen érdekes eredmény annak fényében, hogy
pl. a ciklohexén kettős kötésének hidrogénezése exoterm folyamat (ΔH = -120 kJ/mol), vagyis a ciklohexán
dehidrogénezése ugyanilyen mértékben endoterm. Ennek a jelenségnek az értelmezésére az aromás
szénhidrogének tárgyalásakor fogunk részletesen kitérni (9. fejezet).
4.4. Ciklooktatetraén - fluxionális molekulák
A C8H8 összegképletű, váltakozó egyszeres és kétszeres C,C-kötéseket tartalmazó ciklooktatetraén (COT)
formailag gyűrűben záródó konjugációra (aromás szerkezet kialakítására) képes rendszernek látszik. A
valóságban azonban ez a vegyület normális cikloalkénként reagál: brómot addicionál és kálium-permanganáttal
hidroxilálható.
Ezt elsősorban azzal magyarázhatjuk, hogy a delokalizációhoz rendelkezésre álló elektronok száma nyolc, ami
nem elégíti ki az aromás szerkezet kialakításához megkívánt és később megismerendő (9.2.2. fejezet) Hückel-
féle, 4n + 2 szabályt. A molekula nem is sík alakú, ami tovább csökkenti a π-kötések közötti kölcsönhatások
lehetőségét:
Két elektron felvételével a COT planáris, aromás jellegű dianionná [(8C,10π)2- rendszerré)] alakul. Ekkor
ugyanis már tíz elektron áll rendelkezésre a delokalizált rendszer kialakulásához, ami a Hückel-szabály
értelmében aromás szerkezetet eredményez:
A ciklopentadienid anionhoz hasonlóan ennek az anionnak is ismeretesek fémorganikus szendvics-komplexei
[pl. U(C8H10)2].
Mint láttuk, a ciklooktatetraén lokalizált, alkénes-jellegű kettős kötésekkel rendelkezik. A várakozással
ellentétben mégis általában csak egyféle 1,2-diszubsztituált származékai ismeretesek. Ennek oka az, hogy a két
lehetséges izomer igen gyorsan alakul át egymásba és valójában mindig a kettő egyensúlyi elegyével állunk
szemben:

KONJUGÁLT DIÉNEK ÉS A π-
ELEKTRON DELOKALIZÁCIÓ
107 Created by XMLmind XSL-FO Converter.
Hasonló folyamat természetesen a nem szubsztituált ciklooktatetraénben is lejátszódik:
Ebben az esetben azonban a két izomer egymástól már nem különböztethető meg. Az ilyen izomerizációt
autoizomerizációnak nevezzük és ha a folyamat gyors, a molekula fluxionális viselkedéséről beszélünk. A
fluxionális viselkedés definíciója nem teljesen szabatos, általában azokra a molekulákra használják, amelyeknél
az egyes izomerek átlagos élettartama közönséges hőmérsékleten az NMR spektroszkópia feloldó képességének
határa (≈10-2 sec) alá esik. Ez a gyakorlatban akkor teljesül, ha az izomerizáció aktiválási energiája nem haladja
meg a 60 kJ/mol értéket. A többszörösen telítetlen szénhidrogének, azok ionos származékai és a fémorganikus
komplexek között számos fluxionális molekula ismert.
A COT fluxionális viselkedését élesen meg kell különböztetnünk az aromás gyűrűk π-elektron
delokalizációjától, melyet a következő fejezetben tárgyalunk részletesen. Itt elöljáróban is ki szeretnénk emelni
azonban azt a lényeges elvi különbséget, hogy míg a két COT autoizomer két diszkrét molekula, a benzol
később tárgyalandó Kekulé-szerkezetei csak mezomer határformák, a szemléletességet elősegítő ábrázolások és
nem konkrét molekulaszerkezeteket jelentenek.
Energiaprofilokkal ábrázolva a COT autoizomerek tehát két külön energiaminimumban fekszenek, amelyeket
csak egy viszonylag alacsony energiadomb választ el egymástól (8.5. ÁBRA, A) és ez az energiaprofil reális.
Ugyanakkor a benzolnak viszont csak egyetlen energiaminimuma - azaz szerkezete - van és a 8.5. ÁBRÁN
bemutatott "energiaprofilja" (B) hipotetikus, mivel a lokalizált kettőskötésekkel ábrázolt mezomer
határszerkezeteknek nincs fizikai realitásuk.
8.5. ÁBRA

108 Created by XMLmind XSL-FO Converter.
9. fejezet - AROMÁS SZÉNHIDROGÉNEK (Arének)
A szénhidrogéneknek a szénlánc szerkezete szerinti rendszerezésekor az aromás szénhidrogéneket formailag a
gyűrűs telítetlen szénhidrogének közé kellene besorolnunk. A formai hasonlóság ellenére azonban mégis külön
tárgyaljuk őket, mert - a cikloalkénektől eltérően - reakcióik többségében nem telítetlen kötésrendszerként
viselkednek. Ennek oka az, hogy az aromás szénhidrogének szénvázára jellemző - csak trigonális sp2
szénatomokból felépülő - planáris (többnyire hattagú) gyűrűn a π-elektronok sajátos kapcsolatrendszert, hat π-
elektronból álló zárt konjugációt hoznak létre. Az ezáltal különlegesen stabilissá vált szerkezeten pedig
addíciós reakciók helyett már inkább szubsztitúciós reakciók játszódnak le, vagyis az aromás szénhidrogének
kémiája alapvetően eltér az alkének kémiájától.
Az aromás szénhidrogének legegyszerűbb képviselője ("alapvegyülete") a benzol, C6H6. Elektronszerkezetét a
későbbiekben még részletesen fogjuk tárgyalni. Itt most csak annyit említünk meg, hogy a benzolgyűrűket
tartalmazó molekuláknál az aromás π-elektron-szextett hat elektronjának egyértelmű ábrázolására a
benzolgyűrűnél a Kekulé-féle, lokalizált konjugált kettős kötéses képleteket fogjuk használni (9.1. ÁBRA, A
vagy B; a szénatomokhoz kapcsolódó hidrogénatomokat nem szokás feltüntetni). Tesszük ezt annak ellenére,
hogy tisztában kell lennünk azzal, miszerint az (A) és (B) képletek nem konkrét molekulákat, hanem csak
mezomer határformákat ábrázolnak.
9.1. ÁBRA
Egyes kivételes esetektől eltekintve (lásd pl. 8.5. ÁBRA) tehát kerülni fogjuk a π-elektronok tényleges
delokalizációját valóban jól tükröző (C) ábrázolásmódot (9.1. ÁBRA). Egyrészt azért, mert ez az ábrázolásmód
a kondenzált gyűrűs aromás vegyületekre már amúgy sem használható. A másik, ez ellen az ábrázolásmód ellen
szóló érv pedig az, hogy elnagyoltan jelöli a π-elektronok számát és ezáltal zavarja az aromás gyűrűn lejátszódó
reakciók mechanizmusának megértését.
A benzolt elsőként Faraday különítette el 1825-ben, a benzol nevet Liebig javaslatára fogadták el. A ma
érvényes IUPAC nómenklatúrával ez a név nincs összhangban, mivel az -ol végződés az alkoholok elnevezésére
van fenntartva (12.1c fejezet). A magyar szakirodalomban német hatásra terjedt el. Az angolszász irodalom a
vegyületre - tekintettel alkénes jellegű telítetlenségére - a benzene elnevezést használja. Ez az elnevezés
(Benzen) ma már a német nyelvterületen is terjed.
1. Általános áttekintés
1.1. Az aromás szénhidrogének csoportosítása és elnevezése
-- Egy-gyűrűs (monociklusos) aromás szénhidrogének (benzolhomológok)
A gyűrűbe zárt szénatom(ok)hoz alkil-, alkenil- vagy alkinilcsoportok kapcsolódhatnak.
Egy oldallánc esetén a kapcsolódó alifás csoport neve után tesszük a "benzol" szót. PéIdául: metilbenzol,
etilbenzol, izopropilbenzol, stb. Az egyszerűbb származékoknak gyakran a triviális nevét használjuk: pl.
metilbenzol helyett toluol (toluene), izopropilbenzol helyett kumol (cumene), vinilbenzol helyett sztirol
(styrene):
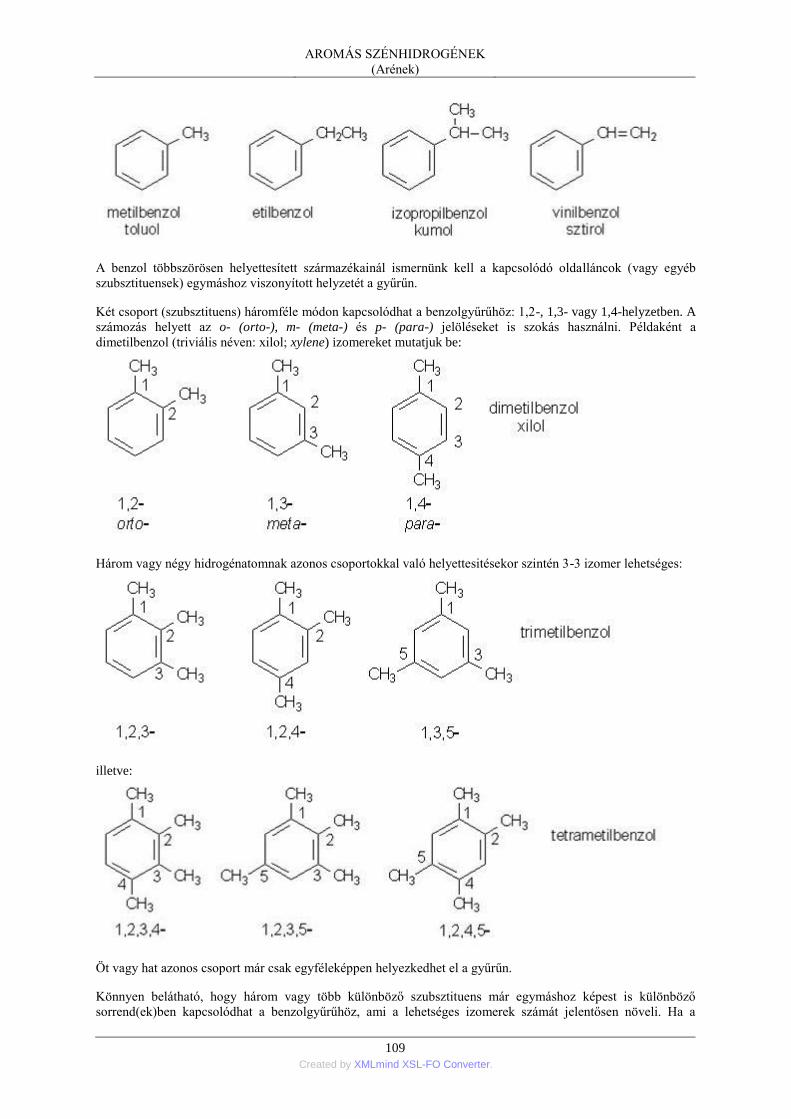
AROMÁS SZÉNHIDROGÉNEK
(Arének)
109 Created by XMLmind XSL-FO Converter.
A benzol többszörösen helyettesített származékainál ismernünk kell a kapcsolódó oldalláncok (vagy egyéb
szubsztituensek) egymáshoz viszonyított helyzetét a gyűrűn.
Két csoport (szubsztituens) háromféle módon kapcsolódhat a benzolgyűrűhöz: 1,2-, 1,3- vagy 1,4-helyzetben. A
számozás helyett az o- (orto-), m- (meta-) és p- (para-) jelöléseket is szokás használni. Példaként a
dimetilbenzol (triviális néven: xilol; xylene) izomereket mutatjuk be:
Három vagy négy hidrogénatomnak azonos csoportokkal való helyettesitésekor szintén 3-3 izomer lehetséges:
illetve:
Öt vagy hat azonos csoport már csak egyféleképpen helyezkedhet el a gyűrűn.
Könnyen belátható, hogy három vagy több különböző szubsztituens már egymáshoz képest is különböző
sorrend(ek)ben kapcsolódhat a benzolgyűrűhöz, ami a lehetséges izomerek számát jelentősen növeli. Ha a

