small supernumerary marker chromosomes (smcs…royanaward.com/files12/2003 starke-ssmc.pdf ·...
TRANSCRIPT

Abstract Small supernumerary marker chromosomes(SMCs) are present in about 0.05% of the human popula-tion. In approximately 30% of SMC carriers (excludingthe ~60% SMC derived from one of the acrocentric chro-mosomes), an abnormal phenotype is observed. The clin-ical outcome of an SMC is difficult to predict as they canhave different phenotypic consequences because of (1)differences in euchromatic DNA-content, (2) different de-grees of mosaicism, and/or (3) uniparental disomy (UPD)of the chromosomes homologous to the SMC. Here, wepresent 35 SMCs, which are derived from all human chro-mosomes, apart from chromosome 6, as demonstrated bythe appropriate molecular cytogenetic approaches, such ascentromere-specific multicolor fluoresence in situ hybrid-ization (cenM-FISH), multicolor banding (MCB), and sub-centromere-specific multicolor FISH (subcenM-FISH). Innine cases without an aberrant phenotype, neither partialproximal trisomies nor UPD could be detected. Abnormalclinical findings, such as psychomotoric retardation and/or craniofacial dysmorphisms, were associated with sevenof the cases in which subcentromeric single-copy probeswere proven to be present in three copies. Conversely, ineight cases with a normal phenotype, proximal euchro-matic material was detected as partial trisomy. UPD wasstudied in 12 cases and subsequently detected in two ofthe cases with SMC (partial UPD 4p and maternal UPD 22in a der(22)-syndrome patient), indicating that SMC carri-ers have an enhanced risk for UPD. At present, smallproximal trisomies of 1p, 1q, 2p, 6p, 6q, 7q, 9p, and 12qseem to lead to clinical manifestations, whereas partial
Heike Starke · Angela Nietzel · Anja Weise · Anita Heller · Kristin Mrasek · Britta Belitz · Christine Kelbova ·Marianne Volleth · Beate Albrecht · Beate Mitulla · Ralf Trappe · Iris Bartels · Sabine Adolph · Andreas Dufke ·Sylke Singer · Markus Stumm · Rolf-Dieter Wegner · Jörg Seidel · Angela Schmidt · Alma Kuechler ·Isolde Schreyer · Uwe Claussen · Ferdinand von Eggeling · Thomas Liehr
Small supernumerary marker chromosomes (SMCs): genotype-phenotype correlation and classification
Hum Genet (2003) 114 : 51–67DOI 10.1007/s00439-003-1016-3
Received: 20 May 2003 / Accepted: 4 August 2003 / Published online: 16 September 2003
ORIGINAL INVESTIGATION
Electronic database information: accession numbers and URLsfor the data in this article are as follows: ENSEMBL-database, http://www.ensembl.org/ National Center for Biotechnology Information (NCBI),http://www.ncbi.nlm.nih.gov/ Genome Database (GDB), http://www.gdb.org/gdb/ OMIM (Online Mendelian Inheritance in Man) Database,http://www.ncbi.nlm.nih.gov/omim
H. Starke · A. Nietzel · A. Weise · A. Heller · K. Mrasek ·A. Kuechler · I. Schreyer · U. Claussen · F. von Eggeling ·T. Liehr (✉)Institut für Humangenetik und Anthropologie, Kollegiengasse 10, 07743 Jena, GermanyTel.: +49-3641-935533, Fax: +49-3641-935502,e-mail: [email protected]
B. BelitzPraxis für Medizinische Genetik, Frankfurter Allee 111, 10247 Berlin, Germany
C. KelbovaPraxis für Medizinische Genetik, Marienstrasse 27, 03046 Cottbus, Germany
M. VollethInstitut für Humangenetik, Leipziger Strasse 44, 39120 Magdeburg, Germany
B. AlbrechtInstitut für Humangenetik, Hufelandstrasse 55, 45122 Essen, Germany
B. MitullaZentralklinikum Suhl, gGmbH, A.-Schweitzer-Strasse 2, 98527 Suhl, Germany
R. Trappe · I. BartelsInstitut für Humangenetik, Zentrum für Hygiene und Humangenetik, Heinrich-Düker-Weg 12, 37073 Göttingen, Germany
S. AdolphOlgahospital, Institut für Klinische Genetik, Bismarkstrasse 3, 70176 Stuttgart, Germany
A. Dufke · S. SingerInstitut für Humangenetik und Anthropologie, Wilhelmstrasse 27, 72074 Tübingen, Germany
M. Stumm · R.-D. WegnerPartnerschaft für Medizinische Genetik, Kurfürstendamm 199, 10719 Berlin, Germany
© Springer-Verlag 2003
J. SeidelKlinik für Kinder- und Jugendmedizin, Kochstrasse 2, 07743 Jena, Germany
A. SchmidtPraxis für Humangenetik, Podbielskistrasse122, 30177 Hannover, Germany
A. KuechlerKlinik für Strahlentherapie, Bachstrasse 18, 07743 Jena, Germany

