structural bioinformatics predicting protein structure
Post on 19-Dec-2015
223 views
TRANSCRIPT


STRUCTURAL BIOINFORMATICS
Predicting Protein structure

What is Structural Bioinformatics?
is the branch of bioinformatics which is related to the analysis and prediction of the three-dimensional structure of biological macromolecules such as proteins, RNA, and DNA.
It deals with generalizations about macromolecular 3D structure such as comparisons of overall folds and local motifs, principles of molecular folding, evolution, and binding interactions, and structure/function relationships,

Structural bioinformatics vs. bioinformatics
DNA mapping DNA and protein
sequence Development of
algorithms for data mining
Determine of 3D structures in biomolecules
Analysis and comparison of biomolecular structures
Prediction of biomolecular structure.
Bioinformatics Structural bioinformatics

Experimental techniques for structure determination
X-ray Crystallography Nuclear Magnetic Resonance
spectroscopy (NMR)

Prediction
Question?
Why is structure prediction is hard..?

Structure Prediction Approaches
1. Homology (Comparative) ModelingBased on sequence similarity with a protein
for which a structure has been solved.
2. Threading (Fold Recognition)Requires a structure similar to a known
structure
3. Ab-initio fold predictionNot based on similarity to a sequence\
structure

Ab-initio fold prediction
Given only the sequence, try to predict the structure based on physico-chemical properties (energy, hydrophobicity etc.)

Fold Recognition(Threading)
Given a sequence and a library of folds, thread the sequence through each fold. Take the one with the highest score.
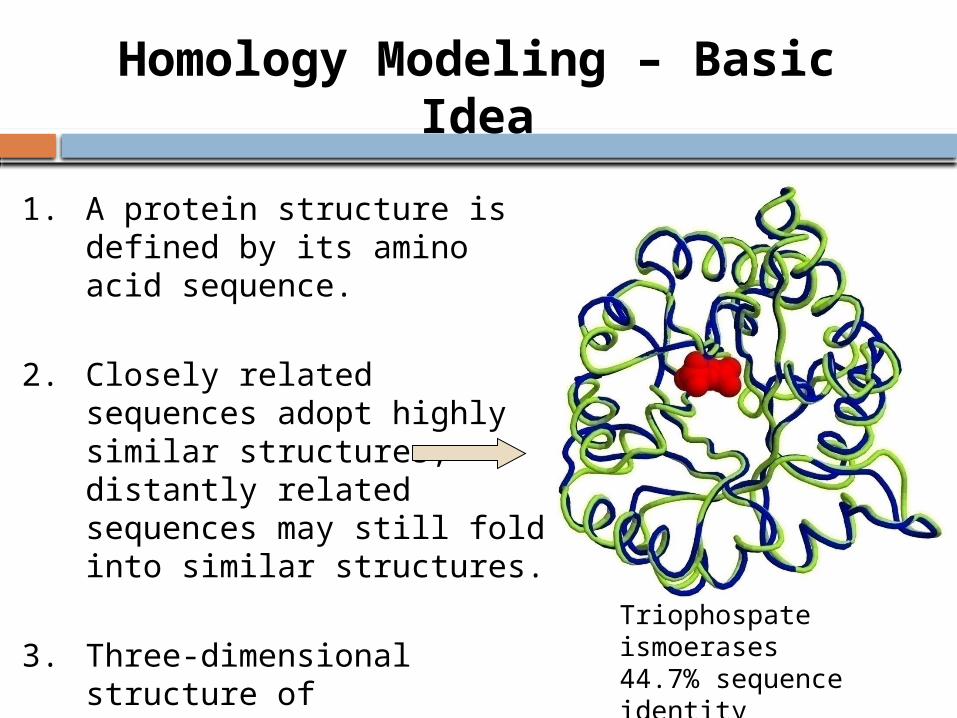
Homology Modeling – Basic Idea
Triophospate ismoerases44.7% sequence identity0.95 RMSD
1. A protein structure is defined by its amino acid sequence.
2. Closely related sequences adopt highly similar structures, distantly related sequences may still fold into similar structures.
3. Three-dimensional structure of proteins from the same family is more conserved than theirprimary sequences.

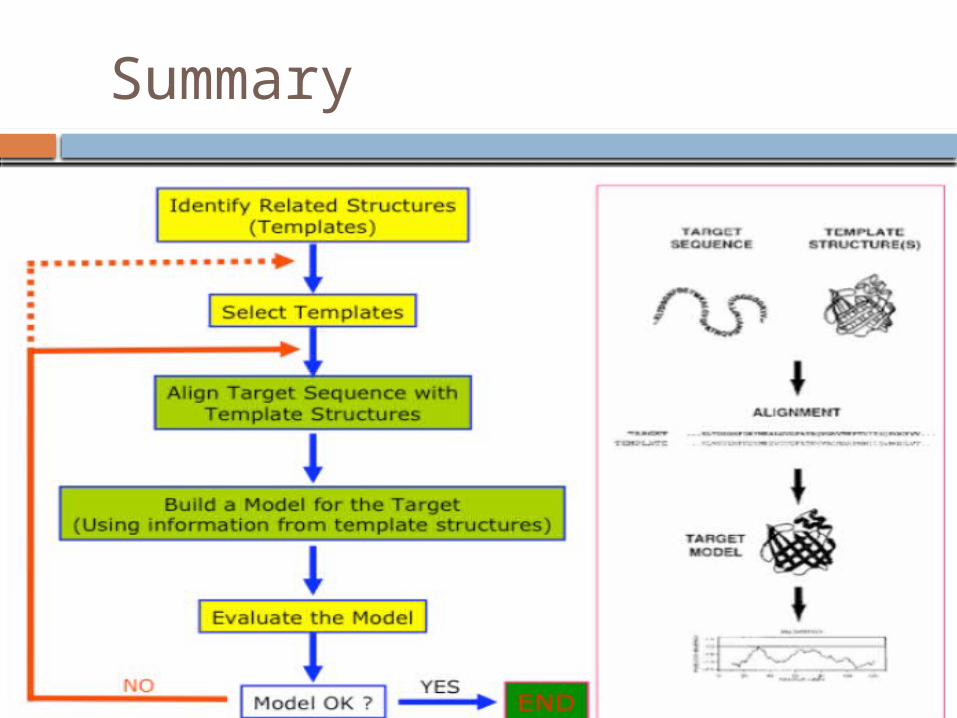
General Scheme1. Searching for structures related to the query
sequence
2. Selecting templates
3. Aligning query sequence with template structures
4. Building a model for the query using information from the template structures
5. Evaluating the modelFiser A et al. Methods in Enzymology 374: 461-491(2004)

General Scheme

1 .Searching For Structures

How to select the right template?
• Close subfamily - phylogenetic tree
• “Environment” similarity

• Two ways to combine multiple templates:
– Global model – alignment with different domain of the target with little overlap between them
– Local model – alignment with the same part of the target
More than one template

3 .Aligning
All comparative modeling programs depend on a target-template alignment.
When the sequence similarity between the template and target proteins is high, simple pairwise alignments are usually fine (e.g. Needleman-Wunsch global alignment).
But some times blast is required.

Sequence alignment algorithms
Examples: the two most used in homology modeling are:
BLAST: General strategy is to optimize the maximal segment pair (MSP) score - BLAST computes similarity, not alignment
FastA (local alignment): searches for both full and partial sequence matches, i.e., local similarity obtained; more sensitive than BLAST, but slower; many gaps may represent a problem

Building the model

5 .Model Evaluation
The accuracy of the model depends on its sequence identity with the template:
Summary

Any Questions ?
Thank you