AROMÁS SZÉNHIDROGÉNEK
(Arének)
110 Created by XMLmind XSL-FO Converter.
gyűrűhöz több, nem azonos oldallánc kapcsolódik, a láncok egymáshoz viszonyított helyzetét számozással
adjuk meg úgy, hogy lehetőleg minél kisebbek legyenek a számok és a szubsztituenseket ABC sorrendben
soroljuk fel: 1-izopropil-3-metilbenzol.
Az aromás szénhidrogénekből egy hidrogénatom elvételével származtatható csoportokat összefoglaló néven
arilcsoportoknak nevezzük (általános jelük: Ar-), két hidrogénatom elvételével pedig az ariléncsoportokat
nyerjük. Így például a benzolból egy hidrogénatom elvételével a fenil- (C6H5- vagy Ph-), két hidrogénatom
elvételével pedig az o-, m- vagy p-feniléncsoport vezethető le. Toluolból már egy hidrogénatom elvételével is
három arilcsoport képezhető: a 2-, 3- és 4-metil-fenilcsoport (vagy o-, m- és p-tolilcsoport).
-- Izolált többgyűrűs aromás szénhidrogének
A benzolgyűrű egy vagy több hidrogénatomját helyettesítheti aromás jellegű (aril-) csoport, valamint nyíltláncú
szénhidrogénhez is kapcsolódhat kettő vagy több arilcsoport. A vegyületek szerkezetére mindkét esetben az
jellemző, hogy az aromás gyűrűk egymástól izoláltak; nem "kondenzálódnak" két közös szénatomon keresztül.
E vegyületek két fő típusát a (poli)fenil-benzolok illetve a polifenil-alkánok képviselik (példaként közöljük a
bifenil és egy terfenil izomer, valamint a trifenilmetán szerkezeti képletét):
-- Kondenzált aromás szénhidrogének
Ha két gyűrűben két egymás melletti szénatom közös, kondenzált gyűrűs vegyületről beszélünk. Funkciós
csoportot nem tartalmazó aromás gyűrűk kondenzálódásával többgyűrűs kondenzált aromás szénhidrogének
alakulnak ki.
Két benzolgyűrű kondenzálódása ("anellálódása") még csak egyféle módon lehetséges: így vezethető le a
naftalin (naphthalene) (C10H8) szerkezete. Három vagy több gyűrű kondenzálódása esetén azonban már más-
más vegyületről van szó aszerint, hogy az anelláció lineáris (a gyűrűk központjait összekötő vonal egyenes),
vagy anguláris (a központok egymással szöget bezáró egyenesekkel köthetők össze). Három benzolgyűrű
lineáris kondenzálódása az antracén, anguláris kondenzálódása pedig a fenantrén szerkezetéhez vezet.
Ha a kondenzált gyűrűs aromás szénhidrogénekből képezünk arilcsoportokat, már egy hidrogénatom elvétele
esetén is kettő vagy több izomer csoport keletkezhet: az aromás gyűrűk egyes szénatomjainak strukturális
helyzete ugyanis a kondenzáció következtében már nem egységes. A naftalinból kettő, az antracénből három, a
fenantrénből pedig öt különböző egyértékű arilcsoport vezethető le. Az így kapott arilcsoportok
megnevezésekor, vagy a kapcsolódó szubsztituens(ek) helyzetének megjelölésekor számozást használunk, vagy

AROMÁS SZÉNHIDROGÉNEK
(Arének)
111 Created by XMLmind XSL-FO Converter.
- az egyszerűbb molekuláknál - görög betűkkel jelöljük a szénvázon a szénatomokat (pl. 1-, vagy α-naftil és 2-,
vagy β-naftil).
Természetesen háromnál több gyűrű is kondenzálódhat egymással: négy gyűrűt tartalmaz például a pirén, ötöt a
benzpirén (pontosabban: benz[a]pirén), hat benzolgyűrű centrális anellációjával pedig a koronén szimmetrikus
szerkezete alakul ki. Az ilyen sokgyűrűs kondenzált aromás szénhidrogének között számos karcinogén
(rákkeltő) hatású vegyület ismeretes, a benz[a]pirén ezek közül az egyik legveszélyesebb (az [a] jelzés arra utal,
hogy az ötödik benzol gyűrű a pirén "a" jelű oldalához anellálódik; ennek a nómenklatúrának az alapjait a
heterociklusos vegyületeknél, a 28.1.2. fejezetben ismertetjük).
A policiklusos aromás szénhidrogének karcinogén hatását elsősorban azoknak a származékoknak tulajdonítják,
amelyek ezekből a vegyületekből - a lebontási folyamat elején - enzimatikus oxidációval keletkeznek. Ilyen pl. a
benz[a]pirén esetében az alábbi dihidro-diol-epoxid:
Hasonló oxidációs származékai miatt maga a benzol is mérgező (májkárosodást okoz), ezért ma már
mennyiségét a motorhajtó benzinekben is erősen korlátozzák. Ugyanakkor a toluol kevésbé káros, mert
enzimatikus oxiációja benzoesavat (19.9.2. fejezet) eredményez, amely vízoldható és a szervezetből kiürül.
-- Cikloalifás gyűrűvel kondenzált aromás szénhidrogének
Aromás és cikloalifás gyűrűs szénhidrogének vegyes kondenzációja szintén változatos szerkezetű - lényegében
aromásnak tekinthető - vegyületekhez vezet. Ezek közül példaképpen említjük meg az indént és fluorént,
valamint a naftalin részlegesen telitett származékát, a tetrahidronaftalint (tetralint). Az 1,2-
ciklopentenofenantrén perhidro- (azaz teljesen hidrogénezett) származéka a később tárgyalandó szteroidok
alapvegyülete.
A benzolgyürü természetesen kondenzálódhat a többé-kevésbé aromás jellegű heterociklusos gyűrűkkel is. Az
így kialakuló vegyületekkel a heterociklusos vegyületek tárgyalásakor fogunk foglalkozni (28. fejezet).

AROMÁS SZÉNHIDROGÉNEK
(Arének)
112 Created by XMLmind XSL-FO Converter.
1.2. Az aromás szénhidrogének fizikai tulajdonságai
A kisebb molekulatömegű aromás szénhidrogének színtelen folyadékok. A kondenzált gyűrűs szénhidrogének
szilárdak, egyszerűbb képviselőik színtelenek. A konjugáció kiterjedésével a vegyületek színessé válnak, illetve
színük fokozatosan mélyül. Például:
A színeltolódás - a nyíltláncú konjugált telítetlen szénhidrogénekhez hasonlóan - itt is a kötő és lazító MO-ok
közötti energiakülönbség fokozatos csökkenésére vezethető vissza. (Részletesen lásd az izoprénvázas
vegyületeknél, 10.4. fejezet.) A kondenzált gyűrűs aromás vegyületek sorában szélső tagként a fekete grafit
szerepelhetne.
Az angulárisan kondenzált aromás szénhidrogének közül megemlitjük a hexahelicént, melynek gyűrűrendszere
már nem fér el egy síkban és ennek folytán bal és jobb "menetes" csavart konfigurációban létezik:
A két forma egymásnak tükörképe, tehát tulajdonképpen enantiomerek és optikai aktivitást mutatnak annak
ellenére, hogy aszimmetrikus szénatomot nem tartalmaznak. Disszimmetrikus molekulákról van szó, melyek
egy darab kétértékű szimmetria-tengellyel rendelkeznek, ez azonban - mint tudjuk - nem zárja ki az optikai
aktivitást (4.4. fejezet).
Figyelemreméltó a hexahelicén rendkívül nagy fajlagos forgatóképessége is (αD20 = 3700°), ami annak
következménye, hogy itt maga a kromofór, a nagy kiterjedésü π-elektronrendszer a disszimmetrikus (vö. 4.7.
fejezet).
Az aromás szénhidrogének vízben nem, a legtöbb szerves oldószerben azonban jól oldódnak. A
benzolhomológok forráspontja általában közel esik az azonos szénatomszámú aliciklusos szénhidrogének
forráspontjához (a benzol forráspontja pl. 80,1 °C, a ciklohexáné 80,7 °C).
2. Az aromás szénhidrogének molekulaszerkezete

AROMÁS SZÉNHIDROGÉNEK
(Arének)
113 Created by XMLmind XSL-FO Converter.
2.1. A benzolgyűrű elektronszerkezete - a VB-megközelítés
A benzol elemi összetételét Faraday 1825-ben CH-nak határozta meg. Az 1800-as évek közepére vált
egyértelművé, hogy a benzol molekulájában hat szénatom kapcsolódik egymáshoz, mindegyik szénatom egy-
egy hidrogénatommal van kapcsolatban és hogy a szénatomok, valamint a hidrogénatomok egymással - külön-
külön - egyenértékűek. A hat szénatom egymáshoz kapcsolódása azonban - az egyenértékűség megtartásával is -
többféle módon képzelhető el. Így Kekulé (1865) szabályos hatszögű gyűrűt feltételezett, és a szén
négyvegyértékűségének fenntartása érdekében a gyűrűben egymást váltogató egyes és kettős C,C-kötésekkel
formulázta a benzol szerkezetét. Ez a "ciklohexatrién" szerkezet azonban nem áll összhangban a benzol kémiai
tulajdonságaival (pl. azzal, hogy a benzol brómot nem addicionál), és a rögzített helyzetű telítetlen kötések
esetén várható kétféle orto-diszubsztituált benzolszármazékok létezését sem támasztotta alá a tapasztalat.
Kekulé ezért azt is feltételezte, hogy a benzolgyűrű kötésrendszere a valóságban két határszerkezet között
oszcillál (vagy a VB-megközelítés modernebb fogalmazásában: a két mezomer határszerkezet
"rezonanciahibridje" ):
Kekulé javaslatát csak fokozatosan fogadta el a tudományos közösség és ezért még más javaslatok is születtek a
benzol szerkezetére. Így Dewar (1867) a benzol szerkezetét keresztkötéseket tartalmazó hattagú gyűrűs
határszerkezetek eredőjeként képzelte el:
Ladenburg (1869) teljesen telített többgyűrűs vázat javasolt, mely azonban nem volt összhangban a csak egyféle
orto- származék létezésével. Azóta ennek, a ma prizmánnak nevezett molekulának több származékát is sikerült
előállítani. Ugyancsak előállították azóta a Dewar által javasolt szerkezetű molekulát is ("dewar-benzol"), amely
azonban nem sík hatszöges szerkezetü, hanem tulajdonképpen [2,2,0]-biciklohexadién:
A benzol szerkezetének további vizsgálata során azután beigazolódott, hogy Kekulé elképzelése alapjában véve
helyes volt, csak azt mai fogalmaink szerint kell értelmeznünk.
Mai felfogásunk szerint a benzolmolekula szénváza hat sp2 szénatom által feszültségmentesen kialakított
szabályos hatszögű, planáris gyűrű, melynek minden szénatomjához radiális irányban, a gyűrű síkjában
kapcsolódik egy-egy hidrogénatom. A hat egyenértékű, egymáshoz páronként egyenletes távolságban
kapcsolódó trigonális szénatom mindegyike egy-egy - a gyűrű síkjára merőlegesen orientált - egy elektronnal
feltöltött p-orbitállal rendelkezik. Ezek a p-orbitálok egymással átfedésben vannak, vagyis a 6 π-elektronnak a
teljes gyűrűre kiterjedt (a gyűrűben "záródó") delokalizációjára van lehetőség. Az így kialakuló (6C,6π) zárt
konjugált rendszer szerkezetét tekintve hasonló a már tárgyalt ciklopentadienid-anionhoz, de annál jóval
stabilisabb. Stabilisabb egyrészt azért, mert nincs elektromos töltése, másrészt mert a szabályos hatszög belső
szöge és a trigonális szénatomok vegyértékszöge közötti egyezés (mindkettő 120°) következtében a gyűrű
feszültségmentes. A planáris szénvázat tehát mindkét oldalán egyenletes eloszlású π-elektronfelhő borítja,
melyben az elektronok tartózkodási valószínűsége minden szénatomon azonos:

AROMÁS SZÉNHIDROGÉNEK
(Arének)
114 Created by XMLmind XSL-FO Converter.
A benzolgyűrűnek ezt a szerkezetét és a hat π-elektron teljes delokalizációját a következő bizonyítékok
támasztják alá:
• Röntgendiffrakcióval igazolható (pl. hexametilbenzol esetén), hogy a hattagú gyűrű planáris és teljesen
szimmetrikus: valamennyi C,C-távolság azonos.
• A C,C-atomtávolságok értéke 139 pm, ami az alifás konjugált diének sp2 szénatomjai közötti egyes kötés (147
pm - lásd butadién) és az izolált C,C kettős kötés (134 pm) értékei közé esik. A 139 pm kötéstávolság
azonban nem a kettő középarányosa, hanem annál kisebb, azaz a benzol C,C-kötései jóval erösebbek annál,
mintha "másfeles" kötéseknek tekintenénk őket. Ez mutatja, hogy a benzol nem egyszerűen két Kekulé-
szerkezet átlaga, hanem olyan konjugált rendszer, amely az egyszerű szuperponálással adódó szerkezetnél
energiaszegényebb (stabilisabb).
• A konjugáció körben záródásából adódó energiacsökkenést (stabilitás-növekedést) jellemző delokalizációs
energia értéke a benzol esetén 124 kJ/mol-nak vehető. Ezt a számot úgy kell értelmeznünk, hogy a benzol
ennyivel energiaszegényebb annál a hipotetikus ciklohexatrién szerkezetnél, amelyet a nyíltláncú konjugált
szerkezeteknél (pl. a butadiénnél) megismert mértékben a π-elektron delokalizáció már bizonyos fokig
stabilizált. A konjugáció körben záródása tehát nagyságrendileg nagyobb energia-nyereséget tesz lehetővé,
mint az "egyszerű" nyíltláncú konjugálódás (vö. butadién: 15 kJ/mol). Ez a magyarázata pl. annak, hogy az
1,3-ciklohexadién dehidrogénezése benzollá exoterm folyamat (ΔH = -24 kJ/mol; lásd 8.4.2. fejezet). Ez a
nagy delokalizációs energiaérték összhangban van azzal a már említett kvalitatív szabállyal is (8.1.2. fejezet),
hogy a delokalizációs energia azokban az esetekben jelentős, amikor a tényleges elektronszerkezetet
szemléltetni kívánó két mezomer határforma azonos.
A benzol delokalizációs energiájának a fentiekben megadott számszerű értéke némileg bizonytalan szám. Azt
ugyanis közvetlenül megmérni nem lehet, mert a lokalizált kettős kötésekkel rendelkező ciklohexatrién nem
létezik és így pl. a butadiénnél követett kísérletes út, a hidrogénezési hők mérése csak közvetett számításokra ad
lehetőséget. Ezzel a kérdéssel részletesebben a benzol hidrogénezése kapcsán, a 9.5.2. fejezetben fogunk
foglalkozni.
2.2. Az aromás kötésrendszer stabilitása - az MO-megközelítés
A benzolgyűrű π-elektroneloszlásának az MO-módszer szerinti értelmezéséből kiindulva lehetőség nyílik arra,
hogy meghatározzuk az aromás jelleg általános kritériumait, vagyis megállapítsuk, milyen feltételek teljesítése
esetén alakulhat ki a molekulában aromás konjugáció.
A jelenség megértéséhez vizsgáljuk meg azt, hogy mi történik a π-molekulapályákkal abban a (hipotetikus)
folyamatban, amikor a nyiltláncú 1,3,5-hexatriénből kialakul a benzolgyűrű. Ehhez nem kell mást tennünk, mint
megvizsgálnunk, milyen energiaváltozással jár az 1,3,5-hexatrién C1 és C6 p-orbitáljainak átfedése
(kölcsönhatása).
Az eredményt szemlélteti a 9.1. TÁBLÁZAT, amelyen feltüntettük az 1,3,5-hexatrién molekulapályáit a
szimmetriatulajdonságokat jól visszaadó atomi pálya kombinációkkal, a gyűrűzáródás folytán bekövetkező MO-
energiaszint-változásokat és az így kialakult benzol π-elektron-molekulapályáit. Ez utóbbinak a hexatriénhez
képest erősen átrendeződött szimmetriaviszonyai tükrözik a benzol teljesen szimmetrikus π-elektroneloszlását.
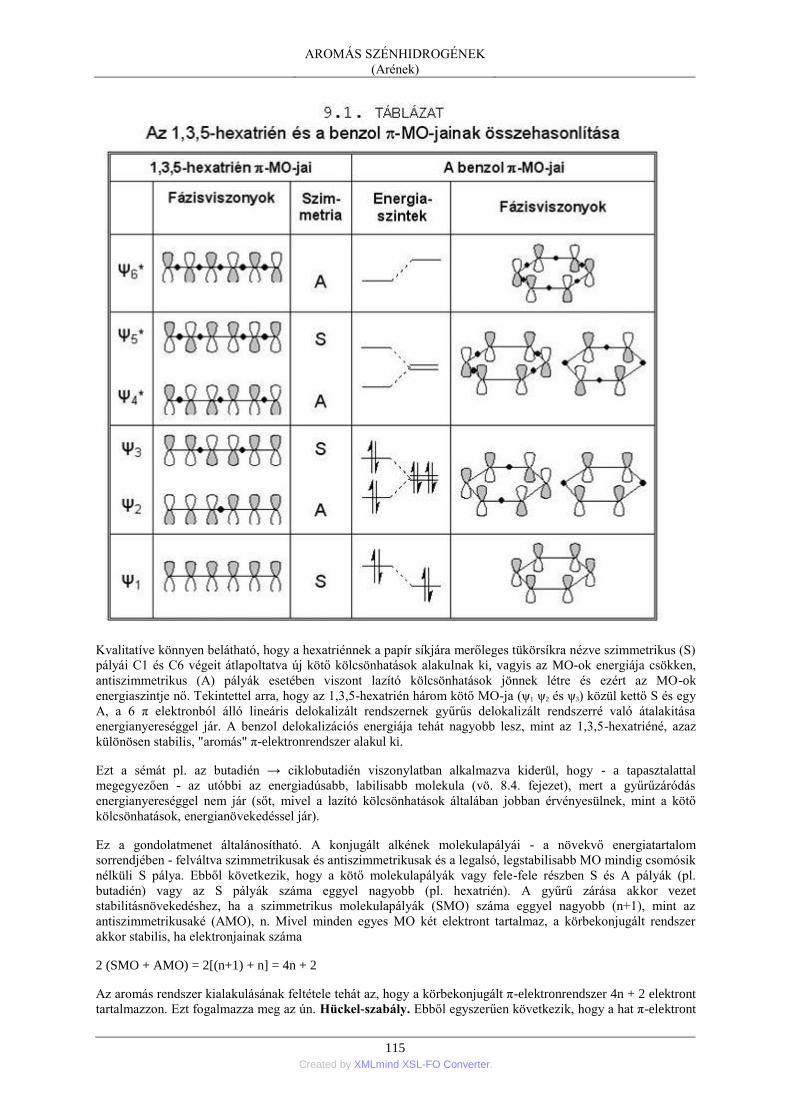
AROMÁS SZÉNHIDROGÉNEK
(Arének)
115 Created by XMLmind XSL-FO Converter.
Kvalitatíve könnyen belátható, hogy a hexatriénnek a papír síkjára merőleges tükörsíkra nézve szimmetrikus (S)
pályái C1 és C6 végeit átlapoltatva új kötő kölcsönhatások alakulnak ki, vagyis az MO-ok energiája csökken,
antiszimmetrikus (A) pályák esetében viszont lazító kölcsönhatások jönnek létre és ezért az MO-ok
energiaszintje nő. Tekintettel arra, hogy az 1,3,5-hexatrién három kötő MO-ja (ψ1 ψ2 és ψ3) közül kettő S és egy
A, a 6 π elektronból álló lineáris delokalizált rendszernek gyűrűs delokalizált rendszerré való átalakitása
energianyereséggel jár. A benzol delokalizációs energiája tehát nagyobb lesz, mint az 1,3,5-hexatriéné, azaz
különösen stabilis, "aromás" π-elektronrendszer alakul ki.
Ezt a sémát pl. az butadién → ciklobutadién viszonylatban alkalmazva kiderül, hogy - a tapasztalattal
megegyezően - az utóbbi az energiadúsabb, labilisabb molekula (vö. 8.4. fejezet), mert a gyűrűzáródás
energianyereséggel nem jár (sőt, mivel a lazító kölcsönhatások általában jobban érvényesülnek, mint a kötő
kölcsönhatások, energianövekedéssel jár).
Ez a gondolatmenet általánosítható. A konjugált alkének molekulapályái - a növekvő energiatartalom
sorrendjében - felváltva szimmetrikusak és antiszimmetrikusak és a legalsó, legstabilisabb MO mindig csomósik
nélküli S pálya. Ebből következik, hogy a kötő molekulapályák vagy fele-fele részben S és A pályák (pl.
butadién) vagy az S pályák száma eggyel nagyobb (pl. hexatrién). A gyűrű zárása akkor vezet
stabilitásnövekedéshez, ha a szimmetrikus molekulapályák (SMO) száma eggyel nagyobb (n+1), mint az
antiszimmetrikusaké (AMO), n. Mivel minden egyes MO két elektront tartalmaz, a körbekonjugált rendszer
akkor stabilis, ha elektronjainak száma
2 (SMO + AMO) = 2[(n+1) + n] = 4n + 2
Az aromás rendszer kialakulásának feltétele tehát az, hogy a körbekonjugált π-elektronrendszer 4n + 2 elektront
tartalmazzon. Ezt fogalmazza meg az ún. Hückel-szabály. Ebből egyszerűen következik, hogy a hat π-elektront

AROMÁS SZÉNHIDROGÉNEK
(Arének)
116 Created by XMLmind XSL-FO Converter.
tartalmazó benzolgyűrűn kívül 10, 14, 18, stb. elektront tartalmazó stabilis aromás szerkezetek is léteznek.
Ilyenek általában több benzolgyűrű anellálódása révén alakulnak ki (pl. a naftalin) de - habár ritkábban -
nagyobb tagszámú gyűrűk is ismertek (pl. ciklooktadekanonaén). Mindezeknél - a 4n+2 szabályon túlmenően -
lényeges feltétel a planáris szerkezet is (ez a p AO-k átlapolásához szükséges).
2.3. Az aromás jelleg kritériumai
Az aromás π-elektron szerkezet kialakulása nincs tisztán sp2 szénatomokból felépülő töltés nélküli
molekulavázhoz kötve: a konjugációban résztvehetnek heteroatomok (O, S, N) szabad elektronpárjai (pl. tiofén,
C5H4S), vagy akár egy sp2 szénatom magános elektronpárja is (pl. a már tárgyalt cíklopentadienid anion, C5H5-),
de kiterjedhet a delokalizáció egy sp2 szénatom üres atomi p pályájára is (pl. cikloheptatrienil kation, C7H7+):
Mint a továbbiakban látni fogjuk, az "aromás jelleg" a gyakorlatban elsősorban bizonyos kémiai
tulajdonságokban, így az elektrofil szubsztitúciós reakciókra való hajlandóságban és az addíciós reakciókkal
szembeni ellenállásban nyilvánul meg. Ezen kívül bizonyos, nem pontosan definiálható stabilitást is
asszociálunk ezzel a fogalommal (pl. termikus igénybevétellel szemben). Míg az MO-elmélet meghatározása
egzakt, de szükségszerüen elvont fogalmakkal operál, ezek a kémiai jellemzők jobban kézzelfoghatóknak
látszanak, de természetesen sokkal kevésbé egyértelműek és nem kvantitatívak.
További lehetőséget kínál az aromás jelleg meghatározására az NMR spektroszkópia. A zárt körbekonjugált π-
elektronrendszerekben, vagyis az aromás molekulákban ugyanis az NMR készülék mágneses terének hatására a
gyűrű alakú π-elektronfelhőben köráram indukálódik (a nyíl a π-elektronok mozgásirányát jelöli), amely a
gyűrűhöz kívülről kapcsolódó protonokra ható mágneses tér erejét növeli (9.2. ÁBRA):