proximal trisomies of 2q, 3p, 3q, 5q, 7p, 8p, 17p, and 18pmay not be associated with significant clinical symptoms.With respect to clinical outcome, a classification of SMCsis proposed that considers molecular genetic and molecu-lar cytogenetic characteristics as demonstrated by presentlyavailable methods.
Introduction
Chromosomal abnormalities, the great majority of whichare numerical, are found in 0.3%–1% of newborns (Buck-ton et al. 1985; Queisser-Luft et al. 2002). The clinicaloutcomes of whole chromosome trisomies and monosomiesare well characterized. However, clinical effects for pa-tients having imbalances of small chromosomal regionsresulting from karyotypes containing small supernumerarymarker chromosomes (SMCs) are less predictable. This isattributable to the lack of rigorous cytogenetic evaluationand good clinical outcome correlation. Small SMCs arereported in 0.01%–0.05% of all prenatally screened fe-tuses. Approximately half of these small SMCs are deriv-atives of chromosome 15 (Crolla 1998; Crolla et al. 1998).Among the remaining cases, there is a great variation inchromosomal origin (Callen et al. 1992; Crolla 1998;Crolla et al. 1998). According to Crolla (1998), 70% ofSMCs derived from non-acrocentric chromosomes are notassociated with clinical symptoms. Because of the vary-ing clinical outcomes attributable to the chromosomal ori-gin of the SMC, their detailed characterization is of inter-est in order to improve post-diagnostic medical care andgenetic counseling. In cases of apparently normal pheno-types, the characterization of SMCs can provide impor-tant information about regions within the human genomethat do not lead to abnormalities in the presence of genedosage imbalances (Sumption and Barber 2001) and/orare subject to gene silencing (for a review, see Li 2002).
The origin of small SMCs is almost impossible to es-tablish by routine cytogenetics alone, whereas fluores-cence in situ hybridization (FISH) methods are highly suitedfor this purpose. SMCs have been successfully character-ized by whole-chromosome-painting (WCP) probes, cen-tromere-specific probes, or combined chromosome micro-dissection and FISH approaches (for a review, see Nietzelet al. 2001). WCP-FISH approaches are well-suited forthe determination of the chromosomal origin of marker orderivative chromosomes providing that they are largerthan 17p, whereas if they are smaller, WCP-FISH is, ingeneral, non-informative (Haddad et al. 1998; case 7, mar1, Starke et al. 1999). As recently reported, for SMCs witha euchromatic content of approximately half of the shortarm of chromosome 17p or more, characterization by themulticolor banding (MCB) technique is possible (Liehr etal. 2002b; Weise et al. 2002). For a fast and easy charac-terization of smaller SMCs, we have recently proposed thecentromere-specific multicolor FISH (cenM-FISH) method(Nietzel et al. 2001). Despite this, analyses of small SMCsto detect the presence of euchromatin have to date beenambiguous and have required further clarification. In the
present paper, we have addressed this problem by utiliz-ing MCB probe-sets and/or a probe-set comprising of 43bacterial or yeast artificial chromosome (BAC or YAC,respectively) clones located in the proximal regions ofeach human chromosome, viz., subcentromeric multicolor-FISH (subcenM-FISH). As reported previously by us(Chudoba et al. 1999; von Eggeling et al. 2002) and oth-ers (for a review, see Kotzot 2002), after identification ofthe origin of the SMC, its normal sister-chromosomesshould be tested for their parental origin to exclude a pos-sible uniparental disomy (UPD). UPD can be tested bymolecular genetic approaches, such as microsatellite analy-sis (Salafsky et al. 2001) or methylation-specific poly-merase chain reaction (PCR; Nietzel et al. 2003).
In this paper, molecular cytogenetics and moleculargenetics have been used for the characterization of SMCsof variable origins in 35 cases, and a correlation of theclinical phenotype with the SMC-associated genotype hasbeen made. Finally, an SMC classification system is pro-posed, which facilitates the characterization and allows aprecise description of SMCs, including their shape, theUPD of their sister-chromosomes, and the degree of mo-saicism.
Materials and methods
Studied cases and cytogenetics
Cytogenetic and molecular cytogenetic studies were performed onchromosomes derived from cultivated amniotic fluid, chorionbiopsy, or peripheral blood cells. Chromosome preparations wereobtained according to standard techniques (Verma and Babu1989).
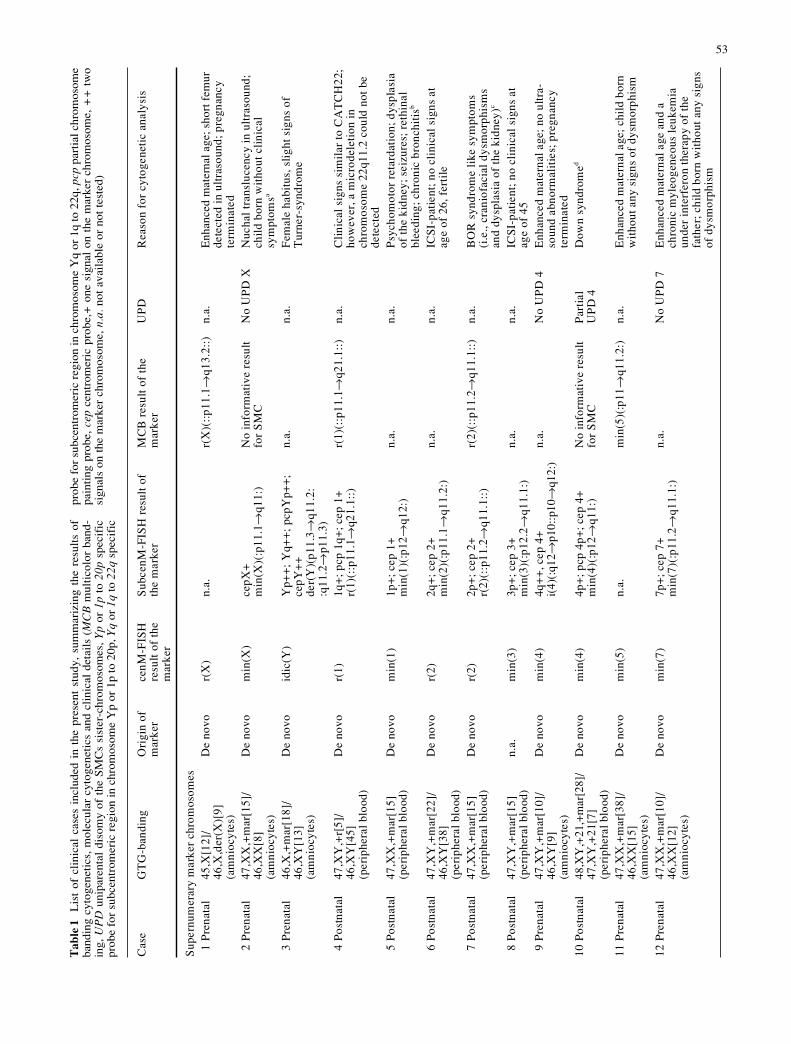
The cases studied, the cytogenetic results, and the clinical de-tails are listed in Table 1. Cases with an additional SMC are num-bered with Arabic numerals (1–35) and those with structurallyaberrant markers have Roman numberals (I–III).
Standard FISH procedures
FISH was performed according to standard procedures (Liehr et al.1995). In some of the cases, 24-color FISH with WCP probes(Senger et al. 1998; case 12), cenM-FISH (Nietzel et al. 2001; allcases apart from I-III), or MCB (Liehr et al. 2002b; cases 1, 2, 4,7, 10, 11, 16, 17, 22, 28, 29) were applied (for technical details, seeSenger et al. 1998; Nietzel et al. 2001; Liehr et al. 2002b). Addi-tionally, in case III, the subtelomeric probe PAC 240G10 specificfor 16q was used (kindly provided by Dr. Lyndal Kearney, Lon-don, UK).
SubcenM-FISH method
The proximal probes included in the subcenM-FISH set (see Table 2)were chosen under the prerequisite that they were the most proxi-mal probes presently mapped in the ENSEMBL database. BAC orYAC DNA (200 ng) was amplified by degenerate oligonucleotide-primed PCR (DOP-PCR; 50-µl volume; Senger et al. 1998). Eachprobe was labeled separately by using a secondary DOP-PCR (20-µlvolume) either with SpectrumGreen or TexasRed as described inNietzel et al. (2001). The probes were derived from C.H.O.R.I.BACPAC Resources (Oakland, USA), Dr. M. Rocchi (Universita’degli Studi di Bari, Sezione di Genetica, Bari, Italy), and Dr. N.Carter (Sanger Centre, Cambridge, UK). For each human chromo-some, a subcenM-FISH probe set was prepared according to the la-
52
53
Cas
eG
TG
-ban
ding
Ori
gin
ofm
arke
rce
nM-F
ISH
resu
lt o
f th
em
arke
r
Sub
cenM
-FIS
H r
esul
t of
the
mar
ker
MC
B r
esul
t of
the
mar
ker
UP
DR
easo
n fo
r cy
toge
neti
c an
alys
is
Sup
ernu
mer
ary
mar
ker
chro
mos
omes
1 P
rena
tal
45,X
[12]
/46
,X,d
er(X
)[9]
(am
nioc
ytes
)
De
novo
r(X
)n.
a.r(
X)(
::p1
1.1Æ
q13.
2::)
n.a.
Enh
ance
d m
ater
nal a
ge; s
hort
fem
urde
tect
ed i
n ul
tras
ound
; pre
gnan
cyte
rmin
ated
2 P
rena
tal
47,X
X,+
mar
[15]
/46
,XX
[8]
(am
nioc
ytes
)
De
novo
min
(X)
cepX
+m
in(X
)(:p
11.1
Æq1
1:)
No
info
rmat
ive
resu
ltfo
r S
MC
No
UP
D X
Nuc
hal
tran
sluc
ency
in
ultr
asou
nd;
chil
d bo
rn w
itho
ut c
lini
cal
sym
ptom
sa
3 P
rena
tal
46,X
,+m
ar[1
8]/
46,X
Y[1
3](a
mni
ocyt
es)
De
novo
idic
(Y)
Yp+
+;
Yq+
+;
pcpY
p++
;ce
pY+
+de
r(Y
)(p1
1.3Æ
q11.
2::q
11.2
Æp1
1.3)
n.a.
n.a.
Fem
ale
habi
tus,
sli
ght
sign
s of
Tur
ner-
synd
rom
e
4 P
ostn
atal
47,X
Y,+
r[5]
/46
,XY
[45]
(per
iphe
ral b
lood
)
De
novo
r(1)
1q+
; pcp
1q+
; cep
1+
r(1)
(::p
11.1
Æq2
1.1:
:)r(
1)(:
:p11
.1Æ
q21.
1::)
n.a.
Cli
nica
l si
gns
sim
ilar
to
CA
TC
H22
;ho
wev
er, a
mic
rode
leti
on in
chro
mos
ome
22q1
1.2
coul
d no
t be
dete
cted
5 P
ostn
atal
47,X
X,+
mar
[15]
(per
iphe
ral b
lood
)D
e no
vom
in(1
)1p
+; c
ep 1
+m
in(1
)(:p
12Æ
q12:
)n.
a.n.
a.P
sych
omot
or r
etar
dati
on; d
yspl
asia
of t
he k
idne
y; s
eizu
res;
ret
hina
lbl
eedi
ng; c
hron
ic b
ronc
hiti
sb
6 P
ostn
atal
47,X
Y,+
mar
[22]
/46
,XY
[38]
(per
iphe
ral b
lood
)
De
novo
r(2)
2q+
; cep
2+
min
(2)(
:p11
.1Æ
q11.
2:)
n.a.
n.a.
ICS
I-pa
tien
t; n
o cl
inic
al s
igns
at
age
of 2
6, f
erti
le
7 P
ostn
atal
47,X
X,+
mar
[15]
(per
iphe
ral b
lood
)D
e no
vor(
2)2p
+; c
ep 2
+r(
2)(:
:p11
.2Æ
q11.
1::)
r(2)
(::p
11.2
Æq1
1.1:
:)n.
a.B
OR
syn
drom
e li
ke s
ympt
oms
(i.e
., cr
anio
faci
al d
ysm
orph
ism
san
d dy
spla
sia
of t
he k
idne
y)c
8 P
ostn
atal
47,X
Y,+
mar
[15]
(per
iphe
ral b
lood
)n.
a.m
in(3
)3p
+; c
ep 3
+m
in(3
)(:p
12.2
Æq1
1.1:
)n.
a.n.
a.IC
SI-
pati
ent;
no
clin
ical
sig
ns a
tag
e of
45
9 P
rena
tal
47,X
Y,+
mar
[10]
/46
,XY
[9]
(am
nioc
ytes
)
De
novo
min
(4)
4q+
+, c
ep 4
+i(
4)(:
q12Æ
p10:
:p10
Æq1
2:)
n.a.
No
UP
D 4
Enh
ance
d m
ater
nal a
ge;
no u
ltra
-so
und
abno
rmal
itie
s; p
regn
ancy
term
inat
ed10
Pos
tnat
al48
,XY
,+21
,+m
ar[2
8]/
47,X
Y,+
21[7
](p
erip
hera
l blo
od)
De
novo
min
(4)
4p+
; pcp
4p+
; cep
4+
min
(4)(
:p12
Æq1
1:)
No
info
rmat
ive
resu
ltfo
r S
MC
Par
tial
UP
D 4
Dow
n sy
ndro
med
11 P
rena
tal
47,X
X,+
mar
[38]
/46
,XX
[15]
(am
nioc
ytes
)
De
novo
min
(5)
n.a.
min
(5)(
:p11
Æq1
1.2:
)n.
a.E
nhan
ced
mat
erna
l age
; ch
ild
born
wit
hout
any
sig
ns o
f dy
smor
phis
m
12 P
rena
tal
47,X
X,+
mar
[10]
/46
,XX
[12]
(am
nioc
ytes
)
De
novo
min
(7)
7p+
; cep
7+
min
(7)(
:p11
.2Æ
q11.
1:)
n.a.
No
UP
D 7
Enh
ance
d m
ater
nal a
ge a
nd a
chro
nic
myl
eoge
neou
s le
ukem
iaun
der
inte
rfer
on t
hera
py o
f th
efa
ther
; chi
ld b
orn
wit
hout
any
sig
nsof
dys
mor
phis
m
Tab
le1
Lis
t of
cli
nica
l ca
ses
incl
uded
in
the
pres
ent
stud
y, s
umm
ariz
ing
the
resu
lts
ofba
ndin
g cy
toge
neti
cs,
mol
ecul
ar c
ytog
enet
ics
and
clin
ical
det
ails
(M
CB
mul
tico
lor
band
-in
g,U
PD
unip
aren
tal
diso
my
of t
he S
MC
s si
ster
-chr
omos
omes
,Y
por
1pto
20p
spec
ific
prob
e fo
r su
bcen
trom
eric
reg
ion
in c
hrom
osom
e Y
p or
1p
to 2
0p,Y
qor
1qto
22q
spec
ific
prob
e fo
r su
bcen
trom
eric
reg
ion
in c
hrom
osom
e Y
q or
1q
to 2
2q,p
cppa
rtia
l chr
omos
ome
pain
ting
pro
be,
cep
cent
rom
eric
pro
be,+
one
sign
al o
n th
e m
arke
r ch
rom
osom
e,+
+tw
osi
gnal
s on
the
mar
ker
chro
mos
ome,
n.a.
not
avai
labl
e or
not
tes
ted)