AROMÁS SZÉNHIDROGÉNEK
(Arének)
117 Created by XMLmind XSL-FO Converter.
9.2. ÁBRA
Ez a hatás az aromás gyűrűhöz kapcsolódó protonok kémiai eltolódását a nem-konjugált alkének sp2
szénatomjaihoz kapcsolódó protonok kémiai eltolódásához képest megnöveli (9.2. TÁBLÁZAT):
Az NMR protonjele egyértelműen mutatja, hogy pl. a ciklooktatetraén nem aromás, hanem alkénes jellegű
vegyület. Az aromás jelleg definiálására tehát az NMR spektrum is alkalmas olyan értelemben, hogy mindazok
a vegyületek, amelyekben a mágneses tér hatására indukált köráram lép fel és ezáltal a gyűrűhöz kapcsolódó
protonok kémiai eltolódása megnő, aromás jellegűeknek tekinthetők. Ezen definíció kétségtelen előnye, hogy
könnyen és egyértelműen mérhető jelenségre támaszkodik. Ennél a módszernél is szükséges azonban a
megfelelően körültekintő értékelés, mert pl. ha a vegyület ionos karakterű, a δ értékek erősen megváltozhatnak.
2.4. A kondenzált gyűrűs aromás szénhidrogének szerkezete
Ezekben a vegyületekben a benzolgyűrű önmagában szimmetrikus elektroneloszlása a kondenzáció
következtében eltorzul: a kondenzáció síkjához közelebb fekvő (pl. 1-, vagy α-helyzetű) szénatomokon az
elektrontöltés felhalmozódik, míg a távolabb eső szénatomok elektronsűrűsége némileg csökken.

AROMÁS SZÉNHIDROGÉNEK
(Arének)
118 Created by XMLmind XSL-FO Converter.
A gyűrűt alkotó szénatomok reakciókészsége tehát a kondenzált gyűrűs rendszerekben már nem egységes. Az
elektrofil reakciókészség például a naftalinban az α-helyzetű, az antracénban és a fenantrénban pedig egyaránt a
9- és 10-helyzetű szénatomokon a legnagyobb.
Az elektroneloszlás torzulásával párhuzamosan megváltoznak a kondenzált gyűrűs rendszerekben a C,C-
atomtávolságok is. A naftalinban például a következő értékeket mérték:
Az egyenlőtlen elektroneloszlást szemléletesen tudjuk értelmezni - legalább is részben - a kondenzált gyűrűs
aromás szénhidrogénekre a VB-szemlélet alapján felirható Kekulé-féle mezomer határszerkezetekkel. Kiderül
ugyanis hogy - a benzolra felírható határszerkezetektől eltérően - ezek nem egyenértékűek. A naftalingyűrű
három határszerkezete például a következő:
Látható, hogy a kettős kötés gyakorisága az 1- (α-) és 2- (β-) szénatomok között nagyobb, mint a 2- és 3-
szénatomok között (ami összhangban van az atomtávolságok közötti eltéréssel). Az is leolvasható a felírt
szerkezetekből, hogy az összesen szereplő hat gyűrűből formálisan csak négy egyértelműen aromás jellegű (A),
kettő pedig ún. kinoidális szerkezetű (K). Ez az elnevezés az orto-benzokinon (17.3.1. fejezet)
elektronszerkezetére utal (a kinonokat nem tekintjük aromás vegyületeknek).
Ezek a mezomer határformák rámutatnak arra, hogy a kondenzált gyűrűs aromás szénhidrogének
elektroneloszlása nem lehet teljesen egyenletes. Ez a tény pedig azzal jár, hogy ezen molekulák "aromás
jellegének" a benzolhoz képest csökkennie kell, vagyis az egy szénatomra jutó delokalizációs energiájuk is
kisebb lesz. Ez a VB-szemlélet alapján is várható, mert a mezomer határszerkezetek - a benzollal ellentétben -
már nem teljesen azonosak. A naftalin esetében mindezen megállapítások összhangban vannak a naftalinnak a
benzolénál - kísérletileg is igazolható - nagyobb reakciókészségével.
Az antracénre felírható négy Kekulé-szerkezet közül kettőben két aromás és egy kinoidális, kettőben pedig egy
aromás és két kinoidális gyűrűt találunk: összesen 12 gyűrűből tehát hat aromás. A fenantrén lehetséges Kekulé-
szerkezeteiben szereplő 15 gyűrű közül tíz aromás szerkezetű. Ezzel összhangban a fenantrén a stabilisabb
vegyület: delokalizációs energiája 385 kJ/mol, az antracén 350 kJ/mol értékével szemben.
2.5. Az izolált gyűrűs aromás szénhidrogének szerkezete
Az izolált gyűrűs aromás szénhidrogének elektronszerkezet és kémiai viselkedés szempontjából általában csak
kevéssé különböznek a benzoltól, illetve a benzol egyszerű alkilszármazékaitól. Új tulajdonságként jelentkezik
azonban ezeknél a vegyületeknél az, hogy viszonylag stabilis szabad gyökök képzésére hajlamosak. Így pl. a
hexafeniletán előállítására irányuló próbálkozások során csak trifenil-metil-gyökök képződnek, amelyek - a
régebbi elképzeléssel ellentétben - nem hexafeniletánná dimerizálódnak, hanem kinoidális szerkezet kialakulása

AROMÁS SZÉNHIDROGÉNEK
(Arének)
119 Created by XMLmind XSL-FO Converter.
mellett az egyik benzolgyűrű para-helyzetén keresztül kapcsolódnak össze. Ez a dimerizálódás egyensúlyi
reakció, az egyensúly erősen a dimer irányába van eltolva:
Azt a megfigyelést, hogy a hexafeniletán sp3-sp3 C,C-kapcsolata nem stabilis, régebben csak a hat fenilcsoport
zsúfoltságával összefüggő sztérikus okokra vezették vissza. Jelenlegi felfogásunk szerint azonban az ilyen
gyökök nagy stabilitásához az is hozzájárul, hogy a párosítatlan elektront hordozó (látszólag sp3 hibridállapotú)
központi szénatom a szomszédos benzolgyűrűk π-elektronrendszereinek hatására gyakorlatilag sp2
hibridállapotba kerül és így ennek közvetítésével a 19 szénatom π-elektronrendszere közösen alkothat egy
kiterjedtebb és ezáltal nagyobb delokalizációs energiával rendelkező konjugált rendszert. (A három benzolgyűrű
sztérikus okok miatt nem tökéletesen egy síkban, hanem lapos propellerhez hasonlóan helyezkedik el a központi
szénatom körül.)
Ugyanezek a megfontolások érvényesek természetesen a trifenil-metil-kationra is, amelynél a központi sp2
szénatom üres p-orbitálja léphet be a konjugációba (különösen stabilis karbéniumion). Ennek a felismerésnek
a fenti egyedi eseten túlmenően az ad általánosabb jelentőséget, hogy segítségével értelmezni tudjuk az
aralkilgyököknek az egyszerű alkilgyököknél nagyobb stabilitását. Így pl. az allilgyökkel és allilkationnal
analóg benzilgyök C6H5CH2• és benzilkation C6H5CH2+ esetén is számolni kell ezzel a fokozott stabilitással. Ez
magyarázza a benzilszármazékok (pl. a benzil-halogenidek) fokozott reakciókészségét.
3. Aromás szénhidrogének előállítása
3.1. Kinyerés kőszénkátrányból
A kőszén magas hőmérsékleten (1000-1200 °C) végzett lepárlásakor tekintélyes mennyiségben keletkeznek
aromás vegyületek, amelyek a lepárlás egyik termékeként kapott kőszénkátrányban dúsulnak fel (a lepárlás
során emellett még világítógáz, gázvíz és koksz keletkezik). A kőszénkátrány aromás tartalmának tekintélyes
része aromás szénhidrogén, elsősorban benzol és homológjai (toluol, xilolok), valamint kondenzált gyűrűs
aromás szénhidrogének (naftalin, antracén, fenantrén, stb.). Ezeket a szokásos elválasztási műveletekkel
(desztilláció, kristályosítás) lehet elkülöníteni és tisztítani. A huszadik század közepéig a kőszénkátrány volt a
szerves vegyipar egyik legfontosabb nyersanyagbázisa.
Ma már azonban az ipar számára elsősorban fontos egy-gyűrűs aromás szénhidrogéneknek (benzol, toluol,
xilolok, az ún. BTX aromások) csak kis része származik kőszénkátrányból; a szükséglet túlnyomó részét most
már a kőolajból kiinduló, dehidrogénezésen, dezalkilezésen és dehidrociklizáláson alapuló eljárásokkal fedezik.
Ezeket a következő fejezetekben tárgyaljuk. A naftalin és antracén fő forrása még ma is a kőszénkátrány, ezen
aromás szénhidrogének jelentősége azonban kisebb.
3.2. Aromatizálás és dehidrociklizálás
A C6 cikloalkánok és cikloalkének dehidrogénező katalizátorok (pl. platina) jelenlétében magasabb
hőmérsékleten (300-500 °C) és kis nyomáson jó hozammal alakulnak át aromás szénhidrogénekké:

AROMÁS SZÉNHIDROGÉNEK
(Arének)
120 Created by XMLmind XSL-FO Converter.
A reakció ciklohexán esetében tulajdonképpen a benzolból való ciklohexán előállítás megforditása.
Az alkánok katalitikus dehidrogénezése és az alkének, aromások katalitikus hidrogénezése egyaránt reverzibilis
folyamatok. Alacsony hőmérséklet és nagy H2 parciális nyomás a hidrogénezés, magas hőmérséklet és kis H2
parciális nyomás a dehidrogénezés irányába tolja el az egyensúlyt. Az utóbbi effektus szélesebb összefüggéseit
tekintve összhangban van azzal a fizikai-kémiai elvvel, hogy a hőmérséklet emelése az entrópianövekedéssel -
vagyis a részecskék számának növekedésével - járó ("bomlási") kémiai folyamatoknak kedvez.
Mivel az alumínium-szilikát hordozóra felvitt platinakatalizátor ilyen reakciókörülmények között izomerizál is,
a metilciklopentán hasonló módon végzett dehidrogénezése is jórészt benzolt szolgáltat, dimetilciklopentánból
és/vagy metilciklohexánból pedig ugyanezen folyamatok révén toluol állítható elő:
A petrolkémiai iparban ezeket a reakciókat az ún. aromatizálási technológiákban hasznosítják úgy, hogy
benzol, toluol és xilolok előállításához C6-C8 cikloalkánokban gazdag, (ún. nafténes) benzinpárlatot kezelnek
alumínium-szilikát hordozós platinakatalizátoron, kb. 500 °C-on és 15 bar hidrogén-nyomáson (katalitikus
reformálás).
Nyíltláncú szénhidrogének is átalakulnak az aromatizálás reakciókörülményei között aromás szénhidrogénekké.
Így például heptánból toluol képződik:
Ez a dehidrociklizálásnak nevezett reakció azonban jóval lassabb és kevésbé szelektív, mint a naftének
aromatizálása, ezért a benzinpárlatok aromatizálása során alárendeltebb szerepet játszik. (A naftén-
dehidrogénezési és a dehidrociklizálási reakciók sebességeinek aránya a katalitikus reformálás
reakciókörülményei között kb. 100:1.)
3.3. Alkil-aromások átalakítása
Az aromatizálási eljárásokkal benzinpárlatokból keletkező aromás szénhidrogének egyedi vegyületeinek aránya
nem felel meg az ipari szükségleteknek, ezért eljárásokat dolgoztak ki az aromás szénhidrogének egymásba való
átalakítására. Ezek közül vázlatosan hármat ismertetünk (a reakciók mechanizmusára a 9.5. fejezetben térünk
vissza).
-- Hidrodezalkilezés
A benzinreformálás során a vegyipari nyersanyagként legnagyobb mennyiségben felhasznált aromás
szénhidrogénből, a benzolból a szükségesnél kevesebb, toluolból viszont a szükségesnél több keletkezik. Ennek
a toluolfeleslegnek egy részét ezért oxidkatalizátorok (Cr2O3, Mo2O3) jelenlétében, 5-600 °C-on és 30-50 bar H2
nyomáson benzollá dezalkilezik:

AROMÁS SZÉNHIDROGÉNEK
(Arének)
121 Created by XMLmind XSL-FO Converter.
Hasonló módon lehetőség van a metilnaftalinnak naftalinná való demetilezésére is.
-- Izomerizálás
Ennek az eljárásnak a xiloloknál van jelentősége, mert a három izomer közül a meta-xilolra nincs jelentős igény,
míg az orto-xilol és a para-xilol a nagy mennyiségben felhasznált ftálsavanhidrid, illetve tereftálsav alapanyaga
(19.9.4. fejezet). A reakció lényegében a reformálás reakciókörülményei között (Pt/Al2O3-SiO2 katalizátor, 4-500
°C és 15 bar H2 parciális nyomás) lejátszódó egyensúlyi reakció. A technológiai körülmények között beáll a
három izomer xilol (és egy kevés etilbenzol) közötti termodinamikai egyensúly, az elegyből a meta-xilolt
elkülönítik és utána recirkuláltatják.
-- Diszproporcionálás és átalkilezés
Savas katalizátorok (pl. zeolitok, vagy AlCl3 + HCl) hatására, megfelelő hőmérsékleten a benzolgyűrűhöz
kapcsolódó metilcsoportok nem csak intramolekulárisan tudnak áthelyeződni (mint a xilol izomerizációnál),
hanem intermolekulárisan is. Így pl. vannak ipari eljárások a toluol diszproporcionálására benzollá és xilolokká,
valamint toluol és polimetilbenzolok elegyének átalakítására xilolok elegyévé:
4. Az aromás szénhidrogének reakciókészsége
4.1. A jellegzetes reakció: az elektrofil szubsztitúció
A benzolgyűrű - stabilis (energiaszegény) szerkezete következtében - csak kis mértékben hajlamos olyan
reakciókra, amelyek az aromás jelleg megszűnésével járnak együtt. Így például halogénaddíció csak különleges
körülmények között játszódik le, az aromás gyűrű aránylag nehezen hidrogénezhető, a gyűrű felszakadásával
járó oxidáció pedig csak erélyes hatásra következik be.
A benzolgyűrű (és általában az aromás vegyületek) legjellegzetesebb reakciója az elektrofil szubsztitució (SE),
melyben a (poláros, vagy polarizálható) reagens elektrofil része kation formában támad a gyűrű elektrondús

AROMÁS SZÉNHIDROGÉNEK
(Arének)
122 Created by XMLmind XSL-FO Converter.
helyén, s az átmenetileg keletkező karbokation protonvesztéssel stabilizálódik. A reakció mechanizmusa
általánosan a következőképpen formulázható (E = a reaktáns elektrofil, Nu = a reaktáns nukleofil része):
Első lépésben a katalizátor hatására kialakul az aromás gyűrűt megtámadó erősen elektrofil kation:
A második lépésben ez a kation addícionálódik az aromás gyűrűre, ezzel megbontva a stabilis π-
elektronszextettet. A kialakult intermedier karbokation nem egy anionos részecske addíciójával stabilizálódik
(így elveszne az igen stabilis aromás szerkezet) hanem ledob egy protont és így szubsztituált aromás vegyület
alakul ki:
Végül a proton felszabítja a katalizátort a komplexből:
Ezzel a mechanizmussal lejátszódó SE reakciók pl. az aromás szénhidrogén-származékok előállítása
szempontjából nagy jelentőségű halogénezés, nitrálás, szulfonálás, valamint a Friedel-Crafts-reakció, melyeknek
segítségével halogént, nitro-, szulfo-, illetve alkil-, vagy acilcsoportot tudunk az aromás gyűrűhöz kapcsolni.
Az intermedierként felírt (5C,4π)+ karbokation képződésére - egyebek között - bizonyíték az, hogy a folyékony
HF-ban oldott aromás szénhidrogének az alábbi reakció következményeként jobban vezetik az elektromos
áramot, mint a HF:
Ez a protonálódási egyensúly annál inkább jobbra tolódik el, minél nagyobb a lehetőség a pozitív töltés
delokalizációjára. A σ-konjugáció révén (5.3.4. fejezet) pl. metilcsoportok fejtenek ki ilyen hatást, mint azt a
9.3. TÁBLÁZAT mutatja.

AROMÁS SZÉNHIDROGÉNEK
(Arének)
123 Created by XMLmind XSL-FO Converter.
4.2. Elektroneltolódások a helyettesített benzolgyűrűben
A benzolgyűrű hidrogénatomjait helyettesítő csoportok megzavarják a gyűrű eredetileg szimmetrikus
elektroneloszlását s így nyilvánvalóan befolyásolják a már helyettesített gyűrű további reakcióit. Ezt a hatást a
gyűrűhöz kapcsolódó szubsztituensek különböző irányú és mértékű induktív és mezomer elektroneltolódási
effektusaik révén (2.4.1. és 2.4.2. fejezetek), valamint esetleges σ-konjugációs kölcsönhatásuk útján (5.3.4.
fejezet) fejtik ki.
-- A szubsztituens hatása a gyűrű reakciókészségére
Az aromás gyűrűhöz kapcsolódó csoportokat többféleképpen osztályozhatjuk attól függően, hogy milyen módon
változtatják meg a gyűrű elektroneloszlását.
Az egyik csoportosításnál csak azt vesszük figyelembe, hogy a szubsztituens - összegezett induktív és mezomer
effektusa révén - növeli vagy csökkenti a gyűrű egészének elektronsűrűségét s ezáltal hogyan befolyásolja
annak reakciókészségét. Ezen felosztás szerint az egyik csoportba azok a szubsztituensek tartoznak, amelyek
ilyen értelmű általános elektrondonor tulajdonságuk következtében megnövelik az aromás gyűrű
elektronsűrűségét. Ilyen pl. a +M és -I effektussal rendelkező fenolos hidroxilcsoport (+M >> -I). A másik
csoportba tartoznak azok a szubsztituensek, amelyek egyértelműen elektronakceptorként viselkednek, vagyis
csökkentik az aromás gyűrű elektronsűrűségét. Ide soroljuk például a -M és -I effektust kifejtő nitro- és
nitrilcsoportot.
Elektrofil szubsztitúcióban a támadó elektrofil csoport annál könnyebben lép reakcióba, minél nagyobb az
elektronsűrűség a gyűrűben. Ha tehát a gyűrűhöz kapcsolódó csoport elektrondonor, a szubsztitúciós reakció
sebessége nagyobb lesz, mint a benzol esetében. Elektronakceptor csoport viszont természetesen csökkenti az SE
reakció sebességét. Elektrondonor csoport tehát növeli, elektronakceptor csoport pedig csökkenti a gyűrű
elektrofil szubsztitucióval szembeni általános reakciókészségét ("aktiválja" illetőleg "dezaktiválja" a gyűrűt).
Nukleofil szubsztitúció (SN) esetén a helyzet fordított. Az ilyen típusú reakciók azonban az aromás gyűrűn
viszonylag ritkák és csak heteroatomokkal szubsztituált származékoknál fordulnak elő.
-- Irányítási szabályok a helyettesített benzolgyűrűben
Az aromás gyűrűhöz kapcsolódó szubsztituensek másik csoportosítása abból a régóta ismert kísérleti tényből
indul ki, hogy szubsztituált benzolszármazékok SE reakciójában a második szubsztituens már nem egyforma
valószínűséggel kapcsolódik a gyűrű szabadon maradt helyeire: az első szubsztituens "irányítja" a második
csoport belépését.
Az irányító hatás szempontjából két szubsztituens-csoportot szokás megkülönböztetni. Az egyik csoportba
tartoznak azok a szubsztituensek, melyek SE reakciókban főleg orto- és para-helyzetbe irányítanak, a másik
csoportba pedig azok, amelyek főleg meta-helyzetbe irányítják a második belépő csoportot.
A tapasztalat szerint orto-, para-irányítók az elektrondonor csoportok és a halogének, meta-irányítók pedig az
elektronakceptorok (a halogének kivételével). Az alkilcsoportok a σ-konjugáció révén az elektrondonor
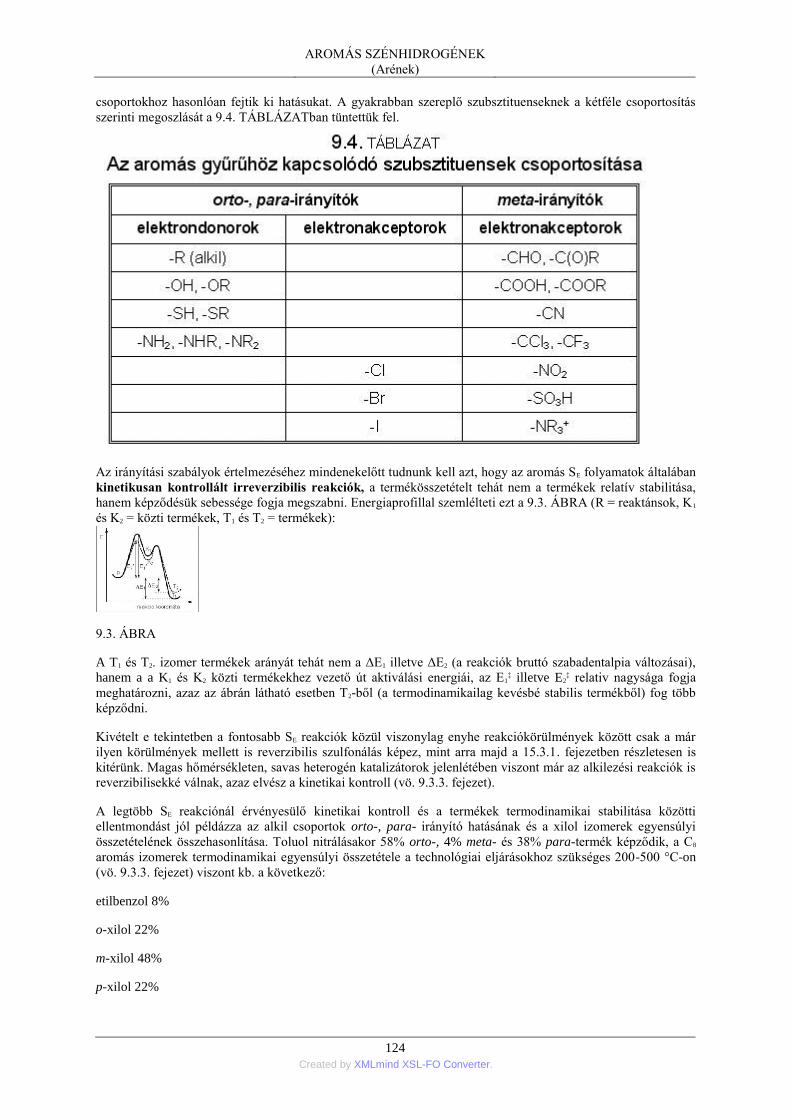
AROMÁS SZÉNHIDROGÉNEK
(Arének)
124 Created by XMLmind XSL-FO Converter.
csoportokhoz hasonlóan fejtik ki hatásukat. A gyakrabban szereplő szubsztituenseknek a kétféle csoportosítás
szerinti megoszlását a 9.4. TÁBLÁZATban tüntettük fel.
Az irányítási szabályok értelmezéséhez mindenekelőtt tudnunk kell azt, hogy az aromás SE folyamatok általában
kinetikusan kontrollált irreverzibilis reakciók, a termékösszetételt tehát nem a termékek relatív stabilitása,
hanem képződésük sebessége fogja megszabni. Energiaprofillal szemlélteti ezt a 9.3. ÁBRA (R = reaktánsok, K1
és K2 = közti termékek, T1 és T2 = termékek):
9.3. ÁBRA
A T1 és T2. izomer termékek arányát tehát nem a ΔE1 illetve ΔE2 (a reakciók bruttó szabadentalpia változásai),
hanem a a K1 és K2 közti termékekhez vezető út aktiválási energiái, az E1‡ illetve E2
‡ relativ nagysága fogja
meghatározni, azaz az ábrán látható esetben T2-ből (a termodinamikailag kevésbé stabilis termékből) fog több
képződni.
Kivételt e tekintetben a fontosabb SE reakciók közül viszonylag enyhe reakciókörülmények között csak a már
ilyen körülmények mellett is reverzibilis szulfonálás képez, mint arra majd a 15.3.1. fejezetben részletesen is
kitérünk. Magas hőmérsékleten, savas heterogén katalizátorok jelenlétében viszont már az alkilezési reakciók is
reverzibilisekké válnak, azaz elvész a kinetikai kontroll (vö. 9.3.3. fejezet).
A legtöbb SE reakciónál érvényesülő kinetikai kontroll és a termékek termodinamikai stabilitása közötti
ellentmondást jól példázza az alkil csoportok orto-, para- irányító hatásának és a xilol izomerek egyensúlyi
összetételének összehasonlítása. Toluol nitrálásakor 58% orto-, 4% meta- és 38% para-termék képződik, a C8
aromás izomerek termodinamikai egyensúlyi összetétele a technológiai eljárásokhoz szükséges 200-500 °C-on
(vö. 9.3.3. fejezet) viszont kb. a következő:
etilbenzol 8%
o-xilol 22%
m-xilol 48%
p-xilol 22%

AROMÁS SZÉNHIDROGÉNEK
(Arének)
125 Created by XMLmind XSL-FO Converter.
Míg tehát a metil csoport egyértelműen orto-, para- irányító, a termodinamikailag legstabilisabb xilol láthatóan
a meta-izomer.
A szubsztituensek tehát általában az E1‡ és E2
‡ relatív nagyságára kifejtett hatásaik alapján irányitanak, mégpedig
a benzolgyűrű elektroneloszlására gyakorolt hatásuk révén. Ez a hatás lehet mind a szubsztituens mezomer
effektusának (M), mind induktív effektusának (I), vagy σ-konjugációs hatásának következménye.
Szemléltetésükre legalkalmasabbak a már tárgyalt mezomer határformák. Ezek segítségével megvizsgálhatjuk
egyrészt a reagáltatandó benzolszármazék elektroneloszlását, másrészt a K1 és K2 közti termékek [az (5C,4π)+
karbokationok] elektronszerkezetét.
Látni fogjuk, hogy a mezomer hatással rendelkező szubsztituenseknél már a kiindulási vegyület
elektroneloszlása is felvilágositást ad arra, hogy melyek a benzolgyűrű elektrondús pontjai, azaz hol tudja az
elektrofil reagens kisebb aktiválási energia befektetése révén megtámadni az aromás szerkezetet. Ezeknél a
szubsztituenseknél az így jelentkező irányító hatás jellege meg fog egyezni a közbenső (5C,4π)+ karbokationok
mezomer határformáinak relatív stabilitásából levonható következtetéssel.
A csak induktív effektussal rendelkező szubsztituenseknél (ilyenek pl. a -CF3 és az -NR3+), vagy a σ-konjugáció
révén ható alkilcsoportoknál a kindulási vegyületnek mezomer határformái nem értelmezhetők, ezért ezeknél
csak a közbenső (5C,4π)+ karbokationok mezomer határformáinak relatív stabilitásából fogunk tudni levonni
következtetéseket.
-- Mezomer effektusuk révén irányító szubsztituensek
Példaképpen először vizsgáljuk meg a mezomer effektusuk révén orto-, para-irányító hidroxilcsoport (+M) és
meta-irányító karbonilcsoport (-M) irányító hatását. Ezen funkciós csoportok elektronszerkezete:
A kiindulási vegyületek, a fenol és a fenil-metil-keton (acetofenon) nem szimmetrikus elektronszerkezetét
tükrözik a következő mezomer határformák:
A határszerkezetekből látható, hogy a mezomer elektroneltolódás hatása a gyűrű meghatározott helyzetű
szénatomjain (az orto- és para-helyeken) jelentkezik. A hidroxilszármazék esetén az orto- és para-helyzetű
szénatomok elektronsűrűsége megnőtt, az elektrofil reaktáns tehát elsősorban ezeken a helyeken fog támadni,
mégpedig a szubsztituálatlan benzol azonos reakciójában mérhetőnél nagyobb sebességgel. A karbonilcsoportot
tartalmazó származék SE reakciójában viszont az elektrofil reaktáns legnagyobb valószinűséggel a meta-helyre
fog belépni, de a szubsztituálatlan benzolhoz viszonyítva kisebb sebességgel (mert a szintén fellépő -I effektus
következtében a meta-helyek elektronsűrűsége is csökkent).
Ezek után vizsgáljuk meg az új E szubsztituens belépése révén kialakuló közbenső karbokationok
elektroneloszlását. Ez az orto-, meta- vagy para-helyekre való kapcsolódás esetén felírható mezomer
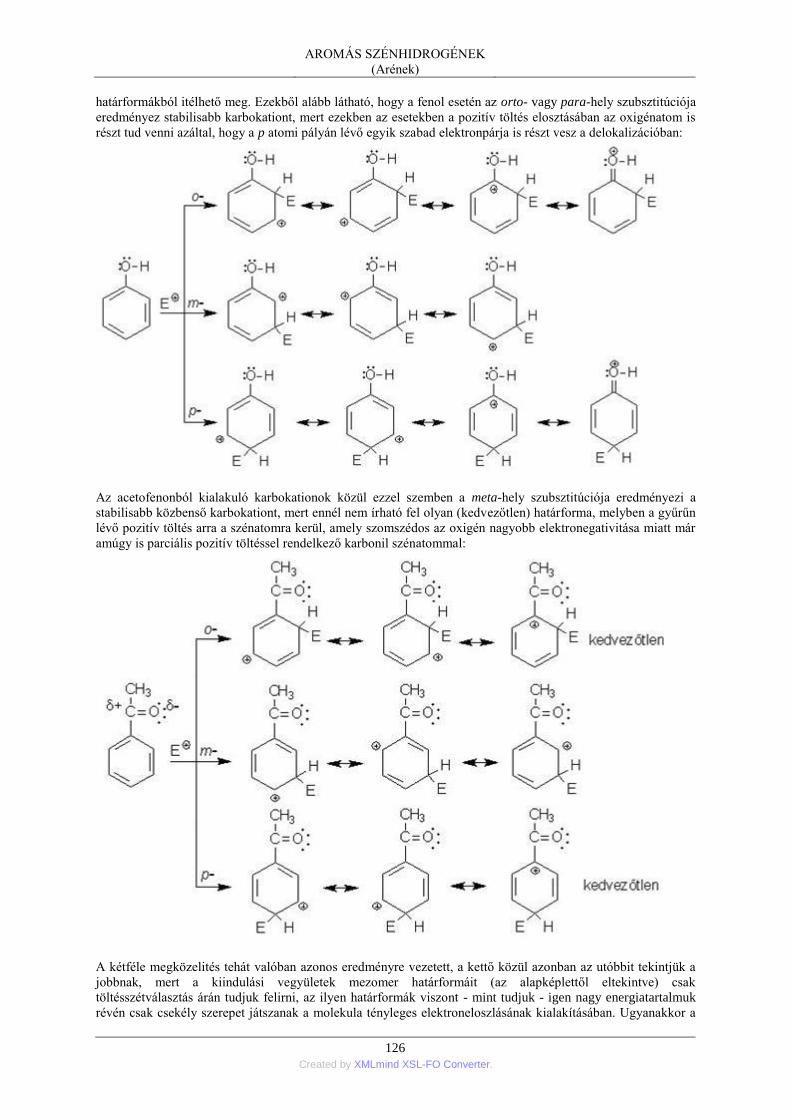
AROMÁS SZÉNHIDROGÉNEK
(Arének)
126 Created by XMLmind XSL-FO Converter.
határformákból itélhető meg. Ezekből alább látható, hogy a fenol esetén az orto- vagy para-hely szubsztitúciója
eredményez stabilisabb karbokationt, mert ezekben az esetekben a pozitív töltés elosztásában az oxigénatom is
részt tud venni azáltal, hogy a p atomi pályán lévő egyik szabad elektronpárja is részt vesz a delokalizációban:
Az acetofenonból kialakuló karbokationok közül ezzel szemben a meta-hely szubsztitúciója eredményezi a
stabilisabb közbenső karbokationt, mert ennél nem írható fel olyan (kedvezőtlen) határforma, melyben a gyűrűn
lévő pozitív töltés arra a szénatomra kerül, amely szomszédos az oxigén nagyobb elektronegativitása miatt már
amúgy is parciális pozitív töltéssel rendelkező karbonil szénatommal:
A kétféle megközelités tehát valóban azonos eredményre vezetett, a kettő közül azonban az utóbbit tekintjük a
jobbnak, mert a kiindulási vegyületek mezomer határformáit (az alapképlettől eltekintve) csak
töltésszétválasztás árán tudjuk felirni, az ilyen határformák viszont - mint tudjuk - igen nagy energiatartalmuk
révén csak csekély szerepet játszanak a molekula tényleges elektroneloszlásának kialakításában. Ugyanakkor a