54
Tab
le 1
(
cont
inue
d)
Cas
eG
TG
-ban
ding
Ori
gin
ofm
arke
rce
nM-F
ISH
resu
lt o
f th
em
arke
r
Sub
cenM
-FIS
H r
esul
t of
the
mar
ker
MC
B r
esul
t of
the
mar
ker
UP
DR
easo
n fo
r cy
toge
neti
c an
alys
is
13 P
ostn
atal
47,X
X,+
mar
[26]
/46
,XX
[14]
(per
iphe
ral b
lood
)
De
novo
r(7)
r(7
;7)
{2 s
ubcl
ones
}∑
7q+
; pc
p 7q
+;c
ep 7
+r(
7)(:
:p11
.1Æ
q11.
21::
)[15
]∑
7q+
+;
pcp
7q+
+;
cep
7++
r(7;
7)(:
:p11
.1Æ
q11.
21:
:p11
.1Æ
q11.
21::
)[4]
∑ 7q
++
++
; pc
p 7q
++
++
;ce
p7+
++
+r(
7;7;
7;7)
(::p
11.1
Æq1
1.21
::p
11.1
Æq1
1.21
::p
11.1
Æq1
1.21
::p
11.1
Æq1
1.21
::)[
1]{3
sub
clon
es}
n.a.
no U
PD
7A
orta
-ins
uffi
cien
cy;
aort
a ro
otec
tasi
a; a
rtia
l se
ptal
def
ect;
sid
eve
tric
les
enla
rged
wit
h pl
exus
cys
t;dy
sacu
sis
on b
oth
ears
14 P
rena
tal
47,X
X,+
mar
[43]
/46
,XX
[7]
(am
nioc
ytes
)
Mat
erna
lr(
8)∑
8p+
; pc
p 8p
+;c
ep 8
+r(
8)(:
:p11
.21Æ
q11.
1::)
n.a.
n.a.
Enh
ance
d m
ater
nal a
ge;
no u
ltra
-so
und
abno
rmal
itie
s; m
othe
r w
itho
utan
y cl
inic
al s
igns
; nor
mal
chi
ld b
orn
15 P
ostn
atal
48,X
XY
,+m
ar[1
5](p
erip
hera
l blo
od)
De
novo
dic(
9)9p
+; c
ep 9
++
dic(
9)(:
p11.
2Æq1
1.1:
:q11
.1Æ
p11.
1:)
n.a.
n.a.
Hyp
ogon
adis
m, g
ynae
com
asti
a,m
enta
l ret
arda
tion
; wea
knes
s of
conn
ecti
ve t
issu
e16
Pos
tnat
al47
,XY
,+m
ar[1
8]/
46,X
Y[2
](b
one
mar
row
)
Acq
uire
dr(
11)
n.a.
r(11
)(::
p11.
2Æq1
3.1:
:q14
::)
n.a.
Aty
pica
l CM
L; S
MC
onl
y in
bon
em
arro
w, n
ot in
per
iphe
ral b
lood
e
17 P
rena
tal
47,X
X,+
mar
[15]
shor
t ter
m c
ultu
re;
47,X
X,+
mar
[3];
46,X
X[3
7] l
ong
term
cult
ure
(am
nioc
ytes
)
De
novo
der(
12)
n.a.
i(12
)(pt
erÆ
q10:
:q10
Æpt
er)
n.a.
Enh
ance
d m
ater
nal a
ge;
incr
ease
dnu
chal
tran
sluc
ency
det
ecte
d in
ultr
asou
nd, p
regn
ancy
ter
min
ated
beca
use
of P
alis
ter-
Kil
lian
synd
rom
e
18 P
ostn
atal
47,X
Y,+
r[21
]/46
,XY
[11]
(per
iphe
ral b
lood
)
De
novo
min
(12)
12q+
; ce
p 12
+m
in(1
2)(:
p11.
1Æq1
2:)
n.a.
n.a.
Psy
chom
otor
ret
arda
tion
;cr
anio
fasc
ial d
ysm
orph
ies
19 P
rena
tal
47,X
Y,+
mar
[12]
/46
,XY
[2]
(am
nioc
ytes
)
De
novo
min
(13o
r21)
cep1
3/21
+ m
in(1
3or2
1)(:
p11.
1Æq1
1.1:
)n.
a.n.
a.E
nhan
ced
mat
erna
l age
; no
ult
ra-
soun
d ab
norm
alit
ies;
chi
ld b
orn
wit
hout
cli
nica
l sym
ptom
s20
Pre
nata
l47
,XY
,+m
ar[1
0](a
mni
ocyt
es)
De
novo
idic
(14)
p++
; ce
p 14
/22+
+id
ic(1
4)(p
terÆ
q11.
1::q
11.1
Æpt
er)
n.a.
n.a.
Enh
ance
d m
ater
nal a
ge;
no u
ltra
-so
und
abno
rmal
itie
s; c
hild
bor
nw
itho
ut c
lini
cal s
ympt
oms
21 P
rena
tal
47,X
Y,+
mar
[10]
(cho
rion
bio
psy)
De
novo
idic
(15)
p++
; ce
p 15
++
idic
(15)
(pte
rÆq1
1.1:
:q11
.1Æ
pter
)
n.a.
No
UP
D 1
5E
nhan
ced
mat
erna
l age
; no
ult
ra-
soun
d ab
norm
alit
ies;
chi
ld b
orn
wit
hout
cli
nica
l sig
ns22
Pos
tnat
al48
,XX
,+2m
ar[1
5](p
erip
hera
l blo
od)
De
novo
i(15
)x2
15q+
+;
p++
; cep
15
++
;pc
p15q
++
(2x
)i(
15)(
pter
Æq1
2–13
::q
12–1
3Æpt
er)x
2
i(15
)(pt
erÆ
q13:
:q13
Æpt
er)x
2no
UP
D 1
5P
osta
xial
pol
ydac
tyly
, mus
cula
rhy
poto
nia,
duc
tus
arte
rios
us a
ndop
en f
oram
en o
vale
, sei
zure
s, s
ever
ede
velo
pmen
tal d
elay
, mic
roce
phal
yf
23 P
ostn
atal
48,X
Y,+
21,+
mar
[15]
(per
iphe
ral b
lood
)D
e no
voi(
15)
p++
, cep
15+
+i(
15)(
pter
Æq1
0::q
10Æ
pter
)n.
a.n.
a.D
own
synd
rom
e

55
24 P
rena
tal
47,X
Y[1
3]/
47,X
Y,+
mar
[16]
(am
nioc
ytes
)
De
novo
min
(16)
cep
16+
min
(16)
(:p1
1.1Æ
q11.
2:)
n.a.
No
UP
D 1
6E
nhan
ced
mat
erna
l age
; no
ult
ra-
soun
d ab
norm
alit
ies;
chi
ld b
orn
wit
hout
cli
nica
l sig
ns25
Pos
tnat
al47
,XY
,+m
ar[1
0]/
46,X
Y[5
](p
erip
hera
l blo
od)
De
novo
min
(17)
17p+
; ce
p 17
+m
in(1
7)(:
p11.
2Æq1
1.1:
)n.
a.n.
a.IC
SI-
pati
ent
of 4
0ye
ars;
pri
mar
yst
eril
ity
beca
use
of a
sthe
nosp
erm
ia
26 P
rena
tal
47,X
Y,+
mar
[15]
(am
nioc
ytes
)D
e no
vom
in(1
8)ce
p 18
+m
in(1
8)(:
p11.
1Æq1
1.1:
)n.
a.N
o U
PD
18
Enh
ance
d m
ater
nal a
ge;
hygr
oma
coll
i de
tect
ed b
y ul
tras
ound
;pr
egna
ncy
term
inat
ed27
Pos
tnat
al47
,XX
,+m
ar[2
3]/
46,X
X[1
7](p
erip
hera
l blo
od)
n.a.
min
(18)
18p+
; pc
p18p
+; c
ep 1
8+m
in(1
8)(:
p11.
21Æ
q11.
1:)
n.a.
n.a.
ICS
I-pa
tien
t of
36
year
s; p
rim
ary
ster
ilit
y; s
he h
ad a
sur
gery
at t
heag
e of
7 b
ecau
se o
f an
art
rial
sep
tal
defe
ct28
Pre
nata
l47
,XY
,+m
ar [
45]/
46,X
Y[1
0](a
mni
ocyt
es)
De
novo
min
(19)
n.a.
min
(19)
(:p1
2Æq1
3.1:
)n.
a.E
nhan
ced
mat
erna
l age
; no
ult
ra-
soun
d ab
norm
alit
ies;
pre
gnan
cyte
rmin
ated
29 P
rena
tal
47,X
Y,+
mar
[15]
(am
nioc
ytes
)P
ater
nal
t(11
;22)
min
(22)
22q+
; ce
p 14
/22+
; cep
22+
der(
22)t
(11;
22)(
q?;q
11.2
1)de
r(22
)t(1
1;22
)(q
23;q
11.2
1)M
ater
nal
UP
D 2
2,no
UP
D 1
1
Enh
ance
d m
ater
nal a
ge;
no u
ltra
-so
und
abno
rmal
itie
s; p
regn
ancy
term
inat
ed b
ecau
se o
f de
r(22
)-sy
ndro
me
30 P
ostn
atal
47,X
Y,+
mar
[20]
(per
iphe
ral b
lood
)D
e no
vom
in(2
2)22
q++
; p+
; cep
14/
22;
cep
22+
,m
in(2
2)de
l(q1
1.2?
3)du
p(q
11.2
1)
n.a.
n.a.
Cat
-eye
syn
drom
e w
ith
psyc
hom
otor
reta
rdat
ion,
cra
niof
asci
al d
ys-
mor
phie
s, a
nal a
tres
ia a
nd i
ris
colo
bom
a31
Pre
nata
l47
,XX
,+m
ar[1
0](a
mni
ocyt
es)
De
novo
idic
(22)
p++
; ce
p 14
/22+
+; c
ep22
++
idic
(22;
22)(
pter
Æq1
1.1:
:q11
.1Æ
pter
)
n.a.
n.a.
Enh
ance
d m
ater
nal a
ge;
chil
d bo
rnw
ith
no d
ysm
orph
ic s
ings
and
norm
al p
sych
omot
oric
dev
elop
men
t
32 P
rena
tal
47,X
Y,+
mar
[10]
(am
nioc
ytes
)M
ater
nal
orig
inid
ic(2
2)p+
+;
cep1
4/22
++
;ce
p22+
+i(
22)(
pter
Æq1
1.1:
:q11
.1Æ
pter
)
n.a.
No
UP
D 2
2A
bnor
mal
tri
ple
test
res
ults
; no
ultr
asou
nd a
bnor
mal
itie
s; m
othe
ran
d m
eanw
hile
bor
n ch
ild
wit
hout
clin
ical
sig
ns33
Pre
nata
l47
,XX
,+m
ar[4
5]/
46,X
X[1
0](a
mni
ocyt
es)
Pat
erna
lor
igin
–in
2 o
f 50
mit
osis
in
peri
pher
albl
ood
∑ id
ic(2
2)∑d
er(2
2){2
sub
clon
es}
∑ 22
q++
; p+
+, c
ep14
/22+
+;
cep
22+
+id
ic(2
2;22
)(pt
erÆ
q11.
21:
:q11
.21Æ
pter
)[12
]∑
cep
14/2
2+; c
ep 2
2+m
in(2
2)(:
p11.
2Æq1
1.1:
)[1]
∑ ce
p 22
+m
in(2
2)(:
p11.
1Æq1
1.1:
)[7]
{3 s
ubcl
ones
}
n.a.
n.a.
Enh
ance
d m
ater
nal a
ge;
no u
ltra
-so
und
abno
rmal
itie
s; p
regn
ancy
term
inat
ed
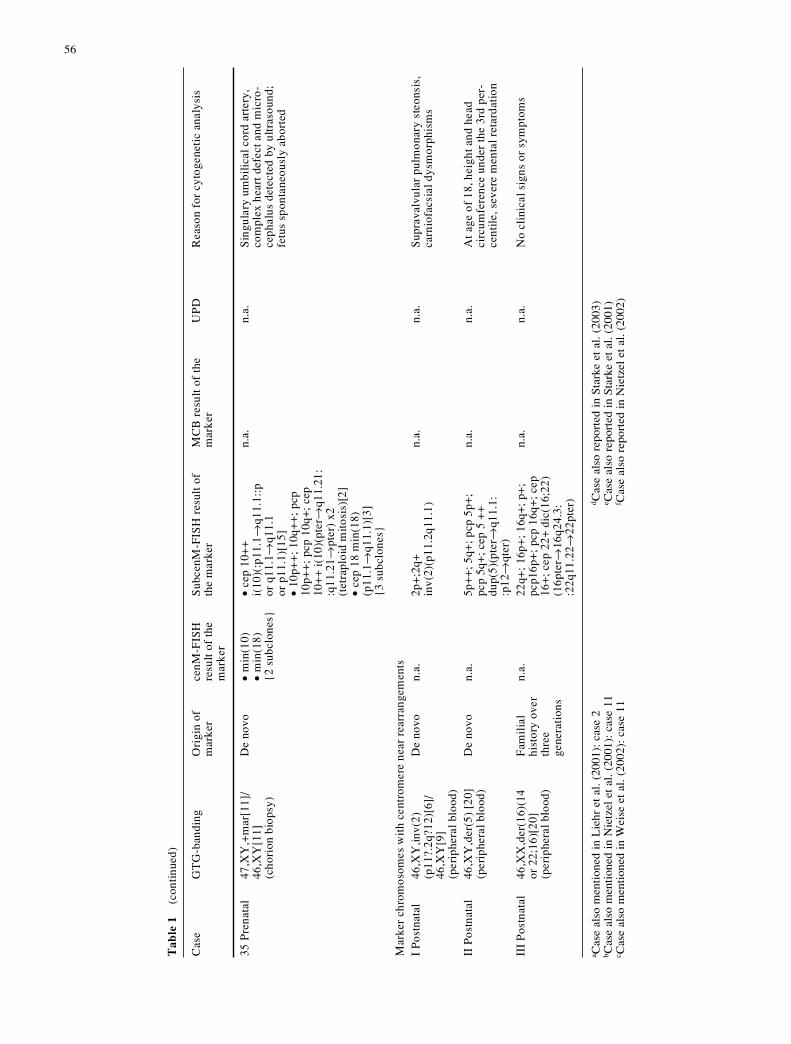
34 P
rena
tal
48,X
X,+
2mar
[12]
/47
,XX
,+m
ar[3
](a
mni
ocyt
es)
De
novo
∑ m
in(9
)∑
min
(9)
and
min
(20)
{2 s
ubcl
ones
}
∑ ce
p 9+
; 20p
+;
cep
20+
min
(9)(
:p11
Æq1
1:)
and
min
(20)
(p11
.2Æ
q11.
1)[1
6]∑
cep
9+m
in(9
)(:p
11Æ
q11:
)[4]
{2 s
ubcl
ones
}
n.a.
n.a.
Abn
orm
al t
ripl
e te
st r
esul
ts;
hygr
oma
coll
i det
ecte
d by
ultr
asou
nd
56
Tab
le 1
(
cont
inue
d)
Cas
eG
TG
-ban
ding
Ori
gin
ofm
arke
rce
nM-F
ISH
resu
lt o
f th
em
arke
r
Sub
cenM
-FIS
H r
esul
t of
the
mar
ker
MC
B r
esul
t of
the
mar
ker
UP
DR
easo
n fo
r cy
toge
neti
c an
alys
is
35 P
rena
tal
47,X
Y,+
mar
[11]
/46
,XY
[11]
(cho
rion
bio
psy)
De
novo
∑ m
in(1
0)∑
min
(18)
{2 s
ubcl
ones
}
∑ ce
p 10
++
i(10
)(:p
11.1
Æq1
1.1:
:por
q11
.1Æ
q11.
1or
p11
.1)[
15]
∑ 10
p++
; 10q
++
; pc
p10
p++
; pc
p 10
q+;
cep
10+
+ i(
10)(
pter
Æq1
1.21
::q
11.2
1Æpt
er)
x2(t
etra
ploi
d m
itos
is)[
2]∑
cep
18 m
in(1
8)(p
11.1
Æq1
1.1)
[3]
{3 s
ubcl
ones
}
n.a.
n.a.
Sin
gula
ry u
mbi
lica
l cor
d ar
tery
,co
mpl
ex h
eart
def
ect a
nd m
icro
-ce
phal
us d
etec
ted
by u
ltra
soun
d;fe
tus
spon
tane
ousl
y ab
orte
d
Mar
ker
chro
mos
omes
wit
h ce
ntro
mer
e ne
ar r
earr
ange
men
tsI
Pos
tnat
al46
,XY
,inv(
2)(p
11?.
2q?1
2)[6
]/46
,XY
[9]
(per
iphe
ral b
lood
)
De
novo
n.a.
2p+
;2q+
inv(
2)(p
11.2
q11.
1)n.
a.n.
a.S
upra
valv
ular
pul
mon
ary
steo
nsis
,ca
rnio
facs
ial d
ysm
orph
ism
s
II P
ostn
atal
46,X
Y,d
er(5
) [2
0](p
erip
hera
l blo
od)
De
novo
n.a.
5p+
+; 5
q+; p
cp 5
p+;
pcp
5q+
; cep
5 +
+du
p(5)
(pte
rÆq1
1.1:
:p12
Æqt
er)
n.a.
n.a.
At
age
of 1
8, h
eigh
t and
hea
dci
rcum
fere
nce
unde
r th
e 3r
d pe
r-ce
ntil
e, s
ever
e m
enta
l ret
arda
tion
III
Pos
tnat
al46
,XX
,der
(16)
(14
or 2
2;16
)[20
](p
erip
hera
l blo
od)
Fam
ilia
lhi
stor
y ov
erth
ree
gene
rati
ons
n.a.
22q+
; 16
p+; 1
6q+
; p+
;pc
p16p
+;
pcp
16q+
; cep
16+
; cep
22+
dic
(16;
22)
(16p
terÆ
16q2
4.3:
:22q
11.2
2Æ22
pter
)
n.a.
n.a.
No
clin
ical
sig
ns o
r sy
mpt
oms
a Cas
e al
so m
enti
oned
in
Lie
hr e
t al
. (20
01):
cas
e 2
b Cas
e al
so m
enti
oned
in
Nie
tzel
et
al. (
2001
): c
ase
11c C
ase
also
men
tion
ed i
n W
eise
et
al. (
2002
): c
ase
11
d Cas
e al
so r
epor
ted
in S
tark
e et
al.
(200
3)e C
ase
also
rep
orte
d in
Sta
rke
et a
l. (2
001)
f Cas
e al
so r
epor
ted
in N
ietz
el e
t al
. (20
02)