AROMÁS SZÉNHIDROGÉNEK
(Arének)
127 Created by XMLmind XSL-FO Converter.
közbenső termékként megjelenő (5C,4π)+ karbokationok mezomer határformái energiatartalom szempontjából
egymáshoz közelálló szerkezetek, így jól írják le a kation elektronszerkezetét.
Az orto-, para-irányító hidroxilcsoport az elektrondonor - tehát a gyűrű SE reakciókkal szembeni
reakciókészségét fokozó - szubsztituensek csoportjába tartozik. Ezért pl. a fenol nitrálásának sebessége kb.
1000-szer akkora, mint a benzolé. Ezt azzal magyaráztuk, hogy az -OH csoportnál a pozitív mezomer effektus
jóval erősebb az elektronegatív oxigénatom következtében szükségszerűen fellépő negatív induktív effektusnál
(+M >> -I). Joggal vetődik fel ezek után az a kérdés, hogy az ugyancsak orto-, para-irányító halogének (Cl, Br,
I) - amelyek elektronegativitása inkább kisebb, mint az oxigéné és ugyanúgy rendelkeznek "mezomériára
alkalmas" szabad elektronpárokkal, mint az oxigén - miért dezaktiválják az aromás gyűrűt, azaz miért tartoznak
mégis az elektronakceptor szubsztituensek közé (lásd 9.4. TÁBLÁZAT). A klórbenzol nitrálásának sebessége
pl. harmincada a benzolénak, vagyis ebben a reakcióban a fenol és a klórbenzol reakciókészségének aránya
30.000:1!
A magyarázat lényege az, hogy a felsorolt halogének szabad elektronpárjai a 3p, 4p, illetve 5p atomi pályákon
helyezkednek el, amelyek - az eltérő méretek következtében - rosszul, vagy gyakorlatilag egyáltalán nem tudnak
kölcsönhatásba kerülni (átfedni) a benzol sp2 szénatomjának 2p pályájával. Ezt az alábbi vázlat igyekszik
szemléltetni:
Ennek következményeként a halogéneknél a mezomer hatás gyenge, vagyis +M < -I, azaz a gyűrű bruttó
reakciókészségének alakításában a negatív induktív effektus dominál, a mezomer effektus csak az irányító
hatásban jelentkezik.
-- Induktív, vagy σ-konjugációs effektusuk révén irányító szubsztituensek
Az alkilcsoportok orto-, para-irányitása, valamint az olyan, mezomer effektussal nem rendelkező csoportok,
mint a -CF3, vagy az -NH3+ meta-irányítása csak a közbenső karbokationok vizsgálata alapján válik érthetővé.
Az alkilcsoportok esetén pl. csak az orto- vagy para-szubsztitúcióval kialakuló közbenső karbokationokra irható
fel olyan mezomer határforma, amelyben a pozitív töltés az alkilcsoporthoz kapcsolódó gyűrű-szénatomra esik.
Ez a határforma az alkilcsoportok σ-konjugációs effektusa folytán stabilisabb a többinél (vö. különböző rendű
karbokationok stabilitása, 5.3.4. fejezet) és így jelentősen megnöveli ezen karbokationok stabilitását. Az alábbi
mezomer határformáknál az alkilcsoport (R) σ-konjugációs hatásának "elektrondonor" jellegét nyíl jelképezi:
AROMÁS SZÉNHIDROGÉNEK
(Arének)
128 Created by XMLmind XSL-FO Converter.
A fentiekkel analóg, de ellenkező hatást fejtenek ki a csupán negatív induktív effektussal rendelkező csoportok,
mint pl. a -CF3:
Ezeknél a szubsztituenseknél a negatív induktív effektus (amelyet nyíllal jelképeztünk) az orto- és para-
szubsztitúcióhoz vezető karbokation intermediereknél a szubsztituenshez kapcsolódó gyűrű-szénatom pozitív
polározottságát tovább fokozza és igen kedvezőtlen mezomer határformákat eredményez. Az SE mechanizmus
szerint lejátszódó reakciók ezért a legkevésbé kedvezőtlen, a meta-termékhez vezető utat választják. A trifluor-
metilbenzol nitrálása pl. igen lassú reakcióban ugyan, de 91%-os szelektivitással meta-terméket szolgáltat.
5. Az aromás szénhidrogének reakciói
5.1. Az aromás gyűrű szubsztitúciós reakciói
Az előző fejezetben részletesen tárgyaltuk az aromás gyűrű legjellegzetesebb reakciójának, az elektrofil
szubsztitúciónak (SE) mechanizmusát, amely az aromás szénhidrogénekkel legtöbbször könnyen és jó
hatásfokkal játszódik le. A kondenzált gyűrűrendszerek reakciókészsége általában nagyobb, mint az izolált
gyűrűké. Most bemutatjuk az aromás elektrofil szubszitúciós reakciók néhány konkrét példáját.
-- Hidrogén-deutérium csere
A legegyszerűbb aromás SE reakció az erős savak hatására könnyen lejátszódó reverzibilis hidrogén-deutérium
csere:
-- A Friedel-Crafts-reakció
A Friedel-Crafts-reakció lényege, hogy aromás szénhidrogént reagáltatunk Lewis-sav katalizátor jelenlétében
elektrofil reagenssel. Az elektrofil reagens jellege szerint a Friedel-Crafts-reakciónak két változatát
különböztetjük meg:

AROMÁS SZÉNHIDROGÉNEK
(Arének)
129 Created by XMLmind XSL-FO Converter.
• alkilezés játszódik le, ha alkil-halogenidből vagy alkénből képződött karbéniumion reagál;
• acilezés játszódik le, ha savkloridból vagy savanhidridből keletkezett aciliumion az elektrofil reagens.
Az első esetben alifás oldalláncot tartalmazó aromás (alkilaromás) szénhidrogén, a második esetben tiszta
aromás vagy vegyes alifás-aromás keton a reakció terméke. A Friedel-Crafts-reakció egyik változata (az
alkilezés) tehát egyszersmind az alkilaromás szénhidrogének egyik előállítási módja is.
A Lewis-savak közül a klasszikus katalizátor a vízmentes alumínium-klorid, de használható katalizátorként
vas(III)-klorid, bór-trifluorid, cink-klorid, foszforsav vagy kénsav is. Külön megemlítjük a legújabb, korszerű
technológiáknál alkalmazott vízmentes hidrogén-fluoridot.
A Friedel-Crafts-alkilezés bruttó reakcióegyenlete alkil-halogenid, mint alkilező ágens alkalmazása esetén:
A reakció mechanizmusának lényege az, hogy a Lewis-sav hatására az alkil-halogenid C,X-kötése
heterolitikusan felszakad, s a keletkező karbéniumion lép elektrofil reakcióba az aromás gyűrűvel. Alumínium-
klorid mint Lewis-sav példáján bemutatva tehát:
A reakcióban keletkező komplex sav elbomlik és a hidrogén-halogenid eltávozik a rendszerből.
A karbokationos mechanizmusból következik, hogy például normál propilcsoport ilyen módon nem vihető be az
aromás gyűrűbe. A propil-kloridból elsődlegesen képződő 1-propilkation ugyanis hidridion-vándorlással
spontán átrendeződik a stabilisabb 2-propilkationná, és ezért a reakció végeredményben izopropil-
származékokat (benzol alkilezése esetén pl. propilbenzol helyett kumolt) szolgáltat.
A Friedel-Crafts-alkilezés reakciókörülményei között - kevés alkil-halogenid jelenlétében - alkének is reagálnak
aromás szénhidrogénekkel. A reakciót az teszi lehetővé, hogy először az alkil-halogenid (általában alkil-klorid)
alkilezi az aromás vegyületet, majd az ennek során keletkező HX az AlCl3-dal az erős H[AlCl3X] komplex savat
képezi, amely az alként protonálással karbéniumionná alakítja:
A petrolkémiai ipar nagy mennyiségben gyárt ezen az úton a megfelelő alkének felhasználásával például
etilbenzolt, kumolt, valamint C10-C14-alkilbenzolokat.
Az etilbenzolt - a műanyagipar számára fontos sztirol alapanyagát - eténből és benzolból alumínium-klorid
katalizátort és kevés etil-klorid promotort alkalmazva állítják elő:
A kumol előállításához SiO2 hordozóra felvitt foszforsav katalizátort használnak (a kumol a fenolgyártás
legfontosabb alapanyaga; 13.4.1. fejezet):

AROMÁS SZÉNHIDROGÉNEK
(Arének)
130 Created by XMLmind XSL-FO Converter.
Az egyenesláncú C10-C14 alkénekkel előállított hosszú szénláncú alkilbenzolok a szintetikus mosószerek fontos
alapanyagai (15.6.3. fejezet).
Ezeknél az alkilezési reakcióknál általában fennáll a polialkilezés veszélye, mivel a gyűrűbe belépő csoport σ-
konjugációs elektrondonor hatása a gyűrű SE reakciókészségét megnöveli. A polialkilezést benzol felesleg
használatával lehet visszaszorítani.
A Friedel-Crafts-acilezéssel egyértelműen állíthatók elő vegyes, vagy tiszta aromás monoketonok, mivel a
belépő -COR acilcsoport elektronakceptor hatása csökkenti a gyűrű további SE reakcióra való készségét. Az
acilezés mechanizmusa analóg az alkilezés mechanizmusával azzal a különbséggel, hogy a elektrofil támadó
ágens a mezoméria-stabilizált, delokalizált elektronszerkezetű acilkation (aciliumion):
Érdekességként megjegyezzük, hogy az így előállított aromás keton megfelelő módon végzett redukcióval (pl. a
Wolff-Kizsnyer, vagy a Clemmensen-módszerrel, lásd 16.3.7. fejezet) alkilaromás szénhidrogénné alakítható át.
Ilyen módon állítható elő például propionil-kloridból kiindulva az egyenesláncú propilbenzol (1-fenilpropán):
-- További elektrofil szubsztitúciós reakciók
Az alkil-aromás vegyületek izomerizálása és diszproporcionálása, amelyeket a 9.3.3. fejezetben ismertettünk,
ugyancsak SE mechanizmusú reakciók. Ezeknél a reakcióknál az benzol gyűrűn lévő alkil (elsősorban metil)
csoportokból a savas heterogén katalizátorok felületén alakulnak ki az alkilkationok, amelyek elektrofil
reagensként támadják meg az aromás gyűrűt.
A halogénezést, nitrálást, szulfonálást és egyéb SE reakciókat részletesen a megfelelő származékok előállitásánál
fogjuk tárgyalni (11.7.1., 15.3. és 24.8.4. fejezetek).
5.2. Az aromás gyűrű addíciós reakciói
-- Hidrogénezés

AROMÁS SZÉNHIDROGÉNEK
(Arének)
131 Created by XMLmind XSL-FO Converter.
Az aromás gyűrű telítésekor az aromás rendszer megbontása a leglassabb (sebességmeghatározó) folyamat.
Benzol katalitikus hidrogénezésekor az első mól hidrogén felvétele energiabefektetést igényel. A hipotetikus
benzol → 1,3-ciklohexadién reakció endoterm (vö. az 1,3-ciklohexadién exoterm dehidrogénezési reakciójával;
8.4.2. fejezet):
A ciklohexán képződéséhez vezető teljes telítés azonban már erősen exoterm folyamat:
A benzol hidrogénezése platina vagy palládium katalizátorral atmoszférikusan és aránylag alacsony
hőmérsékleten (max. 50 °C) elvégezhető. Raney-nikkel alkalmazása esetén 60-100 bar nyomáson és magasabb
hőmérsékleten kell dolgozni. Ciklohexadiént vagy ciklohexént, mint közbenső termékeket a hidrogénezés során
nem lehet kimutatni.
Az a tény, hogy a benzol katalitikus hidrogénezése során közti termékek nem mutathatók ki, érthető, mivel ezen
cikloalkének hidrogéneződése sokkal gyorsabb, mint a benzol reakciója az első mól hidrogénnel és így nem
tudnak felhalmozódni a reakcióelegyben. Heterogén katalitikus folyamatról lévén szó feltehető, hogy ezek a
közti termékek szabad állapotban nem is jelennek meg a reakció folyamán, hanem a katalizátor felületére
adszorbeált ("kemiszorbeált") benzol csak a teljesen hidrogénezett ciklohexán formájában távozik a
katalizátorról.
A kondenzált gyűrűs szénhidrogének enyhe reakciókörülmények között katalitikusan részlegesen is
hidrogénezhetők (naftalin tetrahidro-naftalinná, antracén tetra- vagy oktahidro-antracénné). Erélyesebb
reakciókörülmények között teljes telítődés következik be (dekalin, perhidroantracén, illetve perhidrofenantrén
keletkezik).
Fémnátriummal ammóniás-alkoholos közegben az aromás szénhidrogének dihidro-származékokká
redukálhatók: a kiindulási aréntől függően 1,4-ciklohexadién, 1,4-dihidronaftalin, 9,10-dihidroantracén vagy
9,10-dihidrofenantrén keletkezik. Ennél a Birch-redukciónak nevezett módszernél a redukcióhoz szükséges
elektronokat a fémnátrium, a protonokat az etanol szolgáltatja egy-elektronos lépésekben. Benzol esetében a
bruttó reakcióegyenlet:
-- Mekkora a benzol delokalizációs energiája?
A benzol delokalizációs energiáját a 9.2.1. fejezetben 124 kJ/mol-nak adtuk meg. Ehhez az értékhez a következő
számítások alapján juthatunk:
A ciklohexén hidrogénezésekor 120 kJ/mol, az 1,3-ciklohexadién hidrogénezésekor 230 kJ/mol hő fejlődik.
Ebben az esetben tehát az egy konjugációs kölcsönhatásra bekövetkező π-elektron delokalizáció 2×120 - 230 =
10 kJ/mol energianyereséget jelent (vö. a butadién 15 kJ/mol-os értékével; 8.1. fejezet). Ha ezt a
gondolatmenetet továbbvisszük a hipotetikus 1,3,5-ciklohexatriénre, akkor annak elméleti hidrogénezési hője (a
most már három konjugációs kölcsönhatás folytán) 3×120 - 3×10 = 330 kJ/mol-nak adódik. Mivel a benzol →
ciklohexán reakcióban 206 kJ/mol hő fejlődik, a benzol 330 - 206 = 124 kJ/mol-lal energiaszegényebb, mint a