beling schemes shown in Fig. 1a. The set consisted of the mostproximally available single-copy probes, microdissection-derivedpartial chromosome painting (pcp) probes specific for the long andthe short chromosome arm (Liehr and Claussen 2002), and a cen-tromere-specific probe (Nietzel et al. 2001). For the acrocentricchromosomes, no proximal single-copy probes and no chromo-
some-specific pcp probes for the short arms are available. How-ever, the short arms of the chromosomes 13, 14, 15, 21, and 22 arecovered in the subcenM-FISH probe sets by a probe that stains allp-arms of the acrocentric chromosomes, viz., midi54 (Mrasek et al.2001). For chromosome 22, two centromere-specific probes (cep)were used: D14/22Z1 and D22Z4. In our experience with cenM-FISH experiments, the probe D14/22Z1 is located more distallythan D22Z4 in the p-arm of chromosome 22. However, because ofpolymorphisms, the order can be inverted (e.g., case 34, see be-low).
Microsatellite analysis to exclude UPD
For UPD analysis, genomic DNA was extracted from the peripheralblood lymphocytes of both parents and the patient or from amnio-cytes of the fetus by using standard procedures. The followingmarkers were informative in the 12 studied cases: DXS6807,DXS6810, DXS6810, DXS1047, DXS1001, GATA172D05,DXS6814 (case 2); D4S2639, D4S2633, D4S1627, D4S2623,D4S2394, D4S2431 (case 9); D4S2366, GATA145E01, D4S2633,D4S2639, D4S1627, D4S2623, D4S2431 (case 10); D07S1805,D07S0559, D07S2195, D07S0522 (case 12); D07S1808,D07S1799, D07S0559 (case 13); D15S126; D15S1077 (case 21);D16S422 (case 24); D18S858, D18S046 (case 26); D11S1999,D11S2002, D11S1986, D22S683, D22S689, D22S445 (case 29);D22S420, D22685, D22S683, D22S689 (case 32). Sequence infor-mation and the chromosomal locations of all markers used areavailable from the National Center for Biotechnology Information(NCBI) or the Genome Database (GDB). Twenty-five cycles ofPCR were performed with labeled primers responsive to infrared(IRD800; MWG-Biotech, Germany). Denatured samples were loadedonto a 6% denaturing polyacrylamide gel, separated electrophoret-ically on a Licor DNA 4000 sequencer (LI-COR, Nebraska), anddetected with an infrared laser diode.
Case 22 was tested for UPD by methylation-specific PCR forthe SNRP exon 1 region as described by Nietzel and coworkers(2002).
Results
Molecular cytogenetics
Chromosomal origin of the SMCs
A total of 35 cases (cases 1–35) was included in this partof the study because of the presence of a small SMC. Ofthese, 19 were studied prenatally and 16 postnatally. Theorigin of the SMCs could be clarified in all cases by ap-plication of the cenM-FISH approach. One marker eachwas derived from chromosomes Y, 3, 5, 8, 9, 11, 13/21,14, 16, 17, and 19; two markers each were derivatives ofchromosomes X, 1, 2, 4, 7, 9, 12, or 18; three and fourmarkers were of chromosome 15 and 22 origin, respec-tively. Additionally, two cases are described each showingtwo marker chromosomes of unrelated chromosomal ori-gin: case 34 constitutes derivatives of chromosomes 9 and20 (Fig. 2e1, see also Fig. 3d), and in case 35, the SMCswere derived from chromosomes 10 and 18. In summary,23 of the 24 human chromosomes were involved in theformation of the SMCs in the 35 studied cases (for details,see Table 1).
57
Table 2 Clone localization
Region Localization Clone Comments
Xp Xp11.21 bA465B24Xq Xq12 dJ1065K8Yp Yp11.2 bA75F5Yq Yq11.21 bA489H201p 1p12 bA130B181q 1q21.1 bA35B4 Additional signal
in 1p36.12p 2p11.2 882D52q 2q11.2 774E93p 3p12.1 bA91A153q 3q12.1 bA21I164p 4p12 bA100N214q 4q12 bA535C75p 5p12 bA19F125q 5q11.2 bA160F86p 6p11.2 bA362K18 Additional signal
in 7q326q 6q12 bA387L57p 7p11.2 bA10F117q 7q11.21 bA3N28p 8p11.21 bA64C228q 8q11.21 bA11C209p/q 9p12 bA128P23 Additional signal
in 9q139q 9q13 bA430C1510p 10p11.21 bA232C1310q 10q11.22 bA178A1011p 11p11.21 bA12C1111q 11q12 bA77M712p 12p11.21 bA517B2312q 12q12 bA490D1113q 13q12.1–13q12.2 768a0414q 14q11.2 bA332N615q 15q11.2 bA171C816p 16p11.2 bA67I1016q 16q12.1 bA474B1217p 17p11.2 bA746M117q 17q11.2 bA229K1518p 18p11.21 bA151D11 Additional signal
in 1p22–3118q 18q11.2 bA59E1219p 19p13.1 bA22G1019q 19q13.1 bA14D17 Additional signal
in 6q1420p 20p11.2 bA96L620q 20q11.2 bA243J1621q 21q11.2 bC067E322q 22q11.21 bK115F6

58

Parental origin of the SMC and mosaicism
In five of the 38 clinical cases summarized, markers wereinherited (cases 14, 29, 32, 33, and III). In an additionaltwo of these, the parental origin could not be determinatedbecause of a lack of parental blood samples (cases 8 and27). In one case, the marker was acquired because of amalignancy (case 16), and in the remaining 30 cases, themarkers were de novo.
A mosaicism status of different extent was determinedfor 21 cases (cases 1–4, 6, 9, 10–14, 16–19, 24, 25, 27,33–35; for details, see Table 1).
Presence of euchromatic content on the SMCs
To determine the presence of euchromatic material, theMCB technique was used in those cases where the SMCswere larger than a half of 17p (except cases 3 and 16), i.e.,in cases 1 (Fig. 4a), 4, 11 (Fig. 4b), 17, and 29. In cases 8,22, and 26, the MCB technique was compared with thecorresponding subcenM-FISH probe sets, leading to iden-tical results. In cases 2 and 10, MCB did not provide in-formative results, as the SMCs were smaller than waspracticable for this technique. However, the correspond-ing SMCs were characterized for their presence of eu-chromatin by subcenM-FISH. In case 29, the extent of thechromosome 11 material on the der(22)t(11;22) was deter-mined by MCB (result not shown). In the remaining 28
cases (including case 29), the presence of euchromatin onthe SMC was studied exclusively by the subcenM-FISHapproach. Consequently, the existence of euchromaticmaterial on the SMC could be excluded by the subcenM-probe set for cases 20, 21, 23, 24, 26, 31, 32, 33 (forder(22)), 34 (for der(9); (Fig. 2e2) and 35 (apart from thetetraploid clone; Fig. 2f1, f2). Small partial trisomiescould be detected in cases 5 (Fig. 2a), 6, 8, 10, 12 (Fig.2b), 13 (for r(7)), 14, 18, 25, 27, 29 (for chromosome 22material; Fig. 3a) and 34 (for der(20); Fig. 2e2). Partialtetrasomies of euchromatic material were found in cases3, 9 (Fig. 2c), 13 (for r(7;7)), 15, 30 (for the region22q11.2; Fig. 2d), and 33 (for idic(22)). Partial hexasomywas present in case 22 and in a subclone of case 13 (forr(7;7;7;7)) plus a partial octasomy in a subclone of case35 (for i(10); Fig. 2f1).
Additional potential of the subcenM-FISH technique
For the last mentioned subclone of case 35 and the deriv-ative chromosome Y of case 3, the suitability of the sub-cenM-FISH probe sets becomes obvious, even for largermarker chromosomes. Thus, three cases were included be-cause of the presence of larger marker chromosomes withpericentromeric rearrangements. A small pericentric in-version of chromosome 2 (case I, Fig. 3b), a centromericduplication of a chromosome 5 (case II, Fig. 3c), and a trans-location of the chromosomes 22 and 16 (case III, Fig. 3d)could easily be characterized by the subcenM-FISH tech-nique.
UPD analysis
A total of 12 of the 35 cases with SMCs (cases 2, 9, 10,12, 13, 21–24, 26, 29, and 32) were analyzed to determinethe parental origin of the homologous chromosomes ofthe derivatives (Fig. 5a). UPD could be excluded in 10 ofthe 12 cases studied (Table 1; Fig. 5b), whereas a partialUPD 4p was detected for case 10 (for details, see Starke etal. 2003), and complete maternal UPD 22 and partial pa-ternal UPD 11 was observed in case 29 (Fig. 5c).
Correlation of the molecular and molecular cytogeneticresults with clinical phenotype
A clinical correlation of the results obtained from theFISH and UPD studies could be performed for 30 of the35 cases with SMCs. Because of additional imbalances(case 10, 23, 34, 35) or the finding that the SMC was ac-quired in connection with a malignancy (case 16), thiswas not possible for the remaining cases.
Cases without clinical symptoms
Seventeen of the 35 cases did not show any clinical symp-toms. Seven of these had no detectable euchromatic mate-
59
Fig. 1 a Labeling scheme of the sub-centromere specific multi-color FISH (subcenM-FISH) probe set. In the non-acrocentricchromosomes, the p-arm is labeled with cyanine 5 (Cy5) and the q-arm with diethylaminocoumarin (DEAC). The centromere-nearprobe in the long arm is labeled with SpectrumGreen (Spec.Green)and that in the short arm with Texas Red (TexasRed). In the acro-centric chromosomes, the long arm is stained with Cy5, the shortarm with DEAC, and the centromere-near probe in the long armwith TexasRed. The centromeres are labeled with SpectrumOr-ange (Spec.Orange). b SubcenM-FISH pseudo-color pattern for all24 human chromosomes. Note that, at present, only one chromo-some at a time can be stained; the chromosome figures were as-sembled from 24 different experiments
Fig. 2 a SubcenM-FISH (SubC) result in case 5; the partial tri-somy of 1p12 material has been confirmed (white). The long armof the marker chromosome consists of heterochromatic materialonly. b A partial trisomy 7p11.2 has been detected by the presenceof the SMC in case 12. c In case 9, subcenM-FISH detects thesmallest-ever-described isochromosome, which leads to a partialtetrasomy 4q12 (arrowheads two green signals on the SMC).d Characterization of an unusual “cat eye chromosome” in case 30(arrowheads two white signals on the SMC). e CenM-FISH de-tected two SMCs of different origin in case 34 (arrowheads in e1);subcenM-FISH demonstrated that the SMC 9 had no additional eu-chromatic content, whereas the SMC 20 (e2) leads to a partial tri-somy 20p11.2. In addition to the locus-specifc probe, the partialchromosome painting probe for 20p gave a positive signal on theSMC. f SubcenM-FISH results of case 35, in which cenM-FISH(results not shown) also identified two SMCs of different origin.Neither the SMC derived from chromosome 10 (f1 clone a) northat from chromosome 18 (f2 clone c) had any euchromatic con-tent. In two tetraploid metaphases (f1 clone b), two additionalidentical isochromosomes 10p were detected, plus the four normalchromosomes 10
�

rial (cases 2, 19–21, 24, 31, 32), and in cases 2, 21, 24, and32, a UPD of the sister chromosomes of the SMC addi-tionally could be excluded. Small proximal imbalancescould be detected in 10 of the clinically benign cases (fordetails, see Table 1). UPD could only be excluded for twoof those ten cases (9, 12).
Cases with clinical symptoms
Clinical symptoms such as psychomotor developmentand/or signs of dysmorphisms (for details, see Table 1)were present in 13 of the 35 cases with SMCs. Partialproximal chromosomal imbalances attributable to the pres-
60

ence of the SMC were detected in 12 cases. UPD could bestudied in three of them (13, 22, 29), and a maternal UPD22 combined with a partial paternal UPD 11 was detectedin case 29. Interestingly, in case 26, hygroma colli was de-tected by ultrasound, but neither centromere-near euchro-matic material nor a UPD 18 could be detected.
Discussion
Here, we report 35 cases with SMCs studied in detail fortheir euchromatic centromere-near content and a possibleUPD of the sister chromosomes. So far, this is the largestsuch study of cases with SMCs. Moreover, this is the firststudy to characterize the presence of euchromatin on theSMC and the second large-scale analysis for the occur-rence of UPD in association with an SMC (first study:James et al. 1995). Application of the latter methods en-able real phenotype-genotype correlations to be made. Be-cause of the lack of corresponding data, most case reportsavailable from the literature could not be compared withthe present cases discussed below.
Small SMCs
Ever since 1998 when Crolla summarized all previouslyavailable cases with SMCs, it has been clear that, depend-ing on the chromosomal origin of an SMC, differences
can be observed and expected in clinical outcome. Theexistence of a common “proximal trisomy” syndrome, in-dependent of either the chromosomal origin and the pres-ence of euchromatic content or UPD, can therefore be dis-counted. This has also been confirmed by non-systematicstudies and case reports of small proximal trisomies inconnection with the presence of an SMC and specific clin-ical features (Müller-Navia et al. 1996; Crolla 1998;Crolla et al. 1998; Callen et al. 1999; Giardino et al. 1999,2002; Ostroverkhova et al. 1999; Stankiewicz et al. 2000,2001; Villa et al. 2001; Velagaleti et al. 2002; see alsoTable 3).
The centromeric regions of human chromosomes areknown to be preferential sites for the integration of dupli-cated DNA, which often contains gene fragments (for areview of the corresponding literature, see Bridgland et al.2003). The genetic content and gene density of the re-gions adjacent to the high-, mid-, or low-repetitive cen-tromeric sequences (Horvath et al. 2003) have been stud-ied in detail, to date, only for chromosome 10 (Guy et al.2003). According to the ENSEMBL database, centromericregions contain fewer genes and open reading framesthan, for example, telomeric regions. The detailed analy-sis of cases with SMCs has been advanced by the presentstudy and represents a first step toward the identificationof pericentromeric disease-related genes.
SMC cases with clinical symptoms
Proximal partial chromosomal imbalances associated withsignificant clinical signs are reported in this study for a to-tal of 18 cases. In five of them, either additional imbal-ances or the refractory nature of the SMC hampered aclear clinical correlation (cases 10, 16, 23, 34, 35). Twomore cases could not be introduced into a genotype-phe-notype correlation as they were studied only prenatally(cases 1 and 26). In four additional cases, the SMC couldbe associated with known clinical syndromes, such as ei-ther Turner syndrome (case 3; Cunniff 2002), Pallister-Killian syndrome (case 17; OMIM 601803), inv dup(15)syndrome (case 22; Nietzel et al. 2003), or der(22) syn-drome (case 29; Funke et al. 1999). In the remaining sixcases, psychomotor or mental retardation and/or dysmor-phic signs could be observed in connection with an SMCand the presence of additional euchromatic content de-rived from chromosomes 1p, 1q, 2p, 7q, 9p, or 12q.
For proximal trisomy 9p and 12q, no comparable casesapart from those reported here are currently available. Ob-viously, a detailed clinical correlation cannot be based onone single case only, and clearly, the clinical symptomsdescribed, for example, for proximal trisomy 9p and 12qare distinct from each other (hypogonadism, gynecomas-tia, and weakness of connective tissue are exclusively re-ported for the case with partial trisomy 9p11.2). Proximaltrisomy 2p11.2 has been associated with craniofacial dys-morphisms (case 7 of the present study; see also Ostro-verkhova et al. 1999). However, in addition to the partialtrisomy 2p11.2, a partial trisomy 2q10-q14.1 is also pre-
61
Fig. 3 a In case 29, a typical der(22) syndrome marker was de-tected by the subcenM-FISH set for chromosome 22 (here, the cen-tromeric probe D14/22Z1 was not used in the probe set). The ad-ditional material from chromosome 11 on the SMC did not hy-bridize to the subcenM-FISH probe set. The 24-color FISH analy-sis confirmed that negative material was derived from chromo-some 11. b A small pericentric inversion present in mosaic form incase I was characterized in detail by subcenM-FISH probe set 2(for details see Table 1). A similar case has been described byFryns et al. (1983). c SubcenM-FISH was applied to characterize acentromeric duplication in one chromosome 5 in case II; adup(5)(pter→q11.1::p12→qter) could be defined. d A centromericprobe for chromosome 16 was applied together with a subtelom-eric probe for chromosome 16q and the subcenM-FISH probe setfor chromosome 22. A non-reciprocal translocation with break-points in 16q24.3 and 22q11.22 was found in case III. The sub-telomeric region in 16q is present only in one copy in this family
Fig. 4a, b Multicolor banding (MCB) was applied in case 1 tocharacterize the ring chromosome derived from Xp11.1q13.2 (a)and the derivative chromosome 5p11q11.2 in case 11 (b)
Fig. 5 a SMCs can occur, for example, because of a trisomy,which is recognized by the living cell via unknown mechanisms, inthe zygote. One of the three homologous chromosomes seems tobe selected for degradation by chance, leading to cells with twohomologous chromosomes plus one SMC. Thus, the two sisterchromosomes of an SMC can either be of paternal and maternalorigin, i.e., a “normal situation” is present (“normal”), or both in-tact sister-chromosomes are from only one parent (UPD). b, c UPDwas tested in 11 of the 35 cases with SMCs (m mother, f father,c child). b Example of a “normal situation” (i.e., no UPD de-tected). c A maternal iso-UPD (iUPD) 22 and a paternal partialUPD 11 (two paternal alleles and one maternal allele) were de-tected in case 29
�

sent in the case reported by Ostroverkhova et al. in 1999,obscuring a clear clinical correlation. Psychomotor retar-dation is a common feature in the total of six cases de-scribed with partial trisomy 1p12. In spite of this, the clin-ical findings of the patients are heterogeneous, as mightbe expected with the diversity in the origin and size of theadditional centromere-near euchromatic content (Callenet al. 1999). Indeed, the four ring chromosomes reportedby these authors are different both from each other andfrom those reported here and by Giardino et al. (1999). Asthe clinical features also differ in the two cases availablefor partial trisomy 1q21.1 (present case 4; see also Giar-dino et al. 1999), partial proximal trisomies 1p or 1q canbe deduced to be associated with clinical signs. This is anassumption that can also be made from the available re-ports of partial trisomy 6p (Crolla 1998), 6q (Stankiewiczet al. 2000), and 7q (present case 13; see also Velagaleti etal. 2002).
In case 26, the abnormal clinical phenotype (hygromacolli) could not be correlated with the genotype, as UPD18 and euchromatic content were excluded. Centromere-near euchromatic material, not detectable in this case bythe applied subcenM-FISH probeset for chromosome 18,
may possibly be detected by future analytic methods.Similar to the FISH probe set for the subtelomeric chromo-somal regions initially proposed in 1996/1997 (NationalInstitutes of Health and Institute of Molecular MedicineCollaboration 1996; Knight et al. 1997), the subcenM-FISH probe set may need continual improvement to aidthe identification of further subtleties.
SMC cases without clinical symptoms
No clinical symptoms were associated with the presenceof an SMC in seventeen of the 35 cases. Nonetheless, forcase 27, an operable artrial septal defect was reported, andin four cases (6, 8, 25, 27), the SMC was detected duringpreparation for ICSI. In only one case was primary steril-ity present in the partner of the SMC carrier (case 6). Noproximal trisomies were detected in seven cases (2, 19–21, 24, 31, 32), while being present in the remaining 10 caseswithout clinical abnormalities. For imbalances in the jux-tacentromeric region of 2q, 3p, 4q, 5q, 7p, 8p, 17p, 18p,19p/19q, and 22q, one case each is available (see Table 3).However, cases 11 (5q) and 12 (7p) were newborn chil-
62
Table 3 Summary of cases with small marker mediated partial trisomies; UPD could be excluded for cases 12 and 13 (++ major ab-normalities, – normal phenotype, {} additional euchromatic material present on the SMC, n.a. not available)
SMC Additional material of proximal p Additional material of proximal qderived from chromosome References Clinic References Clinic
X No reports n.a. No reports n.a.Y No reports n.a. No reports n.a.1 Giardino et al. (1999) ++ Giardino et al. (1999) ++
Callen et al. (1999) 4 cases ++ Present study case 4 ++Present study case 5 ++
2 {Ostroverkhova et al. (1999)} ++ Villa et al. (2001) –Present study case 7 ++ Giardino et al. (2002) –
Present study case 6 –3 Present study case 8 – Müller-Navia et al. (1996) –4 No reports n.a. No reports n.a.5 {Stankiewicz et al. (2000)} ++ Present study case 11 –6 Crolla (1998) 3 cases ++ Stankiewicz et al. (2000) ++7 Present study case 12 – Velagaleti et al. (2002) 3x ++
Present study case 13 ++8 Present study case 14 – No reports n.a.9 Present study case 15 ++ No reports n.a.
10 No reports n.a. No reports n.a.11 No reports n.a. No reports n.a.12 No reports n.a. Present study case 18 ++13 No reports n.a.14 No reports n.a.15 For overview, see, e.g., Nietzel et al. (2002)16 No reports n.a. No reports n.a.17 Stankiewicz et al. (2001) 8 cases ++ No reports n.a.
with tris. 17p11.2–12Present study case 25 trisomy 17p11.2 –
18 Present study case 27 – No reports n.a.19 No reports n.a. No reports n.a.20 No reports n.a. No reports n.a.21 No reports n.a.22 No reports n.a.

dren, and no clear prediction can be given about their fu-ture development. Similarly, cases 9 (4q), 28 (19p/19q),and 33 (22q) had no reported ultrasound abnormalitiesand a terminated pregnancy. Excluding the acrocentricchromosomes, case 28 is the only case with minute SMC,which, to our knowledge, currently describes both proxi-mal p and q.
Two cases with proximal trisomy 2q in connectionwith an SMC but without clinical symptoms have beendescribed previously (Villa et al. 2001; Giardino et al.2002). To date, because of problems with distinguishingbetween centromere 1, 5 and 19, it has been difficult tocharacterize small derivatives of chromosome 5 with ahigh degree of confidence. Crolla et al. (1998) report twocases with SMC 5 or 19 with normal development, as pre-sent in case 11 of this study, where the SMC could becharacterized as min(5)(:p11→q11.2:); cases with largerring chromosomes 5 with clinical abnormalities have beensummarized by D’Amato Sizonenko and coworkers (2002).For the remaining proximal trisomies, no additional re-ports are available.
Nonetheless, a possible conclusion that the small prox-imal partial trisomies mentioned above never have clini-cal consequences has to be considered carefully. Differentclinical outcomes are to be expected if the size of the eu-chromatic content differs in SMCs of the same origin (seealso Callen et al. 1999). This is illustrated by case 25 witha partial trisomy 17p11.2, which is the only report withoutclinical symptoms (apart from asthenospermia) out ofnine described cases with small SMC 17 (Stankiewicz etal. 2001). According to this data, the most frequent find-ings in patients with proximal trisomy 17p are psychomo-tor delay and mental retardation, speech delay, feedingdifficulties, seizures, and failure to thrive. In all thosecases with abnormalities, the partial trisomy included theregion 17p12. Thus, SMC 17 containing only 17p11.2 ma-terial and excluding 17p12 seems to be benign, whereaslarger SMCs for chromosome 17 lead to clinical conse-quences. Similar correlations cannot be excluded for theother chromosomes.
Finally, chromatin organization itself, especially of thecentromeres, is known to be important for regulating geneexpression. Regions defined recently as hemichromatic maycause problems, especially in the juxta-centromeric re-gions of chromosomes 1, 9, and 16 (for variants of this re-gion in chromosome 9, see Starke et al. 2002). Experi-ments summarized by Smith (2002) show that creating aneocentromere results in a mechanism reminiscent ofchromatin transcriptional silencing. Thus, possible silenc-ing effects have also to be considered for the cases withpartial proximal trisomies without clinical symptoms.
SMC-related chromosomal mosaics
Mosaicism in connection with SMCs is well known:Crolla (1998) summarizes 144 cases, 54% (i.e., 78) of whichshow mosaic karyotypes. Nevertheless, at least 60% (i.e.,47) of these mosaic-cases have psychomotor developmen-
tal delay and/or dysmorphic stigmata, apparently indepen-dently of the detected fraction of aberrant cells in the pe-ripheral blood. An even lower rate of abnormal clinicalfindings of 40% (27/66 cases) can be observed whenlooking at the non-mosaic cases.
In the present study, mosaicism was observed in 21 ofthe 35 studied cases (i.e., 60%). For the 14 cases includedin Table 3, mosaicsm is present in only two of the patientswith clinical symptoms (cases 13 and 18) and having anSMC in about 65% of their blood-cells. Carriers of SMCswithout clinical symptoms have mosaicism to a muchhigher extent, i.e., in eight of nine cases (cases 4, 6, 11,12, 14, 18, 25, and 27). Here, the SMC is present in 10%to 86% of the peripheral blood cells. To arrive at a finalassessment concerning the real mosaic status of all thosecases, examination of different tissues could be helpful(see, for example, Liehr et al. 1996). In summary, a ten-dency towards mosaicism in cases without clinical symp-toms seems to be evident.
Mosaicism is a phenomenon resulting from SMC for-mation and has not currently attracted enough attention.In three of the studied cases (case 13, 33, and 35), threesubclones instead of one (as suggested after GTG band-ing) were present in the corresponding patients. The pres-ence of a more complex mosaic status than initially sug-gested was established by molecular cytogenetics (i.e.,cenM-FISH and subcenM-FISH). As demonstrated, thisadditional information can help in understanding the pres-ence or absence of clinical symptoms; the severe heartproblems in case 13 could at least in part be explained bythe tetra- and hexasomy 7q11.21 in 25% of the blood cells,in addition to the partial trisomy 7q11.21 in the remainder.Additionally, chromosomal mosaicism can aid the under-standing of the process of SMC formation: in 12/20 meta-phases with SMC in case 33, 22q11.2 material could bedetected twice on an isodicentric chromosome leading toa partial tetrasomy of this region. Furthermore, two addi-tional cell clones could be detected by subcenM-FISHwith derivative chromosomes comprising of D14/22Z1and D22Z4 material or D22Z4 material alone. This can beinterpreted as ongoing instability of the additional chro-mosomal material in the cells. However, the mechanismsor pathways involved in SMC formation remains unclear.
UPD analysis of the SMC related sister chromosomes
UPD, the exceptional inheritance of two homologouschromosomes from only one parent, has been shown pre-viously to appear in cases with SMCs (for reviews, seeKotzot 2002; Shaffer et al. 2001; see also Fig. 5a). UPDfor some chromosomes is without clinical consequences(e.g., von Eggeling et al. 2002) but, for a few chromo-somes, can result in abnormality in the affected individ-ual. The most important chromosomes in connection withUPD are chromosomes 6, 7, 11, 14, 15, and 20 (Chudobaet al. 1999; Shaffer et al. 2001; Kotzot 2002). Nonethe-less, UPD analysis was performed in the present study inany relevant cases in order to combine an exclusion of a
63