AROMÁS SZÉNHIDROGÉNEK
(Arének)
132 Created by XMLmind XSL-FO Converter.
hipotetikus 1,3,5-ciklohexatrién, vagyis ezt tekinthetjük az aromás elektronszerkezet kialakulása révén
felszabadult delokalizációs energiának.
A fenti okfejtés gyenge pontjai nyilvánvalóan egyrészt a ciklohexén → 1,3-ciklohexadién átmenet
energianyereségének extrapolálása az 1,3-ciklohexadién → 1,3,5-ciklohexatrién átmenetre, másrészt az a
felfogás, hogy az "egyszerű" konjugáció révén jelentkező 10 kJ/mol energianyereséget nem tekintjük a benzol
delokalizációs energiája integráns részének. Amennyiben nem extrapolálunk és csak a ciklohexén hidrogénezési
hőjének háromszorosával számolunk, a delokalizációs energia 3×120 - 206 = 154 kJ/mol-nak adódik.
Ismereteink jelenlegi szintje mellett tehát meg kell alkudnunk azzal a ténnyel, hogy a benzol delokalizációs
energiájának nagyságát kísérleti úton, méréssel, nem tudjuk egyértelműen meghatározni.
-- Halogének addíciója
Halogénaddícióra az aromás gyűrű csak különleges reakciókörülmények között hajlamos. A benzol brómot
például csak ultraibolya fény és forralás hatására addícionál. A keletkező aliciklusos származék azonban
hevítéskor hidrogén-bromidot ad le, és tribrómbenzol - tehát aromás vegyület - formájában stabilizálódik.
A benzol klóraddíciójával előállítható hexaklórciklohexán (HCH) fontos rovarirtószer volt, amelynek
alkalmazását azonban ma már környezetkárosító hatása miatt (igen nehezen bomlik le a természetben)
megtiltották. A gyökös mechanizmussal lejátszódó klóraddiciót ultraibolya fény katalizálja:
A kondenzált gyűrűs rendszerek már könnyebben addícionálnak halogént: a fenantrén például szén-tetrakloridos
brómoldat hatására hidegen is fenantrén-9,10-dibromiddá alakul. Ez is azt mutatja, hogy a nem-szimmetrikus
elektroneloszlású kondenzált gyűrűs aromás szénhidrogének egyes szénatomjai már nem teljesen aromás
jellegűek: a fenantrén 9. és 10. szénatomjai közötti kötés mind reakciókészség, mind atomtávolság tekintetében
közelebb áll az alkénes kettőskötéshez, mint az aromás kötéshez. Ezt a fenantrén mezomer határformái is
mutatják (9.2.4. fejezet), mert az öt határforma közül négyben a 9. és 10. szénatomok között kettős kötés van.
5.3. Az aromás gyűrű oxidálása
-- Vegyszeres oxidáció
Vegyszeres oxidációval szemben az aromás szénhidrogének stabilitása eltérő. A jégecetes krómsav például a
benzolt nem támadja meg, a kondenzált gyűrűket viszont kinonokká (konjugált szerkezetű gyűrűs diketonokká)
oxidálja. Például:
Antracénből hasonló módon 9,10-antrakinon, fenantrénből pedig 9,10-fenantrén-kinon keletkezik.
-- Katalitikus oxidáció oxigénnel
Katalitikusan aktivált levegő-oxigénnel benzolból maleinsav-anhidridet, naftalinból ftálsav-anhidridet
állítanak elő ipari méretekben. A katalizátor mindkét esetben vanádium-pentoxid; az alkalmazott hőmérséklet
összhangban van a két gyűrűrendszer reakciókészségével:

AROMÁS SZÉNHIDROGÉNEK
(Arének)
133 Created by XMLmind XSL-FO Converter.
5.4. Az aromás gyűrű komplexképző tulajdonságai
Elektronszegény és elektrondús aromás gyűrűket tartalmazó vegyületek egymással (általában jól kristályosodó
és színes) molekulakomplexeket képeznek, amelyekben az aromás gyűrűk - lapjukkal egymás felé fordulva -
molekulaközi erőkkel kapcsolódnak egymáshoz. Ilyenek pl. a kinhidron (17.3.2. fejezet) és a pikrátok (24.8.5.
fejezet). Ezekkel részletesebben ott foglalkozunk.
Az alkénekhez hasonló módon (6.3.11. fejezet) az aromás gyűrű is képes π-elektronjai révén átmeneti fémekkel
π-komplexeket képezni. Ezek szerkezete az átmenetifém-ciklopentadienil komplexek (metallocének, 8.4.2.
fejezet) szerkezetével analóg. Legegyszerűbb képviselőjük a dibenzol-króm, amely a ferrocénhez hasonlóan
"szendvics" szerkezetű:
5.5. Az alifás oldallánc reakciói
-- Addíció és szubsztitúció az oldalláncban
Az aromás gyűrűhöz kapcsolódó alifás szénhidrogén-láncok általában ugyanúgy reagálnak, mint a szabad alifás
szénhidrogének. A reakciókörülményeket azonban úgy kell megválasztani, hogy az aromás gyűrű lehetőleg ne
lépjen reakcióba.
Így pl. szubsztitúciós reakció meghatározott körülmények között szelektíven lejátszódhat az oldalláncban úgy,
hogy az aromás gyűrű szubsztituciója nem következik be. Példa lehet erre a toluol klórozása: melegen,
katalizátor nélkül végezve azt, gyökös mechanizmussal az oldallánc klórozódik, míg hidegen, katalizátor (pl.
vasreszelék) jelenlétében a klór ionos mechanizmussal az aromás maghoz kapcsolódik (vö. 11.8.1., illetve
11.7.1. fejezetek.).
-- Az oldallánc oxidálása
Oxidációs hatásra az oldallánc érzékenyebben reagál mint az aromás gyűrű. Az oxidáció az alkillánc
magasabbrendű szénatomján (rendszerint a gyűrűhöz közvetlenül kapcsolódó - benzilhelyzetű - szénatomon)
következik be. Különböző hosszuságú vagy szerkezetű oldalláncok közül ennek megfelelően a hosszabb vagy
elágazóbb láncú oxidálódik könnyebben. A m-izopropiltoluol oxidációja például két lépésben játszódik le:

AROMÁS SZÉNHIDROGÉNEK
(Arének)
134 Created by XMLmind XSL-FO Converter.
Ezek az oxidációk vegyszeresen (krómsavval, salétromsavval, kálium-permanganáttal) vagy levegő-oxigénnel
katalitikusan egyaránt végrehajthatók. Végtermékük rendszerint karbonsav.
Speciálisan végzett oxidációval magasabbrendű - elsősorban tercier - C,H-kötések hidroperoxiddá oxidálhatók.
A kumolból így keletkező kumil-hidroperoxidot például ipari méretekben állitják elő, mint a kumolból
kiinduló fenol-szintézis (lásd 13.4.1. fejezet) közbenső termékét:
6. Nem-benzoid aromás szénhidrogének
Az aromás jelleg feltételeinek tárgyalásakor láttuk, hogy a gyűrűt alkotó atomok száma nincs korlátozva, csupán
az a feltétel, hogy a körbekonjugált π-elektronok száma 4n + 2 legyen. Ez teszi lehetővé, az ún. "nem-benzoid"
aromás szénhidrogének kialakulását. Ilyen, nem hattagú gyűrűs, de aromásan konjugált π-elektronszextettet
tartalmazó szénhidrogén-jellegű rendszerek a már tárgyalt ciklopentadienid-anion és az itt ismertetendő
cikloheptatrienilium-kation (triviális nevén tropiliumion):
Mindkettőnek számos stabilis származéka ismeretes. A ciklopentadienid-anion átmenetifém-komplexeivel, a
metallocénekkel már foglakoztunk (8.4. fejezet). A tropiliumion származékai közül megemlítjük az ionos
szerkezetű és stabilis tropilium-bromidot. A tropiliumion stabilitására jellemző, hogy tropiliumionná rendeződik
át az alkilbenzolokból a tömegspektrométerben keletkező PhCH2+ benzilkation.
Ismert a két fentebb említett nem-benzoid aromás rendszer kondenzált származékaként felfogható azulén. Ez a
kék színű, kevéssé stabilis vegyület (10C,10π) delokalizált rendszert tartalmaz. A naftalin izomerje, de
delokalizációs energiája 50 kJ/mol-lal kisebb a naftalinénál. Ezért könnyen a jóval stabilisabb naftalinná
izomerizálható. Az azulén és származékai gyulladásgátló hatással rendelkeznek; előfordulnak a kamillaolajban
és más illóolajokban is.

AROMÁS SZÉNHIDROGÉNEK
(Arének)
135 Created by XMLmind XSL-FO Converter.
7. Polisztirol és származékai
A sztirolt, mint a polisztirol alapanyagát, nagyipari méretekben állítják elő etilbenzol katalitikus
dehidrogénezésével (króm-oxidot és káliumvegyületeket tartalmazó vas(III)-oxid kontakton, 500-600 °C-on). A
sztirol már spontán módon (pl. levegőn, vagy fény hatására) is könnyen polimerizálódik; tároláskor ez a
folyamat inhibitorok (pl. hidrokinon) hozzáadásával lassítható. A sztirol ipari polimerizációját általában
gyökösen (pl. benzoil-peroxiddal) iniciálják. A kialakuló polimer szerkezete:
A polisztirol tiszta állapotban rideg és átlátszó (dekoratív hatású), habosítva csomagolásra, hőszigetelésre
alkalmas. Igen jó műszaki tulajdonságokkal rendelkező műanyag az akrilonitril/butadién/sztirol terpolimer
(ABS); pl. műszerfalak, lökhárítók készülnek belőle.
A sztirolt mintegy 5% divinilbenzollal kopolimerizálva oldószerekben duzzadó, laza térhálós szerkezetű termék
alakul ki, amelyet ioncserélő műgyanták előállítására használnak fel. A kationcserélő műgyantát az aromás
gyűrűk egy részének szulfonálásával (15.3. fejezet) állítják elő:
Vizes oldatok fémion tartalmát a gyanta szulfonsavsó alakjában köti meg és így cseréli ki hidrogén ionokra. Az
anioncserélő műgyanta előállitásához klórmetilezéssel (11.8.1. fejezet), kvaterner ammóniumsó képzéssel és
végül lúgos kezeléssel kvaterner ammónium-hidroxidot (22.1.6. fejezet) alakitanak ki:

AROMÁS SZÉNHIDROGÉNEK
(Arének)
136 Created by XMLmind XSL-FO Converter.
Ez a gyanta a vízben oldott anionokat köti meg és cseréli ki OH- ionokra. A kimerült kationcserélőt erős savval,
az anioncserélőt erős lúggal lehet regenerálni.
Kb. 25% divinilbenzol alkalmazásával merev szerkezetű térhálós polisztirol keletkezik, amelyet szulfonálva
szilárd fázisú, szerves oldószerekben oldhatatlan, de kissé duzzadó terméket kapnak. Ez szerves reakciókban
heterogén savas katalizátorként alkalmazható.