UPD with the exclusion of euchromatic content on theSMC. Additionally, prediction of the phenotypic effectsof UPD is complex as, according to Ledbetter and Engel(1995), three independent factors are involved: (1) effectsof trisomy on the placenta or the fetus; (2) autosomal re-cessive disease attributable to reduction to homozygosity;and (3) imprinted gene effects for some chromosomes. Fi-nally, the situation can also be complicated by the findingthat, when UPD is present, heterodisomy can be com-bined with isodisomy (see, for example, Salafsky et al.2001).
In the present study, analysis for UPD was carried outin 12 of the 35 cases; in the remaining 23 patients, noparental material could be obtained. A partial UPD 4p wasdetected in case 10 (for details, see Starke et al. 2003), andcomplete maternal UPD 22 plus partial paternal UPD 11was determined in case 29 (Fig. 5c). The complete mater-nal UPD 22 plus the partial paternal UPD 11 detected wasto be expected in a case with der(22) syndrome. The pres-ence of a partial UPD 4p in case 10 and the absence of aUPD 15 in case 23 did not lead to any detectable changesin the clinical features of these two patients with Downssyndrome. The exclusion of UPD in cases 13 and 22 isconsistent with their clinical symptoms being caused bythe genomic imbalances associated with the SMCs. In theremaining eight cases, no UPD or clinical symptoms wereobserved. The detection of two out of 12 cases with UPDsuggest that cases with SMC are candidates for the searchfor UPD. This was also established by the first large studyfor UPD in SMC cases; however, the authors only de-tected a single UPD 6 case in 21 studied patients (James etal. 1995).
SMC classification
Detection of an SMC is nearly always unexpected by theclinician and is practically a “by-product” of cytogeneticanalysis. Subsequently, the characterization of the SMCand, especially in prenatal cases, a genotype-phenotypecorrelation according to the literature is needed. The pre-sent study plus a review of the literature has revealed clin-ical correlations in connection with SMC formation andpartial proximal trisomies for 18 of the 43 centromere-near regions of the human genome (see Table 3). It shouldbe mentioned in this respect that a preliminary analysis ofthe small proximal regions concerning their predicted genedensity has not led to any clear correlation of “symptom-atic” or “non symptomatic” regions (http://www.ensembl.org/Homo_sapiens/; data not shown).
To facilitate the description of SMCs according to thenovel molecular cytogenetic possibilities for their charac-terization, we propose here a new SMC classification (seeTable 4). It is based on five main classes that are differen-tiated by the presence or absence of a nucleolar-organizerregion (NOR)/centromeric region and a rearrangement ofthe SMC. In class 1, SMCs derived from acrocentric chro-mosomes are summarized (including all minutes, di-centrics, ring-chromosomes, and other rearrangements,e.g., cases 19–23 and 30–33 of the present study). In class2, SMCs derived from all minute chromosomes apartfrom the acrocentrics are included (as long as they do notcomprise any detectable rearrangements, e.g., cases 2, 5,6, 8, 10–12, 18, 24–28, and 34). Thus, after detection ofan SMC by routine banding cytogenetics including GTG-and CBG-banding and NOR-staining, the overwhelmingmajority of SMCs can be classified into class 1, which en-compasses about 80% of SMCs (Crolla et al. 1998).Known SMCs such as those described in cat eye syn-drome (e.g., Liehr et al. 1992) or the invdup(15) markers(e.g., Nietzel et al. 2003) are also members of class 1. Fur-thermore, the exact origin of the SMC can be subse-quently determined by cenM-FISH (Nietzel et al. 2001) orCM-FISH (Henegariu et al. 2001). Both approaches en-able the one-step identification of all human centromericregions and have been successfully used for the character-ization of SMCs (Nietzel et al. 2001, 2002; von Eggelinget al. 2002; Liehr et al. 2002a, 2002b; Starke et al. 2003).If routine cytogenetics reveals that the SMC belongs inclass 1, acroM-FISH (Langer et al. 2001) or acrocenM-FISH (Trifonov et al. 2003) can be alternatively applied.Following this, the chromosomal origin is clarified, andthe SMC is classified either into class 1 (centric SMC ofacrocentric origin), class 2, 3, or 4 (centric SMC of non-acrocentric origin), or class 5 (SMC with a neocentromere).In general, neocentromeric SMCs are not stained by anyof the four aforementioned approaches; in this case, otherapproaches for their characterization, such as M-FISH(Starke et al. 1999) or microdissection of the SMC (e.g.,Starke et al. 2001) have to be performed. Class 5 includesthe 60 reported cases in the literature with neocentromeres
64
Table 4 Classification scheme for SMC after testing for NOR and chromosomal origin
Class 1 Class 2 Class 3 Class 4 Class 5
SMC derived from an acrocentric chromosome + – – – or + –Chromosome-specific a–satellite present + + + + –Rearrangement on SMC present – or + (if “+” same
chromosome)– + (same chromo-
some as SMC)+ (other chromo-some than SMC)
–
Table 5 Classification scheme for SMC after testing for cen-tromere-near euchromatic content, uniparental disomy (UPD). If,for example, UPD has not been tested but rather the presence ofeuchromatin, the two possible subclasses can be given as a/b or c/d
Sub-classes a b c d
Centromere–near euchromatin – – + +UPD – + – +

(for a review, see Amor and Choo 2002). To distinguishclasses 2, 3, and 4, subcenM-FISH, MCB, or other corre-sponding methods may be needed. Whereas, as mentionedabove, class 2 contains all non-acrocentric and non-re-arranged minute chromosomes, class 3 comprises all casesof non-acrocentric derived SMCs that have rearrange-ments such as isodicentrics, dicentrics, or rings, under theprerequisite that the genetic material is all derived fromonly one chromosome (e.g., cases 1, 3, 4, 7, 9, and 13–17:Callen et al. 1992; case 19: re-examined by Fang et al.1995; case A: Callen et al. 1999; Röthlisberger et al. 2000;Starke et al. 2001). If the SMC comprises material of atleast two different chromosomes (e.g., as in case 29), theseare summarized under class 4, irrespective of whether theyare of acrocentric or non-acrocentric origin.
Furthermore, four subclasses are suggested that allowfor the presence or absence of pericentromeric euchro-matin and/or UPD (see Table 5). Thus, the SMC of cases21 and 31 would be class 1a markers, whereas that of case22 would be a class 1c marker; SMCs of cases 2, 24 and26 are class 2a. A class 2b SMC is described, for example,in the paper of von Eggeling et al. (2002). The min(7) ofcase 12 and the min(4) of case 10 are examples of class 2cand class 2d SMCs, respectively. Cases 9 and 13 haveSMCs of class 3c, and the only representative for class 4(case 29) belongs to subclass 4d. No examples have beendescribed or established so far for classes 1b, 1d, 3a, 3b,3d, 4a–c, and 5a–d. According to Table 6, informationbased on the material studied and the grade of mosaicismpresent therein can be easily included into the proposedSMC nomenclature. The proposed nomenclature may alsoassist the comparison of SMCs in future studies.
Acknowledgements This work was supported by the Robert-Pfleger-Stiftung (Bamberg, Germany), Herbert Quandt Stiftungder VARTA, and the EU (ICA2-CT-2000-10012). We thank Dr.Lisa Shaffer (Spokane, USA), Dr. Jasen Anderson (Brisbane, Aus-tralia), and Dr. Joris Vermeesch (Leuven, Belgium) for helpful dis-cussions. The continuous support of the Carl Zeiss (Jena, Ger-many) is gratefully acknowledged.
References
Amor DJ, Choo KH (2002) Neocentromeres: role in human dis-ease, evolution, and centromere study. Am J Hum Genet 71:695–714
Bridgland L, Footz TK, Kardel MD, Riazi MA, McDermid HE(2003) Three duplicons form a novel chimeric transcriptionunit in the pericentromeric region of chromosome 22q11. HumGenet 112:57–61
Buckton KE, Spowart G, Newton MS, Evans HJ (1985) Forty fourprobands with an additional “marker” chromosome. HumGenet 69:353–370
Callen DF, Eyre H, Yip MY, Freemantle J, Haan EA (1992) Mo-lecular cytogenetic and clinical studies of 42 patients withmarker chromosomes. Am J Med Genet 43:709–715
Callen DF, Eyre H, Fang YY, Guan XY, Veleba A, Martin NJ,McGill J, Haan EA (1999) Origins of accessory small ringmarker chromosomes derived from chromosome 1. J Med Genet36:847–853
Chudoba I, Franke Y, Senger G, Sauerbrei G, Demuth S, BeensenV, Neumann A, Hansmann I, Claussen U (1999) MaternalUPD 20 in a hyperactive child with severe growth retardation.Eur J Hum Genet 7:533–540
Crolla JA (1998) FISH and molecular studies of autosomal super-numerary marker chromosomes excluding those derived fromchromosome 15. II. Review of the literature. Am J Med Genet75:367–381
Crolla JA, Long F, Rivera H, Dennis NR (1998) FISH and molec-ular study of autosomal supernumerary marker chromosomesexcluding those derived from chromosomes 15 and 22. I. Re-sults of 26 new cases. Am J Med Genet 75:355–366
65
Table 6 Classification schemefor SMC after testing for mo-saicism. Studied tissue type:A amniocytes, B bone marrow,C chorion biopsy, F fibroblastsof the skin, H hair root cells,L lymphocytes, M buccal mu-cosa. The degree of mosaicismin percent is indicated inbrackets
Nomenclature for SMC of cases 1–35
Case Class Case Class
1 SMC 3dA(57) 20 SMC 1a/bA(100)2 SMC 2aA(65) 21 SMC 1aC(100)3 SMC 3c/dA(58) 22 SMC 1cL(100)x24 SMC 3c/dL(10) 23 SMC 1a/bL(100)5 SMC 2c/dL(100) 24 SMC 2aA(45)6 SMC 2c/dL(37) 25 SMC 2c/dL(67)7 SMC 3c/dL(100) 26 SMC 2aA(100)8 SMC 2c/dL(100) 27 SMC 2c/dL(58)9 SMC 3cA(53) 28 SMC 2c/dA(82)
10 SMC 2dL(80) 29 SMC 4dA(100)11 SMC 2c/dA(72) 30 SMC 1c/dL(100)12 SMC 2cA(45) 31 SMC 1a/bA(100)13 (subclone 1) SMC 3cL(49) 32 SMC 1aA(100)13 (subclone 2) SMC 3cL(13) 33 (subclone 1) SMC 1c/dA(49)13 (subclone 3) SMC 3cL(3) 33 (subclone 2) SMC 1a/bA(4)14 SMC 3c/dA(86) 33 (subclone 3) SMC 1a/bA(28)15 SMC 3c/dL(100) 34 (SMC 9) SMC 2a/bA(80)16 SMC 3c/dB(90) 34 (SMC20) SMC 2c/dA(20)17 SMC 3c/dA(100) 35 (min 10) SMC 3a/bC(38)18 SMC 2c/dL(66) 35 (i10) SMC 3a/bC(5)19 SMC 1a/bA(86) 35 (min 18) SMC 2a/bC(7)

Cunniff C (2002) Turner syndrome. Adolesc Med 13:359–366D’Amato Sizonenko L, Ng D, Oei P, Winship I (2002) Supernu-
merary marker chromosomes 5: confirmation of a critical re-gion and resultant phenotype. Am J Med Genet 111:19–26
Eggeling F von, Hoppe C, Bartz U, Starke H, Houge G, ClaussenU, Ernst G, Kotzot D, Liehr T (2002) Maternal uniparental di-somy 12 in a healthy girl with a 47,XX,+der(12)(:p11→q11:)/46,XX karyotype. J Med Genet 39:519–521
Fang YY, Eyre HJ, Bohlander SK, Estop A, McPherson E, TragerT, Riess O, Callen DF (1995) Mechanisms of small ring for-mation suggested by the molecular characterization of two smallaccessory ring chromosomes derived from chromosome 4. AmJ Hum Genet 57:1137–1142
Fryns JP, Petit P, Heffinck R, Berghe H van den (1983) Mosaicpericentric inversion of chromosome 2. J Genet Hum 31:157–161
Funke B, Edelmann L, McCain N, Pandita RK, Ferreira J, MerscherS, Zohouri M, Cannizzaro L, Shanske A, Morrow BE (1999)Der(22) syndrome and velo-cardio-facial syndrome/DiGeorgesyndrome share a 1.5-Mb region of overlap on chromosome22q11. Am J Hum Genet 64:747–758
Giardino D, Bettio D, Gottardi G, Rizzi N, Pierluigi M, PerfumoC, Cali A, Dagna Bricarelli F, Larizza L (1999) FISH charac-terization of two supernumerary r(1) associated with distinctclinical phenotypes. Am J Med Genet 84:377–380
Giardino D, Finelli P, Russo S, Gottardi G, Rodeschini O, AtzaMG, Natacci F, Larizza L (2002) Small familial supernumeraryring chromosome 2: FISH characterization and genotype-phe-notype correlation. Am J Med Genet 111:319–323
Guy J, Hearn T, Crosier M, Mudge J, Viggiano L, Koczan D,Thiesen HJ, Bailey JA, Horvath JE, Eichler EE, Earthrowl ME,Deloukas P, French L, Rogers J, Bentley D, Jackson MS (2003)Genomic sequence and transcriptional profile of the boundarybetween pericentromeric satellites and genes on human chro-mosome arm 10p. Genome Res 13:159–172
Haddad BR, Schröck E, Meck J, Cowan J, Young H, Ferguson-Smith MA, Manoir S du, Ried T (1998) Identification of denovo chromosomal markers and derivatives by spectral karyo-typing. Hum Genet 103:619–625
Henegariu O, Bray-Ward P, Artan S, Vance GH, Qumsyieh M,Ward DC (2001) Small marker chromosome identification inmetaphase and interphase using centromeric multiplex fish(CM-FISH). Lab Invest 81:475–481
Horvath JE, Gulden CL, Bailey JA, Yohn C, McPherson JD,Prescott A, Roe BA, De Jong PJ, Ventura M, Misceo D,Archidiacono N, Zhao S, Schwartz S, Rocchi M, Eichler EE(2003) Using a pericentromeric interspersed repeat to recapitu-late the phylogeny and expansion of human centromeric seg-mental duplications. Mol Biol Evol (in press)
James RS, Temple IK, Dennis NR, Crolla JA (1995) A search foruniparental disomy in carriers of supernumerary marker chro-mosomes. Eur J Hum Genet 3:21–26
Knight SJ, Horsley SW, Regan R, Lawrie NM, Maher EJ, CardyDL, Flint J, Kearney L (1997) Development and clinical appli-cation of an innovative fluorescence in situ hybridization tech-nique which detects submicroscopic rearrangements involvingtelomeres. Eur J Hum Genet 5:1–8
Kotzot D (2002) Review and meta-analysis of systematic searchesfor uniparental disomy (UPD) other than UPD 15. Am J MedGenet 111:366–375
Langer S, Fauth C, Rocchi M, Murken J, Speicher MR (2001)AcroM fluorescent in situ hybridization analyses of markerchromosomes. Hum Genet 109:152–158
Ledbetter DH, Engel E (1995) Uniparental disomy in humans: de-velopment of an imprinting map and its implications for prena-tal diagnosis. Hum Mol Genet 4:1757–1764
Li E (2002) Chromatin modification and epigenetic reprogram-ming in mammalian development. Nat Rev Genet 3:662–673
Liehr T, Claussen U (2002) Review: multicolor-FISH approachesfor the characterization of human chromosomes in clinical ge-netics and tumor cytogenetics. Curr Genomics 3:213–235
Liehr T, Pfeiffer RA, Trautmann U (1992) Typical and partial cateye syndrome: identification of the marker chromosome byFISH. Clin Genet 42:91–96
Liehr T, Thoma K, Kammler K, Gehring C, Ekici A, Bathke KD,Grehl H, Rautenstrauss B (1995) Direct preparation of uncul-tured EDTA-treated or heparinized blood for interphase FISHanalysis. Appl Cytogenet 21:185–188
Liehr T, Rautenstrauss B, Grehl H, Bathke KD, Ekici A, Rauch A,Rott HD (1996) Mosaicism for the Charcot-Marie-Tooth dis-ease type 1A duplication suggests somatic reversion. HumGenet 98:22–28
Liehr T, Beensen V, Hauschild R, Ziegler M, Hartmann I, StarkeH, Heller A, Kähler C, Schmidt M, Reiber W, Hesse M,Claussen U (2001) Pitfalls of rapid prenatal diagnosis using theinterphase nucleus. Prenat Diagn 21:419–421
Liehr T, Nietzel A, Rocchi M, Heller A, Starke H, Claussen U,Eggeling F von (2002a) Centromere-specific multicolor-FISH(cenM-FISH) followed by analysis for uniparental disomy – auseful tool in prenatal diagnosis. In: Macek M (ed) Early pre-natal diagnosis, fetal cells and DNA in the mother – presentstate and perpectives. Karolinum Press, Prag, pp 293–300
Liehr T, Nietzel A, Starke H, Heller A, Weise A, Mrasek K,Claussen U (2002b) Characterization of small human markerchromosomes by centromere-specific multicolor-FISH (cenM-FISH) and high resolution multicolor banding (MCB). ECANewsletter 10:3–8
Mrasek K, Heller A, Rubtsov N, Trifonov V, Starke H, Rocchi M,Claussen U, Liehr T (2001) Reconstruction of the female Go-rilla gorilla karyotype using 25-color FISH and multicolorbanding (MCB). Cytogenet Cell Genet 93:242–248
Müller-Navia J, Nebel A, Oehler D, Theile U, Zabel B, Schleier-macher E (1996) Microdissection and DOP-PCR-based reversechromosome painting as a fast and reliable strategy in theanalysis of various structural chromosome abnormalities. Pre-nat Diagn 16:915–922
National Institutes of Health and Institute of Molecular MedicineCollaboration (1996) A complete set of human telomericprobes and their clinical application. Nat Genet 14:86–89
Nietzel A, Rocchi M, Starke H, Heller A, Fiedler W, Wlodarska I,Loncarevic IF, Beensen V, Claussen U, Liehr T (2001) A newmulticolor-FISH approach for the characterization of markerchromosomes: centromere-specific multicolor-FISH (cenM-FISH). Hum Genet 108:199–204
Nietzel A, Heller A, Starke H, Liehr T (2002) Centromere-specificmulticolor-FISH (cenM-FISH). In: Rautenstrauß B, Liehr T(eds) FISH technology, a Springer laboratory manual. Sprin-ger, Berlin Heidelberg New York, pp 425–431
Nietzel A, Albrecht B, Starke H, Heller A, Gillessen-Kaesbach G,Claussen U, Liehr T (2003) Partial hexasomy 15pter→15q13including SNRPN and D15S10: first molecular cytogeneticallyproven case report. J Med Genet 40 E28:1–4
Ostroverkhova NV, Nazarenko SA, Rubtsov NB, Nazarenko LP,Bunina EN (1999) Characterization of a small supernumeraryring marker derived from chromosome 2 by forward and re-verse chromosome painting. Am J Med Genet 87:217–220
Queisser-Luft A, Stolz G, Wiesel A, Schlaefer K, Spranger J (2002)Malformations in newborn: results based on 30,940 infants andfetuses from the Mainz congenital birth defect monitoring sys-tem (1990–1998). Arch Gynecol Obstet 266:163–167
Röthlisberger B, Chrzanowska K, Balmer D, Riegel M, Schinzel A(2000) A supernumerary marker chromosome originating fromtwo different regions of chromosome 18. J Med Genet 37:121–124
Salafsky IS, MacGregor SN, Claussen U, Eggeling F von (2001)Maternal UPD 20 in an infant from a pregnancy with mosaictrisomy 20. Prenat Diagn 21:860–863
Senger G, Chudoba I, Plesch A (1998) Multicolor-FISH – theidentification of chromosome aberrations by 24 colors. Biofo-rum 9:499–503
66

Shaffer LG, Agan N, Goldberg JD, Ledbetter DH, Longshore JW,Cassidy SB (2001) American College of Medical Geneticsstatement of diagnostic testing for uniparental disomy. GenetMed 3:206–211
Smith MM (2002) Centromeres and variant histones: what, where,when and why? Curr Opin Cell Biol 14:279–285
Stankiewicz P, Bocian E, Jakubow-Durska K, Obersztyn E, LatoE, Starke H, Mroczek K, Mazurczak T (2000) Identification ofsupernumerary marker chromosomes derived from chromo-somes 5, 6, 19, and 20 using FISH. J Med Genet 37:114–120
Stankiewicz P, Parka SS, Holder SE, Waters CS, Palmer RW,Berend SA, Shaffer LG, Potocki L, Lupski JR (2001) Trisomy17p10-p12 resulting from a supernumerary marker chromo-some derived from chromosome 17: molecular analysis and de-lineation of the phenotype. Clin Genet 60:336–344
Starke H, Schreyer I, Kähler C, Fiedler W, Beensen V, Heller A,Nietzel A, Claussen U, Liehr T (1999) Molecular cytogeneticcharacterization of a prenatally detected supernumerary minutemarker chromosome 8. Prenat Diagn 19:1169–1174
Starke H, Raida M, Trifonov V, Clement JH, Loncarevic IF, HellerA, Bleck C, Nietzel A, Rubtsov N, Claussen U, Liehr T (2001)Molecular cytogenetic characterization of an acquired minutesupernumerary marker chromosome as the sole abnormality ina case clinically diagnosed as atypical Philadelphia-negativechronic myelogenous leukaemia. Br J Haematol 113:435–438
Starke H, Seidel J, Henn W, Reichardt S, Volleth M, Stumm M,Behrend C, Sandig KR, Kelbova C, Senger G, Albrecht B,Hansmann I, Heller A, Claussen U, Liehr T (2002) Homolo-gous sequences at human chromosome 9 bands p12 andq13–21.1 are involved in different patterns of pericentric re-arrangements. Eur J Hum Genet 10:790–800
Starke H, Mitulla B, Nietzel A, Heller A, Beensen V, GrosswendtG, Claussen U, Eggeling F von, Liehr T (2003) First patientwith trisomy 21 accompanied by an additional der(4)(:p11→q11:)plus partial uniparental disomy 4p15–16. Am J Med Genet116A:26–30
Sumption ND, Barber JC (2001) A transmitted deletion of 2q13 to2q14.1 causes no phenotypic abnormalities. J Med Genet 38:125–127
Trifonov V, Seidel J, Starke H, Prechtel M, Beensen V, Ziegler M,Hartmann I, Heller A, Nietzel A, Claussen U, Liehr T (2003)Enlarged chromosome 13 p-arm hiding a cryptic partial trisomy6p22.2-pter. Prenat Diagn 23:427–430
Velagaleti GV, Jalal SM, Kukolich MK, Lockhart LH, Tonk VS(2002) De novo supernumerary ring chromosome 7: first reportof a non-mosaic patient and review of the literature. Clin Genet61:202–206
Verma RS, Babu A (1989) Human chromosomes – manual of ba-sic technologies, 4th edn. Pergamon, New York
Villa N, Riva P, Colombo D, Sala E, Mariani S, Zorloni C, CrostiF, Dalpra L (2001) Identification of a small supernumerarymarker chromosome, r(2)(p10q11.2), and the problem of deter-mining prognosis. Prenat Diagn 21:801–805
Weise A, Starke H, Heller A, Tonnies H, Volleth M, Stumm M,Gabriele S, Nietzel A, Claussen U, Liehr T (2002) Chromo-some 2 aberrations in clinical cases characterised by high reso-lution multicolour banding and region specific FISH probes. J Med Genet 39:434–439
67